
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient halogen photoelimination from dibromo, dichloro and difluoro tellurophenes†
Elisa I.
Carrera
and
Dwight S.
Seferos
*
Department of Chemistry, University of Toronto, 80 St. George St., Toronto, ON M5S 3H6, Canada. E-mail: dseferos@chem.utoronto.ca; Web: http://www.chem.utoronto.ca/staff/seferos/
First published on 19th August 2014
Abstract
We present the reactivity and photochemistry of 2,5-diphenyltellurophene. A change in oxidation state from Te(II) to Te(IV) occurs by oxidative addition of bromine, chlorine, and fluorine from appropriate halogen sources. Photoreductive halogen elimination is demonstrated using optical absorption spectroscopy and NMR spectroscopy. The photodebromination reaction occurs with 16.9% quantum yield, the highest value for any Te compound. Photoreductive elimination of chlorine and fluorine occurs with quantum yields of 1.6% and 2.3%, respectively, albeit with less efficient halogen trapping when an organic trap is used. Improved fluorine trapping was achieved using water, allowing for much cleaner photodefluorination. This is the first example of photodefluorination from a tellurium compound.
Introduction
Tellurium is a metalloid that undergoes oxidation reactions with halogens and other oxidants.1–4 Organotellurium compounds including tellurium-containing heterocyclic dyes undergo thermally-driven reductive elimination,3,5 however the photochemistry of such compounds has not been extensively studied. In particular, photodehalogenation has been the subject of several recent reports because it is important for catalysis and releasing energy stored in chemical bonds. Gabbaï and coworkers synthesized a dinuclear Te–Pt complex in which Cl2 could be added across the Te and Pt atoms. Light-driven reductive elimination of chlorine was observed for this complex with a 4.4% quantum yield.6 Our group recently reported the first example of photoreductive elimination of bromine and chlorine from a transition metal-free mononuclear organotellurium compound, 2,5-bis[5-(N,N′-dihexylisoindigo)]tellurophene.7 The extended conjugation through tellurophene to isoindigo allows for the use of 505 nm (green) light to drive the photoreaction. However, the quantum yields are low, near 0.2%. To put this in context, dehalogenation quantum yields between 5% and 38% are considered quite good and have been reported for bimetallic Pt–Au, Au–Au, Pt–Rh, and Pt–Pt complexes,8 and recently up to 82% for a monometallic Pt complex.9 Although the primary photoexcitation in the isoindigo-tellurophene system occurs to an excited state that has antibonding Te–X character, it may rapidly decay to a lower-energy excited state that does not have antibonding Te–X character, leading to the relatively poor quantum efficiency. This hypothesis is supported by density functional theory (DFT) calculations. It is therefore important to develop a tellurophene compound where primary photoexcitation occurs to an excited state that has antibonding Te–X character and there are no lower lying excited states. Photodehalogenation from these types of tellurophenes have not been reported by us or others.Herein, we report photoreductive elimination of bromine, chlorine, and fluorine from 2,5-diphenyltellurophene (PT) (Scheme 1). While the dibromo- and dichloro-adducts of PT (PT-Br2 and PT-Cl2) have been previously reported,4,7 the difluoro-adduct (PT-F2) is reported here for the first time. We observe photodehalogenation quantum yields of up to 16.9% for the dibromo-compound at 5 M halogen trap concentration, which is a nearly 100-fold improvement from previous work and is comparable to efficient transition metal based systems.9 The chlorine and even fluorine photoreduction reactions also take place albeit with decomposition reactions when an organic trap is used. Efficient fluorine trapping using water leads to improved photodefluorination reactions with little decomposition. This is the first example of photoreductive defluorination from an organotellurium compound and to the best of our knowledge, the only known photodefluorination reactions occur with organic substrates.10
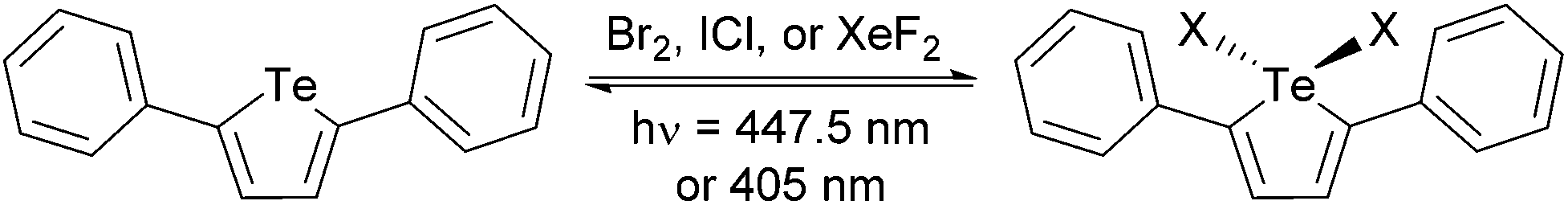 | ||
| Scheme 1 Oxidative addition and photoreductive elimination of halogens. | ||
Results and discussion
DFT calculations were used to determine whether the halogenated compounds (PT-X2) would be good candidates for photoreductive elimination. Geometry optimizations and time dependent (TD-DFT) calculations11 were performed on PT and the halogen adducts using the Gaussian 09 software suite12 at the B3LYP level of theory13 with a split basis set (LANL2DZ for Te,14 and 6-31G(d) for all other atoms15). The TD-DFT calculations predict two strong optical transitions (f = 0.30 and 0.42) for PT, consisting of transitions from the HOMO to the LUMO and from the HOMO to the LUMO+1. The molecular orbital (MO) diagrams indicate that these are π→π* transitions (Fig. 1a). The LUMO is delocalized across the entire molecule. Upon halogen addition, the energy of the HOMO to LUMO gap decreases, which in turn leads to a lower energy calculated optical transition. The shift is less pronounced for PT-F2 and is roughly equal in magnitude for PT-Br2 and PT-Cl2 (Fig. S1, ESI†). Two strong optical transitions are predicted for all three halogenated compounds. The lower energy absorption is dominated by a HOMO→LUMO transition, and the LUMO possesses significant Te–X antibonding character (Fig. 1b). The use of the Stuttgart/Dresden ECP16 for tellurium does not lead to significant differences in the DFT results compared to the LANL2DZ basis set. Since the compounds exhibit charge-transfer character, the calculations were also repeated using the long-range correlation functional CAM-B3LYP17 with the SDD ECP for tellurium. Similar to our previous work,13a CAM-B3LYP is less accurate at predicting energy levels than B3LYP due to an underestimation of the HOMO energy, and an overestimation of the LUMO energy (Fig. S2a, ESI†), resulting in overall higher-energy calculated transitions for PT and PT-Br2. A comparison of the calculated and experimental absorption spectra indicates that the B3LYP functional agrees best with experiment, and changing the basis set from LANL2DZ to SDD does not alter the calculated spectra significantly (Fig. S2b–c, ESI†). It is important to note, however, that the nature of the transitions and the MOs remain the same regardless of the functional used, with significant Te–X antibonding character in the LUMO. Population of this state through photoexcitation should therefore promote halogen dissociation.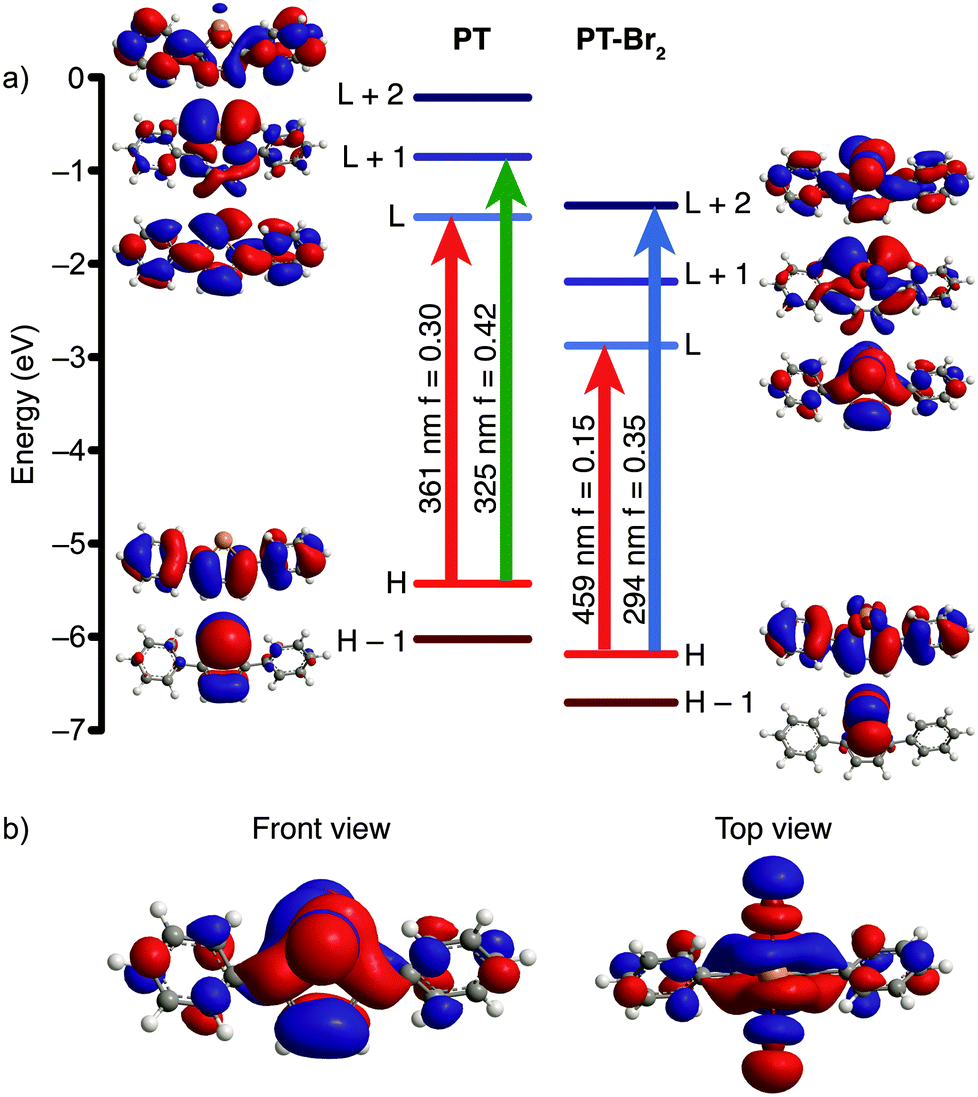 | ||
| Fig. 1 (a) Excited state transitions of PT and PT-Br2 with oscillator strength greater than 0.1 as predicted by TD-DFT, and their associated MOs (isocontour value of 0.02; H refers to HOMO, L refers to LUMO). (b) Front and top view of the calculated LUMO for PT-Br2. | ||
To test this hypothesis, PT was synthesized according to a modified literature method.4 Briefly, tellurium powder was reduced by treatment with sodium borohydride in a refluxing ethanol–water mixture to give sodium telluride. Once the reaction mixture changed from purple to colorless (1.5 h), the temperature was reduced to 80 °C and 1,4-diphenyl-1,3-butadiyne was added and heated for 21 h to give PT in 61% yield after workup. The oxidized tellurophenes PT-Br2, PT-Cl2, and PT-F2 were prepared by reaction with Br2, iodine monochloride (ICl), and xenon difluoride (XeF2), respectively. The identity of the newly synthesized compound PT-F2 was confirmed by NMR spectroscopy and elemental analysis (ESI†).
Optical absorption titration experiments were carried out on dilute solutions of PT (2 × 10−5 M in chloroform) to monitor the conversion to the halogenated species (Fig. 2). For reference, the absorption spectrum of PT consists of a peak at 342 nm (ε = 2.24 × 104 M−1 cm−1) with increasing absorption at wavelengths approaching the high-energy solvent cut-off. Upon halogen addition, the peak at 342 nm decreases while a red-shifted absorption band develops. The magnitude of the observed red-shift increases moving to heavier halogens (PT-F2, λmax = 395 nm, ε = 1.36 × 104 M−1 cm−1; PT-Cl2, λmax = 416 nm, ε = 9.42 × 103 M−1 cm−1; PT-Br2, λmax = 433 nm, ε = 6.34 × 103 M−1 cm−1). For PT-Br2, the magnitude of the red-shift is large enough to observe the fully resolved dual-band absorption. All of this data is consistent with the TD-DFT calculations described above.
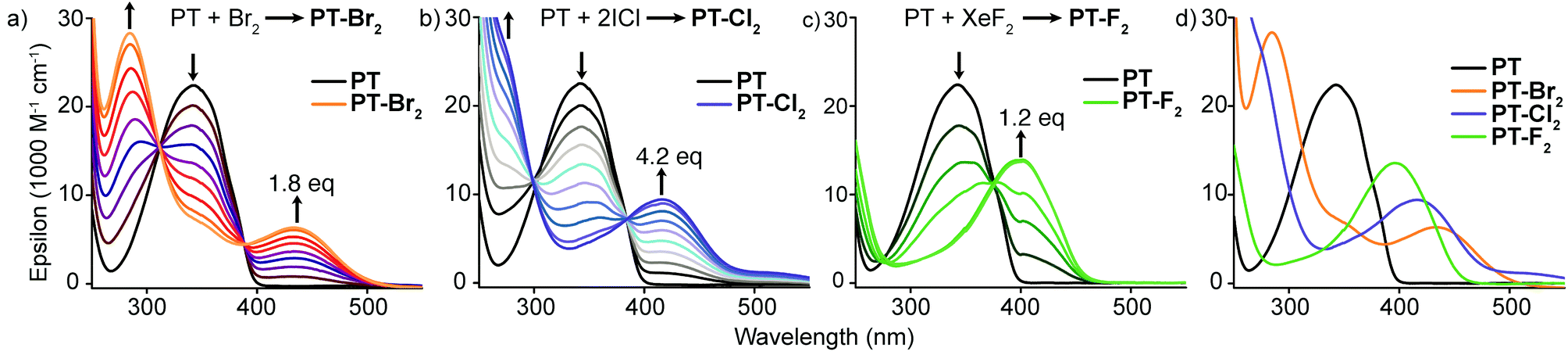 | ||
| Fig. 2 Optical absorption titration experiments with (a) Br2, (b) ICl, and (c) XeF2; (d) comparison of absorption spectra of PT and its halogenated adducts. | ||
At 2 × 10−5 M concentration, conversion of PT to PT-F2, PT-Cl2, and PT-Br2 was complete after the addition of 1.25, 4.2, and 1.8 equivalents of halogen source, respectively. Incomplete conversion after stoichiometric addition of halogen was consistent over several experimental runs. In more concentrated solutions, incomplete conversion was also observed by NMR spectroscopy after stoichiometric halogen addition, however higher conversions were achieved compared to the optical absorption titration experiments. This discrepancy suggests that an excess of halogens is required to drive the reaction forward, especially in dilute solution.
Photolysis experiments were carried out at wavelengths corresponding to the low energy absorption peaks of each compound. A 447.5 nm tri-LED setup was used to irradiate solutions of PT-Br2 (see ESI† for light source specifications). 2,3-Dimethyl,1-3-butadiene (DMBD) was used as a halogen trap. The change in the absorption spectrum during photoexcitation was used to follow the reaction progress (Fig. 3a). A rapid decrease in the low energy absorption is observed along with an increase in absorption around 340 nm. The final trace is obtained within 12 seconds, and matches well with the spectrum of PT. A slight deviation from the high energy isosbestic point suggests minor decomposition.
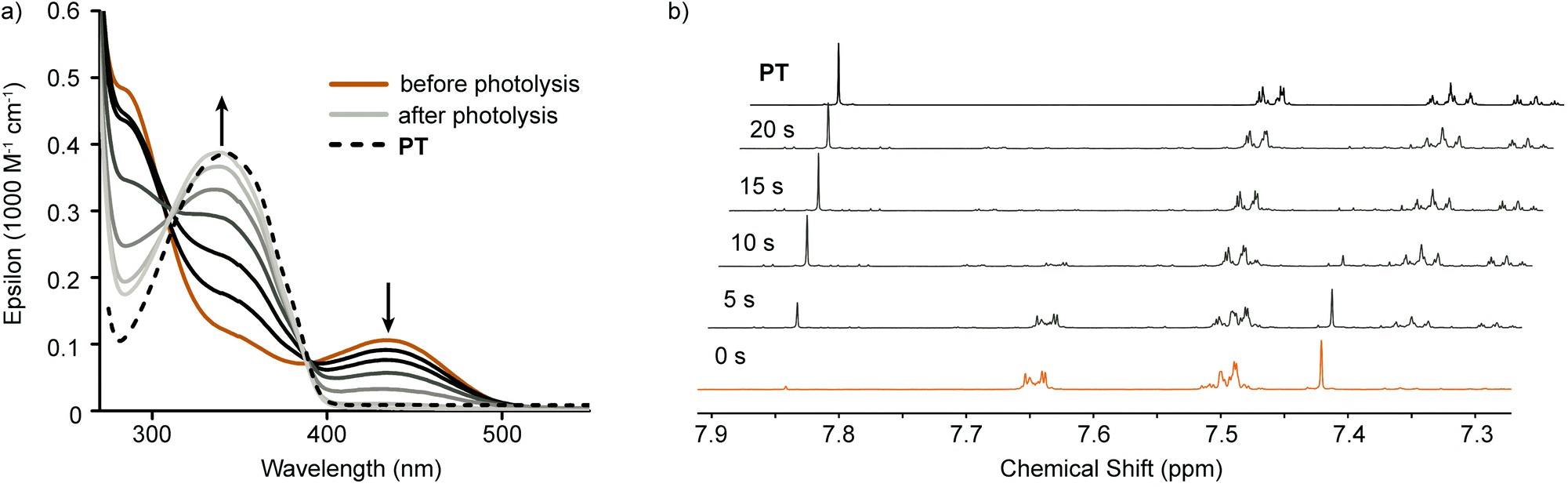 | ||
| Fig. 3 Photolysis of PT-Br2 with 447.5 nm light over time monitored by (a) optical absorption spectroscopy and (b) 1H NMR spectroscopy. | ||
Photolysis reactions were carried out on more concentrated solutions and probed by 1H NMR spectroscopy. The symmetric tellurophene protons give rise to a singlet with satellite peaks arising from proton–tellurium coupling. This signature allows for easy identification of the different tellurophene compounds present in solution. Accordingly, 2.5 mM solutions of PT-Br2 were irradiated in the presence of DMBD (0.9 M) at 5 second time intervals and a spectrum was obtained after each interval (Fig. 3b). During the course of the reaction, the singlet corresponding to the tellurophene protons (7.42 ppm) is depleted and a singlet corresponding to the tellurophene protons of PT develops (7.84 ppm). The changes in chemical shifts of the phenyl proton signals are also consistent with restoration of PT. Evidence of slight decomposition is observed, however these are quite minor and will be discussed below.
Control experiments were carried out to confirm that the reverse reaction is indeed light-driven rather than by other means. Solutions of PT-Br2 in the presence of 1 M DMBD showed no change in the 1H NMR spectrum after 45 hours in the dark. When exposed to room lighting, PT was restored within 45 hours (Fig. S3, ESI†). In the absence of trap, the 1H NMR spectrum of PT-Br2 was unchanged when stored in the dark, but showed slow conversion to PT when exposed to room lighting (30% after 6 days; Fig. S4, ESI†). Finally, a solution of PT-Br2 was irradiated with 447.5 nm light in the absence of trap. The 1H NMR spectrum shows evidence of restored PT, however a significant amount of decomposition occurs (Fig. S5, ESI†). In the presence of trap, most of the liberated bromine reacts with DMBD, however a small amount reacts with the conjugated tellurophene, leading to minor decomposition as evidenced by 1H NMR and optical absorption spectroscopy.
In order to quantify the efficiency of the photoreductive elimination reaction, the photochemical quantum yield (Φ) was determined using potassium ferrioxalate standard actinometry (details provided in the ESI†).18 At 2 M DMBD concentration, the quantum yield is 8.5%, and a linear increase in quantum yield is observed with increasing trap concentration (y = 2.7732x + 2.9707, R2 = 0.9981; Table 1). At the highest experimental DMBD concentration (5 M), the quantum yield of photodebromination is 16.9%. This is the first example of efficient dehalogenation from a compound that does not contain a metal.
Analogous photolysis experiments were carried out on solutions of PT-Cl2 and PT-F2, using a 447.5 nm tri-LED and a 405 nm LED, respectively. Photoreductive elimination of both chlorine and fluorine is successful (ΦPT-Cl2 = 1.6% and ΦPT-F2 = 2.3% as calculated from the loss of halogenated compound in solution); however, decomposition products are observed. The optical absorption spectra after photolysis are skewed to higher energies compared to PT (Fig. S6, ESI†). This suggests that although reductive elimination of chlorine and fluorine is successful, decomposition products are formed in significant proportions. This is further supported by the 1H NMR spectra of photolyzed samples (Fig. S7, ESI†). In these cases, it is likely that the liberated halogens are more reactive toward PT than the trap, leading to a large proportion of decomposition products. Given the extremely high reactivity of fluorine with water to produce HF,19 photodefluorination was carried out on a biphasic mixture of PT-F2 in chloroform and water, in the absence of DMBD. Here we observe less decomposition by 1H NMR spectroscopy due to improved fluorine trapping by water. Fluorine trapping was even further improved when trapping events were no longer limited to the interface between organic and aqueous phases and photolysis experiments were carried out in THF–H2O mixtures (35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :65; see ESI† for Experimental details). As a result, little decomposition of PT is observed by 1H NMR, 19F NMR, and optical absorption spectroscopy (Fig. 4 and Fig. S8, ESI†). Additionally, analysis of the aqueous phase after work-up shows a decrease in pH (qualitatively, using pH paper) and a peak at −129.6 ppm in the 19F NMR spectrum, which is consistent with the presence of F− in aqueous solutions of HF.20 Bulk photolysis on a larger scale led to 77% isolated yield of recovered PT.
:65; see ESI† for Experimental details). As a result, little decomposition of PT is observed by 1H NMR, 19F NMR, and optical absorption spectroscopy (Fig. 4 and Fig. S8, ESI†). Additionally, analysis of the aqueous phase after work-up shows a decrease in pH (qualitatively, using pH paper) and a peak at −129.6 ppm in the 19F NMR spectrum, which is consistent with the presence of F− in aqueous solutions of HF.20 Bulk photolysis on a larger scale led to 77% isolated yield of recovered PT.
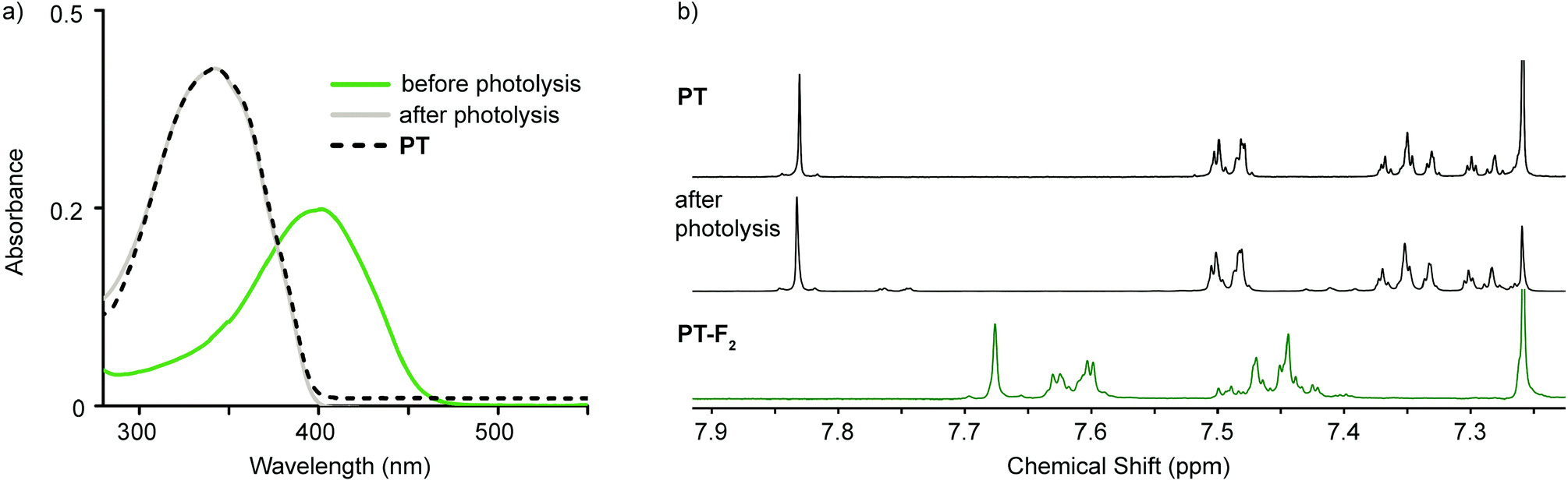 | ||
| Fig. 4 Bulk photolysis of PT-F2 in THF–H2O with 447.5 nm light over time monitored by (a) optical absorption spectroscopy and (b) 1H NMR spectroscopy. | ||
Conclusions
We have shown that 2,5-diphenyltellurophene can be oxidized in the presence of bromine, chlorine, and fluorine and the halogens can be photoreductively eliminated by photoexcitation into the low energy absorption band of the halogenated species. Photoreductive elimination of bromine occurs rapidly with 16.9% quantum efficiency and minimal decomposition using 447.5 nm blue light. The 16.9% quantum yield achieved is comparable to the performance of several transition metal catalysts reported in the literature. Photoreductive elimination of chlorine and fluorine also occurs in the presence of organic trap, albeit with decomposition reactions. However, a significant improvement in fluorine trapping is realized when water is used as a halogen trap, leading to restoration of the reduced tellurophene in 77% isolated yield and very little evidence of decomposition. This is the first example of photoreductive elimination of fluorine from an inorganic compound, and is an unexpected result given the strength of typical M–F bonds. Improving the quantum efficiency of halogen photoreductive elimination reactions is currently under investigation.Acknowledgements
This work was supported by NSERC, the CFI, The Ontario Research Fund, DuPont (for a Young Professor Grant), and the Alfred P. Sloan Foundation (for a Research Fellowship in Chemistry).Notes and references
- M. R. Detty and A. E. Friedman, Organometallics, 1994, 13, 533–540 CrossRef CAS.
- M. R. Detty, A. E. Friedman and M. McMillans, Organometallics, 1994, 13, 3338–3345 CrossRef CAS.
- M. R. Detty and H. R. Luss, Organometallics, 1986, 5, 2250–2256 CrossRef.
- T. M. McCormick, A. A. Jahnke, A. J. Lough and D. S. Seferos, J. Am. Chem. Soc., 2012, 134, 3542–3548 CrossRef CAS PubMed.
- (a) M. R. Detty, Organometallics, 1991, 10, 702–712 CrossRef CAS; (b) M. R. Detty and T. M. Frade, Organometallics, 1993, 12, 2496–2504 CrossRef CAS; (c) A. A. Jahnke, G. W. Howe and D. S. Seferos, Angew. Chem., Int. Ed., 2010, 49, 10140–10144 CrossRef CAS PubMed.
- T.-P. Lin and F. P. Gabbaï, J. Am. Chem. Soc., 2012, 134, 12230–12238 CrossRef CAS PubMed.
- E. I. Carrera, T. M. McCormick, M. J. Kapp, A. J. Lough and D. S. Seferos, Inorg. Chem., 2013, 52, 13779–13790 CrossRef CAS PubMed.
- (a) T. R. Cook, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 28–29 CrossRef CAS PubMed; (b) T. R. Cook, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 28–29 CrossRef CAS PubMed; (c) T. S. Teets and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 7411–7420 CrossRef CAS PubMed; (d) T. S. Teets, D. A. Lutterman and D. G. Nocera, Inorg. Chem., 2010, 49, 3035–3043 CrossRef CAS PubMed; (e) T. R. Cook, B. D. McCarthy, D. A. Lutterman and D. G. Nocera, Inorg. Chem., 2012, 51, 5152–5163 CrossRef CAS PubMed; (f) D. C. Powers, M. B. Chambers, T. S. Teets, N. Elgrishi, B. L. Anderson and D. G. Nocera, Chem. Sci., 2013, 4, 2880–2885 RSC.
- A. R. Karikachery, H. B. Lee, M. Masjedi, A. Ross, M. A. Moody, X. Cai, M. Chui, C. D. Hoff and P. R. Sharp, Inorg. Chem., 2013, 52, 4113–4119 CrossRef PubMed.
- (a) J. Cheng, X. Liang, S. Yang and Y. Hu, Chem. Eng. J., 2014, 239, 242–249 CrossRef CAS PubMed; (b) H. Hidaka, T. Tsukamoto, T. Oyama, Y. Mitsutsuka, T. Takamura and N. Serpone, Photochem. Photobiol. Sci., 2013, 12, 751–759 RSC; (c) J. Burdeniuc, P. E. M. Siegbahn and R. H. Crabtree, New J. Chem., 1998, 22, 503–510 RSC.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- M. J. Frisch, et al., GAUSSIAN 09, (Revision A.1), Gaussian, Inc, Wallingford, CT, 2009 Search PubMed.
- (a) T. M. McCormick, C. R. Bridges, E. I. Carrera, P. M. DiCarmine, G. L. Gibson, J. Hollinger, L. M. Kozycz and D. S. Seferos, Macromolecules, 2013, 46, 3879–3886 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; (c) A. D. Becke, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS PubMed.
- (a) J. S. Gancheff, P. A. Denis and F. E. Hahn, J. Mol. Struct. (THEOCHEM), 2010, 941, 1–9 CrossRef CAS PubMed; (b) M. Dolg and X. Cao, Chem. Rev., 2012, 112, 403–480 CrossRef CAS PubMed.
- (a) T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS PubMed; (b) S. Kupfer, J. Guthmuller and L. González, J. Chem. Theory Comput., 2013, 9, 543–554 CrossRef CAS.
- (a) C. A. Parker, Proc. R. Soc. London, Ser. A., 1953, 220, 104–116 CrossRef; (b) C. G. Hatchard and C. A. Parker, Proc. R. Soc. London, Ser. A., 1956, 235, 518–536 CrossRef CAS; (c) H. J. Kuhn, S. E. Braslavsky and R. Schmidt, Pure Appl. Chem., 2004, 76, 2105–2146 CrossRef CAS.
- (a) F. Fichter and W. Bladergroen, Helv. Chim. Acta, 1927, 10, 549–552 CrossRef CAS; (b) G. H. Cady, J. Am. Chem. Soc., 1935, 57, 246–249 CrossRef CAS.
- J. B. Gerken, J. Fluorine Chem., 2011, 132, 68–70 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, supplementary figures and tables, and computational data. See DOI: 10.1039/c4dt01751j |
| This journal is © The Royal Society of Chemistry 2015 |