
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoporous silicon carbide as nickel support for the carbon dioxide reforming of methane†
C.
Hoffmann
a,
P.
Plate
a,
A.
Steinbrück
a and
S.
Kaskel
*ab
aDepartment of Inorganic Chemistry, Dresden University of Technology, Bergstrasse 66, 01069 Dresden, Germany. E-mail: Stefan.Kaskel@chemie.tu-dresden.de
bFraunhofer Institute for Material and Beam Technology, Winterbergstraße 28, 01277 Dresden, Germany
First published on 17th June 2015
Abstract
Fumed silica is used as a template in the nanocasting approach towards nanoporous silicon carbide, and it can then be applied as a catalyst support. By varying the pyrolysis temperature between 1000 and 1500 °C, the structural parameters of the resulting silicon carbide materials DUT-87 (DUT = Dresden University of Technology) can be controlled. A specific surface of 328 m2 g−1 is obtained. Furthermore, the oxidation behaviour of such nanoporous SiCs is investigated. The materials are distinguished by an impressive thermal stability at 900 °C for at least 12 h, which is allowed by the presence of a passive oxidation even for such highly porous SiCs. Hence, nickel (10 wt%) was supported on the fresh DUT-87 as well as controlled oxidized DUT-87preox samples, and the influence of the different support properties on the characteristics of the catalyst samples which are used in the carbon dioxide reforming of methane was investigated. The SiO2 layer on the SiC for the DUT-87preox samples could prevent the formation of nickel silicide to a large extent at temperatures up to 850 °C. This resulted in higher activities during the dry reforming of methane at 800 °C and the performance of the siliceous supports was significantly exceeded, emphasizing the beneficial effect of SiC. Effective methane reaction rates of 1.2 mmol g−1 s−1 were obtained for KPK1ox which was based on DUT-87 pyrolysed at 1300 °C and oxidative treatment prior to nickel insertion. Furthermore, a stable conversion level was reached over the whole time on stream of 8 h.
Introduction
Bulk silicon carbide is often described as a material with a good stability at high temperatures and resistance to oxidation or chemical corrosion. Furthermore, it has a high mechanical strength and good heat conductivity.1–4 Hence, efforts have been made to introduce porosity into silicon carbide (SiC) to obtain samples with high surface areas. These characteristics are advantageous for the application of SiCs as e.g. catalyst support to disperse active material thereon. Besides shape memory5 and sol–gel routes,6 syntheses were often performed by the nanocasting strategy with siliceous templates. These act as nanoscaled mould to shape a precursor which can be transformed to the desired product, and after SiO2 removal porous SiC is obtained.7–13 Although such preparation methods are known and specific surface areas of several hundred square meters per gram have been reported, SiC has been used as a support only in some applications so far, and usually the materials were not porous and so had low specific surface areas (≪100 m2 g−1).14–26 Nevertheless, a performance increase in the propane oxidative dehydrogenation was reported by Xu and co-workers compared to conventional supports of alumina or silica. These observations were explained by the higher heat conductivity of SiC which avoided a hot spot formation in that exothermic reaction.24 Conversely, cold spot formation might be overcome in endothermic reactions if SiC was used as catalyst support.The carbon dioxide reforming of methane (DRM, eqn (1)) is one highly endothermic reaction (ΔRH° = 247 kJ mol−1) and in principal allows the generation of synthesis gas (a mixture of CO and H2) for e.g. the Fischer–Tropsch process or methanol production.27 Simultaneously, the green-house gases CO2 and CH4 are used as indirect carbon sources which is interesting from an ecological point of view.
| CH4 + CO2 → 2CO + 2H2 | (1) |
Hence, silicon carbide has the potential to be beneficial. Nguyen et al. proved the activity of Ni/SiC systems in the DRM reaction in principle. Their applied supports had specific surface areas below 50 m2 g−1 and lower conversions than the γ-Al2O3 reference system were obtained, but if oxygen was added to the feed at the beginning of the catalytic measurement the activity and stability could be increased.19 The abandonment of precious metals and instead the use of e.g. nickel is favoured because of the lower costs and the higher availability.27
Herein we report the synthesis of nanoporous silicon carbides applying highly abundant fumed silica as a template in the nanocasting approach. Furthermore, the influence of the pyrolysis temperature on the structural parameters of the obtained DUT-87 silicon carbides is investigated (DUT = Dresden University of Technology). Since the thermal stability of a support is interesting in order to achieve a long lifetime, experiments were performed to determine the behaviour in an oxidative environment. The pronounced thermal stability of the obtained nanoporous silicon carbides compared to the bulky ones was demonstrated. Based on the results, the chosen DUT-87 samples were used as supports for 10 wt% Ni and the catalysts were then tested in the carbon dioxide reforming of methane at 800 °C. Besides the fresh SiCs, partial oxidation was performed to generate an oxide layer on the SiC and to investigate the influence of that on the catalytic performance.
Experimental
Syntheses of nanoporous silicon carbides
Preparing the silicon carbide systems via a hard-template route combined with the incipient wetness technique was used as already described earlier.12,13,25 The fumed silica template HDK® H20 (Wacker Chemie AG) and the polymeric silicon carbide precursor SMP-10 (Starfire Systems) were put in a mortar and were mixed until a homogeneous yellow powder was obtained. Typically, 5.0 g of the hydrophobic functionalized SiO2 (white) and 7.5 g SMP-10 (yellow) were used. Preliminary tests had shown that the functionalized HDK® H20 was much easier to handle than non-functionalized fumed silicas which possess exclusively silanol groups at their surface. After impregnation the samples were pyrolysed in a horizontal tube furnace in an argon atmosphere (5.0). Flushing with argon for 2 h was followed by heating at a ramp rate of 120 K h−1 to 300 °C and then holding at this temperature for 5 h. Afterwards the sample was heated to 700 °C (30 K h−1) and finally the maximum pyrolysis temperature Tmax was reached with a ramp of 150 K h−1 and was maintained for 2 h. After cooling down in argon flow the siliceous template was removed by treatment with 250 mL etching solution which consisted of equal volumes of ethanol, water and hydrofluoric acid solution (40 wt%, Merck), over night. The final product was then obtained by filtration and washing with ethanol at least three times. In dependence of the applied temperature Tmax (1000, 1300 and 1500 °C) during pyrolysis, DUT-87 samples have been synthesized and Tmax is given in brackets (e.g. DUT-87(1000) was pyrolysed at 1000 °C).Preparation of the nickel-containing catalyst systems
For supporting nickel, DUT-87(1300), DUT-87(1500) and pre-oxidized variations of both of them should be used. The aim of the oxidative treatment of the SiC before supporting nickel thereon was the formation of a SiO2 layer on the SiC.19 In order to achieve this, DUT-87(1300) and DUT-87(1500) were heated at 300 K h−1 in synthetic air stepwise to 300, 500, 700 and 900 °C. At the first three temperatures the powders were held for 30 min and the last temperature the holding time was extended to 240 min. This procedure gave the passivated silicon carbide systems DUT-87(1300)preox and DUT-87(1500)preox. The syntheses of the catalytic systems were performed by suspending 250 mg of the desired support in a solution of 137.5 mg nickel nitrate (Ni(NO3)2·6H2O, AppliChem) in 20 ml deionised water. After 5 min of mixing the solvent was removed using a rotary evaporator and the samples were further dried at 80 °C.Finally, the samples were calcined at 400 °C in static air for 4 h using a heating time of 3 h. In the same manner the two reference systems were synthesized by using non-porous HDK® N20 (Wacker Chemie AG) or SBA-16 (synthesized as described elsewhere, hydrothermal step at 90 °C) as support.28,29 The aim was to prepare catalysts containing 10 wt% Ni. The resulting systems are denoted as summarized in Table 1.
| Sample | Support | T max [°C] |
|---|---|---|
| KPK1 | DUT-87(1300) | 1300 |
| KPK2 | DUT-87(1500) | 1500 |
| KPK1ox | DUT-87(1300)preox | 1300 |
| KPK2ox | DUT-87(1500)preox | 1500 |
| Ni/PK-SiO2 | HDK® N20 | — |
| Ni/OM-SiO2 | SBA-16 | — |
Characterization
The silicon carbide supports (fresh and pre-oxidized) were characterized using nitrogen physisorption, X-ray diffraction and thermogravimetric analysis. For the catalysts, in addition temperature-programmed reduction was applied. For physisorption experiments a BELSORP Max (Bel Japan Inc.) was used. Before the measurements at 77 K, the samples were activated in vacuum at 150 °C overnight. The specific surface areas SBET were determined using the BET method for relative pressures p/p0 from 0.05–0.3. It was checked that the plot of Vads(1-p/p0) (Vads = specific adsorbed nitrogen volume) against p/p0 for this region showed a monotonic increase.30,31 The pore size distributions were determined according to the BJH method and the total pore volumes VP were quantified at a relative pressure p/p0 of 0.99. SEM was performed with a DSM982 (Zeiss). Powder diffraction data was conducted using a X'Pert PRO (PANalytical) in Bragg-Brentano geometry (CuKα radiation, λ = 0.15405 nm). Thermogravimetric analysis was performed with a STA 409 PC Luxx® (NETZSCH) in synthetic air (100 ml min−1). For each experiment about 60 mg of sample was used and the resulting materials were denoted with the suffix ‘TG’. The temperature program is shown in the results section. An estimation of the time demand for total oxidation tTG,100% was performed by plotting the holding time at 900 °C over the remaining silicon carbide fraction xSiC. This value was calculated assuming the starting materials as stochiometric SiC which is transformed to SiO2 during oxidation. The total oxidation would yield a weight increase of 49.875 wt%. Hence, the remaining fraction xSiC can be calculated as follows: | (2) |
The resulting graph was described using OriginPro 8 fitting function (group: polynomial, function: poly5) with the overall equation:
| y = A0 + A1x + A2x2 + A3x3 + A4x4 + A5x5 | (3) |
The co-domain was fixed in the range of 0 ≤ xSiC ≤ xTG. The value xTG corresponds to the remaining fraction of SiC at the beginning of the holding step at 900 °C (after the stepwise temperature program up to this point) and the value tTG,100% was determined as the intersection of the fitting function with the y-axis, i.e. the point where xSiC became zero. Temperature-programmed reduction (TPR) with a mixture of 5% H2 in argon was performed using a BELCAT-B (Bel Japan Inc.). About 30 mg of the nickel-containing samples were applied and pre-treated at 120 °C in argon for 2 h. The measurements were started at 100 °C and the temperature was raised to 880 °C at a rate of 10 K min−1 while the TCD signal and the sample temperature were monitored. A calibration allowed the determination of the hydrogen consumption xNi(TPR). Furthermore, the stability of the nickel-containing samples was investigated by heating in argon to 850 °C (120 K h−1) or by performing this heating step and following it up by a reduction in hydrogen for 1.5 h.
Catalytic testing in the carbon dioxide reforming of methane
The carbon dioxide or dry reforming of methane (DRM) was performed in a self-made apparatus. It consisted of mass-flow controllers (Bronkhorst) for methane, carbon dioxide and nitrogen, and a switch allowed the application of hydrogen instead of CH4. An appropriate mixing of the gases (74.1 ml min−1 CH4, 92.6 ml min−1 CO2 and 232.5 ml min−1 N2) was achieved with a static mixer inserted into the gas pipe which was build from PFA tubes (Swagelok) with an outer diameter of 6 mm. The reactor was a quartz glass tube with 12 mm outer diameter and 1.5 mm wall thickness. The catalyst (10.0 mg) was diluted with 1 ml SiC powder (1.47 g, F90) and was fixed with quartz wool (9 μm) at restriction points and the reactor was placed in a furnace (RO 50-250, Gero) in vertical alignment. Using a T-shaped connector at the top, the gas line and a quartz glass capillary (closed at the bottom end) which ended in the catalyst bed were fixed at the GL14 screw thread of the reactor. The capillary allowed the insertion of the thermocouple into the catalyst bed to measure and control the temperature at this point. A gauge in the inlet pipe enabled the observation of a potential pressure drop over the catalyst bed. The outlet of the reactor was water-cooled to remove humidity and furthermore was divided into an exhaust and an analytic stream. A gas chromatograph Clarus 500 (PerkinElmer) equipped with a packed (Porapak Q) and a molecular sieve (molecular sieve 5A) column as well as a sampling loop system was used for quantitative analysis. Before measurement the catalyst was pre-reduced at 850 °C in hydrogen (100 ml min−1) for 1.5 h and the temperature was further maintained for 30 min in nitrogen. Afterwards it was cooled down to the reaction temperature of 800 °C. At the beginning of each experiment the gas flows were started sequentially in the order N2, CO2 and CH4 with a delay of 30 s. The chromatographic analysis was started the same time step after the first dosing of methane. Besides inert gas flow, nitrogen acted as an internal standard for volume change determination during all experiments.For comparison, the thermodynamic values were determined using the software Cantera (version 1.7.0) with the service packages Python (version 2.5.4) and Mix-Master (version 1.0) applying the GRI-MECH 3.0 database.32
Results and discussion
Structures and properties of the nanoporous silicon carbides and the oxidative treated SiC
As evidenced by Fig. 1 the resulting silicon carbides show type IV isotherms with H1 hysteresis. This behaviour is characteristic for mesoporous materials with a narrow pore size distribution as was expected from the templates used. The fumed silica can be described as agglomerates of silica sphere aggregates33 and hence, the replica structures should have ink-bottle like pores which on one hand are induced by the primary SiO2 particles and on other hand their contact faces are a consequence of the aggregate formation during their production.34,35 The specific surface areas are between 223 and 328 m2 g−1 and pore volumes in the range of 0.4 to 0.5 cm3 g−1 are reached (Table 2). | ||
| Fig. 1 Nitrogen physisorption isotherms (77 K) of the silicon carbides DUT-87 pyrolysed at 1000, 1300 and 1500 °C (offset: 300 cm3 g−1). | ||
| Sample | T max[°C] | S BET[m2 g−1] | d BJH,Ads[nm] | d BJH,Des[nm] | V P[cm3 g−1] |
|---|---|---|---|---|---|
| DUT-87(1000) | 1000 | 327.6 | 11.1 | 8.2 | 0.54 |
| DUT-87(1300) | 1300 | 223.1 | 18.1 | 8.3 | 0.41 |
| DUT-87(1500) | 1500 | 282.8 | 12.8 | 10.2 | 0.47 |
In accordance with the literature, the sizes of the mesopores formed by the primary SiO2 particles were determined from the adsorption branch applying the BJH method, and the size of the connecting pores was calculated from the desorption branch.7,12 Hence, the first category of pores was in the size range of 11 to 18 nm and those were connected by windows which were 8 to 10 nm in diameter. The results are summarized in Table 2.
The nanoporous character of DUT-87(1500) is further exemplarily shown by SEM (Fig. S1†).
Powder X-ray diffraction revealed the nano-crystalline character of the β-SiC formed during the nanocasting process (Fig. 2). It could be observed that with an increasing pyrolysis temperature from 1000 to 1300 and subsequently 1500 °C, a decreasing peak width for DUT-87(1000), DUT-87(1300) and DUT-87(1500) is present. Applying the Scherrer equation and using the (1 1 1)-signal, crystallite sizes of 1.1, 1.8 and 2.7 nm were determined, respectively. For silicon carbides formed from SMP-10 it is known that they appear in a nano-crystalline manner.7,36,37 Furthermore, Park and co-workers demonstrated the intrinsic sintering stability that can be imparted by the small crystallite sizes resulting at the temperatures applied.7
 | ||
| Fig. 2 Powder X-ray diffraction patterns of the DUT-87 samples pyrolysed at 1000, 1300 and 1500 °C and the reference system β-SiC [29-1129]. | ||
Besides the already mentioned structure parameters, the oxidation behaviour was investigated for the DUT-87 silicon carbides. The stepwise temperature increase yielded mass curves as shown in Fig. 3. Although in all cases a weight loss is observed in the first heating step, it is obvious that the samples behaved differently depending on the pyrolysis temperature Tmax. The mass decrease at the beginning is attributed to surface species which were desorbed. In the following step a mass increase occurred during heating, hence its slope diminished during the holding temperature plateaus of 300, 500 and 700 °C. By plotting the derivation (not shown here), it was clear that in all cases the slopes were smaller at about 600 °C what can be explained by the oxidation of excess carbon which might be present in polymer-derived ceramics and the accompanying loss of mass.38,39 However, the general behaviour can be explained by the transformation of SiC to SiO2 which implies a weight gain. Nevertheless, none of the DUT-87 reached the mass increase of 49.875 wt% which would correspond to the total oxidation of SiC to SiO2. Whilst DUT-87(1000) showed a weight loss at temperatures above 750 °C, at the end the constantly increasing mass for DUT-87(1300) is higher compared to DUT-87(1500). Therefore, the oxidation is accelerated if the pyrolysis temperature Tmax is lowered from 1500 °C to 1300 °C, and if it is further reduced to 1000 °C then a different phenomena is observed. In this case, the chemical composition induces a weight loss. In accordance with the literature, low pyrolysis temperatures resulted in silicon carbide samples with high amounts of hydrogen and oxygen12 which would be released under the TG conditions, as was noticeable for DUT-87(1000) above 750 °C. X-ray diffraction of the samples confirmed the presence of SiC besides that of SiO2 after the TG measurements (Fig. S2†) and the systems can be denoted as SiC@SiO2 because a layer of SiO2 is built on SiC. Hence, Wang et al. used the time-root-law to mathematically describe their TGA curves for polymer-based SiCO samples and to calculate the time needed for complete oxidation,40–42 and in our case this model does not fit. The group observed deviation from the law at higher temperatures as well. Furthermore, the assumptions made by Moene and co-workers were not fulfilled.43 Therefore, the curves of DUT-87(1300) and DUT-87(1500) have been fitted as described in the experimental section and the time demand for complete oxidation tTG,100% can be estimated. The resulting values are 12.5 × 103 h (DUT-87(1300)) and 1.5 × 103 h (DUT-87(1500)). Although DUT-87(1500) had a much lower end mass compared to DUT-87(1300) after the 12 h at 900 °C, the required time for total oxidation tTG,100% is much larger. Therefore, it has to be concluded that the crystalline character as well as the pore structure can play an important role. A similar behaviour was described by Thümmler and Porz for the increased oxide formation at the pore mouths of porous silicon nitrides, and a resulting change in the further oxidation process.44 The results confirm that not only non-porous but also highly porous silicon carbide shows a passive oxidation.45
 | ||
| Fig. 3 Mass curves and temperature profile of the thermogravimetric analysis of the DUT-87 samples. | ||
Nitrogen physisorption could show that the samples of type DUT-87TG were mesoporous (Fig. S3†) and in Table 2 the specific surface areas SBET,TG and pore volumes VP,TG are given. As expected, the values are lower in comparison to the fresh samples. Furthermore, DUT-87(1300) and DUT-87(1500) were passivated selectively, these samples being denoted by the suffix ‘preox’. The treatment parameters were chosen in analogy to the TGA but the holding step was adjusted to 4 h. DUT-87(1300)preox and DUT-87(1500)preox still have specific surface areas of 68 and 121 m2 g−1, respectively (Table 3). Hence, they should be investigated as catalyst supports as well.
Nickel-containing catalyst systems
The investigations of the 10 wt% nickel-containing catalyst systems KPK1, KPK2, KPK1ox and KPK2ox have been focused on their affinity to the nickel silicide formation. The basic reason for this was the possible loss of active material in catalysis due to the reaction of nickel to its silicides.19 Especially, the influence of the application of the passivated supports DUT-87(1300)preox and DUT-87(1500)preox in the catalyst samples KPK1ox and KPK2ox, respectively, against KPK1 and KPK2 was of interest. For this purpose, treatment parameters have been used which were chosen in analogy to the catalyst pre-treatment for the dry reforming of methane and which were followed by XRD measurements as summarized in Fig. 4.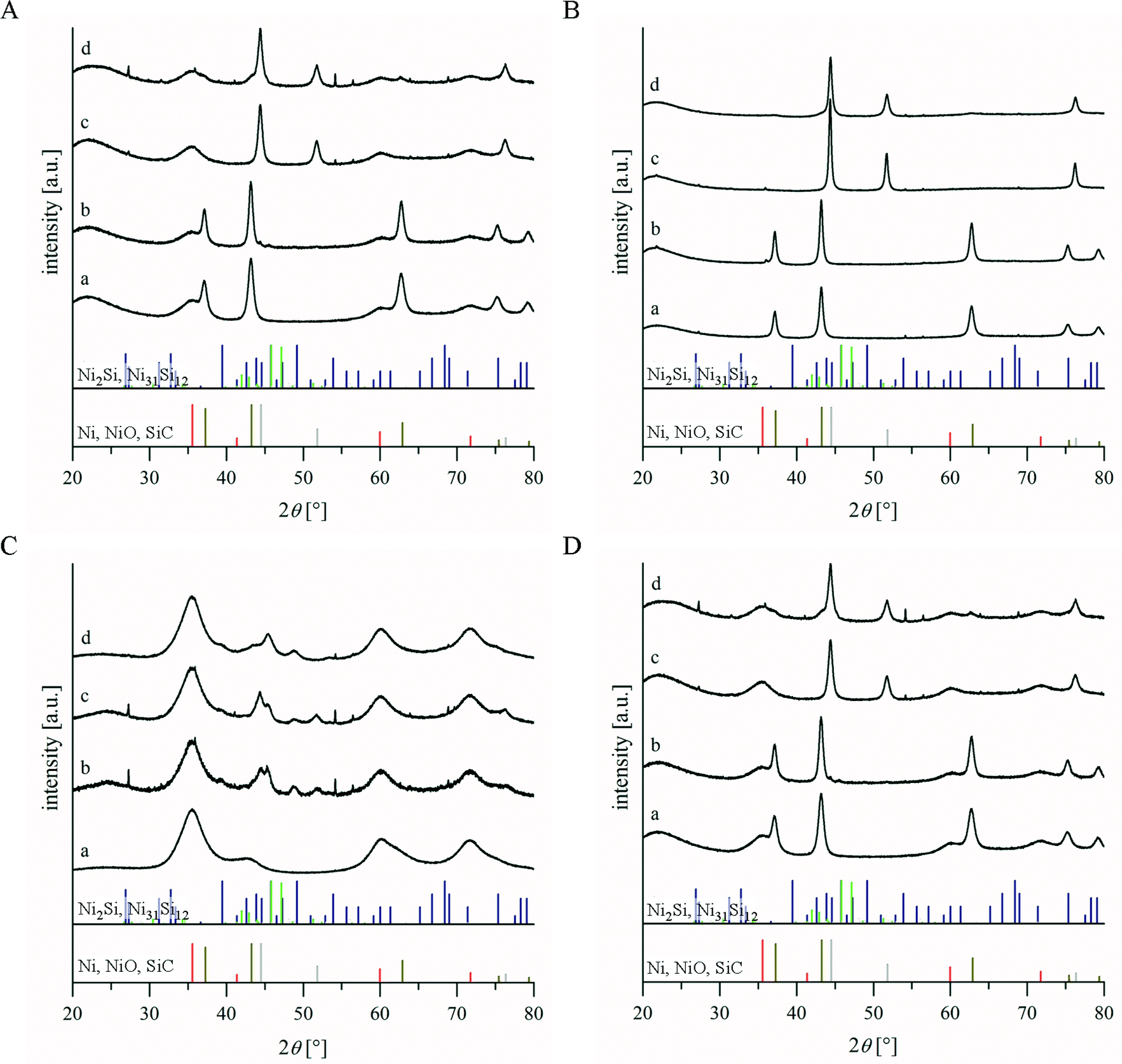 | ||
| Fig. 4 Powder X-ray diffraction results for the catalyst samples: A – KPK1, B – KPK1ox, C – KPK2 and D – KPK2ox as well as references: red – β-SiC [29-1129], olive-green – NiO [4-835], grey – Ni [4–850], blue – Ni2Si [3–943] and green – Ni31Si12 [24–524]. Shown are the patterns of: a) fresh sample, b) after argon treatment up to 850 °C, c) after the same argon treatment and reduction in hydrogen at 850 °C and d) after the temperature-programmed reduction. | ||
The powder patterns depict the presence of nickel(II)-oxide on silicon carbide for all systems even though the peaks for SiC are relatively small, especially for the passivated supports. However, powder X-ray diffraction measurements of the samples DUT-87(1300)TG and DUT-87(1500)TG (Fig. S2†) had confirmed the presence of nano-crystalline β-SiC even after the TGA. Hence, the nickel phase is just more prominent in the XRD of the catalysts and the particles have a size in the ‘nano-range’ as the peak broadening demonstrates. The inert heating to 850 °C resulted in the formation of silicides for KPK1 and KPK2.
The Ni2Si phase could only be found for KPK1, whereas peaks of Ni31Si12 were assigned in both cases. Furthermore, metallic nickel was observable. In contrast, the systems KPK1ox and KPK2ox retained their composition of NiO/SiC@SiO2 without any formation of silicides. If this heating was followed by a reductive step at 850 °C, NiO was completely transformed to Ni0 for KPK1ox and KPK2ox. Although the peak intensity ratio had changed, the complete reduction of the silicide-bound nickel in KPK1 and KPK2 was not possible under these conditions. The changes which were found are probably related to a modified composition of nickel silicides which can be influenced by treatment in hydrogen as well as by longer annealing steps.46 Hence, the applied passivation strategy prohibited the loss of the active material for KPK1ox and KPK2ox as observed after the heating in argon followed by reduction. Additional investigations with TPR measurements resulted in XRD patterns as shown in Fig. 4. These were again characterized by the presence of large amounts of nickel silicides if the supports had not been passivated before the incorporation of the nickel salt during synthesis. However, KPK1ox and KPK2ox allowed the formation of Ni0 under the TPR conditions. The temperature profile for KPK1 shows two maxima at 320 and 390 °C as well as a shoulder at about 490 °C (Fig. 5A). For KPK2 the maximum is located at 470 °C but the signal form suggests that at least two signals are overlapping (Fig. 5B). The groups of Zhang and Moene observed a similar behaviour after supporting nickel from its nitrate on non-porous or rather low specific surface area (31 m2 g−1) silicon carbide.46,47 The signal at about 320 °C can be attributed to bulk nickel(II)-oxide or easily reducible α-NiO (after the classification of Zhang et al.).48,49 Metal–support interactions further affected the reducibility of some of the nickel species and caused the desired higher reduction temperature.47 The decline of the detector signal below the base line at temperatures above 600 °C is obvious and has to be induced by a hydrogen release. It can be assumed that a sequence of chemical reactions (eqn (4) to (6)) during the TPR measurements has an influence.50,51 Thus the reduction of NiO to Ni is followed by a silicide formation with the silicon from the SiC support and hence, carbon is left over. Subsequently, a methanation under hydrogen consumption is possible. This gasification is known for nickel supported on carbon as well as for pure carbon supports.50 Furthermore, the equilibrium between methane pyrolysis and generation was also proven by Snoeck and co-workers whereas results in this field are summarized by Trimm.52,53 The catalytic activity of several metals (e.g. Cu, Pt, Ru) besides nickel was shown. In addition, negative signals during TPR measurements were assigned to spill-over effects of hydrogen to the support which leads to the release at higher temperatures. Also pure carbon supports showed such effects.50,54–56 The TPR curve for a pure silicon carbide (without nickel) gave negative values at temperatures higher than 550 °C as well (not shown here). Accordingly, already small carbon impurities within the porous SiC samples, whose existence was already concluded from the extenuated slope in the TGA at around 600 °C, might be the reason for the observed TPR characteristics.1,12
| NiO + H2 → Ni + H2O | (4) |
| xNi + SiC → NixSi + C | (5) |
| C + 2H2 → CH4 | (6) |
 | ||
| Fig. 5 Results of the temperature-programmed reduction experiments for A – KPK1 and KPK2 as well as B – KPK1ox and KPK2ox; the grey lines depict the baseline. | ||
The determination of the amount of consumed hydrogen up to the baseline intersection xNi(TPR600) resulted in values of 1.84 (KPK1) and 1.55 mmol g−1 (KPK2), which differ from the theoretical value of 1.69 mmol g−1. The reason is the overlapping of hydrogen consuming and releasing processes as discussed in the previous section. If the passivated supports are investigated as for KPK1ox and KPK2ox, the TPR signals behaved the same as is known from Ni/SiO2 (Fig. 5B).57 The signals are located in the temperature range between 300 and 500 °C so that the same nickel species as for KPK1 and KPK2 were present. Furthermore, no intersection with the baseline occurred. Hence, the prevention of contact between nickel and silicon carbide allowed the suppression of carbon formation (compare eqn (4) to (6)) which would induce or reinforce the hydrogen release at higher temperatures. The calcination of NiO/SiC at 1000 °C before TPR measurements described in the literature showed comparable results.46 Furthermore, the hydrogen consumptions of 1.61 (KPK1ox) and 1.49 mmol g−1 (KPK2ox) are much closer to the theoretical value. The slightly lower value for KPK2ox can be attributed to the formation of small amounts of silicide as was confirmed by XRD (Fig. 4). Small NixSi impurities have already been detected after heating in argon and the hydrogen treatment at 850 °C. Probably the structural characteristics of DUT-87(1500) caused an imperfect avoidance of the contact between Ni and SiC. Furthermore, the oxidation behaviour already showed a higher slope of mass increase for this sample at the end which might be a result of the same effects. Applying the Scherrer equation to the (2 0 0)-peak of the powder patterns after H2-TPR yielded Ni crystallite sizes of 14 and 21 nm for KPK1ox and KPK2ox, respectively.
Catalytic performance in the carbon dioxide reforming of methane
How the characteristics of the 10 wt% Ni containing samples discussed in the former section influenced the performance in the dry/carbon dioxide reforming of methane (DRM, eqn (1)) was then investigated. The results for the effective reaction rate of methane reff(CH4) (and the corresponding conversion X(CH4)) are given in Fig. 6 and some characteristic parameters are summarized in Table 4. Besides the silicon carbide (fresh and passivated) supported nickel catalysts, two reference systems were measured as well. Those are nickel on silica which in the first case was the non-porous fumed SiO2 HDK® N20 (Ni/PK-SiO2) and the second was ordered mesoporous oxide SBA-16 (Ni/OM-SiO2).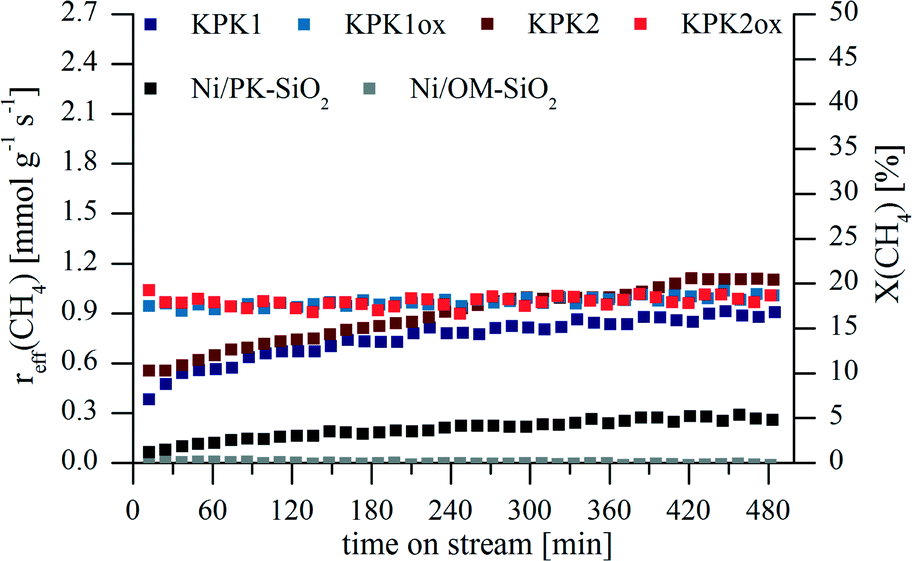 | ||
| Fig. 6 Effective reaction rates (left axis) and accordingly conversions (right axes) of methane during the dry reforming process for KPK1, KPK2, KPK1ox, KPK2ox as well as the two reference systems Ni/PK-SiO2 and Ni/OM-SiO2. | ||
| Catalyst sample | r eff(CH4, 1 h)a[mmol g−1 s−1] | r eff(CH4, 8 h)a[mmol g−1 s−1] |
n
H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nCO(1 h)b[mol mol−1] :nCO(1 h)b[mol mol−1] |
n
H2:nCO(8 h)b[mol mol−1] |
|---|---|---|---|---|
| a Effective reaction rate of methane which was normalized to the catalyst mass of 10.0 mg used after a TOS of 1 h and 8 h. b Molar ratio of hydrogen and carbon monoxide after a TOS of 1 h and 8 h. c Reference systems of nickel on non-porous SiO2 (PK-SiO2 ≡ HDK® N20) and mesoporous SiO2 (OM-SiO2 ≡ SBA-16). | ||||
| KPK1 | 0.6 | 0.9 | 0.42 | 0.44 |
| KPK2 | 0.6 | 1.1 | 0.85 | 0.62 |
| KPK1ox | 0.9 | 1.2 | 0.58 | 0.57 |
| KPK2ox | 1.0 | 1.0 | 0.61 | 0.61 |
| Ni/PK-SiO2c | 0.0 | 0.0 | — | — |
| Ni/OM-SiO2c | 0.1 | 0.3 | 0.36 | 0.38 |
KPK1 and KPK2 reached effective reaction rates for methane of 0.6 mmol g−1 s−1 after 1 h time on stream (TOS). However, those two samples activated over the whole reaction time. The maximum rates for KPK2 outperformed those of KPK1. The powder X-ray diffraction results (Fig. 4) had already shown that treatments analogous to the reduction as pre-treatment for the DRM allowed the generation of Ni0, although nickel silicides were formed as well. Since the peak widths of NiO before and Ni afterwards are larger for KPK2, it can be assumed that nickel was better dispersed which induced higher conversions. Simultaneously, the reverse water gas shift reaction (eqn (7)) was disadvantaged as is confirmed by the higher molar ratio of hydrogen to carbon monoxide. The activation with TOS indicates that structure changes occurred under DRM conditions. Both the variation of the particle sizes and the phase composition are possible. Probably, additional nickel could be extracted out of the silicides during DRM.
The avoidance of the silicide formation during the reductive pre-treatment for KPK1ox and KPK2ox (Fig. 4) could be achieved based on the passivation of the applied DUT-87(1300) and DUT-87(1500) supports. Accordingly, the loss of active material could be diminished which yielded higher effective reaction rates reff(CH4) from the beginning of the DRM experiment. The activities observed were stable over the whole TOS of 8 h.
| H2 + CO2 → CO + H2O | (7) |
The maximum methane conversions obtained are in the range of 16 to 22% which was significantly below the thermodynamic value of 98%. This further promoted the RWGS reaction which is already favoured because of the excess of CO2 used in comparison to the reaction equation, and resulted in lowered H2:CO-ratios compared to the thermodynamically controlled ratio of 0.89 mol mol−1 at 800 °C.
A beneficial effect of a calcination step in an oxygen containing atmosphere before the DRM reaction with Ni/SiC was also described by Nguyen et al. who used low surface area SiC as support.19 But the information given did not allow the calculation of reaction rates and the results were not compared to any other system. Zhang and co-workers reached values of 2.0 mmol g−1 s−1 at 750 °C for Ni/Al2O3 and Ni/La2O3 which were sometimes modified with CaO.58,59 In opposition, rates of 0.19 mmol g−1 s−1 were reported from Guo et al. at similar conditions.60 Hence, the exact preparation conditions and the process for testing is crucial which makes the comparison of the results reported here with the literature difficult. Therefore, nickel supported on silica was tested under identical conditions as the former ones. Hence, SiC (KPK1 and KPK2), SiC@SiO2 (KPK1ox and KPK2ox) and SiO2 (Ni/PK-SiO2 and Ni/OM-SiO2) supports should be compared. The results for the latter supports are given in Fig. 6 and Table 4. Primarily, it was shown by H2-TPR measurements that the NiO could be reduced to Ni0 (Fig. S4†) and the literature-known behaviour for Ni/SiO2 was obtained.57 Nevertheless, the activities are much lower as for all other systems, and in the case of Ni/OM-SiO2 even no conversion could be detected. Probably, this system was already deactivated in the first minutes of the experiment before the determination of the composition of the exhaust gas could be performed. Fast coking and sintering of the active species were described for comparable supports (e.g. SBA-15 and MCM-41).61,62 Pore blocking caused by coke could be a further explanation and might have hindered the mass transport to the active nickel centers.28 For Ni/PK-SiO2 it is likely that the activity increase with TOS was caused by surface nickel silicate species which is known from the literature.63–65 Their reduction under DRM conditions is possible and probably resulted in the availability of higher nickel amounts with TOS.66 The overall low conversion levels for Ni/SiO2 can be explained by large particle sizes and an incomplete nickel(II)-oxide reduction after heating in an inert gas flow.
A performance increase when using SiC compared to silica as the support was already observed by Xu and co-workers in the dehydration of propane.24 In contrast to this highly exothermic reaction the dry reforming of methane is highly endothermic, and it could be shown that SiC is advantageous even in those application fields.
Conclusions
In the presented work, nanoporous silicon carbide was synthesized by a nanocasting approach using hydrophobic functionalized fumed silica as a template. The materials obtained are denoted as DUT-87 (DUT = Dresden University of Technology). Hence, a pore builder was implemented in this pore generating strategy which is highly abundant and very low in cost compared to e.g. ordered mesoporous silica such as SBA-15. The adjustment of the pyrolysis temperature during carbide formation allowed tuning of the structural parameters of the resulting products. Specific surface areas up to 328 m2 g−1 and total pore volumes of 0.54 cm3 g−1 were obtained. Thermogravimetric analysis was used to determine the stability in an oxidative environment and showed that if applying stepwise heating a passive oxidation could be reached for nanoporous silicon carbides which had not been confirmed so far. Even after a treatment at 900 °C for 12 h, a material which can be described as a layer of SiO2 on a SiC core (SiC@SiO2) was stabilized. Hence, it could be shown that even nanoporous silicon carbides are highly stable and that the pore properties and the crystallinity have an influence on the performance. Based on these results, DUT-87 samples were passivated by a controlled oxidation procedure.The use of the fresh SiCs DUT-87(1300) and DUT-87(1500) as supports for 10 wt% Ni resulted in the catalyst samples KPK1 and KPK2, respectively. The substitution of the supports by the passivated carbides gave KPK1ox and KPK2ox.
The application of the KPK catalyst samples in the dry reforming of methane showed that the nickel silicide formation for the KPK1 and KPK2 materials resulted in a loss of active nickel species, and therefore low conversion levels were observed. Much higher reaction rates were achieved if the passivation of the supports was performed and hence, the Ni-SiC contact and so the silicide formation was prevented. The highest effective methane reaction rate was determined for KPK1ox at 1.2 mmol g−1 s−1, correlating to a conversion of 22%. The hydrogen-to-carbon monoxide ratio was 0.6 mol mol−1. Furthermore, stable performances were achieved for the whole investigated TOS of 8 h with KPK1ox as well as KPK2ox.
It has to be emphasized that all catalyst samples based on the DUT-87 supports reached much higher levels of activity than the purely siliceous supports. Furthermore, the passivation of silicon carbides by a deliberate oxidation even enhanced the conversions and stabilities for those nickel-based, precious metal free catalysts. This synthesis strategy allowed an optimization and the differing results for the Ni/SiC@SiO2 (KPKox samples) compared to the Ni/SiO2 samples show the beneficial effect of the SiC supports in the DRM for the first time. Furthermore, these results indicate the importance but at the same time the opportunities of tuning the characteristics of nanoporous SiC as catalyst support for the individual application as desired. Such a controlled synthesis might be successful also for several active materials and in numerous catalytic reactions.
Acknowledgements
Financial support by the German Federal Ministry of Education and Research (BMBF) is gratefully acknowledged (01RC 1006, CO2RRECT).Notes and references
- A. F. Holleman, E. Wiberg and N. Wiberg, Lehrbuch der anorganischen Chemie, de Gruyter, 1995 Search PubMed.
- C. Janiak, T. Klapötke, H. J. Meyer and E. Riedel, Moderne anorganische Chemie, de Gruyter, 2003 Search PubMed.
- W. Kollenberg, Technische Keramik: Grundlagen, Werkstoffe, Verfahrenstechnik, Vulkan-Verlag, 2004 Search PubMed.
- E. Riedel, Anorganische Chemie, de Gruyter, 2004 Search PubMed.
- M. J. Ledoux, S. Hantzer, C. P. Huu, J. Guille and M.-P. Desaneaux, J. Catal., 1988, 114, 176–185 CrossRef CAS.
- X.-Y. Guo and G.-Q. Jin, J. Mater. Sci., 2005, 40, 1301–1303 CrossRef CAS.
- K.-H. Park, I.-K. Sung and D.-P. Kim, J. Mater. Chem., 2004, 14, 3436–3439 RSC.
- Y. F. Shi, Y. Meng, D. H. Chen, S. J. Cheng, P. Chen, H. F. Yang, Y. Wan and D. Y. Zhao, Adv. Funct. Mater., 2006, 16, 561–567 CrossRef CAS PubMed.
- J. Yan, A. Wang and D.-P. Kim, J. Phys. Chem. B, 2006, 110, 5429–5433 CrossRef CAS PubMed.
- P. Krawiec, C. Schrage, E. Kockrick and S. Kaskel, Chem. Mater., 2008, 20, 5421–5433 CrossRef CAS.
- X. Yuan, J. Lü, X. Yan, L. Hu and Q. Xue, Microporous Mesoporous Mater., 2011, 142, 754–758 CrossRef CAS PubMed.
- C. Hoffmann, T. Biemelt, A. Seifert, K. Pinkert, T. Gemming, S. Spange and S. Kaskel, J. Mater. Chem., 2012, 22, 24841–24847 RSC.
- C. Hoffmann, B. Reinhardt, D. Enke and S. Kaskel, Microporous Mesoporous Mater., 2014, 184, 1–6 CrossRef CAS PubMed.
- A. R. de la Osa, A. de Lucas, L. Sánchez-Silva, J. Díaz-Maroto, J. L. Valverde and P. Sánchez, Fuel, 2012, 95, 587–598 CrossRef CAS PubMed.
- B. de Tymowski, Y. Liu, C. Meny, C. Lefèvre, D. Begin, P. Nguyen, C. Pham, D. Edouard, F. Luck and C. Pham-Huu, Appl. Catal., A, 2012, 419–420, 31–40 CrossRef CAS PubMed.
- N. Keller, R. Vieira, J.-M. Nhut, C. Pham-Huu and M. J. Ledoux, J. Braz. Chem. Soc., 2005, 16, 202–209 CAS.
- M. J. Ledoux and C. Pham-Huu, CATTECH, 2001, 5, 226–246 CrossRef CAS.
- B. Lee, H. Koo, M.-J. Park, B. Lim, D. Moon, K. Yoon and J. Bae, Catal. Lett., 2013, 143, 18–22 CrossRef CAS.
- D. L. Nguyen, P. Leroi, M. J. Ledoux and C. Pham-Huu, Catal. Today, 2009, 141, 393–396 CrossRef CAS PubMed.
- P. Nguyen, J.-M. Nhut, D. Edouard, C. Pham, M.-J. Ledoux and C. Pham-Huu, Catal. Today, 2009, 141, 397–402 CrossRef CAS PubMed.
- S. K. Singh, K. M. Parida, B. C. Mohanty and S. B. Rao, React. Kinet. Catal. Lett., 1995, 54, 29–34 CrossRef CAS.
- W.-Z. Sun, G.-Q. Jin and X.-Y. Guo, Catal. Commun., 2005, 6, 135–139 CrossRef CAS PubMed.
- A. Tomoda, D. Mikami, N. Azuma and A. Ueno, J. Adv. Sci., 2001, 13, 414–417 CrossRef CAS.
- J. Xu, Y.-M. Liu, B. Xue, Y.-X. Li, Y. Cao and K.-N. Fan, Phys. Chem. Chem. Phys., 2011, 13, 10111–10118 RSC.
- C. Hoffmann, T. Biemelt, M. R. Lohe, M. H. Rümmeli and S. Kaskel, Small, 2014, 10, 316–322 CrossRef CAS PubMed.
- Y. Liu, O. Ersen, C. Meny, F. Luck and C. Pham-Huu, ChemSusChem, 2014, 7, 1218–1239 CrossRef CAS PubMed.
- M.-S. Fan, A. Z. Abdullah and S. Bhatia, ChemCatChem, 2009, 1, 192–208 CrossRef CAS PubMed.
- F. Kleitz, T. Czuryszkiewicz, L. A. Solovyov and M. Lindén, Chem. Mater., 2006, 18, 5070–5079 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- S. Lowell, J. E. Shields, M. A. Thomas and M. Thommes, Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density, Springer, 2006 Search PubMed.
- F. Rouquerol, J. Rouquerol and K. Sing, in Adsorption by Powders and Porous Solids, Academic Press, London, 1999, ch. 6, pp. 165–189, DOI:10.1016/B978-012598920-6/50007-5.
- G. P. Smith, D. M. Golden, M. Frenklach, N. W. Moriarty, B. Eiteneer, M. Goldenberg, C. T. Bowman, R. K. Hanson, S. Song, J. W. C. Gardiner, V. V. Lissianski and Z. Qin, GRI-MECH 3.0, http://www.me.berkeley.edu/gri_mech/, (accessed 16.05.2014).
- Journal, 2014.
- P. I. Ravikovitch and A. V. Neimark, Langmuir, 2002, 18, 1550–1560 CrossRef CAS.
- P. I. Ravikovitch and A. V. Neimark, Langmuir, 2002, 18, 9830–9837 CrossRef CAS.
- E. Kockrick, R. Frind, M. Rose, U. Petasch, W. Boehlmann, D. Geiger, M. Herrmann and S. Kaskel, J. Mater. Chem., 2009, 19, 1543–1553 RSC.
- P. Krawiec, D. Geiger and S. Kaskel, Chem. Commun., 2006, 2469–2470 RSC.
- L. V. Interrante, K. Moraes, Q. Liu, N. Lu, A. Puerta and L. G. Sneddon, Pure Appl. Chem., 2002, 74, 2111–2117 CrossRef CAS.
- R. Sreeja, B. Swaminathan, A. Painuly, T. V. Sebastian and S. Packirisamy, Mater. Sci. Eng., B, 2010, 168, 204–207 CrossRef CAS PubMed.
- T. Narushima, T. Goto, T. Hirai and Y. Iguchi, Mater. Trans., JIM, 1997, 38, 821–835 CrossRef CAS.
- C. Wagner, Z. Phys. Chem., Abt. B, 1933, 21, 25–41 Search PubMed.
- Y. Wang, H. Li, L. Zhang and L. Cheng, Ceram. Int., 2009, 35, 1129–1132 CrossRef CAS PubMed.
- R. Moene, M. Makkee and J. A. Moulijn, Appl. Catal., A, 1998, 167, 321–330 CrossRef CAS.
- F. Porz and F. Thümmler, J. Mater. Sci., 1984, 19, 1283–1295 CrossRef CAS.
- K. A. Schwetz, in Handbook of Ceramic Hard Materials, ed. R. Riedel, Wiley-VCH, Weinheim, Germany, 2008, ch. 5, pp. 683–748 Search PubMed.
- R. Moene, E. P. A. M. Tijsen, M. Makkee and J. A. Moulijn, Appl. Catal., A, 1999, 184, 127–141 CrossRef CAS.
- G. Zhang, T. Sun, J. Peng, S. Wang and S. Wang, Appl. Catal., A, 2013, 462–463, 75–81 CrossRef CAS PubMed.
- J. Li, C. Wang, C. Huang, W. Weng and H. Wan, Catal. Lett., 2010, 137, 81–87 CrossRef CAS PubMed.
- J. Zhang, H. Xu, X. Jin, Q. Ge and W. Li, Appl. Catal., A, 2005, 290, 87–96 CrossRef CAS PubMed.
- R. Wojcieszak, M. Zieliński, S. Monteverdi and M. M. Bettahar, J. Colloid Interface Sci., 2006, 299, 238–248 CrossRef CAS PubMed.
- Z. Zhang, J. Teng, W. X. Yuan, F. F. Zhang and G. H. Chen, Appl. Surf. Sci., 2009, 255, 6939–6944 CrossRef CAS PubMed.
- J. W. Snoeck, G. F. Froment and M. Fowles, J. Catal., 1997, 169, 240–249 CrossRef CAS.
- D. L. Trimm, Catal. Rev.: Sci. Eng., 1977, 16, 155–189 CAS.
- M. Afzal, F. Mahmood and S. Karim, J. Therm. Anal., 1994, 41, 1119–1128 CrossRef CAS.
- A. B. E. Silva, E. Jordão, M. J. Mendes and P. Fouilloux, Braz. J. Chem. Eng., 1998, 15 Search PubMed.
- Y. J. Zhang, A. Maroto-Valiente, I. Rodriguez-Ramos, Q. Xin and A. Guerrero-Ruiz, Catal. Today, 2004, 93–95, 619–626 CrossRef CAS PubMed.
- L. Znak and J. Zieliński, Appl. Catal., A, 2008, 334, 268–276 CrossRef CAS PubMed.
- Z. Zhang and X. E. Verykios, Appl. Catal., A, 1996, 138, 109–133 CrossRef CAS.
- Z. Zhang, X. E. Verykios, S. M. MacDonald and S. Affrossman, J. Phys. Chem., 1996, 100, 744–754 CrossRef CAS.
- J. Guo, H. Lou, H. Zhao, D. Chai and X. Zheng, Appl. Catal., A, 2004, 273, 75–82 CrossRef CAS PubMed.
- D. Liu, R. Lau, A. Borgna and Y. Yang, Appl. Catal., A, 2009, 358, 110–118 CrossRef CAS PubMed.
- D. Liu, X.-Y. Quek, H. H. A. Wah, G. Zeng, Y. Li and Y. Yang, Catal. Today, 2009, 148, 243–250 CrossRef CAS PubMed.
- P. Burattin, M. Che and C. Louis, J. Phys. Chem. B, 1997, 101, 7060–7074 CrossRef CAS.
- O. Clause, M. Kermarec, L. Bonneviot, F. Villain and M. Che, J. Am. Chem. Soc., 1992, 114, 4709–4717 CrossRef CAS.
- M. Houalla, F. Delannay, I. Matsuura and B. Delmon, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 2128–2141 RSC.
- E. Ruckenstein and Y. Hang Hu, J. Catal., 1996, 162, 230–238 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cy01234h |
| This journal is © The Royal Society of Chemistry 2015 |