
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
P,C-bond cleavage in the ligand sphere of a nickel(II) complex†
Simon P.
Walg
a,
Alexandra D.
Schmidt
a,
Marcus
Schmitz
a,
Saeid
Farsadpour
a,
Johannes
Lang
a,
Mark
Niebergall
a,
Yu
Sun
a,
Peter W.
Roesky
b,
Gereon
Niedner-Schatteburg
a and
Werner R.
Thiel
*a
aTechnische Universität Kaiserslautern, Fachbereich Chemie, Erwin-Schrödinger-Straße 54, D-67663 Kaiserslautern, Germany. E-mail: thiel@chemie.uni-kl.de
bKarlsruher Institut für Technologie (KIT), Institut für Anorganische Chemie, Engesserstraße 15, D-76131 Karlsruhe, Germany
First published on 14th October 2014
Abstract
Reacting nickel(II)perchlorate with a bidentate P,N-ligand in methanol leads to P,C-bond cleavage and gives a five-coordinate nickel complex wherein the nickel(II) site is coordinated by a tridentate P,N,P-ligand and a bidentate N,C-ligand. The carbanion of the latter is the result of the P,C-bond cleaving process. The diamagnetic nickel(II) complex was characterized by means of elemental analysis, NMR spectroscopy, cyclic voltammetry and X-ray structure analysis.
Introduction
Nickel(II) complexes have been established for a long time as catalysts for a series of catalytic transformations such as Kumada-1 and Negishi-type2 cross-coupling or olefin polymerization reactions.3 The broad catalytic applications in combination with the rich variety of catalytically active nickel(II) systems led us to focus on the coordination chemistry of [(2-aminopyrimidin-4-yl)aryl]phosphanes with this metal. In the last few years we have investigated chelating ligands containing 2-aminopyrimidin-4-yl fragments bound to either a second N-donor or a phosphine moiety. In the case of tertiary amino groups being located at the 2-position of the pyrimidine ring, such ligands easily undergo a so-called roll-over metallation4 leading to C–H activation in the 5-position of the pyrimidine ring.5 The resulting metal complexes which are coordinated by a carbanion show good activities in transfer hydrogenation (Ru) and Suzuki–Miyaura coupling reactions (Pd). This inspired us to extend the investigation of this class of ligand to other late transition metals.Herein we report on an unusual P–C bond cleavage performed in the ligand backbone of a nickel(II) complex leading to a stable, diamagnetic, five-coordinate nickel(II) complex. Interestingly this P,C-cleavage occurs at the P,N-ligand [(2-aminopyrimidin-4-yl)aryl]phosphane (1) containing a primary amine (Scheme 1), which is the most unreactive member in the series of primary, secondary and tertiary [(2-aminopyrimidin-4-yl)aryl]phosphanes we had investigated so far.
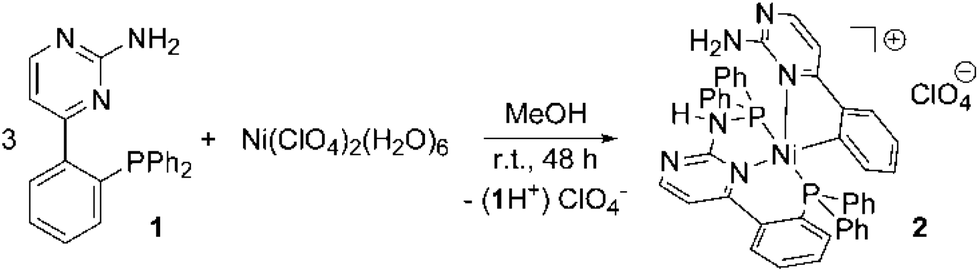 | ||
| Scheme 1 Synthesis of the nickel(II) complex 2. | ||
P,C-cleaving reactions have frequently been described in the literature. There are even some bacteria being able to perform this reaction in the case of alkyl phosphorous species.6 The cleavage of aryl and alkyl phosphorous bonds can be performed at clusters7 or mono- to trinuclear metal complexes8 leading to bridging phosphido ligands. These conversions are mainly following oxidative addition processes at the metal site. According to these findings Hartwig, Bergman and Andersen suggested that the P,C-cleavage occurring at a ruthenium(II) complex also includes an oxidative addition step.9 The most prominent example for a reductive P,C-cleavage is the reaction of PPh3 with lithium to provide Ph2PLi and PhLi,10 which is also reported for other phosphines with different reducing reagents.11 To the best of our knowledge, there is one example, wherein an electrophile (H+) attacks a non-coordinating phosphorous atom in the periphery of a transition metal complex (here: Zr), resulting in a P,C-bond cleavage.12 On the other hand, the attack of a nucleophile to a metal-coordinated phosphorous atom or a phosphorous site possessing good leaving group properties can also lead to P,C-bond cleavage and generate a carbanion, which is further stabilized by coordination to the metal site or by protonation.13
Results and discussion
Ligand 1 is accessible in a few steps starting from diphenyl(trimethylsilyl)phosphane and 1-(2-fluorophenyl)-3-N,N-dimethylaminoprop-2-en-1-one.14 Reacting Ni(ClO4)2(H2O)6 with 1 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio in methanol gives the red-colored, five-coordinate nickel(II) complex 2 in high yields (Scheme 1).
