
Production of alcohols via hydroformylation
Galina Morales
Torres
a,
Robin
Frauenlob
a,
Robert
Franke
bc and
Armin
Börner
*ad
aLeibniz-Institut für Katalyse an der Universität Rostock e.V., A.-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: armin.boerner@catalysis.de
bEvonik Industries AG, Paul-Baumann-Straße 1, 45772 Marl, Germany
cLehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum, Universitätsstraße 150, 44780 Bochum, Germany
dInstitut für Chemie der Universität Rostock e.V., A.-Einstein-Straße 3a, 18059 Rostock, Germany
First published on 3rd November 2014
Abstract
The numerous approaches for the catalytic synthesis of alkyl alcohols using an intermediate hydroformylation step are reviewed. One main strategy is a sequence where hydroformylation and hydrogenation are carried out step by step. More challenging are hydroformylation reactions under reducing conditions. In this regard, the transformation can be assisted by two different catalysts or one single catalyst (tandem reaction). A particular challenge in this respect is the undesired olefin hydrogenation. Sequences where hydroformylation is combined with a subsequent aldol reaction are of huge economic importance. The different performances of the catalytic systems on the basis of rhodium, cobalt, palladium, and ruthenium are described together with typical organic ligands, reaction conditions, and selected applications.
 Galina Morales Torres | Galina Morales Torres studied chemistry at the University Oriente, Cuba. After working for a period as a research assistant in the University of Granma, Cuba in 2004 she joined the group of Prof. Dr. Christian Vogel, Rostock, Germany and completed a PhD on the synthesis of carbohydrates in 2007. Between 2007 and 2014 she has worked as research assistant and professor of organic chemistry at the University of Granma. At present she is part of Prof. Börner's group at the Leibniz-Institute for Catalysis (LIKAT), Germany. Her research focuses on applied homogeneous catalysis and natural products. |
 Robin Frauenlob | Robin Frauenlob received his Ph.D from Trinity College Dublin in 2013. His research focused on transition metal catalysis and combinatorial chemistry. After a postdoctoral position in the group of Prof. Dr. Bettinger where he synthesized modified acenes, he joined the group of Prof. Börner at the Leibniz Institute for Catalysis in Rostock. His current work includes tandem reactions of aromatic compounds. |
 Robert Franke | Robert Franke studied chemistry with focuses on industrial chemistry and theoretical chemistry at Bochum University in Germany. He earned his doctorate degree in 1994 in the field of relativistic quantum chemistry under Prof. Dr. W. Kutzelnigg. After working for a period as a research assistant, in 1998 he joined the process engineering department of the former Hüls AG, a predecessor company of Evonik Industries AG. He is now Director Innovation Management Hydroformylation. He was awarded his professorial research degree (Habilitation) in 2002, since when he has taught at the University of Bochum. In 2011 he was made adjunct professor. His research focuses on homogeneous catalysis, process intensification, and computational chemistry. |
 Armin Börner | Armin Börner studied chemistry at the University of Rostock and finished his Ph.D. thesis about the synthesis of carbohydrates in 1984. Between 1984 and 1992 he was a scientific co-worker at the Academia of Science of the GDR. After finishing his postdoctoral stay in the group of Prof. Dr. H. B. Kagan in Orsay/France in 1993 he went to the Max-Planck-Group for Asymmetric Catalysis where he habilitated in 1995. Since 2000 he is professor of Organic Chemistry at the University of Rostock and head of the department “Asymmetric Catalysis” at LIKAT. He authored ca. 200 papers and reviews and is named as founder in more than 60 patents. About 15 chemical transformations and analytic tools developed in his department have been commercialized or running in an industrial scale. |
1. Introduction
Hydroformylation is the addition of synthesis gas to olefins with the formation of aldehydes.1 The reaction is one of the largest homogeneously catalyzed reactions in industry. The resulting aldehydes are used as intermediates or end products in bulk chemistry and also as components of drugs and for the manufacture of aroma compounds.2 Frequently, the ultimate target of reaction sequences that include a hydroformylation step is the manufacture of alcohols. Alcohols derived from hydroformylation processes are used as organic solvents and detergents or are enormously important as ester components, such as in plasticizers for polymers (PVC). Moreover, elimination of water from alcohols produces olefins, which allows the production of homologue olefins via a hydroformylation–hydrogenation elimination sequence.3 In principle, there are three main approaches for the preparation of alcohols via hydroformylation (Scheme 1):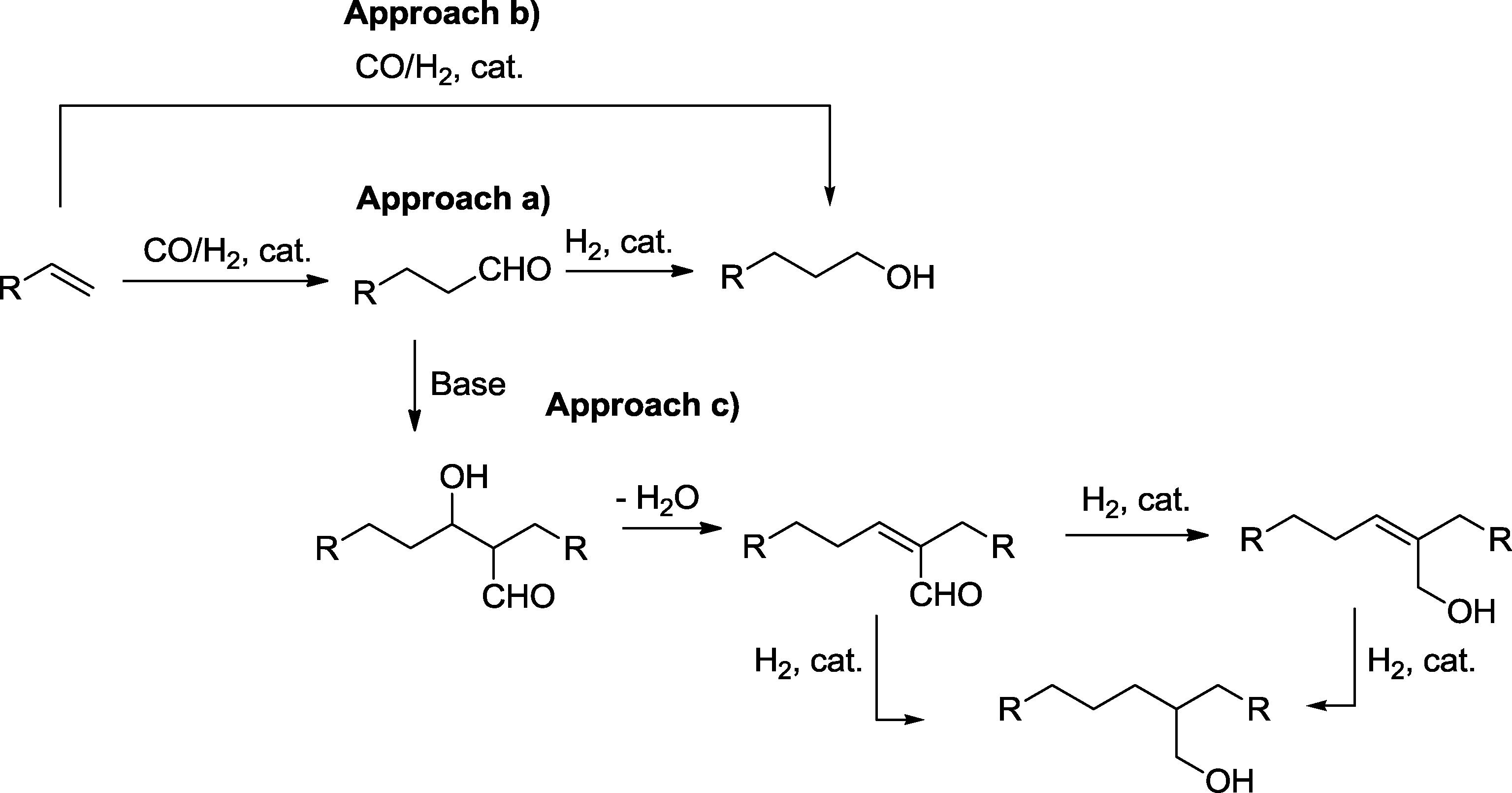 | ||
| Scheme 1 Pathways for the production of alcohols via hydroformylation. | ||
a) A two-step sequence, where hydrogenation directly follows hydroformylation in a separate or in the same reaction vessel.
b) A combined approach with hydroformylation under reducing conditions.
c) A multi-step sequence, where hydroformylation and hydrogenation are separated by another reaction, such as aldol condensation. Final chemoselective reduction of the α,β-unsaturated aldehydes produces either allyl alcohols or 2,2-branched ethanol derivatives.4
Multistep reactions involving a hydroformylation reaction have been addressed several times in the literature.1b,5 These reviews mainly looked at sequences where hydroformylation is part of a so-called tandem reaction. A typical example of this is the hydroformylation–hydrogenation tandem reaction. It should be noted however, that there are several sequential reactions for the production of alcohols using a hydroformylation step, which in a stricter sense cannot be described as tandem reactions.6,7
In this review, the main strategies, problems and achievements will be considered in detail in order to summarize the current state of the art in academic research together with established industrial processes.
2. Production of alcohols in a combined hydroformylation–hydrogenation approach
With the exception of examples where the aldehyde group is indispensable in an intermediate aldol reaction (see below), approaches that directly produce the alcohol from a hydroformylation–hydrogenation protocol are highly desired and have been the focus of past research. When a one-pot reaction is chosen, both reactions can be assisted by two catalysts under the same or under different reaction conditions. In the best case, the requirements for a tandem reaction are fulfilled,6,8 where a single or two catalysts manage both reactions under the same reaction conditions.It should be considered that in general, hydroformylation and hydrogenation require different catalytic conditions, which explains why two-step protocols were preferred in the past. One-pot procedures are better because they streamline the process by lowering reaction time, labor, and overall cost. In principle, we can differentiate between the following approaches for the production of alcohols based on a hydroformylation–hydrogenation approach (Scheme 2):
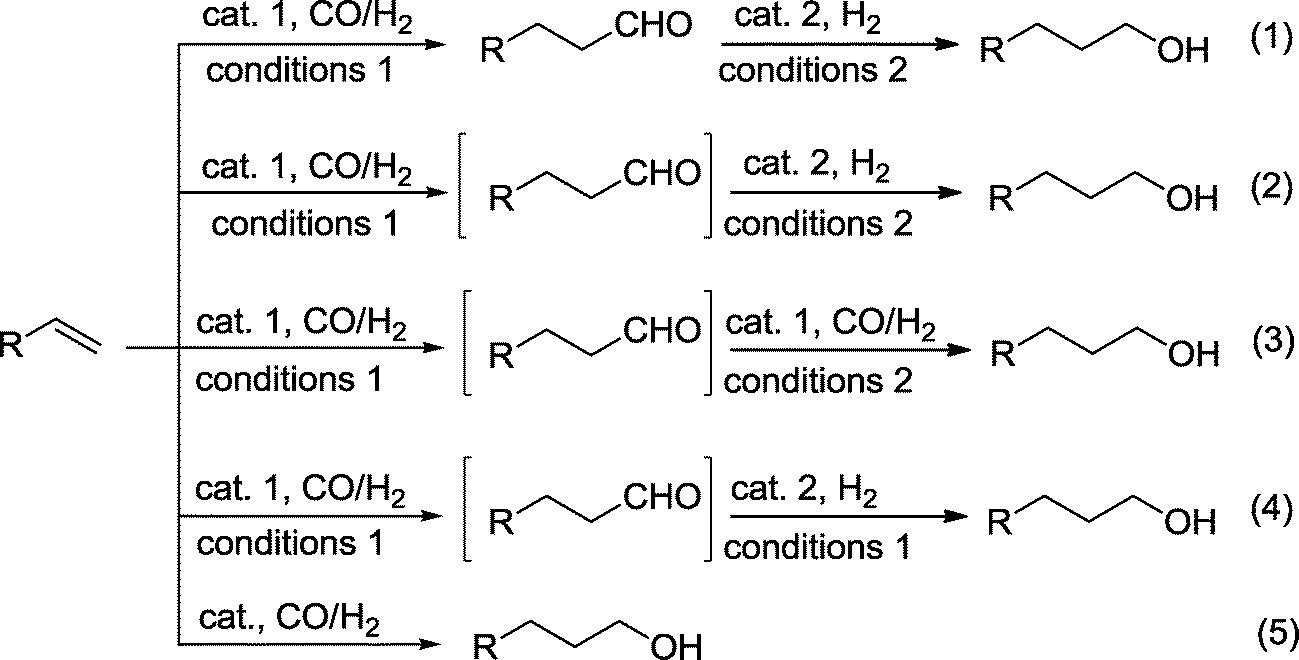 | ||
| Scheme 2 Synthesis of alcohols in a combined hydroformylation–hydrogenation approach. | ||
(1) Two-step process in separate vessels using different catalytic conditions.
(2) One-pot reaction with two catalysts and using different reaction conditions (bicatalytic reaction).
(3) One-pot reaction with a single catalyst using different reaction conditions.
(4) One-pot reaction with two functionally distinct catalysts. They are present from the outset of the reaction. In the best case, the same conditions are used for both reactions (orthogonal tandem reaction).
(5) One-pot reaction with a single catalyst without change of the reaction conditions (auto-tandem reaction).
2.1. Two-step process in separate vessels using different catalytic conditions
This is still one of the preferred operation modes for the production of alcohols on an industrial scale. Usually, hydroformylation is carried out with a homogeneous catalyst, while a heterogeneous hydrogenation catalyst9 is used for the second step. The latter may be located in the same reactor or another reaction zone.10 For both transformations, the individual reaction conditions can be adjusted optimally. In most cases, the hydroformylation catalyst is removed prior to hydrogenation. Hydrogenation of the starting olefin, which is frequently observed during a one-pot hydroformylation–hydrogenation sequence, can be avoided and generally, yields of the isolated aldehydes and product alcohols, respectively, are high. By using this approach, the separated catalysts can be easily recycled.In a typical approach, Oxeno Olefinchemie (now Evonik Industries) patented the hydroformylation of olefins with C6–C16 carbon atoms with a subsequent hydrogenation step (Scheme 3).11 The hydroformylation is catalyzed by a homogeneous cobalt catalyst, which is decomposed with gases containing oxygen after the reaction (“decobalting”).12 After separation of the aqueous phase, containing Co(II)-salts, and the organic phase, unconverted olefins are removed from the aldehydes by distillation. Final hydrogenation of the aldehyde containing distillation residue mediated by Cu, Ni, Cu/Ni, Cu/Cr, Cu/Cr/Ni, Zn/Cr, or Ni/Mo catalysts, yields the corresponding alcohols.
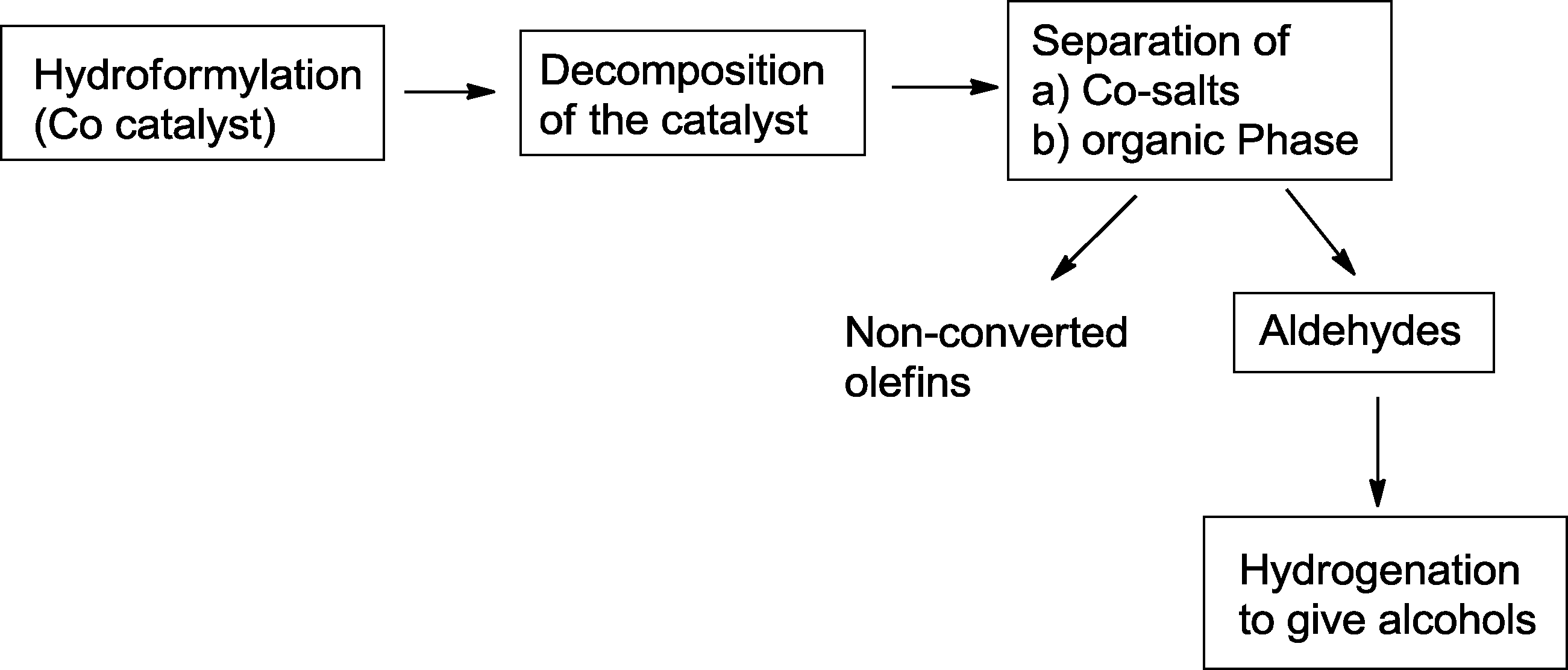 | ||
| Scheme 3 A general two-step approach for the synthesis of alcohols by cobalt catalyzed hydroformylation–hydrogenation.11 | ||
It should be noted that aldehydes formed during hydroformylation may already react further with hydrogen to form alcohols, or with CO and H2 to produce formate esters. Under acidic conditions, alcohols react with aldehydes to produce acetals. This can be avoided by adding water to both the hydroformylation and the subsequent hydrogenation reaction to promote hydrolysis of formate esters and to delay the acetal formation reaction.13 Addition of water and dilution of the substrate also helps avoiding the Canizzaro reaction in the hydrogenation step, which produces acids. Moreover, numerous heterogeneous hydrogenation catalysts are sensitive to traces of sulfur, which may be contained in the olefin feed. Either sulfur is removed before the hydroformylation or a catalyst that is resistant to this impurity (e.g. sulfided Co/Mo, sulfided Ni oxide/WO or sulfided Ni oxide) is used.13 Sometimes, a second hydrogenation process using a different heterogeneous catalyst may be beneficial to further reduce the unwanted methyl ester content.14
BASF claimed the synthesis of a C17 alcohol mixture via an oligomerization–hydroformylation–hydrogenation sequence (Scheme 4).15 The required olefin is prepared by tetramerization of a mixture of i-butene, 1-butene and isomeric 2-butenes (“Raffinate II”) to produce isomeric C16-olefins employing heterogeneous catalysts consisting NiO, TiO2 and Al2O3 as active components. Hydroformylation is carried out in a continuously operating apparatus with the aid of an aqueous cobalt catalyst.16 Each hour, 2.2 metric tons of the olefin were converted into a mixture of isomeric aldehydes. Final hydrogenation in the presence of a heterogeneous Co/Cu/Mo catalyst produced the corresponding mixture of alcohols,17 which were characterized by an iso-index = 3.1.18 These long chain alcohols are particularly suitable for use in detergents.
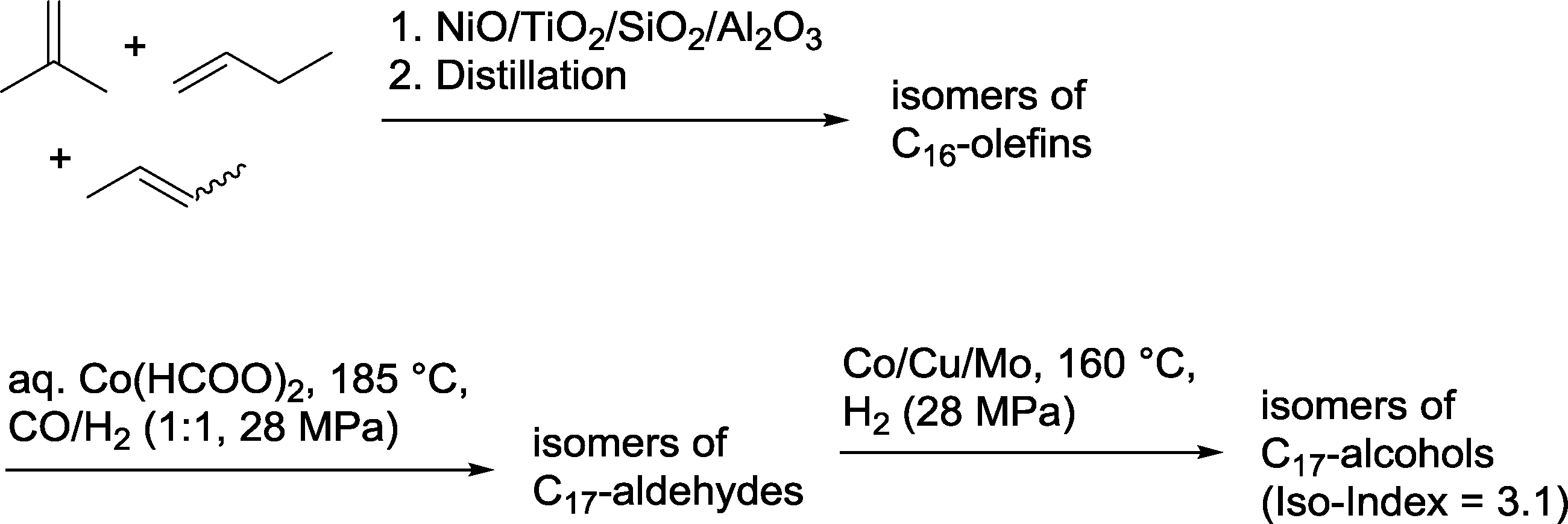 | ||
| Scheme 4 Oligomerization–hydroformylation–hydrogenation sequence for the production of C17 alcohols.15 | ||
When rhodium catalysts, which are modified with ligands containing nitrogen or phosphorus, are used, the catalyst is usually separated from the hydroformylation product by distillation, wherein the rhodium catalyst, together with high-boiling components, remains as a residue. An alternative is liquid/liquid extraction of the aldehyde from the catalyst solution.
A typical example is the Kuraray process, which is used for the chemical production of 1,4-butanediol (BDO) (Scheme 5). In the first step, the rhodium catalyzed hydroformylation of allyl alcohol is carried out in a continuous reactor in the presence of PPh3/PPh2(CH2)4PPh2 (ref. 19) or DIOP20 as ligands in toluene at 65 °C and a syngas pressure of approx. 0.2 MPa. In turn, 4-hydroxybutyraldehyde that forms is extracted with water and subsequently reduced under a pure H2 stream using a RANEY®-Ni catalyst. 1,4-Butanediol is an important starting material for the production of several bulk chemicals, such as tetrahydrofurane and γ-butyrolactone. It is also used as cross-linking agent in polymers (urethanes) and as an alcohol component in polyesters (polybutylene terephthalate (PBT)).21
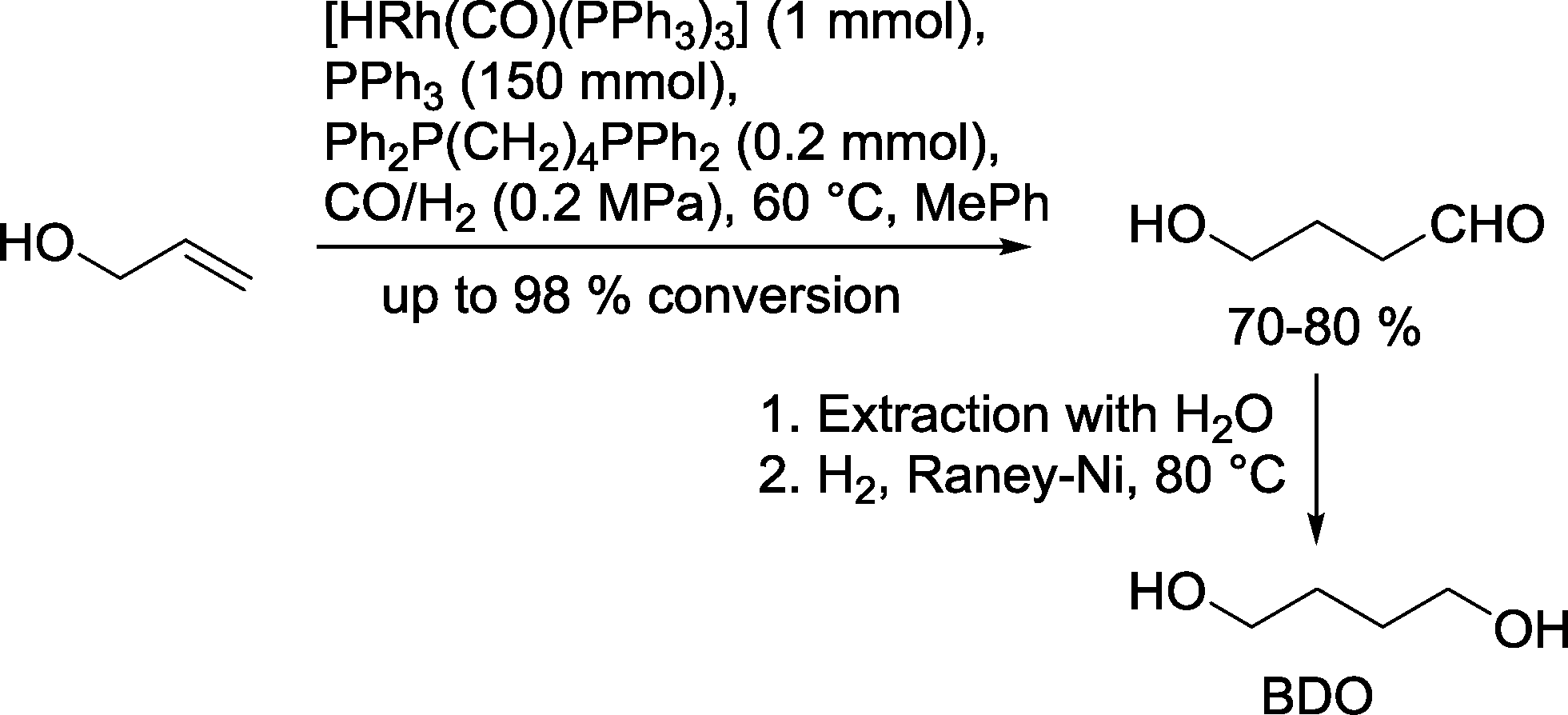 | ||
| Scheme 5 Production of butane-1,4-diol via rhodium catalyzed hydroformylation.19 | ||
2.2. One-pot reaction with two catalysts and using different reaction conditions
This approach can be used only under the condition that both catalysts do not affect each other in their catalytic performance. Diéguez and Masdeu-Bultó developed a one-pot protocol for the preparation of 3-hydroxy-2-methylpropionamide this is starting from acrylamide (Scheme 6).22 Hydroformylation was carried out with a homogeneous PPh3 modified rhodium catalyst. Under optimized conditions, no n-aldehyde was detected and the yield of the branched aldehyde was extremely high. Without isolation, RANEY®-Ni was added to the reaction mixture. Under a pure hydrogen stream, 3-hydroxy-2-methylpropanamide was formed. The product may be used as a starting material for the manufacture of the PMMA-monomer methyl methacrylate.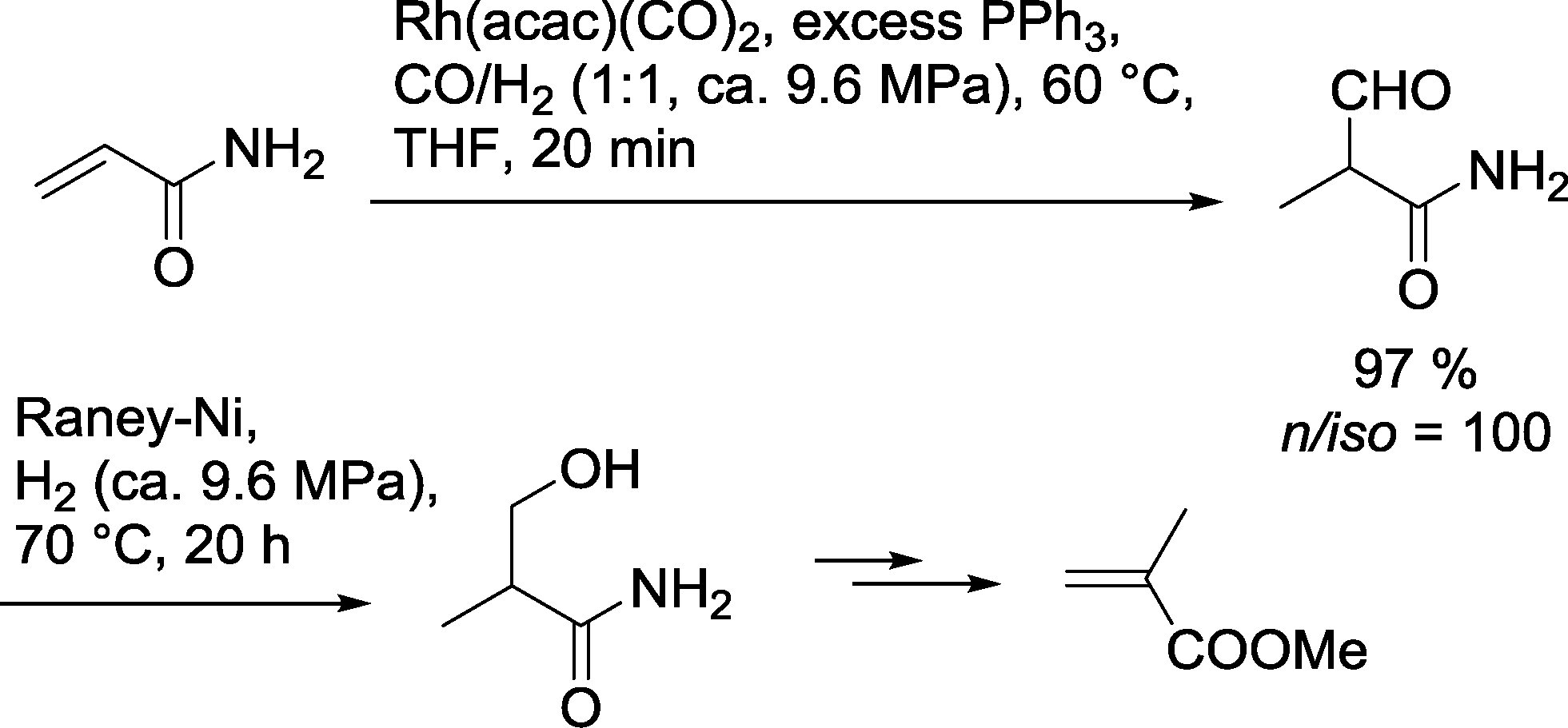 | ||
| Scheme 6 Synthesis of methyl methacrylate using a one-pot hydroformylation–hydrogenation approach.22 | ||
The Bell group investigated one-pot hydroformylation reduction of propene and 1-hexene (Scheme 7).23 After completion of the hydroformylation assisted by a Rh(PPh3) catalyst, the aldehyde products were immediately reduced by means of Ru3(PPh3)3Cl2. At a ratio of PPh3 to Rh or Ru of 103/1, the olefins were almost completely converted to the relevant alcohols. Only traces of isomerized starting olefin were observed, but no formation of the corresponding alkanes. In order to avoid poisoning of the Ru hydrogenation catalyst by CO, almost stoichiometric amounts of CO in relation to the olefin have been recommended. Alternatively, excess CO has been purged from the reactor (multiple rinsing with helium) before hydrogenation was initiated. Hydrogenation benefited from a higher temperature compared to the previous hydroformylation step. The preferred solvent for both reactions is 2-propanol. In particular, n-butanol synthesized by this process is a chemical compound of increasing importance.24 About 2 × 106 metric tons are produced annually for use as a solvent, in plasticizers, and as intermediates for butyl acetate.25 Moreover, it is expected that butanol can replace ethanol as an additive to gasoline.
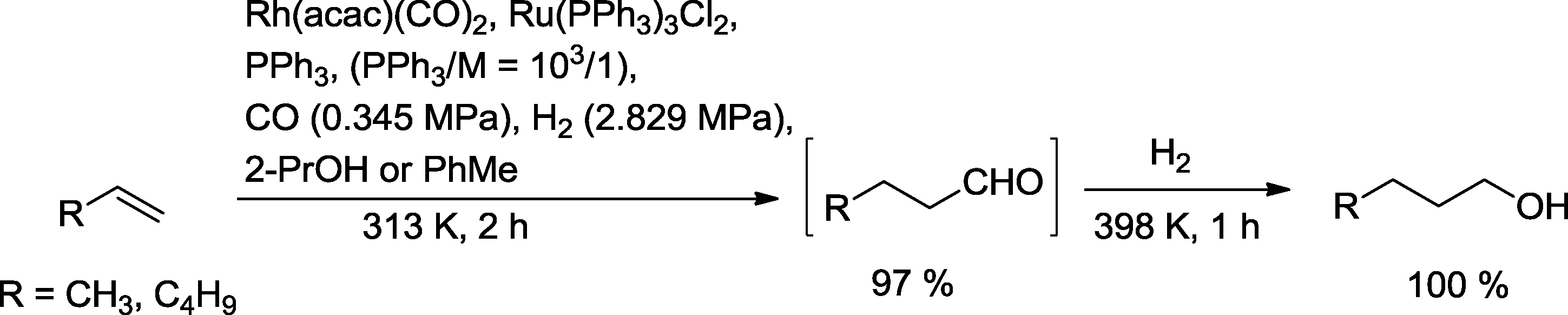 | ||
| Scheme 7 Synthesis of primary alcohols with a one-pot hydroformylation–hydrogenation approach using different gaseous reactants.23 | ||
2.3. One-pot reaction with a single catalyst using different reaction conditions
Sequential one-pot reactions can benefit from a single catalyst, which exhibits different catalytic properties in relation to the reaction conditions. Reek and van Leeuwen designed a silica-supported, switchable, and recyclable hydroformylation–hydrogenation catalyst based on a Xantphos-type ligand (Scheme 8).26 The catalyst for hydroformylation is a neutral rhodium(I) hydride species, whereas the hydrogenation catalyst is its cationic congener. The latter is formed by protonation of the hydride complex with the acidic groups of the polysilicate during the immobilization process. Both catalysts coexist under hydroformylation conditions (CO/H2 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 5 MPa; 80 °C). The neutral complex converted 1-octene with 95% yield into 1-nonanal. Subsequently, the second catalyst reduced nonanal to nonanol with a yield of 97%. When propanol was added, the hydroformylation catalyst could be regenerated and the hydrogenation mode was fully suppressed.
:1, 5 MPa; 80 °C). The neutral complex converted 1-octene with 95% yield into 1-nonanal. Subsequently, the second catalyst reduced nonanal to nonanol with a yield of 97%. When propanol was added, the hydroformylation catalyst could be regenerated and the hydrogenation mode was fully suppressed.
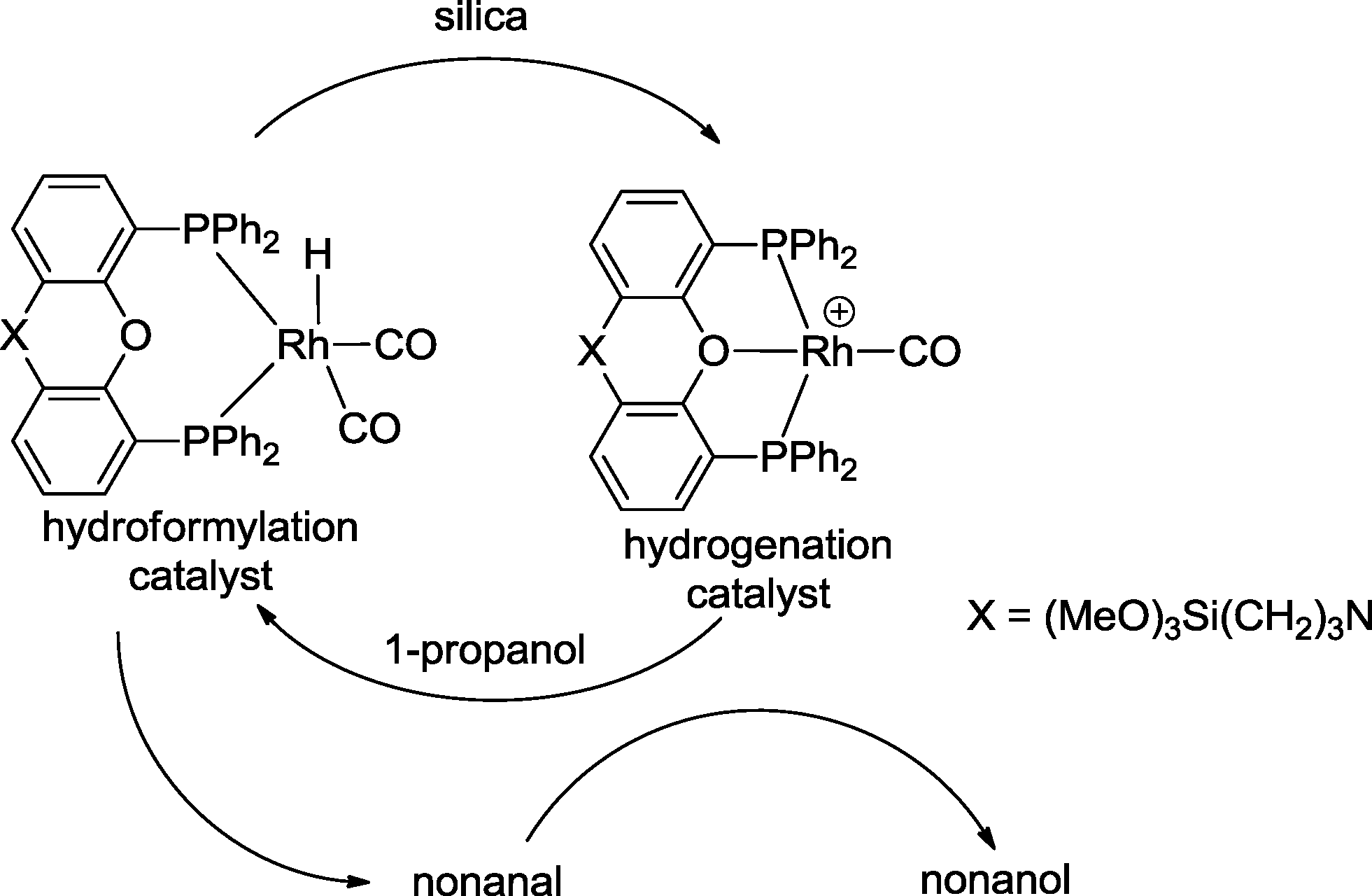 | ||
| Scheme 8 One-pot reaction with a switchable catalyst.26 | ||
2.4. One-pot reaction with two functionally distinct catalysts under uniform reaction conditions
Nozaki and co-workers developed a combination of Rh(Xantphos), Shvo's catalyst and Ru3(CO)12 for the orthogonal hydroformylation–hydrogenation tandem reaction of the fatty alcohol undecanol starting from 1-decene (Scheme 9).27 The rhodium catalyst was originally developed by the van Leeuwen group for the highly n-regioselective hydroformylation of olefins.28 Shvo's catalyst is known for its superior properties in the hydrogenation of several functional groups, including aldehydes.29 When this dual catalytic system was used in DMA (dimethylacetamide) as solvent, an approximately 90% yield of isomeric alcohols with good regioselectivity was achieved. Traces of by-products were decane, acetals, internal olefins, and the corresponding formate. The linear alcohol occurs naturally in many fruits (apples, bananas), butter, eggs, and cooked pork. Due to its floral odor and fatty taste, it is frequently used as a flavor enhancer.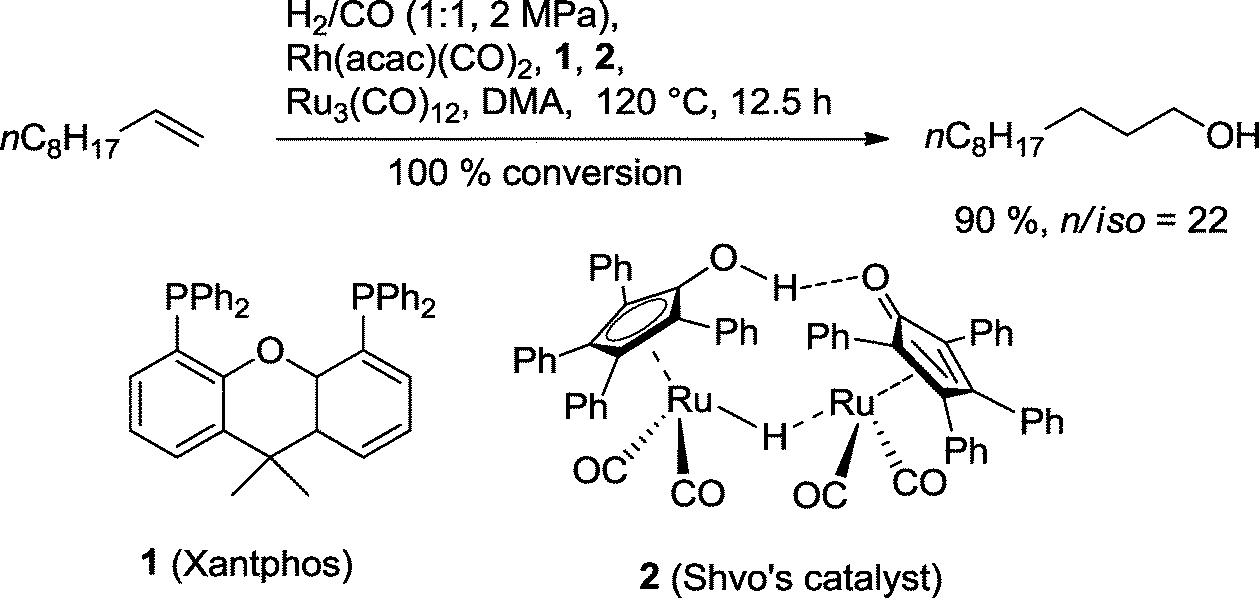 | ||
| Scheme 9 One-pot hydroformylation–hydrogenation of 1-decene with two different catalysts.27 | ||
In a subsequent publication, the range of substrates was extended to a series of functionalized alkenes.30 Allyl alcohol produced only 31% of the desired 1,4-butanediol, but a significant reduction of the starting olefin. Formation of γ-isobutyrolactone was also noted. In contrast, longer alkenyl alcohols yielded up to 95% (e.g. 1,6-hexanediol) of the desired diols. Protection of the alcoholic group (THP, Ac, Bn) had only a slight effect on yields and regioselectivity. The contribution of the individual metal complexes to single steps (olefin isomerization, hydroformylation, hydrogenation) was analyzed in a kinetic study.
Bell and co-workers investigated a similar system with propene as a substrate by using Rh-sulfoxantphos and Shvo's catalyst layered on SiO2.24 It was found that the diphosphine and CO inhibited hydrogenation. However, when the rhodium complex was dissolved in an ionic liquid ([bmim][OctSO4]) and applied to silica (SILP), the bi-layered system converted propene cleanly into butanol with a molar ratio of H2/CO = 10. In this respect, so-called “assisted tandem catalysis” was realized.6
In 2013, Nozaki's group replaced the diphosphine with a diphosphite as an ancillary ligand for rhodium (Scheme 10).31
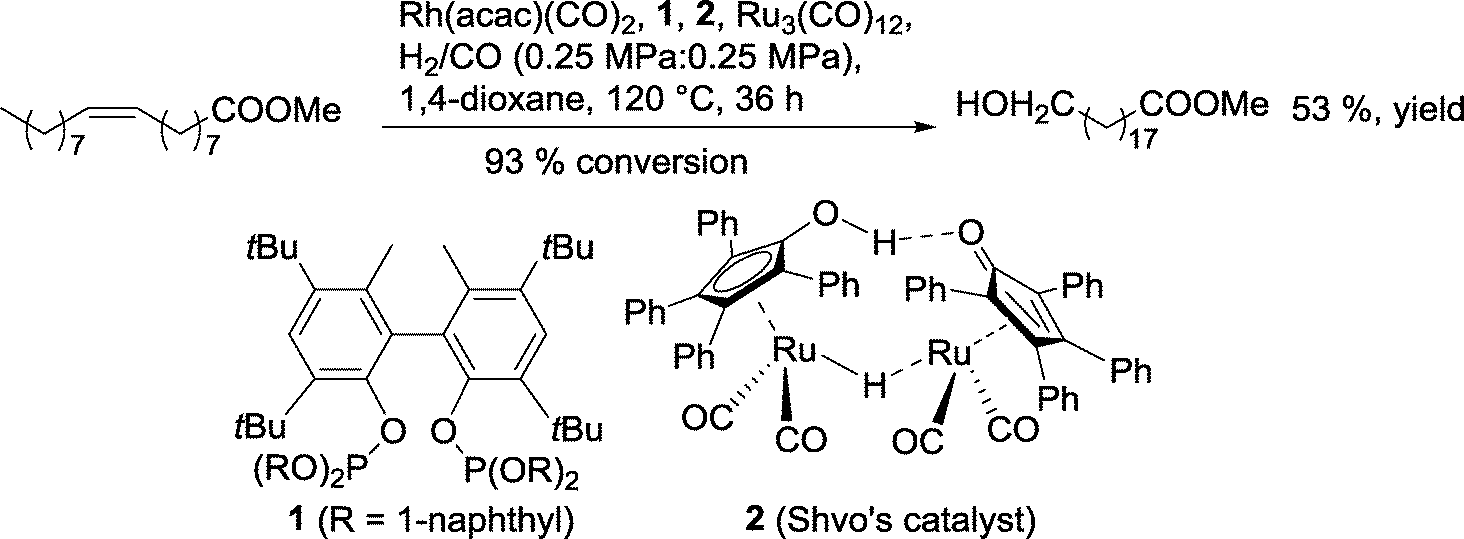 | ||
| Scheme 10 One-pot hydroformylation–hydrogenation of methyl oleate with two different catalysts.31 | ||
With methyl oleate, a 53% yield of the terminal alcohol was observed. With unmodified internal olefins (2-decene, 2-tridecene, 4-octene), even higher regioselectivities in favor of the terminal alcohol were achieved (n/iso up to 12). It was assumed that both rhodium and ruthenium complexes behave in a cooperative manner.
2.5. One-pot reaction with a single catalyst with no change in reaction conditions
Besides the isomerization of the starting olefin,32 hydrogenation of formed aldehydes is one of the most frequently observed side reactions in hydroformylation. This is understandable since several hydroformylation catalysts also exhibit a pronounced hydrogenation activity towards aldehydes.33 Therefore, a single catalyst, which combines a good hydroformylation performance and high activity and chemoselectivity for the hydrogenation of aldehydes, is an ultimate goal. Because of the taxonomy of Fogg and dos Santos, such approaches are named auto-tandem catalysis (Scheme 11).6,8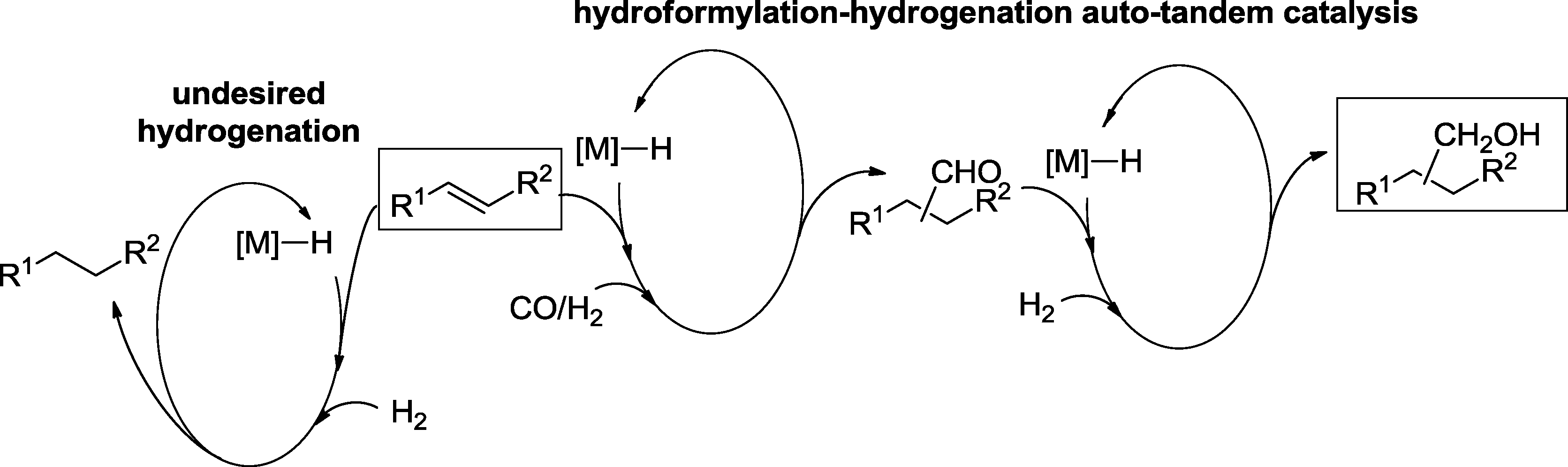 | ||
| Scheme 11 General principle of hydroformylation–hydrogenation auto-tandem catalysis. | ||
It should be noted, however, that sometimes C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds are more easily reduced than CO bonds. As a result, the desired reduction of the aldehyde product may be counterbalanced by hydrogenation of the starting olefin. Moreover, in the presence of an excess of CO, the hydrogenation activity of the catalyst may be blocked. Therefore, in hydroformylation–hydrogenation tandem reactions, the metal catalyst must have highly chemoselective hydrogenation activity without losing its hydroformylation activity and vice versa. This task can be fulfilled with the proper choice of the catalytically active metal and the organic ligand, and by adjusting the reaction conditions.
C bonds are more easily reduced than CO bonds. As a result, the desired reduction of the aldehyde product may be counterbalanced by hydrogenation of the starting olefin. Moreover, in the presence of an excess of CO, the hydrogenation activity of the catalyst may be blocked. Therefore, in hydroformylation–hydrogenation tandem reactions, the metal catalyst must have highly chemoselective hydrogenation activity without losing its hydroformylation activity and vice versa. This task can be fulfilled with the proper choice of the catalytically active metal and the organic ligand, and by adjusting the reaction conditions.
The concomitant hydrogenation of aldehydes to alcohols by the effect of Co(I), Rh(I), Ir(I), Ru(II), Pd(II) and Pt(II) catalyst has been described repeatedly in over 75 years of research into hydroformylation. These observations finally led to the development of highly efficient hydroformylation–hydrogenation auto-tandem protocols, which form almost all the desired alcohols.
On the other hand, Feder and Halpern noticed a strong hydrogenation activity of Co2(CO)8 under hydroformylation conditions, which enables saturation of aromatic hydrocarbons, for example.35 Hydrogenation of multiple unsaturated fatty acid esters could be implemented with the same catalyst under a pure hydrogen stream.36 The competition between undesired hydrogenation of the olefin and hydroformylation depends on the structure of the substrate. Steric hindrance and electron withdrawing groups near the olefin benefit hydrogenation. Thus, the hydrogenation tendency increases in the order 1-heptene > 2- or 3-heptene > EtO(O)C–CHCH2.37 The ratio between hydrogenation of olefin and hydroformylation is dependent on the temperature. In general, hydroformylation dominates at T < 150 °C.38 Thiophene or water may hamper hydrogenation with unmodified Co catalysts.39
Various organic ligands have been tested to modify the intrinsic chemoselectivity of unmodified Co catalysts.40 Only aldehydes were formed with methylidyne ligands in the tetrahedral cobalt clusters MeCCo3(CO)9.41 Also, PPh3 and AsPh3 as ligands at a syngas pressure of 6 MPa (CO/H2 = 1:1) and 80 °C induced solely the formation of aldehydes.42 SbPh3 as a ligand produced the lowest conversion and also produced some alkane derived from hydrogenation of the substrate. Remarkably, with ligands of triphenyl compounds based on elements of the 5th row of the periodic table, an increase in temperature to 195 °C enabled the preferential formation of alcohols for the first time.43
In contrast, phosphites with varying Tolman angles [P(OPh)3, P(O-2,4-tBu-C6H3)3], which are widely used as ligands in rhodium catalyzed hydroformylation, did not exhibit significant activity with cobalt for hydroformylation or for hydrogenation.44 Hydroformylation began only at very high temperatures (190 °C) and, under these conditions, the high chemoselectivity towards the formation of aldehydes was considered as being of particular value.45 In an aqueous two-phase system at 100–130 °C, a cobalt complex based on sulfonated triarylphosphines produced mainly aldehydes.46 However, with these ligands an increase in temperature to 190 °C and a five-fold excess of the catalyst also increased the alcohol yield.46a At 100–110 °C, isomeric fatty aldehydes were formed, whereas at 175–190 °C, the corresponding alcohols were produced. Besides the lower activity compared to phosphines, amines as ligands cause lower chemoselectivities; alkanols as well as alkanes are formed.47
A breakthrough was achieved by Slaugh and Mullineaux at Shell, who discovered the beneficial effect of trialkylphosphines, such as PEt3, P(nBu)3 or P(Cy)3, on the cobalt catalyzed hydroformylation–hydrogenation tandem reaction with several olefins as substrate (1- and 2-pentene, 1-butene, propylene, methyl-pentenes, cyclohexenes, dimethyl-butenes and higher olefins).43,48 The beneficial effect of trialkylphosphines has been explained by an increase in electron density on the metal center, which makes the Co–H bond of HCo(CO)3PR3 more hydridic compared to that of HCo(CO)4.49 Clearly, this property varies with the basicity of the phosphine. Large substituents on the phosphorus, like those present in tris(2-ethylhexyl)phosphine, reduce hydrogenation activity.
It should be noted that [HCo(CO)3(P(nBu)3)] is not only an active hydrogenation catalyst for aldehydes, but also for olefins under a pure hydrogen stream (ca. 2–3 MPa, 40–115 °C) or under hydroformylation conditions (>120 °C, >30 atm CO/H2).50 In general, ligand basicity has only a minor effect on the formation of alkanes.49 An excess of phosphine suppresses hydrogenation activity. Corresponding trialkyl arsenic ligands produced less selective catalysts.51 With arsines, an increase in temperature also increased the yield of the alcohol over the aldehyde. At temperatures of 150–190 °C, almost only alcohols were formed with yields of 80–85%.
Phosphabicyclononanes (“phobanes”)52 or LIM ligands, which have been developed for the same purpose, are more stable and less volatile phosphines than simple trialkylphosphines (Scheme 12).53 Relevant cobalt catalysts typically work at a temperature of 160–185 °C and a syngas pressure of <9 MPa. They are likewise characterized by reduced hydrogenation activity towards the olefin (5–15%).54,55 Alcohols are formed with yields of up to 90%. It is worth noting that in many cases an excess of hydrogen (CO/H2 = 1:2 to 1:10) is used in the syngas mixture. An excess of CO may cause replacement of the phosphine ligand.56 As a result, the unmodified Co complex is formed with its superior reactivity and enhanced tendency to form aldehydes.
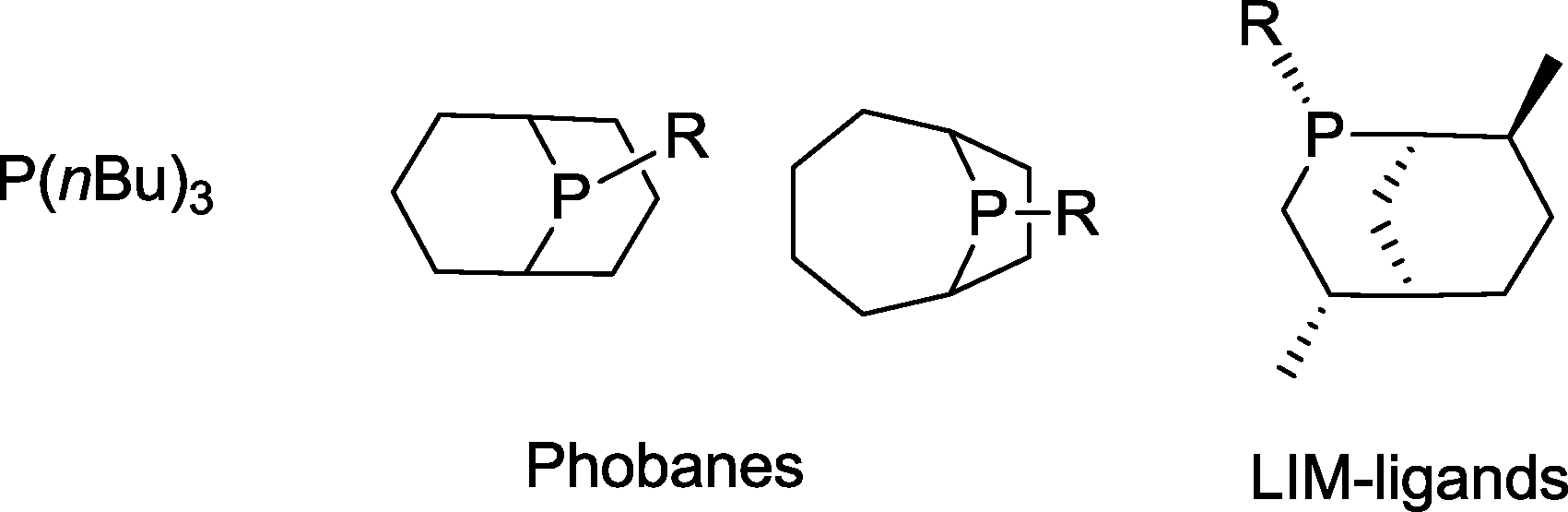 | ||
| Scheme 12 Phosphine ligands suitable for cobalt catalyzed hydroformylation–hydrogenation tandem reactions.52,53 | ||
Auto-tandem catalysts are usually generated by reaction of phosphines with Co2(CO)8 in the presence of syngas, which leads to complexes of the type [HCo(CO)3(PR3)]. In most cases, phosphine ligands are applied in a slight surplus (2:1) compared to the metal. Recently, Bungu and Otto evaluated the activity of Co catalysts containing phosphines with different Tolman cone angles as regards the undesired hydrogenation of starting 1-octene.57 For small cone angles of 132–172° (e.g. P(nBu)3, P(iBu)3, PPh3) the yield of octane varied within a small range of 9–15%, but in contrast, at up to 40%, octane was formed with ligands like PA-C5, PCy3 and PCyp3 displaying cone angles >169° (Scheme 13).
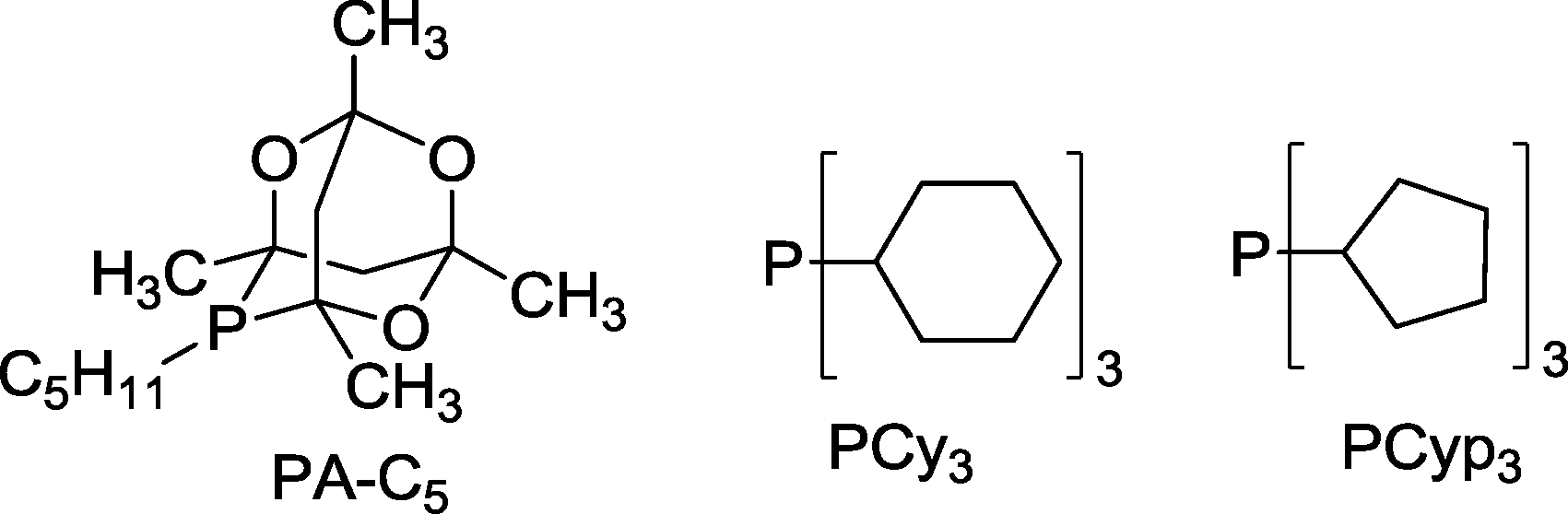 | ||
| Scheme 13 Large cone angle phosphine ligands not suitable for cobalt catalyzed hydroformylation–hydrogenation tandem reaction.57 | ||
In general, an increase of P-ligand concentration decreased the reaction rate, but selectivity in the formation of the alcohol was improved.55 This effect was explained by a shift from the unmodified Co catalysis to the P-ligand modified reaction.53 Catalysts modified in this manner are less active in comparison to the unmodified complex [HCo(CO)4]. Increasing partial pressure of H2 may contribute to the enhancement of the alcohol yield.49 Sometimes, the formation of alcohols is forced by water.58 Polar solvents like DMF can also inhibit hydrogenation of the aldehyde.59
These discoveries ultimately led to the establishment of large scale processes in industry for hydroformylation–hydrogenation of olefins with short and longer alkyl chains (Shell Oxo process).34
A typical benchmark system is the Shell catalyst for reductive hydroformylation of 1-dodecene based on the mixture of phoban-C20 ligands, which operates under a syngas pressure of 8.5 MPa with an excess of H2 in comparison to CO to yield 86.9% isomeric tridecanols (Scheme 14).54 1-Tridecanol is used as a lubricant or as an ingredient in surfactants, solvents, and pesticides.
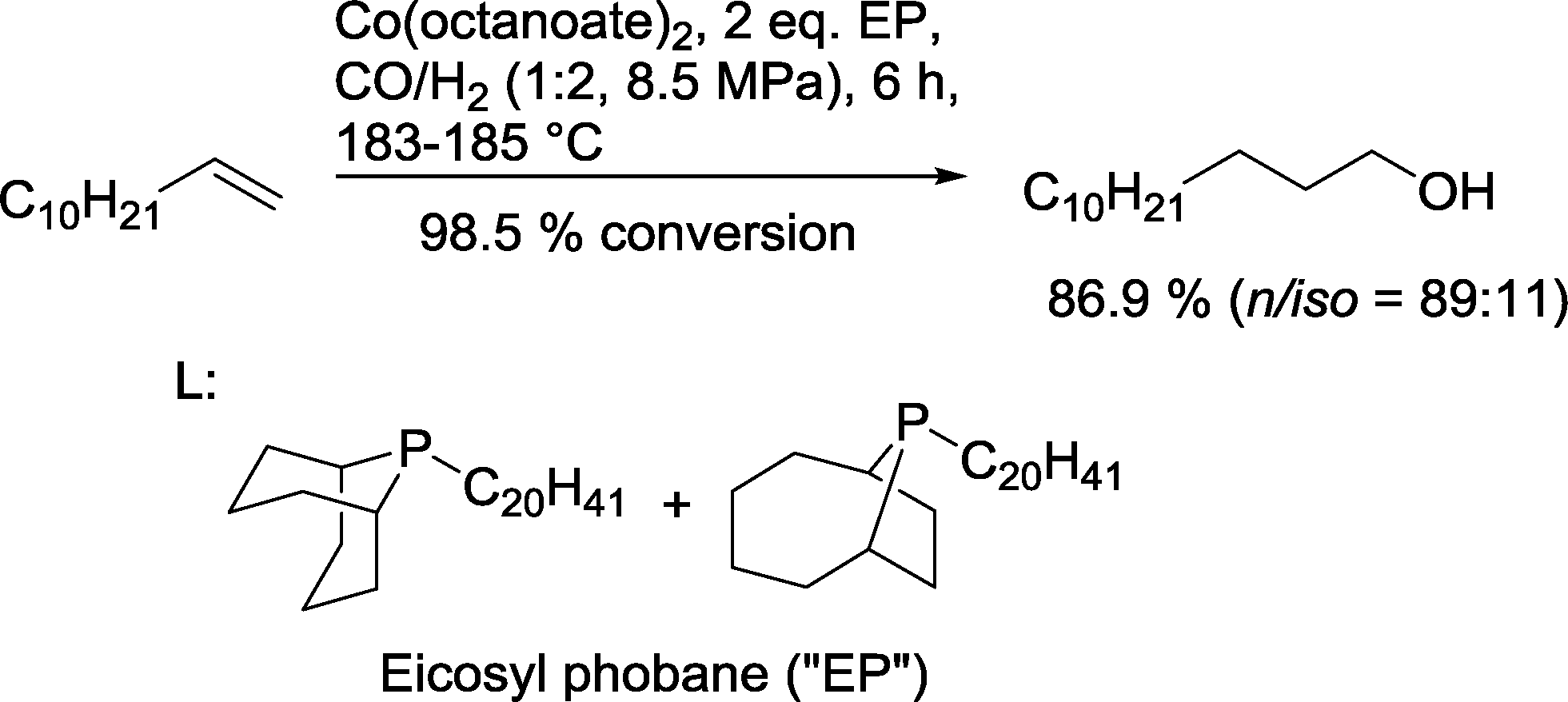 | ||
| Scheme 14 Cobalt catalyzed hydroformylation–hydrogenation tandem reaction.54 | ||
One application of the Co catalyzed hydroformylation–hydrogenation on functionalized olefins is the reaction with 2,7-octadien-1-ol, where with P(nOct)3 as a ligand approximately 50% of the desired diols (n/iso = 9/1) were produced in an excess compared to the corresponding formyl alcohol (Scheme 15).60
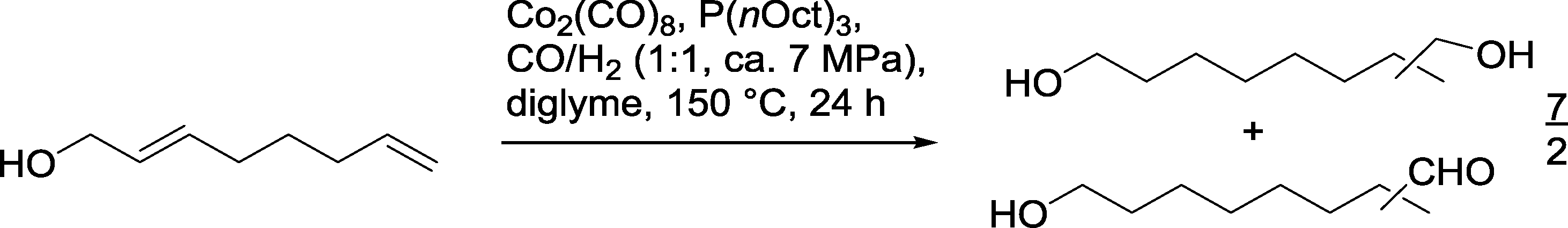 | ||
| Scheme 15 Regioselective cobalt catalyzed monohydroformylation–hydrogenation.60 | ||
2.5.2.1. Unmodified rhodium catalysts. Rhodium catalysts are particularly interesting for highly chemoselective hydroformylation, but a subsequent hydrogenation of aldehydes can also be achieved under hydroformylation conditions. Apart from few heterogeneous catalysts (e.g. (RhVO4)/SiO2),61 mainly homogeneous catalysts, have also been investigated. The hydrogenation activity of simple rhodium carbonyl complexes is low in comparison to their cobalt congeners. Organic ligands enhance hydrogenation activity. Compared to arylphosphines, trialkylphosphines especially induce a much higher electron density on the metal and hence make oxidative additions reactions simpler.62
Among numerous examples in the literature relating to hydroformylation–hydrogenation tandem reactions, there are also a few investigations aimed at assessing the hydrogenation activity of rhodium catalysts. For example, [HRh(CO)(PPh3)3] is a good hydrogenation catalyst for aldehydes in the absence of CO. The hydrogenation activity of unmodified rhodium catalysts is more suppressed by CO compared to cobalt congeners.63
Maruya et al. investigated hydrogenation of aldehydes with catalysts made from [Rh2Cl2(CO)4] or [Rh(acac)(CO)2] and N-methylpyrrolidine or a polymer amine at approx. 3 MPa syngas pressure (CO/H2 = 1:1) and 80 °C.64 A significant inhibition of hydrogenation but not of hydroformylation was discovered with high olefin concentrations. With benzaldehyde as a model substrate, hydrogenation increased with an increase of the H2/CO ratio to approx. 3:1. At higher CO pressures, the hydrogenation rate decreased. This effect was explained by the competition of CO with amine at the metal center, leading to a less active hydrogenation catalyst.
2.5.2.2. Rhodium catalysts modified with nitrogen and other non-phosphorus ligands. Kaneda and Teranishi investigated the hydroformylation–hydrogenation tandem reaction using the water gas shift reaction (WGSR) as source for syngas and an excess of H2. Particularly active catalysts for the reaction with 1-octene were produced with amines as ligands such as N,N,N′,N′-tetramethyl-1,3-propanediamine (TMPDA) (Scheme 16).65 Mainly n-nonanol was formed together with some isomerized olefinic substrate.
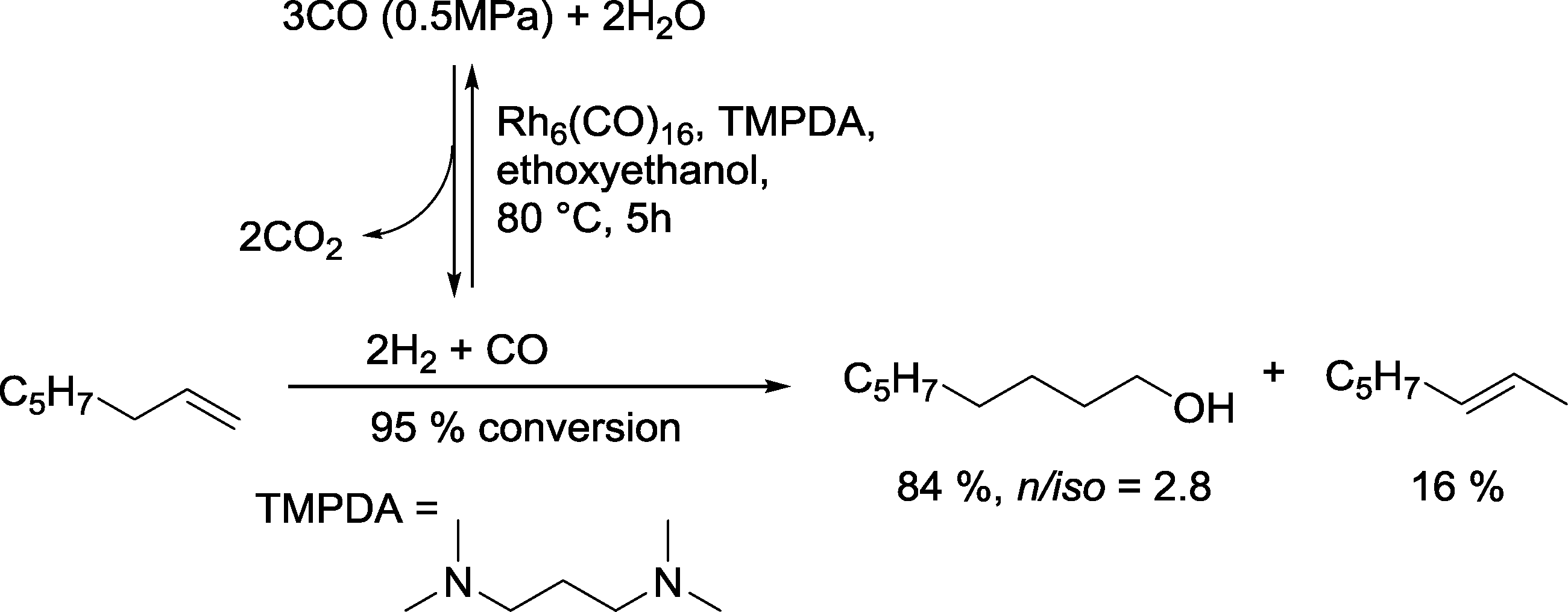 | ||
| Scheme 16 A rhodium catalyst taking benefit from the WGSR in the hydroformylation–hydrogenation tandem reaction.65 | ||
4-Dimethylaminopyridine (4-DMAP) as a ligand was also quite effective, whereas pyridine, N-methyl-piperidine or Et3N induced only a shift of the double bond into the interior of the substrate. A catalyst derived from the Rh6(CO)16 cluster exhibited the highest activity for the tandem reaction, followed by Rh4(CO)12, Rh2(CO)4Cl2, RhCl(PPh3)3 and finally RhCl3·3H2O. In strong contrast to simple α-olefins, α,β-unsaturated carbonyl compounds, like cinnamaldehyde or mesityl oxide were only reduced at the olefinic group.
A similar catalytic system was employed for the reaction with allyl alcohol (Scheme 17).66 With TMPDA as a ligand, 1,4-butanediol was formed with a yield of 72%. When 4-dimethylaminopyridine was used, γ-butyrolactone was the main product.
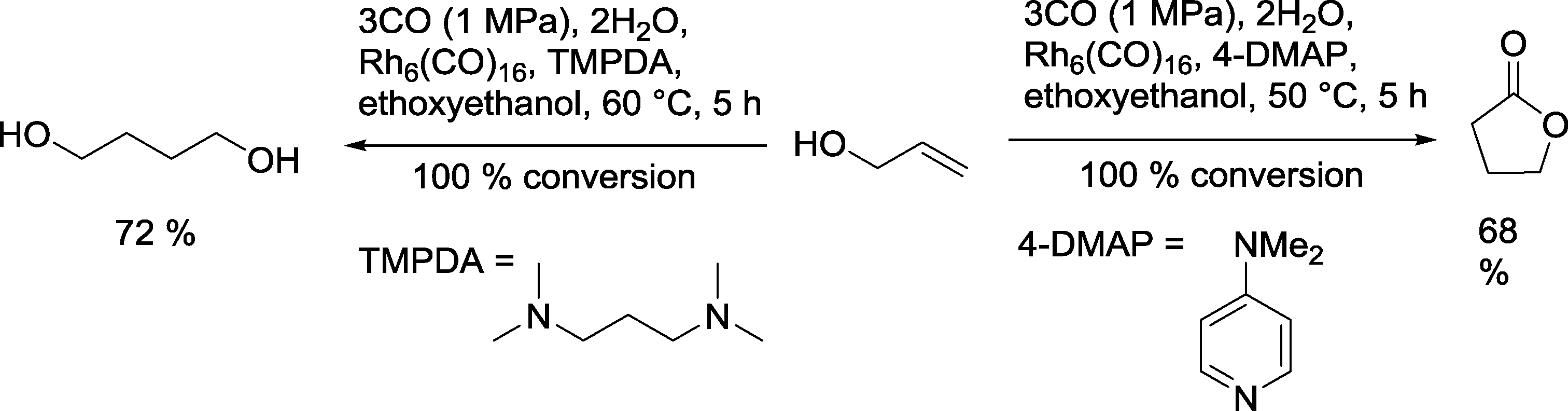 | ||
| Scheme 17 Dependency of the product distribution in the Rh catalyzed hydroformylation on the nature of the N-ligand.66 | ||
Alper's group showed that a variety of aromatic olefins can be transformed with Rh catalysts modified with the bidentate tertiary diamine Me2N(CH2)2NMe2 as a ligand into the desired homologous alcohols (Scheme 18).67 With the exception of allyl benzene, other substrates produced good or high yields of the desired alcohols. As expected for styrene derivatives, mainly branched alcohols were formed. Monodentate ligands like NEt3 reduced the yield. No reaction was observed with the corresponding bidentate secondary amine as a ligand.
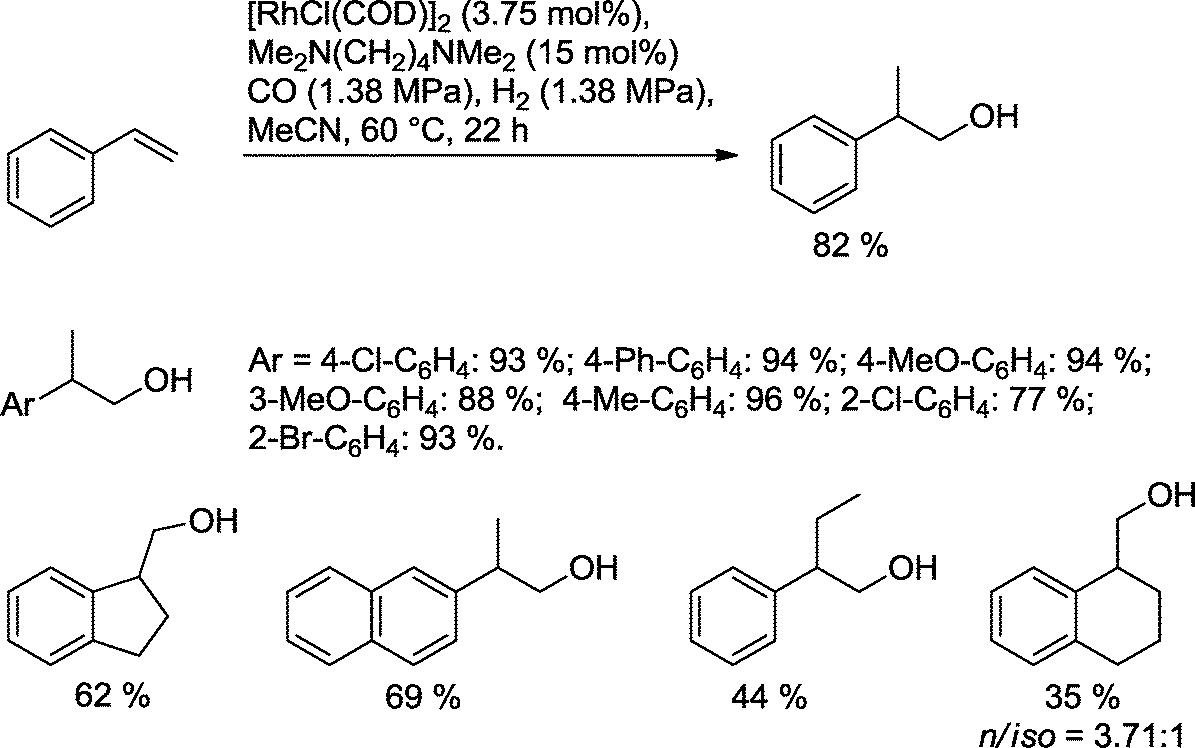 | ||
| Scheme 18 Preparation of 2-aryl-ethanol derivatives via hydroformylation–hydrogenation reaction.67 | ||
Hydrogen in the syngas mixture can be replaced with another hydride source, such as NaBH4. Hence, Zhou and Alper produced alcohols under these conditions with the assistance of a zwitterionic Rh complex (Scheme 19).68
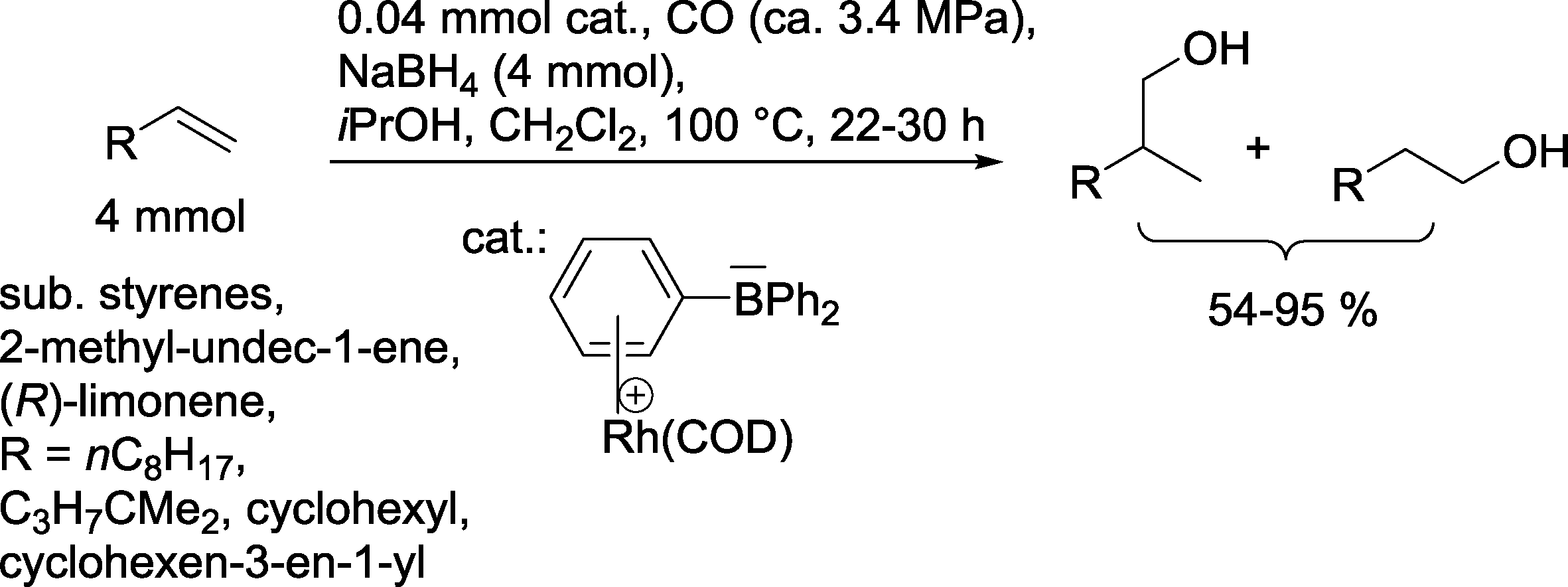 | ||
| Scheme 19 Hydroformylation–hydrogenation approach with NaBH4 as a hydrogen source.68 | ||
The reaction could also be performed in a highly chemoselective manner with PPh3 modified rhodium catalysts ([Rh(COD)(PPh3)2]BPh4 or [HRh(CO)(PPh3)3]), but then the desired n-regioselectivity was negatively affected.
2.5.2.3. Rhodium catalysts modified with phosphorus ligands and solvent effects. Whether the aldehyde or the alcohol is the major product depends largely on the phosphorus ligand and the solvent used for hydroformylation. In several studies, Cole-Hamilton and co-workers found that rhodium catalysts based on trialkylphosphines as ligands, such as [HRh(PEt3)3] or a catalyst derived in situ from the reaction of Rh2(OAc)4 with an excess of PR3 (R = Me, Et, Bu), produce a mixture of alcohol and aldehyde in toluene or THF with 1-hexene.69 In contrast, in methanol, ethanol, and n-butanol only alcohols were formed.70 Surprisingly, isopropanol which is considered to be the most efficient hydrogen donor among these alcohols produced a low yield of alcohol and a similarly high amount of C7 aldehydes. At least two equivalents of PR3 in relation to rhodium must be used, since only complexes accommodating two phosphine ligands produce alcohols.71 Bulky trialkylphosphines, such as PiPr3 or PiBu3, which form rhodium complexes where only a single phosphorus ligand is coordinated to the rhodium center produced mainly aldehydes at 125 °C.72 The chelating dialkylphosphine Me2PCH2CH2PMe2 inhibited the reaction.73 The addition of organic acetates to trialkylphosphine-modified rhodium catalyst promoted the formation of alcohols.74 With phosphites as ligands, alcohols begin to form at about 150 °C.75
Labeling studies with trialkylphosphine-modified rhodium catalysts provided various evidence that aldehydes are not intermediates in the formation of heptanol from 1-hexene, but are a hydroxycarbene-like intermediate (Scheme 20).76 The latter derives from the protonation of the relevant Rh-acyl complex by ethanol and benefits from the high electron density at the metal center, which is caused by trialkylphosphines. The reaction with hydrogen (here D2) delivers the alcohol. A similar mechanism was suggested for the tandem reaction with 2-propen-1-ol as a substrate.77
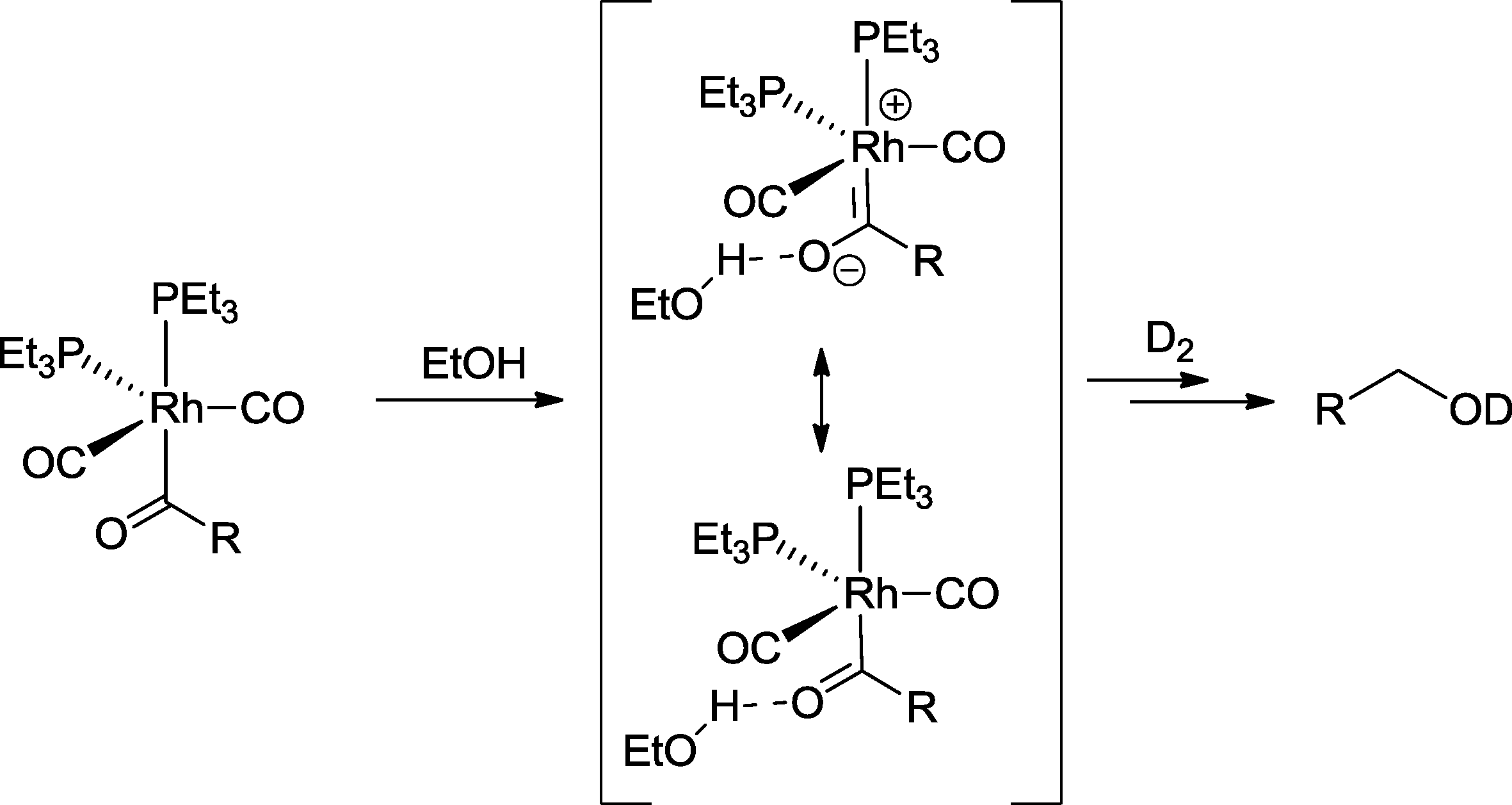 | ||
| Scheme 20 Part of the mechanism of the hydroformylation–hydrogenation with a rhodium catalyst based on trialkylphosphines as ligand in ethanol.76 | ||
If this protocol {[HRh(PEt3)3], CO/H2 = 1:1, ca. 4 MPa; 120 °C, EtOH, 16 h} is applied to other olefins, corresponding homologous alcohols could be produced from ethene, styrene, and 3,3-dimethylbutene in quantitative yields (Scheme 21).73
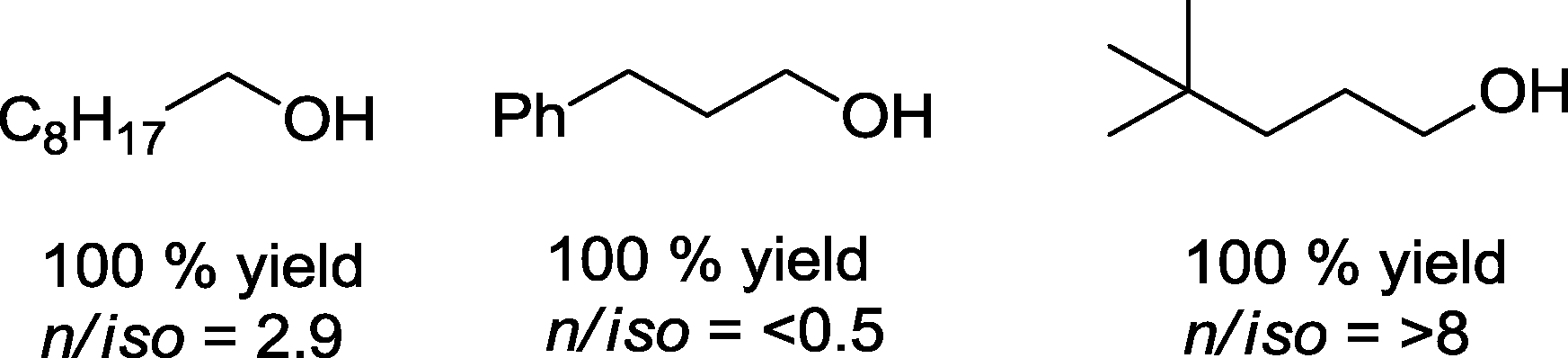 | ||
| Scheme 21 Products of the hydroformylation–hydrogenation with rhodium catalysts based on trialkylphosphines as ligands.73 | ||
The highest rate was observed with ethene (TOF = 54000 h−1). 2-Hexene as a substrate produced a low conversion rate.
When the trialkylphosphine-modified rhodium catalyst was encapsulated into zeolites, the high chemoselectivity was maintained, but the n/iso-ratio of the product alcohol was increased by as much as tenfold.78 Anchoring trialkylphosphines to carbosilane dendrimers based on polyhedral silsesquioxane (POSS) afforded dendrimeric ligands which produced mainly linear alcohols in the hydroformylation–hydrogenation of 1-octene.79 Regioselectivity exceeded that achieved with the low molecular weight ligand.
In another approach, propene was hydroformylated in the presence of a π-cyclopentadienylcarbonyl(tributylphosphine)rhodium catalyst to produce a 1:1 mixture of isomeric butanols (Scheme 22).80 In the best case, a catalyst:substrate ratio of 50000:1 could be achieved.
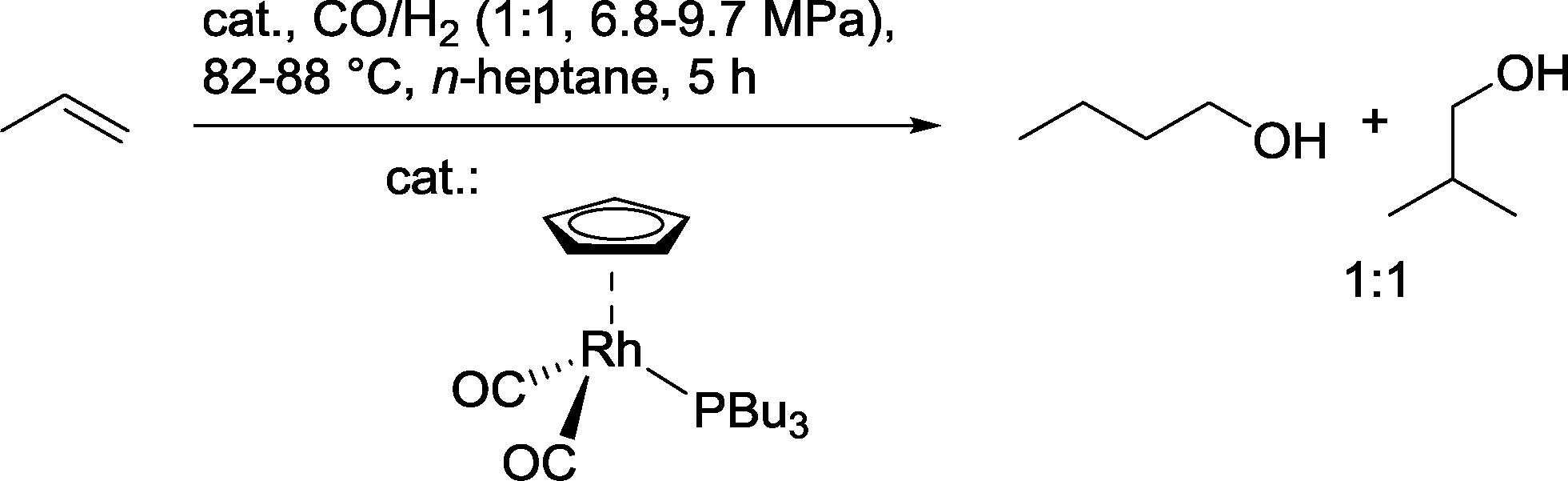 | ||
| Scheme 22 Hydroformylation–hydrogenation of propene with a Cp–Rh(PBu3) catalyst.80 | ||
Rodriguez and Tenn hydroformylated a diluted ethylene feed with rhodium catalysts based on different trialkylphosphines.81 With catalysts containing trioctyl and trihexylphosphine, TOFs of 232–258 h−1 were noted as regards the formation of propanol. In contrast, triphenylphosphine and the more bulky ligands tricyclohexylphosphine and 1,3-dicyclohexylphosphinopropane forced the formation of propanal. The composition of the gas feed had a significant effect on selectivity. Thus, with high ethene concentrations, production of ethane became a serious issue.
Alternatively, paraformaldehyde has been used as syngas surrogate.82 When the reaction of 1-hexene took place in the presence of [H2Rh(O2COH)(PiPr3)3]2 with an excess of paraformaldehyde at 120 °C, mainly the isomeric aldehydes were produced. Raising the temperature to 150 °C resulted in an increase in the yield of the alcohol. Mixed esters were detected as by-products. This indicates that alcohols are formed from the preformed C7-aldehyde and formaldehyde by disproportionation.
Union Carbide claimed selective hydroformylation–hydrogenation of 1,3-butadiene to produce mainly 3- and 4-pentenols by reaction in diglyme (Scheme 23).83 In order to obtain the desired 1,6-hexanediol, 4-pentenol was treated individually under slightly different reaction conditions in ethanol as a solvent to produce a 69% yield of diol.
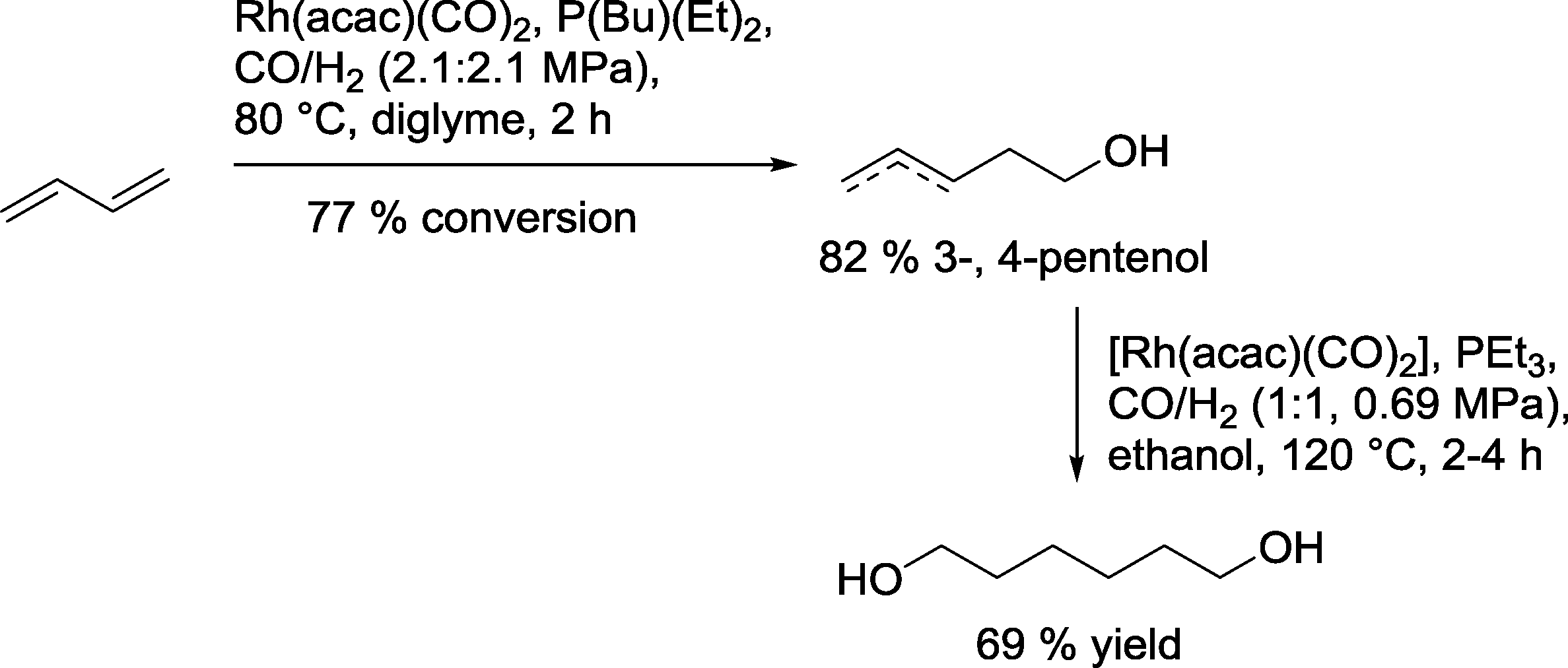 | ||
| Scheme 23 Hydroformylation–hydrogenation of 1,3-butadiene.83 | ||
The results with monodentate phosphines led to the idea to use a bidentate phosphine ligand, such as Xantphos, DIOP or BISBI, together with PEt3 (Scheme 24).84 In a continuously working reactor, higher olefins, like 1-hexene, 1-octene or 1-decene, but also allyl alcohol could be converted into the corresponding alcohols. The n/iso ratios were high and hydrogenation of the starting olefin was less than 5%. A similar performance was noted when the tertiary amine PhCH2NMe2 was added instead of PEt3.
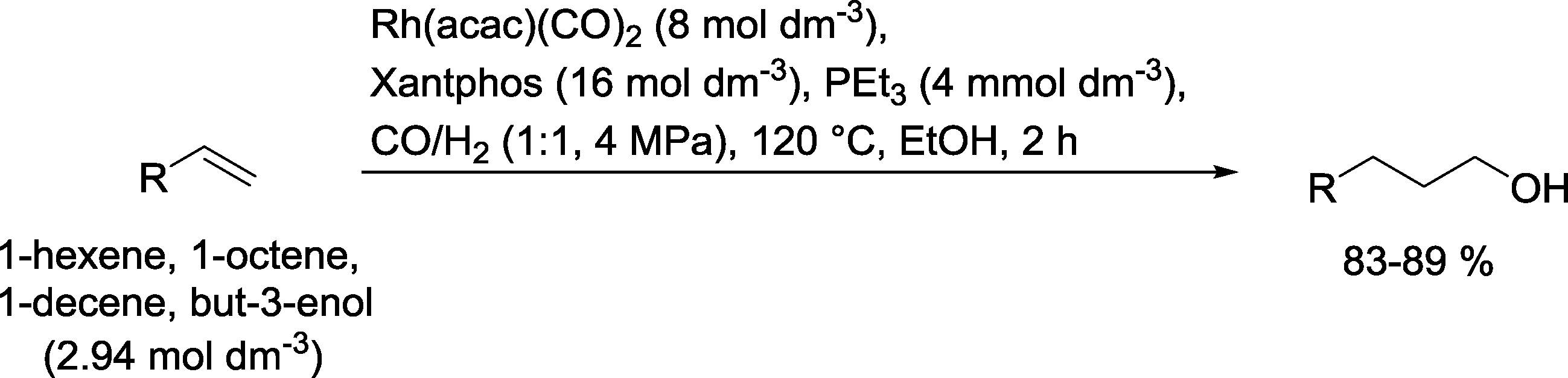 | ||
| Scheme 24 Hydroformylation–hydrogenation of terminal olefins with a Rh(Xantphos) catalyst.84 | ||
Another example was given by Vogt and co-workers.85 Only Xantphos or nBu3P was used as a ligand (Scheme 25). The reaction took place with twice the amount of H2 in relation to CO in an aqueous medium. With the bidentate diphosphine ligand, n/iso ratios of up to 14/1 were achieved. Yields of up to 93% 1-nonanol were obtained from 1-octene. Also internal olefins like trans-2-octene and cis-cyclooctene were successfully converted under these conditions. Hydrogenation of the starting olefin was almost suppressed with short-chain olefins. With 1-decene and 1-dodecene, the corresponding alkanes were also formed but with yields of less than 10%. In contrast to the catalytic system investigated by Cole-Hamilton in detail (see Scheme 20),70 the aldehyde was clearly a reaction intermediate!
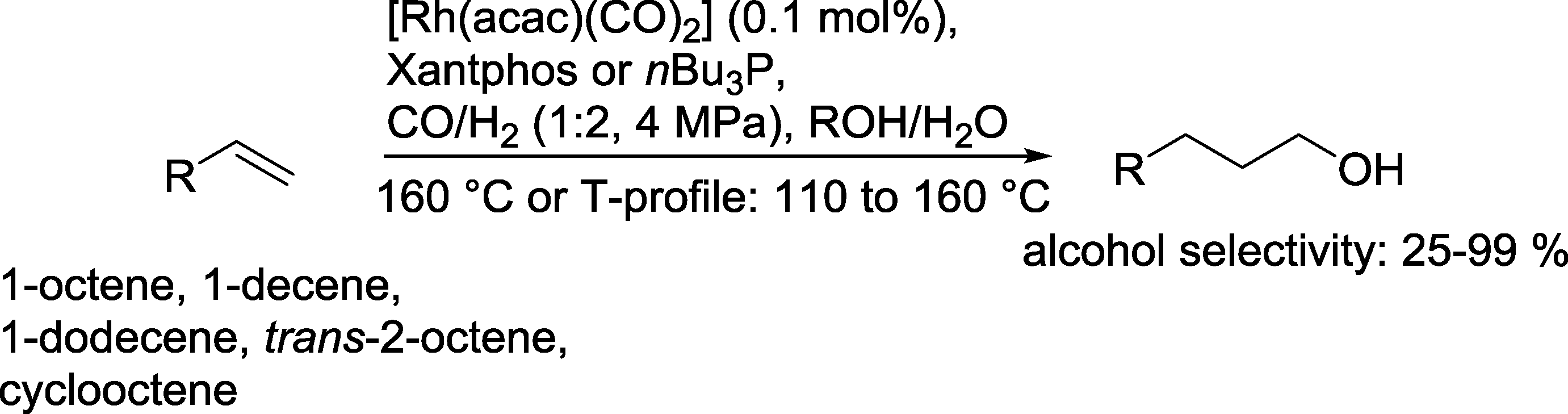 | ||
| Scheme 25 Hydroformylation–hydrogenation of terminal olefins with a Rh(Xantphos) catalyst in water.85 | ||
Alternatively, the Nozaki group used alkyl derivatives of BISBI for the hydroformylation–reduction of 1-decene (Scheme 26).86 Large P-alkyl substituents resulted in a reduced yield of alcohol. The authors assumed therefore that steric effects of the phosphine are more dominant than electronic effects. The use of alcohols as solvents also proved to be essential in these investigations. In some cases, the formation of aldol products inhibited the formation of the desired alcohols. Best results were obtained at 170 °C.
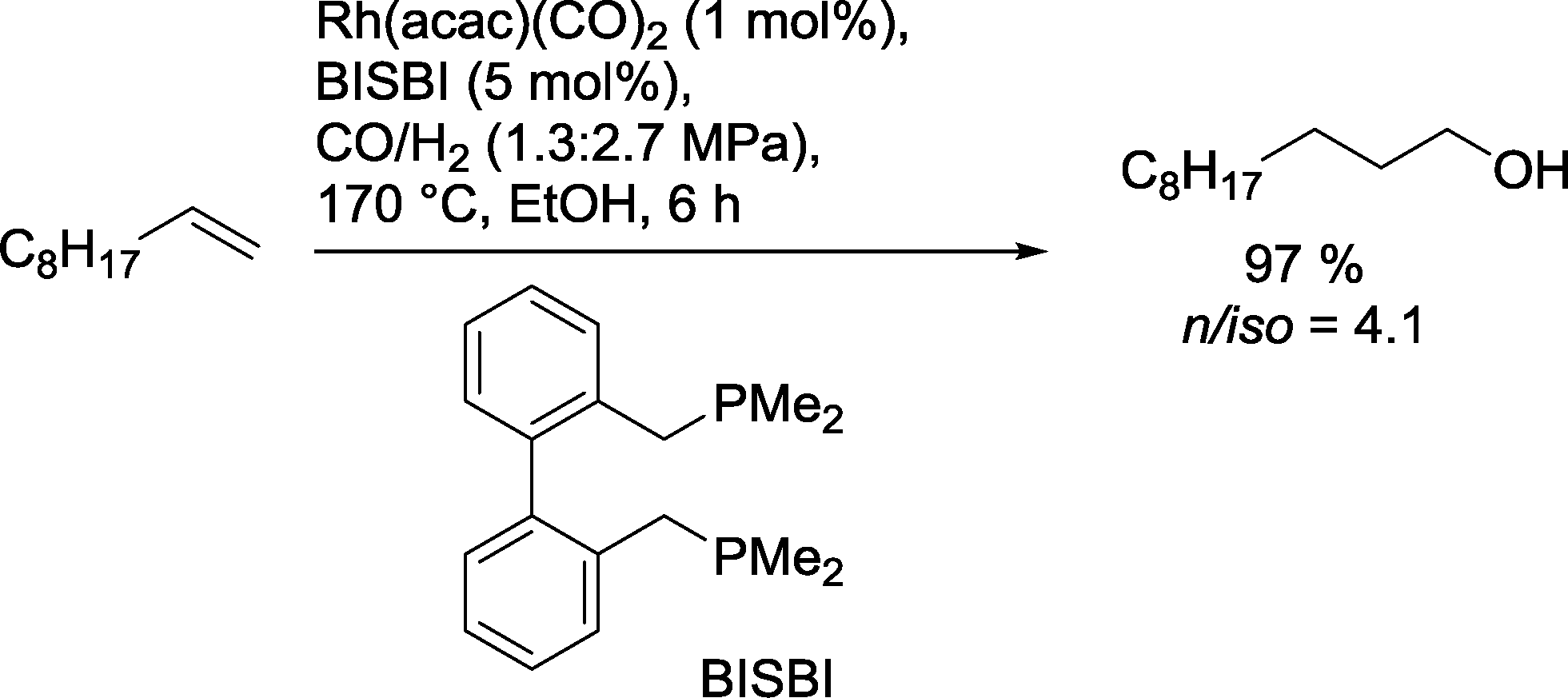 | ||
| Scheme 26 Hydroformylation–hydrogenation of 1-decene with a Rh(BISBI) catalyst in ethanol.86 | ||
2.5.2.4. Ligating groups in the substrate. Perlmutter and co-workers observed an interesting dependency of the chemoselectivity in the hydroformylation of olefins bearing a PPh2-group (Scheme 27).87 For short alkyl chains, n = 1, 2, only isomeric alcohols were formed. In contrast, with longer chains, n = 3, a mixture of alcohols and aldehydes was produced. The substrate with the longest chain, n = 4, produced only isomeric aldehydes. With an unfunctionalized olefin or after oxidation of the steering PPh2-group to P(O)Ph2, the chelate control effect was completely suspended and only the aldehydes were formed.
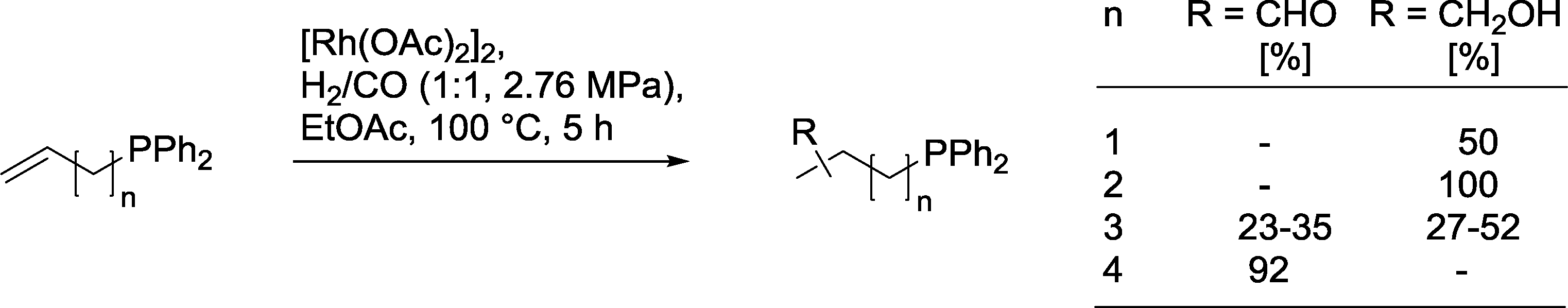 | ||
| Scheme 27 Product distribution in the hydroformylation of ω-phosphinyl alkenes.87 | ||
Similar chelate control was discovered in cyclohexenyl compounds with an exocyclic diphenylphosphinoalkyl group (Scheme 28).88 A steric effect that favored the formation of cis-substituted cyclohexanol derivatives also occurred.
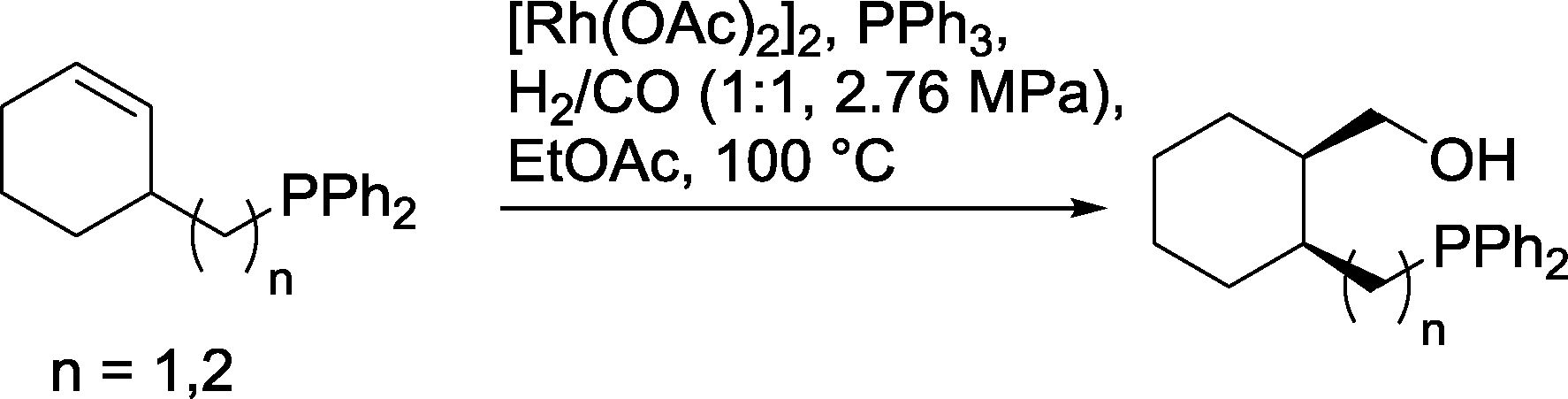 | ||
| Scheme 28 Hydroformylation of cyclohexene derivatives with exocyclic alkyl phosphine groups.88 | ||
Breit and co-workers also demonstrated that secondary binding interactions between the ligand and the substrate may advantageously assist in the formation of the alcohol (Scheme 29).89 As derived from DFT calculations utilizing an analogue pyrrole-based ligand (A) the guanidinium group forms multiple hydrogen bonds with the intermediary aldehyde. This arrangement facilitates the hydrogenation of the carbonyl group by lowering the LUMO energy. Based on this protocol, functionalized and nonfunctionalized olefins could be directly converted into the corresponding alcohols.
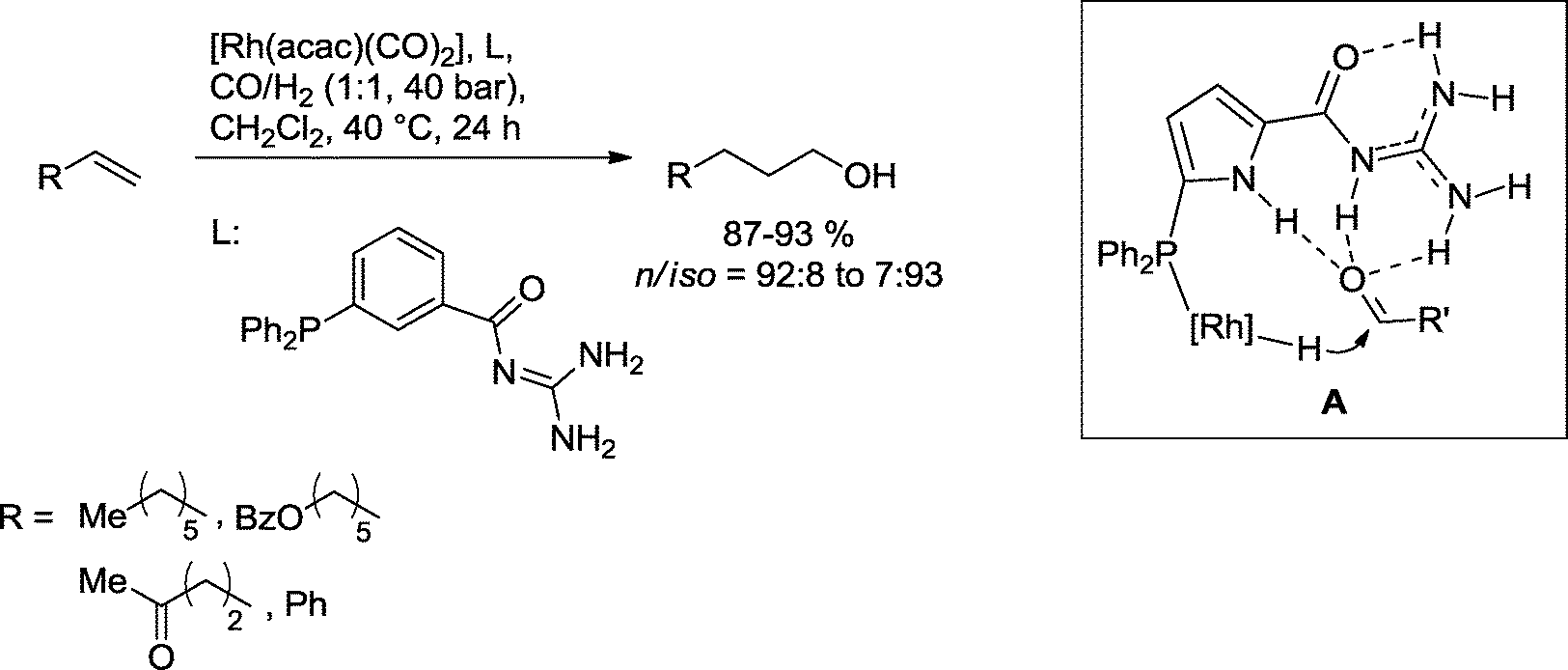 | ||
| Scheme 29 Hydroformylation–hydrogenation reaction assisted by secondary interactions.89 | ||
Recently, this bio-inspired approach was extended to a sequence, where the required olefin is produced in situ by decarboxylation of α,β-unsaturated carboxylic acids.90
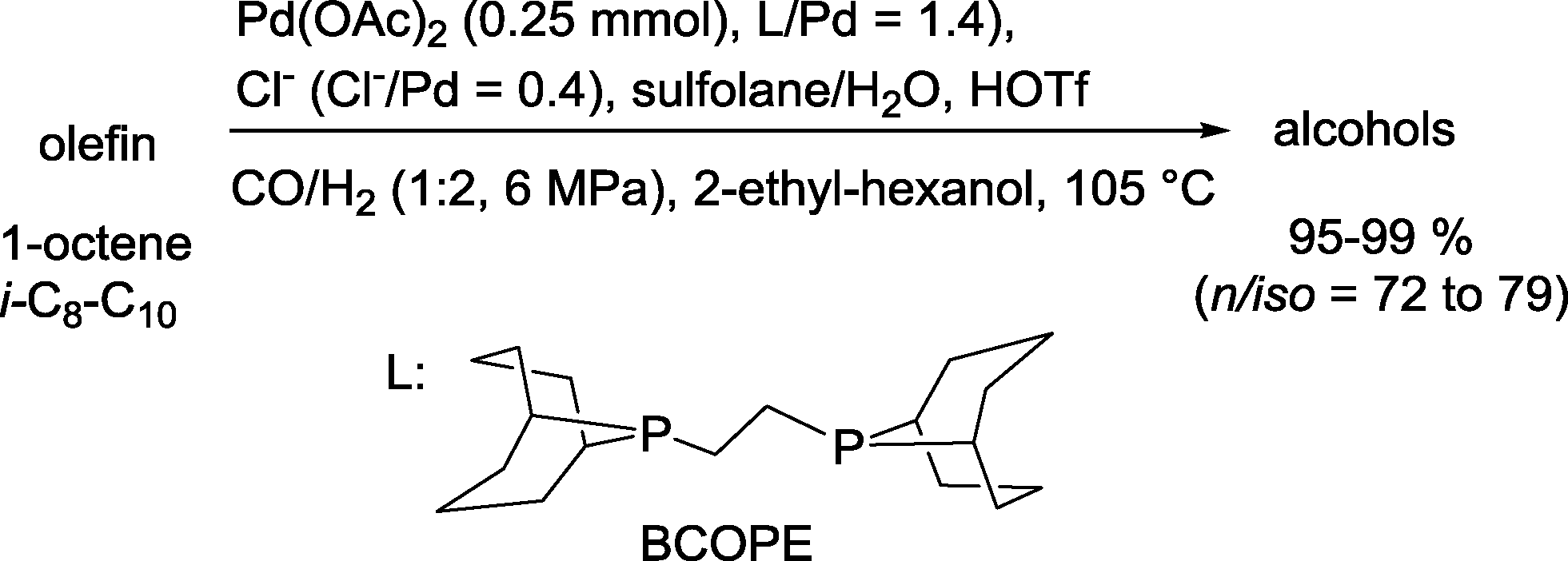 | ||
| Scheme 30 Palladium-catalyzed hydroformylation–hydrogenation tandem reaction.91 | ||
In comparison to the runs in the absence of halide anions, the reaction was accelerated by a factor of 6–7 (chloride/bromide) or 3–4 (iodide). Regioselectivity increased in the reversed order, e.g. iodide > bromide > chloride. No esters were formed. It was argued that the hydrogenolysis of a Pd-acyl intermediate is advantageously influenced by the halide. The latter probably displaces the weakly coordinating triflate anion at this stage (Scheme 31). Hydrogenation of the formed aldehyde takes place in a final step.
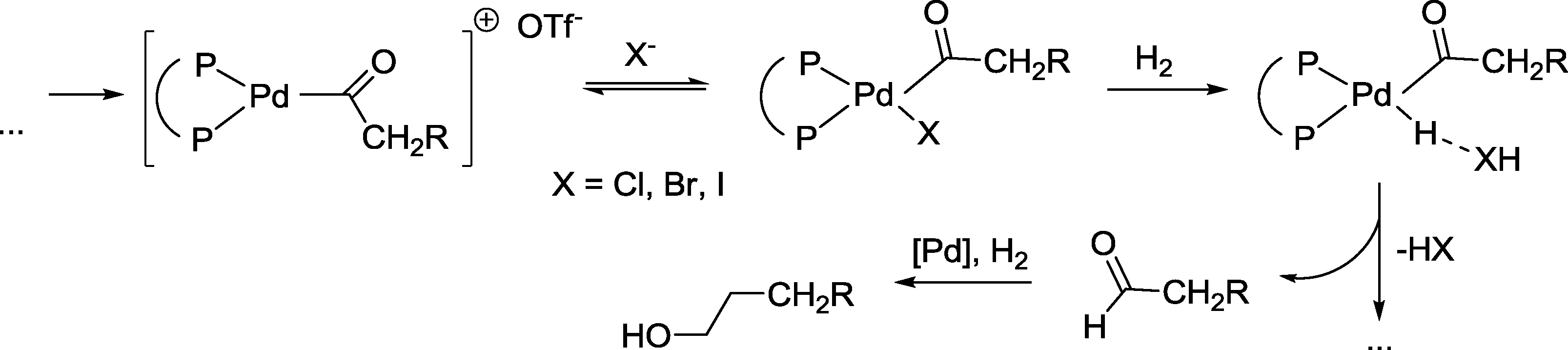 | ||
| Scheme 31 Parts of the mechanism of the palladium catalyzed hydroformylation–hydrogenation tandem reaction.91 | ||
Other bidentate trialkylphosphines produced much lower yields of the desired alcohols, which indicates that besides the halide, the choice of the ligand is also important for the success of the reaction.
:2, 8.3 MPa, 180 °C) mainly into linear alcohols.92
Pakkanen's group published several examples showing that Ru3(CO)12/2,2′-bipyrimidine anchored on silica is active in the reductive hydroformylation of alkenes.93 The outcome and the reproducibility of the reaction are very dependent on the impregnation method.94 The reaction proceeded best in THF. The rate-limiting catalytic step is hydroformylation of the alkene, whereas hydrogenation of the intermediate aldehyde is fast and produces good yields. Formation of the alcohol can be improved by adding NEt3 (ref. 95) or rhodium supported on Al2O3.
The Haukka group analyzed the effect of different 2-substituents on triphenylphosphines on the hydroformylation of 1-hexene.96 Strongly coordinated chelating phosphines with amino or methoxy groups exhibited poor activity. In contrast, weakly coordinating methoxy-substituted phosphines and non-chelating phosphines such as [2-(ethyl)phenyl]-(diphenyl)phosphine induced higher activities. With [RuCl2(CO)2(dmep)2] (dmep = bis(2-methylphenyl)phenylphosphine) as a catalyst at 130 °C and a syngas pressure of 2 MPa (CO/H2 = 1:1) up to 57% heptanols were produced in the hydroformylation of 1-hexene.
In the complex Ru(CO)3(PPh3)2 one phosphine ligand can be easily replaced by CO under UV irradiation.97 The yielded catalyst Ru(CO)4(PPh3) converts ethylene under very smooth conditions (H2/CO = 1:1, 0.07 MPa) into 1-propanol.
Nozaki and co-workers advocated the idea of combining structural features of an n-regioselective hydroformylation catalyst with those of a hydrogenation catalyst (Scheme 32).30 The latter should operate also under syngas.
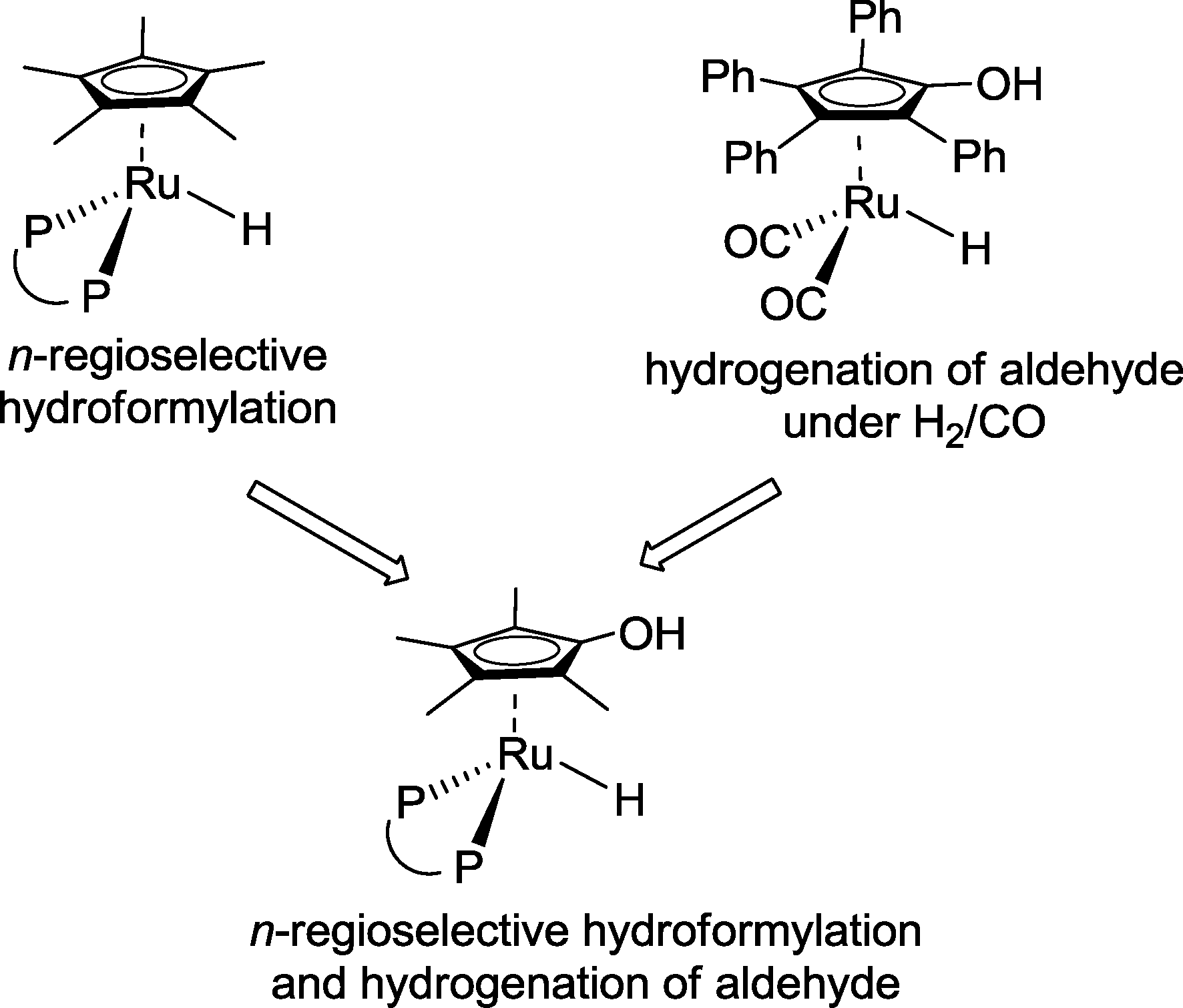 | ||
| Scheme 32 Rational design of a ruthenium-based hydroformylation–hydrogenation auto-tandem catalyst.30 | ||
As a result of this concept, a ruthenium catalyst based on Xantphos was designed which converted 1-decene into 1-undecanol with a 73% yield (Scheme 33). Only 1.2% of the corresponding aldehydes were observed as a by-product.
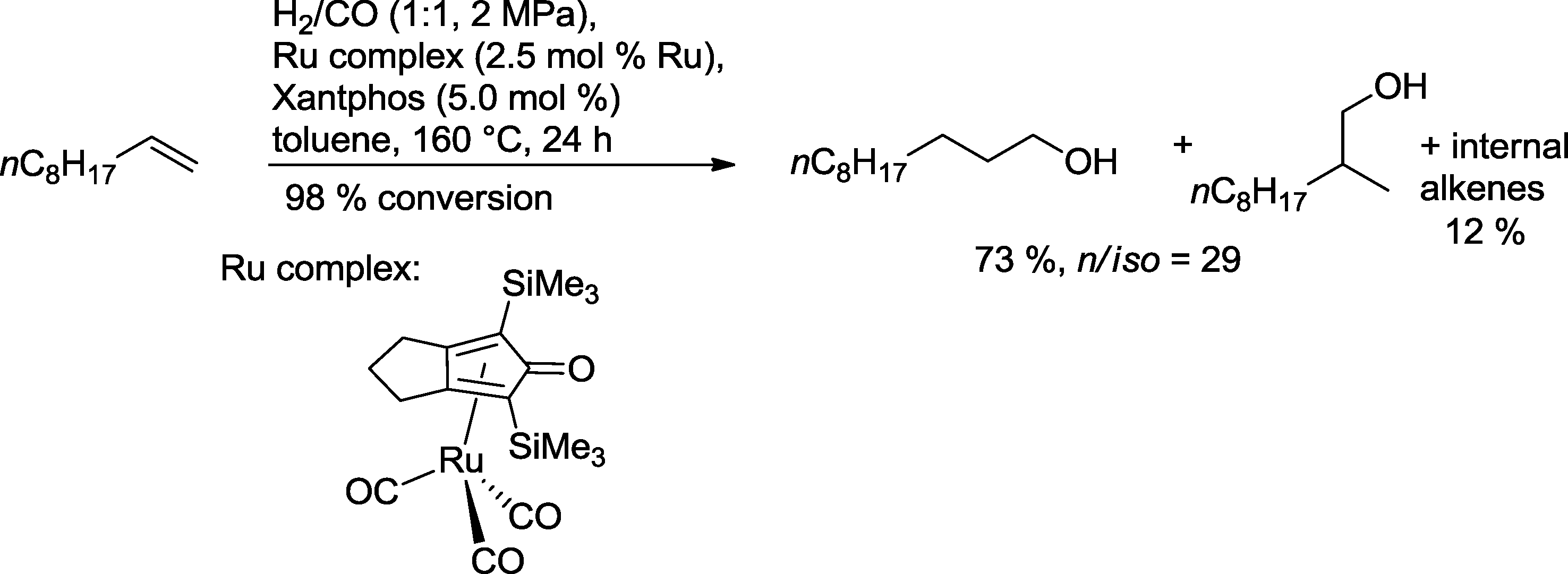 | ||
| Scheme 33 Ruthenium-catalyzed hydroformylation–hydrogenation of 1-decene.30 | ||
Mitsudo and co-workers screened several tertiary and aromatic N-ligands for the ruthenium-catalyzed hydroformylation of styrene (Scheme 34).98 The highest yield of 2-phenylpropanol was observed in the presence of quinuclidine.
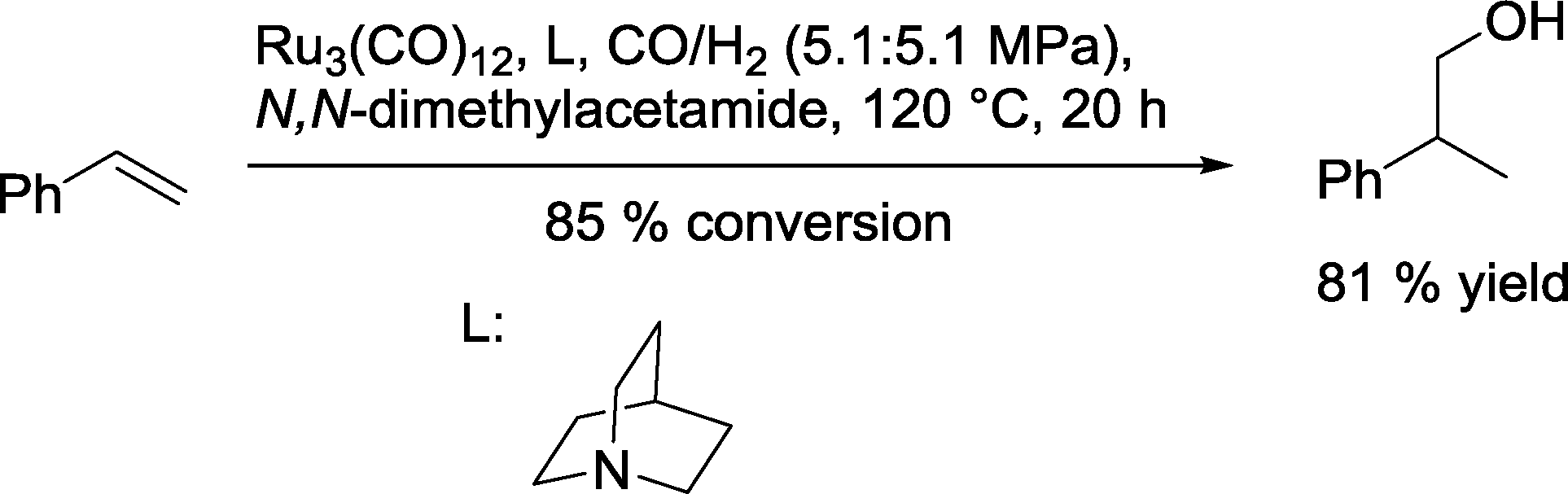 | ||
| Scheme 34 Ruthenium-catalyzed iso-selective hydroformylation–hydrogenation of styrene.98 | ||
Haukka and co-workers demonstrated that replacing pyrazine with 2-substituted pyrazine ligands, such as 2-chloropyrazine, enhances the activity of the parent hydroformylation catalysts and can also contribute to the formation of alcohols.99 In the best case, 40% total yield of isomeric alcohols was achieved. Possible routes for the hydrogenation of the carbonyl group were calculated covering mono- and dihydride ruthenium intermediates.
A range of acyclic and cyclic olefins were cleanly converted by the Beller group into the corresponding alcohols using a Ru catalyst based on imidazole phosphines (Scheme 35).100 The best results were obtained with linear α-olefins as substrates. It is worth noting that styrene and isoprene also produced good yields of the desired alcohols. Methyl α-methylacrylate reacted further to produce the corresponding lactone.
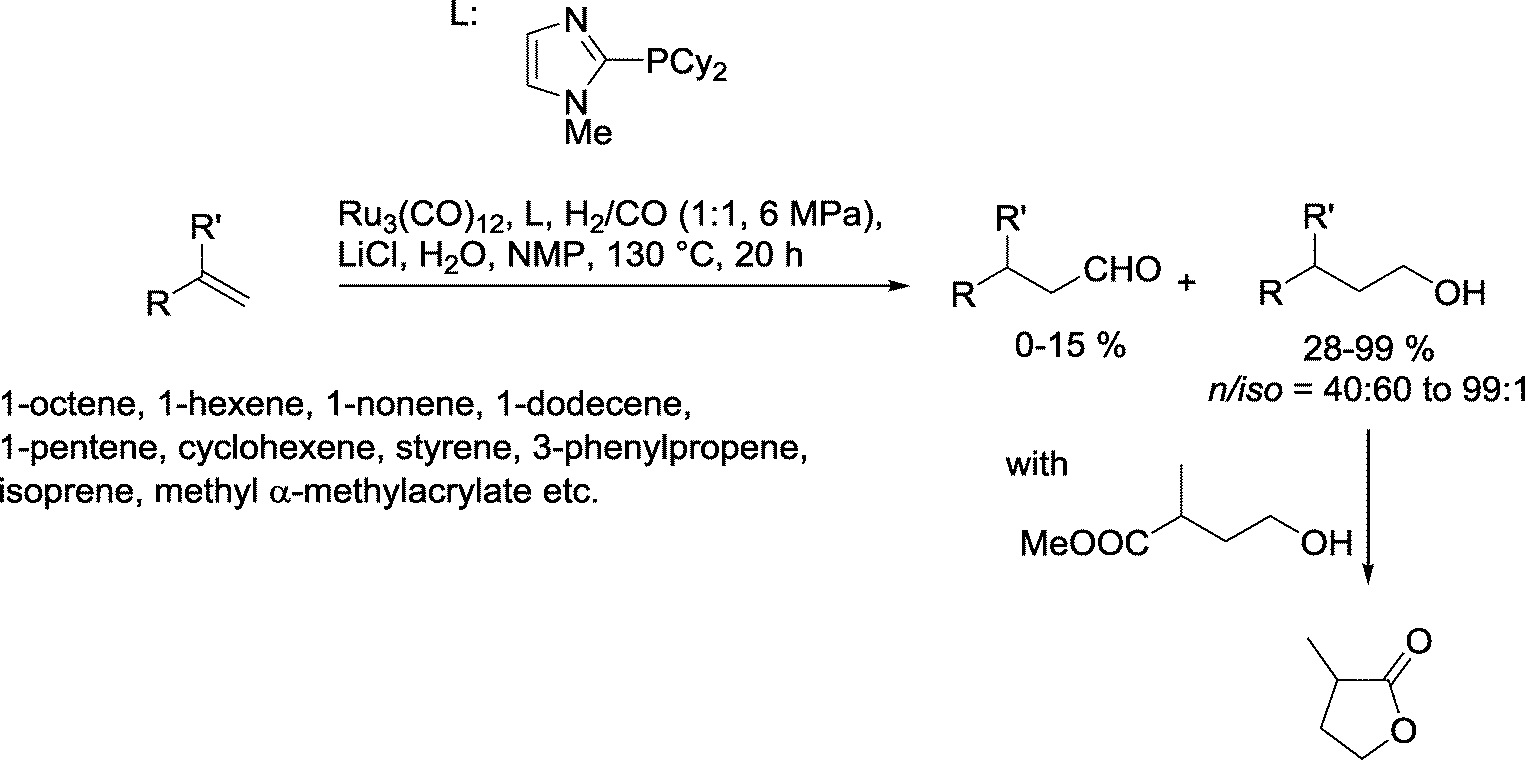 | ||
| Scheme 35 Hydroformylation–hydrogenation with an imidazole phosphine-modified Ru catalyst.100 | ||
A similar catalytic system based on an ether phosphine as a ligand was used for the isomerizing hydroformylation–hydrogenation sequence using an excess of hydrogen (Scheme 36).101,102 Linear 2-olefins produced mainly corresponding C1-prolonged alcohols with n/iso selectivities of up to 86:14. Under these conditions, 2,5-dihydrofuran and 2,3-dihydropyrrol also reacted to the relevant methanol derivatives. The highest selectivity was observed in the reaction with 1-methyl-4-(prop-1-en-2-yl)cyclohex-1-ene, where only the exocyclic double bond reacted.
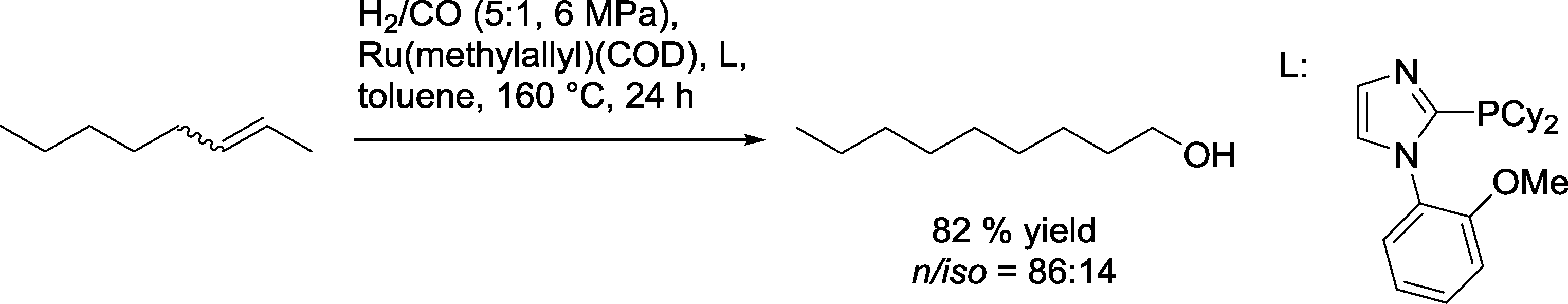 | ||
| Scheme 36 Isomerizing hydroformylation–hydrogenation of 2-octene with an imidazole phosphine-modified Ru catalyst.101 | ||
Homogeneous Ru complexes are particularly useful for adjusting the equilibrium of the reversed water gas shift (RWSG) reaction, which produces CO from CO2. Tominaga and Sasaki used tetranuclear Ru clusters to produce alcohols from olefins based on this approach (Scheme 37).103 Inorganic salts, like LiCl, were essential to suppress undesired hydrogenation of the substrate olefin. The formation of alcohols is favored at 140 °C. Addition of PPh3 completely inhibited the reaction. The highest yield of alcohol was observed with cyclohexene as a substrate (88%). In general, only traces of the aldehydes were observed, while the formation of alkanes was considerable in some cases.
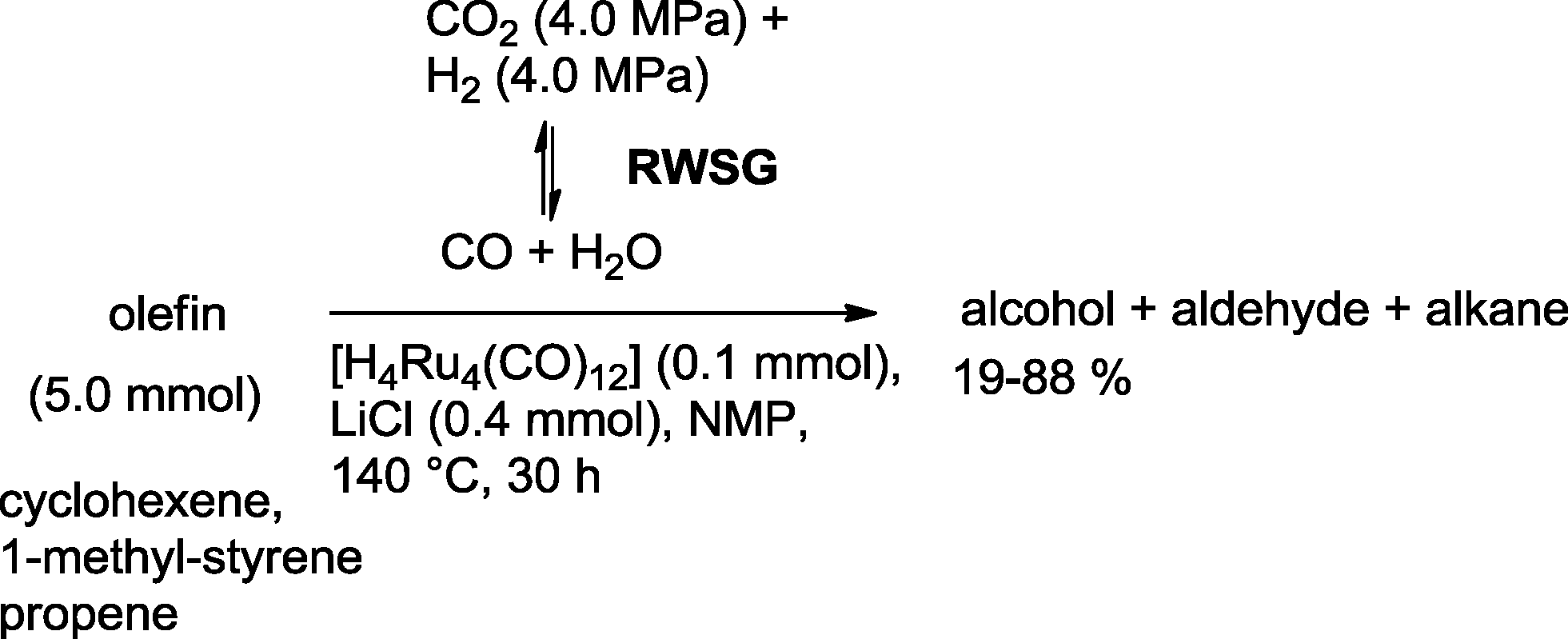 | ||
| Scheme 37 Ruthenium-catalyzed hydroformylation–hydrogenation using reversed water gas shift reaction (RWSG) as a CO source.103 | ||
For the same transformation, the use of the catalytic system [Ru(CO)3Cl2]2/Li2CO3 (ref. 104) or [Ru(CO)4]n/LiCl in DMF also proved to be useful.105 In principle, increasing the total pressure of H2 and CO2 promoted the RWGS and increased the yield of the aldehyde, whereas hydrogenation of the latter benefited from an increase in H2 partial pressure and a decrease in CO2 partial pressure.106
When Co2Rh2(CO)12 or a mixture of Co4(CO)12 and Rh4(CO)12 were impregnated onto organic Dowex® resins, active hydroformylation catalysts were produced.110 With amine groups on the resin or the addition of NEt3 in the hydroformylation of 1-hexene, the corresponding nonanols were formed exclusively with a yield of 95%. The ratio of Rh/Co had a marked effect on product distribution. Best results were observed with a Rh/Co ratio of 2.6–3.6. Other functional groups in the resin did not force the formation of alcohols.
The bimetallic cluster [HRuCo3(CO)12] supported on amorphous carbon also exhibited high chemoselectivity for the formation of alcohols in the continuous hydroformylation–hydrogenation of ethylene (172 °C, C2H4:CO:H2 = 20:20:20 ml min−1, 0.1 MPa) and propylene (203 °C, C3H6:CO:H2 = 20:20:20 ml min−1, 0.1 MPa), respectively.111 A cooperative effect of both metals was assumed, since Co4(CO)12 alone proved to be inactive.
3. Production of alcohols based on a hydroformylation–(aldol-condensation)–hydrogenation approach
Hydroformylation is often followed by an aldol reaction, which produces branched β-hydroxy aldehydes. The C–C-coupling benefits from the activating effect of the newly formed aldehyde group. Alternatively, the aldol reaction can be followed by the elimination of water (aldol condensation), giving rise to unsaturated aldehydes (“enals”). Subsequent hydrogenation of the unsaturated aldehyde produces either allyl alcohols when the reduction is carried out in a chemoselective manner or leads to saturated branched alcohols.112 Especially the latter is carried out in the chemical industry on very large scale.A typical example with huge economic relevance is the manufacture of 2-propyl-heptanol (2-PH), which is for example produced by Evonik Industries as a component of plasticizer alcohols and, to a smaller extent, for use in cosmetics (Scheme 38, Fig. 1).113,114 In the first step, n-valeraldehyde is derived from the rhodium-catalyzed hydroformylation of isomeric butenes. The newly formed aldehyde group is the precondition for the subsequent aldol condensation which takes place in a basic medium. In the final hydrogenation step, the CC-bond and carbonyl group are simultaneously reduced by the effect of a heterogeneous Co or Ni catalyst to produce the desired saturated branched alcohol.115
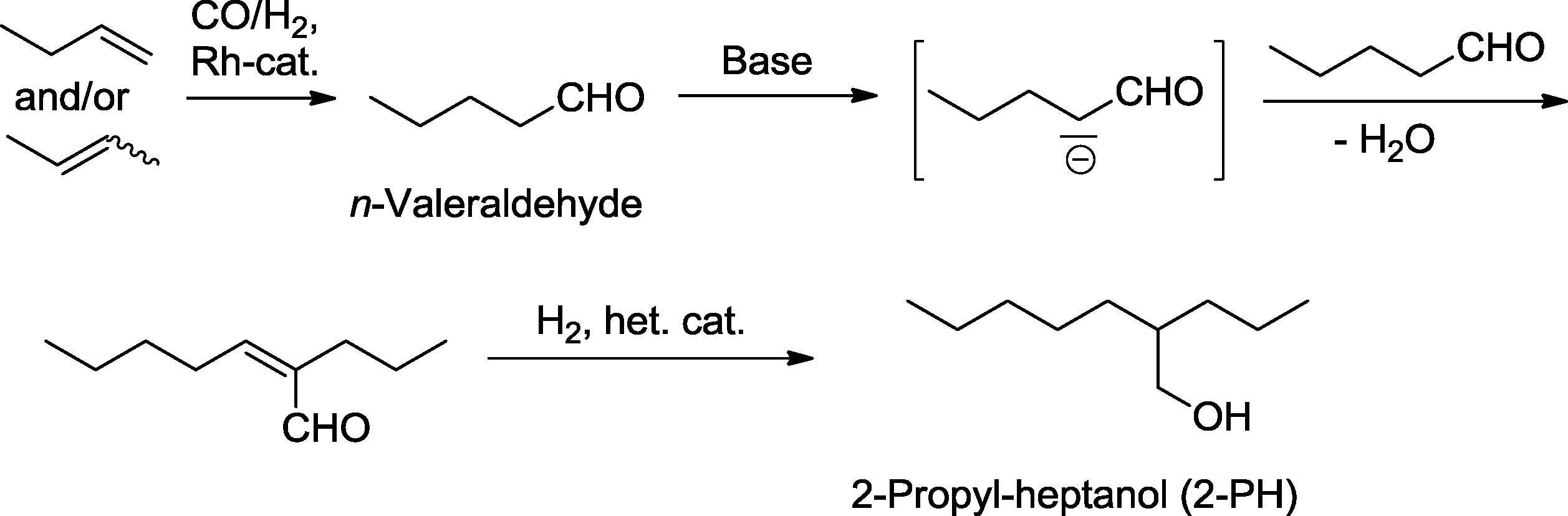 | ||
| Scheme 38 Hydroformylation–(aldol condensation)–hydrogenation approach for large-scale synthesis of 2-propyl-heptanol. | ||
 | ||
| Fig. 1 2-Propyl-heptanol (2-PH) plant of Evonik Industries at Marl (Germany) opened in 2009. | ||
In a similar process, the production of the “workhorse” plasticizer alcohol 2-ethyl-hexanol (2-EH) is carried out, for example, by BASF, Dow, Eastman, and Oxea starting with the hydroformylation of propene using Co at high pressure or using modified Rh catalysts (LP Oxo process).116,117 The mixture of n-butyraldehyde and isobutyraldehyde can be separated by distillation. The former is transformed either into n-butanol or into 2-EH (Scheme 39).
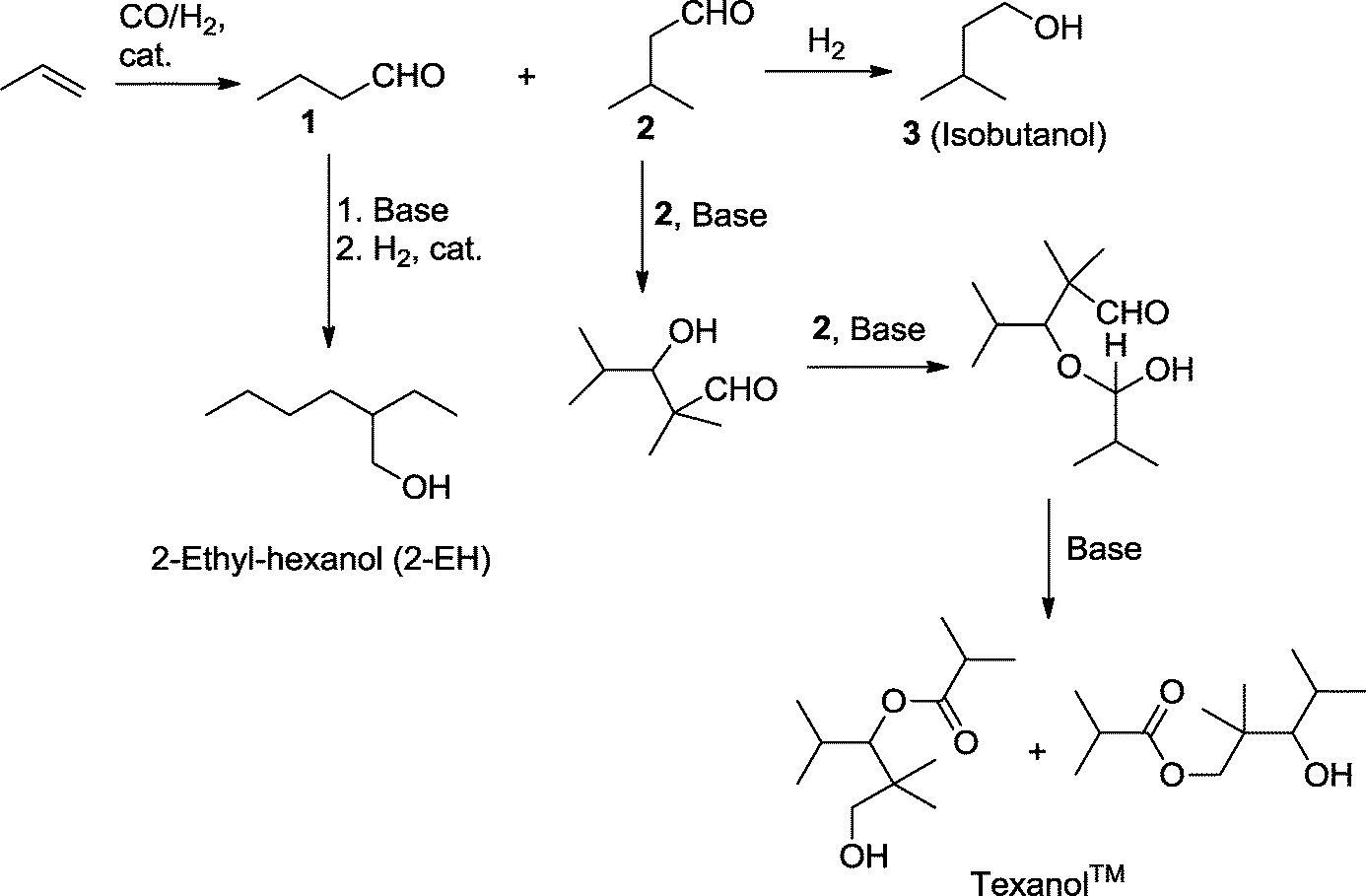 | ||
| Scheme 39 Production of 2-ethyl hexanol and Texanol™ via a hydroformylation step. | ||
Isobutyraldehyde can be reduced to produce isobutanol or it is used to manufacture 2,2,4-trimethyl-1,3-pentanediol monoisobutyrate (Texanol™). The monoester is usually formed as a mixture of two isomers, 1-hydroxy-2,2,4-trimethylpentan-3-yl isobutyrate and 3-hydroxy-2,2,4-trimethylpentyl isobutyrate, when three molecules of isobutyraldehyde are condensed in the presence of a base (NaOH, LiOH).118 Texanol™ is a widely used solvent in the chemical industry. It is also used, for example, as an advanced plasticizer for PVC and as a coalescent for latex paints.119 Since 1962, it has been produced by Eastman Chemical Company and is now also a product of Perstop, for example.
To reduce the costs of constructing plants with separate units, multiple reactions in a single vessel would appear to be interesting. In this respect, Shukla and co-workers investigated the kinetics of 2-EH formation in a one-pot auto-tandem reaction (Scheme 40).120 The heterogeneous catalyst was prepared by impregnation of hydrotalcite Mg1−xAlx(OH2)x+(CO32−)x/n·mH2O with HRh(CO)(PPh3)3. Hydroformylation was carried out at 60 °C. In the optimum case, including subsequent heating to 250 °C to initiate aldol condensation and hydrogenation of the condensation product, gave 2-EH with 18% selectivity after 12 h.
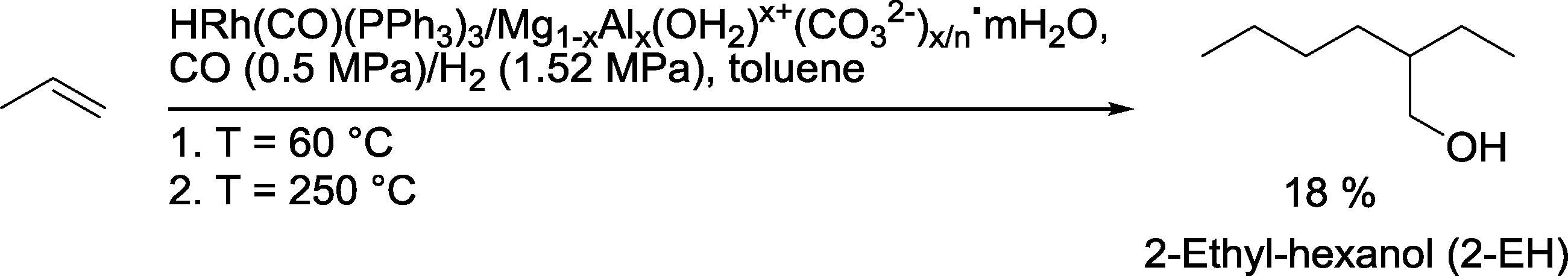 | ||
| Scheme 40 Combined hydroformylation–(aldol condensation)–hydrogenation approach.120 | ||
4. Summary and conclusions
The production of alcohols from olefins using hydroformylation technology is now a well-established procedure. Several strategies have been developed, ranging from separate hydroformylation and hydrogenation steps using different catalysts and reaction conditions to hydroformylation–hydrogenation tandem reactions, which take place in a one-pot reaction using the same catalyst and with uniform reaction conditions. In particular, cobalt and rhodium catalysts have been used. Chemoselectivity of the tandem reaction can be optimized through modification with organic ligands, preferably nitrogen and phosphorus ligands. Other metals, such as palladium and ruthenium have also been tested successfully. To date, no selective iridium catalyst has been described in the literature for this purpose, although relevant homogeneous complexes have been shown to assist hydroformylation of olefins.121 Moreover, hydrogenation of aldehydes with IrH3(PPh3)3 has been known for a long time.122 Besides syngas, other sources for syngas or CO, such as paraformaldehyde or CO2, have also been used to produce alcohols with the assistance of rhodium and ruthenium catalysts. Interestingly, in large scale processes cobalt catalyzed reducing hydroformylation still dominates, whereas rhodium catalysts are first choice for stepwise hydroformylation–hydrogenation processes. The more sophisticated hydroformylation–hydrogenation auto-tandem reactions should be of value especially in fine chemistry. In particular, the use of alternative less expensive metals, such as iron, could be of interest in future research.References
-
(a) M. Beller, B. Cornils, C. D. Frohning and C. W. Kohlpaintner, J. Mol. Catal. A: Chem., 1995, 104, 17 CrossRef CAS
; (b) Rhodium Catalyzed Hydroformylation, ed. P. W. N. M. van Leeuwen and C. Claver, Kluver Academic Publishers, Dordrecht, Netherlands, 2000 Search PubMed
-
(a) G. T. Whiteker and C. J. Cobley, Top. Organomet. Chem., 2012, 42, 35 CrossRef CAS
-
E. Drent, R. van Ginkel and W. W. Jager, Process for producing olefins, (to Shell Internationale Research Maatschappij B. V.), WO Pat., no. 2008/034894, 2008 Search PubMed
-
(a) D. Darensbourg, F. Joo, M. Kannisto, A. Katho and J. H. Reibenspies, Organometallics, 1992, 11, 199 Search PubMed
-
(a) P. Eilbracht, L. Bärfacker, C. Buss, C. Hollmann, B. E. Kitsos-Rzychon, C. L. Kranemann, T. Rische, R. Roggenbuck and A. Schmidt, Chem. Rev., 1999, 99, 3329 CrossRef CAS PubMed
- D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365 CrossRef CAS PubMed
- In ref. 6 a differentiation has been made between: one-pot reactions involving isolated catalytic events, domino catalysis, and tandem catalysis. The latter is subdivided into: orthogonal tandem catalysis, auto-tandem catalysis, and assisted tandem catalysis.
- J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001 CrossRef CAS PubMed
-
S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, John Wiley & Sons, New York, US, 2001, p. 170 Search PubMed
-
(a) See e.g.: R. B. Mason and E. W. S. Nicholson, Sulfactive aldehyde hydrogenation catalyst, (to Esso Research and Engineering Company), US Pat., no. 2813911 A, 1957 Search PubMed
-
W. Tötsch, D. Arnoldi, A. Kaizik and M. Trocha, Process for the production of 7–17 C aliphatic alcohols comprises cobalt catalyzed hydroformylation of 3-16 C olefins whereby the organic phase is extracted with a water containing liquid, (to Oxeno Olefinchemie GmbH), DE Pat., no. 10227995 A1, 2003 Search PubMed
-
(a)
W. F. Ratcliff, Acid decobalting of oxo products, (to Esso Research and Engineering Company), US Pat., no. 2757204 A, 1956 Search PubMed
-
E. T. van Driessche, J.-J. G. Muhls, A. van Vliet, C. R. Beck and C. M. Yarbrough, Plasticiser Alcohol and Production Improvement, (to Exxonmobil Chemical Comp.), US Pat., no. 2010/0312005 A1, 2010 Search PubMed
-
A. V. Grenacher, M. Strohmeyer, H. Hoffmann and H. Elliehausen, Procedure for the preparation of C3-25 alkanols, (to BASF AG), DE Pat., no. 3542595 A1, 1987 Search PubMed
-
J. Rudolph, A. Ulonska, R. Papp, R. Paciello, B. Breitscheidel and K. Faller, Preparation of C17 alcohol mixtures by oligomerization and hydroformylation of olefins, (to BASF SE), WO Pat., no. 2009/124979, 2009 Search PubMed
-
A. V. Grenacher and H. Stepp, Continuous process for hydroformylating olefins with 6 to 20 carbon atoms, (to BASF Aktiengesellschaft), EP Pat., no. 1204624 B1, 2000 Search PubMed
- Inventor not yet nominated, Hydrogenation of organic compounds comprises passing liquid feed through first and second reactors in series and bringing into contact with hydrogen in presence of hydrogenation catalyst, (to BASF AG), DE Pat., no. 10036172 A1, 2001 Search PubMed
- Iso-Index characterizes the average branching degree. It can be determined by 1H NMR spectroscopy using the corresponding carbamates. Usually for the application in detergents and plasticers an Iso-Index = 2.8–3.7 is aimed for.
-
M. Matsumoto, S. Miura, K. Kikuchi, M. Tamaru, H. Kojima, K. Koga and S. Yamashita, High catalyst selectivity to 4-hydroxybutyraldehyde, (to Kuraray Company, Ltd; Daicel Chemical Industries, Ltd.), US Pat., no. 4567305 A, 1986 Search PubMed
-
W. S. Dubner and W. P.-S. Shum, Allyl alcohol hydroformylation, (to ARCO Chemical Technology, L.P.), US Pat., no. 6225509 B1, 2001 Search PubMed
-
http://www.lyondellbasell.com/Products/ByCategory/basic-chemicals/PerformanceChemicalsAndSolvents/1-4-Butanediol/
- L. Garcia, C. Claver, M. Diéguez and A. M. Masdeu-Bultó, Chem. Commun., 2006, 191 RSC
- J. Zakzeski, H. R. Lee, Y. L. Leung and A. T. Bell, Appl. Catal., A, 2010, 374, 201 CrossRef CAS PubMed
- D. G. Hanna, S. Shylesh, P. A. Parada and A. T. Bell, J. Catal., 2014, 311, 52 CrossRef CAS PubMed
- A mixture of n-butyraldehyde and isobutyraldehyde can be separated by distillation. The former is transformed either into n-butanol or into 2-ethyl hexanol. Isobutyraldehyde can be reduced to produce isobutanol as carried out, for example, at Gazprom neftekhim Salavat on a 170 thousand tons scale:
(a) T. G. Shokhova, I. F. Sadretdinov, O. R. Tyhvatyllin and A. S. Alyabev, Oil Gas Bus., 2012, 3, 231 Search PubMed
- A. J. Sandee, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2001, 123, 8468 CrossRef CAS PubMed
- K. Takahashi, M. Yamashita, T. Ichihara, K. Nakano and K. Nozaki, Angew. Chem., Int. Ed., 2010, 49, 4488 CrossRef CAS PubMed
-
(a) M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1995, 14, 3081 CrossRef CAS
-
(a) Y. Shvo, D. Czarkie, Y. Rahamim and D. F. Chodosh, J. Am. Chem. Soc., 1986, 108, 7400 CrossRef CAS
- K. Takahashi, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2012, 134, 18746 CrossRef CAS PubMed
- Y. Yuki, K. Takahashi, Y. Tanaka and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 17393 CrossRef CAS PubMed
- M. Vilches-Herrera, L. Domke and A. Börner, ACS Catal., 2014, 4, 1707 Search PubMed
-
M. L. Clarke and G. J. Roff, Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, Germany, 2007, p. 413 Search PubMed
-
C. D. Frohning and C. W. Kohlpaintner, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, VCH, Weinheim, Germany, 1996, p. 29 Search PubMed
- H. M. Feder and J. Halpern, J. Am. Chem. Soc., 1975, 97, 7186 CrossRef CAS
- E. N. Frankel, E. P. Jones, V. L. Davison, E. Emken and H. J. Dutton, J. Am. Oil Chem. Soc., 1965, 42, 130 CrossRef CAS
- F. Ungvary, Acta Chim. Acad. Sci. Hung., 1982, 111, 117 CAS
-
(a) F. Piacenti, M. Bianchi, P. Frediani, U. Matteoli and A. Lo Moro, J. Chem. Soc., Chem. Commun., 1976, 9, 789 RSC
- C. L. Aldridge and H. B. Jonassen, J. Am. Chem. Soc., 1963, 85, 886 CrossRef CAS
- F. E. Paulik, Catal. Rev., 1972, 6, 49 CAS
- K. Murata and A. Matsuda, Nippon Kagaku Kaishi, 1980, 7, 1077 CrossRef
- V. K. Srivastava, R. S. Shukla, H. C. Bajaj and R. V. Jasra, Appl. Catal., A, 2005, 282, 31 CrossRef CAS PubMed
- L. H. Slaugh and R. D. Mullineaux, J. Organomet. Chem., 1968, 13, 469 CrossRef CAS
-
(a) M. Haumann, R. Meijboom, J. R. Moss and A. Roodt, Dalton Trans., 2004, 1679 RSC
-
J. L. Eisenmann and R. L. Yamartino, Aromatic phosphite catalyst modifiers in the oxo process, (to Diamond Alkali Company), US Pat., no. 3150188 A, 1964 Search PubMed
-
(a) M. Beller and J. G. E. Krauter, J. Mol. Catal. A: Chem., 1999, 143, 31 CrossRef CAS
- T. Mizoroki, M. Kioka, M. Suzuki, S. Sakatani, A. Okumura and K. Maruya, Bull. Chem. Soc. Jpn., 1984, 57, 577 CrossRef CAS
-
(a)
L. H. Slaugh and R. D. Mullineaux, Hydroformylation of olefins, (to Shell Oil Company), US Pat., no. 3239569 A, 1966 Search PubMed
-
(a) E. R. Tucci, Ind. Eng. Chem. Prod. Res. Dev., 1968, 7, 32 CrossRef CAS
-
(a) G. F. Pregaglia, A. Andreetta, G. F. Ferrari and G. Ugo, J. Organomet. Chem., 1971, 30, 387 CrossRef CAS
-
(a) L. Rosi, A. Bini, P. Frediani, M. Bianchi and A. Salvini, J. Mol. Catal. A: Chem., 1996, 112, 367 CrossRef CAS
-
R. F. Mason and J. L. van Winkle, Bicyclic heterocyclic sec- and tert-phosphines, (to Shell Oil Company), US Pat., no. 3400163 A, 1968 Search PubMed
- C. Crause, L. Bennie, L. Damoense, C. L. Dwyer, C. Grove, N. Grimmer, W. Janse van Rensburg, M. M. Kirk, K. M. Mokheseng, S. Otto and P. J. Steynberg, Dalton Trans., 2003, 2036 RSC
-
J. L. Van Winkle, S. Lorenzo, R. C. Morris and R. F. Mason, Single stage hydroformylation of olefins to alcohols, (to Shell Oil Company), US Pat., no. 3420898 A, 1969 Search PubMed
- P. N. Bungu and S. Otto, Dalton Trans., 2007, 2876 RSC
- F. Piacenti, M. Bianchi, E. Benedetti and P. Frediani, J. Organomet. Chem., 1970, 23, 257 CrossRef CAS
- P. N. Bungu and S. Otto, Dalton Trans., 2011, 40, 9238 RSC
- T. Bartik, B. Bartik and B. E. Hanson, J. Mol. Catal., 1993, 85, 121 CrossRef CAS
- G. F. Pregaglia, Chem. Ind., 1969, 51, 1043 CAS
-
P.-K. Wong and A. A. Moxey, Hydroformylation of Alpha-alcohol-diolefins to Dialcohols, (to Shell Oil Company), US Pat., no. 6114588 A, 2000 Search PubMed
- T. Yamagishi, S.-I. Ito, K. Tomishige and K. Kunimori, Catal. Commun., 2005, 6, 421 CrossRef CAS PubMed
- M. Simpson and D. J. Cole-Hamilton, Coord. Chem. Rev., 1996, 155, 163 CrossRef CAS
- D. Evans, J. A. Osborn and G. Wilkinson, J. Chem. Soc. A, 1968, 3133 RSC
- T. Mizoroki, M. Kioka, M. Suzuki, S. Sakatani, A. Okumura and K.-I. Maruya, Bull. Chem. Soc. Jpn., 1984, 57, 577 CrossRef CAS
- K. Kaneda, M. Yasumura, M. Hiraki, T. Imanaka and S. Teranishi, Chem. Lett., 1981, 12, 1763 CrossRef
- K. Kaneda, T. Imanaka and S. Teranishi, Chem. Lett., 1983, 9, 1465 CrossRef
- L. L. W. Cheung, G. Vasapollo and H. Alper, Adv. Synth. Catal., 2012, 354, 2019 CrossRef CAS
- J.-Q. Zhou and H. Alper, J. Chem. Soc., Chem. Commun., 1991, 233 RSC
- J. K. MacDougall and D. J. Cole-Hamilton, J. Chem. Soc., Chem. Commun., 1990, 165 RSC
- J. K. MacDougall and D. J. Cole-Hamilton, Polyhedron, 1990, 9, 1235 CrossRef CAS
- M. C. Simpson, A. W. S. Currie, J.-A. M. Andersen, D. J. Cole-Hamilton and M. J. Green, J. Chem. Soc., Dalton Trans., 1996, 1793 RSC
- P. Cheliatsidou, D. F. S. White and D. J. Cole-Hamilton, Dalton Trans., 2004, 3425 RSC
- J. K. MacDougall, M. C. Simpson, M. J. Green and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 1996, 1161 RSC
-
P. Johnson and M. J. Lawrenson, Improved Hydroformylation Catalyst and process, (to The British Petroleum Company), GB Pat., no. 1181806 A, 1970 Search PubMed
- F. E. Paulik, Catal. Rev., 1972, 6, 49 CAS
- J. K. Macdougall, M. C. Simpson and D. J. Cole-Hamilton, Polyhedron, 1993, 12, 2877 CrossRef CAS
- M. C. Simpson, K. Porteous, J. K. Macdougall and D. J. Cole-Hamilton, Polyhedron, 1993, 12, 2883 CrossRef CAS
- J.-A. M. Andersen and A. W. S. Currie, Chem. Commun., 1996, 1543 RSC
- L. Ropartz, R. E. Morris, D. F. Foster and D. J. Cole-Hamilton, J. Mol. Catal. A: Chem., 2002, 182–183, 99 CrossRef CAS
-
M. J. Lawrenson, Catalytic hydroformylation of olefins to alcohols, (to BP Chemicals Ltd.), GB Pat., no. 1254222 A, 1971 Search PubMed
- B. A. Rodriguez and W. J. Tenn III, Appl. Catal., A, 2012, 421–422, 161 CrossRef CAS PubMed
- T. Okano, T. Kobayashi, H. Konishi and J. Kiji, Tetrahedron Lett., 1982, 23, 4967 CrossRef CAS
-
J. R. Briggs, D. L. Packett, D. R. Bryant, A. G. Phillips, D. J. Schreck, A. S. Guram, K. D. Olson, T. C. Eisenschmid and E. B. Tjaden, Processes For Producing 1,6-hexanediols, (to Union Carbide Chemicals & Plastics Technology Corporation), US Pat., no. 6187970 B1, 2001 Search PubMed
- I. I. F. Boogaerts, D. F. S. White, M. J. Green and D. J. Cole-Hamilton, Chem. Commun., 2010, 46, 2194 RSC
- O. Diebolt, C. Müller and D. Vogt, Catal. Sci. Technol., 2012, 2, 773 CAS
- T. Ichihara, K. Nakano, M. Katayama and K. Nozaki, Chem. – Asian J., 2008, 3, 1722 CrossRef CAS PubMed
- W. R. Jackson, P. Perlmutter and G.-H. Suh, J. Chem. Soc., Chem. Commun., 1987, 724 RSC
- W. R. Jackson, P. Perlmutter, G.-H. Suh and E. E. Tasdelen, Aust. J. Chem., 1991, 44, 951 CrossRef CAS
- L. Diab, T. Šmejkal, J. Geier and B. Breit, Angew. Chem., Int. Ed., 2009, 48, 8022 CrossRef CAS PubMed
- L. Diab, U. Gellrich and B. Breit, Chem. Commun., 2013, 49, 9737 RSC
- D. Konya, K. Q. Almeida Leñero and E. Drent, Organometallics, 2006, 25, 3166 CrossRef CAS
- J. F. Knifton, J. Mol. Catal., 1987, 43, 65 CrossRef CAS
-
(a) L. Alvila, T. A. Pakkanen and O. Krause, J. Mol. Catal., 1993, 84, 145 CrossRef CAS
- M. Haukka, L. Alvila and T. A. Pakkanen, J. Mol. Catal. A: Chem., 1995, 102, 79 CrossRef CAS
- L. Alvila, T. A. Pakkanen, T. T. Pakkanen and O. Krause, J. Mol. Catal., 1992, 73, 325 CrossRef CAS
- M. A. Moreno, M. Haukka, S. Jääskeläinen, S. Vuoti, J. Pursiainen and T. A. Pakkanen, J. Organomet. Chem., 2005, 690, 3803 CrossRef CAS PubMed
- E. M. Gordon and R. Eisenberg, J. Organomet. Chem., 1986, 306, C53 CrossRef CAS
- T.-A. Mitsudo, N. Suzuki, T.-A. Kobayashi and T. Kondo, J. Mol. Catal. A: Chem., 1999, 137, 253 CrossRef CAS
- M. A. Moreno, M. Haukka, A. Turunen and T. A. Pakkanen, J. Mol. Catal. A: Chem., 2005, 240, 7 CrossRef CAS PubMed
- I. Fleischer, K. M. Dyballa, R. Jennerjahn, R. Jackstell, R. Franke, A. Spannenberg and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2949 CrossRef CAS PubMed
- L. Wu, I. Fleischer, R. Jackstell, I. Profir, R. Franke and M. Beller, J. Am. Chem. Soc., 2013, 135, 14306 CrossRef CAS PubMed
- It seems that ether phosphine may force the formation of alcohols: A. Solsona, J. Suades and R. Mathieu, J. Organomet. Chem., 2003, 669, 172 CrossRef CAS
-
(a) K.-I. Tominaga and Y. Sasaki, Catal. Commun., 2000, 1, 1 CrossRef CAS
- S. Jääskeläinen and M. Haukka, Appl. Catal., A, 2003, 247, 95 CrossRef
- M.-L. Kontkanen, L. Oresma, M. A. Moreno, J. Jänis, E. Laurila and M. Haukka, Appl. Catal., A, 2009, 365, 130 CrossRef CAS PubMed
- S.-I. Fujita, S. Okamura, Y. Akiyama and M. Arai, Int. J. Mol. Sci., 2007, 8, 749 CrossRef CAS PubMed
-
(a)
N. E. Kinkade, Catalytic process for the production of alcohols from carbon monoxide, hydrogen and olefins, (to Union Carbide Corp.), US Pat., no. 4590314 A, 1986 Search PubMed
- T. Yamagishi, S.-I. Ito, K. Tomishige and K. Kunimori, Catal. Commun., 2005, 6, 421 CrossRef CAS PubMed
- J. Llorca, B. Barbier, G. A. Martin, J. Sales, P. Ramirez de la Piscina and N. Homs, Catal. Lett., 1996, 42, 87 CrossRef CAS
- L. Alvila, T. A. Pakkanen and T. T. Pakkanen, J. Mol. Catal., 1992, 71, 281 CrossRef CAS
- A. Fukuoka, H. Matsuzaka, M. Hidai and M. Ichikawa, Chem. Lett., 1987, 16, 941 CrossRef
- P. Gallezot and D. Richard, Catal. Rev.: Sci. Eng., 1998, 40, 81 CAS
-
(a)
W. Ahlers, R. Paciello, T. Mackewitz and M. Volland, Method for the production of 2-propylheptanol and hydroformylating catalysts and the further use thereof for carbonylation, hydrocyanation and hydrogenation, (to BASF Aktiengesellschaft), WO Pat., no. 2003018192 A2, 2003 Search PubMed
- Meanwhile, Dow Technology Licensing is licensing together with Davy Process Technology the so-called LP OxoSM Technology for the production of alcohols by low-pressure hydroformylation.
-
(a)
H. Bahrmann, W. Greb, P. Heymanns, P. Lappe, T. Muller, J. Szameitat and E. Wiebus, Hydroformylation of butene mixture, aldol condensation, separation and hydrogenation, (to Hoechst Aktiengesellschaft), US Pat., no. 5463147 A, 1995 Search PubMed
- T. Tudor and M. Ashley, Platinum Met. Rev., 2007, 51, 116 CrossRef
-
(a)
K.-D. Wiese, G. Protzmann, J. Koch and W. Büschken, Catalytic aldol condensation, giving intermediates carboxylic acid or alcohol for plasticizer, detergent or solvent synthesis, involves multiphase reaction in tubular reactor with catalyst in solvent phase and aldehyde in disperse phase, (to Oxeno Olefinchemie GmbH), DE Pat., no. 19957522 A1, 2001 Search PubMed
-
(a)
M. A. Perry and H. J. Hagemayer Jr., Preparation of glycol monoesters by condensation of aldehydes in the presence of a strong inorganic base, (to Eastman Kodak Company), US Pat., no. 3291821 A, 1966 Search PubMed
- M. Cho, D. Ko, Y.-H. Lim, M. Jung, H.-S. Cho and J. H. Ok, Journal of the Korean Society of Magnetic Resonance, 2005, 9, 103 Search PubMed
- S. K. Sharma, R. S. Shukla, P. A. Parikh and R. V. Jasra, J. Mol. Catal. A: Chem., 2009, 304, 33 CrossRef CAS PubMed
- J. Pospech, I. Fleischer, R. Franke, S. Buchholz and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2852 CrossRef CAS PubMed
- R. S. Coffey, Chem. Commun., 1967, 923 Search PubMed
| This journal is © The Royal Society of Chemistry 2015 |