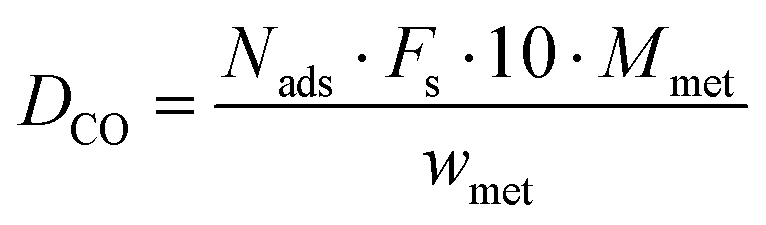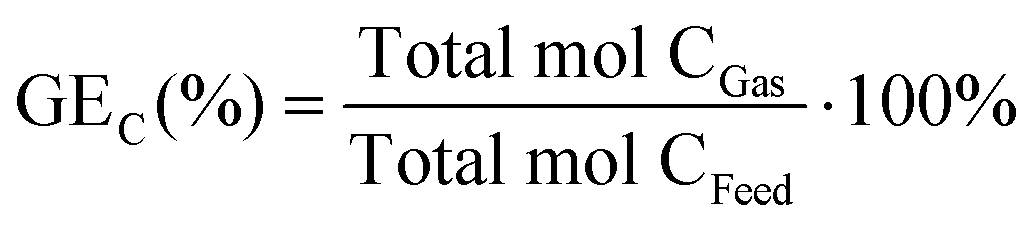Effect of carbon surface functional groups on the synthesis of Ru/C catalysts for supercritical water gasification
Received
9th March 2015
, Accepted 11th May 2015
First published on 12th May 2015
Abstract
A carbon support was treated with HNO3 to create surface functional groups (e.g. –COOH, –OH), which were then characterized by TGA, TPD, CNS elemental analysis, and Boehm titration. HNO3 modified the carbon surface properties by adding a high amount of carboxylic groups, improved the thermal stability of the carbon support, and reduced ca. 50% of the ash. The thermal pre-treatment (723 K under He) following the HNO3 pre-treatment successfully removed the carboxylic groups. 4% Ru/C catalysts were synthesized using the surface-modified carbon supports and characterized by H2-TPR, CO pulse chemisorption, N2-physisorption and HAADF-STEM. The Ru dispersion was increased in the presence of the carboxylic groups. Catalytic supercritical water gasification (CSCWG) of 10 wt.% isopropanol over the 4% Ru/C catalysts was carried out at 723 K and 30 MPa for 50 hours to assess the performance of the catalysts. It was found that the Ru/C catalyst prepared involving a pre-treatment with HNO3 did not exhibit a higher catalytic activity than the catalyst whose carbon support was not pre-treated with HNO3. Hence, the activity and the selectivity during CSCWG were not influenced by the pre-treatment of the catalyst support with HNO3.
1. Introduction
There is a strong motivation for developing renewable alternatives to conventional fossil fuels. Due to the worldwide availability and sustainability of biomass, biomethane production from biomass appears to be an attractive option. The current technologies for biomethane production are conventional gasification/methanation and anaerobic digestion. The conventional gasification/methanation is only able to process dried biomass (water content < 15 wt.%) requiring a drying step with a high energy demand. Although anaerobic digestion is able to treat wet biomass (e.g. microalgae, sewage sludge, biomass residues) avoiding the drying step, it has a low thermal efficiency (25–35%) and requires long residence times (20–33 days).1 Catalytic supercritical water gasification (CSCWG) is an alternative since wet biomass (water content > 60 wt.%) can be processed with a high thermal efficiency (60–70%) avoiding the biomass drying step and offering short residence times (<30 min). For achieving full biomass conversion and a high CH4 selectivity, a catalyst is needed for decomposing the large organic molecules by C–C bond cleavage.2 It has been reported that supported Ru catalysts are the most suitable catalysts for CH4 production due to the high activity and high CH4 selectivity of Ru.3–7 For instance, Osada et al.7 investigated the catalytic performance of various supported metal catalysts (e.g. Ru, Rh, Pt, Pd, and Ni) during CSCWG of lignin at 673 K and 37.1 MPa. The catalytic activity was in the following order: Ru > Pt > Rh > Pd > Ni demonstrating the better catalytic performance of supported Ru catalysts. Elliott et al.8 studied and reported the long-term stability of Ru/rutile-TiO2 (19 weeks), Ru/C (6 weeks) and Ru/ZrO2 (3 weeks) catalysts during catalytic hydrothermal water gasification of 10 wt.% phenol at 623 K and 21 MPa. Waldner et al.6 have compared the catalytic performance of a skeletal nickel catalyst and a Ru/C catalyst during CSCWG of synthetic liquefied wood at 673 K and 30 MPa. They observed that the skeletal nickel catalyst sintered rapidly while the Ru/C catalyst was stable for more than 220 hours. Zöhrer et al.9 tested the catalytic performance of some metal oxides supported Ru catalysts such as Ru/ZrO2 and Ru/TiO2 during CSCWG of glycerol at 673 K and 28.5 MPa. Although the tested catalysts exhibited good catalytic performance, the catalyst deactivation caused by coke deposits was found to be a serious issue. De Vlieger et al.10 found out that a carbon nanotube supported Ru catalyst was highly effective during reforming of acetic acid at 543 K and 23 MPa and also at 673 K and 25 MPa. However, when working in the subcritical region (573–613 K), the high ion product of subcritical water caused a catalyst deactivation by over-oxidation of Ru. Recently, we reported on the gasification of 10 wt.% isopropanol at 723 K and 30 MPa over a period of 96 hours with a Ru/C catalyst at a weight hourly space velocity (WHSVgRu) of 1228 gOrg gRu−1 h−1.11 By working at a higher WHSVgRu (5202 gOrg gRu−1 h−1), we observed that the catalyst lifetime was affected by the decomposition of isopropanol to solid carbon (coke) and hydrogen on the carbon surface. Up to now only few studies12 focused on the catalyst design for CSCWG. There is still a lack of knowledge about the interdependence between the catalyst formulation, its structure, and its catalytic performance. In order to further improve the performance of the Ru/C catalysts, there is a necessity to assess the effect of the most relevant properties of carbon (e.g. surface functional groups, degree of graphitization, impurities) as well as the catalyst preparation method. In a previous study,11 we reported that a higher Ru dispersion and the use of a chloride-free ruthenium salt precursor improved significantly the catalytic activity during CSCWG of isopropanol (723 K, 30 MPa). In this work, we studied the effect of the surface acidity of carbon on the synthesis of Ru/C catalysts by adding different surface functional groups (e.g. –COOH, –OH groups) with HNO3 prior to active phase impregnation, and tested them during CSCWG with 10 wt.% isopropanol in water. It is well known that introducing surface functional groups by acid treatment is beneficial for the metal dispersion of many carbon supported noble metal catalysts.13 The surface functional groups are known to act as “anchoring sites” during the catalyst preparation favoring a better metal dispersion. It was reported that the pre-treatment of the carbon support with HNO3 was able to improve the Pt dispersion from 13% up to 92% compared to the fresh Pt/C catalyst.14 Wang et al.15 reported also the benefit of the HNO3 pre-treatment on the Pt dispersion for multi-walled carbon nanotubes (MWCNT) supported Pt catalysts. Numerous studies16,17 have reported on the benefit of the HNO3 pre-treatment for Ru/C catalysts. For instance, Li et al.16 found that the HNO3 pre-treatment was able to enhance the Ru dispersion from 17% up to 38% in comparison to the fresh catalyst. Zhu et al.17 reported an increase of the Ru dispersion from 24% to 60% after the HNO3 pre-treatment. Gallegos-Suarez et al.18 have recently studied the effect of the surface functional groups of Ru/C catalysts during hydrogenolysis of glycerol and they observed that the HNO3 pre-treatment of the carbon support led to a smaller Ru dispersion. For the fresh 4% Ru/C catalyst they found a Ru dispersion of 19%, whereas the dispersion was 15% for the catalyst prepared with HNO3. According to them, the surface functional groups were responsible for the Ru nanoparticle (NPs) sintering during the thermal treatment. These contradictory observations might be explained by the nature of carbon which is a versatile material containing a variety of impurities as well as exhibiting different surface and physical structure properties. In relation to gaseous fuels production, Wang et al.15,19 studied the effect of the surface functional groups during aqueous phase reforming of ethylene glycol over single-walled carbon nanotubes (SWCNT) and MWCNT supported Pt catalysts. They observed that the surface functional groups negatively affected the catalytic activity. Although the Pt dispersion was improved for the catalyst prepared with HNO3, according to them, the lower catalytic activity was related to the polarity change of the support which caused more adsorption competition on the Pt surface between water and ethylene glycol.
2. Experimental
2.1 Catalyst preparation and characterization
A selected carbon support originating from coconut shells (denoted here as Org10_CO from Desotec) was sieved to yield a fraction of 0.3–0.8 mm, and then pre-treated with HNO3 (30 vol.%) under reflux (363 K) for 5 hours. After filtration, the carbon support pre-treated with HNO3 (CHNO3) was washed with deionized water until neutralization of the filtrate was reached and finally dried at 363 K overnight in an oven. CHNO3 was then treated under He at 723 K for 4 hours in order to remove the less thermally stable surface functional groups. The thermally pre-treated CHNO3 support is denoted as CHT. The samples were impregnated in a pure acetone solution of RuCl3·xH2O (99.9%, Alfa Aesar) during 24 hours, followed by solvent evaporation in a rotary evaporator and washed with 200 mL of pure water during filtration. Finally, the samples were dried in an oven at 363 K overnight. Prior to each experiment, the fresh catalysts were reduced under flowing H2/Ar (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90, 20 mL min−1) at 723 K for 4 hours in order to clean the catalyst from chloride and other carbon deposits. The Ru loading was determined by ICP-OES (Liberty 110, Varian) by measuring the Ru concentration in the spent impregnation solution by taking into account the Ru loss during the washing. The surface functional groups were characterized qualitatively by temperature-programmed oxidation (TPO) and by temperature-programmed desorption (TPD) coupled to a FTIR detector for CO2 and CO detection (NETZSCH STA 449 C). For the TPO, 10 mg of sample was loaded and heated from room temperature (RT) to 383 K under Ar atmosphere for 30 min and then heated up to 1173 K at 10 K min−1 under flowing O2/Ar (10:90, 10 mL min−1). The ash content of the carbon support was determined by TPO. For the TPD, 50 mg of sample was weighted and measured from RT to 1173 K with a ramp of 10 K min−1 under flowing Ar (20 mL min−1). The surface functional groups quantification was performed by Boehm titration following the standardization procedure proposed by Goertzen et al.20,21 This method relies on the existence of oxygen surface groups having different acidities that can be neutralized by bases having different strengths. NaHCO3 (pKa = 6.37), Na2CO3 (pKa = 10.25) and NaOH (pKa = 15.74) were used in this work. As the weakest base, NaHCO3 neutralizes only the carboxylic groups, Na2CO3 neutralizes the lactonic and carboxylic groups, and NaOH neutralizes the phenolic, lactonic, and carboxylic groups. The molar amount of each surface functional group was estimated by difference. 1.5 g of the carbon support was added to 50 mL of one of the three bases: 0.05 M NaHCO3; 0.05 M Na2CO3 and 0.05 M NaOH. Then, the samples were sealed and shaken for 24 hours at RT by a linear shaker. After filtration, an aliquot of 10 mL from the sample solution was taken and neutralized by an excess of 0.05 M HCl and then back-titrated with 0.05 M NaOH. Note that the titration was performed under inert atmosphere (Ar) to avoid dissolution of CO2 from the atmosphere. The endpoint was determined by phenolphthalein. The determination of the moles of carbon surface functionalities (nCSF) was calculated by the following equation according to the back-titration method:
:90, 20 mL min−1) at 723 K for 4 hours in order to clean the catalyst from chloride and other carbon deposits. The Ru loading was determined by ICP-OES (Liberty 110, Varian) by measuring the Ru concentration in the spent impregnation solution by taking into account the Ru loss during the washing. The surface functional groups were characterized qualitatively by temperature-programmed oxidation (TPO) and by temperature-programmed desorption (TPD) coupled to a FTIR detector for CO2 and CO detection (NETZSCH STA 449 C). For the TPO, 10 mg of sample was loaded and heated from room temperature (RT) to 383 K under Ar atmosphere for 30 min and then heated up to 1173 K at 10 K min−1 under flowing O2/Ar (10:90, 10 mL min−1). The ash content of the carbon support was determined by TPO. For the TPD, 50 mg of sample was weighted and measured from RT to 1173 K with a ramp of 10 K min−1 under flowing Ar (20 mL min−1). The surface functional groups quantification was performed by Boehm titration following the standardization procedure proposed by Goertzen et al.20,21 This method relies on the existence of oxygen surface groups having different acidities that can be neutralized by bases having different strengths. NaHCO3 (pKa = 6.37), Na2CO3 (pKa = 10.25) and NaOH (pKa = 15.74) were used in this work. As the weakest base, NaHCO3 neutralizes only the carboxylic groups, Na2CO3 neutralizes the lactonic and carboxylic groups, and NaOH neutralizes the phenolic, lactonic, and carboxylic groups. The molar amount of each surface functional group was estimated by difference. 1.5 g of the carbon support was added to 50 mL of one of the three bases: 0.05 M NaHCO3; 0.05 M Na2CO3 and 0.05 M NaOH. Then, the samples were sealed and shaken for 24 hours at RT by a linear shaker. After filtration, an aliquot of 10 mL from the sample solution was taken and neutralized by an excess of 0.05 M HCl and then back-titrated with 0.05 M NaOH. Note that the titration was performed under inert atmosphere (Ar) to avoid dissolution of CO2 from the atmosphere. The endpoint was determined by phenolphthalein. The determination of the moles of carbon surface functionalities (nCSF) was calculated by the following equation according to the back-titration method:| |  | (1) |
where [B] and VB correspond to the concentration and the volume, respectively, of the reaction base mixed with the carbon support. [HCl] and VHCl are the concentration and the volume of the acid added to the aliquot (Va) previously taken from VB. [NaOH] and VNaOH are related to the concentration and to the volume used in the back-titration that neutralizes the remaining moles of acid. Finally, nHCl/nB is the molar ratio of acid to base allowing to distinguish between monoprotic vs. diprotic reaction bases.
CNS elemental analysis (Vario EL cube, Elementar) was performed to quantify the carbon, nitrogen and sulfur content. The catalysts were characterized by H2-temperature-programmed reduction (H2-TPR) and CO pulse chemisorption in a fully automated instrument (TPD/R/O 1100, Thermo) connected to a TCD. For each H2-TPR measurement, 100 mg of sample was weighted and heated (10 K min−1) from RT to 723 K under flowing H2/Ar (10:90, 20 mL min−1). For the CO pulse chemisorption, the samples were reduced under H2/Ar (10:90, 20 mL min−1) at 723 K for 4 hours in order to clean the ruthenium surface from any deposited carbon species. Then it was flushed by pure He at 723 K for 1.5 hours to remove adsorbed H2 from the catalyst and finally cooled down to RT. The CO pulses were carried out with CO/He (20:80, 40 mL min−1) at RT. The dispersion was calculated by assuming 1 as the stoichiometric factor for CO:Ru. The following formula was used for determining the dispersion:
| |  | (2) |
where
Nads is the amount of gas adsorbed in pulse chemisorption (mmole g
−1);
Fs corresponds to the stoichiometric factor (metal mole per gas mole);
Mmet is the metal atomic weight (g mole
−1) and
wmet is the metal loading of the catalyst (wt.%). The average metal particle sizes were calculated as:
| |  | (3) |
where
dat is the atomic diameter of Ru (
dat = 2.6 Å).
22 The N
2-physisorption measurements were performed with an Autosorb-1 (Quantachrome Instruments) for determining the BET specific surface area and the porosity. The total pore volume was measured at
p/
p0 = 0.99 and the mesopore volume was determined with the
t-plot method. Prior to N
2-physisorption degassing under He at 573 K for 6 hours was carried out for all the samples. The Ru NPs were characterized by Scanning Transmission Electron Microscopy (STEM). The measurements were performed with a spherical aberration corrected STEM microscope (Hitachi HD-2700) with a cold field emission source, operated at an acceleration voltage of 200 kV and equipped with a high angle annular dark field (HAADF) detector. For each sample, different areas were carefully selected in order to have a reliable representation of the average Ru NPs size. The average Ru NPs size (
dp,STEM) and the Ru dispersion (
DSTEM) were calculated as:
| | 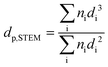 | (4) |
| |  | (5) |
where
ni is the number of particles with diameter
di.22
2.2 Catalytic tests
The CSCWG experiments were carried out in a fixed-bed plug flow (PF) reactor described in detail elsewhere.11 Isopropanol was purchased from VWR BDH Prolabo (99.8%). The mass flow rate (F) was kept at 3 g min−1. The amount of Ru/C catalyst added in the reactor was ca. 0.2 g. WHSVgRu was 1972 gOrg gRu−1 h−1 for all experiments. The reactor was first heated up with water to 723 K under 30 MPa prior to switching to isopropanol (10 wt.% in water). The residence time in the catalyst bed was ca. 0.6 s (pure water density at reaction conditions is 148 kg m−3). The experiments were all carried out for 50 hours.
2.3 Analytical methods for gaseous and liquid effluents
The gas phase was analyzed offline with a gas chromatograph (HP 6890, columns: HP-Plot Q 30 m × 0.53 mm × 40 μm and HP-Plot Molecular Sieve 5A, 30 m × 0.53 mm × 40 μm) with helium as the carrier gas using a Thermal Conductivity Detector (TCD) to detect CO2, CH4, CO and H2 and a Flame Ionization Detector (FID) for higher hydrocarbons (C2H6 and C3Hx). The liquid samples were collected regularly manually and the total organic carbon (TOC) was measured with a TOC analyzer (Vario TOC cube, Elementar).
2.4 Chemical equilibrium composition
The thermodynamic chemical equilibrium calculation was performed using the Aspen Plus® 2006 simulation package by using the Peng–Robinson equation of state. The values for the dry gas composition at the thermodynamic chemical equilibrium (723 K, 30 MPa and 10 wt.% isopropanol) are: CO2 = 24.7 vol.%; CH4 = 65.6 vol.%; H2 = 8.7 vol.%; CO = 0.7 vol.%.
2.5 Terms and definitions
For comparing the catalytic performances based on the Ru amount, the weight hourly space velocity normalized to one gram of Ru (WHSVgRu) is used:| |  | (6) |
The observed activity is defined as the total organic carbon conversion (Xc) from the feed to the liquid effluent:
| |  | (7) |
The carbon gasification efficiency (GEC) is the relation between the total amount of carbon in the gas phase and the total amount of carbon in the feed, defined as:
| |  | (8) |
3. Results and discussion
3.1 Characterization of the carbon support
Some structural properties of the carbon support are listed in Table 1.
Table 1 Structural properties of the carbon support
| Sample |
BET SSA (m2 g−1) |
V
total (cm3 g−1) |
V
mesop. (cm3 g−1) |
V
microp. (cm3 g−1) |
Ash content (wt.%)a |
|
Determined by TPO analysis.
|
| Fresh C |
802 |
0.83 |
0.69 |
0.14 |
11.0 |
| CHNO3 |
781 |
0.63 |
0.45 |
0.18 |
4.9 |
| CHT |
999 |
0.66 |
0.37 |
0.29 |
4.6 |
The physical structure of the carbon support was affected by the HNO3 pre-treatment since a loss of the BET SSA and the total pore volume occurred. This decrease of the porosity was caused either by a collapse of the physical structure or by a physical blockage of the pores by the formation of humic substances during the HNO3 pre-treatment.23–25 After the thermal pre-treatment, the micropore volume increased by almost 30% while the mesopore volume decreased by 35%. Possibly ashes and humic acids trapped in the micropores were removed and led to the increased microporosity. The ash content was reduced by 55% after the HNO3 pre-treatment, showing its efficiency for cleaning the pores as reported by others.17,26
In Table 2, CHONS elemental analysis results show a decrease of the carbon content after the HNO3 pre-treatment due to the increase of the oxygen content, whereas the nitrogen and the hydrogen concentration increased after the oxidation pre-treatment. HNO3 is able to provide a nitronium ion (NO2+) to an aromatic ring (nitration reaction).25,27 After the thermal pre-treatment, a fraction of the nitrogen was released probably by desorption of NO2 that is reported to decompose around 553 K.27 The new increase of the carbon concentration after the thermal pre-treatment reflects the loss of oxygen. Indeed, a considerable fraction of the acidic oxygen-containing functional groups (e.g. carboxylic groups) are removed during the thermal treatment.25 The sulfur concentration was found to decrease slightly after the HNO3 pre-treatment.
Table 2 CHONS elemental analysis of the carbon support
| Sample |
C (wt.%) |
H (wt.%) |
O (wt.%) |
N (wt.%) |
S (wt.%) |
| Fresh C |
85.12 ± 0.05 |
0.75 ± 0.03 |
5.00 ± 0.00 |
0.54 ± 0.01 |
0.26 ± 0.02 |
| CHNO3 |
78.58 ± 0.10 |
0.91 ± 0.01 |
13.00 ± 0.00 |
1.26 ± 0.04 |
0.17 ± 0.01 |
| CHT |
81.88 ± 0.05 |
0.70 ± 0.02 |
10.00 ± 0.00 |
1.08 ± 0.06 |
0.19 ± 0.01 |
In Fig. 1, the first peak at 490 K for the CHNO3 can be assigned to the decomposition of the carboxylic groups.28
 |
| | Fig. 1 TPO analysis of the fresh C, CHNO3, and CHT. | |
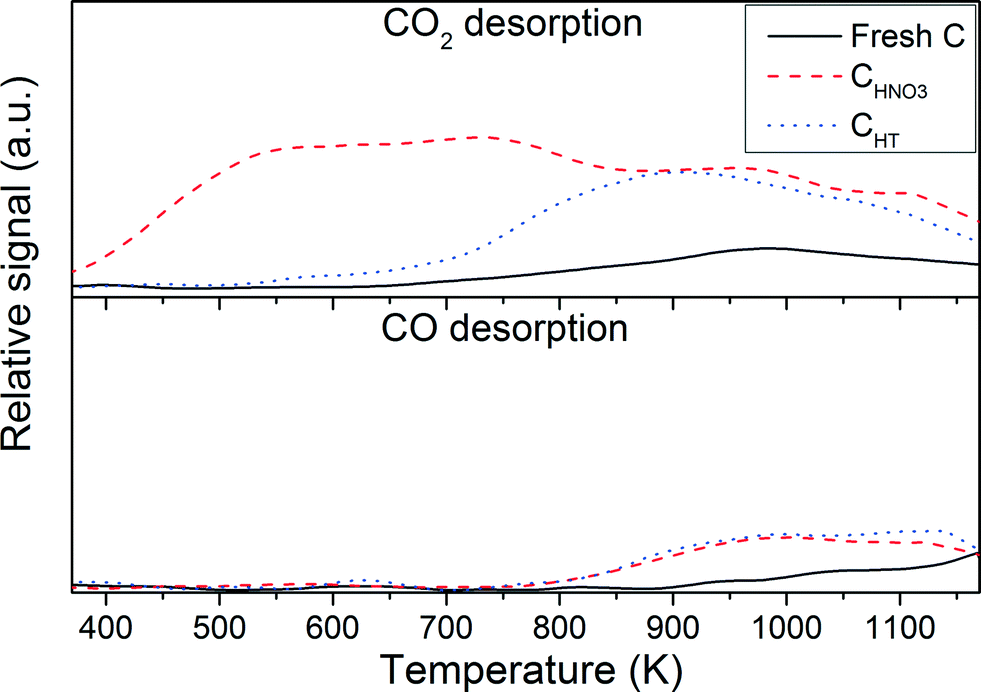
The CHNO3 started to oxidize at a lower temperature than the fresh C and the CHT. The fresh C appears to be fully oxidized at 870 K, whereas the temperature was shifted to 900 K and 940 K for the CHNO3 and CHT, respectively. According to this observation it seems that the HNO3 pre-treatment enhanced the thermal resistance of the carbon support. Moreover, the thermal pre-treatment further increased its thermal stability. Chiang et al.29 have reported that the thermal stability of the MWCNT was improved after the H2SO4/HNO3 pre-treatment. According to them, some existing reactive groups on the support such as CH2 and –CH are decomposed during the acidic pre-treatment, rendering the material more thermally resistant. During the TPD experiments the surface functional groups decompose within a specific temperature range (according to the type of surface functional groups) to produce CO2, CO, and H2O. In Fig. 2, the TPD results for the fresh C support show only a small CO2 desorption peak at 973 K likely corresponding to carboxylic anhydride while the CO signal above 1173 K can be related to carbonyl and/or quinone.30 After the HNO3 pre-treatment the CO2 desorption peaks at ca. 573 K and 723 K can be assigned to carboxylic and lactone, respectively. The CO2 and CO desorption peaks above 973 K are attributed to more thermally stable groups such as carboxylic anhydride, quinone, and carbonyl. The effect of the thermal pre-treatment under He carried up to 723 K removed a large quantity of carboxylic groups, whereas the more thermally stable groups were preserved.
 |
| | Fig. 2 CO2 and CO-TPD analysis of the fresh C, CHNO3, and CHT. | |
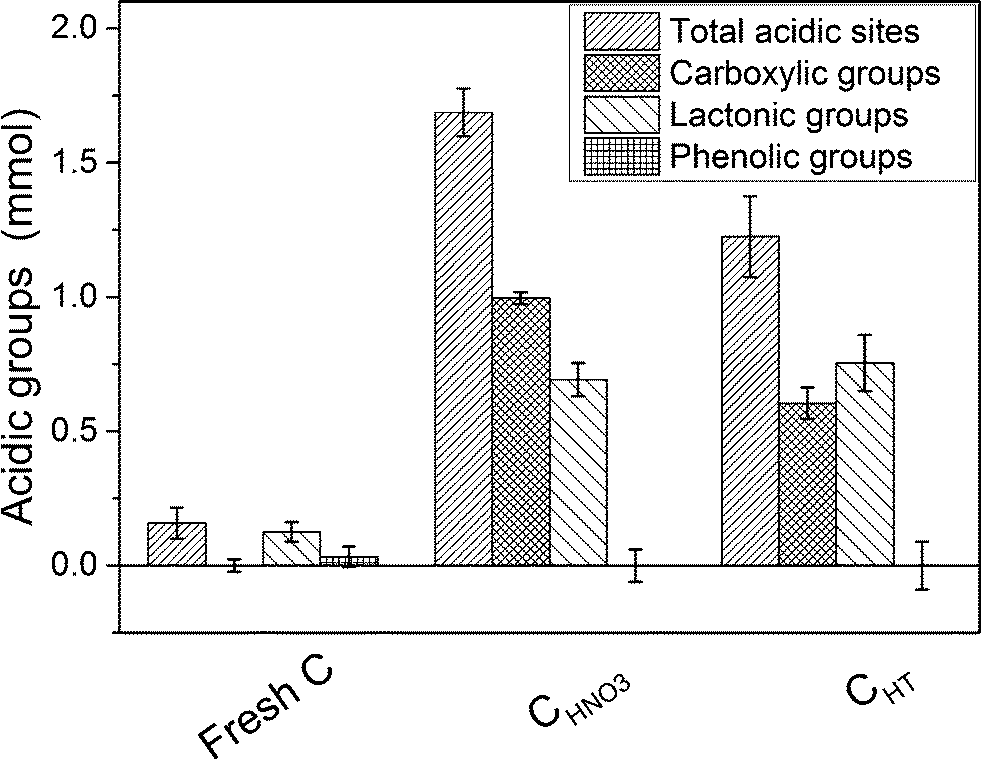
In Fig. 3, the Boehm titration results reveal a good correlation with the TPD results since no carboxylic groups and only a small amount of lactonic groups were detected on the fresh C. After the HNO3 pre-treatment, a considerable increase of the carboxylic and lactonic groups was observed, and the total number of acidic sites was ca. 11 times higher. Hence, the pre-treatment with HNO3 was able to modify the carbon surface properties. As expected, the thermal pre-treatment mainly removed the less thermally stable groups, i.e. the carboxylic groups while the lactonic groups were hardly affected. According to the Boehm titration results, no thermally stable surface functional groups (carbonyl, quinone) were found on the carbon surface. Therefore the pre-treatment with HNO3 rather produced carboxylic and lactonic groups.
 |
| | Fig. 3 Boehm titration results analysis of the fresh C, CHNO3, and CHT. | |
3.2 Catalyst characterization
The characteristics of the Ru/C catalysts are listed in Table 3.
Table 3 Characteristics of the Ru/C catalysts
| Sample |
BET SSA (m2 g−1) |
V
total (cm3 g−1) |
V
microp. (cm3 g−1) |
Ru loading (wt.%) |
H2 consumption (μmol g−1) |
D
CO (−)a |
D
STEM (−)b |
d
p,CO (nm)a |
d
p,STEM (nm)b |
|
Determined by CO pulse chemisorption.
Determined by STEM.
|
| 4% Ru/C |
551 |
0.67 |
0.08 |
4.2 |
790 |
0.09 |
0.26 |
15.3 |
4.5 ± 0.2 |
| 4% Ru/CHNO3 |
728 |
0.70 |
0.14 |
4.0 |
964 |
0.13 |
0.39 |
9.9 |
2.7 ± 0.1 |
| 4% Ru/CHT |
646 |
0.61 |
0.12 |
4.2 |
867 |
0.10 |
0.31 |
12.5 |
3.6 ± 0.1 |
The impregnation with Ru affected the specific surface area significantly (compare with Table 1). Although the three Ru/C catalysts have the same Ru loading, their respective specific surface area differs considerably. According to the Ru NPs measurements, it seems that larger Ru NPs led to a reduction of the specific surface area and of the micropore volume. The blockage of the entrance of some of the micropores by larger Ru NPs may be the reason.
Fig. 4 shows the STEM images of the three Ru/C catalysts where the bright dots represent the Ru NPs. According to the histograms of the particle size distribution, the Ru NPs size distribution of the 4% Ru/C appears to be larger in comparison to the 4% Ru/CHNO3 and 4% Ru/CHT since Ru NPs from 2–10 nm can be seen. The main reason is the lack of anchoring sites which help to obtain smaller Ru NPs during the catalyst preparation.13,16 The larger Ru NPs of the 4% Ru/CHT in comparison to the 4% Ru/CHNO3 reveals that carboxylic groups play an important role for the Ru dispersion improvement. The CO chemisorption results confirmed that the surface functional groups are needed for the Ru dispersion improvement. However, the Ru NPs size based on the CO chemisorption has been overestimated (7–11 nm) for the three Ru/C catalysts. The presence of residual chloride coming from the Ru salt precursor (RuCl3) is responsible for the inhibition of the CO adsorption on the Ru surface.11,16,18,31 For instance, Gallegos-Suarez et al.18 have compared the effect of residual chloride on the Ru NPs size by preparing 4% Ru/C catalysts with a RuCl3·xH2O and a Ru(NO)(NO3)3. The Ru NPs size determined with CO chemisorption was 6.8 nm for the catalyst prepared with RuCl3·xH2O vs. 3 nm for the for the catalyst prepared with Ru(NO)(NO3)3. Interestingly, the Ru NPs were in the range of 3 nm when determined with transmission electron microscopy for both catalysts. Therefore, in presence of residual chloride, the STEM measurements are much more reliable for the determination of the Ru NPs size. In Fig. 5, the H2-TPR results of the three Ru/C catalysts are depicted. After integration of the reduction peaks (see Table 3), the 4% Ru/CHNO3 exhibits the highest H2 consumption, likely due to the reduction of some surface functional groups in the vicinity of the Ru NPs and/or its higher Ru dispersion in comparison to the two other catalysts.
 |
| | Fig. 4 STEM images of the (a) 4% Ru/C, (b) 4% Ru/CHNO3, and (c) 4% Ru/CHT catalysts. | |
 |
| | Fig. 5 H2-TPR profiles of the 4% Ru/C, 4% Ru/CHNO3, and 4% Ru/CHT catalysts. | |
Two distinct reduction peaks can be seen for the Ru/C catalysts. The reduction peaks at 538 K and 563 K can be both attributed to the reduction of RuCl3 to Ru0.32,33 Interestingly, it seems that the presence of the surface functional groups favored the reduction of Ru at higher temperature suggesting a better interaction between Ru and the carbon support. It is likely that Ru was grafted on the oxygen function of the carboxylic groups which have stabilized Ru against the reduction. Gallegos-Suarez et al.18 have also observed a similar shift from 470 K to 520 K when the Ru/C catalyst was pre-treated with HNO3. For the 4% Ru/CHT, the two distinct reduction peaks support the suggestion that the carboxylic groups contribute to the enhancement of the metal–support interaction.
3.3 CSCWG over Ru/C catalysts
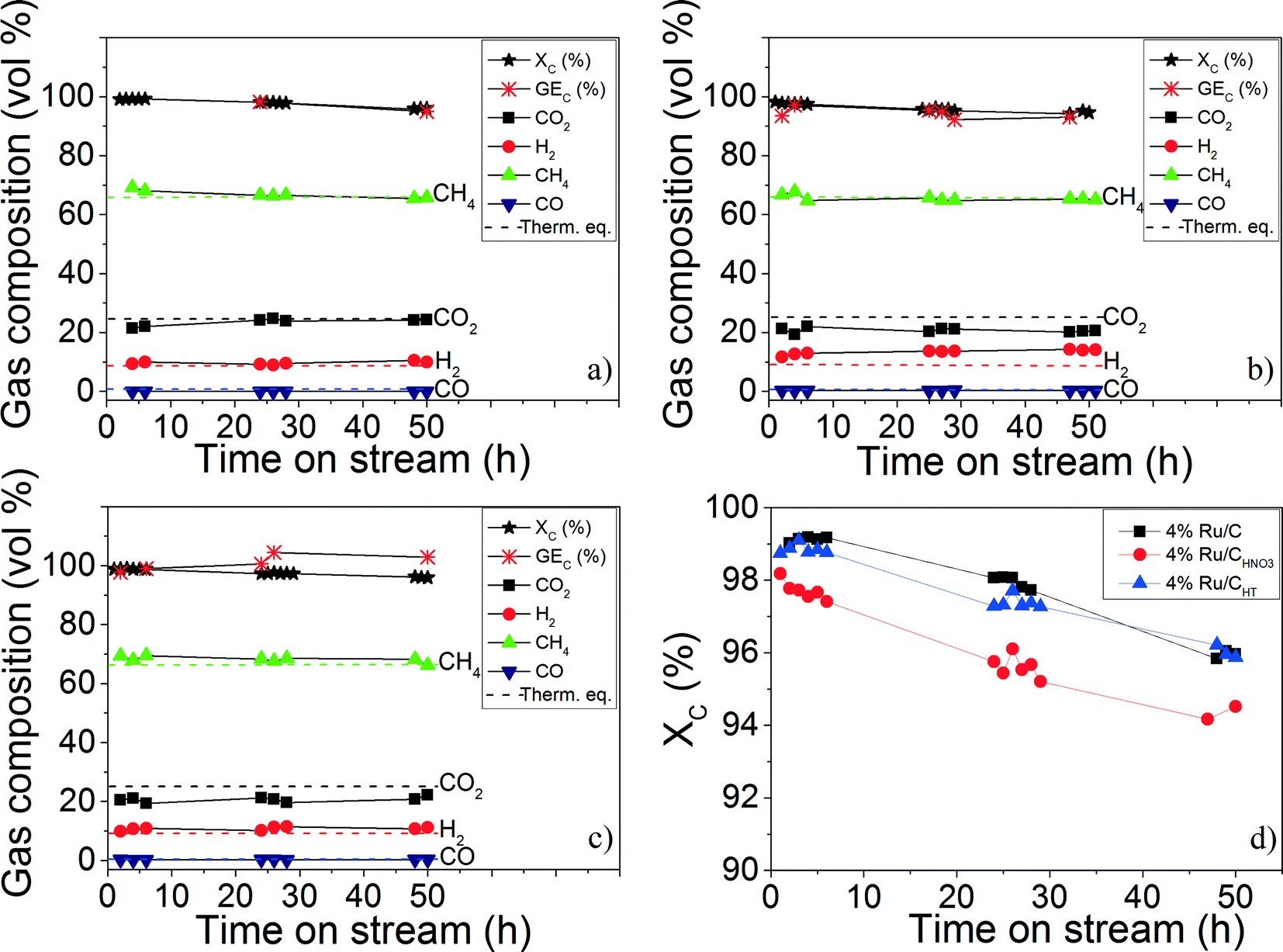
CSCWG of 10 wt.% isopropanol in the absence of a catalyst (blank experiment) at supercritical conditions (723 K and 30 MPa) showed a very low total organic carbon conversion (XC = 4%) indicating the inertness of the reactor wall as well as the stability of isopropanol at these conditions. The catalysts were then tested with 10 wt.% isopropanol at 723 K and 30 MPa. As shown in Fig. 6 and in Table 4, by working at a WHSVgRu = 1972 gOrg gRu−1 h−1, all the three catalysts were able to gasify properly isopropanol to a CH4–rich gas during 50 hours.
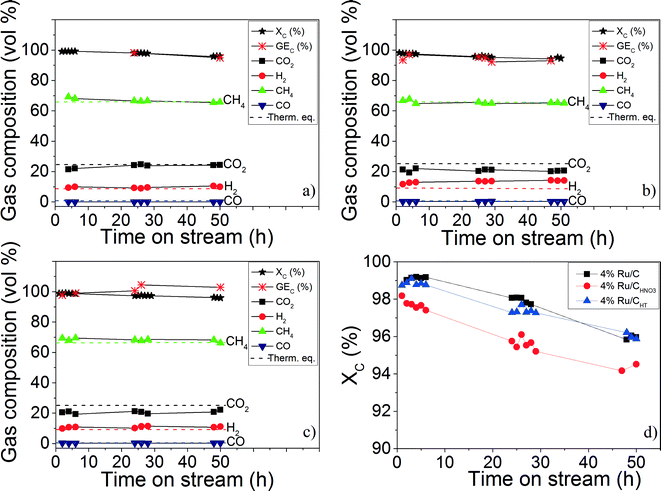 |
| | Fig. 6 CSCWG (723 K, 30 MPa) of 10 wt.% isopropanol over the (a) 4% Ru/C, (b) 4% Ru/CHNO3, (c) and 4% Ru/CHT catalysts during 50 hours with WHSVgRu = 1972 gOrg gRu−1 h−1. (d) Represents the evolution of the total organic carbon conversion (XC) of the three Ru/C catalysts. The dashed lines denote the calculated thermodynamic equilibrium concentrations. | |
Table 4 Results summary after 50 hours of CSCWG (723 K, 30 MPa) of 10 wt.% isopropanol for the Ru/C catalysts performed at WHSVgRu = 1972 gOrg gRu−1 h−1
| Sample |
Time (h) |
X
C (%) |
GEC (%) |
Gas composition (vol.%) |
| CH4 |
CO2 |
H2 |
CO |
C2H6 |
C3Hx |
| 4% Ru/C |
50 |
96.0 |
102.0 |
65.8 |
24.3 |
9.9 |
<0.1 |
<0.1 |
<0.1 |
| 4% Ru/CHNO3 |
50 |
94.5 |
101.2 |
65.0 |
20.6 |
14.1 |
0.3 |
<0.1 |
<0.1 |
| 4% Ru/CHT |
50 |
95.9 |
102.0 |
66.2 |
22.3 |
11.2 |
0.4 |
<0.1 |
<0.1 |
The gas composition was close to the thermodynamic chemical equilibrium indicating the good performance of Ru/C catalysts for enhancing the methanation reaction. Furthermore, the absence of higher hydrocarbons (≥C2+) in the gaseous product is also a reliable indication confirming the high activity of the Ru/C catalysts for C–C bond cleavage. After 50 hours, the activity (XC) of all catalysts dropped from 99% to ca. 95%. This slight decrease was due to a progressive coke deposition on the Ru/C catalyst as reported in a previous study.11 Although the Ru dispersion of the 4% Ru/CHNO3 was higher than the two other catalysts, its catalytic activity was slightly lower (see Fig. 6 (d)). Moreover, the lower CH4 and higher H2 and CO concentration for the 4% Ru/CHNO3 seems to indicate that the methanation reaction was not fully achieved in comparison to the 4% Ru/C. Wang et al.15 have observed the same negative effect of the carboxylic groups during aqueous phase reforming (498 K, 2.7 MPa) of ethylene glycol over Pt/MWCNT catalysts where the catalytic activity of the catalyst prepared with HNO3 was ca. 30% lower than the fresh Pt/MWCNT catalyst. They had also thermally pre-treated (1273 K under He) the carbon support after the HNO3 pre-treatment and reported an activity ca. 15% lower in comparison to the fresh Pt/MWCNT catalyst. They claimed that the main reason for the lower catalytic activity was a change of the carbon surface polarity that inhibited the adsorption of the reactants caused by the carboxylic groups surrounding the Pt NPs. The Ru NPs size effect might also be responsible for the different activity observed. Masini et al.34 observed that the turnover frequency (TOF) was higher for 10 nm Ru NPs than for 4 nm NPs confirming the structure-sensitivity of the methanation reaction. According to them, larger Ru NPs exhibit a higher concentration of under-coordinated sites (e.g. kinks or steps) which are reported to be needed for the improvement of the CO bond dissociation since the latter only takes place on these specific sites.35 The high capability for the CO bond cleavage is determinant since the latter is known to be the rate-determining step of the methanation reaction.35,36
3.4 Characterization of the spent catalysts
The Ru/C catalysts were characterized by N2-physisorption after CSCWG (see Table 5). Although some slight changes of the porosity were noted after 50 hours, the physical structure of the catalysts was well preserved. These results are relevant because they confirm the high stability as well as the robustness of the physical structure of the carbon support during CSCWG. The micropore volume of the spent 4% Ru/C increased to twice the value of the fresh catalyst. A similar result was reported in a previous study.11 Up to now there is no good explanation for such a large increase but it is likely that the washing out of some dust from the pores may form some additional micropores, since the carbon support of the 4% Ru/C was not pre-treated. Although the catalytic activity slightly decreased from 99% to 95%, the porosity was not affected by the coke deposits. In a previous study,11 the porosity of a Ru/C catalyst was also well preserved during CSCWG of 10 wt.% isopropanol although the catalytic activity dropped from 100% to 90%. However, when the catalytic activity dropped up to 10%, most of the porosity was lost due to coke within the catalyst support.
Table 5 Physical structure of the fresh and spent Ru/C catalysts
| Sample |
Time (h) |
BET SSA (m2 g−1) |
V
total (cm3 g−1) |
V
mesop. (cm3 g−1) |
V
microp. (cm3 g−1) |
| 4% Ru/C |
|
551 |
0.67 |
0.59 |
0.08 |
| Spent 4% Ru/C |
50 |
712 |
0.72 |
0.57 |
0.15 |
| 4% Ru/CHNO3 |
|
728 |
0.7 |
0.56 |
0.14 |
| Spent 4% Ru/CHNO3 |
50 |
693 |
0.72 |
0.6 |
0.12 |
| 4% Ru/CHT |
|
646 |
0.61 |
0.49 |
0.12 |
| Spent 4% Ru/CHT |
50 |
680 |
0.64 |
0.51 |
0.13 |
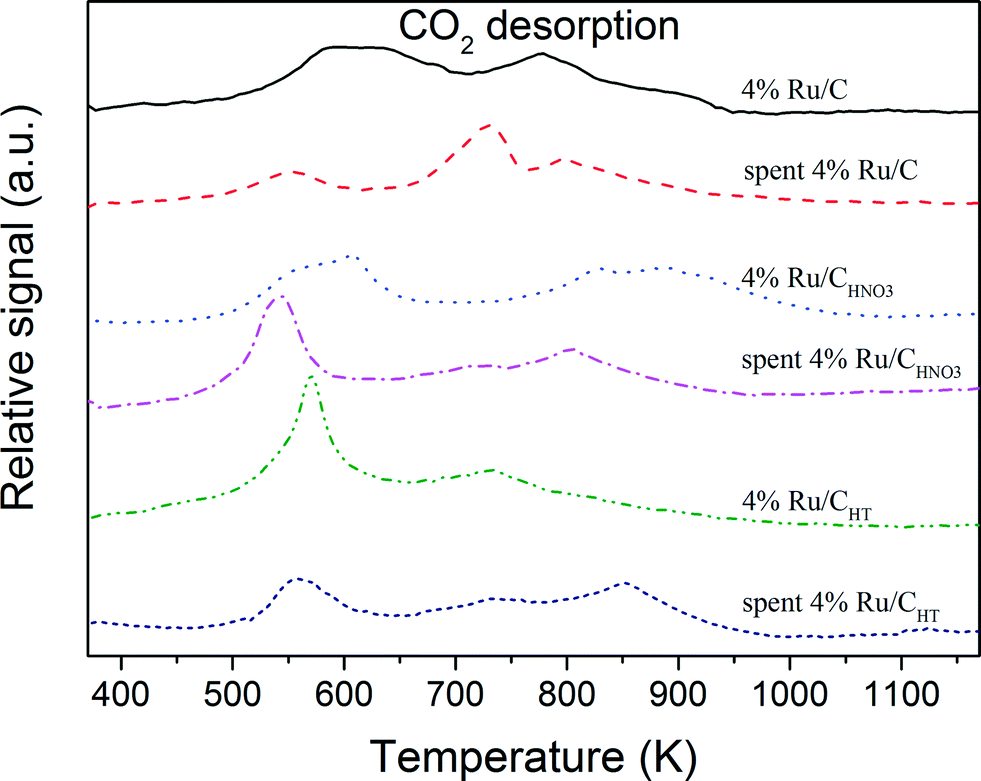
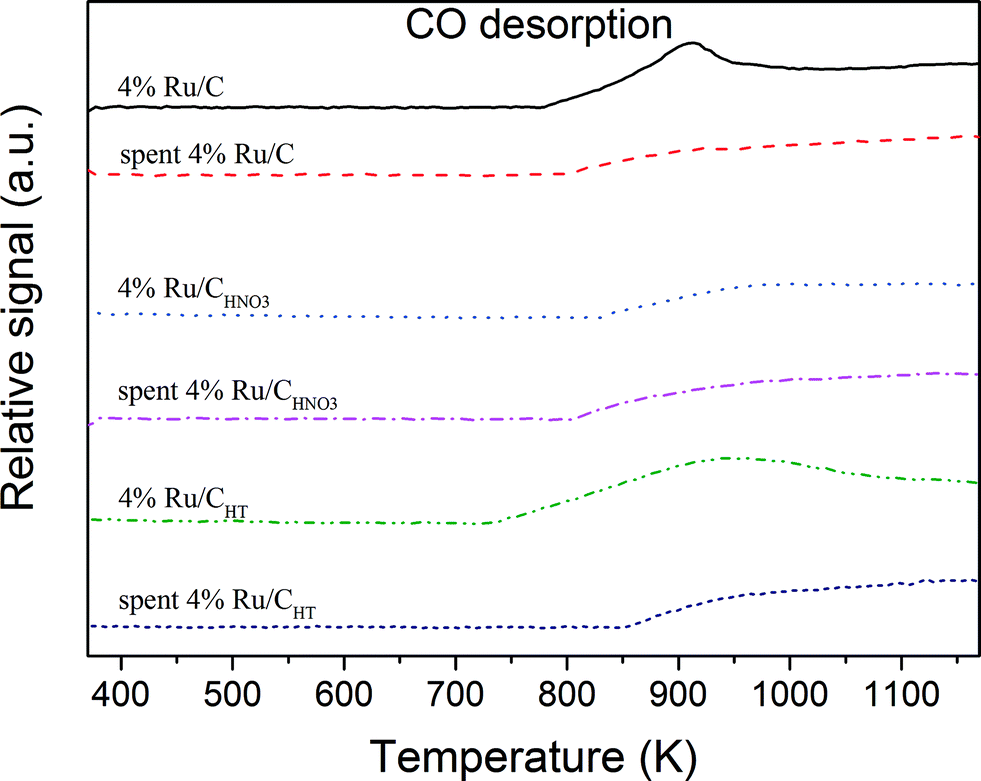
In Fig. 7, three distinct CO2 desorption peaks were observed for the three spent catalysts. Although these peaks look relatively similar, some differences in their intensity and desorption temperature appear. For instance, the intensity of the first desorption peak (540–560 K) increases in the following order: 4% Ru/CHNO3 > 4% Ru/CHT > 4% Ru/C, while the opposite is observed for the desorption peak at 730 K. Interestingly, the CO2 desorption peak at high temperature is shifted to higher temperature for the 4% Ru/CHT (850 K) in comparison to the two other catalysts (800 K). In Fig. 8, a similar trend was observed for the 4% Ru/CHT since the CO desorption started at 850 K while for the other catalysts the latter began at 800 K. At the moment it is not clear what this desorption peak corresponds to. It may be either some CO2/CO species bonded on the Ru surface or even some functional groups in the vicinity of Ru. These peaks might be related to the CO2/CO desorption from different Ru active sites. In fact, each catalyst exhibits specific active sites according to its Ru NPs size distribution.
 |
| | Fig. 7 CO2-TPD analysis of the fresh and spent catalysts. | |
 |
| | Fig. 8 CO-TPD analysis of the fresh and spent catalysts. | |
It is also interesting to discuss the TPD spectra of the fresh Ru/C catalysts. It seems that the Ru incorporation on the carbon support followed by the reduction treatment have both considerably changed the carbon surface functionality (compare with Fig. 2).
For instance, for the 4% Ru/C, three CO2 desorption peaks at 580 K, 640 K, and 780 K are observed, whereas for the fresh C only one desorption peak at 973 K are detected. The same observation can be done for the CO desorption peak at 910 K since the CO desorption occurred above 1100 K for the fresh C. It seems likely that the presence of Ru and the reduction treatment are both responsible for this shift to lower temperature. The desorption of the surface functional groups in the vicinity of the Ru NPs are certainly influenced and facilitated by the interaction with Ru. The similarity of the CO2 desorption peaks for the 4% Ru/CHNO3 with the 4% Ru/C indicates that the carboxylic groups were removed during the reduction treatment. This is a relevant observation confirming that prior to CSCWG the carboxylic groups were fully decomposed. As a consequence, it seems unlikely that the lower catalytic activity for the 4% Ru/CHNO3 was due to a change of the carbon surface acidity but rather due to the formation of smaller Ru NPs exhibiting less under-coordinated sites with the HNO3 pre-treatment.
4. Conclusions
The surface functional groups were needed for the improvement of the Ru dispersion where the carboxylic groups played a role as anchoring groups for the Ru salt precursor. As a result, the Ru NPs size was 2.7 ± 0.1 nm for the Ru/C prepared with HNO3; 3.6 ± 0.1 nm for the Ru/C prepared with HNO3 and thermally pre-treated (723 K under He); and 4.5 ± 0.2 nm for the untreated Ru/C. The pre-treatment of the carbon support with HNO3 was not able to improve the catalytic performance of Ru/C catalyst during CSCWG of isopropanol. The catalytic performance of the Ru/C prepared with HNO3 and thermally pre-treated was similar to the one with the untreated carbon support.
Acknowledgements
This work was financially supported in the frame of the SunCHem project by the Competence Center Energy and Mobility (CCEM-CH) and swisselectric research. The authors would like to thank Albert Schuler for the ICP-OES analyses, Martin Elsener for the CO pulse chemisorption measurements, and Alexander Wokaun for the fruitful discussions.
References
- A. G. Haiduc, M. Brandenberger, S. Suquet, F. Vogel, R. Bernier-Latmani and C. Ludwig, J. Appl. Phycol., 2009, 21, 529–541 CrossRef CAS.
- A. A. Peterson, F. Vogel, R. P. Lachance, M. Froeling, M. J. Antal Jr. and J. W. Tester, Energy Environ. Sci., 2008, 1, 32–65 CAS.
- A. G. Chakinala, J. K. Chinthaginjala, K. Seshan, W. P. M. van Swaaij, S. R. A. Kersten and D. W. F. Brilman, Catal. Today, 2012, 195, 83–92 CrossRef CAS PubMed.
- D. C. Elliott, M. R. Phelps, L. J. Sealock and E. G. Baker, Ind. Eng. Chem. Res., 1994, 33, 566–574 CrossRef CAS.
- D. C. Elliott, L. J. Sealock and E. G. Baker, Ind. Eng. Chem. Res., 1993, 32, 1542–1548 CrossRef CAS.
- M. H. Waldner, F. Krumeich and F. Vogel, J. Supercrit. Fluids, 2007, 43, 91–105 CrossRef CAS PubMed.
- M. Osada, O. Sato, M. Watanabe, K. Arai and M. Shirai, Energy Fuels, 2006, 20, 930–935 CrossRef CAS.
- D. C. Elliott, T. R. Hart and G. G. Neuenschwander, Ind. Eng. Chem. Res., 2006, 45, 3776–3781 CrossRef CAS.
- H. Zöhrer, F. Mayr and F. Vogel, Energy Fuels, 2013, 27, 4739–4747 CrossRef.
- D. J. M. De Vlieger, L. Lefferts and K. Seshan, Green Chem., 2014, 16, 864–874 RSC.
- G. Peng, M. Steib, F. Gramm, C. Ludwig and F. Vogel, Catal. Sci. Technol., 2014, 4, 3329 CAS.
- P. Azadi and R. Farnood, Int. J. Hydrogen Energy, 2011, 36, 9529–9541 CrossRef CAS PubMed.
- Y. Yang, K. Chiang and N. Burke, Catal. Today, 2011, 178, 197–205 CrossRef CAS PubMed.
- A. E. Aksoylu, M. Madalena, A. Freitas, M. F. R. Pereira and J. L. Figueiredo, Carbon, 2001, 39, 175–185 CrossRef CAS.
- X. Wang, N. Li, J. A. Webb, L. D. Pfefferle and G. L. Haller, Appl. Catal., B, 2010, 101, 21–30 CrossRef CAS PubMed.
- Y. Li, C. Pan, W. Han, H. Chai and H. Liu, Catal. Today, 2011, 174, 97–105 CrossRef CAS PubMed.
- H. Zhu, W. Han and H. Liu, Catalysis Letters, 2007, 115, 13–18 CrossRef CAS.
- E. Gallegos-Suarez, M. Perez-Cadenas, A. Guerrero-Ruiz, I. Rodriguez-Ramos and A. Arcoya, Appl. Surf. Sci., 2013, 287, 108–116 CrossRef CAS PubMed.
- X. M. Wang, N. Li, L. D. Pfefferle and G. L. Haller, J. Phys. Chem. C, 2010, 114, 16996–17002 CAS.
- A. M. Oickle, S. L. Goertzen, K. R. Hopper, Y. O. Abdalla and H. A. Andreas, Carbon, 2010, 48, 3313–3322 CrossRef CAS PubMed.
- S. L. Goertzen, K. D. Theriault, A. M. Oickle, A. C. Tarasuk and H. A. Andreas, Carbon, 2010, 48, 1252–1261 CrossRef CAS PubMed.
- A. Borodzinski and M. Bonarowska, Langmuir, 1997, 13, 5613–5620 CrossRef CAS.
- A. Boyano, C. Herrera, M. A. Larrubia, L. J. Alemany, R. Moliner and M. J. Lázaro, Chem. Eng. J., 2010, 160, 623–633 CrossRef CAS PubMed.
- S. Hermans, C. Diverchy, O. Demoulin, V. Dubois, E. M. Gaigneaux and M. Devillers, J. Catal., 2006, 243, 239–251 CrossRef CAS PubMed.
- P. Chingombe, B. Saha and R. J. Wakeman, Carbon, 2005, 43, 3132–3143 CrossRef CAS PubMed.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Orfao, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- I. I. Salame and T. J. Bandosz, J. Colloid Interface Sci., 2001, 240, 252–258 CrossRef CAS PubMed.
- R. R. A. Rios, D. E. Alves, I. Dalmázio, S. F. V. Bento, C. L. Donnici and R. M. Lago, Tailoring activated carbon by surface chemical modification with O, S, and N containing molecules, Mater. Res., 2003, 6(2), 129–135 CrossRef CAS PubMed.
- Y. C. Chiang, W. H. Lin and Y. C. Chang, Appl. Surf. Sci., 2011, 257, 2401–2410 CrossRef CAS PubMed.
- A. Stein, Z. Wang and M. A. Fierke, Adv. Mater., 2009, 21, 265–293 CrossRef CAS PubMed.
- M. Cerro-Alarcon, A. Maroto-Valiente, I. Rodriguez-Ramos and A. Guerrero-Ruiz, Carbon, 2005, 43, 2711–2722 CrossRef CAS PubMed.
- A. Guerrero-Ruiz, P. Badenes and I. Rodriguez-Ramos, Appl. Catal., A, 1998, 173, 313–321 CrossRef CAS.
- S. F. Yin, B. Q. Xu, W. X. Zhu, C. F. Ng, X. P. Zhou and C. T. Au, Catal. Today, 2004, 93–5, 27–38 CrossRef PubMed.
- F. Masini, C. E. Strebel, D. N. McCarthy, A. U. F. Nierhoff, J. Kehres, E. M. Fiordaliso, J. H. Nielsen and I. Chorkendorff, J. Catal., 2013, 308, 282–290 CrossRef CAS PubMed.
- S. B. Vendelbo, M. Johansson, D. J. Mowbray, M. P. Andersson, F. Abild-Pedersen, J. H. Nielsen, J. K. Norskov and I. Chorkendorff, Top. Catal., 2010, 53, 357–364 CrossRef CAS.
- M. P. Andersson, F. Abild-Pedersen, I. N. Remediakis, T. Bligaard, G. Jones, J. Engbæk, O. Lytken, S. Horch, J. H. Nielsen and J. Sehested, J. Catal., 2008, 255, 6–19 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a