
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring the selectivity of glycerol oxidation by tuning the acid–base properties of Au catalysts†
Alberto
Villa
a,
Sebastiano
Campisi
a,
Khaled M. H.
Mohammed
bcg,
Nikolaos
Dimitratos
cef,
Floriana
Vindigni
d,
Maela
Manzoli
d,
Wilm
Jones
ef,
Michael
Bowker
ef,
Graham J.
Hutchings
e and
Laura
Prati
*a
aDipartimento di Chimica, Università degli Studi di Milano, via Golgi 19, I-20133 Milano, Italy. E-mail: Laura.prati@unimi.it
bSchool of Chemistry, Southampton University, SO171BJ, UK
cUniversity College London, 20 Gordon Street, WC1H 0AJ London, UK
dDipartimento di Chimica and NIS Centre of Excellence, Università di Torino, Via P.Giuria 7, 10125 Torino, Italy
eSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF103AT, UK
fUK Catalysis Hub, Research Complex at Harwell (RCaH), Rutherford Appleton Laboratory, Harwell, Oxon, OX11 0FA, UK
gChemistry Department, Faculty of Science, Sohag University, P. O. Box 82524, Sohag, Egypt
First published on 17th October 2014
Abstract
Supported gold nanoparticles are very effective catalysts for the selective oxidation of glycerol which represents an important bio-derived feedstock. In this paper we report that the acid/base properties, especially the acid site density, of these catalysts are the key factor in tuning the selectivity. A range of supported AuPt catalysts have been prepared by sol immobilization using acidic (H-mordenite, SiO2, MCM-41, and sulfated ZrO2) and basic (NiO and MgO) oxides as supports. In particular, using MCM-41 as the support, a high selectivity to glyceraldehyde, an important labile intermediate, was found.
Introduction
Glycerol is a highly functionalized molecule, derived from lipid transesterification in the production of biodiesel, which is recognized as a promising chemical building block for the synthesis of fine chemicals.1 For instance, the selective oxidation of glycerol has been shown to produce valuable products such as glyceraldehyde (GLYALD), glyceric acid (GLYA), dihydroxyacetone (DHA) and tartronic acid (TA).2 Supported noble metal nanoparticles are used as catalysts for this reaction when molecular oxygen is employed as the oxidant even though the nature of the metal and the experimental conditions appear to be of primary importance in determining the activity and directing the selectivity to the desired products.3 In particular gold nanoparticles are both active and selective for this reaction.4 However, the main limitation of gold monometallic catalysts appears to be the requirement for basic conditions.4 From an industrial point of view working under non-basic conditions provides a great advantage and numerous efforts have been devoted to solve this problem. Recently it was found that the catalytic oxidation of glycerol with gold catalysts can be carried out without using a basic environment.5 In particular, by alloying Au with Pt, it is possible to obtain an effective catalytic system in terms of activity and selectivity even in the absence of a base.5a,b,f However, these studies clearly showed that the support greatly affected the activity and the selectivity. Indeed, by using an acidic support, H-mordenite, an enhancement in both the activity and the selectivity to C3 products occurs (glyceric and tartronic acid) compared to similar AuPt particles supported on activated carbon and TiO2.5a More recently, it has been shown that by using basic supports such as MgO and hydrotalcite it is also possible to obtain a high selectivity to C3 products by carefully tuning the reaction conditions, specifically by controlling the reaction temperature.5b,e From these studies it can be concluded that a new generation of bifunctional gold-based catalysts can be prepared, where the acid/base properties of the support play a crucial role, affecting both activity and selectivity of the reaction.The aim of the present study is to clarify the effect of the acid site nature and strength (Lewis and/or Brønsted) and acid site density on the activity and selectivity of Au–Pt nanoparticles supported on acidic supports in base-free glycerol oxidation.
Results and discussion
Four acidic oxides with different acid/base properties, i.e. H-mordenite, SiO2, MCM-41, and sulfated ZrO2 (S-ZrO2), have been used as supports and compared to two basic supports (MgO and NiO). Bimetallic AuPt catalysts were prepared by the sol immobilization technique through a two-step procedure,5a which ensures the formation of alloyed bimetallic particles with controlled particle size. The catalytic tests for the base-free glycerol oxidation at 80 °C were carried out (0.3 M glycerol, glycerol/metal = 500 mol mol−1, 3 atm O2, T = 80 °C). The activity and the selectivity of the catalysts are reported in Table 1.| Catalyst | Activitya (mol molAuPt−1 h−1) | Selectivityb (%) | Adsorbed amount of NH3c (μmol m−2) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| GLYA | GLYALD | TA | DHA | C3 | GLYCA | OXA | FA | C2 + C1 | |||
| Reaction conditions: 0.3 M glycerol, glycerol/metal = 500 mol mol−1, 3 atm O2, T = 80 °C.a Moles of glycerol converted per hour per mole of the metal calculated after 15 min of reaction.b Selectivity calculated at 30% conversion. GLYA = glyceric acid; GLYALD = glyceraldehyde; TA = tartronic acid; DHA = dihydroxyacetone; GLYCA = glycolic acid; OXA = oxalic acid; FA = formic acid; C2 = oxalic acid and glycolic acid; C1 = formic acid.c The amount of NH3 adsorbed under an equilibrium pressure of 5 Torr. The amount of NH3 obtained after subtracting the contribution of Lewis sites. | |||||||||||
| AuPt/MgO | 657 | 21 | 7 | 1 | 12 | 41 | 8 | 6 | 43 | 59 | — |
| AuPt/NiO | 283 | 51 | 10 | 8 | 16 | 85 | 7 | 2 | 8 | 17 | — |
| AuPt/MCM-41 | 228 | 35 | 46 | 1 | 16 | 98 | 1 | 1 | — | 2 | 0.34 |
| AuPt/SiO2 | 156 | 61 | 16 | 1 | 18 | 96 | 2 | 1 | — | 3 | 0.72 |
| AuPt/H-mordenite | 105 | 53 | 19 | — | 23 | 95 | 1 | 2 | 1 | 4 | 2.68 |
| AuPt/S-ZrO2 | 113 | 57 | 19 | 3 | 17 | 96 | 1 | 1 | 1 | 3 | 2.96b |
First of all, the higher activity of the basic supports compared to the acidic ones is not surprising. In terms of activity, as expected, the presence of a basic support such as MgO (isoelectric point of 10.4) increased the activity.5b It is well recognized that the mechanism of glycerol oxidation proceeds via oxidative dehydrogenation. The presence of a base enhances the β-hydride abstraction, which is the limiting step of this reaction.3 It should be noted that AuPt/MgO is able to maintain high selectivity toward glyceric acid even at high conversion only if the temperature is maintained below 25 °C.5b ICP analysis performed on the reaction media evidenced the presence of Mg in solution. Mg could be a sacrificial base, promoting both H-abstraction and the successive desorption of the Mg-glycerate, therefore limiting the deactivation phenomena due to strongly adsorbed byproducts. AuPt NPs on MgO is a very active catalyst, exhibiting an initial activity (657 converted moles of glycerol molAuPt−1 h−1) superior to those observed when the same AuPt NPs are immobilized on NiO (283 moles of glycerol molAuPt−1 h−1) and on the acidic supports (113, 105, 156, and 228 moles of glycerol molAuPt−1 h−1 for S-ZrO2, H-mordenite, SiO2 and MCM41, respectively) (Table 1). However, no direct correlation between the acid/base properties and the catalytic activity was found.
Interestingly, AuPt NPs on MCM41 showed limited deactivation phenomena compared to AuPt supported on S-ZrO2, SiO2 and H-mordenite showing reaction profiles similar to those of AuPt/NiO and AuPt/MgO (S1), determining a sort of breakout line between acid and basic supports. Moreover, recycling experiments carried out just by filtering the catalyst and adding fresh solution of glycerol confirmed the good stability of AuPt/MCM41 at least after six runs in terms of activity and also selectivity (S2). The mesostructure of MCM-41 was maintained after the recycling tests, as confirmed by SAXRD analyses (S3).
From a selectivity point of view, AuPt NPs on acidic supports appear to be the best choice. Indeed, as shown in Table 1, by using acidic supports a higher selectivity to C3 compounds (GLYA + GLYALD + TA + DHA) (~95%) was obtained whereas basic supports, in particular MgO, promoted C–C bond cleavage, forming C2 and C1 products in high amounts (59%).
In addition, the formation of glyceraldehyde, a labile compound, in significant yield was observed with the usage of acidic supports. Particularly AuPt/MCM-41 showed the highest selectivity to glyceraldehyde (46% at 30% conversion). To the best of our knowledge this is the first time that glyceraldehyde has been detected in such a great amount. The presence of glyceraldehyde was also confirmed by 13C-NMR analysis of the reaction mixtures (S4).
It can be noted that the higher selectivity to glyceraldehyde showed by MCM-41 is related to a lower production of glyceric acid compared to the other acidic supports.
By decreasing the reaction temperature from 80 °C to 60 °C and then to 40 °C the activity of the catalyst decreased as expected with an initial activity of 228, 173 and 112 (moles of glycerol molAuPt−1 h−1), respectively (S5). However by decreasing the reaction temperature, the selectivity to glyceraldehyde increased, giving the best result at 40 °C (55% at 30% conversion) (S5).
Focusing on AuPt NPs supported on acidic supports and in order to understand the influence of the nature of the support on the catalytic activity and selectivity, a detailed characterization of the morphology and of the surface properties of the catalysts was performed.
TEM data collected from the samples revealed good AuPt dispersion on all the catalysts (Fig. 1). EDS analysis showed that the AuPt nanoparticles are alloyed particles (S6). Moreover, the size of the AuPt particles is not significantly affected by the support, with a mean diameter of 6.2–7.5 nm (S7). Therefore we can conclude that the impact of the particle size is negligible on the catalytic performance of the AuPt based catalysts tested in this work.
 | ||
| Fig. 1 Representative TEM images of a) AuPt/MCM-41 and b) AuPt/SiO2. | ||
Therefore, we focused on the investigation of the acidic properties of AuPt nanoparticles supported on SiO2, MCM-41, S-ZrO2 and H-mordenite by means of FTIR spectroscopy. 2,6-Dimethylpyridine (2,6-DMP) was used as a probe molecule. The adsorption/desorption of such a probe is suitable because (i) it allows us to identify both Lewis and Brønsted acidic centers present on the surface and above all (ii) it can distinguish Lewis and/or Brønsted sites of different acid strengths.6 Moreover, when the base is probing Brønsted sites, it is adsorbed in its 2,6-dimethylpyridinium form (2,6-DMPH+). This species yields two strong and well recognizable 8a–8b bands (ring stretching modes) centered at 1640–1655 cm−1 and at about 1630 cm−1, respectively. Whereas Lewis bound and H-bonded species will typically give three bands in the 1620–1580 cm−1 range.7 In particular, the ν8a mode, which in the liquid phase appears at 1594 cm−1, is very sensitive and allows the identification of different 2,6-DMP adsorption types on solids. In fact, when the ν8a wavenumber is higher than 1625 cm−1, it characterizes the protonated species (2,6-DMPH+) whereas lower wavenumbers correspond to the coordinated or H-bonded species (DMPL). The 2,6-DMP molecule interacting by H-bonding (a) and adsorbed on Lewis (b) and Brønsted (c) sites is represented in Scheme 1.
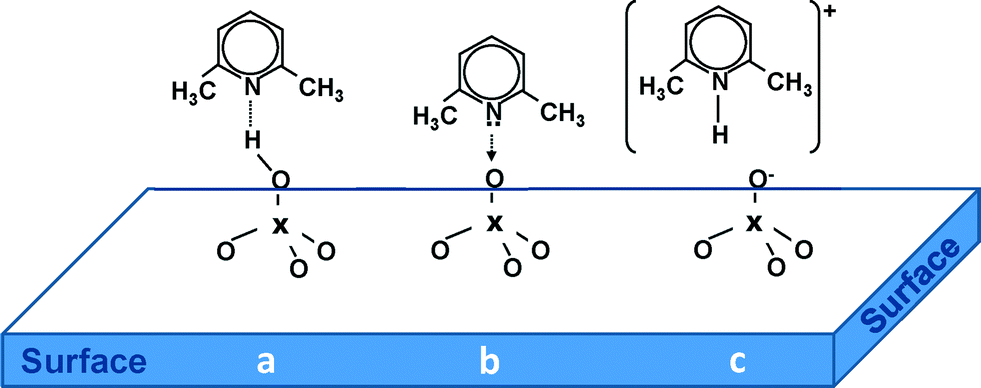 | ||
| Scheme 1 The 2,6-DMP molecule interacting by H-bond (a) and adsorbed on Lewis (b) and Brønsted (c) sites exposed on the surface of the catalysts. | ||
Therefore, to describe the adsorption/desorption of 2,6-DMP, we focus on the bands in the 1650–1550 cm−1 spectral range. The FTIR spectra, normalized with respect to the surface area and collected after interaction with 2 mbar of 2,6-DMP at room temperature (r.t.) and during the outgassing, are shown in Fig. 2.
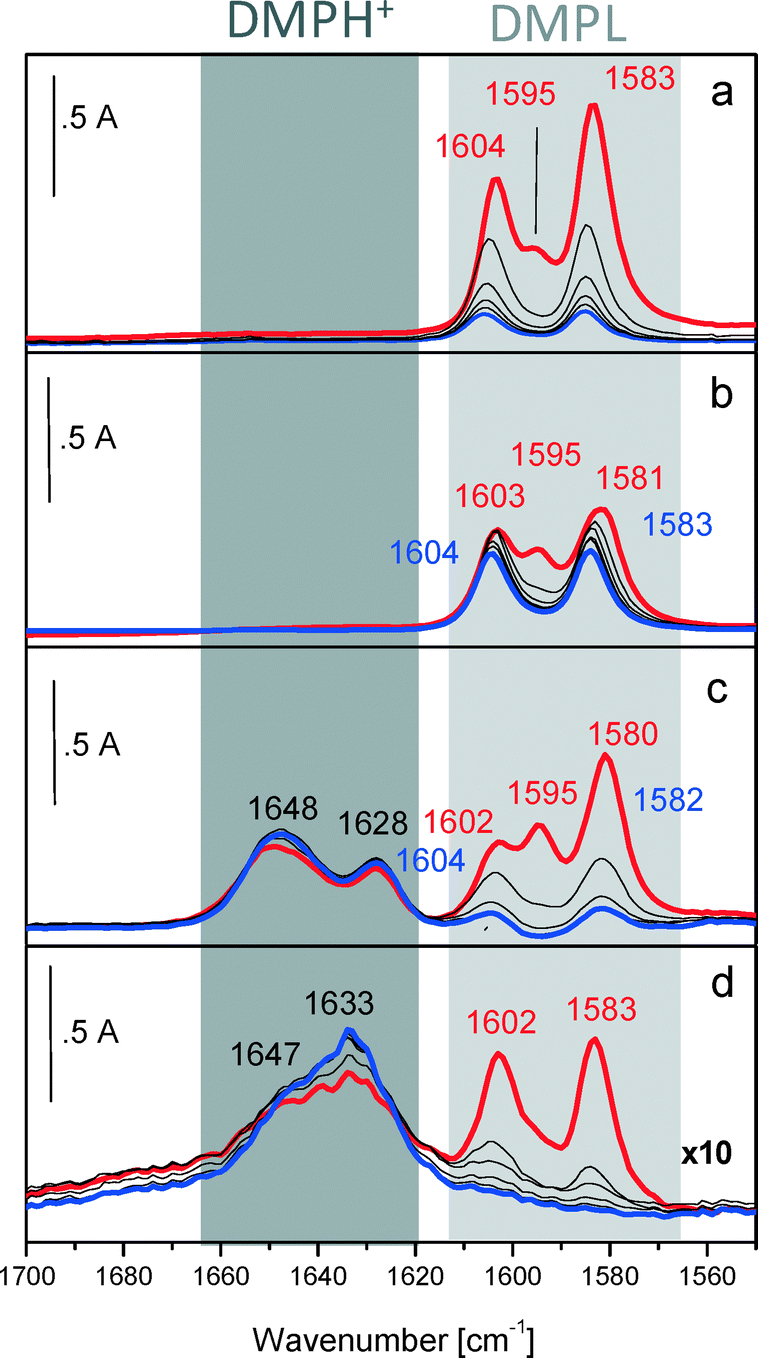 | ||
| Fig. 2 The IR spectral region of the 8a–8b ring modes of 2,6-DMP. FTIR absorbance spectra after interaction with 2 mbar of 2,6-DMP at r.t. (red curve) and during outgassing (black and blue curves) with AuPt NPs on a) SiO2, b) MCM-41, c) S-ZrO2, and d) H-mordenite. | ||
Upon adsorption of 2 mbar of 2,6-DMP at r.t. (red curves), complex bands in the 1605–1580 cm−1 range due to ν8a and ν8b of liquid-like and H-bonded 2,6-DMP are formed on all catalysts. As expected, no Lewis and Brønsted acidity was observed for AuPt/MCM-41 and AuPt/SiO2. Besides the liquid like form, the 2,6-DMP probe also interacts with these systems exclusively through H-bonding with surface silanol groups. Moreover, the ν8a mode is highly blue shifted at 1605 cm−1 (which is a position typically observed when the probe interacts with Lewis sites) confirming the acid character of silanols. The observed bands gradually decreased in intensity after outgassing for 30 min with increasing time at the same temperature (blue curves). The higher stability of the bands related to AuPt/MCM-41, compared with those detected on AuPt/SiO2, is due to the presence of mesopores; in fact the H-bonded 2,6-DMP is gradually removed by prolonging the outgassing time (data not shown for the sake of brevity).
On the other hand, the catalyst supported on sulphated zirconia showed both Lewis (1602 cm−1) and Brønsted acid sites (1628 and 1648 cm−1) whereas for AuPt/H-mordenite we observed almost exclusively Brønsted acidity (bands at 1633 and 1647 cm−1). The apparent absence of Lewis sites on H-mordenite is ascribed to residual water adsorbed on the surface due to the mild pre-treatment of the samples (drying at 80 °C). It should be noted that glycerol oxidation was carried out in water which results in the Lewis sites being masked.
The adsorption/desorption of 2,6-DMP allowed us to identify the different acidic centers present on the surface of the catalysts, however there is no clear correlation between the nature of the sites and the trend observed for the catalytic activity and selectivity. Recently, Stošić et al. reported on the influence of the acid–base properties of zirconia and titania based materials on the dehydration reaction of glycerol.8 In particular, the authors demonstrated that the catalytic activity was dependent not only on the nature of the acid sites but also on the total acidity of the catalysts. Based on these premises, we carried out quantitative studies on the acid sites present on the different samples.
Adsorption microcalorimetry of NH3 molecules was performed to study the strength and the total amount of acid sites. Among the basic molecules, ammonia is a very convenient probe to determine the acid character since it can interact with acidic hydroxyls forming ammonium ions. However, upon the neutralization of the acidic hydroxyls the ammonia molecules added next can react with the ammonium ions (which also act as Brønsted acid sites), forming NH4+×NH3 dimers and as a consequence leading to an overestimation of the amount of Brønsted acid sites.9 IR studies on dimer formation and decomposition within the pores of a TON zeolite demonstrated that the optimal temperature for ammonia adsorption is 400 K because at this temperature only ammonium ions are formed.10
Further, FTIR experiments on NH3 adsorption at 343 K on AuPt/H-mordenite confirmed that no bands related to NH4+×NH3 dimers were observed (S8), therefore the microcalorimetric analyses were performed at 373 K.
In Fig. 3 the calorimetric isotherm curves (section a) and the volumetric isotherm curves (section b) are reported.
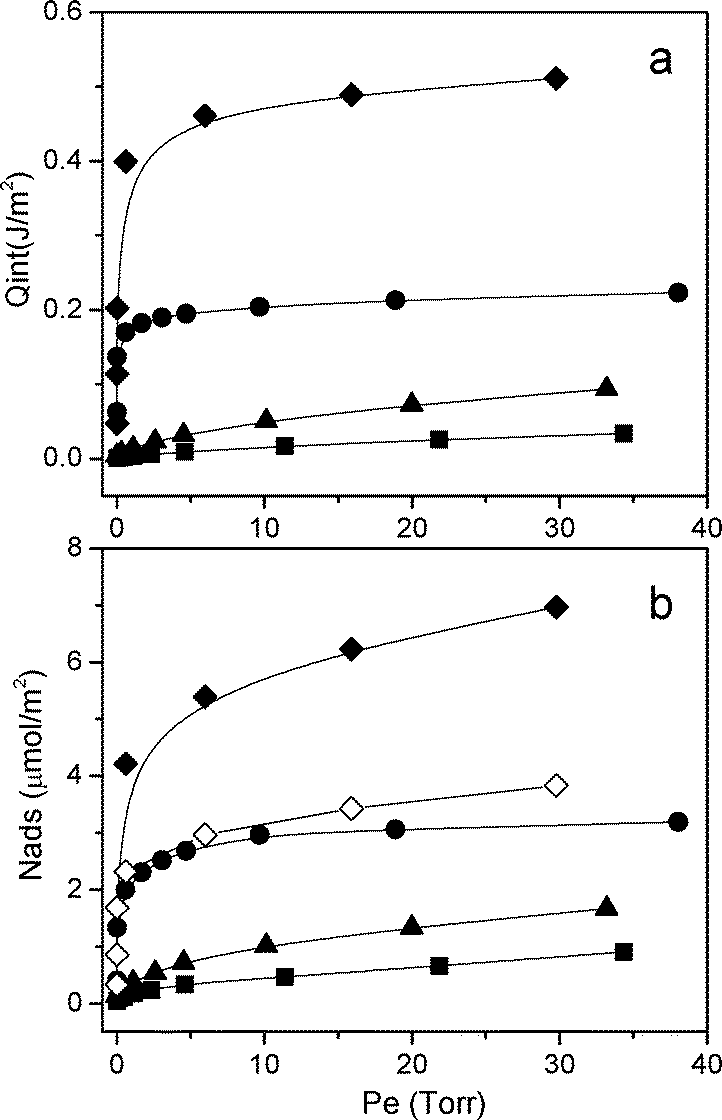 | ||
| Fig. 3 Section a: surface area-normalized calorimetric isotherms (integral heats vs. equilibrium pressure). Section b: surface area-normalized volumetric isotherms (mmol NH3 adsorbed vs. equilibrium pressure). (-■-) AuPt/MCM-41; (-▲-) AuPt/SiO2; (-●-) AuPt/H-mordenite; (-◆-) AuPt/S-ZrO2; and (-◇-) AuPt/S-ZrO2 without Lewis site contribution. | ||
The calorimetric isotherm provides the heat developed upon NH3 adsorption, Qint (J m−2), vs. the equilibrium pressure, Pe (Torr), allowing us to have information on the strength of the interaction between the molecule and the catalyst. In particular, for equilibrium pressures (Pe) up to 0.2 Torr, the developed heats follow the trend: AuPt/SiO2 ≈ AuPt/MCM-41 < AuPt/S-ZrO2 ≈ AuPt/H-mordenite. This finding indicates that AuPt/S-ZrO2 and AuPt/H-mordenite have exposed acid sites that are stronger than those present on AuPt/SiO2 and AuPt/MCM41, in agreement with the FTIR results, revealing the presence of Brønsted acid sites on both catalysts supported on S-ZrO2 and H-mordenite and silanol groups on the silica based systems.
In contrast, for Pe > 0.5 Torr, the Qint follows the sequence: AuPt/S-ZrO2 > AuPt/H-mordenite > AuPt/SiO2 > AuPt/MCM-41. It has been frequently observed that the higher the developed heat, the higher the acidic strength of the site. In our case, the calorimetric isotherms (section a of Fig. 3) display a quite similar trend if compared to that related to the volumetric ones (reported in section b of Fig. 3). Such a feature allows us to state that the differences observed in the developed heats are directly correlated with the different amounts of adsorbed NH3. This is also confirmed by plotting the Qint as a function of the adsorbed NH3 (mmol) (S9).
Moreover, the volumetric isotherms can also provide information on the total amount of acid sites probed by ammonia, and the quantity of sites follows the order: AuPt/S-ZrO2 > AuPt/H-mordenite > AuPt/SiO2 > AuPt/MCM41 (Table 1). However, we have to consider that the total amount of acid sites present on the surface of AuPt/S-ZrO2 encompasses both Lewis and Brønsted sites. Therefore, in order to determine the role of the different kinds of acid sites in the glycerol oxidation reaction, we have to discriminate between Lewis and Brønsted sites and to subtract the contribution due to Lewis sites from the total amount of acid sites. The relative abundance of the Lewis/Brønsted sites can be determined by a quantitative analysis of the IR bands of adsorbed 2,6-DMP on AuPt/S-ZrO2 (red curve in section c of Fig. 2). The deconvolution of the bands was carried out and the amount of acid sites was calculated by applying the Lambert–Beer Law (we used the following integrated molar absorption coefficients: ε(Brønsted) = 6.5 cm mmol−1, ε(Lewis) = 3.4 cm mmol−1).11 It was found that the overall amount of sites exposed at the S-ZrO2 surface was composed of 55% Brønsted sites and 45% Lewis sites. A new volumetric isotherm was constructed by subtracting the contribution of Lewis sites (Fig. 3, section b, (-◇-)). This new curve reveals that on the AuPt/S-ZrO2 and AuPt/H-mordenite catalysts the abundance of Brønsted sites is similar. The similar activity of the two catalysts, reported in Table 1, shows the importance of Brønsted and not of Lewis acid sites.
The characterization studies clearly show that the acidic surface properties of the catalysts affect both activity and selectivity. We also found that by increasing the number of acidic sites, a lower initial activity and more evident deactivation phenomena occur. If we compare the trend in the catalytic activity, AuPt/H-mordenite ≈ AuPt/ZrO2 < AuPt/SiO2 < AuPt/MCM-41 (reported in Table 1), with that in the total amount of acid sites: AuPt/S-ZrO2 > AuPt/H-mordenite > AuPt/SiO2 > AuPt/MCM-41 (data shown in Fig. 3, section b), we find that the higher the total amount of acid sites, the lower the catalytic activity. The AuPt/H-mordenite and AuPt/S-ZrO2 catalysts show similar activity, exposing a comparable abundance of Brønsted sites. In addition, AuPt supported on acidic oxides are more efficient in terms of selectivity to C3 products. Indeed, in all cases a selectivity to C3 > 95% has been observed. Moreover, the C3 product distribution, in particular the amount of glyceraldehyde or glyceric acid detected, seems to be correlated with the amount of acid sites and not with their nature. For AuPt/S-ZrO2, AuPt/H-mordenite and AuPt/SiO2 the main product is glyceric acid (53–61%) with a low amount of aldehyde (16–19%). In contrast, AuPt/MCM-41, which shows the lowest amount of acid sites, is able to stabilize glyceraldehyde, limiting its successive transformation to glyceric acid.
In order to prove this, AuPt/MCM41 and AuPt/H-mordenite, as representatives of the other acidic catalysts, were tested in the transformation of glyceraldehyde to glyceric acid under the same reaction conditions used for glycerol oxidation (0.3 M glyceraldehyde, glyceraldehyde/metal = 500 mol mol−1, 3 atm O2, T = 80 °C). After 6 h of reaction AuPt/H-mordenite completely converted glyceraldehyde to glyceric acid whereas AuPt/MCM-41 showed a conversion of only 25%. These data are in agreement with the well accepted mechanism of aldehyde transformation to a carboxylic acid via acid catalyzed diol formation and the subsequent rapid dehydrogenation to the acid (Scheme 2).3
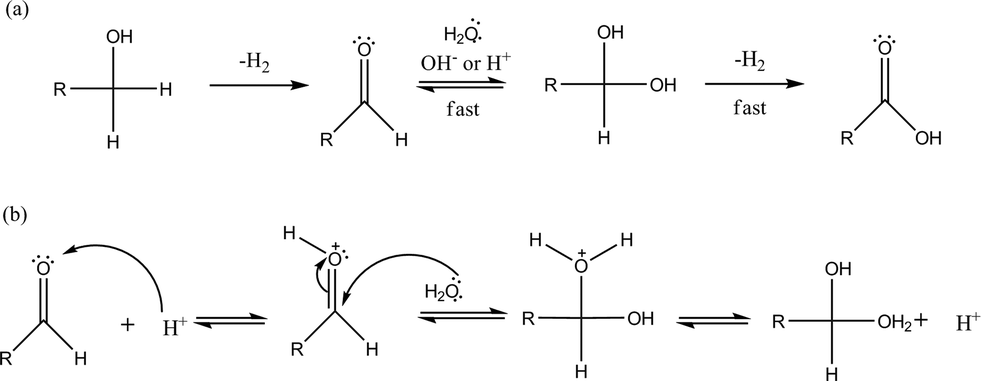 | ||
| Scheme 2 a) Alcohol dehydrogenation mechanism in the presence of water and b) acid catalyzed geminal diol formation. | ||
Experimental
Materials
NaAuCl4·2H2O and K2PtCl4 were from Aldrich (99.99% purity); H-mordenite was from Degussa (SA = 450 m2 g−1); and sulphated zirconia (SA = 78 m2 g−1,), SiO2 (SA = 148 m2 g−1, P.V. 0.78 ml g−1) and MgO (SA = 38 m2 g−1) were from Alfa Aesar. NiO (SA = 210 m2 g−1) and MCM-41 (SA = 980 m2 g−1) were prepared following the procedures reported in ref. 12 and ref. 13, respectively. NaBH4 (purity > 96%) from Fluka and polyvinyl alcohol (PVA) (Mw = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–23000, 87–89% hydrolyzed) from Aldrich were used. A 1 wt% PVA solution in water was prepared. Gaseous oxygen from SIAD was 99.99% pure.
000–23000, 87–89% hydrolyzed) from Aldrich were used. A 1 wt% PVA solution in water was prepared. Gaseous oxygen from SIAD was 99.99% pure.
Catalyst preparation
The MCM-41 mesoporous framework was prepared by a sol–gel method at RT by adopting the procedure described in ref. 13. Typically, C16TAB (5.05 g, 0.014 mol) was dissolved in 100 ml of deionized water with constant stirring (400 r.p.m.). Subsequently, aq. ammonia solution (25% NH3, 37 ml, 0.5 mol) was added dropwise to the previous solution followed by the addition of 152 ml of absolute alcohol as a co-solvent and finally by the addition of 10 ml of TEOS as a silica source. The solution became turbid in one minute and then a bright white colloidal gel was formed. The solution was kept under constant stirring for 2 h and then aged at room temperature for 24 h. The resulting gel was filtered, washed with deionized water and dried at 363 K for 24 h. The template was removed by heating the dried gel to 823 K (1 K min−1) in a tube furnace for 8 h in pure N2 flow. For complete removal of the template, the material was cooled to RT then flushed with pure O2 and heated to 823 K (10 K min−1) for 12 h.NaAuCl4·2H2O (Au: 0.031 mmol) was dissolved in 60 mL of H2O, and PVA (1%, wt%) was added (Au/PVA = 1:0.5 wt/wt). The yellow solution was stirred for 3 min, after which 0.1 M NaBH4 (Au/NaBH4 = 1:4 mol mol−1) was added under vigorous magnetic stirring. The ruby red Au(0) sol was formed immediately. Within a few minutes of sol generation, the gold sol was immobilized by adding the support (acidified to pH 2 by sulphuric acid) under vigorous stirring. The amount of the support was calculated as having a gold loading of 0.60 wt%. After 2 h, the slurry was filtered and the catalyst washed thoroughly with distilled water (neutral mother liquors). The Au/support was dispersed in 40 mL of water, with K2PtCl4 (Pt: 0.021 mmol) and PVA solution (Pt/PVA = 1:0.5 wt/wt) added. H2 was bubbled (50 mL min−1) under atmospheric pressure and room temperature for 2 h. After an additional 18 h, the slurry was filtered and the catalyst washed thoroughly with distilled water. ICP analyses were performed on the filtrate using a Jobin Yvon JV24 instrument to verify the metal loading on the support. The total metal loading was 1 wt%.
Oxidation of glycerol
Reactions were carried out in a thermostatted glass reactor (30 mL) provided with an electronically controlled magnetic stirrer connected to a large reservoir (5000 mL) containing oxygen at 3 atm. The oxygen uptake was followed by the use of a mass flow controller connected to a PC through an A/D board, plotting a flow time diagram. Glycerol was dissolved in 10 mL of water and mixed with the catalyst (final concentration of glycerol: 0.3 M, glycerol/metal = 500 mol mol−1). The reactor was pressurized at 300 kPa of O2 and thermostatted at the appropriate temperature. Once the required temperature (40, 60, and 80 °C) was reached, the gas supply was switched to oxygen and the monitoring of the reaction started. The reaction was initiated by stirring. Samples were removed periodically and analyzed by high-performance chromatography (HPLC) using a column (Alltech OA-10308, 300 mm × 7.8 mm) with UV and refractive index (RI) detectors in order to analyze the mixture of the samples. 0.1% H3PO4 solution was used as the eluent. The identification of the possible products was performed by comparison with the original samples. The presence of glyceraldehyde was confirmed by NMR. 13C NMR spectra were recorded on a Bruker AC 300 NMR spectroscope. The water signal was suppressed using a low power PRESAT pulse in order to minimize signal distortions. 200 μL of 0.3 M glyceraldehyde solution in H2O were added to each sample (final volume 600 μL). The products were recognised by comparison with authentic samples.Recycling test: each run was carried out under the same conditions (0.3 M glycerol, glycerol/metal = 500 mol mol−1, 3 atm O2, T = 80 °C, reaction time = 16 h). The catalyst was recycled in the subsequent run after filtration without any further treatment.
Catalyst characterization
The metal content was checked by ICP analysis of the filtrate on a Jobin Yvon JY24.FTIR spectra were taken on a Perkin-Elmer 2000 spectrometer (equipped with a MCT detector) with the samples in self supporting pellets introduced in the cells allowing thermal treatments in controlled atmospheres and spectrum scanning at room temperature (r.t.) in vacuum or in the presence of probe gases. From each spectrum, the spectrum of the sample before the inlet of the probe was subtracted. The standard IR experiment of 2,6-DMP adsorption/desorption on the various samples, previously activated in vacuum at 393 K, was carried out as follows: (i) admission in the IR cell of an excess dose of 2,6-DMP vapor (~2 Torr) and equilibration at room temperature for 10 min; and (ii) evacuation of the IR cell at r.t. for 15 min.
Microcalorimetric measurements were run on Tian–Calvet heat flow calorimetric equipment (Setaram C80d). Each sample was pretreated in vacuum (10–5 Torr) at 393 K (for IR experiments) and then contacted with successive small doses of NH3 vapour at 373 K. The first adsorption runs (primary isotherms) were stopped at a final equilibrium pressure of 35 Torr. The primary adsorption isotherms were followed by prolonged outgassing at the adsorption temperature and then by a second adsorption run up to the same final NH3 pressure (secondary isotherms).Uptake differences between the primary and secondary isotherms are usually considered to monitor the occurrence and the extent of irreversible adsorption processes.
Samples were prepared for TEM characterisation by dispersing the catalyst powder in high purity ethanol followed by sonication. A drop of this suspension was then evaporated on a holey carbon film supported by a 300 mesh copper TEM grid. The samples were then subjected to bright field diffraction contrast imaging experiments in order to image the particles. The instrument used for this analysis was a Jeol 2100 LaB6 TEM operating at 200 kV. High resolution transmission microscopy (HRTEM) analysis was performed using a side entry Jeol JEM 3010 (300 kV) microscope equipped with a LaB6 filament and fitted with a Link ISIS 200 detector for X-ray EDS analysis. All digital micrographs were acquired by an UltraScan 1000 camera and the images were processed by Gatan DigitalMicrograph. X-Ray powder diffraction data were obtained by using a Rigaku DMax diffractometer with Cu KR radiation operating at 40 keV and 40 mA, with a 0.05° divergence slit; the spectra were recorded in the range 1–8°.
Conclusions
Supported AuPt nanoparticle catalysts were tested for base-free glycerol aqueous phase oxidation, using supports with different acid–base properties. The catalytic behavior was markedly influenced by the support with respect to both activity and selectivity. Basic supports (MgO and NiO) promoted the activity but also increased the C–C bond cleavage reactions, thus decreasing the selectivity to the desired products. In contrast, acidic supports showed a higher selectivity to C3 oxidation products. Spectroscopic and microcalorimetric measurements provided evidence that the catalytic activity and selectivity are not influenced by the nature of the acid sites but by their amount. In particular, both activity and selectivity to glyceraldehyde decreased by increasing the number of acid sites (Brønsted sites and/or silanols). Indeed, the successive transformation of glyceraldehyde to glyceric acid proceeds via an acid catalyzed geminal diol formation and its dehydrogenation to a carboxylic acid. Moreover, Lewis acid sites do not seem to be involved.Acknowledgements
We are grateful to the Research Complex at Harwell for the provision of several of the facilities used in this work and to the EPSRC for studentship funding to WJ.Notes and references
- (a) G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed; (b) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed; (c) P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538 RSC.
- (a) C.-H. Zhou, J. N. Beltramini, Y.-X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527 RSC; (b) B. Katryniok, H. Kimura, E. Skrzyńska, J.-S. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Green Chem., 2011, 13, 1960 RSC; (c) M. Simoes, S. Baranton and C. Coutanceau, ChemSusChem, 2012, 5, 2106 CrossRef CAS PubMed; (d) S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17 RSC.
- (a) M. Bessom and P. Gallezot, Catal. Today, 2000, 57, 127 CrossRef; (b) T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS PubMed; (c) N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Chem. Sci., 2012, 3, 20 RSC.
- (a) S. Carrettin, P. McMorn, P. Johnston, K. Griffin, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1329 RSC; (b) F. Porta and L. Prati, J. Catal., 2004, 224, 397 CrossRef CAS PubMed; (c) S. Demirel-Gulen, M. Lucas and P. Claus, Catal. Today, 2005, 102–103, 166 CrossRef PubMed.
- (a) A. Villa, G. M. Veith and L. Prati, Angew. Chem., Int. Ed., 2010, 49, 4499 CrossRef CAS PubMed; (b) G. L. Brett, Q. He, C. Hammond, P. J. Miedziak, N. Dimitratos, M. Sankar, A. A. Herzing, M. Conte, J. A. Lopez-Sanchez, C. J. Kiely, D. W. Knight, S. H. Taylor and G. J. Hutchings, Angew. Chem., Int. Ed., 2010, 50, 10136 CrossRef PubMed; (c) D. Liang, J. Gao, H. Sun, P. Chen, Z. Hou and X. Zheng, Appl. Catal., B, 2011, 106, 423 CrossRef CAS PubMed; (d) R. Niea, D. Lianga, L. Shenb, J. Gaoa, P. Chena and Z. Hou, Appl. Catal., B, 2012, 127, 212 CrossRef PubMed; (e) D. Tongsakul, S. Nishimura and K. Ebitani, ACS Catal., 2013, 3, 2199 CrossRef CAS; (f) S. A. Kondrat, P. J. Miedziak, M. Douthwaite, G. L. Brett, T. E. Davies, D. J. Morgan, J. K. Edwards, D. W. Knight, C. J. Kiely, S. H. Taylor and G. J. Hutchings, ChemSusChem, 2014, 7, 1326 CrossRef CAS.
- C. Morterra, G. Meligrana, G. Cerrato, V. Solinas, E. Rombi and M. F. Sini, Langmuir, 2003, 19, 5344 CrossRef CAS.
- C. Morterra, G. Cerrato and G. Meligrana, Langmuir, 2001, 17, 7053 CrossRef CAS.
- D. Stošić, S. Bennici, J.-L. Couturier, J.-L. Dubois and A. Auroux, Catal. Commun., 2012, 17, 23 CrossRef PubMed.
- A. Zecchina, L. Marchese, S. Bordiga, C. Pazè and E. Gianotti, J. Phys. Chem., 1997, 101, 1012 Search PubMed.
- J. Datka and K. Góra-Marek, Catal. Today, 2006, 114, 205–210 CrossRef CAS PubMed.
- T. Onfroy, G. Clet and M. Houalla, Microporous Mesoporous Mater., 2005, 82, 99 CrossRef CAS PubMed.
- A. Villa, C. E. Chan-Thaw, G. M. Veith, K. L. More, D. Ferri and L. Prati, ChemCatChem, 2011, 3, 1612 CrossRef CAS.
- K. M. S. Khalil, J. Colloid Interface Sci., 2007, 315, 562–568 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Reaction profiles and temperature effects on glycerol oxidation, NMR characterization, and additional data for calorimetric tests. See DOI: 10.1039/c4cy01246a |
| This journal is © The Royal Society of Chemistry 2015 |