
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deracemisation of profenol core by combining laccase/TEMPO-mediated oxidation and alcohol dehydrogenase-catalysed dynamic kinetic resolution†
Alba
Díaz-Rodríguez
*a,
Nicolás
Ríos-Lombardía
a,
Johann H.
Sattler
b,
Iván
Lavandera
a,
Vicente
Gotor-Fernández
a,
Wolfgang
Kroutil
b and
Vicente
Gotor
*a
aDepartamento de Química Orgánica e Inorgánica, Universidad de Oviedo, Instituto Universitario de Biotecnología de Asturias, Avenida Julián Clavería 8, 33006 Oviedo, Spain. E-mail: alba.x.diaz-rodriguez@gsk.com; vgs@uniovi.es
bDepartment of Chemistry, Organic and Bioorganic Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, 8010 Graz, Austria
First published on 22nd January 2015
Abstract
A mild one-pot methodology has been developed to deracemise rac-2-phenyl-1-propanol by combining the use of non-selective laccase/TEMPO-mediated oxidation with enantioselective bioreduction of the racemic aldehyde intermediate under dynamic conditions. The process was easily scalable and stereocontrollable by selecting the suitable biocatalyst.
The preparation of an enantiomerically pure product in 100% yield from an inexpensive racemate in a one-pot fashion has gained significant attention via biocatalytic deracemisation processes.1 Among the toolbox of enzymes of interest, oxidoreductases occupy a prominent position.2 In the last few years, the number of examples in which these enzymes have been successfully used with synthetic purposes has increased significantly, showing their high versatility.3 In fact, oxidoreductases have been proven useful for a set of transformations even on the industrial scale, especially for the preparation of enantiopure alcohol intermediates.4
Driven by the need to enhance the atom economy of chemical transformations, especial interest has been focused on the development of (bio)catalytic transformations that combine two or more processes in one-pot.5 On the one hand, alcohol dehydrogenases (ADHs) have been successfully employed together with other (bio)catalysts in chemo-enzymatic routes currently employed for the generation of various pharmaceuticals.6 On the other hand, laccases are attracting the attention of synthetic chemists since they consume molecular oxygen to oxidise, e.g. phenolic compounds;7 nevertheless, primary and secondary alcohols are also oxidised in the presence of a mediator. Therefore, laccases have found a number of applications in academia and industry.8
