Uranium-mediated ring-opening polymerization of ε-caprolactone: a comparative study†
Received
23rd July 2015
, Accepted 4th August 2015
First published on 3rd September 2015
Abstract
The ring-opening polymerization (ROP) of the cyclic ester ε-caprolactone was studied using the uranium(IV) complexes [(ImDippN)2U(NMeEt)2] (3), [(C5Me5)2U(NMe2)2] (4) and [(C5Me5)2U(NCMePh)2] (5) as initiators. While the bis(imidazolin-2-iminato) complex 3 displayed a surprisingly high catalytic activity of 1.2 × 107 g (PCL) mol−1 h−1 at room temperature, compounds 4 and 5 exhibited lower catalytic activities even at 90 °C. The activity of the uranium complex 3 was further compared to the imidazolin-2-iminato uranium(IV) complexes [(ImtBuN)4U] (1) and [(ImMesN)3U(NMeEt)] (2), which display catalytic activities of 7.9 × 103 g (PCL) mol−1 h−1, and 5.3 × 103 g (PCL) mol−1 h−1, respectively at an elevated temperature of 90 °C. In order to shed light on the operative mechanisms, kinetic studies were carried out with complexes 3–5.
Introduction
The history of polycaprolactone (PCL) can be traced back to 1934, when Carothers et al. reported the polymerization of ε-caprolactone under heat or by addition of catalytic amounts of potassium carbonate.1 Since then, two main pathways are used for the synthesis of this polymer, which are based either on the free radical ring-opening polymerization of 2-methylene-1-dioxepane2 or on the ring-opening polymerization (ROP) of ε-caprolactone (Scheme 1), which can be achieved by an anionic,3 cationic,3 monomer activated,4 or coordination–insertion mechanism.5
 |
| | Scheme 1 General synthetic methods for polycaprolactone. | |
The great interest in this polymer over the last eight decades can be attributed to its low melting point (59–64 °C), high solubility in a large variety of organic solvents, exceptional miscibility, and mechanical compatibility with a large number of polymers, as well as its biodegradability and biocompatibility.6 In addition, the extensive research carried out during the 1970s and 1980s in the field of biodegradable polymers led to interesting correlations between the molecular weight of the polymer, its biodegradation conditions and degradation kinetics.7 Therefore, the application of PCL in the field of biomedicine is widespread and includes the scaffolds in tissue engineering,8 long-term drug delivery systems7b and contraceptive delivery systems.9 Additionally, PCL is used as a packaging material,9 in microelectronics10 and in adhesives.7e The availability of the monomer ε-caprolactone, and the wide applicability of the corresponding polyester renders PCL an environmentally friendly, low-cost polymer with an increasing demand over the last two decades.11
The polymerization of ε-caprolactone has been investigated with a variety of main group5,12 and transition metals,5,13 as well as with lanthanide catalysts,5,14 affording insights into the mechanistic details, the thermodynamic and kinetic parameters as well as the control of the molecular weight and crystallinity of the resulting PCL. Despite the large variety of metal catalysts examined in the ROP of ε-caprolactone, only a few examples involving actinide-based catalysts can be found in the literature,15 which can be attributed to the high oxophilicity of these elements. The oxophilic nature should result in a decrease in catalytic activity towards oxygen-containing substrates, since a reaction between the actinide centre and the oxygen atom of the substrate can occur, leading to the formation of thermodynamically stable, catalytically inactive actinide-oxo species as reported by Marks et al.16 Since the low catalytic activity of the early actinides towards oxygen-containing substrates is attributed to their high electrophilicity, decreasing the electrophilic nature of the metal should lead to an increased reactivity towards oxygen-containing molecules such as cyclic esters. Our method of choice for making the actinide centre less electrophilic is based on using highly nucleophilic and strongly electron-donating ligands, i.e. the imidazolin-2-iminato motif (ImRN−), which is obtained by the deprotonation of imidazolin-2-imine (ImRNH). This strongly basic and highly nucleophilic ligand class can be considered as 2σ, 4π electron donors towards early transition metals and metals in high oxidation states and therefore as monodentate isolobal analogues to the widely used cyclopentadienyl ligand (Scheme 2).17 Accordingly, the resulting transition metal and lanthanide metal complexes with Ln (Ln = Sc, Y, Gd, Lu),18 Ti,19 Zr,20 V,21 Mo,22 W,22,23 and Re24 usually exhibit short M–N bonds and large, almost linear M–N–C angles.
 |
| | Scheme 2 Resonance structure of imidazolin-2-iminato ligands. | |
Recently, we reported the syntheses and structures of the imidazolin-2-iminato uranium(IV) complexes [(ImtBuN)4U] (1), [(ImMesN)3U(NMeEt)] (2) and [(ImDippN)2U(NMeEt)2] (3).25 This series of complexes was obtained by an acid–base reaction between the homoleptic [U(NMeEt)4] and neutral imidazolin-2-imine ImRNH, which furnished the respective uranium complexes in dependence of the steric demand of the R substituent on the imidazolin-2-imine ligand (Scheme 3). Furthermore, we reported the selective preparation of mono(imidazolin-2-iminato) thorium(IV) and uranium(IV) complexes by a selective protonolysis reaction of actinide metallacycles with neutral imidazolin-2-imines.26 The uranium complexes 1–3 display short U–N bond distances (2.174(11)–2.177(11) Å) and almost linear U–N–C angles (165.0(4)°–172.3(4)°), suggesting a higher bond order of the U–N bond.25
 |
| | Scheme 3 Synthesis of imidazolin-2-iminato uranium(IV) complexes.25 | |
Herein, we report the reactivity of these complexes in the ROP of ε-caprolactone, giving rise to mechanistic, thermodynamic and kinetic details. Moreover, we compare the reactivity and kinetics of the imidazolin-2-iminato complexes 1–3 to two analogous cyclopentadienyl uranium(IV) complexes Cp*2U(NMe2)2427 and Cp*2U(NCMePh)25 (Fig. 1),28 focusing on the differences in reactivities, mechanisms and rates, despite the isolobal analogy between the respective complexes.
 |
| | Fig. 1 Molecular structures of uranium catalysts 3–5. | |
Results and discussion
Cyclopentadienyl uranium(IV) complexes have been investigated in the catalytic ROP of cyclic esters such as L-lactide and ε-caprolactone, exhibiting high activities.15a Due to the high nucleophilicity of imidazolin-2-iminato ligands, we believe that the oxophilicity of the uranium centre should decrease, which should in turn increase the activity of the respective complexes towards oxygen containing substrates. Moreover, in a previous study, a zirconium(IV) imidazolin-2-iminato complex was proven to be suitable for the polymerization of ε-caprolactone.20 Therefore, we decided to investigate the polymerization of ε-caprolactone with complexes 1–3. Surprisingly, these complexes showed markedly different reactivities towards ε-caprolactone. While complexes 1 and 2 only polymerized the cyclic ester at an elevated temperature of 90 °C with moderate activities of 7.9 × 103 g (PCL) mol−1 h−1 and 5.3 × 103 g (PCL) mol−1 h−1 for complexes 1 and 2, respectively, complex 3 polymerizes ε-caprolactone within minutes at room temperature, showing an extraordinary high activity (activity = 1.2 × 107 g (PCL) mol−1 h−1). The polymerization reactions using complexes 1 and 2 as initiators were all carried out using 5 mL of toluene as the solvent, and a catalyst to ε-caprolactone ratio of 1/1000. The lower catalytic activity of complexes 1 and 2 as compared to the bis(imidazolin-2-iminato) uranium compound 3, can probably be attributed to the higher steric encumbrance in complexes 1 and 2, which make the metal centre less accessible for an incoming substrate molecule. The polymerization results using complex 3 are shown in Table 1.
Table 1 Polymerization results for the ROP of ε-caprolactone mediated by complex 3a
| Entry |
Time (min) |
Activity (g mol−1 h−1) |
M
w
(dalton) |
PDI |
Yield (%) |
Polymerization conditions: 5 mL of toluene, r.t., 0.216 μmol of 3, complex 3/ε-CL: 1/60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.
Conditions as in “a” but at 90 °C.
Carried out in THF.
Polymer insoluble in THF; no GPC analysis possible.
The relative calibration of the Mn values was done using polystyrene standards; the Mn values were multiplied by a factor of 0.56 (Mark–Houwink coefficient) and correlated to the actual PCL values.30 000.
Conditions as in “a” but at 90 °C.
Carried out in THF.
Polymer insoluble in THF; no GPC analysis possible.
The relative calibration of the Mn values was done using polystyrene standards; the Mn values were multiplied by a factor of 0.56 (Mark–Houwink coefficient) and correlated to the actual PCL values.30
|
|
1
|
10 |
1.2 × 107 |
21 970 |
1.86 |
28 |
|
2
|
30 |
1.1 × 107 |
23 680 |
1.86 |
78 |
|
3
|
60 |
6.8 × 106 |
30 660 |
2.54 |
99 |
|
4
|
120 |
3.4 × 106 |
223 660 |
2.51 |
99 |
|
5
|
300 |
1.4 × 106 |
327 860 |
3.59 |
99 |
|
6
|
30 |
1.3 × 107 |
355 280 |
2.36 |
98 |
|
7
|
720 |
5.6 × 105 |
|
|
99 |
|
8
|
60 |
2.2 × 106 |
37 890 |
2.03 |
32 |
|
9
|
120 |
3.3 × 106 |
60 110 |
2.29 |
95 |
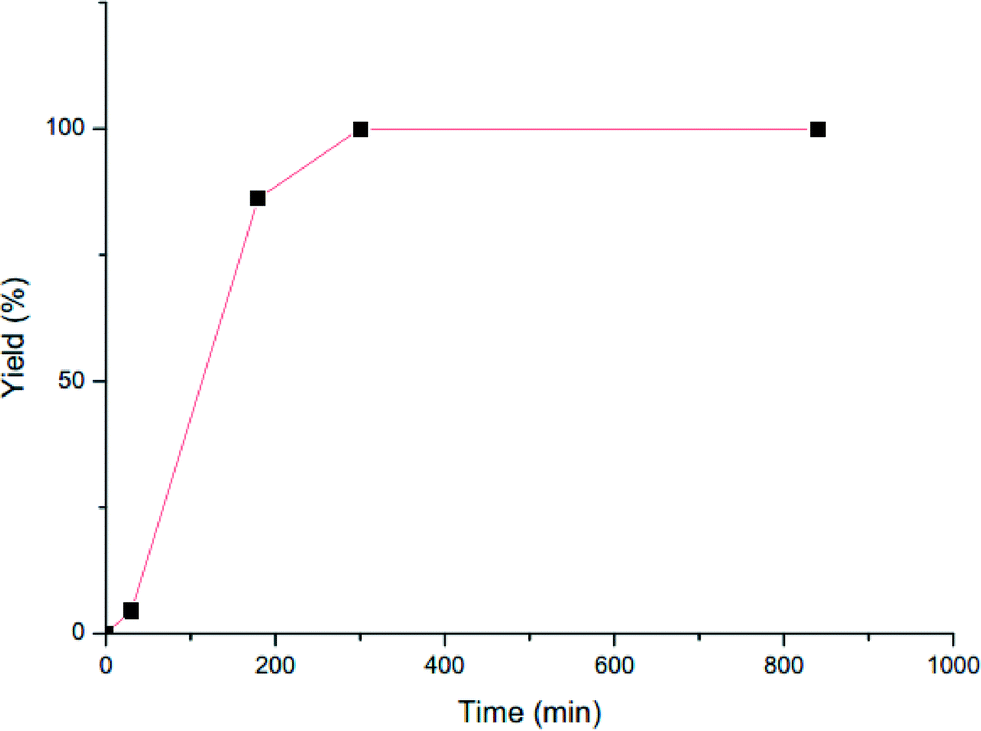
The yield of the obtained polymer increases linearly with time until the monomer is fully consumed after ~60 minutes (Fig. 2), suggesting a living polymerization (expected PDI = 1.0); however, the molecular weights of the polymers do not increase linearly. In addition, the activity of the catalyst remains constant until all the monomer is polymerized. Additional polymerization time reduces the activity almost linearly since there is no additional monomer. Interestingly, after additional time, the molecular weight of the polymer clearly increases, indicating that the complex is able to continue performing a transesterification, which causes also an increase in the PDI (entry 5, Table 1). Hence, the polydispersity of the obtained polymers at the beginning of the polymerization is close to 2, indicative of a single site polymerization mechanism. These results suggest that the polymerization initiated by complex 3 is in a rapid competition with a chain transfer mechanism (transesterification) between the catalytically active species. The transesterification reactions of this type have been previously observed in the ROP of lactides and lactones, as well as in the co-polymerization of these monomers.29
 |
| | Fig. 2 Plot of yield versus time for the polymerization of ε-caprolactone mediated by complex 3. | |
Moreover, the reinsertion of the polymer chain obtained after 720 minutes leads to a polymer with an ultrahigh molecular weight, which is not soluble. When the reaction is carried out at higher temperatures, there is an increase in the activity and in the molecular weight of the polymer. A variation of the solvent to THF resulted in lower activities, suggesting competitive coordination of THF to the active catalytic species, which hampers the coordination of the substrate, ε-caprolactone.
For investigating the mechanism of the polymerization reaction mediated by complex 3, we performed kinetic measurements, which exhibit a first order dependence on ε-caprolactone and the catalyst (eqn (1), Fig. 3).
| |  | (1) |
 |
| | Fig. 3 Plot of rate of polymerization ∂p/∂t versus the concentration of complex 3. | |
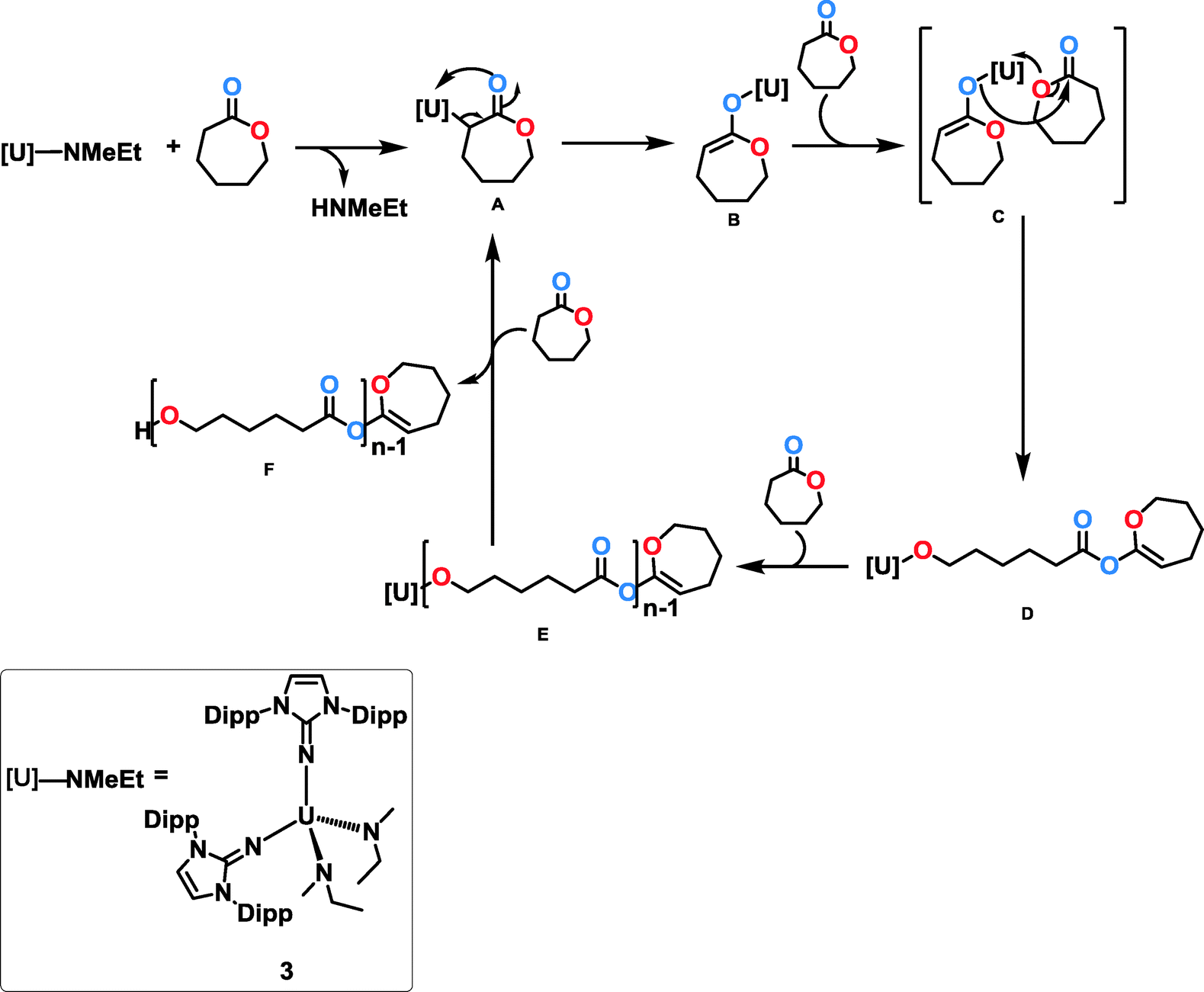
The thermodynamic parameters were determined from the Arrhenius plot (Ea = 12.8(5) kcal mol−1) and the Eyring plot (ΔS‡ = −33.9(8) cal mol−1 K−1, ΔH‡ = 12.2(8) kcal mol−1) which is presented in Fig. 4. A plausible mechanism for the polymerization of ε-caprolactone is shown in Scheme 4. In order to determine, whether both amido groups are active in the polymerization, we performed NMR experiments with stoichiometric amounts of the monomer, which led to the observation that two equivalents of free amines were released per mole of catalyst. After the protonolysis step, the uranium-alkoxo-caprolactonate intermediate B undergoes a reaction with an incoming caprolactone monomer, leading to the open chain intermediate D, which can insert further monomers into the growing polymer chain, leading to the growing polymer chain E. The polymerization is terminated by an additional equivalent of the monomer, ε-caprolactone, leading to the formation of a polymer with caprolactonyl end-group F (see the ESI†) and regeneration of the active catalyst A (Scheme 4).
 |
| | Fig. 4 Arrhenius plot for the polymerization of ε-caprolactone mediated by complex 3. | |
 |
| | Scheme 4 Plausible mechanism for the ROP of ε-caprolactone mediated by complex 3. The second NMeEt unit has been omitted for clarity. | |
