
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of acid–base properties on the Lebedev ethanol-to-butadiene process catalyzed by SiO2–MgO materials†
Carlo
Angelici
,
Marjolein E. Z.
Velthoen
,
Bert M.
Weckhuysen
* and
Pieter C. A.
Bruijnincx
*
Inorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: b.m.weckhuysen@uu.nl; p.c.a.bruijnincx@uu.nl
First published on 25th March 2015
Abstract
The Lebedev ethanol-to-butadiene process entails a complex chain of reactions that require catalysts to possess a subtle balance in the number and strength of acidic and basic sites. SiO2–MgO materials can be excellent Lebedev catalysts if properly prepared, as catalyst performance has been found to depend significantly on the synthesis method. To assess the specific requirements for butadiene production in terms of active sites and to link their presence to the specific preparation method applied, five distinct SiO2–MgO catalysts, prepared by wet-kneading and co-precipitation methods, were thoroughly characterized. The amount and strength of the acidic (pyridine-IR and NH3-TPD) and basic (CDCl3-IR and CO2-TPD) sites of the materials as well as the overall acid/base properties in the liquid phase (Hammett indicators) were determined. The number of acidic and strong basic sites could be correlated with the extent of ethylene and diethyl ether by-product formation. The best performing catalysts are those containing a small amount of strong basic sites, combined with an intermediate amount of acidic sites and weak basic ones. These results thus provide further insight into the relation between the amount and strength of acidic/basic sites, preparation method and catalytic performance.
Introduction
In 1929, Lebedev discovered that butadiene can be synthesized in a one-pot, one-step process from ethanol.1–3 The composition of the catalyst employed by Lebedev was guarded by patents, but Natta later proposed the Lebedev catalyst to be a mixture of different amounts of SiO2 and MgO.4 Various investigations showed (promoted) SiO2–MgO materials to indeed be excellent catalysts for the one-step ethanol-to-butadiene conversion, yet noted a large influence of the preparation method on catalytic performance.4–6 We also previously observed that SiO2–MgO catalysts prepared via different methods gave remarkably different 1,3-butadiene yields and showed large differences in catalytic performance in general.7 This was attributed to the different structural properties that result from the preparation method employed, i.e. wet-kneading, co-precipitating or physically mixing of the two components. The morphological and structural differences observed for the various catalysts point at differences in the number, strength and type of the acidic and basic sites that are needed for the Lebedev conversion. Indeed, even though the actual mechanism of the Lebedev process is still being discussed, the overall reaction necessitates a (complex) interplay between acid- and base-catalyzed elementary steps. Insight into what is actually required for a good Lebedev catalyst in terms of acidity/basicity and to link these requirements to the specific preparation method is therefore an important goal, as such information might also shed further light on the mechanism at hand. Various mechanisms have been suggested over the years and these are discussed in detail in recent reviews.8,9 For example, Gruver and co-workers suggested the Lebedev reaction to proceed via a Prins-like condensation involving acetaldehyde and ethylene, based on the observation of a linear correlation between butadiene and ethylene yields.10 This mechanism was actually first proposed by Natta et al. and subsequently discarded by the same authors based on the observation that the addition of up to 20% ethylene in the feed did not increase butadiene yield.4 It can indeed be said that this mechanism is unlikely to operate as it requires the formation of a primary carbocation in the process. Of the other suggested mechanistic pathways, the one based on aldol condensation of two acetaldehyde molecules (Scheme 1, steps 2, 3), followed by crotonaldehyde reduction (step 4) and finally dehydration (step 5) is currently most widely accepted.11–13 For various catalysts and process conditions, acetaldehyde formation,13 as well as crotonaldehyde formation11,14 and reduction12 have all been suggested to be rate-limiting for this pathway. Interestingly, for the related Guerbet reaction, which entails coupling of ethanol to butanol, an acetaldehyde aldol condensation-based mechanism was also typically assumed; however, Scalbert et al. recently proposed direct condensation of two ethanol molecules to be the main mechanistic pathway to butanol, based on thorough kinetic and thermodynamic studies.15 It should be noted though that Guerbet catalysts, such as the hydroxyapatite used and considered to be purely basic by Scalbert et al., differ from Lebedev ones, which typically require a more complex combination of acidity and basicity. That such a combination is required was further substantiated by Tsuchida et al. who studied ethanol conversion over a number of hydroxyapatites (with different Ca/P ratio, thus tuning the amount of basic and acidic sites) and observed the highest butadiene selectivity (13.8%) for the catalyst having a ratio of basic to acidic sites close to 1; conversely, catalysts with higher basic to acidic site ratios showed low butadiene selectivity (≤1%), yet displayed significantly increased 1-butanol selectivity.16 León and co-workers also observed the selectivity to different C4 compounds to depend on the acid–base properties, in this case of Mg–Al mixed oxides used for ethanol condensation to a mixture of C4 compounds that included butadiene. Catalysts containing the strongest basic sites were more selective towards butanol, while increased acidity correlated with improved selectivity towards butadiene.17 The authors later showed that replacement of Al with Fe reduced acidity significantly, increasing the selectivity towards acetaldehyde and 1-butanol at the expense of ethylene and butadiene production.18 These examples clearly show the influence the acid–base distribution has on the products obtained upon ethanol conversion, possibly even altering the dominant mechanistic route. | ||
| Scheme 1 The commonly reported aldol condensation route for the one-step ethanol to butadiene conversion. | ||
Little has been reported, though, on the acid–base properties of the SiO2–MgO catalysts used typically for the Lebedev process. Niiyama et al. already concluded that both acidic and basic sites are required, based on a study of acidity (measured by titration with Hammett indicators and gravimetric measurements of adsorbed pyridine) and basicity (by gravimetric measurements of adsorbed boron trifluoride) of SiO2–MgO catalysts with different Si/Mg ratios.13 Very recently Janssens et al. reported on the influence of acidity and basicity of Ag-promoted, dry-milled SiO2–MgO catalysts on performance in the Lebedev process.19 Based on pyridine-IR (acidity), CO2-TPD and CO2-IR (basicity), they concluded that either weak basic sites or acid–base pair catalyze the aldol condensation step, while acid sites of different nature were proposed to be involved in steps 3, 4 and 5 of the mechanism depicted in Scheme 1.
