
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient nitrogen-13 radiochemistry catalyzed by a highly stable immobilized biocatalyst†
Eunice S.
da Silva
a,
Vanessa
Gómez-Vallejo
b,
Jordi
Llop
*a and
Fernando
López-Gallego
*cd
aRadiochemistry and Nuclear Imaging, CIC biomaGUNE, Paseo Miramon 182, 20009, San Sebastian, Spain. E-mail: jllop@cicbiomagune.es
bRadiochemistry Platform, Molecular Imaging Unit, CIC biomaGUNE, Paseo Miramon 182, 20009, San Sebastian, Spain
cBiofunctional Nanomaterials Unit, CIC biomaGUNE, Paseo Miramon 182, 20009, San Sebastian, Spain. E-mail: flopez.ikerbasque@cicbiomagune.es
dIKERBASQUE, Basque Foundation for Science, Bilbao, Spain
First published on 23rd February 2015
Abstract
The continuous progress of nuclear imaging techniques demands the development and implementation of efficient, clean and sustainable synthetic routes for the preparation of novel radiotracers. This is especially challenging in the context of radiotracers labelled with short-lived positron emitters such as nitrogen-13. Biocatalysis can offer attractive alternatives to conventional chemistry, because enzymes present exquisite chemical selectivity and high turnover numbers. However, enzymes have been poorly exploited in the area of radiochemistry. Herein, we present the design and fabrication of a heterogeneous biocatalyst suitable for the reduction of [13N]NO3− to [13N]NO2−. A eukaryotic nitrate reductase from Aspergillus niger was immobilized on different carriers and immobilization parameters were determined. Optimal results were obtained for agarose beads activated with a positively charged tertiary amino group (Ag-DEAE). The immobilized preparation was 12-fold more thermostable than the soluble enzyme. Biochemical characterization of the immobilized enzyme showed interesting thermally-induced hyperactivation driven by the interaction between the enzyme and the carrier. The heterogeneous biocatalyst could be re-used up to 7 reaction cycles while preserving its initial activity. Finally, to demonstrate the potential of this heterogeneous biocatalyst in the context of radiochemistry, radiosynthesis of S-[13N]nitrosoglutathione was carried out using the enzymatically produced [13N]NO2− as the labelling agent.
Introduction
Recent advances in Positron Emission Tomography (PET) have encouraged chemists to synthesize novel radiotracers to enable non-invasive diagnosis of a larger variety of diseases and investigation of their molecular basis.1 Due to the short half-life (T1/2) of the most commonly used positron emitters, i.e. fluorine-18 (18F, T1/2 = 109.8 min), carbon-11 (11C, T1/2 = 20.4 min) and nitrogen-13 (13N, T1/2 = 9.97 min), the radiosynthesis of PET tracers requires the development of efficient chemical schemes in order to synthesize and purify the radioactive species in a short period of time.2 Indeed, radiolabelling methods must be fast and highly efficient to perform chemical reactions and downstream processing in an appropriate time scale, thus preventing excessive radioactivity loss while improving the specific activity of the resulting radiotracers.Biocatalysis can offer attractive solutions in the area of radiochemistry because enzymes present exquisite chemical selectivity and high turnover numbers. Enzymes enable fast chemical conversions, and yield highly pure products under extremely mild conditions. However, whereas enzymes have naturally evolved as soluble and labile catalysts, stability and solubility issues often limit their industrial applications. These issues may be circumvented by enzyme immobilization, which turns enzymes into stable and heterogeneous biocatalysts.3 In this context, appropriate selection of the solid carrier and immobilization chemistry is crucial to achieve highly active and stable heterogeneous enzymes that can be readily integrated into continuous processes.
There are a handful of examples where immobilized enzymes have been utilized for the synthesis of different 18F-and 11C-labelled radiotracers.3–7 Nevertheless, enzyme-assisted preparation of radiotracers labelled with shorter-lived positron emitters such as 13N is more challenging and remains rather unexplored.8 In fact, there are only a few examples where immobilized enzymes have been applied to the catalytic incorporation of 13N into L-amino acids.9–11
Usually, 13N is generated via the 16O(p,α)13N nuclear reaction by proton irradiation of pure water. Nitrogen-13 is formed as a mixture of 13N-labelled nitrate ([13N]NO3−), nitrite ([13N]NO2−) and ammonia ([13N]NH3), [13N]NO3− being the major species (~85% of total radioactivity); if a radical scavenger (e.g. 5 mM ethanol) is added to the irradiated water, [13N]NH3 is the major species. As recently shown by our research group, the labelling agent [13N]NO2− is a useful precursor for the preparation of a wide range of labelled compounds such as S-[13N]nitrosothiols,12,13S-[13N]nitrosamines,1413N-labelled azo derivatives15,16 and 13N-labelled azides,17 and further applications are currently being investigated. Unfortunately, direct production of [13N]NO2− in the cyclotron is not feasible and is usually achieved by reduction of [13N]NO3− to [13N]NO2− in a column containing activated cadmium. Besides the inherent toxicity of cadmium, which may hamper the translation of the technology to the clinical arena, this experimental setting presents several technological pitfalls.
Inspired by the biological cycle of nitrogen, we reasoned that eukaryotic nitrate reductase (eNR) may efficiently catalyze the reduction of [13N]NO3− to the more convenient labelling agent [13N]NO2−. eNR is a homodimer which contains three different domains per monomer: first, an N-terminus domain that binds NADPH and FAD+ as redox cofactors (NADPH-domain), second, a cytochrome b domain containing a Fe-heme cofactor (heme-domain) and third, a C-terminus domain that binds a molybdopterin cofactor (MOCO-domain). All these cofactors are required during the catalytic cycle; while FAD+, heme and MOCO cofactors are stably bound to the enzymes, NADPH must be exogenously added to the reaction media. The enzyme mechanism relies on the large conformational change in which the NADPH/FAD+ and heme domains approaches the MOCO domain, allowing electron transfer from NADPH to molybdenum via the heme group to reduce NO3− to NO2−.18 Immobilization of this enzyme is rather challenging since restrictions in the conformational flexibility required for the catalysis may compromise the reducing capacity. This structural complexity may explain the low recovered activity of immobilized eNRs reported elsewhere.19
In the current work, we present the unprecedented use of eNR as an efficient heterogeneous biocatalyst for the reduction of [13N]NO3− to [13N]NO2−. The immobilization chemistry has been optimized to yield active and stable preparations of immobilized eNR on porous agarose beads. As a proof of concept of the applicability of the experimental set up to the preparation of PET tracers, the 13N-labelled nitrosothiol S-[13N]nitrosoglutathione ([13N]GSNO) has been prepared with excellent radiochemical conversion.
Results and discussion
Immobilization of eNR on functionalized porous agarose beads
Eukaryotic nitrate reductase (eNR) from Aspergillus niger was immobilized through different immobilization chemistries on a survey of agarose-based matrices (see Table 1) having the same physical parameters (surface area, pore size and particle diameter) but different functional groups on their surfaces. We screened several immobilization chemistries (reversible and irreversible) in order to find the optimal one in terms of both activity and stability of the resulting heterogeneous biocatalyst.| Carrier | Reactive group | Immobilization chemistry | Immobilized activity Aia (mU g−1) | Immobilization yield ψb (%) | Expressed activity Ae (mU g−1)c/(%)d |
|---|---|---|---|---|---|
| The initial offered activity was always 1000 mU per gram.a Ai = the activity of the enzyme immobilized on 1 gram of carrier after the immobilization process. This activity was calculated as the difference between the offered activity and the activity in the supernatant after incubation with the carrier for 1 h.b Immobilization yield (ψ) = (immobilized activity/offered activity) × 100.c The expressed activity (Ae) is defined as the measured activity of the immobilized enzyme after washing.d Relative expressed activity (Ae) = (expressed activity/immobilized activity) × 100. | |||||
| Ag-CB | Cyanogen bromide | Covalent bonding through the eNR Nt | 964 ± 32 | 96 ± 3 | 26 ± 4/(3) |
| Ag-DEAE | Diethylaminoethyl | Ionic adsorption through the most acidic eNR regions | 995 ± 10 | 99 ± 1 | 210 ± 60/(21) |
| Ag-PEI | Polyethyleneimine | 982 ± 12 | 98 ± 1 | 155 ± 25/(16) | |
| Ag-MANAE | Monoaminoethyl | 820 ± 24 | 82 ± 2 | 192 ± 41/(23) | |
| Ag-DS | Dextran sulphate | Ionic adsorption through the most basic eNR regions | 750 | 75 | 58/(8) |
Table 1 shows that all carriers were able to efficiently immobilize eNR (immobilization yields >75% in all cases), although low values of expressed activity were achieved after immobilization when compared to those of the free enzyme. Irreversible immobilization and cationic exchange on Ag-CB and Ag-DS led to a dramatic decrease in the expressed activity (26 ± 4 and 58 mU g−1, respectively). On the other hand, ionic immobilization of eNR on positively charged carriers yielded values of expressed activity in the range 155–210 mU g−1, resulting in a relative expressed activity of up to 23%. It is noteworthy that although the chemistry that drove the immobilization on Ag-MANAE, Ag-DEAE and Ag-PEI was the same, Ag-MANAE produced lower immobilization yields (82 ± 2%) than Ag-DEAE (99 ± 1%) and Ag-PEI (98 ± 1%). This may be explained by the fact that the Ag-MANAE surface presents a lower density of positively charged groups than Ag-DEAE and Ag-PEI surfaces.
In order to explain the functional discrepancies obtained with the different immobilized preparations, we further studied the eNR surface to understand how the protein orientation on the carrier surface may affect its functionality. Unfortunately, the X-ray structure of eNR from A. niger has not been solved yet; hence its 3D-structural homology model containing the NADPH-, heme- and MOCO-domains was built (Fig. 1A) and the electrostatic surface potential was calculated in order to determine the spatial charge distribution across the enzyme surface (Fig. 1B and C). The analysis of this structural model reveals an acidic belt on the MOCO-domain located at the opposite face with regard to the active site. The model also shows that the heme, FAD+, NAD+ and molybdenum binding sites are surrounded by basic amino acids (Fig. 1C). These structural observations suggest that eNR immobilized on Ag-DS is oriented through its active site (basic region), hampering the accessibility of both NADPH and nitrate to the catalytic pocket with a consequent decrease in catalytic efficiency. Nevertheless, the eNR orientation on the positively charged carriers seems to occur through one acidic region located far away from the binding pockets. As a consequence, active centres remain fairly accessible to the substrates, resulting in significantly higher relative expressed activity values for such immobilized preparations (Fig. 1C). Moreover, such orientation seems to negligibly affect dimer stability because the dimerization domain is not involved in the protein–carrier interaction. In fact, when eNR immobilized on Ag-DEAE was incubated under high ionic strength conditions, the enzyme was quantitatively eluted to the supernatant, demonstrating the ionic character and the reversibility of this immobilization chemistry.
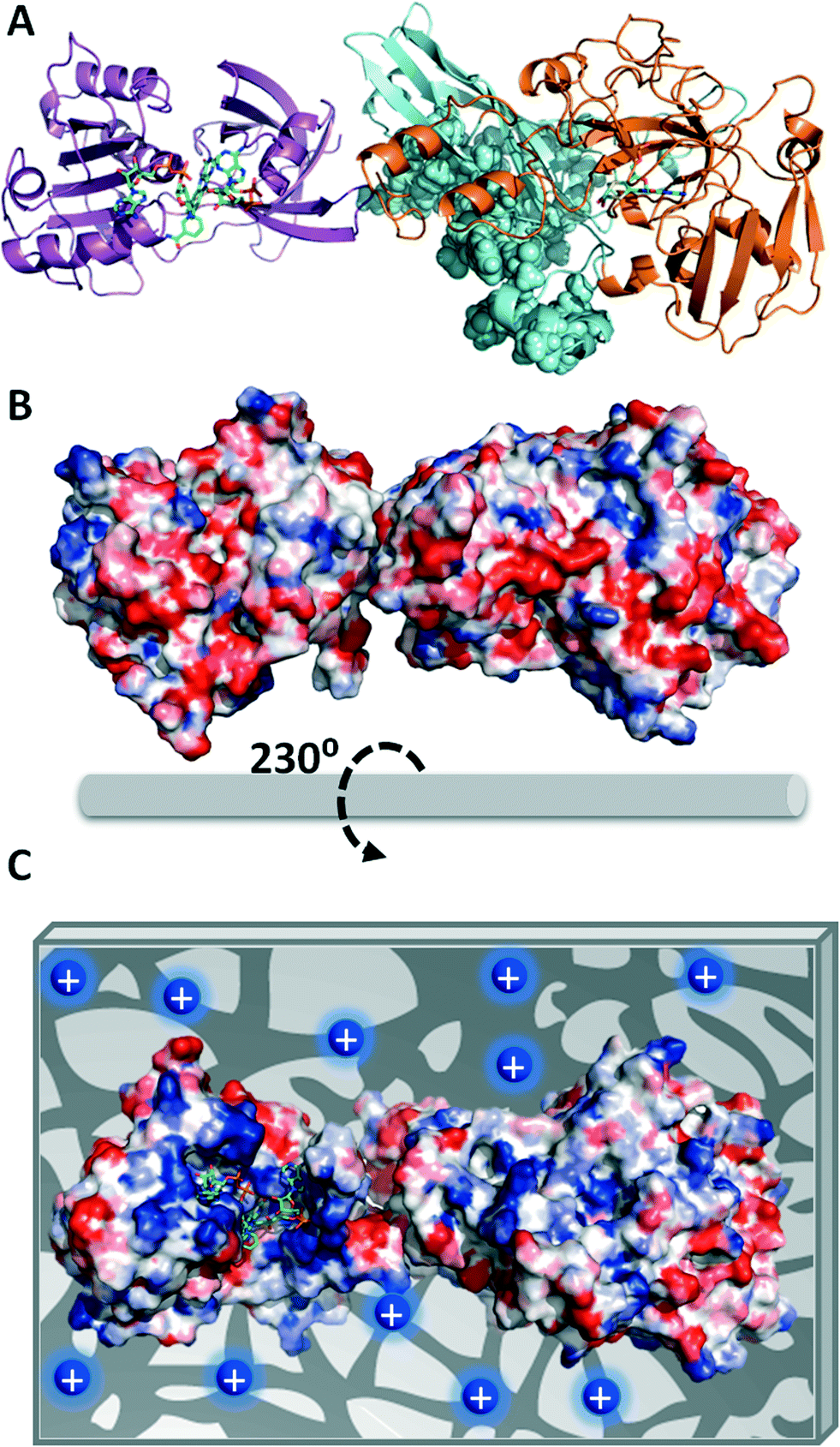 | ||
| Fig. 1 Structural model of a monomer of eNR from Aspergillus niger. (A) Cartoon representation of the 3D-structural homology model using the X-ray structures of eNR from Pichia pastoris and cytochrome B5 from rat complexed with both FAD+ and NAD+ as templates for the molybdenum domain (N-terminus, orange) and the cytochrome domain (C-terminus, purple), respectively. The sphere representation colored in cyan depicts the dimerization domain of eNR. The green sticks represent the cofactors NAD+, FAD+ and molybdopterin. (B) Surface electrostatic potential of eNR calculated by the Bluues server (back face of the active site) and (C) the proposed orientation of the eNR monomer on the porous Ag-DEAE surface. The blue circles represent the tertiary amine groups located at the carrier surface. The surface electrostatic potential is depicted with a red–blue gradient that corresponds to the acid–base gradient. All the images were prepared by using PyMol version 0.99. | ||
The values of expressed activity shown in Table 1 have been determined by using NADPH as a cofactor; such values are relatively high when compared to previous results obtained using immobilization protocols based on porous hydrogels. For example, eNR from Aspergillus niger immobilized on a porous vinyl polymeric matrix expressed less than 2% of its expressed activity by using NADPH as a cofactor.19 The high structural complexity of eNR might be the main reason for the low expressed activity achieved in this previous work. This enzyme requires enough structural flexibility for efficient electron transfer between the cytochrome and molybdenum domains. Hence, immobilization chemistries that highly rigidify the 3D structure of eNR (covalent immobilization) may hinder catalytic conformational changes or promote wrong protein orientations that limit either electron transfer or substrate accessibility, resulting in a low expressed activity of the immobilized preparation.
Effects of temperature on both activity and stability of eNR immobilized on positively charged agarose beads
The thermal stabilities of immobilized and soluble eNR were tested at different temperatures. Fig. 2A shows the inactivation courses of the three different immobilized preparations incubated at pH = 7.5 and 25 °C. The half-life time of eNR immobilized on Ag-DEAE was estimated to be 30-fold and 6-fold higher than that of eNR immobilized on Ag-MANAE and Ag-PEI, respectively, suggesting a dependence of the thermal stability of the immobilized enzymes on the chemical nature of the amine groups located at the surface of the carrier. The surface of Ag-DEAE containing tertiary amine groups seems to establish more favourable interactions with the acidic protein regions than the Ag-MANAE surface, which presents a low density of primary amine groups, and the Ag-PEI surface coated with a polymeric bed with a high density of primary, secondary and tertiary amine groups. Ag-PEI has been suggested as an excellent carrier to stabilize multimeric enzymes, because the basic polymeric bed establishes 3D-protein–polymer interactions20 that attach all the subunits to the carrier, resulting in the stabilization of the quaternary structure of the protein. Unexpectedly, in this study, eNR immobilized on Ag-PEI was less stable than eNR immobilized on Ag-DEAE. This unexpected result may be explained by the fact that the flexibility of the polymer may generate additional interactions with the protein over time. Such enhancement of the spatial reactivity may lead to undesired interactions between both cytochrome and molybdenum domains and the polymeric brushes, reducing the enzyme flexibility required for electron transfer, and consequently decreasing the nitrate reduction activity over time. These negative interactions would not be possible in the much less flexible Ag-DEAE surface formed by a 2D-monolayer of tertiary amine groups. Hence, our results suggest that the interaction between eNR and the positively charged carriers must be strong enough to guarantee the stabilizing immobilization effect and flexible enough to enable the conformational changes required for catalysis.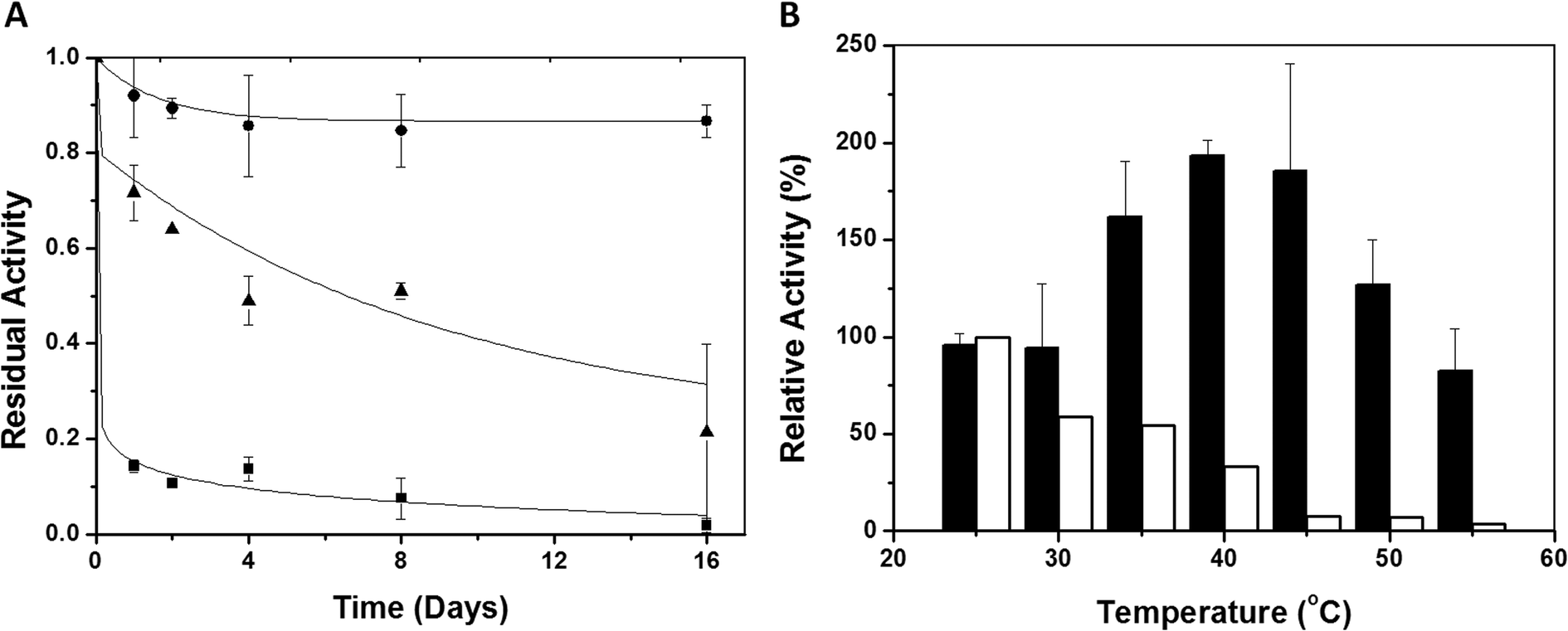 | ||
| Fig. 2 Stability of eNR. (A) Inactivation course of different eNR immobilized preparations at 25 °C and pH 7.5. eNR was immobilized on Ag-DEAE (●), Ag-PEI (▲) and Ag-MANAE (■). Values are normalized to starting activity. (B) Inactivation of soluble eNR (white bars) and eNR immobilized on Ag-DEAE (black bars) at different temperatures. The soluble and immobilized preparations were incubated for 15 minutes at different temperatures and then the activities were assayed at 25 °C. | ||
The Ag-DEAE immobilized enzyme was much more stable than its soluble counterpart over a broad range of temperatures (30–55 °C) (Fig. 2B). Surprisingly, when such an immobilized biocatalyst was incubated for only 15 minutes in the range of 40–45 °C and the enzyme activity was assayed at 25 °C, a 2-fold activity enhancement (hyperactivation) was observed with respect to the non-thermally treated enzyme (Fig. 2B). This effect was not observed with the soluble enzyme, as its initial activity decreased after incubation at T > 25 °C due to thermal inactivation. Interestingly, both the soluble and immobilized enzymes showed similar activity/temperature profiles (Fig. S1†). Altogether, these results suggest that the aforementioned hyperactivation effect can only be attributed to the presence of the carrier and that it is not related to the effect of the temperature on the catalysis itself. Inactivation kinetic studies (pH 7.5 and 40 °C) of the hyperactivated immobilized enzyme proved that the stability was enhanced with respect to its soluble counterpart under the same conditions (Fig. S2†). The immobilized enzyme showed a thermal inactivation constant (Ki) of (1.31 ± 0.63) × 10−5 (s−1) at 40 °C, while the Ki value of the soluble enzyme was (15.3 ± 2.3) × 10−5 (s−1). These values confirm that the immobilization process results in an increased half-life time (T1/2) of eNR up to 883 minutes, which is 12 times longer than the T1/2 of the soluble enzyme.
The thermal-hyperactivation and thermal-stabilization effects observed for eNR immobilized on Ag-DEAE may be explained by some beneficial enzyme–carrier interactions that drive some local structural re-organization in the enzyme, resulting in a more efficient electron transfer between the NADPH and MOCO domains. These interactions may stabilize a more beneficial conformation of eNR, which explains the 2-fold more active and 12-fold more stable nature of the enzyme. These carrier- and temperature-assisted conformational changes were further investigated by fluorescence studies that suggested a relationship between the improved functional properties and structural reorganization (ESI,† Fig. S3). Indeed, changes in the fluorescence spectrum of the immobilized enzyme were observed with increased temperature, while no changes were observed for the soluble enzyme incubated under the same conditions. Here, the temperature seems to act as an external stimulus that induces optimal fitting between the surfaces of the enzyme and the carrier, resulting in a suitable geometric congruence for the catalysis. Despite the nature of this thermally-induced more active conformation being not clear at present, it is worth mentioning that similar effects have been described for other thermostable soluble enzymes.21–23 In any case, we put forth one of the few experimental demonstrations of thermal hyperactivation and stabilization assisted by the carrier surface, which acts not only as a scaffold to stabilize eNR but also as an active surface that induces temperature-triggered positive conformational changes on the enzyme.
Kinetic parameters and loading capacity of eNR immobilized on Ag-DEAE
Based on both the high stability and acceptable catalytic efficiency of eNR immobilized on Ag-DEAE, we selected this heterogeneous biocatalyst to carry out sustainable reduction of [13N]NO3− to [13N]NO2− in aqueous media under mild conditions. However, before applying this biocatalyst to radiochemistry, we carried out its kinetic characterization. To this aim, Michaelis–Menten parameters were determined for both soluble and immobilized eNRs towards both nitrate and NADPH (Table 2).| K m (mM) | k cat (s−1) | k cat/Km (s−1 mM−1) | ||
|---|---|---|---|---|
| a The steady-state kinetic parameters were measured at pH 7.5 and 25 °C (see sections Materials and Methods). | ||||
| NaNO3 | NR (soluble) | 0.27 ± 0.07 | 30.33 | 111.23 |
| NR on Ag-DEAE | 0.06 ± 0.03 | 11.97 | 199.44 | |
| NADPH | NR (free) | 0.07 ± 0.03 | 27.93 | 399.05 |
| NR on Ag-DEAE | 1.86 ± 0.82 | N. D. | N. D. | |
Kinetic studies provided us with interesting data that helped to understand the activity decrease resulting from the immobilization process. At first glance, the immobilized preparation showed a 26-fold higher Km[NADPH] than the soluble enzyme. Contrarily, Km[NO3] was 4.5 times lower for the immobilized eNR than that for the soluble preparation. These results confirm that immobilization of eNR on Ag-DEAE negatively affected the enzyme binding towards NADPH, which explains the low expressed activity after the immobilization process. The high Km[NADPH] values for the immobilized preparation may be due to two main reasons: i) conformational changes induced by the carrier during the immobilization process that diminish the enzyme affinity towards NADPH, and ii) mass transfer issues that hamper NADPH diffusivity from the bulk solution to the active sites of the immobilized eNR molecules.
In order to elucidate which reason contributes more to the low expressed activity, the effect of enzyme loading on both immobilization yield and expressed activity after immobilization was tested. Fig. 3 shows that immobilization was always quantitative regardless of the enzyme load. However, the relative expressed activity presented a strong negative logarithmic correlation with the enzyme load. Under very low enzyme loads (37.5 μgeNR gcarrier−1), the relative expressed activity was 85%, while the value decreased to approximately 20% at loads higher than 75 μgeNR gcarrier−1 (Fig. 3).
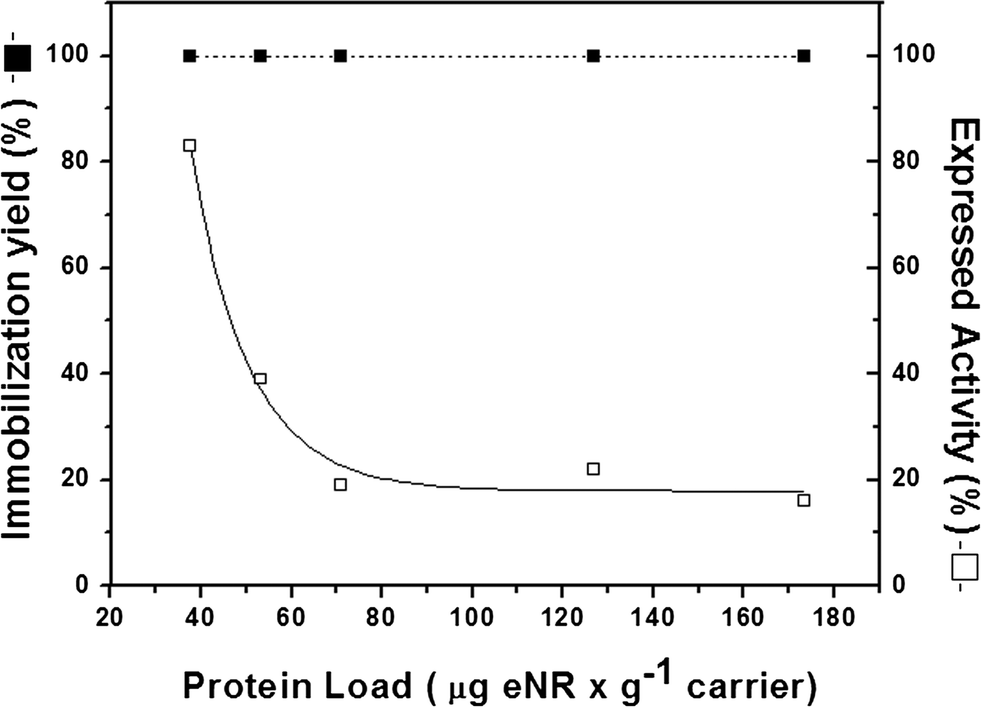 | ||
| Fig. 3 Enzyme-loading capacity of Ag-DEAE. The experiment was carried out by adding different protein concentrations to 200 mg of the carrier at pH 7.5 and 25 °C. Immobilization yield (full square) and relative expressed activity (open square) were determined for each enzyme load and were calculated as previously described in Table 1. | ||
This experiment demonstrates that immobilization chemistry does not promote inactive enzyme conformations that intrinsically diminish the specific activity after the immobilization process. In such a scenario, the expressed activity should be always low regardless of the enzyme load. Therefore, it is plausible to think that the mass transfer limitations of the substrate and the cofactors are the main reason that explains the low expressed activity of the immobilized eNR. Contextualizing these results together with the high Km[NADPH] values for the immobilized preparation, we suggest that the cofactor withstands severe mass transfer limitations to diffuse across the Ag-DEAE microstructure and reach the enzyme active sites. Under high enzyme loads, NADPH does not efficiently diffuse into the porous microstructure of the carrier to saturate all active sites immobilized on such a carrier. On the contrary, under very low enzyme loads, the vast majority of the eNR molecules are saturated with NADPH. These results are in good agreement with previous data,24 which showed that eNR from A. niger covalently immobilized on non-porous particles expresses more than 90% of its specific activity after immobilization. Moreover, they strongly suggest that the low specific activity of eNR immobilized on positively charged agarose beads is due to limited NADPH diffusion rather than inaccurate protein orientation, because positively charged porous carriers seem to immobilize eNR through an optimal orientation according to the protein surface analysis (Fig. 1C).
Reduction of radiolabelled nitrate catalyzed by eNR immobilized on Ag-DEAE and reusability of the immobilized biocatalyst
eNR immobilized on Ag-DEAE was used for the reduction of [13N]NO3− to [13N]NO2−. The reaction was carried out in batch mode and the immobilized biocatalyst was re-used for several reaction cycles (Fig. 4). In the first cycle, the immobilized enzyme only yielded 60% [13N]NO2− in 4 minutes while the soluble enzyme reduced 96% of [13N]NO3− to [13N]NO2−. Surprisingly, in the second cycle the immobilized enzyme reduced up to 93% of [13N]NO3− to [13N]NO2−. We suggest that the differences in NADPH effective concentration inside the carrier pores might cause this intriguing effect.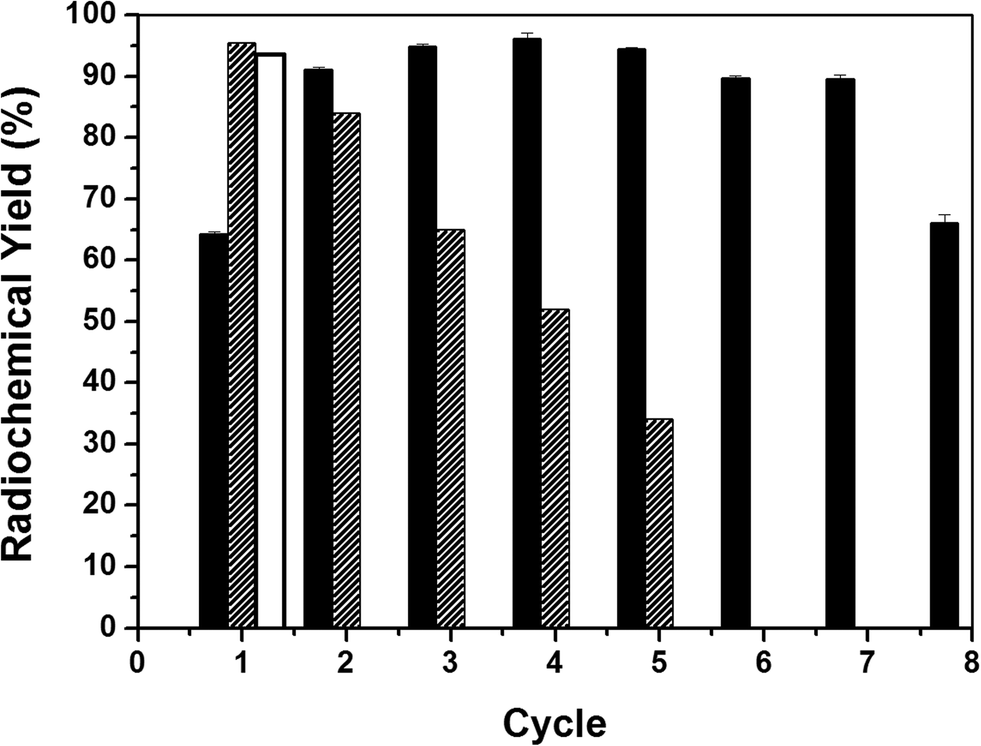 | ||
| Fig. 4 Reusability of eNR immobilized on Ag-DEAE. Soluble eNR (white bar), eNR immobilized on Ag-DEAE without NADPH equilibration (black bars) and eNR immobilized on Ag-DEAE equilibrated with NADPH (striped bars) were used to catalyze the reduction of [13N]NO3− to [13N]NO2− at 25 °C and pH 7.5. The reactions catalyzed by soluble and non-equilibrated immobilized eNR were carried out by adding exogenous NADPH, while the reactions catalyzed by equilibrated immobilized eNR were carried out without adding exogenous NADPH. | ||
We hypothesize that during the first reaction cycle some negatively charged NADPH molecules are ionically absorbed to the positively charged carrier surface. However, such molecules would present an association/dissociation equilibrium that would simultaneously provide one fraction of soluble NADPH available for the enzyme catalysis and a second fraction of NADPH trapped on the carrier surface that would be re-used in the consecutive cycles. In this scenario, the effective concentration of NADPH in the second and successive cycles will be higher, explaining the higher yield observed in the second cycle compared to that in the first one. In order to demonstrate this ionic interaction between NADPH and Ag-DEAE, we equilibrated the eNR immobilized on Ag-DEAE with NADPH in solution before triggering the first reaction cycle. The equilibrated heterogeneous biocatalyst was able to reduce 96% of [13N]NO3− to [13N]NO2− in the first cycle (Fig. 4). Moreover, we directly demonstrated by analyzing the NADPH fluorescence inside the solid particles of the heterogeneous biocatalyst that NADPH was bound to the carrier (Fig. S4†). This experimental evidence confirms that a fraction of NADPH is ionically trapped in the carrier surface, increasing the internal concentration of NADPH that results in higher reduction yields. In fact, when the radiochemical reduction was carried out with the NADPH-equilibrated immobilized enzyme, the heterogeneous biocatalyst was able to catalyze the radiolabelled nitrate reduction for 5 cycles with more than 30% yield without exogenous addition of NADPH (Fig. 4). This experiment provides evidence that NADPH molecules are reversibly bound to the carrier surface providing an internal cofactor concentration that is sufficient for the immobilized eNR to catalyze the radiochemical reduction with the maximum yield during the first cycle and proportionally lower yields in the consecutive cycles. The lower reduction yield throughout the cycles agrees with the fact that NADPH is oxidized to NADP+ decreasing the pool of NADPH trapped in the carrier porous structure. Therefore, throughout the consecutive cycles, immobilized eNR suffers from lower availability of reduced NADPH, explaining the lower yields throughout the operational cycles. Contrarily, when reduction was carried out with addition of exogenous NADPH in each reaction cycle, the maximum yield was maintained for 7 cycles, demonstrating the excellent operational stability of the immobilized biocatalyst (Fig. 4). As far as we know, this is the first evidence of a heterogeneous biocatalyst where the carrier plays an important role in supplying the corresponding cofactor to the enzyme. Furthermore, this optimal heterogeneous biocatalyst offers a clean final product without any significant protein contamination, even during the recycling process (Fig. S5†), confirming once again that ionic interactions between eNR and Ag-DEAE are quite strong in spite of their reversible nature.
Two-step chemo-enzymatic solid-phase synthesis of S-[13N]GSNO
To prove the concept that enzymatically synthesized [13N]NO2− can be directly integrated into radiochemical synthetic cascade reactions, we coupled the optimal eNR immobilized on Ag-DEAE to the two-step chemo-enzymatic synthesis of S-[13N]GSNO (Fig. S6†). [13N]NO3− was enzymatically reduced to [13N]NO2− in the presence of NADPH. The supernatant of such a reaction was passed through an anion exchange cartridge in order to trap the enzymatically produced [13N]NO2−. Then, an acidic solution of S-glutathione (1 mM) was also passed through the same anion exchange cartridge, enabling the solid-phase S-nitrosation of glutathione. The resulting product was finally desorbed from the resin by elution with distilled water. 95% of the enzymatically yielded [13N]NO2− was converted to S-[13N]GSNO, similar to the radiochemical conversion obtained in our previous work using Cd-reduced [13N]NO2−. This result confirms that the redox cofactor needed for the enzymatic reactions does not interfere with the next chemical step. Therefore, the radioactive nitrite produced with this heterogeneous biocatalyst is suitable to be used in radiochemical reactions.Experimental
Materials
The agarose beads activated with a primary amine group (Ag-MANAE),25,26 coated with 25 kDa of polyethyleneimine (Ag-PEI)20 and coated with dextran sulphate (Ag-DS)27 were prepared as described elsewhere. Cyanogen bromide-activated Sepharose® 4B (Ag-CB) was purchased from Sigma-Aldrich Química S.A. (Madrid, Spain) and DEAE-Sepharose was purchased from GE Healthcare (Pittsburgh, USA). Nitrate reductase from Aspergillus niger and reduced β-nicotinamide adenine dinucleotide 2′-phosphate tetrasodium salt (β-NADPH, purity >97%) were purchased from Roche Life Science (Barcelona, Spain). Sodium nitrite (NaNO2, ACS reagent, Ph. Eur.), sodium nitrate (NaNO3, ACS reagent), sodium phosphate dibasic (Na2HPO4, for molecular biology, purity ≥98.5%), sodium phosphate monobasic (NaH2PO4, purity ≥99.0%), glutathione (GSH, purity ≥99%), S-nitrosoglutathione (GSNO, purity ≥97%), hydrochloric acid (HCl, 1 M standard solution), sodium hydroxide (NaOH, ACS reagent), trifluoroacetic acid (CF3COOH, HPLC grade) and acetonitrile (CH3CN, HPLC grade) were purchased from Sigma-Aldrich Química S.A. (Madrid, Spain) and used without further purification. Bio-Rad protein assay dye reagent concentrate was purchased from BIO-RAD (Madrid, Spain). The additive for the ion chromatography mobile phase (P/N 5062-2480) was purchased from Agilent Technologies (Madrid, Spain). All others reagents were of analytical grade unless otherwise specified.Methods
Radiochemistry
Enzymatic reduction of [13N]NO3− to [13N]NO2− was carried out with 6.72 mU of immobilized eNR packed in a plastic column. This insoluble preparation was incubated with 90 μL of cyclotron produced 13N solution (which contained [13N]NO3−) in the presence of 10 μL of 2.1 mM NADPH (0.21 mM final concentration) in 500 mM sodium phosphate buffer (final concentration = 50 mM, pH = 7.5) under mild conditions (25° C and pH = 7.5) for 4 min. Relative amounts of [13N]NH4+, [13N]NO2− and [13N]NO3− were measured by radio-HPLC. An Agilent 1200 series HPLC equipped with a quaternary pump, a multiple wavelength detector and a radiometric detector (Gabi, Raytest) was used. An HP Asahipak ODP-50 (5 μm, 125 × 4 mm, Teknokroma, Spain) was used as the stationary phase, and a solution containing an additive for ion chromatography (15 mL) in a water/acetonitrile mixture (86/14, V = 1 L) basified to pH = 8.6 with 1 M sodium hydroxide solution was used as the mobile phase at a flow rate of 1 mL min−1. Simultaneous UV (λ = 254 nm) and isotopic detection were carried out.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) was used as the mobile phase at a flow rate of 1 mL min−1. Simultaneous UV (λ = 220 nm) and isotopic detection were carried out.
:5) was used as the mobile phase at a flow rate of 1 mL min−1. Simultaneous UV (λ = 220 nm) and isotopic detection were carried out.
Conclusions
Biochemical reactions have been exploited in many different synthetic reactions by chemists; however, radiochemists have not paid enough attention to biocatalysis, and there are only a handful of examples where enzymes have been utilized in radiosynthetic schemes. Such under-exploitation is even more dramatic in the case of immobilized enzymes, despite the recent advances in heterogeneous biocatalysis.Immobilized enzymes are exquisitely selective, highly active and stable, and they simplify the process work-up, potentially yielding pure radiotracers that can be directly used for biomedical purposes. Hence, they can be anticipated as ideal tools for the preparation of radiotracers labelled with short-lived radionuclides. In this work, we have applied for first time one kind of eukaryotic nitrate reductase immobilized on a solid carrier to a radiochemical process. This immobilized enzyme has been able to selectively reduce [13N]NO3− to [13N]NO2− aided by NADPH as a redox cofactor. We have demonstrated that by controlling the immobilization chemistry and physico-chemical properties of the carrier, optimal heterogeneous biocatalysts with high potential in synthetic chemistry can be achieved. It is noteworthy that this work is one of the few examples where an immobilized enzyme has been applied to a radiochemical process and is the first report so far on a two-step chemo-enzymatic route to synthesize model radiotracers starting from a precursor ([13N]NO3−) directly produced in the cyclotron.
We have faced the challenge to immobilize an enzyme whose catalytic mechanism depends on several cofactors and conformational changes, recovering enough activity and increasing its stability to catalyze several reaction cycles. The success of this work will so far open the doors to new applications of enzymes in radiochemistry and will potentiate the use of other chemo-enzymatic designs for the synthesis of novel radiotracers, even with short-lived isotopes. Successful enzyme immobilization will contribute to the development of flow radiochemical processes as well as integration of radiochemical synthesis on-chip.31–34
Acknowledgements
This work has been supported by the RADIOMI project (EU FP7-PEOPLE-2012-ITN-RADIOMI). We would like to thank the IKERBASQUE Foundation for the funding given to Dr. F. López-Gallego. We also acknowledge support from COST Action CM1303 Systems Biocatalysis.Notes and references
- K. Chen and X. Chen, Semin. Oncol., 2011, 38, 70 CrossRef PubMed.
- J. S. Fowler and A. P. Wolf, Acc. Chem. Res., 1997, 30, 181 CrossRef CAS.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885 CrossRef CAS.
- G. Slegers, R. H. Lambrecht, T. Vandewalle, L. Meulewaeter and C. Vandecasteele, J. Nucl. Med., 1984, 25, 338 CAS.
- M. Sasaki, M. Ikemoto, M. Mutoh, T. Haradahira, A. Tanaka, Y. Watanabe and K. Suzuki, Appl. Radiat. Isot., 2000, 52, 199 CrossRef CAS.
- M. E. Sergeev, F. Morgia, M. R. Javed, M. Doi and P. Y. Keng, J. Mol. Catal. B: Enzym., 2013, 92, 51 CrossRef CAS PubMed.
- M. E. Sergeev, F. Morgia, M. R. Javed, M. Doi and P. Y. Keng, J. Mol. Catal. B: Enzym., 2013, 97, 74 CrossRef CAS PubMed.
- V. Gómez-Vallejo, V. Gaja, K. B. Gona and J. Llop, J. Labelled Compd. Radiopharm., 2014, 57, 244 CrossRef PubMed.
- M. B. Cohen, L. Spolter, C. C. Chang, N. S. MacDonald, J. Takahashi and D. D. Bobinet, J. Nucl. Med., 1974, 15, 1192 CAS.
- A. S. Gelbard, R. S. Benua, R. E. Reiman, J. M. McDonald, J. J. Vomero and J. S. Laughlin, J. Nucl. Med., 1980, 21, 988 CAS.
- A. J. Cooper and A. S. Gelbard, Anal. Biochem., 1981, 111, 42 CrossRef CAS.
- J. Llop, V. Gómez-Vallejo, M. Bosque, G. Quincoces and I. Peñuelas, Appl. Radiat. Isot., 2009, 67, 95 CrossRef CAS PubMed.
- V. Gómez-Vallejo, K. Kato, I. Oliden, J. Calvo, Z. Baz, J. I. Borrell and J. Llop, Tetrahedron Lett., 2010, 51, 2990 CrossRef PubMed.
- V. Gómez-Vallejo, K. Kato, M. Hanyu, K. Minegishi, J. I. Borrell and J. Llop, Bioorg. Med. Chem. Lett., 2009, 19, 1913 CrossRef PubMed.
- V. Gaja, V. Gomez-Vallejo, M. Puigivila, C. Perez-Campana, A. Martin, A. Garcia-Osta, T. Calvo-Fernandez, M. Cuadrado-Tejedor, R. Franco and J. Llop, Mol. Imaging Biol., 2014, 16, 538 CrossRef PubMed.
- V. Gomez-Vallejo, J. I. Borrell and J. Llop, Eur. J. Med. Chem., 2010, 45, 5318 CrossRef CAS PubMed.
- S. M. Joshi, V. Gomez-Vallejo and J. Llop, Synthesis of 13N-labelled Phenyl Azide via Dutt–Wormall Reaction, Pamplona, Spain, 2014 Search PubMed.
- K. Fischer, G. G. Barbier, H.-J. Hecht, R. R. Mendel, W. H. Campbell and G. Schwarz, Plant Cell, 2005, 17, 1167 CrossRef CAS PubMed.
- R. B. Mellor, J. Ronnenberg, W. H. Campbell and S. Diekmann, Nature, 1992, 355, 717 CrossRef CAS.
- C. Mateo, O. Abian, R. Fernandez-Lafuente and J. M. Guisan, Biotechnol. Bioeng., 2000, 68, 98 CrossRef CAS.
- F. C. Wedler and F. M. Hoffmann, Biochemistry, 1974, 13, 3207 CrossRef CAS.
- F. Facchiano, R. Ragone, M. Porcelli, G. Cacciapuoti and G. Colonna, Eur. J. Biochem., 1992, 204, 473 CrossRef CAS PubMed.
- J. Rocha-Martin, D. Vega, J. Bolivar, C. Godoy, A. Hidalgo, J. Berenguer, J. Guisan and F. Lopez-Gallego, BMC Biotechnol., 2011, 11, 101 CrossRef CAS PubMed.
- V. Sachdeva and V. Hooda, Talanta, 2014, 124, 52 CrossRef CAS PubMed.
- B. C. Pessela, R. Munilla, L. Betancor, M. Fuentes, A. V. Carrascosa, A. Vian, R. Fernandez-Lafuente and J. M. Guisan, J. Chromatogr. A, 2004, 1034, 155 CrossRef CAS PubMed.
- R. Fernandez-Lafuente, C. M. Rosell, V. Rodriguez, C. Santana, G. Soler, A. Bastida and J. M. Guisan, Enzyme Microb. Technol., 1993, 15, 546 CrossRef CAS.
- M. Fuentes, B. C. C. Pessela, J. V. Maquiese, C. Ortiz, R. L. Segura, J. M. Palomo, O. Abian, R. Torres, C. Mateo, R. Fernández-Lafuente and J. M. Guisán, Biotechnol. Prog., 2004, 20, 1134 CrossRef CAS PubMed.
- K. Arnold, L. Bordoli, J. Kopp and T. Schwede, Bioinformatics, 2006, 22, 195 CrossRef CAS PubMed.
- I. Walsh, G. Minervini, A. Corazza, G. Esposito, S. C. E. Tosatto and F. Fogolari, Bioinformatics, 2012, 28, 2189 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248 CrossRef CAS.
- C. Rensch, S. Lindner, R. Salvamoser, S. Leidner, C. Bold, V. Samper, D. Taylor, M. Baller, S. Riese, P. Bartenstein, C. Wangler and B. Wangler, Lab Chip, 2014, 14, 2556 RSC.
- H. Zhang, M. V. Cantorias, N. Pillarsetty, E. M. Burnazi, S. Cai and J. S. Lewis, Nucl. Med. Biol., 2012, 39, 1182 CrossRef CAS PubMed.
- S. Lu, J.-H. Chun and V. W. Pike, J. Labelled Compd. Radiopharm., 2010, 53, 234 CrossRef PubMed.
- P. Y. Keng, S. Chen, H. Ding, S. Sadeghi, G. J. Shah, A. Dooraghi, M. E. Phelps, N. Satyamurthy, A. F. Chatziioannou, C.-J. C. Kim and R. M. van Dam, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 690 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Thermal stability supporting data, fluorescence studies, scheme of the chemo-enzymatic route used to synthesize [13N]GSNO and SDS-PAGE of both soluble and immobilized enzymes. See DOI: 10.1039/c5cy00179j |
| This journal is © The Royal Society of Chemistry 2015 |