
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual ligand-based fluorescence and phosphorescence emission at room temperature from platinum thioxanthonyl complexes†
Fabian
Geist
,
Andrej
Jackel
and
Rainer F.
Winter
*
Fachbereich Chemie der Universität Konstanz, Universitätsstraße 10, D-78457 Konstanz, Germany. E-mail: rainer.winter@uni-konstanz.de
First published on 22nd October 2014
Abstract
New square-planar platinum(II) complexes of the type trans-[Pt(PEt3)2(Tx)(X)] (X = Br, Cl, I or CN) bearing a σ-bonded thioxanthon-2-yl (Tx) ligand have been prepared and characterised by X-ray crystallography, cyclic voltammetry, and by NMR and electronic absorption and luminescence spectroscopy. The ligand X hardly influences the electronic transitions, which indicates that the relevant molecular orbitals are largely confined to the Pt–Tx chromophore. In agreement with TD-DFT calculations the energetically lowest electronic transition is assigned as the Tx-centred π→π* HOMO → LUMO excitation. All four complexes display dual emission from the σ-bonded Tx ligand at ca. 450 nm and at ca. 510 nm, which are assigned as fluorescence originating from the 1π*-state and as phosphorescence originating from the 3π*-state, respectively. The phosphorescence quantum yield increases with increasing σ-donor strength of the ligand X and reaches a uniquely high value of 18.8% for the chlorido complex Pt–Cl. Switching-on of Tx phosphorescence emission by the Pt(PEt3)2(X) fragment goes along with a reduction of the lifetime of the Tx triplet state from several ms in purely organic derivatives to ca. 2 μs in the complexes.
Introduction
Within the last two decades, complexes of mainly the heavy transition metals have been the focus of intense research activities devoted to the fields of light emitting devices,1–6 bioimaging,5,7–9 sensing5,10–14 or sensitisation.15–17 One major advantage of metal complexes when compared to purely organic emitters is their ability to trigger long-lived phosphorescence at ambient temperature. Platinum, in particular, is a widely applied metal in this field because its heavy atom effect, which emanates from its large spin–orbit coupling constant, promotes high intersystem crossing (ISC) efficiencies.18 The utilisation of triplet emitters in organic light emitting devices can theoretically lead to a fourfold increase in efficiency compared to the 25% efficiency limit of organic fluorescent compounds.19,20In practice, the theoretical limit of unitary quantum efficiency is, however, rarely approached. One possible deactivation pathway for the excited states of square-planar platinum(II) complexes involves distortion towards a more tetrahedral geometry by a twisting of the plane of two donors with respect to that of the other two (the so-called D2d distortion). This pathway is particularly relevant for complexes with only mono- or bidentate ligands. The main reason for low quantum yields and hence increased deactivation rates of emissive states is, however, the thermal activation of non-emissive excited metal d-states which are energetically above the emissive triplet states. A ligand field-induced destabilisation of these excited metal d-states will thus lead to increased quantum yields.18,21 Much effort has therefore been directed to introducing strong-field ligands into the coordination sphere of such complexes. Prominent examples are cyclometalating ligands such as the ubiquitous N,C- or N,N,C-chelating arylpyridines or -bipyridines,18,22–39 simple or cyclometalating carbenes and bis(carbenes),3,6,36,37,40–45 or σ-bonded alkynyl ligands.46–51 Particularly abundant are trans-bis(alkynyl)bis(trialkylphosphine)platinum(II) complexes trans-Pt(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR)2(PR3)2.52,53 Such complexes have, for example been used as constituents of luminescent coordination oligomers and polymers featuring platinum atoms in the conjugated main chain54–63 or for two-photon absorption (TPA).64 Other representatives have organic dyes as the substituents at the alkynyl ligands. These complexes exhibit intense ligand-based π→π*-transitions and usually emit from ligand-centred π*-states with moderate fluorescence or phosphorescence quantum yields in fluid solution at ambient temperature.49,53,65–69 Again, ISC to a ligand-based excited triplet state is triggered by the large heavy-atom effect of the Pt(II) coordination centre. In rare cases, where the excited states are mainly localised at those ligands and spin–orbit coupling is attenuated by small Pt (5d) contribution to the relevant frontier molecular orbitals, dual emission from the 1L and 3L or 3MLCT excited states has been observed.70–72
CR)2(PR3)2.52,53 Such complexes have, for example been used as constituents of luminescent coordination oligomers and polymers featuring platinum atoms in the conjugated main chain54–63 or for two-photon absorption (TPA).64 Other representatives have organic dyes as the substituents at the alkynyl ligands. These complexes exhibit intense ligand-based π→π*-transitions and usually emit from ligand-centred π*-states with moderate fluorescence or phosphorescence quantum yields in fluid solution at ambient temperature.49,53,65–69 Again, ISC to a ligand-based excited triplet state is triggered by the large heavy-atom effect of the Pt(II) coordination centre. In rare cases, where the excited states are mainly localised at those ligands and spin–orbit coupling is attenuated by small Pt (5d) contribution to the relevant frontier molecular orbitals, dual emission from the 1L and 3L or 3MLCT excited states has been observed.70–72
σ-Bonded aryl ligands exert an even stronger ligand-field splitting than alkynyl ones. Despite the large and rapidly growing number of complexes featuring the above-mentioned cyclometalating arylpyridine24 or arylphosphine73 ligands, there are relatively few examples of luminescent Pt(II) complexes possessing simple, non-chelating σ-bonded aryl ligands.74–76 Most of these complexes show exclusively aryl ligand-centred emission from the 1π*-state with lifetimes in the ns regime77 or emit with low quantum yields from the 3π*-state.78 Notable exceptions are trans-Pt(PEt3)2Br-complexes featuring the anthracen-9-yl ligand or some of its brominated derivatives of Sharp et al., which are formed by oxidative addition of the respective 9-bromoanthracenes to Pt(PEt3)4. These complexes were found to undergo P-type delayed fluorescence with quantum yields of close to 90% and lifetimes of a few microseconds.79 This finding clearly indicates that a triplet state is involved in the radiative deactivation process of the initially generated excited state and that the Pt(PEt3)2Br fragment is capable of strongly accelerating the ISC rate from the 1π*- to the 3π*-state. In contrast, attaching the Pt(PEt3)2Br-fragment to the 3-position of perylene or the 9-position of perylene-3,4-dicarboximide (PMI) does not lead to the expected fast ISC, but promotes ligand-based fluorescence with quantum yields of 70 or 30%, respectively. Further modification of these complexes by substituting the bromido ligand by either cyanido, thiocyanato or neutral ligands such as 4-methylpyridine or tbutylisocyanide hardly affects the quantum yields in the case of the perylene complexes (70–80%) whereas those for the PMI analogues were found to vary from 30% for the neutral complexes to 78% for the cationic isocyanide derivative.80
The favourable photophysical properties of thioxanthones have prompted us to prepare and to investigate the complexes trans-Pt(PEt3)2(X)(thioxanthonyl) (X = Cl, Br, I or CN) bearing the σ-bonded thioxanthon-2-yl ligand. The ready formation of the 3ππ*- and 3nπ*-states of aromatic carbonyl compounds after photoexcitation into the 1π→π*- and 1n→π*-bands81–84 renders thioxanthone derivatives useful sensitisers85,86 or initiators for radical photopolymerisations of olefins.87–93 The excited states of thioxanthones involve two singlet states S1 (1ππ*) and S2 (1nπ*) and two corresponding triplet states T1 (3ππ*) and T2 (3nπ*), which readily equilibrate (Fig. 1).94,95 Apolar solvents decrease the energy gap between the two singlet states due to either a stabilisation of the 1nπ*-state or a destabilisation of the 1ππ*-state. The energetical proximity of these two states leads to strong vibronic coupling between them. This so-called proximity effect is responsible for increasing the non-radiative deactivation rate as well as increasing the fluorescence energy. Furthermore, this effect also lowers the quantum yield for triplet formation.96 Two principal pathways for ISC in thioxanthones were found: (a) a very fast direct mechanism leading from the 1nπ*- to the 3ππ*-state and (b) a slow indirect mechanism leading from the 1ππ*- to the 3ππ*-state with the 3nπ*-state as the intermediate.95,97 Although ISC of thioxanthones is rather efficient, they mainly emit through fluorescence because the T1 state with a lifetime of 100 to 140 ms is so long-lived that radiationless deactivation pathways become dominant. Phosphorescence is usually observed in glassy matrices at 77 K but only rarely in fluid solution with much lower quantum yields.89,90,97 It was therefore our hope that attachment of the {Pt(PEt3)2X}-fragment to the thioxanthonyl core should further increase the ISC rate and hence open a pathway for radiative decay of the T1 to the S0 state. We here report our findings on the complexes Pt–Cl to Pt–CN of Scheme 1, including the observation of dual fluorescence and phosphorescence emissions from the thioxanthone chromophore.
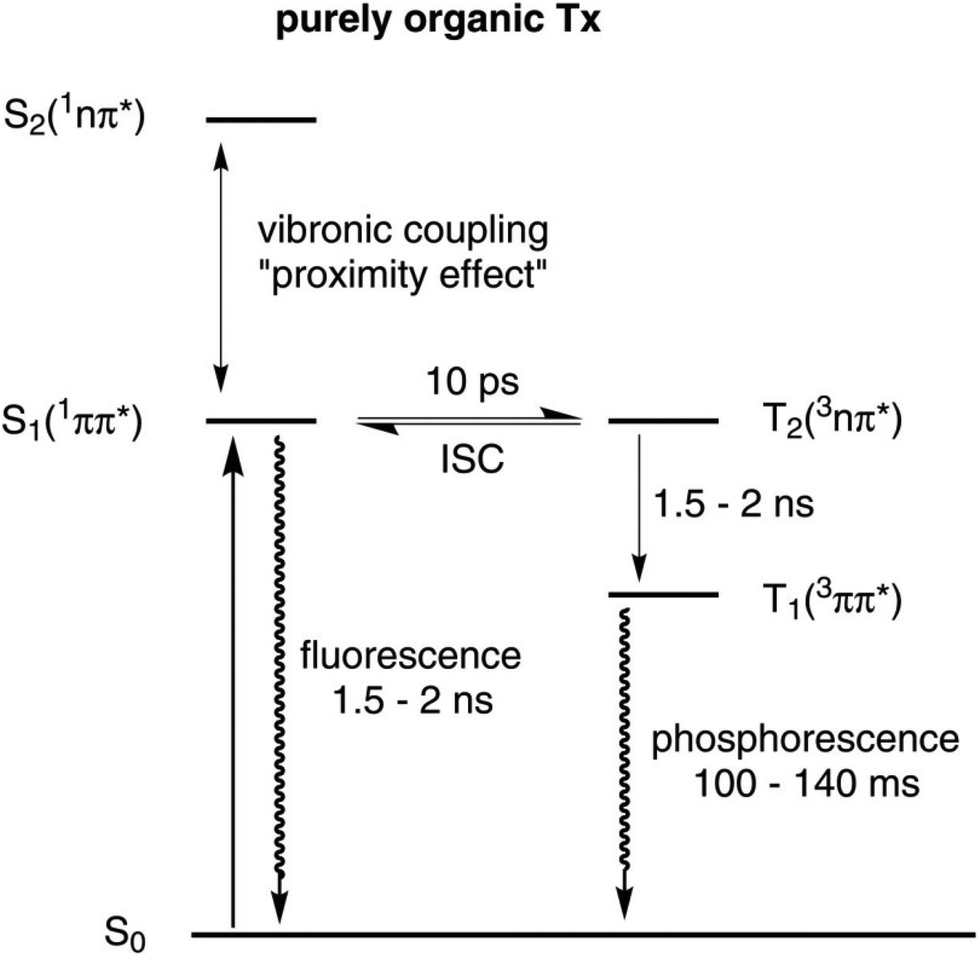 | ||
| Fig. 1 Qualitative Jablonski diagram illustrating the photophysical behaviour of thioxanthones. | ||
 | ||
| Scheme 1 Synthesis of complexes Pt–Br, Pt–CN, Pt–Cl, and Pt–I. | ||
Results and discussion
Synthesis
Oxidative addition of 2-bromo-9H-thioxanthen-9-one (Br–Tx) to Pt(PEt3)4 gave the trans complex Pt–Br (Scheme 1). Monitoring the progress of the reaction via1H and 31P NMR spectroscopy provided no evidence for initial formation of the kinetically favoured cis-isomer at room temperature. Subsequent abstraction of the bromido ligand of Pt–Br with AgBF4 in methanol and addition of NaCl or NaI gave the halogenido complexes Pt–Cl and Pt–I, respectively. The analogous cyanido complex Pt–CN was similarly obtained by treatment of Pt–Br with AgCN. All complexes were purified by recrystallisation from toluene or acetone and were obtained in yields of 63 to 89%. We note that all trans-XPt(PEt3)2(Tx) complexes (X = Br, Cl, I or CN) are much more soluble in polar organic solvents than Br–Tx.Attempts to purify Pt–Br by chromatography on basic alumina with CH2Cl2 as the eluent resulted in halide exchange, putatively via oxidative addition of CH2Cl2 and reductive elimination of CH2BrCl to form complex Pt–Cl.98,99 A second possible reaction sequence is the bromide abstraction by alumina resulting in a cationic complex that subsequently reacts with CH2Cl2 to form Pt–Cl. When basic alumina was added to CD2Cl2 solutions of Pt–Br that were kept in the dark, exchange of the bromido by the chlorido ligand was much slower and occurred only over a period of two weeks as probed by 31P NMR spectroscopy.
Single-crystal X-ray diffraction studies of Pt–Cl and Pt–CN
Slow evaporation of concentrated dichloromethane solutions of Pt–I, Pt–CN, Pt–Cl and Pt–Br gave single crystals in each case. While data sets of Pt–I and Pt–Br were unsatisfactory, needle shaped single-crystals of Pt–Cl and Pt–CN provided data sets of sufficient quality to allow for successful refinement. The ORTEPs of the structures are shown in Fig. 2a for Pt–Cl and in Fig. 2b for Pt–CN while details of the crystallographic measurement, data collection and refinement are provided as Tables S1 and S3 of the ESI.† Complex Pt–Cl crystallises in the monoclinic space group P21/c and Pt–CN in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The Pt atoms of both complexes are in almost ideal square-planar coordination environments with bond angles of 86.67(4) to 93.52(15)° and angle sums of 359.9 and 360.0, respectively (Fig. 2 and 3). While the X–Pt1–C1 (X = Cl1 of Pt–Cl or C14 of Pt–CN) bond angles of 178.98(13) and 178.6(2)° match the expected 180° of a square-planar coordination sphere almost perfectly, the P1–Pt1–P2 bond angles of 175.07(4)° for Pt–Cl and 175.39(5)° for Pt–CN differ by about 5° from the ideal value. This is the result of a slight bending of the PEt3 ligands toward the side of the intracyclic S atom of the thioxanthonyl ligand. The thioxanthonyl ligand is not exactly planar with some puckering about the S1–C11–O1 vector, which amounts to 5.0° for Pt–Cl and to 11.8° for Pt–CN. The best plane through the Tx ligand of the complexes adopts a nearly orthogonal orientation to the platinum coordination plane as is seen by the angles of 85.0° for Pt–Cl and 86.5° for Pt–CN formed between the metal bonded aryl ring of that ligand and the best plane through atoms Pt1, P1, P2, C1 and Cl1 or C14 of the cyanido ligand. The Pt–CN axis of complex Pt–CN is tilted by 1.2° with respect to the plane formed by atoms Pt1, C14, P1, P2 and C1. Pt–C1 bond lengths of 2.010(5) Å for Pt–Cl and of 2.052(5) Å for Pt–CN reflect the trans-effect of the (pseudo)halogenido ligand.
. The Pt atoms of both complexes are in almost ideal square-planar coordination environments with bond angles of 86.67(4) to 93.52(15)° and angle sums of 359.9 and 360.0, respectively (Fig. 2 and 3). While the X–Pt1–C1 (X = Cl1 of Pt–Cl or C14 of Pt–CN) bond angles of 178.98(13) and 178.6(2)° match the expected 180° of a square-planar coordination sphere almost perfectly, the P1–Pt1–P2 bond angles of 175.07(4)° for Pt–Cl and 175.39(5)° for Pt–CN differ by about 5° from the ideal value. This is the result of a slight bending of the PEt3 ligands toward the side of the intracyclic S atom of the thioxanthonyl ligand. The thioxanthonyl ligand is not exactly planar with some puckering about the S1–C11–O1 vector, which amounts to 5.0° for Pt–Cl and to 11.8° for Pt–CN. The best plane through the Tx ligand of the complexes adopts a nearly orthogonal orientation to the platinum coordination plane as is seen by the angles of 85.0° for Pt–Cl and 86.5° for Pt–CN formed between the metal bonded aryl ring of that ligand and the best plane through atoms Pt1, P1, P2, C1 and Cl1 or C14 of the cyanido ligand. The Pt–CN axis of complex Pt–CN is tilted by 1.2° with respect to the plane formed by atoms Pt1, C14, P1, P2 and C1. Pt–C1 bond lengths of 2.010(5) Å for Pt–Cl and of 2.052(5) Å for Pt–CN reflect the trans-effect of the (pseudo)halogenido ligand.
 | ||
| Fig. 2 ORTEPs of Pt–Cl (a) and Pt–CN (b); ellipsoids are drawn at a 40% probability level. H atoms are omitted for clarity except for H6, H7 and H13 of Pt–Cl and H7 and H13 of Pt–CN. Selected bond lengths [Å] and angles [°] of Pt–Cl: Cl1–Pt1 2.4001(13), P1–Pt1 2.3084(11), P2–Pt1 2.3058(11), C1–Pt1 2.010(5), P1–Pt1–P2 175.07(4), C1–Pt1–Cl1 178.98(13), P1–Pt1–Cl1 86.67(4), C1–Pt1–P1 92.63(10), C1–Pt1–P2 87.42(10), P2–Pt1–Cl1 93.21(4); Pt–CN: C14–Pt1 2.048(6), P1–Pt1 2.3098(14), P2–Pt1 2.3049(13), C1–Pt1 2.052(5), P1–Pt1–P2 175.39(5), C1–Pt1–C14 178.6(2), P1–Pt1–C14 88.16(15), C1–Pt1–P1 90.39(14), C1–Pt1–P2 87.92(14), P2–Pt1–C14 93.52(15). | ||
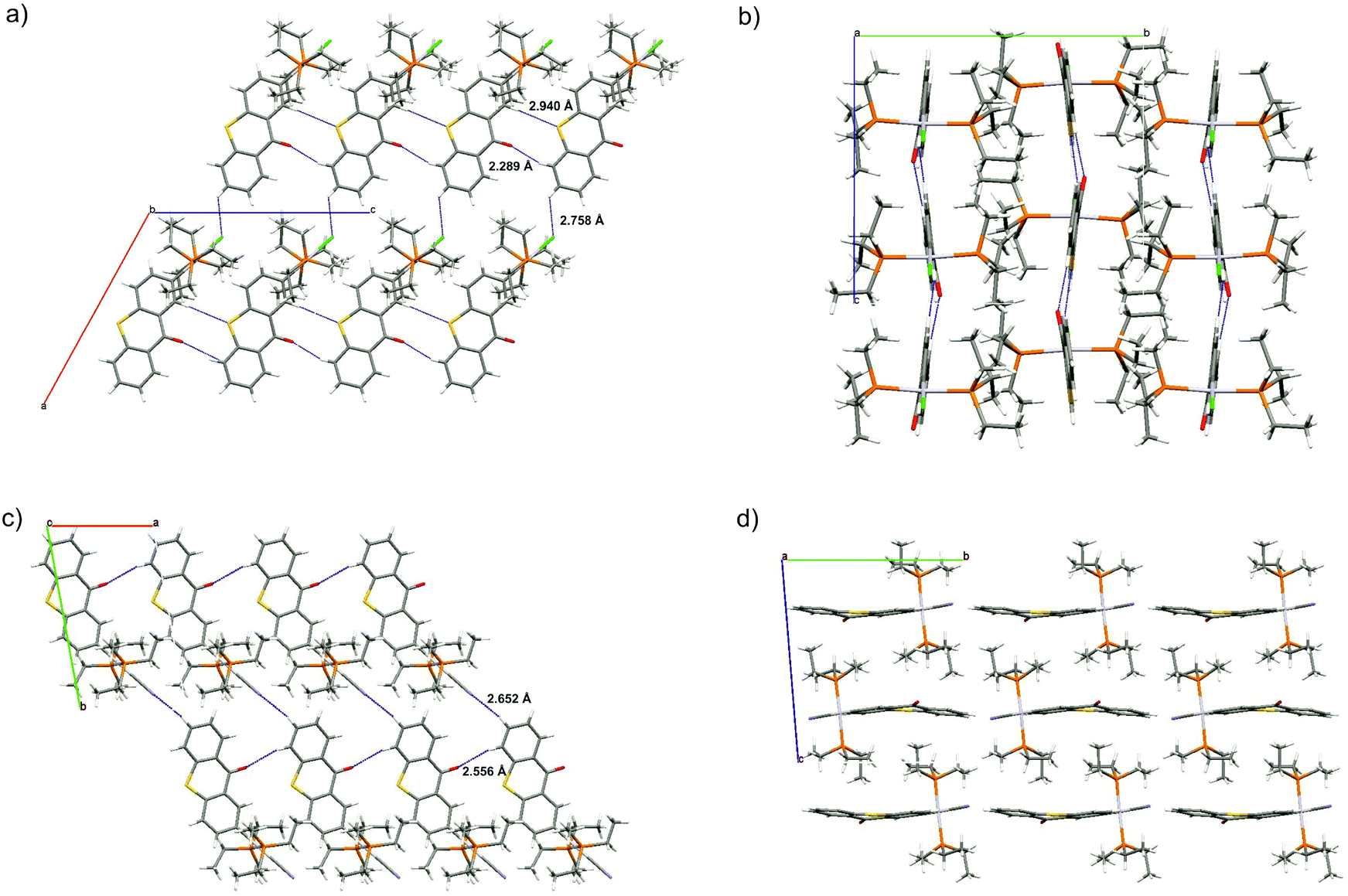 | ||
| Fig. 3 Molecule packing of Pt–Cl and Pt–CN in the crystal. View along the crystallographic b axis (a) and the a axis (b) of Pt–Cl and along the crystallographic c axis (c) and the a axis (d) of Pt–CN. | ||
In the crystal, individual molecules of Pt–Cl associate to 1D chains along the crystallographic c axis by a total of four hydrogen bonds O1–H6a and S1–H13a with O⋯H and S⋯H distances of 2.289 and 2.940 Å for Pt–Cl, respectively, which is 43 and 19 pm shorter than the sum of the van der Waals radii (see Fig. 3a). In contrast, individual molecules of Pt–CN form two hydrogen bonds O1–H13a along the crystallographic a axis with a distance of 2.556 Å, 16 pm shorter than the sum of the van der Waals radii, Fig. 3c. Within each chain of Pt–Cl, neighbouring molecules are tilted with respect to each other by 13.3° with respect to their thioxanthone planes in a slight zigzag pattern (Fig. 3b). Individual chains of both complexes are further connected through hydrogen bonds between H7a and the chlorido or cyanido ligand with H⋯X distances of 2.758 or 2.652 Å, respectively. For Pt–CN, these chains are offset along the bc plane and arrange in a brick wall-type fashion (see Fig. 3d).
Individual molecules of these complexes thus form two-dimensional sheet structures within the ac or ab planes, respectively. Individual chains of Pt–Cl are further offset along the ac plane whereas those of Pt–CN are offset along the bc plane. Each thioxanthonyl ligand of the complexes is surrounded by two phosphine ligands belonging to neighbouring chains, one above and one below the thioxanthone plane. Thus no Pt⋯Pt or π–stacking interactions are observed in the solid state. This may contribute to the much higher solubility of the complexes when compared to the free ligand.
NMR spectroscopic characterisation
The 31P{1H} NMR spectra of Pt–Cl, Pt–Br, Pt–CN and Pt–I all reveal just one sharp singlet flanked by 195Pt satellites (natural abundance 33.8%; Fig. 4a) which testifies to the trans arrangement of the phosphine ligands. The chemical shifts (Table 1) increase in the order I < CN < Br < Cl. For the halogenido ligands the ordering parallels decreasing σ-basicity against platinum, while the cyanido ligand one sticks out because of its strong π-accepting capability.100 Observed coupling constants range from 2524 Hz for Pt–CN to 2725 Hz for Pt–Cl and are unexceptional when compared to other complexes trans-Pt(σ-aryl)(PEt3)2(X).79,80,100 The 1JPt–P coupling constants reflect the s character of the Pt–P and Pt–X σ-bond101,102 and follow the order CN < I < Br < Cl. The 195Pt signal of every complex is split into a triplet by coupling to the two equivalent phosphorus nuclei and the shift follows a similar trend to the 31P chemical shift with the exception of complex Pt–CN, whose 195Pt resonance appears at higher field than that of Pt–I.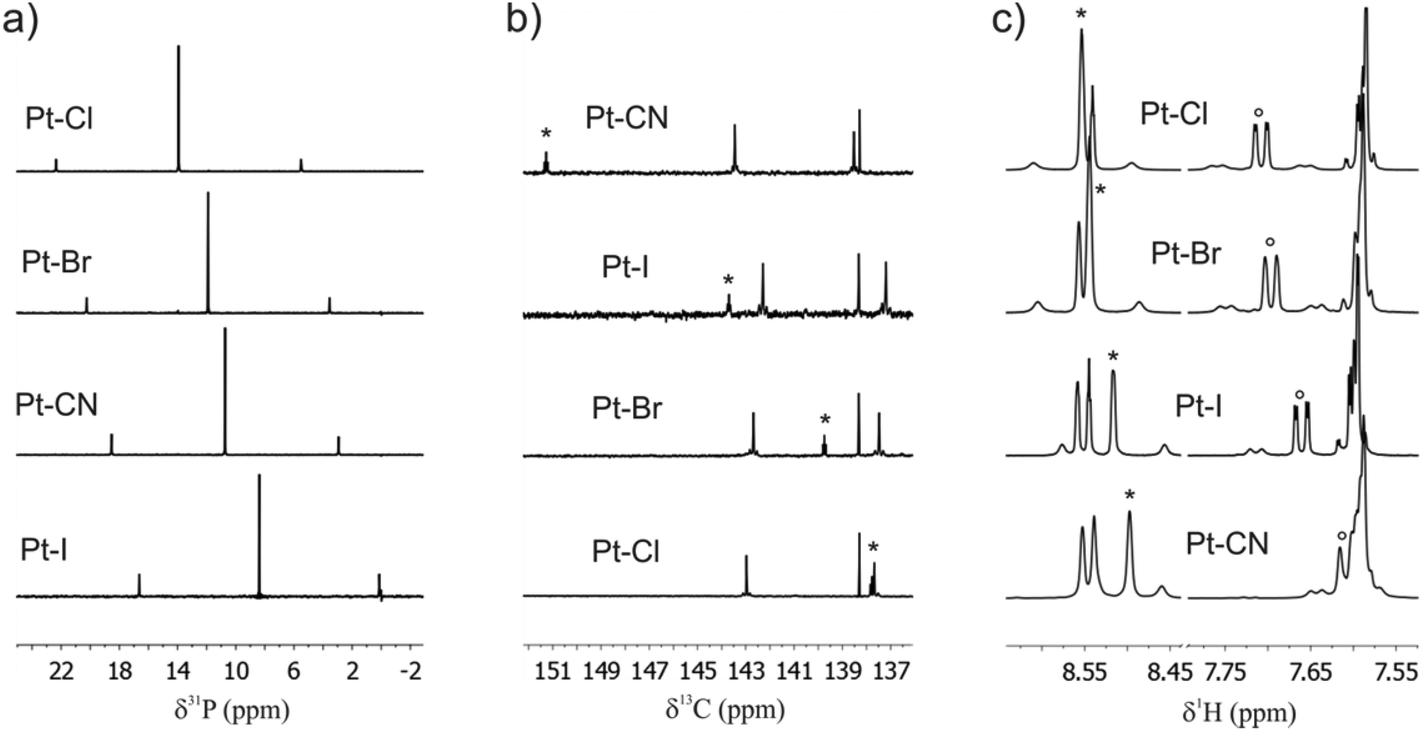 | ||
| Fig. 4 (a) 31P{1H}NMR spectra of Pt–Cl, Pt–Br, Pt–CN and Pt–I. (b) 13C{1H} NMR spectra of Pt–Cl, Pt–Br, Pt–CN and Pt–I. The asterisks indicate the signal of the platinum bonded carbon atom C1. (c) 1H NMR spectra Pt–Cl, Pt–Br, Pt–CN and Pt–I in the spectral region of the ortho protons of the Tx ligand. The asterisks mark the resonance signal atom H13 and the circles that of H2. | ||
| δ 195Pt | δ 31P [ppm]/1JPt–P [Hz] | δ 13C, C1 [ppm]/1JPt–C [Hz]/2JP–C [Hz] | δ 1H, H13 [ppm]/3JPt–H [Hz] | δ 1H, H2 [ppm]/3JPt–H [Hz] | |
|---|---|---|---|---|---|
| Pt–Cl | −4257 | 13.93/2725 | 137.8/960/9.2 | 8.55/69.2 | 7.71/61.9 |
| Pt–Br | −4382 | 11.91/2701 | 139.1/972/9.0 | 8.54/71.3 | 7.70/63.4 |
| Pt–CN | −4674 | 10.73/2524 | 151.3/n.d./10.5 | 8.50/45.1 | 7.61/40.9 |
| Pt–I | −4602 | 8.39/2668 | 143.7/n.d./8.8 | 8.52/71.9 | 7.66/64.0 |
The 13C NMR signal of the metal-bonded C1 atom of every complex is also split into a triplet due to coupling to the two cis positioned phosphorus nuclei with 2JP–C coupling constants ranging from 8.8 Hz for Pt–I to 10.5 Hz for Pt–CN. The chemical shift of the C1 atom clearly depends on the (pseudo)halogenido ligand (Fig. 4b, Table 1 and Experimental section). From the data in Table 1 and Fig. 4b one can infer that the resonance signal of carbon atom C1 dramatically shifts to lower field as the back bonding ability of the trans disposed ligand increases. 195Pt satellites of the C1 13C resonance signal of 960 and 972 Hz, respectively, were detected for complexes Pt–Cl and Pt–Br but not for the other two complexes because of the lower signal to noise ratio of the experimental spectra. For the two complexes Pt–Cl and Pt–Br the 1JPt–C1 coupling constants have an inverse ordering to the 1JPt–P coupling constants, which indicates that these two kinds of ligands compete for the 6s electron density at the Pt atom. The chemical shifts of all other carbon atoms remain almost unchanged along this series. The coupling constants 3JPt–H2 and 3JPt–H13 to the Tx protons in the ortho positions to the platinum atom amount to ca. 70 Hz and 63 Hz, respectively, for the halogenido complexes and to ca. 45 or 41 Hz for the cyanido complex and follow the ordering of the trans-influence of the ligand X (CN ≪ Cl < Br < I).
Electronic absorption spectroscopy
Fig. 5a depicts the room temperature electronic absorption spectra of the four complexes and of the free ligand Br–Tx in CH2Cl2 while relevant data are collected in Table 2. Like for the Br–Tx precursor, the spectra of the platinum complexes feature prominent UV absorption with extinction coefficients ε of the order of 4 × 104 and a much weaker one (ε ≈ 5 × 103) at about 400 nm which is responsible for the yellow colour of Br–Tx and the complexes. Attachment of the {(X)Pt(PEt3)2}-fragment (X = Br, Cl, I or CN) to the thioxanthone core increases the intensity of the UV bands and red shifts the low energy band by 644 cm−1 for the cyanido complex and by ca. 895 cm−1 for the halogenido complexes. Donor substitution of thioxanthones at the 2-position generally red shifts the low energy band owing to a destabilisation of the highest occupied molecular orbital (HOMO). Shifts are in the range of 1740 cm−1 (in acetonitrile) for alkoxy88 and of 410 cm−1 (in benzene) for alkyl87 substituents. Thus, in a first approximation, the {(X)Pt(PEt3)2}-fragment seems to act as a moderate donor towards the thioxanthonyl core, thus lowering the HOMO–LUMO gap.100 A more precise explanation of the red-shift is that the highest occupied platinum d orbital 5dxz is more strongly coupled to the π than to the π* orbital of the thioxanthonyl ligand since the π and the 5dxz orbitals are closer in energy. This leads to a destabilisation of the π orbital while the π* orbital is stabilised by the interaction with the unoccupied Pt 6pz orbital. Thus the energy gap of the π→π*-transition is lowered.78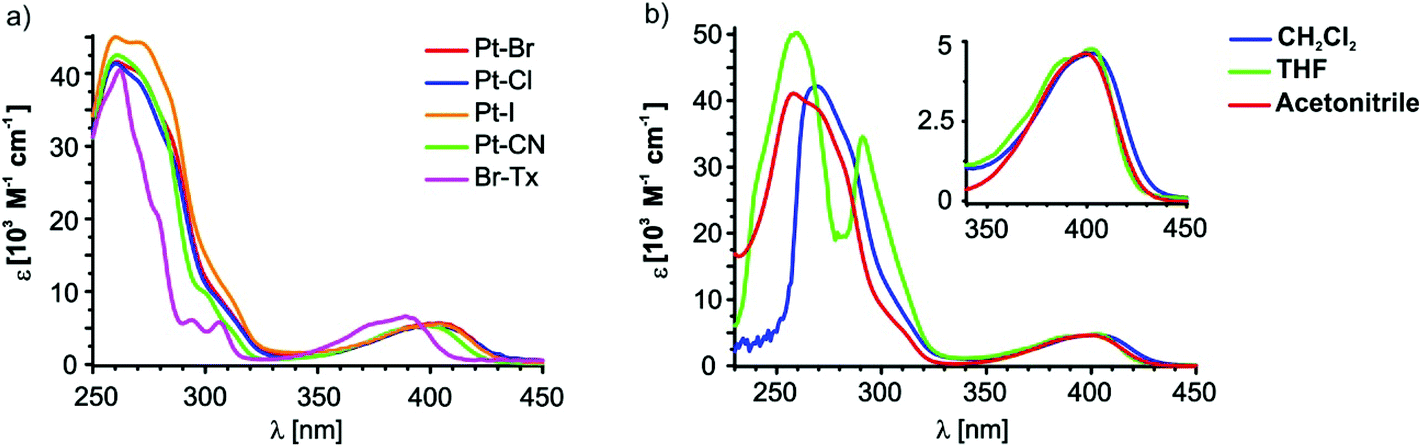 | ||
| Fig. 5 (a) UV-Vis spectra in CH2Cl2 of Pt–Br, Pt–Cl, Pt–I, Pt–CN and the ligand bromothioxanthone (Br–Tx). (b) UV-Vis spectra of Pt–Br in acetonitrile (red), CH2Cl2 (blue) and THF (green). | ||
| Compound | Absorption data | TD-DFT data | |||
|---|---|---|---|---|---|
| λ max [nm] (ε [103 M−1 cm−1]) | λ [nm] | Main configuration | ƒa | Assignment | |
| a Oscillator strength. | |||||
| Pt–Cl | 260 (41.3), 272 (38.2), 286 (28.3), 310 (7.7), 403 (5.2) | 240 | HOMO → L+5 (83%) | 0.19 | MMLLCT |
| 248 | H−7 → LUMO (61%) | 0.29 | ILCT/MMLL′CT | ||
| H−8 → LUMO (16%) | |||||
| 261 | H−5 → LUMO (54%) | 0.63 | MMLL′CT/n→π* | ||
| HOMO → L+1 (19%) | |||||
| 274 | H−5 → LUMO (11%) | 0.23 | MMLL′CT/MLCT/π→π* | ||
| H−4 → LUMO (14%) | |||||
| H−3 → LUMO (19%) | |||||
| HOMO → L+1 (36%) | |||||
| 366 | HOMO → LUMO (97%) | 0.073 | π→π*/MMLL′CT | ||
| Pt–Br | 260 (41.5), 272 (39.5), 285 (31.2), 310 (7.7), 403 (5.7) | 240 | HOMO → L+5 (66%) | 0.12 | ILCT/MMLL′CT |
| 248 | H−8 → LUMO (19%) | 0.32 | ILCT/MMLL′CT | ||
| H−7 → LUMO (57%) | |||||
| 261 | HOMO → L+1 (21%) | 0.73 | MLCT/n→π* | ||
| H−6 → LUMO (57%) | |||||
| 275 | H−1 → L+2 (68%) | 0.21 | σ→σ* | ||
| 365 | HOMO → LUMO (97%) | 0.072 | π→π*/MMLL′CT | ||
| Pt–CN | 260 (42.5), 272 (39.3), 282 (32.5), 301 (9.6), 313 (4.3), 399 (5.3) | 237 | HOMO → L+4 (64%) | 0.21 | π→π*/MLCT |
| H−1 → L+2 (22%) | |||||
| 246 | H−7 → LUMO (80%) | 0.24 | ILCT | ||
| 261 | H−5 → LUMO (48%) | 0.88 | MMLL′CT/ILCT/π→π* | ||
| HOMO → L+1 (27%) | |||||
| H−2 → LUMO (15%) | |||||
| 362 | HOMO → LUMO (97%) | 0.075 | π→π*/MLCT | ||
| Pt–I | 260 (45.0), 273 (44.0), 285 (36.0), 310 (10.0), 403 (5.5) | 248 | H−9 → LUMO (15%) | 0.32 | MMLL′CT/π→π*/ILCT |
| H−8 → LUMO (58%) | |||||
| HOMO → L+4 (12%) | |||||
| 261 | H−6 → LUMO (57%) | 0.86 | MLCT/ILCT/n→π*/MMLL′CT | ||
| HOMO → L+1 (12%) | |||||
| 279 | H−3 → L+2 (74%) | 0.13 | σ→σ* | ||
| 362 | HOMO → LUMO (97%) | 0.072 | π→π*/MMLL′CT | ||
| Tx–Br | 262 (40.3), 270 (29.0), 279 (20.0), 293 (6.1), 306 (5.8), 389 (6.6) | n.c. | |||
As follows from our quantum chemical calculations (vide infra) the (relatively) weak low-energy band corresponds to the HOMO → LUMO transition while several individual transitions contribute to the stronger, structured UV absorption. The energy of the low-energy band remains nearly unchanged upon substitution of the Br− ligand by I− or Cl− while it is shifted by 370 cm−1 to higher energy for the cyanido complex. This blue-shift is a result of the π-accepting ability of the cyanido ligand in trans-position to the Tx ligand, which stabilises the HOMO. This observation is also in accord with the 13C NMR chemical shifts where the strong back bonding ability of the cyanido ligand induces a deshielding of the ipso carbon. Nevertheless, the effect of the ligand X on the HOMO–LUMO gap is rather small. The electrochemical data of the four complexes derived from cyclic voltammetric experiments (0.1 M Bu4NPF6/CH2Cl2v = 400 mV s−1) also show a small effect of the X-ligand since the potentials of the first irreversible oxidation are all in a similar range of 0.94 to 1.09 V (see Fig. S18–S21 of the ESI;† note, however, that the peak potentials of chemically irreversible processes do not strictly correspond to the thermodynamic E0 values but are also affected by the equilibrium and forward rate constants of the chemical follow process).
Fig. 5b shows the electronic absorption spectra of Pt–Br at room temperature in different solvents. The position of the twin band at 400 nm reveals no solvent dependence whereas the onset of the UV band shifts to higher energy with increasing solvent polarity. Of note is also a distinct splitting of this latter band in THF whereas the lower energy components are only a shoulder superimposed on the more intense, higher energy one in the other solvents. From our observation that the split Vis band with individual peaks at ca. 400 nm and 390 nm exhibits no positional solvent dependence and closely resembles the Vis absorption of the free Tx ligand itself, we can infer that both relevant MOs are essentially confined to the σ-bonded Tx ligand and have obviously very similar spatial characteristics. In particular, the underlying transition(s) have no appreciable charge transfer character. Experimental and quantum chemical studies on thioxanthones point to mainly π→π*-character of the underlying transition(s). That assumption is further supported by our present TD-DFT calculations on Pt–Br, Pt–Cl, Pt–I and Pt–CN (Table 2, Fig. 6, and Fig. 25, 27 and 28 of the ESI†). These calculations indicate that the HOMO, the LUMO and the LUMO+1 are almost exclusively located on the thioxanthonyl moiety with major orbital coefficients on the intracyclic sulfur atom for the HOMO, at the thioxanthone carbonyl function for the LUMO and a nodal plane along the S⋯CO axis for the LUMO+1.
 | ||
| Fig. 6 Graphical representations of selected MOs and their TD-DFT energies for complex Pt–Br. Arrows symbolise the main contributors to the respective transitions. | ||
In contrast, the UV bands in the range of 310 to 260 nm possess negative solvatochromism which indicates charge transfer contributions to at least one of the major transitions within the band envelope. According to our TD-DFT calculations, the overall UV absorption comprises four major components: an absorption at a (calculated) energy of 275 nm, which is essentially the HOMO−1 → LUMO+2 transition, one very strong absorption at 261 nm mainly corresponding to the HOMO−6 → LUMO (57%) and HOMO → LUMO+1 (21%) transitions, a band at 248 nm, which is dominated by the HOMO−7 → LUMO (57%) and the HOMO–8 → LUMO (19%) transitions, and one rather weak band at 240 nm of mainly HOMO → LUMO+5 origin. As follows from Fig. 6, the band calculated at 275 nm is a σ→σ* transition of the Br–Pt bond accompanied by an electron density increase at the Br− ligand and the occurrence of a nodal plane between the Pt and the Br atom. The band calculated at 261 nm is a mixture of an intraligand charge-transfer (ILCT) and n→π* transition, both centred at the thioxanthonyl ligand with some shift of electron density from the S and O atoms to the carbon atoms of the thioxanthonyl moiety and a shift of electron density from the Pt bound aryl to the outermost phenyl ring. The 248 nm band comprises a thioxanthone-based intraligand charge transfer (ILCT) and a mixed metal/ligand-to-ligand’ charge-transfer (MLL′CT) from the {(PEt3)2Pt}-unit to the thioxanthonyl ligand owing to appreciable Pt-aryl δ character of the HOMO−8. Of note is also an energy splitting of 0.28 to 0.29 eV between the similar MOs HOMO−8 and HOMO−6, which represent the δ-bonding and -antibonding interactions between the Pt atom and the Pt-bonded aryl ring of the Tx ligand. The band at 240 nm has also some ILCT- and MLL′CT component. Similar results have been obtained for complexes Pt–CN, Pt–Cl and Pt–I (see Fig. S25, S27 and S28 of the ESI†). However, the σ→σ* transition of Pt–Br and Pt–I is absent in Pt–CN and Pt–Cl as a result of the energetically low-lying MOs for the Pt–Cl and Pt–CN σ-bonds.
Luminescence spectroscopy
Fig. 7a displays the room temperature emission spectra of Br–Tx in 10−5 M aerated and of Pt–CN, Pt–I, Pt–Br and Pt–Cl in 10−5 M nitrogen-saturated CH2Cl2 solutions. Br–Tx emits at 417 nm upon irradiation at 370 nm. Excitation at 400 nm into the HOMO → LUMO band of the four complexes results in dual emission at around 450 nm and at ca. 510 nm. Stokes shifts are in the range of 2140 to 2640 cm−1 for the higher energy emission and of 5244 to 5583 cm1 for the one at the lower energy. We note here that the Stokes shifts of the higher energy emission band of the platinum complexes is only mildly larger as that of 1760 cm−1 for the fluorescence emission of Br–Tx itself and it is clearly different from that of Br–Tx in each case. For every complex, the emission band at 450 nm is insensitive against oxygen while that at 510 nm is almost completely quenched in aerated solutions (Fig. 7c). This observation in concert with the large Stokes shifts let us assign the high energy emission as fluorescence and the one at lower energy as phosphorescence, both arising from the σ-bonded Tx ligand. Excitation spectra of Pt–Cl (see Fig. S32 of the ESI†) support this assignment. Emission detection at wavelengths of 450 and 501 nm provide essentially identical spectra with a prominent band 400 nm in exact match with the low-energy π→π* absorption of the Pt-bonded Tx substituent. In further agreement with that hypothesis dual emission from the complexes with attenuated intensities was also observed when Pt–Br was irradiated at 280, 350 or 400 nm (see Fig. S33 of the ESI†). This clearly indicates that both emissions emanate from the same compound.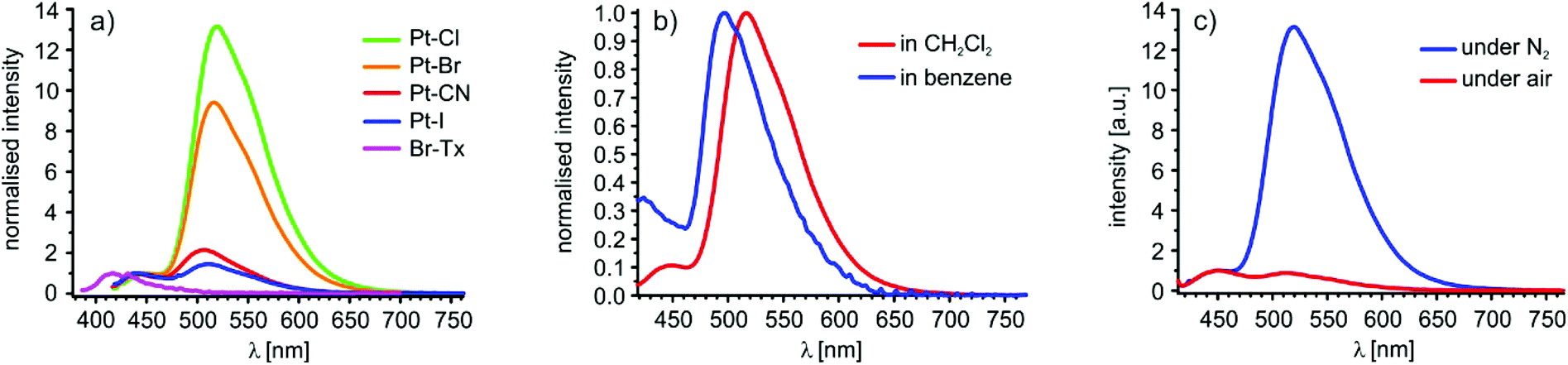 | ||
| Fig. 7 (a) Emission spectra of Pt–Cl (green), Pt–Br (orange), Pt–CN (red), and Pt–I (blue) in nitrogen-saturated and Br–Tx in aerated CH2Cl2solutions. (b) Emission spectra of Pt–Br in CH2Cl2 (red), and benzene (blue). (c) Emission spectra of Pt–Cl under nitrogen atmosphere (blue) and under air (red). | ||
The influence of the ligand X on the emission energies is small, but both bands shift to slightly higher energy as Pt–X back bonding increases (Cl < Br ≪ I < CN). The wavelength of the fluorescence emission of the (pseudo)halogenido complexes thus follows an inverse order as that of the electronic absorption. One possible explanation is that the first excited singlet state undergoes some structural reorientation before emission occurs which leads to a stronger contribution of the (pseudo)halogenido ligand. Such structural reorientation upon irradiation of thioxanthones is not without precedence in the literature96 and also underlies the solvent dependency of Tx emission, very similar to the one that we observe for the investigated complexes (Fig. 7b). Computed structure changes between the T1 and S0 states are, however, rather minor and basically confined to the platinum bonded Tx ligand with a contraction of the Pt–C(Tx) and the C4–S bond lengths of ca. 3 pm and elongation of the Tx C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and the C1–C2 bonds of about 4 pm and 2 pm, respectively (Tables S5 to S8 of the ESI†). The same holds for calculated spin densities as is exemplarily shown for Pt–Cl in Fig. S26 of the ESI.†
O and the C1–C2 bonds of about 4 pm and 2 pm, respectively (Tables S5 to S8 of the ESI†). The same holds for calculated spin densities as is exemplarily shown for Pt–Cl in Fig. S26 of the ESI.†
For the three halogenido complexes the phosphorescence quantum yields increase with increasing σ-basicity of the ligand X against platinum in the order I < CN < Br < Cl from smaller than 2% for Pt–I to close to 19% for Pt–Cl (Table 3). With increasing σ-basicity of the ligand X, the ligand-field splitting also increases which in turn destabilises the non-emissive Pt 5d states. Therefore, thermal excitation from the emissive thioxanthone-centred 3ππ*-states to the Pt 5d states undergoing radiationless decay is diminished.103 Ligand-field arguments would, however, predict that the cyanido complex is the strongest triplet emitter along this series of complexes, which is clearly not the case. Radiationless decay of the excited triplet state may also be triggered by excitation of vibrational quanta, and this decay channel may be particularly efficient in Pt–CN due to the high energy of the CN stretch.104
| λ max,Fl [nm] (St. shift [cm−1]) | λ max,Ph [nm] (St. shift [cm−1]) | ϕ Fl | ϕ Ph | τ [μs] | |
|---|---|---|---|---|---|
| a Measured in benzene at ambient temperature in nitrogen saturated solutions. | |||||
| Br–Tx | 417 (1726) | — | n.m. | — | — |
| Pt–Cl | 451 (2640) | 520 (5583) | 0.006 | 0.188 | 2.17 |
| Pt–Br | 450/421a (2592) | 517/491a (5472) | 0.006 | 0.120/0.031a | n.m. |
| Pt–I | 441 (2138) | 511 (5244) | 0.004 | 0.017 | n.m. |
| Pt–CN | 439 (2283) | 507 (5338) | 0.015 | 0.069 | n.m. |
The phosphorescence quantum yield further increases with increasing polarity of the solvent as is exemplified by complex Pt–Br, which shows a quantum yield of 12% in CH2Cl2 but only one of 3% in benzene. Such behaviour has also been reported for purely organic thioxanthones, which also show strong solvent dependence of the rates of internal conversion and intersystem crossing owing to a modulation of the relative energies of the S1, S2, T1 and T2 states.94–96 This effect is also reflected in the energies of the fluorescence and the phosphorescence bands of Pt–Br that are blue shifted by 1530 cm−1 and 1025 cm−1, respectively, when the solvent is changed from CH2Cl2 to benzene.
We note that the fluorescence quantum yields and, obviously, the lifetimes of the four complexes vary only very little and are also very similar to those of purely organic thioxanthones.88,94,96,105 The rate constant of fluorescence can be estimated as ca. 1.2 to 0.3 ns by the established equation:105
| Φf = kfτf with kf = 5.12 × 107 s−1. |
This shows that attachment of the {Pt(PEt3)X}-fragment has an only limited influence on the already high rate of intersystem crossing (ISC) of the Tx dye. The quantum yield of triplet formation of parent thioxanthone has been determined as 66% for acetonitrile and 84% for benzene solutions.97 Of note is, however, the observation of moderately to rather intense room-temperature phosphorescence from the thioxanthonyl ligand with quantum yields of up to almost 19%. Organic thioxanthones, while being phosphorescent in glassy matrices at 77 K,89,90,94 usually show only very weak emission under these conditions, if any at all.97 Intersystem crossing from the excited S1 to the excited T2 state of thioxanthones is usually very fast (see Fig. 1) and occurs within 10 ps86,95 while the Internal Conversion from the T2 to the T1 state occurs at a rate which is identical to the lifetime of the fluorescence (1.5–2 ns).86 In contrast, the T1 state is very long-lived with half-lives in the range of 100 to 140 ms, making it very susceptible to quenching and photoreactions such as H atom abstraction. The role of the covalently bonded Pt centre is thus to open a pathway for relatively rapid decay of the T1 state to the ground state with the emission of light due to its high spin–orbit coupling constant, which is not available for thioxanthone itself or its organic derivatives. Pt–Cl, the strongest triplet emitter of the present series, has a phosphorescence half-life of 2.17 μs, which is by five orders of magnitude lower compared to that of purely organic thioxanthones. The lifetime clearly proves that the emission band at 510 nm originates from a triplet state and further indicates that phosphorescence emission now becomes competitive to radiationless deactivation pathways.
Conclusions
Four new square-planar complexes trans-Pt(PEt3)2(X)(thioxanthon-2-yl) (X = Cl, Br, CN or I) were successfully synthesised by oxidative addition of 2-bromo-9H-thioxanthen-9-one to Pt(PEt3)4 and subsequent halide exchange. Electronic absorption data in concert with TD-DFT calculations reveal a considerable red-shift of the thioxanthone-centred π→π* (HOMO → LUMO) transition when compared to the free ligand in accordance with the overall donor capabilities of the trans-{Pt(PEt3)2X}-fragment. All four complexes exhibit dual fluorescence and phosphorescence emission in fluid solution at room temperature arising from excited singlet and triplet states located at the thioxanthonyl ligand. While the fluorescence quantum yield is largely independent on X and very similar to that of other thioxanthones, that of the phosphorescence emission is strongly modulated by the ligand X, ranging from 1.7% for Pt–I to a truly exceptional value of 18.8% for Pt–Cl in nitrogen-saturated CH2Cl2 solution. In an intuitive ordering, the phosphorescence quantum yield of the halogenido complexes increases with the ligand-field splitting abilities of ligand X. The lower phosphorescence quantum yield of the strong-field cyanido ligand may be due to its high-energy CN stretch, which probably provides an efficient non-radiative deactivation channel. Key to the observation of dual emission is that the platinum coordination centre makes only limited contribution to the relevant frontier orbitals of the excited states70–72 as is, e.g., seen by the computed spin density distributions of the T1 state. This attenuated Pt contribution has no noticeable influence on the already fast rate of ISC inherent to thioxanthones. It is, however, still sufficient to open a radiative deactivation channel for the usually very long-lived triplet state that is unattainable for thioxanthones themselves, thus diminishing the lifetime of the excited triplet state of the Tx ligand by five orders of magnitude to a value of ca. 2 μs. This allows for thioxanthone-based phosphorescence to be observed in fluid solution with unprecedentedly high quantum yield. We note here that complexes exhibiting dual fluorescence and phosphorescence emission have advantages over two-component sensors for quantitative detection of singlet-oxygen.71 The platinum Tx complexes herein display strong phosphorescence and quantitative quenching of that emission at atmospheric O2 levels while fluorescence emission is fully retained. This makes them promising candidates for such application, and work along these lines is underway.
Experimental section
Materials and general methods
All reactions, except the oxidative addition of 2-bromo-9H-thioxanthen-9-one (Br–Tx) to Pt(PEt3)4, were performed under air with undried solvents. THF was distilled from blue sodium/benzophenone ketyl and was degassed by three freeze–pump–thaw cycles. All other solvents were used without further purification. 2-Bromo-9H-thioxanthen-9-one (Br–Tx)106 and [Pt(PEt3)4]107 were synthesised according to literature procedures.NMR experiments were carried out on a Varian Unity Inova 400, a Bruker Avance III DRX 400 or a Bruker Avance DRX 600 spectrometer. 1H and 13C NMR spectra were referenced to the solvent signal. 31P and 195Pt NMR spectra were referenced to external standard (85% H3PO4 or saturated solution of K2[PtCl6] in D2O, respectively). NMR spectral data are given as follows: chemical shift (δ ppm), multiplicity (m: multiplet, s: singlet, d: doublet, t: triplet, quin: quintet, vm: virtual multiplet,), coupling constant (Hz), integration. In addition to 1D NMR experiments, complete NMR characterisations were performed by 2D NMR experiments (1H,1H gCOSY, 1H,13C gHSQC, 1H,13C gHMBC and 1H,31P gHMBC).
X-ray diffraction analysis was performed at 100 K on a STOE IPDS-II diffractometer equipped with a graphite-monochromated radiation source (λ = 0.71073 Å) and an image plate detection system. A crystal mounted on a fine glass fiber with silicon grease was employed. If not indicated otherwise, the selection, integration and averaging procedure of the measured reflex intensities, the determination of the unit cell dimensions and a least-squares fit of the 2θ values as well as data reduction, LP-correction and space group determination were performed using the X-Area software package delivered with the diffractometer. A semiempirical absorption correction was performed. The structure of Pt–Cl was solved by the heavy-atom methods (SHELXS-97), whereas that of Pt–CN was solved by direct methods (SHELXS-97).118 Solutions of both structures were completed with difference fourier syntheses and refined with full-matrix least-square using SHELXL-97108 minimising ω(F02 − Fc2)2. Weighted R factor (wR2) and the goodness of fit GooF are based on F2. All hydrogen atoms were treated according to a riding model. Molecular structures in this work are plotted with ORTEP 3.109 Hydrogen atoms and solvent molecules are omitted for clarity.
UV-Vis spectra were obtained on a TIDAS fiberoptic diode array spectrometer MCS UV/NIR from j&m in HELLMA quartz cuvettes with 1 cm optical path lengths. Emission spectra were measured on a Perkin Elmer luminescence spectrometer LS50 using HELLMA quartz cuvettes modified with an angle valve from Normag. Time-resolved luminescence spectroscopy was measured on a LP920-KS instrument from Edinburgh Instruments, equipped with a R928 photomultiplier and an iCCD camera from Andor using a Starna quartz cuvette modified with an angle valve from Rotaflow. Luminescence was excited with the frequency-doubled output from a Quantel Brilliant b laser and the pulse length of approximately 10 ns. The concentration of samples was ca. 1.5 × 10−5 M.
Electrochemical investigations in degassed dichloromethane were performed in a vacuum-tight one-compartment cell on a BAS CV50 potentiostat using Pt or glassy carbon disk electrodes from BAS as the working electrode, a platinum counter electrode and a silver pseudoreference electrode. Potentials were calibrated against the ferrocene/ferrocenium couple (E0 Fc/Fc+ = 0.000 V).
Computational details
The ground state electronic structures were calculated by density functional theory (DFT) methods using the Gaussian 09110 program packages. Quantum chemical studies were performed without any symmetry constraints. Open shell systems were calculated by the unrestricted Kohn–Sham approach (UKS).111 Geometry optimization followed by vibrational analysis was made either in vacuum or in solvent media. The quasirelativistic Wood-Boring small-core pseudopotentials (MWB)112,113 and the corresponding optimized set of basis functions114 for Pt and 6-31G(d) polarized double-ζ basis set115 for the remaining atoms were employed together with the Perdew, Burke, Ernzerhof exchange and correlation functional (PBE0).116,117 Solvent effects were described by the polarizable conductor continuum model (PCM)118–121 with standard parameters for CH2Cl2. Absorption spectra and orbital energies were calculated using time-dependent DFT (TD-DFT)122 using the same functional/basis set combination mentioned above. For easier comparison with the experiment, the obtained absorption and emission energies were converted into wavelengths and broadened by a Gaussian distribution (full width at half maximum = 3000 cm−1) using the program GaussSum.123 Molecular orbitals were visualised with the GaussView program.124Acknowledgements
We gratefully acknowledge the Deutsche Forschungsgemeinschaft (DFG) for financial support (grant number Wi1262 10/1). We further thank Dr Inigo Göttker gen. Schnetman and Bernhard Weibert for measurements and solutions of the single crystal X-ray diffraction data, Jun.-Prof. Dominik Wöll for providing a sample of 2-bromo-9H-thioxanthen-9-one to us and Prof. Dr Ulrich Steiner, Konstanz and Dr Stanislav Záliš, Prague, for helpful discussions. We also thank Prof. Oliver Wenger, University of Basel, for making his equipment for measuring the phosphorescence lifetime of complex Pt–Cl available to us and Julia Nomrovski for her help with the measurements. We are indebted to the state of Baden-Württemberg and the Deutsche Forschungsgemeinschaft for providing the computational facilities of the bwUni cluster to us.References
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178–8211 CrossRef CAS PubMed.
- R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- L. F. Gildea and J. A. G. Williams, in Organic Light-Emitting Diodes (OLEDs), ed. A. Buckley, Woodhead Publishing, 2013, pp. 77–113 DOI:10.1533/9780857098948.1.77.
- D.-L. Ma, V. P.-Y. Ma, D. S.-H. Chan, K.-H. Leung, H.-Z. He and C.-H. Leung, Coord. Chem. Rev., 2012, 256, 3087–3113 CrossRef CAS.
- K. Li, G. Cheng, C. Ma, X. Guan, W.-M. Kwok, Y. Chen, W. Lu and C.-M. Che, Chem. Sci., 2013, 4, 2630–2644 RSC.
- D.-L. Ma, H.-Z. He, K.-H. Leung, D. S.-H. Chan and C.-H. Leung, Angew. Chem., Int. Ed., 2013, 52, 7666–7682 CrossRef CAS PubMed.
- K. K.-W. Lo and S. P.-Y. Li, in Molecular Design and Applications of Photofunctional Polymers and Materials, 2012, vol. 2, pp. 130–198 Search PubMed.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- P. K. M. Siu, S.-W. Lai, W. Lu, N. Zhu and C.-M. Che, Eur. J. Inorg. Chem., 2003, 2749–2752 CrossRef CAS.
- V. Guerchais and J.-L. Fillaut, Coord. Chem. Rev., 2011, 255, 2448–2457 CrossRef CAS.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- K. K.-W. Lo, A. W.-T. Choi and W. H.-T. Law, Dalton Trans., 2012, 41, 6021–6047 RSC.
- Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568–1600 RSC.
- H.-B. Xu, L.-X. Shi, E. Ma, L.-Y. Zhang, Q.-H. Wei and Z.-C. Chen, Chem. Commun., 2006, 1601–1603 RSC.
- T. K. Ronson, T. Lazarides, H. Adams, S. J. A. Pope, D. Sykes, S. Faulkner, S. J. Coles, M. B. Hursthouse, W. Clegg, R. W. Harrington and M. D. Ward, Chem. – Eur. J., 2006, 12, 9299–9313 CrossRef CAS PubMed.
- W. T. Eckenhoff and R. Eisenberg, Dalton Trans., 2012, 41, 13004–13021 RSC.
- J. A. G. Williams, Top. Curr. Chem., 2007, 281, 205–268 CrossRef CAS.
- L. J. Rothberg and A. J. Lovinger, J. Mater. Res., 1996, 11, 3174–3187 CrossRef CAS.
- H. Yersin, Top. Curr. Chem., 2004, 241, 1–26 CrossRef CAS.
- P.-T. Chou, Y. Chi, M.-W. Chung and C.-C. Lin, Coord. Chem. Rev., 2011, 255, 2653–2665 CrossRef CAS.
- J. Schneider, P. Du, P. Jarosz, T. Lazarides, X. Wang, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2009, 48, 4306–4316 CrossRef CAS PubMed.
- J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 CrossRef CAS PubMed.
- W. J. Brooks, V. Babayan, S. Lemansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef PubMed.
- S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Inorg. Chem., 2005, 44, 9690–9703 CrossRef CAS PubMed.
- S.-W. Lai, M. C.-W. Chan, T.-C. Cheung, S.-M. Peng and C.-M. Che, Inorg. Chem., 1999, 38, 4046–4055 CrossRef CAS.
- J. H. K. Yip, Suwarno and J. J. Vittal, Inorg. Chem., 2000, 39, 3537–3543 CrossRef CAS PubMed.
- S.-W. Lai and C.-M. Che, Top. Curr. Chem., 2004, 241, 27–63 CrossRef CAS.
- A. S. Ionkin, W. J. Marshall and Y. Wang, Organometallics, 2005, 24, 619–627 CrossRef CAS.
- S. C. F. Kui, I. H. T. Sham, C. C. C. Cheung, C.-W. Ma, B. Yan, N. Zhu, C.-M. Che and W.-F. Fu, Chem. – Eur. J., 2007, 7, 417–435 CrossRef PubMed.
- J. Schneider, P. Du, X. Wang, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2009, 48, 1498–1506 CrossRef CAS PubMed.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- Z. Wang, E. Turner, V. Mahoney, S. Madakuni, T. Groy and J. Li, Inorg. Chem., 2010, 49, 11276–11286 CrossRef CAS PubMed.
- W. Wu, H. Guo, S. Ji and J. Zhao, Inorg. Chem., 2011, 50, 11446–11460 CrossRef CAS PubMed.
- D. P. Rillema, A. J. Cruz, C. Moore, K. Siam, A. Jehan, D. Base, T. Nguyen and W. Huang, Inorg. Chem., 2012, 52, 596–607 CrossRef PubMed.
- A. Tronnier, A. Risler, N. Langer, G. Wagenblast, I. Münster and T. Strassner, Organometallics, 2012, 31, 7447–7452 CrossRef CAS.
- H. Uesugi, T. Tsukuda, K. Takao and T. Tsubomura, Dalton Trans., 2013, 42, 7396–7403 RSC.
- J. Moussa, T. Cheminel, G. R. Freeman, L.-M. Chamoreau, J. A. G. Williams and H. Amouri, Dalton Trans., 2014, 43, 8162–8165 RSC.
- W. Wu, W. Wu, S. Ji, H. Guo, P. Song, K. Han, L. Chi, J. Shao and J. Zhao, J. Mater. Chem., 2010, 20, 9775–9786 RSC.
- Y. Zhang, J. A. Garg, C. Michelin, T. Fox, O. Blacque and K. Venkatesan, Inorg. Chem., 2011, 50, 1220–1228 CrossRef CAS PubMed.
- Y. Zhang, J. Clavadetscher, M. Bachmann, O. Blacque and K. Venkatesan, Inorg. Chem., 2014, 53, 756–771 CrossRef CAS PubMed.
- Y. Unger, D. Meyer, O. Molt, C. Schildknecht, I. Münster, G. Wagenblast and T. Strassner, Angew. Chem., Int. Ed., 2010, 49, 10214–10216 CrossRef CAS PubMed.
- Y. Unger, A. Zeller, M. A. Taige and T. Strassner, Dalton Trans., 2009, 4786–4794, 10.1039/b900655a.
- W. Lu, M. C. W. Chan, Z. Hui, C.-M. Che, N. Zhu and S.-T. Lee, J. Am. Chem. Soc., 2004, 126, 4958–4971 CrossRef CAS PubMed.
- W. Lu, M. C. W. Chan, N. Zhu, C.-M. Che, C. Li and Z. Hui, J. Am. Chem. Soc., 2004, 126, 7639–7651 CrossRef CAS PubMed.
- C. E. Whittle, J. A. Weinstein, M. W. George and K. S. Schanze, Inorg. Chem., 2001, 40, 4053–4062 CrossRef CAS PubMed.
- M. Hissler, W. B. Connick, D. K. Geiger, J. E. McGarrah, D. Lipa, R. J. Lachiotte and R. Eisenberg, Inorg. Chem., 2000, 39, 447–457 CrossRef CAS PubMed.
- T. J. Wadas, R. J. Lachiotte and R. Eisenberg, Inorg. Chem., 2003, 42, 3772–3778 CrossRef CAS PubMed.
- K.-Y. Kim, S. Liu, M. E. Köse and K. S. Schanze, Inorg. Chem., 2006, 45, 2509–2519 CrossRef CAS PubMed.
- F. N. Castellano, I. E. Pomestchenko, E. Shikhova, F. Hua, M. L. Muro and N. Rajapakse, Coord. Chem. Rev., 2006, 250, 1819–1828 CrossRef CAS.
- C.-H. Tao and V. W.-W. Yam, J. Photochem. Photobiol., C, 2009, 10, 130–140 CrossRef CAS.
- R. Saha, M. A. Qaium, D. Debnath, M. Younus, N. Chawdhury, N. Sultana, G. Kociok-Kohn, L.-l. Ooi, P. R. Raithby and M. Kijima, Dalton Trans., 2005, 2760–2765, 10.1039/b505484b.
- W. Wu, J. Zhao, J. Sun, L. Huang and X. Yi, J. Mater. Chem. C, 2013, 1, 705–716 RSC.
- I. Fratoddi, C. Battocchio, A. Furlani, P. Mataloni, G. Polzonetti and M. V. Russo, J. Organomet. Chem., 2003, 674, 10–23 CrossRef CAS.
- K. Gagnon, S. M. Aly, A. Brisach-Wittmeyer, D. Bellows, F.-F. Bérubé, L. Caron, A. S. Abd-El-Aziz, D. Fortin and P. D. Harvey, Organometallics, 2008, 27, 2201–2214 CrossRef CAS.
- L. Liu, D. Fortin and P. D. Harvey, Inorg. Chem., 2009, 48, 5891–5900 CrossRef CAS PubMed.
- D. Bellows, S. M. Aly, C. P. Gros, M. El Ojaimi, J.-M. Barbe, R. Guilard and P. D. Harvey, Inorg. Chem., 2009, 48, 7613–7629 CrossRef CAS PubMed.
- M. S. Khan, M. R. A. A. Al-Mandhary, M. K. Al-Suti, F. R. Al-Battashi, S. Al-Saadi, B. Ahrens, J. K. Bjernemose, M. F. Mahon, P. R. Raithby, M. Younus, N. Chawdhury, A. Köhler, E. A. Marseglia, E. Tedesco, N. Feeder and S. J. Teat, Dalton Trans., 2004, 2377–2385 RSC.
- N. J. Long, C. K. Wong and A. J. P. White, Organometallics, 2006, 25, 2525–2532 CrossRef CAS.
- L. Liu, W.-W. Wong, J.-X. Shi, K.-W. Cheah, T.-H. Lee and L. M. Leung, J. Organomet. Chem., 2006, 691, 4028–4041 CrossRef CAS.
- D. Fortin, S. Clément, K. Gagnon, J.-F. Bérubé, M. P. Stewart, W. E. Geiger and P. D. Harvey, Inorg. Chem., 2009, 48, 446–454 CrossRef CAS PubMed.
- J. M. Keller, K. D. Glusac, E. O. Danilov, S. McIlroy, P. Sreearuothai, A. R. Cook, H. Jiang, J. R. Miller and K. S. Schanze, J. Am. Chem. Soc., 2011, 133, 11289–11298 CrossRef CAS PubMed.
- J. R. Berenguer, E. Lalinde and M. Teresa Moreno, Coord. Chem. Rev., 2010, 254, 832–875 CrossRef CAS.
- J. E. Rogers, J. E. Slagle, D. M. Krein, A. R. Burke, B. C. Hall, A. Fratini, D. G. McLean, P. A. Fleitz, T. M. Cooper, M. Drobizhev, N. S. Makarov, A. Rebane, K.-Y. Kim, R. Farley and K. S. Schanze, Inorg. Chem., 2007, 46, 6483–6494 CrossRef CAS PubMed.
- A. A. Rachford, S. Goeb and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 2766–2767 CrossRef CAS PubMed.
- L. Liu, D. Huang, S. M. Draper, X. Yi, W. Wu and J. Zhao, Dalton Trans., 2013, 42, 10694–10706 RSC.
- H. Guo, M. L. Muro-Small, S. Ji, J. Zhao and F. N. Castellano, Inorg. Chem., 2010, 49, 6802–6804 CrossRef CAS PubMed.
- M.-H. Nguyen and J. H. K. Yip, Organometallics, 2012, 31, 7522–7531 CrossRef CAS.
- M.-H. Nguyen, V. H. Nguyen and J. H. K. Yip, Organometallics, 2013, 32, 7283–7291 CrossRef CAS.
- D. N. Kozhevnikov, V. N. Kozhevnikov, M. Z. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. Gareth Williams, Inorg. Chem., 2011, 50, 3804–3815 CrossRef CAS PubMed.
- Y. Liu, H. Guo and J. Zhao, Chem. Commun., 2011, 47, 11471–11473 RSC.
- Y. Y. Chia and M. G. Tay, Dalton Trans., 2014, 43, 13159–13168 RSC.
- J. Hu, R. Lin, J. H. K. Yip, K.-Y. Wong, D.-L. Ma and J. J. Vittal, Organometallics, 2007, 26, 6533–6543 CrossRef CAS.
- A. Singh and P. R. Sharp, J. Am. Chem. Soc., 2006, 128, 5998–5999 CrossRef CAS PubMed.
- H. B. Lee and P. R. Sharp, Organometallics, 2005, 24, 4875–4877 CrossRef CAS.
- S. Lentijo, G. Aullon, J. A. Miguel and P. Espinet, Dalton Trans., 2013, 42, 6353–6365 RSC.
- M.-H. Nguyen and J. H. K. Yip, Organometallics, 2010, 29, 2422–2429 CrossRef CAS.
- W. Y. Heng, J. Hu and J. H. K. Yip, Organometallics, 2007, 26, 6760–6768 CrossRef CAS.
- B.-Y. Wang, A. R. Karikachery, Y. Li, A. Singh, H. B. Lee, W. Sun and P. R. Sharp, J. Am. Chem. Soc., 2009, 131, 3150–3151 CrossRef CAS PubMed.
- S. Lentijo, J. A. Miguel and P. Espinet, Inorg. Chem., 2010, 49, 9169–9177 CrossRef CAS PubMed.
- W. G. Herkstroeter, A. A. Lamola and G. S. Hammond, J. Am. Chem. Soc., 1964, 86, 4537–4540 CrossRef CAS.
- D. E. Damschen, C. D. Merritt, D. L. Perry, G. W. Scott and L. D. Talley, J. Phys. Chem., 1978, 82, 2268–2272 CrossRef CAS.
- R. W. Anderson Jr., R. M. Hochstrasser, H. Lutz and G. W. Scott, Chem. Phys. Lett., 1974, 28, 153–157 CrossRef.
- R. M. Hochstrasser, H. Lutz and G. W. Scott, Chem. Phys. Lett., 1974, 24, 162–167 CrossRef.
- D. Wöll, J. Smirnova, W. Pfleiderer and U. E. Steiner, Angew. Chem., Int. Ed., 2006, 45, 2975–2978 CrossRef PubMed.
- D. Wöll, S. Laimgruber, M. Galetskaya, J. Smirnova, W. Pfleiderer, B. Heinz, P. Gilch and U. E. Steiner, J. Am. Chem. Soc., 2007, 129, 12148–12158 CrossRef PubMed.
- G. Amirzadeh and W. Schnabel, Makromol. Chem., 1981, 182, 2821–2835 CrossRef CAS.
- M. G. Neumann, M. H. Gehlen, M. V. Encinas, N. S. Allen, T. Corrales, C. Peinado and F. Catalina, J. Chem. Soc., Faraday Trans., 1997, 93, 1517–1521 RSC.
- M. Aydin, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2005, 38, 4133–4138 CrossRef CAS.
- L. Cokbaglan, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2003, 36, 2649–2653 CrossRef CAS.
- T. Corrales, C. Peinado, F. Catalina, M. G. Neumann, N. S. Allen, A. M. Rufs and M. V. Encinas, Polymer, 2000, 41, 9103–9109 CrossRef CAS.
- X. Jiang and J. Yin, Macromol. Chem. Phys., 2008, 209, 1593–1600 CrossRef CAS.
- H. Tar, D. Sevinc Esen, M. Aydin, C. Ley, N. Arsu and X. Allonas, Macromolecules, 2013, 46, 3266–3272 CrossRef CAS.
- J. C. Dalton and F. C. Montgomery, J. Am. Chem. Soc., 1974, 96, 6230–6232 CrossRef CAS.
- C. Ley, F. Morlet-Savary, P. Jacques and J. P. Fouassier, Chem. Phys., 2000, 255, 335–346 CrossRef CAS.
- T.-i. Lai and E. C. Lim, Chem. Phys. Lett., 1980, 73, 244–248 CrossRef CAS.
- X. Allonas, C. Ley, C. Bibaut, P. Jacques and J. P. Fouassier, Chem. Phys. Lett., 2000, 322, 483–490 CrossRef CAS.
- P. K. Monaghan and R. J. Puddephatt, Organometallics, 1985, 4, 1406–1412 CrossRef CAS.
- A. Abo-Amer, M. S. McCready, F. Zhang and R. J. Puddephatt, Can. J. Chem., 2011, 90, 46–54 CrossRef.
- G. W. Parshall, J. Am. Chem. Soc., 1966, 88, 704–708 CrossRef CAS.
- A. Pidcock, R. E. Richards and L. M. Venanzi, J. Chem. Soc. A, 1966, 1707–1710, 10.1039/J19660001707.
- J. A. Pople and D. P. Santry, Mol. Phys., 1964, 8, 1–18 CrossRef CAS.
- T. Sajoto, P. I. Djurovich, A. Tamayo, M. Yousufuddin, R. Bau, M. E. Thompson, R. J. Holmes and S. R. Forrest, Inorg. Chem., 2005, 44, 7992–8003 CrossRef CAS PubMed.
- S. K. Doorn, R. B. Dyer, P. O. Stoutland and W. H. Woodruff, J. Am. Chem. Soc., 1993, 115, 6398–6405 CrossRef CAS.
- D. Burget and P. Jacques, J. Lumin., 1992, 54, 177–181 CrossRef CAS.
- D. W. Wöll, PhD Dissertation, University of Konstanz, 2006.
- T. Yoshida, T. Matsuda, S. Otsuka, G. W. Parshall and W. G. Peet, in Inorganic Syntheses, John Wiley & Sons, Inc., 2007, ch. 32, pp. 122–123 DOI:10.1002/9780470132593.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Solution and Refinement, Universität Göttingen, 1997 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2013, 45, 849–854 CrossRef.
- M. J. Frisch, G. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Blonio, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09, Version 8.0, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- O. Gunnarsson and B. I. Lundqvist, Phys. Rev. B: Solid State, 1976, 13, 4274 CrossRef CAS.
- W. Küchle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7532 CrossRef.
- M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1989, 90, 1730–1734 CrossRef CAS.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- P. H. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Enzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- E. Cancés, B. Menucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- G. Scalamani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110–114124 CrossRef PubMed.
- E. Runge and K. U. G. E, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- T. Keith and J. Millam, GaussView, Version 3, Semichem Inc., Shawnee Mission KS, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Depictions of the 1H, 13C{1H}, 31P{1H} and 195Pt NMR spectra of the complexes; crystal and refinement data as well as ORTEPs and packing diagrams of complexes Pt–Cl and Pt–CN, graphical representations of exemplary cyclic voltammograms and electronic absorption and emission spectra, Tables with structure parameters for the optimised geometries of the complexes, energies and graphical representations of the crucial MOs of the complexes and TD-DFT calculated transitions. CCDC 1017997 (Pt–Cl) and 1017999 (Pt–CN). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt02410a |
| This journal is © The Royal Society of Chemistry 2015 |