
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Specific oxyfunctionalisations catalysed by peroxygenases: opportunities, challenges and solutions
Sebastian Bormann†
a,
Alvaro Gomez Baraibar†
b,
Yan Ni†
b,
Dirk Holtmann
*a and
Frank Hollmann
*b
aDECHEMA Research Institute, Theodor-Heuss-Allee 25, 60486 Frankfurt am Main, Germany. E-mail: holtmann@dechema.de
bDelft University of Technology, Department of Biotechnology, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: f.hollmann@tudelft.nl
First published on 9th January 2015
Abstract
Peroxygenases enable H2O2-driven oxyfunctionalisation of organic compounds such as selective epoxidation, hydroxylation and heteroatom oxygenation. Therefore, peroxygenases are potentially very useful tools for the organic chemist. This contribution reviews the current state of the art in peroxygenases, their availability, engineering and reaction scope. Current challenges and possible solutions are critically discussed.
 Sebastian Bormann | Sebastian Bormann was born in Frankfurt, Germany in 1988. He received his Bachelor's degree in biotechnology from University of Applied Sciences Darmstadt in 2011 and his Master's degree in biotechnology from Braunschweig University of Technology in 2014. Currently, he is a PhD student in the biochemical engineering group of the DECHEMA Research Institute in Frankfurt, Germany. His research is focused on protein engineering of peroxygenases and biocatalysis using such enzymes. |
 Alvaro Gomez Baraibar | Alvaro Gomez Baraibar received his Master's in Biotechnology from the Technical University of Denmark in 2010, with a project dealing with transaminase catalysed reactions. He performed his PhD research at the Research Center Juelich (Germany) on enzymatic process development and characterization of carboligases. In his postdoctoral project in the Biocatalysis group at the Delft University of Technology (Netherlands) he focusses on enzymatic cascades, oxidative enzymes and photocatalysis. |
 Yan Ni | Yan Ni was born in 1986 in Shanghai, China. She obtained her PhD in biochemical engineering in 2013 from the East China University of Science and Technology. Since January 2014 she has been working as a postdoctoral fellow with Dr. Frank Hollmann in the Biocatalysis group at the Delft University of Technology (Netherlands). Her current research focuses on new and efficient (cofactor) regeneration approaches to promote enzymatic reductions, oxidations and specific oxyfunctionalisations. |
 Dirk Holtmann | Dirk Holtmann was born in Bremen, Germany and completed his diploma in chemical engineering/biotechnology in 1999. He obtained his PhD at the Otto von Guericke University of Magdeburg on the electrochemical measurement of the microbial activities. He is head of the biochemical engineering group of the DECHEMA Research-Institute in Frankfurt, Germany. His current research activities concentrate on biocatalysis with P450s, peroxygenases and lipases and the development of electro-enzymatic as well as photo-enzymatic processes. |
 Frank Hollmann | Frank Hollmann studied Chemistry in Bonn, Germany. He received his PhD in Bioelectrocatalysis from the ETH in Zurich, Switzerland. After a postdoctoral stay at the MPI for Coal Research in Mülheim, Germany and some years with Evonik Goldschmidt he joined the Biocatalysis and Organic Chemistry group of TU Delft in 2008 as assistant professor. His research centres on the use of enzymes for organic synthesis. Particularly, he is interested in redoxbiocatalysis for stereospecific reduction and oxyfunctionalisation chemistry using novel biocatalysts and alternative regeneration approaches. |
Introduction
Enzyme-catalysed oxyfunctionalisation certainly is one of the most exciting areas of research both in organic chemistry as well as in enzymology.1–6 Today, oxygenases (E.C. 1.13, monooxygenases and E.C. 1.14, dioxygenases) are the enzyme classes considered for this task. Amongst them, heme-containing monooxygenases (P450 monooxygenases) are the most popular and best-known catalysts giving access to regio- and stereoselective hydroxylation, epoxidation and heteroatom oxygenation reactions.1,2,6–8 Essentially a highly reactive oxyferryl complex embedded in the well-defined three dimensional structure of the protein ligand (Scheme 1) leads to the unique combination of reactivity with selectivity, which is seldom found in ‘biomimetic’ chemical iron complexes.1 | ||
| Scheme 1 Compound I as the active oxyfunctionalisation agent used by P450 monooxygenases and peroxygenases. Shown are some representative oxyfunctionalisation reactions such as sulfoxidation, epoxidation or hydroxylation of activated and non-activated C–H-bonds. | ||
However, the rather complicated molecular architecture of P450 monooxygenases represents a significant challenge with respect to practical application. In P450 monooxygenases the catalytically active compound I is obtained via a sequence of electron transfer to the resting heme group, oxygen binding and further reduction followed by water elimination.7 The reducing equivalents needed for this reaction are delivered from reduced nicotinamide cofactors (NAD(P)H) to the enzymes through sometimes very complicated electron transport chains (Scheme 2, upper). Obviously, such complicated electron transport chains can be very difficult to optimise (both in vivo and in vitro).
 | ||
| Scheme 2 Comparison of P450-catalysed (upper) and peroxygenase-catalysed (lower) oxyfunctionalisation reactions. P450 monooxygenases: generally, the reducing equivalents needed for reductive activation are obtained from reduced nicotinamide cofactors (NAD(P)H), which indirectly deliver two electrons to the P450 monooxygenase via a reductase/mediator cascade. In order to utilize the costly nicotinamide cofactors in catalytic amounts, in situ regeneration is performed most frequently by coupling the P450 monooxygenase cascade (NAD(P)H consuming) to an enzymatic oxidation reaction (NAD(P)H generating). The electron flow is highlighted in blue. In contrast peroxygenases utilise H2O2 directly as the only cosubstrate. | ||
An alternative class of enzymes that circumvents the issues described above and therefore is highly interesting for practical application are the so-called peroxygenases (E.C. 1.11.2).9 These enzymes are structurally related to the aforementioned P450 monooxygenases insofar as they also contain the heme prosthetic group coordinated by a cysteine ligand and therefore, in principle, give access to the same enormous range of reactions and products. In contrast to P450 monooxygenases peroxygenases do not catalyse the reductive activation of molecular oxygen but rather directly use hydrogen peroxide to form the catalytically active oxyferryl species (Scheme 2, lower).
It is important to point out the difference between peroxygenases and peroxidases as well as haloperoxidases originating from different reaction pathways of the oxyferryl species (compound I, Scheme 3) Depending on the enzyme, compound I can return into the resting state by various reactions. The so-called haloperoxidases mediate the oxygenation of halogenide ions (Cl−, Br−, I−) to the corresponding hypohalites (Scheme 3). Peroxidases react by two successive H-atom abstraction reactions (predominantly on phenolic substrates) yielding phenoxyradicals (Scheme 3). Both reactions produce reactive species (hypohalites and radicals) that mostly react further outside of the enzymes' active sites. As a consequence, the selectivity of the subsequent reactions generally is dictated by the chemical reactivity of the reactive species and the starting materials. In contrast, peroxygenases utilize compound I for H-atom abstraction and fast recombination forming new C–O-bonds within the enzyme active site (Scheme 3). As a consequence, the three-dimensional structure of the enzymes' active sites can exert control over the selectivity of the reaction.
 | ||
| Scheme 3 Reactions of compound I in the haloperoxidase pathway (upper), peroxidase pathway (middle) and the peroxygenase pathway (lower). | ||
Structurally, the main difference between peroxidases and peroxygenases appears to be the proximal Fe-ligand: a cysteine ligand (such as in P450 monooxygenases) is crucial for peroxygenase activity (not excluding peroxidase activity of these enzymes) whereas a histidine-ligand is found in peroxidases (only capable of H-atom reactions). It is worth mentioning here that haloperoxidases, in addition the heme-dependent ones, also comprise V-dependent enzymes, which have been discussed elsewhere.10–21
The catalytic cycle of peroxygenases has been studied extensively (Scheme 3).22 In essence it is similar to the so-called peroxide shunt of P450 monooxygenases. Specifically, the oxoferryl porphyrin cation radical intermediate (compound I) is formed from reaction of the resting heme iron with H2O2via heterolytic cleavage of the short lived ferric hydroperoxo intermediate compound 0 (Fe–OOH−).23 At the distal side of the heme, glutamic acid acts as acid–base catalyst promoting compound I formation.24–27 One electron transfer and protonation of the basic oxyferryl intermediate then affords the hydroxyferryl species (compound II) that hydroxylates the substrate radical.28–31 The basicity of the oxo ligand has been described for the chloroperoxidase from Caldariomyces fumago (CfuCPO) and has recently also been elucidated for P450 monooxygenases.28,30 Together with the redox-potential, the high pKa of compound II is the main driving force for oxidation of C–H bonds with high bond-dissociation energies. The heme cysteinate proximal ligand, which is also characteristic for P450 monooxygenases, is believed to increase the basicity of compound II due to electron ‘push’. It is also this high pKa of oxygenating enzymes that allows relatively moderate redox potentials which prevents oxidative self-destruction.30,32
As classified by EC, enzymes with a primary peroxygenase activity are summarized under EC 1.11.2.x which contains unspecific peroxygenase (UPO, EC 1.11.2.1, formerly aromatic peroxygenase), plant seed peroxygenase (EC 1.11.2.3), fatty-acid peroxygenase (EC 1.11.2.4), and myeloperoxidase (EC 1.11.2.2). The latter shows only little peroxygenation activity and is mainly grouped in this sub-class for historical reasons.33 Fatty-acid peroxygenase type enzymes (Cytochrome P450BSβ) are cytosolic bacterial peroxygenases that hydroxylate fatty acid substrates at the α- or β-position. Moreover, they carry out aromatic hydroxylations in the presence of short-chain fatty which supposedly act as acid–base catalysts.34 Plant seed peroxygenase is a membrane-bound heme-thiolate protein with a caleosin-like calcium binding motif that catalyses the oxygenation of unsaturated fatty acids using organic hydroperoxides as cosubstrate.35
Following Hager's seminal publications on the chloroperoxidase from Caldariomyces fumago (CfuCPO) in the 1960ies36 the first wave of exploration of the scope of this and related enzymes generated a lot of promise for organic synthesis. In 2000 van Rantwijk and Sheldon summarised the state of the art then.37 Unfortunately however, the promise of a practical peroxidase-based oxyfunctionalisation chemistry was not fulfilled ever since then. Possibly this is due to the relative poor reactivity of CfuCPO with (non)activated C–H-bonds (vide infra) but certainly also due to difficulties in (heterologous) expression of this enzyme and the resulting high price.
More recently, Hofrichter and coworkers have reported novel peroxygenases with potentially much higher impact in preparative organic synthesis.38–41 The same group have also recently reviewed the phylogenetic relations of peroxygenases33,42 as well as mechanistic aspects.22
In this respect it is interesting to note that N-alkylated flavins can react with H2O2 forming highly reactive flavin(hydro)peroxides capable of certain oxyfunctionalisation reactions (sulfoxidations, epoxidations, Baeyer–Villiger-Oxidations etc.). Recently, Fraaije and coworkers have demonstrated that riboflavin-binding proteins can also accommodate N-alkylated flavins resulting in artificial flavo-peroxygenases.43,44 Very promising first results were reported but further examples e.g. with engineered hybrid enzymes45 will be necessary to assess the full scope of this approach.
The aim of the current contribution therefore was to review the current state of the art in practical application of peroxygenases.
Production of peroxygenases
Fungal peroxidases are frequently produced by submerse (fed-)batch cultivation of the natural host organism in shake flasks or bioreactors. During fermentation the peroxidases are excreted into the culture medium (in accordance with their natural role as e.g. lignin depolymerisation catalysts). Enzyme yields differ significantly from few milligrams per liter to several grams per liter (Table 1). Attempts to express CfuCPO in Escherichia coli, S. cerevisiae, or Pichia pastoris did not yield active enzyme, but eventually heterologous expression was successful in Aspergillus niger, although enzyme levels were significantly lower than with the native host (Table 2).46 Recently, the heterologous expression of UPO from Coprinopsis cinerea (CciUPO) in an industrial A. oryzae production strain has been reported.47 Moreover, the UPO from A. aegerita (AaeUPO) has been heterologously expressed in S. cerevisiae.48 These developments not only pave the way for at scale production of peroxygenases but also represent the first steps towards variation of the enzymes' amino acid composition leading to mutants with improved properties (stability, selectivity, substrate scope etc.).49–51| Organism | Enzyme | Fermentation scale | Max. enzyme concentration [mg L−1] | Ref. |
|---|---|---|---|---|
| SF: shake flask, BR: bioreactor.a Calculated from volumetric activities based on a specific activity of 1400 U mg−1. | ||||
| Caldariomyces. fumago | CfuCPO | SF (0.3 L); BR (1 L) | 715–2310a | 52 and 53 |
| Agrocybe aegerita | AaeUPO | BR (4 L) | 9 | 41 |
| Marasmius rotula | MroUPO | SF (0.2 L) | 445 | 54 |
| Coprinus radians | CraUPO | SF (0.2 L) | 7 | 39 |
It has not been conclusively elucidated why these heme-thiolate peroxygenases can only be heterologously expressed at relatively limited titers. Nonetheless, the increased interest in fungal peroxygenases due to the relatively novel class of unspecific peroxygenases will probably result in more efficient heterologous expression systems. This might also lead to broader availability of these enzymes since to date only CfuCPO is commercially available.
Resolving practical issues
Peroxidases – just like all enzymes – have been optimised to suit their natural host's requirements for survival. These needs not necessarily meet the needs of the organic chemist to perform chemical reactions. Therefore, using peroxygenases ‘as obtained’ is not always practical to achieve high conversions and catalyst use as desired for efficient chemical transformations. However, a range of engineering tools have been developed over the past decades that enable the organic chemist employing peroxygenases according to their needs.Issues to be resolved are (1) the sometimes limited enzyme activity and stability under process conditions and (2) the frequently very low reactant concentrations.
Limited enzyme activity and stability are frequently mentioned as factors limiting the practical usefulness of enzymes. The question however is, how to quantify these values and how to define efficient catalysts from less efficient ones. An enzyme's specific activity can be quantified best in terms of its specific activity (determined in units per milligram) or as turnover frequency (TF, essentially moles of product formed per mole of catalyst per time unit). Obviously, both numbers depend on the reaction conditions and the substrate used therefore.
The question about ‘efficiency’ is much harder to answer. Depending on an author's background and experience, efficiency can mean very different things. From an economic point of view efficiency may be seen as the costs related to the catalyst, i.e. the cost contribution of the catalyst to the overall production costs. In a recent seminal review Tufvesson et al. have provided guidelines to assess such numbers.56 Others have suggested similar numbers for economic evaluation.57 Even though the numbers shown in Table 3 should be seen as rule of thumb they give an impression about the economic feasibility of a given enzyme reaction. Apparently, other factors such as space time yields, downstream processing etc. are at least as important as the catalyst contribution. Also, a seemingly inefficient reaction (in terms of catalyst cost contribution) may lead to very significant simplifications in DSP or give access to cheaper feedstock resulting in an overall more efficient process.
| Product price range [Euro kgProduct−1] | Allowable cost contribution of the catalyst [Euro kgProduct−1] | Minimal productivitya [kgProduct kgCatalyst−1] | Minimal productivityb [TTN] | |
|---|---|---|---|---|
| a Assuming an average catalyst cost of 200 Euro kg−1 (for crude enzyme preparations). b Assuming an enzyme molecular weight of 40 kDa and a product molecular weight of 200 gmol−1. | ||||
| Pharma | >100 | 10 | 20 | 4000 |
| Fine chemical | >15 | 1.5 | 133 | 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 666 666 |
| Speciality chemical | 5 | 0.25 | 800 | 160000 |
| Bulk chemical | 1 | 0.05 | 4000 | 800000 |
Protein engineering
Protein engineering is a powerful tool to address shortcomings of enzymes in terms of stability, reactivity and substrate scope.49,50,58–61 In the case of CfuCPO, only few rational mutants have been reported as of today, since the heterologous expression system A. niger can only be transformed with low efficiency which renders construction of large mutant libraries time consuming, thus obviously limiting the throughput of this screening system. The successful expression of AaeUPO in S. cerevisiae is therefore a milestone for future engineering efforts of fungal peroxygenases as it allows the efficient high-throughput screening of large libraries and will serve as a basis for structure-function research and production of improved enzyme variants.48The two main goals of peroxygenase engineering can be divided between reactivity and stability. Although fungal peroxygenases are dependent on H2O2, they are readily inactivated at elevated peroxide concentrations. While the replacement of easily oxidizable amino acids is a rational strategy for improved oxidative stability, random mutagenesis has been shown to generate further improvements elusive to rational design.62 Other obvious targets for protein engineering include enhanced stability at elevated temperatures and stability in the presence of organic solvents which are often necessary to solubilize highly polar substrates of peroxygenases in aqueous enzymatic reaction systems.
Desired changes concerning the reactivity of peroxygenases focus on the improvement of peroxygenation activity and the concomitant reduction of undesirable one-electron oxidation activity. Structural improvements to increase substrate spectrum and stereospecificity of peroxygenases are also highly desirable.
It has been shown that directed evolution can generate enzyme variants with more than 100-fold improved oxidative and temperature stability in terms of a fungal peroxidase.63 Directed evolution of the peroxygenase AaeUPO resulted in increased enzyme titers, improved catalytic activity (18-fold increase in kcat/Km), and enhanced operational stability in the presence of organic solvents.48,55
Reaction engineering
Immobilization
Immobilization of peroxygenases has been evaluated manifold to increase their stability. For example covalent immobilization of CfuCPO to glass,64 hydrophilic polymers,65 mesoporous silica,66,67 ferromagnetic magnetic beads,68 chitosan membranes,69,70 on agarose gels,71 as cross-linked enzyme aggregates,72,73 or encapsulation in PEG-doped silica matrices,74 have been evaluated. Even though in most of these cases multiple re-use of the immobilized enzyme could be demonstrated, the catalytic properties of the immobilized enzyme fell behind those of the free enzyme. Particularly, the apparent KM value is generally increased by one order of magnitude suggesting diffusion limitations to limit the performance of the immobilized enzymes. Possibly, this also accounts for the seemingly increased robustness against H2O2 reported sometimes. Stabilizing effects have mostly been observed against elevated temperatures or against hostile solvents.Overall, we believe that the benefits and disadvantages of immobilisation have to be carefully evaluated. On the one hand immobilised enzymes tend to be more robust to demanding reaction conditions (mechanical, thermal and solvent-induced stress impairing the tertiary and quarternary structure of the biocatalyst). Furthermore, immobilisation is currently the method of choice for the application of peroxygenases in organic solvents as methods to solubilise peroxygenases in non-aqueous reaction media are scarce.75 On the other hand, immobilisation requires additional efforts in catalyst preparation going hand in hand with increased costs.56 Furthermore, the frequently observed kinetic limitations of immobilised catalysts further reduce their economic and practical attractiveness.
Reaction medium engineering
Recently, Zhai and coworkers reported that small amounts of transition element ions (especially Cr3+) significantly enhance the thermal stability of CfuCPO.76 Indications exist that the metal ions stabilise the enzyme structure. The same authors had previously reported similar beneficial effects by poly saccharides.77,78One of the major limitations of biocatalysis in general lies with the very poor water solubility of many (hydrophobic) reactants of interest. Unfortunately academic researchers tend to circumvent this limitation by adjusting the substrate loading of their reactions to the aqueous solubility of the reagents. The consequences of using such dilute reaction mixtures are:
(1) Poor practical relevance as dilute reaction mixtures are generally economically not feasible (inefficient use of reactor space) and because downstream processing efforts correlate to the volumes of reaction mixtures that have to be handled.
(2) Poor environmental impact as huge volumes of contaminated aqueous waste streams have to be dealt with. As a rule of thumb a 10 mM product solution produces approx. 500 kg of waste water per kg of product (of course depending on the molecular weight of the product).4
Therefore, one major line of research aiming at making peroxygenase-catalysed oxyfunctionalisation reactions more attractive should lie on increasing the overall reactant concentration! Apparently, using water-miscible organic cosolvents represents an interesting step in the right direction. Actually most of the contributions mentioned below make use of such cosolvents to some extent. For example ionic liquids have gained some attention as cosolvent.79–81 Also tertiary butanol has received significant attention as cosolvent and protectant against oxidative stress.82
From a substrate loading point of view (almost) non aqueous reaction media represent the most desirable system. Several groups have demonstrated that various peroxidases are active under non-aqueous reaction conditions. For example van Rantwijk, Sheldon and coworkers have applied immobilised CfuCPO in various organic solvents.83 The specific activity of the immobilised enzyme roughly correlated with the polarity of the solvent applied but no general conclusions could be drawn on the overall performance (in terms of TNs) underlining that our general understanding of enzyme stability in the presence of organic solvents still needs to be improved.84 Klibanov and coworkers have studied the effect of different solvents on HRP-catalysed sulfoxidation of thioanisole demonstrating dramatic effects of the solvent on activity and selectivity of the enzyme.85–88 Using carefully hydrated enzyme preparations Stamatis and coworkers could efficiently use CfuCPO for oxidation/oxyfunctionalisation reactions in organic solvents.89
Two liquid phase systems (2LPSs) represent an interesting compromise between pure aqueous and pure organic solvent-based reaction media (Scheme 4).
 | ||
| Scheme 4 The two liquid phase system (2LPS) approach to achieve overall high substrate loadings. | ||
Such systems generally comprise a hydrophobic, water-inmixible organic phase, which due to its hydrophobicity serves as substrate reservoir and product-sink. Depending on the partitioning coefficient of the reagents the in situ concentration of the reagents in the aqueous, enzyme-containing layer can be controlled and thereby oxidative stress and inhibitory effects of substrate and product be controlled. For example supercritical CO2 (ref. 90) or 1-octanol91 or the substrate itself92 have been reported.
In situ H2O2 generation
As mentioned above, H2O2 has a Janus role in peroxygenase activity. On the one hand, it enables simple access to P450-like reactions by omitting the complicated electron transport chains together with all issues related to it. On the other hand, oxidative inactivation of the heme moiety by H2O2 represents a major challenge to enzyme stability and therewith to the robustness of the chemical transformation. It became clear already at an early stage of development of peroxygenases that simple, stoichiometric addition of H2O2 at once is not practical and that slow addition of H2O2 to maintain its in situ concentration low is necessary. Portionwise addition of H2O2 is rather tedious and also leads to significant volume increases. Manual addition of H2O2 is facilitated by combining H2O2 sensors with H2O2 dosage systems.93,94 Nevertheless, this approach has not really established itself.Also the use of organic hydroperoxides exhibiting a lower reactivity compared to H2O2 could not really establish itself as a practical method.71,83,95–103
Today's state-of-the-art in providing peroxygenases with suitable amounts of H2O2 to enable high reactivity while minimizing their oxidative inactivation is to generate H2O2in situ by partial reduction of molecular oxygen. The reducing equivalents needed for this reaction can be derived from different sources.
Oxidases have been considered very early as co-catalysts to provide peroxygenases with H2O2. Especially the compatibility of both catalysts and the homogeneous nature of the system (avoiding diffusion limitations) makes this approach attractive and popular. Recently Fraaije and coworkers reported an interesting approach of expressing a fusion protein containing oxidase and peroxidase.104 Certainly the most widespread approach is to use glucose oxidase-catalysed oxidation of glucose (Scheme 5).37,105–109
 | ||
| Scheme 5 Glucose oxidase (GOx)-catalysed in situ formation of H2O2 to promote peroxygenase-catalysed oxyfunctionalisations. | ||
Both, the starting material and the catalyst are readily available at very reasonable prices and all reagents are unproblematic. The formation of gluconic acid represents the major disadvantage of this system due to the concomitant pH-change and the additional pH-control needed at larger scale. A further issue, that will probably prevent applications of this system at scale is the high viscosity of concentrated glucose solutions. Furthermore, use of glucose as two-electron donor represents a rather wasteful process (generation of 198 g of by product per mol of H2O2) and thereby does not really comply with the principles of Green Chemistry.110,111 Also ethical issues arise considering that glucose is edible.
Primary alcohols (such as ethanol) represents a somewhat more efficient process (44 g by product per mol H2O2) but is hampered by the stoichiometric accumulation of aldehydes, which is problematic from an enzyme stability- and environmental point-of-view.112,113
Also amino acid oxidases are worth mentioning as in situ H2O2 generation catalysts.114 Very similar to the above-mentioned GOx-scenario, the reagents (amino acids as staring material and α-ketoacid products) are cheap, readily applicable and unproblematic. Interestingly, enough, this approach has not been pursued widely, yet.
Electrochemical reduction of molecular oxygen, from an environmental point-of-view, certainly represents one of the cleanest processes to provide peroxygenases with H2O2 (Scheme 6). Especially if the electrical power is obtained from renewable resources, electrochemical H2O2-generation can be considered to be essentially waste-free. Maybe due to the extra equipment needed or due to a bias amongst some researchers, this promising method is developed and applied by a limited number of research groups.79,115–120
 | ||
| Scheme 6 Electrochemical in situ formation of H2O2 to promote peroxygenase-catalysed oxyfunctionalisations. | ||
Leitner and coworkers90 proposed partial hydrogenation of O2 using Pd-nanoparticle catalysts in an autoclave setting. Even though this method is also appealing from an atom economy point-of-view, issues of equipment (especially to prevent Knallgas reactions) remain to be solved.
We have demonstrated that photocatalytic reduction of O2 (using visible light and simple flavin photocatalysts) may be a viable method for in situ generation of H2O2 (Scheme 7).92,121–123 ETDA however, despite its simplicity to use, yields undesirable formaldehyde as by-product necessitating further improvements. Other sacrificial electron donors such as formate or simple amino acids may represent interesting alternatives to EDTA.
 | ||
| Scheme 7 Photochemical in situ formation of H2O2 to promote peroxygenase-catalysed oxyfunctionalisations. | ||
Finally, also simple and mild reducing agents such as Hantzsch esters or nicotinamide derivatives (Scheme 8)124,125 or organometallic hydrides126 may be interesting methods to provide peroxygenases with suitable amounts of H2O2.
 | ||
| Scheme 8 Chemical in situ formation of H2O2 to promote peroxygenase-catalysed oxyfunctionalisations. | ||
A finite judgment on which in situ H2O2 generation system is ‘the best’ is not yet (and probably will never be) possible. Every system exhibits specific advantages and disadvantages; but the variety offers the practitioner sufficient choices for the specific task given. One criterion may be the turnover number (TN) achievable with the single systems. Table 4 compares the reported TNs of some systems.
| Mode of H2O2 addition | TN | STY (g L−1 d−1) | ee (%) | Ref. | |
|---|---|---|---|---|---|
| CfuCPO | Co-catalyst | ||||
| Step-wise addition | 41000 |
— | 0.7 | 99 | 127 |
| Continuous addition | 109000 |
— | 84 | 99 | 82 |
| GOx/glucose/O2 | 250000 |
n.d. (GOD) | 13 | 99 | 108 |
| Pd/H2/O2 | 6120 | 12.8 (Pd) | 12–67 | 80–94 | 90 |
| Cathode/O2 | 95000 |
— | 30 | 98.5 | 128 |
| 3D electrode/O2 | 145000 |
— | 104 | 99.4 | 129 |
| Flavin/EDTA/hv/O2 | 22400 |
1250 (FMN) | 42 | >98 | 121 |
One major challenge of all methods mentioned above lies with the very poor solubility of molecular oxygen in aqueous media (as a rule of thumb being ca. 0.25 mM at room temperature and atmospheric pressure). Hence, the dissolved oxygen is generally depleted very quickly and diffusion of O2 into the reaction medium becomes overall rate-limiting.
Practical examples
Sulfoxidation
Various peroxidases/peroxygenases have been evaluated as catalysts for the asymmetric oxygenation of prochiral sulphides using thioanisole as standard model substrate. As shown in Table 5, CfuCPO excels amongst the enzymes tested in terms of activity, robustness and enantioselectivity. Therefore it is not very astonishing that CfuCPO has been the peroxygenase of choice for further investigations on the scope of enzymatic sulfoxidation (Table 6).|
|
|||||
|---|---|---|---|---|---|
| Enzymea | TNb | TFc [min−1] | STYd [g L−1 d−1] | ee [%] | Ref. |
| a CfuCPO: chloroperoxidase from C. fumago; V-phytase: vanadate-loaded phytase (from A. ficuum); VBPO: vanadium bromoperoxidase from A. nodosum or C. pilulifera; HRP: horseradish peroxidase; MyG: myoglobin mutant; Lip: lignin peroxidase from P. chrysosporium; TfuDyP: DyP-type peroxidase from T. fusca. b TN: turnover number (moles (product) × moles (catalyst)−1). c TF: turnover frequency (TN × (reaction time)−1). d STY: space time yield. | |||||
| CfuCPO | 109000 |
905 | 84 | 99 (R) | 82 |
| V-phytase | 25000 |
5.5 | 5.3 | 68 (S) | 130 |
| VBPO | 750 | 0.4 | 0.8 | 85 (R) | 19 |
| VBPO | 180 | 0.2 | 0.03 | 55 (S) | 19 |
| HRP | 100 | 1.7 | 3.3 | 60 (S) | 131 |
| MyG | n.d. | 5.5 | n.d. | 97 (R) | 132 |
| Lip | 270 | 4.5 | 0.5 | 37 (S) | 133 |
| TfuDyP | 312 | 0.1 | 0.2 | 61 (R) | 134 |

|
|
||||
|---|---|---|---|---|
| Product | n, Ra | TN | ee (%) | Ref. |
| a n, R refer to the product structures given in the table body. | ||||
 |
n = 0 | 109000 |
99 (R) | 82 |
| n = 1 | 90000 |
99 (R) | 82 | |
| n = 2 | 3300 | 27 (R) | 82 | |
 |
CH3 | 90000 |
99 (R) | 82 |
| OCH3 | 58000 |
99 (R) | 82 | |
| Cl | 85000 |
99 (R) | 82 | |
| Br | 16000 |
99 (R) | 82 | |
| NO2 | 21000 |
99 (R) | 82 | |
 |
— | 3300 | 99 (R) | 82 |
 |
— | 40000 |
99 (R) | 82 |
 |
— | 109000 |
99 (R) | 82 |
 |
R = cyclopentyl; R′ = CH3 | 63000 |
>98 (R) | 135 |
| R = cyclohexyl; R′ = CH3 | 53000 |
85 (R) | 135 | |
| R = ipropyl; R′ = CH3 | 63000 |
>98 (R) | 135 | |
| R = tbutyl; R′ = CH3 | 50000 |
85 (R) | 135 | |
| R = tbutyl; R′ = C2H5 | 19000 |
35 (R) | 135 | |
| R = C5H11; R′ = CH3 | 47000 |
>98 (R) | 135 | |
| R = C8H17; R′ = CH3 | 25000 |
54 (R) | 135 | |
 |
n = 1 | 44000 |
99 (R) | 136 |
| n = 2 | 4400 | 96 (R) | 136 | |
 |
n = 0 | 36000 |
99 (R) | 137 |
| n = 1 | 36000 |
99 (R) | 137 | |
| n = 2 | 9300 | 99 (R) | 137 | |
 |
— | 16000 |
95 (R) | 137 |
 |
— | 36000 |
95 (R) | 137 |
 |
— | 7200 | 95 (R) | 137 |
 |
— | 50 | 82 (R) | 138 |

One feature of CPO-catalysed sulfoxidation is that overoxidation to the corresponding sulfone, which is frequently an issue with chemical sulfoxidation reactions,139 is virtually never observed.37 The substrate scope is fairly broad with sometimes excellent enantioselectivity. Also the catalytic efficiency, expressed as turnover number varies depending on the substrate used. It can, however, be asserted that TNs of CPO in sulfoxidation generally are significantly higher than in other oxygenation reactions (vide infra).
Epoxidation
Until roughly 2010, CfuCPO has been the ‘peroxygenase of choice’ to perform catalytic epoxidation reactions. Table 7 gives a representative overview over the substrate scope of CfuCPO as epoxidation. In some cases excellent enantioselectivities (R) have been observed. But the turnover numbers of CfuCPO in these reactions are far away from being of synthetic interest (even using optimised reaction systems) suggesting that CfuCPO is not a very efficient epoxidation catalyst.|
|
|||||
|---|---|---|---|---|---|
| Product | n | TN | Selectivityb (%) | ee (%) | Ref. |
| a n refers to the product structures given in the table body. b Selectivity: percentage of epoxide of the total product; generally, allylic hydroxylation represents the major side reaction of epoxidation reactions. | |||||
 |
— | 3400 | 100 | 70 (R) | 140 |
 |
n = 1 | 5000 | 61 | 62 (R) | 141 |
| n = 2 | 5000 | 93 | 88 (R) | 141 | |
| n = 3 | 3250 | 89 | 95 (R) | 141 | |
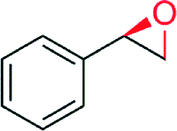 |
— | 840 | 71 | 46 (R) | 141 |
 |
— | 4200 | 100 | 94 (R) | 140 |
 |
— | 4250 | 100 | 94 (R) | 142 |
 |
— | 2100 | 100 | 95 (R) | 143 |
 |
— | 34 | 100 | 10 (S) | 143 |

In contrast, the newly discovered AaeUPO (Table 8) already now shows significantly higher TTNs than CfuCPO. The enantioselectivity of both enzymes appears to be very similar. In those cases where allylic C–H bonds are present in the starting material, epoxidation competes with hydroxylation resulting in sometimes very complex product mixtures.122,144,145
|
|
|||||
|---|---|---|---|---|---|
| Product | n, Ra | TN | Selectivityb (%) | ee (%) | References |
| a n, R refer to the product structures given in the table body. b Selectivity: percentage of epoxide of the total product; generally, allylic hydroxylation represents the major side reaction of epoxidation reactions. | |||||
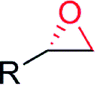 |
R = Me | 308 | 100 | n.d. | 144 |
| R = n-C5H11 | 26 | 85 | 72 (S) | 144 | |
| R = n-C6H13 | 511 | 55 | 66 (S) | 144 | |
 |
5000 | 100 | — | 144 | |
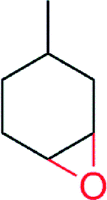 |
5000 | 70 | n.d | 144 | |
 |
— | 2630 | 85 | n.d. | 144 |
 |
— | 6174 | 100 | 7 (R) | 144 |
 |
— | 10600 |
100 | >99 (1R, 2S) | 145 |
 |
— | 10700 |
100 | 29 (n.d.) | 145 |
 |
n = 1 | 10700 |
100 | 2.3 (1R, 2S) | 145 |
| n = 2 | 10600 |
100 | 32 (n.d.) | 145 | |

Hydroxylation
From a preparative point-of-view selective hydroxylation of non-activated C–H-bonds represents one of the most promising applications of peroxygenases for organic synthesis.One of the earliest reactions examined for AaeUPO was the hydroxylation of aromatic compounds. Detailed mechanistic studies revealed that the apparent hydroxylation in fact is an epoxidation followed by spontaneous protonation/rearrangement (Scheme 9).
 | ||
| Scheme 9 Aromatic hydroxylation mediated by AaeUPO as a sequence of epoxidation and spontaneous isomerization.146 | ||
So far, a rather limited amount of aromatic compounds (amongst them many pharmaceutical ingredients) have been evaluated (Table 9). It is interesting to note that in case of alkyl substituted arenes different selectivity patterns have been observed for different peroxygenases. For example, the UPO from Coprinellus radians exhibits preference for benzylic C–H-bonds whereas AaeUPO appears to be selective for aromatic hydroxylation (via epoxidation as described above). Further systematic investigations will be necessary to fully understand (and predict) the UPOs' selectivities; but we believe that a very powerful toolkit for organic synthesis may result here!





The selective hydroxylation of sp3-C–H bonds has been in focus already during the ‘CfuCPO era’ (Table 10). Many research groups have evaluated CfuCPO as catalyst for the hydroxylation of activated benzylic, allylic and propargylic C–H bonds. However, as shown in Table 10, the catalytic efficiency of CfuCPO (in terms of turnover numbers) falls back by some orders of magnitude behind preparative usefulness. Functionalisation of non-activated C–H-bonds such as in linear and cyclic alkanes appears to be out of scope for CfuCPO. Therefore, just as in case of epoxidation, CfuCPO does not seem to be a very useful catalyst for C–H-hydroxylation. However, a different picture evolves with the newly discovered UPOs such as AaeUPO. Already at the present, very early stage of development, this enzyme shows TNs that appear to reach synthetically useful levels in optimised reaction systems.
|
|
||||
|---|---|---|---|---|
| Product | n, R | TN | ee (%) | Ref. |
| AaeAPO | ||||
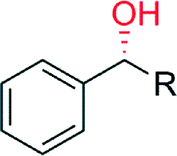 |
R = CH3 | 10600 |
>99 | 145 |
| R = CH2CH3 | 7100 | >99 | 145 | |
| R = (CH2)2CH3 | 5800 | 40 | 145 | |
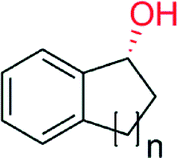 |
n = 1 | 8600 | 87 | 145 |
| n = 2 | 9400 | >99 | 145 | |
 |
— | 22000 |
n.d | 152 |
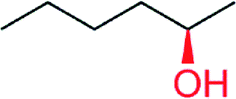 |
— | 1882 | 62.5 (R) | 153 |
 |
17000 |
— | 122 | |
| CfuCPO | ||||
 |
— | 2100 | — | 154 |
| — | 12000 |
— | 91 | |
 |
22000 |
— | 91 | |
 |
R = CH3 | 420 | 97 (R) | 154 |
| R = (CH2)CH3 | 420 | 88 (S) | 154 | |
 |
— | 590 | 94 (n.d) | 155 |
 |
— | 32000 |
— | 156 |
 |
— | 394 | — | 157 |
 |
— | 960 | — | 71 |

Halogenation reactions
Many peroxidases are so-called halo-peroxidases, i.e. they catalyse the H2O2-dependent oxidation of halide ions to the corresponding hypohalous acids (HOCl, HOBr, HOI). Hypohalous acids (and their corresponding bases) are highly reactive agents capable of electrophilic halogenation of a range of starting materials (vide infra). This reaction, however, usually occurs spontaneously (and outside the chiral enzyme environment). Hence, the selectivity of a halo-peroxidase-catalysed halogenation reaction is dominated by the chemical reactivity of the reagents and is not controlled by the enzyme's active site. Maybe this explains the relatively small number of reports dealing with peroxygenase-catalysed halogenation. Nevertheless, some interesting examples of enzyme-initiated halogenation have been reported in the past years. For example Aoun and Barboulene reported CfuCPO – initiated bromohydroxylation of some terminal alkenes (Scheme 10).158 Excellent turnover numbers of up to 300000 were reported. Similarly, Schrader and coworkers reported the halohydroxylation (halogen = Cl, Br, I) of (1S)-(+)-3-carene.159
 | ||
| Scheme 10 CfuCPO-initiated bromohydroxylation of some terminal alkenes. | ||
In addition to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-double bonds also activated aromatic systems such as phenols undergo electrophilic attack by hypohalites, which we recently demonstrated at the halogenation of thymol (Scheme 11).119
C-double bonds also activated aromatic systems such as phenols undergo electrophilic attack by hypohalites, which we recently demonstrated at the halogenation of thymol (Scheme 11).119
 | ||
| Scheme 11 CfuCPO-initiated halogenation of thymol. | ||
Conclusions and outlook
Unspecific peroxygenases form a novel class of heme-thiolate peroxidases with promising potential for organic synthesis. While only few enzymes have been isolated and characterized to date, recent investigations indicate that homologous enzymes are abundant in nature.160 Given the broad substrate spectrum of the already characterized enzymes, and the large peroxygenase sequence space that has yet to be explored, peroxygenases might form a valuable tool for organic chemists for selective and stereospecific oxyfunctionalisation of non-activated C–H bonds. The combination of rational design of these enzymes and adequate reactions system for the in situ production of hydrogen peroxide to further address instability issues92,94,118–122 will probably lead to broader application of the peroxygenases in technical applications.Acknowledgements
Financial support by the European Union (7th Framework Programme, KBBE-2013-7-613549 “INDOX”) is gratefully acknowledged.References
- E. Roduner, W. Kaim, B. Sarkar, V. B. Urlacher, J. Pleiss, R. Gläser, W.-D. Einicke, G. A. Sprenger, U. Beifuß, E. Klemm, C. Liebner, H. Hieronymus, S.-F. Hsu, B. Plietker and S. Laschat, ChemCatChem, 2013, 5, 82–112 CrossRef CAS.
- V. B. Urlacher and M. Girhard, Trends Biotechnol., 2012, 30, 26–36 CrossRef CAS PubMed.
- V. B. Urlacher and S. Eiben, Trends Biotechnol., 2006, 24, 324–330 CrossRef CAS PubMed.
- Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2014, 6, 930–943 CrossRef CAS.
- D. Holtmann, M. W. Fraaije, D. J. Opperman, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2014, 50, 13180–13200 RSC.
- F. Hollmann, I. W. C. E. Arends, K. Buehler, A. Schallmey and B. Buhler, Green Chem., 2011, 13, 226–265 RSC.
- M. K. Julsing, S. Cornelissen, B. Buhler and A. Schmid, Curr. Opin. Chem. Biol., 2008, 12, 177–186 CrossRef CAS PubMed.
- S. Eiben, L. Kaysser, S. Maurer, K. Kuhnel, V. B. Urlacher and R. D. Schmid, J. Biotechnol., 2006, 124, 662–669 CrossRef CAS PubMed.
- A. Chang, M. Scheer, A. Grote, I. Schomburg and D. Schomburg, Nucleic Acids Res., 2009, 37, D588–D592 CrossRef CAS PubMed.
- R. Wever and M. A. van der Horst, Dalton Trans., 2013, 42, 11778–11786 RSC.
- R. Wever, in Vanadium: Biochemical and Molecular Biological Approaches, 2011 Search PubMed.
- R. Wever and R. Renirie, in Peroxidases and Catalases: Biochemistry, Biophysics, Biotechnology, and Physiology, 2010 Search PubMed.
- R. Wever, in Peroxidases and Catalases: Biochemistry, Biophysics, Biotechnology, and Physiology, 2010 Search PubMed.
- R. Renirie, C. Pierlot, R. Wever and J. M. Aubry, J. Mol. Catal. B: Enzym., 2009, 56, 259–264 CrossRef CAS PubMed.
- R. Renirie, C. Pierlot, J.-M. Aubry, A. F. Hartog, H. E. Schoemaker, P. L. Alsters and R. Wever, Adv. Synth. Catal., 2003, 345, 849–858 CrossRef CAS.
- H. B. ten Brink, H. E. Schoemaker and R. Wever, Eur. J. Biochem., 2001, 268, 132–138 CrossRef CAS.
- H. B. ten Brink, H. L. Dekker, H. E. Shoemaker and R. Wever, J. Inorg. Biochem., 2000, 80, 91–98 CrossRef CAS.
- W. Hemrika, R. Renirie, S. Macedo-Ribeiro, A. Messerschmidt and R. Wever, J. Biol. Chem., 1999, 274, 23820–23827 CrossRef CAS PubMed.
- H. B. ten Brink, A. Tuynman, H. L. Dekker, W. Hemrika, Y. Izumi, T. Oshiro, H. E. Schoemaker and R. Wever, Inorg. Chem., 1998, 37, 6780–6784 CrossRef CAS PubMed.
- H. Plat, B. E. Krenn and R. Wever, Biochem. J., 1987, 248, 277–279 CAS.
- E. Deboer, H. Plat, M. G. M. Tromp, R. Wever, M. C. R. Franssen, H. C. Vanderplas, E. M. Meijer and H. E. Schoemaker, Biotechnol. Bioeng., 1987, 30, 607–610 CrossRef CAS PubMed.
- M. Hofrichter and R. Ullrich, Curr. Opin. Chem. Biol., 2014, 19, 116–125 CrossRef CAS PubMed.
- K. Kuhnel, W. Blankenfeldt, J. Terner and I. Schlichting, J. Biol. Chem., 2006, 281, 23990–23998 CrossRef PubMed.
- M. Sundaramoorthy, J. Terner and T. L. Poulos, Structure, 1995, 3, 1367–1377 CrossRef CAS.
- X. W. Yi, A. Conesa, P. J. Punt and L. P. Hager, J. Biol. Chem., 2003, 278, 13855–13859 CrossRef CAS PubMed.
- K. Piontek, E. Strittmatter, R. Ullrich, G. Gröbe, M. J. Pecyna, M. Kluge, K. Scheibner, M. Hofrichter and D. A. Plattner, J. Biol. Chem., 2013, 288, 34767–34776 CrossRef CAS PubMed.
- K. Piontek, R. Ullrich, C. Liers, K. Diederichs, D. A. Plattner and M. Hofrichter, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2010, 66, 693–698 CrossRef CAS PubMed.
- M. T. Green, J. H. Dawson and H. B. Gray, Science, 2004, 304, 1653–1656 CrossRef CAS PubMed.
- J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed.
- T. H. Yosca, J. Rittle, C. M. Krest, E. L. Onderko, A. Silakov, J. C. Calixto, R. K. Behan and M. T. Green, Science, 2013, 342, 825–829 CrossRef CAS PubMed.
- J. T. Groves, Nat. Chem., 2014, 6, 89–91 CrossRef CAS PubMed.
- K. L. Stone and A. S. Borovik, Curr. Opin. Chem. Biol., 2009, 13, 114–118 CrossRef CAS PubMed.
- M. Hofrichter, R. Ullrich, M. J. Pecyna, C. Liers and T. Lundell, Appl. Microbiol. Biotechnol., 2010, 87, 871–897 CrossRef CAS PubMed.
- O. Shoji, T. Fujishiro, H. Nakajima, M. Kim, S. Nagano, Y. Shiro and Y. Watanabe, Angew. Chem., Int. Ed., 2007, 46, 3656–3659 CrossRef CAS PubMed.
- A. Hanano, M. Burcklen, M. Flenet, A. Ivancich, M. Louwagie, J. Garin and E. Blée, J. Biol. Chem., 2006, 281, 33140–33151 CrossRef CAS PubMed.
- D. R. Morris and L. P. Hager, J. Biol. Chem., 1966, 241, 1763–1768 CAS.
- F. van Rantwijk and R. A. Sheldon, Curr. Opin. Biotechnol., 2000, 11, 554–564 CrossRef CAS.
- R. Ullrich and M. Hofrichter, Cell. Mol. Life Sci., 2007, 64, 271–293 CrossRef CAS PubMed.
- D. H. Anh, R. Ullrich, D. Benndorf, A. Svatoś, A. Muck and M. Hofrichter, Appl. Environ. Microbiol., 2007, 73, 5477–5485 CrossRef CAS PubMed.
- R. Ullrich and M. Hofrichter, FEBS Lett., 2005, 579, 6247–6250 CrossRef CAS PubMed.
- R. Ullrich, J. Nüske, K. Scheibner, J. Spantzel and M. Hofrichter, Appl. Environ. Microbiol., 2004, 70, 4575–4581 CrossRef CAS PubMed.
- M. Hofrichter and R. Ullrich, Appl. Microbiol. Biotechnol., 2006, 71, 276–288 CrossRef CAS PubMed.
- G. de Gonzalo and M. W. Fraaije, ChemCatChem, 2012, 5, 403–415 CrossRef.
- G. de Gonzalo, C. Smit, J. F. Jin, A. J. Minnaard and M. W. Fraaije, Chem. Commun., 2011, 47, 11050–11052 RSC.
- M. T. Reetz, M. Rentzsch, A. Pletsch, A. Taglieber, F. Hollmann, R. J. G. Mondière, N. Dickmann, B. Höcker, S. Cerrone, M. C. Haeger and R. Sterner, ChemBioChem, 2008, 9, 552–564 CrossRef CAS PubMed.
- A. Conesa, F. van de Velde, F. van Rantwijk, R. A. Sheldon, C. van den Hondel and P. J. Punt, J. Biol. Chem., 2001, 276, 17635–17640 CrossRef CAS PubMed.
- E. D. Babot, J. C. del Río, L. Kalum, A. T. Martínez and A. Gutiérrez, Biotechnol. Bioeng., 2013, 110, 2323–2332 CrossRef CAS PubMed.
- P. Molina-Espeja, E. Garcia-Ruiz, D. Gonzalez-Perez, R. Ullrich, M. Hofrichter and M. Alcalde, Appl. Environ. Microbiol., 2014, 80, 3496–3507 CrossRef CAS PubMed.
- M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS PubMed.
- M. T. Reetz, Tetrahedron, 2012, 68, 7530–7548 CrossRef CAS PubMed.
- M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138–174 CrossRef CAS PubMed.
- M. Buchhaupt, K. Ehrich, S. Huttmann, J. Guder and J. Schrader, Biotechnol. Lett., 2011, 33, 2225–2231 CrossRef CAS PubMed.
- B.-A. Kaup, K. Ehrich, M. Pescheck and J. Schrader, Biotechnol. Bioeng., 2008, 99, 491–498 CrossRef CAS PubMed.
- G. Grobe, R. Ullrich, M. J. Pecyna, D. Kapturska, S. Friedrich, M. Hofrichter and K. Scheibner, AMB Express, 2011, 1, 31 CrossRef PubMed.
- D. Gonzalez-Perez, P. Molina-Espeja, E. Garcia-Ruiz and M. Alcalde, PLoS One, 2014, 9, e90919 Search PubMed.
- P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2010, 15, 266–274 CrossRef.
- G. W. Huisman, J. Liang and A. Krebber, Curr. Opin. Chem. Biol., 2010, 14, 122–129 CrossRef CAS PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- G. A. Behrens, A. Hummel, S. K. Padhi, S. Schätzle and U. T. Bornscheuer, Adv. Synth. Catal., 2011, 353, 2191–2215 CrossRef CAS.
- N. J. Turner, Nat. Chem. Biol., 2009, 5, 568–574 CrossRef PubMed.
- L. G. Otten, F. Hollmann and I. W. C. E. Arends, Trends Biotechnol., 2010, 28, 46–54 CrossRef CAS PubMed.
- E. Garcia-Ruiz, D. Gonzalez-Perez, F. J. Ruiz-Duenas, A. T. Martinez and M. Alcalde, Biochem. J., 2012, 441, 487–498 CrossRef CAS PubMed.
- J. R. Cherry, M. H. Lamsa, P. Schneider, J. Vind, A. Svendsen, A. Jones and A. H. Pedersen, Nat. Biotechnol., 1999, 17, 379–384 CrossRef CAS PubMed.
- T. A. Kadima and M. A. Pickard, Appl. Environ. Microbiol., 1990, 56, 3473–3477 CAS.
- G. Bayramoglu, B. Altintas, M. Yilmaz and M. Y. Arica, Bioresour. Technol., 2011, 102, 475–482 CrossRef CAS PubMed.
- E. Guerrero, P. Aburto, E. Terres, O. Villegas, E. Gonzalez, T. Zayas, F. Hernandez and E. Torres, J. Porous Mater., 2013, 20, 387–396 CrossRef CAS PubMed.
- S. Águila, R. Vazquez-Duhalt, C. Covarrubias, G. Pecchi and J. B. Alderete, J. Mol. Catal. B: Enzym., 2011, 70, 81–87 CrossRef PubMed.
- G. Bayramoglu, S. Kiralp, M. Yilmaz, L. Toppare and M. Y. Arica, Biochem. Eng. J., 2008, 38, 180–188 CrossRef CAS PubMed.
- L. H. Zhang, C. H. Bai, Y. S. Wang, Y. C. Jiang, M. C. Hu, S. N. Li and Q. G. Zhai, Biotechnol. Lett., 2009, 31, 1269–1272 CrossRef CAS PubMed.
- C. H. Bai, Y. C. Jiang, M. C. Hu, S. N. Li and Q. G. Zhai, Catal. Lett., 2009, 129, 457–461 CrossRef CAS.
- M. Pešić, C. López, G. Álvaro and J. López-Santín, J. Mol. Catal. B: Enzym., 2012, 84, 144–151 CrossRef PubMed.
- D. I. Perez, F. van Rantwijk and R. A. Sheldon, Adv. Synth. Catal., 2009, 351, 2133–2139 CrossRef CAS.
- C. Roberge, D. Amos, D. Pollard and P. Devine, J. Mol. Catal. B: Enzym., 2009, 56, 41–45 CrossRef CAS PubMed.
- L. De Matteis, R. Germani, M. V. Mancini, G. Savelli, N. Spreti, L. Brinchi and G. Pastori, J. Mol. Catal. B: Enzym., 2013, 97, 23–30 CrossRef CAS PubMed.
- P. S. Angerer, A. Studer, B. Witholt and Z. Li, Macromolecules, 2005, 38, 6248–6250 CrossRef CAS.
- H. Y. Li, J. W. Gao, L. M. Wang, X. H. Li, Y. C. Jiang, M. C. Hu, S. N. Li and Q. G. Zhai, Appl. Biochem. Biotechnol., 2014, 172, 2338–2347 CrossRef CAS PubMed.
- C. Li, L. Wang, Y. Jiang, M. Hu, S. Li and Q. Zhai, Appl. Biochem. Biotechnol., 2011, 165, 1691–1707 CrossRef CAS PubMed.
- L. F. Zhi, Y. C. Jiang, Y. S. Wang, M. C. Hu, S. N. Li and Y. J. Ma, Biotechnol. Prog., 2007, 23, 729–733 CrossRef CAS PubMed.
- C. Kohlmann, L. Greiner, W. Leitner, C. Wandrey and S. Lütz, Chem. – Eur. J., 2009, 15, 11692–11700 CrossRef CAS PubMed.
- J. Y. Wu, C. Liu, Y. C. Jiang, M. C. Hu, S. N. Li and Q. G. Zhai, Catal. Commun., 2010, 11, 727–731 CrossRef CAS PubMed.
- C. Chiappe, L. Neri and D. Pieraccini, Tetrahedron Lett., 2006, 47, 5089–5093 CrossRef CAS PubMed.
- M. P. J. van Deurzen, I. J. Remkes, F. van Rantwijk and R. A. Sheldon, J. Mol. Catal. A: Chem., 1997, 117, 329–337 CrossRef CAS.
- F. van de Velde, M. Bakker, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2001, 72, 523–529 CrossRef CAS.
- M. Villela Filho, T. Stillger, M. Müller, A. Liese and C. Wandrey, Angew. Chem., Int. Ed., 2003, 42, 2993–2996 CrossRef CAS PubMed.
- L. H. Dai and A. M. Klibanov, Biotechnol. Bioeng., 2000, 70, 353–357 CrossRef CAS.
- P. K. Das, J. M. M. Caaveiro, S. Luque and A. M. Klibanov, J. Am. Chem. Soc., 2002, 124, 782–787 CrossRef CAS PubMed.
- Y. C. Xie, P. K. Das, J. M. M. Caaveiro and A. M. Klibanov, Biotechnol. Bioeng., 2002, 79, 105–111 CrossRef CAS.
- J. H. Yu and A. M. Klibanov, Biotechnol. Lett., 2006, 28, 555–558 CrossRef CAS PubMed.
- A. A. Tzialla, E. Kalogeris, D. Gournis, Y. Sanakis and H. Stamatis, J. Mol. Catal. B: Enzym., 2008, 51, 24–35 CrossRef CAS PubMed.
- S. K. Karmee, C. Roosen, C. Kohlmann, S. Lütz, L. Greiner and W. Leitner, Green Chem., 2009, 11, 1052–1055 RSC.
- J.-B. Park and D. S. Clark, Biotechnol. Bioeng., 2006, 94, 189–192 CrossRef CAS PubMed.
- E. Churakova, I. W. C. E. Arends and F. Hollmann, ChemCatChem, 2013, 5, 565–568 CrossRef CAS.
- M. P. J. Van Deurzen, K. Seelbach, F. van Rantwijk, U. Kragl and R. A. Sheldon, Biocatal. Biotransform., 1997, 15, 1–16 CrossRef CAS.
- K. Seelbach, M. P. J. van Deurzen, F. van Rantwijk, R. A. Sheldon and U. Kragl, Biotechnol. Bioeng., 1997, 55, 283–288 CrossRef CAS.
- S. Colonna, N. Gaggero, L. Casella, G. Carrea and P. Pasta, Tetrahedron: Asymmetry, 1993, 4, 1325–1330 CrossRef CAS.
- S. Hu and L. P. Hager, Biochem. Biophys. Res. Commun., 1998, 253, 544–546 CrossRef CAS PubMed.
- L. Santhanam and J. S. Dordick, Biocatal. Biotransform., 2002, 20, 265–274 CrossRef CAS.
- S. Hu and J. S. Dordick, J. Org. Chem., 2002, 67, 314–317 CrossRef CAS PubMed.
- D. J. Bougioukou and I. Smonou, Tetrahedron Lett., 2002, 43, 4511–4514 CrossRef CAS.
- D. J. Bougioukou and I. Smonou, Tetrahedron Lett., 2002, 43, 339–342 CrossRef CAS.
- E. Kiljunen and L. T. Kanerva, J. Mol. Catal. B: Enzym., 2000, 9, 163–172 CrossRef CAS.
- B. K. Samra, M. Andersson and P. Adlercreutz, Biocatal. Biotransform., 1999, 17, 381–391 CrossRef CAS.
- E. Kiljunen and L. T. Kanerva, Tetrahedron: Asymmetry, 1999, 10, 3529–3535 CrossRef CAS.
- R. T. Winter, T. E. van den Berg, D. I. Colpa, E. van Bloois and M. W. Fraaije, ChemBioChem, 2012, 13, 252–258 CrossRef CAS PubMed.
- D. Jung, C. Streb and M. Hartmann, Microporous Mesoporous Mater., 2008, 113, 523–529 CrossRef CAS PubMed.
- R. Narayanan, G. Y. Zhu and P. Wang, J. Biotechnol., 2007, 128, 86–92 CrossRef CAS PubMed.
- A. Borole, S. Dai, C. L. Cheng, M. Rodriguez and B. H. Davison, Appl. Biochem. Biotechnol., 2004, 113, 273–285 CrossRef.
- F. van de Velde, N. D. Lourenço, M. Bakker, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2000, 69, 286–291 CrossRef CAS.
- A. D. Ryabov, Y. N. Firsova, V. N. Goral, E. S. Ryabova, A. N. Shevelkova, L. L. Troitskaya, T. V. Demeschik and V. I. Sokolov, Chem. – Eur. J., 1998, 4, 806–813 CrossRef CAS.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- F. Pezzotti and M. Therisod, Tetrahedron: Asymmetry, 2007, 18, 701–704 CrossRef CAS PubMed.
- F. Pezzotti, K. Okrasa and M. Therisod, Tetrahedron: Asymmetry, 2005, 16, 2681–2683 CrossRef CAS PubMed.
- K. Okrasa, A. Falcimaigne, E. Guibé-Jampel and M. Therisod, Tetrahedron: Asymmetry, 2002, 13, 519–522 CrossRef CAS.
- K. Lee and S.-H. Moon, J. Biotechnol., 2003, 102, 261–268 CrossRef CAS.
- C. E. La Rotta, E. D'Elia and E. P. S. Bon, Electron. J. Biotechnol., 2007, 10, 24–37 Search PubMed.
- C. Kohlmann and S. Lütz, Eng. Life Sci., 2006, 6, 170–174 CrossRef CAS.
- T. Krieg, S. Huttmann, K.-M. Mangold, J. Schrader and D. Holtmann, Green Chem., 2011, 13, 2686–2689 RSC.
- L. Getrey, T. Krieg, F. Hollmann, J. Schrader and D. Holtmann, Green Chem., 2014, 16, 1104–1108 RSC.
- D. Holtmann, T. Krieg, L. Getrey and J. Schrader, Catal. Commun., 2014, 51, 82–85 CrossRef CAS PubMed.
- D. I. Perez, M. Mifsud Grau, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2009, 6848–6850 RSC.
- E. Churakova, M. Kluge, R. Ullrich, I. Arends, M. Hofrichter and F. Hollmann, Angew. Chem., Int. Ed., 2011, 50, 10716–10719 CrossRef CAS PubMed.
- I. Zachos, S. Gassmeyer, D. Bauer, V. Sieber, F. Hollmann and R. Kourist, Chem. Commun., 2015 10.1039/c4cc07276f.
- C. E. Paul, E. Churakova, E. Maurits, M. Girhard, V. B. Urlacher and F. Hollmann, Bioorg. Med. Chem., 2014, 22, 5692–5696 CrossRef CAS PubMed.
- C. E. Paul, I. W. C. E. Arends and F. Hollmann, ACS Catal., 2014, 4, 788–797 CrossRef CAS.
- F. Hollmann and A. Schmid, J. Inorg. Biochem., 2009, 103, 313–315 CrossRef CAS PubMed.
- H. Fu, H. Kondo, Y. Ichikawa, G. C. Look and C. H. Wong, J. Org. Chem., 1992, 57, 7265–7270 CrossRef CAS.
- S. Lutz, E. Steckhan and A. Liese, Electrochem. Commun., 2004, 6, 583–587 CrossRef CAS PubMed.
- S. Lutz, K. Vuorilehto and A. Liese, Biotechnol. Bioeng., 2007, 98, 525–534 CrossRef CAS PubMed.
- F. van de Velde, L. Könemann, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2000, 67, 87–96 CrossRef CAS.
- A. Tuynman, H. E. Schoemaker and R. Wever, Chem. Mon., 2000, 131, 687–695 CrossRef CAS.
- S. Ozaki, H. J. Yang, T. Matsui, Y. Goto and Y. Watanabe, Tetrahedron: Asymmetry, 1999, 10, 183–192 CrossRef CAS.
- E. Baciocchi, M. F. Gerini, P. J. Harvey, O. Lanzalunga and S. Mancinelli, Eur. J. Biochem., 2000, 267, 2705–2710 CrossRef CAS.
- E. van Bloois, D. E. T. Pazmino, R. T. Winter and M. W. Fraaije, Appl. Microbiol. Biotechnol., 2010, 86, 1419–1430 CrossRef CAS PubMed.
- S. Colonna, N. Gaggero, G. Carrea and P. Pasta, Chem. Commun., 1997, 439–440 RSC.
- S. G. Allenmark and M. A. Andersson, Chirality, 1998, 10, 246–252 CrossRef CAS.
- R. R. Vargas, E. J. H. Bechara, L. Marzorati and B. Wladislaw, Tetrahedron: Asymmetry, 1999, 10, 3219–3227 CrossRef CAS.
- H. L. Holland, F. M. Brown, D. Lozada, B. Mayne, W. R. Szerminski and A. J. van Vliet, Can. J. Chem., 2002, 80, 633–639 CrossRef CAS.
- G. E. O'Mahony, A. Ford and A. R. Maguire, J. Sulfur Chem., 2013, 34, 301–341 CrossRef.
- A. F. Dexter, F. J. Lakner, R. A. Campbell and L. P. Hager, J. Am. Chem. Soc., 1995, 117, 6412–6413 CrossRef CAS.
- F. J. Lakner, K. P. Cain and L. P. Hager, J. Am. Chem. Soc., 1997, 119, 443–444 CrossRef CAS.
- F. J. Lakner and L. P. Hager, Tetrahedron: Asymmetry, 1997, 8, 3547–3550 CrossRef CAS.
- S. Colonna, N. Gaggero, C. Richelmi and P. Pasta, Trends Biotechnol., 1999, 17, 163–168 CrossRef CAS.
- S. Peter, M. Kinne, R. Ullrich, G. Kayser and M. Hofrichter, Enzyme Microb. Technol., 2013, 52, 370–376 CrossRef CAS PubMed.
- M. Kluge, R. Ullrich, K. Scheibner and M. Hofrichter, Green Chem., 2012, 14, 440–446 RSC.
- M. Kluge, R. Ullrich, C. Dolge, K. Scheibner and M. Hofrichter, Appl. Microbiol. Biotechnol., 2009, 81, 1071–1076 CrossRef CAS PubMed.
- E. Aranda, R. Ullrich and M. Hofrichter, Biodegradable, 2010, 21, 267–281 CrossRef CAS PubMed.
- M. Kinne, M. Poraj-Kobielska, E. Aranda, R. Ullrich, K. E. Hammel, K. Scheibner and M. Hofrichter, Bioorg. Med. Chem. Lett., 2009, 19, 3085–3087 CrossRef CAS PubMed.
- K. Barková, M. Kinne, R. Ullrich, L. Hennig, A. Fuchs and M. Hofrichter, Tetrahedron, 2011, 67, 4874–4878 CrossRef PubMed.
- M. Kinne, R. Ullrich, K. E. Hammel, K. Scheibner and M. Hofrichter, Tetrahedron Lett., 2008, 49, 5950–5953 CrossRef CAS PubMed.
- M. Poraj-Kobielska, M. Kinne, R. Ullrich, K. Scheibner, G. Kayser, K. E. Hammel and M. Hofrichter, Biochem. Pharmacol., 2011, 82, 789–796 CrossRef CAS PubMed.
- M. Kluge, R. Ullrich, K. Scheibner and M. Hofrichter, J. Mol. Catal. B: Enzym., 2014, 103, 56–60 CrossRef CAS PubMed.
- S. Peter, M. Kinne, X. S. Wang, R. Ullrich, G. Kayser, J. T. Groves and M. Hofrichter, FEBS J., 2011, 278, 3667–3675 CrossRef CAS PubMed.
- A. Zaks and D. R. Dodds, J. Am. Chem. Soc., 1995, 117, 10419–10424 CrossRef CAS.
- S. Hu and L. P. Hager, J. Am. Chem. Soc., 1999, 121, 872–873 CrossRef CAS.
- M. P. J. van Deurzen, F. van Rantwijk and R. A. Sheldon, J. Carbohydr. Chem., 1997, 16, 299–309 CrossRef CAS.
- J. Lindborg, A. Tanskanen and L. T. Kanerva, Biocatal. Biotransform., 2009, 27, 204–210 CrossRef CAS.
- S. Aoun and M. Baboulene, J. Mol. Catal. B: Enzym., 1998, 4, 101–109 CrossRef CAS.
- B. A. Kaup, U. Piantini, M. Wust and J. Schrader, Appl. Microbiol. Biotechnol., 2007, 73, 1087–1096 CrossRef CAS PubMed.
- H. Kellner, P. Luis, M. J. Pecyna, F. Barbi, D. Kapturska, D. Kruger, D. R. Zak, R. Marmeisse, M. Vandenbol and M. Hofrichter, PLoS One, 2014, 9 Search PubMed.
Footnote |
| † The authors, listed in alphabetical order, contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |