
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile structure design based on C3N4 for mediator-free Z-scheme water splitting under visible light†
Guixia
Zhao‡
,
Xiubing
Huang‡
,
Federica
Fina
,
Guan
Zhang
and
John T. S.
Irvine
*
School of Chemistry, University of St Andrews, St Andrews, Fife, UK. E-mail: jtsi@st-andrews.ac.uk; Fax: +44 (0)1334463808; Tel: +44 (0)1334463817
First published on 28th April 2015
Abstract
In this work, two photocatalysts (i.e., C3N4 and WO3) were successfully combined into a heterojunction structure by a facile hydrothermal method for mediator-free overall water splitting, analogous to the natural photosynthesis over a two-step photoexcitation Z-scheme system. Hydrogen and oxygen are evolved with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio by irradiating the C3N4-WO3 composites loaded with Pt under visible light (λ > 420 nm) without any redox mediator. Introducing reduced graphene oxide (rGO) into the C3N4-WO3 composites enhances the water splitting efficiency. Through optimizing the mass ratio in the C3N4-WO3 composites, rGO content, amount of loaded Pt and pH value of the reacting system, the highest H2/O2 evolution rates of 2.84 and 1.46 μmol h−1 can be obtained, with a quantum yield of 0.9%. Our findings demonstrate that the hydrothermal method is a promising strategy for constructing intimate heterostructures for Z-scheme water-splitting systems without using any redox mediator, and that rGO can be used to further enhance the performance in optimized conditions.
:1 ratio by irradiating the C3N4-WO3 composites loaded with Pt under visible light (λ > 420 nm) without any redox mediator. Introducing reduced graphene oxide (rGO) into the C3N4-WO3 composites enhances the water splitting efficiency. Through optimizing the mass ratio in the C3N4-WO3 composites, rGO content, amount of loaded Pt and pH value of the reacting system, the highest H2/O2 evolution rates of 2.84 and 1.46 μmol h−1 can be obtained, with a quantum yield of 0.9%. Our findings demonstrate that the hydrothermal method is a promising strategy for constructing intimate heterostructures for Z-scheme water-splitting systems without using any redox mediator, and that rGO can be used to further enhance the performance in optimized conditions.
1. Introduction
With the increasing energy demand and concerns about environmental issues, hydrogen gas is considered as a promising fuel because it produces no carbon emission and other harmful by-products.1 The artificial photosynthesis for hydrogen generation through water splitting under visible light is an important approach, among which, the powdered-type two-step photoexcitation system, i.e., Z-scheme photocatalysis system, is the most similar to natural photosynthesis.2–6 Normally, a Z-scheme system contains two photocatalysts (one for H2 evolution, and the other for O2 evolution) and usually an electron mediator, which transfers the electrons from the excited O2 photocatalyst to the holes generated in the H2 photocatalyst.7It is suggested that electron transfer is the rate-determining process.8 The commonly used electron mediators are ionic redox couples such as IO3−/I−,2,3,9 or Fe3+/Fe2+.4,9,10 Compared with these ionic redox couples, the solid electron shuttle is more advanced for the recycle of the photocatalyst and cleaning of water. The challenges for the efficient H2/O2 evolution lie in the electron-accepting and -donating abilities of the mediator, which should retain a dynamic equilibrium and remain stable during reactions.11 Furthermore, interface contact in the ternary system also plays an important role in maintaining continuous electron flow between the two photocatalysts. Considering the extremely high conductivity and specific surface area of graphene, it may be a good candidate as electron mediator for a Z-scheme system.11 Besides, it is also challenging to choose appropriate photocatalysts for realizing the intimate physical interaction with graphene, the efficient charge carrier separation, the H2/O2 evolution on the H2/O2 photocatalysts and then finally preventing water formation due to the backward reaction of H2 and O2.12,13 In Iwase et al.'s report,11 it has been shown that photoreduced graphene oxide (PRGO) was able to function as a solid electron mediator for Z-scheme overall water splitting, where SrTiO3:Rh was used as H2 evolution photocatalyst, with 0.7 wt% Ru as co-catalyst, and BiVO4 was used as O2 evolution photocatalyst. The PRGO was synthesized by photoreduction, i.e., irradiating the mixture of graphene oxide and the photocatalyst (Ru/SrTiO3:Rh or BiVO4) in the presence of methanol as electron donor. For the combined PRGO/BiVO4 and Ru/SrTiO3:Rh under pH 3.5, the H2 evolution rate is ca. 8 μmol h−1, while under neutral conditions, the performance is much lower, which is considered to be due to the poor physical interaction between PRGO/BiVO4 and Ru/SrTiO3:Rh under neutral conditions. In total, the reported Z-scheme based on PRGO is complicated in preparation as the noble metals (i.e., Ru and Rh) are used as co-catalyst and dopant, respectively, and the performance is dependent on the acidity. Although Sasaki et al.8 reported the interparticle electron transfer without any electron mediator, the water splitting can only proceed efficiently by acidifying the aqueous solution to realize the effective contact through the aggregation of the two photocatalysts, Ru/SrTiO3:Rh and BiVO4. Thus, more sturdy contacts between the H2/O2 evolution phototcatalysts need to be found.
Since the introduction of graphitic carbon nitride (C3N4) into heterogeneous catalysis in 2006,14,15 this yellow polymer consisting of tri-s-triazines interconnected via tertiary amines has attracted increasing interest in materials research especially in H2-evolution photocatalysis under visible light.16–18 The band gap of C3N4 is ca. 2.7 eV, with the CB and VB positions located at ca. −1.1 eV and ca. +1.6 eV vs. normal hydrogen electrode (NHE), respectively.16,19 More importantly, the preparation of C3N4 is low cost since it only consists of two elements (C and N). For O2-evolution photocatalysis from water, tungsten trioxide (WO3) has been widely investigated because of its suitable bandgap, high electron mobility, abundance and low cost.20
Herein, we introduced a novel method to generate a close contact between C3N4 and WO3 by the reaction of Na2WO4·2H2O and C3N4 under hydrothermal conditions. To enhance the performance, rGO was also used to function as electron mediator. By calcining a mixture of rGO and melamine, the graphene layer was able to be homogeneously mixed with C3N4 layers. Then through the hydrothermal method, the C3N4-rGO composites were loaded with WO3 particles, which function as O2 evolution photocatalyst. It is demonstrated that the binary (in the case of no rGO) or ternary (in the case of containing rGO) composites were able to give out H2 and O2 under visible light irradiation (λ > 420 nm) with relatively high performance when loaded with Pt as co-catalyst, and an appropriate amount of rGO will enhance the performance.
2. Experimental section
Synthesis of C3N4-rGO composite and pure C3N4
Graphene oxide was synthesized according to the modified Hummers method as described in our previous work.21 The as-prepared GO was then reduced by the hydrothermal method. 60 mg of GO was dissolved in 30 mL of H2O by moderate sonication and the pH was adjusted to about 7 using NaOH solution before transferring into a Teflon-lined steel autoclave, which was heated in an oven at 140 °C for 18 h. The obtained rGO was collected and washed with distilled water and ethanol several times, and dried at 90 °C for 2 h. C3N4-rGO composites were obtained by calcining the mixtures of melamine and rGO with different mass ratios. These C3N4-rGO composites are noted as C3N4-rGO-x, where x represents the mass ratio of melamine to rGO during the synthesis process. For example, the composite C3N4-rGO-150 was prepared by calcining the mixture of 20 mg of rGO and 3000 mg of melamine at 500 °C for 3 h with a 5 °C min−1 increasing rate. Pure C3N4 was also obtained by calcining melamine at 500 °C for 3 h.Synthesis of C3N4-rGO-WO3 and C3N4-WO3 composites
The ternary composites are noted as C3N4-rGO-x-WO3m–n, m representing the initial reacting mass of C3N4-rGO-x in mg, n representing that of Na2WO4·2H2O. Taking C3N4-rGO-150-WO3 400–260 for example, 400 mg of C3N4-rGO-150 and 260 mg of Na2WO4·2H2O (99%, Aldrich) were mixed in 30 mL of deionized water and kept under stirring at ca. 300 rpm for 12 h. Then the pH value was adjusted to 2 using diluted HCl solution. After further stirring at ca. 300 rpm for another 12 h, the suspension was transferred into a Teflon-lined steel autoclave and kept at 160 °C for 18 h. The C3N4-WO3 composite was obtained by the same method except for using pure C3N4 instead of C3N4-rGO composites. After hydrothermal reaction, the resulted composites were separated by centrifugation at 4000 rpm and washed with distilled water several times before drying at 90 °C for 3 h.Pt-loading on the C3N4-rGO, C3N4-rGO-WO3 and C3N4-WO3 composites
Pt was firstly loaded onto the specimen by impregnation: an appropriate amount of H2PtCl6 solution (ca. 4 mL) was impregnated into the sample powders by evaporating the solvent of the mixed suspension at 90 °C until dry. For the H2-evolution test by C3N4-rGO composites, these Pt-loaded samples were used in the presence of triethanolamine (TEOA) as sacrificial agent. For the overall water splitting test by C3N4-rGO-WO3 and C3N4-WO3 composites, these Pt-loaded samples were further reduced by NaBH4 solution in Ar protection. For example, 200 mg of C3N4-rGO-150-WO3 400–260 loaded with 1 wt% Pt was dissolved in 30 mL of deionized water and purged with Ar for 30 min before adding 5 mg of NaBH4. After 2 h of reduction, the suspension was centrifuged and washed with distilled water several times. The resulted slurry was then used for photocatalytic tests.Material characterization
Specimens were characterized by XRD using a PANalytical Empyrean Reflection diffractometer with a Cu Kα source (λ = 1.541 Å). TEM analysis was performed using a JEM-2011. XPS spectra were obtained using an ESCALAB 250 and all binding energies were referenced to the C 1s peak at 284.6 eV. A UV–vis spectrophotometer (JASCO-V550) was used for obtaining the optical absorbance spectra.Photocatalytic measurement
The measurement was performed with a home-made Teflon reactor with the top window sealed by Pyrex glass. In the H2-evolution test by C3N4-rGO composites loaded with 1 wt% Pt, 200 mg of photocatalyst was dispersed in 100 mL of water and 10 mL of TEOA was added as sacrificial agent. In the O2-evolution test by WO3·1/3H2O, 100 mg of WO3·1/3H2O powder was dispersed in 100 mL of water with 10 mmol L−1 AgNO3 as an electron acceptor. The reactor was firstly purged with pure Ar to remove air from the reaction system, then the solution was irradiated by a 250 W iron doped metal halide UV–vis lamp (UV Light Technology Limited, UK) with a UV cut-off filter (λ > 420 nm) and the amount of H2 or O2 produced was determined by gas chromatography (Agilent 3000 Micro Gas Chromatograph with a TCD detector, using argon carrier gas). The H2- and O2-evolution half reaction results shown in the ESI† demonstrated their activity under visible light. The mediator-free Z-scheme overall water splitting reaction trials were performed in the same top-irradiation system. During these trials, 200 mg of Pt-loaded C3N4-rGO-WO3 or C3N4-WO3 composites was suspended in 100 mL of deionized water, but no TEOA was added. The evolved gas composition in the sealed reactor was measured by gas chromatography.Quantum yield efficiency measurement
The apparent quantum yield (AQY) of the Z-scheme water splitting with a two-photon excitation process was measured using the same reactor with the addition of a band-pass filter (420 nm, Δλ1/2 = 10 nm). The AQY values were calculated using the following equation:22| AQY (%) = [4 × n(H2)]/n(photons) × 100 | (1) |
3. Results and discussion
Fig. 1 shows the typical XRD patterns of WO3 and C3N4-rGO-150-WO3 composites resulting from the hydrothermal reaction of Na2WO4·2H2O without or with different contents of C3N4-rGO-150. The XRD pattern in Fig. 1a denotes the presence of orthorhombic tungsten oxide one third hydrate (WO3·1/3H2O, JCPDS card no. 54-1012), and the characteristic peak from C3N4 located at around 27° increased with the increment of C3N4-rGO-150 content, as shown in Fig. 1b and c. There are no obvious diffraction peaks for WO3·1/3H2O in Fig. 1c, which may be due to the large molar ratio of C3N4 to WO3 in C3N4-rGO-150-WO3 1000–260 (ca. 13.8 calculated from the added amount). If considering the smaller density and bigger particle size of C3N4 than WO3 nanoparticles in C3N4-rGO-150-WO3 1000–260, the XRD diffraction density of C3N4 is much stronger than that of WO3 nanoparticles. Therefore, the diffraction peaks of WO3·1/3H2O properly overlapped with those of C3N4. In addition, the crystallinity of WO3 may be also poor because of the dispersion of C3N4 particles. | ||
| Fig. 1 XRD patterns of pure WO3 (a), C3N4-rGO-150-WO3 400–260 (b) and C3N4-rGO-150-WO3 1000–260 (c). | ||
Fig. 2a and b show the TEM images of C3N4-rGO-150-WO3 400–260 and C3N4-rGO-150-WO3 1000–260, respectively. It is obvious that in the hydrothermal reaction, a higher Na2WO4·2H2O/C3N4-rGO-150 ratio means that more isolated WO3 particles are not loaded on the surface of C3N4-rGO-150, while in C3N4-rGO-150 1000–260, almost no independent WO3 particles exist. By loading Pt as co-catalyst, efficient H2-evolution performance can be realized. Fig. 2c and S1 (see in ESI†) show the TEM images of C3N4-rGO-150 400–260 loaded with 1 wt% Pt after several periods of photocatalytic water splitting tests, indicating that Pt is mainly distributed onto the surface of C3N4 perhaps due to its stronger ability to chelate Pt+. The typical UV–vis light absorbance spectra in Fig. 2d also indicate that there were no obvious differences in the absorbance between the composites with different WO3 contents, while adding rGO indeed increased the absorption toward visible light.
 | ||
| Fig. 2 TEM images of C3N4-rGO-150-WO3 400–260 (a), C3N4-rGO-150-WO3 1000–260 (b), C3N4-rGO-150-WO3 400–260 loaded with 1 wt% Pt after photocatalytic test (c); the UV–vis light absorbance spectra (converted from the corresponding reflectance spectra) of three typical samples loaded with 1 wt% Pt (d). | ||
We used the as-prepared C3N4-rGO-150-WO3 400–260 without loading Pt as photocatalyst for the cycling water-splitting test under visible light (Fig. 3). As shown in Fig. 3a, in the initial four cycles, the performance increased but then decayed on further cycling. The best performance is only about 12 μmol of H2 in 20 hours' irradiation. After loading Pt as co-catalyst, the case is much different, as shown in Fig. 3b. The H2-evolution performance is very stable from the second cycle and the final amount of H2 produced in 20 hours is as high as 35 μmol and the produced O2 is about half in μmol compared with that of H2. It is also noted that without loaded Pt, the photocatalyst underwent an obvious color change during the photo-reaction as shown in Fig. 3c and d, where the milk-white suspension changed into blue after six cycles of test. According to much reported research on the photochromogenic WO3, when irradiated at a photo energy higher than its band gap (2.8 eV), photochromism would occur, in which the photo-generated electron–hole would lead to the reduction of W6+ into blue-colored W5+.23–28 While after being loaded with Pt nanoparticles, the photocatalyst became just a little darker compared with that of the milk-white C3N4-rGO-150-WO3 400–260, and no color change occurred during the photo-reaction, as shown in Fig. 3e. It was discovered that the loaded Pt was able to inhibit the color change, which may be due to the efficient electron capture by the loaded Pt nanoparticles. The gas generation amount dependence on reaction time shown in Fig. S2† for C3N4-rGO-150-WO3 400–260 loaded with 1 wt% Pt indicates that gaseous products evolve linearly for the next 12 h in a ratio of ca. 2:1 (H2 and O2: 1.52 and 0.74 μmol h−1) under visible light.
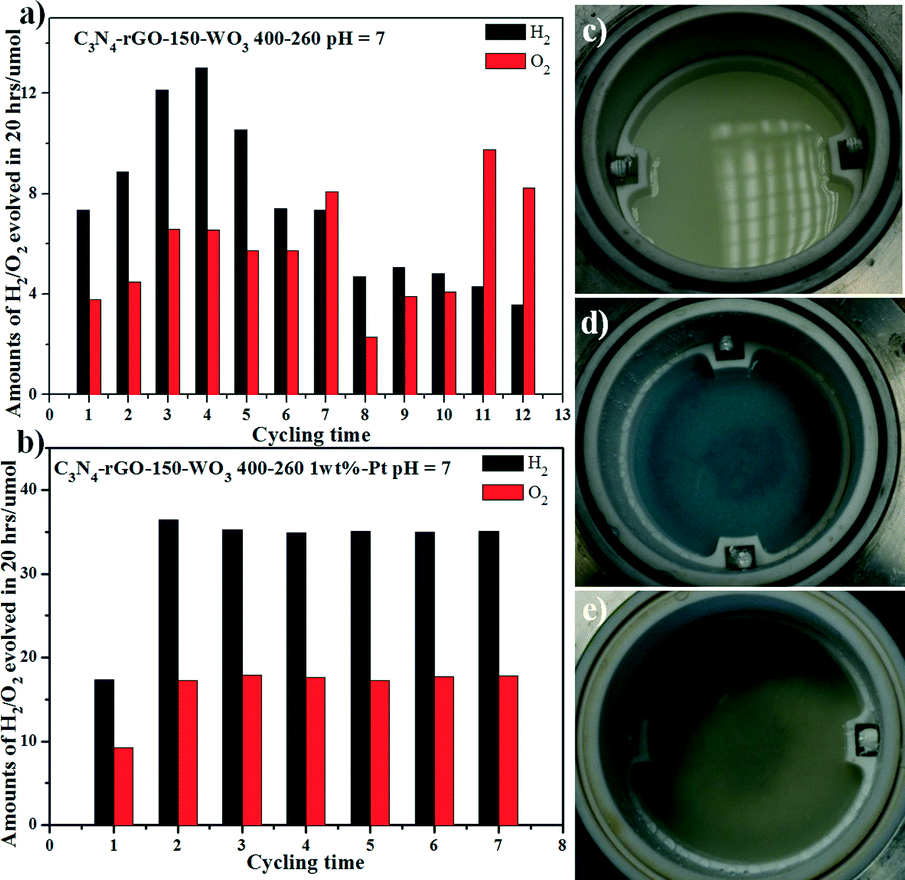 | ||
| Fig. 3 The performance of the cycling test for C3N4-rGO-150-WO3 400–260 without (a) and with (b) Pt under pH = 7; the C3N4-rGO-150-WO3 400–260 without Pt before photo-reaction (c) and after six cycles of photo-reaction (d); the sample C3N4-rGO-150-WO3 400–260 with Pt after six cycles of photo-reaction (e). | ||
To investigate the effects of graphene content, the mass ratio of C3N4 (or C3N4/rGO) and WO3, the pH in the photocatalytic system and the amount of loaded Pt on the water-splitting performance, we tested the H2/O2 evolution amount with different photocatalysts and in different conditions. As indicated by no. 1 and 2 in Table 1, when using only C3N4 loaded with 1 wt% Pt in nonsacrificial conditions, no water splitting occurred, while when using C3N4-rGO-150 composite loaded with 1 wt% Pt, the produced H2 is about 3.3 μmol in 20 hours' irradiation. In Jorge et al.'s work, C3N4 was used for H2 and O2 evolution from water half-splitting with methanol and Ag+ as sacrificial agent, respectively.29 In our photocatalytic reaction test, no. 2, the little water splitting activity should come from the reduction and oxidation of water due to the improved electron–hole separation by graphene. In no. 3–6, it is also shown that different rGO contents in the C3N4-rGO composites have a great effect on the water-splitting activity. Both no rGO and too much rGO in the C3N4-rGO composites give worse performance for the H2/O2 evolution than that in the C3N4-rGO-150 composite (no. 3), even though the C3N4-rGO-150 composite only displays an enhancement of 15.8% for the H2/O2 evolution over the pure C3N4 and WO3 system (no. 6). We have also investigated the rGO effect by testing the H2-evolution performance using different C3N4-rGO composites with TEOA as sacrificial agent and Pt as co-catalyst (more details in Fig. S3 and Table S1 in ESI†). It is implied that rGO has two synergistic effects, one is to prevent the hole–electron recombination by transferring electrons, and the other is to prevent the efficient irradiation due to its opacity and light scattering.30–32 An appropriate amount of graphene in the C3N4-rGO composite would improve the electron transfer and further prevent the charge carrier-recombination,33–35 thus a little enhancement occurred for C3N4-rGO-150 composite with Pt as co-catalyst.
| No. | Hydrothermal conditions | Amount of loaded Ptb | pHc | Activity after 20 h irradiation | |||
|---|---|---|---|---|---|---|---|
| Type of C3N4 or C3N4-rGO | C3N4 or C3N4-rGO (mg) | Na2WO4·2H2O (mg) | H2 (μmol) | O2 (μmol) | |||
| a Conditions: photocatalyst (200 mg in total) in water (100 mL); light source, 250 W iron-doped metal halide ultraviolet–visible lamp, with a cut-off filter (λ > 420 nm); top-irradiation cell with a quartz glass. b Pt was loaded firstly by impregnation of H2PtCl6, then lateral reduction with NaBH4 solution and lastly washing several times before using as photocatalyst in slurry. c The pH of the suspension in the photocatalystic test system. | |||||||
| 1 | Pure C3N4 | 400 | 0 | 1 wt% Pt | 7 | Trace | Trace |
| 2 | C3N4-rGO-150 | 400 | 0 | 1 wt% Pt | 7 | 3.3 | 1.1 |
| 3 | C3N4-rGO-150 | 400 | 260 | 1 wt% Pt | 7 | 35.1 | 18.5 |
| 4 | C3N4-rGO-80 | 400 | 260 | 1 wt% Pt | 7 | 26.1 | 14.1 |
| 5 | C3N4-rGO-50 | 400 | 260 | 1 wt% Pt | 7 | 20.8 | 11.2 |
| 6 | Pure C3N4 | 400 | 260 | 1 wt% Pt | 7 | 30.3 | 14.8 |
| 7 | Pure C3N4 | 1000 | 260 | 1 wt% Pt | 7 | 29.9 | 14.5 |
| 8 | C3N4-rGO-150 | 1000 | 260 | 1 wt% Pt | 7 | 48.1 | 24.5 |
| 9 | C3N4-rGO-50 | 400 | 260 | 3 wt% Pt | 7 | 6.0 | 3.3 |
| 10 | C3N4-rGO-150 | 400 | 260 | 3 wt% Pt | 7 | 11.0 | 5.0 |
| 11 | C3N4-rGO-150 | 1000 | 260 | 1 wt% Pt | 3 | 11.4 | 30.5 |
| 12 | C3N4-rGO-150 | 1000 | 260 | 1 wt% Pt | 5 | 29.0 | 15.1 |
| 13 | C3N4-rGO-150 | 1000 | 260 | 1 wt% Pt | 8 | 53.5 | 26.8 |
| 14 | C3N4-rGO-150 | 1000 | 260 | 1 wt% Pt | 10 | 56.6 | 29.2 |
The results for no. 3 and 8 in Table 1 show that sample C3N4-rGO-150-WO3 1000–260 displays a higher performance by 37% than C3N4-rGO-150-WO3 400–260 under the same conditions (i.e., loaded with 1 wt% Pt in pH = 7), indicating that too much WO3 deposited on the surface of C3N4-rGO-150 will prevent the incident light from reaching the C3N4-rGO-150 surface, which results in the weakened photoexcitation of C3N4-rGO-150.36 However, for the pure C3N4 and WO3 system, there is no obvious change in the water-splitting performance with different mass ratios of C3N4 to WO3 (no. 6 and 7 in Table 1). These results show that the WO3 amount has a more obvious influence on the C3N4-rGO-150 system than the pure C3N4 system, further suggesting that rGO is able to play a significant role in the Z-scheme water-splitting system because of its good conductivity and its function as promising electron mediator for transferring the photo-generated electrons from WO3 to the photo-generated holes in C3N4. The effect of the loaded Pt amount is also revealed from a comparison between no. 5 and 9 or no. 3 and 10. Too much Pt has a negative effect on the gas evolution, as proved by other reports.37–39
According to previous research on the Z-scheme photocatalyst system, to ensure the charge transfer, intimate physical interaction between the two photocatalysts was often realized by the aggregation of the two photocatalysts under some appropriate pH.8,11 In our case, the effect of pH on the water-splitting activity was also investigated using C3N4-rGO-150-WO3 1000–260 as target. It is found that with increasing pH, the gas evolution rate is gradually increased (no. 8, 11–14). When the pH equals 3, the produced H2 is as low as 11.4 μmol in 20 hours. Since WO3 is inherently unstable at high pH, we did not test the performance under higher pH conditions. Thus the pH effect should not come from the aggregation level but from the kinetic rate of the H2 and O2 evolution reactions, which can be expressed as the following:
| 2H+ + 2e− → H2 | (2) |
| 2H2O + 4h+ → O2 + 4H+ | (3) |
It can be inferred that although pH has the conflicting effect for eqn (2) and (3), a higher pH is beneficial to the whole performance. This is despite that under acidic conditions, H2-evolution reaction is relatively favorable, the associated negative effect for the O2 evolution would trade off the little benefit since the water oxidation depends on the adsorbed OH− ions onto the WO3 surface and their following oxidation by multiple photo-generated holes.40 Thus under more alkaline conditions, the final H2/O2-evolution performance turns out to be higher, and these results indicate that the O2-evolution reaction is the rate-determining process.
We also prepared a C3N4-WO3 composite via two other methods, one by calcining WO3 nanoparticles and C3N4 mixture at 350 °C and the other by hydrothermal treatment of WO3 nanoparticles and C3N4 mixture at 160 °C. Both of the as-prepared mixtures were loaded with 1 wt% Pt before the photocatalytic test. And for the two cases, no H2 was detected. It is confirmed that the hydrothermal conditions result in the intimate contact between C3N4 (or C3N4-rGO) and WO3 due to the special high-temperature and high-pressure environment. And the in situ formation of WO3 particles on the C3N4 framework also contributes to the intimate interaction between the two photocatalysts.
It is worthy to note that the photocatalysts used in Table 1 were fresh wet samples from the reduction by NaBH4 without drying. And we also tried to dry the sample C3N4-rGO-150-WO3 1000–260 (loaded with 1 wt% Pt) after several cycling tests with stable performance. Interestingly, after drying in air at 80 °C for 4 hours, the sample showed a lower activity than its stable performance before drying, and only after 3 cycles, was the performance recovered, as shown in Fig. 4. Then the sample was dried again at 80 °C for 30 hours, after which the H2/O2 evolution became even worse, and also recovered after 4 cycles (Fig. 4a). We investigated the XPS of the sample dried in air at 80 °C for 30 hours and the fresh wet sample dried in Ar. The XPS survey spectrum shown in Fig. S4 (see in ESI†) demonstrated the existence of C, N, O, Pt and W, but no Na was observed. Although both of the samples showed a high content of PtOx (indicated by the Pt 4f7/2 and Pt 4f5/2 peaks centered at ca. 73.0 eV and 76.3 eV, respectively),41 the metallic Pt content in the sample dried in Ar is obviously higher than that dried in air (indicated by the Pt 4f7/2 and Pt 4f5/2 peaks centered at ca. 71.0 eV and 74.3 eV, respectively), as shown in Fig. 4b and c.42 Therefore, it is inferred that the evolved H2 would reduce PtOx into Pt metal during the irradiation and the activity of the sample is quite sensitive to the Pt metal content.