Even though limited systematic studies on basicity/acidity are available for the overall Lebedev process, structure–activity relations have been reported for the individual steps thought to be involved in butadiene production. Regardless of the actual mechanism, the stoichiometry of the process shows that both dehydrogenation and dehydration must occur for butadiene generation. First, ethanol can be converted into three species: dehydration leads to ethylene and diethyl ether formation, while dehydrogenation yields acetaldehyde (Scheme 2). The latter process is the first step towards butadiene and is generally considered to be catalyzed, in absence of redox functionalities, by basic sites in an E1cB-type process (unimolecular elimination via the conjugate base, Scheme 2) resulting in heterolytic elimination of H2.16,20,21 The mechanism of alcohol dehydration is more controversial. Generally, dehydration of alcohols occurs over acid sites,22,23 but Díez et al. showed that for isopropanol decomposition over alkali-promoted MgO samples other sites might also be capable of this.21 They considered three different routes for propylene formation, including a purely acid-catalyzed E1 mechanism, as well as an E2 mechanism that would involve a cooperative interaction between acidic and basic sites. Finally, as strongly basic catalysts such as 1 mol% Cs2O/MgO showed significantly increased propylene selectivity,24 an E1cB mechanism different from the one leading to the dehydrogenation product acetaldehyde was considered. In this case, a surface alkoxide is formed first, after which a strong basic site abstracts a proton in β-position to the O atom; O elimination finally yields the alkene. León et al., in turn, ascribed the formation of ethylene during ethanol conversion over hydrotalcite-based mixed oxides to either strong basic sites or strong acidic ones.25 It is worth noting that both the E2 and E1cB mechanisms require cooperative action between acidic and basic sites, meaning that not only active site number and strength, but also proximity is key to selective ethanol conversion. Finally, ether formation also involves cooperative action of acid and basic sites and is thought to proceed via the same E2 mechanism proposed for ethylene formation.17 It has also been proposed that (moderate) acid sites alone catalyze the formation of (diisopropyl) ethers over TiO2, ZrO2 and their mixed oxides.26
 | ||
| Scheme 2 Mechanisms of ethanol conversion. Modified from Ono et al.20 A, B and B represent acidic, basic and strong basic sites, respectively. | ||
The next, productive aldol condensation step of the mechanism shown in Scheme 1 is usually reported to be base-catalyzed,27 but cooperativity between acidic and basic sites has also here been shown to be beneficial. Zeidan and Davis, for instance, showed this for the cross-aldolization of acetone and 4-nitrobenzaldehyde by silica functionalized with amines and acidic functional groups.28 Climent et al. also reported on the beneficial effect of having both acid and basic sites in the aldol condensation of benzaldehyde and heptanal.29
A Meerwein–Ponndorf–Verley (MPV) type reduction of crotonaldehyde by ethanol is typically assumed for step 4.12,30,31 Sushkevich et al., recently studied the MPV of crotonaldehyde with ethanol in detail over ZrO2-supported catalysts.32 They proposed that activity increased with the amount of Lewis acidic Zr sites, suggesting that these sites are responsible for hydride transfer. Aramendía et al. also studied MPV of various α,β-unsaturated aldehydes (including crotonaldehyde) over hydrotalcite(-like) catalysts; they proposed that the substrates could interact only when adsorbed on acid–base sites, while also noting that the best catalyst (MgO/Al2O3) had the highest amount of basic sites.33
As noted above, little information is available on the acid/base requirements for the Lebedev reaction. Such studies of the kind and amount of active sites required for selective butadiene formation are complicated by: 1. the number of steps involved, 2. the absence of agreement on the rate-determining step and 3. the contradictory reports on the active sites needed for each implicated elementary step. Correlating the acid–base properties with catalytic performance is, nonetheless, essential for the synthesis of catalysts with improved butadiene selectivity.
We previously showed that different SiO2–MgO preparation methods (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) resulted in significant structural differences. In particular, TEM analysis showed the morphology of wet-kneaded catalysts to consist of islands of MgO together with SiO2 spheres thought to be covered by a (thin) MgO layer; SiO2–MgO co-precipitated catalysts showed an ill-defined morphology, hinting at the formation of a more mixed oxide-like material. The structural differences, in turn, resulted in significant differences in butadiene yield and product distribution (Fig. 1).7 SiO2–MgO (II–IV) samples prepared via wet-kneading showed significantly higher selectivity towards butadiene than the co-precipitated SiO2–MgO (V–VI) catalysts (see Table 1); furthermore, the co-precipitated ones were found to produce higher amounts of the by-products ethylene and diethyl ether.10,11 Low butanol yields were observed for all catalysts (<5%), implying that all SiO2–MgO materials contain a non-negligible amount of acidic sites. These materials thus provide an excellent set for a systematic acid/base study. Here, we report on the acid/base properties of five SiO2–MgO catalysts characterized using the techniques listed in Table 2. Physically-mixed SiO2–MgO (I) gives very poor ethanol conversion and butadiene yield and was therefore not included here. The various techniques used show that strong basic sites and an excessive amount of acidic sites lead to more dehydration products and poor selectivity to butadiene. Instead, those catalysts with an intermediate amount of acidic sites and a low amount of strong basic sites give the best butadiene yields.
:1 molar ratio) resulted in significant structural differences. In particular, TEM analysis showed the morphology of wet-kneaded catalysts to consist of islands of MgO together with SiO2 spheres thought to be covered by a (thin) MgO layer; SiO2–MgO co-precipitated catalysts showed an ill-defined morphology, hinting at the formation of a more mixed oxide-like material. The structural differences, in turn, resulted in significant differences in butadiene yield and product distribution (Fig. 1).7 SiO2–MgO (II–IV) samples prepared via wet-kneading showed significantly higher selectivity towards butadiene than the co-precipitated SiO2–MgO (V–VI) catalysts (see Table 1); furthermore, the co-precipitated ones were found to produce higher amounts of the by-products ethylene and diethyl ether.10,11 Low butanol yields were observed for all catalysts (<5%), implying that all SiO2–MgO materials contain a non-negligible amount of acidic sites. These materials thus provide an excellent set for a systematic acid/base study. Here, we report on the acid/base properties of five SiO2–MgO catalysts characterized using the techniques listed in Table 2. Physically-mixed SiO2–MgO (I) gives very poor ethanol conversion and butadiene yield and was therefore not included here. The various techniques used show that strong basic sites and an excessive amount of acidic sites lead to more dehydration products and poor selectivity to butadiene. Instead, those catalysts with an intermediate amount of acidic sites and a low amount of strong basic sites give the best butadiene yields.
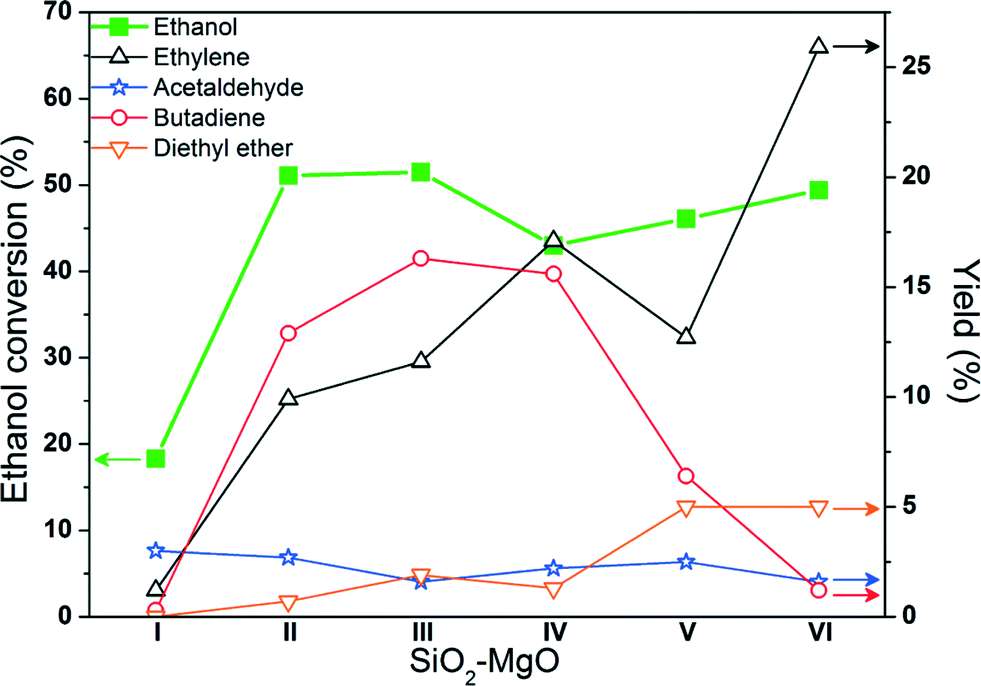 | ||
| Fig. 1 Ethanol conversion and yields of the main (by-) products for the six SiO2–MgO samples after 4 h on stream.7 Conditions: 0.2 g of catalyst, reaction temperature of 698 K, ethanol (gas phase) and nitrogen flow of 2 and 98 mL min−1, respectively. | ||
| Sample name | Technique | SiO2 particle size (nm) | Ethanol conversion (%)d | Butadiene yield (%)d |
|---|---|---|---|---|
| a MgO precursor added 20 min after SiO2 precursor. b MgO and SiO2 precursors added simultaneously. c No silica phase was observed for this sample. d Values after 4 h time on stream. | ||||
| SiO2–MgO (I) | Physical mixture | ~425 | 18.3 | 0.3 |
| SiO2–MgO (II) | Wet-kneading | ~425 | 51.1 | 12.9 |
| SiO2–MgO (III) | Wet-kneading | 30–100 | 51.5 | 16.3 |
| SiO2–MgO (IV) | Wet-kneading | 7–40 | 43.0 | 15.6 |
| SiO2–MgO (V) | Co-precipitationa | ~425 | 46.1 | 6.4 |
| SiO2–MgO (VI) | Co-precipitationb | —c | 49.4 | 1.2 |
| Technique | Sites studied | Type of information |
|---|---|---|
| Hammett indicator | Acidic, basic | Strength, amount |
| Pyridine-IR | Acidic | Strength, nature, (amount) |
| NH3-TPD | Acidic | Strength, amount |
| CDCl3-IR | Basic | Strength, nature, (amount) |
| CO2-TPD | Basic | Strength, amount |
Experimental
Materials
All precursors and chemicals used for the preparation of the SiO2–MgO materials are described in a previous publication.7 For the Hammett indicator study the following chemicals were used: benzene (Sigma-Aldrich, ACS reagent 99.0%) bromothymolblue (ABCR), phenolphthalein (Sigma-Aldrich, ACS reagent), 2,4-dinitroaniline (Acros Organics, 99%), 4-chloro-2-nitroaniline (Sigma-Aldrich purum, ≥98% (HPLC)), 4-nitroaniline (Acros Organics, 99%), 4-chloroaniline (Sigma-Aldrich, 98%) and diphenylmethane (99% Sigma-Aldrich). For IR analysis pyridine (Acros Organics, 99+%, for spectroscopy) and CDCl3 (Cambridge Isotope Laboratories, D, 99.8%) were used as probes.Preparation of SiO2–MgO catalysts
The five SiO2–MgO catalysts, as well as the SiO2 and MgO single oxides, were prepared according to our previously published procedures.7 All samples are prepared with a 1:1 molar ratio of the two components. A summary of the differences in preparation for the various samples is given in Table 1.
Hammett indicator study
All experiments were performed using standard Schlenk techniques. Benzene was dried over molecular sieves and stored under an Ar atmosphere. A solution of each indicator was prepared by dissolving 0.025 g of the indicator in 25 mL of dry benzene. The seven indicators used were (in order of increasing pKa, Table 3): bromothymolblue (pKa = 7.2), phenolphthalein (pKa = 9.3), 2,4-dinitroaniline (pKa = 15.0), 4-chloro-2-nitroaniline (pKa = 17.2), 4-nitroaniline (pKa = 18.4), 4-chloroaniline (pKa = 26.5) and diphenylmethane (pKa = 35.0). Before every experiment, 0.1 g of catalyst was dried at 473 K in vacuo. The flask containing the catalyst was then put under Ar atmosphere, after which 2 mL of dry benzene were added to the sample under investigation; at this point a few drops of the indicator solution were added to the suspension of the catalyst in benzene. After a few min, a color change was observed on the surface of the solid catalyst, indicating that the majority of the indicator molecules, initially in their neutral form, had been deprotonated. The flask was then stored for 24 h, in order to confirm the color change (or its absence).| Indicator | pKa | Color acidic form | Color basic form |
|---|---|---|---|
| Bromothymol blue | 7.2 | Yellow | Blue |
| Phenolphthalein | 9.3 | Colorless | Red |
| 2,4-Dinitroaniline | 15.0 | Yellow | Violet |
| 4-Chloro-2-nitroaniline | 17.2 | Yellow | Orange |
| 4-Nitroaniline | 18.4 | Colorless | Orange |
| 4-Chloroaniline | 26.5 | Colorless | Pink |
| Diphenylmethane | 35.0 | Colorless | Yellow/orange |
Pyridine- and CDCl3-IR spectroscopy
Fourier Transform Infrared (FT-IR) spectroscopy after pyridine adsorption (Pyridine-IR) measurements were taken with 25 scans per spectrum on a Perkin Elmer System 2000 with a DTGS detector and a resolution of 4 cm−1. Approximately 0.015 g of the catalyst were pressed into a pellet and placed into the pyridine cell. The catalyst was first dried in the cell under vacuum using a temperature ramp of 5 K min−1 to reach the desired temperature of 823 K. IR spectra were taken every 25 K in the temperature range 323–823 K. Once 823 K was reached, a spectrum acquired at this temperature was used to study the OH signals of the various SiO2–MgO samples. This temperature was kept constant for 30 min, after which the sample was cooled to 323 K.At this temperature, pyridine was allowed onto the catalyst. After 20 min of equilibration, the catalyst was put under vacuum for 30 min to remove physisorbed pyridine. Subsequently, temperature-programmed desorption was started with a temperature ramp of 5 K min−1 up to 823 K. After every temperature increase of 25 K, the temperature was kept constant for 5 min. IR spectra were taken every 25 K to study thermal pyridine desorption.
A similar procedure was used for FT-IR after deuterated chloroform adsorption (CDCl3-IR), with the main difference being the drying of the catalyst. This was performed by heating the catalyst-containing cell in a constant N2 flow (~243 mL min−1) instead of in vacuo for practical reasons (i.e., to avoid CDCl3 traces to remain as impurities in a setup dedicated to Pyridine-IR analysis). Once 823 K was reached, this temperature was kept for 60 min after which the sample was cooled to 323 K. At this temperature, the flow was adjusted to approximately 99 mL min−1 and, subsequently, N2 was passed through the CDCl3-containing gas bubbler for 30 min, to carry the probe molecule to the IR cell containing the catalyst. Similar to the Pyridine-IR analysis, a TPD procedure was applied by heating them with 5 K min−1 up to 823 K. However, the observed darkening of catalyst pellets (possibly due to the formation of carbonaceous deposits upon thermal treatment, as a result of decomposition of CDCl3 on basic sites)34 discouraged the use of the FT-IR spectra acquired above 323 K.
NH3- and CO2-TPD
Adsorbed ammonia Temperature Programmed Desorption (NH3-TPD) measurements were performed on a Micromeritics ASAP2920 apparatus. 0.2 g of sample dried in-situ in a He flow with a temperature ramp of 5 K min−1 up to 873 K. Subsequently, the sample was cooled to 373 K; at this point, NH3 pulses of 25.3 cm3 min−1 were applied. The sample was then heated to 873 K with a ramp of 5 K min−1 to induce desorption of NH3.A procedure similar to the one described for NH3-TPD was employed in the case of Adsorbed carbon dioxide Temperature Programmed Desorption (CO2-TPD), the main difference being the lower temperature (313 vs. 373 K) at which CO2 pulses are fed to the sample. For the calculation of the number of acidic or basic sites, it was assumed that only one molecule of NH3 or CO2 can adsorb on a single site.
Results and discussion
IR analysis of OH region
The FT-IR spectra of the wet-kneaded catalyst SiO2–MgO (III) and the single oxides MgO and SiO2 show that significant differences between the samples are present in the OH region (3770–3600 cm−1, Fig. 2). The FT-IR band of the hydroxyl groups on magnesia is reported to vary between 3800–3200 cm−1 depending on its thermal history, coordination and, hydrogen bonding interactions. Knözinger et al. identified four different OH groups on the surface of MgO, with peaks at around 3750 and 3700 cm−1 being attributed to isolated OH groups (mono- or multicoordinated, respectively).35 The peak observed at ~3730 cm−1 can thus be ascribed to both kinds of isolated OH groups (a low-intensity shoulder can be observed at ~3750 cm−1). | ||
| Fig. 2 FT-IR spectra of a) SiO2–MgO (III), b) SiO2 and c) MgO. The spectra, taken at 823 K, are offset for clarity. | ||
Silica is reported to show a sharp absorption at ~3745 cm−1 for isolated silanols.36 In our case, despite having been subjected to a pretreatment temperature as high as 823 K, a rather broad band centered at around 3700 cm−1 is seen instead for the Stöber-like SiO2 (spheres of 30–100 nm in diameter), pointing at both isolated and H-bonded silanol groups (Fig. 2).
Notably, the FT-IR spectrum of SiO2–MgO (III) shows three, rather than two local maxima. Based on the above, the peak at 3730 cm−1 was assigned to hydroxyl groups on MgO and the partially obscured peak at 3700 cm−1 to the stretching of silanol groups.
The third maximum at ~3672 cm−1, is not observed in the spectrum of either of the single oxides, and must thus originate from an interaction between the two components. Notably, antigorite, a magnesium silicate mineral with formula Mg3Si2O5(OH)4 and a structure consisting of octahedral Mg(OH)2 layers covalently bonded with tetrahedral silica layers (Fig. 3), shows one single peak in the OH region at 3670 cm−1. This vibration is assigned to OH groups in the octahedral Mg layer that are interacting with silica via H-bonding.37 Interestingly, antigorite is also reported to be active in the Lebedev process.6 Based on this and taking into account the TEM morphology of the wet-kneaded catalysts (a thin MgO layer is seen to be deposited on top of the silica spheres, ESI†Fig. 2), the latter peak was assigned to brucite-like OH groups H-bonded to SiO2 (Mg–(OH)⋯O–Si). The vibration at 3672 cm−1 could thus be tentatively assigned to similar species, keeping in mind that a number of other magnesium silicates also show absorption at a similar spectroscopic position (e.g., talc at 3674 cm−1).38 A similar observation, pointing at magnesium silicate formation, has recently been reported.19
 | ||
| Fig. 3 Schematic representation of the antigorite structure and its FT-IR spectrum as obtained from the RRUFF database.37 | ||
Significant differences among the various SiO2–MgO catalysts are observed in the OH region of the FT-IR spectra (Fig. 4). All catalysts prepared via wet-kneading show the three peaks discussed above, albeit with different relative intensities. In particular, the intensity of the central peak (OH groups on SiO2) decreases upon decreasing the size of the silica (Table 1), with the peak at 3672 cm−1 simultaneously increasing in intensity. Indeed, a smaller SiO2 size means that the MgO phase can interact with a larger portion of the external SiO2 surface, resulting in more Mg–O–H⋯O–Si groups in SiO2–MgO (III) and SiO2–MgO (IV).
 | ||
| Fig. 4 FT-IR spectra of a) SiO2–MgO (II), b) SiO2–MgO (III), c) SiO2–MgO (IV), d) SiO2–MgO (V) and e) SiO2–MgO (VI). The spectra, taken at 823 K in the region 3770–3600 cm−1, are offset for clarity. | ||
For the two co-precipitated catalysts (V–VI), the peak at 3672 cm−1 is notably absent. As a result of the method of precipitation of both components in ethanol, no crystalline MgO phase is formed (ESI,†Fig. 1); furthermore, the ill-defined morphology observed by TEM for SiO2–MgO (VI) (ESI,†Fig. 2) strongly hints at the formation of an amorphous mixed oxide structure and antigorite-like structures are therefore not expected.
Hammett indicator study
The Hammett indicator method assesses the overall acidity and basicity of solid materials with a number of indicators of known pKa and pKb.20 All indicators used in this study are, initially, in the protonated form. Before the experiment, the SiO2–MgO catalyst is treated at 473 K overnight (a temperature lower than those used in FT-IR and TPD analysis; the pretreatment temperature employed is important as it affects the presence of adsorbates, e.g. H2O and CO2, on the surface of the samples). The interaction of the indicator with the catalyst causes the adsorbed indicator molecules to be deprotonated on the condition that H_ (as defined by Tanabe39 as a measure of strength for basic sites of solid materials) of the tested sample is higher than the pKb of the basic form of the indicator. As the experiments are performed in benzene, the indicator molecules once deprotonated can interact with any acidic site, if present, of the solid materials and revert back to their initial form. The observed color therefore probes the contribution of all accessible acidic and basic sites and their strength (ESI,†Scheme 1). The H_ of a catalyst is then determined as the range between the pKa of the last indicator showing color change and the pKa of the first indicator that is not transformed into its deprotonated, basic form.Table 4 shows the results for the color changes for four catalysts and for the single oxides, SiO2 and MgO. SiO2–MgO (V) could not be tested as it showed an intrinsic coloration. As expected, MgO and SiO2 were observed to be the most and the least basic materials, respectively. Of the SiO2–MgO catalysts, SiO2–MgO (II) and SiO2–MgO (III) are the most basic, having a H_ in the range 15.0–17.2; the current set of Hammett indicators does not allow differentiation in basicity of these two samples. SiO2–MgO (VI) is the least basic, with a H_ value between 9.3 and 15.0. Finally, SiO2–MgO (IV) is more basic than phenolphthalein but less than 4-chloro-2-nitroaniline (pKa = 9.3 and 17.2, respectively). When tested against 2,4-dinitroaniline a color between the basic and acidic indicator forms was observed, suggesting a H_ value of ~15.0 for SiO2–MgO (IV). The Hammett indicator study thus showed the overall basicity/acidity to follow the order (from the most to the least basic): MgO > SiO2–MgO (II) = SiO2–MgO (III) > SiO2–MgO (IV) > SiO2–MgO (VI) > SiO2.
| Sample | H_a |
|---|---|
| a The H_ value refers to the strength of solid bases as the pKa range between the last indicator showing color change and the first one for which the color change could not be appreciated. b The color observed is in between that of the neutral (protonated) and deprotonated forms of 2,4-dinitroaniline (pKa = 15.0). c H_ could not be determined due to intrinsic coloration of the material. | |
| MgO | 18.4–25.5 |
| SiO2–MgO (II) | 15.0–17.2 |
| SiO2–MgO (III) | 15.0–17.2 |
| SiO2–MgO (IV) | ~15.0b |
| SiO2–MgO (V) | n.d.c |
| SiO2–MgO (VI) | 9.3–15.0 |
| SiO2 | 7.2–9.3 |
The overall acidity/basicity order of the SiO2–MgO catalysts coincides with catalyst performance; in particular, co-precipitated SiO2–MgO (VI) was found to be less basic than all wet-kneaded catalysts, which agrees well with the highest yields of the ethylene and diethyl ether by-products that are observed for this material. While color changes with Hammett indicators are somewhat subjective, the order given by the Hammett indicator study is confirmed by the pyridine-IR and NH3-TPD results, as discussed below.
Acidity
Pyridine is commonly used to probe acidity and to distinguish between Lewis and Brønsted acid sites.40,41 Before pyridine-IR analysis, the surface of the samples was cleaned by heating the samples to 823 K. No FT-IR peaks were detected at 1550 and 1650 cm−1, which would have been indicative of Brønsted acid sites, for any of the SiO2–MgO catalysts (Fig. 5) or for the two single oxides. Janssens et al., similarly, did not observe Brønsted acid sites for the Ag/MgO–SiO2 nor for the unsupported MgO–SiO2 materials with pyridine-IR.19 Surprisingly, they did consider mildly Brønsted acidic silanol sites, i.e. so weakly acidic that no interaction with pyridine is seen, to be responsible for the dehydration of crotyl alcohol to butadiene. On the other hand, four bands (1445, 1490, 1577 and 1604 cm−1) attributed to pyridine adsorbed on Lewis acidic sites where observed at the same position for all the SiO2–MgO catalysts. Indeed, coordinatively unsaturated Mg2+ cations can behave as Lewis acid sites in these Mg-containing materials. Similar peaks were also observed for SiO2 and MgO (not shown), albeit with much lower intensity and thermal stability. | ||
| Fig. 5 FT-IR spectra of a) SiO2–MgO (II), b) SiO2–MgO (III), c) SiO2–MgO (IV), d) SiO2–MgO (V) and e) SiO2–MgO (VI) after adsorption of pyridine. The spectra, taken at 423 K in the region 1650–1400 cm−1, are offset for clarity (L, Lewis acid sites; B, Brønsted acid sites). | ||
The intensity of the observed FT-IR bands (especially of the most intense one at around 1445 cm−1) can be used to estimate the amount of acidic sites (Fig. 5), giving the following order: SiO2–MgO (VI) > SiO2–MgO (IV) > SiO2–MgO (V) ~ SiO2–MgO (III) > SiO2–MgO (II). The background in the considered region varies considerably, however; in particular for the co-precipitated catalysts, carbonate species present on the catalyst surface hamper a more accurate quantification of the Lewis acid sites.42 After pyridine adsorption, the samples were heated stepwise to 823 K, to probe the strength of the acid sites. The temperatures at which all adsorbed pyridine is lost are: SiO2–MgO (VI) (748 K) > SiO2–MgO (IV) (723 K) > SiO2–MgO (III) (673 K) > SiO2–MgO (II) (623 K) > SiO2–MgO (V) (573 K) > MgO (498 K) > SiO2 (398 K).
The amount of acidic sites of the various materials was further quantified by NH3-TPD with one broad peak of NH3 desorption being observed for all SiO2–MgO catalysts (Fig. 6). The amount of acidic sites is generally in good agreement with the pyridine-IR results: SiO2–MgO (VI) > SiO2–MgO (V) > SiO2–MgO (IV) > SiO2–MgO (III) > SiO2–MgO (II) (Table 5). That SiO2–MgO (V) contains more acidic sites than SiO2–MgO (IV) according to NH3-TPD might be due to the difference in size of the probes or to the difference in background signal in the FT-IR spectra. The co-precipitated samples thus possess a higher amount of acidic sites than the wet-kneaded ones. This again correlates well with the higher amount of dehydration products (i.e., ethylene and diethyl ether) obtained on the former set of catalysts. Furthermore, the wet-kneaded catalysts containing smaller silica particles, i.e. SiO2–MgO (III) and SiO2–MgO (IV), possess a higher amount of acidic sites, again correlating with the higher amounts of dehydration products observed (Fig. 1).
 | ||
| Fig. 6 NH3 temperature-programmed desorption (TPD) profiles for the various SiO2–MgO materials in the range 373–873 K. | ||
Basicity
IR spectroscopy of adsorbed deuterated chloroform (CDCl3) allows for the study of basic sites of solid materials. CDCl3 is preferred over CHCl3, as the interaction of the latter with basic sites would result in peaks in the range 3200–2800 cm−1, a region where the ubiquitous –OH stretching bands are located.43Depending on the kind of basic site CDCl3 is interacting with, different bands are observed originating from C–D stretching vibrations; i.e. bands in the range 2245–2250 cm−1 are ascribed to the interaction with weak basic sites (B1, B2 in Scheme 3), while peaks at 2210–2220 cm−1 are due to strong basic sites (C). For each of these types of sites, a shift towards lower wavenumbers implies relatively stronger basic sites.20
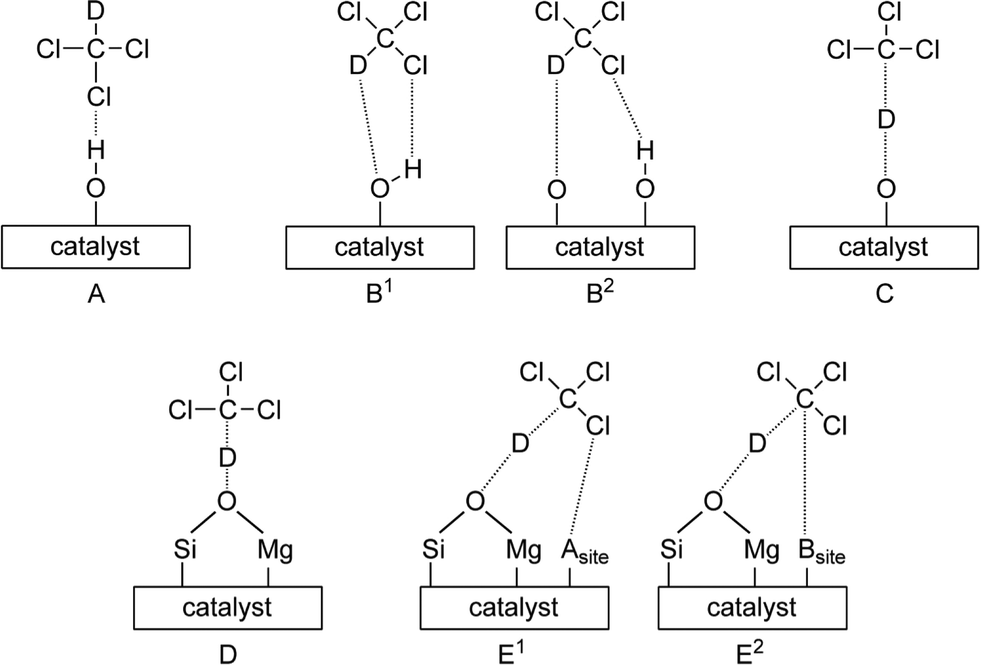 | ||
| Scheme 3 Schematic representation of the interactions for CDCl3 with basic sites of a solid catalyst. Structures A–C are reported by Ono et al.20 Structures D, E1 and E2 are postulated in the present study. Asite and Bsite represent acidic and basic sites, respectively. | ||
The spectra shown for a selected number of SiO2–MgO catalysts and MgO (Fig. 7) were recorded at 323 K. All samples show three similar features at ~2264, 2250 and 2213 cm−1. The low-intensity peak observed for all samples at ~2264 cm−1 has been seen before for SiO2 probed with CDCl3. The similarity in peak position to the C–D stretching vibration of gaseous CDCl3 suggests that CDCl3 interacts through the Cl atom with a OH group of SiO2, thus having a minimally perturbed C–D bond and behaving as a physisorbed species (A).43
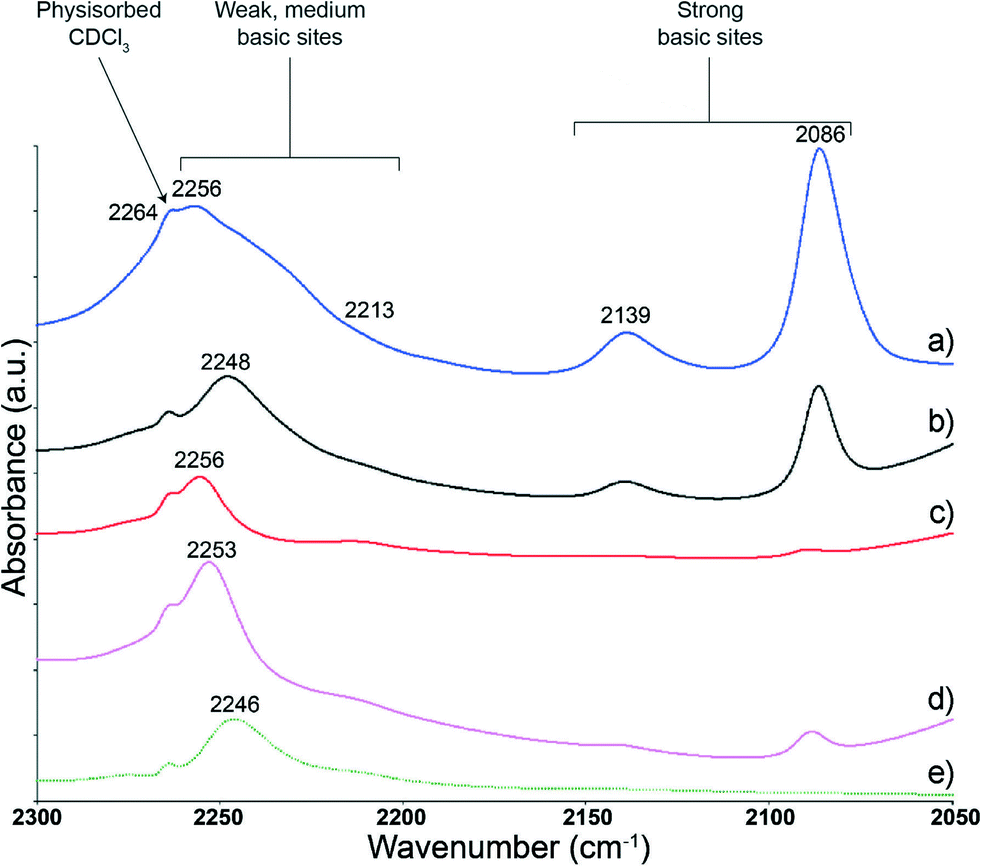 | ||
| Fig. 7 FT-IR spectra of a) SiO2–MgO (VI), b) SiO2–MgO (II), c) SiO2–MgO (III), d) SiO2–MgO (IV) and e) MgO after adsorption of CDCl3. The spectra, taken at 323 K in the region 2300–2050 cm−1, are offset for clarity. | ||
All materials show a weak shoulder at approximately 2213 cm−1 attributed to CDCl3 interaction with strong basic sites. The interpretation of the position and intensity of this spectroscopic feature is hindered by the intense peak for weak basic sites. Indeed, for all materials, the region 2264–2213 cm−1 is dominated by a broad peak resulting from the interaction of CDCl3 on weak basic sites, with the peak maxima being a function of the average strength of these weak basic sites as stated above.
Furthermore, for all SiO2–MgO materials, two additional FT-IR peaks at around 2139 and 2086 cm−1 are found, values that are lower than those usually reported for C–D stretching vibrations. This suggests that the C–D bond is further weakened possibly due to the presence of stronger basic sites. Comparing the basicity of different oxides, Paukshtis et al. observed a band at 2160 cm−1, tailing up to ~2120 cm−1 and assigned this band to adsorption of CDCl3 on CaO on strong basic sites of structure C (the peak was not seen for other oxides, including SiO2 and MgO).43 This assignment to a structure of type C is somewhat disputed, as others have assigned the peak at 2213 cm−1 to structure C instead.20,34 Notably, Lopez and coworkers observed a peak at around 2139 cm−1 for sulfated SiO2–MgO materials and attributed it to the interaction of CDCl3 on oxygen atoms bridged on Si and Mg (D). A low-intensity peak was also observed, but not assigned, at around 2028 cm−1 in the same study.34 The fact that the FT-IR peak at 2139 cm−1 is completely absent in the case of MgO, while being observed, albeit with different intensity, for all the SiO2–MgO catalysts corroborates the assignment to a structure of type D. The peak at ~2086 cm−1 has, to the best of our knowledge, not been reported before. This feature might originate from the presence of very strong basic sites. On the other hand, it could also be the result of two sites in close proximity. In particular, in addition to the well-known interaction of deuterium with a basic site, it is possible that: 1) the partially positive carbon atom of CDCl3 interacts with another basic site or 2) one of the chlorine atoms interacts with an acidic site on the surface. Both interactions (E1 and E2) would cause weakening of the C–D bond. The peaks at 2139 and 2086 cm−1 are highest in intensity for SiO2–MgO (VI). We previously showed with other characterization techniques, such as UV–vis, XRD and TEM, that this sample possesses the highest degree of intimate contact between SiO2 and MgO,7 supporting the assignment to CDCl3 bonded to O(H) bridged between Si and Mg atoms. Further (computational) studies are required for a firm assignment of this peak.
