
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of structural and electronic properties of organomolybdenum(II) complexes of the type [CpMo(CO)3R] and [CpMo(O2)(O)R] (R = Cl, CH3, CF3) on the catalytic olefin epoxidation†
Simone A.
Hauser‡
a,
Robert M.
Reich‡
a,
János
Mink
bc,
Alexander
Pöthig
d,
Mirza
Cokoja
d and
Fritz E.
Kühn
*ae
aMolecular Catalysis, Department of Chemistry and Catalysis Research Center, Technische Universität München, Lichtenbergstraße 4, D-85747 Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de; Fax: + 49 89 289 13473; Tel: +49 89 289 13096
bHungarian Academy of Sciences, Chemical Research Center, Pusztszeri u. 59–67, 1025 Budapest, Hungary
cFaculty of Information Technology, University of Pannonia, Egyetem u, 10 8200 Veszprém, Hungary
dCatalysis Research Center, Technische Universität München, Ernst-Otto-Fischer-Straße 1, D-85747 Garching bei München, Germany
eChair of Inorganic Chemistry, Department of Chemistry and Catalysis Research Center, Technische Universität München, Lichtenbergstraße 4, D-85747 Garching bei München, Germany
First published on 16th January 2015
Abstract
Six compounds of the type [CpMo(CO)3R] (R = Cl (1), CH3 (2), CF3 (3)) and [CpMo(O2)(O)R] (R = Cl (4), CH3 (5), CF3 (6) (Cp = η5-cyclopentadienyl)) have been synthesised and characterised. The crystal structures of [CpMo(CO)3CF3] and [CpMo(O2)(O)CF3] are compared to their literature known chloro and methyl derivatives. The influence of the groups R on the performance as epoxidation catalysts is examined. DFT calculations, IR-spectroscopy and X-ray crystallography help to explain differences in reactivity and enable a rational design of active molybdenum tricarbonyl and oxo-peroxo complexes.
Introduction
Epoxidation of olefins is a key process in industry.1 Epoxides are used e.g. as monomers in polymer chemistry, in electronics, the textile industry, pharmacy and the paper industry.2 Organometallic compounds such as methyltrioxorhenium (MTO) proved to be very active molecular catalysts for the epoxidation of a broad variety of olefins.3,4 It has been postulated that replacement of the CH3-moiety by the electron withdrawing CF3 group should further increase the catalytic activity of such a compound, due to the increased Lewis acidity on the metal centre. However, it was not possible so far to synthesise [CF3ReO3].5–8 Rhenium, a rather rare metal, however is much more expensive than other metals that can be utilised as central atoms in organometallic oxidation catalysts, molybdenum being an example (Re: ca. 3000 € kg−1; Mo: ca. 22 € kg−1 (July 2014)). Therefore and due to their easy derivatisation and better stability compared to most rhenium compounds of formula [RReO3],3,9 molybdenum compounds of the general formula [CpMo(CO)3R] and [CpMo(O2)(O)R] also gained interest as oxidation catalysts. Some of them indeed rival MTO in olefin epoxidation activity.10–12Based on mechanistic considerations the activity of molybdenum-based organometallic epoxidation catalysts appears to be largely governed by the Lewis acidity of the metal centre.13 An electron-withdrawing metal centre is considered of being able to both facilitate the direct coordination of an olefin to the metal, as described in Mimoun-type mechanisms,14,15 and the indirect coordination via oxygen atoms as suggested in Sharpless-type16 mechanisms (Scheme 1).
 | ||
| Scheme 1 Two major mechanistic proposals of olefin epoxidation: Mimoun-type and Sharpless-type mechanisms. | ||
The compound [CpMo(CO)3CF3] has already been applied in oxidation catalysis for cyclooctene, styrene and 1-octene as substrates.17 More recently it was also applied for the industrially important oxidation of propylene.18 However, so far it is unclear why [CpMo(CO)3CF3] appears to be less active than its methylated analogue [CpMo(CO)3CH3] in non-coordinating solvents, in contrast to previous expectations (vide supra). In this work structural and electronic properties and their relation to catalytic activities of compounds [CpMo(CO)3R] and [CpMo(O2)(O)R] R = CF3 and their derivatives, with R = Cl, CH3 are examined. Crystal structures of the fluorinated compounds were determined. In combination with DFT calculations and vibrational spectroscopy data these structures help to provide an explanation for the observed differences in activity.
Results and discussion
Comparison of the crystal structures of [CpMo(CO)3R] R = Cl (1), CH3 (2), CF3 (3)
The structure of the fluorinated molybdenum tricarbonyl derivative 3 has been determined by single crystal X-ray diffraction (Fig. 1). | ||
| Fig. 1 Ortep style drawing of the solid-state structure of 3 [CpMo(CO)3CF3]. The thermal ellipsoids are shown at the 50% probability level. | ||
As its methyl and chloride analogues, 3 crystallises as racemic twin in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . A bond length comparison of the three compounds (listed in Table 1) reveals some interesting features. The atom numbering has been adapted for the Cl and CH3 derivatives according to Fig. 2 in order to allow an easy comparison.
. A bond length comparison of the three compounds (listed in Table 1) reveals some interesting features. The atom numbering has been adapted for the Cl and CH3 derivatives according to Fig. 2 in order to allow an easy comparison.
| Bond lengths [Å] | 1 (ref. 19) | 2 (ref. 20) | 3 |
|---|---|---|---|
| a (For further information see ESI S.1†). | |||
| Mo–C1 | 2.014(2) | 1.984(4) | 2.003(3) |
| Mo–C2 | 1.980(2) | 1.997(4) | 2.015(3) |
| Mo–C3 | 2.008(2) | 1.993(4) | 2.001(3) |
| Mo–R | 2.5030(6) | 2.326(3) | 2.234(3) |
| C1–O1 | 1.138(3) | 1.144(4) | 1.145(4) |
| C2–O2 | 1.145(3) | 1.139(5) | 1.137(3) |
| C3–O3 | 1.136(3) | 1.131(4) | 1.143(3) |
| C6–F1 | — | — | 1.368(5) |
| C6–F2 | — | — | 1.367(5) |
| C6–F3 | — | — | 1.376(5) |
| Bond angles [°] | |||
| C1–Mo–C2 | 78.15(10) | 78.09(17) | 76.80(11) |
| C2–Mo–C3 | 75.80(10) | 78.87(15) | 76.11(11) |
| R–Mo–C1 | 78.15(7) | 72.39(13) | 76.45(10) |
| R–Mo–C3 | 77.86(7) | 72.42(17) | 75.99(10) |
| Mo–C1–O1 | 176.8(2) | 178.9(4) | 176.9(2) |
| Mo–C2–O2 | 177.9(2) | 178.9(4) | 177.6(2) |
| Mo–C3–O3 | 177.9(2) | 177.0(3) | 178.4(2) |
| Mo–R force constants [N cm−1] | 1.28 | 1.53 | 1.88 |
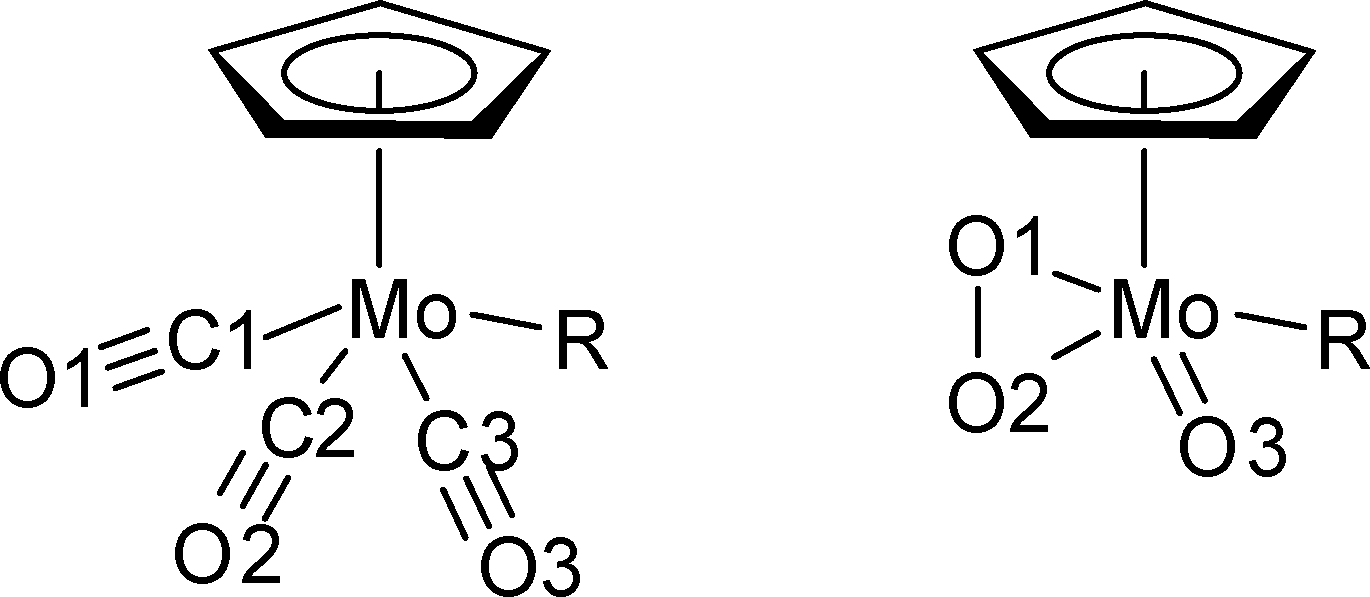 | ||
| Fig. 2 Drawings of the tricarbonyl (R = Cl (1), CH3 (2), CF3 (3), left) and oxoperoxo (R = Cl (4), CH3 (5), CF3 (6), right) complexes with the atom numbering used in Tables 1 and 3. | ||
The Mo–C2 bond, trans-positioned to R, appears to be influenced by the nature of R, however, no clear trend is apparent: it is elongated compared to the cis-carbonyls in complex 3, whilst in complex 1, this bond is shorter than the other two CO bonds. Further, the carbon–oxygen bonds show varying lengths, again with some trans-effect: a considerable elongation is apparent in complex 1, and in complex 3, it is shortened. In complex 2, however, no similar tendency can be observed.
The Mo–C–O bond angles show only small differences. Comparison of complex 3 to complex 2 reveals that in the latter, the angles between the carbonyls and the methyl group are smaller than the angles between the carbonyl ligands, whereas in complex 3, all angles between the three carbonyls and the CF3 group are nearly constant. The Mo–R bond angles show a decrease from R = Cl > CH3 > CF3.
Comparison of the vibrational spectra and force constants of [CpMo(CO)3R] R = Cl (1), CH3 (2) and CF3 (3) complexes
There are several reports on the infrared spectra of [CpMo(CO)3R]-type complexes.21–24 However, to the best of our knowledge (see ESI Table S3.3†), the first comprehensive overview on IR and Raman frequencies of these complexes, together with a complete assignment of the corresponding vibrational modes in accordance with several other reports is presented.25–27 Only a simplified force constant calculation (discussing only CO stretching vibrations and force constants) of the four complexes had been published so far.23 Whilst the fundamental frequencies of the cyclopentadienyl ligand practically do not depend on the ligand R bound to the metal centre, the CO stretching and Mo-ligand vibrations significantly change with the group R.For a better comparison, the characteristic stretching modes of the complexes are summarised in Table 3. The averaged CO stretching frequencies (ν1 + ν2 + ν14) are decreasing in the order 1983 > 1978 ≫ 1945 cm−1 for complex 3, 1 and 2, respectively. It appears that the coordination strength of the CO groups is quite similar in complexes 1 and 3 but different for the CH3 (2) derivative, where the CO groups are more strongly bound. This trend is also reflected in the averaged Mo–C stretching frequencies: 485 ≫ 462 > 455 cm−1 for complexes 2, 1 and 3, respectively. The high MoC stretching frequency leads to a higher MoC stretching force constant, 3.11 N cm−1 in case of complex 2, while smaller values of 2.86 and 2.94 N cm−1 are obtained for complexes 3 and 1.
| Frequencies [cm−1] | 1 | 2 | 3 | Description |
|---|---|---|---|---|
| a All fundamental vibrational frequencies are averaged values of experimental data listed in ESI. b Carbonyl ligand opposite to R group. c Stretch–stretch interaction between two CO groups in “short” distance. d Stretch–stretch interaction between two CO groups in “long” distance. | ||||
| ν 1, A′ | 2041 | 2012 | 2052 | CO sym. stretch |
| ν 14, A′′ | 1963 | 1920 | 1971 | CO asym. stretch |
| ν 2, A′ | 1929 | 1903 | 1927 | CO asym. stretch |
| ν 5, A′ | 468 | 502 | 478 | MoC sym. stretch |
| ν 16, A′′ | 468 | 502 | 478 | MoC asym. stretch |
| ν 7, A′ | 430 | 452 | 431 | MoC asym. stretch |
| ν 8, A′ | 280 | 406 | 250 | MoR stretch |
| ν 9, A′ | 355 | 335 | 351 | MoCp stretch |
| Force constants [N cm−1] | ||||
| K1(CO)b | 14.05 | 13.97 | 14.01 | |
| K2(CO) | 15.30 | 14.40 | 15.38 | |
| F s(CO,CO)c | 0.62 | 0.56 | 0.59 | |
| F l(C′O,CO)d | 0.31 | 0.28 | 0.29 | |
| K(MoC) | 2.94 | 3.11 | 2.86 | |
| K(Mo-R) | 1.28 | 1.53 | 1.88 | |
| K(Mo-Cp) | 3.16 | 3.11 | 3.18 | |
The coordination of the cyclopentadienyl ligand is slightly weaker in the CH3 derivative; the force constant is 3.11 N cm−1, referring to the lower stretching frequency, 335 cm−1 (Table 2). It is interesting to note that the difference between the K1(CO) and K2(CO) stretching force constants are bigger for 3 and 1 (about 1.5 N cm−1, Table 2), while it is only about 0.5 N cm−1 for complex 2.
Considering that the Mo–C stretching force constant value, 3.11 N cm−1, refers to the averaged bond distance of 1.991 Å for complex 2, and that the averaged bond distance for complex 3, 2.006 Å corresponds to a force constant of 2.86 N cm−1, the Mo–C bond length of complex 1 can be estimated to be ca. 2.001 Å by a linear approximation of the bond force constant versus the inverse of the square of the bond length, i.e. within the error range of the averaged experimental bond distance (Table 1).
Values of the Cotton–Kraihanzel CO stretching force constants, K1 and K2 (ref. 27) have been estimated from the CO stretching frequencies.23 The two force constants for [CpMo(CO)3CH3] are 15.49 and 16.45 N cm−1, being about 10% higher than the full calculation results. For the other two complexes the extent of force constant overestimation varies between 7 and 12%. Therefore, it can be concluded that the Cotton–Kraihanzel method strongly overestimates the CO stretching force constants for this type of tricarbonyl complexes.
Epoxidation of cyclooctene catalysed by compounds [CpMo(CO)3R] R = Cl (1), CH3 (2), CF3 (3) and DFT study
There are various DFT studies of half-sandwich complexes of the type [CpMo(O)2R] by Calhorda et al. (R = Me) and Poli et al. (R = Cl).28,29 DFT calculations (see ESI†) of the fluorinated compound 3 show that the introduction of an electron-withdrawing CF3 group enhances the Lewis acidity of the metal centre and should therefore lead to an increased activity of the complex 3 (compared to 1 and 2) towards oxidation catalysis. The polarity difference between metal and ligand is largest for compound 3 (for details see ESI Table S2.1†). This is in accord with our previous studies.30 However, the oxidation of the test substrate cyclooctene to its epoxide seems to reveal that the activity decreases from compound 1 to 3 (Fig. 3). In reality, however, it appears that not the catalytic activity of the active species decreases (vide infra), but the formation of the active species is much slower starting from compound 3 compared to compound 1 or 2. A long induction period is apparent for compound 3 and a yield of only 10% is reached after 240 min, whereas the oxidation of cyclooctene with 1 (99% after 240 min) and 2 (93% after 240 min) reaches (almost) completeness in the same time period. tert-Butylhydroperoxide (TBHP) is the oxidant of choice. The use of hydrogen peroxide as oxidant is not recommendable as its by-product water leads to a decomposition of the catalyst. The exact mechanism of catalyst decomposition in such complexes and the nature of the products has also been recently studied.31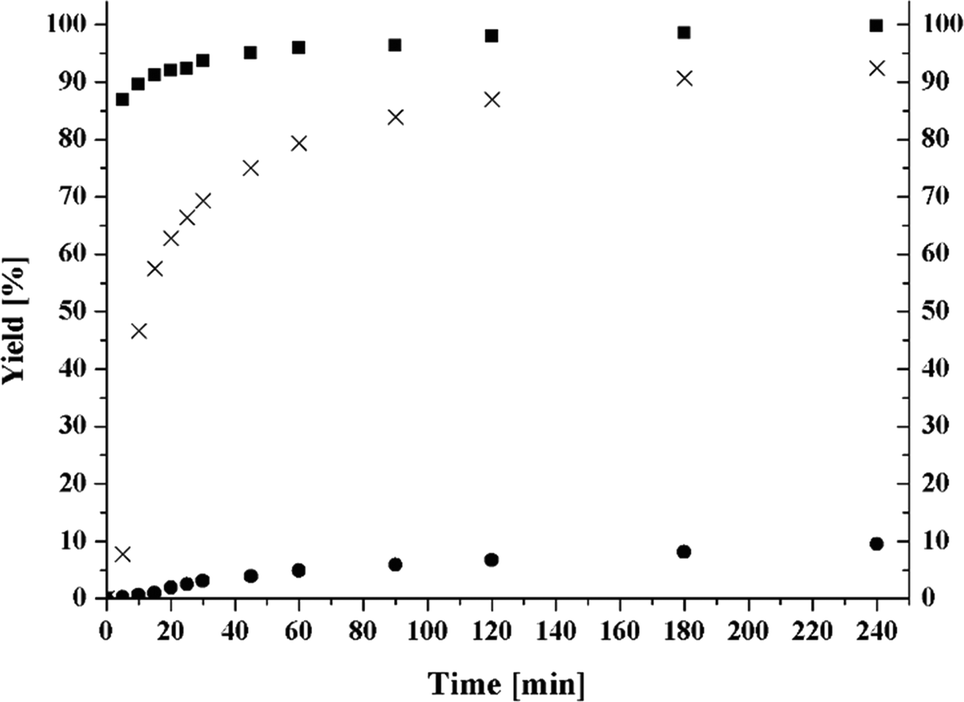 | ||
Fig. 3 Kinetics of the tricarbonyl complexes 1 [CpMo(CO)3Cl] (■), 2 [CpMo(CO)3CH3] (X) and 3 [CpMo(CO)3CF3] (●). Catalyst:cyclooctene:tert-butylhydroperoxide (TBHP); ratio: (0.0025![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.2); T = 25 °C. :1:1.2); T = 25 °C. | ||
The observed activity of the catalytic reactions seems to be in accord with the rate of oxidative decarbonylation. The mechanism of this reaction had been addressed by Gonçalves et al.19 Previous FT-IR spectroscopy studies showed that the decarboxylation reactivity of complex 3 is significantly lower compared to compound 2, where the CO bands have disappeared after a reaction time of 30 min.17
There are various explanations for the observed slow oxidative decarbonylation of compound 3. An indication for a high stability and therefore a slow oxidative decarbonylation of compound 3 is the high melting point (153 °C24). Furthermore, the decrease of the Mo–R bond lengths and increase in the force constants (Table 1) from chloride in 1 to the fluorinated methyl group in 3 (2.234 Å) indicates that the interactions of the metal with the substituent are strongest in 3 (Table 1). The short Mo–R bond in 3 leads to the assumption of a partial Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond character with two short C–F and one elongated C–F bond (Table 1). It is known that the CF3 group, with energetically lower σ* C–F orbitals32 might be able to act as π-acceptor and therefore increase the stability of the complex, resulting in a slow oxidative decarbonylation. DFT-calculations of the HOMOs of the compounds 1–3 were therefore considered as helpful for a better understanding of the Mo – ligands interactions
C double bond character with two short C–F and one elongated C–F bond (Table 1). It is known that the CF3 group, with energetically lower σ* C–F orbitals32 might be able to act as π-acceptor and therefore increase the stability of the complex, resulting in a slow oxidative decarbonylation. DFT-calculations of the HOMOs of the compounds 1–3 were therefore considered as helpful for a better understanding of the Mo – ligands interactions
Apparently, the position of the HOMOs of compounds 1–3 is able to explain these differences in reactivity. The chloride in compound 1 is known to act as σ- and strong π-donor (see orbital on chloride, Fig. 4, left), destabilizing the complex. Its methyl analogue 2 is a pure σ-donor, which cannot accept electron density from the metal (Fig. 4, middle). However the orbitals, which are able to donate electrons are smaller compared to those of 1. Therefore, compound 2 is more stable than complex 1. The HOMO of compound 3 looks quite different (Fig. 4, right).
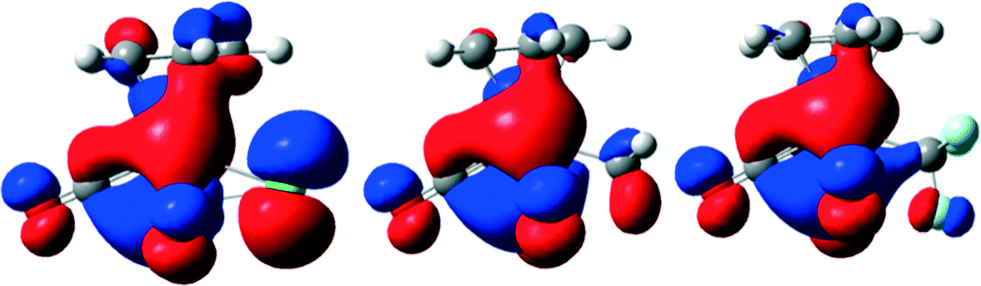 | ||
| Fig. 4 Comparison of the HOMOs of the compounds [CpMo(CO)3R] with R = Cl (1), CH3 (2) and CF3 (3) (from left to right) in gas phase (B3LYP/6-31+G**(d,p) level of theory, for details see ESI†). | ||
The fluorinated ligand seems to be able to accept π-electrons, which increases the stability of the complex due to possible back-donation. This explains the pronounced initiation period for the oxidation of the complex. The electronic situation (see also ESI. 2.1†) confirms the stability of the perfluorinated complex 3 compared to 2 and 1 and is the reason for the previously reported slow oxidative decarbonylation.17 The calculations, which show that in the HOMO of compound 3, one fluorine atom is occupied and two are not, are in agreement with the crystal structure of 3 showing one elongated C–F bond.
Comparison of the crystal structures of the compounds [CpMo(O2)(O)R] R = Cl (4), CH3 (5), CF3 (6)
The treatment of the tricarbonyl complexes 1–3 with excess TBHP leads to a displacement of the carbonyl ligands by an oxo and a η2-peroxo group of the type [CpMo(O2)(O)R] R = Cl (4), CH3 (5), CF3 (6).The structures of 4 (ref. 33) and 5 (ref. 34) are known. It was possible to isolate and crystallise their fluorinated counterpart 6 [CpMo(O2)(O)CF3] (Fig. 5). Bond lengths and angles of the three complexes are compared in Table 3. Catalysis experiments were previously performed with cyclooctene as substrate and TBHP as oxidant at room temperature, applying 4–6 as catalysts. The results indicated a higher activity of the fluorinated oxo-peroxo complex.17 However no convincing explanation for the different activities could be presented. Now it is possible to compare the crystal structures of compounds 4–6 and to derive hints for catalytic activity from structural tendencies.
 | ||
| Fig. 5 Ortep style drawing of the solid-state structure of [CpMo(O2)(O)CF3] (6). The thermal ellipsoids are shown at a 50% probability level. | ||
It is noteworthy that the Mo-peroxo bonds are of comparable length for the three compounds as the confidence intervals overlap (Mo–O1 and Mo–O2, Table 3). Comparison of the oxo-peroxo bonds (O1–O2), however, shows that in the fluorinated compound 6 [CpMo(O2)(O)CF3] this bond is longer than in compound 4 [CpMo(O2)(O)Cl] and 5 [CpMo(O2)(O)CH3]. The confidence intervals of 4 and 5 show a slight overlap but indicate that the O1–O2 peroxo bond length is (slightly) longer for compound 4. The O1–Mo–O2 angles are decreasing in the order CF3 > CH3 > Cl. The O2–Mo–O3 angle has the largest value for the fluorinated compound 6 compared to 5 and 4. Again, within the error range the angles are identical for compound 4 and 5, with the latter being possibly slightly smaller. In contrast to the tricarbonyl complex 3 an elongation of one C–F bond length is not apparent.
Epoxidation of cyclooctene catalysed by compounds [CpMo(O2)(O)R] R = Cl (4), CH3 (5), CF3 (6)
Stoichiometric addition of cyclooctene to the peroxo species [CpMo(O2)(O)R] R = Cl, CH3, CF3 shows that addition of TBHP is essential as no conversion is observed in contrast to the known activity of the (isolated) rhenium bis-peroxo species obtained from MTO during catalytic epoxidation with H2O2.4,35Comparison of catalytic activities in cyclooctene epoxidation shows that the fluorinated compound 6 is more active than compounds 4 and 5 (Fig. 6). This can be explained with the structural differences of the three compounds as longer peroxo bonds (O1–O2, Table 3) enable an easier breaking of the bonds and larger O1–Mo–O2 angles (Table 3) facilitate the activation of the catalyst according to theoretical studies of Thiel et al. where TBHP coordinates to the molybdenum centre (Scheme 2).36,37
 | ||
| Fig. 6 Kinetics of the oxo-peroxo complexes 4 [CpMo(O2)(O)Cl] (■), 5 [CpMo(O2)(O)CH3] (X) and 6 [CpMo(O2)(O)CF3] (●). Catalyst:cyclooctene:TBHP; ratio: (0.0025:1:1.2); T = 25 °C. | ||
 | ||
| Scheme 2 Mechanism of olefin epoxidation by Mo catalyst with TBHP as oxidation agent proposed by Thiel et al.36,37 | ||
This is in accord with previous theoretical calculations concerning the mechanism of the olefin epoxidation with [CpMo(CO)3CH3]29 and [CpMo(CO)3CF3]17 with the intermediates [CpMo(O2)(O)CF3] and [CpMo(O)2CF3]. The activity of the fluorinated oxo-peroxo complex after 30 min is comparable to the activity of MTO with hydrogen peroxide as oxidant for the epoxidation of cyclooctene at 25 °C.38
The lower activity of the oxo-peroxo compounds [CpMo(O2)(O)R] R = Cl (1), CH3 (2) in comparison to the tricarbonyl derivatives [CpMo(CO)3R] R = Cl (4), CH3 (5) suggests that not only the oxo-peroxo species in combination with TBHP is an active species, but also the dioxo species [CpMo(O)2R] (R = Cl, CH3) which forms with excess TBHP from the tricarbonyl precursor, as has been reported before.34
Conclusion
An experimental study, supported by DFT calculations helps to explain the catalytic activities of CpMoR complexes in cyclooctene epoxidation reactions. The determination of the force fields, obtained by normal coordinate analysis (NCA) from experimental spectra, permits a discussion of the frequency assignments and regularities in molecular parameters (bond lengths, force constants) as a function of the ligand R involved.The obtained crystal structure of the tricarbonyl percursor [CpMo(CO)3CF3] (3) allows a comparison with related literature known crystal structures of the compounds [CpMo(CO)3R] R = Cl (1) and CH3 (2).
Combined with DFT calculations, it shows that introduction of a Lewis-acidic substituent R on the tricarbonyl complex does not enhance the epoxidation activity in a “one pot reaction” (where the active catalyst species and epoxide are supposed to form subsequently, based on the assumption that catalyst formation is fast in comparison to the catalytic epoxidation) as its bonding properties might lead to a quite stable tricarbonyl precursor and slow down the oxidative decarbonylation, which needs to take place before the active species are formed.
It was also possible to crystallise the oxo-peroxo complex [CpMo(O2)(O)CF3] (6) allowing a structural comparison with the literature know compounds [CpMo(O2)(O)R] R = Cl (4) and CH3 (5). The structures help to understand the seemingly changed activities, when applying oxo-peroxo species instead of tricarbonyl compounds, from which they are derived. The catalytic activity of the oxo-peroxo species decreases with R = CF3 > Cl > CH3, since bent angles and elongated O–O and Mo-peroxo bonds facilitate the breaking of one of the two peroxo bonds. In addition more space for the oxidation agent to coordinate to the metal centre is provided which goes along with the proposed mechanism of Thiel et al. for the epoxidation of olefins with TBHP.
Accordingly the convenient approach to use a [CpMo(CO)3R] compound as epoxidation catalyst precursor in a “one pot reaction” is not always a good strategy. In some cases it is advisable to apply an already oxidised species in order to obtain a faster catalytic reaction.
Experimental section
General methods
The tricarbonyl complexes [CpMo(CO)3R] (R = Cl,19 CH3/CD3,20 CF3 (ref. 24) and the oxo-peroxo complexes [CpMo(O2)(O)R] (R = Cl,33 CH3,34 CF3 (ref. 17, 39 and 40)) were synthesised according to literature procedures.Mid-IR (350–4000 cm−1, 32 scans, resolution 4 cm−1) absorption spectra were recorded in an N2 purged atmosphere using dynamically aligned Varian Scimitar-2000, and Varian IR-670 spectrometers. Far-IR spectra (700–40 cm−1, 128 scans, resolution 4 cm−1) were recorded with a dedicated Bio-Rad FTS-40 spectrometer equipped with wire-mesh beam splitter, polyethylene-windowed deuterated triglycine sulfate (DTGS) detector, and high-pressure mercury lamp as source.
IR spectra were recorded with a Golden Gate or GladiATR micro attenuated total reflectance (ATR) accessory equipped with a diamond ATR element. Some of the far-IR spectra were obtained also with a GladiATR attachment or in transmittance mode in polyethylene pellet. Raman spectra (50–4000 cm−1, 256 scans, resolution 4 cm−1) were excited with a Spectra-Physics Nd-YAG-laser (1024 nm) and recorded by means of a dedicated Bio-Rad FT-Raman spectrometer equipped with a liquid N2-cooled Ge detector. The laser power at sample position was about 100–200 mW.
Single crystals of [CpMo(CO)3CF3] suitable for XRD-analysis were obtained by slow sublimation in vacuum (10−3 mbar) at 60 °C; single crystals of [CpMo(O2)(O)CF3] were grown from a diethyl ether solution with slow hexane diffusion at −20 °C.
Single-crystal X-ray structure determinations
CCDC 1030682 (3) and CCDC 1030683 (6) contain the supplementary crystallographic data for this paper. For more detailed crystallographic information see ESI.†Catalysis studies
In all catalytic reactions, TBHP (5.5 M in decane, over 4 Å molecular sieves) was used as oxidant and cyclooctene as substrate, unless stated otherwise. The reactions were performed at room temperature and initiated by addition of the oxidant. The kinetic data were collected by using 1H NMR and GC.(a) The reactions monitored by 1H NMR were carried out in C6D6 in a total volume of 0.4–0.8 mL.
(b) The quantitative GC analysis was performed by taking samples of the reaction mixture at specific time intervals and treating them with MnO2 to quench excess peroxide. After filtration, the samples were diluted with an isopropanol solution containing the standards p-xylene and indane (4 mg L−1). The conversion of cyclooctene and the formation of cyclooctene epoxide were calculated from calibration curves (r2 = 0.999) recorded prior to the reaction course.
Force field study
Normal coordinate calculations by means of Wilson's GF matrix method were performed to obtain force constants by optimizing the vibrational frequencies using a symmetrised valence force field. Normal coordinate calculation of the title complexes is not trivial and, therefore, is not carried out routinely.26 In order to carry out a “complete” normal coordinate analysis the Cp ring, CH3, CD3 and CF3 groups were introduced as point masses, using “spectroscopic masses” as 77.1,26 16.5, 19.5 and 79 (ref. 41) atomic mass units for Cp, CH3, CD3 and CF3, respectively. The complexes belong to the Cs point group and the irreducible representation for the simplified structure is 13 A′ + 8 A′′. If we consider the compounds as having planar symmetry (Cs), then all the 21 fundamentals should appear both in the IR and Raman spectra as well. This is in good agreement with our experimental observations (see ESI†). Geometrical parameters were taken from Table S3.1 (ESI†) but we used a uniform CO bond length of 1.14 Å. The starting force fields were adopted from ref. 23 for CO stretching and ref. 26 for other stretching and skeletal coordinates. The calculated results were refined to the experimental frequencies of the complexes. Tables 2, S3.1 and S3.2† outline the results (calculated fundamental frequencies, potential energy distributions, complete and selected force constants).Due to the strong solid-state effect obtained in both in IR and Raman spectra the fundamental frequencies were obtained as averaged frequencies of multiple (generally 2–3 well defined bands or shoulders) features of a certain mode. These fundamental frequencies are listed in Tables 2 and S3.1 in the ESI.†
The calculated frequencies are found to be in very good agreement with the experimental observations (Table S3.1†). The small isotope shifts of the CD3 group are reproduced by the calculation. The Potential Energy Distribution (PED) indicates that the CO stretchings are rather localised modes with small (about 10%) contribution of MoC stretchings. In contrast, the MoC stretching modes (ν5, ν7 and ν16) are more complex vibrations, exhibiting strong interactions with the MoCO linear bending coordinates.
The PC-based program package developed by Mink and Mink was used for the calculations.42
Computational details
All calculations have been performed with Gaussian03.43 The level of theory contains the hybrid DFT functional B3LYP44,45 and the double zeta 6–31+G**46 basis set for all atoms excluding Mo and the Stuttgart 1997 ECP for molybdenum.47 All obtained geometries have been identified via the numbers of negative frequencies as minima (NImag = 0). Free energy differences have been calculated for the gasphase in 298.15 K and 1.0 atm.Acknowledgements
S.A.H. and R.M.R. thank the TUM Graduate School for financial support.Notes and references
- J. T. Jankowiak and M. A. Barteau, J. Catal., 2005, 236, 366–378 CrossRef CAS PubMed.
- Y. C. Kim, N. C. Park, J. S. Shin, S. R. Lee, Y. J. Lee and D. J. Moon, Catal. Today, 2003, 87, 153–162 CrossRef CAS PubMed.
- F. E. Kühn, A. M. Santos and M. Abrantes, Chem. Rev., 2006, 106, 2455–2475 CrossRef PubMed.
- C. C. Romão, F. E. Kühn and W. A. Herrmann, Chem. Rev., 1997, 97, 3197–3246 CrossRef PubMed.
- M. Lein, A. Hammerl, H. L. Hermann and P. Schwerdtfeger, Polyhedron, 2007, 26, 486–492 CrossRef CAS PubMed.
- S. Köstlmeier, O. D. Häberlen, N. Rösch, W. A. Herrmann, B. Solouki and H. Bock, Organometallics, 1996, 15, 1872–1878 CrossRef.
- C. Mealli, J. A. Lopez, M. J. Calhorda, C. C. Romao and W. A. Herrmann, Inorg. Chem., 1994, 33, 1139–1143 CrossRef CAS.
- R. Wiest, T. Leininger, G. H. Jeung and M. Benard, J. Phys. Chem., 1992, 96, 10800–10804 CrossRef CAS.
- N. Grover and F. E. Kühn, Curr. Org. Chem., 2012, 16, 16–32 CrossRef CAS.
- S. A. Hauser, M. Cokoja and F. E. Kühn, Catal. Sci. Technol., 2013, 3, 552–561 CAS.
- P. Altmann, M. Cokoja and F. E. Kühn, Eur. J. Inorg. Chem., 2012, 2012, 3235–3239 CrossRef CAS.
- A. Raith, P. Altmann, M. Cokoja, W. A. Herrmann and F. E. Kühn, Coord. Chem. Rev., 2010, 254, 608–634 CrossRef CAS PubMed.
- S. Huber, M. Cokoja and F. E. Kühn, J. Organomet. Chem., 2014, 751, 25–32 CrossRef CAS PubMed.
- P. Chaumette, H. Mimoun, L. Saussine, J. Fischer and A. Mitschler, J. Organomet. Chem., 1983, 250, 291–310 CrossRef CAS.
- H. Mimoun, I. Seree de Roch and L. Sajus, Tetrahedron, 1970, 26, 37–50 CrossRef CAS.
- K. B. Sharpless, J. M. Townsend and D. R. Williams, J. Am. Chem. Soc., 1972, 94, 295–296 CrossRef CAS.
- S. A. Hauser, M. Cokoja, M. Drees and F. E. Kühn, J. Mol. Catal. A: Chem., 2012, 363–364, 237–244 CrossRef CAS PubMed.
- I. I. E. Markovits, M. H. Anthofer, H. Kolding, M. Cokoja, A. Pöthig, A. Raba, W. A. Herrmann, R. Fehrmann and F. E. Kühn, Catal. Sci. Technol., 2014, 4, 3845–3849 CAS.
- A. Valente, J. Seixas, I. Gonçalves, M. Abrantes, M. Pillinger and C. Romão, Catal. Lett., 2005, 101, 127–130 CrossRef CAS.
- R. B. King and M. B. Bisnette, J. Organomet. Chem., 1967, 8, 287–297 CrossRef CAS.
- D. J. Parker and M. H. B. Stiddard, J. Chem. Soc. A, 1970, 480–490 RSC.
- D. J. Parker, J. Chem. Soc. A, 1970, 1382–1386 RSC.
- R. B. King and L. W. Houk, Can. J. Chem., 1969, 47, 2959–2964 CrossRef CAS.
- R. B. King and M. B. Bisnette, J. Organomet. Chem., 1964, 2, 15–37 CrossRef CAS.
- M. Abrantes, A. M. Santos, J. Mink, F. E. Kühn and C. C. Romão, Organometallics, 2003, 22, 2112–2118 CrossRef CAS.
- É. Bencze, J. Mink, C. Németh, W. A. Herrmann, B. V. Lokshin and F. E. Kühn, J. Organomet. Chem., 2002, 642, 246–258 CrossRef.
- F. A. Cotton and C. S. Kraihanzel, J. Am. Chem. Soc., 1962, 84, 4432–4438 CrossRef CAS.
- A. Comas-Vives, A. Lledós and R. Poli, Chem. – Eur. J., 2010, 16, 2147–2158 CrossRef CAS PubMed.
- P. J. Costa, M. J. Calhorda and F. E. Kühn, Organometallics, 2009, 29, 303–311 CrossRef.
- M. Drees, S. A. Hauser, M. Cokoja and F. E. Kühn, J. Organomet. Chem., 2013, 748, 36–45 CrossRef CAS PubMed.
- N. Grover, A. Pöthig and F. E. Kühn, Catal. Sci. Technol., 2014, 4, 4219–4231 CAS.
- C. Hall and R. N. Perutz, Chem. Rev., 1996, 96, 3125–3146 CrossRef CAS PubMed.
- M. V. Galakhov, P. Gómez-Sal, T. Pedraz, M. A. Pellinghelli, P. Royo, A. Tiripicchio and A. Vázquez de Miguel, J. Organomet. Chem., 1999, 579, 190–197 CrossRef CAS.
- A. M. Al-Ajlouni, D. Veljanovski, A. Capapé, J. Zhao, E. Herdtweck, M. J. Calhorda and F. E. Kühn, Organometallics, 2008, 28, 639–645 CrossRef.
- P. Huston, J. H. Espenson and A. Bakac, Inorg. Chem., 1993, 32, 4517–4523 CrossRef CAS.
- P. Gisdakis, I. V. Yudanov and N. Rösch, Inorg. Chem., 2001, 40, 3755–3765 CrossRef CAS PubMed.
- W. R. Thiel and T. Priermeier, Angew. Chem., Int. Ed. Engl., 1995, 34, 1737–1738 CrossRef CAS.
- P. Altmann and F. E. Kühn, J. Organomet. Chem., 2009, 694, 4032–4035 CrossRef CAS PubMed.
- H. Huang, R. P. Hughes and A. L. Rheingold, Organometallics, 2010, 29, 1948–1955 CrossRef CAS.
- J. D. Koola and D. M. Roddick, Organometallics, 1991, 10, 591–597 CrossRef CAS.
- L. Hajba, J. Mink, F. E. Kühn and I. S. Gonçalves, Inorg. Chim. Acta, 2006, 359, 4741–4756 CrossRef CAS PubMed.
- J. Mink and L. Mink, Available from J. Mink, e-mail: jmink@chemres.hu, 2004.
- M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian03, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- Y. W. C. Lee and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- R. D. J. Hehre and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef PubMed.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1030682–1030683. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cy01604 |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |