
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co-oxidation of octane and benzaldehyde using molecular oxygen with Au–Pd/carbon prepared by sol-immobilisation†
Virginie
Peneau
,
Greg
Shaw
,
Simon J.
Freakley
,
Michael M.
Forde
,
Nikolaos
Dimitratos
,
Robert L.
Jenkins
,
Stuart H.
Taylor
and
Graham J.
Hutchings
*
Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: Hutch@cf.ac.uk; Fax: +44 (0) 2090874 030; Tel: +44 (0) 2920874059
First published on 14th April 2015
Abstract
The selective oxidation of linear alkanes with molecular oxygen under mild conditions remains a challenging topic in the field of catalysis. In this study we investigate the co-oxidation of C–H bonds in substrates with different relative reactivities, the aim being to couple the oxidised products in situ to form the corresponding esters. Initial attempts were made to co-oxidise octane with toluene to form octyl benzoate using Au–Pd catalysts. During the study the oxidation of octane in the presence of benzaldehyde, an oxidation product of toluene, was also investigated in order to demonstrate the potential feasibility of the reaction. This work summarises our attempts to show whether a co-oxidation system could be an effective way to oxidise linear alkanes.
Introduction
The selective oxidation of alkanes to more valuable functionalised products, such as alcohols, aldehydes, carboxylic acids and derivatives has been well studied. C–H bonds in alkanes and aromatic molecules are difficult to activate selectively using only molecular oxygen as the oxidant1,2 without generating CO2. A problem associated with the selective oxidation of primary C–H bonds to oxygenates, e.g. an alcohol, is that the C–H bond in the corresponding alcohol is easier to activate than that of the alkane. Therefore over-oxidation to undesired products remains a problem.3,4 The second challenge is that the primary C–H bond of an alkane is more difficult to activate compared to the secondary C–H bond making terminal oxidation a challenge.One of the most promising routes for alkane oxidation is the use of heterogeneous catalysts in the liquid phase, which can be carried out under relatively mild conditions.5–10 Appreciable alkane oxidation rates have been achieved when using hydrogen peroxide as the oxidant,11–13 however, the use of molecular oxygen or even air is highly desirable due to their lower environmental impact and cost.14 Gold catalysis has shown potential for the oxidation of alcohols, such as benzyl alcohol, and the direct synthesis of hydrogen peroxide from molecular hydrogen and oxygen.15–20 Many studies have shown a synergistic effect upon palladium addition to gold supported catalysts for oxidation reactions. For example, Prati and co-workers observed an improved activity using AuPd bimetallic catalysts rather than the corresponding monometallic catalysts for the oxidation of glycerol. The presence of Pd also shows a difference in the selectivity of the products. Subsequently, the same set of catalysts was investigated for the oxidation of a range of alcohols, revealing a positive synergistic effect for AuPd.21
The preparation of catalysts by sol immobilisation, allows control of the particle diameter between 2 to 4 nm.22–24 These materials have been shown to be highly active for both alcohol oxidation and the primary C–H bond activation in toluene.25 It has been previously shown that Au–Pd catalysts prepared by sol immobilisation on a carbon support are particularly effective for the oxidation of toluene. Kesavan et al. achieved high toluene conversions (94%) and high selectivity towards benzyl benzoate (~95%), at 160 °C in the presence of oxygen with no initiator present.24 The same catalyst was also active for toluene oxidation in the presence of TBHP at 80 °C via a surface bound radical mechanism.26
The AuPd supported catalysts prepared by sol immobilisation are also effective for the oxidation of methane using H2 and O2 to generate in situ hydrogen peroxide under mild conditions, and production of methanol was even observed at 2 °C. Although reported productivities were low, that work showed that it is feasible to activate and oxidise primary C–H bonds using Au–Pd systems.27–30
In the current work, we speculated that the activation of toluene by an AuPd catalyst could initiate the oxidation of the primary C–H bond of octane (used as a model for linear alkanes) in the presence of oxygen or air. Benzaldehyde, an oxidation product of toluene, has previously been used in studies as a sacrificial aldehyde to oxidise various substrates. When used in excess, benzaldehyde is able to initiate the oxidation of other molecules by the formation of a peroxy radical species, which is also an intermediate in the oxidation of benzaldehyde to benzoic acid.31,32 It has been shown that the oxidation of toluene can proceed through a radical mechanism with benzaldehyde as a primary product.33 In principle, the peroxy radical species formed during the oxidation of toluene to benzaldehyde should be able to activate the C–H bonds in octane. The mixture of toluene radical species with octane could form octane-based radical species or coupling products though cross-propagation and cross termination reactions respectively.34
In this study the feasibility of using co-oxidation as a strategy to achieve to oxidation of linear alkanes in the presence of a molecule which is more readily oxidised by molecular oxygen is investigated. Unlike the sacrificial aldehyde system, the co-oxidant used in the current work was introduced in low concentrations in order to utilise it as an initiator for the oxidation reaction.
Experimental
Catalyst preparation
The 0.5 wt% Au–0.5 wt% Pd catalyst supported on carbon was prepared using a sol-immobilisation method. Aqueous solutions of PdCl2 (Johnson Matthey) and HAuCl4·3H2O (Johnson Matthey), of the desired concentration were prepared. Subsequently, polyvinyl alcohol (PVA) solution (1 wt% aqueous solution, Aldrich, MW = 10 kDa, PVA/(Au + Pd) (wt/wt) = 1.2) was freshly prepared and added as a stabilizer. A freshly prepared NaBH4 solution (Sigma Aldrich, 0.1 M aqueous solution, NaBH4/(Au + Pd) (mol/mol) = 5) was added to form a dark-brown sol. After 30 min of sol generation, the colloid was immobilized by adding the carbon (Darco GC60, Sigma Aldrich), acidified to pH 1 with H2SO4 (Fischer Scientific), under vigorous stirring conditions. The amount of support material required was calculated so as to have a total final metal loading of 1 wt%. After 2 h the slurry was filtered, the catalyst washed thoroughly with distilled water and then dried (120 °C, 16 h).Hydrocarbon oxidation
Chemicals used: toluene (>99.9%, Sigma-Aldrich), octane (>99%, Sigma-Aldrich), decane (99+%, Alfa Aesar), dodecane (99+%, Aldrich), cyclooctane (>99%, Aldrich), benzaldehyde (98+%, Acros organic), formaldehyde (37% wt in H2O, Sigma-Aldrich), propanal (97%, Sigma-Aldrich), butanal (redistilled, 99.5%, Aldrich).All reactions were performed in a stainless-steel autoclave (Parr reactor) containing a Teflon liner vessel with total volume of 50 mL (working volume of 35 mL). For all reactions the stirrer was set at 1500 rpm; the reaction mixture was raised to the required temperature and reacted for 20 h unless otherwise stated. Typically, the vessel was charged with an alkane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aldehyde mixture (10 ml, 9:1 molar) and catalyst (50 mg). The autoclave was purged 5 times with nitrogen and 2 times with oxygen leaving the reactor with 20 bar O2 pressure.
:aldehyde mixture (10 ml, 9:1 molar) and catalyst (50 mg). The autoclave was purged 5 times with nitrogen and 2 times with oxygen leaving the reactor with 20 bar O2 pressure.
The subsequent final reaction mixtures were dissolved in 5 ml isopropanol before analysis due to the formation of solid products during reaction. The identification and analysis of the products was carried out using GC-MS (a Waters GCT Premier and a HP 6890 N with a 30 m Agilent DB-5 ms column) for the liquid phase and a GC (Varian 450-GC equipped with FID & TCD detectors, methaniser and CP-SiL5CB column (50 m, 0.33 mm diameter, He carrier gas)) for the liquid and gas phases. For quantification of the amount of reactants (alkanes and aldehydes) consumed an external calibration method was utilised with 1,3,5-trifluorotoluene (Sigma Aldrich) as a standard.
Results and discussion
Initially, the effect of the addition of octane was investigated into a reaction carried out under conditions previously reported for the oxidation of toluene (160 °C, 10 bar O2, 20 h) using a 1% AuPd/C catalyst (0.2 g)24 prepared by sol immobilisation35 was investigated. This catalyst was characterised previously by STEM-HAADF and XPS. Using microscopy the mean particle size was determined to be 3.7 nm with a median size of 3.3 nm. X-ray photoelectron spectroscopy (XPS) analysis of the Au–Pd catalysts immobilized on carbon showed that the surface Pd/Au ratio was 2.1, and confirmed that the Pd was predominantly in the metallic state.24 It was proposed that radical compounds from toluene activation could initiate the activation of octane and, in the presence of oxygen, form oxygenated C8 products. Ketones, aldehydes and alcohols as well as products from C–C scission of octane were expected. Four possible octyl alcohols (octan-1-ol, octan-2-ol, octan-3-ol and octan-4-ol) could be formed from the activation of different C–H bonds in the molecule and subsequent coupling with benzoic acid could form four octyl benzoates (Scheme 1 – shows the desired primary activation and trapping of octane). For the reaction with octane and toluene only, the reaction products from toluene oxidation (benzaldehyde, benzoic acid, and benzyl benzoate) were detected using GCMS, identified by comparison with standards, but not quantified. No oxygenated products were detected as a result of octane oxidation (Fig. S1†). | ||
| Scheme 1 Proposed scheme for the coupling of octane with benzaldehyde. | ||
We revised our initial concept after considering that the rate of oxidation of benzaldehyde, the proposed activator molecule, was probably too fast and so activation of C–H bond in octane had low probability in the toluene–octane system. Therefore used benzaldehyde as a co-oxidant for the oxidation of octane, considering that the formation of peroxy radicals from benzaldehyde oxidation is likely to initiate the activation of octane.
Octane oxidation reactions were performed in the absence of benzaldehyde or catalyst present, at a range of temperatures between 50 and 120 °C under an oxygen pressure of 20 bar. The result (Fig. 1) indicates that the auto-oxidation of octane occurs at temperatures above 80 °C, with no oxidation products detected below this temperature. At 100 °C the conversion of octane is approximately 1%, increasing to 6.4% at 120 °C. At these temperatures cracking products from octane along with the octyl alcohols and ketones were detected. With the addition of benzaldehyde to the reaction we observed the formation of oxidation products even at 50 °C (Fig. 1). At 50 °C, we observed octane conversion of 1.5% of octane, increasing to 6.8% at 120 °C. This shows that the addition of benzaldehyde to the reaction at low temperatures (50 to 80 °C) initiates the activation of octane to form various oxygenated products even in the absence of a catalyst. At higher temperatures (100 to 120 °C), the addition of benzaldehyde suppresses C–C scission and the auto-oxidation of octane.
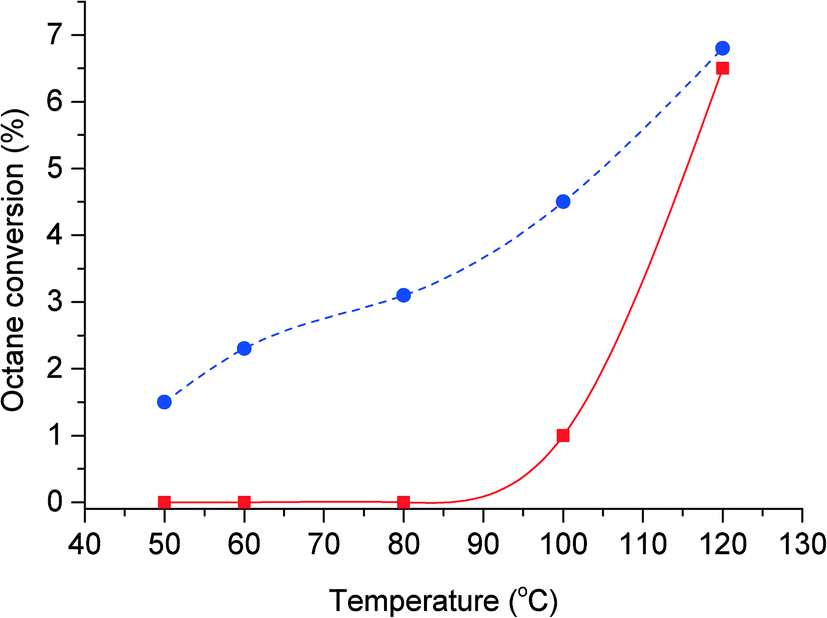 | ||
Fig. 1 Effect of reaction temperature on octane conversion with ( ) and without ( ) and without ( ) benzaldehyde in the absence of catalyst. Reaction conditions: octane (60 mmol), oxygen, ratio octane (mol)/benzaldehyde (mol) = 9:1, no catalyst, 20 h, 1500 rpm. ) benzaldehyde in the absence of catalyst. Reaction conditions: octane (60 mmol), oxygen, ratio octane (mol)/benzaldehyde (mol) = 9:1, no catalyst, 20 h, 1500 rpm. | ||
Fig. 2 shows that the formation of octanol and octanone increases with temperature. In the presence of benzaldehyde, 0.4% octane is converted to octanone/octanol at 50 °C, rising to 3% at 120 °C. The formation of octyl benzoate is first observed at 100 °C in the system and increases with increasing temperature. At lower temperatures the formation of the ester would be slower due to the lower concentration of octanol produced. The presence of benzaldehyde and its oxidation intermediates/products are able to activate octane in the presence of O2. However, selectivities are independent of benzaldehyde. Reaction in the absence of benzaldehyde at 120 °C shows similar selectivities to those in the presence of benzaldehyde for the same temperature and conversion.
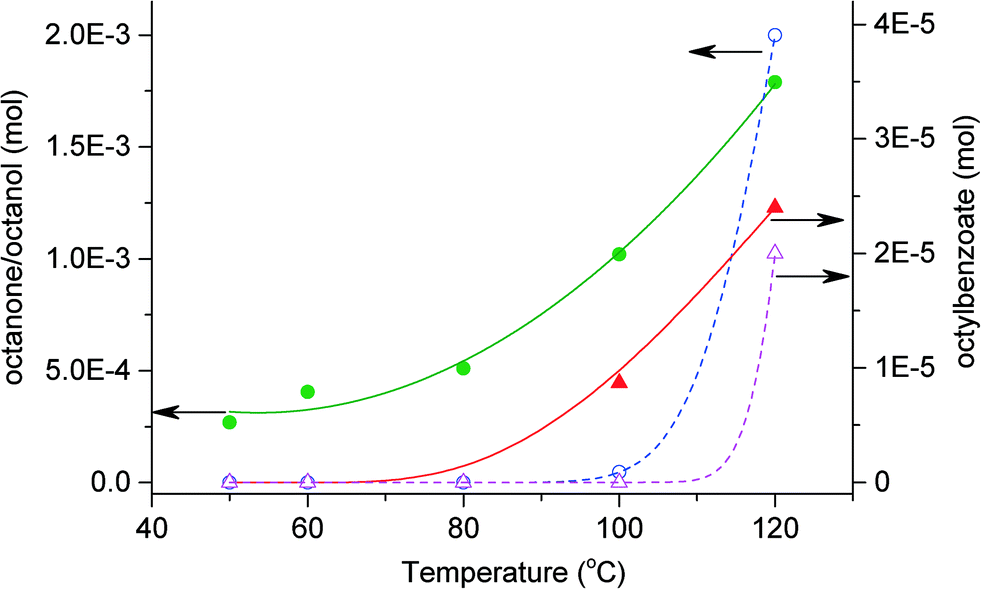 | ||
Fig. 2 Effect of reaction temperature on the oxidation of octane in the presence or absence of benzaldehyde. In the presence of benzaldehyde (full line): octanone–octanol ( ) and octylbenzoate ( ) and octylbenzoate ( ). In the absence of benzaldehyde (dash line): octanone–octanol ( ). In the absence of benzaldehyde (dash line): octanone–octanol ( ) and octylbenzoate ( ) and octylbenzoate ( ). Reaction conditions: octane (60 mmol), oxygen, ratio octane (mol)/benzaldehyde (mol) = 9:1, no catalyst, 20 h, 1500 rpm. ). Reaction conditions: octane (60 mmol), oxygen, ratio octane (mol)/benzaldehyde (mol) = 9:1, no catalyst, 20 h, 1500 rpm. | ||
The oxidation of octane with the addition of benzaldehyde was also performed at 80 °C in the presence of the 1% AuPd/C catalyst. At this temperature without catalyst we observed the formation of oxygenated products, but not the formation of octyl benzoate. The addition of 1% AuPd/C increases the formation of oxygenated products (octanone and octanol) compared to the blank reaction without catalyst. The conversion of octane increases to 5% compared to 3% in the absence of a catalyst and octyl benzoate is formed (Table 1).
:1, 1% AuPd/C (50 mg), time 20 h, 80 °C, 1500 rpm
| Octane conversion (%) | Octylbenzoate selectivity (%) | Octanol/octanone selectivity (%) | |
|---|---|---|---|
| No catalyst | 3.1 | — | 29 |
| 1% AuPd/C | 5 | 0.7 | 58 |
A reaction in the absence of benzaldehyde but with catalyst present was carried out to confirm the role of benzaldehyde. No products were detected, confirming that both benzaldehyde and the catalyst are necessary to oxidise octane at 80 °C. Furthermore, adding a catalyst increases the conversion and more importantly the formation of oxygenated products and octylbenzoate.
The reaction parameters were subsequently investigated, including the effect of variation in catalyst mass, oxygen pressure, reaction time and reaction temperature, in order to gain a greater understanding of this reaction. Typically the standard conditions were: 20 bar O2, 80 °C, 20 h and 50 mg catalyst. The effect of pressure is shown in Fig. 3. When decreasing the pressure to 10 bar the octane conversion remains similar, while at 30 bar the conversion increases to 8.4%. However, at higher pressure (>20 bar) GCMS analysis show the presence of lighter products arising from oxidative breaking of C–C bonds. These products increase while the selectivity to C8 oxygenated products decreased, suggesting that the breaking of C–C bonds occur also for C8 oxygenate products.
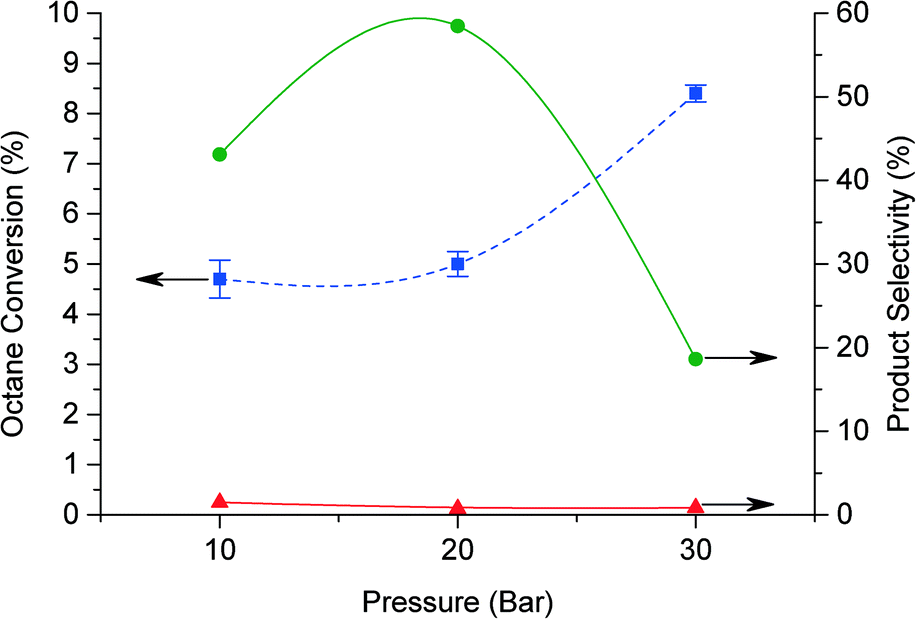 | ||
Fig. 3 Effect of pressure for octane oxidation. Conversion of octane ( ) and product selectivity. Octanone/octanol ( ) and product selectivity. Octanone/octanol ( ) and octylbenzoate ( ) and octylbenzoate ( ). Reaction conditions: octane (60 mmol), oxygen (10 to 30 bar), ratio octane (mol)/benzaldehyde (mol) = 9:1, 1% AuPd/C (50 mg), 20 h, 80 °C, 1500 rpm. ). Reaction conditions: octane (60 mmol), oxygen (10 to 30 bar), ratio octane (mol)/benzaldehyde (mol) = 9:1, 1% AuPd/C (50 mg), 20 h, 80 °C, 1500 rpm. | ||
The conversion of octane increases from 5% to 7.7% as the mass of catalyst is increased from 0.05 g to 0.2 g as shown in Fig. 4. In the absence of catalyst, octanol and octanone are observed with 29% selectivity. The selectivity towards C8 oxygenates increases to 60% on addition of the catalyst. With further addition of catalyst the selectivity decreases, while the formation of lighter alkanes and oxygenated products increases with increasing catalyst mass.
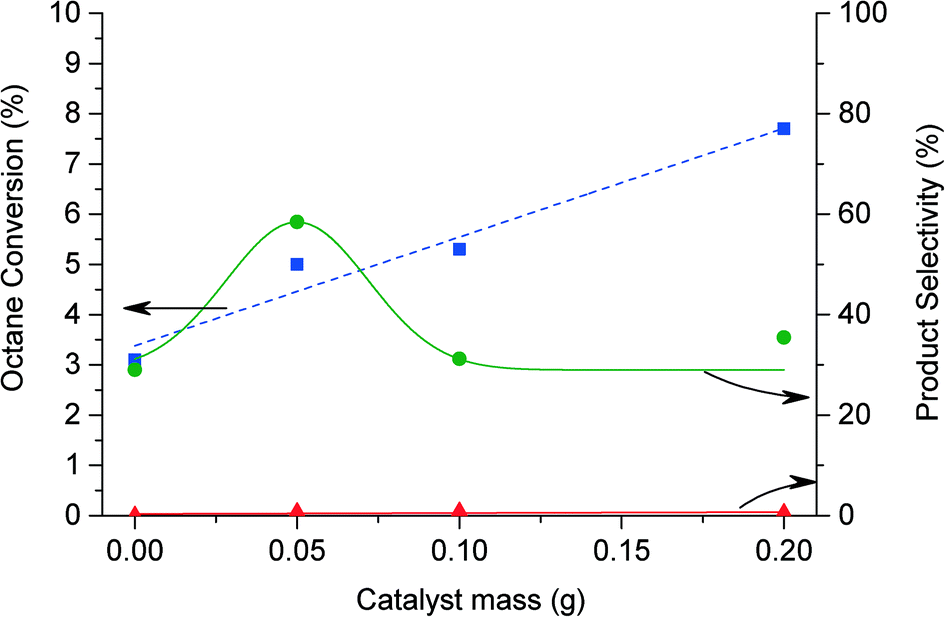 | ||
Fig. 4 Effect of 1% AuPd/C mass for octane oxidation. Conversion of octane ( ) and product selectivity. Octanone/octanol ( ) and product selectivity. Octanone/octanol ( ) and octylbenzoate ( ) and octylbenzoate ( ). Reaction conditions: octane (60 mmol), oxygen, ratio octane (mol)/benzaldehyde (mol) = 9:1, 20 h, 80 °C, 1500 rpm. ). Reaction conditions: octane (60 mmol), oxygen, ratio octane (mol)/benzaldehyde (mol) = 9:1, 20 h, 80 °C, 1500 rpm. | ||
The effect of reaction time was subsequently investigated using a catalyst loading of 50 mg. Fig. 5 shows that octane conversion increased until 48 h reaching a maximum of c.a. 6.5%. The rate of increase of conversion is not linear as reaction times are extended indicating that the rate of reaction is decreasing over time. Interestingly, it is apparent that the change of rate is occurring when benzaldehyde is consumed. This indicates there is an important role of benzaldehyde in the initiation of octane conversion. The formation of octanol, octanone and octylbenzoate increases up to 24 h reaction. Increasing reaction time further decreases the selectivity to octanone and octanol, whilst a greater amount of octylbenzoate is formed. At extended reaction times the amount of cracking products increases relative to the amount of C8 oxygenated products, indicating subsequent cracking of the initially formed C8 oxygenated products. Benzaldehyde conversion and benzoic acid selectivity are also shown at increasing reaction times for these experiments (Fig. 5). The conversion of benzaldehyde reached a maximum of 98% after 7 h reaction, with high selectivity (ca. 80%) to benzoic acid as expected, although 100% selectivity to benzoic acid is not reached as some benzyl species are used to form ester molecules.
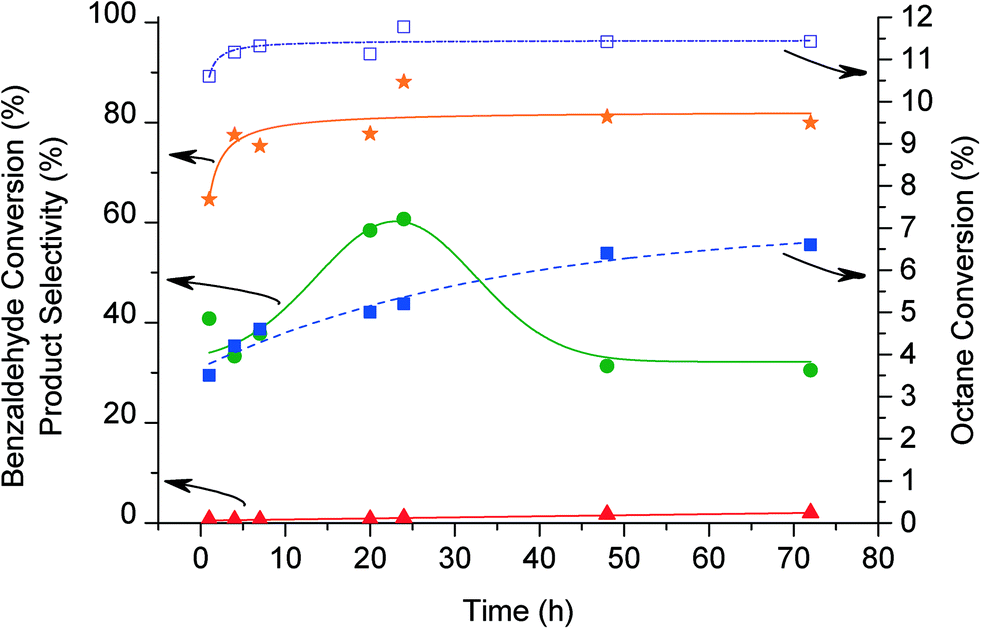 | ||
Fig. 5 Effect of reaction time for benzaldehyde conversion. Conversion of octane ( ), octanone/octanol selectivity ( ), octanone/octanol selectivity ( ), octylbenzoate ( ), octylbenzoate ( ). Conversion of benzaldehyde ( ). Conversion of benzaldehyde ( ) and benzoic acid selectivity ( ) and benzoic acid selectivity ( ). Reaction conditions: octane (60 mmol), oxygen, ratio octane (mol)/benzaldehyde (mol) = 9:1, 1% AuPd/C (50 mg), 80 °C, 1500 rpm. ). Reaction conditions: octane (60 mmol), oxygen, ratio octane (mol)/benzaldehyde (mol) = 9:1, 1% AuPd/C (50 mg), 80 °C, 1500 rpm. | ||
The effect of temperature was also investigated (Fig. 6). The conversion increases to a maximum of 24% at 140 °C. This increase is considered to be due to a high contribution of octane autoxidation at the higher temperature. At higher temperatures, 160 °C, the presence of the catalyst may not promote the conversion of octane; hence there is no further increase in conversion. However, the presence of the catalyst permits control over the autoxidation, contributing to an increase in selectivity to primary oxidation products when benzaldehyde is present. C–C scissions increased at higher temperature, corresponding to a dramatic decrease in selectivity to C8 oxygenate products from 58% to 15%. However at temperatures higher than 140 °C the selectivity for octylbenzoate increased to 12%. Table 2 shows the selectivity for each alcohol and aldehyde. These results are representative for octanol and octanone selectivity for each condition. As expected the selectivity toward products with carbon 2, 3 and 4 oxidised are higher than products arising from oxidation of the first carbon. However, when there is a decrease of oxygenate selectivity all species follow the same trend. At low selectivity towards oxygenate no 1-octanol is produced. The highest selectivity for 1-octanol (ca. 1.3%) was obtained after 48 h reaction.
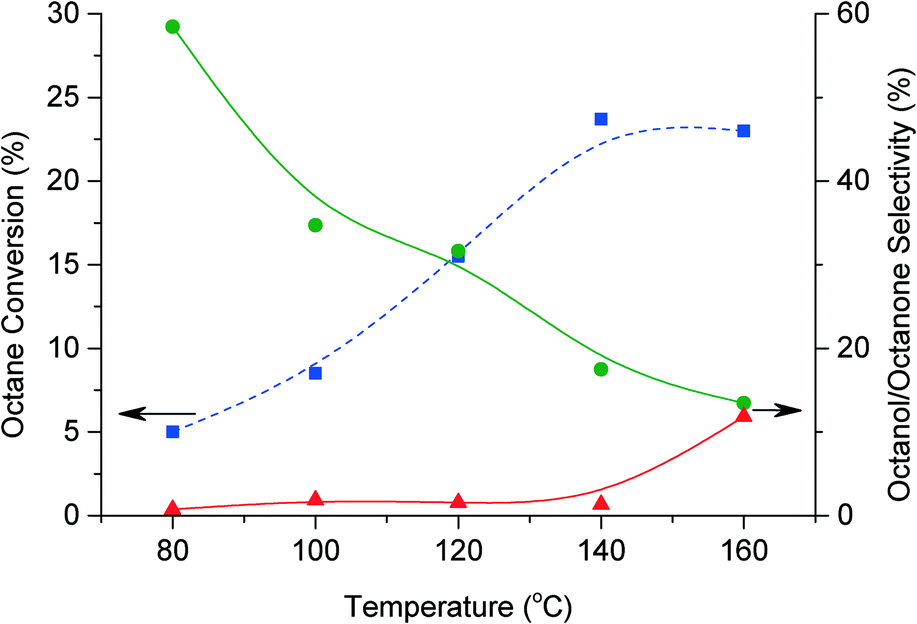 | ||
Fig. 6 Effect of temperature on octane oxidation. Conversion of octane ( ) and products selectivity. Octanone/octanol ( ) and products selectivity. Octanone/octanol ( ) and octylbenzoate ( ) and octylbenzoate ( ). Reaction conditions: octane (60 mmol), oxygen, ratio octane (mol)/benzaldehyde (mol) = 9:1, 1% AuPd/C (50 mg), 20 h, 1500 rpm. ). Reaction conditions: octane (60 mmol), oxygen, ratio octane (mol)/benzaldehyde (mol) = 9:1, 1% AuPd/C (50 mg), 20 h, 1500 rpm. | ||
:1, 1% AuPd/C (50 mg), 20 h, 1500 rpm
| Selectivity (%) | |||||||
|---|---|---|---|---|---|---|---|
| T (°C) | 1-Octanol | 2-Octanol | 3-Octanol | 4-Octanol | 2-Octanone | 3-Octanone | 4-Octanone |
| 80 | 0.6 | 1.1 | 7.3 | 10.2 | 15.7 | 8.8 | 14.8 |
| 100 | 0.3 | 0.6 | 4.3 | 6.0 | 9.3 | 5.2 | 8.8 |
| 120 | 0.1 | 0.2 | 4.2 | 5.3 | 6.1 | 7.4 | 8.3 |
| 140 | 0.6 | 0.9 | 1.4 | 1.4 | 4.6 | 4.0 | 4.6 |
| 160 | 0.0 | 0.3 | 0.4 | 0.9 | 3.2 | 3.9 | 4.6 |
Overall, the variation of reaction conditions (pressure, catalyst mass, temperature and time) led to an increase of octane conversion. The formation of octylbenzoate demonstrates that the proposed coupling between octanol and benzoic acid occurs. This coupling prevents over oxidation of the alcohol to aldehyde or ketone by blocking the alcohol site. A subsequent hydrolysis of the ester, octylbenzoate in this case, would generate the desired alcohol. However, with an increase of conversion the selectivity towards target products decreases while light oxygenates, alkyl ester and benzyl ester are detected. It is assumed that formation of sequential products will subsequently initiate further reactions leading to the change of selectivity toward undesirable products.
It can be concluded that benzaldehyde plays two roles: (i) it can initiate the oxidation of octane, (ii) it can be oxidised to benzoic acid which subsequently couples with octanol to form octylbenzoate, thus protecting the alcohol from over-oxidation and C–C scission.
The initial experiments of this study were based on the assumption that the oxidation of toluene using 1% AuPd/C at 160 °C was proceeding through the formation of benzaldehyde and benzoic acid. However, this study shows that is not the case and benzyl benzoate is formed through another pathway. This possibility is supported by our recent study where we have shown that the formation of benzoic acid from benzaldehyde oxidation with oxygen was quenched in the presence of benzyl alcohol by inhibiting the formation of perbenzoic radical.36 Hence, we consider that when using toluene as co-oxidant the oxidation of octane could not be initiated due to the presence of benzyl alcohol in the reaction mixture.
Consequently, other aldehydes (formaldehyde, butanal and propanal) were investigated under the same conditions to ascertain if they could have the same effect. In the case of formaldehyde no products were detected. It is considered that formaldehyde did not initiate the oxidation of octane, possibly due to the aldehyde being in the gas phase under the reaction conditions utilised. The boiling point of formaldehyde is −19 °C, and at 80 °C even under 20 bar pressure formaldehyde will be in the gas phase when octane is in the liquid phase, not allowing any significant interaction between the aldehyde and octane to initiate the oxidation. In the presence of propanal and butanal, oxidation products from octane were observed. The formation of the esters, octylpropanoate and octylbutanoate were detected. These experiments demonstrate that an aldehyde can initiate octane oxidation in the liquid phase.
Other alkanes were also investigated with and without the presence of benzaldehyde in order to demonstrate the general applicability of the co-oxidant. Higher alkanes were investigated based on safety considerations, since with lower alkanes under these conditions the alkane/oxygen mixture would have been within the explosive limits. Hence, decane, dodecane and cyclooctane were tested. In every instance the GC-MS chromatogram showed the presence of oxidation products which were not present in the absence of benzaldehyde. All these reactions show that in the presence of aldehyde the oxidation of the alkane can be promoted. It is thought the aldehyde is acting as an initiator. There are a number of studies that support this hypothesis. Oxidation of various molecules has been achieved using an aldehyde and O2 system. The combination of an aldehyde and O2 in the presence of a catalyst has been shown to lead to the formation of a peroxy acid. The peroxy acid can then act as an oxidant in systems such as oxidative desulfurisation using cobalt or manganese salt catalysts32,37 and epoxidation using heteropolyoxometalate.38 In the current paper, we propose that an aldehyde and O2 result in the formation of peroxy acid, which would be used to oxidise alkanes to alcohol and ketones as presented in Scheme 2. Simultaneously the corresponding carboxylic acid will be formed and used to capture the alcohol to form the ester.
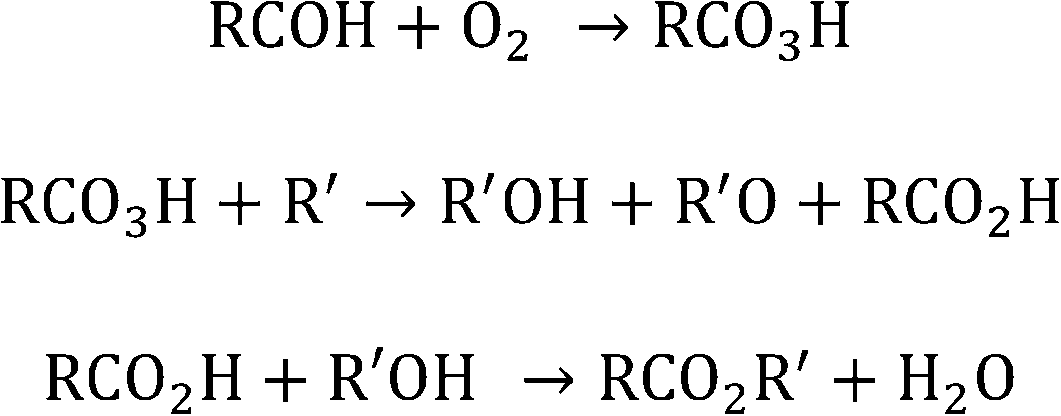 | ||
| Scheme 2 Proposed scheme for the oxidation of octane to an octyl ester using aldehyde/O2 system. | ||
Conclusions
The addition of octane as a co-reactant in toluene oxidation has been investigated but no success was observed for coupling the potential oxidation products to form octylbenzoate. Benzaldehyde was, however, found to facilitate octane activation through the formation of peroxy radicals. Reactions carried out in the absence of benzaldehyde have shown that the autoxidation of octane occurs at temperatures >80 °C. With the addition of benzaldehyde the oxidation products can be observed at 50 °C and the formation of octylbenzoate occurs at 100 °C. The conversion and the selectivity towards oxygenates can be enhanced by the addition of 1% AuPd/C at 80 °C. We show that octane can be oxidized at 80 °C in the presence of benzaldehyde and 1% AuPd/C with 5% conversion, 58% selectivity for the alcohols and ketones and 0.7% of octylbenzoate (coupling between octane and benzaldehyde). Changing the reaction conditions, in terms of reaction time and temperature, increased the conversion but also the formation of products resulting from carbon–carbon bond scission. At 160 °C the conversion was observed to be 24% with 12% selectivity for octyl benzoate.This reaction was investigated with other aldehydes, with propanal and butanal both showing the same capacity to oxidize octane and formed the ester. Using this concept, the oxidation of decane, dodecane and cyclooctane was successfully achieved on addition of benzaldehyde.
We consider that this study demonstrates that the oxidation of an alkane can be initiated in the presence of an aldehyde to form alcohols and ketones. Moreover, this study shows the activation of the primary C–H bond for long alkane chain is possible using this methodology. Hence we consider this strategy could be extended in order to activate the oxidation of lower alkane such as methane with appropriate catalyst development.
Acknowledgements
We thank the President's scholarship from Cardiff University for financial support of VP. We thank Dr Peter Miedziak for assistance with the graphical abstract.Notes and references
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2934 CrossRef CAS PubMed.
- C. J. Jones, D. Taube, V. R. Ziatdinov, R. A. Periana, R. J. Nielsen, J. Oxgaard and W. A. Goddard, Angew. Chem., 2004, 116, 4726–4729 CrossRef PubMed.
- B. Michalkiewicz, J. Catal., 2003, 215, 14–19 CrossRef CAS.
- B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, J. Am. Chem. Soc., 1995, 28, 154–162 CAS.
- O. Younes-Metzler, J. Svagin, S. Jensen, C. H. Christensen, O. Hansen and U. Quaade, Appl. Catal., A, 2005, 284, 5–10 CrossRef CAS PubMed.
- J. A. Labinger, Fuel Process. Technol., 1995, 42, 325–338 CrossRef CAS.
- D. Wolf, Angew. Chem., Int. Ed., 1998, 37, 3351–3353 CrossRef CAS.
- Y. Seki, J. S. Min, M. Misono and N. Mizuno, J. Am. Chem. Soc., 2000, 104, 5940–5944 CAS.
- Z.-A. Qiao, P. Zhang, S.-H. Chai, M. Chi, G. M. Veith, N. C. Gallego, M. Kidder and S. Dai, J. Am. Chem. Soc., 2014, 136, 11260–11263 CrossRef CAS PubMed.
- P. Zhang, Y. Gong, H. Li, Z. Chen and Y. Wang, Nat. Commun., 2013, 4, 1593 CrossRef PubMed.
- H. H. Monfared and Z. Amouei, J. Mol. Catal. A: Chem., 2004, 217, 161–164 CrossRef CAS PubMed.
- S. Das, T. Bhowmick, T. Punniyamurthy, D. Dey, J. Nath and M. K. Chaudhuri, Tetrahedron Lett., 2003, 44, 4915–4917 CrossRef CAS.
- C. Hammond, N. Dimitratos, R. L. Jenkins, J. A. Lopez-Sanchez, S. A. Kondrat, M. Hasbi ab Rahim, M. M. Forde, A. Thetford, S. H. Taylor, H. Hagen, E. E. Stangland, J. H. Kang, J. M. Moulijn, D. J. Willock and G. J. Hutchings, ACS Catal., 2013, 3, 689–699 CrossRef CAS.
- P. Gallezot, Catal. Today, 1997, 37, 405–418 CrossRef CAS.
- G. C. Bond, C. Louis and D. T. Thompson, Catalysis by gold, Imperial College Press, London, 1st edn, 2006, pp. 217–243 Search PubMed.
- C. Mohr, H. Hofmeister, M. Lucas and P. Claus, Chem. Eng. Technol., 2000, 23, 324–328 CrossRef CAS.
- J. E. Bailie and G. J. Hutchings, Chem. Commun., 1999, 2151–2152 RSC.
- D. E. De Vos and B. F. Sels, Angew. Chem., Int. Ed., 2004, 44, 30–32 CrossRef PubMed.
- M. Sankar, E. Nowicka, P. J. Miedziak, G. L. Brett, R. L. Jenkins, N. Dimitratos, S. H. Taylor, D. Knight, D. Bethell and G. J. Hutchings, Faraday Discuss., 2010, 145, 341 RSC.
- J. K. Edwards, E. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2009, 48, 8512–8515 CrossRef CAS PubMed.
- N. Dimitratos, A. Villa, D. Wang, F. Porta, D. Su and L. Prati, J. Catal., 2006, 244, 113–121 CrossRef CAS PubMed.
- R. C. Tiruvalam, J. C. Pritchard, N. Dimitratos, J. A. Lopez-Sanchez, J. K. Edwards, A. F. Carley, G. J. Hutchings and C. J. Kiely, Faraday Discuss., 2011, 152, 63–86 RSC.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS PubMed.
- J. A. Lopez-Sanchez, N. Dimitratos, N. Glanville, L. Kesavan, C. Hammond, J. K. Edwards, A. F. Carley, C. J. Kiely and G. J. Hutchings, Appl. Catal., A, 2011, 391, 400–406 CrossRef CAS PubMed.
- G. J. Hutchings, Catal. Today, 2008, 138, 9–14 CrossRef CAS PubMed.
- M. I. bin Saiman, G. L. Brett, R. Tiruvalam, M. M. Forde, K. Sharples, A. Thetford, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, D. M. Murphy, D. Bethell, D. J. Willock, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5981–5985 CrossRef CAS PubMed.
- A. F. Carley, N. Dimitratos, G. J. Hutchings, R. L. Jenkins, H. A. B. Rahim, J. A. Lopez-Sanchez, S. H. Taylor and D. J. Willock, Hydrocarbon selective oxidation with heterogeneous gold catalysts, PCT/GB2009/051461, 2011.
- X. Guo, P. Brault, G. Zhi, A. l. Caillard, J. Guoqiang, C. Coutanceau, S. Baranton and X. Guo, J. Phys. Chem. C, 2011, 115, 11240–11246 CAS.
- S. M. Lang and T. M. Bernhardt, Faraday Discuss., 2011, 152, 337 RSC.
- M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock, D. M. Murphy, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2013, 52, 1280–1284 CrossRef CAS PubMed.
- T. V. Rao, B. Sain, S. Kafola, B. R. Nautiyal, Y. K. Sharma, S. M. Nanoti and M. O. Garg, Energy Fuels, 2007, 21, 3420–3424 CrossRef CAS.
- S. Murata, K. Murata, K. Kidena and M. Nomura, Energy Fuels, 2004, 18, 116–121 CrossRef CAS.
- J. A. A. Hoorn, P. L. Alsters and G. F. Versteeg, Int. J. Chem. React. Eng., 2005, 3, A6 Search PubMed.
- T. Mill and D. G. Hendry, Comprehensive Chemical Kinetics, Elsevier Scientific, Amsterdam, 1st edn, 1980, p. 64 Search PubMed.
- L. Kesavan, R. Tiruvalam, M. H. Ab Rahim, M. I. bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195–199 CrossRef CAS PubMed.
- M. Sankar, E. Nowicka, E. Carter, D. M. Murphy, D. W. Knight, D. Bethell and G. J. Hutchings, Nat. Commun., 2014, 3332 Search PubMed.
- V. Dumont, L. Oliviero, F. Mauge and M. Houalla, Catal. Today, 2008, 130, 195–198 CrossRef CAS PubMed.
- M. Hamamoto, K. Nakayama, Y. Nishiyama and Y. Ishii, J. Org. Chem., 1993, 58, 6421–6425 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy00453e |
| This journal is © The Royal Society of Chemistry 2015 |