
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing the effect of reducing agents on the selective catalytic reduction of NOx over Ag/Al2O3 catalysts
Carmine
D'Agostino
*a,
Sarayute
Chansai
b,
Isabelle
Bush
a,
Chensong
Gao
a,
Mick D.
Mantle
a,
Christopher
Hardacre
*b,
Stuart L.
James
b and
Lynn F.
Gladden
*a
aDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Pembroke Street, Cambridge, CB2 3RA, UK. E-mail: cd419@cam.ac.uk; lfg1@cam.ac.uk; Tel: +44(0)1223 761629 Tel: +44(0)1223 334762
bCentre for the Theory and Application of Catalysis, CenTACat, School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AG, UK. E-mail: C.Hardacre@qub.ac.uk; Tel: +44(0)28 9097 4592
First published on 30th October 2015
Abstract
The selective catalytic reduction (SCR) of NOx in the presence of different reducing agents over Ag/Al2O3 prepared by wet impregnation was investigated by probing catalyst activity and using NMR relaxation time analysis to probe the strength of surface interaction of the various reducing agent species and water. The results reveal that the strength of surface interaction of the reducing agent relative to water, the latter present in engine exhausts as a fuel combustion product and, in addition, produced during the SCR reaction, plays an important role in determining catalyst performance. Reducing agents with weak strength of interaction with the catalyst surface, such as hydrocarbons, show poorer catalytic performance than reducing agents with a higher strength of interaction, such as alcohols. This is attributed to the greater ability of oxygenated species to compete with water in terms of surface interaction with the catalyst surface, hence reducing the inhibiting effect of water molecules blocking catalyst sites. The results support the observations of earlier work in that the light off-temperature and maximum NOx conversion and temperature at which that occurs are sensitive to the reducing agent present during reaction, and the proposal that improved catalyst performance is caused by increased adsorption strength of the reducing agent, relative to water, at the catalyst surface. Importantly, the NMR relaxation time analysis approach to characterising the strength of adsorption more readily describes the trends in catalytic behaviour than does a straightforward consideration of the polarity (i.e., relative permittivity) of the reducing agents studied here. In summary, this paper describes a simple approach to characterising the interaction energy of water and reducing agent so as to aid the selection of reducing agent and catalyst to be used in SCR conversions.
Introduction
The selective catalytic reduction (SCR) of NOx to N2 has drawn considerable attention in the past ten years1,2 due to its effectiveness in removing NOx from various exhausts in the presence of excess oxygen, a condition typically found in diesel engine exhausts.3 Metal nanoparticles supported on porous oxides are perhaps the most widely used type of catalysts;2 of these, Ag/Al2O3 catalysts have shown promising results due to their ability to reduce NOx in both laboratory and full-scale tests.4One important aspect of the SCR reaction is the choice of the reducing agents and how this affects the catalytic performances. Indeed, the nature of the reducing agent can greatly affect SCR catalyst activity. Hydrocarbons, like those typically presents in fuel mixtures, are perhaps the most common reducing agents.2,3,5 However, the use of other reducing agents such as alcohols, has also been investigated.6 Alcohols appear to be better reducing agents, showing much lower light-off temperatures (i.e., the temperature at which 50% of conversion is achieved) relative to hydrocarbons.2 This is thought to be due to the greater ability of alcohols relative to hydrocarbons to compete with water for adsorption sites on the catalyst.2
Shimizu7 suggested that adsorption properties of the reducing agent affect the rate at which surface acetates, important intermediate species during the SCR reaction, are formed. Therein, the importance of competitive adsorption with water was also highlighted, suggesting that the use of reducing agents with a greater enthalpy of adsorption results in lesser inhibition of reducing agent adsorption by competitive water adsorption, which leads to higher water tolerance and greater reactivity.3
It is, therefore, clear that adsorption properties of reducing agent molecules over the catalyst surface are of significant importance for SCR reactions. However, it is our understanding that a detailed experimental study of surface interactions between different reducing agents and SCR catalysts, aiming at validating the current hypothesis, has not yet been reported. The issue of characterising competitive adsorption processes is recognised as being of importance in understanding SCR2,8 processes, as well as the wider field of heterogeneous catalysis and surface science.9,10 It is clear that the nature of the reducing agent has a strong effect on SCR catalytic activity. In the literature it has been reported2,3,11–17 that oxygenated molecules, such as alcohols, give improved catalyst performances compared to hydrocarbons, which is thought to originate from competitive adsorption effects between reducing agent molecules and water, the latter being able to inhibit the catalyst sites.2,3,18 This is indeed a plausible explanation, although it has to be said that, according to our knowledge, there is little experimental evidence to support it.
NMR relaxation time analysis provides a mean of probing surface interactions between adsorbate/adsorbent by probing changes in molecular dynamics of molecules due to the proximity of a solid surface.19,20 In particular, the T1/T2 ratio, T1 being the NMR spin–lattice relaxation time and T2 the NMR transverse spin relaxation time, is an indicator of the strength of surface interaction.19–22 This ratio has been recently used to understand catalytic performances in several heterogeneous catalytic processes21,23 and was also used in our previous work to understand the effect of ball milling on the SCR reaction of n-octane over Ag/Al2O3 catalysts.24 Therein, it was shown that surface modifications due to ball milling of the catalyst may increase the catalytic performance by reducing the competitive adsorption of water relative to the hydrocarbon, which highlighted the importance of competitive adsorption with water during the SCR reaction.
In the current work, we focus on investigating the strength of surface interaction of different reducing agents and see how this reflects on catalytic performances. We have studied the SCR of NOx in the presence of various reducing agents, namely toluene, n-octane and ethanol, over Ag/Al2O3 prepared by standard wet impregnation. NMR T1 and T2 relaxation measurements, from which T1/T2 ratios were also calculated, were used to quantify the strength of surface interaction of the different reducing agents and water on the catalyst in order to understand to what extent this parameter affects the overall catalyst activity. In earlier work25 we have shown that T1/T2 gives a characterisation of the strength of a molecule–surface interaction and this value can be related, following appropriate calibration, directly to the adsorption energy as determined by temperature-programmed desorption analysis.
Experimental
Materials and chemicals
Toluene (≥99.5%), ethanol (≥99.5%) and n-octane (≥99%) were purchased from Sigma Aldrich and were used without any pre-treatment. Deionised water was obtained from a laboratory water purification system (ELGA DV 25). The wet impregnated 2% Ag/Al2O3 catalyst was prepared using a procedure previously reported.26Activity tests
The catalytic activity tests were performed as reported elsewhere.27 For completeness, the essential details are also given here. Typically, the catalytic activity tests over Ag/Al2O3 catalyst were carried out in a fixed-bed flow reactor system, consisting of a quartz reactor tube. The catalyst was held in place between plugs of quartz wool and a K-type thermocouple was placed in the centre of the catalyst bed. Each of the gases in the feed system was controlled individually by mass flow controllers, while the hydrocarbon and water vapour were introduced to the system by means of separate saturators with Ar as a carrier gas. The hydrocarbon used was supplied using a saturator placed in an ice/water bath. The H2O saturator temperature was controlled using a thermostatic bath. All the lines following the water saturator were trace-heated to prevent condensation. A feed gas stream consisting of 720 ppm NO; 542 ppm n-C8H18, or 620 ppm C7H8, or 2170 ppm CH3CH2OH; 4.3% O2, 7.2% H2O; 7.2% CO2; and Ar balance was introduced to the reactor, which was heated from 150 to 600 °C and then back down to 150 °C stepwise at 50 °C intervals dwelling at each temperature for 40 min in order to obtain steady-state conditions. The C1 concentration of the reducing agent (i.e., concentration as total carbon) was 4340 ppm and the C1/NO ratio was kept at 6 for all catalytic activity tests. Three reducing agents, i.e. n-octane, toluene, and ethanol were used. The total gas flow rate was 276 cm3 min−1 over 276 mg of catalyst, which had been sieved to obtain particle sizes in the range 250–450 μm. The space velocity for all catalytic tests was 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm3 g−1 h−1 (calculated using the total gas flow rate divided by the amount of the catalyst used in the activity test). The inlet and outlet NOx concentrations were determined by a Signal 4000VM series chemiluminescence detector. The oxidation of the hydrocarbon was measured online using a Bruker Tensor 27 IR spectrometer, fitted with a gas cell of volume 190 cm3. All the activity data was measured during the decreasing temperature ramp. Using this method, the activity remained constant once the desired temperature had been reached and the conversions were calculated from an average of outlet NOx readings at each temperature.
000 cm3 g−1 h−1 (calculated using the total gas flow rate divided by the amount of the catalyst used in the activity test). The inlet and outlet NOx concentrations were determined by a Signal 4000VM series chemiluminescence detector. The oxidation of the hydrocarbon was measured online using a Bruker Tensor 27 IR spectrometer, fitted with a gas cell of volume 190 cm3. All the activity data was measured during the decreasing temperature ramp. Using this method, the activity remained constant once the desired temperature had been reached and the conversions were calculated from an average of outlet NOx readings at each temperature.
NMR experiments
NMR experiments were performed on a Bruker DMX 300 operating at a frequency of 300.13 MHz. The powdered catalyst was pressed into 10 mm tablets before being broken into grains of typically 2–3 mm in dimension. This procedure makes the samples easier to handle and, by reducing the amount of liquid in the inter-particle space, it makes the NMR measurement more sensitive to the liquid–surface interaction (i.e., the amount of signal from bulk liquid is minimised). The catalyst grains were then dried in an oven at 110 °C for 12 h and then immersed in the liquid of interest for at least 24 h. The wet catalyst grains were then removed from the liquid, placed onto a pre-soaked filter paper in order to remove excess external liquid and finally placed into a 5 mm NMR tube. To ensure a saturated atmosphere in the NMR tube, hence minimising errors due to evaporation of volatile liquids, a small piece of adsorbent filter paper was impregnated with the liquid under investigation. This was then placed under the cap of the NMR tube, which was finally sealed with parafilm. All the NMR measurements were performed at atmospheric pressure and room temperature.NMR data analysis
Proton NMR relaxation times T1 and T2 were measured using the standard inversion recovery and CPMG (Carr-Purcell-Meiboom-Gill) techniques,28 respectively, which are schematically shown in Fig. 1. Experimental data were fitted using single exponential functions. The T1 relaxation time constant was obtained by fitting the experimental data to the equation: | (1) |
 | ||
| Fig. 1 (a) T1 inversion recovery and (b) T2 CPMG pulse sequences used in this work. In the inversion recovery pulse sequence (a) the NMR signal is acquired after a time delay τ for a list of different time delays. In the CPMG pulse sequence (b) the NMR signal is acquired after a series of n echoes, which correspond to a 2 × τ × n total time delay for a list of different time delays. | ||
The T2 relaxation time constant was obtained by fitting the experimental data to the equation:
 | (2) |
In eqn (1) and (2), S represents the NMR signal intensity and t the time. Note that the NMR signal intensity was calculated by integrating the whole NMR spectrum of the species confined within the catalyst. In this way, the calculated NMR relaxation times are representative of the whole molecular species adsorbed over the catalyst surface.
Results and discussion
SCR catalyst testing
The data for the catalytic activity of NOx reduction is summarised in Table 1 measured via the light-off temperature, T50%, at which 50% of conversion is achieved, and the maximum conversion of NOx at the temperature at which such conversion is achieved.27 A higher light-off temperature indicates a poorer catalytic performance.| Compound | T 50% [°C] | Max NOx conversion [%] (T [°C]) |
|---|---|---|
| Toluene | 520 | 69 (600) |
| n-Octane | 390 | 80 (478) |
| Ethanol | 295 | 98 (354) |
It is clear that both toluene and n-octane give T50% values significantly higher than when ethanol is used as the reducing agent; further, toluene has a higher light-off temperature than n-octane. Moreover, this ranking of catalyst performance with reducing agent is furthermore supported on comparing the maximum NOx conversion and temperature at which it occurs. We also report the T50% values relative to the reducing agent conversion, which are shown in Table 2. The observed reactivity trend for the reducing agent is similar to that observed for the NOx, with ethanol showing a significantly higher reactivity, hence a lower T50% compared to the two other hydrocarbons.
| Compound | T 50% [°C] | Max reducing agent conversion [%] (T [°C]) |
|---|---|---|
| Toluene | 435 | 99 (560) |
| n-Octane | 425 | 99 (525) |
| Ethanol | 265 | 99 (400) |
Probing strength of surface interaction
Fig. 2 and 3 show, respectively, the T1 and the T2 experimental data acquired for the different reducing agents and water in Ag/Al2O3. The quality of the data and the fittings to eqn (1) and (2) for T1 and T2, respectively, are excellent. The T1 and T2 values obtained for these data are reported in Table 3. Table 3 also reports the T1/T2 ratio calculated from the individual T1 and T2 values. | ||
| Fig. 2 T 1 plots for different reducing agents and water within Ag/Al2O3 catalyst. T1 is measured using the inversion recovery pulse sequence. Solid lines are fitting to eqn (1). | ||
 | ||
| Fig. 3 T 2 plots for different reducing agents and water within Ag/Al2O3 catalyst. T2 is measured using the CPMG pulse sequence. Solid lines are fitting to eqn (2). | ||
| Compound | T 1 [ms] | T 2 [ms] | T 1/T2 [–] |
|---|---|---|---|
| a For the pure bulk liquids, T1 ~ T2 with the following values: 4240 ms for toluene, 2500 ms for n-octane, 2630 ms for ethanol, 2700 ms for water. | |||
| Toluene | 1390 | 85 | 16 |
| n-Octane | 1204 | 66 | 18 |
| Ethanol | 522 | 13 | 40 |
| Water | 212 | 3 | 71 |
As it is seen from Table 3, the oxygenated molecules have significantly higher values of T1/T2 ratio, indicating a much stronger interaction with the catalyst surfaces than the n-octane and toluene, which is in agreement with the current hypothesis reported in the literature.2,7 The stronger interaction of the oxygenated species with the surface is likely to arise from hydrogen bonding with the solid surface, which acts to enhance surface interactions.29
Effect of reducing agent
By inspection of Table 3, it can be observed that the different species have very different relaxation properties. The reducing agents have higher values of T1 and T2 compared to water. In ethanol and particularly water, the decrease of T2 relative to T1 is much more significant compared to the two hydrocarbons, resulting in greater values of the T1/T2 ratio observed, particularly for water, the latter showing a T1/T2 = 71, significantly higher than ethanol, T1/T2 = 40, and substantially higher than n-octane, T1/T2 = 18, and toluene, T1/T2 = 16. These values can be explained in terms of molecules adsorbed onto the surface and exhibiting modified rotational and translational dynamics at the pore surface.19 In particular, a decrease of T2 values relative to T1 values can be related to a slower translational dynamics over the surface, due to stronger surface interactions.19,30 As a result, the T1/T2 ratio increases. Such a ratio can be considered to be an analogous of a surface interaction energy31 as we have recently demonstrated.25 It has to be noted that paramagnetic species may also affect the values or NMR relaxation times but this is not relevant to our current work for reasons previously explained in detail elsewhere.24,25In summary, from the NMR relaxation time results it can be inferred that water shows the greatest strength of surface interaction, significantly greater than that of all the reducing agents, as it can be inferred by the large values of its T1/T2 ratio, which are also plotted in Fig. 4 for clarity. As for the reducing agents, toluene shows the lowest strength of surface interaction (i.e., lowest T1/T2 value), followed closely by n-octane, which shows slightly higher values; ethanol shows considerably higher strength of surface interaction compared to toluene and n-octane and its T1/T2 values are significantly closer to water compared to those measured for the two hydrocarbons. The results clearly suggest that, compared to toluene and n-octane, ethanol has a greater ability to compete with water for adsorption when compared to toluene and n-octane. This has been speculated in the literature2,18 and the current results give a clear experimental evidence of this effect.
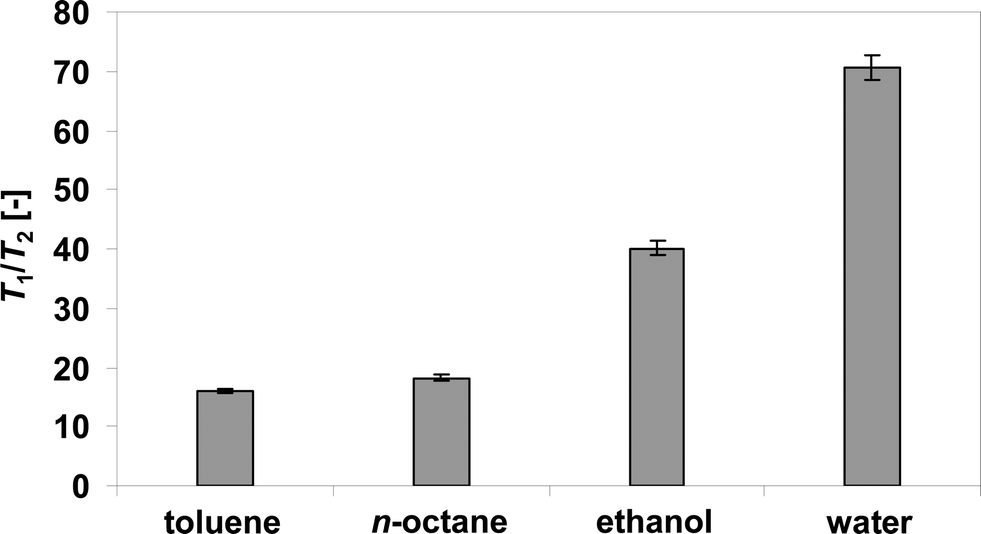 | ||
| Fig. 4 T 1/T2 values for water and reducing agents within Ag/Al2O3 catalyst. Errors bars are also reported. | ||
The current results seem to explain well the catalytic performances reported in the literature in terms of type of reducing agent.2,32 It is now of interest to compare the results gathered from NMR relaxation measurements to the catalytic performance of these samples, in order to see to what extent surface interactions and catalyst activity are inter-related.
Catalyst activity and strength of surface interaction
There is a general agreement in the literature2–4,7,33–36 that the SCR of NO with hydrocarbons reaction begins with the oxidation of NO by O2 to adsorbed NOx species and with the oxidation of reductant to form partially oxidized hydrocarbon species over the catalyst surface, which have been reported to reduce adsorbed NOx species via organo-nitro and/or organo-nitrito adsorbed species to yield gaseous N2.Comparing the SCR catalytic tests, summarised in Tables 1 and 2, and the results from NMR relaxation time measurements, summarised in Table 3 and Fig. 4, it is clear that the reactivity trend reflects well, at least qualitatively, the trend in surface interactions obtained from NMR relaxometry measurements of T1/T2. A reducing agent of higher strength of surface interaction, such as ethanol, will have greater ability to reach the surface and react, hence enhancing the catalyst performances. This is consistent with several catalytic studies previously reported on SCR reactions in the presence of oxygenated reducing agents.2,6,37,38 The large variation in catalytic performances observed in this work by changing the reducing agent strongly suggests that its adsorption strength is important for the overall catalytic process. The use of reducing agents of higher adsorption strength can indeed have several implications that will ultimately affect the whole SCR process, as this will reduce the competitive adsorption of water, improving the surface coverage of the reducing agent molecules, as previously suggested.24 In addition, a higher strength of surface interaction of the reducing agent, associated with its higher surface concentration, would increase its ease of partial oxidation, which is important in order to form partially oxidized organic species over the catalyst surface that reduce adsorbed NOx species via organo-nitro and/or organo-nitrito adsorbed species and ultimately yield N2.
It is interesting to note that the trend in T1/T2 matches qualitatively the trend in both T50% for NOx and reducing agent. In particular, it can be observed a much closer correlation between T1/T2 of reducing agents (see Table 3) and T50% of reducing agents (see Table 2). Indeed, the difference in T1/T2 values between n-octane and toluene is not as large as that observed between these two hydrocarbons and ethanol. The trend in T50% of reducing agent leads to the same conclusion. This strengthens the hypothesis that reducing agents with higher strength of surface interactions increase its ease of partial oxidation, which is important in order to reduce adsorbed NOx species.24
In general, for the species studied in this work, we observe that the trend of T1/T2 in Table 3 reflects to some extent the polarity of the molecules. The trend for relative permittivity is εwater = 80.4 > εethanol = 24.5 > εtoluene = 2.38 ~ εn-octane = 2, which is similar to the trend in T1/T2. However, based solely on polarity, one might expect toluene to give slightly better SCR catalytic performances than n-octane, which is not the case. The T1/T2 values reveal that, despite its slightly greater polarity compared to n-octane, toluene has a slightly lower strength of surface interaction with the catalyst surface, which agrees qualitatively with the reaction data.
In summary, the current results show that the adsorption strength of the reducing agent plays an important role in determining the catalyst activity in the SCR reaction. NMR relaxation time measurements allow us to quantify this by means of T1/T2 values, hence offering a tool to optimise and rationalise the selection of different reducing agents.
Conclusions
The SCR reaction of NOx in the presence of toluene, n-octane and ethanol over a wet impregnated Ag/Al2O3 catalyst has been investigated by assessing the catalyst activity, using reaction studies, and adsorption phenomena, using NMR relaxation time analysis. The trend in strength of surface interaction, obtained by NMR relaxation measurements, explains the reactivity trend. Reducing agents with weaker strength of surface interaction relative to water, such as hydrocarbons, show poorer activity compared to reducing agents with stronger strength of interaction, such as ethanol. This is likely to be due to the greater ability of the reducing agents with higher interaction strength to reduce the inhibitory effect of water molecules blocking the catalytic sites, hence improving the conditions for the surface formation of partially oxidized organic species needed for the SCR reaction to proceed further. The results serve, therefore, as an experimental validation of the hypothesis on competitive adsorption previously speculated in the literature. In summary, the adsorption strength of the reducing agent over the catalyst surface is a very important parameter to take into account when investigating SCR reaction and NMR relaxation can be used as a valid tool to probe such adsorption phenomena, hence rationalising the choice of catalysts and reducing agents.Acknowledgements
We gratefully acknowledge funding for this work from the EPSRC CASTech grant (EP/G012156/1). Carmine D'Agostino would like to acknowledge Wolfson College, Cambridge, for supporting his research activities. The authors would also like to thank Dr Jonathan Mitchell for useful discussions.References
- J. P. Breen and R. Burch, Top. Catal., 2006, 39, 53–58 CrossRef CAS.
- R. Burch, J. P. Breen and F. C. Meunier, Appl. Catal., B, 2002, 39, 283–303 CrossRef CAS.
- K. Shimizu, A. Satsuma and T. Hattori, Appl. Catal., B, 2000, 25, 239–247 CrossRef CAS.
- K. Eranen, F. Klingstedt, K. Arve, L. E. Lindfors and D. Y. Murzin, J. Catal., 2004, 227, 328–343 CrossRef.
- Y. Kintaichi, H. Hamada, M. Tabata, M. Sasaki and T. Ito, Catal. Lett., 1990, 6, 239–244 CrossRef CAS.
- H. Hamada, Y. Kintaichi, M. Sasaki, T. Ito and T. Yoshinari, Appl. Catal., A, 1992, 88, L1–L7 CrossRef CAS.
- K. Shimizu, J. Shibata, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2001, 3, 880–884 RSC.
- Y. H. Hu, K. Griffiths and P. R. Norton, Surf. Sci., 2009, 603, 1740–1750 CrossRef CAS.
- A. Ruban, B. Hammer, P. Stoltze, H. L. Skriver and J. K. Norskov, J. Mol. Catal. A: Chem., 1997, 115, 421–429 CrossRef CAS.
- C. H. Christensen and J. K. Norskov, J. Chem. Phys., 2008, 128, 8 CrossRef PubMed.
- T. Miyadera, Appl. Catal., B, 1993, 2, 199–205 CrossRef CAS.
- T. Miyadera and K. Yoshida, Chem. Lett., 1993, 1483–1486 CrossRef CAS.
- S. Kameoka, Y. Ukisu and T. Miyadera, Phys. Chem. Chem. Phys., 2000, 2, 367–372 RSC.
- J. H. Lee, S. J. Schmieg and S. H. Oh, Appl. Catal., A, 2008, 342, 78–86 CrossRef CAS.
- H. Dong, S. Shuai, R. Li, H. Wang, X. Shi and H. He, Chem. Eng. J., 2008, 135, 195–201 CrossRef CAS.
- C. Zhang, H. He, S. Shuai and J. Wang, Environ. Pollut., 2007, 147, 415–421 CrossRef CAS PubMed.
- K. Arve, K. Eranen, M. Snare, F. Klingstedt and D. Y. Murzin, Top. Catal., 2007, 42–43, 399–403 CrossRef.
- K. Shimizu and A. Satsuma, Phys. Chem. Chem. Phys., 2006, 8, 2677–2695 RSC.
- J. Mitchell, L. M. Broche, T. C. Chandrasekera, D. J. Lurie and L. F. Gladden, J. Phys. Chem. C, 2013, 117, 17699–17706 CAS.
- D. Weber, J. Mitchell, J. McGregor and L. F. Gladden, J. Phys. Chem. C, 2009, 113, 6610–6615 CAS.
- C. D'Agostino, G. L. Brett, P. J. Miedziak, D. W. Knight, G. J. Hutchings, L. F. Gladden and M. D. Mantle, Chem. – Eur. J., 2012, 18, 14426–14433 CrossRef PubMed.
- P. J. McDonald, J. Mitchell, M. Mulheron, P. S. Aptaker, J. P. Korb and L. Monteilhet, Cem. Concr. Res., 2007, 37, 303–309 CrossRef CAS.
- C. D'Agostino, T. Kotionova, J. Mitchell, P. J. Miedziak, D. W. Knight, S. H. Taylor, G. J. Hutchings, L. F. Gladden and M. D. Mantle, Chem. – Eur. J., 2013, 19, 11725–11732 CrossRef PubMed.
- K. Ralphs, C. D'Agostino, R. Burch, S. Chansai, L. F. Gladden, C. Hardacre, S. L. James, J. Mitchell and S. F. R. Taylor, Catal. Sci. Technol., 2014, 4, 531–539 CAS.
- C. D'Agostino, J. Mitchell, M. D. Mantle and L. F. Gladden, Chem. – Eur. J., 2014, 20, 13009–13015 CrossRef PubMed.
- U. Kamolphop, S. F. R. Taylor, J. P. Breen, R. Burch, J. J. Delgado, S. Chansai, C. Hardacre, S. Hengrasmee and S. L. James, ACS Catal., 2011, 1, 1257–1262 CrossRef CAS.
- K. L. Ralphs, S. Chansai, C. Hardacre, R. Burch, S. Taylor and S. L. James, Catal. Today, 2014, 246, 198–206 CrossRef.
- E. Fukushima and S. W. Roeder, Experimental pulse NMR, Addison-Weslkey, Reading, US, 1981 Search PubMed.
- A. Ignatchenko, D. G. Nealon, R. Dushane and K. Humphries, J. Mol. Catal. A: Chem., 2006, 256, 57–74 CrossRef CAS.
- P. J. McDonald, J. P. Korb, J. Mitchell and L. Monteilhet, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 9 Search PubMed.
- S. Godefroy, M. Fleury, F. Deflandre and J. P. Korb, J. Phys. Chem. B, 2002, 106, 11183–11190 CrossRef CAS.
- H. He, Y. B. Yu, Y. Li, Q. Wu, X. L. Zhang, C. B. Zhang, X. Y. Shi and X. P. Song, Chin. J. Catal., 2010, 31, 491–501 CAS.
- J. P. Breen, R. Burch, C. Hardacre, C. J. Hill and C. Rioche, J. Catal., 2007, 246, 1–9 CrossRef CAS.
- P. Sazama, L. Capek, H. Drobna, Z. Sobalik, J. Dedecek, K. Arve and B. Wichterlova, J. Catal., 2005, 232, 302–317 CrossRef CAS.
- F. C. Meunier, J. P. Breen, V. Zuzaniuk, M. Olsson and J. R. H. Ross, J. Catal., 1999, 187, 493–505 CrossRef CAS.
- F. C. Meunier, V. Zuzaniuk, J. P. Breen, M. Olsson and J. R. H. Ross, Catal. Today, 2000, 59, 287–304 CrossRef CAS.
- M. Tatsuo, Appl. Catal., B, 1993, 2, 199–205 CrossRef.
- M. Tabata, H. Tsuchida, K. Miyamoto, T. Yoshinari, H. Yamazaki, H. Hamada, Y. Kintaichi, M. Sasaki and T. Ito, Appl. Catal., B, 1995, 6, 169–183 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |