
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new class of luminescent Cu(I) complexes with tripodal ligands – TADF emitters for the yellow to red color range†
Timo
Gneuß
a,
Markus J.
Leitl
b,
Lars H.
Finger
a,
Nicholas
Rau
a,
Hartmut
Yersin
*b and
Jörg
Sundermeyer
*a
aPhilipps-Universität Marburg, Materials Science Centre and Fachbereich Chemie, Hans-Meerwein-Straße 4, 35032 Marburg, Germany. E-mail: jsu@staff.uni-marburg.de; Fax: +49 (0)6421 28-25711; Tel: +49 (0)6421 28-25693
bUniversität Regensburg, Institut für Physikalische Chemie, Universitätsstr. 31, 93053, Regensburg, Germany. E-mail: hartmut.yersin@ur.de; Fax: +49 (0)941 943-4488; Tel: +49 (0)941 943-4464
First published on 12th November 2014
Abstract
A new class of emissive and neutral Cu(I) compounds with tripodal ligands is presented. The complexes were characterized chemically, computationally, and photophysically. Under ambient conditions, the powders of the compounds exhibit yellow to red emission with quantum yields ranging from about 5% to 35%. The emission represents a thermally activated delayed fluorescence (TADF) combined with a short-lived phosphorescence which represents a rare situation and is a consequence of high spin–orbit coupling (SOC). In the series of the investigated compounds the non-radiative rates increase with decreasing emission energy according to the energy gap law while the radiative rate is almost constant. Furthermore, a well-fit linear dependence between the experimental emission energies and the transition energies calculated by DFT and TD-DFT methods could be established, thus supporting the applicability of these computational methods also to Cu(I) complexes.
Introduction
In the last few decades, luminescent copper(I) complexes have been intensely studied due to their structural and photophysical diversity.1–12 Research in this field has gained additional momentum recently, since such complexes can be highly attractive for application in organic-light emitting diodes (OLEDs) or light-emitting electrochemical cells (LEECs).13–27 This is because Cu(I) complexes often exhibit a thermally activated delayed fluorescence (TADF),28–37 which allows utilizing all injected excitons for the generation of light by making use of the singlet harvesting effect.28–32,34–37 In this regard, Cu(I) complexes can provide a low-cost alternative to expensive emitters based on 3rd row transition metals, such as platinum or iridium.34,35,38–53For Cu(I) compounds, different structure motifs have been in the focus of research. In particular, extensive photophysical studies were conducted for copper(I) complexes that are pseudo-tetrahedrally coordinated by two bidentate chelating ligands. Homoleptic complexes with N^N (bisimine)6,9,10,54–60 or P^P (bisphosphine)54,61–63 ligands as well as heteroleptic complexes with N^N/P^P (bisimine/bisphosphine)10,28,31,32,54,64–66 ligands were investigated. Typically, for such complexes optical or electrical excitation induces a metal-to-ligand charge transfer (MLCT) by which Cu(I) is formally oxidized to Cu(II).1,5,6,9,31,32,56,67 Such a transition is accompanied by a flattening distortion towards a more planar geometry that promotes non-radiative deactivation pathways and as a consequence results in a reduction of the emission quantum yield.2,10,28,31,32,57–59
Also, several three-coordinate, trigonal Cu(I) complexes with one monodentate and one bidentate ligand have been studied.20,37,68–73 These complexes exhibit structural reorganization on excitation as well, which is represented by the so-called Y→T shape distortion.20,70,71 Similarly to the complexes with two bidentate ligands, these distortions result in an increased non-radiative deactivation.
Besides mononuclear complexes, dinuclear Cu(I) complexes and Cu(I) clusters have also been studied.4,11,27,29,30,74–79
In this work, a new class of luminescent mononuclear copper(I) halide complexes with tripodal ligands has been investigated. Compounds with this structural motif have been much less explored for their photophysical behavior. To our knowledge, only a few tripodally coordinated Cu(I) complexes have been investigated accordingly.80,81 The complexes studied in this contribution are based on five different tripodal ligands, tris(2-pyridyl)phosphine oxide (tpypo) L1, tris(2-pyridyl)phosphine sulfide (tpyps) L2, tris(2-pyridyl)phosphine selenide (tpypse) L3, tris(2-pyridyl)arsine oxide (tpyaso) L4, and tris(2-pyridyl)methane (tpym) L5. By reacting these ligands with the respective copper halide CuX (X = Cl, Br, I), the complexes [CuCltpypo] C1, [CuBrtpypo] C2, [CuItpypo] C3, [CuCltpyps] C4, [CuBrtpyps] C5, [CuItpyps] C6, [CuItpypse] C7, [CuItpyaso] C8, and [CuItpym] C9 were created. For these, the structures were determined using NMR spectroscopy, mass spectrometry, elemental analysis, and X-ray analysis. Moreover, photophysical studies and characterizations as well as quantum chemical calculations were carried out for this series of Cu(I) complexes.
Results and discussion
Synthesis of the ligands
The three ligands tpypo L1, tpyps L2, and tpypse L3 were prepared in a two-step synthesis (Scheme 1). Tris(2-pyridyl)phosphine (tpyp) was synthesized from 2-bromopyridine and phosphorus trichloride via the classical method.82,83 For this purpose 2-lithiopyridine was generated with nBuLi at −78 °C. Further reaction with phosphorus trichloride under salt elimination at −100 °C gave the desired product. Meanwhile, other synthetic methods are known to prepare tpyp, for example, with 2-bromopyridine and red phosphorus.84–86 In the second step, the oxidized ligand tpypo L1 was synthesized according to a previously published method, which was slightly modified. In the literature,87–90 hydrogen peroxide solution was used to oxidize tpyp, whereas in our procedure tBuOOH (80% in DTBP) was used. The other two ligands tpyps L2 and tpypse L3 could be easily prepared by refluxing tpyp with elemental sulfur or gray selenium in toluene.87,88 The progress of the reaction could be monitored via31P-NMR spectroscopy. An excess of sulfur or selenium in the reaction mixture was separated by filtration via a syringe filter. Yields between 45% and 94% were reached in the oxidation reactions.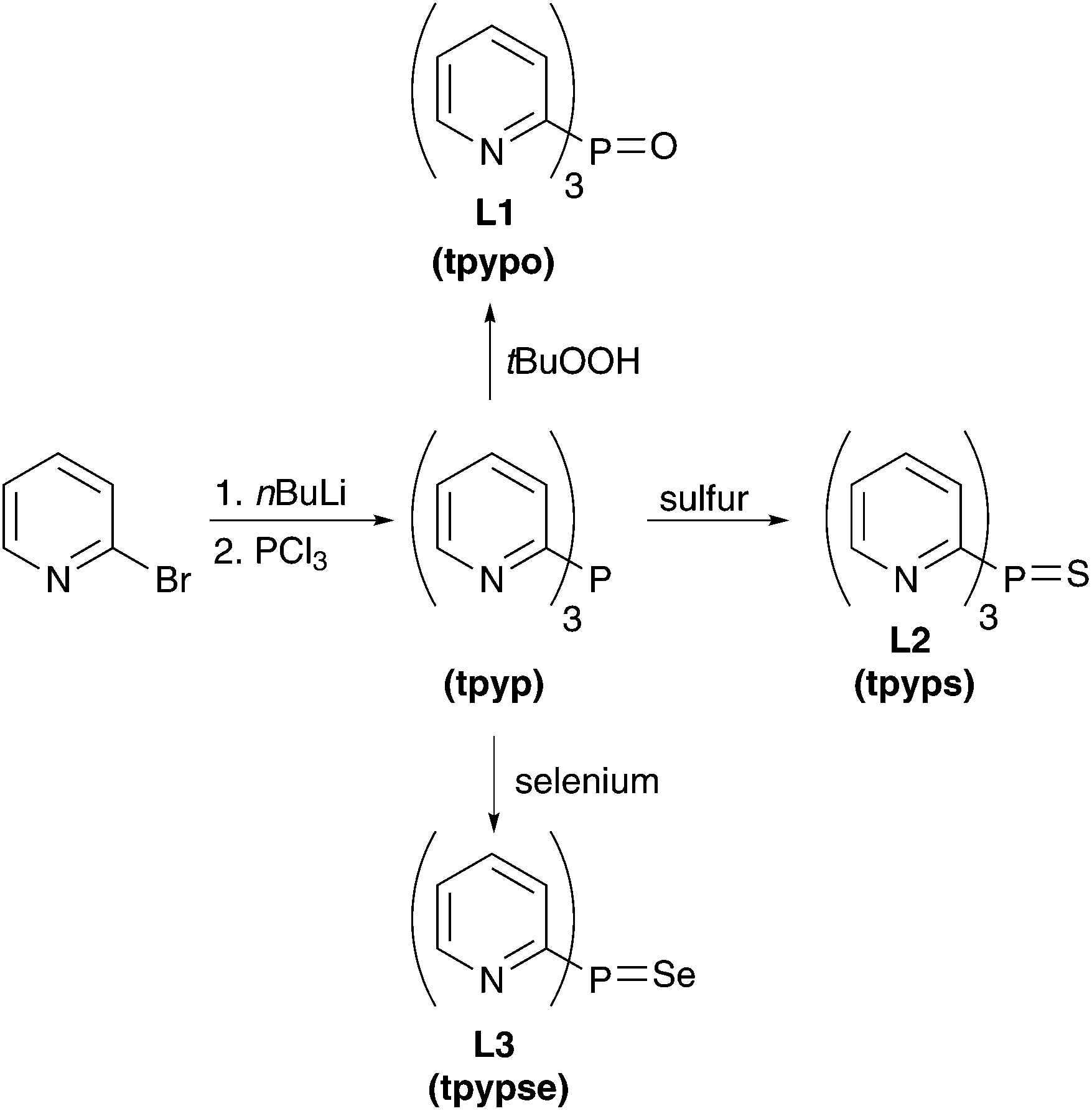 | ||
| Scheme 1 Synthetic route to the ligands tpypo L1, tpyps L2, and tpypse L3. | ||
The synthesis of the arsine ligand tpyaso L4 was carried out analogously to tpypo L1. First, the arsine compound tpyas is formed followed by oxidation with tBuOOH solution to tpyaso L4 (Scheme 2). In contrast to tpyp, the arsine compound tpyas is stable in air.
 | ||
| Scheme 2 Synthetic route to the arsine ligand tpyaso L4. | ||
The methane ligand tpym L5 was prepared by a literature-known method.91 First, 2-picoline is lithiated with nBuLi followed by reaction with 2-fluoropyridine under salt elimination (Scheme 3). The obtained mixture of the desired tpym L5 and the by-product di(2-pyridyl)methane dpym was separated by distillation.
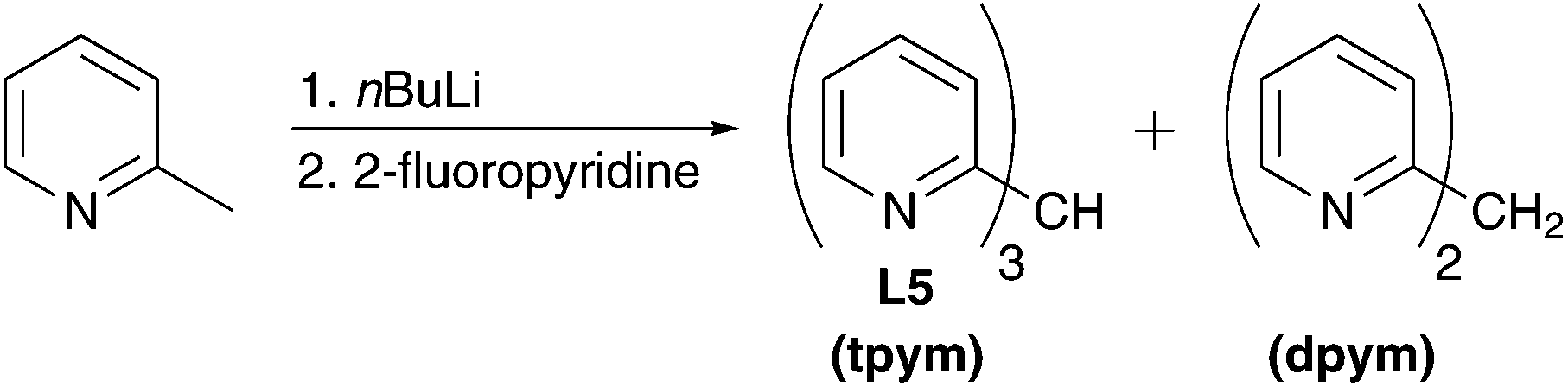 | ||
| Scheme 3 Synthesis of the methane derivative tpym L5. | ||
Synthesis of the copper(I) complexes
In this section the syntheses of nine new copper(I) complexes are presented (Scheme 4). To obtain the complexes, the ligand was dissolved in acetonitrile and reacted with a copper(I) halide (CuCl, CuBr, and CuI) at ambient temperature. A colored precipitate was formed, which was collected by centrifugation. The yields of the different Cu(I) complexes varied significantly from 17% to 75%, depending on the solubility of the respective complex in the solvent.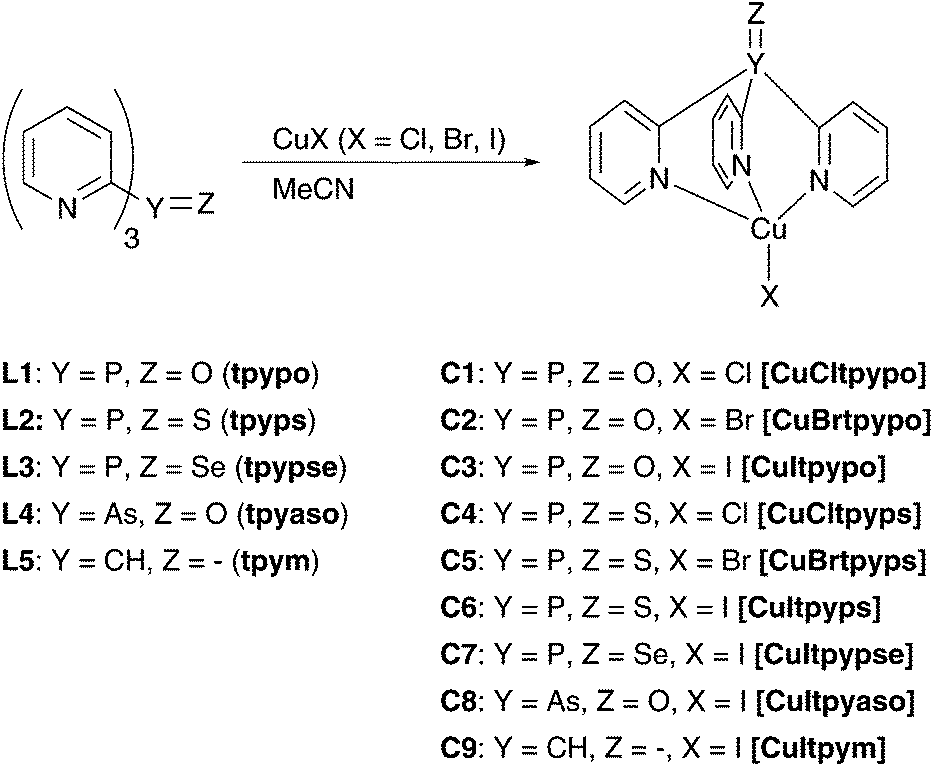 | ||
| Scheme 4 Synthesized emissive copper(I) complexes. | ||
We have also studied the coordination compounds of the phosphine tpyp with Cu(I) halide. Since the compounds are insoluble in common organic solvents, they could not be adequately characterized. It is assumed that the compounds are coordination polymers involving P–Cu interactions.
X-ray crystal structures
Crystal structures of tpyas and of the copper(I) complexes C1–C9 have been determined using X-ray diffraction measurements. The crystallographic data and structure refinement details are summarized in Tables 1 and 2. Selected bond distances and angles are listed for ligand tpyas in Fig. S1† and for complexes C1–C9 in Tables 3 and 4. The crystal structure of tpyas is discussed only in the ESI,† since no complexes have been made with this ligand.| tpyas | C1 | C2 | C3 | C4 | |
|---|---|---|---|---|---|
| Habitus | Block | Needle | Needle | Needle | Needle |
| Color | Colourless | Red | Orange | Red | Red |
| Formula | C15H12AsN3 | C16H13.50ClCuN3.50OP | C16.50H14.25BrCuN3.75OP | C16.50H14.25CuIN3.75OP | C18H16.50ClCuN4.50PS |
| Fw [g mol−1] | 309.20 | 400.76 | 455.49 | 502.48 | 457.88 |
| Crystal size [mm] | 0.230 × 0.230 × 0.220 | 0.660 × 0.260 × 0.190 | 0.450 × 0.190 × 0.130 | 0.210 × 0.040 × 0.040 | 0.250 × 0.040 × 0.040 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P21/c | Pbca | P21/n | P21/n | Pccn |
| a [Å] | 9.1529(9) | 8.4558(6) | 8.5684(10) | 8.6959(3) | 15.0776(12) |
| b [Å] | 9.1102(6) | 27.6843(13) | 28.964(3) | 29.6332(11) | 30.280(2) |
| c [Å] | 16.0342(16) | 28.3435(16) | 14.501(2) | 14.5579(6) | 8.6832(4) |
| α [°] | 90 | 90 | 90 | 90 | 90 |
| β [°] | 100.982(8) | 90 | 106.175(5) | 105.894(3) | 90 |
| γ [°] | 90 | 90 | 90 | 90 | 90 |
| Cell volume [Å3] | 1312.5(2) | 6635.0(7) | 3456.3(7) | 3608.0(2) | 3964.3(4) |
| Z | 4 | 16 | 8 | 8 | 8 |
| D calc [Mg m−3] | 1.565 | 1.605 | 1.751 | 1.850 | 1.534 |
| Abs. coeff. [mm−1] | 2.578 | 1.582 | 3.679 | 3.021 | 1.434 |
| F(000) | 624 | 3248 | 1812 | 1956 | 1864 |
| T [K] | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) |
| λ [Å] | 0.71073 | 0.71069 | 0.71069 | 0.71069 | 0.71069 |
| Reflns collected | 8036 | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 305 305 |
21656 |
33130 |
13790 |
| Indep. reflns | 2763 | 7307 | 7647 | 7632 | 4187 |
| Obs. reflns [I > 2(I)] | 2318 | 5432 | 4427 | 4265 | 1823 |
| Reflns used for refin. | 2763 | 7307 | 7647 | 7632 | 4187 |
| Abs. Correction | Semi-empirical | Semi-empirical | Semi-empirical | Analytical | Semi-empirical |
| GOF | 0.964 | 1.037 | 1.031 | 0.689 | 0.715 |
| wR2 | 0.0666 | 0.1677 | 0.1068 | 0.0492 | 0.0703 |
| R 1 [I > 2σ(I)] | 0.0254 | 0.0658 | 0.0610 | 0.0300 | 0.0417 |
| C5 | C6 | C7 | C8 | C9 | |
|---|---|---|---|---|---|
| Habitus | Plate | Block | Needle | Needle | Plate |
| Color | Orange | Orange | Orange | Yellow | Yellow |
| Formula | C20H19.50BrCuN5.50PS | C47H39Cu3I3N10P3S3 | C20H19.50CuIN5.50PSe | C16.50H14.25AsCuIN3.75O | C16H13CuIN3 |
| Fw[g mol−1] | 543.39 | 1504.35 | 637.28 | 546.43 | 437.73 |
| Crystal size [mm] | 0.200 × 0.130 × 0.050 | 0.310 × 0.280 × 0.180 | 0.440 × 0.110 × 0.040 | 0.280 × 0.050 × 0.040 | 0.490 × 0.250 × 0.100 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/a | Pc21b | P21/a | P21/n | P21/n |
| a [Å] | 8.5838(3) | 13.2413(5) | 8.7685(3) | 8.7081(4) | 14.7528(4) |
| b [Å] | 15.2046(7) | 15.7217(6) | 15.6075(5) | 29.7666(14) | 13.4193(3) |
| c [Å] | 17.8716(6) | 25.7987(11) | 18.0144(8) | 14.5936(9) | 16.0010(4) |
| α [°] | 90 | 90 | 90 | 90 | 90 |
| β [°] | 103.386(3) | 90 | 104.011(3) | 105.559(4) | 90.266(2) |
| γ [°] | 90 | 90 | 90 | 90 | 90 |
| Cell volume [Å3] | 2269.11(15) | 5370.7(4) | 2392.00(16) | 3644.2(3) | 3167.72(14) |
| Z | 4 | 4 | 4 | 8 | 8 |
| D calc [Mg m−3] | 1.591 | 1.860 | 1.770 | 1.992 | 1.836 |
| Abs. coeff. [mm−1] | 2.904 | 3.152 | 3.809 | 4.705 | 3.324 |
| F(000) | 1092 | 2920 | 1236 | 2100 | 1696 |
| T [K] | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) |
| λ [Å] | 0.71069 | 0.71069 | 0.71069 | 0.71069 | 0.71069 |
| Reflns collected | 17036 |
18930 |
13242 |
35607 |
47871 |
| Indep. reflns | 4804 | 10265 |
5056 | 35607 |
6720 |
| Obs. reflns [I > 2(I)] | 3562 | 6869 | 3816 | 16911 |
5801 |
| Reflns used for refin. | 4804 | 10265 |
5056 | 35607 |
6720 |
| Abs. correction | Semi-empirical | Semi-empirical | Semi-empirical | Integration | Semi-empirical |
| GOF | 1.085 | 0.747 | 0.865 | 0.764 | 1.098 |
| wR2 | 0.1196 | 0.0822 | 0.0613 | 0.1465 | 0.0745 |
| R 1 [I > 2σ(I)] | 0.0522 | 0.0376 | 0.0298 | 0.0580 | 0.0274 |
| C1 | C2 | C3 | C4 | C5 |
|---|---|---|---|---|
| Cu1–N1 2.067(3) | Cu1–N1 2.072(4) | Cu1–N1 2.062(3) | Cu1–N1 2.078(3) | Cu1–N1 2.043(4) |
| Cu1–N2 2.047(4) | Cu1–N2 2.063(5) | Cu1–N2 2.060(3) | Cu1–N2 2.063(3) | Cu1–N2 2.052(4) |
| Cu1–N3 2.047(3) | Cu1–N3 2.049(5) | Cu1–N3 2.049(4) | Cu1–N3 2.027(3) | Cu1–N3 2.048(4) |
| Cu1–Cl1 2.212(1) | Cu1–Br1 2.344(1) | Cu1–I1 2.499(1) | Cu1–Cl1 2.230(1) | Cu1–Br1 2.335(1) |
| P1–O1 1.489(3) | P1–O1 1.479(4) | P1–O1 1.477(3) | P1–S1 1.942(2) | P1–S1 1.944(2) |
| P1–C1 1.828(4) | P1–C1 1.830(6) | P1–C1 1.820(4) | P1–C1 1.837(4) | P1–C1 1.835(5) |
| P1–C6 1.820(4) | P1–C6 1.812(6) | P1–C6 1.829(4) | P1–C6 1.827(4) | P1–C6 1.827(5) |
| P1–C11 1.827(4) | P1–C11 1.818(6) | P1–C11 1.820(5) | P1–C11 1.817(4) | P1–C11 1.825(5) |
| Cl1–Cu1–N1 113.8(1) | Br1–Cu1–N1 111.9(1) | I1–Cu1–N1 112.6(1) | Cl1–Cu1–N1 114.9(1) | Br1–Cu1–N1 121.7(1) |
| Cl1–Cu1–N2 116.8(1) | Br1–Cu1–N2 118.7(1) | I1–Cu1–N2 119.7(1) | Cl1–Cu1–N2 117.5(1) | Br1–Cu1–N2 122.6(1) |
| Cl1–Cu1–N3 131.1(1) | Br1–Cu1–N3 131.5(1) | I1–Cu1–N3 129.7(1) | Cl1–Cu1–N3 129.6(1) | Br1–Cu1–N3 117.8(1) |
| N1–Cu1–N2 97.3(1) | N1–Cu1–N2 98.3(2) | N1–Cu1–N2 98.5(1) | N1–Cu1–N2 95.1(1) | N1–Cu1–N2 95.8(2) |
| N1–Cu1–N3 95.3(1) | N1–Cu1–N3 95.9(2) | N1–Cu1–N3 95.6(1) | N1–Cu1–N3 96.7(1) | N1–Cu1–N3 96.4(2) |
| N2–Cu1–N3 96.1(1) | N2–Cu1–N3 94.1(2) | N2–Cu1–N3 94.5(1) | N2–Cu1–N3 96.1(2) | N2–Cu1–N3 96.5(2) |
| O1–P1–C1 113.2(2) | O1–P1–C1 113.8(2) | O1–P1–C1 113.6(2) | S1–P1–C1 113.5(2) | S1–P1–C1 113.3(2) |
| O1–P1–C6 113.4(2) | O1–P1–C6 113.2(3) | O1–P1–C6 112.6(2) | S1–P1–C6 114.0(1) | S1–P1–C6 113.8(2) |
| O1–P1–C11 112.9(2) | O1–P1–C11 113.0(3) | O1–P1–C11 113.3(2) | S1–P1–C11 112.5(2) | S1–P1–C11 113.5(2) |
| P1–Cu1–Cl1 168.9(0) | P1–Cu1–Br1 168.1(1) | P1–Cu1–I1 169.8(0) | P1–Cu1–Cl1 170.5(0) | P1–Cu1–Br1 176.7(0) |
| C6 | C7 | C8 | C9 |
|---|---|---|---|
| Cu1–N1 2.069(9) | Cu1–N1 2.054(3) | Cu1–N1 2.058(9) | Cu1–N1 2.091(2) |
| Cu1–N2 2.068(10) | Cu1–N2 2.043(3) | Cu1–N2 2.063(10) | Cu1–N2 2.074(2) |
| Cu1–N3 2.056(9) | Cu1–N3 2.046(3) | Cu1–N3 2.055(10) | Cu1–N3 2.057(2) |
| Cu1–I1 2.512(1) | Cu1–I1 2.486(1) | Cu1–I1 2.510(2) | Cu1–I1 2.465(0) |
| P1–S1 1.935(3) | P1–Se1 2.104(1) | As1–O1 1.636(8) | |
| P1–C1 1.846(12) | P1–C1 1.840(4) | As1–C1 1.936(12) | C16–C1 1.523(3) |
| P1–C6 1.816(12) | P1–C6 1.843(4) | As1–C6 1.935(12) | C16–C6 1.524(3) |
| P1–C11 1.841(11) | P1–C11 1.838(4) | As1–C11 1.936(13) | C16–C11 1.524(3) |
| I1–Cu1–N1 119.9(3) | I1–Cu1–N1 117.8(1) | I1–Cu1–N1 111.1(3) | I1–Cu1–N1 117.9(1) |
| I1–Cu1–N2 122.5(3) | I1–Cu1–N2 120.0(1) | I1–Cu1–N2 118.4(3) | I1–Cu1–N2 126.8(1) |
| I1–Cu1–N3 122.6(3) | I1–Cu1–N3 124.2(1) | I1–Cu1–N3 126.6(3) | I1–Cu1–N3 129.5(1) |
| N1–Cu1–N2 93.2(4) | N1–Cu1–N2 96.4(1) | N1–Cu1–N2 101.5(4) | N1–Cu1–N2 91.1(1) |
| N1–Cu1–N3 96.9(4) | N1–Cu1–N3 96.2(1) | N1–Cu1–N3 97.3(4) | N1–Cu1–N3 89.7(1) |
| N2–Cu1–N3 94.8(4) | N2–Cu1–N3 96.3(1) | N2–Cu1–N3 97.4(4) | N2–Cu1–N3 90.7(1) |
| S1–P1–C1 113.4(4) | Se1–P1–C1 113.1(1) | O1–As1–C1 114.1(5) | H16–C16–C1 107.1 |
| S1–P1–C6 113.9(4) | Se1–P1–C6 112.9(1) | O1–As1–C6 112.6(5) | H16–C16–C6 107.1 |
| S1–P1–C11 113.5(4) | Se1–P1–C11 114.2(1) | O1–As1–C11 114.2(5) | H16–C16–C11 107.1 |
| P1–Cu1–I1 178.4(1) | P1–Cu1–I1 176.0(0) | As1–Cu1–I1 170.6(1) | C16–Cu1–I1 172.7(0) |
Suitable single crystals of copper complexes C1–C9 could be obtained from a saturated acetonitrile solution (at ambient temperature) after storing for a few days at 4 °C. The complexes crystallize either in the monoclinic or orthorhombic crystal system. As a representative example of all the copper compounds investigated here, the molecular structures of the three complexes [CuItpypo] C3, [CuItpyaso] C8, and [CuItpym] C9 are shown in Fig. 1. The copper centers are coordinated by the halide anion and the three N atoms of the tripodal ligand in a distorted tetrahedral configuration. In each case, the halide atom deviates slightly from the Y–Cu axis (Y = P, As, C), which is due to packing effects that result from interactions with neighboring molecules (see Fig. S2–S4† for the three complexes C1–C3 exhibiting the most pronounced bending of the halide). This is supported by quantum chemical calculations performed on the complex [CuCltpypo] C1 which predict that the energy of the molecule is minimal if the chloride is lying on the P–Cu axis corresponding to an angle of P1–Cu1–Cl1 = 180°. However, bending the chloride away from this axis by 10°, corresponding to an angle of P1–Cu1–Cl1 = 170°, results only in a minor energy increase of 1 kJ mol−1. This indicates that the potential energy surface describing the halide bending is very flat. For the three complexes [CuCltpypo] C1, [CuBrtpypo] C2, and [CuItpypo] C3 the halide bending is most pronounced, with a bending angle of about 10° (P1–Cu1–X1 ≈ 170°) away from the P–Cu axis (see the angle Y(P, As, C)–Cu–X) in Tables 3 and 4). For the two complexes [CuBrtpyps] C5 and [CuItpyps] C6 with bonding angles of P1–Cu1–Br1 = 176.7(0)° and P1–Cu1–I1 = 178.4(1)°, respectively, the halide bending is relatively small.
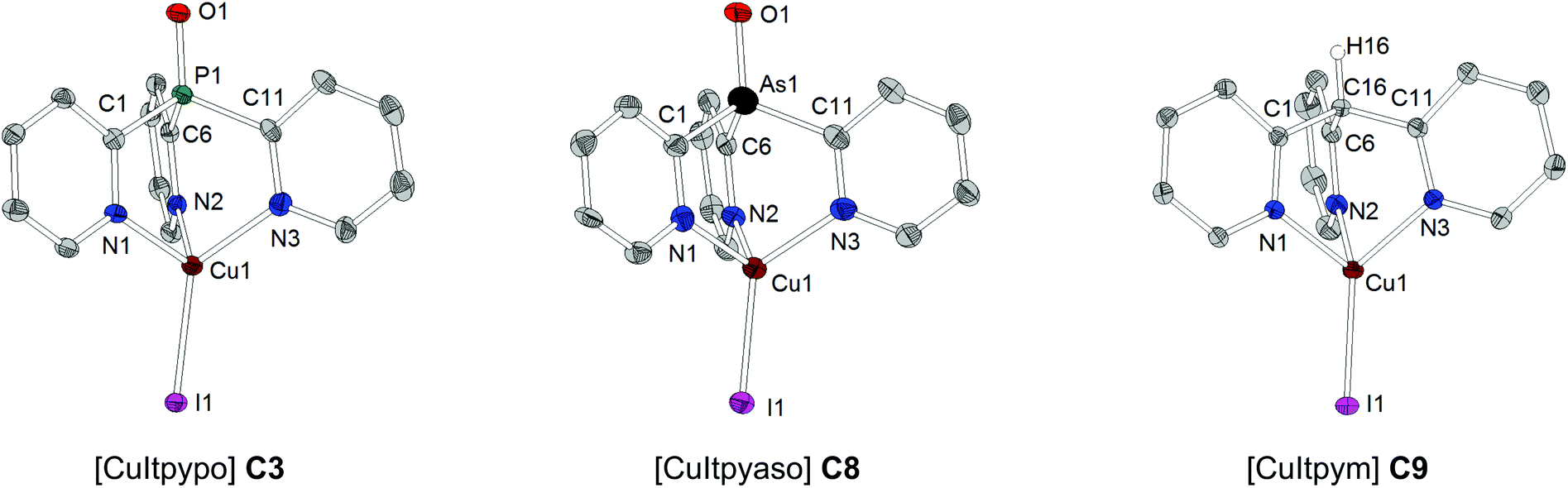 | ||
| Fig. 1 Molecular structures of [CuItpypo] C3, [CuItpyaso] C8, and [CuItpym] C9 (thermal ellipsoids with 50% probability) resulting from X-ray analyses. Hydrogen atoms (except for H16 of C9) and solvent molecules are omitted for clarity. | ||
The bond lengths of the various Cu–N bonds vary only slightly within the range of 2.027(3) Å to 2.091(2) Å for all investigated copper complexes. The bite angle N–Cu–N for the complexes with the tripodal phosphine ligands range from 93.2(4)° to 98.5(1)°. The bite angle is slightly larger for [Cul-typaso] C8 with the tripodal arsine ligand, with angles of N1–Cu1–N2 = 101.5(4)°, N1–Cu1–N3 = 97.3(4)°, and N2–Cu1–N3 = 97.4(4)°. In [CuItpym] C9 with the tripodal methane ligand the bite angle is significantly smaller, with angles of N1–Cu1–N2 = 91.1(1)°, N1–Cu1–N3 = 89.7(1)°, and N2–Cu1–N3 = 90.7(1)°.
The Cu–X bond length increases from chloride to iodide due to the increasing atomic size of the halide, for example, in the series [CuXtpypo] from Cu1–Cl1 = 2.212(1) Å to Cu1–Br1 = 2.344(1) Å to Cu1–I1 = 2.499(1) Å. The bond length of the P–O bond is about 1.48 Å and that of the P–S bond is about 1.94 Å. Thereby they are in the range of the corresponding bond lengths of triphenylphosphine oxide (P–O 1.479(2) Å) and triphenylphosphine sulfide (P–S 1.950(3) Å).92,93
Computational studies
To learn more about the tripodally coordinated copper complexes, quantum chemical calculations have been carried out for complexes C1–C3 and C6–C9, using density functional theory (DFT) and time-dependent density functional theory (TD-DFT) with the functional B3LYP and the basis set def2-TZVPP. It has been shown that this method gives good results for Cu(I) complexes, especially for a description of the transition energies.94We want to focus the discussion on [CuItpypo] C3. For this compound, the optimized ground state geometry and the first excited triplet state geometry were calculated. The results are displayed in Fig. 2. In the ground state, the halide is lying on the axis that is defined by the P1–Cu1 atoms (Fig. 2a). However, in the triplet state geometry, the halide is bent away from this axis (Fig. 2b). This is clearly displayed by the three I–Cu–N angles. In the ground state geometry, all three angles I–Cu–N are nearly equivalent to values of about 123°. In the triplet state, the angle I1–Cu1–N1, amounting to 110.3°, is significantly smaller while the two other angles, I1–Cu1–N2 amounting to 127.4° and I1–Cu1–N3 amounting to 128.3°, are slightly larger than in the ground state (compare with Table S1†). Moreover, it can be seen that in the triplet state geometry two Cu–N bonds are distinctly shortened, i.e. from the ground state value of 2.145 Å to 1.986 Å (Cu1–N2) and from 2.144 Å to 1.985 Å (Cu1–N3), respectively, while the Cu1–N1 bond length increases from 2.146 Å to 2.265 Å. This shows that two Cu–N bonds are strengthened and one Cu–N bond is weakened in the triplet state.
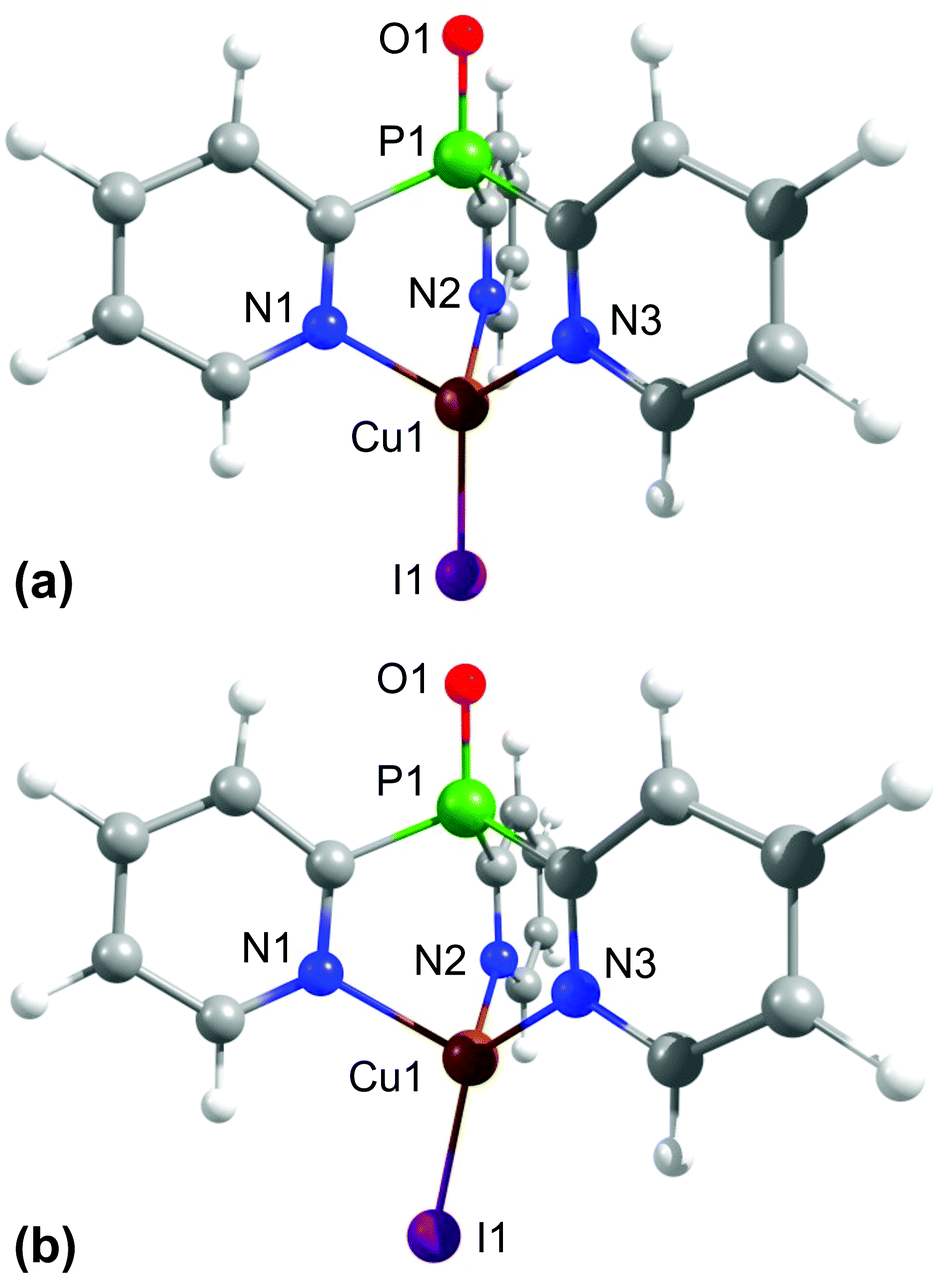 | ||
| Fig. 2 Optimized ground state (a) and first excited triplet state (b) geometries of [CuItpypo] C3. Calculations were performed on the B3LYP/def2-TZVPP level of theory. | ||
The occurrence of such a distortion in the excited state can be understood when the composition of the frontier orbitals is analyzed (Fig. 3). For [CuItpypo] C3, the HOMO is mainly localized on the copper atom and the halide, whereas the LUMO is mainly distributed over two of the three pyridine moieties of the tripod ligand. TD-DFT calculations reveal that transitions between these frontier orbitals largely determine the first excited singlet state S1 and triplet state T1, especially in the triplet state geometry. Therefore, these states can be classified to be of (metal + halide)-to-ligand charge transfer (1,3(M + X)LCT) character. To quantify the amount of charge that is transferred on excitation the natural charges for the copper and iodine atoms were calculated for the ground and the first excited triplet state. They amount to +0.321 (Cu) and −0.562 (I) for the ground state (S0 geometry) and to +0.665 (Cu) and −0.324 (I) for the first excited triplet state (T1 geometry). This nicely confirms the occurrence of a pronounced charge transfer away from the metal and halide upon excitation. Especially, the transfer of charge away from the copper center, in a rather crude approximation representing a formal oxidation of Cu(I) to Cu(II), has an important consequence. As Cu(I) prefers a tetrahedral coordination environment, whereas Cu(II) prefers a planar one, significant structural reorganizations occur upon excitation. In the case of [CuItpypo] C3 these reorganizations are mainly represented by the bending of the halide away from the Cu–P axis and by changes of the Cu–N binding distances as described above.
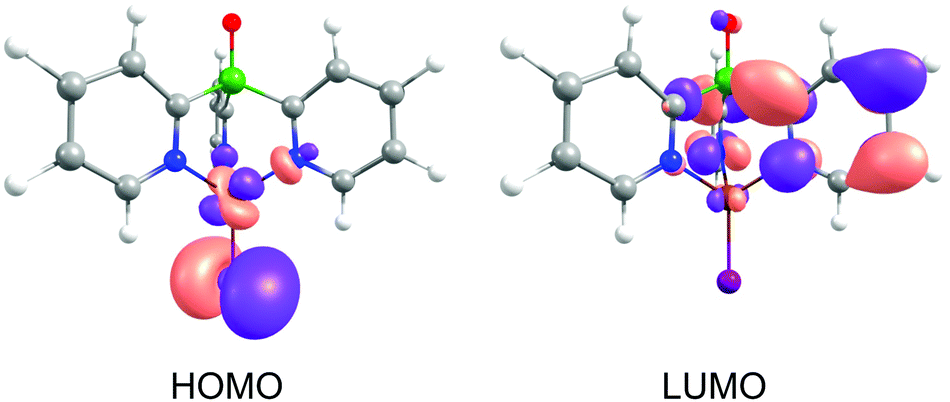 | ||
| Fig. 3 Contour surfaces of HOMO and LUMO for [CuItpypo] C3 calculated for the ground state geometry. Calculations were performed on the B3LYP/def2-TZVPP level of theory. The contour value of the MOs amounts to 0.04. | ||
Interestingly, a comparison of the two tripodal copper complexes, K[Cu(SC6F5)(HB(3,5-iPr2pz)3)], representing a Cu(I) complex, and [Cu(SC6F5)(HB(3,5-iPr2pz)3)], representing a Cu(II) complex, supports the results that are predicted from our calculations.95,96 Compared to the Cu(I) complex, two of the three Cu–N bonds are shorter for the Cu(II) homologue (Cu(I): Cu–N11 = 2.188(7) Å, Cu–N31 = 2.062(7) Å; Cu(II): Cu–N11 = 2.037(9) Å, Cu–N31 = 1.930(9) Å), whereas the third Cu–N bond is longer (Cu(I): Cu–N21 = 2.065(7) Å; Cu(II): Cu–N21 = 2.119(8) Å). Moreover, the bending of the thiolate away from the B–Cu axis is by about 7° larger for the Cu(II) complex than for the Cu(I) homologue (Cu(I): B–Cu–S = 174°, Cu(II): B–Cu–S = 167°).
For completeness it is remarked that similar geometry distortions for the triplet state geometry were also found for all other complexes presented in this study. This is shown in Table S1† also for the two complexes [CuItpyaso] C8 and [CuItpym] C9.
In the scope of our calculations, the energy gaps between HOMO and LUMO show only slight variations between about 2.5 and 2.7 eV (data for all complexes are summarized in Table S2†). For an estimate of the transition energies, a comparison of the HOMO–LUMO gaps is, however, not sufficient. Instead, TD-DFT calculations have been performed.
The TD-DFT transition energies of the different compounds, as calculated for the optimized T1 state geometry, differ significantly from the experimental emission energies (see Table S3†). However, it is not expected that these calculated gas phase data can exactly reproduce the experimental data, since (i) TD-DFT results have the tendency to underestimate the transition energies that correspond to charge-transfer states94,97,98 and since (ii) especially for Cu(I) complexes, the experimental emission energies depend strongly on the environment and, in particular, on its rigidity,28,31,32 which is not taken into account in our calculations. Nevertheless, the calculated transition energies nicely reproduce the trend of the emission energies as determined for the Cu(I) complexes doped into amorphous PMMA (polymethylmethacrylate) (Fig. 4). It is remarked that the experimental trend is not reproduced by TD-DFT calculations performed for the ground state geometry.
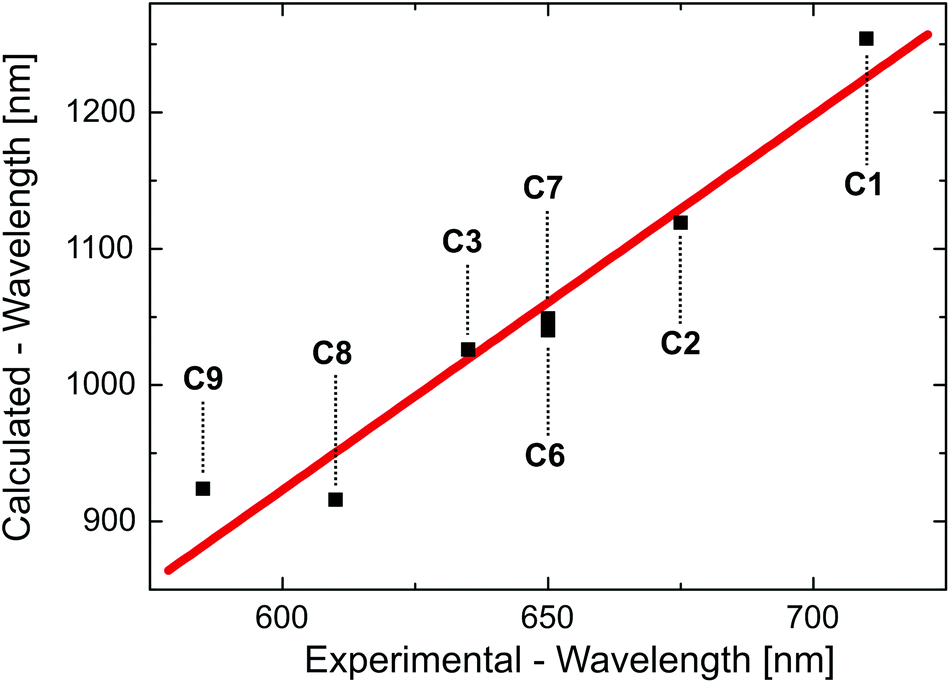 | ||
| Fig. 4 Calculated T1→S0 transition wavelength versus experimentally determined emission wavelengths for different Cu(I) complexes as defined in Scheme 4. The calculated values result from TD-DFT calculations performed for the optimized triplet state geometry. The experimental values represent the emission maxima as found for compounds doped into PMMA. The red line represents a linear fit to the data points. The coefficient of determination for this fit amounts to R2 = 0.92. | ||
Furthermore, our calculations suggest that the energy separations ΔE(S1–T1) between the T1 and S1 states are rather small, lying between about 1000 and 1350 cm−1 (compare Table S3†). For such small energy splittings the occurrence of a thermally activated delayed fluorescence is expected. A more detailed discussion of this aspect is presented in the next section.
Photophysical studies
Again, the discussion will focus on [CuItpypo] C3. Fig. 5 shows the absorption spectrum of this compound and of the tpypo L1 ligand recorded in dichloromethane. The absorption bands observed in the range between about 230 nm and 300 nm are present in both the complex and the ligand. Therefore, they are assigned to result from ligand centered transitions. In contrast, in the range between about 300 and 500 nm, the ligand does not absorb, but for the complex two separate absorption bands are observed. This allows us to assign these bands to charge transfer transitions in agreement with the results obtained from DFT and TD-DFT calculations that predict low-lying (M + X)LCT states.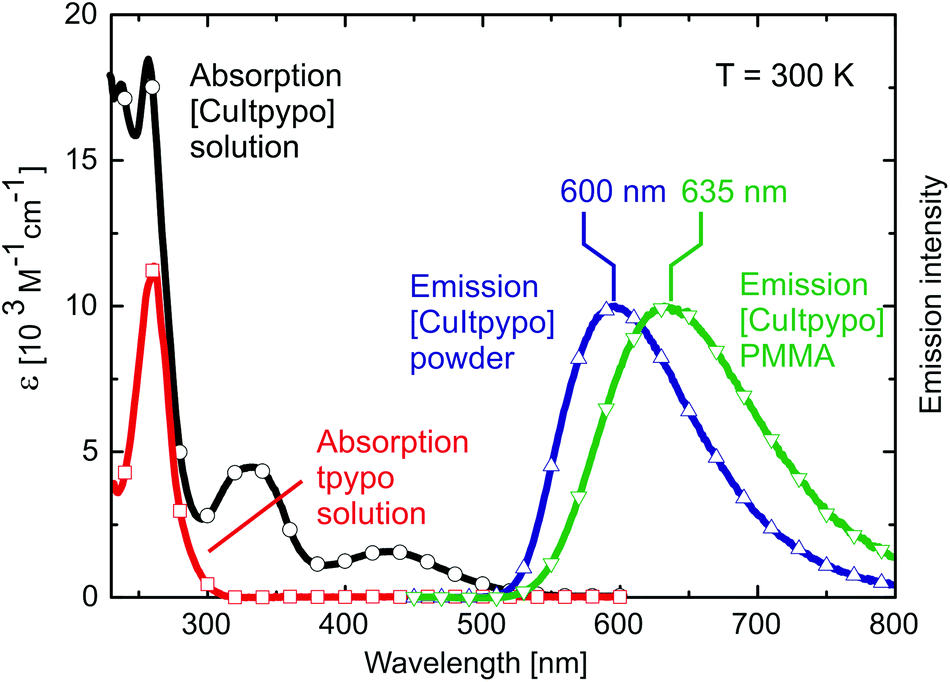 | ||
| Fig. 5 Absorption spectra of the ligand tpypo L1 and the corresponding complex [CuItpypo] C3 recorded in a dichloromethane solution. Emission spectra are displayed for [CuItpypo] C3 as a powder and doped into a PMMA matrix, respectively. All measurements were recorded under ambient conditions. | ||
In fluid solution, an emission of [CuItpypo] C3 is not observed at ambient temperature even after oxygen was carefully removed by at least five freeze–pump–thaw cycles. In contrast, when doped into a PMMA matrix, the complex exhibits a red emission with a maximum at λmax = 635 nm and an emission quantum yield of ΦPL = 16%. As powder, the emission is orange with a maximum at λmax = 600 nm and a quantum yield of ΦPL = 20% (Fig. 5).
The emission of [CuItpypo] C3 in PMMA compared to the neat powder is red shifted by 35 nm. This can be attributed to differences in matrix rigidity. With decreasing rigidity the complex can undergo more distinct distortions upon (M + X)LCT excitation resulting in an energy stabilization of the emitting state(s). In addition to this red shift, the distortions cause an increase of non-radiative deactivations to the ground state due to increased Franck Condon factors that couple the excited state to the ground state.99,100 As a consequence, the quantum yield decreases with decreasing rigidity of the environment from 20% (powder) to 16% (PMMA) to ≪1% (solution).
The emission decay time and its temperature dependence show further important information. As for [CuItpypo] C3 doped into PMMA the decay strongly deviates from a mono-exponential behavior (due to distinct inhomogeneity effects),101 we focus on the emission properties of the powders.102 At ambient temperature, an emission decay time of τ(300 K) = 4 μs and an emission quantum yield of ΦPL(300 K) = 20% are found for [CuItpypo] C3. When the powder sample is cooled to T = 77 K, the decay time increases to τ(77 K) = 24 μs and the emission quantum yield to ΦPL(77 K) = 63%. Calculation of the radiative and non-radiative rates according to kr = ΦPLτ−1 and knr = (1 − ΦPL)τ−1, respectively, reveals that the radiative rate increases by a factor of about two from kr(77 K) = 3 × 104 s−1 to kr(300 K) = 5 × 104 s−1, whereas the non-radiative rate increases tenfold from knr(77 K) = 2 × 104 s−1 to knr(300 K) = 2 × 105 s−1. Furthermore, the powder sample shows a slight blue-shift of the emission maximum from λmax(77 K) = 610 nm to λmax(300 K) = 600 nm.
An increase of the radiative rate and a blue-shift of the emission on increasing the temperature from T = 77 K to 300 K has often been reported for Cu(I) complexes and can be explained by the occurrence of a thermally activated delayed fluorescence (TADF).28–32,34,35 Accordingly, at low temperature, only emission from the lowest excited triplet state T1 occurs. With temperature increase, a thermal population of the energetically only slightly higher lying first excited singlet state S1 becomes possible. As the S1→S0 transition is significantly more allowed than the spin-forbidden T1→S0 transition, an effective reduction of the emission decay time results. Moreover, as the S1 state lies energetically higher than the T1 state, a blue shift of the emission is expected to occur with increasing temperature.
However, for the compound [CuItpypo] C3, only a moderate increase of the radiative rate by a factor of about 2 was found to occur on heating. This is much less than what has been reported for other Cu(I) complexes. For example, the compounds presented in ref. 29 (Cu(I) halide dimers with different chelating aminophosphane ligands) experience a radiative rate increase by a factor of 40 to 150 and the compound discussed in ref. 36 (Cu(I) chloride dimer with a chelating diphosphane ligand) even shows a much higher factor of 490. The small radiative rate increase found for [CuItpypo] C3 can be rationalized by taking into account an additional and efficient emission decay path from the triplet state to the singlet ground state. The compounds discussed in ref. 29 and 36 exhibit long triplet decay times between 250 μs and 2200 μs. In contrast, the triplet decay time of [CuItpypo] C3 amounts to only 24 μs (T = 77 K value). As a consequence, a further shortening of the emission decay time by involving the TADF mechanism at higher temperature is much less effective. Further support for this interpretation is given in ref. 37 (three-coordinate Cu(I) carbene complex) where a Cu(I) complex with a similarly short triplet decay time of 34 μs is investigated. This compound also displays only a moderate increase of the radiative rate by a factor of about 3 when the TADF process is activated.
Obviously, the only moderate increase of the radiative rate upon heating can be rationalized by a significant contribution of the triplet state emission even at ambient temperature. Accordingly, both the singlet state S1 and the triplet state T1 contribute to the overall emission at ambient temperature. Consequently, the expected blue-shift of the emission maximum with increasing temperature is not as clearly displayed for the studied complexes as for TADF-only emitters (compare ref. 28–32, 34–36). However, the corresponding blue-shift is displayed on the high-energy flanks of the spectra measured at T = 77 K and 300 K, respectively (compare with Table 5).
| Temp. [K] | λ max [nm] | λ 50% [nm] | τ [μs] | Φ PL [%] | k r [104 s−1] | k nr [104 s−1] | |
|---|---|---|---|---|---|---|---|
| [CuCltpypo] C1 | 300 | 645 | 595 | 3 | 8 | 3 | 30 |
| 77 | 645 | 610 | 13 | 14 | 1 | 7 | |
| [CuBrtpypo] C2 | 300 | 620 | 575 | 4 | 18 | 5 | 20 |
| 77 | 620 | 585 | 20 | 36 | 2 | 3 | |
| [CuItpypo] C3 | 300 | 600 | 550 | 4 | 20 | 5 | 20 |
| 77 | 610 | 555 | 24 | 63 | 3 | 2 | |
| [CuItpyps] C6 | 300 | 595 | 555 | 8 | 34 | 4 | 8 |
| 77 | 595 | 565 | 24 | ||||
| [CuItpypse] C7 | 300 | 640 | 595 | 2 | 4 | 2 | 50 |
| 77 | 675 | 630 | 9 | ||||
| [CuItpyaso] C8 | 300 | 600 | 550 | 4 | 12 | 3 | 20 |
| 77 | 610 | 565 | 23 | ||||
| [CuItpym] C9 | 300 | 550 | 510 | 5 | 28 | 6 | 10 |
| 77 | 550 | 530 | 22 |
Fig. 6 shows the emission spectra of the compounds C1–C3 and C6–C9. The complexes [CuCltpypo] C1 and [CuBrtpypo] C2 exhibit the same trends in the emission behavior as those described for [CuItpypo] C3. For all the other investigated compounds corresponding trends are expected to occur. An overview of the respective emission parameters is given in Table 5. The complexes [CuCltpyps] C4 and [CuBrtpyps] C5 exhibit very similar emission properties to complexes [CuCltpypo] C1 and [CuBrtpypo] C2, respectively. Therefore, these are not explicitly discussed here.
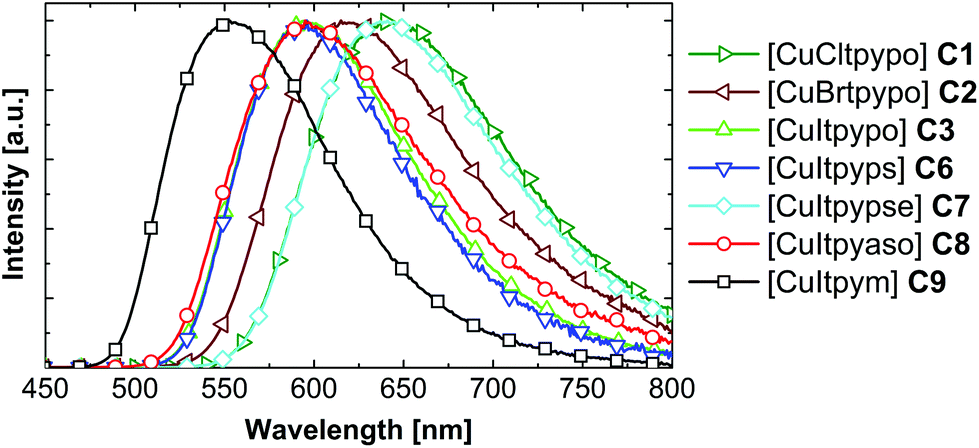 | ||
| Fig. 6 Emission spectra of the investigated complexes as powders at ambient temperature. The samples were excited at λexc = 350 nm. | ||
An analysis of the radiative and non-radiative rates for the studied series of complexes reveals an interesting trend. At ambient temperature, powders of all compounds show similar radiative rates kr ranging from 3 × 104 to 6 × 104 s−1. In contrast, the non-radiative rates knr increase drastically with increasing emission wavelength. Such a trend is not unexpected and can be described by the energy gap law. In its simplest form it can be written as103
| knr ∼ exp[−γΔE/(ħωn)] | (1) |
 | ||
| Fig. 7 Radiative kr and non-radiative rates knr of the investigated compounds versus emission energy. Data are given for the powders of the complexes at ambient temperature. The curves are displayed as a guide for the eye. | ||
Conclusion and outlook
Emitter materials that exhibit a thermally activated delayed fluorescence (TADF) or a phosphorescence are highly attractive for use in OLEDs as they can convert all injected excitons into light. For applications, the emitter should also exhibit a short (radiative) decay time to minimize saturation effects.104 The complexes investigated in this study are interesting in this regard; when compared to other Cu(I) complexes they exhibit relatively short phosphorescence (T1→S0 emission) decay times, for example, amounting to only about 24 μs (radiative decay time 38 μs) in the case of [CuItpypo] C3. Thus, it can be concluded that spin–orbit coupling is particularly effective when compared to other Cu(I) complexes for which triplet decay times of several hundred microseconds or longer are not unusual.29,32,36 Besides this already effective radiative deactivation process via phosphorescence, an additional radiative TADF path becomes important at ambient temperature for the investigated compounds. The combined emission paths of phosphorescence and TADF result in a distinct increase of the overall radiative rate when compared to TADF-only emitters. This property is highly attractive when the compounds are applied as emitters in OLEDs, in particular, to reduce saturation effects.Another interesting observation was made for the investigated compounds. In this series, the non-radiative rate increases strongly with decreasing emission energy following the energy gap law. As a consequence, the emission quantum yields decrease towards the red range of the spectrum from 28% for [CuItpym] C3 to 4% for [CuItpypse] C7. Accordingly, shifting the emission to the blue using methods of chemical engineering will result in a significant reduction of the non-radiative rates and will therefore lead to an increase of the emission quantum yield. An extrapolation of the data presented to shorter wavelengths reveals that for an emission wavelength of 460 nm, an emission quantum yield greater than 70% would be expected.
Experimental
General remarks
The syntheses and handling of air- and moisture-sensitive substances were carried out using standard Schlenk and glovebox techniques. Solvents were dried using standard procedures105 and stored over Al2O3/molecular sieves 3 Å/R3-11G catalyst (BASF).The starting materials were obtained from commercial sources (Sigma-Aldrich, Merck, Acros Organics, Alfa Aesar) and used as received. The following materials were prepared according to literature procedures: tris(2-pyridyl)phosphine82,83 (tpyp), tris(2-pyridyl)methane91 (tpym) L5, copper(I) chloride106, and copper(I) iodide.107
NMR spectra were recorded at 300 K on a Bruker DPX 250, Bruker ARX 300, Bruker DRX 400, Bruker ARX 500, or Bruker DRX 500 using CDCl3 or DMSO-d6 as the solvent. Chemical shifts are given with respect to tetramethylsilane (1H, 13C) and phosphoric acid (31P). Calibration of 1H and 13C NMR spectra was accomplished with the solvent signals, and 31P spectra were calibrated externally.
The numbering of the hydrogen and carbon atoms is shown for the three ligands tpyp, tpyas, and tpym in Fig. 8.
 | ||
| Fig. 8 Example of the numbering of compounds. | ||
Electrospray ionization (ESI) mass spectra were recorded on a Thermo Fisher Scientific LTQ FT Ultra using methanol, acetonitrile, or dichloromethane as the solvent. IR spectra were recorded on a Bruker Alpha FT-IR spectrometer using powder samples at ambient temperature. Elemental analysis was done using an Elementar vario MICRO cube. UV-Vis absorption measurements were carried out using a Varian Cary 300 double beam spectrometer. Emission spectra were recorded with a Fluorolog 3-22 (Horiba Jobin Yvon) spectrophotometer which was equipped with a cooled photo-multiplier (RCA C7164R). For the decay time measurements, the same photomultiplier was used in combination with a FAST ComTec multichannel scaler PCI card with a time resolution of 250 ps. As the excitation source for the decay time measurements, a pulsed diode laser (Picobrite PB-375L) with an excitation wavelength of λexc = 378 nm and a pulse width <100 ps was used. For absolute measurements of photoluminescence quantum yields at ambient temperature and at 77 K, a Hamamatsu Photonics (C9920-02) system was applied. Doping of polymethylmethacrylate (PMMA) films was performed by dissolving the respective complex (<1 wt%) and the polymer in dichloromethane. After this, the solution was spin-coated onto a quartz-glass plate. All calculations were carried out with Gaussian09.108 As the functional, B3LYP was used and as the basis set, def2-TZVPP was used. As the starting geometry, the structures obtained from X-ray measurements were used. No symmetry constraints were applied.
The data collection for the single crystal structure determinations was performed on a Stoe IPDS-II or IIT or a Bruker D8 QUEST diffractometer by the X-ray service department of the Fachbereich Chemie, University of Marburg. The Stoe IPDS-II and IIT devices are equipped with a Mo-Kα X-ray source (λ = 0.71073 Å), a graphite mono-chromator and an active imaging plate. Stoe IPDS software (X-AREA) was used for data collection, cell refinement and data reduction, respectively.109 The D8-QUEST is equipped with a Mo-Kα X-ray micro source (Incotec), a fixed chi goniometer and a PHOTON 100 CMOS detector. Bruker software (Bruker Instrument Service, APEX2, SAINT) was used for data collection, cell refinement and data reduction.110 The structures were solved with SIR-97111 or SHELXS-97,112 refined with SHELXL-2014112 and finally validated using PLATON113 software, all within the WinGX114 software bundle. Absorption corrections were either applied with WinGX (multi-scan115 or analytical116) or beforehand with the APEX2 software (multi-scan).117 Compound C8 was described as a non-merohedral twin. An HKLF5 absorption correction using Stoe X-Red32 (TwinAbs)109 was applied. Graphic representations were created using Diamond 3.118 C-bound H-atoms were constrained to the parent site. In all graphics the displacement ellipsoids are shown for the 50% probability level, and hydrogen atoms are shown with arbitrary radius. CCDC 1021437–1021446 contain the supplementary crystallographic data (excluding structure factors) for the structures reported in this paper.
Synthesis of the ligands
General procedure for the copper complexes
The ligand tpypo, tpyps, tpypse, tpyaso or tpym was dissolved in a minimal amount of acetonitrile and CuCl, CuBr or CuI was added. After stirring for 1 d at room temperature the precipitated product was isolated by centrifugation. The supernatant liquid was used for the preparation of single crystals. After a few days of storage at 4 °C, single crystals were obtained.Acknowledgements
The authors thank the German Ministry of Education and Research (BMBF) and the German Association of Chemical Industry (Verband der chemischen Industrie, VCI) for financial support.Notes and references
- C. Kutal, Coord. Chem. Rev., 1990, 99, 213–252 CrossRef CAS.
- O. Horváth, Coord. Chem. Rev., 1994, 135–136, 303–324 CrossRef.
- V. W.-W. Yam, K. K.-W. Lo, W. K.-M. Fung and C.-R. Wang, Coord. Chem. Rev., 1998, 171, 17–41 CrossRef CAS.
- P. C. Ford, E. Cariati and J. Bourassa, Chem. Rev., 1999, 99, 3625–3647 CrossRef CAS PubMed.
- V. W.-W. Yam and K. K.-W. Lo, Chem. Soc. Rev., 1999, 28, 323–334 RSC.
- D. V. Scaltrito, D. W. Thompson, J. A. O'Callaghan and G. J. Meyer, Coord. Chem. Rev., 2000, 208, 243–266 CrossRef CAS.
- P. G. Graham, R. D. Pike, M. Sabat, R. D. Bailey and W. T. Pennington, Inorg. Chem., 2000, 39, 5121–5132 CrossRef CAS.
- A. Vogler and H. Kunkely, Top. Curr. Chem., 2001, 213, 143–182 CrossRef CAS.
- N. Armaroli, Chem. Soc. Rev., 2001, 30, 113–124 RSC.
- N. Armaroli, G. Accorsi, F. Cardinali and A. Listorti, Top. Curr. Chem., 2007, 280, 69–115 CrossRef CAS.
- K. Tsuge, Chem. Lett., 2013, 42, 204–208 CrossRef CAS.
- G. Blasse and D. R. McMillin, Chem. Phys. Lett., 1980, 70, 1–3 CrossRef CAS.
- Q. Zhang, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Mater., 2004, 16, 432–436 CrossRef CAS.
- N. Armaroli, G. Accorsi, M. Holler, O. Moudam, J.-F. Nierengarten, Z. Zhou, R. T. Wegh and R. Welter, Adv. Mater., 2006, 18, 1313–1316 CrossRef CAS.
- G. Che, Z. Su, W. Li, B. Chu, M. Li, Z. Hu and Z. Zhang, Appl. Phys. Lett., 2006, 89, 103511 CrossRef PubMed.
- Q. Zhang, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Funct. Mater., 2006, 16, 1203–1208 CrossRef CAS.
- A. Tsuboyama, K. Kuge, M. Furugori, S. Okada, M. Hoshino and K. Ueno, Inorg. Chem., 2007, 46, 1992–2001 CrossRef CAS PubMed.
- Q. Zhang, J. Ding, Y. Cheng, L. Wang, Z. Xie, X. Jing and F. Wang, Adv. Funct. Mater., 2007, 17, 2983–2990 CrossRef CAS.
- J. C. Deaton, S. C. Switalski, D. Y. Kondakov, R. H. Young, T. D. Pawlik, D. J. Giesen, S. B. Harkins, A. J. M. Miller, S. F. Mickenberg and J. C. Peters, J. Am. Chem. Soc., 2010, 132, 9499–9508 CrossRef CAS PubMed.
- M. Hashimoto, S. Igawa, M. Yashima, I. Kawata, M. Hoshino and M. Osawa, J. Am. Chem. Soc., 2011, 133, 10348–10351 CrossRef CAS PubMed.
- Z. Liu, M. F. Qayyum, C. Wu, M. T. Whited, P. I. Djurovich, K. O. Hodgson, B. Hedman, E. I. Solomon and M. E. Thompson, J. Am. Chem. Soc., 2011, 133, 3700–3703 CrossRef CAS PubMed.
- A. Wada, Q. Zhang, T. Yasuda, I. Takasu, S. Enomoto and C. Adachi, Chem. Commun., 2012, 48, 5340–5342 RSC.
- Q. Zhang, T. Komino, S. Huang, S. Matsunami, K. Goushi and C. Adachi, Adv. Funct. Mater., 2012, 22, 2327–2336 CrossRef CAS.
- S. Igawa, M. Hashimoto, I. Kawata, M. Yashima, M. Hoshino and M. Osawa, J. Mater. Chem. C, 2013, 1, 542–551 RSC.
- X.-L. Chen, R. Yu, Q.-K. Zhang, L.-J. Zhou, X.-Y. Wu, Q. Zhang and C.-Z. Lu, Chem. Mater., 2013, 25, 3910–3920 CrossRef CAS.
- Z. Liu, J. Qiu, F. Wei, J. Wang, X. Liu, M. G. Helander, S. Rodney, Z. Wang, Z. Bian, Z. Lu, M. E. Thompson and C. Huang, Chem. Mater., 2014, 26, 2368–2373 CrossRef CAS.
- D. M. Zink, D. Volz, T. Baumann, M. Mydlak, H. Flügge, J. Friedrichs, M. Nieger and S. Bräse, Chem. Mater., 2013, 25, 4471–4486 CrossRef CAS.
- R. Czerwieniec, K. Kowalski and H. Yersin, Dalton Trans., 2013, 42, 9826–9830 RSC.
- M. J. Leitl, F.-R. Küchle, H. A. Mayer, L. Wesemann and H. Yersin, J. Phys. Chem. A, 2013, 117, 11823–11836 CrossRef CAS PubMed.
- D. M. Zink, M. Bächle, T. Baumann, M. Nieger, M. Kühn, C. Wang, W. Klopper, U. Monkowius, T. Hofbeck, H. Yersin and S. Bräse, Inorg. Chem., 2013, 52, 2292–2305 CrossRef CAS PubMed.
- C. L. Linfoot, M. J. Leitl, P. Richardson, A. F. Rausch, O. Chepelin, F. J. White, H. Yersin and N. Robertson, Inorg. Chem., 2014, 53, 10854–10861 CrossRef CAS PubMed.
- R. Czerwieniec, J. Yu and H. Yersin, Inorg. Chem., 2011, 50, 8293–8301 CrossRef CAS PubMed.
- C. A. Parker and C. G. Hatchard, Trans. Faraday Soc., 1961, 57, 1894–1904 RSC.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS PubMed.
- H. Yersin, A. F. Rausch and R. Czerwieniec, Organometallic Emitters for OLEDs: Triplet Harvesting, Singlet Harvesting, Case Structures, and Trends, in Physics of Organic Semiconductors, ed. W. Brütting and C. Adachi, Wiley-VCH Verlag, 2012, pp. 371–424 Search PubMed.
- H. Yersin, M. J. Leitl and R. Czerwieniec, Proc. SPIE, 2014, 9183, 22–21 Search PubMed.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032–16038 CrossRef CAS PubMed.
- P.-T. Chou and Y. Chi, Chem. – Eur. J., 2007, 13, 380–395 CrossRef CAS PubMed.
- S. Kappaun, C. Slugovc and E. J. W. List, Int. J. Mol. Sci., 2008, 9, 1527–1547 CrossRef CAS PubMed.
- J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef PubMed.
- C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418–4441 CrossRef CAS.
- Y. You and S. Y. Park, Dalton Trans., 2009, 1267–1282 RSC.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC.
- J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS PubMed.
- T. Hu, L. He, L. Duan and Y. Qiu, J. Mater. Chem., 2012, 22, 4206–4215 RSC.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 124, 8300–8334 CrossRef.
- C.-L. Ho and W.-Y. Wong, New J. Chem., 2013, 37, 1665–1683 RSC.
- C.-L. Ho, H. Li and W.-Y. Wong, J. Organomet. Chem., 2014, 751, 261–285 CrossRef CAS PubMed.
- Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- A. Bossi, A. F. Rausch, M. J. Leitl, R. Czerwieniec, M. T. Whited, P. I. Djurovich, H. Yersin and M. E. Thompson, Inorg. Chem., 2013, 52, 12403–12415 CrossRef CAS PubMed.
- J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef CAS PubMed.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- A. Barbieri, G. Accorsi and N. Armaroli, Chem. Commun., 2008, 2185–2193 RSC.
- D. Felder, J.-F. Nierengarten, F. Barigelletti, B. Ventura and N. Armaroli, J. Am. Chem. Soc., 2001, 123, 6291–6299 CrossRef CAS PubMed.
- Z. A. Siddique, Y. Yamamoto, T. Ohno and K. Nozaki, Inorg. Chem., 2003, 42, 6366–6378 CrossRef CAS PubMed.
- V. Kalsani, M. Schmittel, A. Listorti, G. Accorsi and N. Armaroli, Inorg. Chem., 2006, 45, 2061–2067 CrossRef CAS PubMed.
- A. Robertazzi, A. Magistrato, P. de Hoog, P. Carloni and J. Reedijk, Inorg. Chem., 2007, 46, 5873–5881 CrossRef CAS PubMed.
- A. Lavie-Cambot, M. Cantuel, Y. Leydet, G. Jonusauskas, D. M. Bassani and N. D. McClenaghan, Coord. Chem. Rev., 2008, 252, 2572–2584 CrossRef CAS PubMed.
- L.-Y. Zhou, M.-S. Ma, Z.-L. Zhang, H. Li, Y.-X. Cheng and A.-M. Ren, Org. Electron., 2012, 13, 2627–2638 CrossRef PubMed.
- O. Moudam, A. Kaeser, B. Delavaux-Nicot, C. Duhayon, M. Holler, G. Accorsi, N. Armaroli, I. Séguy, J. Navarro, P. Destruel and J.-F. Nierengarten, Chem. Commun., 2007, 3077–3079 RSC.
- R. Venkateswaran, M. S. Balakrishna, S. M. Mobin and H. M. Tuononen, Inorg. Chem., 2007, 46, 6535–6541 CrossRef CAS PubMed.
- G. Accorsi, N. Armaroli, B. Delavaux-Nicot, A. Kaeser, M. Holler, J.-F. Nierengarten and A. D. Esposti, J. Mol. Struct. (THEOCHEM), 2010, 962, 7–14 CrossRef CAS PubMed.
- S.-M. Kuang, D. G. Cuttell, D. R. McMillin, P. E. Fanwick and R. A. Walton, Inorg. Chem., 2002, 41, 3313–3322 CrossRef CAS PubMed.
- K. Saito, T. Arai, N. Takahashi, T. Tsukuda and T. Tsubomura, Dalton Trans., 2006, 4444–4448 RSC.
- I. Andrés-Tomé, J. Fyson, F. B. Dias, A. P. Monkman, G. Iacobellis and P. Coppo, Dalton Trans., 2012, 41, 8669–8674 RSC.
- D. R. McMillin, J. R. Kirchhoff and K. V. Goodwin, Coord. Chem. Rev., 1985, 64, 83–92 CrossRef CAS.
- T. S. Teets, D. V. Partyka, A. J. Esswein, J. B. Updegraff III, M. Zeller, A. D. Hunter and T. G. Gray, Inorg. Chem., 2007, 46, 6218–6220 CrossRef CAS PubMed.
- K. J. Lotito and J. C. Peters, Chem. Commun., 2010, 46, 3690–3692 RSC.
- V. A. Krylova, P. I. Djurovich, M. T. Whited and M. E. Thompson, Chem. Commun., 2010, 46, 6696–6698 RSC.
- V. A. Krylova, P. I. Djurovich, J. W. Aronson, R. Haiges, M. T. Whited and M. E. Thompson, Organometallics, 2012, 31, 7983–7993 CrossRef CAS.
- M. Osawa, Chem. Commun., 2014, 50, 1801–1803 RSC.
- V. A. Krylova, P. I. Djurovich, B. L. Conley, R. Haiges, M. T. Whited, T. J. Williams and M. E. Thompson, Chem. Commun., 2014, 50, 7176–7179 RSC.
- P. Aslanidis, P. J. Cox, S. Divanidis and A. C. Tsipis, Inorg. Chem., 2002, 41, 6875–6886 CrossRef CAS PubMed.
- H. Araki, K. Tsuge, Y. Sasaki, S. Ishizaka and N. Kitamura, Inorg. Chem., 2005, 44, 9667–9675 CrossRef CAS PubMed.
- T. Tsukuda, A. Nakamura, T. Arai and T. Tsubomura, Bull. Chem. Soc. Jpn., 2006, 79, 288–290 CrossRef CAS.
- A. Tsuboyama, K. Kuge, M. Furugori, S. Okada, M. Hoshino and K. Ueno, Inorg. Chem., 2007, 46, 1992–2001 CrossRef CAS PubMed.
- H. Araki, K. Tsuge, Y. Sasaki, S. Ishizaka and N. Kitamura, Inorg. Chem., 2007, 46, 10032–10034 CrossRef CAS PubMed.
- S. B. Harkins, N. P. Mankad, A. J. M. Miller, R. K. Szilagyi and J. C. Peters, J. Am. Chem. Soc., 2008, 130, 3478–3485 CrossRef CAS PubMed.
- A. Acosta, J. I. Zink and J. Cheon, Inorg. Chem., 2000, 39, 427–432 CrossRef CAS.
- V. Pawlowski, G. Knör, C. Lennartz and A. Vogler, Eur. J. Inorg. Chem., 2005, 3167–3171 CrossRef CAS.
- F. R. Keene, M. R. Snow, P. J. Stephenson and E. R. T. Tiekink, Inorg. Chem., 1988, 27, 2040–2045 CrossRef CAS.
- K. Kurtev, D. Ribola, R. A. Jones, D. J. Cole-Hamilton and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1980, 55–58 RSC.
- S. F. Malysheva, A. O. Korocheva, N. A. Belogorlova, A. V. Artemév, N. K. Gusarova and B. A. Trofimov, Dokl. Chem., 2012, 445, 164–165 CrossRef CAS.
- S. F. Malysheva, A. V. Artemév, N. A. Belogorlova, A. O. Korocheva, N. K. Gusarova and B. A. Trofimov, Russ. J. Gen. Chem., 2012, 82, 1307–1308 CrossRef CAS.
- B. A. Trofimov, N. K. Gusarova, A. V. Artemév, S. F. Malysheva, N. A. Belogorlova, A. O. Korocheva, O. N. Kazheva, G. G. Alexandrov and O. A. Dyachenko, Mendeleev Commun., 2012, 22, 187–188 CrossRef CAS PubMed.
- F. G. Mann and J. Watson, J. Org. Chem., 1948, 13, 502–531 CrossRef CAS.
- A. N. Kharat, A. Bakhoda, T. Hajiashrafi and A. Abbasi, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185, 2341–2347 CrossRef CAS.
- Y. Uchida, K. Matsuoka, R. Kajita, Y. Kawasaki and S. Oae, Heteroat. Chem., 1997, 8, 439–449 CrossRef CAS.
- F. R. Keene, P. J. Stephenson and E. R. T. Tiekink, Inorg. Chim. Acta, 1991, 187, 217–220 CrossRef CAS.
- A. Maleckis, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 6618–6625 CrossRef CAS PubMed.
- K. A. Al-Farhan, J. Crystallogr. Spectrosc., 1992, 22, 687–689 CrossRef CAS.
- P. W. Codding and K. A. Kerr, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3785–3787 CrossRef.
- A. Jesser, M. Rohrmüller, W. G. Schmidt and S. Herres-Pawlis, J. Comput. Chem., 2014, 35, 1–17 CrossRef CAS PubMed.
- N. Kitajima, K. Fujisawa, M. Tanaka and Y. Moro-oka, J. Am. Chem. Soc., 1992, 114, 9232–9233 CrossRef CAS.
- K. Fujisawa, K. Fujita, T. Takahashi, N. Kitajima, Y. Moro-oka, Y. Mastunaga, Y. Miyashita and K. Okamoto, Inorg. Chem. Commun., 2004, 7, 1188–1190 CrossRef CAS PubMed.
- K. L. McCall, J. R. Jennings, H. Wang, A. Morandeira, L. M. Peter, J. R. Durrant, L. J. Yellowlees, J. D. Woollins and N. Robertson, J. Photochem. Photobiol., A, 2009, 202, 196–204 CrossRef CAS PubMed.
- A. Vlček Jr. and S. Záliš, Coord. Chem. Rev., 2007, 251, 258–287 CrossRef PubMed.
- W. Siebrand, J. Chem. Phys., 1967, 46, 440–447 CrossRef CAS PubMed.
- G. W. Robinson and R. P. Frosch, J. Chem. Phys., 1963, 38, 1187–1203 CrossRef CAS PubMed.
- A. F. Rausch, M. E. Thompson and H. Yersin, Inorg. Chem., 2009, 48, 1928–1937 CrossRef CAS PubMed.
- Typically, powder samples exhibit strong concentration quenching effects, for example, due to energy transfer to defect sites. However, in Cu(I) compounds typically a geometry distortion occurs on excitation, resulting in the trapping of the excitation on one molecule.28,29,32 This effect seems to prevent concentration quenching.
- J. S. Wilson, N. Chawdhury, M. R. A. Al-Mandhary, M. Younus, M. S. Khan, P. R. Raithby, A. Köhler and R. H. Friend, J. Am. Chem. Soc., 2001, 123, 9412–9417 CrossRef CAS PubMed.
- C. Murawski, K. Leo and M. C. Gather, Adv. Mater., 2013, 25, 6801–6827 CrossRef CAS PubMed.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Elsevier, Burlington, 1996 Search PubMed.
- R. N. Keller, H. D. Wycoff and L. E. Marchi, Inorg. Synth., 1946, 2, 1–4 CrossRef.
- G. B. Kauffman, L. Y. Fang, N. Viswanathan and G. Townsend, Inorg. Synth., 1983, 22, 101–103 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.02), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Stoe & Cie, Stoe & Cie GmbH, Darmstadt, Germany, 2011.
- Bruker, Bruker AXS Inc., Madison, Wisconsin, USA, 2012.
- A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- R. H. Blessing, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 33–38 CrossRef.
- N. W. Alcock, in Cryst. Computing, ed. F. R. Ahmed, S. R. Hall and C. P. Huber, Munksgaard, Copenhagen, 1970, pp. 271–276 Search PubMed.
- Bruker, Bruker AXS Inc., Madison, Wisconsin, USA, 2012.
- K. Brandenburg and H. Putz, Crystal Impact GbR, Bonn, Germany, 2012.
Footnote |
| † Electronic supplementary information (ESI) available: Discussion of the molecular structure of tpyas, packing diagrams of complexes C1, C2 and C3, and details of quantum chemical calculations and emission spectra. CCDC 1021437–1021446. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt02631d |
| This journal is © The Royal Society of Chemistry 2015 |