
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of novel MgAl layered double oxide grafted TiO2 cuboids and their photocatalytic activity on CO2 reduction with water vapor†
Cunyu
Zhao
a,
Lianjun
Liu
a,
Guiying
Rao
a,
Huilei
Zhao
b,
Luhui
Wang
c,
Jinye
Xu
a and
Ying
Li
*b
aUniversity of Wisconsin-Milwaukee, Mechanical Engineering Department, Milwaukee, WI 53211, USA
bTexas A&M University, Department of Mechanical Engineering, College Station, TX 77843, USA. E-mail: yingli@tamu.edu; Fax: +1 979 845 3081; Tel: +1 979 862 4465
cZhejiang Ocean University, Chemical Engineering Department, Zhejiang, PR China
First published on 8th April 2015
Abstract
A series of magnesium/aluminum (MgAl) layered double oxide (LDO) grafted TiO2 cuboids (MgAl-LDO/TiO2) with various molar ratios of (Mg + Al) to Ti were synthesized by a combination of hydrothermal and coprecipitation methods, in which the growth of MgAl-LDO platelets was controlled. The MgAl-LDO/TiO2 composite materials were used for photocatalytic CO2 reduction with water vapor under UV light irradiation in a continuous-flow reactor. CO was found to be the main product from CO2. At near room temperature (e.g., 50 °C), MgAl-LDO/TiO2 did not significantly enhance CO2 reduction compared with pure TiO2 cuboids. At a moderately elevated reaction temperature (e.g., 150 °C), the MgAl-LDO/TiO2 sample with an optimum 10 wt.% MgAl-LDO loading demonstrated CO2 reduction activity five times higher than that of bare TiO2 cuboids. The photo-induced electrons on TiO2 may migrate to the MgAl-LDO/TiO2 interfacial sites to promote CO2 reduction. Findings in this work may lead to a new area of hybrid adsorbent/photocatalyst materials that are capable of sequential CO2 capture and photocatalytic conversion.
1. Introduction
Emission of carbon dioxide (CO2) from consumption of fossil fuels is one of the main causes of global climate change. Photocatalytic reduction of CO2 with water under sunlight has been considered as a promising way to lower the CO2 level in the atmosphere, meanwhile producing alternative fuels such as carbon monoxide (CO), methane (CH4) and methanol.1,2 Among the various photocatalyst materials, TiO2 is widely studied due to its suitable band positions, environmentally benign nature, low cost and easy availability.3–6 However, photocatalytic CO2 reduction with water using TiO2 as the photocatalyst typically has low energy conversion efficiency.1,2,7,8 This is because of the major obstacles including the fast recombination rate of photo-generated electron–hole (e–h) pairs, the wide band gap of TiO2 (3.2 eV for anatase), and the fast backward reactions.5,9 Approaches such as loading noble metal or metal oxides and doping with non-metal elements have been applied to improve the CO2 photoreduction activity of TiO2.1,10,11 In addition to these well-known challenges, there are other factors that hinder the photocatalytic activity but are rarely studied in the literature, such as the weakened CO2 adsorption on TiO2 at the solid–gas interface in the presence of water vapor and limited desorption of reaction products or intermediates from the catalyst surface.12,13 As a result, enhancing the CO2 adsorption on the photocatalyst, a prior step to photoreduction, is important to improve the CO2 photoreduction efficiency of hydrocarbon fuels.Our previous studies have reported MgO–TiO2 composites as hybrid adsorbent/photocatalyst materials for enhanced CO2 photoreduction.12,13 MgO was chosen as the CO2 adsorbent because of its good CO2 adsorption capability that is boosted in the presence of H2O vapor.14–16 We found that MgO–TiO2 possessed much higher activity and more stable performance than pristine TiO2, particularly at a medium temperature range (around 150 °C), which may be due to the easier desorption of reaction intermediates at a higher temperature and the enhanced CO2 adsorption by MgO compared with bare TiO2. We also reported that the concentration and dispersion of MgO on the MgO/TiO2 composite strongly influenced the CO2 photoreduction activity and 5% MgO was the optimum loading on the TiO2 surface. However, since both MgO and TiO2 were in the form of nanoparticles in the MgO/TiO2 composites, it was difficult to distinguish the two components by microscopic analysis and to correlate the catalytic activity with the materials morphology or structure. In addition, MgO has been reported to have relatively low kinetics in CO2 adsorption at moderate temperatures (250–400 °C) possibly due to the changing of basic sites or –OH groups.17,18 Hence, other medium temperature CO2 sorbents with different morphologies and faster adsorption kinetics may be of greater interest to serve as the adsorbent component of the hybrid material for enhanced CO2 photoreduction.
Layered double hydroxides (LDHs), also known as hydrotalcite-like compounds, and their post-calcination product, layered double oxides (LDOs), have been investigated as CO2 sorbents.19–21 LDHs can be chemically expressed by general formula M1−x2+Mx3+(OH)2Ax·nH2O, where M2+ and M3+ are metal ions and A stands for exchangeable anions (Cl−, NO3−, CO32−, or SO42−).22–25 The characteristics of LDHs provide the LDO properties with a large number of Brønsted basic sites, and thus LDOs are considered as promising candidates for CO2 adsorption,26 and activated MgAl-LDOs are found to have a high sorption capacity for CO2.19,21,27 It is reported that MgAl-LDOs can capture much more CO2 than MgO at the medium temperature range 150–450 °C.28,29 Hence, MgAl-LDO could be a good candidate as a replacement of MgO to combine with the TiO2 photocatalyst to improve CO2 photoreduction operated at a medium temperature (~150 °C).
The objective of this work is to design a novel hybrid adsorbent/photocatalyst material by grafting MgAl-LDOs (as the CO2 adsorbent component) onto the surface of micrometer size TiO2 cuboids (as the photocatalyst component) and to investigate the catalytic activity of CO2 photoreduction in correlation with the materials properties. To obtain such a hybrid material structure, it is desirable to have micrometer size TiO2 for MgAl-LDO grafting because MgAl-LDOs are reported to be micrometer or sub-micrometer size platelets. The platelet-shape MgAl-LDOs can be distinct from the micrometer-size TiO2 cuboids, and thus, the morphology and concentration of the two components can be easily manipulated.
2. Experimental
2.1. Synthesis of MgAl-LDO grafted TiO2 cuboids
The MgAl-LDH grafted TiO2 cuboids were synthesized by a two-step process that has not been reported before. In the first step hydrogenated titanate (H2Ti3O7) nanobelts were produced and in the second step, MgAl-LDH was formed by coprecipitation with the H2Ti3O7 nanobelts that were transformed into a cuboid shape along the coprecipitation process. H2Ti3O7 nanobelts (micrometers in length) were prepared via a hydrothermal method that has been widely reported in the literature.30–32 Typically, 1.0 g of TiO2 (Aeroxide P90) was dispersed in 60 ml of 10 M NaOH aqueous solution with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O/ethanol volume ratio. The solution was kept stirring for 30 min and then transferred to a Teflon-lined autoclave. The sodium titanate obtained after 16 h of hydrothermal conditions at 180 °C was washed with 0.1 M HCl aqueous solution until pH 3–4 and then washed with deionized (DI) water until pH 7, forming H2Ti3O7 nanobelts.
:1 H2O/ethanol volume ratio. The solution was kept stirring for 30 min and then transferred to a Teflon-lined autoclave. The sodium titanate obtained after 16 h of hydrothermal conditions at 180 °C was washed with 0.1 M HCl aqueous solution until pH 3–4 and then washed with deionized (DI) water until pH 7, forming H2Ti3O7 nanobelts.
In the second step, the MgAl-LDH grafted TiO2 cuboids were prepared by a coprecipitation method. The as-prepared H2Ti3O7 nanobelts were re-dispersed in 30 ml of 3.0 M urea solution to form solution A. Mg(NO3)2·6H2O and Al(NO3)3·9H2O were dissolved in 30 ml of DI water with [Mg2+] + [Al3+] = 0.15 M, n(Mg)/n(Al) = 2:1 to obtain solution B. Solution B was dropwise added into solution A under stirring. The mixture was transferred to a round bottom flask in an oil bath. The flask was equipped with a water condenser with cooling water. The temperature for the mixture was set to be ~95 °C and was kept under continuous magnetic stirring for 12 h. The obtained precipitate was washed with DI water until pH 7 and vacuum dried at 80 °C overnight to form MgAl-LDH grafted TiO2. Finally, the powder was calcined at 400 °C for 3 hours to form MgAl-LDO grafted TiO2. To find the optimum MgAl-LDO concentration in the composites to obtain the best photocatalytic CO2 reduction activity, the samples with different mass ratios of MgAl-LDO to TiO2 were prepared. The samples were denoted as x% MgAl-LDO/TiO2, in which x represents the measured weight percentage of MgAl-LDO in the sample. Pure TiO2 cuboids were also synthesized as the control.
2.2. Materials characterization
The crystal structures of the MgAl-LDO/TiO2 samples were identified by X-ray diffraction (XRD, Scintag XDS 2000) using Cu Kα irradiation at 45 kV and a diffracted beam monochromator at 40 mA. The optical properties were examined by UV-vis diffuse reflectance spectroscopy using a UV-vis spectrometer (Ocean Optics) with BaSO4 as the background. Scanning electron microscopy (SEM) (Hitachi S4800) was used to investigate the catalyst morphology. The dispersion of elements (Mg, Al, Ti, O) on MgAl-LDO/TiO2 was analyzed by X-ray elemental mapping. The real concentration of MgAl-LDO in the sample was calculated by the difference of the weight of MgAl-LDO/TiO2 before and after washing and drying with 1 M HCl solution.The Brunauer–Emmett–Teller (BET) specific surface area of the composites was measured by nitrogen adsorption at 77 K on a surface area and porosity analyzer (Micrometrics ASAP 2020). Before each adsorption measurement, approximately 0.10 g of the sample was degassed at 180 °C for 6 hours. The BET surface area was determined by a multipoint BET method using the adsorption data in the relative pressure (P/P0) range 0.05–0.3. The thermal stability of the catalyst materials was carried out using a thermogravimetric analyzer (TGA-DAT-2960 SDT) at a heating rate of 20 °C min−1 from 25 to 700 °C in air.
2.3. Measurement of CO2 photoreduction activity
The photocatalytic reduction of CO2 with H2O vapor was conducted using a home-made quartz tube photoreactor operating in a continuous flow mode, as shown in Fig. S1.† For each test, 100 mg of the catalyst was used and evenly dispersed onto a rectangular glass-fiber filter that was placed alongside the wall of the quartz tube and facing the UV light illumination. The catalyst loading process is also shown in Fig. S1.† CO2 cylinder gas (99.999%, Praxair) continuously passed through a DI water bubbler bringing a gas mixture of CO2 + H2O (with 2.3 vol.% H2O) into the photoreactor. A high gas flow rate was used initially to purge out air inside the reactor for 2 h, which in the meantime ensured CO2 adsorption reaching equilibrium on the catalyst surface. Then the flow rate was lowered and maintained at 2.0 sccm during photoreaction. A 100 W mercury vapor lamp or a 450 W Xe lamp with a 400 nm UV cut-off filter was used as the UV-vis light and visible light source, respectively, and the light spectra are shown in Fig. S2.† For the mercury vapor lamp, the light intensity was measured to be about 10 mW cm−2 in the UV region (λ < 390 nm, centered at 365 nm). As a reference, the UV intensity in the sunlight (AM1.5G) is around 5 mW cm−2. Thus, the UV intensity from the mercury vapor lamp applied in this study was equivalent to approximately 2 suns. For the Xe lamp with a UV filter, the light intensity in the visible region (400–700 nm) was about 84 mW cm−2, again close to 2-sun conditions. To reach and maintain a reaction temperature at 150 °C, the optimum temperature found in our previous work for MgO–TiO2 catalysts,13 a 250 W infrared lamp was used to heat up the photoreactor and the temperature can be adjusted by varying the distance between the IR lamp and the reactor. The gaseous products in the reactor effluent were continuously analyzed at a 15 min interval using a gas chromatograph (GC, Agilent 7890A) equipped with an automated gas valve and a thermal conductivity detector (TCD) and flame ionization detector (FID).3. Results and discussion
3.1. Crystal structure of MgAl-LDO/TiO2
Fig. 1 shows the XRD patterns for MgAl-LDH, MgAl-LDO, 10% MgAl-LDH/TiO2, 10% MgAl-LDO/TiO2 and TiO2 cuboid samples. The diffraction peaks in both MgAl-LDH and 10% MgAl-LDH/TiO2 patterns were indexed to MgAl-LDH19,23,33 and weaker peaks were observed for the 10% MgAl-LDH/TiO2 sample compared with MgAl-LDH. No TiO2 characteristic peaks were observed for 10% MgAl-LDH/TiO2 indicating that the TiO2 crystal phase was not formed without calcination. For TiO2 cuboids, all the diffraction peaks were attributed to the anatase phase (JCPDS no. 21-1272).2,7,8 For the MgAl-LDO sample, all the diffraction peaks were indexed to MgO.12,19,34 For the 10% MgAl-LDO/TiO2 samples, only TiO2 anatase diffraction peaks are observed, and no MgO diffraction peaks are seen. The Scherrer equation was applied to calculate the crystallite size of TiO2. The diffraction peaks of TiO2 anatase in 10% MgAl-LDO/TiO2 composites are narrow compared with TiO2 cuboids, suggesting a larger crystallite size of TiO2 due to MgAl-LDO addition. The average crystallite size of TiO2 anatase in TiO2 cuboids was 15 nm. By comparison, the average crystallite size of TiO2 anatase in 10% MgAl-LDO/TiO2 composites was 32 nm. A possible reason is that the addition of Mg(NO3)2 and Al(NO3)3 consumes urea during the second step of materials synthesis and helps preserve some of the H2Ti3O7 nanobelts that are later transformed to crystalline TiO2 anatase.32 | ||
| Fig. 1 XRD patterns of samples: (a) MgAl-LDH; (b) MgAl-LDO; (c) 10% MgAl-LDH/TiO2; (d) 10% MgAl-LDO/TiO2 and (e) TiO2 cuboids (* represents MgAl-LDH characteristic peaks, A represents TiO2 anatase, and ◆ represents MgO characteristic peaks). | ||
3.2. Morphology, textural structure and optical property of MgAl-LDO/TiO2
Fig. 2 shows the SEM images of H2Ti3O7 titanate nanowires (precursor to TiO2 cuboids), MgAl-LDOs and TiO2 cuboids. MgAl-LDOs consist of platelets around 2 μm in size. The length of the TiO2 cuboids is in the micrometer size range with a high aspect ratio. The length of TiO2 cuboids pretty much agrees with that of the titanate nanowires, suggesting that the cuboids are indeed transformed from nanowires during the second step of materials synthesis. | ||
| Fig. 2 SEM images of (a) H2Ti3O7 titanates, (b) MgAl-LDOs and (c) TiO2 cuboids. | ||
The textural properties are characterized by BET analysis and the results are summarized in Table 1. Bare TiO2 cuboids displayed a specific surface area of 109 m2 g−1 and a pore volume of 0.48 cm3 g−1, where the pores are probably attributed to inter-nanoparticle spacing. The MgAl-LDO had a specific surface area of 180 m2 g−1, greater than that of TiO2 cuboids, possibly due to the smaller interplanar spacing between the layered oxides, which is also reflected by the smaller pore size. Notably, the 10% MgAl-LDO/TiO2 sample possessed a high specific surface area (175 m2 g−1) close to the bare MgAl-LDO, although the concentration of MgAl-LDO was not high. This indicates certain interaction between the MgAl-LDH nanoflakes and TiO2 nanoparticles during the formation of the hybrid material.
| Sample | BET specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|
| TiO2 cuboids | 109 | 0.48 | 16 |
| 10% MgAl-LDO/TiO2 | 175 | 0.39 | 9 |
| MgAl-LDO | 180 | 0.28 | 8 |
The thermogravimetric analysis (TGA) result of the 10% MgAl-LDH/TiO2 sample is given in Fig. S3,† which shows two main steps of weight loss which are consistent with the literature.22 The adsorbed water in LDH was released at a relatively low temperature to 200 °C where it was mostly in the form of interlamellar water. Carbonate ions of the LDH sample were decomposed at higher temperatures from 200 to 500 °C in parallel with the water loss. The weight loss almost ceased at above 500 °C indicating the completion of the decarbonation process, i.e., LDH was fully converted to LDO.
Fig. 3 shows the SEM images of MgAl-LDO/TiO2 samples with different compositions. The lower magnification SEM images in Fig. 3a, c, and e show that the three samples were composed of MgAl-LDOs grafted on micrometer size TiO2 cuboids. The platelet shape of MgAl-LDOs on the composites after calcination was almost the same as the as-prepared uncalcined composites MgAl-LDHs (SEM images not shown here). In most of the literature reports, the morphology of MgAl-LDH platelets cannot be maintained after calcination.25,35 The successful grafting of MgAl-LDO platelets on micrometer-size TiO2 cuboids in this work demonstrates a novel approach in synthesizing such composite materials with the desired morphology. The higher magnification SEM images in Fig. 3b, d, and f show that both the size and the coverage of MgAl-LDO platelets grafted on TiO2 cuboids increased as the MgAl-LDO loading increased.
 | ||
| Fig. 3 SEM images of the MgAl-LDO/TiO2 composites: (a, b) 8% MgAl-LDO/TiO2, (c, d) 10% MgAl-LDO/TiO2, and (e, f) 12% MgAl-LDO/TiO2. | ||
The distributions of Mg, Al, Ti and O elements in the MgAl-LDO/TiO2 composites were analyzed by X-ray elemental mapping, and the results are shown in Fig. 4. The elemental mapping images demonstrated that the cuboid skeletons were mainly composed of the Ti element and the grafted platelets were composed of Mg and Al elements that were evenly distributed on the cuboid surface. The O element is distributed on both the cuboids and the platelets, agreeing with the composition of mixed oxides for this composite material.
 | ||
| Fig. 4 X-ray elemental mapping images of the MgAl-LDO/TiO2 composites: (a) 8% MgAl-LDO/TiO2, (b) 10% MgAl-LDO/TiO2, and (c) 12% MgAl-LDO/TiO2. | ||
Diffuse reflectance UV-vis spectra were recorded to investigate the influence of MgAl-LDO on the optical property of TiO2, and the plots of Kubelka–Munk function were made to determine the band gap values, as shown in Fig. 5. The absorption edge of the TiO2 is around 390 nm, corresponding to a band gap of about 3.2 eV, which agrees with most reported literature data on the band gap of TiO2 anatase. The MgAl-LDO alone shows no light adsorption in the wavelength range measured. The incorporation of MgAl-LDOs on TiO2 cuboids leads to a slight blue-shift in the adsorption edge and a reduced absorption in the UV region, probably due to the increased surface roughness resulting in more reflection.
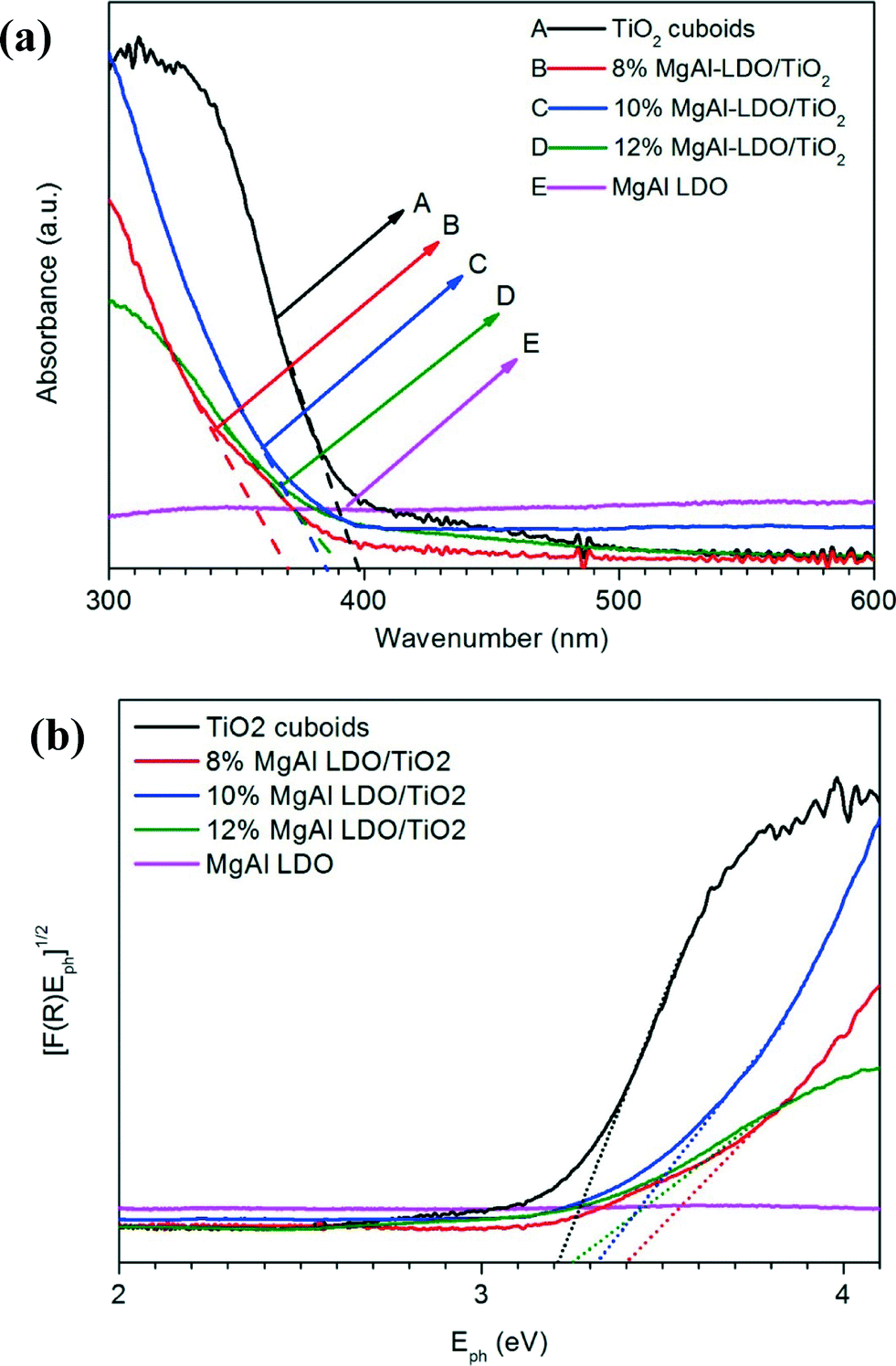 | ||
| Fig. 5 (a) UV-vis diffuse reflectance spectra and (b) plots of the square root of Kubelka–Munk function versus the photon energy for the catalyst materials of TiO2 cuboids, MgAl-LDO, and MgAl-LDO/TiO2 composites with different MgAl-LDO concentrations. | ||
To further confirm possible structural changes, UV/Vis adsorption spectra of H2Ti3O7 and 10% MgAl-LDH/TiO2 were recorded as shown in Fig. S4.† By comparing TiO2 cuboids with H2Ti3O7, it is observed that TiO2 cuboids have a red-shift in the absorption edge, which indicates compositional changes due to the calcination of H2Ti3O7. By comparing 10% MgAl-LDH/TiO2 with 10% MgAl-LDO/TiO2, difference in the absorption shift is also observed due to the conversion of LDH to LDO. By comparing MgAl-LDO/TiO2 with TiO2 cuboids, it is observed that there is less absorption in the UV region for the MgAl-LDO/TiO2, which indicates possible interaction of MgAl-LDO with TiO2.36 From the XRD results, there is no obvious change of TiO2 diffraction peaks or appearance of new peaks, suggesting that the extent of Mg atom incorporation into the TiO2 structure, if any, is not significant.
3.3. Photocatalytic activities of CO2 reduction
Photocatalytic activity tests were carried out under UV light irradiation at 50 °C for 4 h (Fig. 6a) and subsequently at 150 °C for another 8 h (Fig. 6b). CO was found to be the major product with a minor CH4 concentration that was one or two orders of magnitude lower than CO. Thus, only CO production results were shown in Fig. 6 to compare the activities of the different catalysts. Fig. 6a shows that bare TiO2 cuboids had a CO production rate of around 1.0 μmol g−1 h−1 at 50 °C while pure MgAl-LDO had no activity for CO production. The addition of MgAl-LDO on TiO2 cuboids had no significant improvement in the CO production rate except for the 10% MgAl-LDO/TiO2 sample that has a doubled activity compared with bare TiO2 cuboids. The main reason for such insignificant improvement is maybe because MgAl-LDOs has weak CO2 adsorption at low temperatures.26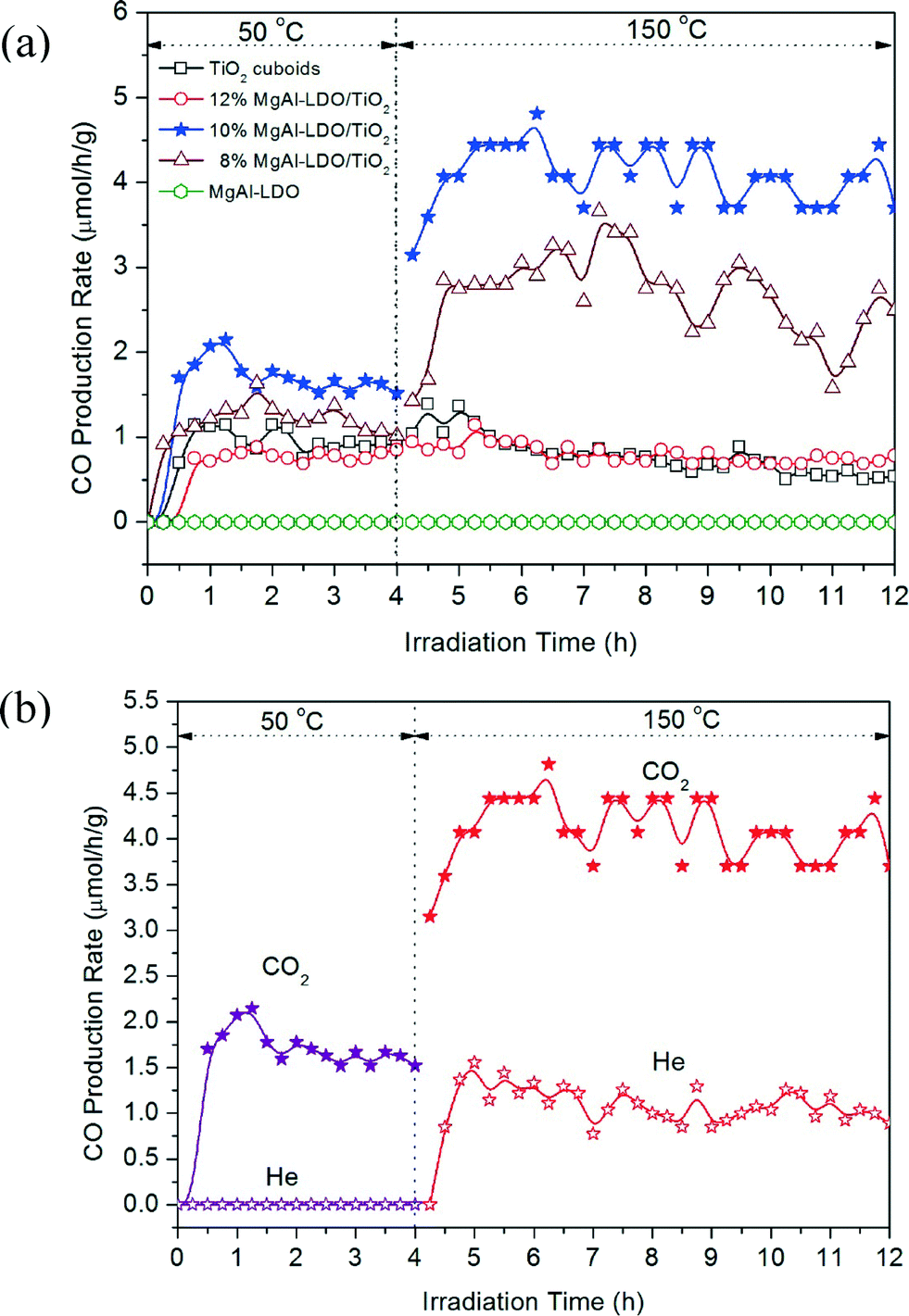 | ||
| Fig. 6 The rate of CO production from CO2 photoreduction under UV light irradiation at 50 °C for 4 h and subsequently at 150 °C for 8 h using (a) TiO2 cuboids, MgAl-LDO and three MgAl-LDO/TiO2 composites in the CO2 + H2O vapor atmosphere, and (b) 10% MgAl-LDO/TiO2 in the He + H2O vapor atmosphere and CO2 + H2O vapor atmosphere, respectively. | ||
For the following 8 h test under UV light at 150 °C (Fig. 6b), the CO production rate of bare TiO2 cuboids was almost the same as the first 4 hours at 50 °C. Pure MgAl-LDOs still showed no activity of CO2 reduction. Both 8% MgAl-LDO/TiO2 and 10% MgAl-LDO/TiO2 exhibited an obvious enhancement in CO production, reaching an average of about 2.8 and 4.3 μmol g−1 h−1, respectively, compared with about 0.7 μmol g−1 h−1 for bare TiO2 cuboids at 150 °C. In other words, 10% MgAl-LDO/TiO2 was five times more active than bare TiO2 cuboids. Moreover, the activities of both 8% MgAl-LDO/TiO2 and 10% MgAl-LDO/TiO2 at 150 °C were much higher than those at 50 °C. In addition, the downward trend in the CO production rate for the 8% MgAl-LDO/TiO2 sample indicates that it is inferior to the 10% MgAl-LDO/TiO2 sample in terms of stability. Interestingly, the activity of 12% MgAl-LDO/TiO2 did not show any enhancement compared with bare TiO2 even at 50 °C or 150 °C. The above results indicate that at an optimum MgAl-LDO loading (10% in this study), the CO2 reduction activity can be significantly increased at a higher temperature (150 °C). It also agrees with our hypothesis that the CO2 adsorption capability of MgAl-LDOs could promote the CO2 photoreduction capability by TiO2. It is likely that the photo-induced electrons on TiO2 could migrate to the adjacent CO2 adsorption sites at the interface of TiO2 and MgAl-LDO and thus promote CO2 reduction. However, the exact mechanism is not clear so far and will be investigated in our future research. A very high loading of MgAl-LDO (12% in this case) did not promote CO2 photoreduction or may have detrimental effect, probably because of the following two reasons: (1) the MgAl-LDOs may have covered all the TiO2 cuboid surfaces (Fig. 3f) so that the contact between TiO2 and adsorbed CO2 is limited, and (2) the light absorption capacity is significantly reduced at a high MgAl-LDO loading (Fig. 5).
The CO2 photoreduction with H2O by the 10% MgAl-LDO/TiO2 sample under visible light was also carried out using a Xe lamp equipped with a 400 nm cut-off filter, and the result is shown in Fig. S5.† At 50 °C under visible light, the catalyst exhibited no activity and at 150 °C, the CO production rate reached around 1.0 μmol g−1 h−1. Apparently, the material is more active under UV than under visible light, which agrees with the band gap of the material as shown in Fig. 5.
To examine the cycling capability of the catalyst, an additional experiment was conducted using 10% MgAl-LDO/TiO2 under the mercury vapor lamp irradiation at 150 °C for two on-and-off cycles for a total period of 12 h. The results are shown in Fig. S6.† The CO production rate was around 4.0 μmol g−1 h−1 when light was on. When the light was switched off, the CO production rate decreased rapidly and became zero within one hour. Turning the light back on obviously re-activated the photoreduction and the CO production rate bounced back to 3.6 μmol g−1 h−1, almost the same level as in the first cycle. Switching off the light again led to a stop in the photoreduction. This on-and-off cycling test result clearly indicates that CO production was indeed from CO2 photoreduction with water activated by the UV light irradiation. The result that the CO production rate in the 2nd cycle can be recovered to the similar level in the 1st cycle demonstrates a good cycling capability of the catalyst.
Because the MgAl-LDO may still contain a small amount of carbonate species even after the calcination process, we have conducted another set of experiments to understand the possible interference of carbonate species on the catalyst to the reduction of CO2 from the gas phase. Tests were carried out by comparing the photocatalytic activity of 10% MgAl-LDO/TiO2 under two gas environments: (1) helium (He) + H2O vapor and (2) CO2 + H2O vapor, and the results are shown in Fig. 6b. There was no CO production in the He + H2O atmosphere at 50 °C, but when the temperature was increased to 150 °C, CO was produced at a rate of about 1.0 μmol g−1 h−1. This result indicates that carbonates in MgAl-LDOs were stable at low temperatures but were activated at higher temperatures and reduced to CO through photocatalytic reactions. By contrast, when CO2 was present, the CO production rate was much higher, 1.5 μmol g−1 h−1 at 50 °C and 4.3 μmol g−1 h−1 at 150 °C. Comparison of these results suggests that carbonate residues in the MgAl-LDO/TiO2 composites did not have significant contribution to the CO production and most of these were derived from gas-phase CO2 reduction. To further verify the source of CO production, additional experiments were conducted to measure the photocatalytic activity of H2Ti3O7 and TiO2 cuboids in the He/H2O atmosphere, and the result is shown in Fig. S7.† No CO production was observed for H2Ti3O7 and TiO2 cuboids at either 50 °C or 150 °C. Only 10% MgAl-LDO/TiO2 showed some CO production at 150 °C. This result further proved that the small amount of CO produced in the He/H2O atmosphere was attributed to carbonate residues in the MgAl-LDO component of the composite and not from carbon contamination of TiO2.
On the other hand, the above interesting finding suggests that if we pre-load carbonates on the composite catalyst by capturing CO2 first and then exposing the material under UV light or sunlight, it is possible that we can convert the captured CO2 into CO in the second step, and thus we can separate the CO2 capture and conversion process. A benefit of doing this is to achieve separation of CO2 conversion products (e.g., CO) from the bulk CO2, an approach advantageous to most CO2 photoreduction processes reported in the literature where the products are mixed with unreacted CO2. We will conduct further research to investigate this novel idea.
4. Conclusions
This work has demonstrated the successful synthesis of novel hybrid materials, MgAl-LDO platelets grafted on TiO2 cuboids, with a controlled mass ratio of MgAl-LDO to TiO2. The surface coverage of MgAl-LDO on TiO2 and the reaction temperature are two important parameters that influence the activity of CO2 photocatalytic reduction to CO. The hybrid MgAl-LDO/TiO2 showed a CO production rate five times higher than that of bare TiO2 at the optimum 10 wt.% MgAl-LDO loading and at 150 °C. The possible reason for the enhancement is that the grafted MgAl-LDO functions as a CO2 adsorbent and its CO2 capture ability increases with increasing temperature (below 200 °C) and that the photoinduced electrons generated on TiO2 may migrate to the interfacial sites and promote CO2 reduction. At too high a loading of MgAl-LDO when the platelets completely cover the TiO2 cuboid surface, the light absorption ability is impaired and the contact between TiO2 and CO2 is blocked, and thus no improvement in CO2 photoreduction is observed. The carbonate residues on the MgAl-LDO/TiO2 have a minor contribution to the CO production under photoirradiation, which inspires a new process of sequential CO2 capture and photocatalytic conversion to fuels using this novel hybrid material. In this case, the products can be effectively separated from unreacted CO2. This interesting idea will be validated in our future work.Acknowledgements
The authors acknowledge the financial support from National Science Foundation (NSF) Early Faculty CAREER Award (CBET-1254709).References
- C. Y. Zhao, A. Krall, H. L. Zhao, Q. Y. Zhang and Y. Li, Int. J. Hydrogen Energy, 2012, 37, 9967–9976 CrossRef CAS PubMed.
- L. J. Liu, D. T. Pitts, H. L. Zhao, C. Y. Zhao and Y. Li, Appl. Catal., A, 2013, 467, 474–482 CrossRef CAS PubMed.
- L. J. Liu, H. L. Zhao, J. M. Andino and Y. Li, ACS Catal., 2012, 2, 1817–1828 CrossRef CAS.
- Y. Li, W. N. Wang, Z. L. Zhan, M. H. Woo, C. Y. Wu and P. Biswas, Appl. Catal., B, 2010, 100, 386–392 CrossRef CAS PubMed.
- Q. Y. Zhang, Y. Li, E. A. Ackerman, M. Gajdardziska-Josifovska and H. L. Li, Appl. Catal., A, 2011, 400, 195–202 CrossRef CAS PubMed.
- C. L. Yu, Q. Z. Fan, Y. Xie, J. C. Chen, Q. Shu and J. C. Yu, J. Hazard. Mater., 2012, 237, 38–45 CrossRef PubMed.
- C. Y. Zhao, L. J. Liu, Q. Y. Zhang, J. Wang and Y. Li, Catal. Sci. Technol., 2012, 2, 2558–2568 CAS.
- H. L. Zhao, L. J. Liu, J. M. Andino and Y. Li, J. Mater. Chem. A, 2013, 1, 8209–8216 CAS.
- I. H. Tseng, J. C. S. Wu and H. Y. Chou, J. Catal., 2004, 221, 432–440 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- Y. Kohno, H. Hayashi, S. Takenaka, T. Tanaka, T. Funabiki and S. Yoshida, J. Photochem. Photobiol., A, 1999, 126, 117–123 CrossRef CAS.
- L. J. Liu, C. Y. Zhao, H. L. Zhao, D. Pitts and Y. Li, Chem. Commun., 2013, 49, 3664–3666 RSC.
- L. Liu, C. Zhao, D. Pitts, H. Zhao and Y. Li, Catal. Sci. Technol., 2014, 2014, 1539–1546 Search PubMed.
- Y. Kohno, H. Ishikawa, T. Tanaka, T. Funabiki and S. Yoshida, Phys. Chem. Chem. Phys., 2001, 3, 1108–1113 RSC.
- Q. Y. Li, L. L. Zong, C. Li and J. J. Yang, Appl. Surf. Sci., 2014, 314, 458–463 CrossRef CAS PubMed.
- S. J. Xie, Y. Wang, Q. H. Zhang, W. Q. Fan, W. P. Deng and Y. Wang, Chem. Commun., 2013, 49, 2451–2453 RSC.
- G. Xiao, R. Singh, A. Chaffee and P. Webley, Int. J. Greenhouse Gas Control, 2011, 5, 634–639 CrossRef CAS PubMed.
- A. Chakradhar and U. Burghaus, Surf. Sci., 2013, 616, 171–177 CrossRef CAS PubMed.
- Y. S. Gao, Z. Zhang, J. W. Wu, X. F. Yi, A. M. Zheng, A. Umar, D. O'Hare and Q. Wang, J. Mater. Chem. A, 2013, 1, 12782–12790 CAS.
- J. W. Wang, L. A. Stevens, T. C. Drage and J. Wood, Chem. Eng. Sci., 2012, 68, 424–431 CrossRef CAS PubMed.
- M. Dadwhal, T. W. Kim, M. Sahimi and T. T. Tsotsis, Ind. Eng. Chem. Res., 2008, 47, 6150–6157 CrossRef CAS.
- T. Bujdoso, A. Patzko, Z. Galbacs and I. Dekany, Appl. Clay Sci., 2009, 44, 75–82 CrossRef CAS PubMed.
- K. Teramura, S. Iguchi, Y. Mizuno, T. Shishido and T. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 8008–8011 CrossRef CAS PubMed.
- M. Q. Zhao, Q. Zhang, J. Q. Huang and F. Wei, Adv. Funct. Mater., 2012, 22, 675–694 CrossRef CAS PubMed.
- Y. Kuang, L. N. Zhao, S. A. Zhang, F. Z. Zhang, M. D. Dong and S. L. Xu, Materials, 2010, 3, 5220–5235 CrossRef CAS PubMed.
- M. K. R. Reddy, Z. P. Xu, G. Q. Lu and J. C. D. Da Costa, Ind. Eng. Chem. Res., 2006, 45, 7504–7509 CrossRef.
- M. Leon, E. Diaz, S. Bennici, A. Vega, S. Ordonez and A. Auroux, Ind. Eng. Chem. Res., 2010, 49, 3663–3671 CrossRef CAS.
- J.-I. Yang and J.-N. Kim, Korean J. Chem. Eng., 2006, 23, 77–80 CrossRef CAS.
- X. P. Wang, J. J. Yu, J. Cheng, Z. P. Hao and Z. P. Xu, Environ. Sci. Technol., 2008, 42, 614–618 CrossRef CAS.
- Z. Yang, G. Du, Z. Guo, X. Yu, Z. Chen, T. Guo and H. Liu, J. Mater. Chem., 2011, 21, 8591–8596 RSC.
- Q. Li, J. Zhang, B. Liu, M. Li, S. Yu, L. Wang, Z. Li, D. Liu, Y. Hou, Y. Zou, B. Zou, T. Cui and G. Zou, Cryst. Growth Des., 2008, 8, 1812–1814 CAS.
- Q. Li, J. Zhang, B. Liu, M. Li, R. Liu, X. Li, H. Ma, S. Yu, L. Wang, Y. Zou, Z. Li, B. Zou, T. Cui and G. Zou, Inorg. Chem., 2008, 47, 9870–9873 CrossRef CAS PubMed.
- J. Wang, D. D. Li, X. A. Yu, M. L. Zhang and X. Y. Jing, Colloid Polym. Sci., 2010, 288, 1411–1418 CAS.
- N. C. S. Selvam, R. T. Kumar, L. J. Kennedy and J. J. Vijaya, J. Alloys Compd., 2011, 509, 9809–9815 CrossRef CAS PubMed.
- Y. S. Zhao, J. G. Li, F. Fang, N. K. Chu, H. Ma and X. J. Yang, Dalton Trans., 2012, 41, 12175–12184 RSC.
- Š. Paušová, J. Krýsa, J. Jirkovský, G. Mailhot and V. Prevot, Environ. Sci. Pollut. Res., 2012, 19, 3709–3718 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy00216h |
| This journal is © The Royal Society of Chemistry 2015 |