:3 ratio in methanol gives the red-colored, five-coordinate nickel(II) complex 2 in high yields (Scheme 1).
Single crystals of 2 suitable for an X-ray structure determination were grown from a concentrated solution in methanol. The nickel(II) complex 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two molecules of methanol in the asymmetric unit. One methanol molecule is distorted over two positions, the perchlorate counter anion is distorted as well. Fig. 1 shows the molecular structure of the cation. Characteristic bond parameters are listed in the caption.
with two molecules of methanol in the asymmetric unit. One methanol molecule is distorted over two positions, the perchlorate counter anion is distorted as well. Fig. 1 shows the molecular structure of the cation. Characteristic bond parameters are listed in the caption.
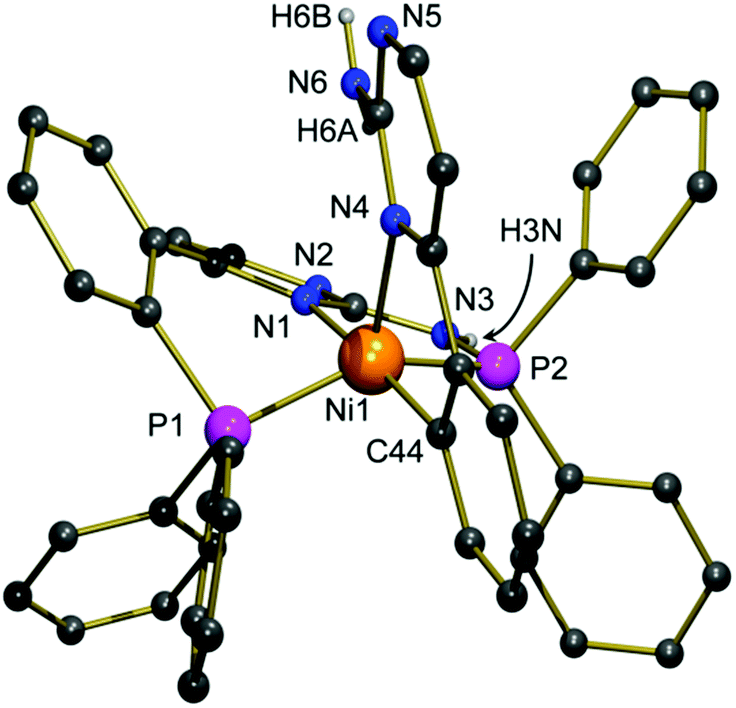 | ||
| Fig. 1 Molecular structure of the cation of compound 2 in the solid state; carbon bound hydrogen atoms, the perchlorate anion and both co-crystallizing methanol molecules are omitted for clarity. Characteristic bond lengths [Å], bond angles [°], and hydrogen bond parameters: Ni1–P1 2.1968(9), Ni1–P2 2.1367(9), Ni1–N1 1.978(3), Ni1–N4 2.135(3), Ni1–C44 1.903(3), P1–Ni1–P2 149.25(4), P1–Ni1–N1 89.12(8), P1–Ni1–N4 106.20(7), P1–Ni1–C44 90.58(9), P2–Ni1–N1 86.71(8), P2–Ni1–N4 104.42(7), P2–Ni1–C44 90.53(10), N1–Ni1–N4 102.94(10), N1–Ni1–C44 174.06(12), N4–Ni1–C44 82.84(12); hydrogen bonds (O1 from perchlorate): N3–H3N 0.86(3), H3N⋯N2 2.07(3), N3⋯N2 2.929(5), N3–H3N⋯N2 179(4), N6–H6A 0.87(3), H6A⋯O1 2.58(4), N6⋯O1 3.129(5), N6–H6A⋯O1 122(3), N6–H6A 0.87(3), H6A⋯N1 2.51(3), N6⋯N1 3.150(4), N6–H6A⋯N1 131(3), N6–H6B 0.87(3), H6B⋯N5 2.14(3), N6⋯N5 3.013(4), N6–H6B⋯N5 179(5). | ||
The determination of the molecular structure of the nickel(II) complex 2 proves that a PPh2 moiety has been split from one of the [(2-aminopyrimidin-4-yl)aryl]phosphanes and has been attached to the NH2 group of the second ligand. The nickel(II) ion is now coordinated in a distorted square-pyramidal manner by two phosphorous atoms, two nitrogen atoms and one carbon atom. Pentacoordinate nickel complexes are not uncommon. However, they are mainly observed in a trigonal-bipyramidal geometry with nitrogen and sulphur donors.15 Since the angle P1–Ni1–P2 is close to 140° and the angles P1–Ni1–N4 and P2–Ni1–N4 are larger than 100°, the compound adopts a geometry, which is in between a square-pyramidal and a trigonal-bipyramidal coordination mode. This coordination mode is caused by the bending in the six-membered ring (N1–C–C–C–P1–Ni1) which prevents the donor atoms of the tridentate P,N,P-ligand from being located in one plane with the nickel(II) site. The Ni–P bond lengths differ only slightly. In contrast, there is a big difference in the Ni–N bond lengths: Ni–N1 (1.978 Å) is about 15 pm shorter than Ni1–N4 (2.135 Å), although the nitrogen atom N1 is located in the trans-position to the carbanion site C44, exhibiting a strong trans-influence. This observation is frequently made for rigid tridentate ligands: due to steric restrictions, the M–L distance to the inner donor site is generally found to be considerably shortened compared to the outer ones.16 The reason why C44 is found in the trans-position to N1 is probably an intramolecular hydrogen bond that exists between H6A and N1 (2.51 Å). As expected, the Ni–C44 (1.903 Å) bond is the shortest of all M–L bonds, thus approx. 23 pm shorter than the Ni–N4 bond, reflecting the anionic nature of this carbon atom and the resulting very strong Ni–C bond. The ligand backbone of compound 2 contains several sites that can act as proton donors or acceptors in intermolecular hydrogen bonds, resulting in the formation of a zig-zag chain (see the ESI†) generated by linkages between H3N and N2 as well as H6B and N5. The perchlorate anion interacts via a hydrogen bond with H6A. The Ni–C-bond is neither hydrolysed by the protic amino group of the molecule nor by the protic solvent methanol as a result of the almost perfect shielding by the two diphenylphosphino moieties. These are located in the trans-positions of the distorted square pyramidal coordination environment (see the ESI†).
The 31P{1H} NMR spectrum of 2 shows two sharp resonances for the phosphorous atoms P1 (13.9 ppm) and P2 (47.2 ppm) with a 2JPP coupling of 231.6 Hz. The large coupling is consistent with two different phosphorous sites being located in the trans-position to each other. Since there is no structurally related nickel(II) complex in the literature, we took diamagnetic, four-coordinate complexes as makeshifts to assign the 31P resonance of compound 2. The 31P resonance of trans-(PPh3)2Ni(Ph)Cl is reported to appear at approx. 21 ppm.17 Kirchner et al. investigated a square-planar nickel(II) pincer complex with two trans-orientated arylamino(diphenyl)phosphine units and found a 31P resonance at 77.8 ppm.18 These values allow to assign the resonance at 13.9 ppm to the triaryl-substituted phosphorous atom and the resonance at 47.2 to the diphenylphosphine site carrying one amino group. The general shift of the 31P resonances of 2 (18 VE system) to lower field compared to the model systems (16 VE systems) is due to the increased electron density. Hey-Hawkins et al. found the homoleptic nickel(0) complex Ni(Ph2P-NHPh)2 with a 31P signal at 16.6 ppm.19 Due to the completely asymmetric structure of 2, resulting in four different phenyl groups, and the multiple P,H-couplings, the complete interpretation of the 1H NMR spectrum is difficult. Nevertheless by means of a H,H-COSY experiment, the two sets of AB spin systems (8.54/7.12 ppm, 2JHH = 5.05 Hz; 8.19/7.33 ppm, 2JHH = 5.38 Hz) being affiliated to the two pyrimidine rings can easily be identified. Since the resonance at 8.54 ppm shows a second coupling of 1.20 Hz (either to the NH-group or the N–P phosphorous atom), it can be assigned to the pyrimidine proton next to the ring nitrogen atom in the tridentate P,N,P-ligand. Furthermore there are the signals of two ABCD spin systems (7.84/7.65/7.58/7.50 and 7.62/6.75/6.35/6.26 ppm) standing for the two ortho-substituted phenylene rings. The latter one is considerably deshielded and can therefore be assigned to the bidentate ligand with the Ni–C bond. Examination of the course of the generation of 2 by 31P NMR spectroscopy failed due to the formation of insoluble (HL+ClO4−). However, no intermediate could be detected even at the beginning of the reaction either due to a very rapid transformation of this intermediate into 2 or due to a paramagnetic nature of the intermediate.
ESI-MS measurements further confirm the molecular composition of the cation of 2 (m/z = 767 amu with respect to 58Ni, see the ESI†). In the infrared spectrum of 2, one would expect to generally find the bands of three N–H stretching vibrations, one for the NH–PPh2 unit and the symmetric and the asymmetric vibration of the NH2 group. In fact, there are two bands, one at 3437 and a slightly stronger band at 3354 cm−1. We assign the latter one to the overlapped bands of the N–H stretching vibration10 and the symmetric NH2 stretching vibration.20
Cyclic voltammetry was carried out to get an insight into the redox behaviour of compound 2. The nickel(II) complex is irreversibly oxidized at a peak potential of 0.77 V in acetonitrile solution with respect to the SCE (for a graphic see the ESI†). No reduction could be observed up to a potential of −1.50 V. Electron rich square planar PCP-type pincer complexes of nickel(II) show similar oxidation potentials depending on the nature of the C-donor site,21 while e.g. for less electron rich complexes such as (PPh3)2Ni(NCS)2 no oxidation was found, but irreversible reduction processes.22 This shows the electron-rich nature of the five-coordinate nickel(II) complex 2.
We suggest an intramolecular mechanism for the P,C-bond cleavage and the P,N-bond formation. The nucleophilic character of the amino group of aminopyrimidines is not very strong. In the nucleophilic solvent methanol the formation of methoxy(diphenyl)phosphine as the main product should occur in an intermolecular reaction. We propose, that in the first step, the formation of the dicationic nickel(II) complex A takes place (Scheme 2, top). In A the two P,N-donors are coordinated in a square planar mode. This compound might undergo loose interaction with the perchlorate anions (not drawn). Provided that the P,N bond formation takes place in an intramolecular way, the two nitrogen resp. and the two phosphorous sites have to be oriented trans to each other, a situation which would also prevent steric hindrance of the two diphenylphosphino units.
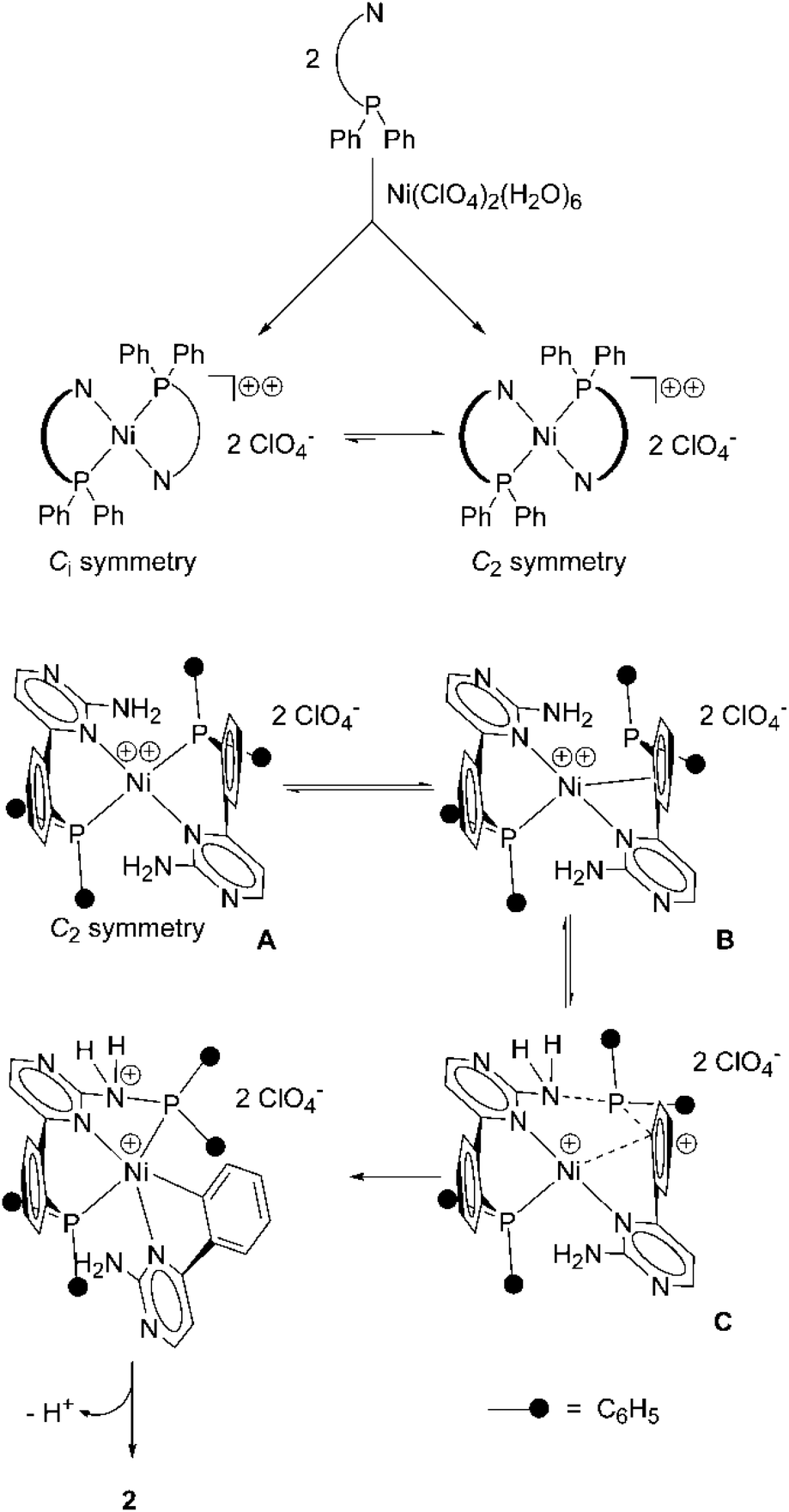 | ||
| Scheme 2 Generation of the nickel(II) complex 2 by P,C-bond cleavage and subsequent P,N-bond formation. | ||
We know from a X-ray structure analysis of the palladium(II) complex (1)PdCl2,5 wherein the palladium centre is cis-coordinated by the phosphorous and the nitrogen atom, that the six-membered ring including P, Pd, and N is severely bent. Due to the bending of the P,N units, compound A might exist in two isomeric forms (Scheme 2, top), wherein the bridging phenylene units either point in opposite directions (Ci symmetry) or in the same direction (C2 symmetry). According to preliminary quantum chemical calculations on the mechanism, the C2 symmetric isomer is about 9 kcal mol−1 lower in energy than the Ci symmetric one. After Ni–P cleavage has occurred in the first step of the mechanism, the detached phosphorous atom has to move towards the NH2 group of the second ligand. This is, in our opinion, strongly favoured for the C2 symmetric isomer (Scheme 2, bottom), since the nickel site can only in this case undergo an interaction with the bridging phenylene unit, that will lead to an electrophilic attack at the phosphorous substituted carbon atom (B). This allows a neighboring amino group to perform a nucleophilic attack at the phosphorous atom which cleaves the P,C-bond and forms the P,N-bond (C). Finally a third equivalent of ligand 1, which is necessary to obtain high yields of the product, takes over the proton from the nitrogen atom and the aminophosphine coordinates to the nickel centre resulting in the formation of compound (2).
This kind of P,C-bond cleavage reaction is up to now limited to nickel(II) and ligand 1. We have carried out the same reaction with manganese(II), iron(II), cobalt(II), copper(II) and zinc(II) perchlorates which do not show any reaction with the ligand. The same was observed for other triphenylphosphine functionalized tertiary aminopyrimidines and pyrazoles, which we frequently use in our group. In none of these cases, P,C-bond cleavage could be found. Instead, expected coordination compounds with intact ligand structures have been observed. The behaviour of these ligands might be explained by steric and electronic considerations: pyrazoles are poor nucleophiles since the N-σ orbital is occupied for the N–H bond, while the N-π orbital is conjugated in the 6-electron ring structure. For [(2-aminopyrimidin-4-yl)aryl]phosphanes carrying a tertiary amino group –NR2, steric hindrance of the groups R will prevent the attack of the phosphorous atom at the amino group.
Experimental
General remarks
All reaction steps were carried out under an argon atmosphere using Schlenk techniques. Nickel(II) perchlorate hexahydrate was purchased from Sigma Aldrich and used without further purification. Ligand 1 was synthesized according to a published procedure.5 All solvents were degassed according to standard techniques before use. 1H, 13C and 31P NMR spectra were recorded on a Bruker Spectrospin Avance 400 device, ESI mass spectra were recorded on a Bruker Esquire 6000 equipment.Synthesis of the nickel(II) complex 2
Nickel(II) perchlorate hexahydrate (373 mg, 1.02 mmol) was dissolved in methanol (20 ml) and 1 (1.11 g; 3.11 mmol) was added to the green solution. The resulting green suspension was stirred for 48 h at room temperature whereby its colour changed to red. The solvent was removed in vacuum and dichloromethane (20 ml) was added, leading to a red solution and a white precipitate, which was filtered off. After evaporation of the solvent from the filtrate, the remaining reddish-brown solid was stirred with toluene (15 ml) at room temperature for 16 h to dissolve residues of the free ligand 1, then filtered off and dried under vacuum. To obtain crystals suitable for an X-ray structure analysis, the compound was recrystallized from a minimum amount of methanol. Yield: 778 mg (896 μmol; 88%) of a reddish-brown solid. Elemental analysis for C44H35ClN6NiO4P2 (867.91): calcd C 60.89%, H 4.06%, N 9.68%; found C 61.19%, H 4.24%, N 9.48%.Cyclic voltammetry
Electrochemical experiments were performed at room temperature in 0.2 M solution of NBu4ClO4 in acetonitrile using a potentiostat/galvanostat 273 A of Princeton Applied Research, a platinum foil as the working electrode, a platinum net as the counter electrode and a saturated calomel electrode as the reference electrode. The scan rate was 100 mV s−1. The ferrocene/ferrocenium redox couple served as the internal reference (+0.42 V vs. SCE).X-ray structure analysis of 2
Crystal data and refinement parameters for compound 2 are collected in Table 1. The structure was solved using a direct method (SIR9223), completed by subsequent difference Fourier syntheses, and refined by full-matrix least-squares procedures.24 A semi-empirical absorption correction from equivalents (Multiscan) was carried out.25 The molecule was found co-crystallized with two equivalents of methanol, one of which being disordered. The hydrogen atoms bound to the nitrogen atoms N3 and N6 were located in the difference Fourier synthesis and were refined semi-freely with the help of a distance restraint, while constraining their U-values to 1.2 times the U(eq) values of the corresponding nitrogen atoms. All the other hydrogen atoms were placed in calculated positions and refined using a riding model. CCDC-1014112 contains the supplementary crystallographic data for this paper.| 2 | |
|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|, ωR2 = [∑ω(Fo2 − Fc2)2/∑ωFo2]1/2. b GooF = [∑ω(Fo2 − Fc2)2/(n − p)]1/2. | |
| Empirical formula | C46H43ClN6NiO6P2 |
| Formula weight | 931.96 |
| Crystal size [mm] | 0.16 × 0.10 × 0.06 |
| T [K] | 150(2) |
| λ [Å] | 1.54184 |
| Crystal system | Triclinic |
| Space group |
P |
| a [Å] | 11.6115(7) |
| b [Å] | 13.4918(9) |
| c [Å] | 14.9171(10) |
| α [°] | 87.203(5) |
| β [°] | 75.689(6) |
| γ [°] | 70.567(6) |
| V [Å3] | 2134.0(2) |
| Z | 2 |
| ρ calcd. [g cm−3] | 1.450 |
| μ [mm−1] | 2.417 |
| θ-range [°] | 3.48–62.85 |
| Refl. coll. | 14868 |
| Indep. refl. | 6800 [Rint = 0.0275] |
| Data/restr./param. | 6800/122/612 |
| Final R indices [I > 2σ(I)]a | 0.0525, 0.1340 |
| R indices (all data) | 0.0610, 0.1419 |
| GooFb | 1.027 |
| Δρmax/min (e Å−3) | 1.307/−1.224 |
Conclusions
The reaction of [(2-aminopyrimidin-4-yl)aryl]phosphane with nickel(II)perchlorate hexahydrate leads to a novel five-coordinate nickel(II) complex in high yield wherein the nickel(II) site is coordinated by a tridentate P,N,P′-ligand and a bidentate, carbanionic N,C-ligand. Both ligand systems are formed from the bidentate[(2-aminopyrimidin-4-yl)aryl]phosphane which undergoes a P,C-bond cleavage. The pyridinyl amino group acts as a nucleophile and takes over a PPh2 unit in an intramolecular process from the neighbouring ligand moiety.Acknowledgements
Financial support from the DFG-funded transregional collaborative research centre SFB/TRR 88 “Cooperative effects in homo- and heterometallic complexes (3MET)” is gratefully acknowledged.Notes and references
- K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374 CrossRef CAS; R. J. P. Corriu and J. P. Masse, J. Chem. Soc., Chem. Commun., 1972, 144 RSC; J. Terao, H. Watanabe, A. Ikumi, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2002, 124, 4222 CrossRef PubMed; J. Terao, A. Ikumi, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2003, 125, 5646 CrossRef PubMed; L.-C. Liang, P.-S. Chien, J.-M. Lin, M.-H. Huang, Y.-L. Huang and J.-H. Liao, Organometallics, 2006, 25, 1399 CrossRef; O. Vechorkin and X. Hu, Angew. Chem., Int. Ed., 2009, 48, 2937 CrossRef PubMed; O. Vechorkin, Z. Csok, R. Scopelliti and X. Hu, Chem. – Eur. J., 2009, 15, 3889 CrossRef PubMed; S. Gu and W. Chen, Organometallics, 2009, 28, 909 CrossRef.
- A. Devasagayaraj, T. Stüdemann and P. Knochel, Angew. Chem., Int. Ed. Engl., 1995, 34, 2723 CrossRef CAS; R. Giovannini and P. Knochel, J. Am. Chem. Soc., 1998, 120, 11186 CrossRef; A. E. Jensen, W. Dohle and P. Knochel, Tetrahedron, 2000, 56, 4197 CrossRef; C. E. Tucker and J. G. de Vries, Top. Catal., 2002, 19, 111 CrossRef; J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 12527 CrossRef PubMed; J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 14726 CrossRef PubMed; A. Gavryushin, C. Kofink, G. Manolikakes and P. Knochel, Org. Lett., 2005, 7, 4871 CrossRef PubMed; A. Gavryushin, C. Kofink, G. Manolikakes and P. Knochel, Tetrahedron, 2006, 62, 7521 CrossRef PubMed; E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738 CrossRef PubMed.
- W. Keim, B. Hoffmann, R. Lodewick, M. Peuckert and G. Schmitt, J. Mol. Catal., 1979, 6, 79 CrossRef CAS; M. Peuckert and W. Keim, Organometallics, 1983, 2, 594 CrossRef; A. Behr, V. Falbe, U. Freudenberg and W. Keim, Isr. J. Chem., 1986, 27, 277 CrossRef; G. Wilke, Angew. Chem., Int. Ed. Engl., 1988, 27, 185 CrossRef; N. A. Kolhatkar, A. M. Monfette, S. Lin and M. J. Miri, J. Polym. Sci., 2002, 50, 986 CrossRef; J. N. L. Dennett, A. L. Gillon, K. Heslop, D. J. Hyett, J. S. Fleming, C. E. Lloyd-Jones, A. G. Orpen, P. G. Pringle, D. F. Wass, J. N. Scutt and R. H. Weatherhead, Organometallics, 2004, 23, 6077 CrossRef; G. Noël, J. C. Röder, S. Dechert, H. Pritzkow, L. Bolk, S. Mecking and F. Meyer, Adv. Synth. Catal., 2006, 348, 887 CrossRef; F.-S. Liu, H.-B. Hu, Y. Xu, L.-H. Guo, S.-B. Zai, K.-M. Song, H.-Y. Gao, L. Zhang, F.-M. Zhu and Q. Wu, Macromolecules, 2009, 42, 7789 CrossRef.
- G. Nord, A. C. Hazell, R. G. Hazell and O. Farver, Inorg. Chem., 1983, 22, 3429 CrossRef CAS; P. J. Spellane, R. J. Watts and C. J. Curtis, Inorg. Chem., 1983, 22, 4060 CrossRef; P. S. Braterman, G. H. Heat, A. J. Mackenzie, B. C. Noble, R. D. Peacock and L. J. Yellowlees, Inorg. Chem., 1984, 23, 3425 CrossRef; E. C. Tyo, A. W. Castleman Jr., D. Schröder, P. Milko, J. Roithova, J. M. Ortega, M. A. Cinellu, F. Cocco and G. Minghetti, J. Am. Chem. Soc., 2009, 131, 13009 CrossRef PubMed; G. Minghetti, S. Stoccoro, M. A. Cinellu, G. L. Petretto and A. Zucca, Organometallics, 2008, 27, 3415 CrossRef.
- S. Farsadpour, L. Taghizadeh Ghoochany, Y. Sun and W. R. Thiel, Eur. J. Inorg. Chem., 2011, 4603 CrossRef CAS; S. Farsadpour, L. Taghizadeh Ghoochany, S. Shylesh, G. Dörr, A. Seifert, S. Ernst and W. R. Thiel, ChemCatChem, 2012, 4, 395 CrossRef; L. Taghizadeh Ghoochany, C. Kerner, S. Farsadpour, Y. Sun, F. Menges, G. Niedner-Schatteburg and W. R. Thiel, Eur. J. Inorg. Chem., 2013, 4305 CrossRef.
- C. M. Chen, Q. Z. Ye, Z. M. Zhu, B. L. Wanner and C. T. Walsh, J. Biol. Chem., 1990, 265, 4461 Search PubMed; J. W. McGrath, G. B. Wisdom, G. McMullan, M. J. Larkin and J. P. Quinn, Eur. J. Biochem., 1995, 234, 225 Search PubMed; A. Inoue, H. Shinokubo and K. Oshima, J. Am. Chem. Soc., 2003, 125, 1484 CrossRef CAS PubMed.
- M. Randles, A. C. Willis, M. P. Cifuentes and M. G. Humphrey, J. Organomet. Chem., 2007, 692, 4467 CrossRef CAS PubMed; W. H. Watson, G. Wu and M. G. Richmond, Organometallics, 2006, 25, 930 CrossRef; S. M. Waterman, V.-A. Tolhurst, M. G. Humphrey, B. W. Skelton and A. H. White, J. Organomet. Chem., 1996, 515, 89 CrossRef; A. J. Deeming, M. K. Shinhmar, A. J. Arce and Y. De Sanctis, J. Chem. Soc., Dalton Trans., 1999, 1153 RSC.
- N. M. Doherty, G. Hogarth, S. A. R. Knox, K. A. Macpherson, F. Melchior, D. A. V. Morton and A. G. Orpen, Inorg. Chim. Acta, 1992, 198–200, 257 CrossRef CAS; W.-D. Wang and R. Eisenberg, J. Am. Chem. Soc., 1990, 112, 1833 CrossRef; H. Wachtler, W. Schuh, K.-H. Ongania, K. Wurst and P. Peringer, Organometallics, 1998, 17, 5640 CrossRef; P. E. Garrou, Chem. Rev., 1985, 85, 171 CrossRef.
- J. F. Hartwig, R. G. Bergman and R. A. Andersen, J. Organomet. Chem., 1990, 394, 417 CrossRef CAS.
- P. H. M. Budzelaar, J. A. van Doorn and N. Meijboom, Recl. Trav. Chim. Pays-Bas, 1991, 110, 420–432 CrossRef CAS.
- M. F. Cain, S. C. Reynolds, B. J. Anderson, D. S. Glueck, J. A. Golen, C. E. Moore and A. L. Rheingold, Inorg. Chim. Acta, 2011, 369, 55 CrossRef CAS PubMed; S. A. Reiter, S. D. Nogai and H. Schmidbaur, Z. Anorg. Allg. Chem., 2005, 631, 2595 CrossRef.
- Y. El Harouch, V. Cadierno, A. Igau, B. Donnadieu and J.-P. Majoral, J. Organomet. Chem., 2004, 689, 953 CrossRef CAS PubMed.
- A. Caballero, F. A. Jalón, B. R. Manzano, G. Espino, M. Pérez-Manrique, A. Mucientes, F. J. Poblete and M. Maestro, Organometallics, 2004, 23, 5694 CrossRef CAS; A. Inoue, H. Shinokubo and K. Oshima, J. Am. Chem. Soc., 2003, 125, 1484 CrossRef PubMed; T. J. Geldbach and P. S. Pregosin, Eur. J. Inorg. Chem., 2002, 1907 CrossRef; S.-C. Tsai, Y.-S. Fu, J.-H. Liao and S. J. Yu, Helv. Chim. Acta, 2006, 89, 3007 CrossRef.
- A. Reis, D. Dehe, S. Farsadpour, I. Munstein, Y. Sun and W. R. Thiel, New J. Chem., 2011, 35, 2488 RSC; D. Dehe, I. Munstein, A. Reis and W. R. Thiel, J. Org. Chem., 2011, 76, 1151 CrossRef CAS PubMed.
- J. K. Stalick and J. A. Ibers, Inorg. Chem., 1969, 8, 1084 CrossRef CAS; J. K. Stalick and J. A. Ibers, Inorg. Chem., 1969, 8, 1090 CrossRef; P. S. Shetty and Q. Fernando, J. Am. Chem. Soc., 1970, 92, 3964 CrossRef; T. E. Kokina, L. A. Glinskaya, R. F. Klevtsova and S. V. Larionov, J. Struct. Chem., 2002, 43, 312 CrossRef; S. V. Larionov, T. E. Kokina, L. A. Glinskaya and R. F. Klevtsova, Russ. J. Coord. Chem., 2002, 28, 560 CrossRef.
- (a) F. Bachechi, Struct. Chem., 2003, 14, 263 CrossRef CASH. Glas, K. Köhler, P. Maas, E. Herdtweck, M. Spiegler and W. R. Thiel, Eur. J. Inorg. Chem., 2001, 4, 2075 CrossRef.
- A. Zeller, E. Herdtweck and T. Strassner, Eur. J. Inorg. Chem., 2003, 1802 CrossRef CAS; H. A. Bronstein and C. K. Luscombe, J. Am. Chem. Soc., 2009, 131, 12894 CrossRef PubMed.
- D. Benito-Garagorri, V. Bocokić, K. Mereiter and K. Kirchner, Organometallics, 2006, 25, 3817 CrossRef CAS.
- O. Kühl, P. C. Junk and E. Hey-Hawkins, Z. Anorg. Allg. Chem., 2000, 626, 1591 CrossRef.
- (a) L. Varga, T. Nagy, I. Kövesdi, J. Benet-Buchholz, G. Dormán, L. Ürge and F. Darvas, Tetrahedron, 2003, 59, 655 CrossRef CASA. Alberola, C. Andrés, A. González Ortega, R. Pedrosa and M. Vicente, Synth. Commun., 1987, 17, 1309 CrossRef CAS; X.-Y. Yu, P.-G. Yi, D.-H. Ji, B.-R. Zeng, X.-F. Li and X. Xu, Dalton Trans., 2012, 41, 3684 RSC.
- A. Castonguay, A. L. Beauchamp and D. Zargarian, Organometallics, 2008, 27, 5723 CrossRef CAS.
- K. Ramalingam, R. Thiruneelakandan, G. Bocelli and L. Righi, Transition Met. Chem., 2012, 37, 265 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- CrysAlisPro, Agilent Technologies, Version 1.171.36.24, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data, additional structure plots. CCDC-1014112. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt02158d |
| This journal is © The Royal Society of Chemistry 2015 |