Herein, we report a multienzymatic approach for the deracemisation of the β-chiral alcohol 2-phenyl-1-propanol rac-1, the key core of NSAID drugs, by combining two redox processes in a stepwise or one-pot fashion. Other methodologies have been achieved for the deracemisation of mainly secondary alcohols using whole cells or enantiocomplementary ADHs.2c,9 Here, the designed redox sequence involves the non-stereoselective oxidation of rac-1 by a laccase/TEMPO system10 to the chiral labile aldehyde rac-2, followed by selective reduction with an ADH in a dynamic kinetic resolution process (DKR, Scheme 1). While the DKR of rac-2 has been performed in the presence of alcohol dehydrogenases such as horse liver ADH (HLADH)11 or Sulfolobus solfataricus ADH (SsADH),12 these previous approaches start with the aldehyde although racemic alcohol rac-1 is more accessible and cheaper than the corresponding aldehyde. Moreover, aldehyde 2 is unstable and has been proven to undergo decarboxylation.13 It is very important to note that in previous reports, only the enantiomer (S)-1 was obtained; therefore the development of a methodology that could afford both enantiomers is of interest.14 Additionally, the previous example that used HLADH with this substrate was performed at 0.5 mM concentration, which is not convenient for synthetic purposes.11b
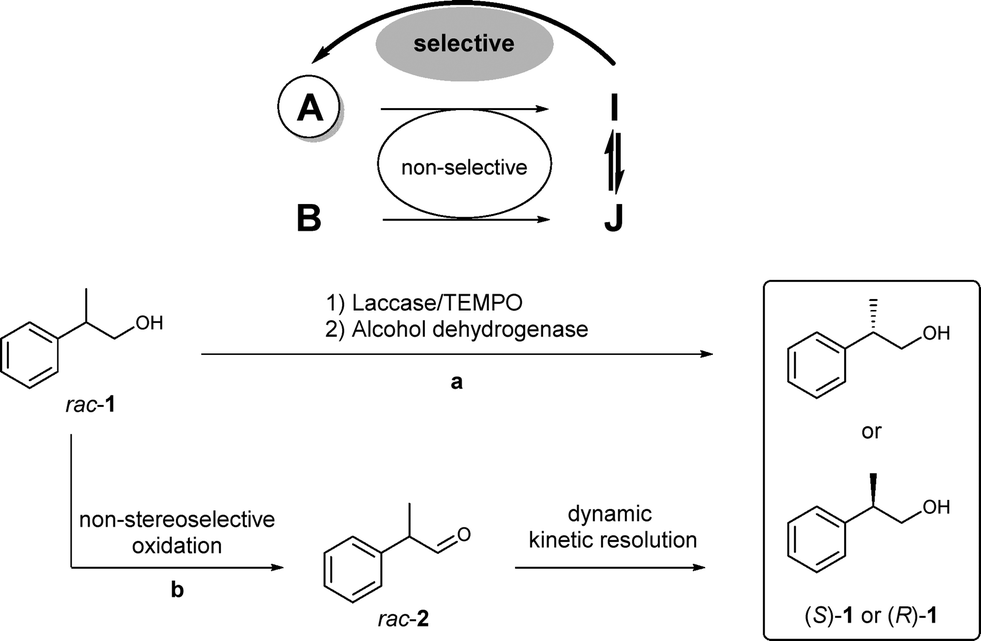 | ||
| Scheme 1 General strategy for the deracemisation of profenol 1 in (a) a two-step one-pot fashion involving a change in pH to favour the ADH-catalysed bioreduction process; (b) a two-step two-pot fashion involving the isolation of the aldehyde intermediate after an extraction protocol. A and B are substrate enantiomers; I and J are intermediate enantiomers. Arrows in bold shows the dynamic process. | ||
The laccase/TEMPO system,10,15e.g. Trametes versicolor laccase/TEMPO, has already been described for the selective transformation of racemic 2-phenyl-1-propanol (rac-1, Scheme 2) to 2-phenylpropionaldehyde (rac-2) in a very good yield (85%).15h Nevertheless, aldehyde 2 was obtained as a racemate.
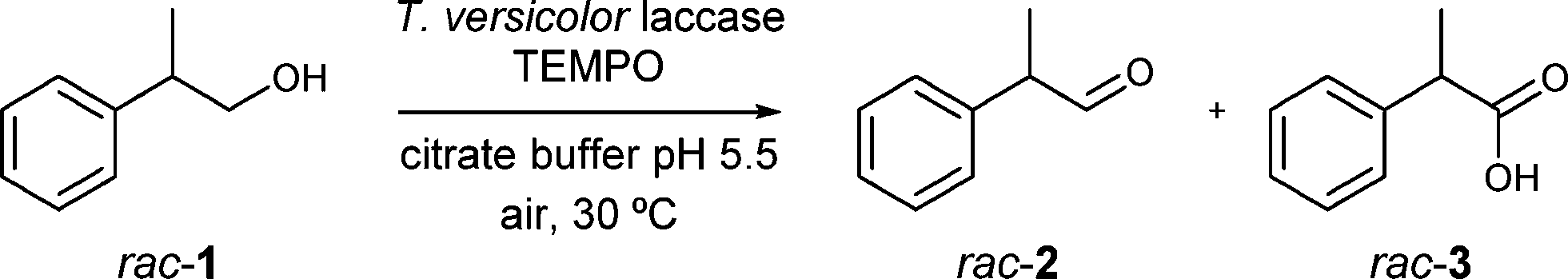 | ||
| Scheme 2 Oxidation of 2-phenyl-1-propanol (rac-1) using the laccase/TEMPO catalytic system. | ||
First, we studied the laccase-catalysed process in detail (see the ESI,† Tables S1 and S2). At a substrate concentration of 30 mM, the oxidation of rac-1 gave rac-2 in 85% yield after 2 h employing laccase from T. versicolor and TEMPO (Table S2†). Under these conditions, we also observed the formation of racemic 2-phenylpropionic acid rac-3 (15%, Scheme 2) which was tried to be minimised. Thus, the oxidation reaction was run at a more elevated substrate concentration (90 mM), leading to a decrease in the undesired carboxylic acid (<7%, Table S2†).
Focusing now on the enantioselective reduction step (Scheme 3), we tested a number of ADHs available in our laboratory to find the best candidates for the deracemisation protocol. Initial experiments showed that ADH from E. coli (ADH-P)16 and HLADH11a were the best options for obtaining (S)-1 with 94% and 90% ee, respectively (entries 1–2, Table 1). On the other hand, Evo-1.1.200 (ref. 17) led to the other antipode (R)-1 although with moderate selectivity (entry 8, Table 1). In all these cases, ethanol or 2-propanol was employed as a cosubstrate for cofactor recycling in a “coupled-substrate” mode.
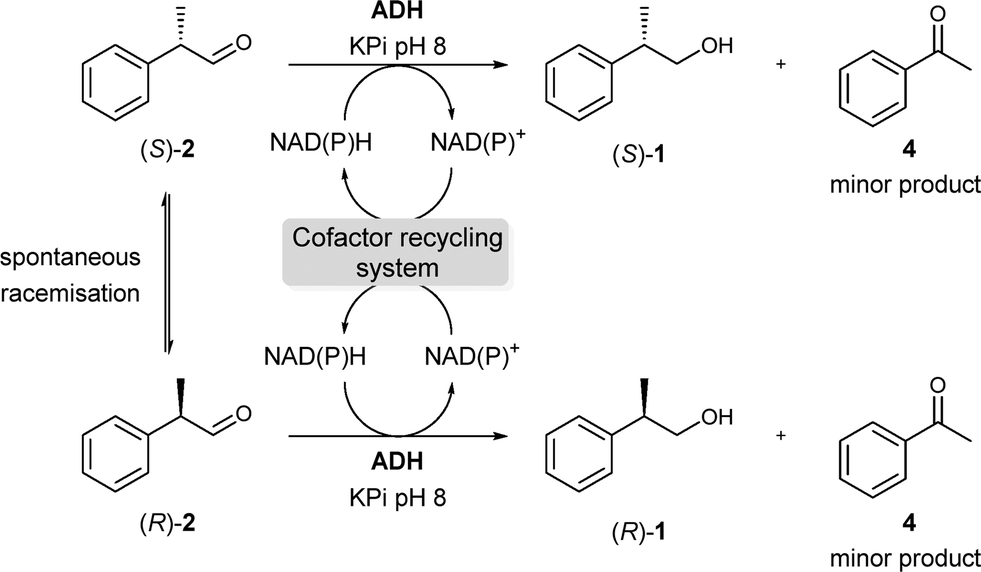 | ||
| Scheme 3 ADH-catalysed reduction of rac-2 to obtain either (S)-1 or (R)-1. | ||
| Entry | ADH | c (%) | ee (%) |
|---|---|---|---|
| a Reactions were performed at 30 mM substrate concentration, pH 8 and 30 °C, conversions were determined by GC and enantiomeric excesses were determined by chiral HPLC. | |||
| 1 | HLADH | >97 | 90 (S) |
| 2 | ADH-P | >97 | 94 (S) |
| 3 | ADH-T | >97 | 8 (S) |
| 4 | LBADH | 96 | 48 (S) |
| 5 | SyADH | >97 | 50 (S) |
| 6 | RasADH | >97 | 10 (S) |
| 7 | ADH-A | 95 | 6 (S) |
| 8 | Evo-1.1.200 | >97 | 68 (R) |
Then, additional experiments were carried out in order to enhance the enantiomeric excess of (R)-1 (entries 1–4, Table 2). A considerable improvement was observed with Evo-1.1.200 when the bioreduction was performed at pH 9 (≥90% (R), entries 1 and 2, Table 2). In fact, (R)-selective Evo-1.1.200 showed an improved enantioselection with increasing temperature or/and pH. Remarkably, despite the increase in ee for the desired alcohol (R)-1, the formation of acetophenone 4 as a by-product13 was also detected under more drastic conditions (entries 2–4, Table 2). Blank reactions using aldehyde 2 indicated that this compound was not stable at elevated temperatures or pH values. Thus, pH 9 and a temperature of 30 °C were chosen for Evo-1.1.200 (entry 1, Table 2). Similar experiments were performed for HLADH (entries 5–8, Table 2) and ADH-P (ESI,† Table S4). However, these bioprocesses did not show any improvement at higher pH values or temperatures so a pH 8 and 30 °C were further used for HLADH (entry 5, Table 2).
| Entry | ADH | Conditions | c (%) | eeb (%) |
|---|---|---|---|---|
| a Conversions were determined by GC and the percentage of acetophenone appears in brackets. b Enantiomeric excesses were determined by chiral HPLC. | ||||
| 1 | Evo-1.1.200 | pH 9, 30 °C | >97 (5) | 90 (R) |
| 2 | Evo-1.1.200 | pH 9, 60 °C | >97 (10) | 94 (R) |
| 3 | Evo-1.1.200 | pH 10, 30 °C | 97 (6) | 88 (R) |
| 4 | Evo-1.1.200 | pH 10, 45 °C | 90 (10) | >97 (R) |
| 5 | HLADH | pH 8, 30 °C | >97 (5) | 90 (S) |
| 6 | HLADH | pH 8, 60 °C | >97 (16) | 94 (S) |
| 7 | HLADH | pH 9, 30 °C | >97 (14) | 88 (S) |
| 8 | HLADH | pH 10, 30 °C | >97 (14) | 94 (S) |
After these initial studies, a stepwise procedure was investigated. Thus, after the laccase-mediated oxidation, the bioreduction was carried out without isolating the aldehyde intermediate, thus the extracted crude product from the oxidation was directly used in the reduction. In general, the overall transformation of rac-1 into enantioenriched (R)- or (S)-1 proceeded similarly well with HLADH and Evo-1.1.200, resulting in high conversions (>95%) and good ee values (86–87%, entries 1–2, Table 3). Remarkably, only traces of the carboxylic acid were observed in these two-step processes. For ADH-P, this reaction did not run to completion (entry 3).
| Entry | ADH | 1 | 2 | 4 | ee (%) |
|---|---|---|---|---|---|
| a Conversions were determined by GC and enantiomeric excesses were determined by chiral HPLC. b Isolated yields appear in brackets. c Stepwise transformation. d One-pot transformation. | |||||
| 1c | Evo-1.1.200 | 75 | 5 | 20 | 86 (R) |
| 2c | HLADH | 87 | <1 | 12 | 87 (S) |
| 3c | ADH-P | 79 | 10 | 10 | 88 (S) |
| 4d | Evo-1.1.200 | 85 (72) | 7 | 8 | 86 (R) |
| 5d | HLADH | 84 (71) | <1 | 16 | 82 (S) |
Consequently, these transformations were carried out in one-pot. As the initial experiments indicated that both reactions could proceed on a preparative scale, the deracemisations were performed on a 150 mg scale. For this purpose, the non-stereoselective oxidation of 1 (90 mM) with laccase/TEMPO was left for 3.5 h in citrate buffer at pH 5.5 and, after that time, the solution was diluted with a phosphate buffer and the pH was adjusted (pH 8 for HLADH and pH 9 for Evo.1.1.200; final substrate concentration = 30 mM). Since the laccase was not active at pH 8, overoxidation of the enantioenriched aldehyde was negligible. Finally, the NADH cofactor (1 mM) and cosubstrate (5% v/v) were added. The reaction products were isolated in 71 and 72% yield and ee = 82–86% for both enantiomers (entries 4–5, Table 3). Significantly, the optical purity of the resulting alcohol 1 decreased in comparison with the values obtained from the single bioreduction reaction (Table 2), which can be caused by side-oxidation reactions18 during the sequence and the loss of the ADH efficiency at prolonged times.
In an attempt to broaden the scope of this methodology, ibuprofenol and naproxenol were also tried as substrates under laccase/TEMPO-catalysed oxidative conditions. Unfortunately, we found some solubility issues for both alcohols, obtaining mixtures of the corresponding aldehydes and carboxylic acids to a similar extent in the Trametes versicolor laccase-mediated transformation. Using a plain buffer as a solvent or different quantities of a cosolvent such as methyl tert-butyl ether or acetonitrile did not overcome this limitation (data not shown). Medium engineering studies will be necessary to select more appropriate oxidative conditions and will be reported in due course.
Conclusions
In summary, a new strategy to deracemise 2-phenyl-1-propanol was described by combining a chemoenzymatic approach for the non-selective oxidation of this alcohol, followed by dynamic reductive kinetic resolution of the aldehyde intermediate. The cascade reaction consisted of aerobic oxidation mediated by laccase from Trametes versicolor and TEMPO, while the stereoselective reduction of the corresponding racemic aldehyde intermediate was mediated by an ADH under dynamic conditions. These intermediates have been proven to be unstable, so the use of a sequential or one-pot deracemisation procedure starting from the stable racemic alcohol is highly desirable. Using this methodology, both enantiomers were accessible by using two stereocomplementary ADHs, mainly HLADH and Evo-1.1.200. This strategy was successfully carried out on a preparative scale (150 mg), therefore representing a powerful synthetic route for the preparation of optically active profenol-like compounds.Acknowledgements
We thank Prof. Martina Pohl for the generous donation of HLADH plasmid. This research is part of the BIONEXGEN project (grant agreement 266025) sponsored by the European Union inside the 7th Framework Programme (FP7 2007-2013). Financial support from MICINN (projects CTQ2011-24237 and CTQ2013-44153-P) is also gratefully acknowledged. I.L. thanks the Spanish MICINN for personal funding (Ramón y Cajal Program).Notes and references
- (a) K. Faber, Chem. – Eur. J., 2001, 7, 5004–5010 CrossRef CAS; (b) N. J. Turner, Curr. Opin. Chem. Biol., 2010, 14, 115–121 CrossRef CAS PubMed; (c) M. Rachwalski, N. Vermue and F. P. J. T. Rutjes, Chem. Soc. Rev., 2013, 42, 9268–9282 RSC.
- (a) C. C. Gruber, I. Lavandera, K. Faber and W. Kroutil, Adv. Synth. Catal., 2006, 348, 1789–1805 CrossRef CAS; (b) F. Hollmann, I. W. C. E. Arends, K. Buehler, A. Schallmey and B. Bühler, Green Chem., 2011, 13, 226–265 RSC; (c) F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285–2313 RSC; (d) K. Robins and A. Osorio-Lozada, Catal. Sci. Technol., 2012, 2, 1524–1530 RSC.
- (a) T. Matsuda, R. Yamanaka and K. Nakamura, Tetrahedron: Asymmetry, 2009, 20, 513–517 CrossRef CAS PubMed; (b) G. W. Huisman, J. Liang and A. Krebber, Curr. Opin. Chem. Biol., 2010, 14, 122–129 CrossRef CAS PubMed; (c) M. M. Musa and R. S. Phillips, Catal. Sci. Technol., 2011, 1, 1311–1323 RSC; (d) M. Hall and A. S. Bommarius, Chem. Rev., 2011, 111, 4088–4110 CrossRef CAS PubMed; (e) Y. Ni and J.-H. Xu, Biotechnol. Adv., 2012, 30, 1279–1288 CrossRef CAS PubMed; (f) Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH, Weinheim, 3rd edn, 2012 Search PubMed.
- (a) S. M. A. de Wildeman, T. Sonke, H. E. Schoemaker and O. May, Acc. Chem. Res., 2007, 40, 1260–1266 CrossRef CAS PubMed; (b) K. Götz, L. Hilterhaus and A. Liese, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH, Weinheim, 3rd edn, 2012, pp. 1205–1223 Search PubMed; (c) A. S. Rowan, T. S. Moody, R. M. Howard, T. J. Underwood, I. R. Miskelly, Y. He and B. Wang, Tetrahedron: Asymmetry, 2013, 24, 1369–1381 CrossRef CAS PubMed.
- (a) A. Bruggink, R. Schoevaart and T. Kieboom, Org. Process Res. Dev., 2003, 7, 622–640 CrossRef CAS; (b) E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS; (c) J. H. Schrittwieser, J. Sattler, V. Resch, F. G. Mutti and W. Kroutil, Curr. Opin. Chem. Biol., 2011, 15, 249–256 CrossRef CAS PubMed; (d) F. R. Bisogno, I. Lavandera and V. Gotor, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Hoboken, 2011, pp. 1–20 Search PubMed; (e) H. Pellissier, Tetrahedron, 2013, 69, 7171–7210 CrossRef CAS PubMed; (f) I. Oroz-Guinea and E. García-Junceda, Curr. Opin. Chem. Biol., 2013, 17, 236–249 CrossRef CAS PubMed; (g) C. A. Denard, J. F. Hartwig and H. Zhao, ACS Catal., 2013, 3, 2856–2864 CrossRef CAS.
- H. Gröger and W. Hummel, Curr. Opin. Chem. Biol., 2014, 19, 171–179 CrossRef PubMed.
- (a) E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2605 CrossRef CAS PubMed; (b) H. Claus, Arch. Microbiol., 2003, 179, 145–150 CAS; (c) S. G. Burton, Curr. Org. Chem., 2003, 7, 1317–1331 CrossRef CAS; (d) H. Claus, Micron, 2004, 35, 93–96 CrossRef CAS PubMed; (e) P. Astolfi, P. Brandi, C. Galli, P. Gentili, M. F. Gerini, L. Greci and O. Lanzalunga, New J. Chem., 2005, 29, 1308–1317 RSC; (f) S. Camarero, D. Ibarra, M. J. Martínez and A. T. Martínez, Appl. Environ. Microbiol., 2005, 71, 1775–1784 CrossRef CAS PubMed; (g) L. Quintanar, C. Stog, A. B. Taylor, P. J. Hart, D. J. Kosman and E. I. Solomon, Acc. Chem. Res., 2007, 40, 445–452 CrossRef CAS PubMed.
- For reviews dealing with applications of laccases, see: (a) S. Riva, Trends Biotechnol., 2006, 24, 219–226 CrossRef CAS PubMed; (b) S. R. Couto and J. L. T. Herrera, Biotechnol. Adv., 2006, 24, 500–513 CrossRef PubMed; (c) S. Witayakran and A. J. Ragauskas, Adv. Synth. Catal., 2009, 351, 1187–1209 CrossRef CAS; (d) T. Kudanga, G. S. Nyanhongo, G. M. Guebitz and S. Burton, Enzyme Microb. Technol., 2011, 48, 195–208 CrossRef CAS PubMed; (e) Z. Yang, Synlett, 2012, 23, 2574–2575 CrossRef PubMed; (f) M. Mogharabi and M. A. Faramarzi, Adv. Synth. Catal., 2014, 356, 897–927 CrossRef CAS.
- (a) C. V. Voss, C. C. Gruber, K. Faber, T. Knaus, P. Macheroux and W. Kroutil, J. Am. Chem. Soc., 2008, 130, 13969–13972 CrossRef CAS PubMed; (b) C. V. Voss, C. C. Gruber and W. Kroutil, Synlett, 2010, 991–998 CAS; (c) C. E. Paul, I. Lavandera, V. Gotor-Fernández, W. Kroutil and V. Gotor, ChemCatChem, 2013, 5, 3875–3881 CrossRef CAS.
- K. Kędziora, A. Díaz-Rodríguez, I. Lavandera, V. Gotor-Fernández and V. Gotor, Green Chem., 2014, 16, 2448–2453 RSC.
- (a) J. Grunwald, B. Wirz, M. P. Scollar and A. M. Klibanov, J. Am. Chem. Soc., 1986, 108, 6732–6734 CrossRef CAS; (b) D. Giacomini, P. Galletti, A. Quintavalla, G. Gucciardo and F. Paradisi, Chem. Commun., 2007, 4038–4040 RSC; (c) P. Galletti, E. Emer, G. Gucciardo, A. Quintavalla, M. Pori and D. Giacomini, Org. Biomol. Chem., 2010, 8, 4117–4123 RSC; (d) S. Kara, D. Spickermann, J. H. Schrittwieser, C. Leggewie, W. J. H. van Berkel, I. W. C. E. Arends and F. Hollmann, Green Chem., 2013, 15, 330–335 RSC.
- J. A. Friest, Y. Maezato, S. Broussy, P. Blum and D. B. Berkowitz, J. Am. Chem. Soc., 2010, 132, 5930–5931 CrossRef CAS PubMed.
- (a) D. P. Riley, D. P. Getman, G. R. Beck and R. M. Heintz, J. Org. Chem., 1987, 52, 287–290 CrossRef CAS; (b) C. S. Fuchs, M. Hollauf, M. Meissner, R. C. Simon, T. Besset, J. N. H. Reek, W. Riethorst, F. Zepeck and W. Kroutil, Adv. Synth. Catal., 2014, 356, 2257–2265 CrossRef CAS.
- Up to now, there is just one report about an (R)-selective enzyme applied to this substrate from Thermus thermophilus HB27, although the alcohol was obtained with moderate ee (71%), see: J. Rocha-Martín, D. Vega, J. M. Bolivar, A. Hidalgo, J. Berenguer, J. M. Guisán and F. López-Gallego, Bioresour. Technol., 2012, 103, 343–350 CrossRef PubMed.
- (a) M. Fabbrini, C. Galli, P. Gentili and D. Macchitella, Tetrahedron Lett., 2001, 42, 7551–7553 CrossRef CAS; (b) A. Barilli, F. Belinghieri, D. Passarella, G. Lesma, S. Riva, A. Silvani and B. Danieli, Tetrahedron: Asymmetry, 2004, 15, 2921–2925 CrossRef CAS PubMed; (c) M. Marzorati, B. Danieli, D. Haltrich and S. Riva, Green Chem., 2005, 7, 310–315 RSC; (d) D. Monti, A. Candido, M. M. Cruz Silva, V. Krěn, S. Riva and B. Danieli, Adv. Synth. Catal., 2005, 347, 1168–1174 CrossRef CAS; (e) I. W. C. E. Arends, Y.-X. Li, R. Ausan and R. A. Sheldon, Tetrahedron, 2006, 62, 6659–6665 CrossRef CAS PubMed; (f) A. Díaz-Rodríguez, I. Lavandera, S. Kanbak-Aksu, R. A. Sheldon, V. Gotor and V. Gotor-Fernández, Adv. Synth. Catal., 2012, 354, 3405–3408 CrossRef; (g) J. Gross, K. Tauber, M. Fuchs, N. G. Schmidt, A. Rajagopalan, K. Faber, W. M. F. Fabian, J. Pfeffer, T. Haas and W. Kroutil, Green Chem., 2014, 16, 2117–2121 RSC; (h) A. Díaz-Rodríguez, L. Martínez-Montero, I. Lavandera, V. Gotor and V. Gotor-Fernández, Adv. Synth. Catal., 2014, 356, 2321–2329 CrossRef.
- A. Karlsson, M. El-Ahmad, K. Johansson, J. Shafqat, H. Jörnvall, H. Eklund and S. Ramaswamy, Chem.-Biol. Interact., 2003, 143–144, 239–245 CrossRef CAS.
- ADH commercially available from Evocatal GmbH, see: R. C. Simon, E. Busto, N. Richter, F. Belaj and W. Kroutil, Eur. J. Org. Chem., 2014, 111–121 CrossRef CAS.
- P. Könst, H. Merkens, S. Kara, S. Kochius, A. Vogel, R. Zuhse, D. Holtmann, I. W. C. E. Arends and F. Hollmann, Angew. Chem., Int. Ed., 2012, 51, 9914–9917 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical methods and NMR spectra. See DOI: 10.1039/c4cy01351d |
| This journal is © The Royal Society of Chemistry 2015 |