The large discrepancy between the activity of the isolobal cyclopentadienyl uranium(IV) complex Cp*2UMe2 (ref. 15) and complex 3 towards ε-caprolactone raised the question, whether the high activity of 3 could be attributed to the replacement of the cyclopentadienyl moiety by imidazolin-2-iminato ligands or to the replacement of the methyl ligands by amido groups. Therefore, we synthesized the respective isolobal complex Cp*2U(NMe2)2 (4),27 and compared the kinetic data and reactivity with 3. The polymerization results are shown in Table 2.
Table 2 Polymerization results for the ROP of ε-caprolactone mediated by complex 4a
| Entry |
Time (min) |
Activity (g mol−1 h−1) |
M
w
(dalton) |
PDI |
Yield (%) |
|
Polymerization conditions: 5 mL of toluene, 90 °C, 4.08 μmol of complex 4, complex 4/ε-CL:1/1000.
Conditions as in “a” but at r.t.
The relative calibration of the Mn values was done using polystyrene standards; the Mn values were multiplied by a factor of 0.56 (Mark–Houwink coefficient) and correlated to the actual PCL values.30
|
|
1
|
30 |
4.6 × 103 |
41 040 |
2.78 |
2 |
|
2
|
60 |
3.9 × 104 |
58 680 |
1.39 |
34 |
|
3
|
120 |
2.6 × 104 |
73 520 |
2.60 |
45 |
|
4
|
180 |
3.2 × 104 |
97 190 |
1.65 |
85 |
|
5
|
300 |
2.2 × 104 |
99 840 |
1.48 |
98 |
|
6
|
840 |
7.9 × 103 |
148 540 |
1.92 |
98 |
|
7
|
840 |
978 |
9000 |
1.32 |
12 |
In comparison to complex 3, the cyclopentadienyl analogue 4 displays lower activity at 90 °C and almost no activity at room temperature. The molecular weights of the polymers obtained are lower than those obtained for complex 3. An increase in the average molecular weight of the polymer can be observed as a function of time (Fig. 5), and the polydispersity values indicate a single-site polymerization process. As with complex 3, when the polymerization is complete, additional time reduces linearly the activity; thus, the catalyst is able to perform a transesterification, as indicated by the larger molecular weight and increased PDI.
 |
| | Fig. 5 Plot of yield versus time for the polymerization of ε-caprolactone mediated by complex 4. | |
The kinetic measurements performed with complex 4 show a first order dependence on monomer and catalyst (eqn (2); Fig. 6).
| |  | (2) |
 |
| | Fig. 6 Plot of rate of polymerization ∂p/∂t versus the concentration of complex 4. | |
NMR experiments with stoichiometric amounts of the substrate, confirmed an intermolecular mechanism, initiated by the amido ligands (Scheme 5). However, the metal centre does not react with the acidic hydrogen atom in the α-position to the carbonyl leading to the release, of free amine. Instead, the uranium centre reacts with the oxygen atom of the carbonyl group over intermediate B, (Scheme 5) and nucleophilic attack at the carbonyl carbon atom leads to an amido end-group in the first polymer chain F, generating catalytically active uranium-alkoxocaprolate species G. Intermediate G can now react with a further equivalent of ε-caprolactone, leading to the formation of the open-chain intermediate H. After insertion of additional ε-caprolactone monomers into the growing polymer chain of H, the reaction is terminated by an incoming monomer, yielding a polymer with a caprolactonyl end-group (I) (see the ESI†) and regenerating the active catalyst G.
 |
| | Scheme 5 Plausible mechanism for the ROP of ε-caprolactone mediated by complex 4. The second NMe2 unit has been omitted for clarity. | |
The energy of activation for the ROP of ε-caprolactone mediated by complex 4 was determined from the Arrhenius plot (Fig. 7) with a value of Ea = 19.0(7) kcal mol−1. In comparison to the activation barrier of the polymerization catalysed with complex 3, compound 4 has a much higher barrier of activation, which explains the high temperature required for the polymerization. The entropy of activation is comparable and just slightly larger than for complex 3 (ΔS‡ = −27.8(8) cal mol−1 K−1).
 |
| | Fig. 7 Arrhenius plot for the polymerization of ε-caprolactone mediated by complex 4. | |
The structural and electronic similarity of the imidazolin-2-iminato ligands to the ketimido ligand reported as ancillary ligands in actinide complexes by Kiplinger et al.28 should also result in a comparable reactivity. Due the π-character attributed to the U–N bond in the latter, the ketimido ligand similar to imidazolin-2-iminato ligands should not initiate the polymerization by promoting a nucleophilic attack on the substrate, in contrast to the amido moieties in 3 and 4. Therefore, complex 5 should in theory exhibit a very low activity in the ROP of ε-caprolactone, if the U–N bond displays a higher bond order. Therefore, the uranium(IV) bis(ketimido) complex 5 was synthesized and its reactivity towards the ROP of ε-caprolactone was studied. The results are summarized in Table 3. Similar to 4, complex 5 showed only a catalytic activity at higher temperatures. The activities obtained were higher than those of complex 4, but lower than those found for 3. The isolated polyester exhibits high molecular weights which increase over time, and narrow polydispersities (~2.0) indicating a single-site catalyst mechanism. When the polymerization was carried out at room temperature or in THF, no product was obtained. For elucidating the mechanism of this reaction, an NMR scale reaction with stoichiometric amounts of ε-caprolactone was carried out. Neither the ketimido ligands nor the cyclopentadienyl ligands could be observed as a free ketimine, or cyclopentadiene, respectively, which suggests a Lewis acid-catalysed mechanism.
Table 3 Results for the polymerization of ε-caprolactone mediated by complex 5a
| Entry |
Time (min) |
Activity (g mol−1 h−1) |
M
w
(dalton) |
PDI |
Yield (%) |
|
Polymerization conditions: 5 mL of toluene, 90 °C, 3.36 μmol of complex 5; complex 5/ε-CL 1/1000.
Conditions as in “a” but at r.t.
Couldn't be determined due to low conversion.
The relative calibration of the Mn values was done using polystyrene standards; the Mn values were multiplied by a factor of 0.56 (Mark–Houwink coefficient) and correlated to the actual PCL values.30
|
|
1
|
30 |
4.6 × 103 |
|
|
<2 |
|
2
|
60 |
6.6 × 104 |
84 140 |
1.87 |
58 |
|
3
|
120 |
5.1 × 104 |
156 580 |
1.80 |
89 |
|
4
|
300 |
2.1 × 104 |
270 050 |
1.72 |
94 |
|
5
|
840 |
7.9 × 103 |
108 700 |
2.54 |
97 |
|
6
|
840 |
0 |
|
|
<1 |
The mechanism presented in Scheme 6 involves an activation of the monomer by the Lewis acidic metal complex, which was previously observed with other main group and transition metals, followed by a nucleophilic attack of an incoming monomer unit B, leading to the growing polymer chain D.30 The polymerization process is terminated by an additional equivalent of ε-caprolactone, leading to the formation of a polymer with a caprolactonyl end-group (E) (see the ESI†) under regeneration of the active catalyst A.
 |
| | Scheme 6 Plausible mechanism for the ROP of ε-caprolactone mediated by complex 5. | |
Kinetic and thermodynamic NMR studies have shown a first order dependence on the monomer and the catalyst (eqn (3), Fig. 8).
| | 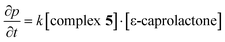 | (3) |
 |
| | Fig. 8 Plot of rate of polymerization ∂p/∂t versus the concentration of complex 5. | |
The energy of activation (Ea = 23.55 kcal mol−1) was determined as described previously from the Arrhenius plot (Fig. 9); the enthalpy of activation (ΔH‡ = 22.8(5) kcal mol−1) and the entropy of activation (ΔS‡ = −15.0(9) cal mol−1 K−1) were determined from the Eyring plot. The large value for the energy of activation is reflected in the high temperatures required and provides an explanation for the lack of reactivity at room temperature.
 |
| | Fig. 9 Arrhenius plot for the polymerization of ε-caprolactone mediated by complex 5. | |
Conclusions
A series of imidazolin-2-iminato and pentamethyl)cyclopentadienyl uranium(IV) complexes (1–5) were studied as initiators in the ring-opening polymerization (ROP) of the cyclic ester ε-caprolactone. Due to the high nucleophilicity of the imidazolin-2-iminato ligands, the U–N bond in complexes 1–3 displays a bond order higher than one. Hence, these complexes should display a slightly decreased oxophilicity and therefore a higher catalytic activity toward oxygen-containing molecules. The activity of complex 3 in the ROP of ε-caprolactone was investigated, leading to an extraordinarily high activity. Mechanistic studies confirmed a coordination–insertion mechanism, in which both amido moieties were found to be active in the polymerization reaction, yielding two equivalents of free amine in the first step of the catalytic cycle. Kinetic NMR studies showed a first order dependence in the monomer and the catalyst. Because of the isolobal analogy between the imidazolin-2-iminato and cyclopentadienyl ligands,17 the reactivity of complex 427 towards the ROP of ε-caprolactone was also investigated. The pentamethylcyclopentadienyl complex 4 was found to be only active at high temperatures; the activity and rates were both lower than that for 3. Mechanistic studies sustain a coordination–insertion mechanism, which is slightly different from the mechanism for the ROP mediated by complex 3. Although in both cases, the coordination of the metal centre to the substrate initiates the polymerization reaction; in the case of complex 4, the amido moieties are not eliminated as free amine, but can be found as an end group in the first polymer chain that is introduced in the first step of the catalytic cycle. Surprisingly, the protonolysis reaction observed for complex 3 was not observed in the activation step for complex 4, although the resulting N-dimethylamine and N-ethylmethylamine display very similar pKa values. However, no dissociation of the pentamethylcyclopentadienyl moiety was observed. Therefore, we synthesized the uranium(IV) ketimido complex 5,28 in which the two ketimido moieties display similar bonding properties as the imidazolin-2-iminato ligands in 3 and should not dissociate upon addition of the substrate. Due to the strong bonding of the pentamethylcyclopentadienyl and the ketimido ligands in 5, a coordination–insertion mechanism is not likely, and a lower activity is expected, due to the steric encumbrance of the ligands, rendering the uranium(IV) centre less accessible for an incoming monomer. Mechanistic studies confirmed that all four ligands stay coordinated to the uranium centre upon addition of a stoichiometric amount of ε-caprolactone, suggesting a cationic mechanism, in which the uranium complex 5 acts as a Lewis acid. The ketimido complex 5 exhibits low activity at elevated temperatures (90 °C) and no activity at room temperature, which was further sustained by the low rates of polymerization found by kinetic NMR measurements. Complex 3 displayed the lowest activation barrier, which explains its extremely high activity at low temperatures as compared not just to actinides but to any other reported metal-induced polymerization.
Experimental section
All manipulations of air sensitive materials were performed with the rigorous exclusion of oxygen and moisture in flamed Schlenk-type glassware on a high vacuum line (10−5 torr), or in nitrogen filled Vacuum Atmospheres glovebox with a medium capacity recirculator (1–2 ppm oxygen). Argon and nitrogen were purified by passage through a MnO oxygen removal column and a Davison 4 Å molecular sieve column. Analytically pure solvents were dried and stored with Na/K alloy and degassed by three freeze–pump–thaw cycles prior to use (THF, hexane, toluene, benzene-d6, toluene-d8). [U(NMeEt)4],25 [(ImtBuN)4],25 [(ImMesN)3U(NMeEt)2],25 [(ImDippN)2U(NMeEt)2],25 [Cp*2U(NMe2)2],27 and [Cp*2U(NCMePh)2],28 were synthesized according to published procedures. ε-caprolactone (Sigma Aldrich) was distilled under reduced pressure from CaH2 and stored in the glovebox prior to use. The NMR spectra were recorded on DPX 200, Avance 300 and Avance 500 Bruker spectrometers. The chemical shifts for 1H NMR and 13C NMR are reported in ppm and referenced using residual proton or carbon signals of the deuterated solvent relative to tetramethylsilane. GPC measurements were carried out on a Waters Breeze system with a styrogel RT column and with THF (HPLC grade, T.G. Baker) as the mobile phase at 30 °C. Relative calibration was done with polystyrene standards (Aldrich, 2000–1800000 range). The Mn values were multiplied by a factor of 0.56 and correlated to actual PCL values.31
Catalytic polymerization of ε-caprolactone
A sealable glass tube, equipped with a magnetic stirring bar, was loaded with the required amount of the uranium complex from a stock solution, the respective amount of ε-caprolactone (in a ratio of catalyst to ε-caprolactone of 1/1000 for complexes 4 and 5, or 1/60000 for complex 3) and 5 mL of dry toluene inside the glove box. The polymerization was carried out under rapid stirring for the required amount of time and at the respective temperature. Then, the reaction was quenched by the addition of methanol. After removing the solvent under reduced pressure, the polymer was precipitated from cold methanol, isolated by filtration, washed with three portions of cold methanol (50 mL each) and dried overnight under vacuum. The activity was determined as PCL (g)/mol (cat)·time (h). A sample of the isolated PCL (40 mg) was dissolved in THF and used for the determination of the Mn, Mw and PDI values.
For the kinetic 1H NMR studies, a J-Young NMR tube was loaded with the respective amount of catalyst from a stock solution, ε-caprolactone and toluene-d8 inside the glove box, then the tube was subsequently sealed, and the reaction mixture was frozen at the liquid nitrogen temperature until the start of the 1H NMR measurements. The sample was heated (if required) in the NMR spectrometer.
Acknowledgements
This work was supported by the German Israel Foundation GIF under Contract I-1264-302.5/2014
References
- J. Van Natta, J. W. Hill and W. H. Carothers, J. Am. Chem. Soc., 1934, 56, 455–459 CrossRef.
- W. J. Bailey, Z. Ni and S.-R. Wu, J. Polym. Sci., Part A: Polym. Chem., 1982, 20, 3012–3030 Search PubMed.
- K. M. Stridsberg, M. Ryner and A.-C. Albertsson, Adv. Polym. Sci., 2002, 157, 41–65 CrossRef CAS.
- M. S. Kim, K. S. Seo, G. Khang and H. B. Lee, Macromol. Rapid Commun., 2005, 26, 643–648 CrossRef CAS.
- A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801–826 RSC.
-
(a) V. R. Sinha, K. Bausal, R. Kaushik, R. Kumria and A. Trehan, Int. J. Pharm., 2004, 278, 1–23 CrossRef CAS PubMed;
(b) M. Labet and W. Thielmans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC;
(c) M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
-
(a) R. A. Gross and B. Karla, Science, 2002, 297, 803–807 CrossRef CAS PubMed;
(b) R. Chandra and R. Rustgi, Prog. Polym. Sci., 1998, 23, 1273–1335 CrossRef CAS;
(c) D. R. Chen, J. Z. Bei and S. G. Wang, Polym. Degrad. Stab., 2000, 67, 455–459 CrossRef CAS;
(d) C. De Kesel, C. V. Wauven and C. David, Polym. Degrad. Stab., 1997, 55, 107–113 CrossRef CAS;
(e) P. Joshi and G. Madras, Polym. Degrad. Stab., 2008, 93, 1901–1908 CrossRef CAS;
(f) J. Peña, T. Corrales, I. Izquierdo-Barba, A. L. Doadrio and M. Vallet-Regí, Polym. Degrad. Stab., 2006, 91, 1424–1432 CrossRef.
-
(a) C. X. Lam, S. H. Teoh and D. W. Hutmacher, Polym. Int., 2007, 718–728 CrossRef CAS;
(b) M. J. Jenkins, K. L. Harrison, M. M. C. G. Silva, M. J. Whitaker, K. M. Shakesheff and S. M. Howdle, Eur. Polym. J., 2006, 42, 3145–3151 CrossRef CAS;
(c) D. W. Hutmacher, T. Schanz, I. Zein, K. W. Ng, S. Hin, T. Kim and C. Tan, J. Biomed. Mater. Res., 2001, 55, 203–216 CrossRef CAS PubMed.
- Y. Ikada and H. Tsuji, Macromol. Rapid Commun., 2000, 21, 117–132 CrossRef CAS.
- J. L. Hedrick, T. Magbitang, E. F. Connor, T. Glauser, W. Volksen, C. J. Hawker, V. Y. Lee and R. D. Miller, Chem. – Eur. J., 2002, 8, 3308–3319 CrossRef CAS.
-
(a)
M. Minami and S. Kozaki, US Pat., 2003/0023026 A1, 2003 Search PubMed;
(b) F. Majoumo-Mbe, E. Smolensky, P. Lönnecke, D. Shpasser, M. S. Eisen and E. Hey-Hawkins, J. Mol. Catal. A: Chem., 2005, 240, 91–98 CAS;
(c) O. Coulembier, P. Degeé, J. L. Hendrick and P. Dubois, Prog. Polym. Sci., 2006, 31, 723–747 CrossRef CAS.
-
(a) T. Biela, A. Duda and S. Penczek, Macromol. Symp., 2002, 183, 1–10 CrossRef CAS;
(b) J. A. Castro-Osma, C. Alonso-Moreno, J. C. García-Martinez, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Lara-Sánchez and A. Otero, Macromolecules, 2013, 46, 6388–6394 CrossRef CAS.
-
(a) M. Z. Chen, H.-M. Sun, W.-F. Li, Z. G. Wang, Q. Shen and Y. Zhang, J. Organomet. Chem., 2006, 691, 2489–2494 CrossRef CAS;
(b) L. Pastigo, J. Sanchez-Nieves, P. Royo and M. E. G. Mosquera, Dalton Trans., 2009, 3756–3765 RSC.
-
(a) N. Nomura, A. Taira, T. Tomioka and M. Okada, Macromolecules, 2000, 33, 1497–1499 CrossRef CAS;
(b) I. Palard, A. Soum and S. M. Guillaume, Chem. – Eur. J., 2004, 10, 4054–4062 CrossRef CAS PubMed;
(c) Y. Fugen, L. Tingling, L. Li and Z. Yuan, J. Rare Earths, 2012, 30, 753–756 CrossRef;
(d) L. Fang, Y. Yao, Y. Zhang, Q. Shen and Y. Wang, Z. Anorg. Allg. Chem., 2013, 639, 2324–2330 CrossRef CAS.
-
(a) E. Barnea, D. Moradove, J.-C. Berthet, M. Ephritikhine and M. S. Eisen, Organometallics, 2006, 25, 320–322 CrossRef CAS;
(b) E. Rabinovich, S. Aharonovich, M. Botoshansky and M. S. Eisen, Dalton Trans., 2010, 39, 6667–6676 RSC;
(c) A. Walshe, J. Fang, L. Maron and R. J. Baker, Inorg. Chem., 2013, 52, 9077–9086 CrossRef CAS PubMed;
(d) C. E. Hayes, Y. Sarazin, M. J. Katz, J.-F. Carpentier and D. B. Lenznoff, Organometallics, 2013, 32, 1183–1192 CrossRef CAS;
(e) W. Ren, N. Zhao, L. Chen and G. Zi, Inorg. Chem. Commun., 2013, 30, 26–28 CrossRef CAS;
(f) I. S. R. Karmel, T. Elkin, N. Fridman and M. S. Eisen, Dalton Trans., 2014, 43, 11376–11387 RSC;
(g) I. S. R. Karmel, N. Fridman and M. S. Eisen, Organometallics, 2015, 34, 636–643 CrossRef CAS;
(h) I. S. R. Karmel, N. Fridman, M. Tamm and M. S. Eisen, Organometallics, 2015, 34, 2933–2942 CrossRef CAS.
- Z. Lin and T. J. Marks, J. Am. Chem. Soc., 1987, 109, 7979–7985 CrossRef CAS.
-
(a) N. Kuhn, M. Göhner, M. Grathwohl, J. Wiethoff, G. Freking and Y. Chen, Z. Anorg. Allg. Chem., 2003, 629, 793–802 CrossRef CAS;
(b) X. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116–138 CrossRef CAS.
-
(a) A. G. Trambitas, T. K. Panda and M. Tamm, Z. Anorg. Allg. Chem., 2010, 636, 2156–2171 CrossRef CAS;
(b) A. G. Trambitas, T. K. Panda, J. Jenter, P. W. Roesky, C. Daniliuc, C. G. Hrib, P. G. Jones and M. Tamm, Inorg. Chem., 2010, 49, 2435–2446 CrossRef CAS PubMed;
(c) T. K. Panda, C. G. Hrib, P. G. Jones and M. Tamm, J. Organomet. Chem., 2010, 695, 2768–2773 CrossRef CAS;
(d) A. G. Trambitas, J. Yang, D. Melcher, C. G. Daniliuc, P. G. Jones, Z. Xie and M. Tamm, Organometallics, 2011, 30, 1122–1129 CrossRef CAS;
(e) A. G. Trambitas, D. Melcher, L. Hartenstein, P. W. Roesky, C. G. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2012, 51, 6753–6761 CrossRef CAS PubMed.
-
(a) M. Tamm, S. Randoll, T. Bannenberg and E. Herdtweck, Chem. Commun., 2004, 876–877 RSC;
(b) K. Nomura, H. Fukuda, W. Apisuk, A. Trambitas, B. Kitiyanan and M. Tamm, J. Mol. Catal. A: Chem., 2012, 363–364, 501–511 CrossRef CAS;
(c) M. Sharma, H. S. Yameen, B. Tumanskii, S.-A. Filimon, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2012, 134, 17234–17244 CrossRef CAS PubMed;
(d) W. Apisuk, A. G. Trambitas, B. Kitiyanan, M. Tamm and K. Nomura, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2575–2580 CrossRef CAS;
(e) D. Shoken, M. Sharma, M. Botoshansky, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2013, 135, 12592–12595 CrossRef CAS PubMed.
- A. Glöckner, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2012, 51, 4368–4378 CrossRef PubMed.
- S. Zhang, M. Tamm and K. Nomura, Organometallics, 2011, 30, 2712–2720 CrossRef CAS.
- B. Haberlag, X. Wu, K. Brandhorst, J. Grunenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem. – Eur. J., 2010, 16, 8868–8877 CrossRef CAS PubMed.
-
(a) H. S. Beer, C. G. Hrib, P. G. Jones, K. Brandhorst, J. Grunenberg and M. Tamm, Angew. Chem., Int. Ed., 2007, 46, 8890–8894 CrossRef PubMed;
(b) S. Beer, K. Brandhorst, J. Grunenberg, C. G. Hrib and P. G. Jones, Org. Lett., 2008, 10, 981–984 CrossRef CAS PubMed;
(c) S. Lysenko, C. G. Daniliuc, P. G. Jones and M. Tamm, J. Organomet. Chem., 2013, 744, 7–14 CrossRef CAS.
- M. Tamm, S. Beer and E. Herdtweck, Z. Naturforsch., B: J. Chem. Sci., 2004, 59, 1497–1504 CAS.
- I. S. R. Karmel, M. Botoshansky, M. Tamm and M. S. Eisen, Inorg. Chem., 2014, 53, 694–696 CrossRef CAS PubMed.
- I. S. R. Karmel, N. Fridman, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2014, 136, 17180–17192 CrossRef CAS PubMed.
- P. J. Fagan, J. M. Manriquez, S. H. Vollmer, C. S. Day and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 2206–2220 CrossRef CAS.
- K. C. Jantunen, C. J. Burns, I. Castro-Rodriguez, R. E. Da Re, J. T. Golden, D. E. Morris, B. L. Scott, F. L. Taw and J. L. Kiplinger, Organometallics, 2004, 23, 4682–4692 CrossRef CAS.
-
(a) K. Ito, Y. Hashizuka and Y. Yamashita, Macromolecules, 1977, 10, 821–824 CrossRef CAS;
(b) F. Chabot, M. Vert, S. Chapelle and P. Gragner, Polymer, 1983, 24, 53–59 CrossRef CAS;
(c) H. R. Kricheldorf, M. Berl and N. Scharnagl, Macromolecules, 1988, 21, 286–293 CrossRef CAS;
(d) M. Bero, J. Kaspercyk and Z. J. Jelinski, Makromol. Chem., 1990, 191, 2287–2296 CrossRef CAS;
(e) J. Kaspercyk and M. Bero, Makromol. Chem., 1993, 194, 913–925 CrossRef;
(f) T. J. J. Whiteborne and F. Schaper, Can. J. Chem., 2014, 9, 206–214 CrossRef.
-
(a) P. Kubisa and S. Penczek, Prog. Polym. Sci., 1999, 24, 1409–1437 CrossRef CAS;
(b)
A. Duda and A. Kowalski, Handbook of Ring Opening Polymerization, Wiley-VCH Verlag GmbH & Co. KG 2009, Weinheim, 2009, ch. 1, pp. 1–53 Search PubMed;
(c) B. G. Belen'kaya, Y. B. Lyudvig, A. L. Izyuminkov and Y. I. Kul'velis, Polym. Sci. U.S.S.R., 1982, 24, 306–313 CrossRef;
(d) E. B. Ludvig and B. G. Belenkaya, J. Macromol. Sci., Part A: Pure Appl.Chem., 1974, 8, 819–828 CrossRef CAS.
- A. Duda, Z. Florjanczyk, A. Hofman, S. Slomkowski and S. Penczek, Macromolecules, 1990, 23, 1640–1646 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy01162k |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence