
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterization and solid-state photoluminescence studies of six alkoxy phenylene ethynylene dinuclear palladium(II) rods†
João
Figueira‡
a,
Wojciech
Czardybon§
a,
José Carlos
Mesquita
a,
João
Rodrigues
 *a,
Fernando
Lahoz
*b,
Luca
Russo¶
c,
Arto
Valkonen||
c and
Kari
Rissanen
*c
*a,
Fernando
Lahoz
*b,
Luca
Russo¶
c,
Arto
Valkonen||
c and
Kari
Rissanen
*c
aCQM – Centro de Química da Madeira, MMRG, Universidade da Madeira, Campus Universitário da Penteada, 9000-390 Funchal, Portugal. E-mail: joaor@uma.pt; Web: http://www.uma.pt/jrmmrg Fax: +351 291705149/249; Tel: +351 291705108
bDepartamento de Física Fundamental y Experimental, Electrónica y Sistemas, Faculdad de Física, Universidad de La Laguna, 38206 La Laguna, Spain. E-mail: flahoz@ull.es
cDepartment of Chemistry, NanoScience Center, University of Jyväskylä, PO. Box 35, 40014 JYU, Finland. E-mail: kari.t.rissanen@jyu.fi
First published on 8th January 2015
Abstract
A rare family of six discrete binuclear [PdCl(PEt3)2] phenylene ethynylene rods with alkoxy side chains (methoxy, ethoxy and heptoxy) have been developed, and their solid-state photoluminescence results have been presented and discussed. The shorter bridging ligands are of the general formula H–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–C6H2(R)2–CC–H, where R = H, OCH3, OC2H5, and OC7H15, whereas the longer ones are based on H–CC–C6H4–CC–C6H2(R)2–CC–C6H4–CC–H, where R = OCH3, OC2H5. These ligands display increasing length in both the main dimension (backbone length) as well as the number of carbons in the side chains (R, alkoxide side chain) that stem from the central phenylene moiety. The X-ray crystal structures of two of the prepared complexes are reported: one corresponds to a shorter rod, 1,4-bis[trans-(PEt3)2ClPd-CC]-2,5-diethoxybenzene (6c), while the second one is associated with a longer rod, the binuclear complex 1,4-bis[trans-(PEt3)2ClPd-4-(–CC–C6H4–CC)]-2,5-diethoxybenzene (7c). All new compounds were characterized by NMR spectroscopy (1H, 13C{1H} and 31P{1H}) as well as ESI-MS(TOF), EA, FTIR, UV-Vis, cyclic voltammetry and solid-state photoluminescence. Our work shows the influence of the alkoxy side chains on the electronic structure of the family of binuclear Pd rods by lowering its oxidation potential. In addition to this, the increase of the length of the bridge results in a higher oxidation potential. Solid state photoluminescence results indicate that Pd complexes are characterized by a marked decrease in both the emission intensity and the fluorescence lifetime values as compared to their ligands. This behaviour could be due to some degree of ligand-to-metal charge transfer.
C–C6H2(R)2–CC–H, where R = H, OCH3, OC2H5, and OC7H15, whereas the longer ones are based on H–CC–C6H4–CC–C6H2(R)2–CC–C6H4–CC–H, where R = OCH3, OC2H5. These ligands display increasing length in both the main dimension (backbone length) as well as the number of carbons in the side chains (R, alkoxide side chain) that stem from the central phenylene moiety. The X-ray crystal structures of two of the prepared complexes are reported: one corresponds to a shorter rod, 1,4-bis[trans-(PEt3)2ClPd-CC]-2,5-diethoxybenzene (6c), while the second one is associated with a longer rod, the binuclear complex 1,4-bis[trans-(PEt3)2ClPd-4-(–CC–C6H4–CC)]-2,5-diethoxybenzene (7c). All new compounds were characterized by NMR spectroscopy (1H, 13C{1H} and 31P{1H}) as well as ESI-MS(TOF), EA, FTIR, UV-Vis, cyclic voltammetry and solid-state photoluminescence. Our work shows the influence of the alkoxy side chains on the electronic structure of the family of binuclear Pd rods by lowering its oxidation potential. In addition to this, the increase of the length of the bridge results in a higher oxidation potential. Solid state photoluminescence results indicate that Pd complexes are characterized by a marked decrease in both the emission intensity and the fluorescence lifetime values as compared to their ligands. This behaviour could be due to some degree of ligand-to-metal charge transfer.
Introduction
Conjugated one-dimensional rods have been the centre of attention of several recent studies related to the development of molecular electronic devices such as wires, switches or insulators.1–7 It is well known that the incorporation of transition metal centers enables even finer tuning of the properties of these rods.8–11Transition metal alkynyl complexes, with their ability to perturb electronic properties through metal alkynyl dπ–pπ interactions,12,13 are primary candidates for the preparation of metallomolecular devices.14 The added rigid framework provided by the phenylene ethynylene ligand, as well as the easy processability, improves its potential for molecular device preparation using the building block approach, which allows for the fine tuning of magnetic, electronic and photoluminescence properties.9–11,14,15 The introduction of side chains into the aromatic moieties has the major advantage of not only increasing solubility,16,17 but also it facilitates the preparation of thin films, improves interface with other co-polymers,18 contributes to electroluminescence enhancement,19 inhibits chain-to-chain interactions and also gives more regular polymerizations products,20 refines emission color shifting in electroluminescent materials,21 helps improve and tune the formation of thin films for photovoltaic cells22 as well as increase delocalization and consequently inter-metal communication in binuclear complexes.23–26
Moreover, the decrease of the oxidation potential as a result of methyl (donor) β-substitution in thiophene,27 as well as the decrease in the Eg (energy gap, EHOMO–ELUMO) yielded by methoxy (also donor) substitution in phenylene vinylene oligomers, was previously reported.28
If Pd-σ-alkynyls are often used for the preparation of homometallic3,4,29–33 or heterometallic34–36 (with the incorporation of Ru, Fe or Ni metal centers) organometallic polymers, the synthesis of discrete binuclear Pd rods is comparatively much more scarce and is not so often found in the literature.5,37–39
Previous studies showed that [PdCl2(PEt3)2] compounds form square planar systems with the bridging oligophenylene ligands.15 Consequently, an increase of the π delocalization from the ring to the metal center would be expected, which could improve the conducting properties of the rod. This co-planarity between the metal center and the π system of the ligand is dictated by the ligand as Onitsuka and colleagues reported for 1,4-diethynylbenzene palladium complexes.40 This report shows that the metal centers are slightly twisted out of the plane because of steric hindrance caused by the phosphane groups of the metal center. As such, choosing a bridging ligand that, in addition to a solid conjugate backbone, also promotes co-planarity and processability is of great importance. As a matter of fact, the structure and geometry of the target systems greatly influence their properties, resulting in an insulator or a conductor molecule.41–48
The luminescence properties of organometallic compounds in solution are well-documented.9–11,49–59 The inclusion of transition metal moieties gives access to efficient spin–orbit coupling, which, in turn, enables the population of excited states of triplet character and phosphorescence characteristics of organometallic compounds. As such the coordination of the organic compounds to metal centers can enrich the emission properties by enabling access to new excited-state species.57 Yam and co-workers52 reported the carbazole bridged dinuclear Pd and Pt rods which were non-emissive in solution (CH2Cl2), whereas it showed luminescence in the solid state (77 K) with a decay time of about 2.9 μs. In the case of the platinum analogues, the luminescence was strongly quenched in CH2Cl2 solutions. This was evidenced by the short lifetime observed in the solution (<0.1 μs) as compared to that measured in the solid state, which was similar to the palladium counterparts. Moreover, when testing glass samples of this series (77 K, EtOH–MeOH 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), the luminescence intensity increased drastically, and the lifetime was augmented to 50 μs.
:1, v/v), the luminescence intensity increased drastically, and the lifetime was augmented to 50 μs.
Previous results from our group and others showed the importance of solid-state photoluminescence studies in the characterization of new photonic hybrid waveguides,60–62 and prompted us to systematically study the effect of substituents attached to bridging ligands on the electronic communication between the two metal centers and on the solid-state photoluminescence efficiency of a new family of binuclear [PdCl(PEt3)2] phenylene ethynylene rods with alkoxy side chains (methoxy, ethoxy and heptoxy). As such, a family of palladium terminated dinuclear rods (Scheme 1, 6a–d, 7b–c), based on 1,4-diethynylbenzene derivatives (Scheme 2, 1a–d, 4b–c), was prepared and characterized.
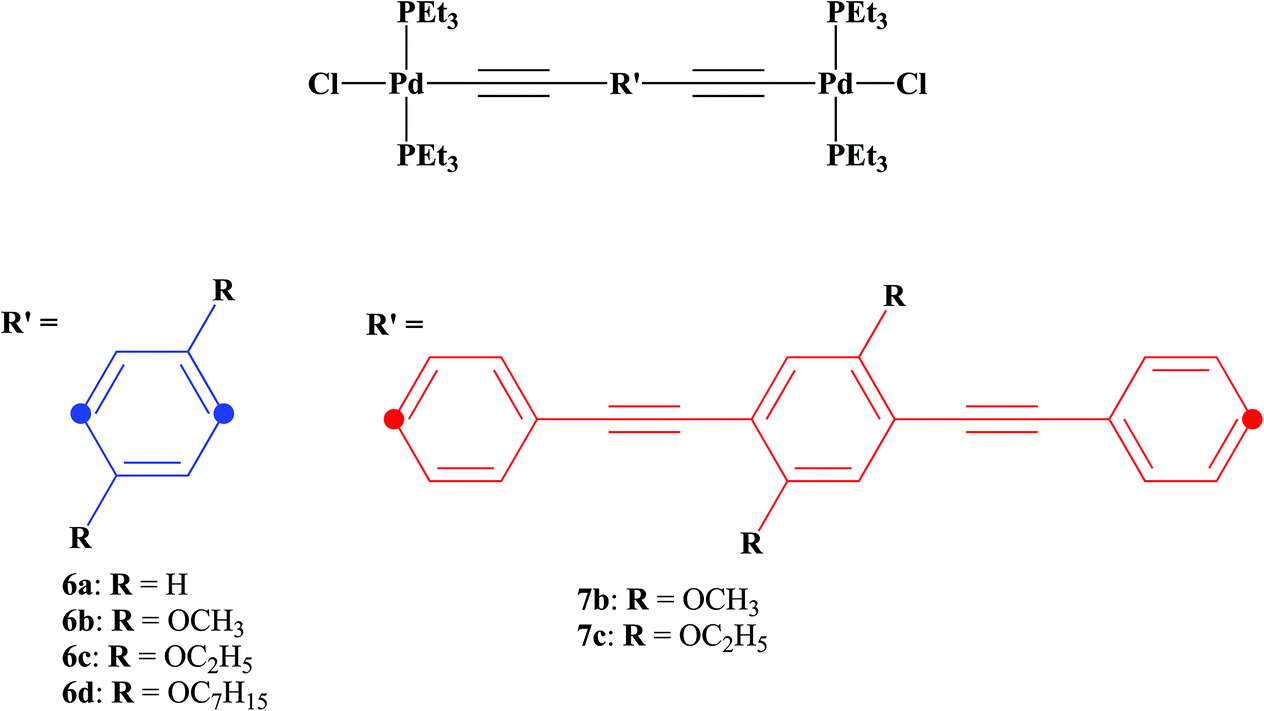 | ||
| Scheme 1 The series of trans-[PdCl(PEt3)2]+ rods prepared in this work. | ||
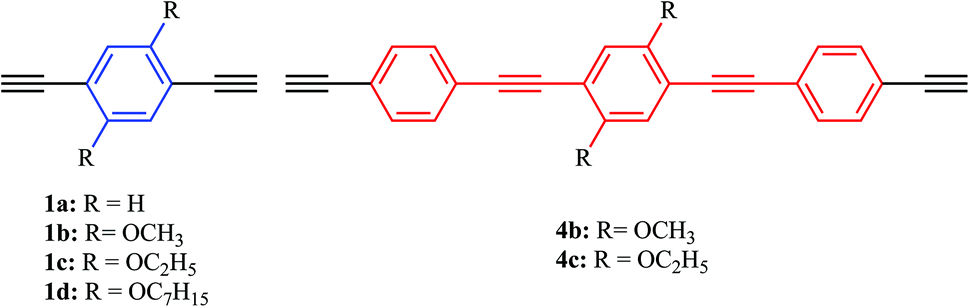 | ||
| Scheme 2 The 1,4-diethynylbenzene derivatives used as bridging ligands. | ||
Results and discussion
Synthesis
We have synthesized phenylene ethynylene based bridging ligands of one or three benzene aromatic rings (compounds 1a–d and 4b–c, respectively, Scheme 3). In the case of the longer bridging ligands (4b–c), the Sonogashira–Hagihara coupling conditions were used (Scheme 3).19,41,48,62–69 Intermediary products as well as the final products previously reported (1a–d)34,62,63 were confirmed at least by 1H and 13C (see ESI†). The yields for these reactions are in the range of those reported in the literature (i.e. 50–76%). The tris ringed analogues of 1a and 1d (R = H and OC7H15 respectively 4a and 4d) were not prepared by us due to isolation and purification problems encountered in the synthesis of these ligands. Nevertheless, the numbering was done taking into account these compounds to help the direct comparison of compounds 1b/6b/7b and 1c/6c/7c.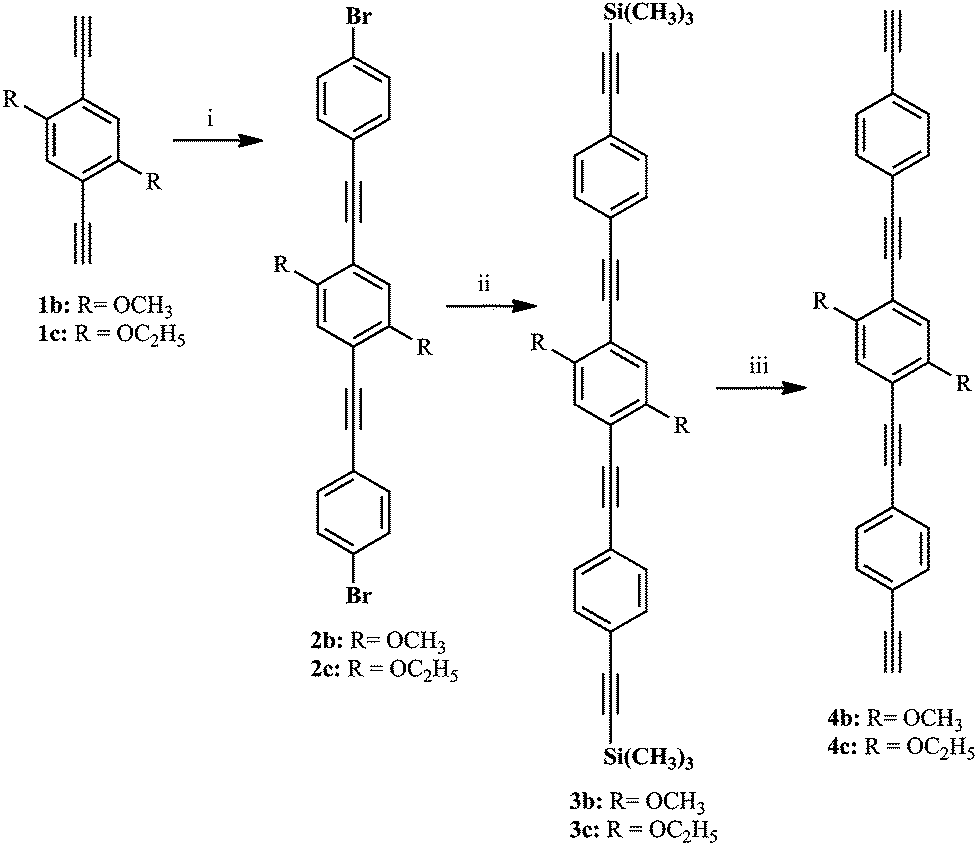 | ||
| Scheme 3 Preparation of the longer bridging ligands (4b–c); i. Sonogashira–Hagihara coupling, 1-bromo-4-iodobenzene, 45 °C; ii. Sonogashira–Hagihara coupling, trimethylsilylacetylene, 55 °C; iii. Degassed CH2Cl2–CH3OH (1:1 mixture), KF, room temp. | ||
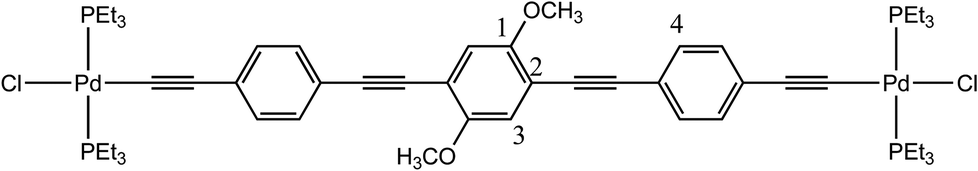
The preparation of the palladium rods (6a–d, 7b–c, Scheme 1) was performed with good yields (60–80%) by following the methodology reported in the literature40 and using trans-[PdCl2(PEt3)2] (5). These rods are sufficiently stable to be washed with water, and two new X-ray structures were obtained for a shorter rod (6c) and a longer rod (7c) both with the same 1,4-diethoxybenzene central moiety.
Characterization
The new compounds were characterized by NMR spectroscopy (1H, 13C{1H}, and 31P{1H}) as well as ESI-MS(TOF), EA, FTIR, UV-Vis, cyclic voltammetry and solid-state photoluminescence (see the Experimental section and ESI†). Briefly, the FTIR studies for the new palladium rods (6a–d, 7b–c) show the characteristic shift of the νCC band with the coordination to the metal. This was also observed by the shift, albeit small, of the singlet observed in the 31P NMR. Furthermore the Pd rod complexes display (in comparison with the free ligands), in the 1H spectrum, mostly a shielding of the aromatic (no more than 0.5 ppm) and side chain protons (ca. 0.1 ppm only). The aromatic protons show very similar chemical shifts in comparison with the free ligands (e.g.6b, OCH3 6.69 vs. 6.98 ppm in the free ligand). The same observation can be made for the first protons of the side chains (e.g.6b, OCH3 3.74 vs. 3.87 ppm in the free ligand). Furthermore, the values for the equivalent protons across Pd rods are relatively closer. This can be observed for the side chains as well as for the aromatic protons. The palladium rods (6a–d, 7b–c) all show 31P NMR singlets at around 18 ppm which is only about 1 ppm more than the free palladium starting material trans-[Pd(PEt3)2Cl2] (5). There is no significant change in this value throughout the series. The main difference in the NMR data of the palladium rods comes from the virtual coupling observed in the 13C NMR. This C–P coupling70 was found at around 15 ppm in the 13C spectra for all the rods.
The Pd complexes were also characterized by ESI-TOF. An [M − Cl]+ peak was found for most complexes, except for complex 6d, for which a peak at 2240.51 m/z [2M + Na + H]+ was observed. Furthermore, [M + Na]+ ion peaks were also observed for all the studied compounds. For 6a, an ion peak of 727.0 m/z was found, which corresponds to [M − Cl − PEt3 + H]+. Several other ions were visible in the case of the longer complexes. For example, for 7b, 881.3 m/z presents a peak with the formal composition [M − (Pd(PEt3)2Cl) + PEt3]+. Similar fragmentation and cleavage have been observed in the literature.35
UV-Vis studies
Significant information about the influence of the chemical structure on the optoelectronic properties of the compounds can be obtained from the analysis of the UV-Vis absorption spectra.71The absorption is dominated by spin allowed π→π* transitions of the delocalized electronic charge of the ethynylbenzene moieties, which are rich in conjugated bonds. Two main bands are observed for the tris ringed ligands 4b and 4c at about 316 and 376 nm. However, the absorption bands of the single ring ligand 1b are notably blue shifted, showing a maximum at 346 nm (Fig. 1). It should be mentioned that the absorption bands of the tris ringed ligands show a red shift when compared with a similar 1,4-diphenylethynylbenzene reported by Nakatsuji,72 with a peak at 322 nm. Nevertheless, Nakatsuji and co-workers performed this study in a CHCl3 solution which might account for the high ε that was observed (62.0 × 103 M−1 cm−1). No significant change was observed in the position of the absorption bands of the 4b and 4c compounds. Probably this observation is due to the small difference in the length of side chains (OCH3 to OC2H5) of the prepared compounds.
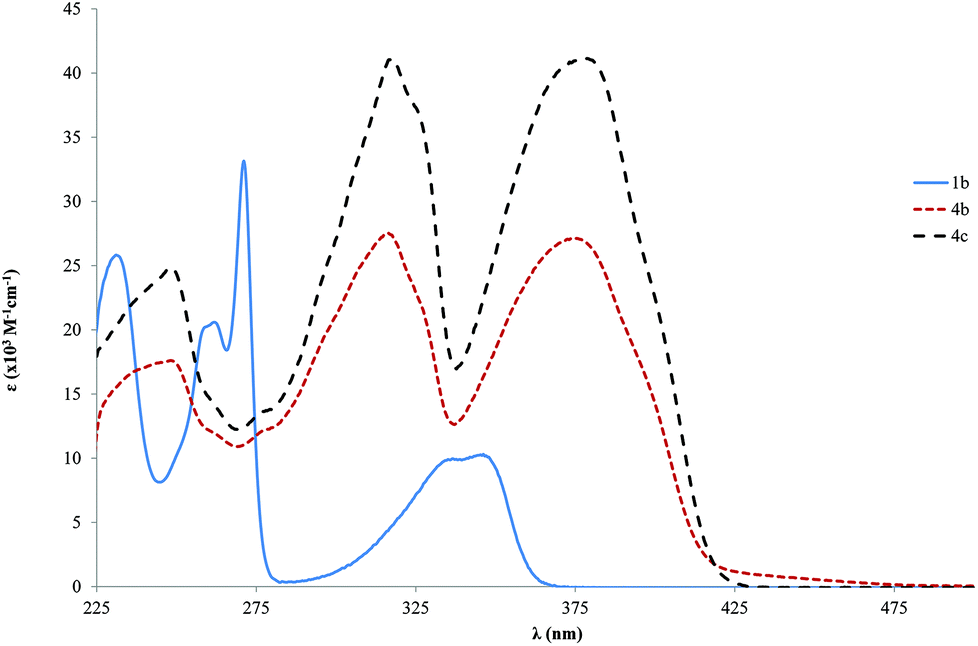 | ||
| Fig. 1 Absorption spectra (ε vs. λ) for the shorter (1b) and longer free ligands (4b–c) in CH2Cl2. | ||
Concerning the palladium complexes, the spectra of the palladium rod (6a) based on the single ringed free ligand without any side chains (1a) are presented and compared to the starting materials in Fig. 2. The bands of the absorption spectrum of 6a can be clearly assigned to those of the starting materials as they show similar positions with a slight blue shift. It seems that the coordination of the bridge to the palladium termini has little effect on the π→π* absorption bands.
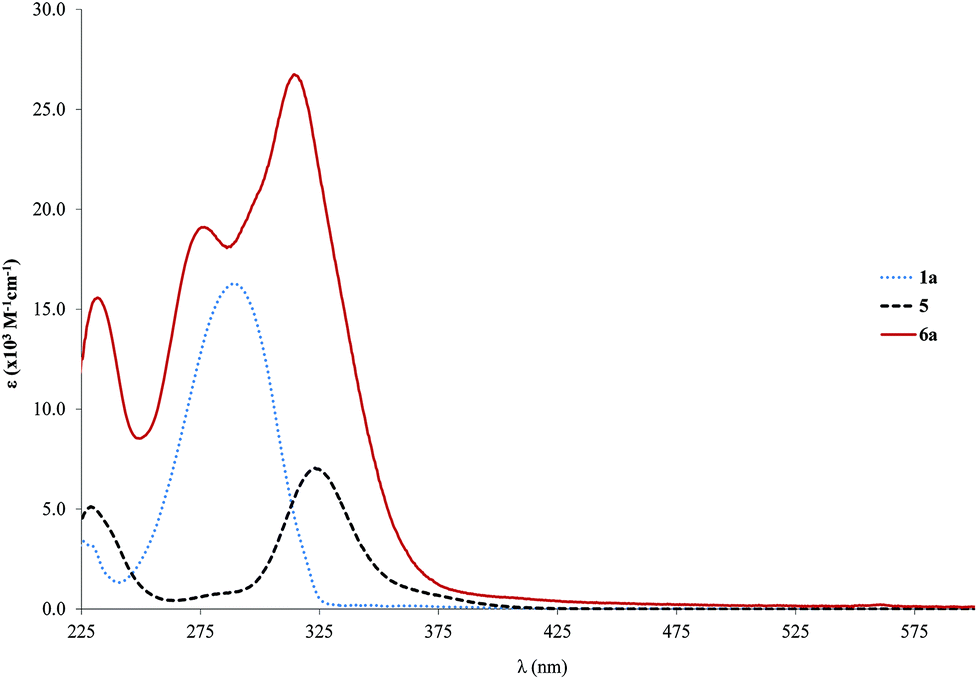 | ||
| Fig. 2 Absorption spectra (ε vs. λ) for the palladium rod 6a as well as the respective free ligand (1a) and starting complex 5 in CH2Cl2. | ||
In the case of the other palladium rods, the shift is in the expected low energy direction (red shift) as further conjugation is brought about by the coordination. Moreover, the high similarity between the absorption of the starting ligand and the resulting complex is an indication of the predominantly ligand character of the transitions (π→π*) responsible for the absorption spectrum of the complex. This observation was also reported by several other authors32,73–79 who additionally admitted some degree of MLCT (metal-to-ligand charge transfer) for the low energy bands (dπ→π*(CC–R)) in similar compounds (ClL2Pd–CC–C6H4–R, where R = (CC–C6H4–CC–PdLCl)2 or analogous Pt complexes).
We have analyzed the effect of the length of the ring backbone on the absorption properties. A table which summarizes the position of the relevant absorption bands for both the ligands and the Pd complexes is available in the ESI (Table S1†). In the case of 1b to 6b (shorter bridging ligands), the variation is from 346 to 362 nm, and for 4b to 7b (longer bridging ligands), it changes from 375 to 385 nm which is even lower that the single ringed case. The increase of ε is also lower in the case of the tris ringed rods (7b–c, Fig. S2,† about twofold relative to free ligands) when compared to the single ringed counterparts (6b–d, about 3 fold in relation to the free ligands). This can point to lesser conjugation, which might be due to trapped electron density on the ligand. Furthermore, the increase in side chain length is mostly inconsequential after a length of 6c (OC2H5), in the particular case of the studied alkoxy chains, as only slight variations in the range of 1–5 nm are observed when changing only the alkyl length. Nevertheless, with respect to ε, a marked decrease is observed from 6c to 6d (31–25 × 103 M−1 cm−1, Fig. 3). Finally, the significant differences found in both the positions as well as the intensities of the absorption bands of the Pd-complexes compared to their associated free ligands are directly related to π→π* electronic transition properties. It might be an indication of different electronic conduction properties, which are important for optoelectronic applications. Moreover, this point will be further analyzed in the solid-state fluorescence decay studies section.
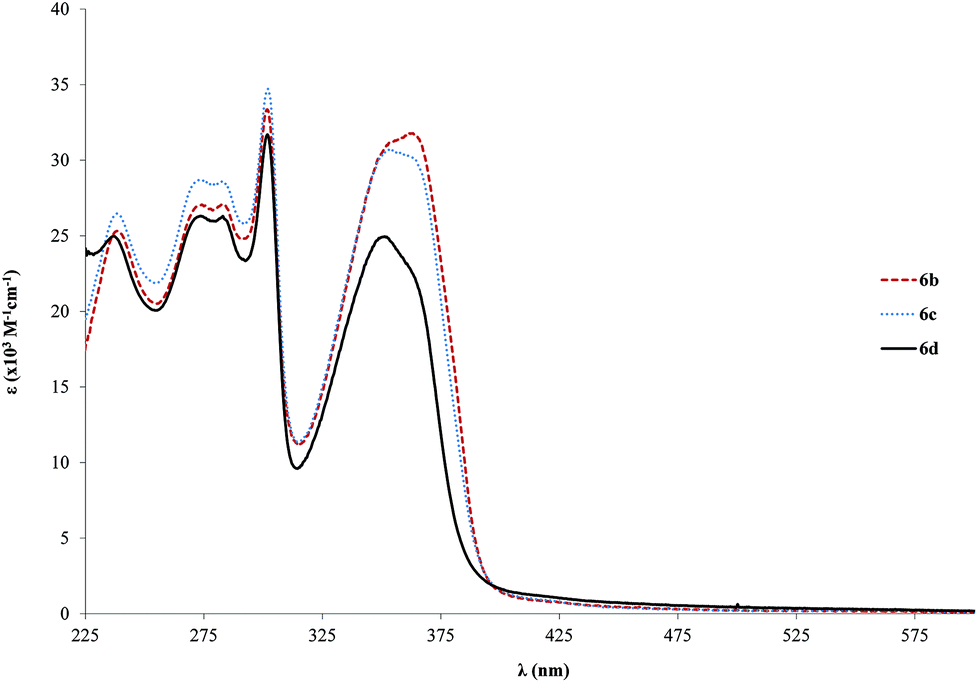 | ||
| Fig. 3 Electronic spectra (ε vs. λ) for the palladium rods (6b–d) in CH2Cl2. | ||
Electrochemistry studies
The free ligands only present an irreversible oxidation wave in the working potential intervals when the side chain is introduced, i.e., the 1a ligand does not show any oxidation wave, but 1b–d and 4b–c, all present high potential oxidations (ca. 1.6 V for the shorter – 1a–d – and 1.4 V for the longer bridging ligands – 4b–c). This wave is most likely the one-electron oxidation of the phenyl ring which shifts to lower energies by the electron donating effect of the alkoxy side chains. Other authors,80 working with similar compounds, have also performed the same attribution.As for the shorter rods (Fig. 4, 6a–d), it is possible to observe the existence of two electrode processes. The first one corresponds to a reversible process while the second one is probably irreversible. Reduction for the first process is only observable at 100 mV s−1 in the compound with the longer side chain (6d) but is relatively weak. However, when the scan rate is increased, the reduction is observed more intensely, indicating a following chemical reaction step. The first oxidation wave width of the compounds with alkoxy side chains (6b–d) suggests that this conversion is 1e−, which can indicate the formation of the PdII/PdIII pair.81 A following conversion to PdIII/PdIV could characterize the process at higher potentials, but once again, the similarity to the free ligands (as observed in the UV studies), as well as the region where it is found, is close to the electrochemical process of the solvent making it difficult to characterize. The first process observed for the rods with smaller side chains (6a–b) tends to irreversibility.
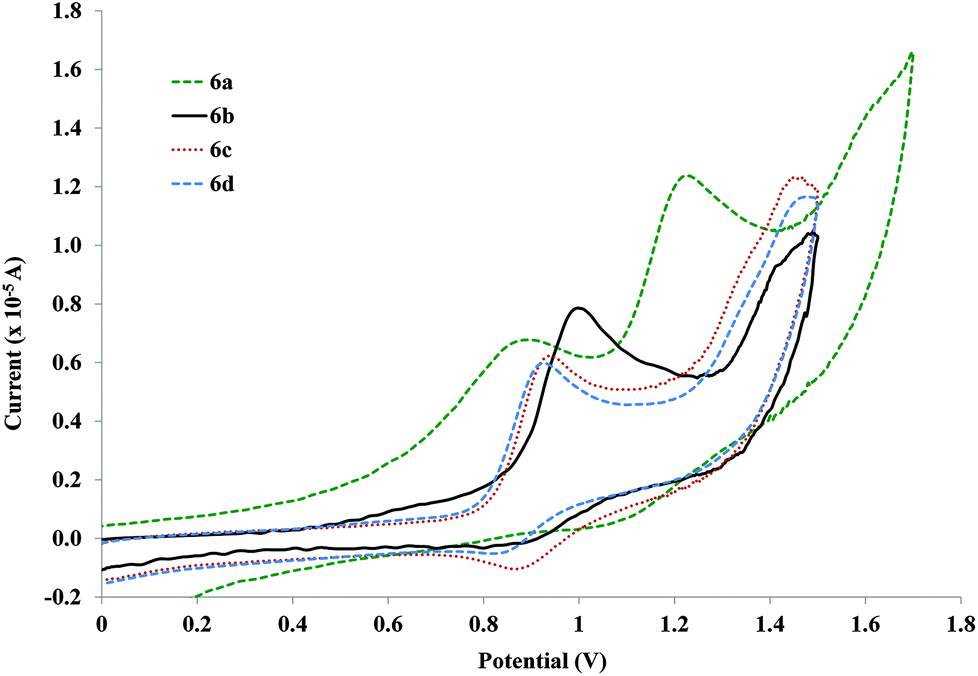 | ||
| Fig. 4 Cyclic voltammograms for the shorter palladium rods without (6a) and with alkoxy side chains (6b–d) at 100 mV s−1vs. Ag/AgCl (KCl saturated) in CH2Cl2. | ||
The most striking aspect of the aggregated voltammograms in Fig. 4 is that there is a shift to higher anodic potentials when the side chain is introduced (890 (6a) to ca. 996 mV for 6b). This value then shifts back down with the increase of the side chain's length (6c and 6d). This potential appears to stabilize after a side chain of OC2H5 (6c) since for 6d (side chain of OC7H15), the values are very close (945 and 927 mV respectively). Nevertheless, this decrease from OCH3 to OC2H5 side chains is not very pronounced when observing the longer palladium rods, since the anodic potentials for these oxidation processes are already very close (1.31 V for both compounds 7a–b, Fig. 5) and actually higher than that of the 6a (with no side chain). Moreover, they are very close to the ones observed for the free ligands (4b–c). This observation further cements the expected low delocalization not only through the palladium centre but also through these more extended ligands.82
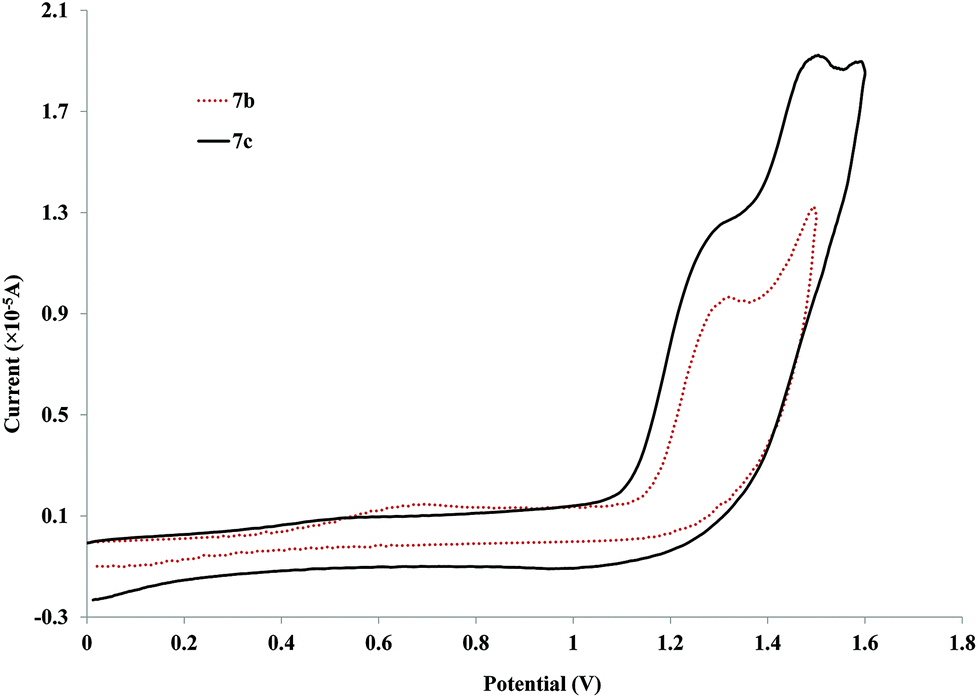 | ||
| Fig. 5 Cyclic voltammograms for the longer palladium rods (7b–c) with alkoxy side chains at 100 mV s−1vs. Ag/AgCl (KCl saturated) in CH2Cl2. | ||
X-ray crystal structure analysis
Two X-ray structures were solved for the prepared Pd rods. One is related with the shorter rod (6c) and the other to the analogous longer palladium rod (7c). Their ORTEP-383 plots are presented in Fig. 6 and 7, respectively. A search of the Cambridge Structural Database (CSD)84 reported only three similar structures.37,85 These are related to PdCl(PR3)2 (where R = butyl, ethyl) rods bridged by 1,4-diethynylbenzene,37,85 as well as another one37 with the central decorated phenyl ring with OC8H19 side chains similar to the shorter Pd rods reported in this work (6b–d). Apart from a published example bearing gold complex termini,86 no X-ray structure of a dinuclear rod bridged by a tris ringed ligand was found. Table 1 summarizes the crystallographic data for the two reported structures (selected bond angle and distance values are available in the ESI†).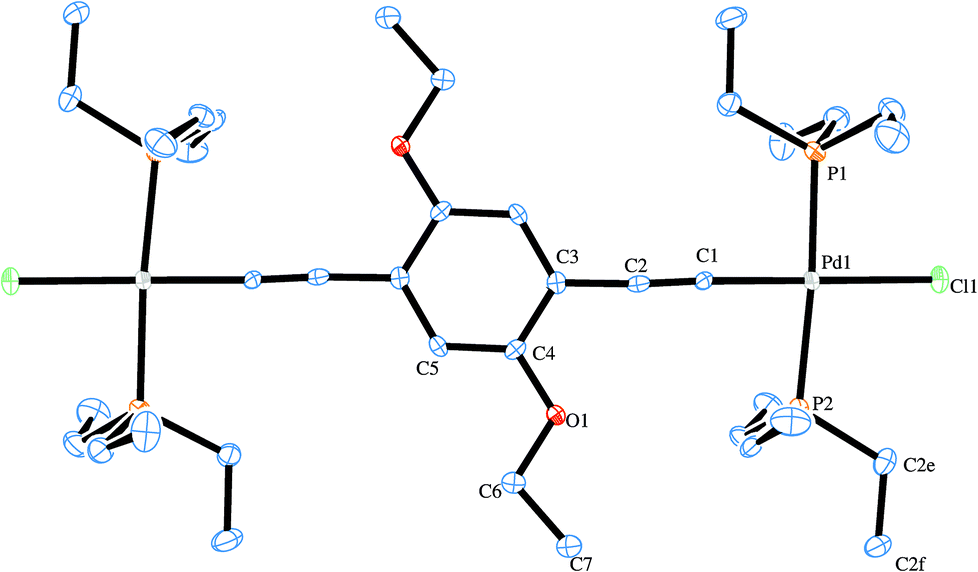 | ||
| Fig. 6 ORTEP-383 plot of (6c). H atoms are excluded for clarity. | ||
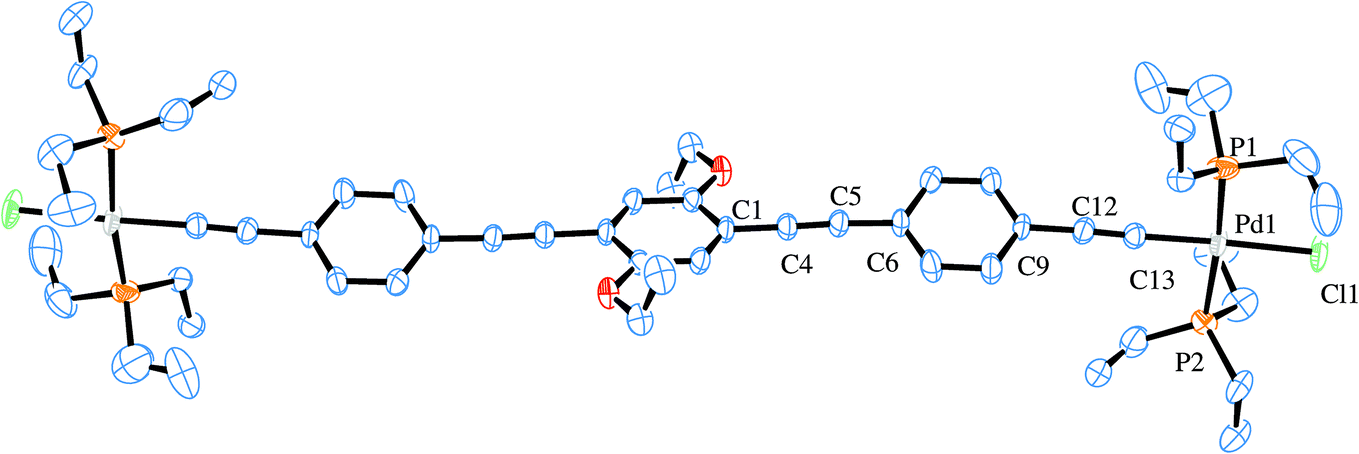 | ||
| Fig. 7 ORTEP-383 plot of the major component in 7c. H atoms and disorder are excluded for clarity. | ||
| 6c | 7c | |
|---|---|---|
| CCDC Depository | 882324 | 882325 |
| Crystallization solvents | CH2Cl2–diethyl ether | CH2Cl2–diethyl ether |
| Empirical formula | C38H72Cl2O2P4Pd2 | C54H80Cl2O2P4Pd2 |
| T (K) | 173(2) | 123(2) |
| λ (Å) | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n |
| a (Å) | 12.093(2) | 13.4614(3) |
| b (Å) | 15.652(3) | 11.7059(3) |
| c (Å) | 13.128(3) | 18.7397(4) |
| α (°) | 90 | 90 |
| β (°) | 107.02(3) | 103.7050(10) |
| γ (°) | 90 | 90 |
| V (Å3) | 2376.0(8) | 2868.89(12) |
| Z | 2 | 2 |
| ρ calc (g cm−3) | 1.348 | 1.353 |
| μ (mm−1) | 1.032 | 0.868 |
| Total reflections | 19588 |
9254 |
| Unique reflections | 5367 | 5186 |
| R int | 0.1385 | 0.0292 |
| Crystal size (mm) | 0.4 × 0.02 × 0.01 | 0.13 × 0.6 × 0.06 |
| Colour | Orange | Yellow |
| Habit | Flat needle | Plate |
| F(000) | 996 | 1212 |
| thetamin–thetamax (°) | 3.46–27.52 | 2.34–25.25 |
| Data | 5367 | 5186 |
| Restraints | 0 | 421 |
| Parameters | 217 | 424 |
| Goodness-of-fit (GOF) on F2 | 1.077 | 1.055 |
| R indices (all data) | ||
| R 1 | 0.1425 | 0.0765 |
| wR2 | 0.1003 | 0.1461 |
| Final R indices (I > 2σ(I)) | ||
| R 1 | 0.0709 | 0.0591 |
| wR2 | 0.0814 | 0.1343 |
| Largest difference in peak and hole (e Å−3) | 0.658 and −0.711 | 1.304 and −0.957 |
The structure of 6c (Fig. 6) contains half a molecule in the asymmetric unit and, in a first look, there appears to be a co-planarity of the phenylene ring with the palladium center, which has the expected square planar geometry. Nevertheless and although the ring is not twisted in relation to the Pd centre, the coordination plane around the Pd intersects the plane formed by the phenyl ring with an angle of 9.8(3)°. The ethoxy side chain does not run across the same plane formed by the phenylene ring, bending slightly to one side due to, apparently, packing short contacts. In contrast, the free ligand62 shows co-planar side chains.
As for bond distances for this structure, CC, is 1.176(9) Å and Pd–C is 1.964(7) Å which are the typical values for metal-σ-acetylide moieties.36,37,40,85,87 Furthermore Pd–P is 2.320(2) Å and Pd–Cl is 2.362(2) Å. Regarding the bridging ligand, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–O are 1.385(8)–1.411(9) and 1.385(8) Å long, respectively, which are also similar to other binuclear Pd rods.37,85 The Pd–CC angle is 175.9(7)° which is in agreement with the expected linear geometry. The P–Pd–Cl angles are 88.17(7) and 96.13(7)° and the P–Pd–C is 177.2(2)° which consequently means a slight deviation from fully square planar geometry. The values for these angles and the corresponding distortion are in good agreement with those previously reported by other authors.32,37,88 Intermolecular interactions commence through short-contacts between the terminal chlorides and the H-atoms from the phosphanes and the ethoxy side chains. The observed packing was very similar to that observed by Lo Sterzo and co-workers for [Cl(PBu3)2Pd–CC]2-1,4-benzene.37 The same authors reported a sheet-like packing when the alkoxy side chain was the longer octyloxy which clearly drives the formation of the lattice.37 It is noteworthy that this highly oriented lattice comes at the cost of a loss of co-planarity between the Pd centre and the phenylene ring. This could be regarded as a disadvantage for some applications,89–92 As described by Mayor and co-workers,93 who found, in a family of biphenyl compounds, conductance values thirty times lower when a full torsion (90°) is seen. Further explanation for this observation could also be based on the lack of co-planarity between the central phenyl ring and the two Pd coordination planes.
C and C–O are 1.385(8)–1.411(9) and 1.385(8) Å long, respectively, which are also similar to other binuclear Pd rods.37,85 The Pd–CC angle is 175.9(7)° which is in agreement with the expected linear geometry. The P–Pd–Cl angles are 88.17(7) and 96.13(7)° and the P–Pd–C is 177.2(2)° which consequently means a slight deviation from fully square planar geometry. The values for these angles and the corresponding distortion are in good agreement with those previously reported by other authors.32,37,88 Intermolecular interactions commence through short-contacts between the terminal chlorides and the H-atoms from the phosphanes and the ethoxy side chains. The observed packing was very similar to that observed by Lo Sterzo and co-workers for [Cl(PBu3)2Pd–CC]2-1,4-benzene.37 The same authors reported a sheet-like packing when the alkoxy side chain was the longer octyloxy which clearly drives the formation of the lattice.37 It is noteworthy that this highly oriented lattice comes at the cost of a loss of co-planarity between the Pd centre and the phenylene ring. This could be regarded as a disadvantage for some applications,89–92 As described by Mayor and co-workers,93 who found, in a family of biphenyl compounds, conductance values thirty times lower when a full torsion (90°) is seen. Further explanation for this observation could also be based on the lack of co-planarity between the central phenyl ring and the two Pd coordination planes.
The crystal structure of 7c shows disorder in all groups coordinated to the Pd atom, except for the acetylide group (see ESI†). The major component (60% population) is presented in Fig. 7. The central and terminal phenyl rings in 7c, which also contains half a molecule in the asymmetric unit, do not show too much twisting between each other, with the angle formed between the corresponding planes being only 23.3(2)°. Nevertheless, there is an 86.7(2)° angle (major, 84.2(2)° for the minor component) formed between the central phenyl moiety and the coordination plane around Pd. The bond distances Pd–P of 2.340(4) and 2.283(7) Å as well as Pd–Cl of 2.346(6) Å in 7c (values for the minor component in ESI†) are in agreement with the literature reported distances of 2.306 Å for Pd–P and 2.334 Å for Pd–Cl.37,85 The acetylide to palladium distance, Pd–CC, is of 1.947(6) Å, which is also close to reported values of 1.939 Å. The acetylenic bond lengths are 1.184(8) Å for the Pd coordinated and 1.200(8) Å for the isolated CC bonds. These are in accordance with the reported values (1.198 Å)37,85 and only slightly shorter when coordinated to the Pd. Finally, the CAr–O of 1.360(8) Å with the CAr–O–CH2 angle of 118.6(5)° are similar to the literature.37,85 The coordination around the Pd atoms, viz., the angle values for P–Pd–Cl, is of 96.5(2)° and 89.1(2)° with P–Pd–P 171.1(2) and C–Pd–Cl 177.8(2)°, as expected, close to the previously reported values for related systems.37,85Fig. 8 shows the packing diagram of complex 7c (along the a-axis) where it is possible to observe the layer orientation of the molecules.
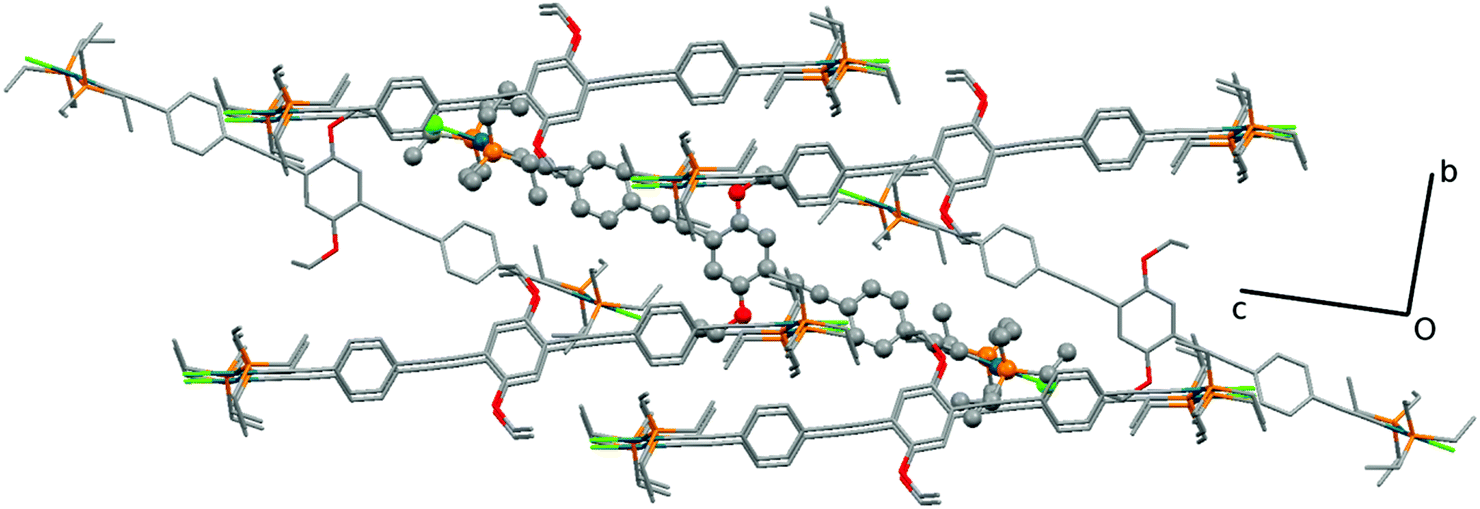 | ||
| Fig. 8 Packing diagram of 7c (along the a-axis), which shows the different orientation of the molecules between the layers. The molecule in the asymmetric unit is emphasized (ball-and-stick). The disorder and hydrogen atoms are removed for clarity. | ||
Solid-state fluorescence decay studies
Since a potential application of these rods would be as solid-state electronic devices, solid-state fluorescence studies would help to determine the conjugation properties of these materials.94 We have studied the decay of the fluorescence when the powder samples were excited with a pulsed laser (80 ps pulse width) at 405 nm. The results obtained for the free ligands and their respective Pd rods are shown in Fig. 9 and 10, respectively. The fluorescence decay was detected at about the maximum of the emission bands. A non-exponential decay was observed in all the cases, which is very common for non-diluted solid state samples. The non-exponential behavior of the decay curves can be due to ligand-to-ligand or ligand-to-metal interactions, as well as to the contribution of a distribution of environments, which is typical in powder samples. The photoluminescence decays are consistent with a multi-exponential equation, which can be useful to estimate an average lifetime that represents the decay rate of the complex.37 Good agreement was found for a three exponential decay curve (eqn (1)).| I(t) = B1e(−t/τ1) + B2e(−t/τ2) + B3e(−t/τ3) | (1) |
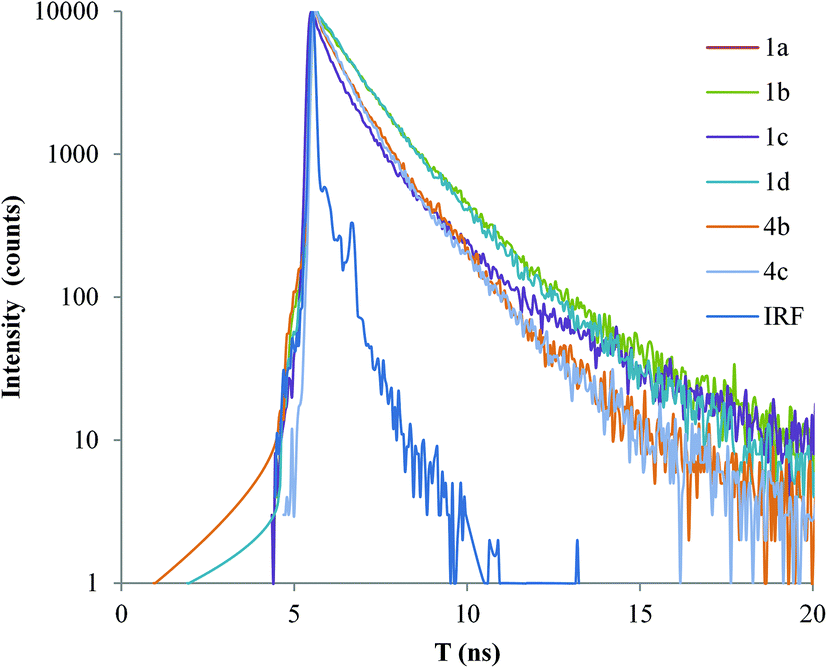 | ||
| Fig. 9 Decay of the fluorescence of the free ligands (1a–d, 4b–c) IRF. | ||
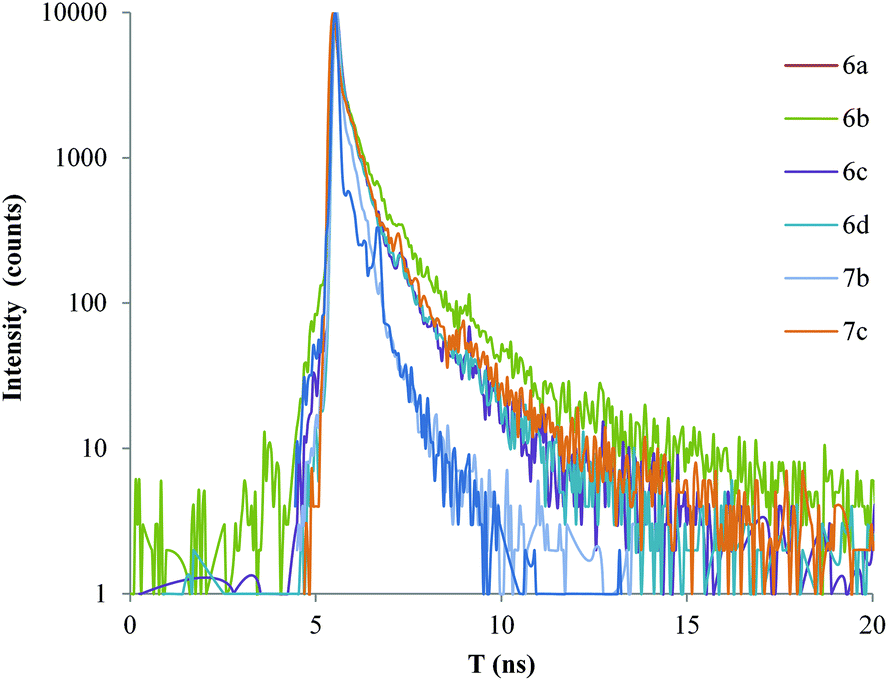 | ||
| Fig. 10 Decay of the fluorescence for the Pd rods (6a–d, 7b–c). | ||
The fitting was done using an IRF (instrumental response function) reconvolution analysis with F900 software by Edinburgh Instruments. The average lifetime is then calculated using the following equation (eqn (2)):95
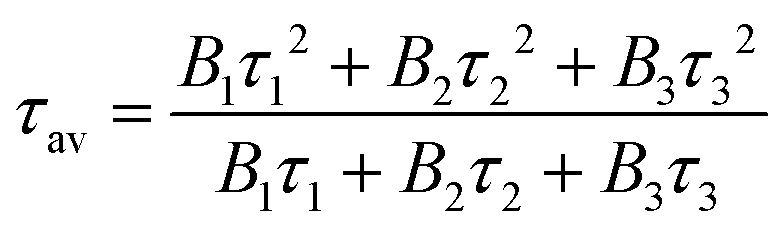 | (2) |
The results for the free ligands and for the Pd complexes are given in Table 2.
| Bridge | λ/nm | τ/ns | Rod | λ/nm | τ/ns |
|---|---|---|---|---|---|
| 1a | 510 | 0.80 | 6a | 454 | 0.24 |
| 1b | 510 | 1.00 | 6b | 510 | 0.60 |
| 1c | 510 | 0.70 | 6c | 510 | 0.30 |
| 1d | 510 | 0.90 | 6d | 510 | 0.30 |
| 4b | 510 | 1.20 | 7b | 450 | 0.05 |
| 4c | 490 | 0.95 | 7c | 510 | 0.40 |
The free ligands (1a–d, 4b–c) show average lifetime values comparable to those found in the literature for the new hybrid composites of poly(2-methoxy-5-(2′-ethylhexyloxy)-1,4-phenylenevinylene) (MEH-PPV) and clay (montmorillonite), purposely prepared for use in optoelectronic devices (like OLEDs).96 These nanocomposites show increased lifetimes when the concentration is higher, but values are still on the picosecond scale. Moreover, lasing capabilities were reported97 for p-(phenylene ethynylene) polymers, but in solution phase. Reports show98 that fluorescence lifetime values of 2 ns were obtained for phenylene ethynylenes compounds similar to the studied here. These reported values are nevertheless for chloroform solutions. Furthermore, this report98 also shows similar lifetime values for the fluorescence when comparing a tris ringed with its twisted (by a secondary chain) counterpart, even though a significantly lower (21 fold) quantum yield was found for the twisted compound. The authors98 do not account for this discrepancy and they attribute it to the equipment's fast decay.
When comparing the values presented in Table 2, higher lifetime values are found for the longer ligands (4b–c). Furthermore, a clear trend is observed for all the complexes that were analyzed (6a–d, 7b–c), the average lifetime decreases in the Pd compounds as compared to their respective free ligands. Moreover, a decrease in the emission intensity of the Pd complexes of one or two orders of magnitude was detected when compared to their associated free ligands. This could be accounted for by the charge distribution (from ligand to metal centre) reducing available electronic density that would induce a non-radiative relaxation path, which would decrease the lifetime and radiative emission quantum yield.
In particular, for the two compounds for which X-ray data are available, 6c and 7c, their average lifetimes can be compared with those of their respective free-ligand molecules, 1c and 4c, respectively. In the case of the single ring sample, it reduces from 0.7 ns, in the free ligand 1c, to 0.3 ns for the Pd coordinated complex, 6c. The lifetime diminishes from the 0.95 ns found in the tris ringed free ligand 4c to 0.4 ns in the corresponding metal complex, 7c. This represents a similar reduction of lifetime of 43% and 42% for the single and tris ringed systems. If we consider an important structural parameter obtained from the X-ray analysis, such as the co-planarity angle between the central phenyl and the Pd coordination plane, it changes from 10.96° in the single ring complex, 6c, to 86.56° for the tris ringed metal compound, 7c. One could expect a higher overlapping of the π delocalized electronic density to the metal center for a small co-planarity angle. However, the similar lifetime reduction observed in both compounds seems to indicate that a significant ligand to metal charge transfer exists in both cases, despite the low co-planarity exhibited in the 7c complex. This can be due to the partial tilting between the central and terminal phenyl rings (23.29°), and also to the strong electronegativity of the Pd center.
Experimental section
Materials
Chemicals were used as received unless otherwise noted. All reactions were carried out under a dry nitrogen atmosphere using standard Schlenk techniques. Solvents were freshly distilled under nitrogen. Triethylamine and diethylamine were distilled over potassium hydroxide; n-hexane and dichloromethane over calcium hydride; tetrahydrofuran, diethyl ether and toluene over sodium/benzophenone ketyl. Dimethylsulfoxide and glacial acetic acid were used as received and degassed under nitrogen (with stirring or sonication). Methanol was degassed under argon (with sonication). Solvents used in column chromatography were used as received. CuI and CuCl were pre-dried at 120 °C for at least 2 h (usually overnight) before usage. Compound 5, trans-[PdCl2(PEt3)2], was used as received from Acros Organics taking care to store it at 4 °C.Physical measurements
The infra-red spectra were obtained using a Bruker Vertex 70 FTIR spectrophotometer equipped with an ATR (attenuated total reflectance) diamond add-on. Samples were collected at a resolution of 2 cm−1 using a spectral range of 4000 to 400 cm−1. NMR spectra were recorded with a Bruker 400 MHz NMR (Bruker Avance II+). 1H, 13C{1H} and 31P{1H} NMR spectra were collected at 400 MHz, 101 MHz and 161 MHz respectively. The chemical shifts (δ) are reported in ppm and referenced to residual solvent peaks99 for 1H and 13C. 31P NMR is referenced to external 85% H3PO4. Melting points were determined with an Electrothermal 9200 melting point apparatus, using sealed capillaries without calibration. Mass spectra were obtained using a Micromass LCT spectrometer ESI-TOF spectrometer. Elemental analyses were carried out using a VariolEL instrument from Elementar Analysensysteme. Cyclic voltammetry was performed using a PARSAT advanced electrochemical equipment with tetra-N-butylammonium hexafluorophosphate 0.1 M as the supporting electrolyte in CH2Cl2 with an analyte concentration of 1 × 10−3 M. The reference electrode used was the Ag/AgCl (saturated KCl). Platinum was used as the working and secondary electrodes. The potentials were calibrated against the ferrocene/ferrocenium couple. Absorption spectra were recorded using a GBC-Cintra 40 in dry and degassed (argon) CH2Cl2 at 1 × 10−5 M for the free ligands (1a–d, 4b–c) and trans-[PdCl2(PEt3)2] (5) and at 1 × 10−6 M for the rods (6a–d, 7b–c). All solution measurements were performed using dry and degassed solvent and at room temperature unless otherwise noted. Lifetime fluorescence studies were carried out by measuring the samples in the form of powder between two microscope slides. The measurements were then performed using a LifeSpec II Spectrometer from Edinburg Instruments, with a pulsed 405 nm diode laser with 80 ps pulse duration as the excitation source. The Instrumental Response Function (IRF) of the spectrometer has been measured in order to correct the lifetime data. As the powder samples did not present all the same particle size and the sample size was different, integrated fluorescence spectra can only be compared by orders of magnitude of difference. Accordingly, samples are compared to their average lifetimes, which is independent of the factors referred above.Synthesis of the bridging ligands
The new 1,4-diethynylbenzene single ringed derivatives (Scheme 1) were prepared according to methods previously described in the literature (1a,341b63 and 1c–d62). The longer bridging ligands (4b–c) were prepared by following the Sonogashira–Hagihara procedures19,41,62,65–69 by coupling the shorter analogues (1b–c, respectively) with 1-iodo-4-bromobenzene, followed by another coupling to insert the terminal acetylenic moiety (Scheme 3). Cleavage of the trimethylsilyl protecting group was achieved with KF or K2CO3.100 The preparation of the intermediaries (2b–c, 3b–c) is detailed in the ESI.†:1), and K2CO3 (211 mg, 1.52 mmol) was added into one portion. The reaction mixture was stirred for 16 h at room temperature and then filtered. The resulting solution was evaporated under low pressure and purified by column chromatography (neutral alumina, petroleum ether (40–60 °C) and 5% diethyl ether). Compound 4b was obtained as a yellow powder (200 mg, 58.8%). Mp = 170–172 °C. 1H NMR (400 MHz, CDCl3): δ 3.18 (s, 2H, CC–H), 3.91 (s, 6H, OCH3), 7.03 (s, Ar–H, central ring), 7.47 (d, 4H, Ar–H outer ring, JH,H = 8.20 Hz), 7.52 (d, 4H, Ar–H outer ring, JH,H = 8.28 Hz). 13C{1H} NMR (101 MHz, CDCl3): δ 56.9 (s, OCH3), 79.3, 83.7, 88.0, 94.9 (s, CC, internal and external), 113.8, 116.0, 122.4, 124.1, 131.9, 132.4 (s, Ar. central and outer rings), 154.4 (s, Ar. C–O–CH3). FTIR (cm−1): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3271 (m, νCC−H), 2102 (vw, νCC), 655 (s, δCC–H), 1510 (w), 1500 (w), 1491.79 (w), 1461 (w), 1936 (m) (ar. νCC). TOF-MS (ESI+) of C28H18O2: m/z = calc. 409.12, found 409.12 [M + Na]+; calc. 441.15, found 441.13 [M + Na + CH3OH]+; calc. 493.10, found 493.13 [M + K + Cl + CH3OH + H]+; calc. 795.25, found 795.23 [2M + Na]+. Anal. Calcd for C28H18O2·2H2O (422.48): C, 79.60; H, 5.25 found C, 66.5; H, 5.17.
:1) and K2CO3 (15 mg, 0.10 mmol) was added at once. The reaction mixture was stirred for 16 h at room temperature and then filtered. The final solution was evaporated under low pressure and purified by column chromatography (neutral alumina, petroleum ether (40–60 °C) and 5% diethyl ether). The target compound 4c was obtained as a yellow powder (14 mg, 75.5%). Mp = 212–215 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (t, 6H, JH,H = 6.97 Hz), 3.17 (s, 2H, CC–H), 4.11 (s, 6H, OCH3), 7.01 (s, Ar–H, central ring), 7.47 (d, 4H, Ar–H outer ring, JH,H = 8.56 Hz), 7.49 (d, 4H, Ar–H outer ring, JH,H = 8.60 Hz). 13C{1H} NMR (101 MHz, CDCl3): δ 15.1 (s, OCH2CH3), 65.5 (s, OCH2CH3), 79.1, 83.5, 88.1, 94.6 (s, CC, internal and external) 117.4, 122.1, 124.1, 131.6, 132.2 (s, Ar. central and outer rings), 153.7 (s, Ar. C–O–CH2CH3). FTIR (cm−1): = 3275 (m, νCC–H), 2105 (vw, νCC), 661 (s, δCC–H), 1516 (m), 1499 (w), 1489 (w), 1474 (w), 1417 (m), 1403 (m), 1392 (m) (ar. νCC). TOF-MS (ESI+) of C30H22O2: m/z = calc. 437.15, found 437.20 [M + Na]+; calc. 469.18, found 469.22 [M + Na + CH3OH]+; calc. 851.31, found 851.37 [2M + Na]+. Anal. Calcd for C30H22O2·0.45 H2O (422.2): C, 85.26; H, 5.46; found C, 85.2; H, 5.56.
= 3271 (m, νCC−H), 2102 (vw, νCC), 655 (s, δCC–H), 1510 (w), 1500 (w), 1491.79 (w), 1461 (w), 1936 (m) (ar. νCC). TOF-MS (ESI+) of C28H18O2: m/z = calc. 409.12, found 409.12 [M + Na]+; calc. 441.15, found 441.13 [M + Na + CH3OH]+; calc. 493.10, found 493.13 [M + K + Cl + CH3OH + H]+; calc. 795.25, found 795.23 [2M + Na]+. Anal. Calcd for C28H18O2·2H2O (422.48): C, 79.60; H, 5.25 found C, 66.5; H, 5.17.
:1) and K2CO3 (15 mg, 0.10 mmol) was added at once. The reaction mixture was stirred for 16 h at room temperature and then filtered. The final solution was evaporated under low pressure and purified by column chromatography (neutral alumina, petroleum ether (40–60 °C) and 5% diethyl ether). The target compound 4c was obtained as a yellow powder (14 mg, 75.5%). Mp = 212–215 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (t, 6H, JH,H = 6.97 Hz), 3.17 (s, 2H, CC–H), 4.11 (s, 6H, OCH3), 7.01 (s, Ar–H, central ring), 7.47 (d, 4H, Ar–H outer ring, JH,H = 8.56 Hz), 7.49 (d, 4H, Ar–H outer ring, JH,H = 8.60 Hz). 13C{1H} NMR (101 MHz, CDCl3): δ 15.1 (s, OCH2CH3), 65.5 (s, OCH2CH3), 79.1, 83.5, 88.1, 94.6 (s, CC, internal and external) 117.4, 122.1, 124.1, 131.6, 132.2 (s, Ar. central and outer rings), 153.7 (s, Ar. C–O–CH2CH3). FTIR (cm−1): = 3275 (m, νCC–H), 2105 (vw, νCC), 661 (s, δCC–H), 1516 (m), 1499 (w), 1489 (w), 1474 (w), 1417 (m), 1403 (m), 1392 (m) (ar. νCC). TOF-MS (ESI+) of C30H22O2: m/z = calc. 437.15, found 437.20 [M + Na]+; calc. 469.18, found 469.22 [M + Na + CH3OH]+; calc. 851.31, found 851.37 [2M + Na]+. Anal. Calcd for C30H22O2·0.45 H2O (422.2): C, 85.26; H, 5.46; found C, 85.2; H, 5.56.
Synthesis of the palladium complexes
The bridged palladium complexes (Scheme 1) were obtained modifying the method reported by Onitsuka,40 coordinating the described ligands (1a–d, 4b–c) to the trans-[PdCl2(PEt3)2] (5) moiety in diethylamine (solvent and base) using copper (I) (CuCl 5% molar equiv.) as a catalyst.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C]benzene, 6a.
Compound 1,4-bis[trans-(PEt3)2ClPd–CC]benzene (6a) was first prepared by us, but Mukherjee and co-workers85 were able to publish previously its preparation, with only minor differences. For that reason and because its preparation was confirmed by FTIR, NMR and MS spectroscopy, no EA analysis was performed in that case. Compound 6a is prepared in the same way as the other short Pd rods (6b–d). Briefly, the starting materials trans-[PdCl2(PEt3)2] (5, 200 mg, 0.48 mmol) and 1a (41 mg, 0.32 mmol) were dissolved in NHEt2 (20 mL). CuCl (1 mg, 0.01 mmol) was added into one portion and the mix was stirred at room temperature for 14 h. It was then evaporated and redissolved in CH2Cl2. The resulting solution was washed with water, dried over Na2SO4 and evaporated under reduced pressure. A yellow powder was obtained (142 mg, 49.6%). Mp = 119 °C (decomp.). 1H NMR (400 MHz, CDCl3): δ 1.51 (t, J = 21 Hz, 36H, PCH2–CH3), 1.92 (q, J = 8 Hz, 24H, PCH2), 7.07 (s, 4H, Ar–H). 13C{1H} NMR (101 MHz, CDCl3): δ 8.3 (PCH2CH3), 15.4 (vt, JC–P = 13.80 Hz, PCH2CH3), 125.1, 130.4; 31P{1H} NMR (161 MHz, CDCl3): δ 18.1. FTIR (cm−1): = 2118 (w, νCC); 1498 (m), 1452 (m), 1409 (m), 1378 (m) (ar. νCC). TOF-MS (ESI+) of C34H64Cl2P4Pd2: m/z = calc. 727.1, found 727.0 [M − Cl − PEt3 + H]+, calc. 845.2, found 845.1 [M − Cl]+, 903.1 [M + Na]+.
C]-2,5-dimethoxybenzene, 6b.
The starting materials trans-[PdCl2(PEt3)2] (5, 200 mg, 0.48 mmol) and 1b (40 mg, 0.12 mmol) were dissolved in NHEt2 (20 mL). CuCl (1 mg, 0.01 mmol) was added into one portion and the mixture was stirred at room temperature for 14 h. It was then evaporated and redissolved in CH2Cl2. The resulting solution was washed with water, dried over Na2SO4 and evaporated under reduced pressure. Complex 6b was obtained as a yellow powder (128 mg, 62.9%). Mp = 153 °C (decomp.) 1H NMR (400 MHz, CDCl3): δ 1.21 (t, J = 21 Hz, 36H, PCH2–CH3), 2.02 (q, J = 8 Hz, 24H, PCH2), 3.75 (s, OCH3), 6.69 (s, 2H, Ar–H). 13C{1H} NMR (101 MHz, CDCl3): δ 8.0 (PCH2CH3), 15.3 (t, JC–P = 13.80 Hz, PCH2CH3), 30.9 (Pd–CC), 42.2 (Pd–CC), 56.2 (C1), 115.7 (C3), 154.3 (C2). 31P{1H} NMR (161 MHz, CDCl3): δ 18.1. FTIR (cm−1): = 2115 (s, νCC), 1493 (m), 1454 (m), 1409 (m), 1385 (m) (ar. νCC). TOF-MS (ESI+) of C36H68O2Cl2P4Pd2: m/z = 907.21, found 907.70 [M − Cl + 2H]+; calc. 963.15, found 963.09 [M + Na]+. Anal. Calcd for C36H68Cl2O2P4Pd2·1.05H2O (969.46) C, 45.07; H, 7.36; found C, 44.74; H, 7.03.
C]-2,5-diethoxybenzene, 6c.
The starting materials trans-[PdCl2(PEt3)2] (5, 250 mg, 0.60 mmol) and 1c (58 mg, 0.27 mmol) were dissolved in NHEt2 (20 mL). CuCl (1 mg, 0.01 mmol) was added into one portion, and the mixture was stirred at room temperature for 14 h. It was then evaporated and redissolved in CH2Cl2. The resulting solution was washed with water, dried over Na2SO4 and evaporated under reduced pressure. The final product was obtained as a yellow powder (214 mg, 76.3%). Orange crystals were obtained by diffusion of diethyl ether in a saturated dichloromethane solution at −20 °C. Mp = 145 °C (decomp.). 1H NMR (400 MHz, CDCl3): δ 1.19 (t, J = 20 Hz, 36H, PCH2–CH3), 1.46 (t, J = 18 Hz, 6H, OCH2–CH3), 2.01 (q, J = 18 Hz, 24H, PCH2), 3.97 (q, J = 18 Hz, 4H, OCH2), 6.69 (s, 2H, Ar–H). 13C{1H} NMR (101 MHz, CDCl3): δ 8.7 (PCH2CH3), 15.5 (s, OCH2CH3), 15.7 (t, JC–P = 13.97 Hz, PCH2CH3), 64.8 (s, OCH2CH3), 100.1 (t, JC–P = 16.06 Hz, Pd–CC), 102.9 (t, JC–P = 5.83 Hz, Pd–CC), 116.0 (s, C1), 117.4 (s, Ar.), 153.2 (s, Ar. C–O–CH3). 31P{1H} NMR (161 MHz, CDCl3): δ 18.0. FTIR (cm−1): = 2113 (w, νCC), 1498 (m), 1478 (m), 1409 (m), 1389 (m, ar. νCC). TOF-MS (ESI+) of C38H72Cl2O2P4Pd2: m/z = calc. 933.24, found 933.14 [M − Cl + H]+; calc. 991.18, found 991.10 [M + Na]+. Anal. Calcd for C38H72Cl2O2P4Pd2·0.75CH2Cl2·0.35H2O (1038.59): C, 44.81; H, 7.20; found: C, 44.85; H, 7.24.
C]-2,5-diheptoxybenzene, 6d.
The starting materials trans-[PdCl2(PEt3)2] (5, 250 mg, 0.60 mmol) and 1d (96 mg, 0.27 mmol) were dissolved in NHEt2 (20 mL). CuCl (1 mg, 0.01 mmol) was added into one portion and the mixture was stirred at room temperature for 14 h. It was then evaporated and redissolved in CH2Cl2. The resulting solution was washed with water, dried over Na2SO4 and evaporated under reduced pressure. Compound 6d was obtained as a yellow powder (193 mg, 62.5%). Mp = 98.9 °C (decomp.). 1H NMR (400 MHz, CDCl3): δ 0.89 (t, J = 6.86 Hz, 6H, O(CH2)6CH3), 1.19–1.31 (m, 52H, PCH2–CH3, O(CH2)2(CH2)4CH3), 1.48–1.93 (m, OCH2CH2(CH2)4CH3), 3.89 (t, J = 6.82 Hz, 4H, OCH2(CH2)5CH3), 2.03 (q, J = 7.4 Hz, 24H, PCH2), 6.69 (s, 2H, Ar–H). 13C{1H} NMR (101 MHz, CDCl3): δ 8.0 (PCH2CH3), 15.3 (t, JC–P = 13.80 Hz, PCH2CH3), 5.7, 13.8, 22.6, 26.1, 29.2, 29.7, 31.8 (s, (OCH2)6CH3), 99.6 (s, Pd–CC, JC–P = 16.13 Hz), 102.7 (Pd–CC, JC–P = 5.50 Hz), 115.6, 117.1 (s, Ar. C), 152.8 (s, Ar. C–O–CH2). 31P{1H} NMR (161 MHz, CDCl3): δ 18.5. FTIR (cm−1): = 2113 (w, νCC), 1498 (m), 1478 (m), 1404 (m), 1389 (m) (ar. νCC). TOF-MS (ESI+) of C48H92Cl2O2P4Pd2: m/z = calc. 1131.34, found 1131.34 [M + Na]+; calc. 2240.69, found 2240.51 [2M + Na + H]+; Anal. Calcd for C48H92Cl2O2P4Pd2·2H2O·0.6CH2Cl2 (1192.74): C, 48.81; H, 8.19; found: C, 48.83; H, 8.21.
C–C6H4–CC)]-2,5-dimethoxybenzene, 7b.
C]benzene, 6a.
Compound 1,4-bis[trans-(PEt3)2ClPd–CC]benzene (6a) was first prepared by us, but Mukherjee and co-workers85 were able to publish previously its preparation, with only minor differences. For that reason and because its preparation was confirmed by FTIR, NMR and MS spectroscopy, no EA analysis was performed in that case. Compound 6a is prepared in the same way as the other short Pd rods (6b–d). Briefly, the starting materials trans-[PdCl2(PEt3)2] (5, 200 mg, 0.48 mmol) and 1a (41 mg, 0.32 mmol) were dissolved in NHEt2 (20 mL). CuCl (1 mg, 0.01 mmol) was added into one portion and the mix was stirred at room temperature for 14 h. It was then evaporated and redissolved in CH2Cl2. The resulting solution was washed with water, dried over Na2SO4 and evaporated under reduced pressure. A yellow powder was obtained (142 mg, 49.6%). Mp = 119 °C (decomp.). 1H NMR (400 MHz, CDCl3): δ 1.51 (t, J = 21 Hz, 36H, PCH2–CH3), 1.92 (q, J = 8 Hz, 24H, PCH2), 7.07 (s, 4H, Ar–H). 13C{1H} NMR (101 MHz, CDCl3): δ 8.3 (PCH2CH3), 15.4 (vt, JC–P = 13.80 Hz, PCH2CH3), 125.1, 130.4; 31P{1H} NMR (161 MHz, CDCl3): δ 18.1. FTIR (cm−1): = 2118 (w, νCC); 1498 (m), 1452 (m), 1409 (m), 1378 (m) (ar. νCC). TOF-MS (ESI+) of C34H64Cl2P4Pd2: m/z = calc. 727.1, found 727.0 [M − Cl − PEt3 + H]+, calc. 845.2, found 845.1 [M − Cl]+, 903.1 [M + Na]+.
C]-2,5-dimethoxybenzene, 6b.
The starting materials trans-[PdCl2(PEt3)2] (5, 200 mg, 0.48 mmol) and 1b (40 mg, 0.12 mmol) were dissolved in NHEt2 (20 mL). CuCl (1 mg, 0.01 mmol) was added into one portion and the mixture was stirred at room temperature for 14 h. It was then evaporated and redissolved in CH2Cl2. The resulting solution was washed with water, dried over Na2SO4 and evaporated under reduced pressure. Complex 6b was obtained as a yellow powder (128 mg, 62.9%). Mp = 153 °C (decomp.) 1H NMR (400 MHz, CDCl3): δ 1.21 (t, J = 21 Hz, 36H, PCH2–CH3), 2.02 (q, J = 8 Hz, 24H, PCH2), 3.75 (s, OCH3), 6.69 (s, 2H, Ar–H). 13C{1H} NMR (101 MHz, CDCl3): δ 8.0 (PCH2CH3), 15.3 (t, JC–P = 13.80 Hz, PCH2CH3), 30.9 (Pd–CC), 42.2 (Pd–CC), 56.2 (C1), 115.7 (C3), 154.3 (C2). 31P{1H} NMR (161 MHz, CDCl3): δ 18.1. FTIR (cm−1): = 2115 (s, νCC), 1493 (m), 1454 (m), 1409 (m), 1385 (m) (ar. νCC). TOF-MS (ESI+) of C36H68O2Cl2P4Pd2: m/z = 907.21, found 907.70 [M − Cl + 2H]+; calc. 963.15, found 963.09 [M + Na]+. Anal. Calcd for C36H68Cl2O2P4Pd2·1.05H2O (969.46) C, 45.07; H, 7.36; found C, 44.74; H, 7.03.
C]-2,5-diethoxybenzene, 6c.
The starting materials trans-[PdCl2(PEt3)2] (5, 250 mg, 0.60 mmol) and 1c (58 mg, 0.27 mmol) were dissolved in NHEt2 (20 mL). CuCl (1 mg, 0.01 mmol) was added into one portion, and the mixture was stirred at room temperature for 14 h. It was then evaporated and redissolved in CH2Cl2. The resulting solution was washed with water, dried over Na2SO4 and evaporated under reduced pressure. The final product was obtained as a yellow powder (214 mg, 76.3%). Orange crystals were obtained by diffusion of diethyl ether in a saturated dichloromethane solution at −20 °C. Mp = 145 °C (decomp.). 1H NMR (400 MHz, CDCl3): δ 1.19 (t, J = 20 Hz, 36H, PCH2–CH3), 1.46 (t, J = 18 Hz, 6H, OCH2–CH3), 2.01 (q, J = 18 Hz, 24H, PCH2), 3.97 (q, J = 18 Hz, 4H, OCH2), 6.69 (s, 2H, Ar–H). 13C{1H} NMR (101 MHz, CDCl3): δ 8.7 (PCH2CH3), 15.5 (s, OCH2CH3), 15.7 (t, JC–P = 13.97 Hz, PCH2CH3), 64.8 (s, OCH2CH3), 100.1 (t, JC–P = 16.06 Hz, Pd–CC), 102.9 (t, JC–P = 5.83 Hz, Pd–CC), 116.0 (s, C1), 117.4 (s, Ar.), 153.2 (s, Ar. C–O–CH3). 31P{1H} NMR (161 MHz, CDCl3): δ 18.0. FTIR (cm−1): = 2113 (w, νCC), 1498 (m), 1478 (m), 1409 (m), 1389 (m, ar. νCC). TOF-MS (ESI+) of C38H72Cl2O2P4Pd2: m/z = calc. 933.24, found 933.14 [M − Cl + H]+; calc. 991.18, found 991.10 [M + Na]+. Anal. Calcd for C38H72Cl2O2P4Pd2·0.75CH2Cl2·0.35H2O (1038.59): C, 44.81; H, 7.20; found: C, 44.85; H, 7.24.
C]-2,5-diheptoxybenzene, 6d.
The starting materials trans-[PdCl2(PEt3)2] (5, 250 mg, 0.60 mmol) and 1d (96 mg, 0.27 mmol) were dissolved in NHEt2 (20 mL). CuCl (1 mg, 0.01 mmol) was added into one portion and the mixture was stirred at room temperature for 14 h. It was then evaporated and redissolved in CH2Cl2. The resulting solution was washed with water, dried over Na2SO4 and evaporated under reduced pressure. Compound 6d was obtained as a yellow powder (193 mg, 62.5%). Mp = 98.9 °C (decomp.). 1H NMR (400 MHz, CDCl3): δ 0.89 (t, J = 6.86 Hz, 6H, O(CH2)6CH3), 1.19–1.31 (m, 52H, PCH2–CH3, O(CH2)2(CH2)4CH3), 1.48–1.93 (m, OCH2CH2(CH2)4CH3), 3.89 (t, J = 6.82 Hz, 4H, OCH2(CH2)5CH3), 2.03 (q, J = 7.4 Hz, 24H, PCH2), 6.69 (s, 2H, Ar–H). 13C{1H} NMR (101 MHz, CDCl3): δ 8.0 (PCH2CH3), 15.3 (t, JC–P = 13.80 Hz, PCH2CH3), 5.7, 13.8, 22.6, 26.1, 29.2, 29.7, 31.8 (s, (OCH2)6CH3), 99.6 (s, Pd–CC, JC–P = 16.13 Hz), 102.7 (Pd–CC, JC–P = 5.50 Hz), 115.6, 117.1 (s, Ar. C), 152.8 (s, Ar. C–O–CH2). 31P{1H} NMR (161 MHz, CDCl3): δ 18.5. FTIR (cm−1): = 2113 (w, νCC), 1498 (m), 1478 (m), 1404 (m), 1389 (m) (ar. νCC). TOF-MS (ESI+) of C48H92Cl2O2P4Pd2: m/z = calc. 1131.34, found 1131.34 [M + Na]+; calc. 2240.69, found 2240.51 [2M + Na + H]+; Anal. Calcd for C48H92Cl2O2P4Pd2·2H2O·0.6CH2Cl2 (1192.74): C, 48.81; H, 8.19; found: C, 48.83; H, 8.21.
C–C6H4–CC)]-2,5-dimethoxybenzene, 7b.
The starting materials trans-[PdCl2(PEt3)2] (5, 300 mg, 0.72 mmol) and 4b (126 mg, 0.32 mmol) were dissolved in NHEt2 (40 mL). CuCl (1 mg, 0.01 mmol) was added at once and the mixture was stirred at room temperature for 14 h. It was then evaporated and redissolved in CH2Cl2. The resulting solution was washed with water, dried over Na2SO4 and evaporated under reduced pressure, giving 7b as a yellow powder (273 mg, 74.4%). Mp = 155 °C (decomp.). 1H NMR (400 MHz, CDCl3): δ 1.22 (t, J = 8.18 Hz, 36H, PCH2–CH3), 1.98 (q, J = 3.61 Hz, 24H, PCH2), 3.90 (s, 6H, OCH3), 7.01 (s, 2H, Ar–H), 7.21 (d, 4H, 3JH,H = 8.08 Hz), 7.42 (d, 4H, 3JH,H = 8.12 Hz). 13C{1H} NMR (101 MHz, CDCl3): δ 8.7 (s, PCH2CH3), 15.8 (t, JC–P = 13.97 Hz, PCH2CH3), 56.7 (s, OCH3), 86.8, 95.8 (s, C
 C, internal), 99.5 (t, Pd–CC, JC–P = 16.13 Hz), 107.1 (t, Pd–CC, JC–P = 5.87 Hz), 113.9, 116.0, 120.5, 128.3, 130.9 (s, Ar. central and outer rings), 154.3 (s, Ar. C–O–CH3). 31P{1H} NMR (161 MHz, CDCl3): δ 18.4. FTIR (cm−1): = 2110 (w, νCC), 1501 (m), 1488 (m), 1457 (m), 1397 (m) (ar. νCC). TOF-MS (ESI+) of C54H80Cl2O2P4Pd2: m/z = calc. 763.19, found 763.19 [M − (PdCl(PEt3)2) + H]+; calc. 881.22, found 881.26 [M − (PdCl(PEt3)2) + H + PEt3]+; calc. 987.18, found 987.15 [M − Cl − PEt3 + H]+; calc. 1105.26, found 1105.26 [M − Cl]+. Anal. Calcd for C52H76Cl2O2P4Pd2·0.35H2O (1147.09) C, 54.45; H, 6.74; found: C, 54.33; H, 6.59.
C–C6H4–CC)]-2,5-diethoxybenzene, 7c.
C, internal), 99.5 (t, Pd–CC, JC–P = 16.13 Hz), 107.1 (t, Pd–CC, JC–P = 5.87 Hz), 113.9, 116.0, 120.5, 128.3, 130.9 (s, Ar. central and outer rings), 154.3 (s, Ar. C–O–CH3). 31P{1H} NMR (161 MHz, CDCl3): δ 18.4. FTIR (cm−1): = 2110 (w, νCC), 1501 (m), 1488 (m), 1457 (m), 1397 (m) (ar. νCC). TOF-MS (ESI+) of C54H80Cl2O2P4Pd2: m/z = calc. 763.19, found 763.19 [M − (PdCl(PEt3)2) + H]+; calc. 881.22, found 881.26 [M − (PdCl(PEt3)2) + H + PEt3]+; calc. 987.18, found 987.15 [M − Cl − PEt3 + H]+; calc. 1105.26, found 1105.26 [M − Cl]+. Anal. Calcd for C52H76Cl2O2P4Pd2·0.35H2O (1147.09) C, 54.45; H, 6.74; found: C, 54.33; H, 6.59.
C–C6H4–CC)]-2,5-diethoxybenzene, 7c.
The starting materials trans-[PdCl2(PEt3)2] (5, 250 mg, 0.60 mmol) and 4c (100 mg, 0.24 mmol) were dissolved in NHEt2 (30 mL). CuCl (1 mg, 0.01 mmol) was added into one portion and the mixture was stirred at room temperature for 14 h. It was then evaporated and redissolved in CH2Cl2. The resulting solution was washed with water, dried over Na2SO4 and evaporated under reduced pressure. The target compound 7c was obtained as a yellow powder (241 mg, 85.9%). Yellow crystals were obtained by diffusion of diethyl ether in a saturated dichloromethane solution at −20 °C. Mp = 150 °C (decomp.). 1H NMR (400 MHz, CDCl3): δ 1.22 (t, J = 8.02 Hz, 36H, PCH2–CH3), 1.48 (t, 6H, OCH2CH3) 1.98 (q, J = 3.61 Hz, 24H, PCH2), 4.10 (s, 4H, OCH2CH3), 6.95 (s, 2H, Ar–H), 7.21 (d, 4H, 3JH,H = 8.44 Hz), 7.40 (d, 4H, 3JH,H = 8.44 Hz). 13C{1H} NMR (101 MHz, CDCl3): δ 8.7 (s, PCH2CH3), 15.3 (s, OCH2CH3), 15.8 (t, JC–P = 1393 Hz, PCH2CH3), 5.7 (s, OCH3), 87.1, 95.6 (s, C
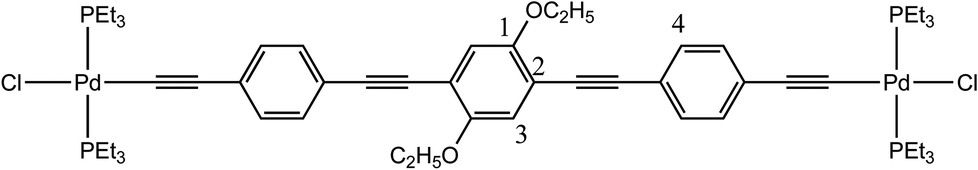 C, internal), 99.4 (t, Pd–CC, JC–P = 15.80 Hz), 107.1 (t, Pd–CC, JC–P = 5.53 Hz), 114.6, 117.6, 120.5, 128.2, 130.9 (s, Ar. central and outer rings), 153.8 (s, Ar. C–O–CH3). 31P{1H} NMR (161 MHz, CDCl3): δ 18.1 ppm. FTIR (cm−1): = 2112 (w, νCC), 1510 (m), 1487 (m), 1454 (m), 1413 (m), 1377 (m) (ar. νCC). TOF-MS (ESI+) of C54H80Cl2O2P4Pd2: m/z = calc. 1133.29 found 1133.33 [M − Cl]+. Anal. Calcd for C54H80Cl2O2P4Pd2 (1168.83): C, 55.49; H, 6.90; found: C, 56.10; H, 6.74.
C, internal), 99.4 (t, Pd–CC, JC–P = 15.80 Hz), 107.1 (t, Pd–CC, JC–P = 5.53 Hz), 114.6, 117.6, 120.5, 128.2, 130.9 (s, Ar. central and outer rings), 153.8 (s, Ar. C–O–CH3). 31P{1H} NMR (161 MHz, CDCl3): δ 18.1 ppm. FTIR (cm−1): = 2112 (w, νCC), 1510 (m), 1487 (m), 1454 (m), 1413 (m), 1377 (m) (ar. νCC). TOF-MS (ESI+) of C54H80Cl2O2P4Pd2: m/z = calc. 1133.29 found 1133.33 [M − Cl]+. Anal. Calcd for C54H80Cl2O2P4Pd2 (1168.83): C, 55.49; H, 6.90; found: C, 56.10; H, 6.74.
Crystallographic data collection and structure determination
X-ray data for compounds 6c and 7c were collected using a Bruker-Nonius KappaCCD diffractometer with an APEX-II detector at 173(2) and 123(2) K, respectively, using MoKα radiation (λ = 0.71073 Å). The structures were solved by direct methods with SIR-2004101 and refined by least squares using SHELXL-97.102 Hydrogen atoms were included at calculated positions and refined as riding atoms with isotropic displacement parameters coupled to those of the parent atoms. The non-hydrogen atoms were refined using anisotropic parameters. ORTEP-383 plots have been drawn with 30% probability ellipsoids. For disordered 7c, a large number of restraints were applied to stabilize the structure of major and minor components. The Pd–Cl, Pd–P and P–C bond distances were restrained to be equal (s = 0.02) in both components. Also, all C–C distances in ethyl groups bonded to P atoms as well as all P–methyl distances over two bonds were restrained to be equal (s = 0.02). Anisotropic displacement parameters for Cl, P and disordered C atoms were restrained to be equal (s = 0.01 for P and methylene C, and s = 0.02 for Cl and methyl C). Finally, two atoms of the minor component of disordered 7c, were restrained to improve the isotropicity of this structure's shape.Conclusions
The design and synthesis of novel molecular non-metallated or metallated systems is currently a hot topic of research, since the potential integration of these functional molecular structures within electronic devices could extend the life of silicon technology. In this work, we have reported the preparation, structural characterization and electrochemical properties of a rare family of [PdCl(PEt3)2]+ rods based on derivatives of 1,4-diethynylbenzene (phenylene ethynylene rods with alkoxy side chains: methoxy, ethoxy and heptoxy). We also systematically studied the effect of substituents attached to bridging ligands on the electronic communication between the two metal centers and on the solid-state photoluminescence efficiency of this family of binuclear [PdCl(PEt3)2] phenylene ethynylene rods. Preparation of bridging ligands was done using Sonogashira–Hagihara coupling, and two new ligands (4b–c) were obtained in good yields as well as the resulting bimetallic rods. The structures of 1,4-bis[trans-(PEt3)2ClPd–CC]-2,5-diethoxybenzene (6c) and the longer rod 1,4-bis[trans-(PEt3)2ClPd-4-(–CC–C6H4–CC)]-2,5-diethoxybenzene (7c) were unambiguously confirmed by single crystal X-ray crystallography. Although the usage of the side chains does allow better manipulation (solubility) of the materials and induces lower oxidation potentials in regard to the undecorated rods, our results show no significant differences as the length of these chains increases. It was observed that the increase of the length of the new bridging ligands brings about a higher oxidation potential in relation to the shorter counterparts. Fluorescence lifetime values are found to be longer in the ligands (4b–c) than those in the studied Pd complexes (6a–d, 7b–c), which exhibit a marked decrease in both the emission intensity and the fluorescence lifetime values. This observation could be due to some degree of ligand-to-metal charge transfer. Based on the current results, the development of new hybrid molecular/semiconductor systems formed by non-metallated or metallated π-conjugated oligo(phenylene ethynylene)s molecular wires, covalently grafted onto not previously functionalized porous silicon substrates is currently underway in our laboratory, in view of their possible application as waveguides.
Conflict of Interest
The authors declare no competing financial interest.Acknowledgements
We thank the Fundação para a Ciência e a Tecnologia (FCT-IP) for the generous support of our work through the network contracts PTNMR-2013, RNEM-2013, as well as the research project PTDC/CTM/098451/2008, the PhD grant SFRH/BD/29325/2006 (JF), the post-doc grant SFRH/BPD/20695/2004/7Y55 (WC) and CQM pluri-annual funding (PEst-OE/QUI/UI0674/2011-2013). The Academy of Finland is gratefully acknowledged for funding (KR grant no. 122350, 140718, 265328 and 263256). Support from Agencia Canaria de Investigación, Innovación y Sociedad de la Información, Gobierno de Canarias, through project SolSubC200801000088 is also acknowledged. We gratefully acknowledge the continued support of our work by Vidamar Madeira Resort. We are grateful to Dr César Fernandes and Laboratório Regional de Engenharia Civil for the usage of the ATR-FTIR equipment.References
- D. Taher, B. Walfort and H. Lang, Inorg. Chem. Commun., 2004, 7, 1006–1009 CrossRef CAS PubMed.
- M. Mayor, C. von Hanisch, H. B. Weber, J. Reichert and D. Beckmann, Angew. Chem., Int. Ed., 2002, 41, 1183–1186 CrossRef CAS.
- D. Taher, B. Walfort and H. Lang, Inorg. Chim. Acta, 2006, 359, 1899–1906 CrossRef CAS PubMed.
- D. Taher, B. Walfort, G. van Koten and H. Lang, Inorg. Chem. Commun., 2006, 9, 955–958 CrossRef CAS PubMed.
- H. Lang, D. Taher, B. Walfort and H. Pritzkow, J. Organomet. Chem., 2006, 691, 3834–3845 CrossRef CAS PubMed.
- K. Döring, D. Taher, B. Walfort, M. Lutz, A. L. Spek, G. P. M. van Klink, G. van Koten and H. Lang, Inorg. Chim. Acta, 2008, 361, 2731–2739 CrossRef PubMed.
- L. Sun, Y. A. Diaz-Fernandez, T. A. Gschneidtner, F. Westerlund, S. Lara-Avila and K. Moth-Poulsen, Chem. Soc. Rev., 2014, 43, 7378–7411 RSC.
- K. Liu, X. Wang and F. Wang, ACS Nano, 2008, 2, 2315–2323 CrossRef CAS PubMed.
- W.-Y. Wong and C.-L. Ho, Acc. Chem. Res., 2010, 43, 1246–1256 CrossRef CAS PubMed.
- C.-C. Ko and V. Wing-Wah Yam, J. Mater. Chem., 2010, 20, 2063–2070 RSC.
- G.-J. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2011, 40, 2541–2566 RSC.
- D. L. Lichtenberger, S. K. Renshaw, A. Wong and C. D. Tagge, Organometallics, 1993, 12, 3522–3526 CrossRef CAS.
- D. L. Lichtenberger, S. K. Renshaw and R. M. Bullock, J. Am. Chem. Soc., 1993, 115, 3276–3285 CrossRef CAS.
- P. J. Low, Coord. Chem. Rev., 2013, 257, 1507–1532 CrossRef CAS PubMed.
- C. Battocchio, I. Fratoddi, M. V. Russo, V. Carravetta, S. Monti, G. Iucci, F. Borgatti and G. Polzonetti, Surf. Sci., 2007, 601, 3943–3947 CrossRef CAS PubMed.
- M. Moroni, J. Le Moigne, T. A. Pham and J. Y. Bigot, Macromolecules, 1997, 30, 1964–1972 CrossRef CAS.
- Y. Liu, M. S. Liu, X.-C. Li and A. K. Y. Jen, Chem. Mater., 1998, 10, 3301–3304 CrossRef CAS.
- D. A. M. Egbe, A. Wild, E. Birckner, U.-W. Grummt and U. S. Schubert, Macromol. Symp., 2008, 268, 25–27 CrossRef CAS.
- C. Weder and M. S. Wrighton, Macromolecules, 1996, 29, 5157–5165 CrossRef CAS.
- T. A. Skotheim, Handbook of Conducting Polymers, M. Dekker, New York, 1986 Search PubMed.
- U. Mitschke and P. Bäuerle, J. Mater. Chem., 2000, 10, 1471–1507 RSC.
- B. Friedel, C. R. McNeill and N. C. Greenham, Chem. Mater., 2010, 22, 3389–3398 CrossRef CAS.
- H. Detert and E. Sugiono, Synth. Met., 2000, 115, 89–92 CrossRef CAS.
- Y. Matsuura, Y. Tanaka and M. Akita, J. Organomet. Chem., 2009, 694, 1840–1847 CrossRef CAS PubMed.
- M. Akita, Y. Tanaka, C. Naitoh, T. Ozawa, N. Hayashi, M. Takeshita, A. Inagaki and M.-C. Chung, Organometallics, 2006, 25, 5261–5275 CrossRef CAS.
- P. Hamon, F. Justaud, O. Cador, P. Hapiot, S. Rigaut, L. Toupet, L. Ouahab, H. Stueger, J.-R. Hamon and C. Lapinte, J. Am. Chem. Soc., 2008, 130, 17372–17383 CrossRef CAS PubMed.
- J. Bargon, S. Mohmand and R. J. Waltman, IBM J. Res. Dev., 1983, 27, 330–331 CrossRef CAS.
- F. Meyers, A. J. Heeger and J. L. Brédas, J. Chem. Phys., 1992, 97, 2750–2759 CrossRef CAS PubMed.
- S. Takahashi, M. Kariya, T. Yatake, K. Sonogashira and N. Hagihara, Macromolecules, 1978, 11, 1063–1066 CrossRef CAS.
- S. Takahashi, H. Morimoto, E. Murata, S. Kataoka, K. Sonogashira and N. Hagihara, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 565–573 CrossRef CAS.
- S. Takahashi, E. Murata, M. Kariya, K. Sonogashira and N. Hagihara, Macromolecules, 1979, 12, 1016–1018 CrossRef CAS.
- V. W.-W. Yam, L. Zhang, C.-H. Tao, K. Man-Chung Wong and K.-K. Cheung, J. Chem. Soc., Dalton Trans., 2001, 1111–1116 Search PubMed.
- J. Lewis, N. J. Long, P. R. Raithby, G. P. Shields, W.-Y. Wong and M. Younus, J. Chem. Soc., Dalton Trans., 1997, 4283–4288 RSC.
- O. Lavastre, M. Even, P. H. Dixneuf, A. Pacreau and J.-P. Vairon, Organometallics, 1996, 15, 1530–1531 CrossRef CAS.
- W. Weng, T. Bartik, M. Brady, B. Bartik, J. A. Ramsden, A. M. Arif and J. A. Gladysz, J. Am. Chem. Soc., 1995, 117, 11922–11931 CrossRef CAS.
- W.-Y. Wong, W.-K. Wong and P. R. Raithby, J. Chem. Soc., Dalton Trans., 1998, 2761–2766 RSC.
- A. La Groia, A. Ricci, M. Bassetti, D. Masi, C. Bianchini and C. Lo Sterzo, J. Organomet. Chem., 2003, 683, 406–420 CrossRef CAS PubMed.
- H. Lang, K. Döring, D. Taher, U. Siegert, B. Walfort, T. Rüffer and R. Holze, J. Organomet. Chem., 2009, 694, 27–35 CrossRef CAS PubMed.
- H. Ogawa, K. Onitsuka, T. Joh, S. Takahashi, Y. Yamamoto and H. Yamazaki, Organometallics, 1988, 7, 2257–2260 CrossRef CAS.
- K. Onitsuka, S. Yamamoto and S. Takahashi, Angew. Chem., Int. Ed., 1999, 38, 174–176 CrossRef CAS.
- X. H. Wu, S. Jin, J. H. Liang, Z. Y. Li, G.-A. Yu and S. H. Liu, Organometallics, 2009, 28, 2450–2459 CrossRef CAS.
- J. M. Tour, Chem. Rev., 1996, 96, 537–554 CrossRef CAS PubMed.
- M. A. Reed, Science, 1997, 278, 252–254 CrossRef CAS.
- J. M. Seminario, A. G. Zacarias and J. M. Tour, J. Am. Chem. Soc., 1998, 120, 3970–3974 CrossRef CAS.
- D. J. Wold and C. D. Frisbie, J. Am. Chem. Soc., 2000, 122, 2970–2971 CrossRef CAS.
- X. D. Cui, Science, 2001, 294, 571–574 CrossRef CAS PubMed.
- D. J. Wold and C. D. Frisbie, J. Am. Chem. Soc., 2001, 123, 5549–5556 CrossRef CAS PubMed.
- O. Lavastre, J. Plass, P. Bachmann, S. Guesmi, C. Moinet and P. H. Dixneuf, Organometallics, 1997, 16, 184–189 CrossRef CAS.
- T.-Y. Dong, S.-W. Chang, S.-F. Lin, M.-C. Lin, Y.-S. Wen and L. Lee, Organometallics, 2006, 25, 2018–2024 CrossRef CAS.
- T.-Y. Dong, M.-C. Lin, S.-W. Chang, C.-C. Ho, S.-F. Lin and L. Lee, J. Organomet. Chem., 2007, 692, 2324–2333 CrossRef CAS PubMed.
- T.-Y. Dong, H.-Y. Lin, S.-F. Lin, C.-C. Huang, Y.-S. Wen and L. Lee, Organometallics, 2008, 27, 555–562 CrossRef CAS.
- C.-H. Tao, N. Zhu and V. W.-W. Yam, J. Photochem. Photobiol., A, 2009, 207, 94–101 CrossRef CAS PubMed.
- C.-H. Tao and V. W.-W. Yam, J. Photochem. Photobiol., C, 2009, 10, 130–140 CrossRef CAS PubMed.
- E. G. Tennyson and R. C. Smith, Inorg. Chem., 2009, 48, 11483–11485 CrossRef CAS PubMed.
- C. K. M. Chan, C.-H. Tao, H.-L. Tam, N. Zhu, V. W.-W. Yam and K.-W. Cheah, Inorg. Chem., 2009, 48, 2855–2864 CrossRef CAS PubMed.
- Z. Ji, S. Li, Y. Li and W. Sun, Inorg. Chem., 2010, 49, 1337–1346 CrossRef CAS PubMed.
- V. W.-W. Yam and K. K.-W. Lo, in Encyclopedia of Inorganic and Bioinorganic Chemistry, 2011 Search PubMed.
- K.-Q. Wu, J. Guo, J.-F. Yan, L.-L. Xie, F.-B. Xu, S. Bai, P. Nockemann and Y.-F. Yuan, Dalton Trans., 2012, 41, 11000–11008 RSC.
- H. Li, J. Li, J. Ding, W. Yuan, Z. Zhang, L. Zou, X. Wang, H. Zhan, Z. Xie, Y. Cheng and L. Wang, Inorg. Chem., 2014, 53, 810–821 CrossRef CAS PubMed.
- F. Lahoz, N. Capuj, C. J. Oton and S. Cheylan, Chem. Phys. Lett., 2008, 463, 387–390 CrossRef CAS PubMed.
- C. J. Oton, D. Navarro-Urrios, N. E. Capuj, M. Ghulinyan, L. Pavesi, S. González-Pérez, F. Lahoz and I. R. Martín, Appl. Phys. Lett., 2006, 89, 011107–011109 CrossRef PubMed.
- J. Figueira, J. Rodrigues, L. Russo and K. Rissanen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, o33–o36 CAS.
- S. M. Dirk, D. W. Price, S. Chanteau, D. V. Kosynkin and J. M. Tour, Tetrahedron, 2001, 57, 5109–5121 CrossRef CAS.
- S. K. Park, J. Photochem. Photobiol., A, 2000, 135, 155–162 CrossRef CAS.
- U. H. F. Bunz, Chem. Rev., 2000, 100, 1605–1644 CrossRef CAS PubMed.
- K. L. Chandra, S. Zhang and C. B. Gorman, Tetrahedron, 2007, 63, 7120–7132 CrossRef CAS PubMed.
- J. M. Tour, Molecular Electronics: Commercial Insights, Chemistry, Devices, Architecture, and Programming, World Scientific, River Edge, N.J., 2003 Search PubMed.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- A. Nagy, Z. Novák and A. Kotschy, J. Organomet. Chem., 2005, 690, 4453–4461 CrossRef CAS PubMed.
- H. Günther, NMR Spectroscopy: Basic Principles, Concepts, and Applications in Chemistry, Wiley, Chichester, New York, 2nd edn, 1995 Search PubMed.
- A table which summarizes the position of the relevant absorption bands for both the ligands and the Pd complexes is available in the Supplementary Information (Table S1†).
- S. I. Nakatsuji, K. Matsuda, Y. Uesugi, K. Nakashima, S. Akiyama and W. Fabian, J. Chem. Soc., Perkin Trans. 1, 1992, 755–758 RSC.
- E. Pretsch, P. Bühlmann and M. Badertscher, Structure Determination of Organic Compounds: Tables of Spectral Data, Springer, Berlin, 4th edn, 2009 Search PubMed.
- C. L. Choi, Y. F. Cheng, C. Yip, D. L. Phillips and V. W.-W. Yam, Organometallics, 2000, 19, 3192–3196 CrossRef CAS.
- H. Masai, K. Sonogashira and N. Hagihara, Bull. Chem. Soc. Jpn., 1971, 44, 2226–2230 CrossRef CAS.
- V. W.-W. Yam, C.-H. Tao, L. Zhang, K. M.-C. Wong and K.-K. Cheung, Organometallics, 2001, 20, 453–459 CrossRef CAS.
- W. M. Kwok, D. L. Phillips, P. K.-Y. Yeung and V. W.-W. Yam, Chem. Phys. Lett., 1996, 262, 699–708 CrossRef CAS.
- W. M. Kwok, D. L. Phillips, P. K.-Y. Yeung and V. W.-W. Yam, J. Phys. Chem. A, 1997, 101, 9286–9295 CrossRef CAS.
- G. F. Manbeck, W. W. Brennessel, R. A. Stockland and R. Eisenberg, J. Am. Chem. Soc., 2010, 132, 12307–12318 CrossRef CAS PubMed.
- Y. Suenaga, Y. Hirano, Y. Umehata, K. Kamei and M. Maekawa, RIST, Sci. Technol., 2011, 2011/2012, 23–33 Search PubMed.
- L. M. Mirica and J. R. Khusnutdinova, Coord. Chem. Rev., 2013, 257, 299–314 CrossRef CAS PubMed.
- R. F. Winter, Organometallics, 2014, 33, 4517–4536 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- CSD version 5.34 (February 2014 update) Search PubMed.
- A. K. Bar, S. Shanmugaraju, K.-W. Chi and P. S. Mukherjee, Dalton Trans., 2011, 40, 2257–2267 RSC.
- W. M. Khairul, M. A. Fox, P. A. Schauer, D. S. Yufit, D. Albesa-Jové, J. A. K. Howard and P. J. Low, Dalton Trans., 2010, 39, 11605–11615 RSC.
- K. Onitsuka, H. Ogawa, T. Joh, S. Takahashi, Y. Yamamoto and H. Yamazaki, J. Chem. Soc., Dalton Trans., 1991, 1531–1536 RSC.
- C. J. Cardin, D. J. Cardin, M. F. Lappert and K. W. Muir, J. Chem. Soc., Dalton Trans., 1978, 46–50 RSC.
- L. Venkataraman, J. E. Klare, C. Nuckolls, M. S. Hybertsen and M. L. Steigerwald, Nature, 2006, 442, 904–907 CrossRef CAS PubMed.
- D. Vonlanthen, A. Mishchenko, M. Elbing, M. Neuburger, T. Wandlowski and M. Mayor, Angew. Chem., Int. Ed., 2009, 48, 8886–8890 CrossRef CAS PubMed.
- J. Rotzler, D. Vonlanthen, A. Barsella, A. Boeglin, A. Fort and M. Mayor, Eur. J. Org. Chem., 2010, 1096–1110 CrossRef CAS.
- D. Vonlanthen, J. Rotzler, M. Neuburger and M. Mayor, Eur. J. Org. Chem., 2010, 120–133 CrossRef CAS.
- A. Mishchenko, D. Vonlanthen, V. Meded, M. Bürkle, C. Li, I. V. Pobelov, A. Bagrets, J. K. Viljas, F. Pauly, F. Evers, M. Mayor and T. Wandlowski, Nano Lett., 2010, 10, 156–163 CrossRef CAS PubMed.
- L.-J. Fan, Y. Zhang, C. B. Murphy, S. E. Angell, M. F. L. Parker, B. R. Flynn and W. E. Jones, Coord. Chem. Rev., 2009, 253, 410–422 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- C. J. Jing, L. S. Chen, Y. Shi and X. G. Jin, Chin. Chem. Lett., 2005, 16, 1519–1522 CAS.
- W. Holzer, A. Penzkofer, S. H. Gong, A. P. Davey and W. J. Blau, Opt. Quantum Electron., 1997, 29, 713–724 CrossRef CAS.
- H. Nakayama and S. Kimura, J. Phys. Chem. A, 2011, 115, 8960–8968 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- P. G. Gassman and T. L. Guggenheim, J. Am. Chem. Soc., 1982, 104, 5849–5850 CrossRef CAS.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381–388 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 64, 112–122 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C, 31P NMR, ESI-MS, FTIR and UV-Vis spectra as well as the remainder of the cyclic voltammograms can be found in the ESI. CCDC 882324 and 882325. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00493k |
| ‡ Present address: Clinical Neuroscience Unit, Department of Pharmacology and Clinical Neuroscience, Umeå University, 90187 Umeå, Sweden. |
| § Present address: Selvita S.A. ul. Bobrzyńskiego 1430-348 Kraków, Poland. |
| ¶ Present address: Rigaku Europe SE, Am Hardtwald 11, 76275 Ettlingen, Germany. |
| || Present address: Tampere University of Technology, Department of Chemistry and Bioengineering, P.O. Box 541, 33101 Tampere, Finland. |
| This journal is © The Royal Society of Chemistry 2015 |