The two catalysts having the highest butadiene selectivity, i.e. SiO2–MgO (III–IV) are shown to have a significantly lower amount of strong basic sites as compared with SiO2–MgO (II). In addition, the weak basic sites of SiO2–MgO (II) are relatively stronger than those observed for the other two wet-kneaded catalysts. As discussed in the introduction, Climent et al. observed that weak basic and acidic sites are preferred over strong ones in catalysts for the aldol condensation of heptanal with benzaldehyde.29 The higher butadiene yield observed for SiO2–MgO (III–IV) as compared with SiO2–MgO (II) can thus be ascribed to a more beneficial ratio and strength of acid and basic sites for the self-aldolization of acetaldehyde (step 2, Scheme 1). This is corroborated by the observation that a relatively larger amount of acetaldehyde is observed for SiO2–MgO (II), implying that the latter catalyst is not able to perform the aldol condensation step as efficiently as the other wet-kneaded SiO2–MgO samples. SiO2–MgO (II) might furthermore not be sufficiently acidic for the promotion of the dehydration steps involved in the mechanism (steps 3 and 5), adding to the reduced performance.
Finally, CO2-TPD was used for the quantification of the basic sites of the various SiO2–MgO samples. Desorption was monitored over a temperature range of 313–873 K (Fig. 8), after pretreating the samples by bringing them to a temperature of 873 K with 5 K min−1 in He flow. The amounts of basic sites probed by CO2 (Table 5) follows the order: SiO2–MgO (II) > SiO2–MgO (III) > SiO2–MgO (IV) > SiO2–MgO (V) > SiO2–MgO (VI). The catalysts prepared via wet-kneading thus show a higher amount of basic sites than the co-precipitated ones.
 | ||
| Fig. 8 CO2 desorption for the various SiO2–MgO catalysts in the range 313–873 K. | ||
The CO2-TPD basicity order follows the order of the Hammett indicator studies, but the absolute amounts of basic sites are unexpectedly low. Indeed, the amount of basic sites calculated with CO2-TPD is actually 10-fold lower than the amount of acidic sites observed with NH3-TPD. Ordomskiy and co-workers previously reported a similar discrepancy for SiO2, 2 wt% ZrO2/SiO2 and 2 wt% MgO/SiO2 materials used for the condensation of acetaldehyde to crotonaldehyde.44 NH3- and CO2-TPD showed SiO2 to possess almost no active (basic or acidic) sites, while promotion with MgO increased the amount of the basic sites (from ~8 to 37 μmol g−1), and acidic sites (from ~30 to 116 μmol g−1). This significant increase in the amount of acidic sites for the MgO/SiO2 sample was attributed to low-coordination Mg atoms. The MgO–SiO2 materials, prepared by dry milling and subsequently exposed to water, reported by Janssens et al. contained a similar amount of basic sites (0.022–0.042 mmol g−1) as our materials.19 No quantification of the acid sites was reported for these samples, precluding further comparison.
Clearly, the Hammett indicator results show our SiO2–MgO materials to be prevalently basic. In addition, the basicity order obtained by CDCl3-IR study is quite different from the CO2-TPD one, with the former technique showing the co-precipitated catalyst SiO2–MgO (VI) to be more basic than all wet-kneaded ones. These observations suggest that CO2-TPD, under the applied experimental conditions, is unable to probe all (catalytically) relevant basic sites. It should furthermore be noted that the interaction of CO2 with certain basic sites can lead to the formation of carbonate species of extremely high thermal stability.42 Moreover, it is known that the presence or absence of H2O can significantly alter the nature and stability of such adsorbed carbonate species.45 In our case, any H2O present would be desorbed (together with CO2) during thermal pretreatment; CO2 is then introduced on a heavily modified catalyst surface (dry and, probably, significantly dehydroxylated). For this reason, carbonates are formed that differ in chemical nature from those originally found on the catalyst surface as a result of adsorption of atmospheric CO2. Indeed, the CO2 desorption seen in Fig. 8 upon further heating the samples from 673 K to temperatures higher than 873 K suggests that very strong basic sites are present. Significant CO2 desorption peaks centered at around 1000 K (ESI,† Fig. S3) were indeed observe upon further heating. The CO2 desorbed at these high temperatures should, however, not be included in the quantification of basic sites, as significant structural changes will occur at these elevated temperatures. While the TPD results thus do support the strong basic sites proposed based on the CDCl3-IR results, we do not consider CO2-TPD a reliable method for the quantification of the basic sites on our samples. The TPD results also suggest that carbonates might be present during catalysis. Indeed, as the carbonates formed by adsorption of atmospheric CO2 upon exposure to air can also be thermally very stable, the exact conditions of the thermal pretreatment employed prior to a catalytic run or characterization to remove such adsorbates, both in terms of atmosphere and temperature used, can be critical.
Conclusion
Systematic characterization of the acid–base properties of SiO2–MgO catalysts prepared by different methods provided new insights into how catalyst preparation influences catalytic performance. Such a characterization study for SiO2–MgO catalysts for the Lebedev process is essential to establish a structure-activity relationship. For the series of catalysts studied here, the co-precipitated ones proved to contain a higher amount of acidic and strong basic sites than those prepared by wet-kneading. As a result, higher selectivity towards ethylene and diethyl ether and lower butadiene yields are obtained.Certain acidity is required, though, as illustrated by the fact that the catalyst with the lowest amount of acid sites also gave the lowest butadiene yield of the wet-kneaded catalysts. The best catalysts were those with a limited amount of strong basic sites and an intermediate amount of acidic ones. The aldol condensation step that is key to butadiene formation is known to benefit from cooperativity between weak acid and basic sites. Based on the spectroscopic data, it is therefore proposed that the best catalysts are those that can perform the aldol condensation step most efficiently, as a result of the right amount and proximity of acidic and basic sites of moderate strength. A graphical summary of the consequences of preparation method on morphology, acid–base properties and performance of SiO2–MgO catalysts is given in Fig. 9.
 | ||
| Fig. 9 Graphical summary of the morphology, acid–base properties and consequences for catalysis for the wet-kneaded and co-precipitated catalysts. | ||
It is clear that, regardless of the actual mechanism, the elementary steps leading to butadiene formation require a subtle balance in acid–base sites. Fundamental understanding of the acid–base properties required for the Lebedev process (as a whole and for the individual elementary steps) is therefore expected to ultimately result in the design of new and improved catalysts for the Lebedev process.
Acknowledgements
This project has been performed in the framework of the CatchBio program. The authors gratefully acknowledge the support of the Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science.Notes and references
- S. V. Lebedev, British Pat. 331, 402, 1929 Search PubMed.
- S. V. Lebedev, J. Gen. Chem., 1933, 3, 698–708 CAS.
- S. V. Lebedev, Chem.-Ztg., 1936, 60, 313–316 Search PubMed.
- G. Natta and R. Rigamonti, Chim. Ind., 1947, 29, 195–201 CAS.
- R. Ohnishi, T. Akimoto and K. Tanabe, J. Chem. Soc., Chem. Commun., 1985, 1613–1614 RSC.
- S. Kvisle, A. Aguero and R. P. A. Sneeden, Appl. Catal., 1988, 43, 117–131 CrossRef CAS.
- C. Angelici, M. E. Z. Velthoen, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2014, 7, 2505–2515 CrossRef CAS PubMed.
- C. Angelici, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2013, 6, 1595–1614 CrossRef CAS PubMed.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degrève, P. A. Jacobs and B. F. Sels, Chem. Soc. Rev., 2014, 43, 7917–7953 RSC.
- V. Gruver, A. Sun and J. J. Fripiat, Catal. Lett., 1995, 34, 359–364 CrossRef CAS.
- W. M. Quattlebaum, W. J. Toussaint and J. T. Dunn, J. Am. Chem. Soc., 1947, 69, 593–599 CrossRef CAS.
- S. K. Bhattacharyya and S. K. Sanyal, J. Catal., 1967, 7, 152–158 CrossRef CAS.
- H. Niiyama, S. Morii and E. Echigoya, Bull. Chem. Soc. Jpn., 1972, 45, 655–659 CrossRef CAS.
- B. B. Corson, H. E. Jones, C. E. Welling, J. A. Hinckley and E. E. Stahly, Ind. Eng. Chem., 1950, 42, 359–373 CrossRef CAS.
- J. Scalbert, F. Thibault-Starzyk, R. Jacquot, D. Morvan and F. Meunier, J. Catal., 2014, 311, 28–32 CrossRef CAS PubMed.
- T. Tsuchida, J. Kubo, T. Yoshioka, S. Sakuma, T. Takeguchi and W. Ueda, J. Catal., 2008, 259, 183–189 CrossRef CAS PubMed.
- M. León, E. Díaz and S. Ordóňez, Catal. Today, 2011, 164, 436–442 CrossRef PubMed.
- M. León, E. Díaz, A. Vega, S. Ordóñez and A. Auroux, Appl. Catal., B, 2011, 102, 590–599 CrossRef PubMed.
- W. Janssens, E. V. Makshina, P. Vanelderen, F. De Clippel, K. Houthoofd, S. Kerkhofs, J. A. Martens, P. A. Jacobs and B. F. Sels, ChemSusChem, 2015, 8, 994–1008 CrossRef CAS PubMed.
- Y. Ono and H. Hattori, Solid Base Catalysis, Tokyo Institute of Technology Press/Springer, 2010 Search PubMed.
- V. K. Díez, C. R. Apesteguía and J. I. Di Cosimo, Catal. Today, 2000, 63, 53–62 CrossRef.
- M. Perissinotto, M. Lenarda, L. Storaro and R. Ganzerla, J. Mol. Catal. A: Chem., 1997, 121, 103–109 CrossRef CAS.
- J. Macht, C. D. Baertsch, M. May-Lozano, S. L. Soled, Y. Wang and E. Iglesia, J. Catal., 2004, 227, 479–491 CrossRef CAS PubMed.
- A. Gervasini, J. Fenyvesi and A. Auroux, Catal. Lett., 1997, 43, 219–228 CrossRef CAS.
- M. León, E. Díaz, A. Vega, S. Ordóñez and A. Auroux, Appl. Catal., B, 2011, 102, 590–599 CrossRef PubMed.
- M. E. Manríquez, T. López, R. Gómez and J. Navarrete, J. Mol. Catal. A: Chem., 2004, 220, 229–237 CrossRef PubMed.
- J. N. Chheda and J. A. Dumesic, Catal. Today, 2007, 123, 59–70 CrossRef CAS PubMed.
- R. K. Zeidan and M. E. Davis, J. Catal., 2007, 247, 379–382 CrossRef CAS PubMed.
- M. J. Climent, A. Corma, V. Fornés, R. Guil-Lopez and S. Iborra, Adv. Synth. Catal., 2002, 344, 1090–1096 CrossRef CAS.
- M. D. Jones, C. G. Keir, C. Di Iulio, R. A. M. Robertson, C. V. Williams and D. C. Apperley, Catal. Sci. Technol., 2011, 1, 267–272 CAS.
- H.-J. Chae, T.-W. Kim, Y.-K. Moon, H.-K. Kim, K.-E. Jeong, C.-U. Kim and S.-Y. Jeong, Appl. Catal., B, 2014, 150–151, 596–604 CrossRef CAS PubMed.
- V. L. Sushkevich, I. I. Ivanova, S. Tolborg and E. Taarning, J. Catal., 2014, 316, 121–129 CrossRef CAS PubMed.
- M. A. Aramendía, V. Borau, C. Jiménez, J. M. Marinas, J. R. Ruiz and F. Urbano, Appl. Catal., A, 2003, 249, 1–9 CrossRef.
- J. C. Lavalley, Catal. Today, 1996, 27, 377–401 CrossRef CAS.
- E. Knözinger, K.-H. Jacob, S. Singh and P. Hofmann, Surf. Sci., 1993, 290, 388–402 CrossRef.
- B. A. Morrow and A. J. McFarlan, J. Phys. Chem., 1992, 96, 1395–1400 CrossRef CAS.
- http://rruff.info/Antigorite .
- http://rruff.info/talc .
- K. Tanabe, Solid Acids and Bases, Kodansha/Elsevier, 1989 Search PubMed.
- S. Damyanova, M. A. Centeno, L. Petrov and P. Grange, Spectrochim. Acta, Part A, 2001, 57, 2495–2501 CrossRef CAS.
- A. Travert, A. Vimont, A. Sahibed-Dine, M. Daturi and J.-C. Lavalley, Appl. Catal., A, 2006, 307, 98–107 CrossRef CAS PubMed.
- C. Angelici, B. M. Weckhuysen and P. C. A. Bruijnincx, submitted for publication.
- E. A. Paukshtis, N. S. Kotsarenko and L. G. Karakchiev, React. Kinet. Catal. Lett., 1979, 12, 315–319 CrossRef CAS.
- V. V. Ordomsky, V. L. Sushkevich and I. I. Ivanova, J. Mol. Catal. A: Chem., 2010, 333, 85–93 CrossRef CAS PubMed.
- J. V. Evans and T. L. Whateley, Trans. Faraday Soc., 1967, 63, 2769–2777 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy00200a |
| This journal is © The Royal Society of Chemistry 2015 |