 | ||
| Fig. 4 The cycling performance of the sample (C3N4-rGO-150-WO3 1000–260 loaded with 1 wt% Pt) dried after several photo-test cycles as fresh slurry (a); XPS of the sample dried in air for 30 hours (b); XPS of the fresh wet sample dried in Ar (c). | ||
The evolution of H2 from water splitting was further confirmed by using D2O as the solvent in the suspension of photocatalyst. The evolved gas mixture was then detected by the mass spectra and the results are shown in Fig. S5 (see in ESI†). The extra signal located at 4 amu is due to evolved D2, which is absent in that of the background gas. We also measured the apparent quantum yield efficiency under monochromatic light at 420 nm. The sample C3N4-rGO-150-WO3 1000–260 (loaded with 1 wt% Pt) under neutral conditions shows an AQY efficiency of ca. 0.9% while the homologous sample without rGO shows a smaller value (i.e., 0.7%), further demonstrating the positive effect of rGO.
Although Martin et al.10 recently reported a similar Z-scheme water splitting on the basis of C3N4 and WO3 (or BiVO4), their performance is strongly dependent on the redox mediator type (i.e., I−/IO3−) and the pH. However, in our C3N4-rGO (or C3N4) and WO3 composite system, no redox mediator is needed for the water-splitting photocatalytic test, which further infers that the intimate contact resulting from hydrothermal conditions is of benefit to the H2/O2 evolution in the two-step photoexcitation Z-scheme system. Fig. 5 shows a tentatively proposed mechanism for the H2/O2 evolution by the C3N4-WO3 composite with or without the mediation of rGO. According to a previous study, the CB and VB positions of WO3 are located at about +0.41 eV and +3.18 eV,43 while those of C3N4 are about −1.13 eV and +1.57 eV.19 It is not difficult to infer that under irradiation, O2 could be evolved from the WO3 surface and H2 from the C3N4 surface. Considering the Pt function as discussed above, H2 is more readily evolved from the surface of Pt than that of C3N4. Therefore, water is oxidized by the photo-induced holes in the VB of WO3 and reduced by the electrons on Pt trapped from the CB of C3N4. The electrons on CB of WO3 are able to combine with the holes in VB of C3N4 at the interface due to the intimate contact. rGO successfully functions as a positive electron mediator because of its good conductivity for electron migration, its sturdy contact with C3N4 resulted from its calcination with the precursor (i.e., melamine), and its good contact with WO3 particles under the hydrothermal conditions.
 | ||
| Fig. 5 Proposed mechanism for the H2/O2 evolution by C3N4-WO3 composite with or without the mediation of rGO. | ||
4. Conclusions
In summary, we have introduced a mediator-free Z-scheme system design based on C3N4 by utilizing WO3 as O2 photocatalyst for the first time, where C3N4 and WO3 are combined as composites through a facile hydrothermal method. Successful water splitting under visible light (λ > 420 nm) can be realized without any redox mediator when Pt is loaded on the surface of the composites. More importantly, rGO is introduced into these composites to form C3N4-rGO-WO3 composites and this shows a positive effect on the performance in an optimized amount. The highest H2/O2 evolution rates are about 2.84 and 1.46 μmol h−1 under visible light (λ > 420 nm) with a quantum yield of 0.9% at 420 nm. Although the resulting performance is not in the highest for water splitting, the newly proposed methodology for constructing heterostructures through the hydrothermal method will pave the way for more efficient overall water splitting with two-step photoexcitation Z-scheme systems in the absence of a redox mediator.Acknowledgements
The authors gratefully thank the Engineering and Physical Sciences Research Council (EPSRC) platform grant EP/K006800/1, EP/K036769/1 and EP/K022237/1 for financial support.Notes and references
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed.
- R. Abe, T. Takata, H. Sugihara and K. Domen, Chem. Commun., 2005, 3829–3831 RSC.
- M. Tabata, K. Maeda, M. Higashi, D. Lu, T. Takata, R. Abe and K. Domen, Langmuir, 2010, 26, 9161–9165 CrossRef CAS PubMed.
- H. Kato, M. Hori, R. Konta, Y. Shimodaira and A. Kudo, Chem. Lett., 2004, 33, 1348–1349 CrossRef CAS.
- R. M. Navarro, M. C. Alvarez-Galvan, J. A. Villoria de la Mano, S. M. Al-Zahrani and J. L. G. Fierro, Energy Environ. Sci., 2010, 3, 1865–1882 CAS.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 CAS.
- Y. Miseki, S. Fujiyoshi, T. Gunji and K. Sayama, Catal. Sci. Technol., 2013, 3, 1750–1756 CAS.
- Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536–17542 CAS.
- M. Higashi, R. Abe, K. Teramura, T. Takata, B. Ohtani and K. Domen, Chem. Phys. Lett., 2008, 452, 120–123 CrossRef CAS PubMed.
- D. J. Martin, P. J. T. Reardon, S. J. A. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568–12571 CrossRef CAS PubMed.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS PubMed.
- K. Yamaguti and S. Sato, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 1237–1246 RSC.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Chem. Commun., 2006, 4530–4532 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- S. C. Yan, S. B. Lv, Z. S. Li and Z. G. Zou, Dalton Trans., 2010, 39, 1488–1491 RSC.
- H. Kominami, K.-i. Yabutani, T. Yamamoto, Y. Kera and B. Ohtani, J. Mater. Chem., 2001, 11, 3222–3227 RSC.
- G. Zhao, J. Li, X. Ren, C. Chen and X. Wang, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, S. S. K. Ma, Y. Li and K. Domen, Chem. Mater., 2014, 26, 4144–4150 CrossRef CAS.
- C. Bechinger, G. Oefinger, S. Herminghaus and P. Leiderer, J. Appl. Phys., 1993, 74, 4527–4533 CrossRef CAS PubMed.
- J. G. Zhang, D. K. Benson, C. E. Tracy, S. K. Deb, A. W. Czanderna and C. Bechinger, J. Electrochem. Soc., 1997, 144, 2022–2026 CrossRef CAS PubMed.
- Y. He, Z. Wu, L. Fu, C. Li, Y. Miao, L. Cao, H. Fan and B. Zou, Chem. Mater., 2003, 15, 4039–4045 CrossRef CAS.
- W. Morales, M. Cason, O. Aina, N. R. de Tacconi and K. Rajeshwar, J. Am. Chem. Soc., 2008, 130, 6318–6319 CrossRef CAS PubMed.
- G. Hodes, D. Cahen and J. Manassen, Nature, 1976, 260, 312–313 CrossRef CAS PubMed.
- M. A. Butler, R. D. Nasby and R. K. Quinn, Solid State Commun., 1976, 19, 1011–1014 CrossRef CAS.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Corà, S. Firth, J. A. Darr and P. F. McMillan, J. Phys. Chem. C, 2013, 117, 7178–7185 CAS.
- W. Ho, J. C. Yu, J. Lin, J. Yu and P. Li, Langmuir, 2004, 20, 5865–5869 CrossRef CAS.
- J. Yu, Y. Hai and M. Jaroniec, J. Colloid Interface Sci., 2011, 357, 223–228 CrossRef CAS PubMed.
- J. Yu, X. Zhao and Q. Zhao, Thin Solid Films, 2000, 379, 7–14 CrossRef CAS.
- X.-H. Li, J.-S. Chen, X. Wang, J. Sun and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 8074–8077 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS.
- Y. Li, H. Zhang, P. Liu, D. Wang, Y. Li and H. Zhao, Small, 2013, 9, 3336–3344 CAS.
- P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920–4935 CrossRef CAS PubMed.
- M. Hara, J. Nunoshige, T. Takata, J. N. Kondo and K. Domen, Chem. Commun., 2003, 3000–3001 RSC.
- N. Bao, L. Shen, T. Takata and K. Domen, Chem. Mater., 2007, 20, 110–117 CrossRef.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940–4947 CAS.
- Y. Zhong, K. Ueno, Y. Mori, X. Shi, T. Oshikiri, K. Murakoshi, H. Inoue and H. Misawa, Angew. Chem., 2014, 126, 10518–10522 CrossRef PubMed.
- G. M. Bancroft, I. Adams, L. L. Coatsworth, C. D. Bennewitz, J. D. Brown and W. D. Westwood, Anal. Chem., 1975, 47, 586–588 CrossRef CAS.
- G. Johansson, J. Hedman, A. Berndtsson, M. Klasson and R. Nilsson, J. Electron Spectrosc. Relat. Phenom., 1973, 2, 295–317 CrossRef CAS.
- S. J. Hong, S. Lee, J. S. Jang and J. S. Lee, Energy Environ. Sci., 2011, 4, 1781–1787 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: H2 evolution performance by C3N4-rGO composites with TEOA as sacrificial agent under visible light, rGO contents in different C3N4-rGO composites, and the mass spectra results using D2O. See DOI: 10.1039/c5cy00379b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |