
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of highly dispersed Pd nanoparticles supported on multi-walled carbon nanotubes and their excellent catalytic performance for oxidation of benzyl alcohol
Vijay M.
Shinde
,
Emmanuel
Skupien
and
Michiel
Makkee
*
Catalysis Engineering, Chemical Engineering Department, Delft University of Technology, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: m.makkee@tudelft.nl; Fax: +31(0) 15 2785006; Tel: +31(0) 15 2781391
First published on 12th June 2015
Abstract
Narrow sized and highly homogeneous dispersed Pd nanoparticles have been synthesized on nitric acid-functionalized multi-walled carbon nanotubes (CNTs) without a capping agent. The TEM images show that the extremely small Pd nanoparticles with an average size of about 1.5 nm were homogeneously dispersed on the surface of the CNTs. The characterization results indicate that the pretreatment with nitric acid not only improved the dispersion of Pd, but also enhanced the strong interaction between the Pd nanoparticles and the CNTs, thereby preventing their agglomeration and leaching in the liquid phase. On pretreatment with HNO3, it is possible to generate more acidic groups on the surface of CNTs without a significant change in textural properties. The catalytic performance of the aforementioned material was investigated for selective oxidation of benzyl alcohol. Pd/CNTs exhibits high activity (~98% conversion) and selectivity (~90%) to benzaldehyde with excellent reusability. The high activity of the catalyst was attributed to the small size, high dispersion of Pd nanoparticles and higher accessibility of reactants. A careful analysis of the kinetic data suggests that there are different sites for the disproportionation and oxidation reactions. The excellent reusability of Pd/CNT catalysts makes this material a promising candidate for selective benzyl oxidation. Further, the results of the present study show that it is possible to synthesize uniformly dispersed Pd nanoparticles on various carbon supports without a capping agent.
1. Introduction
Noble metal nanoparticles supported on carbon nanotubes as heterogeneous catalysts have gained increasing interest due to their one-dimensional morphology and synergistic effects.1 It has been reported that the noble metal nanoparticles supported on carbon nanotubes exhibit an improved catalytic activity. The catalytic properties of the nanoparticles are highly sensitive to the size, morphology, composition, and dispersion of the noble metal particles.2–4 For the synthesis of highly dispersed and stable catalyst, a strong interaction between the metal precursor and the support is essential.5 However, the uniform dispersion of the noble metal nanoparticles on carbon support is difficult due to its chemical inertness. High temperature treatment with concentrated acid or doping with heteroatoms is usually used to functionalize the surfaces of CNTs.6–8 These pretreatments can often control the hydrophobicity of CNTs and introduce active sites on the CNT surface.9–11 The concentration, distribution, and nature of the functional groups also influence the dispersion of noble metals. Furthermore, the functionalization of supports can prevent the sintering of supported nanoparticles.12 In addition, functional groups on the support render a strong metal–support interaction which averts the problem of leaching of the active phase in liquid phase reactions.There are several methods to synthesize noble metal nanoparticle/CNT composites, including impregnation, the colloidal chemistry method, and the polyol method.13–16 Among these methods, the polyol method is a promising alternative to synthesize nanoparticles due to its ability to control particle size and size distribution, high dispersion, and precisely tunable bimetallic composition. However, it has been shown that there is a large variation of the resulting mean particle size with the final composition of catalyst.17 In general, the ultrafine Pd nanoparticles are vulnerable to serious aggregation18,19 therefore, the capping agents (surfactants or polymeric stabilizers) are commonly used to stabilize the nanoparticles.20,21 However, in most of the cases, the removal of the capping agent is required to activate the catalyst. This is usually accomplished by washing and/or heat treatment.20,21 However, most of the time, the complete removal of the capping agents is difficult due to either a strong interaction between the nanoparticles and the capping agent or due to the aggregation tendency of the nanoparticles at high temperature(s). Therefore, it remains a great challenge to synthesize nanoparticles without any capping agent.22 An improved method has been later developed without the addition of a capping agent which involves refluxing of the carbon nanotubes in a mixture of concentrated nitric and sulfuric acids to create surface functional groups, such as carbonyl (–CO), hydroxylic (–COH) and carboxylic (–COOH). The acid treated CNTs were then used for the deposition of metal nanoparticles. Ethylene glycol was used as a solvent and reducing agent to reduce the metal precursor23,24 at 120 °C or by heating with microwave radiation.25,26 Although no capping agent was used during the synthesis, it was difficult to remove the oxidation products of ethylene glycol at elevated temperatures and it remained adsorbed on the Pd nanoparticles.
Recently, a facile method for the synthesis of nanoparticles using CO as a gaseous surfactant has been reported for a Pt–Ni bimetallic alloy27 in which the unique role of the CO molecule, as a size confining molecular adsorbate due to its size-dependent coverage and chemisorption energy in the restriction of the mean particle size of the alloy at 4 nm regardless of the Ni content, has been illustrated. Therefore, it has been anticipated that the direct reduction of a Pd precursor in the presence of gaseous molecules such as CO or H2, due to their strong adsorption on Pd metal, should be able to control the growth of Pd nanoparticles. A physical adsorption technique was used for the deposition of Ru nanoparticles on CNTs in which the Ru precursor was firstly impregnated upon CNTs and then was reduced to metallic Ru nanoparticles.28 The resulting metal nanoparticles had a mean size in the range of 3–7 nm and metal loadings were lower than 0.2 wt%. To the best of our knowledge, a simple synthesis method for a highly dispersed Pd nanoparticle on CNTs at high metal loading without a capping agent has not been reported in the literature.
The selective oxidation of alcohols to their corresponding carbonyl compounds is a subject of growing interest. Noble metal-supported catalysts such as Pd, Pt, Ru, and Au-supported catalysts are active for the selective oxidation of alcohols.29–31 Among these noble metal-supported catalysts, Pd-supported catalysts show relatively high activity and selectivity.29 These studies reveal that the aldehyde selectivity is close to 60–80% over the Pd-supported catalysts.32 In particular, the addition of Au to Pd catalysts improves not only the catalytic activity, but also the selectivity to the aldehyde.33 Thus, the bimetallic catalysts are promising candidates for transformation and offer greater flexibility not only in chemical composition but also in interatomic arrangement compared to monometallic catalysts.34 However, the bimetallic catalysts need a very careful synthesis procedure in order to avoid any segregation or inhomogeneity that weakens the synergistic effects of the bimetallic catalysts.35 In this paper, we have described a convenient approach to synthesize highly and uniformly dispersed Pd nanoparticles on CNTs without a capping agent. The CNTs were firstly functionalized with nitric acid to create the surface O-functional groups for the deposition of metal nanoparticles and then the Pd precursor, PdCl2, was impregnated on the surface of CNTs through drying and a subsequent reduction of metal precursor in the presence of H2. The results showed that the functionalization of the CNTs demonstrated a uniform deposition of Pd nanoparticles on CNTs at relatively high loading of 3 wt%. The synthesized Pd/CNTs exhibited an average size of 1.5 nm and a narrow particle size distribution. Pd catalysts supported on the various form of carbons such as carbon black, activated carbon, and graphite were also synthesised using the aforementioned approach. The catalytic performance of all these materials was investigated for the selective oxidation reaction of benzyl alcohol into benzaldehyde. The Pd/CNTs showed a better activity, selectivity, and reusability than the Pd nanoparticles supported on the other form of carbons.
2. Experimental
2.1. Synthesis and characterization
Four carbon supports, namely: multi-walled CNTs (Cheap Tubes Inc., USA), carbon black (Cabot Corporation, USA), activated carbon (Fluka Analytical, Netherlands), and graphite (Alfa Aesar, Germany) were used. All the Pd-supported catalysts were prepared using an impregnation–reduction method. The supports were firstly functionalized with a pretreatment of concentrated nitric acid. Typically, 2 g of CNTs were suspended in 150 ml of concentrated HNO3 (68 wt%) and refluxed at 120 °C for 6 h. After cooling to room temperature, the mixture was filtered and washed with demineralized water for several times until the pH of filtrate reached 7, and was subsequently dried in air oven at 120 °C for 2 h. The required amount of PdCl2 (Sigma Aldrich, Netherlands) for 3 wt% Pd loading was dissolved in 20 ml of demineralized water (a small amount of HCl was added to ensure the complete solubility of PdCl2). After stirring for 30 min, the CNTs were dispersed into the above solution and stirred for 6 h at room temperature until a paste-like mixture was obtained due to evaporation of water. Finally, the mixture was dried at 120 °C and subsequently reduced in 5% of H2 in N2 flowing at 100 ml min−1 at 350 °C in a flow reactor for 2 h. The same procedure was followed to synthesize all other Pd-supported catalysts.X-ray diffraction (XRD) patterns were recorded on a Phillips X'Pert diffractometer using Co Kα radiation (k = 0.1788 nm) operated at 40 kV and 30 mA. The scans were recorded at a rate of 0.5° min−1. The peaks were identified with reference to the JCPDS database. The transmission electron microscopy (TEM) images were recorded on a JEOL JEM-2010F instrument operated at 200 kV. The sample was dispersed in ethanol and dried on a carbon-coated copper grid (300 mesh). The TEM images were recorded in different regions of the Cu grid. The size distribution of the Pd nanoparticles were obtained by measuring around 150–200 particles in arbitrarily chosen areas of the image. X-ray photoelectron spectroscopy (XPS) was used to examine the electronic properties of the catalysts. The XPS spectra were recorded on a Thermo Scientific Multilab equipped with an Al anode (Al K-alpha = 1486.6 eV). The XPS spectra were calibrated using a binding energy of C 1s observed at 284.6 eV. The background due to inelastic process was corrected using the Shirley method. A fixed Gaussian–Lorentzian shape function was used to deconvolute the overlapping peaks. The normalized peak intensities (corrected with the atomic sensitivity factors) were used to estimate the relative concentration of the Pd0 and Pd2+ states on the various carbon supports.
Temperature programmed desorption (TPD) of the catalysts was performed to study surface oxygen mobility in a fixed bed reactor connected to a mass spectrometer. 100 mg of a sample was heated from room temperature to 900 °C at a heating rate of 10 °C min−1 in He flowing at 30 ml min−1. The mass spectrometer was calibrated using calcium oxalate as a reference. The evaluation of the species such as CO and CO2 was measured as function of temperature.
N2 adsorption–desorption isotherms were measured with a Quantachrome Autosorb-6B unit gas adsorption analyzer (Micromeritics, USA) using a continuous adsorption procedure. The BET method was used to calculate the specific surface areas of the sample. Prior to N2 adsorption, the sample was degassed at 200 °C for 12 h in vacuum. Pd content of the carbon catalysts was determined by ICP-OES analyses (Micro Analytical Laboratory Kolbe, Germany).
2.2. Catalytic activity measurements
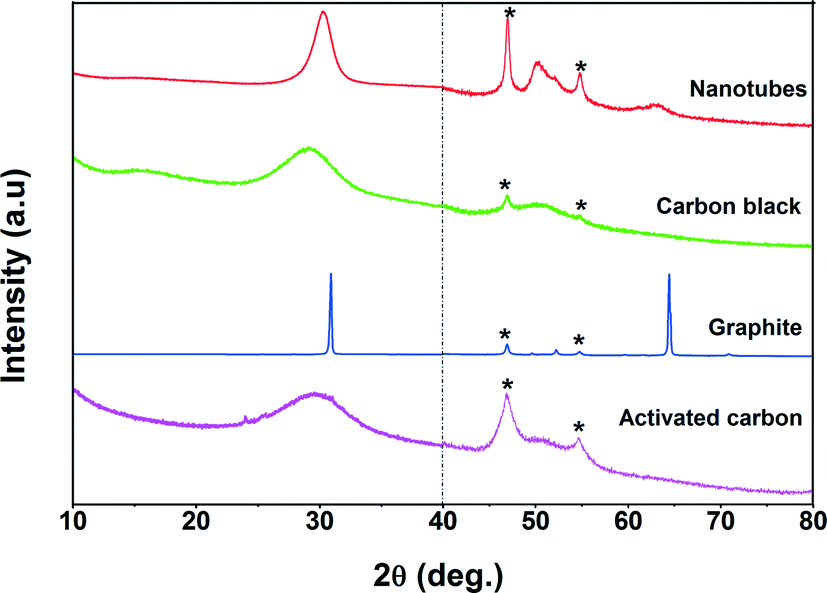
The benzyl alcohol oxidation reaction was carried out in a 100 ml round-bottom stirred vessel equipped with a condenser. In a typical experiment, 50 ml of xylene (a mixture of isomers) and 300 mg of catalyst were introduced into the reactor. Toluene, frequently used as a solvent in the literature, was not used, since toluene is or could be one of the (oxidation) products of the reaction. The vessel was heated to 90 °C using an oil bath. The reaction mixture was stirred at 900 rpm and an air flow rate of 100 ml min−1 was continuously bubbled through the reaction mixture. After stabilization of the reactor temperature at 90 °C, 2.4 g of benzyl alcohol was introduced using a syringe (t = 0 min). The samples were collected in a regular interval and analyzed by GC (Varian CP-3380 with a 50 m CP-Sil 52 CB column), equipped with an FID detector. The initial temperature of the GC oven was programmed from 150 °C (held for 6 min) to 350 °C at a heating rate of 20 °C min−1. An external standard was used to quantify the amount of reactants consumed and products formed during the reaction. It was observed that no reaction occurred either in the absence of Pd catalyst or in the presence of the support. The rate of reaction was linearly proportional to the catalyst amount and independent of the speed of stirring, indicating that mass transport limitations were absent. For the reusability test, the catalyst was recovered after the test by vacuum filtration and washed with xylene at room temperature. The conversion of benzyl alcohol, the selectivity to benzaldehyde, and the quasi-turnover frequency are defined as follows: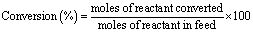 | (1) |
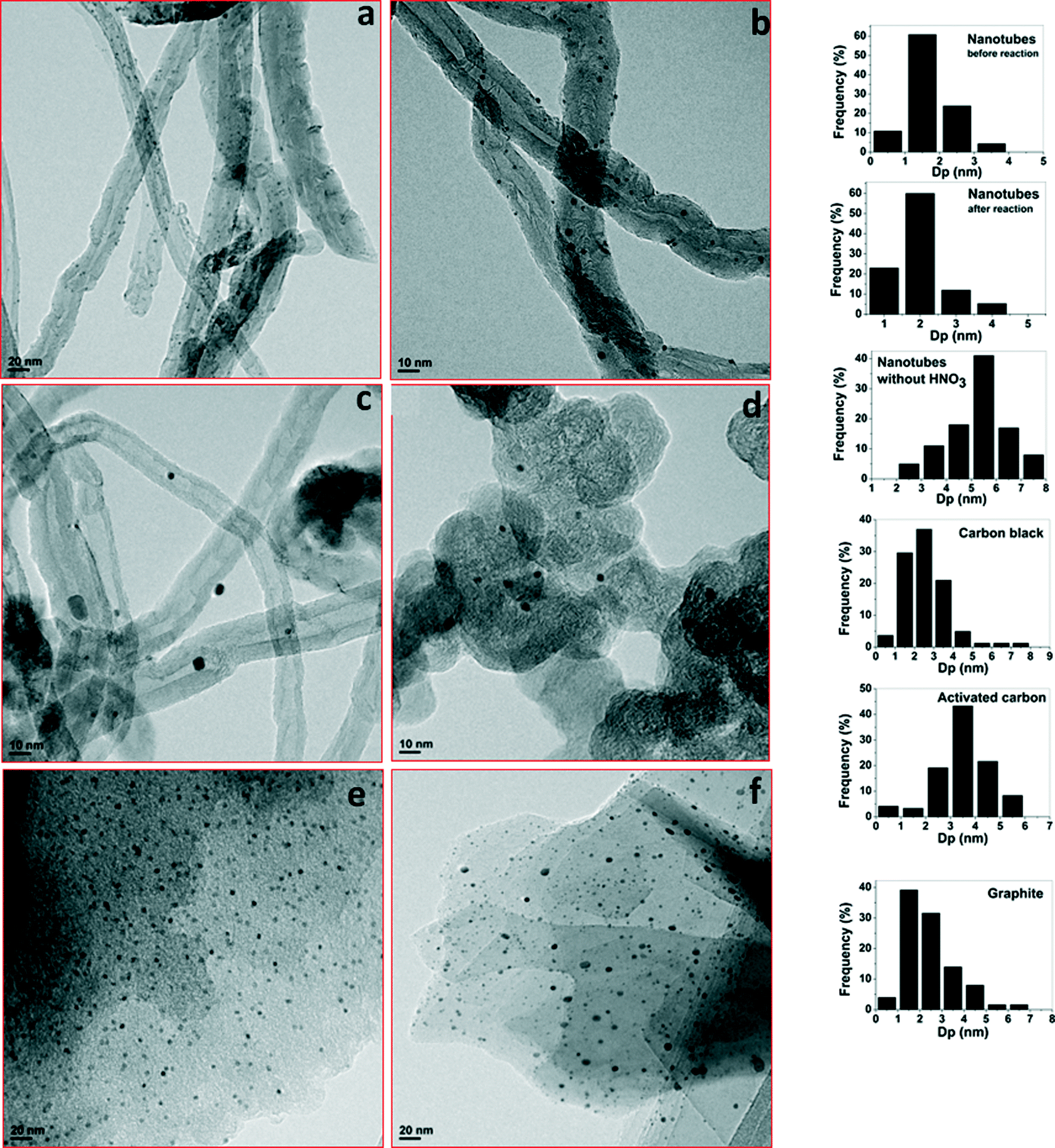
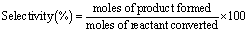 | (2) |
 | (3) |
3. Results and discussion
3.1. Structural studies
Table 1 summarizes the results of textural analysis of HNO3 treated and non-acid treated Pd/CNTs. There was no dramatic change observed in the textural properties of CNTs after HNO3 treatment. The BET surface area and microporous volume of the sample are slightly modified after the acid pretreatment which is probably due to the removal of impurities blocking the pore channels of CNTs. Fig. 1 shows the XRD patterns of various Pd-supported catalysts. A broad diffraction peak at 31° was observed for all carbon-supported catalysts which is a characteristic peak of “graphitic” carbon.36 Two specific features of graphitic carbon were observed at 31° and 64.6° for the graphite-supported catalyst, while a broad peak was observed at 31° for all other carbon-supported catalysts. The diffraction peaks at 46.9°, 54.7°, and 81.1° are indexed to the palladium face-centered cubic (fcc) phase indicating the formation of metallic Pd particles. The crystalline size of Pd was calculated using the Scherrer formula and found to be in the range of 4–6 nm. Since the crystallite size calculated using the Scherrer formula is influenced by many factors and may be misleading for nanoparticles, the particle sizes were also measured by TEM.37| Catalyst | Pd loading (%)a | Pore volume (cm3 g−1) | Pd cluster size (nm)b | Surface area (m2 g−1) | CO/CO2 ratioc | Pd0 (%)d |
|---|---|---|---|---|---|---|
| a Determined by ICP. b Average particle size observed by TEM. c Measured by TPD. d Measured by XPS. | ||||||
| CNTs | 2.39 | 0.553 | 1.5 | 150 | 0.99 | 85–90 |
| CNTs without HNO3 | 2.39 | 0.5 | 5.5 | 131 | 3.27 | 75 |
| Carbon black | 2.84 | 0.026 | 2.6 | 160 | 2.07 | 80 |
| Graphite | 2.87 | — | 1.5–2.5 | 5 | 2.21 | 85 |
| Activated carbon | 3.08 | 0.632 | 3.5 | 1551 | 1.73 | 80 |
 | ||
| Fig. 1 XRD patterns of the various Pd-supported catalysts. | ||
Fig. 2(a) and (b) display the TEM images of acid treated and non-acid treated Pd/CNTs along with their particle size distributions. It can be seen from the TEM images that the Pd nanoparticles are spherical and uniformly distributed over acid treated Pd/CNTs. The particle size distribution shows that the monodispersed ultrafine Pd particles are deposited on CNTs. The average size of the Pd nanoparticles was found to be 1.5 nm for the acid pretreated sample. The comparison between acid treated and non-acid treated Pd catalysts were also performed in order to understand the effect of acid functionalization on the particle size distribution. It has been observed from the TEM images that there was a significant difference in the Pd particle size between the acid treated and the non-acid treated samples. The relatively large and less dispersed Pd clusters were observed on the surface of CNTs without HNO3 pretreatment. The average size of the Pd particles was found to be 5.5 nm for the non-acid treated sample. The XPS results (see Fig. 4) suggest that the surface carboxylic acid groups were generated during acid pretreatment and these sites probably act as the anchoring sites which prevent agglomeration of Pd nanoparticles. It is well-known that the oxidizing treatments can functionalize the carbon surface mainly through the formation of carboxylic acids and phenolic groups. These functional groups often decrease the hydrophobicity of the support and increase the adsorption capacity of organic compounds.38,39 Since the Pd nanoparticles were uniformly dispersed over Pd/CNTs, it seems that the surface carboxylic acid groups are homogeneously formed throughout the surface of CNTs. The hydrogen ion of the carboxyl group can exchange with various kinds of metal cation complexes. It has been reported that there is a strong correlation between the Pt deposited and the concentration of surface carboxylic groups on Pt/carbon black.40 In the present study, the interaction between the surface carboxylic acid groups and Pd precursor are more likely to affect the dispersion of the Pd nanoparticles over CNTs and this is indeed observed after acid pretreatment. Therefore, the acid pretreatment in liquid phase favors the formation of carboxylic groups which control and stabilize the growth of Pd nanoparticles on CNTs.
 | ||
| Fig. 2 Bright field images and particle size distribution for various Pd-supported catalysts: (a) nanotubes with HNO3 pretreatment, (b) nanotubes after the reaction (with HNO3 pretreatment) (c) nanotubes without HNO3 pretreatment, (d) carbon black (e) activated carbon, and (f) graphite. | ||
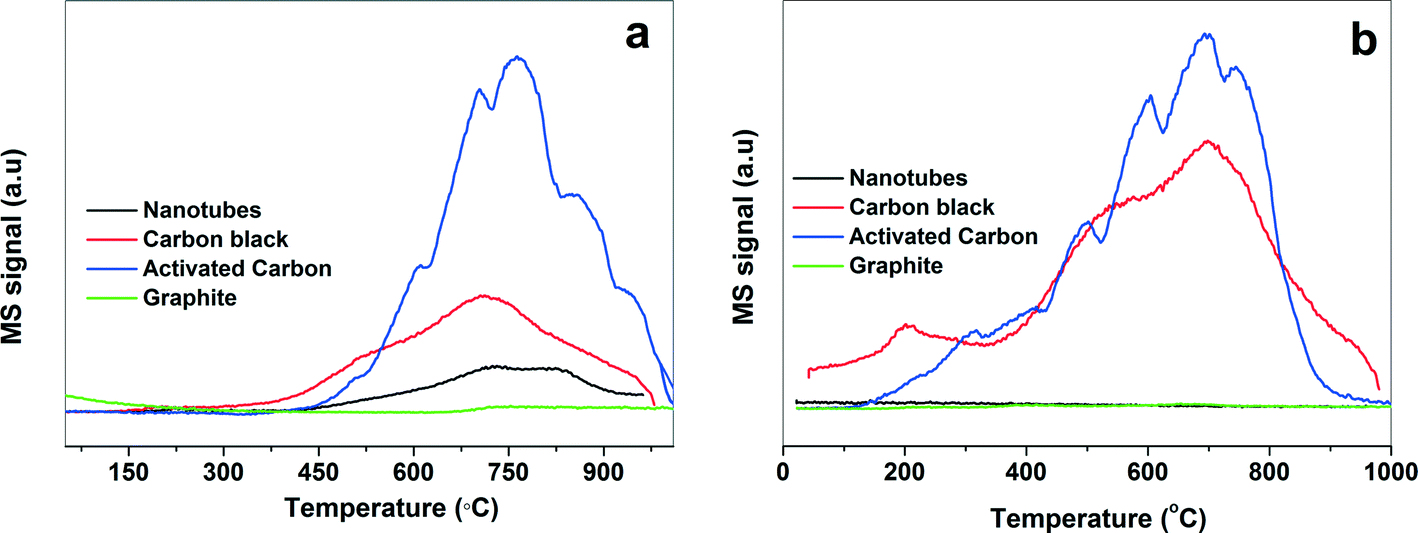 | ||
| Fig. 3 TPD for (a) CO and (b) CO2 over various Pd-supported catalysts. | ||
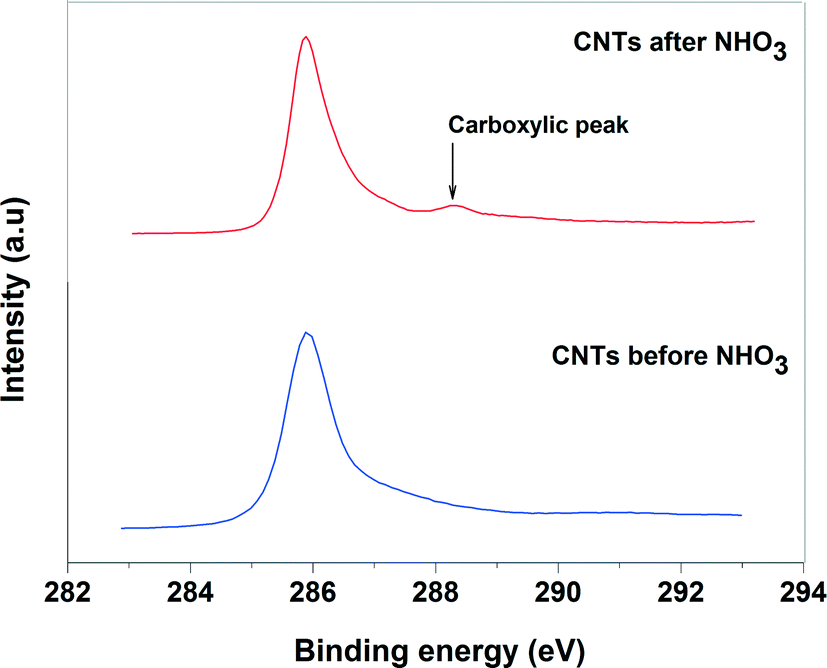 | ||
| Fig. 4 XPS spectra of C 1s for CNTs before and after HNO3 treatment. | ||
In order to emphasize the importance of the artificially generated functional groups in stabilizing Pd nanoparticles, the functionalization was also extended to synthesize Pd supported on various carbon forms. Fig. 2(c)–(e) display the TEM images of Pd supported on various forms of carbon along with their particle size distributions. All catalysts exhibited very narrow particle size distribution irrespective of the support used. The average size of Pd nanoparticle for various catalysts is given in Table 1. The average particle size of Pd supported on nanotubes, carbon black, and graphite are similar and smaller than the average size of Pd supported on activated carbon. This indicates that there is no correlation between the particle size and surface area of the support but rather the nature of the surface functional groups on the support influences the particle sizes and their distribution.41–43 Therefore, a simple impregnation–reduction method with the functionalization of the carbon support can effectively be used to synthesize ultrafine Pd particles on the carbon supports.
In general, the acid treatment introduces the oxygen/nitrogen functionality which improves the metal–support interactions and hence the activity and stability of the catalyst.44 There is a correlation between surface oxygen availability and activity. The higher the surface oxygen content, the higher the activity of the catalyst. Therefore, TPD was used to study the surface chemistry of carbon supports45–47 by monitoring the CO and CO2 desorption as a function of the temperature. It also provides the information about the type and amount of O-containing functional groups present on the carbon support.45–47 The origin of CO2 evolution at low temperature (150–450 °C) is due to the decomposition of carboxylic acids, while at high temperature (600–800 °C) it is due to the decomposition of lactones. The decomposition of carboxylic anhydrides produce both CO and CO2 (400–650 °C), whereas the decomposition of phenols (600–800 °C), carbonyls and quinones (750–1000 °C) result in the formation of CO.45–47 The TPD profiles for CO and CO2 for the various Pd-supported catalysts are shown in Fig. 3. The area under the curve of CO and CO2 were used to calculate the amount of oxygen-containing functional groups and it follows the order: activated carbon > carbon black > nanotubes > graphite. In the case of graphite, very small peaks for CO and CO2 were observed indicating that the concentration of oxygen-containing functional groups is very small. In case of the nanotubes, a small CO desorption peak between 400 and 900 °C indicated the existence of phenol and carbonyl/quinone functional groups on the surface. The CO peak observed between 350 °C and 940 °C for carbon black corresponds to the decomposition of anhydrides and carbonyls/quinones and the weak CO2 signal at 610 °C and 860 °C is assigned to the dissociation of lactones. For the activated carbon, the CO desorption peak above 750 °C is due to the presence of the carbonyl/quinone groups, while the CO2 desorption below 400 °C is attributed to the presence of carboxylic acids. The simultaneous desorption of CO and CO2 around 700 °C also indicates the presence of the carboxylic anhydrides. However, the presence of CO2 desorption peak around 600 °C shows that the contribution due to some lactones cannot be ruled out. The ratio of CO/CO2 is usually used as an indicator of acid–base properties of the carbonaceous material.48,49 A high CO/CO2 ratio indicates a higher basic character of the carbon support. Table 1 shows that the nanotubes are more acidic while the graphite is the least acidic in nature.
The XPS was used to identify the nature of functional groups on acid pretreatment and the oxidation state of Pd-supported catalysts. Fig. 4 shows the C 1s spectra of the acid pretreated and non-acid pretreated CNTs. For the acid pretreated sample, a small peak at 290 eV was observed in addition to the main peak of carbon at 284.6 eV. This small peak can be assigned to the oxygen-containing groups such as the carboxylic groups. The absence of such peak for non-acid pretreated CNTs confirms that the oxygen was incorporated into the surface of the oxidized CNTs and the role of the carboxylic groups in the improvement of the Pd dispersion cannot be ruled out.
Fig. 5 shows the XPS spectra of the Pd supported on the various carbon supports before the reaction. The Pd spectra for all catalysts were wide indicating either multiple oxidation states of the Pd species or several different interactions of the oxidized carbon support. The binding energies of Pd 3d observed at 335.5 and 337 eV correspond to metallic Pd0 and PdO species, respectively. The spectra were also deconvoluted to obtain the contribution of the individual oxidation states and the relative concentration of Pd0 for each catalyst is given in Table 1. The small amount of the Pd2+ state was observed for all catalysts before the reaction. The XPS spectra of the catalyst after the reaction were also analyzed to observe if there is any change in the oxidation state of Pd. The XPS spectra of the used catalyst show that there is a large amount of Pd2+ (~67%) present after the reaction. However, the activity of the used catalyst (without washing of the catalyst) is very similar to that of the fresh catalyst. Therefore, the observed difference in the activity of catalysts cannot be attributed to the oxidation state of Pd.
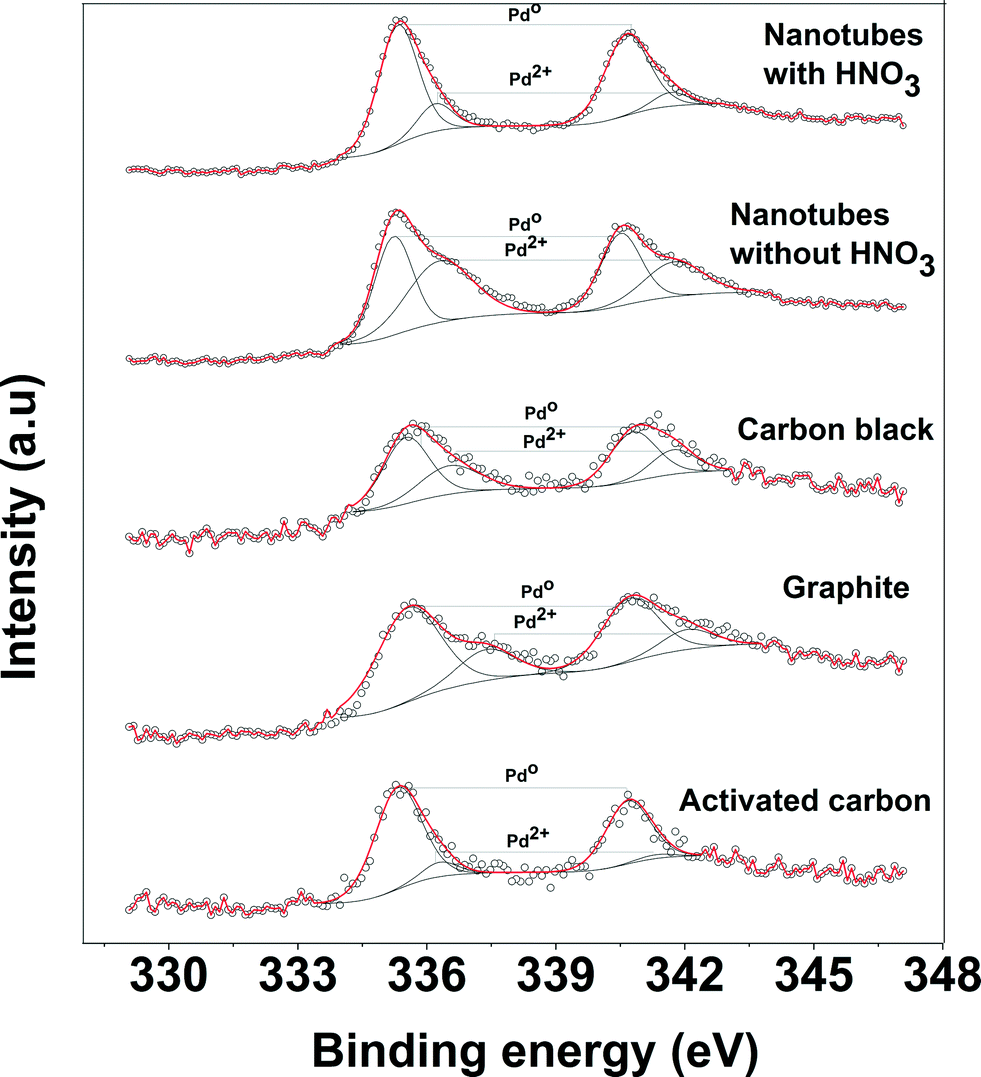 | ||
| Fig. 5 XPS spectra of Pd 3d for various Pd-supported catalysts. | ||
It is well-known that the highly dispersed Pd0 nanoparticles are susceptible to re-oxidation on contact with air (or oxygen).50 The XPS measurement was performed to determine the oxidation state of Pd in the samples after reduction at 350 °C and subsequent storage in air at room temperature. The deconvolutions of the XPS spectra show that 75% of Pd was present as Pd0 in the non-acid treated Pd/CNT sample, whereas 85–90% of Pd was present as Pd0 in the HNO3 pretreated Pd/CNTs sample. The high amount of metallic Pd in the acid treated Pd/CNTs is also consistent with the highly dispersed nanoparticles observed by TEM. Thus, the XPS results show that the introduction of oxygen-containing functional groups on the carbon support stabilize the Pd nanoparticles and prevent them from oxidation upon exposure to the air at room temperature. The selected preparation method leads to reproducible, almost monodisperse Pd nanoparticles with an average particles size of 1.5 nm.
3.2. Alcohol oxidation activity
The evolution of the catalytic performance as a function of time was studied over Pd supported on various carbon-supported catalysts and the online profiles of benzyl alcohol conversion are depicted in Fig. 6. The blank experiments without Pd loading exhibits a negligible benzyl alcohol conversion (<7% at 90 °C and 6 h reaction time) for all studied carbon supports. However, in the presence of the Pd catalyst, benzyl alcohol conversion increases monotonically with time. The benzyl alcohol conversion over nanotubes, carbon black, and activated carbon reaches nearly complete conversion within 2 h, while the conversion over the graphite-supported catalyst increases continuously (50% conversion after 6 h) with time. This shows that the Pd/CNT-supported catalyst was highly active, while the Pd/graphite-supported catalyst was least active under similar experimental conditions. Table 2 shows the catalytic performance of various Pd-supported catalysts in the selective oxidation of benzyl alcohol. The order of the reaction rate is: nanotubes > carbon black > activated carbon > graphite. The final product mixtures, for all catalysts, consist of benzaldehyde (>89%), toluene (<10%) and minor side products (<1%) such as benzene, benzoic acid and benzyl ester for all catalysts. The main oxidation product, benzaldehyde, is formed by oxidation of benzyl alcohol and the major side product, toluene, is mainly initially formed by disproportionation of benzyl alcohol into toluene and benzaldehyde. It should be noted that during the experiment, toluene is oxidized to benzyl alcohol at a much slower rate than that of oxidation of benzyl alcohol to benzaldehyde. The selectivity to benzaldehyde is high for all catalysts and it slightly decreases with time due to subsequent oxidation of benzaldehyde into benzoic acid. The highest production rate of benzaldehyde was observed for CNTs, while a low rate of benzyl alcohol was found for the graphite catalyst. The present finding contradicts the results reported in the literature where it has been reported that the Pd/CNTs showed lower activity than that of Pd/activated carbon which was attributed to the lower metal dispersion on CNTs.44 However, our results demonstrate that it is possible to tune the surface chemistry of the support by specific chemical treatment and, thereby, change the activity of the catalytic surface.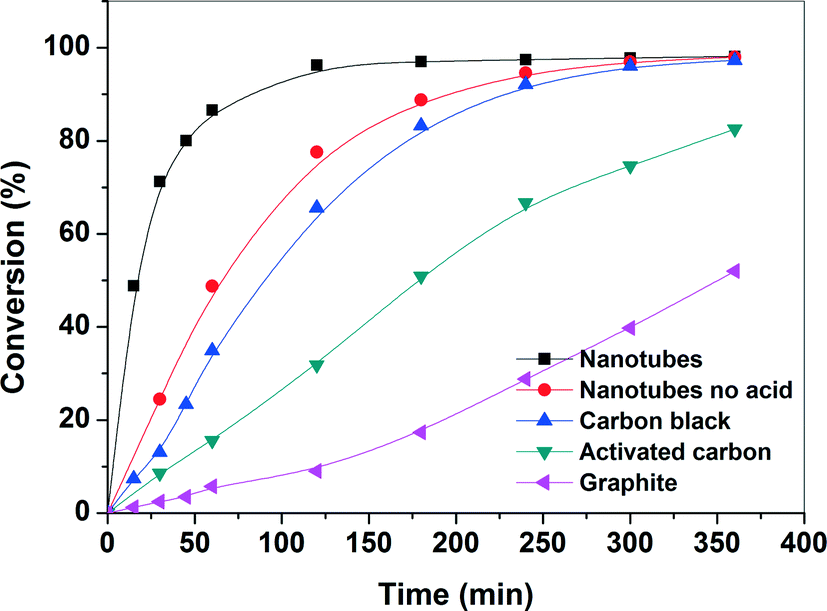 | ||
| Fig. 6 Conversion profile for benzyl oxidation with various Pd-supported catalysts. | ||
| Catalyst | Rate of reaction (mol g−1 of Pd min−1) | Conversion (%) | TOFa | Aldehyde selectivity (%) | Toluene selectivity (%) |
|---|---|---|---|---|---|
| Reaction conditions: 300 mg of catalyst; 2.4 g of benzyl alcohol in 50 mL of xylene; air flow rate of 100 mL min−1; 90 °C; 6 h reaction time; carbon balance 98.5%.3.a TOF was calculated after 15 min. | |||||
| Pd/CNTs | 1.618 | 98 | 2552 | 89 | 8 |
| Pd/CNTs without HNO3 | 0.422 | 95 | 1256 | 88 | 7 |
| Pd/carbon black | 0.227 | 97 | 628 | 89 | 6 |
| Pd/activated carbon | 0.073 | 83 | 445 | 92 | 5 |
| Pd/graphite | 0.044 | 51 | 125 | 96 | 4 |
In order to understand the high catalytic activity of the CNTs, several factors need to be considered. Firstly, the effect of support material on particle size distribution and/or the mode of anchoring of Pd nanoparticles onto the support should be taken into account. The TEM images and the particle size distribution data show that there is no large difference in sizes of the Pd nanoparticles, which is in the range from 1.5 to 4 nm. Despite having similar metal loading and Pd particle size, particularly for carbon black and activated carbon-supported catalysts, a large variation in the catalytic activity for the oxidation of benzyl alcohol was observed. The slight variation in overall particle-size distribution cannot be accounted for the observed catalytic behavior. Further, the TEM images also show that the dispersion of Pd nanoparticles over the activated carbon and graphite supports is relatively higher than that of the CNTs. However, their catalytic performance for the benzyl oxidation reaction is very poor compared to that of Pd/CNT catalyst. The enhanced activity of Pd/CNTs could be attributed to a unique structure of CNTs which significantly enhanced the diffusion and the accessibility of the reactants to the catalytic sites. Secondly, the presence of the surface oxygen species, such as carboxylic acid and phenolic groups on the carbon support could be considered because the catalytic activities of the carbon support are closely related to the presence of such oxygen-containing species. Frequently, the availability of the surface oxygen species is used to explain a superior catalytic performance of carbon-supported catalysts.51,52 Our TPD results showed that the total amount of O-containing functional groups for CNTs is much less than that of the activated carbon. This observation is, however, also not in line with the activity order implying that the amount of oxygen-containing functional groups are not the determining factor for the activity of Pd-supported CNT catalyst. Nevertheless, the role of O-functionality such as carboxylic acid groups cannot be ruled out in improving the metal dispersion and the stabilization of the nanoparticles. In order to understand the role of the carboxylic acid groups, Pd/CNT catalysts were also synthesized without nitric acid pretreatment. The Pd/CNTs without acid pretreatment showed lower reaction rate than those of the acid treated Pd/CNT catalysts (see Fig. 6 and Table 2). This indicates that the O-containing functional groups, mainly carboxylic groups, render a strong metal–support interaction which increases the dispersion of Pd nanoparticles. Therefore, the physical and chemical properties of the carbon support play an important role in determining the catalytic activity. From the activity trend observed for the various Pd-supported carbon catalysts, it is expected that several interlocked factors such as structure and size, support and steric hindrance for reactants determine the overall catalytic activity53 and contribution due of each individual factor is difficult to analyze. Thus, the higher activity of CNTs can be collectively attributed mainly to the higher accessibility of CNTs in combination with the narrow range Pd nanoparticles.
3.3. Reusability of the catalyst
The recycling of the catalyst is very important for industrial application. If the active sites are not properly anchored onto the support, they leach during the reaction, resulting in loss of activity in the subsequent runs. In addition, a strong inhibition due to adsorption of the products on the surface of the catalyst was observed in our previous study and the catalyst showed no activity in subsequent runs.54 Therefore, the reusability of the catalyst for the aerobic oxidation of benzyl alcohol was evaluated, and the results are shown in Fig. 7(a). The catalyst was reused with and without solvent washing. After completion of the reaction, fresh benzyl alcohol was injected into the reaction mixture (without washing of the catalyst). Similar conversion of benzyl alcohol and yield of benzaldehyde were observed with the absence of the initial disproportionation of benzyl alcohol into toluene and benzaldehyde. This confirms that the inhibition due to the products is most likely absent. Furthermore, in order to check the leaching of Pd (if any) in the liquid phase, the catalyst from the previous run was washed with xylene and dried in vacuum at room temperature for 6 h before the next run (washing of the catalyst). A similar reaction rate for benzyl alcohol up to ~98% conversion was observed. This shows a strong metal–support interaction which prevents the leaching of Pd nanoparticles during the reaction. A long term recyclability of the catalyst was also investigated for six successive runs. Each time the fresh benzyl alcohol was injected into the reaction mixture after the completion of reaction without washing of the catalyst. The catalyst exhibited high selectivity for benzyl alcohol oxidation even after the fifth run. However, there is a minor decrease in the initial activity after the catalyst in the fifth run (Fig. 7c). Nevertheless, it is interesting to note that the catalyst showed more than 90% conversion after 4 h in all runs. Further, the catalyst was also regenerated to understand loss of initial activity. The used catalyst was calcined at 300 °C and reused again under similar experimental conditions. The catalyst showed an increase in the activity in the sixth run compared to that of the fifth run. However, the activity of the catalyst in the sixth run was still in some extent lower than that of the fresh catalyst. The increase in the activity of the catalyst in the sixth run (after calcination) confirms that the organic species were adsorbed onto the surface of catalyst thereby inhibiting the active sites during recycle test. Thus, the calcination treatment has a positive effect on the regeneration of the catalyst, as the active sites were cleaned by removal of the strongly adsorbed species. Furthermore, the TEM analysis of spent catalyst shows that the size and particle size distribution were similar to that of the fresh catalyst and no aggregation of the Pd nanoparticles were observed after the reaction (Fig. 2b). Therefore, the above results demonstrate that Pd/CNTs is a promising catalytic material in the benzyl alcohol oxidation reaction with high activity, selectivity, and stability.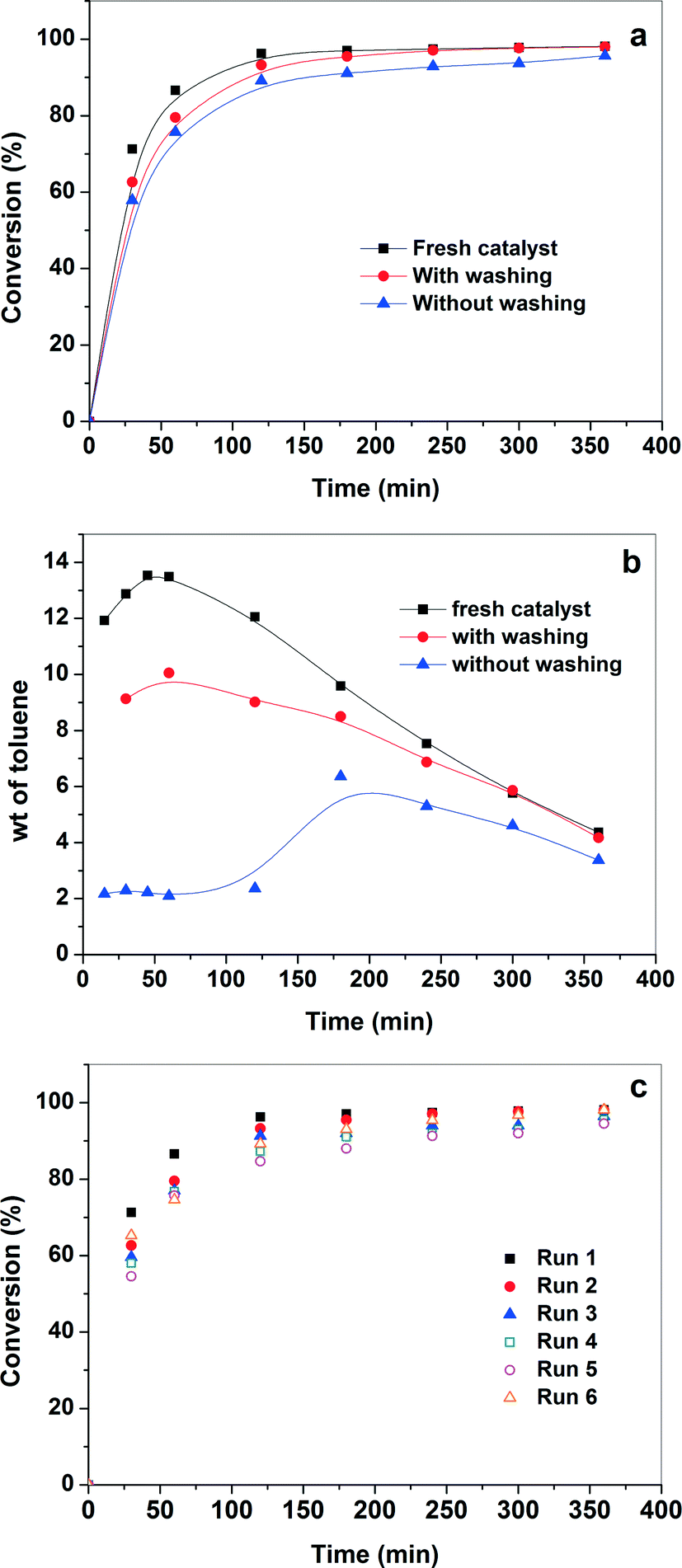 | ||
| Fig. 7 (a) Catalytic activity and (b) selectivity for Pd/CNTs with and without washing of catalyst by solvent (c) catalytic activity for six consecutive cycles. | ||
The selectivity to toluene for each run is also plotted in Fig. 7(c) to understand if the nature of the active sites changes in the subsequent runs. Fig. 7(c) shows that the initial rate of disproportionation of benzyl alcohol is high and it goes through a maximum. The maximum in toluene concentration is explained by further oxidation of toluene into benzyl alcohol and benzyl aldehyde. It also should be noted that the rate of disproportionation of benzyl alcohol for fresh catalyst is higher than the used catalyst (with washing). However, the rate of benzaldehyde formation is similar for both the catalysts (not shown). This means that there are different active sites for the disproportionation and the oxidation reactions. The disproportionation sites are easily poisoned by the presence of the products. This observation is further confirmed by the fact that practically no toluene was observed initially over the used catalyst (without washing). To distinguish between the nature of these two active sites in the present study is not possible and was not a part of the scope of this investigation. The understanding of the nature of these sites helps to develop a more active, selective, and stable catalyst for the selective oxidation of benzyl alcohol into benzaldehyde.
4. Conclusions
A controlled design of self-assembling Pd nanoparticles on the surface of functionalized CNTs without a capping agent is reported. The synthesized Pd/CNTs has a uniform dispersion and narrow size distribution of 1.5 nm. The synthesized Pd/CNTs show an excellent catalytic activity towards the selective oxidation of benzyl alcohol which is collectively attributed to a highly dispersed, homogeneous and nanosized Pd nanoparticles on CNTs. In addition, the peculiar morphology of the carbon nanotubes also significantly improves the diffusion of the reactant and product(s) and the accessibility to the catalytic sites. The complete reusability of the catalyst without Pd leaching shows that the functionalization of the carbon support rendered a strong metal–support interaction. Meanwhile, this synthesis approach was not limited only to the structural form of carbon (nanotube), but it can also be used to synthesize a wide variety of carbon-supported catalysts with controlled Pd nanoparticle size distribution. The catalytic activities significantly depend on the nature of the supports in the selective aerobic oxidation of benzyl alcohol, which followed the order: nanotubes > carbon black > activated carbon > graphite with high selectivity to benzaldehyde (>89%). Further, this study showed that there is no correlation between the amount of oxygen-containing functional groups and the activity of Pd-supported catalysts. The excellent activity and reusability of Pd/CNTs, demonstrated in this study, make it a promising candidate for the selective oxidation of benzyl alcohol. On the fresh and thoroughly washed catalysts a significant disproportionation was observed, indicating a two-site reaction mechanism, one of disproportionation and one of oxidation. The disproportionation sites are very rapidly deactivated by the reaction products.Acknowledgements
The authors thank Mrs. Willy Rook for the BET surface measurements and CO chemisorption studies, Kevin Mouthaan for GC analyses, and Bart van der Linden for TPD studies.References
- Y. Xing, J. Phys. Chem. B, 2004, 108, 19255–19259 CrossRef CAS.
- C. C. de Paula, A. Garcia Ramos, A. C. da Silva, E. Cocchieri Botelho and M. C. Rezende, Carbon, 2002, 40, 787–788 CrossRef CAS.
- B. Yoon and C. M. Wai, J. Am. Chem. Soc., 2005, 127, 17174–17175 CrossRef CAS PubMed.
- J.-P. Tessonnier, L. Pesant, G. Ehret, M. J. Ledoux and C. Pham-Huu, Appl. Catal., A, 2005, 288, 203–210 CrossRef CAS PubMed.
- Y. Zhang, Y. Liu, G. Yang, Y. Endo and N. Tsubaki, Catal. Today, 2009, 142, 85–89 CrossRef CAS PubMed.
- Z. Jian, P. Liu, F. Li, P. He, X. Guo, M. Chen and H. Zhou, Angew. Chem., Int. Ed., 2014, 53, 442–446 CrossRef CAS PubMed.
- S. Akbayrak and S. Özkar, ACS Appl. Mater. Interfaces, 2012, 4, 6302–6310 CAS.
- C. Chen, J. Zhang, B. Zhang, C. Yu, F. Peng and D. Su, Chem. Commun., 2013, 49, 8151–8153 RSC.
- R. Singh and A. Singh, Carbon, 2009, 47, 271–278 CrossRef CAS PubMed.
- J. Xu and T. Zhao, J. Power Sources, 2010, 195, 1071–1075 CrossRef CAS PubMed.
- T. Koh, H. M. Koo, T. Yu, B. Lim and J. W. Bae, ACS Catal., 2014, 4, 1054–1060 CrossRef CAS.
- A. Tavasoli, R. M. M. Abbaslou, M. Trepanier and A. K. Dalai, Appl. Catal., A, 2008, 345, 134–142 CrossRef CAS PubMed.
- S. Wang, X. Wang and S. P. Jiang, Langmuir, 2008, 24, 10505–10512 CrossRef CAS PubMed.
- T. Fujigaya and N. Nakashima, Adv. Mater., 2013, 25, 1666–1681 CrossRef CAS PubMed.
- S.-A. Park, D.-S. Kim, T.-J. Kim and Y.-T. Kim, ACS Catal., 2013, 3, 3067–3074 CrossRef CAS.
- X. Chen, Y. Hou, H. Wang, Y. Cao and J. He, J. Phys. Chem. C, 2008, 112, 8172–8176 CAS.
- A. Quintanilla, V. Butselaar-Orthlieb, C. Kwakernaak, W. Sloof, M. Kreutzer and F. Kapteijn, J. Catal., 2010, 271, 104–114 CrossRef CAS PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- Z.-P. Sun, X.-G. Zhang, H. Tong, R.-L. Xue, Y.-Y. Liang and H.-L. Li, Appl. Surf. Sci., 2009, 256, 33–38 CrossRef CAS PubMed.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS PubMed.
- W. Li, X. Zhao and A. Manthiram, J. Mater. Chem. A, 2014, 2, 3468–3476 CAS.
- V. Lordi, N. Yao and J. Wei, Chem. Mater., 2001, 13, 733–737 CrossRef CAS.
- R. Yu, L. Chen, Q. Liu, J. Lin, K.-L. Tan, S. C. Ng, H. S. Chan, G.-Q. Xu and T. A. Hor, Chem. Mater., 1998, 10, 718–722 CrossRef CAS.
- W. X. Chen, J. Y. Lee and J. Z. Liu, Chem. Commun., 2002, 2588–2589 RSC.
- Z. Liu, L. Hong, M. P. Tham, T. H. Lim and H. Jiang, J. Power Sources, 2006, 161, 831–835 CrossRef CAS PubMed.
- C. Cui, L. Gan, M. Neumann, M. Heggen, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 4813–4816 CrossRef CAS PubMed.
- J. Planeix, N. Coustel, B. Coq, V. Brotons, P. Kumbhar, R. Dutartre, P. Geneste, P. Bernier and P. Ajayan, J. Am. Chem. Soc., 1994, 116, 7935–7936 CrossRef CAS.
- X. Wang, G. Wu, N. Guan and L. Li, Appl. Catal., B, 2012, 115–116, 7–15 CrossRef CAS PubMed.
- A. Abad, A. Corma and H. Garcia, Chem. – Eur. J., 2008, 14, 212–222 CrossRef CAS PubMed.
- C. Zhou, Y. Chen, Z. Guo, X. Wang and Y. Yang, Chem. Commun., 2011, 47, 7473–7475 RSC.
- A. Villa, M. Plebani, M. Schiavoni, C. Milone, E. Piperopoulos, S. Galvagno and L. Prati, Catal. Today, 2012, 186, 76–82 CrossRef CAS PubMed.
- J. Feng, C. Ma, P. Miedziak, J. K. Edwards, G. L. Brett, D. Li, Y. Du, D. J. Morgan and G. J. Hutchings, Dalton Trans., 2013, 42, 14498–14508 RSC.
- N. Dimitratos, A. Villa, D. Wang, F. Porta, D. Su and L. Prati, J. Catal., 2006, 244, 113–121 CrossRef CAS PubMed.
- A. Villa, N. Janjic, P. Spontoni, D. Wang, D. S. Su and L. Prati, Appl. Catal., A, 2009, 364, 221–228 CrossRef CAS PubMed.
- X. Yu, Y. Huo, J. Yang, S. Chang, Y. Ma and W. Huang, Appl. Surf. Sci., 2013, 280, 450–455 CrossRef CAS PubMed.
- E. Antolini, F. Cardellini, L. Giorgi and E. Passalacqua, J. Mater. Sci. Lett., 2000, 19, 2099–2103 CrossRef CAS.
- J. Rangel-Mendez and M. Streat, Water Res., 2002, 36, 1244–1252 CrossRef CAS.
- Y. Li, C. Lee and B. Gullett, Fuel, 2003, 82, 451–457 CrossRef CAS.
- K. Yasuda and Y. Nishimura, Mater. Chem. Phys., 2003, 82, 921–928 CrossRef CAS PubMed.
- A. Villa, G. M. Veith, D. Ferri, A. Weidenkaff, K. A. Perry, S. Campisi and L. Prati, Catal. Sci. Technol., 2013, 3, 394–399 CAS.
- E. G. Rodrigues, S. A. Carabineiro, J. J. Delgado, X. Chen, M. F. Pereira and J. J. Órfão, J. Catal., 2012, 285, 83–91 CrossRef CAS PubMed.
- N. E. Kolli, L. Delannoy and C. Louis, J. Catal., 2013, 297, 79–92 CrossRef CAS PubMed.
- A. Villa, D. Wang, P. Spontoni, R. Arrigo, D. Su and L. Prati, Catal. Today, 2010, 157, 89–93 CrossRef CAS PubMed.
- J. L. Figueiredo, M. F. Pereira, M. M. Freitas and J. J. Órfão, Ind. Eng. Chem. Res., 2007, 46, 4110–4115 CrossRef CAS.
- E. G. Rodrigues, M. F. Pereira, X. Chen, J. J. Delgado and J. J. Órfão, J. Catal., 2011, 281, 119–127 CrossRef CAS PubMed.
- J. Figueiredo, M. Pereira, M. Freitas and J. Orfao, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- Y.-C. Chiang, W.-H. Lin and Y.-C. Chang, Appl. Surf. Sci., 2011, 257, 2401–2410 CrossRef CAS PubMed.
- C. Moreno-Castilla, M. Lopez-Ramon and F. Carrasco-Marın, Carbon, 2000, 38, 1995–2001 CrossRef CAS.
- A. F. Lee, S. F. Hackett, J. S. Hargreaves and K. Wilson, Green Chem., 2006, 8, 549–555 RSC.
- J. L. Figueiredo and M. F. R. Pereira, Catal. Today, 2010, 150, 2–7 CrossRef CAS PubMed.
- J. Zhu, S. A. Carabineiro, D. Shan, J. L. Faria, Y. Zhu and J. L. Figueiredo, J. Catal., 2010, 274, 207–214 CrossRef CAS PubMed.
- D. Wang, A. Villa, D. Su, L. Prati and R. Schlögl, ChemCatChem, 2013, 5, 2717–2723 CrossRef CAS PubMed.
- E. Skupien, R. J. Berger, V. P. Santos, J. Gascon, M. Makkee, M. T. Kreutzer, P. J. Kooyman, J. A. Moulijn and F. Kapteijn, Catalysts, 2014, 4, 89–115 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |