
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A luminescence line-narrowing spectroscopic study of the uranium(VI) interaction with cementitious materials and titanium dioxide†
Jan
Tits
*a,
Clemens
Walther‡
b,
Thorsten
Stumpf§
b,
Nathalie
Macé¶
a and
Erich
Wieland
a
aLaboratory for Waste Management, Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland. E-mail: jan.tits@psi.ch
bInstitute for Nuclear Waste Disposal, Karlsruhe Institute of Technology, D-76344 Eggenstein-Leopoldshafen, Germany
First published on 4th November 2014
Abstract
Non-selective luminescence spectroscopy and luminescence line-narrowing spectroscopy were used to study the retention of UO22+ on titanium dioxide (TiO2), synthetic calcium silicate hydrate (C-S-H) phases and hardened cement paste (HCP). Non-selective luminescence spectra showed strong inhomogeneous line broadening resulting from a strongly disordered UO22+ bonding environment. This problem was largely overcome by using luminescence line-narrowing spectroscopy. This technique allowed unambiguous identification of three different types of UO22+ sorbed species on C-S-H phases and HCP. Comparison with spectra of UO22+ sorbed onto TiO2 further allowed these species to be assigned to a surface complex, an incorporated species and an uranate-like surface precipitate. This information provides the basis for mechanistic models describing the UO22+ sorption onto C-S-H phases and HCP and the assessment of the mobility of this radionuclide in a deep geological repository for low and intermediate level radioactive waste (L/ILW) as this kind of waste is often solidified with cement prior to storage.
Introduction
Cementitious materials are commonly used worldwide for the solidification of low- and intermediate level radioactive waste (L/ILW) prior to storage in surface or deep geological repositories.1,2 For an accurate prediction of the long term fate of this radioactive waste, a comprehensive understanding of the chemical interactions of the radionuclides present in the waste with the solidification material is essential. In the past, radionuclide retention by cementitious materials has been typically attributed to adsorption on the surfaces of cement minerals (e.g. Evans3 and references therein). However, other immobilization processes, such as incorporation into the solid matrix, may take place. Incorporation processes give rise to the isolation of the radionuclide from the pore solution and may result in an irreversible immobilization in the solid matrix. The release of incorporated radionuclides is controlled by slow processes such as recrystallization and dissolution rather than by rapid surface desorption reactions. Calcium silicate hydrates (C-S-H) are major constituents of cementitious materials. They have a tobermorite-like layered structure consisting of Ca–O sheets linked on each side to silicate chains in a “dreierkette” arrangement.4 These calcium silicate layers carry a net negative charge neutralised by either protons or Ca2+ cations in the interlayer. C-S-H phases are further characterized by high recrystallization rates making them an ideal system for radionuclide incorporation. Recently, several studies have found experimental evidence for the incorporation of trivalent and tetravalent actinides and lanthanides by substitution for Ca in the interlayers and the Ca–O layer of C-S-H phases.5–9 It is still an open question whether or not larger actinyl ions such as UO22+ exhibit similar incorporation behaviour.Uranium is an important component of intermediate-level radioactive waste and the isotopes of this radionuclide contribute significantly to its long-term dose.10,11 Under the typical redox conditions prevailing in the alkaline environment of the cementitious near-field of an ILW repository (Eh = −0.23 V),12 uranium exists predominantly in the valence state +VI as the linear dioxo uranyl ion, UO22+.
Sorption of hexavalent actinides (UO22+, NpO22+) onto cementitious materials has been subject of several studies including macroscopic wet chemistry investigations as well as spectroscopic investigations: Batch sorption studies on C-S-H phases revealed fast sorption kinetics as steady state was reached within a period of approximately 5–10 days.13–15 Furthermore, sorption was found to be linear over a wide concentration range and highly dependent on the pH and the C-S-H composition. Indeed, measured solid–liquid distribution ratios (Rd) varied between 106 L kg−1 at pH = 10.0 and 102 L kg−1 at pH = 13.3.14,15 Tits et al.14 further demonstrated that, in the case of NpO22+, the observed dependence of Rd values on the C-S-H composition can almost entirely be explained by assuming competition between the sorption process and the formation of non-sorbing highly hydrolysed anionic NpO2(OH)42− species in solution.14
Several attempts have been made to decipher the local coordination environment of UO22+ sorbed onto cementitious materials with the help of X-ray absorption spectroscopy (XAS)16–20 and luminescence spectroscopy.15 These studies revealed (1) the existence of at least two types of UO22+ sorbed species, (2) a sorbed UO22+ structural environment dominated by O, Si and Ca atoms arranged in a similar way as in calcium uranyl silicates. The weak back-scattering contributions from neighbouring Si and Ca atoms of the C-S-H structure in the XAS spectra, however, prevented an unequivocal identification of the coordination environment and thus the determination of the dominant sorption process (surface complexation or incorporation, respectively). In addition, if more than one type of species is present, XAS data can only provide an averaged coordination environment. Hence, there is a need for complementary spectroscopic information about the coordination environment of sorbed UO22+ in C-S-H phases with the aim of providing conclusive evidence for the significance of incorporation as an immobilization process for hexavalent actinides in cementitious materials.
Conventional non-selective luminescence spectroscopy is commonly applied to study the speciation of UO22+ in both crystalline and amorphous solids (e.g., ref. 15, 21–28). However, this technique often gives rise to severe spectral inhomogeneous broadening effects, thus hiding valuable spectroscopic information from electronic and vibronic transitions.15,29,30 Inhomogeneous broadening occurs when UO22+ ions occupy sites in amorphous host matrices or at the surface of solids.15,21,26,27 In both cases the local coordination environment of the luminescing species is ill-defined resulting in a significant variation of the energies of states of the sorbed UO22+ ions. Although the electronic transition line for each single sorbed UO22+ ion may be sharp, the position of the electronic transition line and the spacings between the vibronic lines will vary among non-equivalent sorbed UO22+ ions, which results in inhomogeneously broadened overall electronic and vibronic transition bands composed of all the sharp lines of each single sorbed UO22+ ion. Inhomogeneous line broadening can be reduced by applying site-selective excitation.29–34 This well-established technique is called luminescence line-narrowing spectroscopy (LLNS) and has been successfully applied for the detection and characterization of organic molecules and biomolecular systems (e.g. Jankowiak35 and references therein). A narrow-band tunable laser is used to selectively excite a small subset of a set of UO22+ luminescence sites in a sample, at a frequency within the inhomogeneously broadened absorption band. The excited subset of UO22+ ions decays radiatively to produce a well-resolved narrow-band spectrum allowing extraction of the spectroscopic information from the electronic and vibronic transitions. By scanning the laser source over the frequency range of the inhomogeneously broadened absorption band, all subsets of luminescing UO22+ ions can be probed individually and the variation of the optical properties within the set of luminescing ions can be evaluated.
The aim of the present study is to determine the speciation of UO22+ taken up by C-S-H phases in cementitious materials. To achieve this goal, the optical properties of UO22+ sorbed on C-S-H phases and hardened cement paste (HCP) on the one hand side and of UO22+ adsorbed onto the surface of titanium dioxide (TiO2) on the other hand side, were investigated using LLNS. UO22+ is known to sorb on TiO2 through the formation of inner-sphere surface complexes.25,36–38 Furthermore, TiO2 is a solid phase known to be stable under alkaline conditions and, in contrast to C-S-H phases, it is characterized by a low solubility and a low recrystallization rate.39 Therefore, UO22+ incorporation into the structure of this mineral is unlikely, thus making it an ideal solid to study the optical properties of UO22+ surface complexes under alkaline conditions. Differences in the luminescence spectra of UO22+ sorbed on TiO2, C-S-H phases and HCP will enable us to determine whether or not this cation is incorporated in the C-S-H structure.
