
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in g-C3N4 based low cost photocatalytic system: activity enhancement and emerging applications
Shengming
Yin
a,
Jianyu
Han
ab,
Tianhua
Zhou
ac and
Rong
Xu
*a
aSchool of Chemical & Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive, Singapore 637459. E-mail: rxu@ntu.edu.sg; Fax: +65 67947553; Tel: +65 67906713
bEnergy Research Institute @ NTU, Nanyang Technological University, 50 Nanyang Drive, Singapore 637553
cSinBeRISE CREATE, National Research Foundation, CREATE Tower Level 11, 1 Create way, Singapore 138602
First published on 15th July 2015
Abstract
Graphitic C3N4 (g-C3N4) has continuously attracted attention since it was reported as a metal-free semiconductor for water splitting. However, its ability to evolve hydrogen from water is significantly dependent on the use of noble metal co-catalyst, mainly Pt. In recent years, good progress has been achieved in developing co-catalysts containing earth abundant elements only for constructing low cost and efficient g-C3N4 based photocatalytic systems. Besides, exfoliation of bulk g-C3N4 into two dimensional g-C3N4 nanosheets offers large surface area and exposed active sites, which are beneficial for activity enhancement. Furthermore, oxygen evolution and CO2 photoreduction over g-C3N4 have gained increasing interests due to the demand to achieve overall water splitting and conversion of CO2 into chemicals and fuels. In this mini-review, we will briefly summarize the latest research works on g-C3N4 based photocatalytic systems during the last three years with emphasis on the progress achieved in enhancing the hydrogen evolution activity of g-C3N4 by loading noble metal free co-catalysts, exfoliating bulk g-C3N4 into nanosheets, and applying the g-C3N4 system in photocatalytic O2 evolution and CO2 reduction.
 Shengming Yin | Shengming Yin received his bachelor and master's degree in Materials Science and Engineering at Huazhong University of Science and Technology, China in 2010 and 2013. He is now pursuing his PhD degree under the supervision of Prof. Xu at Nanyang Technological University. His research interest focuses on the design of noble-metal free photocatalytic systems for water splitting. |
 Jianyu Han | Jianyu Han received his bachelor's degree in Chemistry from Nanjing University, China in 2011. As a graduate student in Nanyang Technological University, he concentrates on solar fuel production from photocatalytic water splitting and photocatalytic carbon dioxide reduction. |
 Tianhua Zhou | Dr. Tianhua Zhou received his PhD at Fuzhou University, China in 2008 under the supervision of Prof. Jian-Xi Zhao. Then he joined Prof. Jiang-Gao Mao's group as a research assistant professor. Since 2012, he has been a research fellow in Prof. Rong Xu's group at Nanyang Technological University. His current research interests focus on the development of new photo-(electro) catalysts for water oxidation, hydrogen evolution, and carbon dioxide conversion. |
 Rong Xu | Prof. Rong Xu received her PhD degree in Chemical Engineering (2004) from the National University of Singapore. She joined the School of Chemical & Biomedical Engineering at Nanyang Technological University in 2004 and she is currently an Associate Professor there. Her main research interests include the development of semiconductors and molecular photocatalysts for solar energy conversion, and fabrication of nanostructured materials for biomedical and environmental applications. |
1. Introduction
Energy and environmental problems have become more and more severe over the recent years due to the overuse of fossil fuels and uncontrolled CO2 emission from the combustion of fossil fuels. It is of utmost urgency to find green technology to address these concerns.1 One promising method is the utilization of solar energy with the help of semiconductor photocatalysts. A process known as photocatalytic water splitting converts water into H2 which is considered to be a promising clean energy source to replace fossil fuels;2 another process called artificial photosynthesis is focused on synthesizing hydrocarbon molecules from CO2 which is a mimic of the natural photosynthesis in green plants.3Since Fujishima reported a photoelectrochemical (PEC) water splitting process by using a TiO2 photoanode, researchers have spent efforts on improving the conversion efficiency of this process.4 Various semiconductors, e.g. oxide, (oxy)nitride, and (oxy)sulfide, have been shown capable of splitting water under light irradiation with suitable co-catalysts deposited on the surface.5–7 Unfortunately, the relatively low efficiency and noble metal-containing materials of these systems make them unfavourable for practical usage. The price of H2 by this method is still much higher than expected. Further improving the solar to hydrogen (STH) efficiency and reducing the cost are of particular significance to realize the potential of this technology. Compared to water splitting, the study of CO2 photoreduction is still in its infancy partially due to the fact that it is energetically more difficult to reduce CO2 than proton. CO2 reduction requires a much larger driving force than water reduction (Table 1).8 In addition, CO2 reduction is a multiple electron process, which makes it kinetically harder to proceed. The selectivity to products is also an important aspect in CO2 reduction. After all, the development of photocatalytic water splitting and CO2 reduction depends on the efficient utilization of solar power and the enhancement of catalytic conversion of water and CO2 into fuels and chemicals. These serve as the criteria for the selection of suitable photocatalysts.
| Reaction | E°′ (V) vs. SCE |
|---|---|
| CO2 + 2H+ + 2e− → HCO2H | −0.85 |
| CO2 + 2H+ + 2e− → CO + H2O | −0.77 |
| CO2 + 4H+ + 4e− → C + 2H2O | −0.44 |
| CO2 + 4H+ + 4e− → HCHO + H2O | −0.72 |
| CO2 + 6H+ + 6e− → CH3OH + H2O | −0.62 |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.48 |
Graphitic carbon nitride (g-C3N4), also known as “melon”, is the most stable allotrope among different carbon nitride materials. It can be synthesized via thermal condensation of low cost nitrogen-rich precursors such as cyanamide, dicyandiamide, melamine, thiourea and urea. It has a graphene-like structure consisting of two-dimensional frameworks of tri-s-triazine connected via tertiary amines. This unique structure and high degree of condensation make g-C3N4 stable at elevated temperatures as high as 600 °C and in different chemical environments (acid, base or organic solvent). Furthermore, this tri-s-triazine ring structure makes it an indirect semiconductor with a band gap of ~2.7 eV, corresponding to an optical wavelength of 460 nm in the visible light range.9,10 In 2009, Wang et al. reported that photocatalytic water splitting can be achieved using this metal-free polymeric g-C3N4.11 Since then, enormous attention has been drawn to g-C3N4 because it is ideal for the construction of low cost photocatalysts. Nevertheless, the photoactivity of g-C3N4 suffers from several main drawbacks. Firstly, the relatively large band gap (2.7 eV) limits its effective utilization of visible light of longer wavelengths. It was estimated that a photocatalyst with a band gap as narrow as 1.8–2.0 eV and suitable band positions would be desirable from the viewpoint of both solar energy harvesting and surface kinetics of water splitting reactions.12 Secondly, the low charge carrier mobility inhibits the separation and transportation of electrons and holes. It was revealed by both first principle calculations and experimental results that the charge carrier mobility of the pristine g-C3N4 can be enhanced by doping non-metal elements which widen the valence band (VB) of g-C3N4.13–16 Furthermore, the surface inertness of g-C3N4 due to the nature of the covalent bond leads to the low reaction rates of hydrogen and oxygen evolution half reactions.9 The surface area of bulk g-C3N4 is generally small since it is prepared by condensation of organic precursor compounds at high temperature. There also exist rich grain boundary defects on the surface of g-C3N4 resulting from incomplete condensation, which may cause the recombination of excited charges.17
As a result, the efforts that have been made to address these issues include modification of the electronic structure of g-C3N4 by integrating certain organic molecules into its ring structure,18,19 doping metal or non-metal ions in the bulk structure of g-C3N4 to form an impurity level in its forbidden band to improve its visible light absorption properties,14,15,20–23 and suppression of electron–hole recombination by forming composites of g-C3N4 and other semiconductors with suitable band alignment or carbon materials to improve the charge transportation.24–28 Sensitization of the pristine g-C3N4 by organic dyes or quantum dots is also efficient to enhance its performance under visible light.29,30 Besides, morphology control of g-C3N4 to increase the specific surface area as well as to expose the active sites for photocatalytic reaction is another strategy to achieve photocatalytic activity improvement. Mesoporous g-C3N4 (mpg-C3N4) is commonly prepared using SiO2 nanospheres as the template.31 In addition, g-C3N4 with other morphologies such as spherical nanoparticles,32 nanorods,33–35 hollow spheres,36,37 and ordered 3D structures38 have been prepared using template or non-template methods. There are many literature reports demonstrating the use of such modifications to improve the performance of the g-C3N4 system, which has been shown as a promising material for solar fuel production.
In this mini review, we intend to summarize the recent progress in the development of efficient and low cost g-C3N4 systems on a few important aspects including deposition of noble metal free cocatalysts on g-C3N4, exfoliation of bulk g-C3N4 into g-C3N4 nanosheets and application of g-C3N4 in water oxidation, overall water splitting and CO2 photoreduction. These challenging research areas have drawn increasing interests from many research groups including our own. Some review articles have been recently published focusing on the fabrication, chemical modification, bandgap engineering and heterostructure formation of g-C3N4 based photocatalysts.9,10,17,28,39 We believe that this review would be a good complement to the literature on g-C3N4 based photocatalyst systems.
2. Noble metal free g-C3N4 system
Depositing a suitable cocatalyst on the surface of a semiconductor is of great importance to improve its efficiency. Cocatalysts significantly enhance the surface reaction rate by lowering the activation energy and facilitate the separation of electrons and holes by forming a Schottky junction between the semiconductor and the cocatalyst. The role of the cocatalyst in photocatalysis and photoelectrocatalysis has been well reviewed by Yang et al.40Pt is so far the most efficient and commonly used cocatalyst of g-C3N4 for hydrogen production. Despite that, a series of non-noble metal containing hydrogen evolution reaction (HER) electrocatalysts have been explored; however, only a few of them are successfully deposited on g-C3N4 as cocatalysts.41 To further reduce the cost of g-C3N4 based photocatalysts and make them available for industrial usage, noble metal free cocatalysts are highly needed.
As a pioneering work in this area, Dong et al. reported in 2012 that a molecular cocatalyst Ni(TEOA)2Cl2 can be used in the system of g-C3N4 to replace Pt for stable hydrogen evolution.42 In this system, g-C3N4 plays the main role of light absorber. Ni(TEOA)2Cl2 is formed via self-assembly of Ni2+ ions and triethanolamine (TEOA) which is the sacrificial reagent in solution. A similar nickel-containing complex, nickel–thiourea–triethylamine (Ni–TU–TETN), formed in situ during photocatalytic reaction, was also reported to be an active cocatalyst on the surface of g-C3N4 for hydrogen production.43 But the quantum efficiency (QE) obtained using Ni–TU–TETN/g-C3N4 is only 0.2% at 400 nm irradiation, which is much smaller than 1.51% at 400 nm reported for Ni–TEOA/g-C3N4. Co(dmgH)2pyCl has been studied as a electrocatalytic HER catalyst and was used for photocatalytic hydrogen production in a homogeneous system as well.44 When coupled with g-C3N4 in TEOA solution, it was able to produce hydrogen under visible light. The highest QE of this system was 0.62% under 365 nm irradiation.45 However, the activity decreased dramatically after 8 h of photocatalytic reaction due to the decomposition of Co(dmgH)2pyCl. To overcome this, a more stable Ni(dmgH)2 HER catalyst was used to construct a Ni(dmgH)2/g-C3N4 photocatalyst. Ni(dmgH)2 sub-microcrystals were grown on g-C3N4via a simple precipitation method. Its activity remained almost the same after three recycle runs, indicating the enhanced stability of this system although the activity needs to be further improved.46
Besides the transition metal containing complexes, some inorganic compounds have also been coupled with g-C3N4 for the construction of noble metal free g-C3N4 photocatalysts. For example, our group has successfully deposited NiS on g-C3N4via a facile hydrothermal method.47 An aqueous solution of nickel acetate (NiAc) and thioacetamide was used for the synthesis of NiS in the presence of mpg-C3N4, allowing Ni2+ and the sulfur precursor to enter into the pores of mpg-C3N4 for the formation of a nanosized and well-disperse NiS cocatalyst. The formation of NiS nanoparticles was confirmed by high resolution transmission electron microscopy (HRTEM) analysis, element mapping and X-ray photoelectron spectroscopy (XPS). The typical HRTEM images and elemental mapping results are shown in Fig. 1A. The loading of NiS has a significant effect on the activity and the optimum NiS loading was found to be 1.25 wt% (Fig. 1B). For comparison, the activity of a mixture of mpg-C3N4 and pre-synthesized NiS was much lower than that of NiS/mpg-C3N4, which is due to the poor interface between NiS and mpg-C3N4 and the relatively larger particle size of pre-synthesized NiS. A high QE of 1.9% at 440 nm was obtained over the optimized NiS/mpg-C3N4 photocatalyst. The same photocatalyst demonstrates good stability with 84% of the initial activity retained after 4 runs of photoreaction. Subsequently, Chen et al. reported the preparation of NiS/g-C3N4via ion exchange that converts Ni2+/g-C3N4 to NiS/g-C3N4 using Na2S.48 Another type of nickel sulfide supported on g-C3N4, NiS2/g-C3N4, was prepared via a hydrothermal method using NiAc and thiourea in the presence of g-C3N4.49 The activity of the optimized NiS2/g-C3N4 system is 4 times that of Pt/g-C3N4. However, the activity dropped to 60% of the initial value after five cycles. All these results indicate that nickel sulfides represent promising cocatalysts for the construction of noble metal free g-C3N4 photocatalysts for hydrogen evolution. In addition, cobalt sulfide has been evaluated as a cocatalyst for mpg-C3N4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 47,50 and exhibited lower activities than NiS under the same conditions.47
47,50 and exhibited lower activities than NiS under the same conditions.47
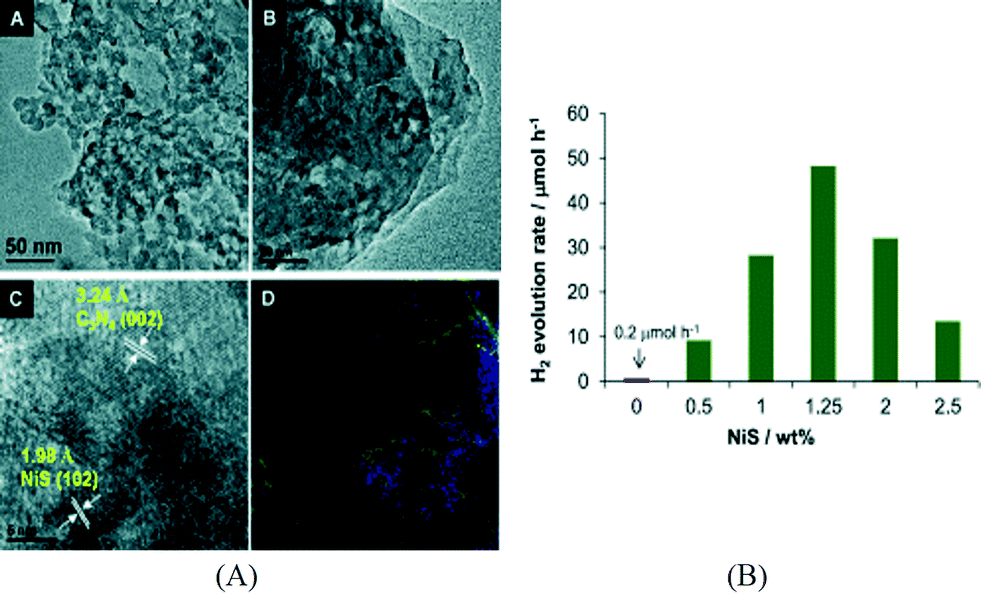 | ||
| Fig. 1 (A) TEM, HRTEM and element mapping images of NiS/mpg-C3N4; (B) effect of NiS cocatalyst loading on hydrogen production.47 Copyright 2013 WILEY-VCH Verlag GmbH & Co. KGaA Weinheim. | ||
In the family of metal sulfides, MoS2 and WS2 nanosheets have received a lot of interests as efficient noble metal free HER catalysts for both electrocatalytic and photocatalytic reactions.51 MoS2 and WS2 have also been loaded on g-C3N4 to act as cocatalysts. It is expected that the nanosheet morphology of these catalysts can facilitate charge transfer between g-C3N4 and the catalyst. Furthermore, the thin layer structure of the nanosheets can minimize the light blocking effect by cocatalysts in the aggregated form. Both MoS2/mpg-C3N4 and WS2/mpg-C3N4 were prepared by an impregnation method.52,53 As schematically shown in Fig. 2A, (NH4)2MoS4 or (NH4)2WS4 was impregnated on mpg-C3N4 and the subsequent sulfidation was carried out in H2S or H2S/H2 atmosphere at an elevated temperature. MoS2 nanosheets were observed in HRTEM images (Fig. 2B). As can be seen in Fig. 2C, the activity of the optimized MoS2/mpg-C3N4 was found to be higher than that of Pt/mpg-C3N4 and a QE of 2.1% was obtained at 400 nm. The stability of this system is also good with 80% of the initial activity maintained after recycling for four runs. Compared to MoS2/mpg-C3N4, the activity of WS2/mpg-C3N4 is relatively lower and decreased dramatically after recycle runs.
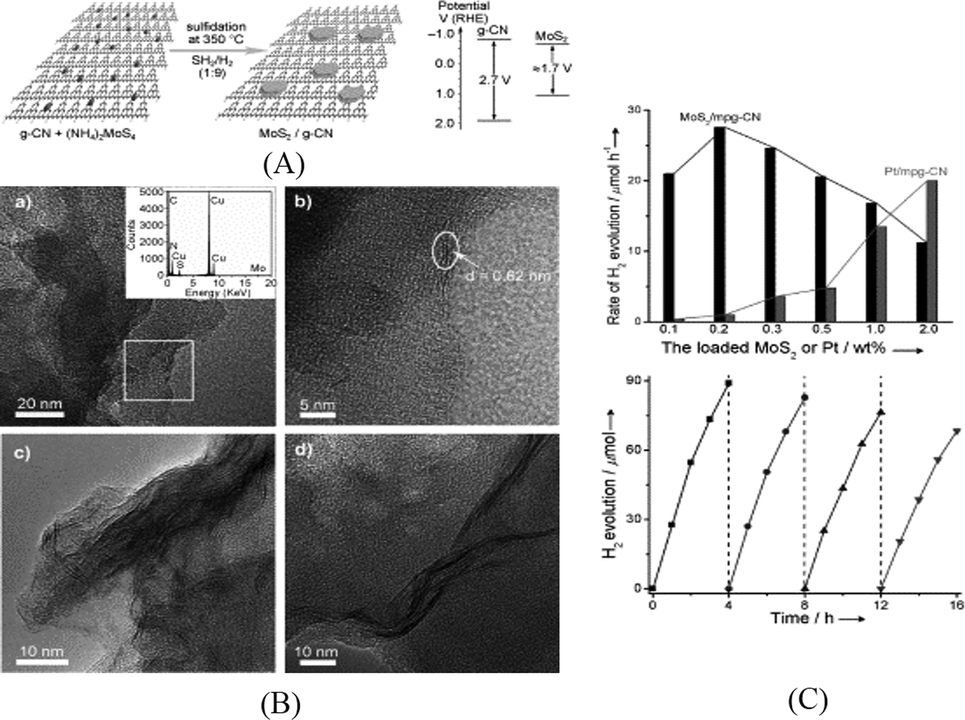 | ||
| Fig. 2 (A) Schematic of the MoS2/mpg-C3N4 nanojunction and the band energy diagram; (B) TEM and HRTEM images of MoS2/mpg-C3N4; (C) hydrogen production rate of mpg-C3N4 loaded with MoS2 or Pt and recycling behavior for 0.2 wt% MoS2/mpg-C3N4.52 Copyright 2013 WILEY-VCH Verlag GmbH & Co. KGaA Weinheim. | ||
Hydroxides such as Ni(OH)2 and Cu(OH)2 were also reported to be capable of evolving hydrogen when coupled with g-C3N4.54,55 The preparation method of hydroxide/g-C3N4 was simply precipitating metal cations by NaOH with pre-added g-C3N4 followed by aging for a certain period of time. Both Ni(OH)2/g-C3N4 and Cu(OH)2/g-C3N4 were found to be stable and efficient for photocatalytic hydrogen production. A relatively high QE of 1.1% was obtained over Ni(OH)2/g-C3N4 at 420 nm. XPS study for the hydroxide/g-C3N4 samples before and after photoreaction revealed that the in situ formed Ni0 or Cu0 cluster via reduction of metal ions by photo electrons is the key to the hydrogen production process.
Very recently, Zhang et al. prepared a core–shell structured Ni/NiO loaded on g-C3N4 by an impregnation method for hydrogen production.56 Ni2+ on g-C3N4 was first reduced to Ni nanoparticles in H2 flow and then the surface of the Ni nanoparticles was oxidized to NiO after being annealed in air leading to the formation of core–shell nanostructure. Ni/NiO-g-C3N4 showed improved activity for photocatalytic hydrogen production compared with the pristine g-C3N4, Ni-g-C3N4 and NiO-g-C3N4 due to the enhanced charge transfer across the interface. The overpotential for HER was also reduced for Ni/NiO-g-C3N4, confirming that the Ni/NiO core/shell structure is a suitable catalyst for HER.
In short, various noble metal free g-C3N4 based photocatalysts have been investigated. It is promising to find that the activities of some photocatalysts approach or surpass that of Pt/g-C3N4. However, it is still challenging to prepare stable and efficient noble metal free g-C3N4 photocatalysts with their activities comparable or better than other semiconductor based systems. The photoelectric current and photocatalytic efficiency are still low due to the drawbacks mentioned before. One of the possible solutions could be the development of new preparation methods to enhance the interface between the cocatalysts and g-C3N4. Furthermore, similar to the case of graphene, the electronic and morphological properties of g-C3N4 itself could be tuned by exfoliation to enhance the photocatalytic performances, which is discussed in the next section.
3. Generation of g-C3N4 nanosheets by exfoliation toward enhanced photocatalytic activity
Among various methods for control and modification of the morphology of g-C3N4, exfoliation is a simple and facile process which has been shown effective to enhance the photocatalytic performance of g-C3N4. Bulk g-C3N4 has a graphite-like structure which contains elementary layers built up from the ring structure of carbon nitride and van der Waals interaction between the layers. This unique structure makes g-C3N4 possible to be exfoliated into graphene-like single layered nanosheets. By exfoliating bulk g-C3N4 into nanosheets, the specific surface area can be increased and the catalytic centers are exposed. Furthermore, due to the quantum confinement effect, the conduction band (CB) position of g-C3N4 nanosheets can be shifted to more negative values than that of their bulk compartment, which provides a larger driving force for photocatalytic reaction.The exfoliation methods of g-C3N4 can be classified as thermal exfoliation and liquid exfoliation. Niu et al. reported the preparation of graphene-like g-C3N4 nanosheets by thermal etching of bulk g-C3N4.57 After the g-C3N4 sample was thermally oxidized in air at 500 °C for 2 h, g-C3N4 nanosheets with a thickness ranging from 1.62 nm to 2.62 nm were obtained corresponding to 4 to 7 carbon nitride layers. Compared to bulk g-C3N4, the optical absorption of g-C3N4 nanosheets exhibited a 20 nm blue shift which is due to quantum confinement. The surface area of g-C3N4 nanosheets increased from 50 m2 g−1 to 306 m2 g−1. The synergistic effect of the higher surface area and the more negative CB position results in a significant increase in their hydrogen production activity. Xu et al. further modified this thermal exfoliation method by firstly preparing an NH4Cl/g-C3N4 composite with NH4Cl intercalated in the interlayer space via hydrothermal reaction. Then the composite was annealed in N2 to exfoliate g-C3N4 into nanosheets by the evolved gaseous NH3.58 The preparation method is schematically illustrated in Fig. 3A. The surface area of g-C3N4 nanosheets is 10 times that of the bulk g-C3N4 prepared using dicyandiamide as the precursor. As shown in Fig. 3B, the average thickness of the as-prepared g-C3N4 nanosheets is around 2.8 nm, measured by atomic force microscopy (AFM). Although thermal exfoliation is considered to be a low cost, large scale and environmentally friendly method for preparing g-C3N4 nanosheets, the highest yield of the nanosheets obtained so far is 6 wt% of the starting bulk g-C3N4.57
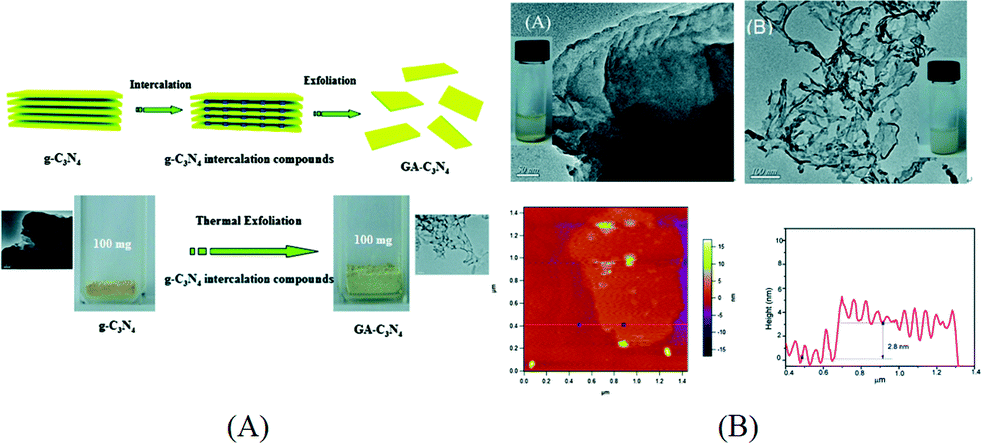 | ||
| Fig. 3 (A) Schematics of thermal exfoliation of g-C3N4 into nanosheets and optical images of g-C3N4 before and after exfoliation; (B) typical TEM images of bulk g-C3N4 and g-C3N4 nanosheets, AFM image and thickness profile of exfoliated nanosheets. Reproduced from ref. 58 with permission from The Royal Society of Chemistry. | ||
Liquid exfoliation is a more commonly used method and is done by ultrasonication of bulk g-C3N4 in a suitable solvent. After this process, the exfoliated nanosheets can be easily separated from the remaining bulk g-C3N4 by centrifugation. It is considered as a facile method for the preparation of 2D materials. Zhang et al. firstly reported the preparation of g-C3N4 nanosheets using water as the solvent during ultrasonication.59 As there is dangling hydrogen in the carbon nitride layer of g-C3N4, it was supposed that a polar solvent like H2O is effective for the swelling and exfoliation of g-C3N4. The swelling and exfoliation process is shown in Fig. 4A. Besides H2O, formamide, dimethylformamide, ethanol and methanol were used as exfoliation solvents. Only H2O was found effective and the concentration of the resulting nanosheet suspension was measured to be 0.15 mg mL−1. The change in UV-vis spectrum and photoluminescence (PL) spectrum before and after exfoliation can be attributed to the enlarged bandgap induced by quantum confinement (Fig. 4B). As shown in Fig. 4C, the thickness of the as-prepared g-C3N4 nanosheets is ~2.5 nm, corresponding to 7 atomic layers, and the lateral length of the nanosheets ranges from 70 nm to 140 nm. The as-prepared nanosheets were used for bioimaging due to their high PL quantum yield and biocompatibility. Inspired by this result, researchers have adopted the liquid exfoliation method for the preparation of g-C3N4 nanosheets and expanded the application of g-C3N4 nanosheets to other areas. For example, Sun and co-workers applied the same method and used the resulting nanosheets as an efficient fluoro-sensor for detection of Cu2+.60 Cheng et al. exfoliated g-C3N4 in water and loaded Au nanoparticles on the exfoliated g-C3N4 nanosheets via photodeposition. The composite was found to be able to efficiently degrade methyl orange under visible light.61 In addition, Zhang et al. reported that the reaction pathway for photocatalytic phenol degradation is changed when exfoliated nanosheets were used as the photocatalyst.62 In this case, g-C3N4 nanosheets with a thickness of 2 nm were prepared by sonication in water. The change in bandgap and CB position was measured by the UV-vis spectrum and Mott–Schottky plot. It was found that oxygen was reduced to H2O2 on g-C3N4 nanosheets via a two-electron transfer process while ˙O2− was formed on the surface of bulk g-C3N4via a one-electron transfer process. This was attributed to the formation of 1,4-endoperoxide species on the surface of g-C3N4 nanosheets. The two-electron transfer pathway promoted the efficient separation of excited electrons and holes and facilitated the formation of reactive species which resulted in the enhanced photocatalytic activity for phenol degradation.
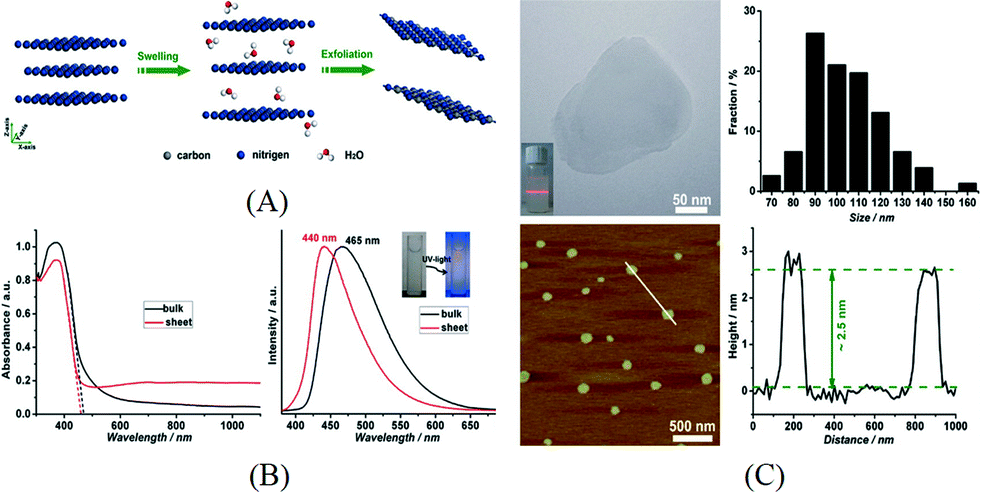 | ||
| Fig. 4 (A) Schematics of the swelling and exfoliation of g-C3N4 by H2O; (B) UV-vis spectrum and PL spectrum for bulk g-C3N4 and nanosheets; (C) typical TEM image and AFM image showing the lateral dimensions and thickness of g-C3N4 nanosheets. Reprinted with permission from ref. 59. Copyright 2015 American Chemical Society. | ||
Besides water, other polar solvents or mixed solvents have also been investigated for the exfoliation of g-C3N4. For instance, isopropanol alcohol (IPA) was found to be another suitable solvent for exfoliation of g-C3N4 into nanosheets due to the surface energy matching of IPA and g-C3N4.63 An extended sonication duration of 10 h yielded g-C3N4 nanosheets with an average thickness of 2 nm which is thinner than those prepared using H2O as the solvent. Similarly, the exfoliated g-C3N4 nanosheets exhibited a high surface area of 384 m2 g−1. The electrochemical impedance spectrum (EIS) results indicated an enhanced charge separation and transfer ability. Compared to bulk g-C3N4, the electron transfer resistance of g-C3N4 nanosheets decreased by 75%. The photocatalytic hydrogen production activity of g-C3N4 nanosheets is 10 times that of the bulk counterpart and is also higher than that of mpg-C3N4. In another work, She et al. prepared g-C3N4 nanosheets via sonication in 1,3-butanediol (1,3-BUT).64 Due to the polarity and surface energy matching, 1,3-BUT can swell and exfoliate g-C3N4 into nanosheets with a thickness of 0.9–2.1 nm. Mixed solvents have also been used for exfoliation of g-C3N4 such as the mixture of ethanol and water.65 Interestingly, it was found that the ratio of the two solvents affects the yield and the highest concentration of nanosheets at 3 mg mL−1 was obtained with an ethanol/water volume ratio of 1:3. This concentration is much higher than that obtained in the single solvent. Furthermore, it is usual that a g-C3N4 monolayer was obtained using such mixed solvents. Monolayer g-C3N4 nanosheets can also be produced via a combination of heat treatment and liquid exfoliation and the resultant nanosheets exhibited efficient photocatalytic disinfection activity for Escherichia coli.66Fig. 5 shows the EIS curves and transient photoresponse of the bulk and single-layered g-C3N4, indicating the enhanced charge transportation properties of the g-C3N4 nanosheets.
Although facile and simple, the disadvantage of liquid exfoliation method is obvious. It usually takes long sonication time and the yield is generally lower than 15%. In our recent work, the exfoliation of mpg-C3N4 in ethanol was carried out using a probe sonicator.67 The probe sonicator with a higher power intensity is directly immersed into the suspension and hence is much more effective than a bath sonicator. It is widely used in the exfoliation of layered metal chalcogenides like MoS2.68 The resultant yield (25.8%) for exfoliated mpg-C3N4 nanosheets was much higher than previously reported values. As shown in Fig. 6A, the thickness of such mpg-C3N4 nanosheets was 2–3 nm in average, corresponding to 5–8 carbon nitride layers. After depositing Pt or Co3O4 on the surface of mpg-C3N4 nanosheets as a cocatalyst (Fig. 6B), the photocatalytic activities for hydrogen evolution or degradation of Rhodamine B, respectively, were greatly enhanced compared to those of the bulk g-C3N4 (Fig. 6C and D). It is remarkable that the hydrogen evolution rate is 26 times that of the bulk g-C3N4.
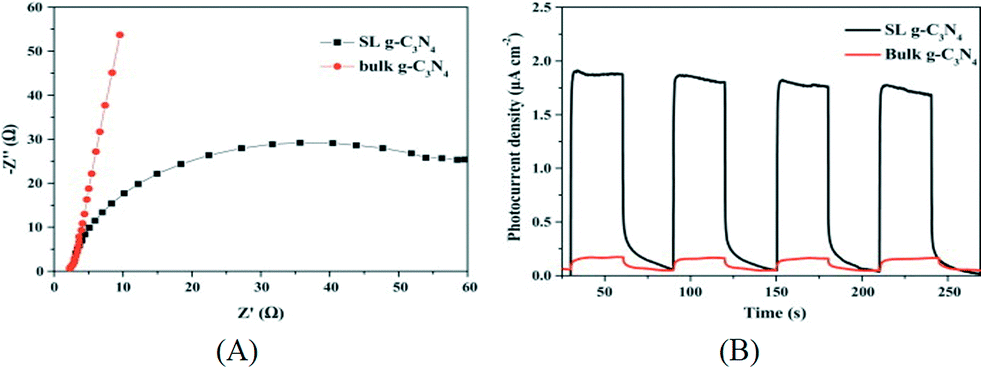 | ||
| Fig. 5 (A) EIS of the bulk g-C3N4 and single-layered g-C3N4; (B) transient photoresponse of bulk g-C3N4 and single-layered g-C3N4. Reprinted from ref. 65, copyright 2015 with permission from Elsevier. | ||
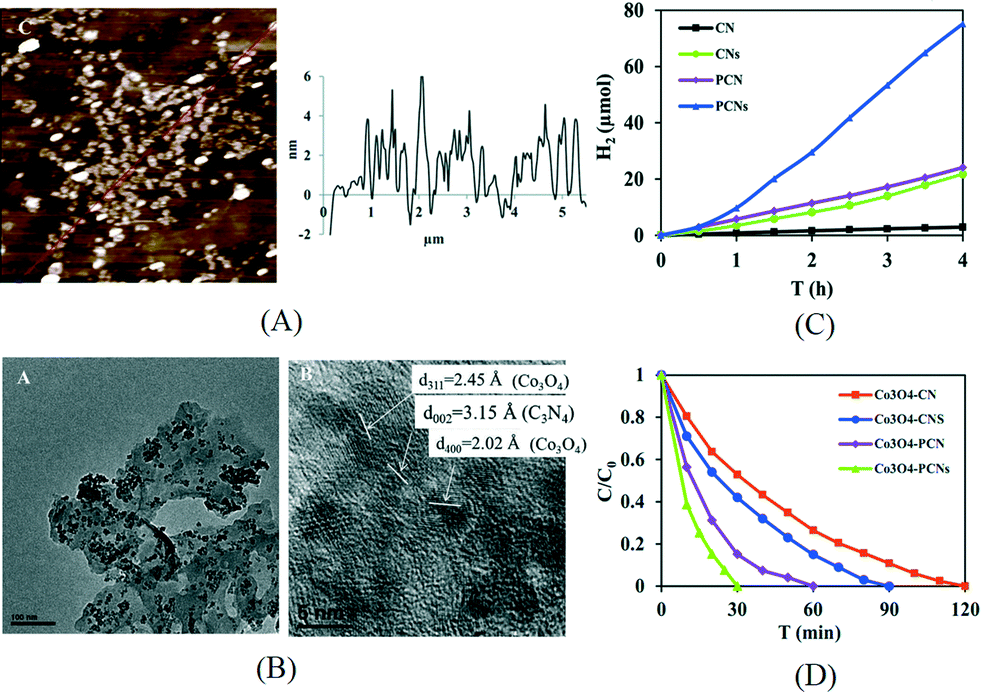 | ||
| Fig. 6 (A) AFM image and thickness profile of mpg-C3N4 nanosheets exfoliated using a probe sonicator; (B) TEM and HRTEM images of Co3O4/porous g-C3N4 nanosheets; (C) hydrogen production over Pt loaded g-C3N4 samples under visible light; (D) degradation of Rhodamine B over Co3O4 loaded g-C3N4 samples under UV-vis light (CN: g-C3N4, CNs: exfoliated g-C3N4 nanosheets, PCN: mesoporous g-C3N4, PCNs: exfoliated mesoporous g-C3N4 nanosheets). Reproduced from ref. 67 with permission from The Royal Society of Chemistry. | ||
Inspired by the Hummers method for the exfoliation of graphite into graphene, Zhu et al. developed a chemical etching method for the preparation of single atomic layered g-C3N4 nanosheets.69 It was found that only nanoparticles were obtained in the presence of KMnO4 since g-C3N4 is not stable enough against oxidation by KMnO4.57 H2SO4 (98%) was firstly intercalated into the interlayer space of g-C3N4. Then g-C3N4 nanosheets were obtained by the rapid heating effect of H2SO4 (98%) mixed with water. The yield of g-C3N4 nanosheets reached as high as 30%. AFM observation revealed that the thickness of 60% of the exfoliated nanosheets was around 0.4 nm, which is close to the theoretical thickness of a single carbon nitride layer. Correspondingly, the surface area was significantly increased from 4.3 m2 g−1 to 205.8 m2 g−1 after exfoliation. After loading Pt on the surface of the as-prepared g-C3N4 nanosheets, they exhibited an enhancement factor of 2.6 for hydrogen evolution compared to bulk g-C3N4. A subsequent research work demonstrated that the surface charge of the nanosheets prepared using this chemical etching method can be tuned by adding a different charge guest. The electrostatic assembly of the g-C3N4 nanosheets with CdS or BiOBr was prepared and investigated for the photocatalytic degradation of methyl orange (MO) and aminobenzoic acid.70 The CdS/g-C3N4 nanosheet composite exhibited superior photocatalytic activity. However, before the nanosheets can be used for photocatalytic reaction, the suspension should be carefully and thoroughly washed to fully remove the acid.
Besides the abovementioned research efforts, the exfoliated g-C3N4 nanosheets have also been coupled with other materials such as carbon nanotubes, reduced graphene oxide and WO3 arrays via electrostatic interactions for potential application in electrocatalysis or PEC reactions.71–74 In summary, g-C3N4 can be effectively exfoliated into monolayer or few layered nanosheets in polar solvents including water, some organic solvents and acid solution, or by a simple thermal exfoliation. The exfoliated nanosheets present enhanced photocatalytic or PEC activities due to the larger specific surface area, enhanced charge separation and transfer ability and more negative CB. The exfoliation process is simple but time consuming and the yield of nanosheets is generally low. Another main disadvantage of the sonication assisted exfoliation method is that boundary defects may be induced during the treatment which may lead to charge recombination. So far, we are unable to synthesize larger domain sizes with the current synthetic scheme.12 Further development of efficient and economic preparation methods for g-C3N4 nanosheets is needed for their practical applications.
4. g-C3N4 based photocatalysts for oxygen evolution and overall water splitting
Water oxidation, the other half reaction in overall water splitting, is a four-electron transfer process and considered to be the rate-limiting step in overall water splitting. Most of the research efforts on g-C3N4 based photocatalysts have been focused on the water reduction half reaction, which proceeds in the presence of a hole scavenger. There are only limited studies on water oxidation by the g-C3N4 system, not to mention overall water splitting. Despite the energetic barrier and sluggish kinetics, water oxidation by g-C3N4 also suffers from the self-oxidation of the catalyst which leads to the evolution of N2 similar to the case of (oxy)nitrides e.g. Ta3N5 and LaTiO2N.75 An efficient cocatalyst and a protection layer need to be developed to mitigate this problem.When g-C3N4 was first reported to be active for water splitting, its oxygen evolution activity was studied using AgNO3 as an electron acceptor.11 With RuO2 loaded as a water oxidation catalyst (WOC), g-C3N4 was shown to be able to oxidize water into O2 with a low reaction rate. Maeda et al. showed that photocorrosion can be significantly inhibited by loading RuO2 as an efficient WOC. Otherwise the evolution of N2 is significant.76 When the loading amount of RuO2 was optimized to 3 wt%, the highest oxygen evolution rate was obtained (12 μmol h−1). McMillan et al. studied the effect of the precursor and reaction parameters during g-C3N4 preparation on its oxygen evolution activity with RuO2 as the cocatalyst.77
Besides the aforementioned drawbacks of g-C3N4, one of the main factors limiting its water oxidation activity is its VB edge position at about 1.4 V vs. NHE.11,14,15 Compared to the VB position of widely studied oxides such as WO3 (2.7 V vs. NHE), BiVO4 (2.8 V vs. NHE) and Fe2O3 (2.2 V vs. NHE), g-C3N4 can only provide moderate water oxidation ability.78 Wang and co-workers used trithiocyanuric acid as the precursor to prepare sulfur-mediated g-C3N4 (CNS).79 After condensation, the sulfur amount in the final product was less than 1 wt%. The release of sulfur species during synthesis altered the connectivity pattern as well as the topology of g-C3N4 and lowered the VB position by ca. 0.2 V. It showed a 4-fold increase in O2 production compared to the pristine g-C3N4. However, the oxygen production rate of CNS was only 2.5 μmol h−1 under visible light irradiation since the photocatalytic experiment was conducted without loading a suitable WOC.
Then, the same group loaded cobalt species as WOC on CNS by an impregnation method and constructed an efficient noble metal free photocatalysis system for water oxidation.80 The structure of the cocatalyst was confirmed to be mainly Co3O4 by XPS and HRTEM. Fluorescence quenching study indicated efficient charge transfer from CNS to Co3O4. The PEC study showed that Co3O4/CNS lowered the onset potential of anodic photocurrent by 120 mV and also enhanced the photocurrent response. Using AgNO3 as an electron acceptor, the optimized Co3O4/CNS exhibited an oxygen evolution rate of 25.1 μmol h−1 under visible light. However, the oxygen evolution rate gradually decreased after 5 h which could be due to the deposition of Ag nanoparticles which blocked the catalytic centers as well as light absorption. Recently, Zhang et al. reported the deposition of another cobalt-based cocatalyst, Co(OH)2, on the surface of g-C3N4via simple precipitation (Fig. 7A). The PL quenching study in Fig. 7B revealed the efficient charge transfer from g-C3N4 to Co(OH)2. During the first hour, the oxygen evolution rate obtained was 27.4 μmol h−1 under UV-vis light irradiation (Fig. 7C).81 After calcination in air, Co(OH)2/g-C3N4 can be converted to Co3O4/g-C3N4, which is less active. Furthermore, the electrocatalytic oxygen evolution reaction (OER) onset potential of Co(OH)2/g-C3N4 was found to be lower than that of Co3O4/g-C3N4 by the linear sweep voltammogram shown in Fig. 7D.
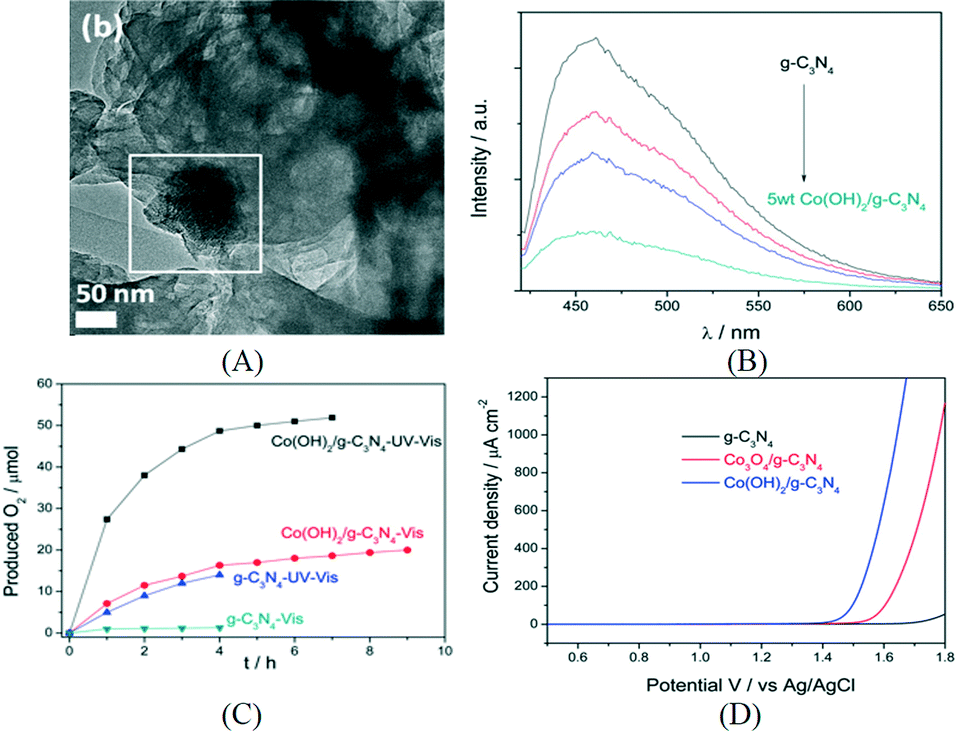 | ||
| Fig. 7 (A) TEM image of Co(OH)2/g-C3N4; (B) PL quenching of g-C3N4 and Co(OH)2/g-C3N4; (C) photocatalytic oxygen production over g-C3N4 and Co(OH)2/g-C3N4 under different conditions; (D) linear sweep voltammogram of different samples. Reprinted with permission from ref. 81. Copyright 2015 American Chemical Society. | ||
In the area of overall water splitting by g-C3N4 based systems, only limited literature has been found reporting the successful construction of g-C3N4 photocatalysts that can evolve H2 and O2 simultaneously at a stoichiometric ratio. Lee et al. decorated a cobalt phosphate (CoPi) catalyst on the surface of mpg-C3N4via direct photodeposition of Co2+ ions in a phosphate buffer solution or first deposition of metallic cobalt nanoparticles and then conversion of metallic nanoparticles to CoPi on mpg-C3N4.82 The as-prepared CoPi/mpg-C3N4 is active for both HER and OER half reactions in the presence of a suitable hole and electron scavenger, respectively. CoPi, a widely studied OER catalyst, was found to be converted in situ to Co-oxo/hydroxo-phosphate which is an active electrocatalyst for HER when a hole scavenger exists. In the absence of any charge scavenger, CoPi/mpg-C3N4 was found to be able to split water into H2 and O2 at a stoichiometric ratio in phosphate buffer solution with H2 and O2 produced at 13.6 μmol g h−1 and 6.6 μmol g−1 h−1, respectively. The activity for overall water splitting is stable after three runs. Though the reaction rate is still very low, this is the first example of overall water splitting by a g-C3N4 based photocatalyst.
Besides the single particulate system described earlier, using a Z-scheme photocatalyst which contains two photon systems is another way to achieve overall water splitting. Inspired by natural photosynthesis, the Z-scheme system is composed of a H2-evolving photocatalyst, an O2-evolving photocatalyst and an electron mediator. Photocatalysts that are only active for half reactions can be employed to construct Z-scheme systems, which extends the choice of photocatalyst for overall water splitting. However, the activity of a Z-scheme system depends highly on a suitable combination of its components. So far the most active Z-scheme system employs Ru/SrTiO3:Rh and BiVO4 as the H2-evolving photocatalyst and the O2-evolving photocatalyst, respectively.83 Other photocatalysts such as metal oxynitrides and metal sulfides are less active or inactive for the Z-scheme system. Tang and his co-workers constructed for the first time a Z-scheme system employing g-C3N4 as the H2-evolving photocatalyst.84 The mechanism is illustrated in Fig. 8A. Both BiVO4 and WO3 were used as the O2-evolving photocatalyst and soluble Fe3+/Fe2+ and IO3−/I− pairs were used as the electron mediator. With such a scheme, stoichiometric H2 and O2 were produced. As shown in Fig. 8, g-C3N4–NaI–WO3 was the most efficient in this Z-scheme configuration with production rates of H2 and O2 at 74 μmol g−1 h−1 and 37 μmol g−1 h−1, respectively, under full arc irradiation. The stability of this system is also confirmed to be good as stoichiometric H2 and O2 are evolved after three recycling runs and the gas production rate is almost the same as the initial run (Fig. 8D). Very recently, an effort was made on the construction of a mediator free Z-scheme overall water splitting system using g-C3N4 and WO3 as the HER and OER photocatalysts, respectively.85 The g-C3N4–WO3 composite was synthesized by in situ growth of WO3 on the surface of g-C3N4 in a hydrothermal reaction. In addition, they used reduced graphene oxide (rGO) as the electron mediator which is expected to enhance the performance. After loading Pt as the cocatalyst, the photoactivity of g-C3N4–WO3 and g-C3N4/rGO-WO3 was measured in pure water without redox couples such as I−/IO3−. Stoichiometric H2 and O2 can be stably evolved under visible light irradiation. A QE of 0.9% was obtained with the optimized sample at 420 nm. These results indicate that the intimate contact between two photocatalysts and improved electron transfer ability may benefit the H2/O2 evolution in the two step photoexcitation Z-scheme system. Further improvement of the activity of these systems is still needed.
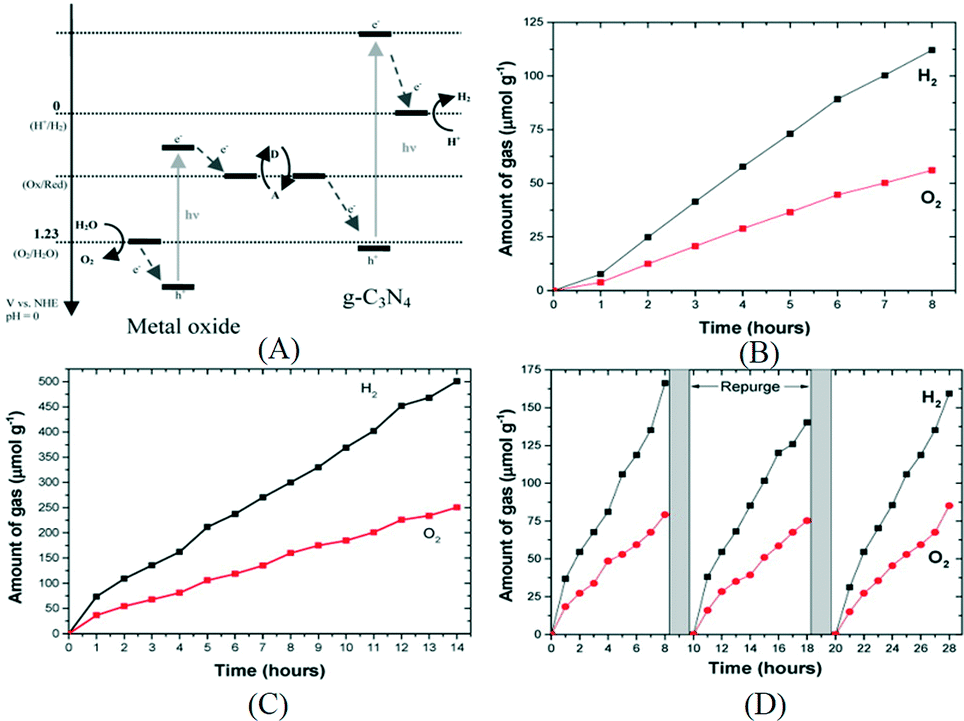 | ||
| Fig. 8 (A) Schematics of Z-scheme photocatalysts constructed using g-C3N4 as the H2-evolving photocatalyst and metal oxides as the O2-evolving photocatalyst; overall water splitting over (B) g-C3N4–FeCl2–BiVO4 under full arc irradiation, (C) g-C3N4–NaI–WO3 under full arc irradiation, (D) g-C3N4–NaI–WO3 under visible light irradiation. Reprinted with permission from ref. 84. Copyright 2015 American Chemical Society. | ||
Recently, Kang et al. constructed a metal free g-C3N4 based composite photocatalyst by coupling g-C3N4 with carbon dots (CDs).86 The solar to hydrogen (STH) efficiency reached 2% for the optimized CDs/g-C3N4 system, which is much higher than the thus reported values for photocatalytic systems except that of CoO (5.1%) which is self-corroded within 1 h.87 Such an STH efficiency reaches not far from the value of 5% set by the U.S. Department of Energy, which corresponds to the H2 production cost of $2.3 per kg. Furthermore CD/g-C3N4 exhibited long term stability for 200 days with the catalyst being separated, dried and reused after each day. The high stability can be attributed to its chemical and structure stability. A two-step OER mechanism including H2O2 production and subsequent decomposition of H2O2 to O2 was found to be key to the high STH efficiency. This two-step pathway was verified by electrochemical study and shown to be faster than the conventional four-electron process. Thus the STH efficiency was increased by accelerating the rate-limiting step of OER.
In summary, the non-noble metal containing g-C3N4 based system is capable of efficiently evolving O2 and is promising in overall water splitting. The metal free feature of g-C3N4 makes it a promising platform for the construction of low cost photocatalytic systems to further lower the cost of solar fuel production.
5. CO2 reduction using g-C3N4 based photocatalysts
CO2 reduction is a rapidly developing research area as this technology provides possible solutions to the environmental and energy problems we are facing. Many studies and reviews have been published showing the promising findings in reduction of CO2 into value added products.1,3,8,88–91 Similar to the situation in water oxidation and overall water splitting, there have not been extensive studies on CO2 photoreduction by g-C3N4 based systems. Nevertheless, during the last few years, CO2 photoreduction using g-C3N4 has received increasing attention due to the interesting properties of g-C3N4 that may offer some opportunities in this area.In 2012, Dong and co-workers prepared g-C3N4 and porous g-C3N4 by heating melamine or melamine hydrochloride and investigated their photocatalytic CO2 reduction activity in the presence of water vapor under visible light.92 Under these reaction conditions, CO was obtained as the reduction product. Subsequently, Mao et al. reported the use of g-C3N4 with different microstructures for CO2 photoreduction in NaOH solution without depositing any cocatalysts.93 In this work, urea and melamine were used for the preparation of g-C3N4. It was found that the specific surface area of u-g-C3N4 (39.5 m2 g−1) is much higher than that of m-g-C3N4 (3.7 m2 g−1), which leads to more efficient surface adsorption, better charge separation and improved photoactivity. The CO2 reduction products over u-g-C3N4 were CH3OH and C2H5OH while only C2H5OH was produced over m-g-C3N4. It is interesting that hydrocarbons were produced by CO2 reduction over the g-C3N4 system instead of gaseous CO possibly because the reaction was conducted in aqueous solution. However, CH3OH or C2H5OH may be oxidized on the surface of g-C3N4 by in situ evolved O2 during long photoreaction, as indicated by the decreased reaction rate after irradiation beyond 9 h. Tuning the electronic structure of g-C3N4 by doping S into the pristine g-C3N4 can enhance its optical adsorption as well as the CO2 reduction activity. S doped g-C3N4 was prepared by the condensation of thiourea and the CH3OH yield over S doped g-C3N4 is 1.4 times that over the pristine g-C3N4.94 To construct more efficient systems, one strategy is to couple the highly active homogeneous catalyst with g-C3N4. For instance, a Ru complex, cis,trans-[Ru{4,4′-(CH2PO3H2)2-2,2′-bipyridine}(CO)2Cl2]) (Ru) was adsorbed on the surface of mpg-C3N4 with a high surface area of 180 m2 g−1.95 The Ru/mpg-C3N4 was able to reduce CO2 into formic acid under visible light while a small amount of H2 and CO was also detected in acetonitrile in the presence of TEOA as the sacrificial reagent. Isotopic measurement results indicated that formic acid entirely came from CO2 reduction while 77% of the evolved CO originated from the carbonyl ligand unit of the Ru catalyst. The detachment of the carbonyl ligand from Ru was a slow process and has also been reported in other papers including homogeneous and heterogeneous systems.96,97 However, the photocatalytic activity of Ru/mpg-C3N4 for formic acid production was found to stay almost unchanged even after 5 h of reaction. The Ru catalyst after carbonyl ligand detachment was expected to be the active species for CO2 reduction. Subsequently, the same group studied the effect of the pore-wall structure of mpg-C3N4 and the effect of the Ru complex structure on CO2 photoreduction.98,99 Based on these results, it is understood that the photoactivity is sensitive to the specific surface area of mpg-C3N4 and is not related to the pore size and volume. Furthermore, introduction of too much meso-porosity results in the shrinkage of mpg-C3N4 walls and leads to activity drop. With –PO3H2 used as the linker group, RuP/mpg-C3N4 efficiently reduced CO2 to HCOOH under visible light in N,N-dimethylacetamide with TEOA as the sacrificial reagent. This hybrid material gave a high turnover number (TON) of larger than 1000 and a QE of 5.7% at 400 nm. In another study, Lin et al. prepared a Co(bpy)3Cl2/g-C3N4 hybrid material by self-assembly as the photocatalyst for the reduction of CO2 in acetonitrile under visible light in the presence of TEOA.100 CO and H2 were the main products. A TON of 4.3 with a relatively high selectivity of 88.4% for CO production was obtained by the optimized hybrid system. The surface of g-C3N4 or mpg-C3N4 was also modified with cobalt species as oxidative promoters to enhance CO2 photoreduction. Co(bpy)3Cl2/CoOx/mpg-C3N4 gave the highest TON of 13 and the selectivity of CO to H2 is 78.5%. Noble metal cocatalyst-loaded g-C3N4 has also been used for CO2 photoreduction. Under UV-vis light, CO2 can be reduced to hydrocarbons (mainly CH4, CH3OH and HCHO) using a Pt/g-C3N4 photocatalyst.101 Pt acts as an electron sink to enrich the surface of g-C3N4 with electrons for efficient CO2 reduction. The maximum yield can be obtained when the loading amount of Pt was 0.75%. However, Pt can also act as a catalyst for oxidation of HCHO over time. Pt/g-C3N4 was also prepared via a polyol process and used for photoreduction of CO2 in the presence of water vapor under day light lamp irradiation.102 CH4 was the main product for CO2 reduction and a 5.1 fold enhancement of CH4 production was obtained after 2% Pt was loaded on g-C3N4.
The composites of g-C3N4 and metal oxides have been investigated by various research groups for CO2 photoreduction. As in the case of hydrogen production, coupling g-C3N4 with a suitable semiconductor enhances the charge separation via band alignment, which leads to increased activity. For example, the composite of NaNbO3 nanowires and g-C3N4 was prepared by annealing the mixture of NaNbO3 nanowires and melamine at 520 °C in air.103 In this case, g-C3N4 was expected to grow on the surface of NaNbO3 and thus to make a good interface, which is revealed by the SEM and TEM images in Fig. 9B and C. The band energy diagram of NaNbO3 and g-C3N4 is shown in the schematics in Fig. 9A. The suitable band alignment between NaNbO3 and g-C3N4 facilitates the charge separation in the composite. After photodeposition of 0.5% Pt, the composite was capable to reduce CO2 to CH4 and the activity was much higher than those of the individual components loaded with Pt. Cao et al. prepared an In2O3/g-C3N4 photocatalyst by a solvothermal method in dimethyl sulfoxide.104 In2O3 nanocrystals were grown on the surface of sheet-like g-C3N4. In2O3/g-C3N4 exhibited similar optical adsorption properties to the pristine g-C3N4 but the transient photoresponse showed an increased photocurrent for In2O3/g-C3N4. After loading Pt as an electron sink over 10% In2O3/g-C3N4, 159.2 ppm CH4 can be evolved for 4 h. Transient PL decay indicated the inhibited charge recombination in In2O3/g-C3N4 due to enhanced charge separation at the interface. In another study, a g-C3N4/TiO2 heterojunction was prepared by an in situ growth method.105 The surface area of the composite increased with the percentage of TiO2 in the composite. When the photoreduction of CO2 was carried out with water vapor without a cocatalyst under UV-vis irradiation, CO was found to be the main product although a small amount of CH4 was also produced. Bi2WO6 was previously prepared by a hydrothermal method and shown active for the reduction of CO2 into CO under visible light.106 A solvothermal process was used to grow Bi2WO6in situ to form a g-C3N4/Bi2WO6 composite.107 The measured CB and VB positions of g-C3N4 and Bi2WO6 were used to explain the possible mechanism for the photoreduction of CO2 to CO.Compared to the pure Bi2WO6 prepared using the hydrothermal or solvothermal method, the activity of the composite was largely enhanced. ZnO, a large band gap semiconductor, was also used to make a composite with g-C3N4 by an impregnation method for CO2 reduction. The charge separation and transportation were promoted by the suitable band alignment between g-C3N4 and ZnO, which leads to an enhanced activity.108 Besides oxides, carbon materials have also been coupled with g-C3N4 for CO2 photoreduction. For example, a sandwich-like graphene/g-C3N4 hybrid nanostructure was fabricated using graphene oxide as a structure directing agent.109 The hybrid material shows enhanced activity for the conversion of CO2 to CH4 in the presence of water vapor under a daylight lamp. The enhanced photoactivity was attributed to the improved electron transfer induced by graphene.
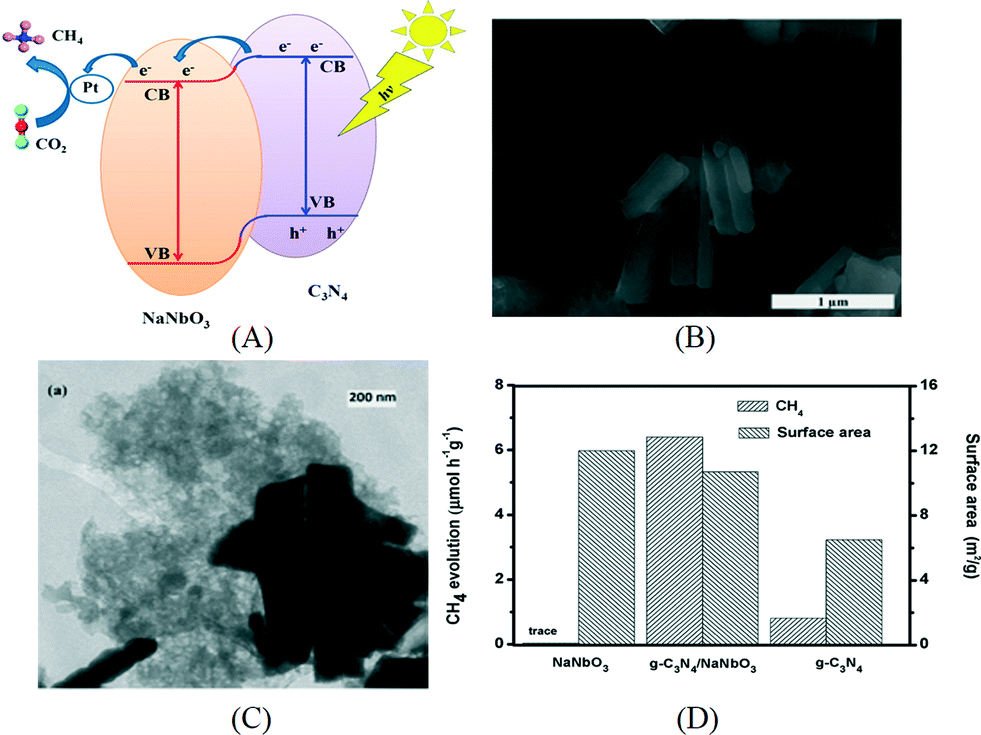 | ||
| Fig. 9 (A) Schematics of energy diagram of NaNbO3/g-C3N4; (B) SEM image and (C) TEM image of NaNbO3/g-C3N4; (D) photocatalytic CO2 reduction and surface area of NaNbO3/g-C3N4. Reprinted with permission from ref. 103. Copyright 2015 American Chemical Society. | ||
Besides the electronic and catalytic aspects, modifying g-C3N4 with a component for CO2 adsorption and enrichment has been found effective in enhancing the performance of the overall system. For example, Wang et al. coupled g-C3N4 with a Co-containing zeolitic imidazole framework (Co-ZIF).110 Co-ZIF-9 has a high CO2 adsorption capacity of 2.7 mmol g−1 and affords a high microporous surface area of 1607 m2 g−1. As a result, Co-ZIF-9 can capture and concentrate CO2 in its pores. After an electron mediator, bipyridine, was added into the reaction solution; the photoexcited electrons can be transferred from g-C3N4 to Co-ZIF-9 for CO2 reduction as revealed by PL quenching study. CO was the main product in this system and a QE of 0.9% can be obtained, even without the loading of a cocatalyst. Our group reported the construction of a hybrid system using g-C3N4 and an anionic clay called layered double hydroxide (LDH) which has high affinity for CO2.111 The structure of the LDH/g-C3N4 composite is shown in Fig. 10A. Mg–Al-LDH with a positive surface charge was assembled with oppositely charged g-C3N4 by electrostatic interactions. In the TEM image shown in Fig. 10B, the hexagonal-shaped LDH nanosheets can be seen on the surface of g-C3N4. Due to its unique layered structure, CO2 molecules can be intercalated in the interlayer space of LDH in the form of CO32− anions. After loading Pd as the cocatalyst, CH4 was the main product for CO2 reduction using this photocatalyst under UV-vis light. Isotopic measurement and blank control experiment indicated that most of the evolved CH4 was from CO2 instead of other carbon-containing species. The photocatalytic activity of the assembly system is 4 times that of the control sample without LDH. Among the LDHs intercalated with dominant NO3−, dominant CO32− and the mixture of the two, LDH-CO32− shows superior performance (Fig. 10C), suggesting that the intercalated CO32− anions from dissolved CO2 in water can act as a carbon source and are easily reduced since LDH is in close contact with both g-C3N4 and Pd. As shown in Fig. 10D, LDHs with different metal compositions were compared and Mg–Al-LDH exhibited the best performance for CO2 reduction which correlates with the highest CO2 adsorption capacity of Mg–Al-LDH among all LDHs. The highest QE of 0.093% was obtained at 440 nm over the optimized Pd/LDH/g-C3N4 assembly. The QE is still low but this example shows that the concept of coupling g-C3N4 with a CO2 capturing material is promising in CO2 reduction.
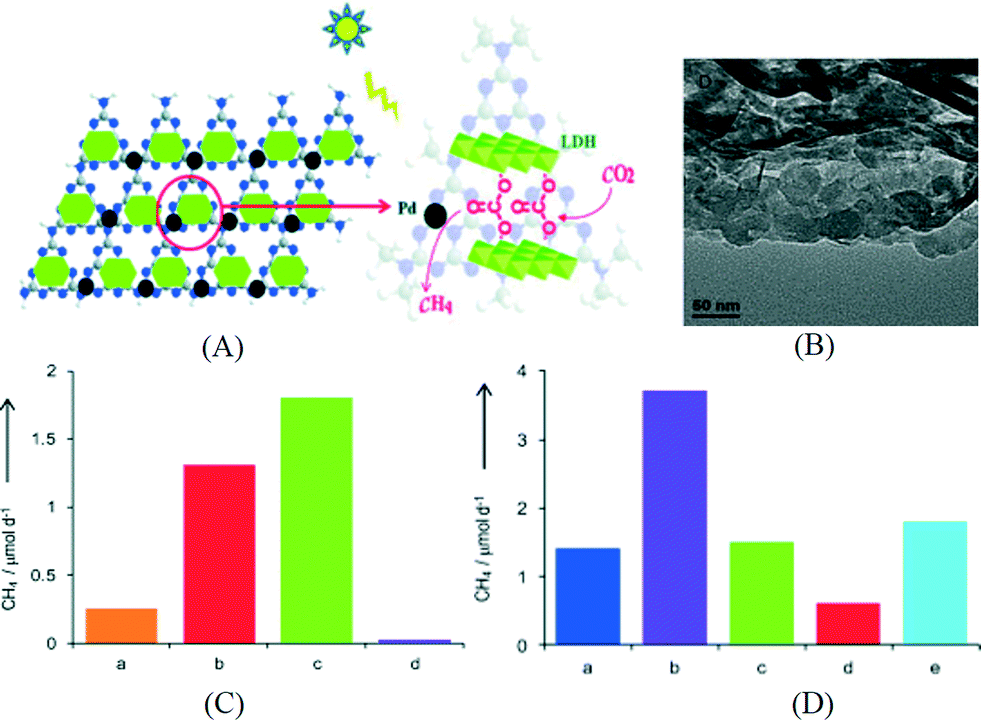 | ||
| Fig. 10 (A) Schematics of the structure of Mg–Al-LDH/g-C3N4; (B) TEM image of Mg–Al-LDH/g-C3N4; (C) CO2 reduction activity of Pd loaded photocatalysts: a. LDH-NO3−/g-C3N4, b. LDH/g-C3N4, c. LDH-CO32−/g-C3N4, d. g-C3N4; (D) CO2 reduction activity of Pd loaded photocatalysts: a. g-C3N4, b. Mg–Al-LDH/g-C3N4, c. Zn–Al-LDH/g-C3N4, d. Ni–Al-LDH/g-C3N4, e. Zn–Cr-LDH/g-C3N4.111 Copyright 2014 WILEY-VCH Verlag GmbH & Co. KGaA Weinheim. | ||
In summary, the study of g-C3N4 based photocatalysts for CO2 reduction is still at its early stage, but has received increasing interests in recent years. Different g-C3N4 based photocatalysts were synthesized including organic–inorganic hybrids, metal deposited g-C3N4, nanocomposites of g-C3N4 with oxides or carbon materials, and composites of g-C3N4 with CO2 adsorbing materials. The photocatalytic activity of g-C3N4 based photocatalysts depends on many factors and continuous efforts are needed for the development of more efficient and stable photocatalysts for CO2 reduction. It is also worth noting that the reported experimental setups, reaction conditions and product analysis methods vary from one paper to another. This makes it difficult to directly compare the reported activity data from different research groups. While it is challenging to adopt the same experimental setup by different groups, it is important to use reliable methods to accurately quantify various products of CO2 reduction. Several review articles including ours published in the past few years on this topic can provide useful practical guidelines to researchers in this field.112–114
6. Conclusion and outlook
This mini review summarizes the latest research efforts on the development of g-C3N4 based photocatalyst systems with the emphasis on the development of non-noble metal cocatalysts for g-C3N4, exfoliation of g-C3N4 to nanosheets with enhanced photoactivity, and application of g-C3N4 based photocatalysts for water oxidation, overall water splitting and CO2 reduction. The study of non-noble metal cocatalyst loaded g-C3N4 has already achieved some breakthroughs. The activities of a few photocatalysts like MoS2/g-C3N4 and Ni(OH)2/g-C3N4 were reported to approach or surpass that of Pt/g-C3N4. Further improvement of efficiency and stability is still in need. Exfoliation is a promising method to improve the photocatalytic activity of g-C3N4. Facile and simple liquid exfoliation can be conducted in water, certain organic solvents or acid solutions. Single layered or few layered g-C3N4 nanosheets can be obtained after exfoliation. The larger surface area, improved charge transfer ability and enhanced charge separation contribute to the superior photocatalytic activities of g-C3N4 nanosheets. The performance can be further improved by preparing porous g-C3N4 nanosheets. However, the current exfoliation method suffers from long sonication time and low yield. It is necessary to develop more efficient exfoliation methods. Water oxidation can be achieved by g-C3N4 based photocatalysts such as cobalt oxide loaded g-C3N4, although there are only limited research works on water oxidation over g-C3N4. The performance and stability of current g-C3N4 water oxidation photocatalysts are quite poor. Meanwhile, the study of overall water splitting by g-C3N4 based photocatalysts is in its early stage although some systems including single particulate systems and Z-schemes have been shown capable of evolving stoichiometric H2 and O2 from water. Further development of efficient g-C3N4 based photocatalysts for water oxidation is thus needed for their application in overall water splitting. There have been increasing research interests in CO2 photoreduction using g-C3N4 due to its suitable electronic band structure. Drawbacks including moderate optical adsorption properties, low charge carrier mobility, inert surface, low surface area and rich grain boundary defects need to be overcome to improve its photocatalytic activity. Some innovative ways and more systematic work are probably required to develop g-C3N4 to efficient and stable photocatalysts, which is a challenging task.Acknowledgements
This work was supported by Nanyang Technological University. S. M. Yin acknowledges the research scholarship from Nanyang Technological University.Notes and references
- D. Kim, K. K. Sakimoto, D. Hong and P. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294 RSC.
- K. Maeda and K. Domen, MRS Bull., 2011, 36, 25 CrossRef CAS.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983 CrossRef CAS PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150 CrossRef CAS PubMed.
- Y. Wang, X. C. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- Y. Moriya, T. Takata and K. Domen, Coord. Chem. Rev., 2013, 257, 1957 CrossRef CAS.
- X. Ma, Y. Lv, J. Xu, Y. Liu, R. Zhang and Y. Zhu, J. Phys. Chem. C, 2012, 116, 23485 CAS.
- Y. Zhang, T. Mori, J. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294 CrossRef CAS PubMed.
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. M. Lu and H. Cheng, J. Am. Chem. Soc., 2010, 132, 11642 CrossRef CAS PubMed.
- G. Dong, K. Zhao and L. Zhang, Chem. Commun., 2012, 48, 6178 RSC.
- Y. Zhang, T. Mori and J. Ye, Sci. Adv. Mater., 2012, 4, 282 CrossRef CAS.
- Z. Lin and X. Wang, Angew. Chem., Int. Ed., 2013, 52, 1735 CrossRef CAS PubMed.
- M. Zhang and X. Wang, Energy Environ. Sci., 2014, 7, 1902 CAS.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609 CrossRef CAS.
- G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin and X. Wang, Adv. Mater., 2014, 26, 805 CrossRef CAS PubMed.
- J. Hong, X. Xia, Y. Wang and R. Xu, J. Mater. Chem., 2012, 22, 15006 RSC.
- P. Martin-Ramos, J. Martin-Gil, R. Dante, F. Vaquero, R. Navarro and J. Fierro, Int. J. Hydrogen Energy, 2015, 40, 7273 CrossRef CAS.
- Y. Zhang, T. Mori, L. Niu and J. Ye, Energy Environ. Sci., 2011, 4, 4517 CAS.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355 CAS.
- A. Du, S. Sanvito, Z. Li, D. Wang, Y. Jiao, T. Liao, Q. Sun, Y. H. Ng, Z. Zhu, R. Amal and S. C. Smith, J. Am. Chem. Soc., 2012, 134, 4393 CrossRef CAS PubMed.
- X. Wang, J. Chen, X. Guan and L. Guo, Int. J. Hydrogen Energy, 2015, 40, 7546 CrossRef CAS.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15 RSC.
- Y. Wang, J. Hong, W. Zhang and R. Xu, Catal. Sci. Technol., 2013, 3, 1703 CAS.
- L. Ge, F. Zuo, J. Liu, Q. Ma, C. Wang, D. Sun, L. Bartels and P. Feng, J. Phys. Chem. C, 2012, 116, 13708 CAS.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680 CrossRef CAS PubMed.
- J. Zhang, M. Zhang, C. Yang and X. Wang, Adv. Mater., 2014, 26, 4121 CrossRef CAS PubMed.
- Y. Cui, Z. Ding, X. Fu and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 11814 CrossRef CAS PubMed.
- X. Li, X. Wang and M. Antonietti, Chem. Sci., 2012, 3, 2170 RSC.
- X. Bai, L. Wang, R. Zong and Y. Zhu, J. Phys. Chem. C, 2013, 117, 9952 CAS.
- D. Zheng, C. Pang, Y. Liu and X. Wang, Chem. Commun., 2015, 51, 9706 RSC.
- J. H. Sun, J. S. Zhang, M. W. Zhang, M. Antonietti, X. Z. Fu and X. C. Wang, Nat. Commun., 2012, 3, 1139 CrossRef.
- J. Zhang, F. Guo and X. Wang, Adv. Funct. Mater., 2013, 23, 3008 CrossRef CAS.
- S. Cao and J. Yu, J. Phys. Chem. Lett., 2014, 2101 CrossRef CAS PubMed.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900 CrossRef CAS PubMed.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787 RSC.
- J. Dong, M. Wang, X. Li, L. Chen, Y. He and L. Sun, ChemSusChem, 2012, 5, 2133 CrossRef CAS PubMed.
- D. Wang, Y. Zhang and W. Chen, Chem. Commun., 2014, 50, 1754 RSC.
- J. Huang, K. L. Mulfort, P. Du and L. X. Chen, J. Am. Chem. Soc., 2012, 134, 16472 CrossRef CAS PubMed.
- S. Cao, X. Liu, Y. Yuan, Z. Zhang, J. Fang, S. C. J. Loo, J. Barber, T. C. Sum and C. Xue, Phys. Chem. Chem. Phys., 2013, 15, 18363 RSC.
- S. Cao, Y. Yuan, J. Barber, S. C. J. Loo and C. Xue, Appl. Surf. Sci., 2014, 319, 344 CrossRef CAS.
- J. Hong, Y. Wang, Y. Wang, W. Zhang and R. Xu, ChemSusChem, 2013, 6, 2263 CrossRef CAS PubMed.
- Z. Chen, P. Sun, B. Fan, Z. Zhang and X. Fang, J. Phys. Chem. C, 2014, 118, 7801 CAS.
- L. Yin, Y. Yuan, S. Cao, Z. Zhang and C. Xue, RSC Adv., 2014, 4, 6127 RSC.
- Y. Zhu, Y. Xu, Y. Hou, Z. Ding and X. Wang, Int. J. Hydrogen Energy, 2014, 39, 11873 CrossRef CAS.
- H. Vrubel, D. Merki and X. Hu, Energy Environ. Sci., 2012, 5, 6136 CAS.
- Y. Hou, A. B. Laursen, J. Zhang, G. Zhang, Y. Zhu, X. Wang, S. Dahl and I. Chorkendorff, Angew. Chem., Int. Ed., 2013, 52, 3621 CrossRef CAS PubMed.
- Y. Hou, Y. Zhu, Y. Xu and X. Wang, Appl. Catal., B, 2014, 156–157, 122 CrossRef CAS.
- J. Yu, S. Wang, B. Cheng, Z. Lin and F. Huang, Catal. Sci. Technol., 2013, 3, 1782 CAS.
- X. Zhou, Z. Luo, P. Tao, B. Jin, Z. Wu and Y. Huang, Mater. Chem. Phys., 2014, 143, 1462 CrossRef CAS.
- G. Zhang, G. Li and X. Wang, ChemCatChem, 2015 DOI:10.1002/cctc.201500069.
- P. Niu, L. Zhang, G. Liu and H. Cheng, Adv. Funct. Mater., 2012, 22, 4763 CrossRef CAS.
- H. Xu, J. Yan, X. She, L. Xu, J. Xia, Y. Xu, Y. Song, L. Huang and H. Li, Nanoscale, 2014, 6, 1406 RSC.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2012, 135, 18 CrossRef PubMed.
- J. Tian, Q. Liu, A. M. Asiri, A. O. Al-Youbi and X. Sun, Anal. Chem., 2013, 85, 5595 CrossRef CAS PubMed.
- N. Cheng, J. Tian, Q. Liu, C. Ge, A. H. Qusti, A. M. Asiri, A. O. Al-Youbi and X. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 6815 CAS.
- H. Zhang, L. Guo, L. Zhao, B. Wan and Y. Yang, J. Phys. Chem. Lett., 2015, 958 CrossRef CAS PubMed.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452 CrossRef CAS PubMed.
- X. She, H. Xu, Y. Xu, J. Yan, J. Xia, L. Xu, Y. Song, Y. Jiang, Q. Zhang and H. Li, J. Mater. Chem. A, 2014, 2, 2563 CAS.
- Q. Lin, L. Li, S. Liang, M. Liu, J. Bi and L. Wu, Appl. Catal., B, 2015, 163, 135 CrossRef CAS.
- H. Zhao, H. Yu, X. Quan, S. Chen, Y. Zhang, H. Zhao and H. Wang, Appl. Catal., B, 2014, 152–153, 46 CrossRef CAS.
- J. Hong, S. Yin, Y. Pan, J. Han, T. Zhou and R. Xu, Nanoscale, 2014, 6, 14984 RSC.
- K. Chang, M. Li, T. Wang, S. Ouyang, P. Li, L. Liu and J. Ye, Adv. Energy Mater., 2015, 5, 1402279 Search PubMed.
- J. Xu, L. Zhang, R. Shi and Y. Zhu, J. Mater. Chem. A, 2013, 1, 14766 CAS.
- F. Cheng, H. Wang and X. Dong, Chem. Commun., 2015, 51, 7176 RSC.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2015, 9, 931 CrossRef CAS PubMed.
- Y. Hou, F. Zuo, A. P. Dagg, J. Liu and P. Feng, Adv. Mater., 2014, 26, 5043 CrossRef CAS PubMed.
- Y. Zhao, F. Zhao, X. Wang, C. Xu, Z. Zhang, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2014, 53, 13934 CrossRef CAS PubMed.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2014, 53, 7281 CrossRef CAS PubMed.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, J. Am. Chem. Soc., 2012, 134, 8348 CrossRef CAS PubMed.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940 CAS.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Corà, S. Firth, J. A. Darr and P. F. McMillan, J. Phys. Chem. C, 2013, 117, 7178 CAS.
- M. Walter, E. Warren, J. McKone, S. Boettcher, Q. Mi, E. Santori and N. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- J. Zhang, J. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. Fu and X. Wang, Energy Environ. Sci., 2011, 4, 675 CAS.
- J. Zhang, M. Grzelczak, Y. Hou, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Chem. Sci., 2012, 3, 443 RSC.
- G. Zhang, S. Zang and X. Wang, ACS Catal., 2015, 941 CrossRef CAS.
- R. Lee, P. D. Tran, S. S. Pramana, S. Y. Chiam, Y. Ren, S. Meng, L. H. Wong and J. Barber, Catal. Sci. Technol., 2013, 3, 1694 CAS.
- A. Kudo, MRS Bull., 2011, 36, 32 CrossRef CAS.
- D. J. Martin, P. J. T. Reardon, S. J. A. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568 CrossRef CAS PubMed.
- G. Zhang, X. Huang, F. Fina, G. Zhang and J. Irvine, Catal. Sci. Technol., 2015, 5, 3416 Search PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970 CrossRef CAS PubMed.
- L. Liao, Q. Zhang, Z. Su, Z. Zhao, Y. Wang, Y. Li, X. Lu, D. Wei, G. Feng, Q. Yu, X. Cai, J. Zhao, Z. Ren, H. Fang, F. Robles-Hernandez, S. Baldelli and J. Bao, Nat. Nanotechnol., 2013, 9, 69 CrossRef PubMed.
- Q. Xiang, B. Cheng and J. Yu, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201411069.
- K. Li, X. An, L. Park, M. Khraisheh and J. Tang, Catal. Today, 2014, 224, 3 CrossRef CAS.
- L. Yuan and Y. Xu, Appl. Surf. Sci., 2015, 342, 154 CrossRef CAS.
- X. Li, J. Wen, J. Low, Y. Fang and J. Yu, Sci. China Mater., 2014, 57, 70 CrossRef.
- G. Dong and L. Zhang, J. Mater. Chem., 2012, 22, 1160 RSC.
- J. Mao, T. Peng, X. Zhang, K. Li, L. Ye and L. Zan, Catal. Sci. Technol., 2013, 3, 1253 CAS.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal., B, 2015, 176–177, 44 CrossRef CAS.
- K. Maeda, K. Sekizawa and O. Ishitani, Chem. Commun., 2013, 49, 10127 RSC.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596 CrossRef CAS PubMed.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673 CrossRef CAS PubMed.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406 CrossRef CAS PubMed.
- K. Maeda, R. Kuriki, M. Zhang, X. Wang and O. Ishitani, J. Mater. Chem. A, 2014, 2, 15146 CAS.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2014, 2, 353 CrossRef CAS.
- J. Yu, K. Wang, W. Xiao and B. Cheng, Phys. Chem. Chem. Phys., 2014, 16, 11492 RSC.
- W. Ong, L. Tan, S. Chai and S. Yong, Dalton Trans., 2015, 44, 1249 RSC.
- H. Shi, G. Chen, C. Zhang and Z. Zou, ACS Catal., 2014, 4, 3637 CrossRef CAS.
- S. Cao, X. Liu, Y. Yuan, Z. Zhang, Y. Liao, J. Fang, S. C. J. Loo, T. C. Sum and C. Xue, Appl. Catal., B, 2014, 147, 940 CrossRef CAS.
- S. Zhou, Y. Liu, J. Li, Y. Wang, G. Jiang, Z. Zhao, D. Wang, A. Duan, J. Liu and Y. Wei, Appl. Catal., B, 2014, 158–159, 20 CrossRef CAS.
- Z. Sun, Z. Yang, H. Liu, H. Wang and Z. Wu, Appl. Surf. Sci., 2014, 315, 360 CrossRef CAS.
- M. Li, L. Zhang, X. Fan, Y. Zhou, M. Wu and J. Shi, J. Mater. Chem. A, 2015, 3, 5189 CAS.
- Y. He, Y. Wang, L. Zhang, B. Teng and M. Fan, Appl. Catal., B, 2015, 168–169, 1 CAS.
- W. Ong, L. Tan, S. Chai and S. Yong, Chem. Commun., 2015, 51, 858 RSC.
- S. Wang, J. Lin and X. Wang, Phys. Chem. Chem. Phys., 2014, 16, 14656 RSC.
- J. Hong, W. Zhang, Y. Wang, T. Zhou and R. Xu, ChemCatChem, 2014, 6, 2315 CrossRef CAS.
- J. Hong, W. Zhang, J. Ren and R. Xu, Anal. Methods, 2012, 1086 Search PubMed.
- A. Corma and H. Garcia, J. Catal., 2013, 308, 168 CrossRef CAS.
- J. A. Herron, J. Kim, A. A. Upadhye, G. W. Huber and C. T. Maravelias, Energy Environ. Sci., 2015, 8, 126 CAS.
| This journal is © The Royal Society of Chemistry 2015 |