
Synergistic catalysis-induced ring-opening reactions of 2-substituted 3,4-dihydropyrans with α-oxoketene dithioacetals†
Changhui
Liu
a,
Amir
Taheri
a,
Bingbing
Lai
a and
Yanlong
Gu
*ab
aKey Laboratory for Large-Format Battery, Materials and System, Ministry of Education, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology (HUST), 1037 Luoyu Road, Hongshan District, Wuhan 430074, China. E-mail: klgyl@hust.edu.cn; Fax: (0)86 (0)27 87 54 45 32
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Lanzhou, PR China 730000
First published on 14th October 2014
Abstract
According to the conventional catalytic activation mode, the difficulty of using a less reactive substrate can be alleviated by means of employing a strong catalyst. Because of the susceptibility of the reaction product to strong acid, ring-opening Friedel–Crafts reactions of 2-substituted 3,4-dihydropyrans with a less reactive nucleophile, α-oxoketene dithioacetal, could not be performed by using strong acid as a catalyst. In order to find a suitable catalyst system for this reaction, a co-catalyst, CuBr2, that can increase the reactivity of α-oxoketene dithioacetal was used in conjunction with a moderate Lewis acid, MnCl2·4H2O. The mechanism of synergistic catalysis was also studied with the aid of spectroscopic investigation. It was found for the first time that CuBr2-induced disintegration of a super-conjugation system exists in α-oxoketene dithioacetal and is responsible for the increase of its reactivity. An intramolecular Michael addition of the ring-opening product was also developed, which provided a densely substituted cyclohexane derivative in good yield. Finally, a hitherto unreported S,N-doped eleven-membered heterocycle was synthesized on the basis of the developed reactions.
Introduction
Organic synthesis with the aid of synergistic catalysis has gained great attention. As a definition of synergistic catalysis given by Allen and MacMillan, a nucleophile and an electrophile are simultaneously activated by two separate catalysts to afford a single chemical transformation.1 In this concept, two catalysts and two catalytic cycles work in concert to accomplish a reaction.2 Because this concept enabled researchers to develop some organic reactions that are either difficult or unattainable with a conventional system, a great deal of attention has been paid, in the past decade, to exploring suitable catalyst pairs in order to take as much benefit from this strategy as possible.3Recently, our group has become interested in the use of 2-substituted 3,4-dihydropyran as a building block in organic synthesis.4 These compounds can react with nucleophiles, generating some valuable aliphatic or heterocyclic compounds by a process that involves a ring opening of the endocyclic enone.5 However, only a few active nucleophiles have been successfully applied so far in this unique ring-opening reaction, such as indole, thiophenol and 1,3-cyclohexanedione. Many other nucleophiles that have relatively low reactivity cannot be used. According to the conventional catalytic activation mode, the use of a stronger catalyst can alleviate, to some extent, the difficulty of using a less reactive substrate. Unfortunately, because the generated product contains a fragment of β-ketoester or 1,3-diketone, which is rather susceptible toward strong acid due to the intrinsically high reactivity in either transesterification or decarboxylation,4d the use of a strong acid as a catalyst will result in a loss of the selectivity. Therefore, only weak acids, such as MnCl2·4H2O and LiBr·H2O, have proven to be suitable catalysts in the previous reports.4
In order to use a less reactive nucleophile in the ring-opening reaction of the title compound dihydropyran, we turned to establishing a system based on the concept of synergistic catalysis. In an envisioned system, two catalysts work simultaneously. The first one (acid catalyst) activates the dihydropyran; meanwhile, the other one increases the reactivity of the nucleophile, thus allowing successful trapping of the reaction intermediate with the less reactive nucleophile. Herein, we disclose the successful outcome of this endeavor in which a catalyst system composed of MnCl2·4H2O and CuBr2 worked synergistically for the ring-opening reaction of 2-substituted 3,4-dihydropyran with a less reactive nucleophile, α-oxoketene dithioacetal.
Results and discussion
Initially, the ring-opening reaction of dihydropyran 1a with α-oxoketene dithioacetal 2a was investigated. We chose to use α-oxoketene dithioacetal as a nucleophile because this type of compound was proved to be an invaluable building block for organic synthesis.6 The reaction was performed in nitromethane at 50 °C. As shown in Table 1, with the MnCl2·4H2O catalyst, only a trace amount of the desired product 3a was detected, and both of the starting materials remained unchanged at the end of the reaction (entry 1). Increasing the reaction temperature to 110 °C was not successful in initiating the reaction (entry 2). Under identical conditions, MnCl2·4H2O was shown to be an effective catalyst for the ring-opening reaction of 1a with indole.4a The observed unreactivity of these reactions might result from the poor reactivity of 2a.7 Many Lewis and Brønsted acids were then examined in this reaction; however, no good result was obtained (see the ESI,† Table S1). Like the case of using indole as a nucleophile,4a when strong acids such as PTSA, In(OTf)3, Sc(OTf)3, I2 and FeCl3 were employed as catalysts, the ring-opening reactions always suffered from poor selectivity, although dihydropyran 1a was completely consumed. However, in the case of using weak acids such as LiBr·H2O and ZnCl2 as catalysts, no reaction occurred. Obviously, the model reaction was plagued by the following two inconsistent factors: (i) intrinsically low reactivity of α-oxoketene dithioacetal 2a, which forces the use of a strong catalyst, and (ii) susceptibility of the ring-opening product to strong acid, which precludes the possibility of using a strong catalyst in this reaction.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Co-catalyst | Temp (°C) | Solvent | Time (h) | Yield (%) |
| a 1a: 0.20 mmol, 2a: 0.24 mmol, acid catalyst: 0.03 mmol, co-catalyst: 0.01 mmol, solvent: 1.0 ml. b 1a was consumed at the end of the reaction. c Reaction scale: 20 mmol, yield: 80%. d Anhydrous HBr was generated by adding 50% sulfuric acid to 40% aqueous HBr and dried by anhydrous CaCl2. | ||||||
| 1 | MnCl2·4H2O | — | 50 | CH3NO2 | 10 | Trace |
| 2 | MnCl2·4H2O | — | 110 | CH3NO2 | 10 | Trace |
| 3 | MnCl2·4H2O | CuBr2 | 50 | CH3NO2 | 10 | 83(80)c |
| 4 | — | CuBr2 | 50 | CH3NO2 | 10 | 47 |
| 5 | — | CuBr2/H2O | 50 | CH3NO2 | 10 | 58 |
| 6 | MnCl2·4H2O | CuCl2 | 50 | CH3NO2 | 10 | 45 |
| 7 | MnCl2·4H2O | CuSO4 | 50 | CH3NO2 | 10 | 11 |
| 8 | MnCl2·4H2O | NaBr | 50 | CH3NO2 | 10 | Trace |
| 9 | MnCl2·4H2O | TBAB | 50 | CH3NO2 | 10 | Trace |
| 10 | MnCl2·4H2O | CuBr | 50 | CH3NO2 | 10 | Trace |
| 11 | ZnCl2 | CuBr2 | 50 | CH3NO2 | 10 | 52 |
| 12 | ZnI2 | CuBr2 | 50 | CH3NO2 | 10 | 57 |
| 13 | Alum | CuBr2 | 50 | CH3NO2 | 10 | 69 |
| 14 | LiBr·H2O | CuBr2 | 50 | CH3NO2 | 10 | 62 |
| 15 | MnBr2 | CuBr2 | 50 | CH3NO2 | 10 | 55 |
| 16 | MnCl2 | CuBr2 | 50 | CH3NO2 | 10 | 60 |
| 17 | HBr (5 mol%, 36% aqu.) | — | 50 | CH3NO2 | 2 | <5 |
| 18 | HBr (5 mol%, 36% aqu.) | MnCl2 | 50 | CH3NO2 | 2 | <5 |
| 19 | HBr (0.5 mol%, 46% aqu.) | — | 50 | CH3NO2 | 2 | Trace |
| 20 | HBrd (5 mol%, anhydrous gas) | MnCl2 | 50 | CH3NO2 | 2 | Trace |
| 21 | MnCl2·4H2O | CuBr2 | rt | CH3NO2 | 24 | 12 |
| 22 | MnCl2·4H2O | CuBr2 | 80 | CH3NO2 | 5 | 53b |
| 23 | MnCl2·4H2O | CuBr2 | 50 | DCE | 10 | 26 |
| 24 | MnCl2·4H2O | CuBr2 | 50 | CH3CN | 10 | 39 |
| 25 | MnCl2·4H2O | CuBr2 | 50 | EtOH | 10 | NR |
| 26 | MnCl2·4H2O | CuBr2 | 50 | Toluene | 10 | NR |
| 27 | MnCl2·4H2O | CuBr2 | 50 | — | 10 | NR |
| 28 | MnCl2·4H2O (5 mol%) | CuBr2 | 50 | CH3NO2 | 10 | 66 |
| 29 | MnCl2·4H2O (9 mol%) | CuBr2 (3 mol%) | 50 | CH3NO2 | 10 | 75 |
| 30 | MnCl2·4H2O | CuBr2 | 50 | CH3NO2 | 5 | 58 |

In order to solve the reactivity problem and also to ensure good selectivity, we then tried to add an activator into the reaction system with the hope of increasing the nucleophilicity of 2a without using a strong acid. Wang and Liu8 have reported that CuBr2 can somehow increase the nucleophilicity of the α-carbon atom of ketene dithioacetals. Inspired by this report, we then added CuBr2 into the reaction system. To our great delight, 3a was obtained in 83% yield after 10 hours of reaction at 50 °C (entry 3). When CuBr2 alone was used as a catalyst, the yield of 3a reached only 47% (entry 4). In the presence of water, the efficiency of the CuBr2 catalyst was slightly improved, but the yield of 3a was still far less than that of the MnCl2·4H2O/CuBr2 system (entry 5). These results indicated that an effect of synergistic catalysis indeed existed in the MnCl2·4H2O/CuBr2 system.
In this system, when CuBr2 was replaced by CuCl2 or CuSO4, yields of 3a were decreased to 45% and 11%, respectively (entries 6 and 7). Only a trace amount of 3a was detected when NaBr and TBAB were used instead of CuBr2 (entries 8 and 9). An attempt to perform the reaction in a system composed of MnCl2·4H2O and CuBr led only to recovery of unreacted starting materials (entry 10). All of these results indicated that the use of CuBr2 was indispensable in this system in order to obtain an evident synergistic effect. The reaction was also carried out in the CuBr2/nitromethane system in the presence of different acid catalysts, such as ZnCl2, ZnI2, alum, LiBr·H2O and also MnBr2 (entries 11 to 15). MnCl2·4H2O was superior to all the other catalysts tested. It should be noted that lattice water in MnCl2·4H2O is also critical for the model reaction as only a moderate yield was obtained when anhydrous MnCl2 was used in this system (entry 16).
Wang and Liu9 have observed a unique catalytic activity of HBr in the electrophilic alkylation of aldehydes with α-oxoketene dithioacetals, which led them to suggest that the good catalytic activity of CuBr2 can be ascribed to the formation of HBr due to decomposition of CuBr2. In order to figure out which one is the real catalyst, we also scrutinized this catalyst in our reaction. It was found that dihydropyran 1a was consumed within 2 hours by using an aqueous solution of HBr as a catalyst in the presence or absence of MnCl2. However, in these cases, a complex messy mixture was obtained in which the desired product 3a was hardly observed by TLC detection (entries 17 and 18). Incidentally, the formation of these side products cannot be avoided by decreasing the amount of HBr (entry 19). When anhydrous HBr was used instead of an aqueous solution of HBr, the result obtained is the same (entry 20). Because the reaction over the CuBr2 catalyst proceeded with good selectivity, the generation of HBr is not involved in the mechanism of the catalysis.9 Further investigation revealed that the reaction was also affected by many parameters, such as the amount of CuBr2, the ratio of CuBr2/MnCl2·4H2O, solvent, temperature and reaction time. The optimal conditions were determined to be as follows: 5 mol% of CuBr2, MnCl2·4H2O/CuBr2 ratio of 3/1, nitromethane solvent, 50 °C and 10 hours (entries 21 to 29).
With the optimized procedure in hand, we probed the scope of the reaction with respect to both the dihydropyrans and the α-oxoketene dithioacetals. As shown in Fig. 1, a variety of dihydropyrans and α-oxoketene dithioacetals could be used in the ring-opening reaction with the aid of synergistic catalysis between CuBr2 and MnCl2·4H2O, and the corresponding products were obtained in good to excellent yields. Particularly, α-oxoketene dithioacetals substituted by electron-withdrawing groups, such as methoxycarbonyl and nitro, can also be used to react with 2-aryl-3,4-dihydropyrans under the optimal conditions. A reaction on the preparative scale (20 mmol) was also investigated, and it was found that the reaction proceeded uneventfully, indicating the effectiveness of this method for practical synthesis (Table 1, entry 3).
 | ||
| Fig. 1 Ring-opening reaction of 2-aryl-3,4-dihydropyrans with α-oxoketene dithioacetals. | ||
In the literature, another commonly used α-oxoketene dithioacetal is 3,3-bis(methylthio)-1-phenyl-2-propen-1-one 2b, which sometimes had displayed different reactivity compared with that of 2a. For that reason, we then examined this α-oxoketene dithioacetal in the title ring-opening reaction. As shown in Fig. 2, the CuBr2/MnCl2·4H2O system displayed also a distinct synergistic effect in catalyzing the reaction of this α-oxoketene dithioacetal. All the methylthio, cyano and ether functionalities in the starting materials were successfully delivered into the ring-opening products.
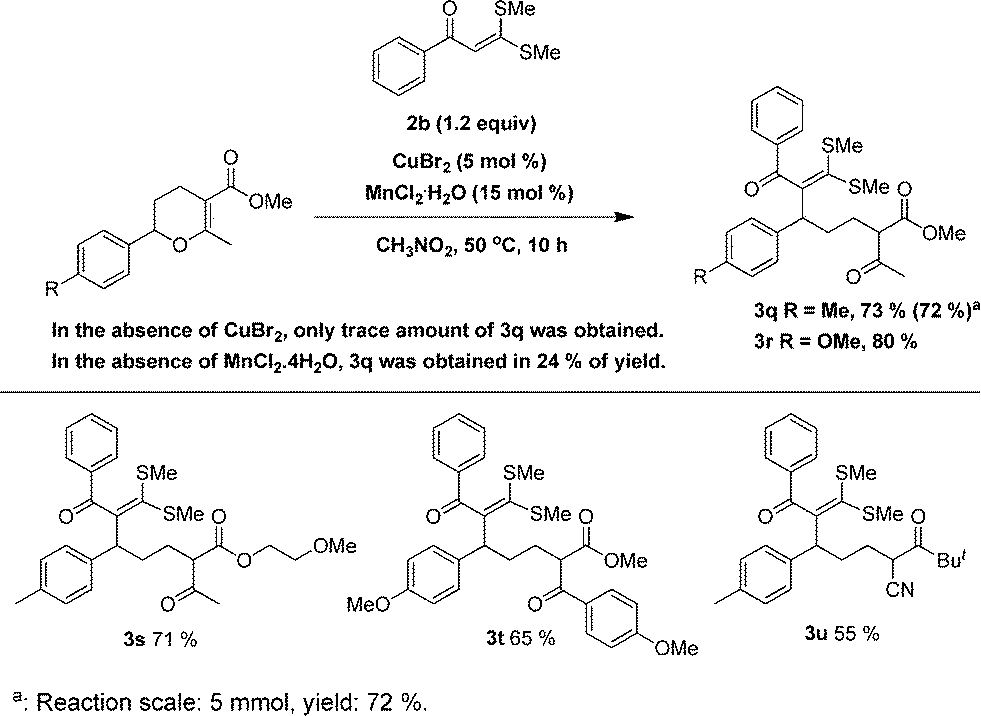 | ||
| Fig. 2 Ring-opening reaction of 2-aryl-3,4-dihydropyrans with 2b-like α-oxoketene dithioacetals. | ||
In order to shed light on the mechanism, 2b was treated in the presence of CuBr2 in nitromethane-d3. As shown in Fig. 3, after 30 minutes of stirring at 50 °C, the α-hydrogen of 2b (the single peak at 6.79 ppm) nearly disappeared (2). Accordingly, in the high-resolution mass spectrum of the reaction solution, a peak at 248.0291 m/z was observed, which can be ascribed to the formation of [M(2b) – H + D + Na]+ (Fig. S2†). These results indicated that hydrogen–deuterium exchange occurred in the presence of CuBr2. In the absence of CuBr2, the α-hydrogen of 2b was not deuterated (Fig. 3, (1)). When CuBr2 was replaced by CuCl2 or NaBr, no hydrogen–deuterium exchange was observed (Fig. 3, (3) and (4)). Fig. 3 also showed that while the peaks of the two SMe groups separated very well (2.54 and 2.58 ppm) in the absence of copper salt,10 they began to coalesce (2.57 ppm) when CuBr2 or CuCl2 was added. We suspect that (i) the oxygen of the ketocarbonyl in 2b may have a deshielding effect on the adjacent SMe group, which gave rise to the discrimination of these two SMe groups in terms of their chemical environment, resulting in the observation of two peaks in the 1H NMR profile, and (ii) the addition of a copper(II) salt decreased the deshielding effect, diminishing to some extent the distinction between the two SMe groups.
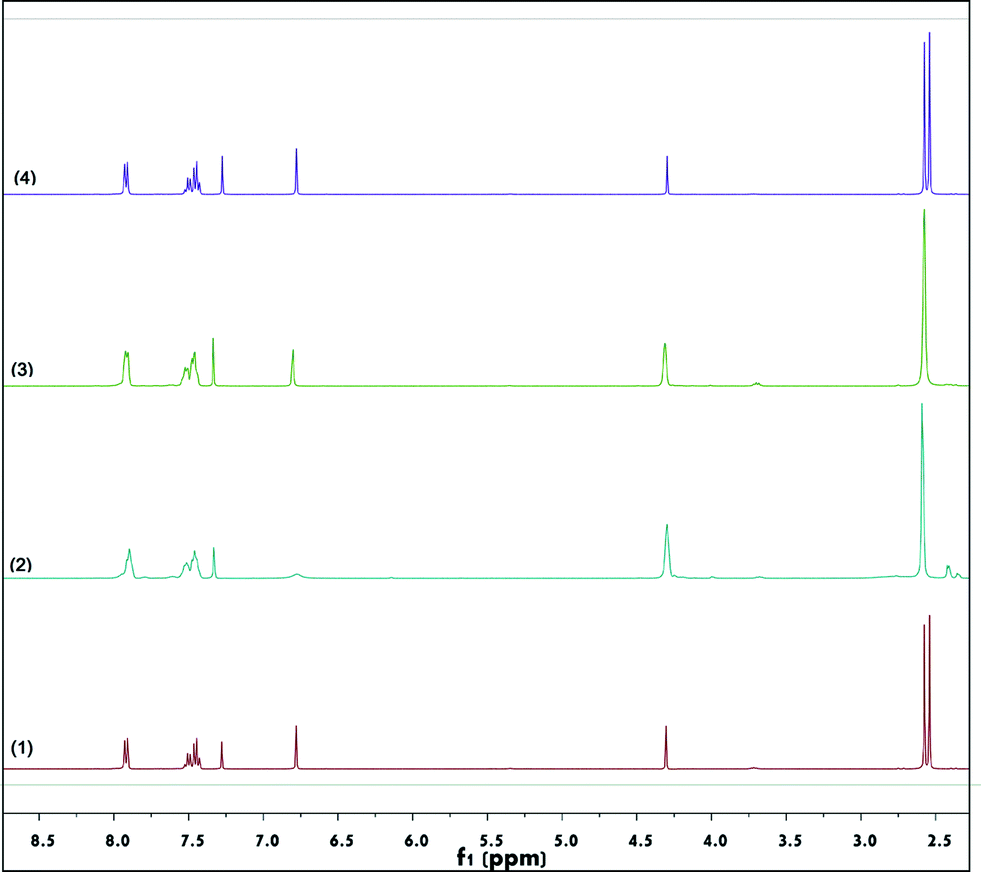 | ||
| Fig. 3 Hydrogen–deuterium exchange reaction of 2b with CD3NO2. (1) Without catalyst, (2) treated with CuBr2, (3) treated with CuCl2, and (4) treated with NaBr. | ||
DFT calculation revealed that in the geometry-optimized structure of 2b, there was a conjugated system that involved not only the double bond and ketocarbonyl group but also the phenyl ring and the two SMe fragments (see Fig. 4, structure (I), and also the ESI). Indeed, in the UV-vis spectrum of 2b, the maximal absorbance was observed at 342 nm, which indicated the existence of a conjugated system in this molecule.11 Intriguingly, when CuBr2 was added, the maximal absorbance decreased significantly (Fig. 5). As the maximal absorbance of CuBr2, which appeared at 268 nm, was also decreased substantially in this case, we suspect that there was a strong interaction between 2b and CuBr2. This can also be verified by characterization with in situ IR technology. In the spectra obtained from a nitromethane solution of 2b and CuBr2, the strength of the characteristic absorption of the carbonyl stretching vibration of 2b decreased gradually upon addition of CuBr2 (see the ESI,† Fig. S3). When CuCl2 was added in the solution of 2b, the UV absorbance at 342 nm was increased, which was ascribed to superposition of these two species. These results implied that the bromide anion in CuBr2 plays a key role in the disintegration of the conjugated system. To support this supposition, TBAB (tetrabutylammonium bromide) was also added to the system of CuCl2 and 2b. As we expected, the maximal absorbance at 342 nm decreased dramatically. However, in the UV spectrum of an acetonitrile solution of CuCl2 and TBAB, the characteristic absorption of CuBr2 was observed as well (see the ESI,† Fig. S4). Therefore, the absorbance decrease of the characteristic peak of 2b observed after adding CuCl2 and TBAB can be linked to the formation of CuBr2.
 | ||
| Fig. 4 Schematic illustration of the structure change of 2b associated with addition of CuBr2. Red = O, yellow = S, dark grey = C, French grey = H. | ||
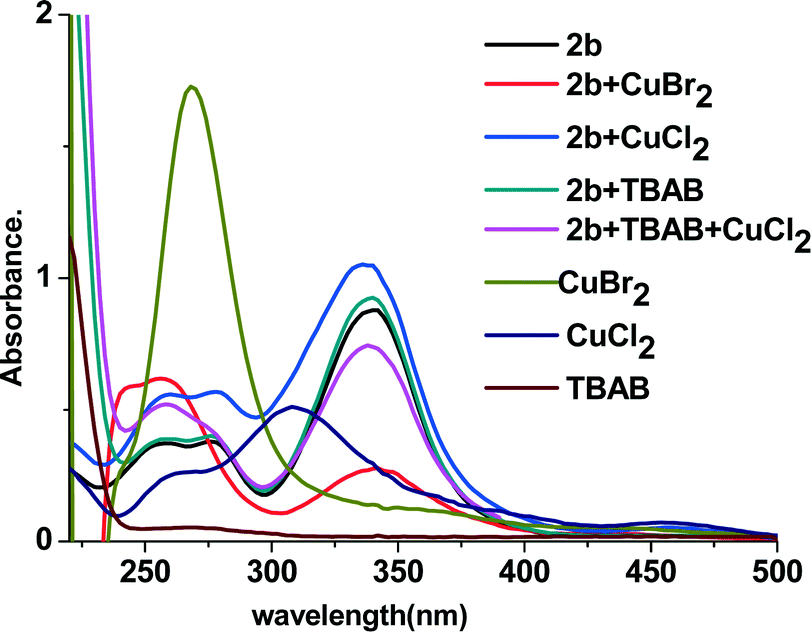 | ||
| Fig. 5 UV-vis spectra of 2b under different conditions. | ||
On the basis of the above-mentioned results, a plausible mechanism was proposed. As shown in Fig. 6, it should be noted that for α-oxoketene dithioacetal 2b, the nucleophilicity was influenced by two factors: (i) the electron-donating effect of the two methylthio groups, which tended to increase the electron density of the α-carbon, which in turn increased the nucleophilicity, and (ii) the conjugation effect of the phenyl ring, the ketocarbonyl group and the double bond, which tended to decrease the electron density, which in turn decreased the nucleophilicity. In the reaction system, the copper(II) salt interacted with α-oxoketene dithioacetal 2b through coordination,12 generating a carbenium ylide intermediate (III). This interaction has the following two effects: (i) it weakened the deshielding effect of the oxygen in the ketocarbonyl group toward the adjacent SMe group, thus allowing the 1H NMR peaks of two SMe groups to get closer, and (ii) it also enables the bromide anion to interact smoothly with a part of 2b, most likely the SMe group,9a somehow resulting in the disintegration of the super-conjugation system in this molecule. As a result, a sulfur-stabilized carbonium ylide was presumably formed. As the α-proton of 2b was adjacent to the two electron-withdrawing groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and MeS+ = CSMe), its acidity was significantly increased.13 Meanwhile, a carbonium intermediate (IV) was generated by means of an activation of the dihydropyran 1a with MnCl2, which can be trapped with the ylide intermediate (III), thus affording the desired product 3q. In view of the fact that with the CuBr2 catalyst alone, 3a and 3q could be obtained in 47% and 24% yields, respectively (Fig. 2); we therefore deduced that CuBr2 can promote, to some extent, the generation of the intermediate (IV), but its efficiency is far less than that of MnCl2·4H2O. In this mechanism, the intermediate (III) seems to be relatively stable, thus ensuring the trapping of the intermediate (IV) in a timely and effective manner. We have also attempted to track the formation of (III) with ESI-MS. However, when CuBr2 was used, owing to a rapid disproportionation of CuBr2 under the operational conditions in this analysis,14 we failed to obtain the expected evidence. Fortunately, when Cu(OTf)2 was used, which is relatively stable, we were able to observe an evident formation of (III) (see the ESI,† Fig. S5). The role of the bromide anion in this reaction is apparently very important. It is most likely involved in the charge delocalization of sulfur-stabilized ylide.9,15 This point deserves further investigation.
O and MeS+ = CSMe), its acidity was significantly increased.13 Meanwhile, a carbonium intermediate (IV) was generated by means of an activation of the dihydropyran 1a with MnCl2, which can be trapped with the ylide intermediate (III), thus affording the desired product 3q. In view of the fact that with the CuBr2 catalyst alone, 3a and 3q could be obtained in 47% and 24% yields, respectively (Fig. 2); we therefore deduced that CuBr2 can promote, to some extent, the generation of the intermediate (IV), but its efficiency is far less than that of MnCl2·4H2O. In this mechanism, the intermediate (III) seems to be relatively stable, thus ensuring the trapping of the intermediate (IV) in a timely and effective manner. We have also attempted to track the formation of (III) with ESI-MS. However, when CuBr2 was used, owing to a rapid disproportionation of CuBr2 under the operational conditions in this analysis,14 we failed to obtain the expected evidence. Fortunately, when Cu(OTf)2 was used, which is relatively stable, we were able to observe an evident formation of (III) (see the ESI,† Fig. S5). The role of the bromide anion in this reaction is apparently very important. It is most likely involved in the charge delocalization of sulfur-stabilized ylide.9,15 This point deserves further investigation.
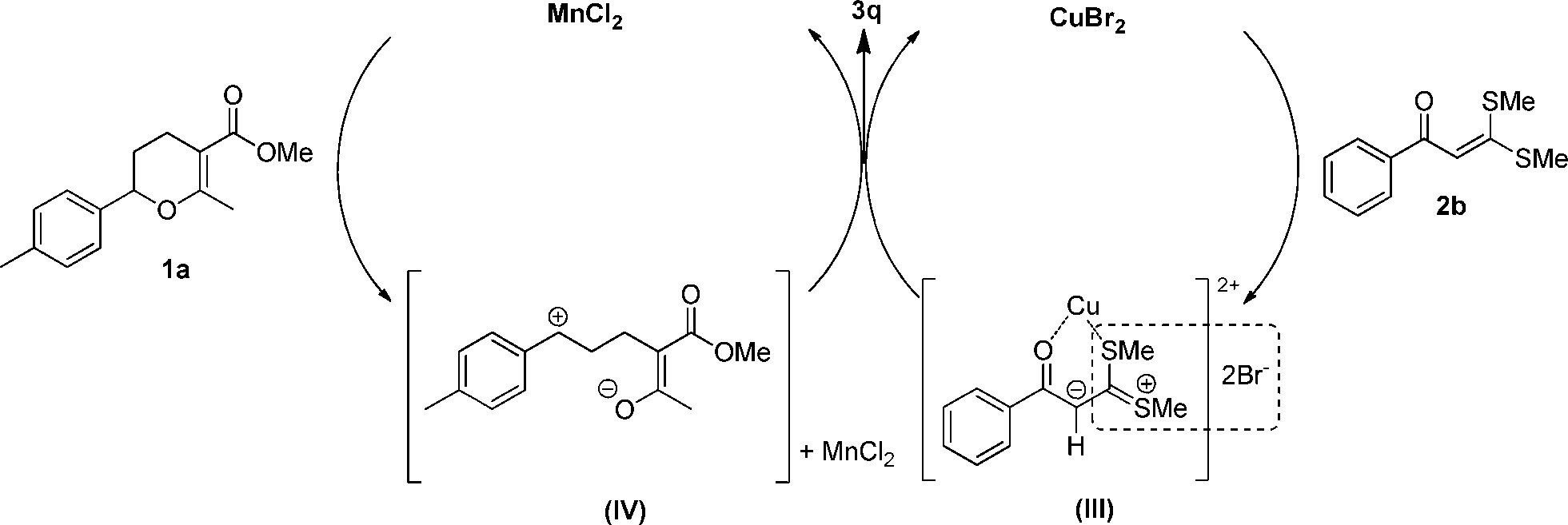 | ||
| Fig. 6 Proposed mechanism. | ||
2-Butoxy-3,4-dihydropyran 4a can also react with α-oxoketene dithioacetal 2a with the aid of the CuBr2/MnCl2 system. Interestingly, this reaction produced a hitherto unreported densely substituted buta-1,3-diene 5a in 92% yield with exclusive selectivity for the trans isomer. The synergistic effect between CuBr2 and MnCl2 catalysts is evident as the yields obtained over each metal chloride salt alone are rather low (Fig. 7). The reaction might proceed through in the same electrophilic ring-opening pathway. Due perhaps to the steric hindrance of the intermediate, 2a cannot trap the reaction intermediate, and as a result, formation of the proton elimination product 5a is thermodynamically more favorable. Many other 2-butoxy-3,4-dihydropyrans and α-oxoketene dithioacetals could be used in this reaction, producing the expected buta-1,3-dienes in good to excellent yields (Fig. 7). In all the cases, only trans products were obtained. It should be noted that the reaction in Fig. 7 showed the first reaction sequence of an acetal-containing 3,4-dihydropyran involving (i) electrophilic ring-opening alkylation and (ii) double bond formation through a proton elimination reaction pathway.
 | ||
| Fig. 7 Ring-opening reactions of 2-butoxy-3,4-dihydropyrans with α-oxoketene dithioacetals. | ||
Because the formed product contains not only a fragment of 1, 3-dicarbonyl compound but also the skeleton of a ketene dithioacetal, it would not be unreasonable to expect that these products could be useful for organic synthesis. Interestingly, we found that in the presence of the CuBr2 catalyst, one of the obtained ring-opening products, 3d, can be easily converted into a densely substituted cyclohexane 6a (Scheme 1). This type of intramolecular Michael reaction of α-oxoketene dithioacetal has not been reported yet.16 An attempt to synthesize 6a directly from the dihydropyran and α-oxo ketene dithioacetal 2a without isolating 3d was not successful. It is worth noting that the produced 6a-like compounds in this suitable precursor for the synthesis of 3-aryl-cyclohexanones are valuable intermediates for organic synthesis.17 In addition, an eleven-membered S,N-containing heterocyclic compound 9a has also been synthesized successfully by using 3q as the starting material through a two-step method (Scheme 2). The first step was isomerization of 3q in the presence of I2 and NaHCO3, which afforded 7a in an almost quantitative yield. The second step was condensation of 7a with o-aminothiophenol with the aid of Ni(ClO4)2·6H2O, which produced 9a in 95% yield. It should be noted that such an S- and N-doped cyclic molecule has not been reported. Because many S- and N-containing heterocyclic compounds have been used in organic synthesis, some of them showing interesting biological activities,18 it is not unreasonable to expect that 9a-type molecules might be valuable for both synthetic and medicinal chemistry. All of these examples demonstrated that the title ring-opening reaction is indeed useful for organic synthesis.
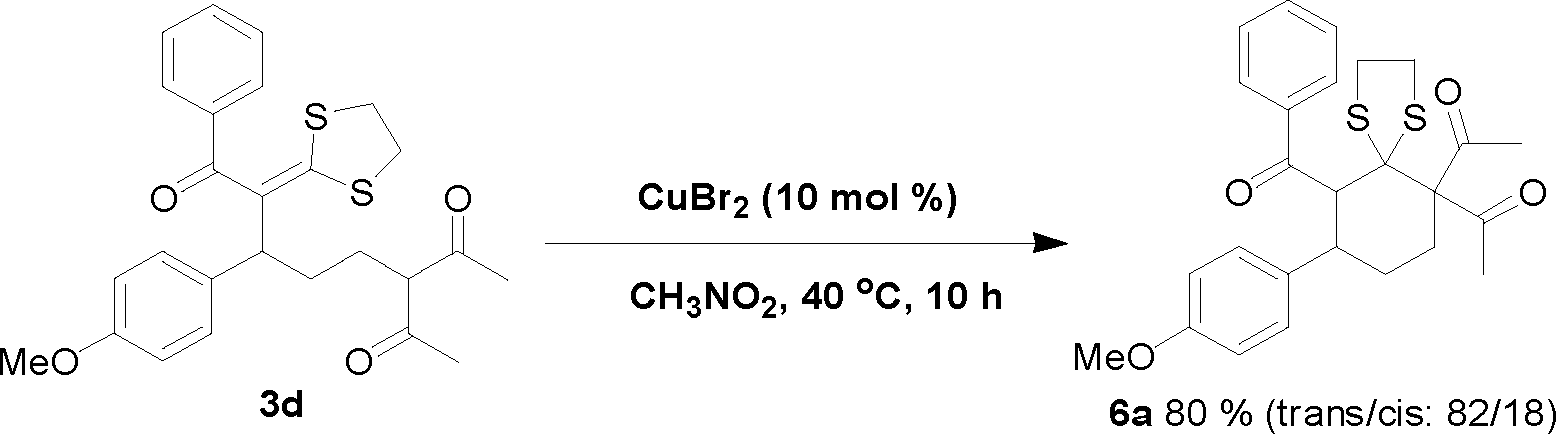 | ||
| Scheme 1 Intramolecular Michael addition of 3d. | ||
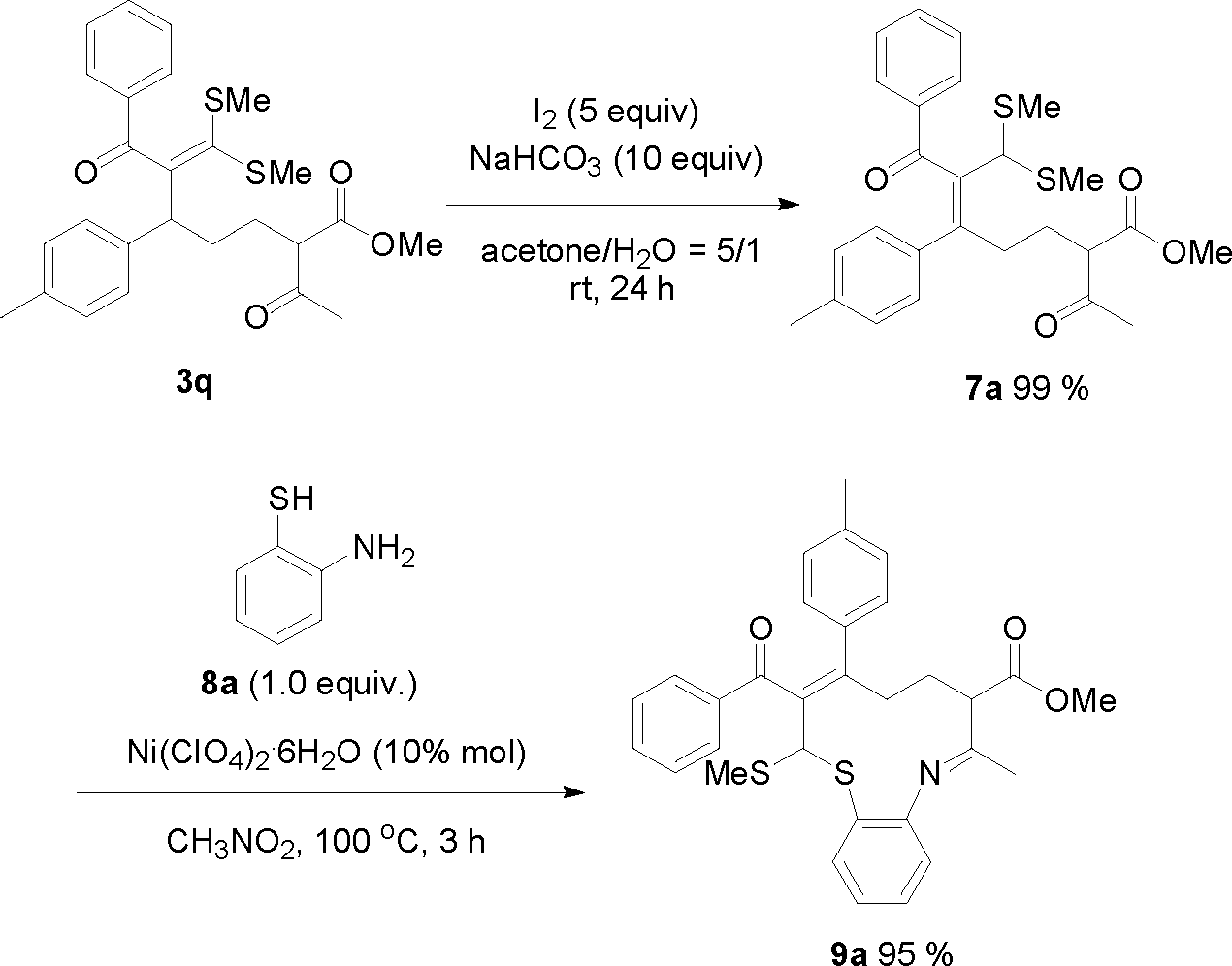 | ||
| Scheme 2 Synthesis of 9a from 3q. | ||
Conclusion
In conclusion, with the aid of a unique synergistic effect between CuBr2 and MnCl2·4H2O catalysts, we have successfully accomplished the electrophilic ring-opening reaction of 2-substituted 3,4-dihydropyrans with α-oxoketene dithioacetals. An interesting CuBr2-induced disintegration of a conjugated system in α-oxoketene dithioacetal was described, which can be linked to the activation of this nucleophile. This example not only demonstrated the great usefulness of the concept of synergistic catalysis in overcoming the difficulties of organic synthesis but also enabled us to access a specific class of molecules that are valuable for organic synthesis. When 2-aryl-3,4-dihydropyrans were used, a Friedel–Crafts ring-opening product was generated with the aid of MnCl2·4H2O/CuBr2. Employing 2-alkoxy-3,4-dihydropyrans as substrates in this system offered an efficient method to access some densely substituted buta-1,3-dienes. Finally, the generated products were successfully used in two synthetic reactions, which clearly manifested the great usefulness of the model ring-opening reaction.Experimental section
General information
Melting points were determined by using a microscopic melting point meter and were uncorrected. IR spectra were recorded with a FT-IR Bruker (EQUINOX 55) spectrometer using KBr pellets or neat liquid. 1H and 13C NMR spectra were recorded on a Bruker AV-400. Chemical shifts were expressed in parts per million relative to Me4Si in solvent. All chemicals used were of reagent grade and were used as received without further purification. 2-Substituted 3,4-dihydropyrans were synthesized according to the literature.19 α-Oxoketene dithioacetals were prepared according to previously reported methods.20 All reactions were conducted in a 10 mL V-type flask equipped with a triangular magnetic stirring bar.Spectroscopic data of new products
Methyl 2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-oxo-7-phenyl-5-(p-tolyl)heptanoate (3a) (0.17 mmol, 78 mg, 83%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.54 (d, J = 8.0 Hz, 2H), 7.44 (t, J = 7.4 Hz, 1H), 7.31 (t, J = 7.5 Hz, 2H), 7.18 (d, J = 6.7 Hz, 2H), 7.02 (d, J = 7.7 Hz, 2H), 3.99 (t, J = 7.6 Hz, 1H), 3.69 (d, J = 9.6 Hz, 3H), 3.47 (dd, J = 15.0, 7.5 Hz, 1H), 3.32 (t, J = 3.9 Hz, 2H), 3.21 (t, J = 5.9 Hz, 2H), 2.26 (s, 3H), 2.18 (s, 3H), 2.15–2.06 (m, 1H), 2.03–1.95 (m, 1H), 1.98–1.86 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.1, 195.9, 170.2, 138.5, 138.4, 138.3, 136.0, 132.2, 129.2, 129.0, 128.3, 128.1, 59.7, 59.5, 52.3, 52.1, 39.0, 37.1, 30.8, 26.8, 21.0 ppm. IR(KBr) v: 2959, 2865, 1725, 1710, 1640, 1575, 1449, 1149, 1022, 732 cm−1. HRMS-ESI (m/z) calcd for C26H28NaO4S2, [M + Na]+ 491.1327, found 491.1328.Ethyl 2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-oxo-7-phenyl-5-(p-tolyl)heptanoate (3b) (0.16 mmol, 76 mg, 79%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.54 (dd, J = 7.7, 2.9 Hz, 2H), 7.44 (t, J = 7.2 Hz, 1H), 7.31 (t, J = 7.1 Hz, 2H), 7.19 (dd, J = 7.9, 3.7 Hz, 2H), 7.02 (d, J = 6.7 Hz, 2H), 4.21–4.08 (m, 2H), 4.00 (t, J = 7.2 Hz, 1H), 3.44 (q, J = 7.1 Hz, 1H), 3.34–3.30 (m, 2H), 3.21 (t, J = 5.6 Hz, 2H), 2.26 (s, 3H), 2.18 (s, 3H), 2.12–1.98 (m, 2H), 1.99–1.81 (m, 2H), 1.28–1.17 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.3, 195.8, 169.7, 149.5, 138.4, 136.0, 132.1, 129.2, 129.0, 128.3, 128.1, 128.1, 61.3, 59.9, 52.1, 39.0, 37.1, 30.8, 28.7, 26.8, 21.0, 14.1 ppm. IR(KBr) v: 3054, 2924, 1739, 1713, 1645, 1512, 1449, 1273, 1149, 912 cm−1. HRMS-ESI (m/z) calcd for C27H30NaO4S2, [M + Na]+ 505.1483, found 505.1484.
6-Acetyl-2-(1,3-dithiolan-2-ylidene)-1-phenyl-3-(p-tolyl)octane-1,7-dione (3c) (0.14 mmol, 62 mg, 69%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.54 (t, J = 8.7 Hz, 2H), 7.44 (t, J = 7.4 Hz, 1H), 7.31 (t, J = 7.6 Hz, 2H), 7.21–7.14 (m, 2H), 7.03 (t, J = 6.3 Hz, 2H), 3.99 (t, J = 7.4 Hz, 1H), 3.69 (dd, J = 30.7, 13.8 Hz, 1H), 3.34 (dd, J = 10.1, 5.8 Hz, 2H), 3.25–3.18 (m, 2H), 2.38–2.27 (m, 2H), 2.26 (s, 3H), 2.14 (s, 3H), 2.08 (s, 3H), 2.00–1.86 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 204.6, 191.3, 149.0, 138.2, 136.1, 132.3, 129.3, 129.0, 128.4, 128.0, 125.8, 68.8, 52.2, 39.0, 37.1, 31.1, 29.3, 28.9, 22.9, 21.0 ppm. IR (KBr) v: 3045, 2924, 1724, 1699, 1645, 1576, 1511, 1449, 1275, 731 cm−1. HRMS-ESI (m/z) calcd for C26H28NaO3S2, [M + Na]+ 475.1378, found 475.1379.
6-Acetyl-2-(1,3-dithiolan-2-ylidene)-3-(4-methoxyphenyl)-1-phenyloctane-1,7-dione (3d) (0.18 mmol, 86 mg, 92%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.58–7.50 (m, 2H), 7.44 (t, J = 7.3 Hz, 1H), 7.31 (t, J = 7.5 Hz, 2H), 7.20 (d, J = 8.5 Hz, 2H), 6.75 (d, J = 8.5 Hz, 2H), 3.97 (t, J = 7.5 Hz, 1H), 3.73 (s, 3H), 3.65 (t, J = 6.4 Hz, 1H), 3.40–3.31 (m, 2H), 3.22 (t, J = 5.6 Hz, 2H), 2.38–2.21 (m, 2H), 2.11 (d, J = 22.0 Hz, 6H), 1.99–1.86 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 204.6, 196.1, 191.3, 158.2, 148.5, 138.3, 133.3, 132.3, 129.3, 129.1, 128.6, 128.4, 113.7, 68.8, 55.2, 51.8, 39.0, 37.1, 31.2, 29.3, 26.8, 22.9 ppm. IR (KBr) v: 2998, 2927, 1725, 1700, 1646, 1578, 1513, 1449, 1360, 1248, 1179, 1033, 831 cm−1. HRMS-ESI (m/z) calcd for C26H28NaO4S2, [M + Na]+ 491.1327, found 491.1339.
Methyl-2-acetyl-6-(1,3-dithiolan-2-ylidene)-5-(4-methoxyphenyl)-7-oxo-7-phenylheptanoate (3e) (0.14 mmol, 70 mg, 72%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.53 (d, J = 7.9 Hz, 2H), 7.44 (t, J = 7.3 Hz, 1H), 7.31 (t, J = 7.4 Hz, 2H), 7.21 (d, J = 7.1 Hz, 2H), 6.75 (d, J = 8.0 Hz, 2H), 3.98 (t, J = 7.4 Hz, 1H), 3.73 (s, 3H), 3.70 (d, J = 9.8 Hz, 3H), 3.47 (dd, J = 15.0, 7.5 Hz, 1H), 3.35–3.31 (m, 2H), 3.21 (t, J = 5.8 Hz, 2H), 2.19 (s, 3H), 2.11 (t, J = 10.9 Hz, 1H), 1.99–1.86 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.2, 196.0, 170.2, 158.2, 149.0, 148.8, 138.4, 133.4, 132.2, 129.2, 128.3, 127.8, 113.6, 59.7, 55.2, 52.4, 51.7, 39.0, 37.1, 30.9, 28.7, 26.8 ppm. IR (KBr) v: 2952, 2930, 1741, 1711, 1612, 1512, 1448, 1357, 1249, 1180, 1034, 831, 733 cm−1. HRMS-ESI (m/z) calcd for C26H28NaO5S2, [M + Na]+ 507.1276, found 507.1270.
2-Methoxyethyl-2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-oxo-7-phenyl-5-(p-tolyl)heptanoate (3f) (0.16 mmol, 81 mg, 79%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.54 (d, J = 7.9 Hz, 2H), 7.43 (t, J = 7.4 Hz, 1H), 7.31 (t, J = 7.6 Hz, 2H), 7.18 (d, J = 7.2 Hz, 2H), 7.02 (d, J = 7.5 Hz, 2H), 4.31–4.22 (m, 2H), 4.00 (t, J = 7.5 Hz, 1H), 3.59–3.51 (m, 2H), 3.49 (dd, J = 15.3, 7.6 Hz, 1H), 3.33 (s, 3H), 3.33–3.30 (m, 2H), 3.20 (t, J = 5.6 Hz, 2H), 2.25 (s, 3H), 2.19 (d, J = 1.3 Hz, 3H), 2.15–1.98 (m, 2H), 2.00–1.83 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.1, 195.8, 169.7, 149.4, 138.3, 136.0, 132.1, 129.2, 129.2, 129.0, 128.3, 128.1, 70.2, 64.1, 59.5, 58.9, 52.1, 39.0, 37.1, 30.9, 28.7, 26.8, 21.0 ppm. IR (KBr) v: 3053, 2925, 1740, 1714, 1645, 1513, 1449, 1274, 1029, 912, 732 cm−1. HRMS-ESI (m/z) calcd for C28H32NaO5S2, [M + Na]+ 535.1589, found 535.1379.
Methyl-2-acetyl-5-(4-(tert-butyl)phenyl)-6-(1,3-dithiolan-2-ylidene)-7-oxo-7-phenylheptanoate (3g) (0.15 mmol, 78 mg, 76%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.46 (t, J = 8.0 Hz, 2H), 7.40 (t, J = 7.4 Hz, 1H), 7.26 (t, J = 7.5 Hz, 2H), 7.23–7.13 (m, 4H), 4.02 (t, J = 7.3 Hz, 1H), 3.71 (d, J = 2.6 Hz, 3H), 3.50 (dt, J = 10.8, 7.1 Hz, 1H), 3.39–3.33 (m, 2H), 3.23 (t, J = 5.8 Hz, 2H), 2.20 (d, J = 1.7 Hz, 3H), 2.17–2.09 (m, 1H), 2.03–1.89 (m, 3H), 1.24 ppm (s, 9H). 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.2, 196.1, 170.2, 149.4, 138.2, 132.1, 132.0, 129.2, 129.1, 128.2, 127.6, 127.6, 125.2, 59.8, 59.5, 52.4, 38.9, 37.1, 34.3, 31.3, 30.8, 28.8, 26.8 ppm. IR(KBr) v: 3025, 2957, 1742, 1716, 1647, 1451, 1273, 1151, 729 cm−1. HRMS-ESI (m/z) calcd for C29H34NaO4S2, [M + Na]+ 533.1796, found 533.1787.
Ethyl-6-(1,3-dithiolan-2-ylidene)-2-(4-methoxybenzoyl)-5-(4-methoxyphenyl)-7-oxo-7-phenylheptanoate (3h) (0.15 mmol, 90 mg, 76%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.91 (dd, J = 21.8, 8.9 Hz, 2H), 7.52 (dd, J = 6.9, 5.0 Hz, 2H), 7.42 (dd, J = 13.6, 6.3 Hz, 1H), 7.34–7.25 (m, 2H), 7.23 (d, J = 7.4 Hz, 2H), 6.89 (dd, J = 16.6, 8.9 Hz, 2H), 6.74 (dd, J = 8.6, 4.8 Hz, 2H), 4.33–4.23 (m, 1H), 4.16–4.06 (m, 2H), 4.01 (t, J = 7.4 Hz, 1H), 3.85 (d, J = 3.7 Hz, 3H), 3.73 (d, J = 4.7 Hz, 3H), 3.30 (dd, J = 7.2, 4.3 Hz, 2H), 3.19 (dd, J = 12.0, 5.8 Hz, 2H), 2.23–1.92 (m, 4H), 1.20–1.10 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 196.0, 193.8, 170.0, 163.8, 158.1, 149.3, 138.5, 133.7, 132.1, 131.0, 129.3, 129.3, 129.2, 129.1, 128.9, 128.8, 128.8, 128.3, 113.9, 113.8, 113.6, 113.6, 61.2, 55.5, 55.1, 54.0, 51.6, 39.0, 37.1, 31.2, 27.7, 14.0 ppm. IR (KBr) v: 3059, 2958, 2930, 1733, 1675, 1644, 1600, 1511, 1458, 1249, 1176, 1031, 911, 732 cm−1. HRMS-ESI (m/z) calcd for C33H34NaO6S2, [M + Na]+ 613.1694, found 613.1708.
Methyl-2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-oxo-5,7-di-p-tolylheptanoate (3i) (0.20 mmol, 95 mg, 98%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.47 (dd, J = 8.2, 2.1 Hz, 2H), 7.19 (dd, J = 8.0, 1.8 Hz, 2H), 7.12 (d, J = 7.7 Hz, 2H), 7.02 (d, J = 7.7 Hz, 2H), 3.96 (t, J = 7.6 Hz, 1H), 3.69 (d, J = 8.7 Hz, 3H), 3.47 (dd, J = 16.0, 7.3 Hz, 1H), 3.32 (t, J = 5.1 Hz, 2H), 3.20 (t, J = 5.8 Hz, 2H), 2.35 (s, 3H), 2.25 (s, 3H), 2.17 (d, J = 9.7 Hz, 3H), 2.15–2.07 (m, 1H), 2.02–1.85 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.3, 195.7, 170.2, 147.2, 143.2, 138.4, 136.0, 135.6, 129.6, 129.1, 129.0, 128.8, 128.7, 128.1, 128.1, 59.5, 52.3, 39.0, 37.1, 31.0, 29.1, 28.7, 26.8, 21.7, 21.0 ppm. IR (KBr) v: 2998, 2923, 1741, 1714, 1643, 1603, 1512, 1437, 1274, 1150, 911, 733 cm−1. HRMS-ESI (m/z) calcd for C27H30NaO4S2, [M + Na]+ 505.1483, found 505.1467.
Methyl-2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-(4-methoxyphenyl)-7-oxo-5-(p-tolyl)heptanoate (3j) (0.17 mmol, 86 mg, 86%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.56 (dd, J = 8.8, 3.0 Hz, 2H), 7.17 (dd, J = 8.0, 2.3 Hz, 2H), 7.01 (d, J = 7.5 Hz, 2H), 6.79 (dd, J = 8.9, 1.9 Hz, 2H), 3.97–3.90 (m, 1H), 3.81 (s, 3H), 3.69 (d, J = 7.2 Hz, 3H), 3.48 (dd, J = 16.7, 6.7 Hz, 1H), 3.33 (t, J = 5.3 Hz, 2H), 3.19 (t, J = 9.1 Hz, 2H), 2.25 (s, 3H), 2.18 (d, J = 2.0 Hz, 3H), 2.14–2.08 (m, 1H), 2.00–1.88 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.2, 194.9, 170.2, 163.3, 138.3, 136.0, 132.0, 130.6, 129.0, 128.8, 128.0, 113.6, 59.7, 59.4, 55.4, 52.3, 38.9, 37.1, 31.4, 28.7, 26.8, 21.0 ppm. IR (KBr) v: 3003, 2927, 1742, 1714, 1641, 1596, 1509, 1439, 1256, 1150, 1028, 911, 732 cm−1. HRMS-ESI (m/z) calcd for C27H30NaO5S2, [M + Na]+ 521.1432, found 521.1435.
Ethyl-2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-(4-fluorophenyl)-7-oxo-5-(p-tolyl)heptanoate (3k) (0.15 mmol, 74 mg, 74%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.58–7.52 (m, 2H), 7.16 (dd, J = 7.8, 4.3 Hz, 2H), 7.04–6.94 (m, 4H), 4.19–4.08 (m, 2H), 3.97 (t, J = 7.6 Hz, 1H), 3.45 (dd, J = 13.9, 7.0 Hz, 1H), 3.38–3.32 (m, 2H), 3.26–3.20 (m, 2H), 2.25 (s, 3H), 2.19 (d, J = 2.4 Hz, 3H), 2.16–2.08 (m, 1H), 1.99–1.86 (m, 3H), 1.28–1.21 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.1, 194.5, 171.1, 169.7, 167.8, 166.6, 138.2, 138.1, 136.1, 132.0, 131.9, 129.1, 128.0, 127.9, 115.5, 115.3, 61.3, 60.4, 59.6, 52.5, 38.9, 37.1, 31.0, 26.7, 21.0, 14.2 ppm. 19F NMR (377 MHz, CDCl3, 25 °C) δ −106.1, −106.1 ppm. IR (KBr) v: 2928, 2926, 1738, 1711, 1647, 1595, 1506, 1274, 1511, 1020, 847, 733, 599 cm−1. HRMS-ESI (m/z) calcd for C27H29FNaO4S2, [M + Na]+ 523.1389, found 523.1382.
Ethyl-2-acetyl-7-(4-chlorophenyl)-6-(1,3-dithiolan-2-ylidene)-7-oxo-5-(p-tolyl)heptanoate (3l) (0.15 mmol, 75 mg, 73%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.46 (dd, J = 8.4, 4.8 Hz, 2H), 7.27 (dd, J = 7.7, 2.7 Hz, 2H), 7.15 (dd, J = 7.9, 4.1 Hz, 2H), 7.02 (d, J = 6.4 Hz, 2H), 4.21–4.10 (m, 2H), 3.97 (t, J = 7.6 Hz, 1H), 3.45 (dd, J = 13.5, 7.1 Hz, 1H), 3.36–3.33 (m, 2H), 3.25–3.21 (m, 2H), 2.26 (s, 3H), 2.20 (d, J = 2.2 Hz, 3H), 2.15–2.06 (m, 1H), 2.03–1.86 (m, 3H), 1.26–1.22 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.2, 194.6, 169.7, 149.0, 138.6, 138.6, 138.2, 138.1, 136.7, 136.7, 136.2, 130.6, 129.1, 128.6, 128.0, 127.9, 61.3, 59.6, 58.5, 52.3, 38.9, 37.1, 26.7, 21.0, 18.4, 14.1 ppm. IR (KBr) v: 2954, 2924, 1741, 1714, 1646, 1579, 1494, 1227, 1151, 1005, 811, 732 cm−1. HRMS-ESI (m/z) calcd for C27H29ClNaO4S2, [M + Na]+ 539.1093, found 539.1089.
Ethyl-2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-(4-iodophenyl)-7-oxo-5-(p-tolyl)heptanoate (3m) (0.15 mmol, 92 mg, 76%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C) δ 7.66 (dd, J = 8.5, 1.9 Hz, 2H), 7.23 (dd, J = 8.4, 5.2 Hz, 2H), 7.15 (dd, J = 7.9, 4.3 Hz, 2H), 7.02 (d, J = 6.5 Hz, 2H), 4.19–4.10 (m, 2H), 3.97 (t, J = 7.5 Hz, 1H), 3.44 (dd, J = 13.1, 7.1 Hz, 1H), 3.36–3.32 (m, 2H), 3.25–3.22 (m, 2H), 2.26 (s, 3H), 2.19 (d, J = 2.0 Hz, 3H), 2.15–2.07 (m, 1H), 2.02–1.84 (m, 3H), 1.27–1.24 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.2, 195.1, 169.7, 149.6, 138.1, 138.1, 137.8, 137.7, 137.6, 136.2, 130.6, 129.1, 128.0, 127.9, 61.4, 59.6, 58.4, 52.2, 38.9, 37.1, 28.7, 21.0, 18.4, 14.1 ppm. IR (KBr) v: 2956, 2922, 1737, 1714, 1647, 1585, 1511, 1275, 1089, 1015, 819, 546 cm−1. HRMS-ESI (m/z) calcd for C27H29INaO4S2, [M + Na]+ 631.0450, found 631.0467.
Methyl-4-(2-(1,3-dithiolan-2-ylidene)-6-(methoxycarbonyl)-7-oxo-3-(p-tolyl)octanoyl)benzoate (3n) (0.15 mmol, 77 mg, 73%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.96 (d, J = 8.1 Hz, 2H), 7.52 (d, J = 8.2 Hz, 2H), 7.15 (d, J = 7.9 Hz, 2H), 7.02 (d, J = 7.8 Hz, 2H), 4.01 (t, J = 7.6 Hz, 1H), 3.91 (s, 3H), 3.71 (d, J = 7.7 Hz, 3H), 3.47 (q, J = 7.3 Hz, 1H), 3.37–3.31 (m, 2H), 3.23 (t, J = 6.0 Hz, 2H), 2.26 (s, 3H), 2.20 (s, 3H), 2.15–1.99 (m, 2H), 1.97–1.85 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.0, 194.9, 170.1, 166.4, 152.7, 152.3, 142.6, 142.5, 138.0, 136.2, 132.7, 129.5, 129.1, 128.7, 128.1, 127.9, 59.7, 59.5, 52.3, 51.8, 39.0, 37.1, 30.5, 29.1, 26.8, 21.0 ppm. IR (KBr) v: 2953, 2926, 1722, 1648, 1512, 1437, 1278, 1111, 1017, 913, 734 cm−1. HRMS-ESI (m/z) calcd for C28H30NaO6S2, [M + Na]+ 549.1381, found 549.1383.
Methyl-2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-(4-nitrophenyl)-7-oxo-5-(p-tolyl)heptanoate (3o) (0.16 mmol, 84 mg, 82%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C) δ 8.12 (d, J = 8.8 Hz, 2H), 7.54 (d, J = 7.7 Hz, 2H), 7.11 (d, J = 8.0 Hz, 2H), 7.02 (d, J = 7.9 Hz, 2H), 4.03 (t, J = 7.6 Hz, 1H), 3.72 (d, J = 4.6 Hz, 3H), 3.48 (dd, J = 13.6, 6.9 Hz, 1H), 3.41–3.35 (m, 2H), 3.28 (t, J = 6.0 Hz, 2H), 2.27 (s, 3H), 2.21 (d, J = 1.6 Hz, 3H), 2.13–2.00 (m, 2H), 1.98–1.86 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 202.8, 193.6, 170.0, 155.1, 149.3, 144.5, 137.8, 136.4, 129.5, 129.4, 129.2, 127.7, 123.5, 59.4, 52.5, 51.6, 39.0, 37.1, 30.4, 28.9, 26.8, 21.0 ppm. IR (KBr) v: 2953, 2925, 1741, 1715, 1647, 1600, 1523, 1438, 1348, 1273, 1149, 912, 853, 733 cm−1. HRMS-ESI (m/z) calcd for C26H27NNaO6S2, [M + Na]+ 536.1177, found 536.1180.
Ethyl-2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-(4-nitrophenyl)-7-oxo-5-(p-tolyl)heptanoate (3p) (0.15 mmol, 81 mg, 77%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 8.12 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 8.0 Hz, 2H), 7.11 (d, J = 7.7 Hz, 2H), 7.02 (d, J = 7.3 Hz, 2H), 4.21–4.16 (m, 2H), 4.04 (t, J–7.6 Hz, 1H), 3.46 (dd, J = 12.7, 7.2 Hz, 1H), 3.40–3.36 (m, 2H), 3.28 (t, J = 6.0 Hz, 2H), 2.27 (s, 3H), 2.21 (d, J = 1.0 Hz, 3H), 2.16–2.00 (m, 2H), 1.98–1.85 (m, 2H), 1.28–1.23 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.0, 193.5, 169.6, 155.2, 154.8, 149.3, 144.4, 137.1, 136.4, 129.1, 127.8, 123.5, 61.4, 59.8, 51.6, 39.0, 37.1, 30.5, 28.8, 26.7, 21.0, 14.1 ppm. IR (KBr) v: 2957, 2927, 1737, 1714, 1648, 1600, 1523, 1449, 1348, 1274, 1150, 1018, 855, 734 cm−1. HRMS-ESI (m/z) calcd for C27H29NNaO6S2, [M + Na]+ 550.1334, found 550.1336.
Methyl-2-acetyl-6-benzoyl-7,7-bis(methylthio)-5-(p-tolyl)hept-6-enoate (3q) (0.15 mmol, 69 mg, 73%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.44–7.39 (m, 2H), 7.36 (t, J = 6.8 Hz, 1H), 7.19 (t, J = 7.6 Hz, 2H), 6.99 (dd, J = 8.1, 2.1 Hz, 2H), 6.90 (d, J = 7.6 Hz, 2H), 4.63 (dt, J = 11.5, 7.3 Hz, 1H), 3.71 (d, J = 2.4 Hz, 3H), 3.51 (dd, J = 12.1, 6.6 Hz, 1H), 2.38 (d, J = 2.5 Hz, 3H), 2.22 (d, J = 12.5 Hz, 3H), 2.18 (s, 3H), 2.03–1.96 (m, 1H), 1.95 (s, 3H), 1.93–1.82 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.2, 196.0, 170.2, 149.0, 137.6, 137.0, 136.3, 134.3, 132.3, 129.0, 128.8, 128.2, 128.2, 127.8, 59.6, 59.3, 52.4, 49.2, 30.8, 28.7, 26.4, 20.9, 16.8 ppm. IR (KBr) v: HRMS-ESI (m/z) calcd for C26H30KO4S2, [M + K]+ 509.1223, found 509.1230.
Methyl-2-acetyl-6-benzoyl-5-(4-methoxyphenyl)-7,7-bis(methylthio)hept-6-enoate (3r) (0.16 mmol, 78 mg, 80%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ = 7.45–7.41 (m, 2H), 7.35 (t, J = 7.3 Hz, 1H), 7.21 (t, J = 6.6 Hz, 2H), 7.03 (dd, J = 8.7, 2.3 Hz, 2H), 6.63 (d, J = 7.3 Hz, 2H), 4.66–4.57 (m, 1H), 3.71 (s, 3H), 3.67 (s, 3H), 3.51 (dd, J = 12.7, 6.5 Hz, 1H), 2.38 (d, J = 2.6 Hz, 3H), 2.22 (d, J = 8.8 Hz 3H), 2.03–1.97 (m, 1H), 1.96 (s, 3H), 1.92–1.80 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.2, 196.1, 170.2, 158.3, 149.2, 137.5, 134.1, 132.4, 132.1, 129.3, 128.8, 127.9, 113.8, 59.6, 59.3, 55.2, 52.4, 48.8, 28.7, 26.4, 16.8, 16.4 ppm. IR (KBr) v: 2954, 2923, 1741, 1715, 1656, 1610, 1511, 1448, 1359, 1249, 1179, 1034, 910, 732 cm−1. HRMS-ESI (m/z) calcd for C26H30NaO5S2, [M + Na]+ 509.1432, found 509.1432.
2-Methoxyethyl-2-acetyl-6-benzoyl-7,7-bis(methylthio)-5-(p-tolyl)hept-6-enoate (3s) (0.14 mmol, 73 mg, 71%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.46–7.39 (m, 2H), 7.35 (t, J = 7.3 Hz, 1H), 7.23–7.15 (m, 2H), 7.00 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 6.9 Hz, 2H), 4.63 (dd, J = 17.3, 7.4 Hz, 1H), 4.31–4.23 (m, 2H), 3.56–3.52 (m, 3H), 3.33 (d, J = 6.4 Hz, 3H), 2.38 (d, J = 2.9 Hz, 3H), 2.22 (d, J = 11.3 Hz, 3H), 2.17 (s, 3H), 2.01–1.96 (m, 1H), 1.95 (d, J = 1.4 Hz, 3H), 1.90–1.84 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 203.1, 195.7, 169.9, 149.2, 137.6, 137.0, 136.3, 134.3, 132.3, 129.0, 128.8, 128.2, 127.8, 70.2, 64.1, 59.7, 58.9, 49.3, 30.9, 28.5, 26.2, 20.7, 16.8, 16.4 ppm. IR (KBr) v: 2921, 1740, 1713, 1656, 1512, 1448, 1356, 1243, 1128, 1029, 707, 550 cm−1.HRMS-ESI (m/z) calcd for C28H34NaO5S2, [M + Na]+ 537.1745, found 537.1748.
Ethyl-6-benzoyl-2-(4-methoxybenzoyl)-5-(4-methoxyphenyl)-7,7-bis(methylthio)hept-6-enoate (3t) (0.13 mmol, 77 mg, 65%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C) δ 7.94 (dd, J = 11.8, 8.9 Hz, 2H), 7.44 (d, J = 7.8 Hz, 2H), 7.34 (t, J = 6.9 Hz, 1H), 7.20 (t, J = 7.0 Hz, 2H), 7.07–7.02 (m, 2H), 6.91 (dd, J = 8.8, 4.3 Hz, 2H), 6.62 (dd, J = 8.4, 6.6 Hz, 2H), 4.69–4.61 (m, 1H), 4.36–4.28 (m, 1H), 4.11 (dd, J = 12.1, 7.0 Hz, 2H), 3.85 (s, 3H), 3.65 (d, J = 5.9 Hz, 3H), 2.33 (d, J = 3.3 Hz, 3H), 2.11–2.01 (m, 4H), 1.94 (d, J = 5.4 Hz, 3H), 1.29–1.19 ppm (m, 3H). 13C NMR (100 MHz, CDCl3, 25 °C): δ 196.1, 193.8, 170.3, 163.7, 158.6, 149.2, 137.6, 133.9, 132.3, 131.1, 129.4, 128.8, 127.9, 113.6, 60.9, 55.5, 55.2, 54.3, 48.5, 31.2, 27.3, 16.8, 16.4, 14.1 ppm. IR (KBr) v: 2957, 2922, 1732, 1674, 1660, 1599, 1510, 1458, 1250, 1174, 1029, 840, 706 cm−1. HRMS-ESI (m/z) calcd for C33H36NaO6S2, [M + Na]+ 615.1851, found 615.1859.
6-Benzoyl-7,7-bis(methylthio)-2-pivaloyl-5-(p-tolyl)hept-6-enenitrile (3u) (0.11 mmol, 53 mg, 55%): yellowish oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.41 (d, J = 7.8 Hz, 2H), 7.36 (t, J = 7.4 Hz, 1H), 7.20 (t, J = 7.7 Hz, 2H), 6.99 (t, J = 6.9 Hz, 2H), 6.91 (d, J = 7.6 Hz, 2H), 4.69–4.56 (m, 1H), 3.95–3.83 (m, 1H), 2.39 (d, J = 6.5 Hz, 3H), 2.19 (s, 3H), 2.13–1.97 (m, 2H), 1.95 (s, 3H), 1.92–1.81 (m, 2H), 1.21 (d, J = 1.5 Hz, 9H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 205.8, 196.0, 148.2, 137.5, 136.7, 135.0, 132.4, 129.2, 128.9, 128.1, 127.9, 117.4, 58.5, 48.7, 45.5, 37.1, 29.7, 26.1, 20.9, 16.8, 16.5 ppm. IR (KBr) v: 2961, 2923, 2243, 1720, 1656, 1596, 1476, 1451, 1265, 1236, 911, 733 cm−1. HRMS-ESI (m/z) calcd for C28H33NNaO2S2, [M + Na]+ 502.1850, found 502.1850.
(E)-Ethyl 2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-oxo-7-phenylhept-4-enoate (5a) (0.18 mmol, 72 mg, 92%): yellowish oil, 1H NMR (600 MHz, CDCl3, 25 °C): δ 7.71–7.65 (m, 2H), 7.47 (d, J = 7.4 Hz, 1H), 7.38 (t, J = 7.6 Hz, 2H), 6.36 (d, J = 15.7 Hz, 1H), 5.47–5.20 (m, 1H), 4.14 (dd, J = 9.5, 7.2 Hz, 2H), 3.48–3.31 (m, 5H), 2.57 (t, J = 7.3 Hz, 2H), 2.16 (d, J = 16.2 Hz, 3H), 1.23 (t, J = 7.1 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 202.2, 193.0, 168.9, 138.3, 131.8, 130.6, 129.7, 129.5, 128.1, 123.9, 61.5, 59.1, 38.5, 36.6, 31.6, 29.3, 14.1 ppm. IR (KBr) v: 2961, 2923, 1736, 1713, 1646, 1597, 1449, 1279, 1174, 1020, 806, 696 cm−1. HRMS-ESI (m/z) calcd for C20H22NaO4S2, [M + Na]+ 413.0857, found 413.0843.
(E)-Ethyl 2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-oxo-7-(p-tolyl)hept-4-enoate (5b) (0.18 mg, 73 mg, 90%): yellowish oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.62 (d, J = 8.0 Hz, 2H), 7.19 (d, J = 7.9 Hz, 2H), 6.36 (dd, J = 15.7, 1.2 Hz, 1H), 5.29 (ddd, J = 14.4, 7.8, 6.5 Hz, 1H), 4.19–4.08 (m, 2H), 3.46–3.31 (m, 5H), 2.58 (t, J = 7.3 Hz, 2H), 2.39 (s, 3H), 2.16 (d, J = 1.0 Hz, 3H), 1.26–1.19 (m, 3H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 202.3, 193.2, 168.9, 142.8, 135.3, 130.8, 129.8, 129.1, 128.8, 61.5, 59.2, 38.5, 36.7, 31.7, 29.4, 21.7, 14.1 ppm. IR (KBr) v: 2959, 2925, 1738, 1716, 1658, 1605, 1456, 1363, 1280, 1176, 1020, 962, 845, 764 cm−1. HRMS-ESI (m/z) calcd for C21H24NaO4S2, [M + Na]+ 427.1014, found 427.0997.
(E)-Ethyl 2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-(4-methoxyphenyl)-7-oxohept-4-enoate (5c) (0.17 mmol, 72 mg, 86%): yellowish oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.74 (d, J = 8.9 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 6.38 (d, J = 15.7 Hz, 1H), 5.41–5.18 (m, 1H), 4.17–4.12 (m, 2H), 3.86 (s, 3H), 3.45–3.33 (m, 5H), 2.60 (t, J = 7.3 Hz, 2H), 2.18 (s, 3H), 1.23 (t, J = 7.2 Hz, 3H) ppm. 13C NMR (150 MHz, CDCl3, 25 °C): δ 202.3, 192.7, 168.9, 163.0, 132.1, 130.7, 128.5, 113.5, 61.5, 59.2, 55.4, 38.4, 36.9, 31.7, 29.3, 14.1 ppm. IR (KBr) v: 2960, 2926, 1735, 1716, 1652, 1598, 1508, 1462, 1257, 1167, 1026, 962, 776, 605 cm−1. HRMS-ESI (m/z) calcd for C21H24NaO5S2, [M + Na]+ 443.0963, found 443.0958.
(E)-Ethyl 2-acetyl-7-(4-chlorophenyl)-6-(1,3-dithiolan-2-ylidene)-7-oxohept-4-enoate (5d) (0.16 mmol, 66 mg, 78%): yellowish oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.62 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 6.34 (d, J = 15.8 Hz, 1H), 5.30 (td, J = 15.4, 7.7 Hz, 1H), 4.18 (ddd, J = 17.8, 14.5, 7.3 Hz, 2H), 3.49–3.30 (m, 5H), 2.58 (t, J = 7.1 Hz, 2H), 2.17 (s, 3H), 1.24 (t, J = 7.1, 3H) ppm. 13C NMR (150 MHz, CDCl3, 25 °C): δ 202.1, 191.5, 168.9, 138.0, 136.7, 131.0, 130.5, 130.2, 129.3, 128.8, 128.4, 123.4, 107.9, 61.5, 59.1, 38.6, 36.6, 31.6, 29.2, 14.1 ppm. IR (KBr) v: 2962, 2926, 1737, 1715, 1617, 1588, 1480, 1280, 1172, 1090, 1011, 965, 849, 792 cm−1. HRMS-ESI (m/z) calcd for C20H21ClNaO4S2, [M + Na]+ 447.0467, found 447.0462.
(E)-Ethyl 2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-(4-iodophenyl)-7-oxohept-4-enoate (5e) (0.13 mmol, 69 mg, 67%): yellowish oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.74 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 6.33 (d, J = 15.8 Hz, 1H), 5.35–5.23 (m, 1H), 4.20–4.12 (m, 2H), 3.40 (s, 5H), 2.58 (t, J = 7.3 Hz, 2H), 2.17 (s, 3H), 1.25 (dd, J = 8.6, 5.7 Hz, 3H) ppm. 13C NMR (150 MHz, CDCl3, 25 °C): δ 202.1, 191.7, 168.9, 137.3, 131.1, 130.5, 130.3, 128.8, 61.6, 59.0, 38.6, 36.6, 31.6, 29.3, 14.1 ppm. IR (KBr) v: 2960, 2925, 1737, 1716, 1580, 1451, 1362, 1279, 1176, 1003, 846, 649 cm−1. HRMS-ESI (m/z) calcd for C20H21INaO4S2, [M + Na]+ 538.9824, found 538.9812.
(E)-Ethyl 2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-(4-fluorophenyl)-7-oxohept-4-enoate (5f) (0.18 mmol, 72 mg, 88%): yellowish oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.82–7.64 (m, 2H), 7.07 (t, J = 8.6 Hz, 2H), 6.35 (d, J = 15.8 Hz, 1H), 5.45–5.12 (m, 1H), 4.14 (qd, J = 7.1, 3.6 Hz, 2H), 3.54–3.29 (m, 5H), 2.59 (t, J = 6.8 Hz, 2H), 2.17 (s, 3H), 1.24 (t, J = 7.1 Hz, 3H) ppm. 13C NMR (150 MHz, CDCl3, TMS, 25 °C) δ = 202.1, 191.6, 168.9, 134.4, 132.2, 132.1, 130.6, 129.8, 123.7, 115.3, 115.1, 61.5, 59.1, 38.5, 36.7, 31.6, 29.2, 14.1 ppm. 19F NMR (565 MHz, CDCl3) δ = −100.00 ppm. IR (KBr) v: 2962, 2928, 1738, 1716, 1618, 1597, 1453, 1280, 1227, 1155, 853, 776, 596 cm−1. HRMS-ESI (m/z) calcd for C20H21FNaO4S2, [M + Na]+ 431.0763, found 431.0754.
(E)-Ethyl 2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-(4-nitrophenyl)-7-oxohept-4-enoate (5g) (0.13 mmol, 56 mg, 64%): yellowish oil, 1H NMR (600 MHz, CDCl3, 25 °C): δ 8.23 (d, J = 8.7 Hz, 2H), 7.75 (d, J = 8.8 Hz, 2H), 6.31 (d, J = 15.8 Hz, 1H), 5.39–5.27 (m, 1H), 4.18–4.08 (m, 2H), 3.50–3.39 (m, 4H), 3.36 (t, J = 7.3 Hz, 1H), 2.56 (td, J = 7.2, 1.1 Hz, 2H), 2.17 (s, 3H), 1.24 (t, J = 5.9 Hz, 3H) ppm. 13C NMR (150 MHz, CDCl3, 25 °C): δ 201.8, 196.4, 168.8, 144.5, 131.8, 130.9, 130.2, 128.8, 123.2, 61.6, 58.9, 38.9, 36.4, 30.6, 19.2, 14.1 ppm. IR (KBr) v: 2959, 2922, 1717, 1647, 1597, 1523, 1449, 1348, 1281, 1220, 1016, 1074, 858, 797 cm−1. HRMS-ESI (m/z) calcd for C20H21NNaO6S2, [M + Na]+ 458.0708, found 458.0700.
(E)-6-Acetyl-2-(1,3-dithiolan-2-ylidene)-1-phenyloct-3-ene-1,7-dione (a mixture of enol and ketone form) (5h) (0.19 mmol, 68 mg, 94%): yellowish oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 16.68 (s, 0.5 H), 7.65 (t, J = 7.8 Hz, 2H), 7.45 (dd, J = 10.9, 7.3 Hz, 1H), 7.37 (dt, J = 14.8, 7.5 Hz, 2H), 6.34 (d, J = 15.8 Hz, 0.5H), 6.21 (d, J = 15.9 Hz, 0.5H), 5.41 (dt, J = 15.9, 5.3 Hz, 0.5H), 5.31–5.18 (m, 0.5H), 3.57 (t, J = 7.3 Hz, 0.5H), 3.40 (s, 4H), 2.94 (d, J = 4.0 Hz, 1H), 2.57 (t, J = 7.2 Hz, 1H), 2.08 (s, 3H), 2.03 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3, 25 °C): δ 218.5, 203.3, 191.5, 138.3, 132.3, 131.8, 131.8, 130.7, 130.7, 129.5, 129.5, 129.3, 129.3, 128.3, 128.3, 128.1, 128.1, 128.0, 128.0, 123.8, 67.9, 38.7, 38.5, 36.7, 36.7, 36.4, 36.4, 31.8, 30.9, 29.3, 23.0 ppm. IR (KBr) v: 2958, 2923, 1724, 1700, 1614, 1452, 1358, 1281, 1249, 1152, 960, 695, 633 cm−1. HRMS-ESI (m/z) calcd for C19H20NaO3S2, [M + Na]+ 383.0752, found 383.0740.
(E)-Methyl 2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-oxo-7-phenylhept-4-enoate (5i) (0.18 mmol, 69 mg, 92%): yellowish oil, 1H NMR (600 MHz, CDCl3, 25 °C): δ 7.67 (d, J = 8.2 Hz, 2H), 7.47 (t, J = 7.4 Hz, 1H), 7.38 (t, J = 7.5 Hz, 2H), 6.35 (d, J = 15.8 Hz, 1H), 5.32–5.24 (m, 1H), 3.72–3.66 (m, 3H), 3.43–3.36 (m, 5H), 2.60–2.52 (m, 2H), 2.15–2.12 (m, 3H) ppm. 13C NMR (150 MHz, CDCl3, 25 °C): δ 202.1, 193.0, 169.4, 157.4, 138.3, 131.8, 130.7, 129.6, 129.5, 128.1, 123.9, 58.9, 52.5, 38.5, 36.6, 31.7, 29.4 ppm. IR (KBr) v: 2952, 2925, 1742, 1715, 1617, 1450, 1358, 1279, 1249, 1152, 965, 695, 633 cm−1. HRMS-ESI (m/z) calcd for C19H20NaO4S2, [M + Na]+ 399.0701, found 399.0700.
(E)-Ethyl 6-(1,3-dithiolan-2-ylidene)-2-(4-methoxybenzoyl)-7-oxo-7-phenylhept-4-enoate (5j) (0.15 mmol, 70 mg, 73%): yellowish oil, 1H NMR (600 MHz, CDCl3, 25 °C): δ 7.91 (d, J = 8.9 Hz, 2H), 7.64 (d, J = 7.1 Hz, 2H), 7.39 (t, J = 7.4 Hz, 1H), 7.30 (t, J = 7.7 Hz, 2H), 6.91 (dd, J = 9.1, 5.2 Hz, 2H), 6.38 (d, J = 15.7 Hz, 1H), 5.46–5.25 (m, 1H), 4.08 (dtd, J = 18.0, 7.1, 3.6 Hz, 2H), 3.91–3.83 (m, 5H), 3.37 (s, 3H), 2.76–2.68 (m, 2H), 1.14 (t, J = 7.2 Hz, 3H) ppm. 13C NMR (150 MHz, CDCl3, 25 °C): δ 198.5, 195.3, 163.9, 131.7, 131.1, 130.3, 129.5, 128.0, 113.9, 100.0, 61.4, 55.5, 53.6, 38.5, 36.6, 32.6, 14.0 ppm. IR (KBr) v: 2962, 2924, 1733, 1673, 1600, 1511, 1455, 1314, 1262, 1173, 1027, 846, 729 cm−1. HRMS-ESI (m/z) calcd for C26H26NaO5S2, [M + Na]+ 505.1119, found 505.1148.
(E)-Allyl 2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-oxo-7-phenylhept-4-enoate (5k) (0.18 mmol, 72 mg, 89%): yellowish oil, 1H NMR (600 MHz, CDCl3, 25 °C): δ 7.67 (d, J = 7.1 Hz, 2H), 7.47 (t, J = 7.4 Hz, 1H), 7.38 (t, J = 7.6 Hz, 2H), 6.36 (d, J = 15.8 Hz, 1H), 5.86 (ddt, J = 16.4, 10.5, 5.8 Hz, 1H), 5.39–5.18 (m, 3H), 4.65–4.49 (m, 2H), 3.44–3.33 (m, 5H), 2.59 (t, J = 7.2 Hz, 2H), 2.15 (s, 3H) ppm. 13C NMR (150 MHz, CDCl3, 25 °C): δ 202.0, 193.0, 168.6, 157.3, 138.3, 131.8, 131.4, 130.8, 129.6, 129.5, 128.1, 123.9, 119.1, 66.0, 59.0, 38.5, 36.6, 31.7, 29.4 ppm. IR (KBr) v: 2956, 2924, 1741, 1715, 1617, 1576, 1451, 1359, 1249, 1151, 965, 723, 695, 633 cm−1. HRMS-ESI (m/z) calcd for C21H22NaO4S2, [M + Na]+ 425.0857, found 425.0847.
(E)-Prop-2-yn-1-yl 2-acetyl-6-(1,3-dithiolan-2-ylidene)-7-oxo-7-phenylhept-4-enoate (5l) (0.17 mmol, 68 mg, 85%): yellowish oil, 1H NMR (600 MHz, CDCl3, 25 °C): δ 7.67 (d, J = 7.8 Hz, 2H), 7.48 (t, J = 7.4 Hz, 1H), 7.39 (t, J = 7.6 Hz, 2H), 6.36 (dd, J = 15.8 Hz, 6.3, 1H), 5.29 (ddd, J = 14.5, 7.2, 3.6 Hz, 1H), 4.67 (dd, J = 7.1 Hz, 2.4, 1H), 3.68 (s, 1H), 3.43–3.37 (m, 5H), 2.58 (dd, J = 16.2, 7.5 Hz, 2H), 2.48 (t, J = 2.4, 1H), 2.17 (d, J = 4.3 Hz, 3H) ppm. 13C NMR (150 MHz, CDCl3, 25 °C): δ 201.4, 193.0, 168.2, 138.3, 131.8, 131.0, 130.7, 129.6, 129.5, 129.3, 128.1, 123.8, 75.5, 58.8, 52.7, 52.5, 38.5, 36.6, 31.6, 29.4 ppm. IR (KBr) v: 2957, 2924, 1744, 1715, 1616, 1450, 1277, 1149, 963, 806, 695, 633 cm−1. HRMS-ESI (m/z) calcd for C21H20NaO4S2, [M + Na]+ 423.0701, found 423.0686.
1,1′-(10-Benzoyl-9-(4-methoxyphenyl)-1,4-dithiaspiro[4.5]decane-6,6-diyl)diethanone (6a) (0.16 mmol, 75 mg, 80%): yellowish oil, 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.52 (t, J = 7.4 Hz, 2H), 7.44 (t, J = 7.3 Hz, 1H), 7.31 (t, J = 7.6 Hz, 2H), 7.20 (d, J = 8.6 Hz, 2H), 6.75 (d, J = 8.5 Hz, 2H), 4.31 (t, J = 6.7 Hz, 0.2H), 4.01 (t, J = 7.6 Hz, 1H), 3.73 (s, 3H), 3.35 (td, J = 11.4 Hz, 5.3, 2H), 3.23 (t, J = 5.6 Hz, 2H), 2.40 (s, 3H), 2.36 (s, 3H), 2.33–2.20 (m, 3H), 2.19–2.06 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3, 25 °C): δ 200.6, 200.0, 196.0, 158.2, 149.3, 138.3, 133.1, 132.3, 129.2, 129.1, 128.4, 113.7, 75.2, 55.2, 51.6, 39.0, 37.1, 35.1, 29.2, 26.8, 26.7 ppm. IR(KBr) v: 2957, 2926, 1713, 1644, 1611, 1579, 1511, 1448, 1357, 1278, 1248, 1180, 1034, 912, 733, 695 cm−1. HRMS-ESI (m/z) calcd for C26H28NaO4S2, [M + Na]+ 491.1327, found 491.1316.
Methyl 2-acetyl-6-benzoyl-7,7-bis(methylthio)-5-(p-tolyl)hept-5-enoate (7a, E/Z = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), (0.5 mmol, 233 mg, 99%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C) δ 7.47–7.30 (m, 3H), 7.18 (dt, J = 15.6, 7.9 Hz, 2H), 6.98 (dd, J = 15.5, 8.0 Hz, 2H), 6.88 (t, J = 8.5 Hz, 2H), 4.61 (dt, J = 15.2, 7.2 Hz, 1H), 4.29 (d, J = 52.0 Hz, 1H), 3.78 (d, J = 19.0 Hz, 3H), 2.41 (d, J = 7.8 Hz, 3H), 2.37 (s, 1.5H), 2.31–2.22 (m, 1H), 2.20 (s, 1.5H), 2.17 (d, J = 5.7 Hz, 3H), 2.14–2.00 (m, 1H), 1.95 (d, J = 1.7 Hz, 3H), 1.93–1.77 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3, TMS, 25 °C) δ = 205.2, 204.9, 196.1, 195.9, 171.4, 171.1, 149.2, 148.8, 137.6, 137.5, 137.0, 136.9, 136.3, 134.6, 134.3, 132.3, 129.0, 129.0, 128.9, 128.8, 128.3, 128.1, 127.8, 127.8, 84.1, 84.0, 53.4, 53.2, 49.4, 49.3, 33.4, 33.3, 26.9, 26.6, 24.8, 24.7, 20.9, 16.8, 16.7, 16.5, 16.5 ppm. IR (KBr) v: 2954, 2923, 2854, 1723, 1720, 1657, 1596, 1579, 1513, 1448, 1359, 1312, 1261, 1127, 909, 707 cm−1. HRMS-ESI (m/z) calcd for C26H30KO4S2, [M + K]+ 509.1223, found 509.1226.
:1), (0.5 mmol, 233 mg, 99%): light yellow oil, 1H NMR (400 MHz, CDCl3, 25 °C) δ 7.47–7.30 (m, 3H), 7.18 (dt, J = 15.6, 7.9 Hz, 2H), 6.98 (dd, J = 15.5, 8.0 Hz, 2H), 6.88 (t, J = 8.5 Hz, 2H), 4.61 (dt, J = 15.2, 7.2 Hz, 1H), 4.29 (d, J = 52.0 Hz, 1H), 3.78 (d, J = 19.0 Hz, 3H), 2.41 (d, J = 7.8 Hz, 3H), 2.37 (s, 1.5H), 2.31–2.22 (m, 1H), 2.20 (s, 1.5H), 2.17 (d, J = 5.7 Hz, 3H), 2.14–2.00 (m, 1H), 1.95 (d, J = 1.7 Hz, 3H), 1.93–1.77 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3, TMS, 25 °C) δ = 205.2, 204.9, 196.1, 195.9, 171.4, 171.1, 149.2, 148.8, 137.6, 137.5, 137.0, 136.9, 136.3, 134.6, 134.3, 132.3, 129.0, 129.0, 128.9, 128.8, 128.3, 128.1, 127.8, 127.8, 84.1, 84.0, 53.4, 53.2, 49.4, 49.3, 33.4, 33.3, 26.9, 26.6, 24.8, 24.7, 20.9, 16.8, 16.7, 16.5, 16.5 ppm. IR (KBr) v: 2954, 2923, 2854, 1723, 1720, 1657, 1596, 1579, 1513, 1448, 1359, 1312, 1261, 1127, 909, 707 cm−1. HRMS-ESI (m/z) calcd for C26H30KO4S2, [M + K]+ 509.1223, found 509.1226.
(3E,8E)-Methyl 3-benzoyl-8-methyl-2-(methylthio)-4-(p-tolyl)-2,5,6,7-tetrahydrobenzo[b][1,4]thiaazacycloundecine-7-carboxylate (9a), (0.38 mmol, 212 mg, 95%): light yellow oil, 1H NMR (600 MHz, CDCl3, 25 °C) δ 7.41 (dd, J = 12.4, 7.7 Hz, 2H), 7.35 (td, J = 7.3, 3.7 Hz, 1H), 7.19 (dt, J = 12.5, 6.2 Hz, 2H), 7.06–6.84 (m, 6H), 6.73 (dd, J = 15.3, 7.7 Hz, 2H), 5.06 (d, J = 62.6 Hz, 1H), 4.63 (dt, J = 15.7, 7.6 Hz, 1H), 3.79 (d, J = 53.7 Hz, 3H), 2.35 (d, J = 2.0 Hz, 3H), 2.33–2.20 (m, 2H), 2.18 (d, J = 9.7 Hz, 3H), 2.14–1.99 (m, 2H), 1.94 (d, J = 11.2 Hz, 3H) ppm. 13C NMR (150 MHz, CDCl3, 25 °C) δ = 195.9, 173.6, 173.3, 149.1, 146.1, 137.6, 137.1, 136.4, 134.4, 132.3, 132.3, 129.0, 128.9, 128.8, 128.2, 128.2, 127.9, 127.8, 126.0, 125.9, 125.6, 125.5, 121.1, 121.0, 120.9, 111.6, 111.4, 100.0, 78.1, 78.0, 53.4, 53.2, 49.2, 49.1, 38.8, 38.6, 28.4, 28.2, 20.9, 16.8, 16.8, 16.5, 16.5 ppm. IR (KBr) v: 2952, 2921, 2852, 1736, 1657, 1595, 1581, 1512, 1470, 1448, 1313, 1254, 1178, 1121, 910, 740 cm−1. HRMS-ESI (m/z) calcd for C31H31NNaO3S2, [M + Na]+ 552.1643, found 552.1655.
Acknowledgements
The authors thank the National Natural Science Foundation of China for the financial support (21173089 and 21373093). The authors are also grateful to the Analytical and Testing Centre of HUST and Prof. Aiwen Lei, Wuhan University, for his kind help with using the in situ IR instrument. Chutian Scholar Program of the Hubei Provincial Government and the Cooperative Innovation Center of Hubei Province are also acknowledged. This work was also supported by the fundamental research funds for the central universities in China (2014ZZGH019).Notes and references
- E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC.
- See some reviews: (a) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222–234 CrossRef CAS PubMed; (b) D. H. Paull, C. J. Abraham, M. T. Scerba, E. Alden-Danforth and T. Lectka, Acc. Chem. Res., 2008, 41, 655–663 CrossRef CAS PubMed; (c) W. Tang, S. Johnston, J. A. Iggo, N. G. Berry, M. Phelan, L. Lian, J. Bacsa and J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 1668–1672 CrossRef CAS PubMed; (d) Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC; (e) Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337–1378 RSC; (f) Z. Yu, W. Jin and Q. Jiang, Angew. Chem., Int. Ed., 2012, 51, 6060–6072 CrossRef CAS PubMed; (g) L. Stegbauer, F. Sladojevich and D. J. Dixon, Chem. Sci., 2012, 3, 942–958 RSC; (h) A. Gualandi, L. Mengozzi, C. M. Wilson and P. G. Cozzi, Chem. – Asian J., 2014, 9, 984–995 CrossRef CAS PubMed.
- See some recent examples: (a) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 4986–4987 CrossRef CAS PubMed; (b) M. Ikeda, Y. Miyake and Y. Nishibayashi, Angew. Chem., Int. Ed., 2010, 49, 7289–7293 CrossRef CAS PubMed; (c) G. Bergonzini, S. Vera and P. Melchiorre, Angew. Chem., Int. Ed., 2010, 49, 9685–9688 CrossRef CAS PubMed; (d) S. Zhou, S. Fleischer, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5120–5124 CrossRef CAS PubMed; (e) C. Li, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2009, 131, 6967–6969 CrossRef CAS PubMed; (f) D. E. A. Raup, B. Cardinal-David, D. Holte and K. A. Scheidt, Nat. Chem., 2010, 2, 766–771 CrossRef CAS PubMed; (g) F. M. Koch and R. Peters, Angew. Chem., Int. Ed., 2007, 46, 2685–2689 CrossRef CAS PubMed.
- (a) M. Li, C. Tang, J. Yang and Y. Gu, Chem. Commun., 2011, 47, 4529–4531 RSC; (b) M. Li, H. Li, T. Li and Y. Gu, Org. Lett., 2011, 13, 1064–1067 CrossRef CAS PubMed; (c) M. Li and Y. Gu, Tetrahedron, 2011, 67, 8314–8320 CrossRef CAS; (d) M. Li, J. Yang and Y. Gu, Adv. Synth. Catal., 2011, 353, 1551–1564 CrossRef CAS.
- (a) N. Zanatta, L. da S. Fernandes, F. M. Nachtigall, H. S. Coelho, S. S. Amaral, A. F. C. Flores, H. G. Bonacorso and M. A. P. Martins, Eur. J. Org. Chem., 2009, 1435–1444 CrossRef CAS; (b) N. Zanatta, L. S. da Fernandes, S. München, H. S. Coelho, S. S. Amaral, L. Fantinel, H. G. Bonacorso and M. A. P. Martins, Synthesis, 2010, 2348–2354 CrossRef CAS.
- See recent reviews: (a) L. Pan, X. Bi and Q. Liu, Chem. Soc. Rev., 2013, 42, 1251–1286 RSC; (b) L. Wang, W. He and Z. Yu, Chem. Soc. Rev., 2013, 42, 599–621 RSC; (c) See some examples: H. Yu, W. Jin, C. Sun, J. Chen, W. Du, S. He and Z. Yu, Angew. Chem., Int. Ed., 2010, 49, 5792–5797 CrossRef CAS PubMed; (d) Y. Liu, J. Liu, M. Wang, J. Liu and Q. Liu, Adv. Synth. Catal., 2012, 354, 2678–2682 CrossRef CAS; (e) S. Kumar, H. Ila and H. Junjappa, J. Org. Chem., 2009, 74, 7046–7051 CrossRef CAS PubMed; (f) E. R. Anabha, K. N. Nirmala, A. Thomas and C. V. Asokan, Synthesis, 2007, 428–432 CAS; (g) X. Liu, L. Pan, J. Dong, X. Xu, Q. Zhang and Q. Liu, Org. Lett., 2013, 15, 6242–6245 CrossRef CAS PubMed; (h) Y. Dong, B. Liu, P. Chen, Q. Liu and M. Wang, Angew. Chem., Int. Ed., 2014, 53, 3442–3446 CrossRef CAS PubMed.
- Direct comparison of indole and α-oxoketene dithioacetal in terms of reactivity as a nucleophile has been rarely reported. See an example in which indole displayed better reactivity than α-oxoketene dithioacetal: (a) C. Xu, J. Liu, W. Ming, Y. Liu, J. Liu, M. Wang and Q. Liu, Chem. – Eur. J., 2013, 19, 9104–9109 CrossRef CAS PubMed; (b) Indirect evidence can be found in the electrophilic alkylation of aldehyde with these two nucleophiles. While the reaction of indole can be performed under catalyst-free conditions, a catalyst has to be used in the similar reaction of α-oxoketene dithioacetal: F. He, P. Li, Y. Gu and G. Li, Green Chem., 2009, 11, 1767–1773 RSC; (c) Q. Zhang, Y. Liu, M. Wang, Q. Liu, J. Hu and Y. Yin, Synlett, 2006, 3009–3014 CAS.
- Y. Liu, M. Wang, H. Yuan and Q. Liu, Adv. Synth. Catal., 2010, 352, 884–892 CrossRef CAS.
- (a) H. Yuan, M. Wang, Y. Liu, L. Wang, J. Liu and Q. Liu, Chem. – Eur. J., 2010, 16, 13450–13457 CrossRef CAS PubMed; (b) H.-J. Yuan, M. Wang, Y.-J. Liu and Q. Liu, Adv. Synth. Catal., 2009, 351, 112–116 CrossRef CAS.
- The peak splitting has also been observed, see L. A. Smyth, T. P. Matthews, P. N. Horton, M. B. Hursthouse and I. Collins, Tetrahedron, 2010, 66, 2843–2854 CrossRef CAS.
- The maximum UV-absorption of acrylopheone skeletons appeared generally in the range from 250 nm to 300 nm. W. B. Black and R. E. Lutz, J. Am. Chem. Soc., 1955, 77, 5134–5140 CrossRef CAS.
- α-Oxoketene dithioacetal 2b has been used as a ligand to coordinate with Cu(II) halide, see: (a) L. Beyer, R. Kirmse, J. Stach, R. Szargan and E. Hoyer, Z. Anorg. Allg. Chem., 1981, 476, 7–15 CrossRef CAS; (b) G. Dorange and J. E. Guerchais, Bull. Soc. Chim. Fr., 1971, 43–50 CAS.
- (a) A. A. Isab and M. I. M. Wazeer, J. Coord. Chem., 2005, 58, 529–537 CrossRef CAS; (b) S. Ahmad, A. A. Isab and A. P. Arnold, J. Coord. Chem., 2003, 56, 539–544 CrossRef CAS; (c) T. Okauchi, T. Tanaka and T. Minami, J. Org. Chem., 2001, 66, 3924–3929 CrossRef CAS PubMed.
- The disproportionation of CuBr2 was accelerated under the conditions of ESI-MS analysis because only a complex of Cu+ was detected when CuBr2 was injected. In the disproportionation of CuBr2, CuBr and Br2 were produced simultaneously. However, using a combination of CuBr, Br2 and MnCl2·4H2O as the catalyst in the model reaction gave only 11% of 3a, implying that the catalytic ability of CuBr2 cannot be ascribed to its disproportionation..
- M. Gonsior and I. Krossing, Chem. – Eur. J., 2004, 10, 5730–5736 CrossRef CAS PubMed.
- Intermolecular Michael reactions using α-oxoketene dithioacetal as the Michael acceptor have been reported, see: (a) M. Abass and A. Khodairy, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 287–297 CrossRef CAS; (b) A. K. El-Shafie, A. M. Soliman, A. A. Sultan and E. Abd-Alla, J. Heterocycl. Chem., 2006, 43, 133–137 CrossRef CAS.
- (a) A. G. Schultz and R. E. Harrington, J. Am. Chem. Soc., 1991, 113, 4926–4931 CrossRef CAS; (b) T. Hampel and R. Brueckner, Org. Lett., 2009, 11, 4842–4845 CrossRef CAS PubMed.
- (a) M. S. C. Pedras and Y. Yu, Bioorg. Med. Chem., 2008, 16, 8063–8071 CrossRef CAS PubMed; (b) S. W. Rabkin and S. S. Klassen, Eur. J. Pharmacol., 2007, 562, 174–182 CrossRef CAS PubMed.
- (a) Y. Gu, R. De Sousa, G. Frapper, C. Bachmann, J. Barrault and F. Jérôme, Green Chem., 2009, 11, 1968–1972 RSC; (b) Y. Gu, J. Barrault and F. Jérôme, Adv. Synth. Catal., 2009, 351, 3269–3278 CrossRef CAS.
- G. K. Verma, R. K. Verma, G. Shukla, N. Anugula, A. Srivastava and M. S. Singh, Tetrahedron, 2013, 69, 6612–6619 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c4cy01212g |
| This journal is © The Royal Society of Chemistry 2015 |