Experimental
Materials
All solutions and suspensions were prepared using reagent-grade chemicals and deionized, decarbonated water (Milli-Q water) generated by a Milli-Q Gradient A10 water purification system (Millipore Billerica, USA). Teflon containers and centrifuge tubes used for the preparation of the samples, were washed, left overnight in a solution of 0.1 M HCl, and thoroughly rinsed with deionized water prior to use. Sample preparation was carried out in a glovebox under an inert nitrogen atmosphere (CO2 and O2 <2 ppm).Analytical techniques
Solution compositions were determined using an Applied Research Laboratory ARL 3410D inductively coupled plasma optical emission spectrometer (ICP-OES) or a Perkin Elmer ELAN 6100 inductively coupled plasma mass spectrometer (ICP-MS). A combination glass pH electrode (Metrohm, Switzerland) calibrated against dilute standard pH buffers (pH = 7.0–11.0) was used to determine the pH.Sample preparation
An artificial cement pore water (ACW-I) was prepared based on an estimate of the NaOH and KOH concentration in a Portland cement porewater before any degradation of the material had occurred.40 It contains 0.18 M KOH and 0.114 M NaOH and has a pH of 13.3. A C-S-H phase with a Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Si mol ratio (C:S) of 1.07 was synthesized in ACW-I using a method adapted from Atkins et al.:41 Silica fume (AEROSIL 300, Degussa-Huls AG, Baar Switzerland) was mixed with CaO in a polyethylene bottle at a weight ratio of 1:1. To this, ACW-I was added to achieve a solid to liquid (S:L) ratio of 2.5 × 10−2 kg L−1. A detailed characterization of this C-S-H phase is given in Tits et al.42 After an ageing period of 4 weeks, the C-S-H suspension was ready for further use in sorption experiments.
:Si mol ratio (C:S) of 1.07 was synthesized in ACW-I using a method adapted from Atkins et al.:41 Silica fume (AEROSIL 300, Degussa-Huls AG, Baar Switzerland) was mixed with CaO in a polyethylene bottle at a weight ratio of 1:1. To this, ACW-I was added to achieve a solid to liquid (S:L) ratio of 2.5 × 10−2 kg L−1. A detailed characterization of this C-S-H phase is given in Tits et al.42 After an ageing period of 4 weeks, the C-S-H suspension was ready for further use in sorption experiments.
A sulfate-resistant Portland cement (CEM I 52.5 N HTS) provided by Lafarge (France), was used to prepare the HCP samples. A modified ACW (ACW-II)40 with pH = 13.3 and simulating the composition of a pore water in equilibrium with fresh HCP ([OH] = 0.3 M, [K]t = 0.18 M, [Na]t = 0.114 M, [Ca]t = 1.6 × 10−3 M, [Al]t = 5 × 10−5 M, [S]t = 2 × 10−3 M, [Si]t = 5 × 10−5 M) was added to the solid material to achieve the final S:L of 2.5 × 10−2 kg L−1.
TiO2 was obtained from Materion Advanced Chemicals Inc., Milwaukee, USA. This material is a mixture of 90% rutile and 10% anatase and has a specific surface area of 5 ± 1 m2 g−1.43 TiO2 suspensions were prepared by mixing the appropriate amounts of TiO2 powder with ACW-I to obtain a S:L ratio of 2.5 × 10−2 kg L−1. The suspensions were equilibrated for 1 day, centrifuged at 4000 rpm for 1 hour, and the supernatant solution replaced with fresh ACW-I. This washing procedure was repeated several times until the composition of the supernatant solution analysed by ICP-OES was constant and the TiO2 was in equilibrium with the ACW-I solution.
0.1 mL of a 1.08 × 10−2 M uranyl nitrate solution in 10−3 M HNO3 was added to 40 mL of the C-S-H, HCP and TiO2 suspensions. The UO22+ doped suspensions were shaken end-over-end for 30 days. After this equilibration period, phase separation of the solid and liquid phase was carried out by centrifugation at 4000 rpm for 1 hour. The resulting wet pastes were transferred to a self constructed copper sample holder with a sapphire window, sealed with a Teflon disk and cooled to <15 K by a helium refrigerated cryostat (CTI-cryogenics, USA) prior to luminescence measurements to reduce homogeneous spectral broadening. Determination of the UO22+ concentration in the supernatant solution by ICP-MS showed that >95% of the UO22+ added to the C-S-H suspension, was retained on the solid phase resulting in a UO22+ loading of ∼250 ppm (∼10−3 mol kg−1).
UO22+ luminescence spectroscopy
UO22+ luminescence is induced via charge-transfer excitation involving the transfer of an electron from the σu orbitals (mainly of the axial O2−) to 5fδ and 5fϕ orbitals of the U6+ ion.44–46 The subsequent radiative relaxation gives rise to an electronic transition line, E, having a frequency of radiation, νE, in the range 21000 cm−1 < νE < 19000 cm−1. UO22+ emission spectra are further characterized by a pronounced vibronic structure originating from the resolution of the vibrational energy levels in the ground state. In general, the totally symmetric stretch vibration (νs) of the UO22+ moiety is the most pronounced vibration mode and it appears as a series of homologous lines, S1–4, (vibronic progression) superimposed on E (e.g.Fig. 2, spectrum a and b in the results section). The spacing (Δ) between E and the first line of this series, S1, results in values for νs in the ground state as follows: νs = ∼870 cm−1 for the UO22+ aquo ion47,48 and 700 cm−1 < νs < 800 cm−1 for uranium minerals (silicates, carbonates) and UO22+ sorbed species.26,28,49
UO22+ luminescence spectra are usually measured after indirect UO22+ excitation raising the molecule from the ground state to one of the upper electronic excited states. De-excitation follows in two steps: (1) radiationless de-excitation through internal conversion and vibrational relaxation to the lowest vibrational level of the first excited state and (2) relaxation from the first excited state to the ground state with the emission of light. In case of UO22+ ions occupying a large range of sorption sites with slightly different bonding environments, this technique leads to the excitation of all UO22+ luminescence sites within the inhomogeneously broadened absorption band resulting in poorly resolved broadened luminescence spectra. To reduce the effect of inhomogeneous broadening, LLN spectroscopy has been applied. The Jablonski diagram in Fig. 1 adapted from Jankowiak,35 illustrates the principle of this technique applied to UO22+. In this diagram, the electronic ground state (0,0) and the first electronic excited state (1,0) of UO22+ in an ill-defined coordination environment (e.g. an amorphous matrix) are shown. Each of these electronic states supports several vibrational energy levels (vibronic states) while, to simplify the diagram, only the first of them is shown for each electronic state (0,1) and (1,1). The variation of the energies of the excited state levels within the inhomogeneously broadened absorption band is represented by the slope, Γinh (Fig. 1).
 | ||
| Fig. 1 Energy-level diagram illustrating resonant LLN E(1)) and non-resonant LLN (E(2)) after site-selective laser excitation at an energy, νex (diagram adapted from Jankowiak35). See text for a detailed explanation. | ||
Two different cases can be identified50: (1) Resonant luminescence line-narrowing (red color in Fig. 1): A small subset of UO22+ ions (A1) is directly excited to the lowest vibrational level of the first excited state (1,0) using an energy, νex, in the inhomogeneously broadened absorption band. These excited ions decay radiatively directly to the ground state. The frequency, νE(1), of the resulting narrow electronic transition line (E(1)), is identical to the frequency of the exciting laser light (νex = νE1). (2) Non resonant line-narrowing (blue color in Fig. 1): Using the same excitation energy, νex, another small subset of UO22+ ions (A2) may be excited to a higher-lying vibrational level of the first excited state (1,1). From there, these ions decay non-radiatively to the lowest vibrational level of the first excited state by vibrational relaxation, followed by radiative decay to the ground state. The resulting narrow electronic line (E(2)) has a frequency, νE(2), lower than νE(1). It can be deduced from Fig. 1 that the spacing between E(1) and E(2) gives a value for the frequency of the totally symmetric stretch vibration of this second small subset of UO22+ ions in the excited state, ν′S(2). Non-resonant line-narrowing occurs when consecutive excited vibronic states of UO22+ overlap due to inhomogeneous broadening.
In case UO22+ clusters are formed in the sample, LLN spectra may be disturbed by homo-resonance energy transfer processes from the directly excited UO22+ ions (donor ions) to nearby unexcited ions (acceptor ions) with slightly different transition frequencies (concentration quenching).51,52 This process is possible provided that the emission spectrum of the donor UO22+ ion overlaps the absorption spectrum of the acceptor UO22+ ion so that several vibronic transitions in the donor have similar energies as the corresponding transitions in the acceptor; i.e. the transitions are in resonance. This energy transfer results from long-range dipole–dipole interactions between donor and acceptor and not from the transfer of a photon. Following energy transfer, the acceptor ions may decay radiatively themselves or transfer their excitation energy to other nearby unexcited ions. The energy transfer efficiency depends on the extent of overlap between the donor emission spectrum and the acceptor absorption spectrum and on the donor–acceptor distance with an inverse 6th power law. Homo-resonance energy transfer occurs in samples with high concentrations of luminescence centers and is effective at donor–acceptor distances up to 10 nm for systems with large absorption coefficients (ε) and high luminescence quantum efficiencies (Φ0).52 Absorption coefficients and quantum yields for UO22+ luminescence strongly depend on the surrounding matrix, but they are generally quite low resulting in much smaller maximum distances for resonance energy transfer. Resonance energy transfer processes result in broadening of the transition lines. In extreme cases (e.g., the formation of UO22+ precipitates), the resonant electronic transition line in LLN spectroscopy may completely disappear.
The UO22+ luminescence lifetime depends partially on the presence of quenching ligands near the UO22+ moiety. Shorter lifetimes are an indication for increased quenching. E.g. the luminescence of UO22+ clusters exhibiting concentration quenching will typically be characterized by a shorter luminescence lifetime. Excited UO22+ species typically decay mono-exponentially and multi-exponential luminescence decay is often an indication for the presence of more than one luminescing species in a sample.
Luminescence spectroscopy was performed using a pulsed OPO laser system (Spectra Physics MOPO HF). Indirect excitation of UO22+ was performed at an excitation energy of 24390 cm−1. The range used for the direct excitation was 21270 cm−1 < νex < 17857 cm−1. The luminescence signal was detected by an optical multichannel analyzer consisting of a Czerny Turner 300 mm polychromator (Jobin Yvon, HR 320 with gratings ranging from 300 to 1200 lines mm−1) and an intensified, gated photodiode array (Spectroscopy instruments, ST 180, IRY 700G) for the experiments with C-S-H phases and HCP, or a Czerny Turner spectrometer (Andor Shamrock with gratings ranging 150/mm–2400/mm) coupled to an intensified ICCD (Andor Technologies) camera optimized for high on off ratio by use of a PROXITRONIC image intensifier for the experiments with TiO2. The laser light and Rayleigh and Raman scattering are filtered from the luminescence emission spectra by using a pulsed laser source and applying a minimum gate delay of 10 μs between the laser pulse and camera gating.
For the determination of the luminescence lifetime, luminescence decay profiles were recorded with a delay time step of 5 μs or 25 μs and a total of 90 steps were collected for each decay profile.
Results and discussion
Non-selective luminescence spectroscopy of UO22+ doped C-S-H phases
Fig. 2c shows the normalized luminescence spectrum of uranyl ions sorbed onto C-S-H phases with a C:S ratio of 1.07 in ACW at a pH of 13.3 after indirect laser excitation at νex = 24390 cm−1. This spectrum is compared with the spectra of the free UO22+ ion in 0.1 M HNO3 and an uranyl solution in 0.5 M tetramethyl ammonium hydroxide (TMAOH) at pH = 13.7. Thermodynamic calculations performed using the NEA chemical thermodynamic database53 showed that the speciation of U(VI) in ACW is dominated by the strongly hydrolyzed UO2(OH)42− complex. The uranyl-doped C-S-H phases exhibit weakly structured spectra similar to those reported in an earlier paper.15 The UO22+ luminescence spectra are red shifted upon hydrolysis. This red-shift is even more pronounced for UO22+ sorbed on C-S-H phases. It is caused by a significant weakening of the axial U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds upon sorption due to the increased electron-donating abilities of the ligands in the equatorial plane of the sorbed UO22+ ions.15,48,54
O bonds upon sorption due to the increased electron-donating abilities of the ligands in the equatorial plane of the sorbed UO22+ ions.15,48,54
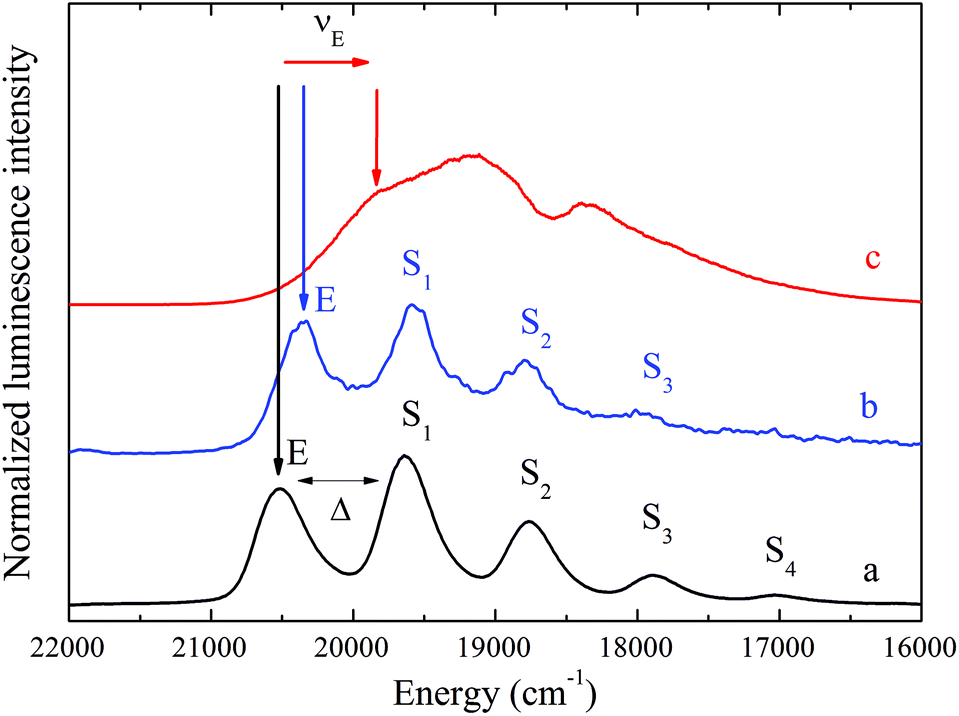 | ||
| Fig. 2 Non-selective luminescence spectra recorded at 15 K of (a) 10−5 M UO22+ in 0.1 M HNO3, pH = ∼1.0, (b) 10−5 M UO22+ in 0.5 M TMAOH, pH = 13.7, (c) UO22+ sorbed onto C-S-H phases (C:S = 1.07, in ACW at pH = 13.3); loading = 10−3 mol kg−1. νex = 24390 cm−1. Delay time = 10 μs, gate width = 1.0 ms. | ||
The broad luminescence bands indicate strong variation in the bonding environment of the sorbed UO22+ ions.
LLN spectroscopy of UO22+ doped C-S-H phases
For the study of the sorption of UO22+ onto C-S-H phases with LLN spectroscopy, three experimental parameters were varied: (1) The excitation energy (νex): Variation of νex allows individual excitation of different subsets of UO22+ luminescence centers within the inhomogeneously broadened electronic transition band, (2) the gate width (time period over which the luminescence signal is recorded) of the photodiode array: A change in the spectral shape with varying gate width is an indication for the presence of species with different lifetimes, (3) the delay time after the laser pulse: Luminescence signals recorded after increasing delay times after the laser pulse can be used for the construction of decay profiles allowing the determination of the lifetime of the excited state of a luminescing UO22+ species.Excitation spectra and LLN spectra of UO22+ sorbed on C-S-H phases in ACW-I at pH 13.3 were obtained by varying νex within the range of the inhomogeneously broadened electronic transition band (νE) observed in the non-selective emission spectrum (21000 cm−1–18000 cm−1; Fig. 2, spectrum c). At each excitation energy, a full emission spectrum was recorded and the luminescence intensity was integrated over the emission energy range between 22000 cm−1 and 16000 cm−1. The excitation spectra are poorly resolved due to the effect of inhomogeneous broadening and do not provide much spectral information. In the following, the excitation spectra of UO22+ doped samples will therefore only be shown for completeness of the datasets, but they will not be considered for the spectral analysis.
Two series of luminescence spectra were recorded (Fig. 3). A gate delay time of 10 μs was used in both series to filter out the emission interference originating from Rayleigh and Raman scattering. The gate width of the camera was fixed to 10 μs in the first series and 10 ms in the second series. UO22+ species with a shorter luminescence lifetime will dominate the spectra recorded with the small gate width whereas UO22+ species with a longer lifetime will prevail in the spectra recorded with large gate width. The spectra of the first series are again characterized by broad, weakly resolved luminescence bands without any pronounced structure (Fig. 3a). The absence of a sharp luminescence line resonant with the energy of the laser excitation in these LLN spectra, is a strong indication for the presence of homo resonance energy transfer processes between adjacent sorbed UO22+ moieties. The Förster distance (the distance between donor and acceptor at which the energy transfer efficiency is 50%) for UO22+ in C-S-H phases was estimated using the calculation procedure described by Hink et al.55 and resulted in a value of 1.1 nm (see ESI†). This observation suggests that at least one luminescent UO22+ species in the UO22+ doped C-S-H phases is present as a (surface) precipitate with neighbouring UO22+ ions at distances closer than ∼1.1 nm. Calculations using the NEA thermodynamic data for uranium53 completed with solubility data for alkali uranates of Yamamura et al.56 and CaUO4(s) of Moroni et al.57 indicate that in ACW-I, Na2U2O7 should be the solubility controlling phase. The UO22+ concentration in solution in equilibrium with the UO22+ doped C-S-H phase was measured to be 7.0 × 10−7 M, which is a factor of 10 below the solubility limit of Na2U2O7 in ACW (6 × 10−6 M). Hence, Na2U2O7 precipitation in the UO22+ doped C-S-H sample is not very probable and UO22+ cluster formation must be catalyzed by the C-S-H phase. On the other hand side, U–U distances in uranates are in the range 3.5 Å < RU–U < 6.6 Å;58i.e., close enough for efficient homo resonance energy transfer.
 | ||
| Fig. 3 Excitation spectra (dotted lines) and selected LLN spectra of UO22+ sorbed on a C-S-H phase (C:S = 1.07) in ACW at pH = 13.3. UO22+ loading = 10−3 mol kg−1. Excitation energy: 21008 cm−1 < νex < 18868 cm−1. Delay time = 10 μs. Gate width = 10 μs (a) and 10 ms (b). The intensities of the excitation spectra in Fig. 3a and 3b are divided by a factor 50 and 40, respectively. | ||
The second series of luminescence spectra recorded with a larger gate width of 10 ms are characterized by a series of sharp luminescence bands in the frequency region 20500 cm−1 < ν < 19000 cm−1. These spectra indicate the presence of at least one more UO22+ sorbed species with a longer lifetime.
Furthermore, the spectra of this UO22+ sorbed species appear to be less disturbed by energy transfer processes suggesting a more homogenous distribution of this UO22+ sorbed species in the C-S-H sample. The first sharp line in each of these spectra is the luminescence line resonant with the laser excitation energy. It represents the electronic transition line of a subset of UO22+ ions directly excited by the applied laser energy. The position of this line shifts with νex. The other sharp transition lines represent either non-resonant electronic transitions or vibronic transitions coupled to the electronic transitions.
A detailed spectral analysis was undertaken with the help of three typical emission spectra selected from the second series of spectra recorded with a large gate width (Fig. 4). The spectra were selected to include the spectrum measured with a high λex, a spectrum measured with an intermediate νex (arbitrarily chosen) and a spectrum measured with a low νex having sufficient intensity to give an exploitable spectrum. The bands in each spectrum are assigned based upon their energy relative to the resonance line. The interpretation is based upon the Jablonski diagramme described in Fig. 1 and the assignment of the lines is summarized in Table 1.
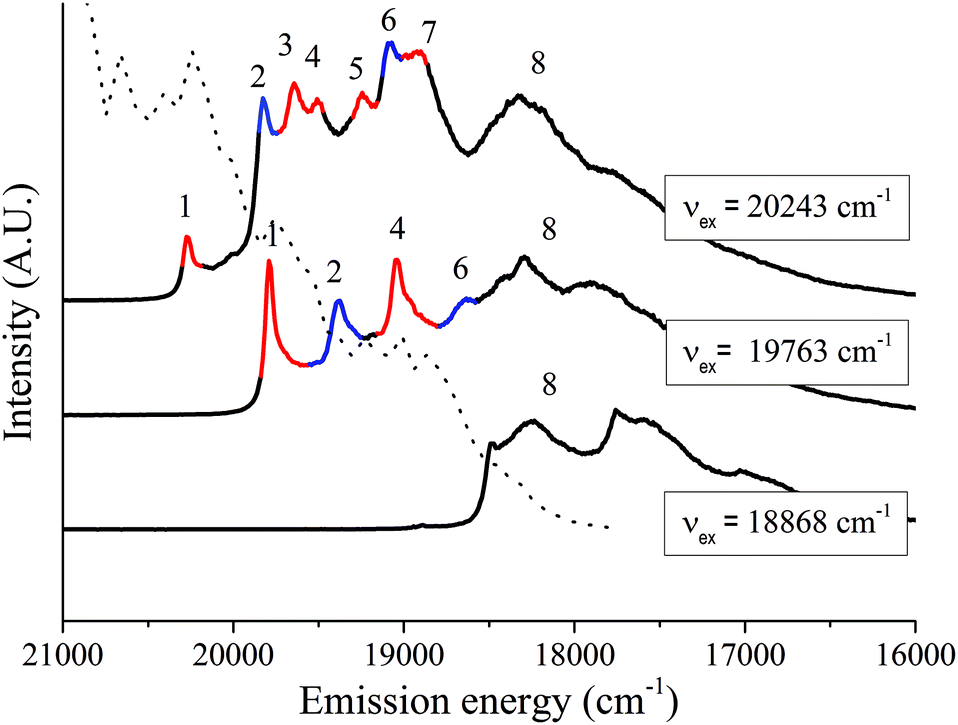 | ||
| Fig. 4 Selected LLN spectra of UO22+ sorbed on a C-S-H phase (C:S = 1.07) in ACW at pH = 13.3 recorded at three different excitation energies (νex = 20243 cm−1, 19763 cm−1, and 18868 cm−1). Delay time = 10 μs. Gate width = 10 ms. The numbered transition lines are described in the text and in Table 1. Red = lines assigned to species U1, blue = lines assigned to species U2. | ||
| Number | Description |
|---|---|
| (1) | ν E(1): Resonant electronic transition line |
| (2) | ν E(2): Non-resonant electronic transition line of species U1 |
| (3) | ν E(3): Non-resonant electronic transition line of species U2 |
| (4) | First line of vibronic progression on νE(1) |
| (5) | Could not be assigned |
| (6) | First line of vibronic progression on νE(2) |
| (7) | First line of vibronic progression on νE(3) |
| (8) | Structureless spectrum of UO22+ precipitate |
The first line (1) is always the luminescence line resonant with the laser energy and represents the electronic transition (E(1)) of a small subset of UO22+ sorbed species, U1. A series of homologous lines (vibronic progression) having a spacing, Δ, between 700 and 800 cm−1, is associated with this electronic transition. These vibronic lines result from the coupling between the electronic transition and the totally symmetric stretch vibration in the ground state, νs(1), of this subset. Indeed, the first line of this homologous series appears to be line (4) at a spacing Δ1–4 = 758 cm−1 (Table 1). The other lines of this series are not resolved. Lines (2) and (3) in the spectra must represent non-resonant electronic transition lines (E(2) and E(3)) of two more subsets of UO22+ sorbed species. The first members of the vibronic progressions on each of these electronic transitions are the lines (6) and (7), with spacings Δ2–6 = 744 ± 10 cm−1 and Δ3–7 = 758 ± 10 cm−1, respectively. There are two possible interpretations for the presence of the two non-resonant electronic transitions lines (2) and (3): both subsets belong to the same set of UO22+ sorbed species or both subsets belong to two entirely different sets of UO22+ sorbed species.
(1) In the first case, the non-resonant lines (2) and (3) result from excitations to the second and third vibrational level of the first excited state (1,1) and (1,2), respectively, and the spacings Δ1–2 and Δ2–3, representing the frequencies of the totally symmetric stretch vibrations in the excited state (ν′s(2) and (ν′s(3)), should be similar (see Fig. 1). The values of Δ1–2 and Δ2–3, however, were found to differ significantly; i.e., 406 ± 10 cm−1 and 226 ± 10 cm−1, respectively, indicating that this interpretation is wrong.
(2) Both subsets belong to two entirely different sets of UO22+ sorbed species (U1 and U2) with clearly different coordination environments. In this case, the non-resonant lines (2) and (3) result from excitations to the second vibrational level of the first excited state of a subset of U1 and a subset of U2, respectively. The spectral characteristics of these two species are listed in Table 2. The main spectral difference between these two sets of UO22+ sorbed species is the frequency of the symmetric stretch vibration in the excited state, ν′s, being 406 ± 10 cm−1 (Δ1–2) and 632 ± 10 cm−1 (Δ1–3) for U1 and U2, respectively.
243 cm−1, 19763 cm−1 and 18868 cm−1 (Fig. 4). Comparison with the spectral characteristics of the UO22+ aquo ion and UO2(OH)42− (literature data)
| Type of UO22+ sorbed species | ν s/cm−1 | ν ′s/cm−1 |
|---|---|---|
| U1 | Δ 1–4 = Δ3–7 = 758 ± 10 | Δ 1–3 = 632 ± 10 |
| U2 | Δ 2–6 = 744 ± 10 | Δ 1–2 = 406 ± 10 |
| U3 | — | — |
The values of νs and ν′s found for the sorbed species U1 and U2 are significantly smaller than the values found for the UO22+ aquo ion and the UO2(OH)42− hydroxo species indicating a strong weakening of the axial UO bond54 in agreement with the red-shift of the electronic transition line observed in the non-selective luminescence spectra in Fig. 2. This axial UO bond weakening results from increased σ and π donating abilities of the ligands coordinated to the U atom in the equatorial plane indicating strong bonding of the UO22+ upon sorption.54
The LLN spectrum recorded at an excitation energy of 18868 cm−1 exhibits a completely different shape (Fig. 4). Similarly to the first series of LLN spectra recorded with short gate width (Fig. 3a), this spectrum is characterized by a few broad, weakly resolved luminescence bands without any pronounced structure. Furthermore, it does not contain a resonant electronic transition line. These spectral features are again a strong indication for the occurrence of energy transfer processes suggesting the presence of a (surface) precipitated UO22+ species, U3. This observation confirms the assumption made for the interpretation of the first series of LLN spectra recorded with a short gate width. The strong red-shift of the spectrum of this surface precipitate is in agreement with luminescence spectra of alkaline-earth uranates described in the literature.59,60 The poorly resolved transition bands of this spectrum don't allow the spectral characteristics of this species, νs and ν′s to be determined. The spectral characteristics of the three sorbed UO22+ species, U1, U2 and U3, deduced from the LLN spectra, are summarized in Table 2.
In Fig. 5, the values of νs and ν′s for U1 and U2 obtained from the spacings (Δ) between the different transition lines in all the luminescence spectra recorded after excitation with increasing νex, are plotted against νex. This plot reveals that the frequencies of νs and ν′s for U1 and U2 decrease with increasing νex. This means that the UO axial distances of the subsets of U1 and U2, measured at higher excitation energies, are longer than the UO axial distances of the subsets measured at lower excitation energies. This observation is in agreement with the decreasing values of νE for these subsets of U1 and U2 with increasing νex and is an expression of the variability of the bonding environments within the two types of species, U1 and U2.
 | ||
| Fig. 5 Values for the UO22+ totally symmetric stretch vibration in the ground state, νs, and in the excited state, ν′s, for subsets of the sorbed UO22+ species U1 and U2, as function of the excitation energy, νex. | ||
In order to further check the existence of the postulated three different types of sorbed UO22+ species U1, U2 and U3, the lifetimes of the different transition lines in the luminescence spectrum obtained after excitation at an energy of 19763 cm−1 were determined. Transition lines with the same lifetime are attributed to the same type of UO22+ sorbed species, whereas transition lines with different lifetimes are attributed to another type of sorbed species. Decay profiles measured for the three sharp lines (1, 2, and 4 in Fig. 4) and the broad band (8 in Fig. 4) at higher energy are very similar; i.e., they can be fitted with a bi-exponential decay model suggesting the presence of at least two species with lifetimes of 108 ± 31 μs and 275 ± 31 μs, respectively (Fig. 6a, Table 3).
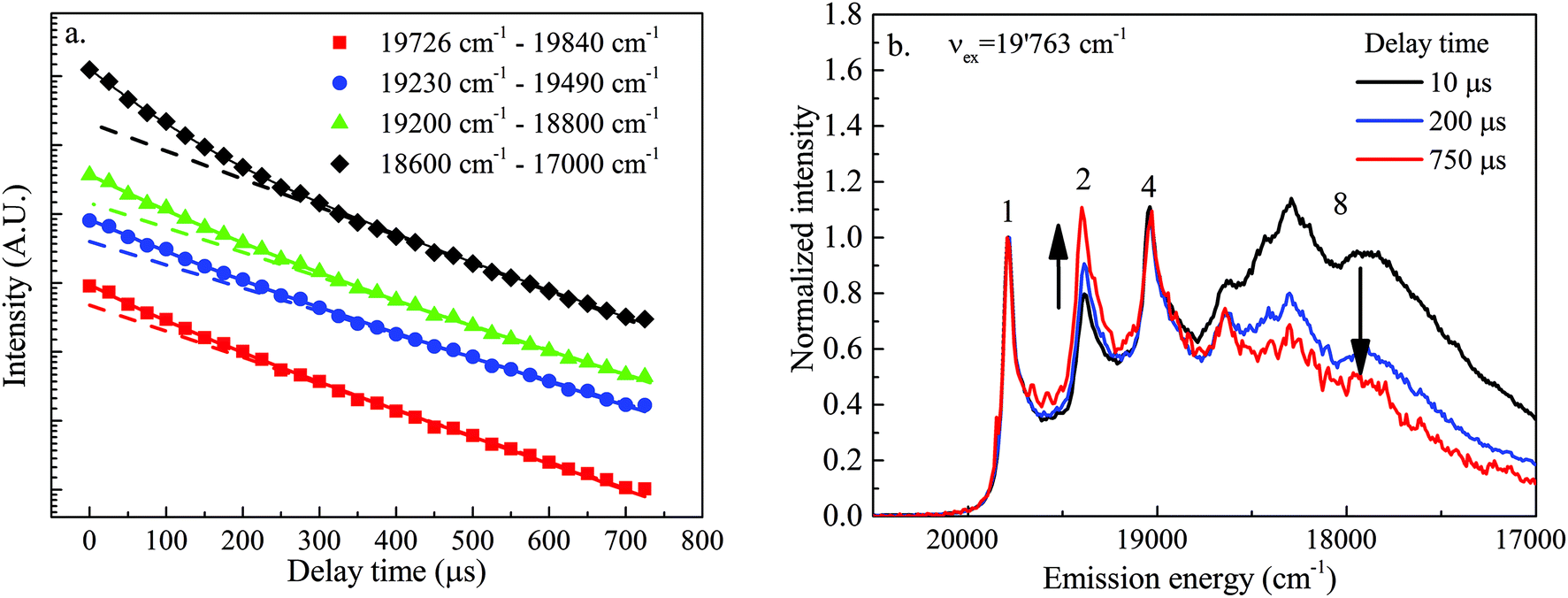 | ||
| Fig. 6 (a) Luminescence lifetimes of different emission lines after direct excitation at νex = 19763 cm−1. (b) LLN spectra recorded after excitation at νex = 19763 cm−1 using three different delay times. Gate width = 10 ms. | ||
763 cm−1. Gate width = 10 ms
| Line number: | t 1 (μs) | t 2 (μs) |
|---|---|---|
| 1 (19726 cm−1–19840 cm−1) |
106 | 263 |
| 2 (19230 cm−1–19490 cm−1) |
120 | 297 |
| 4 (19200 cm−1–18800 cm−1) |
125 | 286 |
| 8 (18600 cm−1–17000 cm−1) |
81 | 256 |
| Mean and 95% confidence interval using “Student's t” numbers | 108 ± 31 | 275 ± 31 |
The short lifetime is attributed to the uranium (surface) precipitate, U3, as this species is also dominantly present in the series of spectra recorded with a short gate width (Fig. 3a). This lifetime corresponds very well with the lifetime of 100 μs measured at 4.2 K by De Jong et al.60 for MgUO4(s), an uranate having a structure very similar to the structure of the Ca–uranate (CaUO4(s)) expected to precipitate at the surface of C-S-H phases in the present system.
Unfortunately, the difference in lifetimes of the two other species is too small to be detected with a decay profile analysis. However, a close inspection of the spectra recorded at different delay times, normalized to the resonant luminescence line, clearly shows that line (1) and line (4) decay at exactly the same rate, whereas line (2) exhibits a slower decay (the line rises relative to the first line with increasing delay time) (Fig. 6b). Furthermore, the broad band (8) at lower frequency exhibits a faster decay (the band decreases relative to the first line with increasing delay time). This observation clearly confirms that transition lines (1) and (4) belong to the same set of UO22+ sorbed species (U1) whereas transition line (2) belongs to an additional set of species (U2) with a longer luminescence lifetime relative to that of U1. The longer lifetime indicates that non-radiative de-excitation processes for this set of species are less efficient due to the presence of a lower number of quenchers (e.g. H2O molecules) in its proximity. The broad transition band at higher frequencies belongs to the (surface) precipitate, U3, with a shorter luminescence lifetime relative to that of U1.
In summary, LLN spectroscopy of UO22+ sorbed on a C-S-H phase in ACW at pH = 13.3 allowed three different sets of species, U1, U2 and U3 to be identified. One of these species (U3) is a (surface) precipitate. This conclusion is in agreement with the results from previous time-resolved non-selective luminescence studies on a large number of C-S-H phases with varying compositions.15
Comparison with UO22+ surface complexation on TiO2 and with UO22+ sorbed on HCP: evidence for UO22+ incorporation in C-S-H phases
LLN spectra of UO22+ sorbed on TiO2 (loading = 10−3 mol kg−1) in the presence of ACW at pH = 13.3 were recorded with a delay time of 10 μs and using a gate width of 10 ms (Fig. 7a). Reducing the gate width to 10 μs did not have a significant effect on the shape of the LLN spectra. This is in contrast to the observations made with samples containing UO22+ sorbed onto C-S-H phases for which a reduction of the gate width resulted in the disappearance of the resonance transition line and in a significant loss of spectral resolution caused by energy transfer between neighbouring UO22+ ions due to the formation of an uranate-like (surface) precipitate. This observation suggests that such a precipitate with a short-lived luminescence signal did not form in the TiO2 sample although the aqueous chemistry and the total UO22+ concentrations in the samples are identical with those in the C-S-H sample. Hence, it appears that an uranate-like (surface) precipitate is only formed on the surface of C-S-H phases. The LLN spectra of UO22+ sorbed on TiO2 contain resonant electronic transition lines albeit much broader than in the case of UO22+ sorbed onto C-S-H phases. Furthermore, the other transition bands observed in the LLN spectra are also much broader. Both observations suggest the presence of a significant homo resonance energy transfer from excited UO22+ ions to neighbouring unexcited ions with slightly different bonding environments. Note that the presence of closely neighbouring UO22+ atoms is not due to the formation of a UO22+ precipitate as the spectral features of such a precipitate (i.e., the short-lived broad structureless peak at 18500 cm−1 > νE > 17500 cm−1) is absent. A UO22+ loading of 10−3 mol kg−1 on a TiO2 solid phase having a specific surface area of ∼5 m2 g−1 (ref. 43) means that approximately 12 UO22+ atoms are adsorbed on a surface area of 10 nm2. If the sorbed UO22+ is homogeneously distributed over the TiO2 surface, the distance between adjacent sorbed UO22+ ions is ∼3 nm. A rough estimate of the Förster distance in this system gives a value of ∼0.7 nm (see ESI†), significantly shorter than the distance of 3 nm calculated based upon the UO22+ loading on the solid. Hence, homo resonance energy transfer cannot explain the line broadening observed in this sample. We don't have an explanation for this observation at present.
 | ||
| Fig. 7 LLN spectra of UO22+ sorbed on TiO2 (a) and on HCP (b) in the presence of ACW at pH = 13.3. Loading = 10−3 mol kg−1. Excitation energy: 21008 cm−1 < νex < 18868 cm−1. Delay time = 10 μs. Gate width = 10 ms. The intensities of the excitation spectra in Fig. 7a and 7b are divided by a factor 500 and 200, respectively. | ||
At low excitation energy (18868 cm−1) only the laser line is visible in the luminescence spectra. In contrast to the sample with UO22+ sorbed on C-S-H phases, no luminescence signal is detected after excitation at this energy, confirming the absence of an uranate-like precipitate.
LLN spectra of UO22+ sorbed on HCP again exhibit better resolved spectra (Fig. 7b). A clear electronic transition line can be identified in each spectrum, followed by several additional well-resolved narrow bands. At high excitation energy (18868 cm−1) a luminescence spectrum similar to that for the UO22+ loaded C-S-H sample is observed indicating the presence of an uranate-like (surface) precipitate. For further interpretation of the bands in these two samples, LLN spectra of UO22+–C-S-H, UO22+–TiO2 and UO22+–HCP recorded after excitation at 506 nm are compared in Fig. 8. The numbers used for the transition lines are the same as given in Fig. 4. Species U1 (red vertical lines) is found in the spectra of all three samples, although the spacing, Δ1–4, is slightly larger for the U1 species on TiO2 compared to the C-S-H sample and the HCP sample. We thus conclude that species U1 is a surface complex. Species U2 (blue vertical lines) is only present in the spectra of the C-S-H sample and the HCP sample and completely absent in the spectra of the TiO2 sample. The bands are sharp and well resolved indicating that energy transfer is not taking place so that this species must be homogeneously distributed in the C-S-H/HCP matrix. The absence of this sorbed species in the TiO2 sample, its homogeneous distribution in the C-S-H/HCP matrix and its longer lifetime suggesting a lower number of quenchers in close proximity, leads to the conclusion that these bands must belong to a UO22+ species incorporated in the C-S-H structure in both samples.
 | ||
| Fig. 8 LLN spectra of UO22+ sorbed on TiO2, a C-S-H phase (C:S = 1.07), and on HCP in ACW at pH = 13.3. UO22+ loading = 10−3 mol kg−1. Excitation energy: νex = 19763 cm−1. Delay time = 10 μs. Gate width = 10 ms. Numbered transition lines are described in the text. | ||
The broad band typical for the uranate-like precipitate species, U3, in the frequency range, 18500 cm−1 < ν < 17000 cm−1 is only visible in the C-S-H and HCP samples and is completely absent in the TiO2 sample. This uranate-like solid phase must therefore be a Ca-uranate-like surface precipitate forming only on the Ca-rich C-S-H surfaces which are present in both the C-S-H and HCP samples.
Conclusions
LLN spectroscopy allows the unambiguous identification of three different types of sorbed UO22+ species in C-S-H phases and in HCP.Comparison of LLN spectra obtained for UO22+ sorbed on C-S-H phases and HCP with LLN spectra obtained for UO22+ surface complexes on TiO2 further allows the different types of sorbed species to be assigned to a surface complex, an incorporated species and a surface precipitate, respectively. Inhomogeneous broadening of absorption bands and luminescence bands after non-selective excitation indicates that all three sorbed UO22+ species are characterized by a significant variation in their bonding environment.
LLN spectroscopy indicates that UO22+ is retained in cement not only through adsorption on the surface of the solid cement components but also through incorporation into the structure of C-S-H phases. Similar incorporation processes were already observed in the past for trivalent and tetravalent actinides. Here, an unambiguous proof for the incorporation of the bulky UO22+ cations in the C-S-H structure is presented. The study shows that UO22+ sorbed on cement phases becomes isolated from the cement porewater. This suggests that the subsequent release from the cement phases is governed by dissolution or recrystallization processes of the C-S-H phases rather than by rapid surface desorption.
Acknowledgements
The authors would like to thank Dr M. Marques Fernandes (PSI) for the many helpful discussions. Thanks are extended to S. Büchner (Institute for Nuclear Waste Disposal, Karlsruhe Institute of Technology) for his invaluable assistance with the laser equipment. Partial financial support by the Swiss National Cooperative for the Disposal of Radioactive Waste (Nagra) is kindly acknowledged.Notes and references
- T. W. Hicks, T. D. Baldwin, P. J. Hooker, P. J. Richardson, N. A. Chapman, I. G. McKinley and F. B. Neall, Concepts for the geological disposal of intermediate-level radioactive waste, Galson Scienses Ltd report 0736-1, Version 1.1 on behalf of UK NDA, 2008.
- F. P. Glasser, in Handbook of advanced radioactive waste conditioning technologies, ed. M. I. Ojovan, Woodhead, Oxford, UK, 2011, pp. 67–135 Search PubMed.
- N. D. M. Evans, Cem. Concr. Res., 2008, 38, 543–553 CrossRef CAS.
- I. G. Richardson, Cem. Concr. Res., 2008, 38, 137–158 CrossRef CAS.
- T. Stumpf, J. Tits, C. Walther, E. Wieland and T. Fanghänel, J. Colloid Interface Sci., 2004, 276, 118–124 CrossRef CAS.
- J. Tits, T. Stumpf, T. Rabung, E. Wieland and T. Fanghänel, Environ. Sci. Technol., 2003, 37, 3568–3573 CrossRef CAS.
- P. Mandaliev, T. Stumpf, J. Tits, R. Dähn, C. Walther and E. Wieland, Geochim. Cosmochim. Acta, 2011, 75, 2017–2029 CrossRef CAS.
- P. Mandaliev, E. Wieland, R. Dähn, J. Tits, S. V. Churakov and O. Zaharko, Appl. Geochem., 2010, 25, 763–777 CrossRef CAS.
- M. Schlegel, I. Pointeau, N. Coreau and P. Reiller, Environ. Sci. Technol., 2004, 38, 4423–4431 CrossRef CAS.
- D. McGinnes, Model radioactive waste inventory for reprocessing waste and spent fuel, Nagra Technical Report NTB 01-01, Nagra, Wettingen, Switzerland, 2002.
- NAGRA, Project Opalinus Clay. Safety report. Demonstration of disposal feasibility for spent fuel, vitrified high-level waste and long-lived intermediate level waste (Entsorgungsnachweis), Nagra Technical Report NTB 02-05, Nagra, Wettingen, Switzerland, 2002.
- P. Wersin, L. H. Johnson, B. Schwyn, U. Berner and E. Curti, Redox conditions in the near-field of a repository for SF/HLW and ILW in Opalinus Clay, Nagra Technical Report NTB 02-13, Nagra, Wettingen, Switzerland, 2003.
- I. Pointeau, C. Landesman, E. Giffault and P. Reiller, Radiochim. Acta, 2004, 92, 645–650 CrossRef CAS.
- J. Tits, X. Gaona, A. Laube and E. Wieland, Radiochim. Acta, 2014, 102, 385–400 CrossRef CAS.
- J. Tits, G. Geipel, N. Macé, M. Eilzer and E. Wieland, J. Colloid Interface Sci., 2011, 359, 248–256 CrossRef CAS.
- E. R. Sylwester, P. G. Allen, P. Zhao and B. E. Viani, Mater. Res. Soc. Symp. Proc., 2000, 608, 307–312 CrossRef CAS.
- P. Zhao, P. G. Allen, E. R. Sylwester and B. E. Viani, Radiochim. Acta, 2000, 88, 729–736 CAS.
- M. Harfouche, E. Wieland, R. Dähn, T. Fujita, J. Tits, D. Kunz and M. Tsukamoto, J. Colloid Interface Sci., 2006, 303, 195–204 CrossRef CAS.
- N. Macé, E. Wieland, R. Dähn, J. Tits and A. Scheinost, Radiochim. Acta, 2013, 101, 379–389 CrossRef.
- X. Gaona, E. Wieland, J. Tits, A. Scheinost and R. Dähn, Appl. Geochem., 2013, 28, 109–118 CrossRef CAS.
- C. J. Chisholm-Brause, J. M. Berg, K. M. Little, R. A. Matzner and D. E. Morris, J. Colloid Interface Sci., 2004, 277, 366–382 CrossRef CAS.
- H.-S. Chang, G. V. Korshin, Z. Wang and J. Zachara, Environ. Sci. Technol., 2006, 40, 1244–1249 CrossRef CAS.
- C. Götz, G. Geipel and G. Bernhard, J. Radioanal. Nucl. Chem., 2011, 287, 961–969 CrossRef.
- G. Geipel, Coord. Chem. Rev., 2006, 250, 844–854 CrossRef CAS.
- J. Vandenborre, R. Drot and E. Simoni, Inorg. Chem., 2007, 46, 1291–1296 CrossRef CAS.
- Z. Wang, J. Zachara, J.-F. Boily, Y. Xia, T. C. Resch, D. A. Moore and C. Liu, Geochim. Cosmochim. Acta, 2011, 75, 2965–2979 CrossRef CAS.
- A. Kowal-Fouchard, R. Drot, E. Simoni and J. J. Ehrhardt, Environ. Sci. Technol., 2004, 38, 1399–1407 CrossRef CAS.
- Z. Wang, J. M. Zachara, P. L. Gassman, C. Liu, O. Qafoku, W. Yantasee and J. G. Catalano, Geochim. Cosmochim. Acta, 2005, 69, 1391–1403 CrossRef CAS.
- C. D. Flint, P. A. Tanner, R. Reisfeld and H. Tzehoval, Chem. Phys. Lett., 1983, 102, 249–253 CrossRef CAS.
- G. K. Liu, H. Z. Zhuang, J. V. Beitz, C. W. Williams and V. S. Vikhnin, Phys. Solid State, 2002, 44, 1433–1439 CrossRef CAS.
- M. Marques Fernandes, M. Schmidt, T. Stumpf, C. Walther, D. Bosbach, R. Klenze and T. Fanghänel, J. Colloid Interface Sci., 2008, 321, 323–331 CrossRef CAS.
- K. L. Lam and A. F. Leung, J. Non-Cryst. Solids, 1977, 23, 385–400 CrossRef CAS.
- A. F. Leung, J. Non-Cryst. Solids, 1976, 21, 41–46 CrossRef CAS.
- M. Marques Fernandes, T. Stumpf, B. Baeyens, C. Walther and M. H. Bradbury, Environ. Sci. Technol., 2010, 44, 921–927 CrossRef.
- R. Jankowiak, in Shpol'skii spectroscopy and other site-selection methods, ed. C. Gooijer, F. Ariese and J. Hofstraat, Wiley-Interscience, New York, 2000, pp. 235–271 Search PubMed.
- C. Den Auwer, R. Drot, E. Simoni, S. D. Conradson, M. Gailhanou and J. Mustre de Leon, New J. Chem., 2003, 27, 648–655 RSC.
- M. J. Comarmond, T. E. Payne, J. J. Harrison, S. Thiruvoth, H. Wong, R. D. Augterson, G. R. Lumpkin, K. Müller and H. Foerstendorf, Environ. Sci. Technol., 2011, 45, 5536–5542 CrossRef CAS.
- G. Lefèvre, J. Kneppers and M. Fédoroff, J. Colloid Interface Sci., 2008, 327, 15–20 CrossRef.
- J. Schmidt and W. Vogelsberger, J. Solution Chem., 2009, 38, 1267–1282 CrossRef CAS.
- E. Wieland, J. Tits, A. Ulrich and M. H. Bradbury, Radiochim. Acta, 2006, 94, 29–36 CrossRef CAS.
- M. Atkins, F. P. Glasser and A. Kindness, Cem. Concr. Res., 1992, 22, 241–246 CrossRef CAS.
- J. Tits, E. Wieland, C. J. Müller, C. Landesman and M. H. Bradbury, J. Colloid Interface Sci., 2006, 300, 78–87 CrossRef CAS.
- K. Müller, H. Foerstendorf, T. Meusel, V. Brendler, G. Lefèvre, M. J. Comarmond and T. E. Payne, Geochim. Cosmochim. Acta, 2012, 76, 191–205 CrossRef.
- C. Görller-Walrand, S. De Houwer, L. Fluyt and K. Binnemans, Phys. Chem. Chem. Phys., 2004, 6, 3292–3298 RSC.
- R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS.
- S. P. McGlynn and J. K. Smith, J. Mol. Spectrosc., 1961, 6, 164–187 CrossRef CAS.
- C. Nguyen-Trung, D. A. Palmer, G. M. Begun, C. Pfeiffert and R. E. Mesmer, J. Solution Chem., 2000, 29, 101–129 CrossRef CAS.
- F. Quilès, C. Nguyen-Trung, C. Carteret and B. Humbert, Inorg. Chem., 2011, 50, 2811–2823 CrossRef.
- Z. Wang, J. Zachara, J. P. McKinley and S. C. Smith, Environ. Sci. Technol., 2004, 39, 2651–2659 CrossRef.
- G. F. Imbush, Phys. Scr., 1987, T19, 354–362 CrossRef.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science+Business Media LLC, New-York, USA, 3rd edn, 2006 Search PubMed.
- B. Valeur, Molecular fluorescence: principles and applications, Wiley-VCH Verlag GmbH, Weinheim, 2001 Search PubMed.
- R. Guillaumont, T. Fanghänel, J. Fuger, I. Grenthe, V. Neck, D. A. Palmer and M. H. Rand, Chemical Thermodynamics Vol. 5. Update on the Chemical Thermodynamics of Uranium, Neptunium, Plutonium, Americium and Technetium, Elsevier, North Holland, Amsterdam, The Netherlands, 2003 Search PubMed.
- S. Tsushima, Dalton Trans., 2011, 40, 6732–6737 RSC.
- M. A. Hink, N. V. Visser, J. W. Borst, A. Van Hoek and A. J. W. Visser, J. Fluoresc., 2003, 13, 185–188 CrossRef CAS.
- T. Yamamura, A. Kitamura, A. Fukui, S. Nishikawa, T. Yamamoto and H. Moriyama, Radiochim. Acta, 1998, 83, 139–146 CAS.
- L. P. Moroni and F. P. Glasser, Waste Manage., 1995, 15, 243–254 CrossRef CAS.
- D. M. Krol, P. M. Ros and A. Roos, J. Phys. Chem. A, 1980, 73, 1521–1526 CrossRef CAS.
- G. Blasse, Inorg. Chim. Acta, 1987, 129 Search PubMed.
- K. P. De Jong, D. M. Krol and G. Blasse, J. Lumin., 1978, 20, 241–248 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt02172j |
| ‡ Present address: Institut für Radioökologie und Strahlenschutz, Leibnitz Universität Hannover, D-30419 Hannover, Germany. |
| § Present address: Helmholtz-Zentrum Dresden-Rossendorf, Institute of Resource Ecology, D-01314 Dresden, Germany. |
| ¶ Present address: CEA Saclay, F-91191 Gif-sur-Yvette CEDEX France. |
| This journal is © The Royal Society of Chemistry 2015 |