
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFactors influencing the catalytic oxidation of benzyl alcohol using supported phosphine-capped gold nanoparticles†
Rohul H.
Adnan
ab,
Gunther G.
Andersson
c,
Matthew I. J.
Polson
a,
Gregory F.
Metha
d and
Vladimir B.
Golovko
*ae
aDepartment of Chemistry, University of Canterbury, Christchurch, 8041, New Zealand. E-mail: vladimir.golovko@canterbury.ac.nz
bChemistry Department, University of Malaya, Kuala Lumpur, 50603, Malaysia
cFlinders Centre for Nanoscale Science and Technology, Flinders University, Adelaide, SA 5001, Australia
dDepartment of Chemistry, University of Adelaide, Adelaide, SA 5005, Australia
eThe MacDiarmid Institute for Advanced Materials and Nanotechnology, PO Box 600, Wellington, 6140, New Zealand
First published on 20th November 2014
Abstract
Two phosphine-stabilised gold clusters, Au101(PPh3)21Cl5 and Au9(PPh3)8(NO3)3, were deposited and activated on anatase TiO2 and fumed SiO2. These catalysts showed an almost complete oxidation of benzyl alcohol (>90%) within 3 hours at 80 °C and 3 bar O2 in methanol with a high substrate-to-metal molar ratio of 5800 and turn-over frequency of 0.65 s−1. Factors influencing catalytic activity were investigated, including metal–support interaction, effects of heat treatments, chemical composition of gold clusters, the size of gold nanoparticles and catalytic conditions. It was found that the anions present in gold clusters play a role in determining the catalytic activity in this reaction, with NO3− diminishing the catalytic activity. High catalytic activity was attributed to the formation of large gold nanoparticles (>2 nm) that coincides with partial removal of ligands which occurs during heat treatment and catalysis. Selectivity towards the formation of methyl benzoate can be tuned by selection of the reaction temperature. The catalysts were characterised using transmission electron microscopy, UV-vis diffuse reflectance spectroscopy and X-ray photoelectron spectroscopy.
1 Introduction
For centuries, gold was considered to be chemically inert and catalytically inactive, making it valuable in jewellery and as coinage metal but worthless for applications in catalysis. However, this perception has changed since the early report by Haruta et al. that small gold particles were catalytically active in CO oxidation at low temperature1 and the discovery by Hutchings et al. that a gold-based catalyst could be used in the hydrochlorination of acetylene.2 These discoveries triggered a revolution in gold-based catalysis, with numerous follow-up papers demonstrating the superiority of gold-based catalysts over the more expensive platinum group metals in various catalytic processes, with improvements in activity, selectivity and stability against deactivation paving a pathway to numerous commercially-oriented patents.3,4 The number of studies on nanoparticulate gold-based catalysts for a broad range of bulk and fine chemical synthesis processes has grown exponentially.1,5–8Oxidation reactions, particularly alcohol oxidation, are among the most important and useful reactions used by the chemical industry and academia.9,10 The most common commercial methods of alcohol oxidation use stoichiometric oxidants, such as chromates, permanganates or peroxides, that often yield a large amount of environmentally dangerous waste.11 The use of harmful organic solvents (e.g. chlorinated solvents) may also have a negative environmental impact.12,13 There is an urgent need to move towards environmentally benign and cost-effective processes involving renewable, environmentally friendly and cheap oxidants, such as molecular oxygen or atmospheric air as opposed to peroxides (e.g. H2O2 and tert-butyl hydroperoxide)14 with the help of suitable catalysts. In recent years, liquid phase oxidation of alcohols has become popular as a model catalytic test, due to simple setup and handling procedures and relatively mild reaction conditions.15,16 Supported platinum group catalysts are widely used in the oxidation of alcohols, yet they often require a secondary metal as a promoter to increase the selectivity and stability of such catalysts.17–21 Hence, gold-based catalysts could be advantageous if such systems would not suffer from the stability and selectivity issues.22
A systematic study of the nature of the active sites is crucial in improving the activity and selectivity of catalysts towards the formation of the desired partial oxidation products. In the case of heterogeneous Au-based catalysts, the following factors are often considered to define catalytic activity and selectivity: gold particle size and morphology, oxidation state of gold species, metal–support interaction and specific parameters of catalytic testing conditions. The nature of the active site in the liquid phase oxidation of alcohols is currently still unclear, with both ultra-small particles (<2 nm)23,24 and larger particles (>2 nm)25 reported as active catalysts. Tsukuda et al. reported that as the size of gold nanoparticles reduced below 2 nm, the turnover frequency (TOF) for the aerobic oxidation of p-hydroxybenzylalcohol increased significantly.26 The authors proposed that the catalytic activity of these small gold nanoparticles was due to the increased electron density on the gold core and also charge transfer from the poly(N-vinyl-2-pyrrolidone) ligands. However, Haider et al. observed that smaller size was not necessarily responsible for higher activity – the authors reported that the optimum size of the gold nanoparticles for ethanol oxidation is ca. 7 nm, irrespective of the supports.25 Zheng et al. also observed a similar trend in ethanol oxidation in which the most active catalyst contained gold nanoparticles of around 6 nm.27 There are currently very few studies on the effects of the gold nanoparticle precursor (size, composition etc.) on the performance of resulting catalysts since the majority of reports in the literature are focused predominantly on the morphology and size of gold nanoparticles.23,28–30 Only recently, several studies on the effects of ligands, such as citrate, polyvinylpyrrolidone and polyvinyl alcohol, on the performance of gold-based catalysts have been reported in a wide range of catalytic reactions – glycerol oxidation,31 hydrogenation of cinnamaldehyde32 and reduction of p-nitrophenol.33 These findings clearly suggest that the nature of ligands plays a critical role in determining the performance of the catalytically active gold particles in a particular reaction, perhaps, similarly to the role of support.34 Hence, it is important to study the effect of the composition (size of the gold core, neutral organic ligands and anionic ligands or counter-ions) of precursors used in fabrication of gold-based catalysts in order to design catalysts with high activity and improved selectivity.
Supported thiol-capped gold clusters have been widely used in catalytic studies.35–37 For example, Jin et al. supported Au25(SR)18, Au38(SR)24 and Au99(SPh)42 clusters on various metal oxides and tested them in selective oxidation and hydrogenation reactions.38–41 Tsukuda et al. used Au10(SG)10, Au18(SG)14, Au25(SG)18 and Au39(SG)24 deposited on hydroxyapatite in styrene oxidation.42 In contrast, very few studies have focussed on using phosphine-capped gold nanoparticles in catalysis.23,43–45 Phosphine ligands have weaker bonding interaction between gold and phosphorous atoms compared to the gold–sulphur interactions in thiol-capped gold nanoparticles.46
Building on our recent X-ray spectroscopy studies of supported triphenylphosphine-stabilised gold nanoparticles,47,48 we hereby aim to study the catalytic performance of materials derived from selected phosphine-capped gold clusters, supported and activated on titania and silica using liquid phase benzyl alcohol oxidation as a model catalytic test. Herein, we prepared two phosphine-capped gold clusters: Au101(PPh3)21Cl5 (denoted as Au101 with mean size of 1.6 ± 0.3 nm) and Au9(PPh3)8(NO3)3 (denoted as Au9 with mean size of 0.8 nm). These clusters were then deposited and activated on TiO2 (anatase) and SiO2 (fumed) using sol-immobilisation method; a well-established method used by Rossi,10 Jin39 and Hutchings49 for immobilising pre-synthesized gold nanoparticles or clusters onto supports. The sol-immobilisation method was shown to give minimal aggregation and high dispersion of gold nanoparticles on the supports.50 The catalysts were then calcined under different atmospheres: oxygen (O2) and oxygen and subsequently hydrogen (O2–H2) at 200 °C to dislodge the phosphine ligands and expose the metal core adsorbed on the support to facilitate access by the substrate.47
2 Experimental
2.1 Materials
All reactants were analytical reagent grade and used without further purification. Benzyl alcohol (>99%), benzaldehyde (>99%), methyl benzoate (99%), benzoic acid (99.7%), and anisole (>99.5% anhydrous) were purchased from Sigma Aldrich. HPLC grade methanol (99.9%, Fisher Scientific) was degassed (N2) and dried over pre-calcined alumina prior to use. Tetrachloroauric acid was prepared using 99.99% pure gold following the procedure detailed by Brauer.51 Triphenylphosphine-stabilised gold nanoparticles with an average estimated formula Au101(PPh3)21Cl5 (denoted as Au101) have been synthesised according to procedure reported earlier by Weare et al.52 while Au9(PPh3)8(NO3)3 was made according to the protocol reported by Wen et al.53 Anatase (99.5%, ca. 70 m2 g−1, 10–30 nm particles) was purchased from SkySpring Nanomaterials. Instrument grade H2 (99.98% with <20 ppm H2O, BOC) and O2 (99.2% with <10 ppm H2O, Southern Gas Services Ltd.) were used for the calcination procedures.2.2 Preparation of catalysts
The clusters were deposited onto the support using the protocol reported by Zhu et al.38 For the purpose of this study we focused on materials with 0.17 wt% Au (i.e. ratio of the weight of the Au due to presence of clusters relative to the weight of support) on titania and 0.5 wt% Au on silica to give equivalent surface density of gold particles.47First, the calculated amount of support (silica or titania) was dried in the Schlenk tube under vacuum at 200 °C with stirring overnight (12 h). Upon cooling to room temperature, 5.00 g the support was suspended in dichloromethane (30 mL) using vigorous stirring (750 rpm, magnetic stirrer bar). A solution of calculated amount of gold cluster (e.g. 11.2 mg of Au101) in dichloromethane (10 mL) was added to the suspension of support under vigorous stirring at room temperature and the mixture was left stirring overnight (12 h). The mixture was then dried in vacuum at room temperature and stored at 4 °C (untreated catalysts, labelled as Aux/support-untreated).
Two heat treatments were employed in this work: a) calcination under pure O2 atmosphere at 200 °C for 2 h and b) calcination under pure O2 at 200 °C for 2 h followed by calcination in H2 at 200 °C for 2 h.47 All cluster deposition and activation experiments were performed using glassware wrapped in Al foil to avoid exposure of materials to light. All prepared catalysts were stored in vials wrapped in Al foil in a fridge (4 °C). For the purpose of following discussion catalysts are labelled in the following manner: “x wt% Auy/support-treatment”, where x corresponds to the weight % of pure Au and y = 9 or 101.
2.3 Catalytic testing
The catalytic oxidation of benzyl alcohol was performed in stainless steel autoclaves (total internal volume of 57 mL) equipped with Teflon liners and magnetic stirrers, with temperature control of the reaction achieved by using a hotplate stirrer controlled by a thermocouple immersed into the reaction mixture. Typically, a mixture of 0.270 g, 2.50 mmol benzyl alcohol and 0.135 g, 1.25 mmol anisole (internal standard) in methanol (25 mL) were charged into a Teflon liner. Then, 0.345 g, 2.50 mmol K2CO3 and 50 mg of catalyst were added, giving benzyl alcohol to Au molar ratio of 5800. A purge–vent cycle was completed 3 times with pure oxygen to ensure high purity of the atmosphere before the autoclave was pressurised with 5 bar of oxygen and heated to a specific temperature for the desired reaction time. At the end of a catalytic test, the reaction was stopped by cooling the autoclave to 3 °C in an ice-bath. The reaction mixture was centrifuged at 5000 rpm for 15 minutes to separate product mixture and the catalyst. The catalyst was washed with methanol (3 × 15 mL) and dried in vacuo prior to recycling. Each catalytic test was repeated at least in triplicate giving lower than 3% differences in conversion and selectivity.Product mixtures were analysed using high performance liquid chromatography (HPLC) Dionex Ultimate 300 system equipped with UV detector and fitted with a Luna 5μ C18(2) (250 × 4.60 mm) reversed phase column. The products were eluted using a mixture of 0.05 v/v% trifluoroacetic acid in water (70%) and acetonitrile (30%). Gold content was measured via atomic absorption spectroscopy (AAS) using a Varian SpectraAA 220 FS with ASL hollow cathode lamp for the Au element.
2.4 Catalyst characterization
High resolution transmission electron microscopy (HRTEM) was performed using a Philips CM-200 system operating at 200 kV. Samples were deposited as a suspension in methanol onto Cu (300 mesh) grids coated with a holey carbon film and dried in vacuum immediately prior to TEM study. Statistical evaluation of at least 250 gold particles per sample was undertaken to estimate mean particle sizes and construct size distribution histograms. X-ray photoelectron spectroscopy (XPS) study was performed at the soft X-ray beamline at the Australian Synchrotron using a SPECS Phoibos 150 hemispherical electron analyser with the photon energy set to 690 eV. The irradiation spot size was ca. 600 × 600 μm, providing an X-ray flux of ca. 1012 photon mm−2 s−1.47 XPS samples were prepared by suspending catalysts in dichloromethane at a concentration of ca. 1 mg mL−1. A 10 μL drop of each sample was deposited onto a clean 6 × 6 mm silicon (Si) wafer and dried in vacuum (in the dark) immediately before analysis. UV-vis diffuse reflectance spectroscopy (UV-vis DRS) spectra were recorded using a Cintra 404 (GBC Scientific Equipment) spectrophotometer. Thermogravimetric analysis (TGA) was performed using a TGA-DSC Q6000 Universal Analyser. 31P NMR spectra were recorded by taking a small aliquot of the reaction mixture, dissolving it in CDCl3 solvent and collecting data using an Agilent Technologies 400 Hz NMR system.3 Results and discussion
3.1 Catalyst characterization
Au101 nanoparticles and Au9 clusters are smaller than 2 nm, are highly monodisperse, have a high surface area to volume ratio, and do not have a localised surface plasmon resonance (LSPR) due to their non-metallic surface state.46,52 We prepared heterogeneous gold catalysts derived from Au101 and Au9 clusters deposited onto the surface of TiO2 (anatase) and SiO2 (fumed) nanopowders. The 0.17 wt% Au101/TiO2-untreated and 0.17 wt% Au9/TiO2-untreated catalysts made via the sol-immobilisation method show minimal cluster aggregation (Fig. 1a, 2a). Imaging ultra-small clusters on support, such as Au9 on TiO2, was very difficult due to poor contrast of the ultra-small gold particles over support material, as was emphasised by Hutching et al.54 Hence, no statistical analysis of the particle size distribution was provided for 0.17 wt% Au9/TiO2-untreated. Although, in the case of Au9 cluster poor contrast did not allow precise particle size determination, the absence of larger, easily visible in HRTEM aggregates is evident (Fig. 2a). Our earlier study using synchrotron XPS and NEXAS also confirmed the presence of a significant fraction of supported Au9 clusters on TiO2.47 The gold loading of 0.17 wt% on TiO2 was chosen to minimise the aggregation of gold nanoparticles, yet still to be able to obtain discernible signals in X-ray photoelectron spectroscopy (XPS) and UV-vis diffuse reflectance spectroscopy (UV-vis DRS) studies. In the case of clusters supported on fumed SiO2, the Au loading was normalised based on the surface area (to be 0.5 wt%) in order to obtain equivalent (to TiO2) surface coverage by clusters.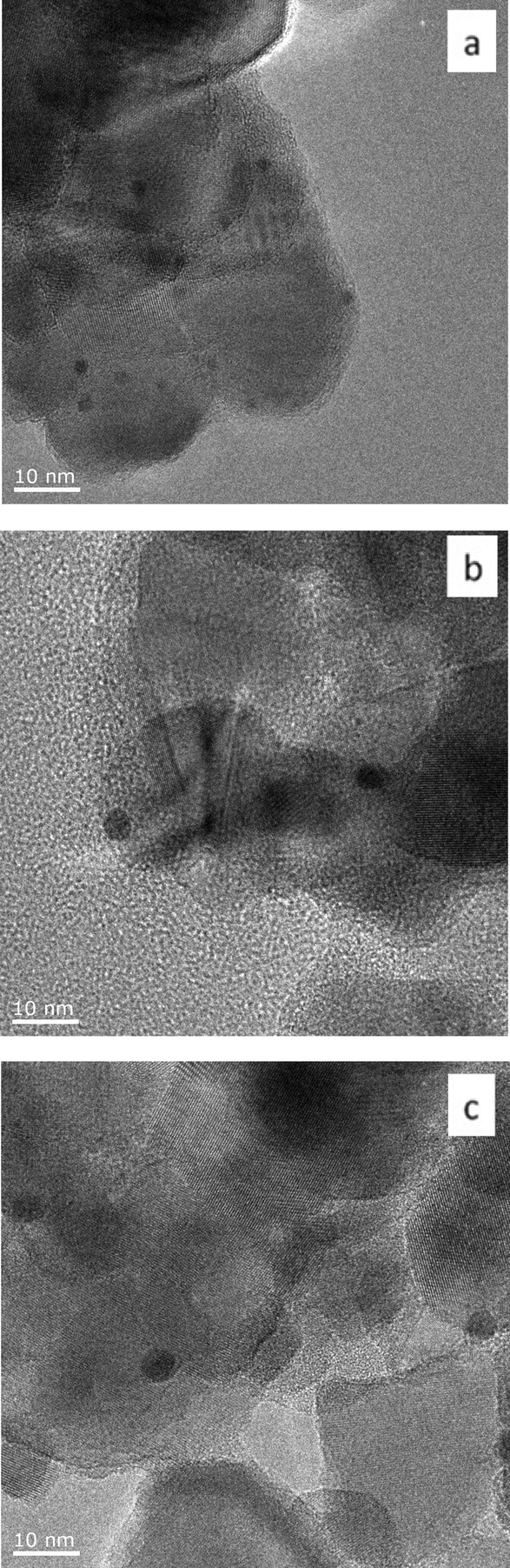 | ||
| Fig. 1 Representative TEM images of 0.17 wt% Au101/TiO2 catalysts before catalytic reaction, a) Au101/TiO2-untreated, b) Au101/TiO2–O2, and c) Au101/TiO2–O2–H2. | ||
 | ||
| Fig. 2 Representative TEM images of 0.17 wt% Au9/TiO2 catalysts before catalytic reaction, a) Au9/TiO2-untreated, b) Au9/TiO2–O2, and c) Au9/TiO2–O2–H2. | ||
A careful heat-treatment was employed in order to remove the capping ligands and expose the gold core to the substrate, yet to minimise aggregation. Here, calcination of the catalysts was done at 200 °C under different environments: under pure oxygen (O2) and oxygen followed by hydrogen (O2–H2).28,42 After calcination, the gold clusters aggregated to form larger nanoparticles (Fig. 1 and 2).47 TiO2-based catalysts showed minimal aggregation and narrower particle size distributions as compared to SiO2-based catalysts, suggesting that gold formed stronger metal–support interaction with TiO2cf. SiO2 (Fig. 1 and 3). UV-vis DR spectra (Fig. 4 and 5) showed the absence of LSPR bands in the cases of untreated catalysts allows us to infer that the gold nanoparticles were predominantly with sizes below 2 nm and retained their non-metallic state. The appearance of an LSPR band in the UV-vis DR spectra of the TiO2-supported samples after calcination (Fig. 4 and 5) indicated formation of plasmonic gold nanoparticles which is indicative of the increase in particle size to greater than 2 nm.55–57 It was previously reported that the position of the maximum of the LSPR band moves to longer wavelength as the size of gold particles increases, although in some cases this general trend broke down due to the effect of the dielectric constant of the surrounding environment.58–61 Interestingly, the 0.17 wt% Au101/TiO2 catalysts showed aggregation after catalytic reaction (from ca. 2.0 to ca. 3.6 nm; Table 1). Particle size analysis based on the statistical evaluation of numerous TEM images (Table 1) confirms that the increase in particle size after heat-treatments and after catalytic reactions for Aux/TiO2 catalysts (x = 9, 101) was consistent with the red shift in the peak maxima position of the LSPR bands observed in UV-vis DR spectra.
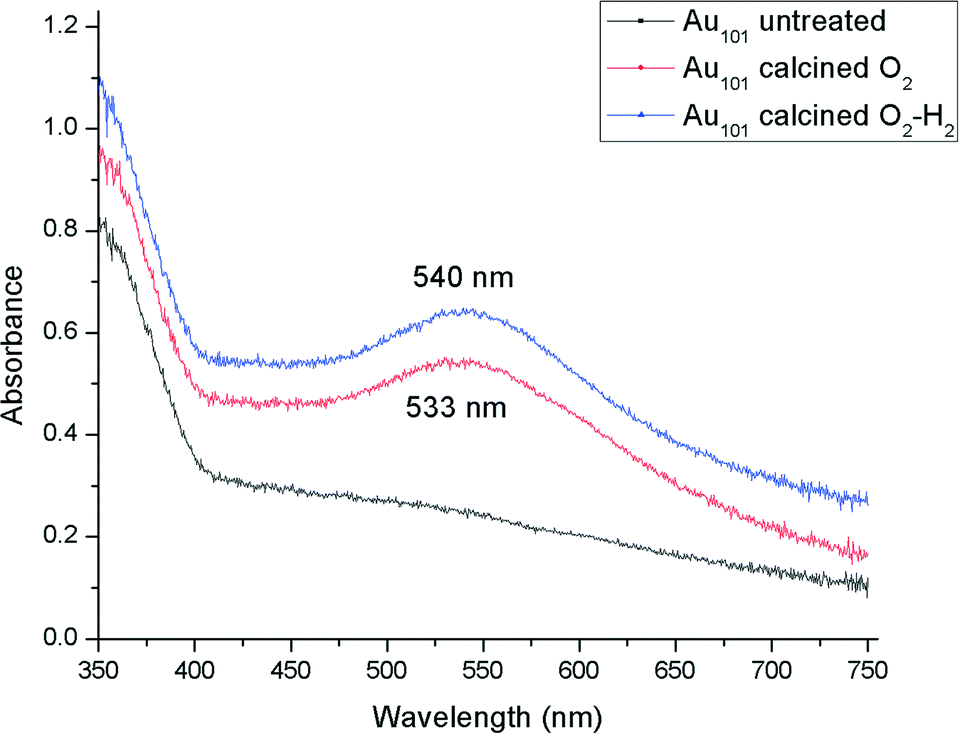 | ||
| Fig. 3 UV-vis DR spectra of 0.17 wt% Au101/TiO2 catalysts. | ||
 | ||
| Fig. 4 UV-vis DR spectra of 0.17 wt% Au9/TiO2 catalysts. | ||
 | ||
| Fig. 5 Representative TEM images of 0.5 wt% Au101/SiO2 catalysts before catalytic reaction, a) Au101/SiO2-untreated, b) Au101/SiO2–O2, and c) Au101/SiO2–O2–H2. | ||
| Entry | Catalyst | Condition | Sizea (nm) | LSPR peak maximumb (nm) |
|---|---|---|---|---|
| a Measured using TEM. b Measured using UV-vis DRS. c Estimated based on the absence of LSPR peak in the UV-vis DR spectra. | ||||
| 1 | 0.17% Au101/TiO2-untreated | Before reaction | 2.0 ± 0.4 | No peak |
| 2 | 0.17% Au101/TiO2–O2 | Before reaction | 3.5 ± 0.8 | 533 |
| 3 | 0.17% Au101/TiO2–O2–H2 | Before reaction | 4.4 ± 1.2 | 540 |
| 4 | 0.17% Au101/TiO2-untreated | After reaction | 3.6 ± 1.2 | 540 |
| 5 | 0.17% Au101/TiO2–O2 | After reaction | 3.9 ± 1.0 | 541 |
| 6 | 0.17% Au101/TiO2–O2–H2 | After reaction | 4.6 ± 1.5 | 546 |
| 7 | 0.17% Au101/TiO2-untreated | After recycle | 4.6 ± 2.5 | 541 |
| 8 | 0.17% Au101/TiO2–O2 | After recycle | 4.2 ± 1.2 | 547 |
| 9 | 0.17% Au101/TiO2–O2–H2 | After recycle | 5.0 ± 1.5 | 551 |
| 10 | 1.3% Au101/TiO2-untreated | Before reaction | 2.7 ± 0.6 | 530 |
| 11 | 0.17% Au9/TiO2-untreated | Before reaction | <2c | No peak |
| 12 | 0.17% Au9/TiO2–O2 | Before reaction | 2.4 ± 0.5 | 555 |
| 13 | 0.17% Au9/TiO2–O2–H2 | Before reaction | 2.9 ± 0.9 | 558 |
| 14 | 0.17% Au101/SiO2-untreated | Before reaction | <2c | No peak |
| 15 | 0.17% Au101/SiO2–O2 | Before reaction | 3.6 ± 2.7 | 529 |
| 16 | 0.17% Au101/SiO2–O2–H2 | Before reaction | 4.4 ± 2.5 | 524 |
| 17 | 0.5% Au101/SiO2-untreated | Before reaction | 3.6 ± 1.2 | 537 |
| 18 | 0.5% Au101/SiO2–O2 | Before reaction | 4.7 ± 1.6 | 525 |
| 19 | 0.5% Au101/SiO2–O2–H2 | Before reaction | 5.7 ± 2.9 | 518 |
For 0.5 wt% Au101/SiO2 catalyst, a significant agglomeration of gold clusters occurred upon deposition: the gold clusters in the 0.5 wt% Au101/SiO2-untreated catalyst aggregated to form gold nanoparticles of mean size 3.6 ± 1.2 nm. The observed particle size is almost twice the size of pristine Au101 cluster and substantially larger than the size of the gold particles (2.0 ± 0.4 nm) in the case of Au101 immobilized on TiO2 at the same surface coverage. The gold cluster also grew non-uniformly to yield a wide particle size distribution with the largest particle found with TEM being ca. 8 nm and the smallest being ca. 2 nm (Fig. 5a and S9a ESI†). After calcination under O2 and O2 followed by H2 at 200 °C, the agglomeration progressed further resulting in the formation of larger gold nanoparticles with wider particle size distributions (Fig. 5(b and c) and S9(b and c) ESI†). A summary of supported gold particle mean sizes and positions of the LSPR peak maxima are given in Table 1.
Our recent XPS study of phosphine-capped Au clusters on TiO2 showed that for untreated Au9/TiO2 and Au101/TiO2 a significant fraction of phosphine ligands were dislodged from the gold cores and formed phosphine oxide-like species by interaction with oxygen on the TiO2 surface, leaving a tiny amount of phosphine ligands bound to the gold core.47 Calcination under O2 atmosphere detached the majority of phosphine ligands from the gold core by forming phosphine-oxide species. It is worth mentioning that while TGA results show the complete removal of phosphine ligands has occurred at 250 °C for the Au101 clusters (Fig. S3, ESI†) and at 240 °C for the Au9 cluster (Fig. S6, ESI†), the phosphine ligands attached to supported Au clusters were not removed as phosphoric acid (H3PO4), but as phosphine-oxide species probably due to the interaction with the oxygen from TiO2 surfaces (e.g. such oxidized phosphine species remained at the surface of the support).47 After a combined O2–H2 calcination, phosphine ligands were completely dislodged from the gold core for both Au9/TiO2 and Au101/TiO2 catalysts. Our first XPS study of a wider range of ultra-small gold phosphine clusters suggests that the nature of the anionic species (Cl− and NO3−) did not significantly influence the position of the Au 4f7/2 peak (84.7 ± 0.1 eV for Au11 cluster containing Cl−vs. 85.1 ± 0.1 eV for both Au8 and Au9 clusters containing NO3−) whereas the size of the cluster has a more pronounced effect (83.9 ± 0.1 eV for Au11 cluster containing Cl−).48
The leaching of metal particles into solution is a common problem for metal supported heterogeneous catalysts. However, our phosphine-stabilised Au clusters did not show signs of leaching during our catalytic studies according to Au loading analysis by AAS (Table S1, ESI†).
3.2 Catalytic testing
The liquid phase oxidation of benzyl alcohol using gold nanoparticle-based catalysts reported here yielded benzoic acid and methyl benzoate as the major products, which were identified using HPLC-MS and quantified using HPLC-UV (using an internal standard and reference compound solutions of known concentration for calibration). The untreated catalysts with 0.17 wt% gold loading were inactive, showing no conversion while heat treated catalysts showed almost complete (>92%) conversion of benzyl alcohol after 4 hours (see Table 2). The most active catalysts gave very high catalytic activity with a molar substrate to metal ratio of 5800 and turnover frequency up to 0.65 s−1 (which is orders of magnitude greater than in any of the related earlier reports, Table S2 ESI†). There were several factors contributing to the high activity of our catalysts. Firstly, the water soluble base (K2CO3) is known to be a promoter in liquid phase oxidation of alcohols.62,63 Rossi proposed that alcohols cannot adsorb directly onto gold and a base was required to deprotonate the hydroxyl group forming a metal alkoxide prior to adsorption onto gold.22 The indispensable role of the base was shown when no conversion of benzyl alcohol was achieved in its absence (Table 1, entries 4–6). There are studies of benzyl alcohol oxidation using supported-gold catalysts under base-free conditions, nevertheless, in those studies, high conversions (>90%) were rarely achieved.15,16,64 In some cases, high conversions of benzyl alcohol under base-free conditions were also reported, for example, Su et al. reported that gold nanoparticles supported on the binary mesostructured Ga–Al mixed oxide were able to catalyse benzyl alcohol under base-free conditions at 80 °C with a high conversion (98%).65 Abad et al. employed Au/CeO2 in the oxidation of various alcohols and observed high conversions under mild conditions.66 Those authors suggested that the base was not required to deprotonate alcohols because the support contained active sites that were able to form cerium alkoxide, assisting the oxidation of alcohols.| Entry | Catalyst | Size (nm) | Time (h) | Conversion (%) | Selectivity to methyl benzoate (%) | Selectivity to benzoic acid (%) | TOF (s−1) |
|---|---|---|---|---|---|---|---|
| Reaction conditions: 50 mg catalyst, 2.5 mmol benzyl alcohol, 25 mL methanol (solvent), 1.25 mmol anisole (internal standard), 2.5 mmol K2CO3, 5 bar O2 pressure, 80 °C.a Without gold catalyst.b Without base (K2CO3).c Recycled catalysts.d 850 mg of a catalyst was used to retain the same total amount of gold as in the case of 0.17 wt% Au catalyst.e The size of gold nanoparticle was estimated from the absence of LSPR band in UV-vis DR spectrum.f KNO3 (or KCl) was added according to equivalent molar percentage by impregnation method. | |||||||
| 1 | Blanka,b | n/a | 4 | 0 | 0 | 0 | 0 |
| 2 | Anatasea,b | n/a | 4 | 0 | 0 | 0 | 0 |
| 3 | Anatasea | n/a | 4 | 0 | 0 | 0 | 0 |
| 4 | 0.17% Au101/TiO2-untreatedb | 2.0 ± 0.4 | 4 | 0 | 0 | 0 | 0 |
| 5 | 0.17% Au101/TiO2–O2b | 3.5 ± 0.8 | 4 | 0 | 0 | 0 | 0 |
| 6 | 0.17% Au101/TiO2–O2–H2b | 4.4 ± 1.2 | 4 | 0 | 0 | 0 | 0 |
| 7 | 0.17% Au101/TiO2-untreated | 2.0 ± 0.4 | 4 | 0 | 0 | 0 | 0 |
| 8 | 0.17% Au101/TiO2–O2 | 3.5 ± 0.8 | 4 | 96 | 79 | 21 | 0.51 |
| 9 | 0.17% Au101/TiO2–O2–H2 | 4.4 ± 1.2 | 4 | 97 | 75 | 23 | 0.51 |
| 10 | 0.17% Au101/TiO2-untreatedc | 3.6 ± 1.2 | 4 | 29 | 65 | 15 | 0.12 |
| 11 | 0.17% Au101/TiO2–O2c | 3.9 ± 1.0 | 4 | 96 | 73 | 20 | 0.51 |
| 12 | 0.17% Au101/TiO2–O2–H2c | 4.6 ± 1.5 | 4 | 98 | 70 | 23 | 0.51 |
| 13 | 0.01% Au101/TiO2-untreatedd | <2e | 4 | 0 | 0 | 0 | 0 |
| 14 | 0.01% Au101/TiO2–O2d | <2e | 4 | 0 | 0 | 0 | 0 |
| 15 | 0.01% Au101/TiO2–O2–H2d | <2e | 4 | 0 | 0 | 0 | 0 |
| 16 | 1.3% Au101/TiO2-untreated | 2.7 ± 0.6 | 4 | 99 | 87 | 13 | 0.05 |
| 17 | 0.17% Au101/TiO2–O2 | 3.5 ± 0.8 | 3 | 93 | 78 | 22 | 0.65 |
| 18 | 0.17% Au101/TiO2–O2–H2 | 4.4 ± 1.2 | 3 | 92 | 76 | 24 | 0.65 |
| 19 | 0.17% Au9/TiO2-untreated | <2e | 4 | 0 | 0 | 0 | 0 |
| 20 | 0.17% Au9/TiO2–O2 | 2.4 ± 0.5 | 4 | 0 | 0 | 0 | 0 |
| 21 | 0.17% Au9/TiO2–O2–H2 | 2.9 ± 0.9 | 4 | 20 | 23 | 18 | 0.08 |
| 22 | 0.17% Au101/SiO2-untreated | <2e | 4 | 0 | 0 | 0 | 0 |
| 23 | 0.17% Au101/SiO2–O2 | 3.6 ± 2.7 | 4 | 69 | 72 | 24 | 0.25 |
| 24 | 0.17% Au101/SiO2–O2–H2 | 4.4 ± 2.5 | 4 | 69 | 70 | 26 | 0.26 |
| 25 | 0.5% Au101/SiO2-untreated | 3.6 ± 1.2 | 4 | 63 | 74 | 16 | 0.09 |
| 26 | 0.5% Au101/SiO2–O2 | 4.7 ± 1.6 | 4 | 95 | 63 | 31 | 0.13 |
| 27 | 0.5% Au101/SiO2–O2–H2 | 5.7 ± 2.9 | 4 | 99 | 65 | 32 | 0.14 |
| 28 | 0.17% Au101/TiO2–O2 + KNO3f | 3.5 ± 0.8 | 4 | 85 | 78 | 21 | 0.46 |
| 29 | 0.17% Au9/TiO2–O2 + KClf | 2.4 ± 0.5 | 4 | 0 | 0 | 0 | 0 |
Secondly, the nature of the support and metal–support interaction dictated the performance of the catalysts. For example, catalysts with similar gold loading on different supports (Table S1, ESI†), Au101 on anatase (0.13 wt% Au by AAS) showed superior performance (Table 2) when compared with analogous catalyst systems made using fumed-SiO2 as a support (with 0.18 wt% Au by AAS). Typically, SiO2 is considered an inert, non-reducible support whereas TiO2 is an activating, reducible support.67 The higher catalytic activity of Au/TiO2 catalysts over Au/SiO2 was also observed in CO oxidation in many studies.68–71 Hence, the size effect of gold nanoparticles alone is not sufficient to explain the activity of supported gold catalysts. Metal–support interactions (MSI) play crucial role in defining reactivity and selectivity of supported gold catalysts. Haruta et al. and Baiker et al. hypothesized that the oxygen activation occurs at the perimeter (i.e. interface) of the gold nanoparticles and metal oxide supports.72–74 This hypothesis could explain the lower activity of Au101/SiO2 catalysts as compared to Au101/anatase in benzyl alcohol oxidation observed in this study, since silica based catalysts with much larger particles will have much smaller surface area corresponding to the gold–support interface. The high activity of heat treated catalysts can be attributed to stronger metal–support interaction established during calcination. Haruta et al. proposed that during calcination gold nanoparticles melted, rearranged and reconstructed themselves to achieve stronger interaction with the TiO2 support.75 This hypothesis is in line with the report by Buffat and Borel that 2 nm gold nanoparticles melt around 600 K,76 which is significantly lower than the melting point of the bulk gold (1337 K). Wu et al. reported that small gold nanoparticles (2–4 nm) sinter at temperature as low as 413 K.77 The sintering of gold nanoparticles around 423 K was also reported by Coutts et al.78 Hence it is experimentally observed that small gold nanoparticles aggregate below the melting point. Analysis of supported-gold particle sizes as measured by TEM (Table 1) showed that the particle size distribution of gold nanoparticles was wider (indicated by larger standard deviation) on SiO2 support as compared to TiO2 for the same Au loading (target 0.17 wt%), suggesting that weaker metal–support interaction in Au101/SiO2 catalysts facilitates aggregation resulting in wider particle size distributions despite the significantly higher surface area of SiO2 support.
For the same Au loading (0.17 wt%) and support (TiO2), Au101 cluster-based catalysts showed much higher catalytic activity compared to Au9-based catalysts even though the gold particles size were smaller in the case of Au9/TiO2 (cf. Au101/TiO2 analogues, Table 2, entries 19–21 cf. entries 4–6). We hypothesize that the presence of anionic species within the gold cluster (Cl− in Au101vs. NO3− in Au9) strongly affect the catalytic activity. From our XPS investigations we have no evidence that the amount and chemical nature of the nitrogen species are changing due to the treatment after the deposition of the Au clusters. To identify the role of the component of the cluster precursor responsible for quenching of the catalytic activity, we investigated the role of such an anionic component in benzyl alcohol oxidation. Firstly, we added KCl, with the amount according to the weight percentage of Cl− in Au101(PPh3)21Cl5 (0.08 wt%) to mimic the presence of Cl− anion, to the catalyst and calcined it under O2 to resemble Au101/TiO2–O2; we found no increase in the catalytic activity (Table 2, entry 29 cf. entry 20). When KNO3 (amount estimated to mimic the amount of NO3− from Au9, 4.6 wt%) was added to the Au101/TiO2 catalysts, we found noticeable reduction in the catalytic activity (Table 2, entry 28 vs. 8). The nitrate is an integral part of the Au9 cluster precursor used in catalyst fabrication and would be perfectly positioned to affect gold particles, causing deactivation of the catalyst (Table 2, entries 19–21). For the Au101 based catalyst (Table 2, entry 28), the nitrate was introduced after the catalyst had been formed, which could limit the efficiency of the interaction of the nitrate with the gold particles and result in the moderate drop in activity. Hence, it appears that the presence of NO3 lowers the catalytic activity of Au101-based catalysts and could be the reason for the significantly lower activity of Au9-based analogues. To the best of our knowledge this is the first report of the effect of NO3− on activity of gold-based heterogeneous catalysts. While numerous studies report that ultra-small gold nanoparticles (<2 nm) are the key active site,23,26,79 our results demonstrate that the size of gold nanoparticles alone is not sufficient to explain the catalytic activity of catalysts made using phosphine-capped Au nanoparticles as it is considerably affected by the type of anions present in the supported gold cluster catalysts, type of support and activation treatment protocols. Quintanilla et al. investigated the effect of N-based compounds (dodecylamine vs. polyvinylpyrrolidone) as stabilizing agents for gold nanoparticles deposited onto γ-Al2O3 in benzyl alcohol oxidation.80 The authors found that the polyvinylpyrrolidone-capped gold nanoparticles had higher catalytic activity than the dodecylamine-capped gold nanoparticles due to the reduced steric hindrance of the gold surface by the polymer, allowing access of the substrate to the surface of gold nanoparticles. However, in our case, the N-based compound is present as a counter anion (NO3−), not as a stabilizing agent. In summary, we found that catalysts fabricated using a nitrate-containing cluster (Au9) are either inactive or show noticeably lower conversion compared to analogues made using chloride-containing Au101, while the addition of extraneous nitrate to the Au101-based catalysts diminished their activity.
The effect of different calcination conditions is negligible for Au101-based catalysts but has significant impact on Au9/TiO2 catalysts (see Table 2). Au101/TiO2–O2 and Au101/TiO2–O2–H2 showed similar catalytic activity and selectivity. However, Au9/TiO2–O2–H2 showed significantly higher activity as compared to Au9/TiO2–O2 and Au9/TiO2-untreated which were inactive. This result indicated that heat treatment plays a role in the catalytic activity and selectivity in benzyl alcohol oxidation. Yuan et al. prepared Au9/TiO2 for CO oxidation.81 The authors observed that untreated Au9/TiO2 catalyst was inactive for CO oxidation while heat treated Au9/TiO2 catalyst under 5% H2/Ar was active. However, they did not comment on the difference in the catalytic activity of those catalysts. In the case of Au nanoparticles containing residual Cl− species, calcination under H2 atmosphere could remove Cl− in the form of HCl, as proposed by Haruta et al.82 However, at this stage we could not conclude the fate of NO3− species after combined O2–H2 calcination because the available XPS data provide no evidence about how the and nature of the nitrogen species changes after such calcination.
The proposed reaction pathway of benzyl alcohol oxidation in methanol in the presence of base is shown in Fig. 6. Benzyl alcohol is first oxidized to benzaldehyde, which serves as an intermediate in this catalytic system. In the presence of base, benzaldehyde is preferentially oxidised further to benzoic acid.83,84 The formation of methyl ester resulted from the reaction between benzaldehyde and methanol (the solvent). We carried out a control reaction using benzaldehyde and benzoic acid as substrates (instead of benzyl alcohol). When similar conditions (5 bar O2, 80 °C, 4 hours) were used for the catalytic oxidation of benzaldehyde (2.5 mmol) in methanol (25 mL) using Au101/anatase catalysts, a complete transformation of benzaldehyde to benzoic acid and methyl benzoate with the same distribution of products (i.e. benzoic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methyl benzoate of 70:30) was observed. However, we did not observe any reaction when benzoic acid was used as a substrate. Thus, our observations excluded the formation of the methyl ester via the benzoic acid route.
:methyl benzoate of 70:30) was observed. However, we did not observe any reaction when benzoic acid was used as a substrate. Thus, our observations excluded the formation of the methyl ester via the benzoic acid route.
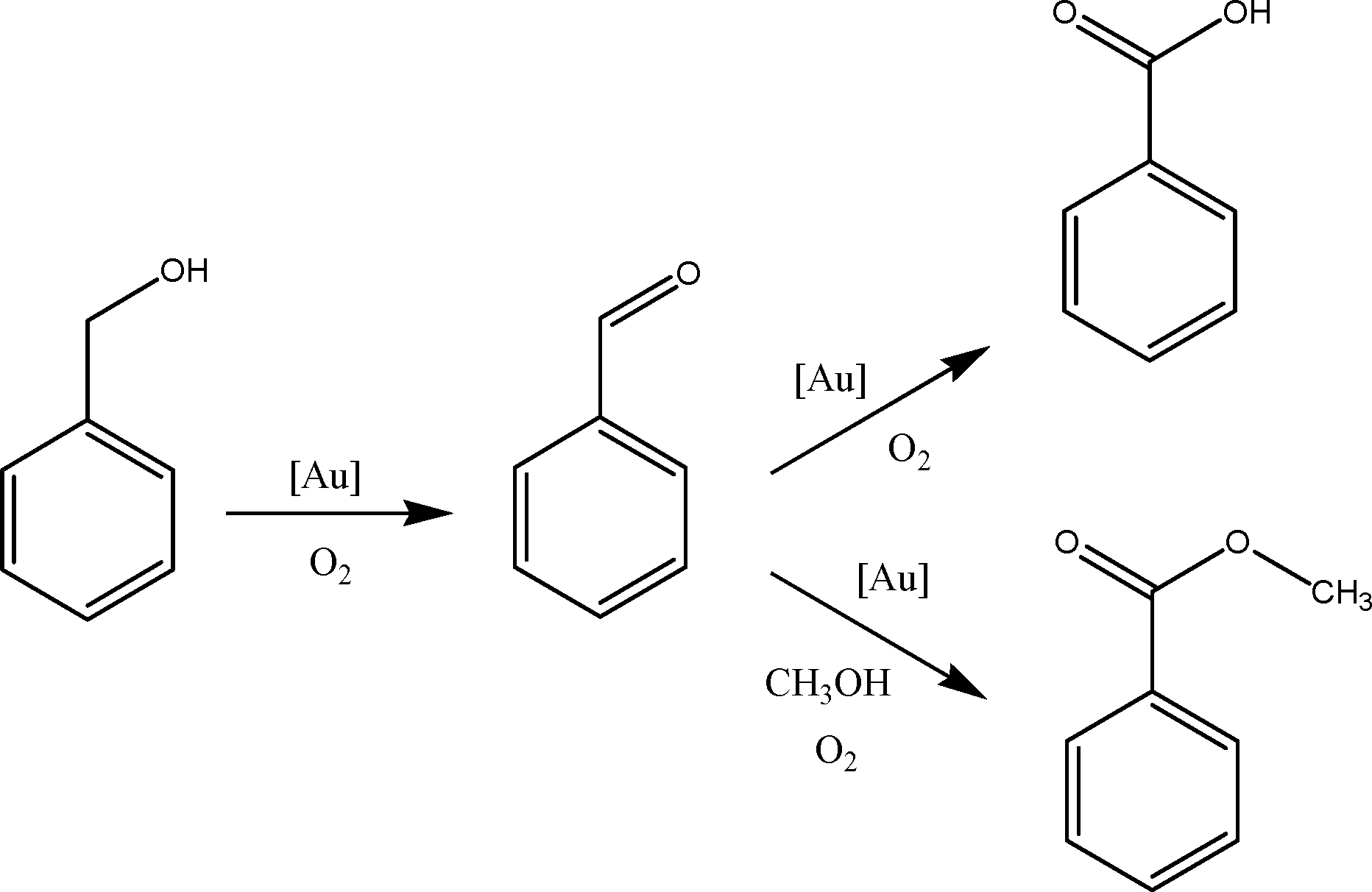 | ||
| Fig. 6 Proposed reaction pathway of oxidation of benzyl alcohol in methanol. | ||
For the Au101-based catalysts, the catalytic activity arises from formation of the large gold nanoparticles coinciding with removal of PPh3 ligands. All untreated Au101/TiO2 (0.01 and 0.17 wt%) and Au101/SiO2 (0.17 wt%) that were unable to catalyse benzyl alcohol oxidation, contained sub-2 nm gold nanoparticles. In trying to explain this observation one has to consider effects of metal core properties and the possibility that a significant proportion of an otherwise reactive gold surface could be blocked by protective phosphine ligands. Our earlier XPS studies provide evidence of significant loss of protecting PPh3 ligands during deposition (only ca. 10% of P XPS signal intensity remained, due to residual PPh3 ligands) in the case of 0.17 wt% Au101/TiO2.47 Although it is impossible to outright dismiss the argument that the residual 10% of ligands could completely deactivate the catalysts, one should attempt to find an alternative explanation. An earlier report by Valden et al. demonstrated an important transition from non-metallic to metallic properties occurring for ca. Au300 clusters on TiO2.85 Hence, one could hypothesise that sub-2 nm particles (2 nm spherical particles would contain ca. 250 atoms)86 could be inactive, as observed in our reactions, due to their non-metallic state. The presence of non-metallic particles in these catalysts is further confirmed by our UV-vis DRS results (Table 1, entries 1 and 14). However, untreated 1.3 wt% Au101/TiO2 (mean gold particle size of 2.7 ± 1.6 nm) and untreated 0.5% Au101/SiO2 (mean gold particle size of 3.6 ± 1.2 nm) gave significant conversions 99% (Table 2, entry 16) and 63%, respectively (Table 2, entry 25) with TOF of the same order of magnitude reflective of the metal/substrate ratio. This observation implies that in the case of highly metal-loaded Au101-derived catalysts, the formation of larger, metallic nanoparticles (which is likely to coincide with even more pronounced loss of phosphine ligands during aggregation) could be responsible for the observed activity. The gold nanoparticles in Au101/TiO2 catalysts agglomerated slightly after the catalytic tests (Table 2). The 31P NMR spectrum of the reaction mixture showed the presence of triphenylphosphine oxide, which was formed from the phosphine ligands dislodged from the gold core, which then oxidised during the catalytic reaction and dissolved in the catalytic reaction mixture. The Au101/TiO2-untreated catalyst was active upon recycling (i.e. in the 2nd consecutive catalytic test) and showed noticeably increased conversion (29%, Table 2, entry 10) compared to inactive fresh catalyst, and formed considerable amounts of benzaldehyde as compared to other catalytic tests in this study (Fig. S11†). Again, the increase in activity of the 0.17 wt% Au101/TiO2-untreated samples upon recycling could be attributed to an increase in size of the gold nanoparticles and the simultaneous loss of phosphine ligands from the gold core during the first catalytic test. However, comparison between the two catalysts with very similar ca. 3.5 nm particle sizes – 0.17 wt% Au101/TiO2 recycled (Table 2, entry 10) and activated by calcination under O2 (Table 2, entry 5) shows that particle size is clearly not the only factor. It should be emphasized that the TEM imaging does not allow easy quantification of the density/number of Au particles per particle of support and such TEM-based sizing alone could miss the presence of an inactive population of TEM-invisible smaller particles. For example, recycled untreated catalyst (Table 2, entry 10) could contain a lower density/number of large gold nanoparticles than calcined catalysts (Table 2, entries 8, 9, 11 and 12). Yet, a possibly more important factor in explaining the observed differences could be the effect of the higher-temperature treatment under O2 atmosphere which could form a better contact between the gold particles and the support, while PPh3 removal from the gold core is even more pronounced when compared to the recycled catalysts. For example, Haruta et al. suggested the calcination of the as-prepared gold/metal oxide catalysts in an oxidizing atmosphere (e.g. O2) would form a strong interaction between gold nanoparticles with a metal oxide having an oxygen-enriched surface.87 Both heat treated Au101/TiO2 catalysts under O2 and O2–H2 showed no significant loss of catalytic activity and selectivity after the first catalytic test.
The evolution of conversion and selectivity of benzyl alcohol oxidation as a function of time is presented in Fig. 7. The high conversion of benzyl alcohol (80–90%) was observed after the first two to three hours. The selectivity towards the formation of methyl benzoate and benzoic acid are independent of reaction time but primarily dependent on the reaction temperature, as shown in Fig. 8. A considerable amount (ca. 20%) of benzaldehyde was formed after one hour and then depleted over time (Fig. 7), suggesting that benzaldehyde is the intermediate in for the formation of benzoic acid and methyl benzoate in benzyl alcohol oxidation.88–90 The effect of the temperature in this reaction was studied from room temperature (25 °C) up to 80 °C (Fig. 8). Reduction in the temperature of the catalytic reaction results in the reduction of the catalytic activity. However, the product selectivity shows non-linear behaviour. The highest selectivity towards methyl benzoate (93%) was observed at 50 °C with 53% conversion of benzyl alcohol, suggesting the selective formation of methyl benzoate is possible by manipulating the reaction temperature.
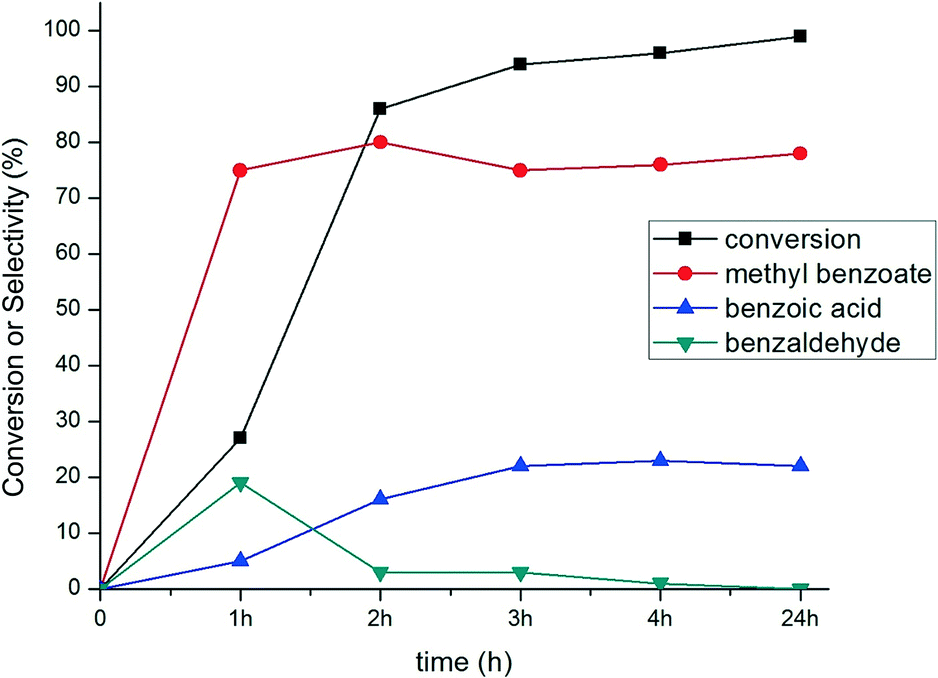 | ||
| Fig. 7 Time-dependent profile of benzyl alcohol oxidation using 0.17% Au101/TiO2–O2. Reaction conditions: 2.5 mmol benzyl alcohol, 25 mL methanol, 1.25 mmol anisole, 2.5 mmol K2CO3, 5 bar O2, 80 °C. | ||
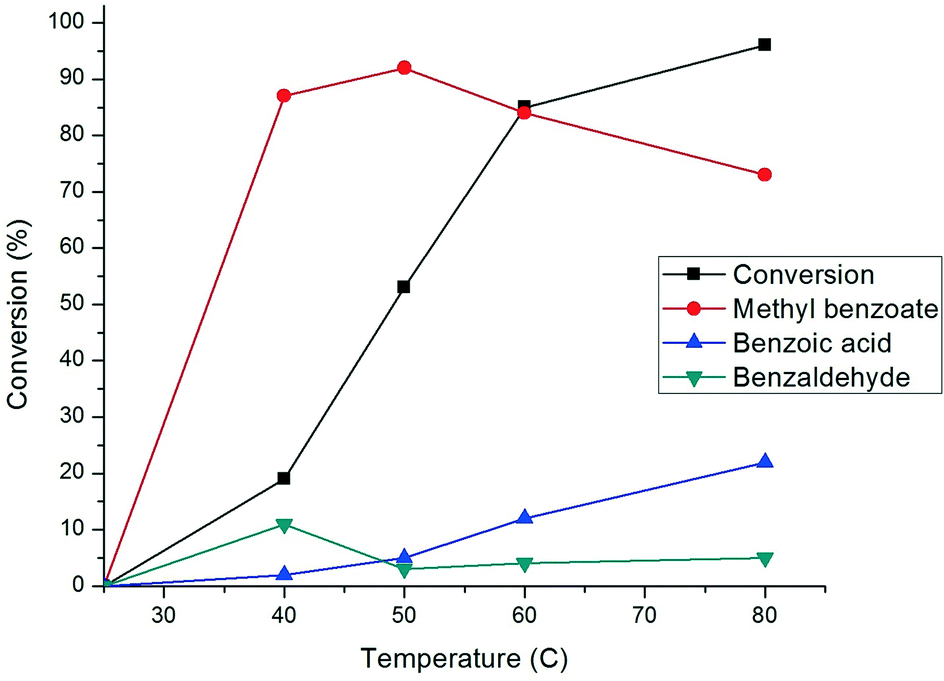 | ||
| Fig. 8 The effect of temperature on the conversion and profile of benzyl alcohol oxidation products using 0.17% Au101/TiO2–O2. Reaction conditions: 2.5 mmol benzyl alcohol, 25 mL methanol, 1.25 mmol anisole, 2.5 mmol K2CO3, 5 bar O2, and 4 hours. | ||
4 Conclusion
In summary, we report that high catalytic activity (conversions of >90%) is achieved using Au101-based catalysts whereas Au9-based catalysts are barely active. In this study, we find that the catalytic performance in benzyl alcohol oxidation is determined by gold particle size, the type of the support and the counter anions present in the gold cluster composition. For Au101-based catalysts on both TiO2 and SiO2, we observe that the catalytic activity appears with the formation of large (>2 nm) gold nanoparticles which coincides with the partial removal of phosphine ligands. We also observe the higher catalytic activity of TiO2-supported catalysts as compared to that of SiO2-supported catalysts, which could be due to the stronger metal–support interaction effect as proposed by Haruta. The effect of heat treatment under different atmospheres on catalytic performance is less pronounced in the case of Au101/TiO2 catalysts, but significantly affects Au9/TiO2 catalysts with noticeable catalytic activity (20% conversion) observed only for Au9/TiO2–O2–H2. The selectivity toward the formation of methyl benzoate can be tuned by manipulating the reaction temperature. The highest selectivity towards methyl benzoate was achieved at 50 °C with 93%.Acknowledgements
The authors would like to thank Professor Milo Kral and Mike Flaws for their help with TEM, Alistair Duff for AAS measurements of the gold content in catalysts, Dr. Meike Holzenkaempfer and Dr. Marie Squire for development of the HPLC methodology, and Professor Bryce Williamson for valuable discussions and Dr. Sedigheh Ghadamgahi for help with initial optimization of catalytic test conditions. This work was supported by the MacDiarmid Institute, University of Canterbury and University of Malaya, Kuala Lumpur (UM High Impact Research Grant UM-MOHE UM.C/HIR/MOHE/SC/11) and Australian Synchrotron (AS112/SXR/4641) for XPS study on phosphine gold nanoparticles).References
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405–408 CrossRef.
- B. Nkosi, M. D. Adams, N. J. Coville and G. J. Hutchings, J. Catal., 1991, 128, 378–386 CrossRef CAS.
- M. Haruta and M. Daté, Appl. Catal., A, 2001, 222, 427–437 CrossRef CAS.
- M. Haruta, Angew. Chem., Int. Ed., 2014, 53, 52–56 CrossRef CAS PubMed.
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS PubMed.
- T. Mallat and A. Baiker, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 11–28 CrossRef CAS PubMed.
- N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Chem. Sci., 2012, 3, 20–44 RSC.
- C. H. Christensen and J. K. Nørskov, Science, 2010, 327, 278–279 CrossRef PubMed.
- S. Mandal, K. K. Bando, C. Santra, S. Maity, O. O. James, D. Mehta and B. Chowdhury, Appl. Catal., A, 2013, 452, 94–104 CrossRef CAS PubMed.
- C. Della Pina, E. Falletta and M. Rossi, J. Catal., 2008, 260, 384–386 CrossRef CAS PubMed.
- H. Miyamura, T. Yasukawa and S. Kobayashi, Green Chem., 2010, 12, 776–778 RSC.
- V. Augugliaro and L. Palmisano, ChemSusChem, 2010, 3, 1135–1138 CrossRef CAS PubMed.
- C. P. Vinod, K. Wilson and A. F. Lee, J. Chem. Technol. Biotechnol., 2011, 86, 161–171 CrossRef CAS.
- J. Kilmartin, R. Sarip, R. Grau-Crespo, D. Di Tommaso, G. Hogarth, C. Prestipino and G. Sankar, ACS Catal., 2012, 2, 957–963 CrossRef CAS.
- V. R. Choudhary, A. Dhar, P. Jana, R. Jha and B. S. Uphade, Green Chem., 2005, 7, 768–770 RSC.
- V. R. Choudhary, R. Jha and P. Jana, Green Chem., 2007, 9, 267–272 RSC.
- T. Mallat and A. Baiker, Catal. Today, 1994, 19, 247–283 CrossRef CAS.
- P. Gallezot, Catal. Today, 1997, 37, 405–418 CrossRef CAS.
- R. Garcia, M. Besson and P. Gallezot, Appl. Catal., A, 1995, 127, 165–176 CrossRef CAS.
- J. H. J. Kluytmans, A. P. Markusse, B. F. M. Kuster, G. B. Marin and J. C. Schouten, Catal. Today, 2000, 57, 143–155 CrossRef CAS.
- M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127–141 CrossRef CAS.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552–560 CrossRef CAS.
- Y. Liu, H. Tsunoyama, T. Akita and T. Tsukuda, Chem. Lett., 2010, 39, 159–161 CrossRef CAS.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374–9375 CrossRef CAS PubMed.
- P. Haider, B. Kimmerle, F. Krumeich, W. Kleist, J.-D. Grunwaldt and A. Baiker, Catal. Lett., 2008, 125, 169–176 CrossRef CAS.
- H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086–7093 CrossRef CAS PubMed.
- N. F. Zheng and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 14278–14280 CrossRef CAS PubMed.
- Y. Liu, H. Tsunoyama, T. Akita, S. Xie and T. Tsukuda, ACS Catal., 2010, 1, 2–6 CrossRef.
- U. Hartfelder, C. Kartusch, M. Makosch, M. Rovezzi, J. Sa and J. A. van Bokhoven, Catal. Sci. Technol., 2013, 3, 454–461 CAS.
- Q. Zhang, W. Deng and Y. Wang, Chem. Commun., 2011, 47, 9275–9292 RSC.
- A. Villa, D. Wang, D. S. Su and L. Prati, ChemCatChem, 2009, 1, 510–514 CrossRef CAS.
- R.-Y. Zhong, X.-H. Yan, Z.-K. Gao, R.-J. Zhang and B.-Q. Xu, Catal. Sci. Technol., 2013, 3, 3013–3019 CAS.
- K. Y. Lee, Y. W. Lee, J.-H. Lee and S. W. Han, Colloids Surf., A, 2010, 372, 146–150 CrossRef CAS PubMed.
- A. Kulkarni, R. J. Lobo-Lapidus and B. C. Gates, Chem. Commun., 2010, 46, 5997–6015 RSC.
- X. Nie, C. Zeng, X. Ma, H. Qian, Q. Ge, H. Xu and R. Jin, Nanoscale, 2013, 5, 5912–5918 RSC.
- G. Li, C. Liu, Y. Lei and R. Jin, Chem. Commun., 2012, 48, 12005–12007 RSC.
- G. Li, H. Qian and R. Jin, Nanoscale, 2012, 4, 6714–6717 RSC.
- Y. Zhu, H. Qian and R. Jin, Chem. – Eur. J., 2010, 16, 11455–11462 CrossRef CAS PubMed.
- Y. Zhu, H. Qian, B. A. Drake and R. Jin, Angew. Chem., Int. Ed., 2010, 49, 1295–1298 CrossRef CAS PubMed.
- P. Huang, G. Chen, Z. Jiang, R. Jin, Y. Zhu and Y. Sun, Nanoscale, 2013, 5, 3668–3672 RSC.
- G. Li, C. Zeng and R. Jin, J. Am. Chem. Soc., 2014, 136, 3673–3679 CrossRef CAS PubMed.
- Y. Liu, H. Tsunoyama, T. Akita and T. Tsukuda, Chem. Commun., 2010, 46, 550–552 RSC.
- V. G. M. Turner, P. Abdulkin, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981–983 CrossRef PubMed.
- B. G. Donoeva, D. S. Ovoshchnikov and V. B. Golovko, ACS Catal., 2013, 3, 2986–2991 CrossRef CAS.
- D. S. Ovoshchnikov, B. G. Donoeva, B. E. Williamson and V. B. Golovko, Catal. Sci. Technol., 2014, 4, 752–757 CAS.
- G. H. Woehrle, L. O. Brown and J. E. Hutchison, J. Am. Chem. Soc., 2005, 127, 2172–2183 CrossRef CAS PubMed.
- D. P. Anderson, R. H. Adnan, J. F. Alvino, O. Shipper, B. Donoeva, J.-Y. Ruzicka, H. Al Qahtani, H. H. Harris, B. Cowie, J. B. Aitken, V. B. Golovko, G. F. Metha and G. G. Andersson, Phys. Chem. Chem. Phys., 2013, 15, 14806–14813 RSC.
- D. P. Anderson, J. F. Alvino, A. Gentleman, H. A. Qahtani, L. Thomsen, M. I. J. Polson, G. F. Metha, V. B. Golovko and G. G. Andersson, Phys. Chem. Chem. Phys., 2013, 15, 3917–3929 RSC.
- V. Peneau, Q. He, G. Shaw, S. A. Kondrat, T. E. Davies, P. Miedziak, M. Forde, N. Dimitratos, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2013, 15, 10636–10644 RSC.
- A. Villa, D. Wang, G. M. Veith, F. Vindigni and L. Prati, Catal. Sci. Technol., 2013, 3, 3036–3041 CAS.
- Handbook of Preparative Inorganic Chemistry, ed. G. Brauer, Academic Press, NY, 1963, vol. 1 Search PubMed.
- W. W. Weare, S. M. Reed, M. G. Warner and J. E. Hutchison, J. Am. Chem. Soc., 2000, 122, 12890–12891 CrossRef CAS.
- F. Wen, U. Englert, B. Gutrath and U. Simon, Eur. J. Inorg. Chem., 2008, 2008, 106–111 CrossRef.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- R. Balasubramanian, R. Guo, A. J. Mills and R. W. Murray, J. Am. Chem. Soc., 2005, 127, 8126–8132 CrossRef CAS PubMed.
- P. V. Kamat, J. Phys. Chem. B, 2002, 106, 7729–7744 CrossRef CAS.
- S. Link and M. A. El-Sayed, Annu. Rev. Phys. Chem., 2003, 54, 331–366 CrossRef CAS PubMed.
- E. A. Coronado, E. R. Encina and F. D. Stefani, Nanoscale, 2011, 3, 4042–4059 RSC.
- R. Fenger, E. Fertitta, H. Kirmse, A. F. Thunemann and K. Rademann, Phys. Chem. Chem. Phys., 2012, 14, 9343–9349 RSC.
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef CAS.
- C. Yang, Y. Zhou, G. An and X. Zhao, Opt. Mater., 2013, 35, 2551–2555 CrossRef CAS PubMed.
- J. Yang, Y. Guan, T. Verhoeven, R. van Santen, C. Li and E. J. M. Hensen, Green Chem., 2009, 11, 322–325 RSC.
- N. Zheng and G. D. Stucky, Chem. Commun., 2007, 3862–3864 RSC.
- T. Ishida, M. Nagaoka, T. Akita and M. Haruta, Chem. – Eur. J., 2008, 14, 8456–8460 CrossRef CAS PubMed.
- F.-Z. Su, Y.-M. Liu, L.-C. Wang, Y. Cao, H.-Y. He and K.-N. Fan, Angew. Chem., Int. Ed., 2008, 47, 334–337 CrossRef CAS PubMed.
- A. Abad, P. Concepción, A. Corma and H. García, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef CAS PubMed.
- M. M. Schubert, S. Hackenberg, A. C. van Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113–122 CrossRef CAS.
- S. D. Lin, M. Bollinger and M. A. Vannice, Catal. Lett., 1993, 17, 245–262 CrossRef CAS.
- H. Zhu, Z. Ma, J. C. Clark, Z. Pan, S. H. Overbury and S. Dai, Appl. Catal., A, 2007, 326, 89–99 CrossRef CAS PubMed.
- S. H. Overbury, L. Ortiz-Soto, H. Zhu, B. Lee, M. Amiridis and S. Dai, Catal. Lett., 2004, 95, 99–106 CrossRef CAS.
- L. Delannoy, N. El Hassan, A. Musi, N. N. Le To, J.-M. Krafft and C. Louis, J. Phys. Chem. B, 2006, 110, 22471–22478 CrossRef CAS PubMed.
- M. Haruta, Chem. Rec., 2003, 3, 75–87 CrossRef CAS PubMed.
- M. Daté, M. Okumura, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 2129–2132 CrossRef PubMed.
- J.-D. Grunwaldt and A. Baiker, J. Phys. Chem. B, 1999, 103, 1002–1012 CrossRef CAS.
- S. Tsubota, T. Nakamura, K. Tanaka and M. Haruta, Catal. Lett., 1998, 56, 131–135 CrossRef CAS.
- P. Buffat and J. P. Borel, Phys. Rev. A: At., Mol., Opt. Phys., 1976, 13, 2287–2298 CrossRef CAS.
- Y. Wu, Y. Li, P. Liu, S. Gardner and B. S. Ong, Chem. Mater., 2006, 18, 4627–4632 CrossRef CAS.
- M. J. Coutts, M. B. Cortie, M. J. Ford and A. M. McDonagh, J. Phys. Chem. C, 2009, 113, 1325–1328 CAS.
- Y. Liu, H. Tsunoyama, T. Akita and T. Tsukuda, J. Phys. Chem. C, 2009, 113, 13457–13461 CAS.
- A. Quintanilla, V. C. L. Butselaar-Orthlieb, C. Kwakernaak, W. G. Sloof, M. T. Kreutzer and F. Kapteijn, J. Catal., 2010, 271, 104–114 CrossRef CAS PubMed.
- Y. Yuan, K. Asakura, H. Wan, K. Tsai and Y. Iwasawa, Catal. Lett., 1996, 42, 15–20 CrossRef CAS.
- M. Haruta, CATTECH, 2002, 6, 102–115 CrossRef CAS.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17–45 RSC.
- C. Bianchi, F. Porta, L. Prati and M. Rossi, Top. Catal., 2000, 13, 231–236 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS.
- C. L. Cleveland, Phys. Rev. Lett., 1997, 79, 1873–1876 CrossRef CAS.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- C. Marsden, E. Taarning, D. Hansen, L. Johansen, S. K. Klitgaard, K. Egeblad and C. H. Christensen, Green Chem., 2008, 10, 168–170 RSC.
- A. B. Powell and S. S. Stahl, Org. Lett., 2013, 15, 5072–5075 CrossRef CAS PubMed.
- W. Cui, M. Jia, W. Ao and B. Zhaorigetu, React. Kinet., Mech. Catal., 2013, 110, 437–448 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental details about synthesis of the gold clusters and the characterisations are available. See DOI: 10.1039/c4cy01168f |
| This journal is © The Royal Society of Chemistry 2015 |