
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lithiated sulfoxides: α-sulfinyl functionalized carbanions†
Gerd
Ludwig
a,
Tobias
Rüffer
b,
André
Hoppe
a,
Till
Walther
a,
Heinrich
Lang
b,
Stefan G.
Ebbinghaus
a and
Dirk
Steinborn
*a
aInstitute of Chemistry, Martin Luther University Halle-Wittenberg, Kurt-Mothes-Straße 2, D-06120 Halle, Germany. E-mail: dirk.steinborn@chemie.uni-halle.de
bInstitute of Chemistry, Chemnitz University of Technology, Straße der Nationen 62, D-09107 Chemnitz, Germany
First published on 26th September 2014
Abstract
Reactions of alkyl aryl sulfoxides H–CRR′S(O)Ar with n-BuLi–TMEDA (TMEDA = N,N,N′,N′-tetramethylethylenediamine) afforded α-sulfinyl functionalized alkyl aryl lithium compounds of the type [Li2{CRR′S(O)Ar}2(TMEDA)2] (1, R/R′ = H/H, Ar = Ph; 2, R/R′ = H/H, Ar = p-Tol; 3, R/R′ = Me/Me, Ar = Ph; 4, R/R′ = H/Ph, Ar = Ph; 5, R/R′ = Me/Ph, Ar = Ph). The compounds were characterized by 1H, 13C and 7Li NMR spectroscopy and, except for 5, by single-crystal X-ray diffraction analyses. In crystals of 1, 2, 3 and 4·Et2O dinuclear molecules with four-membered Li2O2 rings were found. There are no Li⋯Cα contacts, thus, “free” carbanions are the main structural feature. Reactions of 1–6 (6, R/R′ = H/Me, Ar = Ph) with benzaldehyde and benzophenone afforded the corresponding sulfoxides of the type ArS(O)CRR′CHPhOH (1a–6a) and ArS(O)CRR′CPh2OH (1b–6b), respectively. The reactions yielding 1a/3a and 4b/6b proceeded with high diastereoselectivities. By X-ray diffractometry it has been shown that in the case of 3a and 4b the diastereomers consisting of the two enantiomers SSRC and RSSC were formed.
Introduction
Sulfur is one of the most employed elements in organic synthesis1–4 and three typical sulfur containing functionalities are sulfides, sulfoxides and sulfones.5–7 An important property of the sulfur atom is its ability to activate an α-hydrogen atom of an attached alkyl group, with an increased activation in the order –S– < –S(O)– < –S(O)2–. Thus, sulfanyl-, sulfinyl-, and sulfonyl alkyl carbanions can be readily generated by lithiation of the corresponding sulfides, sulfoxides and sulfones, respectively.8–11 α-Sulfinyl functionalized alkyl aryl lithium compounds of the type Li[CRR′S(O)Ar] are of special interest, because on the one hand they possess a Lewis-basic heteroatom (nonbonding electron pair at the sulfur atom) and on the other hand a dipole stabilized heteroatomic center.12 Generally, sulfinyl functionalized alkyl aryl lithium compounds are good synthons for enantioselective and diastereoselective syntheses.13–17 Thus, Durst et al. found that methylation and deuteration of lithiated sulfoxides proceeded with good diastereoselectivities.18,19 On the basis of these and further findings,20–31 it was stated that the diastereofacial differentiation in reactions of α-sulfinyl functionalized alkyl aryl lithium compounds is determined by the chiral sulfinyl group as follows: electrophiles with an oxygen-containing group like D2O, benzaldehyde, benzophenone or CO2 tend to attack the anionic C atom on the side of the S–O bond due to an attractive interaction of the electrophile with the countercation Li+. In the case of CH3I the electrophile approaches from the opposite side, because of the lack of an attractive interaction with the countercation Li+ as shown in Fig. 1.32,33 On that basis and under consideration of quantum chemical calculations of Wolfe et al., the “ion-pair model” for α-sulfinyl functionalized alkyl aryl lithium compounds of the type Li[CRR′S(O)Ar] was established (cf.Fig. 1).7,34,35 The first X-ray crystal structure analysis of a lithiated sulfoxide, [Li2{CMePhS(O)Ph}2(TMEDA)2], was reported by Boche et al. in 1986.36 The lithium compound crystallized in a dimer fashion with a Li2O2 four-membered ring as main structural feature. Furthermore, this lithium compound is characterized by a “free” carbanion, meaning that there are no interactions between Li+ and the carbanionic C atom. Thus, the formerly postulated “ion-pair model” was found to be in accordance with the crystal structure determination of Boche et al. Since then no further crystal structure analyses of lithiated sulfoxides have been described. Here, we report on the synthesis, characterization and solid state structures of lithiated sulfoxides of the type [Li2{CRR′S(O)Ar}2(TMEDA)2]. Furthermore, the stereoselective C–C bond formation of the lithiated sulfoxides with benzaldehyde and benzophenone was investigated under the aspect of a diastereofacial differentiation.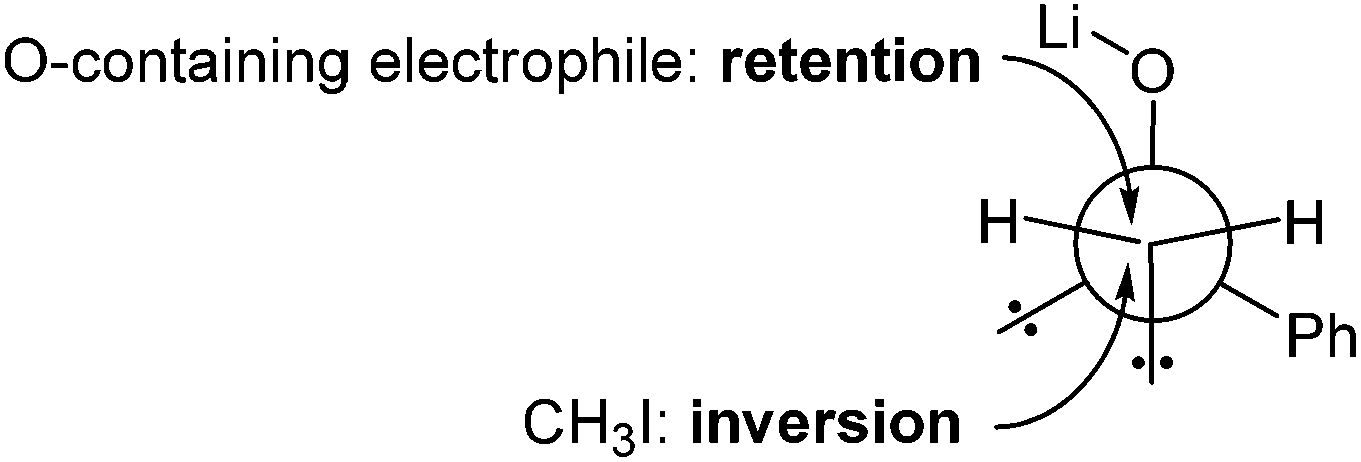 | ||
| Fig. 1 Diastereofacial differentiation for reactions of lithiated sulfoxides (S atom is hidden by the α-C atom) with electrophiles according to the “ion-pair model” (adapted from ref. 7). | ||
Results and discussion
Synthesis of lithiated alkyl aryl sulfoxides
Lithiated alkyl aryl sulfoxides of the type [Li2{CRR′S(O)Ar}2(TMEDA)2] (1–5) were prepared by metallation of the corresponding sulfoxide with n-BuLi/TMEDA in diethyl ether/n-pentane according to Scheme 1. The products were obtained as strongly moisture- and oxygen-sensitive yellowish crystals in yields between 61 and 84%. All complexes were characterized by NMR spectroscopy (1H, 13C, 7Li) and, with exception of 5, by single-crystal X-ray structure analyses. Selected NMR spectroscopic parameters of complexes 1–5 are given in Table 1. The 1H and 13C NMR spectra of 1–5 give proof of the selective metallation of the α-C atoms of the alkyl aryl sulfoxides. Thus, an ortho-metallation of the aryl rings can be fully excluded. The 13C chemical shifts of the α-C atoms are in direct relation to their substitution pattern: the resonances of the unsubstituted α-C atoms in 1 and 2 were found at about 35 ppm, whereas those of the substituted ones (3–5) are located between 54 and 65 ppm. On the other hand, in all lithiated compounds 1–5 the 7Li resonances are in a narrow range (–0.25 to 0.55 ppm). The virtually identical TMEDA resonances in 1–5 might indicate that in solution the TMEDA ligand is partially cleaved off (dynamic coordination/decoordination) as also observed in other cases.37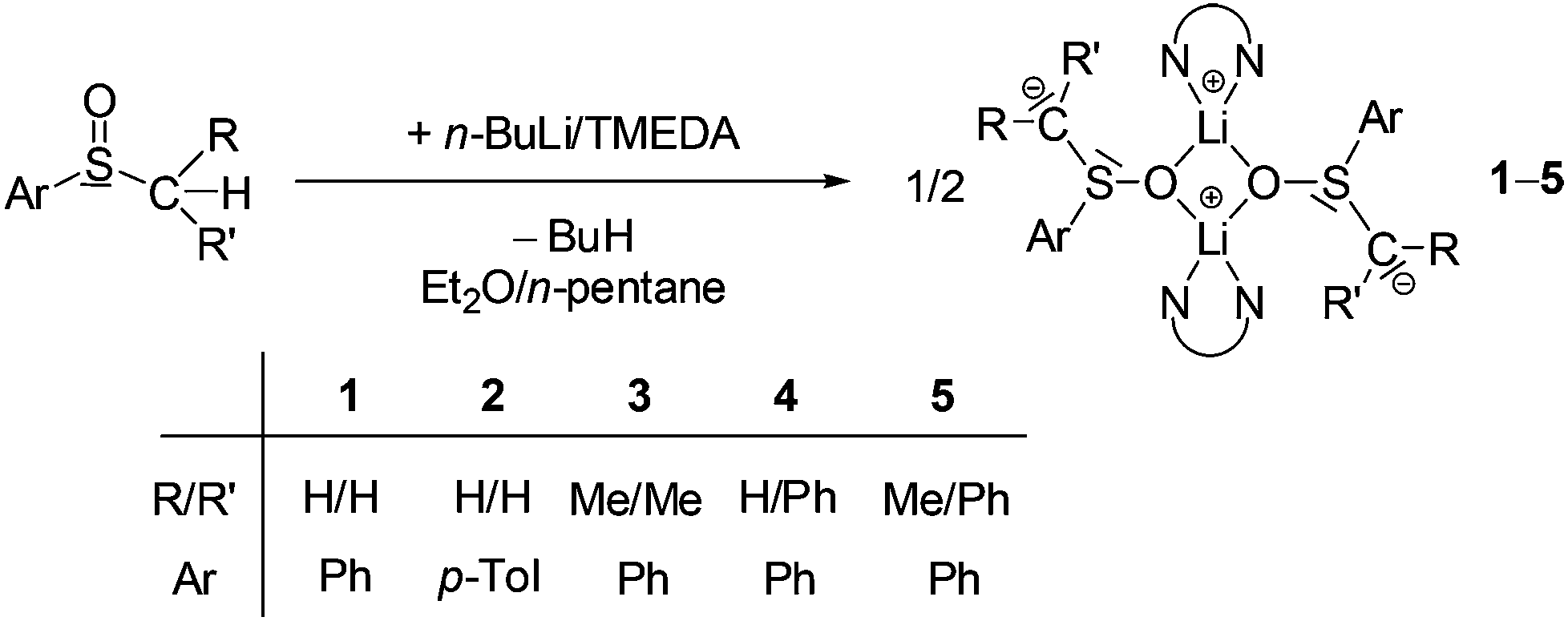 | ||
| Scheme 1 Synthesis of lithiated sulfoxides [Li2{CRR′S(O)Ar}2(TMEDA)2](1–5). | ||
| R/R′ | Ar | δ α-H | δ α-C | δ Li | |
|---|---|---|---|---|---|
| 1 | H/H | Ph | 1.96 | 34.7 | 0.10 |
| 2 | H/H | p-Tol | 1.88 | 36.3 | 0.55 |
| 3 | Me/Me | Ph | — | 53.8 | −0.10 |
| 4 | H/Ph | Ph | 3.77 | 65.3 | −0.25 |
| 5 | Me/Ph | Ph | — | 62.2 | −0.20 |
Solid-state structures of lithiated alkyl aryl sulfoxides
Crystals of [Li2{CH2S(O)Ph}2(TMEDA)2] (1), [Li2{CH2S(O)p-Tol}2(TMEDA)2] (2), [Li2{CMe2S(O)Ph}2(TMEDA)2] (3) and [Li2{CHPhS(O)Ph}2(TMEDA)2]·Et2O (4·Et2O) suitable for X-ray diffraction analyses were obtained from diethyl ether solutions at −7 °C. All compounds crystallized as isolated dinuclear molecules without unusual intermolecular interactions (shortest distance between non-hydrogen atoms: 3.606(2) Å, C14⋯C17′, 1; 3.003(6) Å, C19⋯C9′, 2; 3.646(3) Å, C9⋯C9′, 3; 3.362(2) Å, C32⋯C14B′, 4·Et2O). The asymmetric unit of 1 contains two symmetrically independent molecules with very similar structures. The dinuclear compound 3 exhibits crystallographically imposed C2 symmetry. The molecular structures are presented in Fig. 2–5. Selected structural parameters are given in the respective figure captions, although, in respect of the quality of the structure solutions, this discussion must not be exaggerated.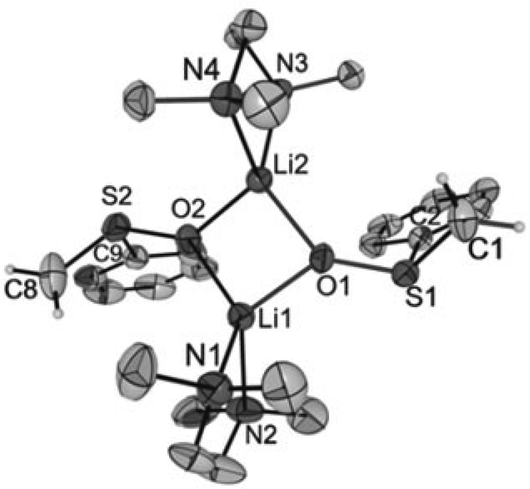 | ||
| Fig. 2 Structure of one of the two symmetrically independent molecules [Li2{CH2S(O)Ph}2(TMEDA)2] in crystals of 1 (thermal ellipsoids at 30%; only the major occupied position of disordered atoms is displayed). For reasons of clarity, only the hydrogen atoms of the α-C atoms are shown. Selected distances (in Å) and angles (in °) (values of the two symmetrically independent molecules are given separated by a slash): Li1–O1 1.878(8)/1.909(8), Li2–O2 1.882(8)/1.900(8), Li1⋯Li2 2.614(1)/2.634(1), S1–O1 1.576(3)/1.568(3), S2–O2 1.574(3)/1.573(3), S1–C1 1.660(5)/1.611(8), S2–C8 1.606(7)/1.645(6), S1–C2 1.809(5)/1.811(5), S2–C9 1.817(5)/1.819(5), Li1⋯C8 3.775(1)/3.907(1), Li2⋯C1 3.889(9)/3.898(1), O1–S1–C1 116.9(3)/118.3(4), O2–S2–C8 117.9(3)/117.3(3), O1–S1–C2 98.6(2)/99.0(2), O2–S2–C9 97.9(2)/98.0(2), C1–S1–C2 101.3(2)/101.6(4), C8–S2–C9 102.0(3)/102.1(3), O1–Li1–O2 90.9(3)/90.8(3), O1–Li1⋯Li2–O2 159.1(5)/160.0(5). | ||
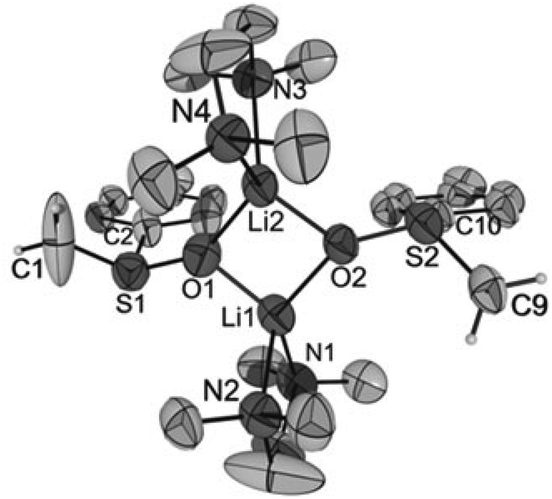 | ||
| Fig. 3 Molecular structure of [Li2{CH2S(O)p-Tol}2(TMEDA)2] in crystals of 2 (thermal ellipsoids at 30%; only the major occupied position of disordered atoms is displayed). For reasons of clarity, only the hydrogen atoms of the α-C atoms are shown. Selected distances (in Å) and angles (in °): Li1–O1 1.843(1), Li2–O2 1.878(9), Li1⋯Li2 2.558(1), S1–O1 1.583(4), S2–O2 1.581(4), S1–C1 1.572(1), S2–C9 1.665(7), S1–C2 1.844(4), S2–C10 1.821(3), Li1⋯C9 4.007(1), Li2⋯C1 4.039(2), O1–S1–C1 120.9(7), O1–S1–C2 97.2(2), C1–S1–C2 103.4(6), O1–Li1–O2 93.9(5), O1–Li1⋯Li2–O2 163.9(6). | ||
 | ||
| Fig. 4 Molecular structure of [Li2{CMe2S(O)Ph}2(TMEDA)2] in crystals of 3 (thermal ellipsoids at 30%). H atoms have been omitted for clarity. Selected distances (in Å) and angles (in °): C1–C2 1.499(3), C1–C3 1.499(3), Li1–O1 1.890(4), Li1⋯Li1′ 2.609(7), S1–O1 1.583(1), S1–C1 1.646(2), S1–C4 1.809(2), Li1⋯C1 4.144(4), S1–C1–C2 115.2(1), S1–C1–C3 123.5(1), C2–C1–C3 116.4(2), O1–S1–C1 119.6(9), O1–S1–C4 99.0(8), C1–S1–C4 102.4(1), O1–Li1–O1′ 92.0(2), O1–Li1⋯Li1′–O1′ 162.4(2). | ||
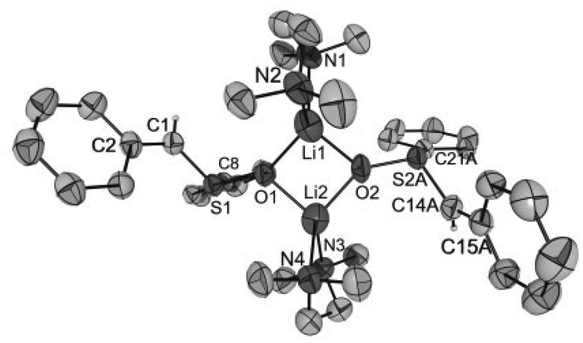 | ||
| Fig. 5 Molecular structure of [Li2{CHPhS(O)Ph}2(TMEDA)2] (4) in crystals of 4·Et2O (thermal ellipsoids at 30%; only the major occupied position of disordered atoms is displayed). For reasons of clarity, only the hydrogen atoms of the α-C atoms are shown. Selected distances (in Å) and angles (in °): C1–C2 1.447(4), C14A–C15A 1.448(7), Li1–O1 1.903(6), Li1–O2 1.902(6), Li1⋯Li2 2.646(8), S1–O1 1.565(2), S2A–O2 1.573(2), S1–C1 1.641(3), S2A–C14A 1.666(5), S1–C8 1.795(3), S2A–C21A 1.826(5), Li1⋯C1 3.888(6), Li2⋯C14A 3.769(7), S1–C1–C2 122.2(2), S2A–C15A–C14A 123.3(4), O1–S1–C1 115.7(1), O1–S1–C8 100.8(1), C1–S1–C8 102.3(1), O1–Li1–O2 91.0(3), O1–Li1⋯Li2–O2 162.7(3). | ||
As for the structure described by Boche,36 the central building blocks of the dinuclear molecules of 1–4 are four-membered Li2O2 rings with Li–O bond lengths between 1.843(1) Å and 1.917(9) Å and Li1⋯Li2 (respectively, Li1⋯Li1′ for 3) distances between 2.558(1) and 2.646(8) Å. The dihedral angles O1–Li1⋯Li2–O2 (respectively, O1–Li1⋯Li1′–O1′ for 3) between 159.1(5)° and 163.9(6)° show that the rings are folded. Thus, the Li2O2 rings in 1–4 are structurally similar to those in other lithium compounds (Li–O: median 1.917 Å, lower/upper quartile 1.851/1.984 Å; Li⋯Li′: median 2.607 Å, lower/upper quartile 2.481/2.759 Å; O–Li⋯Li′–O′: median 170.5°, lower/upper quartile 163.4/174.9°, n = 398).38 Two nitrogen atoms of the chelating TMEDA ligands complete the distorted coordination tetrahedrons around the lithium atoms. The shortest Li⋯Cα distance (3.769(7) Å) was found in 4·Et2O. Thus, in no compound is an evidence for an intramolecular interaction between Li+ and the carbanionic α-C atom.
For a more detailed description of the molecular structures of the dinuclear lithium compounds 1–4 quantum-chemical calculations on the DFT level of theory were performed using the B3LYP functional and high-quality basis sets for all atoms (details see Experimental). In all calculations solvent effects (THF) were considered according to Tomasi's polarized continuum model.39–41 Comparison of the solid-state structures [Li2{CRR′S(O)Ar}2(TMEDA)2] (1–4) with the analogous calculated structures 1*–4*‡ revealed a good agreement including the conformation of the Li2O2 rings and the Li⋯Cα distances (cf. Table S1†). Of special interest is the degree of pyramidalization of the carbanionic α-C atoms, which can be evaluated by the sum of angles around the α-C atom and its deviation Δd from the plane spanned by its substituents (S, C/H, C/H) (cf. Table S2†). For 1*–3* (R/R′ = H/H, Me/Me) slightly pyramidalized α-C atoms were found (sum of angles: 347.0–354.7°; Δd = 0.210–0.260 Å), as also found in crystals of 3 (sum of angles: 355.0°; Δd = 0.208 Å). On the other hand, the sulfinyl substituted benzyl carbanion in 4* (R/R′ = H/Ph) is almost trigonal-planar (sum of angles: 359.5°; Δd = 0.059 Å), as also in the compound with R/R′ = Me/Ph (sum of angles: 357.0°; Δd = 0.120 Å).36 This planarization can be traced back to a π bond between the p orbitals of the carbanions and the phenyl rings. Similar conclusions were drawn from 13C NMR spectroscopic data of Li[CHPhS(O)Me], Li[CHPhS(O)t-Bu] and Li[CHPhS(O)2t-Bu].42,43
Furthermore, in all compounds the S–Cα bond lengths, both the experimental and the calculated values, were found to be significantly shorter (1.572(1)–1.666(5)/1.677–1.687 Å, 1–4/1*–4*) than the S–Ci bond lengths (1.795(3)–1.844(4)/1.826–1.834 Å, 1–4/1*–4*), albeit the Ci atoms are clearly sp2 hybridized. An analysis of the Wiberg bond indices (WBI) of the S–C bonds in the lithiated sulfoxides 1*–4* and the corresponding nonlithiated (neutral) sulfoxides H–CRR′S(O)Ar exhibited – as expected – extraordinarily strong S–Cα bonds in 1*–4* (WBI = 1.24–1.35) compared to those in the neutral sulfoxides (WBI = 0.85–0.91) and also to the S–Ci bonds (1*–4*: WBI = 0.89–0.91; neutral sulfoxides: WBI = 0.90), cf. Table S3.† The strengthening of the S–Cα bonds can be attributed to a stabilizing interaction between the nonbonding orbital on the α-carbon atoms and the S–O antibonding σ* orbital in the sense of a negative hyperconjugation.31,44 The negative hyperconjugation can be reasonably assumed because the nonbonding electron pairs at the α-C atoms are bent only 16.4 to 18.4° away from the antiperiplanar position to the S–O bond (cf. Table S2†).
Reactions of lithiated alkyl aryl sulfoxides with benzaldehyde and benzophenone
Reactions of the racemic lithiated sulfoxides 1–5 and of [Li2{CHMeS(O)Ph}2(TMEDA)2] (6; prepared as described above but without isolation) with benzaldehyde and benzophenone generated the corresponding sulfoxides of the type ArS(O)CRR′CHPhOH (1a–6a; Scheme 2, pathway A) and ArS(O)CRR′CPh2OH (1b–6b; Scheme 2, pathway B), respectively. The products were isolated as colorless crystals in yields between 61 and 89% and characterized by NMR spectroscopy (1H, 13C) and, in part, by single-crystal X-ray structure analyses. Most of the products obtained exist as diastereomers; the diastereomeric excess (de) of the reactions ranged between 14 and 94% (cf.Table 2).§ Recrystallization from diethyl ether–n-pentane of the diastereomeric mixtures of 3a (>94% de), 4a (60% de) and 4b (>94% de) resulted in crystals of PhS(O)CMe2CHPhOH (3a′), PhS(O)CHPhCHPhOH (4a′) and PhS(O)CHPhCPh2OH (4b′) suitable for X-ray diffraction analyses. The molecular structures of 3a′, 4a′ and 4b′ are presented in Fig. S1–S3,† selected structural parameters are given in the respective figure captions. In crystals of 3a′ the molecules are connected by intermolecular O2–H⋯O1′ hydrogen bonds (O2⋯O1′ 2.727(1) Å, O2–H⋯O1′ 170°), resulting in formation of six-membered chains (graph set: C(6)).45 In crystals of 4a′ the molecules are connected by O1–H⋯O2′ hydrogen bonds (O1⋯O2′ 2.710(1) Å, O1–H⋯O2′ 170°) such that centrosymmetric dinuclear units are formed (graph set: R22(12)). In contrast to 3a′ and 4a′, in molecules of 4b′ intramolecular O1–H⋯O2 hydrogen bonds (O1⋯O2 2.710(1) Å, O1–H⋯O2 158°) exist thus forming six-membered rings (graph set: S(6)). According to the distance criterion46,47 all these hydrogen bonds can be characterized as moderately strong. Since 3a′, 4a′ and 4b′ crystallize in the centrosymmetric space group P21/c the crystals are racemic and, hence, two enantiomers (SSRC and RSSC, 3a′; RSSCSC and SSRCRC, 4a′; SSRC and RSSC, 4b′) are present.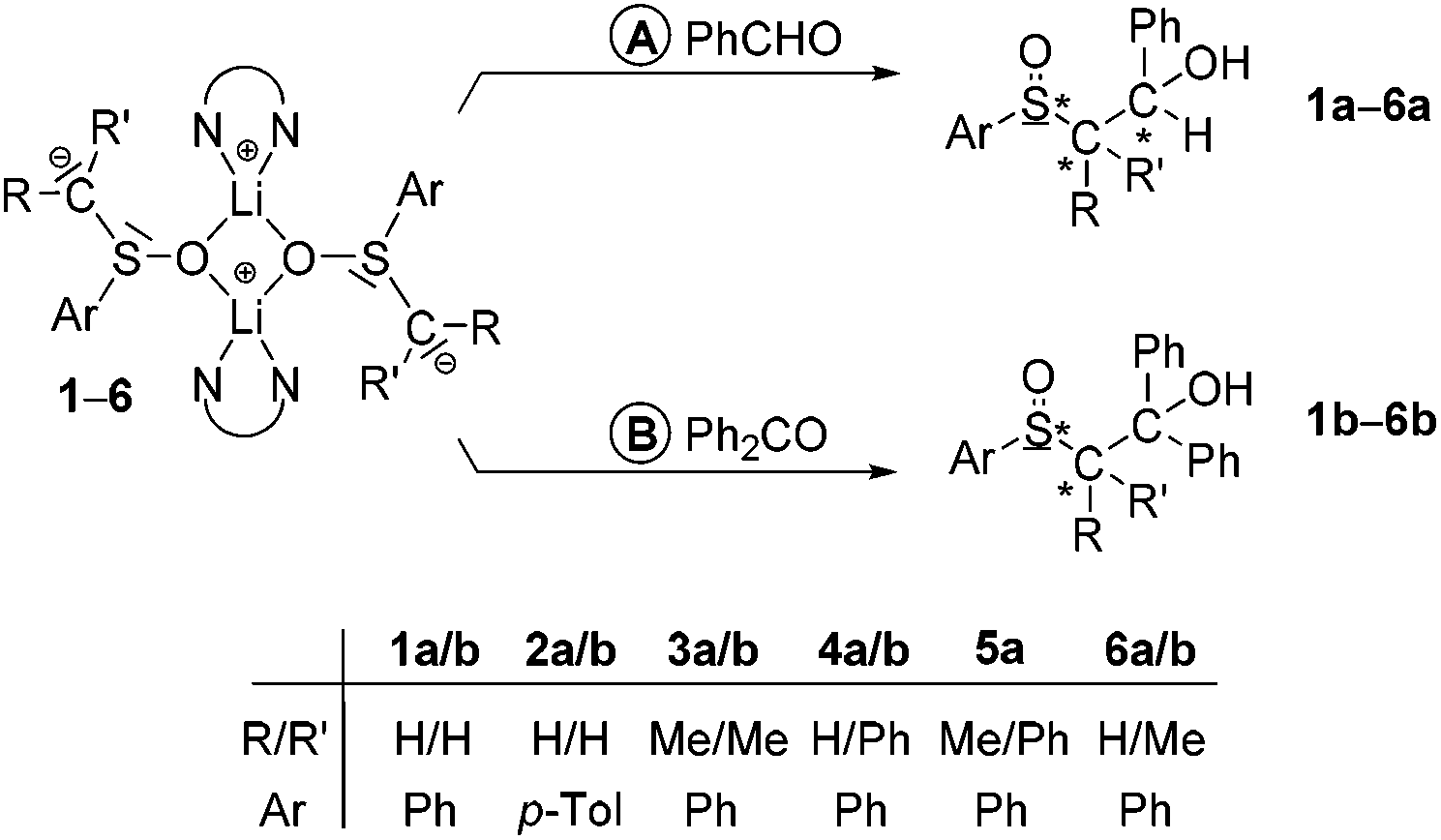 | ||
| Scheme 2 Reaction of lithiated sulfoxides with benzaldehyde (A) and benzophenone (B). | ||
| Benzaldehyde (1a–6a) | Benzophenone (1b–6b) | |||||
|---|---|---|---|---|---|---|
| R/R′ | Ar | no. c.c.a | de in % | no. c.c.a | de in % | |
| a Number of chiral centers in the product. b n/a = not applicable. c For unknown reasons, isolation of 5b as desired product failed. | ||||||
| 1a/b | H/H | Ph | 2 | >94 | 1 | n/ab |
| 2a/b | H/H | p-Tol | 2 | 20 | 1 | n/a |
| 3a/b | Me/Me | Ph | 2 | >94 | 1 | n/a |
| 4a/b | H/Ph | Ph | 3 | 60 | 2 | >94 |
| 5a/b | Me/Ph | Ph | 3 | 14 | 2 | |
| 6a/b | H/Me | Ph | 3 | 62 | 2 | >94 |
About the diastereoselectivity
Regarding the diastereoselection in the asymmetric C–C bond formation of the lithium compounds 1–6 with benzaldehyde and benzophenone, two reaction types have to be distinguished, namely that of an achiral carbanion center with a prochiral electrophile (type I) and vice versa (type II). The following discussion is restricted to the highly diastereoselective reactions where the configuration of the products is known from single-crystal X-ray measurements, see the formation of 3a and 4b for a type I and type II reaction, respectively (Scheme 3). Exemplarily, for 4b it has been shown that the single crystal measured is identical with the bulk material by measuring an X-ray powder diffractogram of it and comparing it with the pattern simulated from the X-ray single crystal data, see Fig. S4.† The nearly perfect match of the two diffractograms gives proof that only the 4b-SSRC/RSSC diastereomer is formed in the reaction. | ||
| Scheme 3 Nucleophilic addition of [Li2{CMe2S(O)Ph}2(TMEDA)2] (3) to the prochiral benzaldehyde (type I) and of [Li2{CHPhS(O)Ph}2(TMEDA)2] (4) to benzophenone (type II), resulting in the formation of diastereomerically pure 3a-SSRC/RSSC and 4b-SSRC/RSSC (shown with the SSRC enantiomers as example), respectively. The six-membered cyclic transition states ts1 and ts2 are explicitly shown. | ||
To understand the preferential formation of the diastereomers (3a-SSRC/RSSC and 4b-SSRC/RSSC) in these reactions, two assumptions have to be made: first, according to the “ion-pair model” (Fig. 1)7 for these two electrophiles (PhCHO and Ph2CO) a precoordination of the carbonyl O atom at the lithium center can be assumed. Second, in accordance with other reactions of lithiated sulfoxides24,48–50 and also with aldol reactions51,52 the formation of six-membered cyclic transition states as given in Scheme 3 can be assumed. Based on the experimental findings, the diastereoselectivities observed in this work can be explained in the following way: the formation of the SSRC- and RSSC-configured products 3a and 4b suggests that the two reactions proceed via the transition state ts2.
Conclusions and summary
Lithiated sulfoxides of the type [Li2{CRR′S(O)Ar}2(TMEDA)2] (1–5) were prepared and their constitution was unequivocally confirmed by NMR and single crystal X-ray diffraction studies. The lithium compounds crystallize in a dimer fashion with Li2O2 four-membered rings and “free” carbanions as main structural features. As additionally revealed by quantum-chemical calculations the carbanionic center is stabilized by a negative hyperconjugation31,44 and remains to be slightly pyramidalized with exception of the benzylic one in 4.C–C bond formation in reactions of 1–3 having an achiral α-C atom with a prochiral electrophile (PhCHO, type I) and of 4 and 6 having a chiral α-C atom with an achiral electrophile (Ph2CO, type II) proved to proceed with high diastereoselectivities (de >94%) with one exception (formation of 2a). In two selected cases (type I: 3 + PhCHO; type II: 4 + Ph2CO) it has been shown that only the SSRC/RSSC diastereomers were formed and not the SSSC/RSRC diastereomers. The stereoselection of analogous reactions (lithiated methyl 1-naphtyl sulfoxide with aromatic ketones) has been traced back to stabilizing π⋯π interactions between the aromatic rings26 which might also be the case in the systems described here. On the other hand, the combination of both a chiral α-C atom and a chiral electrophile led to the formation of three stereogenic centers and to de values between 14 and 62% only.
Thus, the present investigations allow a deeper insight into the molecular structures of lithiated sulfoxides and their reactivity against electrophiles in asymmetric C–C bond formation reactions.
Experimental part
General comments
Organolithium compounds were prepared and handled under purified argon using standard Schlenk techniques. Solvents (diethyl ether, n-pentane, tetrahydrofuran) were dried over Na/benzophenone and freshly distilled prior to use. NMR spectra (1H, 13C, 7Li) were recorded, if not otherwise stated, at 27 °C on Varian Gemini 200 and VXR 400 spectrometers. 1H and 13C chemical shifts are relative to solvent signals (THF-d8, δH 1.72, δC 67.21; CDCl3, δH 7.26, δC 77.16) as internal references; 7Li NMR spectra were referenced to a solution of LiCl in D2O (external). The preparative centrifugally accelerated thin layer chromatography was performed using a Chromatotron (Harrison Research). Sulfoxides were prepared according to literature procedures.531 (R/R′ = H/H, Ar = Ph). Yield: 220 mg (84%). 1H NMR (400 MHz, THF-d8): δ 1.96 (s, br, 2H, LiCH2), 2.16 (s, 12H, 2 × N(CH3)2, TMEDA), 2.31 (s, 4H, 2 × CH2, TMEDA), 7.18–7.22 (m, 1H, p-H, SOPh), 7.28–7.32 (m, 2H, m-H, SOPh), 7.92–7.94 (m, 2H, o-H, SOPh). 13C NMR (100 MHz, THF-d8): δ 34.7 (s, LiCH2), 46.2 (s, N(CH3)2, TMEDA), 58.8 (s, CH2N, TMEDA), 127.8 (s, p-C, SOPh), 127.9 (s, o-C, SOPh), 128.0 (s, m-C, SOPh), 155.0 (s, i-C, SOPh). 7Li NMR (194 MHz, THF-d8): δ 0.1 (s, Li).
2 (R/R′ = H/H, Ar = p-Tol). Yield: 188 mg (68%). 1H NMR (400 MHz, THF-d8): δ 1.88 (s, br, 1H, LiCH2), 2.14 (s, 12H, 2 × N(CH3)2, TMEDA), 2.29 (s, 3H, CH3), 2.31 (s, 4H, 2 × CH2, TMEDA), 7.09–7.11 (m, 2H, m-H, SOTol), 7.80–7.82 (m, 2H, o-H, SOPh). 13C NMR (100 MHz, THF-d8): δ 22.0 (s, CH3), 36.3 (s, LiCH2), 47.2 (s, N(CH3)2, TMEDA), 59.7 (s, CH2N, TMEDA), 126.9 (s, p-C, SOTol), 129.1 (s, o-C, SOTol), 129.7 (s, m-C, SOTol), 138.3 (s, i-C, SOTol). 7Li NMR (194 MHz, THF-d8): δ 0.55 (s, Li).
3 (R/R′ = Me/Me, Ar = Ph). Yield: 200 mg (69%). 1H NMR (400 MHz, THF-d8): δ 1.11 (m, 6H, 2 × CH3), 2.15 (s, 12H, 2 × N(CH3)2, TMEDA), 2.30 (s, 4H, 2 × NCH2, TMEDA), 7.25–7.80 (m, 5H, SOPh). 13C NMR (100 MHz, THF-d8): δ 12.1/15.5 (s/s, 2 × CH3), 45.2 (s, N(CH3)2, TMEDA), 53.8 (s, C(CH3)2), 57.9 (s, CH2N, TMEDA), 124.5 (s, p-C, SOPh), 128.4 (s, m-C, SOPh), 130.1 (s, o-C, SOPh), 143.8 (s, i-C, SOPh). 7Li NMR (194 MHz, THF-d8): δ −0.1 (s, Li).
4 (R/R′ = H/Ph, Ar = Ph). Yield: 206 mg (61%). 1H NMR (400 MHz, THF-d8): δ 2.15 (s, 12H, 2 × N(CH3)2, TMEDA), 2.31 (s, 4H, 2 × NCH2, TMEDA), 3.77 (s, 1H, LiCHPh), 6.15 (m, 1H, p-H, CHPh), 6.70 (m, 4H, o-/m-H, CHPh), 7.20 (t, 3JH,H = 7.1 Hz, 1H p-H, SOPh), 7.28 (t, 3JH,H = 7.4 Hz, 2H, m-H, SOPh), 7.77 (d, 3JH,H = 7.6 Hz, 2H, o-H, SOPh). 13C NMR (100 MHz, THF-d8): δ 45.2 (s, N(CH3)2, TMEDA), 57.9 (s, CH2N, TMEDA), 65.3 (s, LiCHPh), 112.5/115.2 (s/s, 2 × p-C, SOPh + CHPh), 125.4–127.5 (s/s/s/s, 2 × o-C, 2 × m-C, SOPh + CHPh), 148.4/149.5 (s/s, 2 × i-C, SOPh + CHPh). 7Li NMR (194 MHz, THF-d8): δ −0.25 (s, Li).
5 (R/R′ = Me/Ph; Ar = Ph). Yield: 254 mg (72%). 1H NMR (400 MHz, THF-d8): δ 1.46 (s, 3H, CH3), 2.15 (s, 12H, 2 × N(CH3)2, TMEDA), 2.31 (s, 4H, 2 × NCH2, TMEDA), 6.15 (t, 3JH,H = 7.0 Hz, 1H, p-H, CCH3Ph), 6.79 (t, 3JH,H = 7.6 Hz, 2H, m-H, CCH3Ph), 6.93 (d, 3JH,H = 7.5 Hz, 2H, o-H, C6H5C), 7.20 (t, 3JH,H = 7.1 Hz, 1H p-H, SOPh), 7.31 (t, 3JH,H = 7.4 Hz, 2H, m-H, SOPh), 7.62 (d, 3JH,H = 7.5 Hz, 2H, o-H, SOPh). 13C NMR (100 MHz, THF-d8): δ 11.9 (s, CH3), 45.2 (s, N(CH3)2, TMEDA), 57.8 (s, CH2N, TMEDA), 62.2 (s, CCH3Ph), 112.2/115.5 (s/s, 2 × p-C, SOPh + CCH3Ph), 126.6–127.5 (s/s/s/s, 2 × o-C, 2 × m-C, SOPh + CCH3Ph), 148.6/149.7 (s/s, 2 × i-C, SOPh + CCH3Ph). 7Li NMR (194 MHz, THF-d8): δ −0.2 (s, Li).
Reaction of lithiated sulfoxides with benzaldehyde
At −78 °C, to a stirred suspension of [Li2{CRR′S(O)Ar}2(TMEDA)2] (1–6; 0.5 mmol; 6, R/R′ = H/Me; Ar = Ph, has been prepared as 1–5 but without isolation) in diethyl ether (10 mL), PhCHO (1.0 mmol) was added rapidly. Then, the reaction mixture was slowly warmed up to room temperature and stirred for another hour. At 0 °C, an aqueous solution of NH4Cl (ca. 25 mL; 30%) was added slowly. After phase separation, the aqueous phase was extracted with diethyl ether. The combined organic phases were dried (Na2SO4) and the solvents were removed in vacuo. The crude product was purified by centrifugally accelerated thin layer chromatography (eluent: diethyl ether–n-hexane 1/1) yielding pure colorless crystals of ArS(O)CRR′CHPhOH.1a (R/R′ = H/H, Ar = Ph). Yield: 150 mg (61%). Major diastereomer (>94%): 1H NMR (400 MHz, CDCl3): δ 2.97–3.40 (m, 2H, CH2), 3.75 (s, 1H, OH), 5.72, (s, 1H, CHOH), 7.21–7.82 (m, 10H, HPh). 13C NMR (100 MHz, CDCl3): δ 64.2 (s, CH2), 75.6 (s, COH), 124.1/127.2/127.7/128.7/129.9/130.9, (s/s/s/s/s/s, 2 × p-C, 2 × m-C, 2 × o-C, SOPh + Ph), 142.0/145.3 (s/s, 2 × i-C, SOPh + Ph). Minor diastereomer not detectable.
2a (R/R′ = H/H, Ar = p-Tol). Yield: 166 mg (64%). Major diastereomer (60%): 1H NMR (400 MHz, CDCl3): δ 2.42 (s, 3H, CH3), 2.83 (m, 1H, CH2), 3.22 (m, 1H, CH2), 4.05 (br, 1H, OH), 5.26 (s, 1H, CHOH), 7.23–7.56 (m, 9H, HAr). 13C NMR (100 MHz, CDCl3): δ 21.4 (s, CH3), 63.4 (s, CH2), 68.9 (s, COH), 124.0/125.6/127.9/128.6/130.1/139.4, (s/s/s/s/s/s, 2 × p-C, 2 × m-C, 2 × o-C, SOTol + Ph), 142.0/142.1 (s/s, 2 × i-C, SOTol + Ph).
Minor diastereomer (40%): 1H NMR (400 MHz, CDCl3): δ 2.42 (s, 3H, CH3), 2.93 (m, 1H, CH2), 3.22 (m, 1H, CH2), 4.05 (br, 1H, OH), 5.36 (s, 1H, CHOH), 7.23–7.56 (m, 9H, HAr).
3a (R/R′ = Me/Me, Ar = Ph). Yield: 244 mg (89%). Major diastereomer (>94%): 1H NMR (400 MHz, CDCl3): δ 0.82/1.13 (s/s, 3H, CH3), 4.20 (s, 1H, OH), 5.13 (s, 1H, CHOH), 7.26–7.69 (m, 10H, SOPh + Ph). 13C NMR (100 MHz, CDCl3): δ 12.3/18.3 (s/s, CH3/CH3), 62.7 (s, C(CH3)2), 78.8 (s, CHOH), 126.5–131.7 (m, 2 × p-C, 2 × o-C, 2 × m-C, SOPh + Ph), 138.9/139.1 (s/s, 2 × i-C, 2 × i-C, SOPh + Ph). Minor diastereomer not detectable.
4a (R/R′ = H/Ph, Ar = Ph). Yield: 222 mg (69%). Major diastereomer (80%): 1H NMR (400 MHz, CDCl3): δ 3.92 (d, 3JH,H = 9.6 Hz, 1H, CHSO), 5.66 (d, 3JH,H = 9.6 Hz, 1H, CHOH), 6.72 (m, 1H, COH), 6.95–7.54 (m, 15H, HPh). 13C NMR (100 MHz, CDCl3): δ 77.6 (s, CHSO), 78.1 (s, CHOH), 124.8–142.0 (m, CPh).
Minor diastereomer A (13%): 1H NMR (400 MHz, CDCl3): δ 3.59 (d, 3JH,H = 8.3 Hz, 1H, CHSO), 5.52 (d, 3JH,H = 8.3 Hz, 1H, CHOH), 6.72 (m, 1H, COH), 6.95–7.54 (m, 15H, HPh).
Minor diastereomer B (7%): 1H NMR (400 MHz, CDCl3): δ 3.65 (d, 3JH,H = 2.7 Hz, 1H, CHSO), 5.70 (d, 3JH,H = 2.7 Hz, 1H, CHOH), 6.72 (m, 1H, COH), 6.95–7.54 (m, 15H, HPh).
5a (R/R′ = Me/Ph, Ar = Ph). Yield: 259 mg (77%). Major diastereomer (57%): 1H NMR (400 MHz, CDCl3): δ 0.89 (s, 3H, CH3), 5.01 (d, 1H, CH), 6.42, (m, 1H, COH), 6.75–7.38 (m, 15H, HPh). 13C NMR (100 MHz, CDCl3): δ 12.9 (s, CH3), 72.0 (s, CCH3), 75.5 (s, COH), 125.9–141.6 (CPh).
Minor diastereomer (43%): 1H NMR (400 MHz, CDCl3): δ 1.27 (s, 3H, CH3), 5.69 (d, 1H, CH), 6.41, (m, 1H, COH), 6.75–7.38 (m, 15H, HPh).
6a (R/R′ = H/Me, Ar = Ph). Yield: 174 mg (67%). Major diastereomer (81%): 1H NMR (400 MHz, CDCl3): δ 0.97 (d, 3H, 3JH,H = 6.9 Hz, CH3), 2.81 (m, 1H, CH), 3.46 (br, 1H, OH), 5.45, (s, 1H, CHOH), 7.16–7.72 (m, 10H, HPh). 13C NMR (100 MHz, CDCl3): δ 3.4 (s, CH3), 64.7 (s, CH), 73.9 (s, CHOH), 124.3/125.9/127.8/128.5/129.1/130.9 (s/s/s/s/s/s, 2 × p-C, 2 × m-C, 2 × o-C, SOPh + Ph), 141.2/141.3 (s/s, 2 × i-C, SOPh + Ph).
Minor diastereomer (19%): 1H NMR (400 MHz, CDCl3): δ 0.97 (m, 3H, CH3), 2.72 (m, 1H, CH), 3.46 (br, 1H, OH), 5.34, (s, 1H, CHOH), 7.16–7.72 (m, 10H, HPh).
Reaction of lithiated sulfoxides with benzophenone
The reactions of the lithiated sulfoxides (0.5 mmol) with Ph2CO (1.0 mmol) were performed analogously to the reactions with PhCHO (section 4.3), to afford colorless crystals of ArS(O)CRR′CPh2OH.1b (R/R′ = H/H, Ar = Ph). Yield: 238 mg (74%). 1H NMR (400 MHz, CDCl3): δ 3.33–3.57 (m, 2H, CH2), 5.72 (s, 1H, OH), 7.14–7.67 (m, 15H, HPh).
13C NMR (100 MHz, CDCl3): δ 68.8 (s, CH2), 81.4 (s, CPh2OH), 123.9/125.5/126.6/127.6/129.5/131.5/144.0/145.9 (s/s/s/s/s/s/s/s, 2 × p-C, 2 × m-C, 2 × o-C, 2 × i-C, SOPh + CPh2OH).
2b (R/R′ = H/H, Ar = p-Tol). Yield: 231 mg (69%). 1H NMR (400 MHz, CDCl3): δ 2.35 (s, 3H, CH3), 3.92 (m, 2H, CH2), 6.32 (s, 1H, OH), 7.16–7.55 (m, 14H, HAr). 13C NMR (100 MHz, CDCl3): δ 21.3 (s, CH3), 70.4 (s, CH2), 76.8 (s, CPh2OH), 124.5–147.0 (CAr).
3b (R/R′ = Me/Me, Ar = Ph). Yield: 280 mg (80%). 1H NMR (400 MHz, CDCl3): δ 1.20 (s, 3H, CH3), 3.57 (s, 3H, CH3), 5.95 (s, 1H, OH), 7.18–7.95 (m, 15H, HPh). 13C NMR (100 MHz, CDCl3): δ 16.9 (s, CH3), 21.6 (s, CH3), 64.7 (s, CSO), 83.3 (s, CPh2OH), 126.9–145.1 (CPh).
4b (R/R′ = H/Ph, Ar = Ph). Yield: 271 mg (68%). Major diastereomer (>94%): 1H NMR (400 MHz, CDCl3): δ 4.63 (s, 1H, CHPh), 5.97 (s, 1H, COH), 6.93–7.96 (m, 20H, HPh). 13C NMR (125 MHz, CDCl3): δ 74.4 (s, CHPh), 82.0 (s, COH), 124.2–146.3 (CPh). Minor diastereomer not detectable.
6b (R/R′ = H/Me, Ar = Ph). Yield: 252 mg (75%). Major diastereomer (>94%): 1H NMR (400 MHz, CDCl3): δ 0.99 (d, 3JH,H = 6.9 Hz, 3H, CHCH3), 3.65 (q, 3JH,H = 6.8 Hz 1H, CHCH3), 4.85 (s, 1H, OH), 7.14–7.74 (m, 15H, HPh). 13C NMR (125 MHz, CDCl3): δ 5.4 (s, CH3), 64.4 (s, CHCH3), 80.5 (s, COH), 124.2/124.9 (s/s, 2 × m-C, CPh2OH), 125.8 (s, m-C, SOPh), 126.9/127.3 (s/s, 2 × p-C, CPh2OH), 128.4/128.6 (s/s, 2 × o-C, CPh2OH), 129.2 (s, o-C, SOPh), 130.8 (s, p-C, SOPh), 141.1 (s, i-C, SOPh), 145.1/145.9 (s/s, 2 × i-C, CPh2OH). Minor diastereomer not detectable.
X-ray crystallography
Data for X-ray diffraction analyses of single crystals were collected on a Stoe IPDS 2 T diffractometer at 200 K (1, 4·Et2O, 3a′, 4a′, 4b′) and an Oxford Gemini S diffractometer at 115 K (3) using Mo-Kα radiation (λ = 0.71073 Å, graphite monochromator). Data for X-ray diffraction analysis of 2 were collected on an Oxford Gemini S diffractometer at 110 K using Cu-Kα radiation (λ = 1.54184 Å, graphite monochromator). A summary of the crystallographic data, the data collection parameters and the refinement parameters is given in Tables S4 and S5.† Multiscan absorption corrections were applied using the PLATON program package54,55Tmin/Tmax: 0.59/1.49, 1; 0.74/0.99, 4·Et2O; 0.88/0.97, 3a′; 0.81/0.99, 4a′; 0.87/1.00, 4b′) and SCALE3 ABSPACK56 (0.88/1.00, 3), respectively. The structures were solved with direct methods using SHELXS-97 and SHELXS-201357 and refined using full-matrix least-square routines against F2 with SHELXL-97 and SHELXL-2013.58 Hydrogen atoms were placed in calculated positions according to the riding model except those of the O–H⋯O′ hydrogen bonds in compounds 3a′, 4a′ and 4b′ which were located in the electron density maps. All non-hydrogen atoms were refined with anisotropic displacement parameters and hydrogen atoms with isotropic ones. Specific features of the refinement procedures, as used restraints, disorder of atoms/fragments etc., are given below Table S4.† CCDC 1014086–1014092 contains the supplementary crystallographic data for this paper.Powder X-ray diffraction measurements were performed at room temperature on a Bruker D8-Advance diffractometer operating with Cu-Kα radiation. To avoid intensity variations due to texture effects (preferred orientation of the crystallites) the sample powder was mixed with an amorphous diluting agent (volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). The diffraction pattern based on single crystal structure results was simulated using the software PowderCell.59
:2). The diffraction pattern based on single crystal structure results was simulated using the software PowderCell.59
Computational details
DFT calculations were performed with the Gaussian09 program package60 using the functional B3LYP.61 The 6-31+G(d,p) basis sets as implemented in Gaussian09 were employed for all atoms. All systems were fully optimized without any symmetry restrictions. The resulting geometries were characterized as equilibrium structures by the analysis of the force constants of normal vibrations. Solvent effects (THF) were considered according to Tomasi's polarized continuum model as implemented in Gaussian09.39–41Acknowledgements
G. L. gratefully acknowledges financial support from the Graduate Scholarship of the State Saxony-Anhalt.References
- T. Toru and C. Bolm, Organosulfur Chemistry in Asymmetric Synthesis, Wiley, Hoboken, 1st edn, 2008 Search PubMed.
- R. J. W. Cremlyn, An Introduction to Organosulfur Chemistry, Wiley, Chichester, 1st edn, 1996 Search PubMed.
- E. Block, Organic Chemistry: Reactions of Organosulfur Compounds, ed. A. T. Blomquist and H. H. Wasserman, Academic Press, New York, 1978, p. 37 Search PubMed.
- K. Ogura, Comprehensive Organic Synthesis, Selectivity, Strategy, Efficiency in Modern Organic Chemistry, 1991, 1, 505 Search PubMed.
- E. Schaumann, Top. Curr. Chem., 2007, 247, 1 CrossRef.
- S. Patai and Z. Rappoport, The Chemistry of Sulphur-Containing Functional Groups, Wiley, Chichester, 1993 Search PubMed.
- G. Boche, Angew. Chem., Int. Ed., 1989, 28, 277 CrossRef.
- R. Luisi and V. Capriati, Lithium Compounds in Organic Synthesis: From Fundamentals to Application, Wiley-VCH, Weinheim, 1st edn, 2014 Search PubMed.
- G. L. Edwards, in Comprehensive Organic Functional Groups Transformations 1, ed. A. R. Katritzky, O. Meth-Cohn and C. W. Rees, Elsevier, Oxford, 1995, p. 105 Search PubMed.
- F. G. Bordwell, N. R. Vanier, W. S. Matthews, J. B. Hendrickson and P. L. Skipper, J. Am. Chem. Soc., 1975, 97, 7160 CrossRef CAS.
- F. G. Bordwell, J. C. Branca, C. R. Johnson and N. R. Vanier, J. Org. Chem., 1980, 45, 3884 CrossRef CAS.
- A. Krief, Tetrahedron, 1980, 36, 2531 CrossRef CAS.
- H. Pellissier, Tetrahedron, 2006, 62, 5559 CrossRef CAS PubMed.
- G. Solladié, Synthesis, 1981, 185 CrossRef.
- A. J. Walker, Tetrahedron: Asymmetry, 1992, 3, 961 CrossRef CAS.
- H. L. Holland, Chem. Rev., 1988, 88, 473 CrossRef CAS.
- K. Nakamura, M. Higaki, S. Adachi, S. Oka and A. Ohno, J. Org. Chem., 1987, 52, 1414 CrossRef CAS.
- T. Durst, R. R. Fraser, M. R. McClory, R. B. Swingle, R. Viau and Y. Y. Wigfield, Can. J. Chem., 1970, 48, 2148 CrossRef CAS.
- T. Durst, R. Viau and M. R. McClory, J. Am. Chem. Soc., 1971, 93, 3077 CrossRef CAS.
- J. F. Biellmann and J. J. Vicens, Tetrahedron Lett., 1974, 15, 2915 CrossRef.
- J. F. Biellmann and J. J. Vicens, Tetrahedron Lett., 1978, 19, 467 CrossRef.
- G. Chassaing, R. Lett and A. Marquet, Tetrahedron Lett., 1978, 19, 471 CrossRef.
- G. Demailly, C. Greck and G. Solladie, Tetrahedron Lett., 1984, 25, 4113 CrossRef CAS.
- S. G. Pyne and G. Boche, J. Org. Chem., 1989, 54, 2663 CrossRef CAS.
- T. Sato, T. Itoh and T. Fujisawa, Tetrahedron Lett., 1987, 28, 5677 CrossRef CAS.
- H. Sakuraba and S. Ushiki, Tetrahedron Lett., 1990, 31, 5349 CrossRef CAS.
- M. Higaki, M. Goto and A. Ohno, Heteroat. Chem., 1990, 1, 181 CrossRef CAS.
- A. Ohno, M. Higaki and M. Okamura, Heteroat. Chem., 1992, 3, 513 CrossRef CAS.
- S. Kusuda, Y. Ueno and T. Toru, Tetrahedron, 1994, 50, 1045 CrossRef CAS.
- S. Nakamura, H. Takemoto, Y. Ueno, T. Toru, T. Kakumoto and T. Hagiwara, J. Org. Chem., 2000, 65, 469 CrossRef CAS PubMed.
- E. Cadoni, M. Arca, M. Usai, C. Fattuoni, E. Perra, M. G. Cabiddu, S. De Montis and S. Cabiddu, Tetrahedron, 2008, 64, 6349 CrossRef CAS PubMed.
- K. Nishihata and M. Nishio, J. Chem. Soc., Perkin Trans. 2, 1972, 1730 RSC.
- K. Nishihata and M. Nishio, Tetrahedron Lett., 1976, 17, 1695 CrossRef.
- S. Wolfe, A. Stolow and L. A. LaJohn, Tetrahedron Lett., 1983, 24, 4071 CrossRef CAS.
- S. Wolfe, A. Stolow and L. A. LaJohn, Can. J. Chem., 1984, 62, 1470 CrossRef CAS.
- M. Marsch, W. Massa, K. Harms, G. Baum and G. Boche, Angew. Chem., Int. Ed. Engl., 1986, 25, 1011 CrossRef.
- P. C. Andrews, N. D. R. Barnett, R. E. Mulvey, W. Clegg, P. A. O'Neil, D. Barr, L. Cowton, A. J. Dawson and B. J. Wakefield, J. Organomet. Chem., 1996, 518, 85 CrossRef CAS.
- Cambrigde Structural Database (ConQuest), Version 1.11, Crystallographic Data Centre, University Chemical Laboratory, Cambridge, UK, 2009 Search PubMed.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS PubMed.
- M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Chem. Phys. Lett., 1998, 286, 253 CrossRef CAS.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151 CrossRef CAS PubMed.
- R. Lett, G. Chassaing and A. Marquet, J. Organomet. Chem., 1976, 111, C17 CrossRef CAS.
- G. Chassaing and A. Marquet, Tetrahedron, 1978, 34, 1399 CrossRef CAS.
- P. v. R. Schleyer and A. J. Kos, Tetrahedron, 1983, 39, 1141 CrossRef CAS.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- M. Casey, I. Mukherjee and H. Trabsa, Tetrahedron Lett., 1992, 33, 127 CrossRef CAS.
- C. A. Kingsbury, J. Org. Chem., 1972, 37, 102 CrossRef CAS.
- L. Colombo, C. Gennari, C. Scolastico, G. Guanti and E. Narisano, J. Chem. Soc., Chem. Commun., 1979, 591 RSC.
- C. H. Heathcock, Mod. Synth. Methods, 1992, 6, 1 CAS.
- C. Palomo, M. Oiarbide and J. M. Garcia, Chem. – Eur. J., 2002, 8, 36 CrossRef CAS.
- F. Shi, M. K. Tse, H. M. Kaiser and M. Beller, Adv. Synth. Catal., 2007, 349, 2424 Search PubMed.
- MULscanABS, PLATON for Windows Taskbar v1.15, University of Glasgow, 2008 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- SCALE3 ABSPACK, Empirical Absorption Correction, CrysAlis–Software package, Oxford Diffraction Ltd., 2006 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, University of Göttingen, Göttingen, 1998 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, 1997 Search PubMed.
- W. Kraus and G. Nolze, PowderCell for Windows, Federal Institute For Materials Research And Testing, Berlin, Germany, 2000 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Protocols of the quantum chemical calculations, structural data and Wiberg bond indices of 1–4 and 1*–4*, respectively, tables and figures of molecular structures of 3a′, 4a′ and 4b′. CCDC 1014086–1014092. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt02238f |
| ‡ Here and in the following calculated structures are marked with a star. |
| § de Values were determined by 1H NMR spectroscopy (% de = % major diastereomer – % minor diastereomer). The signal-to-noise ratios allowed to detect impurities ≥3%. With the assumption that this amount of the minor diastereomer remains to be undetected, an upper limit for de values of 94% is given. |
| This journal is © The Royal Society of Chemistry 2015 |