
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistic effects of Ni and Cu supported on TiO2 and SiO2 on photocatalytic H2 evolution with an electron donor–acceptor linked molecule†
Yusuke
Yamada
*a,
Shinya
Shikano
a,
Tomoki
Akita
b and
Shunichi
Fukuzumi
*a
aDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; yamada@chem.eng.osaka-u.ac.jp
bNational Institute of Advanced Industrial Science and Technology (AIST), Ikeda, Osaka 563-8577, Japan
First published on 2nd October 2014
Abstract
Synergistic effects of Ni and Cu supported on metal oxides on their catalytic activity for hydrogen evolution were observed in photocatalytic hydrogen evolution with 2-phenyl-4-(1-naphthyl)quinolinium ion (QuPh+–NA) and β-dihydronicotinamide adenine dinucleotide (NADH) as a photocatalyst and an electron donor, respectively. Among the catalysts of Ni and Cu supported on TiO2, SiO2, SiO2–Al2O3 and CeO2, Ni and Cu supported on TiO2 and SiO2 exhibited high catalytic activity at a wide range of Ni contents [Ni/(Ni + Cu)] from 30% to 70%, while Ni or Cu solely supported on TiO2 and SiO2 showed insignificant catalytic activity. The catalytic activity of Ni and Cu supported on TiO2 and SiO2 depends on the preparation methods of the catalysts. The catalysts prepared by a co-impregnation method, in which a precursor solution containing both Ni(NO3)2 and Cu(NO3)2 was used for the impregnation, showed high catalytic activity, whilst catalysts prepared by a sequential impregnation method, in which Ni(NO3)2 and Cu(NO3)2 were loaded and calcined successively, exhibited low catalytic activity. TEM observations with energy-dispersive X-ray spectroscopy (EDS) elemental mapping of these catalysts revealed that Ni and Cu were closely located on the support surfaces of a catalyst prepared by the co-impregnation method, whereas Ni and Cu were separated in the catalyst prepared by the sequential impregnation method. These results suggest that the close location of Ni and Cu is necessary to exhibit the high catalytic activity. Such a synergistic effect among base metals and metal oxide supports would be a key to develop active catalysts for hydrogen evolution without using platinum group metals.
Introduction
Hydrogen (H2) production utilising solar energy attracts much attention from the viewpoint of storage of natural energy as chemical energy.1–11 A photocatalytic H2 evolution system, which mimics natural photosynthesis, can be constructed by using an electron donor, a photosensitiser, an electron mediator and an H2 evolution catalyst.12–20 For example, H2 evolution can be observed by the photoirradiation of a solution containing ethylenediaminetetraacetic acid disodium salt, [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine), methyl viologen and Pt particles as an electron donor, a photosensitiser, an electron mediator and a hydrogen evolution catalyst, respectively.21 This type of H2 evolution systems requires a sacrificial electron donor; however, a high quantum efficiency and rational improvement in catalysis are achievable by replacing each component after modification.8,22,23 As a component of an H2 evolution catalyst, Pt particles have been most widely used because of their very low overpotential for proton reduction.24–26 However, avoiding or reducing the use of Pt metal is strongly demanded because of its high cost and limited supply.22,27Alternative to Pt nanoparticles (PtNPs), Ru nanoparticles (RuNPs) and Ni nanoparticles (NiNPs) have been demonstrated to act as H2 evolution catalysts in reaction systems using organic electron donor–acceptor linked dyads as photocatalysts.28,29 RuNPs exhibit virtually the same activity as PtNPs; however, the catalytic activity of NiNPs was lower than those of PtNPs and RuNPs.28–30 For the improvement of Ni catalysis, concomitant use of other metals such as Cu, Pd and Co is promising as reported for various catalytic reactions.31–33 For example, Ni–Cu alloy nanoparticles have been reported to exhibit catalytic activity for thermal H2 evolution by hydrolysis of sodium borohydride higher than nanoparticles solely composed of Ni or Cu.34 Such a synergistic effect between Ni and Cu has also been reported for supporting catalysts using SiO2,35 Al2O3,36–38 CeO2,39 ZrO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 40 and TiO241 for high-temperature reactions such as ethanol steam reforming. In general, supporting catalytically active metals on a metal oxide is beneficial for improvements not only in the catalytic activity by metal–support interaction but also in the durability of the metal nanoparticles. However, there have been no systematic studies on the synergistic and support effects of Ni and Cu catalysts on photocatalytic H2 evolution.
40 and TiO241 for high-temperature reactions such as ethanol steam reforming. In general, supporting catalytically active metals on a metal oxide is beneficial for improvements not only in the catalytic activity by metal–support interaction but also in the durability of the metal nanoparticles. However, there have been no systematic studies on the synergistic and support effects of Ni and Cu catalysts on photocatalytic H2 evolution.
We report herein the H2 evolution catalysis of Ni–Cu/TiO2 and Ni–Cu/SiO2 in an efficient photocatalytic H2 evolution system composed of a donor–acceptor linked dyad, 2-phenyl-4-(1-naphthyl)quinolinium ion (QuPh+–NA), as a photocatalyst and β-dihydronicotinamide adenine dinucleotide (NADH) as a sacrificial electron donor. The chemical structure of the organic photocatalyst used in this study and the overall reaction scheme are shown in Scheme 1. Upon photoexcitation of QuPh+–NA, electron transfer from the NA moiety to the singlet excited state of the QuPh+ moiety occurs to produce the electron-transfer state (QuPh˙–NA˙+).42,43 Then, electron transfer from NADH to QuPh˙–NA˙+ occurs to produce NADH˙+ and QuPh˙–NA.28 NADH˙+ undergoes deprotonation, releasing one proton to afford NAD˙ that can transfer an electron to QuPh+–NA to produce NAD+ and QuPh˙–NA.28 Two equivalents of QuPh˙–NA thus produced can inject two electrons to Ni–Cu catalysts to evolve H2 from two protons.28 It should be noted that no electron mediator, which is frequently used in homogeneous photocatalytic H2 production, is required in the present photocatalytic system because the electron-transfer state of QuPh+–NA has a sufficient lifetime for the oxidation of NADH and also for electron injection to Ni–Cu catalysts.25,28–30 In this reaction system, effects of the Ni/Cu ratio, preparation methods, the surface area and morphology of SiO2, and the surface area and crystal structure of TiO2 on the catalysis of Ni–Cu/TiO2 and Ni–Cu/SiO2 for H2 evolution were examined to clarify the conditions to achieve the synergistic and support effect, which would be a key to develop active catalysts alternative to platinum group metals.
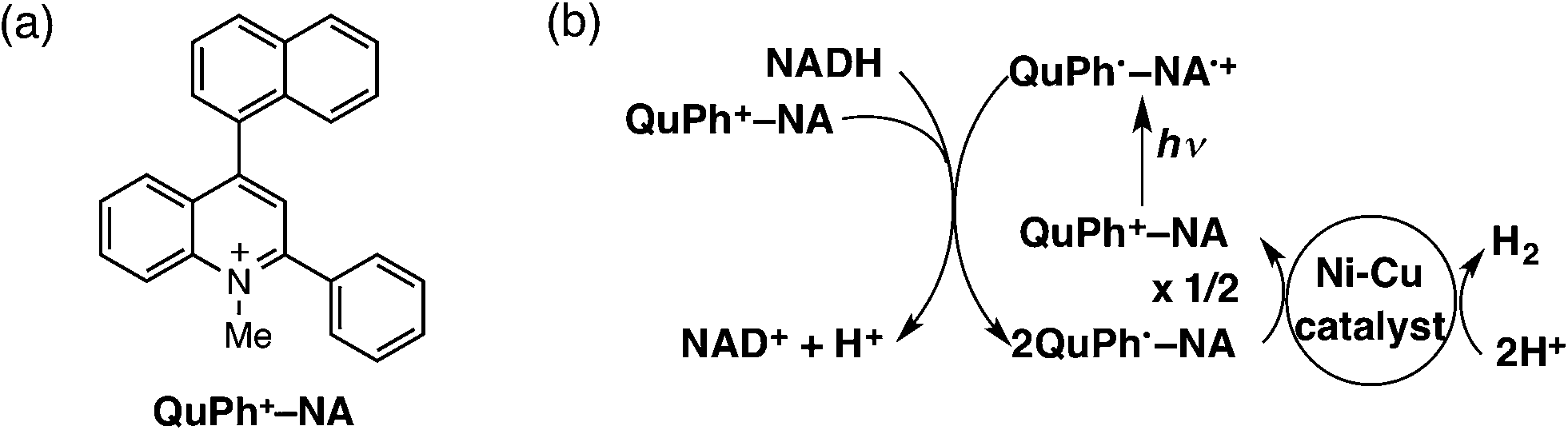 | ||
| Scheme 1 (a) Structure of QuPh+–NA and (b) the overall photocatalytic cycle for H2 evolution using QuPh+–NA and an Ni–Cu catalyst. | ||
2. Results and discussion
2.1 Photocatalytic H2 evolution with Ni and a transition metal supported on metal oxides
The effect of concomitant use of Ni with another metal, Cu, Co or Fe, supported on TiO2 or SiO2 was examined on the H2 evolution catalysis of Ni. These catalysts were prepared by a co-impregnation method. Photocatalytic H2 evolution was performed by photoirradiation (λ > 340 nm) of a mixed suspension (2.0 mL) of MeCN and a phthalate buffer (pH 4.5) [1:1 (v/v)] containing QuPh+–NA (8.8 × 10−4 M), NADH (1.0 × 10−3 M) and TiO2 or SiO2 supporting Ni and another metal (M) (100 mg L−1, M = Cu, Co or Fe), in which the loading amount was 1.5 wt% for each of Ni and M, as shown in Fig. 1.
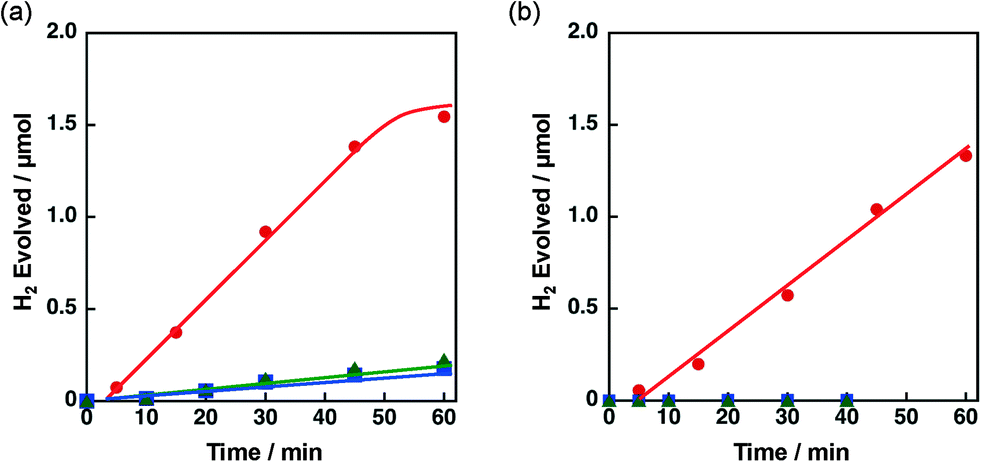 | ||
| Fig. 1 Time courses of H2 evolution by photoirradiation (λ > 340 nm) of a mixed suspension (2.0 mL) of a phthalate buffer (pH 4.5) and MeCN [1:1 (v/v)] containing QuPh+–NA (8.8 × 10−4 M), NADH (1.0 × 10−3 M) and (a) 3 wt% Ni–M/TiO2 or (b) 3 wt% Ni–M/SiO2 (100 mg L−1, M = Cu (red circle), Co (green triangle) and Fe (blue square)). | ||
A significant amount of H2 evolution was observed only for Ni–Cu/TiO2. TiO2 is a well-known photocatalyst; however, no H2 evolution was confirmed for a reaction suspension without QuPh+–NA under the present reaction conditions (Fig. S1a†). From the reaction suspension using Ni–Cu/TiO2 as an H2 evolution catalyst in the presence of QuPh+–NA, continuous H2 evolution was observed after a short induction period for ca. 5 min. This induction period is for the reduction of NiO, NiCuO2 and/or Cu2O species to be catalytically active metallic species because the presence of NiO (JCPDS card no. 78-0643) species in as-prepared Ni–Cu/TiO2 was confirmed by the diffraction peak around 2θ = 43.4° in the powder X-ray diffraction (Fig. 2). This peak may overlap with the diffraction peaks from NiCuO2 (JCPDS card no. 6-720) and Cu2O (JCPDS card no. 05-0667), which have been reported to provide the diffraction peaks at 2θ = 43.8° and 42.5°, respectively, because no diffraction peak was observed at 2θ = 35.6°, which indicates formation of CuO (JCPDS card no. 05-0661). Powder XRD measurements were also performed for the Ni–Cu/TiO2 after the photocatalytic reaction. No diffraction peak indicating formation of Ni0 or Cu0 species was clearly observed in the peak that appeared around 43° (Fig. S2†).
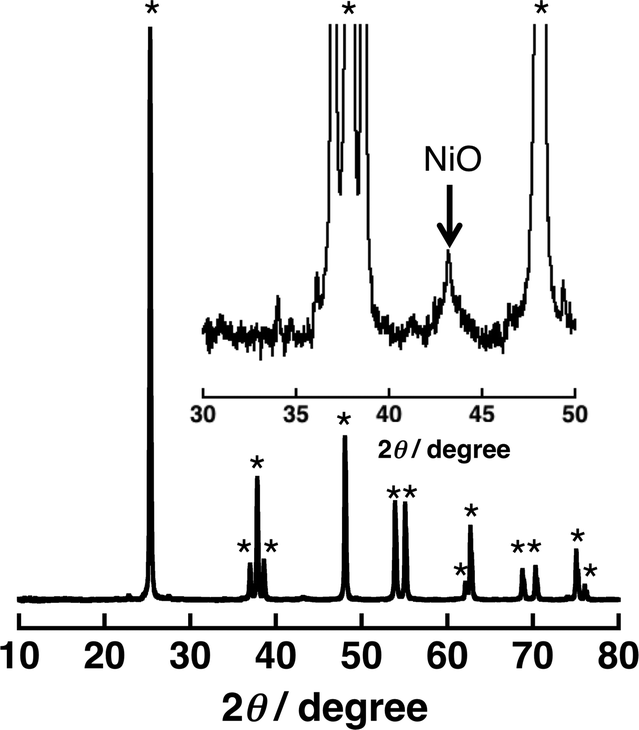 | ||
| Fig. 2 Powder X-ray diffraction patterns of 3 wt% Ni–Cu/TiO2 [Ni/Cu = 1:1 (w/w)]. The peaks with the asterisk symbol (*) originate from TiO2 (anatase). | ||
The initial H2 evolution rate for the reaction system using Ni–Cu/TiO2 was determined to be 1.7 μmol h−1 from the initial (45 min) slope of the reaction system. The apparent quantum yield obtained under photoirradiation (λ = 350 ± 10 nm) for 1 h was 1.1%, where the photon flux determined by using an actinometer was 2.89 × 10−9 einstein s−1. The rate was 10 times slower compared with the H2 evolution rate determined for the reaction system using Pt nanoparticles instead of Ni–Cu/TiO2, in which the weight of used Pt was the same as that of Ni–Cu in the Ni–Cu/TiO2; however, the maximum H2 yield based on the amount of NADH reached as high as 87% (1.7 μmol) without using precious metals. Ceasing H2 evolution by cutting off the light in the course of the reaction ensures that the H2 evolution proceeds photocatalytically (Fig. S1b†). In addition, a photocatalytic system using a reduced amount of QuPh+–NA (0.11 mM) repeatedly evolved H2 five times by the addition of NADH to the reaction solution after ceasing H2 evolution (Fig. S3†), and the turnover number based on QuPh+–NA reached 21. On the other hand, the amount of H2 evolution was less than 0.1 μmol from the suspensions containing TiO2 supporting Ni–Fe and Ni–Co as H2 evolution catalysts by photoirradiation for 60 min (Fig. 1a, green triangle and blue square, respectively). Similarly, a negligible amount of H2 evolution was observed from the reaction suspensions containing Ni/TiO2 and Cu/TiO2 as the H2 evolution catalysts (Fig. S4a†). Thus, Cu acts as the only suitable counterpart of Ni to achieve synergistic effects on the H2 evolution catalysis.
Similar synergistic effects of Ni and Cu on the photocatalytic H2 evolution were observed for Ni–Cu/SiO2 as shown in Fig. 1b, in which the maximum H2 yield reached 77% (1.5 μmol) with a H2 evolution rate of 1.3 μmol h−1 determined from the initial (60 min) slope. No H2 evolution was confirmed for Ni–Fe/SiO2 and Ni–Co/SiO2 by photoirradiation for 40 min. An insignificant amount of H2 evolution was observed from the reaction suspensions employing Ni/SiO2 and Cu/SiO2 as the H2 evolution catalysts (Fig. S4b†). These results obviously indicate that addition of Cu is effective to improve the catalysis of Ni supported on both TiO2 and SiO2, although the addition of Fe or Co was unprofitable.
Support effects on the catalytic activity of Ni–Cu catalysts were also examined by employing TiO2, SiO2, SiO2–Al2O3 and CeO2 as supports. Ni and Cu were loaded on the supports by a co-impregnation method where the loading amount was 1.5 wt% for each Cu and Ni. The Brunauer–Emmett–Teller (BET) surface areas of TiO2, SiO2, SiO2–Al2O3, and CeO2 were determined to be 6.8, 52, 118 and 162 m2 g−1, respectively. The surface area of the TiO2 is significantly low compared to those of others; however, the effect of surface area on the catalytic activity of Ni–Cu/TiO2 is limited (vide infra, Fig. 12). Powder X-ray diffraction patterns of these metal oxides suggested that SiO2 and SiO2–Al2O3 are amorphous in nature, whilst TiO2 and CeO2 are crystalline in the anatase and fluorite structures, respectively (Fig. S5†). These metal oxide supports were chosen from those often used for supporting Ni for various reactions.39–41 As mentioned above, both TiO2 and SiO2 supporting Ni–Cu showed high catalytic activity in terms of both H2 yields and H2 evolution rates (dashed lines in Fig. 3) in the photocatalytic H2 evolution. On the other hand, Ni–Cu supported on SiO2–Al2O3 (red triangle) and CeO2 (blue inverted triangle) showed no significant activity for the photocatalytic H2 evolution as shown in Fig. 3. Although the origin of the support effect is still unclear, the readily reducible nature may be beneficial to produce active sites at the perimeter between Ni–Cu species and the supports. The small negative standard enthalpies of formation for TiO2 and SiO2 (−940 and −910 kJ mol−1, respectively) compared to those for CeO2 and Al2O3 (−1089 and −1676 kJ mol−1, respectively) suggest that TiO2 and SiO2 are more readily reducible than CeO2 and SiO2–Al2O3.44 Thus, SiO2 and TiO2 could be suitable supports to achieve Ni–Cu species that are active for photocatalytic H2 evolution.
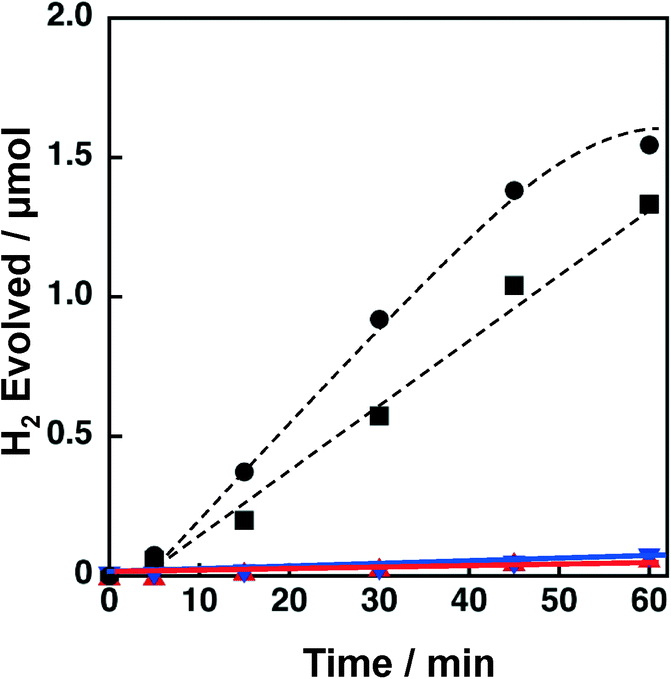 | ||
| Fig. 3 Time courses of H2 evolution by photoirradiation (λ > 340 nm) of a mixed suspension (2.0 mL) of a phthalate buffer (pH 4.5) and MeCN [1:1 (v/v)] containing QuPh+–NA (8.8 × 10−4 M), NADH (1.0 × 10−3 M) and 3 wt% Ni–Cu/MOx (100 mg L−1, MOx = TiO2, circle; SiO2, square; SiO2–Al2O3, red triangle; CeO2, blue inverted triangle). | ||
2.2 Effect of preparation methods on H2 evolution catalysis of Ni–Cu supported on TiO2 and SiO2
The catalytic activity of Ni–Cu/TiO2 and Ni–Cu/SiO2 was highly influenced by preparation methods. When Ni–Cu/TiO2 prepared by a co-impregnation method using a solution containing both Cu(NO3)2 and Ni(NO3)2 was examined for the photocatalytic H2 evolution, a fast H2 evolution rate was observed (Fig. 1a, 1.7 μmol h−1). On the other hand, the H2 evolution rates were as low as 0.5 and 0.3 μmol h−1 with catalysts prepared by a sequential impregnation method, in which Ni/TiO2 was impregnated with a Cu(NO3)2 solution or Cu/TiO2 with a Ni(NO3)2 solution (red circle and blue square in Fig. 4a, respectively). Similar influence of preparation methods was also observed for Ni–Cu/SiO2 as shown in Fig. 4b. The H2 evolution rate obtained for Ni–Cu/SiO2 prepared by co-impregnation was significantly larger (Fig. 1b, 1.3 μmol h−1) than those obtained for Ni–Cu/SiO2 catalysts prepared by the sequential impregnation method, 0.1 and 0.2 μmol h−1.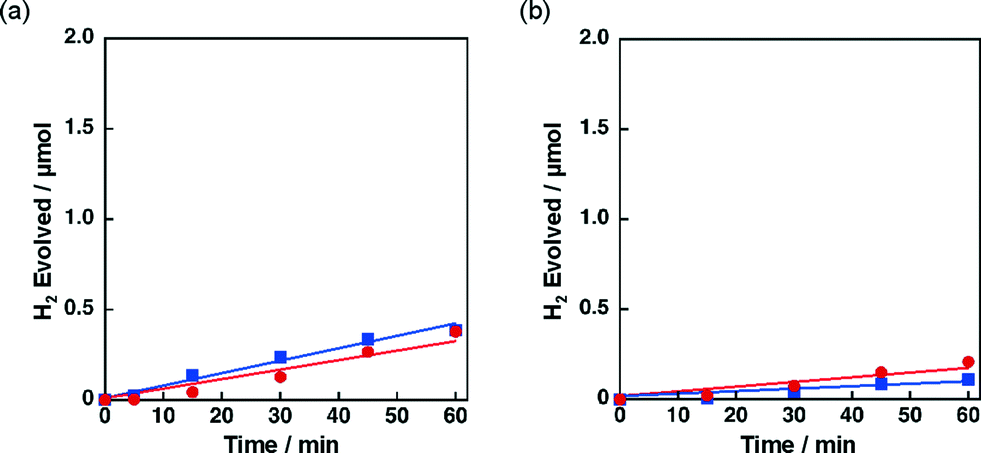 | ||
| Fig. 4 Time courses of H2 evolution by photoirradiation (λ > 340 nm) of a mixed suspension (2.0 mL) of a phthalate buffer (pH 4.5) and MeCN [1:1 (v/v)] containing QuPh+–NA (8.8 × 10−4 M), NADH (1.0 × 10−3 M) and (a) 3 wt% Ni–Cu/TiO2 (100 mg L−1) prepared by a sequential impregnation method (Cu loaded on Ni/TiO2, red circle, and Ni loaded on Cu/TiO2, blue square) and (b) 3 wt% Ni–Cu/SiO2 catalysts (100 mg L−1) prepared by a sequential impregnation (Cu loaded on Ni/SiO2, red circle, and Ni loaded on Cu/SiO2, blue square). | ||
2.3 Characterisation of Ni–Cu supported on TiO2 and SiO2 by UV-vis and TEM with EDS elemental mapping
The Ni–Cu/TiO2 and Ni–Cu/SiO2 catalysts prepared by co-impregnation and sequential impregnation methods were characterised by diffuse reflectance UV-vis spectroscopy (DRS). The DRS results of Ni–Cu/TiO2 catalysts prepared by different procedures are shown in Fig. 5a. Ni–Cu/TiO2 prepared by the co-impregnation method (red) showed strong absorption in the visible region (400–800 nm) compared with Ni–Cu/TiO2 catalysts prepared by the sequential impregnation method, with the Cu loaded on Ni/TiO2 (blue) or Ni loaded on Cu/TiO2 (green). The peaks that appeared around 600–800 nm in the DRS of Ni–Cu/TiO2 catalysts differ from superposition of DRS of Ni/TiO2 and Cu/TiO2 depicted in Fig. S6a,† suggesting that a part of Ni and Cu species have electronic interaction on the TiO2 surfaces.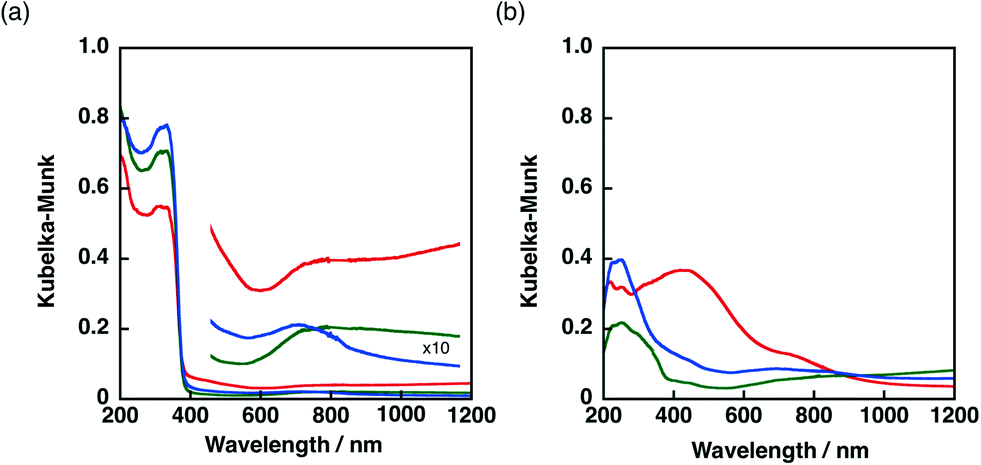 | ||
| Fig. 5 Diffuse reflectance UV-vis spectra of (a) Ni–Cu/TiO2 and (b) Ni–Cu/SiO2 catalysts prepared by co-impregnation (red) and sequential impregnation methods (Cu loaded on Ni/MO2, blue, and Ni loaded on Cu/MO2, green. M = Ti or Si). | ||
A more obvious difference in DRS was observed among Ni–Cu/SiO2 catalysts prepared by different methods as shown in Fig. 5b. The DRS results of Ni/SiO2 and Cu/SiO2 are depicted in Fig. S6b.† The DRS of the Ni–Cu/SiO2 catalyst prepared by the co-impregnation method (red) showed absorption maxima around 480 nm, whereas a small shoulder around 400 nm was observed for the Ni–Cu/SiO2 catalysts prepared by the sequential impregnation method. The difference in the absorption at the visible region provides the difference in colour. The origin of the brownish colour of the Ni–Cu/SiO2 catalyst prepared by the co-impregnation method can be ascribed to the formation of NiCuO2 on SiO2 surfaces, which has been proposed for Ni–Cu supported on cordierite in the literature.45
The difference in the structures of the Ni–Cu/SiO2 catalysts prepared by the co-impregnation and the sequential impregnation methods was confirmed by TEM measurements in the atomic level with the energy-dispersive X-ray spectroscopy (EDS) elemental mapping for Si, Cu and Ni. Fig. 6 shows a high-angle annular dark-field scanning TEM (HAADF-STEM) image of particles 20 nm in size formed on Ni–Cu/SiO2 prepared by the co-impregnation method. The EDS elemental mapping for Si (blue), Cu (red) and Ni (green) indicates that the particle observed in the HAADF-STEM image is composed of both Cu and Ni. On the other hand, the particles on Ni–Cu/SiO2 prepared by the sequential method, in which Cu was loaded on Ni/SiO2, showed separated deposition of Cu and Ni. As shown in Fig. 7, comparison of EDS elemental mapping for Cu (red) and Ni (green) with the HAADF-STEM image indicates that the brightest part of the HAADF-STEM image is the Cu-rich region, and the small particles that appeared in the upper middle are Ni rich or Ni only. These observations suggest that suitable selection of preparation methods is necessary to achieve close location of Ni and Cu on the surfaces of Ni–Cu/SiO2.
 | ||
| Fig. 6 HAADF-STEM and energy-dispersive X-ray spectroscopy (EDS) elemental mapping of Ni–Cu/SiO2 prepared by the co-impregnation method for Si, Cu and Ni. | ||
 | ||
| Fig. 7 HAADF-STEM and EDS elemental mapping of Ni–Cu/SiO2 prepared by the sequential impregnation method (Ni loaded on Cu/SiO2) for Si, Cu and Ni. | ||
2.4 Optimisation of Ni/Cu ratio, loading amount of Ni–Cu and structure of TiO2 and SiO2 supports
The catalytic activity of Ni–Cu/SiO2 with different loading amounts of Ni–Cu [1:1 (w/w)] ranging from 2 to 5 wt% was examined for the photocatalytic H2 evolution. The H2 evolution rates determined from the slopes of time courses of H2 evolution (Fig. 8a) at the reaction time from 5 to 30 min were 0.8, 1.2, 1.3 and 1.0 μmol h−1 for Ni–Cu/SiO2 with the loading amounts of Ni–Cu being 2, 3, 4 and 5 wt%, respectively. The total H2 yield obtained for 3 wt% Ni–Cu/SiO2 was 77%, which is higher than those for 2, 4 and 5 wt% Ni–Cu/SiO2 (28%, 54% and 45%, respectively). Thus, the loading amount of Ni–Cu around 3–4 wt% is optimum to achieve high catalytic activity for Ni–Cu/SiO2.
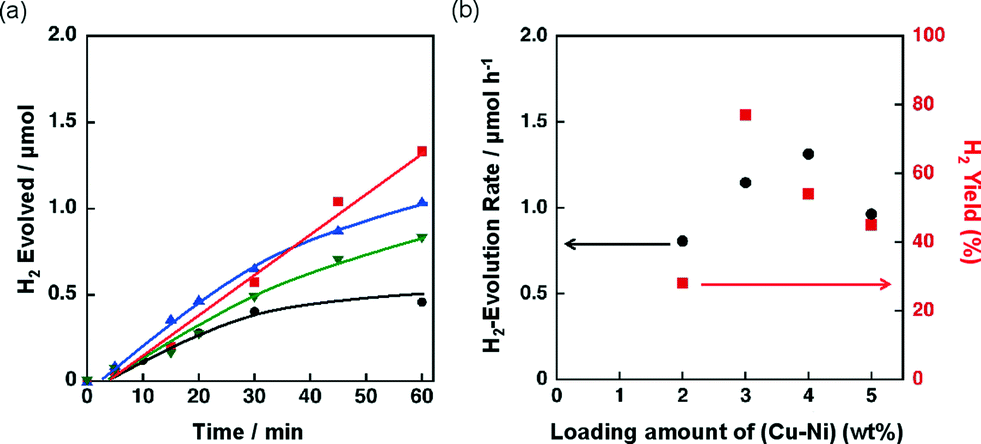 | ||
| Fig. 8 (a) Time course of H2 evolution by photoirradiation (λ > 340 nm) of a mixed suspension (2.0 mL) of a phthalate buffer (pH 4.5) and MeCN [1:1 (v/v)] containing QuPh+–NA (8.8 × 10−4 M), NADH (1.0 × 10−3 M) and Ni–Cu/SiO2 [Ni/Cu = 1/1 (w/w)] (100 mg L−1; 2 wt%, black circle; 3 wt%, red square; 4 wt%, blue triangle; 5 wt%, inverted triangle) prepared by co-impregnation. (b) Effect of the loading amount of Cu and Ni on the initial (30 min) H2 evolution rate and H2 yield. | ||
The effect of the ratio between Ni and Cu on the catalytic activity of 3 wt% Ni–Cu/SiO2 was also examined. Fig. 9a shows the time courses of H2 evolution in the photocatalytic H2 evolution using Ni–Cu/SiO2 catalysts with Ni contents, the Ni/(Ni + Cu) ratio, from 0% to 100%. H2 evolution rates higher than 0.8 μmol h−1 were observed for the catalysts with Ni contents from 25% to 67% as shown in Fig. 9b. The effect of Cu on the reducibility of NiO on SiO2 has been discussed by temperature-programmed reaction with flowing H2/Ar in the literature.35c–e The reduction temperature of NiO on SiO2 around 580 °C decreased to below 300 °C for Ni–Cu/SiO2 under 10% flowing H2/Ar.35c The enhanced reducibility of NiO on Ni–Cu/SiO2 catalysts resulted in high catalytic activity for ethanol steam reforming.35c The promotional effect of Cu(O) on the reduction of NiO can also be expected for the present photocatalytic H2 evolution system, although the reaction temperature was as low as room temperature.
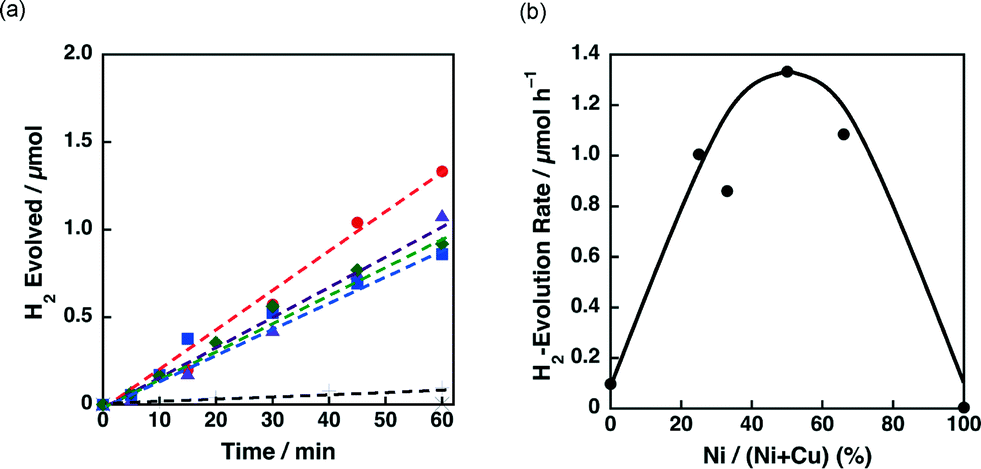 | ||
| Fig. 9 (a) Time courses of H2 evolution by photoirradiation (λ > 340 nm) of a mixed suspension (2.0 mL) of a phthalate buffer (pH 4.5) and MeCN [1:1 (v/v)] containing QuPh+–NA (8.8 × 10−4 M), NADH (1.0 × 10−3 M) and 3 wt% Ni–Cu/SiO2 (100 mg L−1) with various Ni contents prepared by the co-impregnation method [Ni/(Ni + Cu) = 0% (plus), 25% (green diamond), 33% (blue square), 50% (red circle), 67% (purple triangle) and 100% (inverted triangle)]. (b) Plot of the initial H2 evolution rate vs. Ni contents. | ||
Effects of the morphologies and the surface areas of SiO2 supports on the catalysis of Ni–Cu/SiO2 for H2 evolution were also examined by employing two additional SiO2 supports, which have a spherical morphology with the BET surface area similar to that of the SiO2 support [unshaped, low surface area (unshaped LS)] used in the above experiments and an unshaped morphology with a higher BET surface area. The morphologies of SiO2 supports were confirmed by TEM observations as shown in Fig. S7,† and the BET surface areas were determined to be 59 m2 g−1 and 420 m2 g−1 for the SiO2 (spherical) and the SiO2 [unshaped, high surface area (unshaped HS)], respectively. With these SiO2 supports, 3 wt% Ni–Cu/SiO2 catalysts [Ni/Cu = 1:1 (w/w)] were prepared by the co-impregnation method and used as the H2 evolution catalysts in the photocatalytic H2 evolution system. The time courses of the H2 evolution are shown in Fig. 10a. The H2 evolution rates were 0.4 and 0.8 μmol h−1 for Ni–Cu/SiO2 catalysts with spherical and unshaped (high surface area) morphologies, respectively. The H2 evolution rate for Ni–Cu/SiO2 (spherical) being significantly lower than that for Ni–Cu/SiO2 [unshaped LS (52 m2 g−1)] (1.3 μmol h−1) indicates that the morphology of SiO2 affects the catalysis of Ni–Cu/SiO2. The lower catalytic activity obtained by employing SiO2 (spherical) as a support has been reported for Ru/SiO2 in the photocatalytic H2 evolution.30 The smooth surfaces of the SiO2 (spherical) more readily promote the adhesive interaction among the SiO2 particles through supporting metals than the rough surfaces of the SiO2 (unshaped).30 Thus, a part of the supporting metals acting as the adhesive among SiO2 particles hardly work as the H2 evolution catalyst. A similar mechanism is applicable to the present Ni–Cu/SiO2 (spherical) catalysts. For the effects of surface areas, the catalytic activity of Ni–Cu/SiO2 (unshaped LS) was higher than that of Ni–Cu/SiO2 (unshaped HS). This result may suggest that the lower surface area of the SiO2 is favourable to closely locate Ni and Cu to each other. Thus, both morphologies and surface areas of SiO2 supports affect the catalysis of Ni–Cu/SiO2.
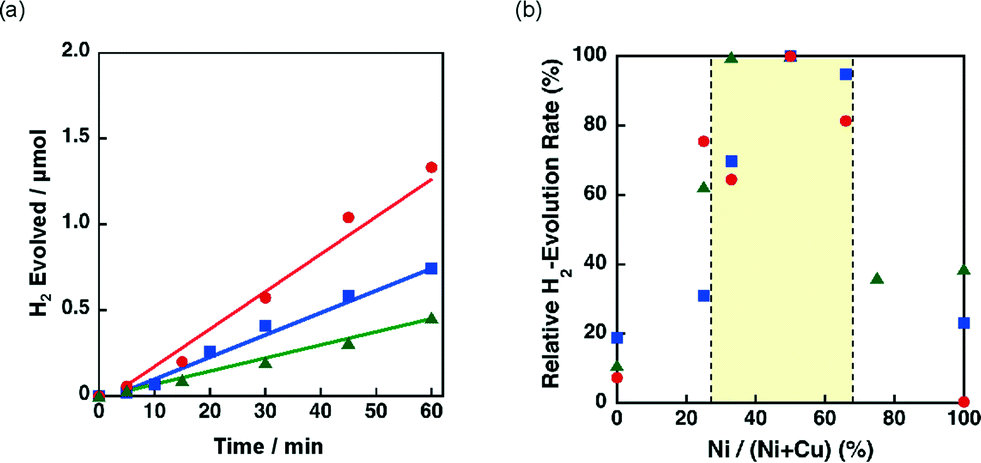 | ||
| Fig. 10 (a) Time course of H2 evolution by photoirradiation (λ > 340 nm) of a mixed suspension (2.0 mL) of a phthalate buffer (pH 4.5) and MeCN [1:1 (v/v)] containing QuPh+–NA (8.8 × 10−4 M), NADH (1.0 × 10−3 M) and 3 wt% Ni–Cu/SiO2 (100 mg L−1). (a) SiO2 with a spherical morphology (green triangle) and unshaped LS (red circle) and unshaped HS (blue square). (b) Plot of the initial H2 evolution rate vs. Ni contents. | ||
The effect of the ratio between Ni and Cu was also investigated for 3 wt% Ni–Cu/SiO2 (spherical) and 3 wt% Ni–Cu/SiO2 (unshaped HS) in the photocatalytic H2 evolution. The time courses of the H2 evolution in the photocatalytic H2 evolution are shown in Fig. S8.†Fig. 10b shows the relative H2 evolution rates depending on the Ni/(Ni + Cu). High catalytic activity was achieved at the Ni contents between 30% and 67% as observed for Ni–Cu/SiO2 (unshaped LS) shown in Fig. 9b.
The catalytic activity of Ni–Cu/TiO2 for the H2 evolution dependent on the Ni contents was also investigated in the photocatalytic H2 evolution. Fig. 11a demonstrates the time courses of H2 evolution in the photocatalytic reaction systems using Ni–Cu/TiO2 with different Ni contents. H2 evolution rates higher than 1.5 μmol h−1 were obtained for the Ni–Cu/TiO2 catalysts with 33–67% of Ni contents. The origin of the synergistic effect of Ni and Cu on TiO2 has been discussed by H2-TPR measurements in the literature.41a The reduction temperature of NiO on TiO2 was lowered from ca. 500 °C to 300 °C in the presence of CuO by increasing the content of CuO.41a Although the photocatalytic H2 evolution was performed at room temperature, the readily reducible nature of NiO influenced by the addition of Cu would be beneficial to form metallic Ni species on the TiO2 support.
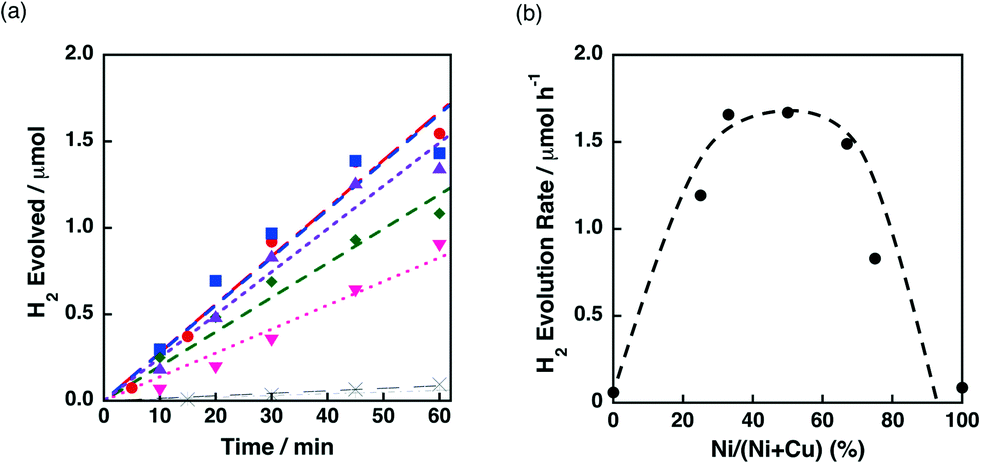 | ||
| Fig. 11 (a) Time course of H2 evolution by photoirradiation (λ > 340 nm) of a mixed suspension (2.0 mL) of a phthalate buffer (pH 4.5) and MeCN [1:1 (v/v)] containing QuPh+–NA (8.8 × 10−4 M), NADH (1.0 × 10−3 M) and 3 wt% Ni–Cu/TiO2 (100 mg L−1) with various Ni contents prepared by co-impregnation [Ni/(Ni + Cu) = 0% (plus), 25% (green diamond), 33% (blue square), 50% (red circle), 67% (purple triangle), 75% (pink inverted triangle) and 100% (cross)]. (b) Plot of the initial H2 evolution rate vs. Ni contents. | ||
Effects of crystal structures and the BET surface areas of TiO2 supports were examined by using a series of 3 wt% Ni–Cu/TiO2 catalysts in the photocatalytic H2 evolution. The crystal structures confirmed by XRD (Fig. S9†) and BET surface areas of the TiO2 supports were rutile with a high surface area (rutile HS, 250 m2 g−1), rutile with a low surface area (rutile LS, 0.3 m2 g−1) and anatase with a high surface area (anatase HS, 46 m2 g−1). The TiO2 support used in the above-mentioned experiments has an anatase structure with a low surface area (anatase LS, 6.8 m2 g−1) (vide supra). With these TiO2 supports, 3 wt% Ni–Cu/TiO2 [Ni/Cu = 1:1 (w/w)] catalysts were prepared by the co-impregnation method. Fig. 12 shows the time courses of H2 evolution in the photocatalytic H2 evolution performed with these Ni–Cu/TiO2 catalysts (rutile HS, rutile LS, and anatase HS) as the H2 evolution catalysts. The H2 evolution rates determined from the initial (60 min) slopes were 1.5, 1.3 and 1.3 μmol h−1 for Ni–Cu/TiO2 (rutile HS, rutile LS and anatase HS, respectively), which are comparable to that for Ni–Cu/TiO2 (anatase LS) (1.7 μmol h−1). Although the surface area of TiO2 (rutile LS) is only ca. 1/1000 that of TiO2 (rutile HS), the catalytic activity of Ni–Cu on these TiO2 supports is comparable. These results suggest that neither the crystal structures nor the BET surface areas of the TiO2 supports are insignificant factors to determine the catalytic activity of Ni–Cu/TiO2.
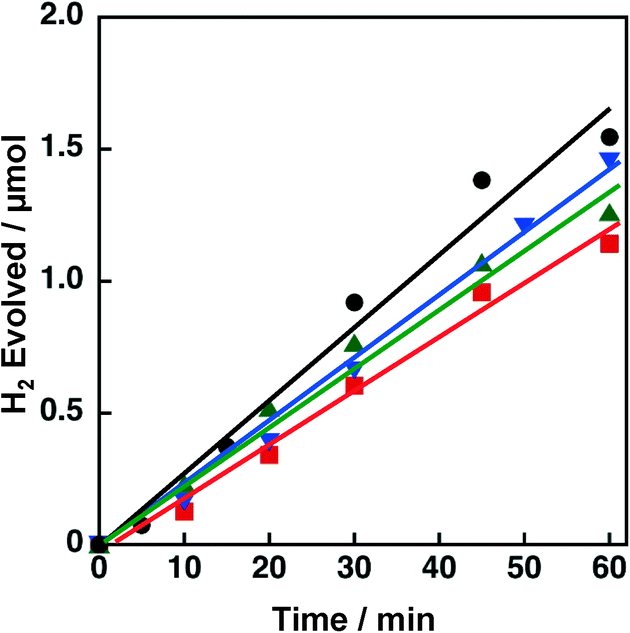 | ||
| Fig. 12 Time course of H2 evolution by photoirradiation (λ > 340 nm) of a mixed suspension (2.0 mL) of a phthalate buffer (pH 4.5) and MeCN [1:1 (v/v)] containing QuPh+–NA (8.8 × 10−4 M), NADH (1.0 × 10−3 M) and 3 wt% Ni–Cu supported on TiO2 (100 mg L−1) with different crystal structures (anatase LS, black circle; rutile HS, blue inverted triangle; anatase HS, red square; rutile LS, green triangle). | ||
3. Conclusions
The catalytic activity of Ni–Cu/TiO2 and Ni–Cu/SiO2 for H2 evolution was greatly enhanced by the synergistic effects of Ni and Cu on the photocatalytic H2 evolution system using 2-phenyl-4-(1-naphthyl)quinolinium ion and NADH as a photocatalyst and an electron donor, respectively. Such a synergistic effect between Ni and Cu was not observed for Ni–Fe and Ni–Co catalysts and for Ni–Cu/SiO2–Al2O3 and Ni–Cu/CeO2. TEM observation and EDS elemental mapping of Ni–Cu/SiO2 prepared by the co-impregnation and the sequential impregnation methods suggest that the close location of Ni and Cu is important to achieve the high catalytic activity. The synergistic effect between Ni and Cu was observed in the wide range of Ni contents, Ni/(Ni + Cu), from 30% to 70%. This study has demonstrated that suitable choices of additives, supports, and catalyst preparation methods are all important to achieve highly active H2 evolution catalysts composed of base metals with synergistic effects.4. Experimental section
4.1 Materials
All chemicals were obtained from chemical companies and used without further purification. SiO2 (unshaped, low surface area), TiO2 (anatase, low surface area) and TiO2 (anatase, high surface area) were purchased from Sigma-Aldrich. SiO2 (unshaped, high surface area) was obtained from Merck. Iron nitrate, copper nitrate, nickel nitrate, 2-propanol, hydrochloric acid (37%), sodium aluminate and titanium(IV) chloride were obtained from Wako Pure Chemical Industries. Aqueous ammonia (28%) and β-dihydronicotinamide adenine dinucleotide disodium salt (reduced form) (NADH) were obtained from Tokyo Chemical Industry. Tetraethyl orthosilicate (TEOS) was delivered by Shin-Etsu Chemical. Acetonitrile was obtained from Nacalai Tesque. CeO2 was provided by Daiichi Kigenso Kagaku Kogyo Co., Ltd. 2-Phenyl-4-(1-naphthyl)quinolinium perchlorate (QuPh+–NA),42 SiO2–Al2O3,46 spherical SiO2,47 and TiO2 (rutile, high surface area)48 were prepared by literature methods. Purified water was provided by using a Millipore DIRECT-Q UV3 water purification system (18.2 MΩ cm).4.2 Preparation of Ni–Cu supported on metal oxides by a co-impregnation method
A typical procedure for the preparation of metal oxides supporting Ni and Cu by a co-impregnation method is as follows: metal oxide (350 mg) was soaked in an ethanol solution (30 mL) containing Ni(NO3)2·6H2O (26.8 mg, 92.2 mmol) and Cu(NO3)2·3H2O (20.6 mg, 85.3 mmol) and sonicated for 30 min. The obtained catalyst precursor was dried at 60 °C in an oven and calcined at 350 °C (ramp rate: 5 °C min−1) for 4 h in air. The obtained powder was soaked in an ethanol solution containing NaBH4 to reduce oxides of Cu and Ni before catalysis evaluation.4.3 Preparation of Ni–Cu supported on TiO2 and SiO2 by a sequential impregnation method
A typical procedure for the preparation of TiO2 and SiO2 catalysts supporting Cu and Ni by a sequential impregnation method is as follows: TiO2 or SiO2 (350 mg) was immersed in an ethanol solution (30 mL) containing Cu(NO3)2·3H2O (20.6 mg, 85.3 mmol) and sonicated for 30 min. The obtained catalyst precursor was dried at 60 °C in an oven and calcined at 350 °C (ramp rate: 5 °C min−1) for 4 h in air. Then, the calcined sample was immersed in an ethanol solution (30 mL) containing Ni(NO3)2·6H2O (26.8 mg, 92.2 mmol). The obtained slurry was dried and calcined. The obtained powder was soaked in an ethanol solution containing NaBH4 to reduce oxides of Cu and Ni before catalysis evaluation.4.4 Preparation of Ni–Co and Ni–Fe supported on TiO2 and SiO2 by a co-impregnation method
These catalysts were prepared by a co-impregnation method as follows: TiO2 or SiO2 (350 mg) was soaked in an ethanol solution (30 mL) containing Ni(NO3)2·6H2O (26.8 mg, 92.2 mmol) and Co(NO3)2·6H2O (26.8 mg, 92.1 mmol) or Fe(NO3)3·9H2O (37.2 mg, 92.1 mmol) and sonicated for 30 min. The obtained catalyst precursor was dried at 60 °C in an oven and calcined at 350 °C (ramp rate: 5 °C min−1) for 4 h in air. The obtained powder was soaked in an ethanol solution containing NaBH4 to reduce oxides of Cu and Ni before catalysis evaluation.4.5 Preparation of SiO2–Al2O3
SiO2 was suspended in an aqueous solution (50 mL) containing sodium aluminate (1.3 g, 16 mmol) and stirred for 20 h at room temperature. The precipitate was collected by filtration and dried at 120 °C. The dried sample was calcined at 550 °C (ramp rate: 5 °C min−1) for 6 h in air.4.6 Preparation of spherical silica
TEOS (18 mL, 80 mmol) was dissolved in a mixture solution of water (31 mL), ethanol (300 mL) and 28% ammonia solution (5.4 mL) and stirred for 4 h at room temperature. After the reaction, the obtained white precipitate was collected by centrifugation (9000 rpm, 10 min) and washed three times with water. The obtained powder was dried at 70 °C in an oven and calcined at 350 °C (ramp rate: 5 °C min−1) for 4 h in air to remove residual ammonia.4.7 Preparation of TiO2 (rutile, high surface area)48
Titanium(IV) chloride (5.7 g, 30 mmol) was dissolved in a mixture solution of water (100 mL), 2-propanol (200 mL) and hydrochloric acid (10 mL) and refluxed for 20 h at 90 °C. After the solution was cooled to room temperature, the suspension was basified to pH 9 with 28% ammonia solution. The white precipitate was collected by centrifugation (9000 rpm, 10 min) and washed three times with water.4.8 Preparation of TiO2 (rutile, low surface area)
TiO2 (rutile, high surface area) was calcined at 1000 °C (ramp rate: 10 °C min−1) for 3 h in air.4.9 Transmission electron microscopy (TEM)
The sizes and shapes of SiO2 supports were determined from bright-field images using a JEOL JEM-2100 that has a thermal field emission gun with an accelerating voltage of 200 kV. The observed samples were prepared by dropping a dispersion of catalysts onto the support and allowing the solvent to evaporate and then scooping up with an amorphous carbon supporting film on a Cu grid. High-angle annular dark-field scanning TEM (HAADF-STEM) images and the energy-dispersive X-ray spectroscopy (EDS) elemental mapping of Ni–Cu/SiO2 were obtained by using an FEI Titan G2 60-300 with an accelerating voltage of 300 kV. The observed samples were directly put on Mo meshes.4.10 Catalyst characterisation by powder X-ray diffraction, diffused reflectance UV-vis spectroscopy and dynamic laser scattering
X-ray diffraction patterns were recorded with a Rigaku MiniFlex 600. Incident X-ray radiation was produced by using a Cu X-ray tube operating at 40 kV and 15 mA with a Cu Kα radiation of 1.54 Å. The scanning rate was 2° min−1 from 5° to 80° in 2θ. The diffuse reflectance UV-vis spectra were recorded with a Jasco V-670 spectrophotometer equipped with an integrating sphere module (SIN-768). Reflectance obtained for each sample was converted to f(R∞) values according to the Kubelka–Munk theory, f(R∞) = (1 − R∞)2/2R∞, where R∞ is the reflectance of the sample layer. BaSO4 was used for background spectra measurements. Dynamic light scattering (DLS) measurements were performed with a Zetasizer Nano ZS instrument (Malvern Instruments Ltd., USA).4.11 N2 adsorption for BET surface area determination
Nitrogen adsorption–desorption at 77 K was performed with a Belsorp-mini (BEL Japan, Inc.) within a relative pressure range from 0.01 to 101.3 kPa. A sample mass of ~100 mg was used for adsorption analysis after pretreatment at 120 °C for 1 h under vacuum conditions and kept in N2 atmosphere until N2 adsorption measurements. The samples were exposed to a mixed gas of He and N2 with a programmed ratio, and an adsorbed amount of N2 was calculated from the change in pressure in a cell after reaching the equilibrium (at least 5 min). The surface area of each catalyst was determined by the Brunauer–Emmett–Teller (BET) method for multiple N2 adsorption amounts under the conditions of partial pressure less than 0.3.4.12 Photocatalytic H2 evolution
A mixed solution (2.0 mL) of a phthalate buffer (pH 4.5) and MeCN [1:1 (v/v)] containing QuPh+–NA (0.88 mM), NADH (1.0 mM) and an H2 evolution catalyst was flushed with N2 gas. The solution was then irradiated for a certain time with a xenon lamp (Ushio Optical, Model X SX-UID 500X AMQ) through a colour filter glass (Asahi Techno Glass L39) transmitting λ > 340 nm at room temperature. After 1 min of stirring in the dark, the gas in the headspace was analysed by Shimadzu GC-14B gas chromatography (detector: TCD, column temperature: 50 °C, column: active carbon with the particle size of 60–80 mesh, carrier gas: N2 gas) to determine the amount of evolved H2.
Acknowledgements
This work was supported by grants-in-aid (no. 24350069 and 25600025) for Scientific Research from the Japan Society for the Promotion of Science (JSPS), an ALCA project from the Japan Science and Technology Agency (JST). We sincerely acknowledge the Research Centre for Ultra-Precision Science & Technology, Osaka University, for TEM measurements.Notes and references
- H. B. Gray, Nat. Chem., 2009, 1, 7–7 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- (a) Z. Han and R. Eisenberg, Acc. Chem. Res., 2014, 47, 2537–2544 CrossRef CAS PubMed; (b) K. S. Joya, Y. F. Joya, K. Ocakoglu and R. van de Krol, Angew. Chem., Int. Ed., 2013, 52, 10426–10437 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- (a) T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 3, 1486–1503 Search PubMed; (b) K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS; (c) K. Maeda and K. Domen, Chem. Mater., 2010, 22, 612–623 CrossRef CAS.
- R. Abe, in New and Future Developments in Catalysis: Solar Photocatalysis, Elsevier B.V., 2013, pp. 341–370 Search PubMed.
- (a) A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC; (b) Y. Miseki and A. Kudo, ChemSusChem, 2011, 4, 245–251 CAS.
- (a) H. Hagiwara, M. Watanabe, T. Daio, S. Ida and T. Ishihara, Chem. Commun., 2014, 50, 12515–12518 RSC; (b) H. Hagiwara, T. Inoue, S. Ida and T. Ishihara, Phys. Chem. Chem. Phys., 2011, 13, 18031–18037 RSC.
- (a) K. Mori, J. Aoyama, M. Kawashima and H. Yamashita, Dalton Trans., 2014, 43, 10541–10547 RSC; (b) T. Kamegawa, S. Matsuura, H. Seto and H. Yamashita, Angew. Chem., Int. Ed., 2013, 52, 916–919 CrossRef CAS PubMed.
- (a) Y. Horiuchi, T. Toyao, M. Takeuchi, M. Matsuoka and M. Anpo, Phys. Chem. Chem. Phys., 2013, 15, 13243–13253 RSC; (b) T. Toyao, M. Saito, S. Dohshi, K. Mochizuki, M. Iwata, H. Higashimura, Y. Horiuchi and M. Matsuoka, Chem. Commun., 2014, 50, 6779–6781 RSC.
- S. Fukuzumi and Y. Yamada, ChemSusChem, 2013, 6, 1834–1847 CrossRef CAS PubMed.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC.
- (a) J. J. Concepcion, R. L. House, J. M. Papanikolas and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15560–15564 CrossRef CAS PubMed; (b) A. K. Vannucci, L. Alibabaei, M. D. Losego, J. J. Concepcion, B. Kalanyan, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20918–20922 CrossRef CAS PubMed.
- A. J. Cowan and J. R. Durrant, Chem. Soc. Rev., 2013, 42, 2281–2293 RSC.
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed.
- M. D. Kärkäs, E. V. Johnston, O. Verho and B. Åkermark, Acc. Chem. Res., 2014, 47, 100–111 CrossRef PubMed.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- (a) H. Ozawa and K. Sakai, Chem. Commun., 2011, 47, 2227–2242 RSC; (b) M. Kobayashi, S. Masaoka and K. Sakai, Angew. Chem., Int. Ed., 2012, 51, 7431–7434 CrossRef CAS PubMed.
- (a) J. P. Prieto, EPA Newsl., 2013, 84, 85–86 Search PubMed; (b) J. M. Thomas, Energy Environ. Sci., 2014, 7, 19–19 RSC.
- (a) M. Wang, L. Chen and L. Sun, Energy Environ. Sci., 2012, 5, 6763–6778 RSC; (b) N. Wang, M. Wang, L. Chen and L. Sun, Dalton Trans., 2013, 42, 12059–12071 RSC.
- (a) M. Grätzel, Acc. Chem. Res., 1981, 14, 376–384 CrossRef; (b) J. Kiwi, K. Kalyanasundaram and M. Grätzel, Struct. Bonding, 1982, 49, 37–125 CrossRef.
- S. Fukuzumi, D. Hong and Y. Yamada, J. Phys. Chem. Lett., 2013, 4, 3458–3467 CrossRef CAS.
- S. Fukuzumi, Y. Yamada, T. Suenobu, K. Ohkubo and H. Kotani, Energy Environ. Sci., 2011, 4, 2754–2766 CAS.
- (a) H. Kotani, R. Hanazaki, K. Ohkubo, Y. Yamada and S. Fukuzumi, Chem. – Eur. J., 2011, 17, 2777–2785 CrossRef CAS PubMed; (b) H. Kotani, K. Ohkubo, Y. Takai and S. Fukuzumi, J. Phys. Chem. B, 2006, 110, 24047–24053 CrossRef CAS PubMed; (c) H. Kotani, T. Ono, K. Ohkubo and S. Fukuzumi, Phys. Chem. Chem. Phys., 2007, 9, 1487–1492 RSC.
- (a) Y. Yamada, T. Miyahigashi, K. Ohkubo and S. Fukuzumi, Phys. Chem. Chem. Phys., 2012, 14, 10564–10571 RSC; (b) Y. Yamada, H. Tadokoro and S. Fukuzumi, RSC Adv., 2013, 3, 25677–25680 RSC; (c) Y. Yamada, A. Nomura, H. Tadokoro and S. Fukuzumi, Catal. Sci. Technol., 2014 10.1039/C4CY01005A.
- S. Harinipriya and M. V. Sangaranarayanan, Langmuir, 2002, 18, 5572–5578 CrossRef CAS.
- P. S. Bassi, Gurudayal, L. H. Wong and J. Barber, Phys. Chem. Chem. Phys., 2014, 16, 11834–11842 RSC.
- Y. Yamada, T. Miyahigashi, H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 16136–16145 CrossRef CAS PubMed.
- Y. Yamada, T. Miyahigashi, H. Kotani, K. Ohkubo and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 6111–6118 CAS.
- Y. Yamada, S. Shikano and S. Fukuzumi, J. Phys. Chem. C, 2013, 117, 13143–13152 CAS.
- S. K. Beaumont, S. Alayoglu, V. V. Pushkarev, Z. Liu, N. Kruse and G. A. Somorjai, Faraday Discuss., 2013, 162, 31–44 RSC.
- (a) B. R. Greenhalgh, S. M. Kuznicki and A. E. Nelson, Appl. Catal., A, 2007, 327, 189–196 CrossRef CAS PubMed; (b) D. Gulkova and M. Zdrazil, Collect. Czech. Chem. Commun., 1999, 64, 735–746 CrossRef CAS; (c) J. Hermannsdoerfer, M. Friedrich, N. Miyajima, R. Q. Albuquerque, S. Kuemmel and R. Kempe, Angew. Chem., Int. Ed., 2012, 51, 11473–11477 CrossRef CAS PubMed; (d) M. P. M. Kaninski, S. M. Miulovic, G. S. Tasic, A. D. Maksic and V. M. Nikolic, Int. J. Hydrogen Energy, 2011, 36, 5227–5235 CrossRef PubMed; (e) M. A. Keane, S. Gomez-Quero, F. Cardenas-Lizana and W. Shen, ChemCatChem, 2009, 1, 270–278 CrossRef CAS; (f) S. A. Nikolaev and V. V. Smirnov, Catal. Today, 2009, 147, 336–341 CrossRef PubMed; (g) A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 3000–3004 CrossRef CAS; (h) J. Zhang, J.-O. Mueller, W. Zheng, D. Wang, D. Su and R. Schlögl, Nano Lett., 2008, 8, 2738–2743 CrossRef CAS PubMed; (i) Q.-L. Zhu, J. Li and Q. Xu, J. Am. Chem. Soc., 2013, 135, 10210–10213 CrossRef CAS PubMed.
- S.-Z. Kang, L. Chen, X. Li and J. Mu, Appl. Surf. Sci., 2012, 258, 6029–6033 CrossRef CAS PubMed.
- (a) Y. Zhang, W. Huang, S. E. Habas, J. N. Kuhn, M. E. Grass, Y. Yamada, P. Yang and G. A. Somorjai, J. Phys. Chem. C, 2008, 112, 12092–12095 CrossRef CAS; (b) H. Yen and F. Kleitz, J. Mater. Chem. A, 2013, 1, 14790–14796 RSC.
- (a) S. A. Khromova, A. A. Smirnov, O. A. Bulavchenko, A. A. Saraev, V. V. Kaichev, S. I. Reshetnikov and V. A. Yakovlev, Appl. Catal., A, 2014, 470, 261–270 CrossRef CAS PubMed; (b) J. Ashok, P. S. Reddy, G. Raju, M. Subrahmanyam and A. Venugopal, Energy Fuels, 2009, 23, 5–13 CrossRef CAS; (c) L.-C. Chen and S. D. Lin, Appl. Catal., B, 2011, 106, 639–649 CrossRef CAS PubMed; (d) S. K. Saraswat and K. K. Pant, J. Nat. Gas Sci. Eng., 2013, 13, 52–59 CrossRef CAS PubMed; (e) A. J. Vizcaino, A. Carrero and J. A. Calles, Int. J. Hydrogen Energy, 2007, 32, 1450–1461 CrossRef CAS PubMed.
- S. Ma, Y. Tan and Y. Han, J. Ind. Eng. Chem., 2011, 17, 723–726 CrossRef CAS PubMed.
- Y. Echegoyen, I. Suelves, M. J. Lázaro, R. Moliner and J. M. Palacios, J. Power Sources, 2007, 169, 150–157 CrossRef CAS PubMed.
- M. Kang, M. W. Song, T. W. Kim and K. L. Kim, Can. J. Chem. Eng., 2002, 80, 63–70 CrossRef CAS.
- (a) B. M. Reddy, K. N. Rao and P. Bharali, Ind. Eng. Chem. Res., 2009, 48, 8478–8486 CrossRef CAS; (b) Y. Li, Q. Fu and M. Flyzani-Stephanopoulos, Appl. Catal., B, 2000, 27, 179–191 CrossRef CAS.
- P. López, G. Mondragón-Galicia, M. E. Espinosa-Pesqueira, D. Mendoza-Anaya, M. E. Fernández, A. Gómez-Cortés, J. Bonifacio, G. Martínez-Barrera and R. Pérez-Hernández, Int. J. Hydrogen Energy, 2012, 37, 9018–9027 CrossRef PubMed.
- (a) H.-J. Kim, L. Lu, J.-H. Kim, C.-H. Lee, T. Hyeon, W. Choi and H.-I. Lee, Bull. Korean Chem. Soc., 2001, 22, 1371–1374 CAS; (b) M. Lazaro, Y. Echegoyen, C. Alegre, I. Suelves, R. Moliner and J. Palacios, Int. J. Hydrogen Energy, 2008, 33, 3320–3329 CrossRef CAS PubMed; (c) P. Li, J. Liu, N. Nag and P. A. Crozier, J. Catal., 2009, 262, 73–82 CrossRef CAS PubMed; (d) N. Riaz, F. K. Chong, B. K. Dutta, Z. B. Man, M. S. Khan and E. Nurlaela, Chem. Eng. J., 2012, 185–186, 108–119 CrossRef CAS PubMed; (e) N. Riaz, F. K. Chong, Z. B. Man, M. S. Khan and B. K. Dutta, Ind. Eng. Chem. Res., 2013, 52, 4491–4503 CrossRef CAS.
- H. Kotani, K. Ohkubo and S. Fukuzumi, Faraday Discuss., 2012, 155, 89–102 RSC.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- D. D. Wagman, W. H. Evans, V. B. Parker, R. H. Schumm, I. Halow, S. M. Bailey, K. L. Churney and R. L. Nuttall, J. Phys. Chem. Ref. Data, 1982, 11, 1–392 CrossRef PubMed.
- (a) H. G. El-Shobaky, Appl. Catal., A, 2004, 278, 1–9 CrossRef CAS PubMed; (b) H. G. El-Shobaky and Y. M. Fahmy, Appl. Catal., B, 2006, 63, 168–177 CrossRef CAS PubMed.
- M. L. D. A. Martinez, F. A. L. D'Amicis, A. R. Beltramon, M. B. G. Costa and O. A. Anunziata, Mater. Res. Bull., 2011, 46, 1011–1021 CrossRef CAS PubMed.
- W. Stöber and A. Fink, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- B. I. Lee, X. Wang, R. Bhave and M. Hu, Mater. Lett., 2006, 60, 1179–1183 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Time courses of H2 evolution (Fig. S1, S4 and S8), powder X-ray diffraction (Fig. S2, S5 and S9), the amount of H2 evolved in the repetitive experiments (Fig. S3), diffused reflectance spectra (Fig. S6), and TEM images (Fig. S7). See DOI: 10.1039/c4cy01128g |
| This journal is © The Royal Society of Chemistry 2015 |