
Hydrodeoxygenation of dibenzofuran over SiO2, Al2O3/SiO2 and ZrO2/SiO2 supported Pt catalysts
Lei
Wang
,
Huihui
Wan
,
Shaohua
Jin
,
Xiao
Chen
,
Chuang
Li
and
Changhai
Liang
*
Laboratory of Advanced Materials and Catalytic Engineering, School of Chemical Engineering, Dalian University of Technology, Dalian 116012, China. E-mail: changhai@dlut.edu.cn; Fax: +86 411 84986353; Tel: +86 411 84986353
First published on 29th August 2014
Abstract
The surface of silica with mesopores (SiO2) was post-modified by the deposition of highly dispersed Al2O3 or ZrO2 oxides. Loading of Pt on the modified silica supports yielded higher metal dispersion with respect to the parent silica based on the CO chemisorption and transmission electron microscopy results. Hydrodeoxygenation (HDO) of dibenzofuran (DBF) over the as-prepared Pt catalysts mainly proceeds via the hydrogenation of the aromatic rings, which is followed by the cleavage of the C–O bond to produce oxygen-free hydrocarbons. Preferable hydrogenation of the aromatic rings is observed over the smaller Pt nanoparticles. The relatively strong acidic properties of Al2O3/SiO2 or ZrO2/SiO2, revealed by the NH3-TPD profiles, promote the selective C–O bond cleavage of hexahydrodibenzofuran to alter the HDO reaction pathway. The small sized Pt nanoparticles supported on the Al2O3 or ZrO2 modified silica supports show superior HDO performance with enhanced deoxygenation ability to those over unmodified silica. According to the pseudo-first-order kinetics analysis, the fitted HDO rate constant follows the order: Pt/Al2O3/SiO2 > Pt/ZrO2/SiO2 > Pt/SiO2 under a constant temperature, which is attributed to the cooperative function of the dispersed metal particles and the acidic sites of supports.
1 Introduction
Currently, there is an increasing demand for renewable energy production due to rapid energy consumption and global climate change. Renewable energy from biomass transformation is of major interest in reducing the dependence on traditional fossil fuels.1–3 The composition of the bio-oils derived from biomass includes phenols, furans, organic acids, ketones, aldehydes, etc.4 As expected, the bio-oils contain high oxygen content, which results in high viscosity, low heating value, and poor thermal stability.5 There is still much upgrading work to be done before practical use of these bio-oils.Catalytic HDO is an alternative process to upgrade the bio-oils to hydrocarbon fuels as reported throughout the literature.6–8 Oxygen can be removed by cleavage of the C–O bond to produce water as a byproduct. The HDO process is usually operated under high pressure of hydrogen and moderate temperatures, which is also applicable to the conventional hydrodesulfurization (HDS), hydrodenitrogenation (HDN) and hydrodemetallization (HDM) processes.9 Furan compounds are reported as typical kinds of biomass derived molecules used to test the catalyst activities in HDO reactions.6,10 DBF has a furan structure and is considered as a lignin model compound containing the β-5 linkage which is often found in a five-membered ring linking two aromatic structures via both a C–C bond and a C–O bond.11 DBF is formed as an important intermediate in the biomass gasification with a weight percent of 13.19%, which is almost twice as much as other kinds of major benzene products (7.13%).12 In the past few years, several groups have reported the HDO of benzofuran (claimed as another typical β-5 linkage model) or DBF as a model to study the conversion of the β-5 linkage in lignin.13–16 Due to the relatively larger molecular size and lower reactivity compared to benzofuran, the HDO of DBF as a β-5 linkage model allows the determination of catalytic selectivity toward either aromatic ring hydrogenation or ether linkage cleavage.4,16–18 The HDO product selectivity is closely related to the hydrogenation ability of the active metal sites and cleavage of the C–O bond promoted by acidic sites in the HDO reactions.
For the rational design of HDO catalysts, the right choice of support is very important. In the last few decades, zeolites,19,20 silica,21,22 carbon,23 and metal oxides11,24–26 have been demonstrated to be applicable as supports for HDO catalysts. Previously, metal oxides, such as Al2O3, ZrO2, TiO2, etc., showed good stability in the HDO reactions in the presence of water.27,28 Metal catalysts supported over the metal oxides usually show enhanced metal dispersion.29 It is claimed that metal oxides could activate the phenolic compounds and influence their adsorption modes in the HDO reactions, which leads to a better selectivity towards the target HDO products.30,31 However, metal oxides commonly have small surface areas, therefore the adsorption and concentration of reactants around the active sites is very limited. For this reason, the synthesis of mesoporous materials supported metal oxides has been investigated as an efficient way to make up for the referred deficiency.32 Deposition of metal oxides onto supports with a large surface area (e.g. ordered mesoporous silica) not only takes advantage of mesoporous materials with specific surface area and pore volume, but also results in materials which exhibit properties not found in the bulk metal oxides alone.33,34 For instance, nanocrystalline Al2O3, ZrO2 or TiO2 was embedded inside the pores of SBA-15. The incorporation of metal oxides enhances the interaction of the support with NiMo catalysts with better dispersion of the active phase and improves its catalytic performance in the HDS of dibenzothiophene and its derivatives.35,36
In the present work, we modified the surface of silica with mesopores by the deposition of metal oxides Al2O3 or ZrO2 to prepare a series of novel supported Pt catalysts. It is apparent that the deposited metal oxides on the surface of silica promoted the dispersion of Pt nanoparticles. The catalytic performance of such Pt/Al2O3/SiO2, Pt/ZrO2/SiO2 and Pt/SiO2 catalysts in the HDO of DBF was investigated in order to reveal the influence of the support on the HDO reaction network and kinetics. The superior hydrogenation and deoxygenation ability of Pt/Al2O3/SiO2 and Pt/ZrO2/SiO2 catalysts was attributed to both the higher dispersion of metal Pt particles and the unique acidic properties of the supports.
2 Experimental
2.1 Catalyst preparation
The surface of silica with mesopores was post-modified by the coating of alumina or zirconia to obtain Al2O3/SiO2 or ZrO2/SiO2 supports according to the NH3/water vapor-induced internal hydrolysis method.37–39 All of the supports were calcined under static air at 773 K in a furnace before further use. A supported catalyst with nominal 0.50 wt% Pt loading was prepared by the following steps. Firstly, a certain amount of metal precursor H2PtCl6·6H2O was dissolved in 70 mL methanol at room temperature, then 1.00 g calcined support was added under vigorous stirring until the formation of a homogenous slurry. The slurry was attached to a rotary evaporator, using the water bath to maintain the flask at 308 K. Methanol was removed after 30 min and the slightly damp powder was allowed to air dry at 333 K for 6 h prior to reduction. Lastly, the prepared catalysts were heated to 673 K at a rate of 2 K min−1 and reduced for 2 h under 40 mL min−1 H2.2.2 Catalyst characterization
X-ray diffraction (XRD) analysis of the samples was carried out using a Rigaku D/Max-RB diffractometer with a Cu Kα monochromatized radiation source (λ = 1.54178 Å), operated at 40 kV and 100 mA. Raman measurements were obtained using a Thermo Scientific DXR Raman Microscope with a 532 nm Ar laser. The elemental contents of the catalysts, including the metal Al or Zr in the oxides and the precious metal Pt, were detected either by inductively coupled plasma atomic emission spectroscopy (ICP-AES) (Perkin-Elmer Optima 2000DV), X-ray fluorescence (XRF) (SRS-3400 Sequential X-ray Spectrometer System) or energy dispersive X-ray spectroscopy (EDS) (FEI QUANTA 450). Transmission electron microscopy (TEM) images were taken on an FEI Tecnai G20 at an acceleration voltage of 300 kV.In the temperature programmed desorption of ammonia (NH3-TPD) experiments, the calcined supports were outgassed in He at 573 K for 2 h, and then saturated at 373 K in a 10% NH3/He stream (50 mL min−1) for 1 h. After removing most of the weakly physisorbed NH3 by flowing He (50 mL min−1), the chemisorbed NH3 was determined with a thermal conductivity detector (TCD) by heating at 10 K min−1 up to 973 K under the same flow of He.
Nitrogen adsorption and desorption isotherms at 77 K were measured using an Autosorb IQ surface area and pore size analyser. The specific surface area was calculated by the BET (Brunauer–Emmett–Teller) method and the pore volume was calculated from the volume of liquid nitrogen at p/p0 = 0.99. The non-local density functional theory (NLDFT) method considering sorption of nitrogen at 77 K in cylindrical silica pores was used to determine the pore size distribution by using the adsorption branch.
CO chemisorption analysis was performed in the same Autosorb IQ apparatus under static volumetric conditions. Prior to measurement, the ex situ reduced catalysts were activated in situ in H2 at 573 K for 2 h and evacuated for 2 h, then the furnace was cooled to 303 K. The chemisorption isotherms were obtained by measuring the amount of CO adsorbed at pressures varying from 80 to 560 mm Hg at 303 K. After completing the initial analysis, the reversibly adsorbed gas was evacuated and the analysis repeated to determine the chemisorbed molecules alone. A stoichiometry of CO/metal = 1 was accordingly taken to estimate the number of metal active sites.
2.3 Catalytic tests
Typically, the HDO of DBF experiments were performed at 553 K and 3.0 MPa total pressure in a continuous-flow fixed-bed reactor over 50 mg catalyst diluted with 5.0 mL 60–80 mesh quartz sands. Before the HDO experiments, the as-prepared Pt catalysts were activated in situ with 40 mL min−1 H2 at 3.0 MPa and 573 K for 1 h. Then, the temperature was adjusted to the reaction temperature. The liquid reactants were composed of 3.0 wt% DBF, 1.0 wt% n-dodecane (as internal standard for gas chromatography (GC) analysis) and 96.0 wt% n-decane (as inert solvent). The experimental data were collected at different weight times until the fresh catalyst reached steady state. The weight time is defined as τ = Wcat/nfeed, where Wcat denotes the catalyst weight and nfeed denotes the total molar flow feed to the reactor. The variation of weight time is achieved by changing either the flow rate of the liquid reactants or the catalyst weight. After being condensed in a trap at room temperature, the reaction products were collected and analyzed using an Agilent GC 7890A with a flame ionization detector and a 0.25 μm × 0.32 mm × 30 m HP-5 capillary column. Product identifications were conducted on an Agilent 6890N with 5973 MSD and a 0.25 μm × 0.32 mm × 30 m HP-5MS capillary column.The conversion (X) and selectivity (S) in the HDO of DBF were used to present the activities of the as-prepared Pt catalysts, which were calculated as
| X = (n0 − nDBF)/n0 × 100% | (1) |
| S = ni/∑ni × 100% | (2) |
| TOF = (F/W) × X/M | (3) |
3 Results and discussion
3.1 Characterization of supports and as-prepared Pt catalysts
The surface of silica with mesopores was modified post-synthesis by the deposition of Al2O3 or ZrO2 species. From the wide-angle XRD patterns in Fig. 1a, it is apparent that the surface modified samples Al2O3/SiO2 or ZrO2/SiO2 do not exhibit any distinct peaks associated with metal oxides as compared to the original SiO2 support. After loading of 0.50 wt% Pt on the supports, no clear Pt peaks around 2θ = 40 ° are shown in the wide-angle XRD patterns. This is ascribed to either the small size of the loaded Pt nanoparticles or the low metal loading. The small-angle XRD patterns in the Fig. 1a inset are flat without typical diffraction peaks related to the ordered mesoporous structure of SiO2. The Raman spectra of the supported Al2O3/SiO2 and ZrO2/SiO2 samples are presented in Fig. 1b and scarcely exhibit any new significant spectral features with respect to the parent SiO2. The deposition of alumina or zirconia on the silica support causes a decrease in the intensity of the band at 970 cm−1 of the Si–OH vibration, which indicates the consumption of the surface Si–OH groups.40,41 The post-synthesis modification of the SiO2 surface by deposited metal oxides leads to direct interaction with the silica surface hydroxyl groups, resulting in the broadening of the 970 cm−1 band. Therefore, the Al2O3 and ZrO2 oxides anchor to the silica at the Si–OH sites to create a surface metal oxide layer with residual exposed Si–OH species present.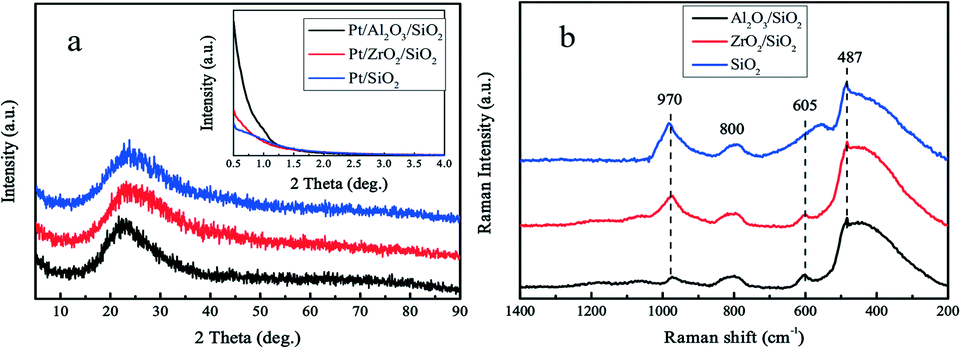 | ||
| Fig. 1 Small-angle (inset) and wide-angle XRD patterns (a) of Pt/Al2O3/SiO2, Pt/ZrO2/SiO2, and Pt/SiO2 catalysts with 0.50 wt% Pt loading. (b) Raman spectra of Al2O3/SiO2, ZrO2/SiO2, and SiO2 supports using 532 nm excitation energy. | ||
In Fig. 2a, the typical type IV nitrogen adsorption–desorption isotherms of the adopted supports indicate that the porous structure of SiO2 is preserved after the deposition of ZrO2 or Al2O3 species over the surface. The BET surface areas, pore volumes, and average pore diameters are summarized in Table 1. It is obvious that the BET surface areas and pore volumes decrease sharply after the deposition of metal oxides, from 405 to 221 m2 g−1 and 1.01 to 0.78 cm3 g−1, respectively. A relatively broad pore size distribution with a pore diameter of around 11.6 nm is observed from Fig. 2b. The decrease of surface area and expansion of pore diameter (8.1 nm vs. 11.6 nm) is mostly ascribed to the slight etching of the surface silica by ammonium hydroxide during the post-synthesis modification. The elemental analysis by XRF and EDS confirms the successful grafting of an ideal amount of metal oxides to the silica surface. The actual amount of supported metal oxides on the SiO2 largely exceeds the nominal loaded 0.50 wt% Pt, which is believed to be helpful for the dispersion of Pt particles.42 The acidity of the studied supports was determined by NH3-TPD and the desorption profiles are shown in Fig. 3. All of the three supports exhibit similar TPD profiles with two main broad peaks centered at around 500 K and 700 K. These two peaks could be associated with weak acidic sites and strong acidic sites.24,43 With regard to the TPD peaks, it is clear that the total acidity of the supports decreases in the order: Al2O3/SiO2 > ZrO2/SiO2 > SiO2, which correlates well with previous work.24
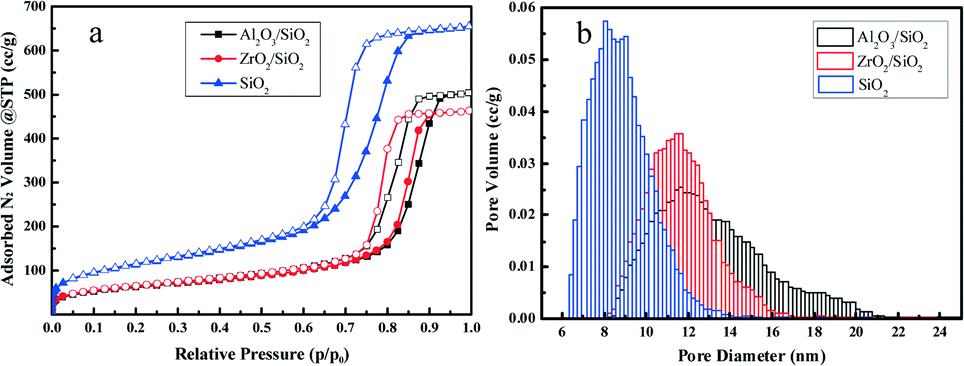 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms (a) and pore size distributions (b) of Al2O3/SiO2, ZrO2/SiO2, and SiO2 supports. | ||
| Sample | S BET (m2 g−1) | V p (cm3 g−1) | d p (nm) | Metal compositionb (wt%) | |
|---|---|---|---|---|---|
| XRF | EDS | ||||
| a BET surface area (SBET), pore volume (Vp), and average pore diameter (dp). b The weight percent of Al or Zr in the modified SiO2 supports. | |||||
| Al2O3/SiO2 | 221 | 0.78 | 11.6 | 1.75 | 1.26 |
| ZrO2/SiO2 | 224 | 0.72 | 11.6 | 5.47 | 3.76 |
| SiO2 | 405 | 1.01 | 8.1 | — | — |
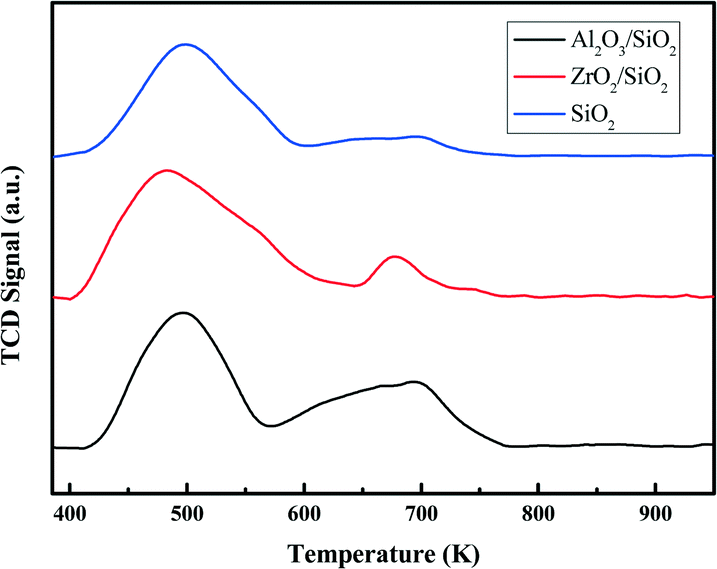 | ||
| Fig. 3 NH3-TPD profiles of the Al2O3/SiO2, ZrO2/SiO2, and SiO2 supports. | ||
Fig. 4 displays the TEM images together with histograms of the particle size of the reduced Pt catalysts. The Pt nanoparticles are highly dispersed with no apparent aggregation observed. The size of the Pt nanoparticles from statistical analysis is centered at 2.5, 2.3, and 4.5 nm for the Pt/Al2O3/SiO2, Pt/ZrO2/SiO2, and Pt/SiO2, respectively. It is clear that the supported metal oxides Al2O3 or ZrO2 promote the dispersion of Pt nanoparticles during the impregnation. The metal dispersion and Pt particle size were also estimated from the CO chemisorption and the corresponding results are listed in Table 2. Both the Pt/Al2O3/SiO2 and Pt/ZrO2/SiO2 catalysts show more than twice the amount of CO uptake compared to Pt/SiO2. The calculated Pt dispersion for the Pt/Al2O3/SiO2 or Pt/ZrO2/SiO2 catalyst reaches 44% with respect to 21% for the Pt/SiO2 catalyst. Pt particle size measured from CO chemisorption shows a good agreement with the statistical results from the TEM images. The actual Pt content in the as-prepared Pt catalysts was measured by ICP-AES. All of the catalysts actually contained at least 0.41 wt% Pt, which is slightly less than the nominal 0.50 wt% Pt loading. Hence, the difference of Pt particle size over the studied catalysts is hardly due to the amount of metal loading. It is strongly suggested that the Al2O3 and ZrO2 particles coated on the surface of SiO2 has a positive effect on the dispersion of Pt nanoparticles during the catalyst preparation. As explained by most of the previous works, the metal oxide support would interact strongly with the precious metal such as platinum at an elevated temperature under hydrogen.44,45 The strong interaction between platinum and metal oxides suppresses the migration and aggregation of Pt nanoparticles on the surface of metal oxides.41 Therefore, the Pt catalysts impregnated on the Al2O3/SiO2 or ZrO2/SiO2 surface show higher metal dispersion than those on the unmodified SiO2 surface.
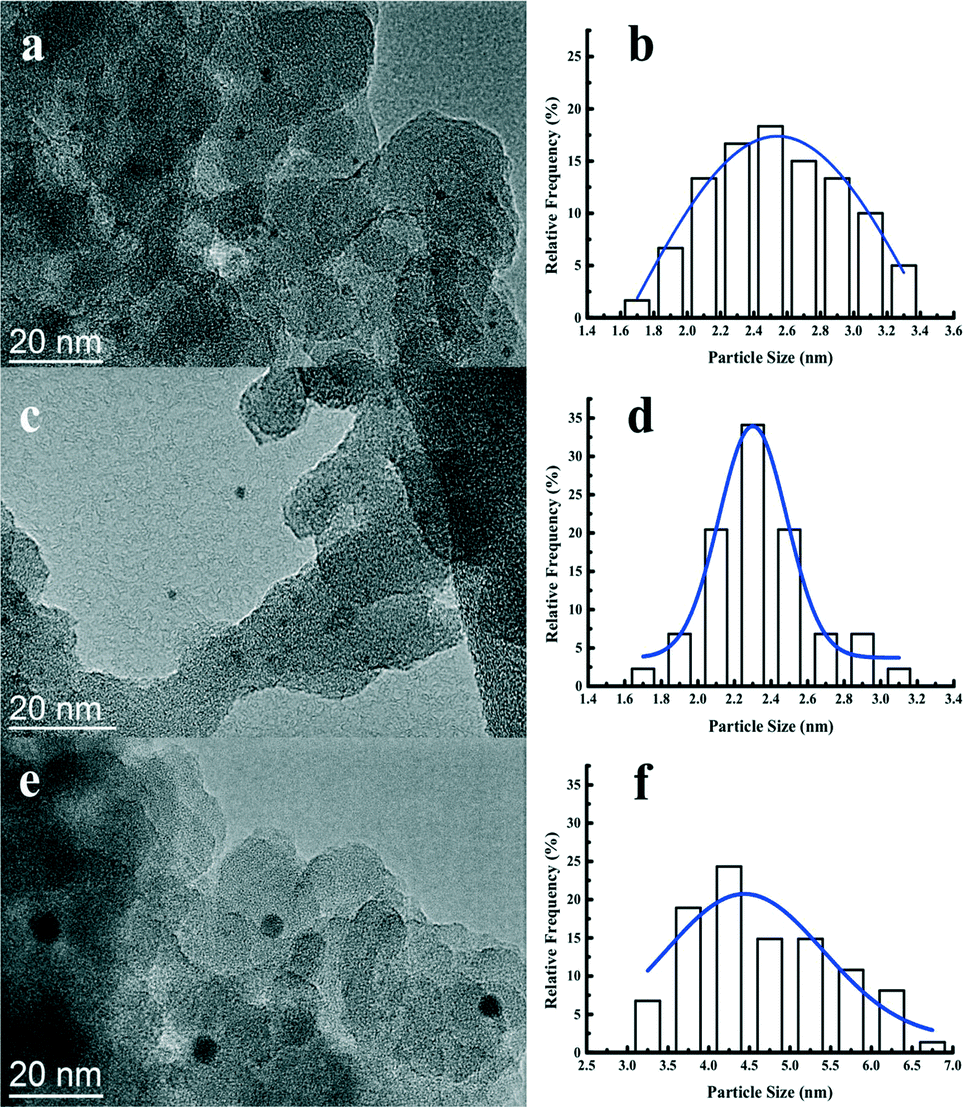 | ||
| Fig. 4 TEM images and the corresponding Pt particle size distribution of the Pt/Al2O3/SiO2 (a, b), Pt/ZrO2/SiO2 (c, d), and Pt/SiO2 (e, f) catalysts. | ||
3.2 Product distribution of HDO over the as-prepared Pt catalysts
The HDO of DBF over the as-prepared Pt/Al2O3/SiO2, Pt/ZrO2/SiO2 and Pt/SiO2 catalysts goes through the hydrogenation reaction route as shown in the proposed HDO reaction network (Scheme 1). Briefly, hydrogenation of the aromatic rings of DBF takes place first, and then oxygen removal is conducted by hydrogenolysis or dehydration–hydrogenation of the saturated C–O bond to obtain bicyclohexane (BCH) and its isomer cyclopentylmethylcyclohexane (iso-BCH). Trace single-ring product cyclohexane (CH) and ring-opening products are detected as the further cracking of those bicyclo-hydrocarbons occurs. The formation of these products and intermediates has been previously discussed in the literature.4,17,46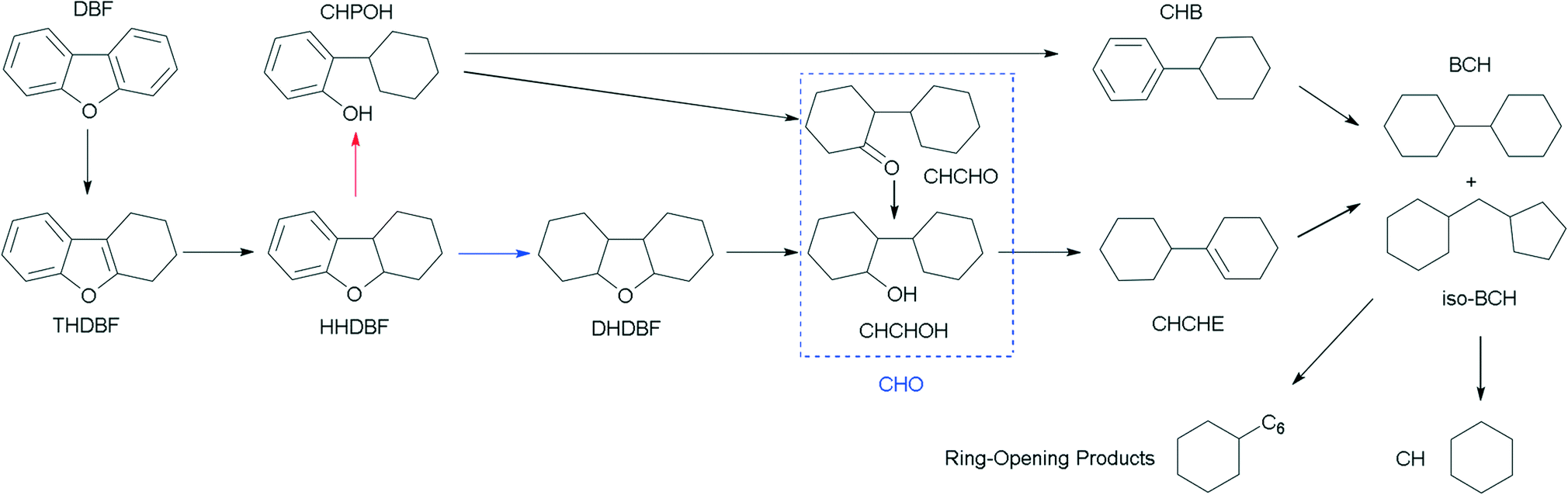 | ||
| Scheme 1 Proposed reaction network of the HDO of DBF over the as-prepared Pt catalysts. | ||
Fig. 5 shows the HDO product distribution and selectivity over Pt/Al2O3/SiO2 as a function of weight time at 280 °C and 3.0 MPa total pressure. Several main products in the HDO reaction of DBF are chosen to reveal the overall HDO reaction process. Tetrahydrodibenzofuran (THDBF), hexahydrodibenzofuran (HHDBF) and dodecahydrodibenzofuran (DHDBF) are obtained by the partial or total hydrogenation of the aromatic rings of DBF. Hydrogenolysis of the C–O bond of HHDBF directly produces 2-cyclohexylphenol (CHPOH). The cleavage of the saturated C–O bond of DHDBF results in the production of 2-cyclohexylcyclohexanol (CHCHOH). The abbreviation CHO is defined as the sum of CHCHO and CHCHOH in the HDO products. The deoxygenation of CHPOH or CHCHOH leads to the production of BCH and iso-BCH. Finally, the C–C bond scission of these bicyclo-hydrocarbons takes place to produce small amounts of CH. The hydrogenolysis of the phenolic hydroxyl group in CHPOH results in CHB. At low weight time stages (e.g. 0.40 g min mol−1), the reactant DBF mainly transforms to THDBF and HHDBF through the partial hydrogenation of a single aromatic ring of the DBF molecule. Less than 10% product distribution is achieved for both THDBF and HHDBF. With increasing the weight time to 0.80 g min mol−1, CHPOH and THDBF percentage in the product distribution increases with a value close to 10%. Further hydrogenation of THDBF to HHDBF becomes preferable, resulting in reduced product distribution of THDBF. However, the hydrogenolysis of the saturated C–O bond in the HHDBF leads to more of the oxygen-containing intermediate CHPOH with 11% product distribution and 18% selectivity. As the weight time increased, it was noticeable that totally deoxygenated products BCHs (BCH and iso-BCH), with BCH taking more than 90% percent, were the dominant species in the product distribution. The selectivity toward BCHs exceeds 70% at weight times higher than 1.14 g min mol−1. Meanwhile, a measurable amount of CH at a weight time higher than 0.57 g min mol−1 is explained by the further cracking of BCHs. In the whole HDO process over Pt/Al2O3/SiO2, DHDBF, CHO and CHB are detected in small amounts with around 2% product distribution and less than 5% selectivity in this study. The CHCHE and ring-opening products were confirmed by the GC-MS instrument with trace amounts. Pt/ZrO2/SiO2 and Pt/SiO2 catalysts were also tested in the HDO of DBF under the same reaction conditions to investigate the activity and reaction network of DBF. The variation trend of the product distribution and selectivity are similar to those obtained over the Pt/Al2O3/SiO2 catalyst with 0.50 wt% Pt loading. It is worth mentioning that the C–C bond scission of BCHs scarcely occurs over the Pt/ZrO2/SiO2 and Pt/SiO2 catalysts with less than 1% CH yielded at a weight time of 1.77 g min mol−1, whereas 7% CH is obtained over the Pt/Al2O3/SiO2 catalyst under the same HDO reaction conditions. This comparison implies that the acidic characters of modified SiO2 supports introduced by the deposition of oxides promote the scission of C–C bonds with the help of metal active sites under such conditions. However, CH is hardly detected under the selected low weight time stages over the as-prepared Pt catalysts in this work. It is ascribed to either the relatively weak acidic properties of the modified supports with limited hydrocracking ability or the short contact between the catalyst and the deoxygenated BCHs species.
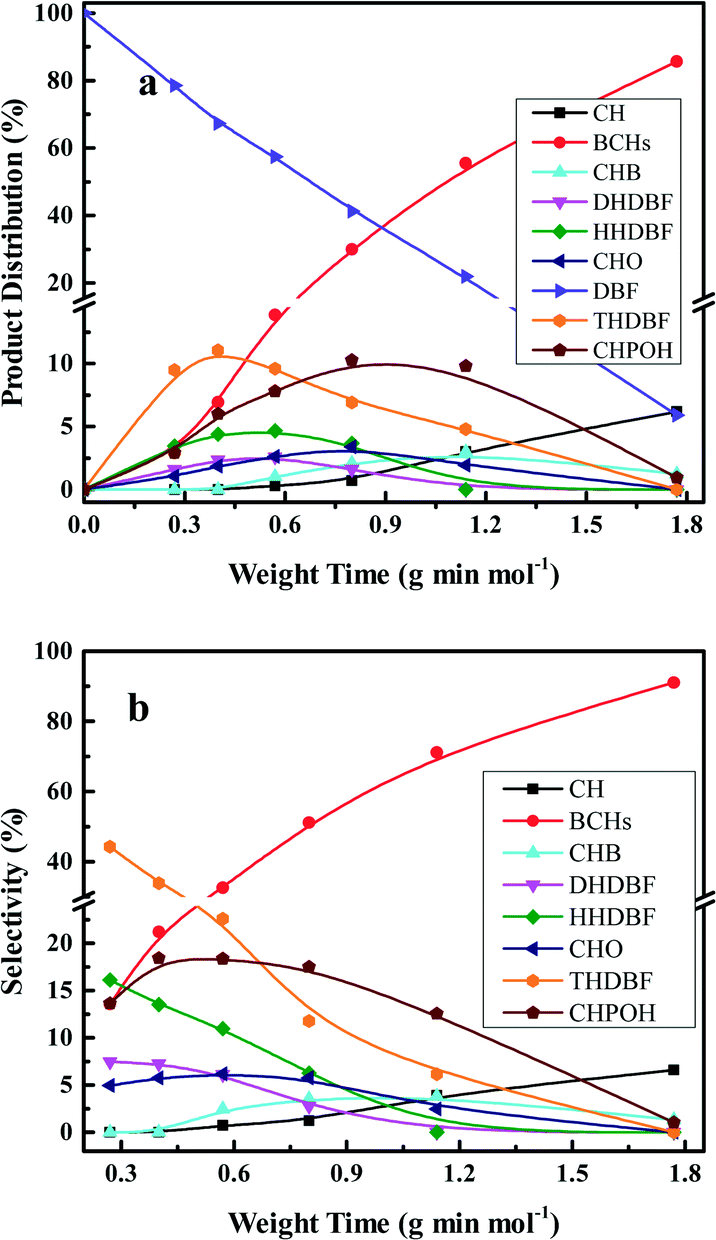 | ||
| Fig. 5 Product distributions (a) and selectivities (b) of the HDO of DBF over 0.50 wt% Pt/Al2O3/SiO2 as a function of weight time at 280 °C and 3.0 MPa total pressure. | ||
3.3 Comparison of HDO over the as-prepared Pt catalysts
In order to further reveal the HDO performance of the prepared Pt catalysts, the deoxygenation degree in the HDO of DBF is compared at various HDO conversions. From Fig. 6a, it is clear that the totally deoxygenated products accumulate rapidly with increasing the HDO conversion. Hydrogenation of the aromatic rings of DBF has a higher contribution under lower HDO conversion (i.e. <40%). However, deoxygenation of the hydrogenated DBFs or the oxygen-containing compounds, cycloalcohols or cycloketones, becomes favored under higher HDO conversion, which directly leads to the high yield of oxygen-free high carbon number hydrocarbon fuels. The Pt/Al2O3/SiO2 and Pt/ZrO2/SiO2 catalysts exhibit quite similar deoxygenation abilities at high HDO conversion (>60%) stages, whereas the Pt/Al2O3/SiO2 is superior for deoxygenation compared with Pt/ZrO2/SiO2 at low conversion stages. Both the Pt/Al2O3/SiO2 and Pt/ZrO2/SiO2 catalysts show better deoxygenation performances in comparison with Pt/SiO2 under the studied HDO conversions.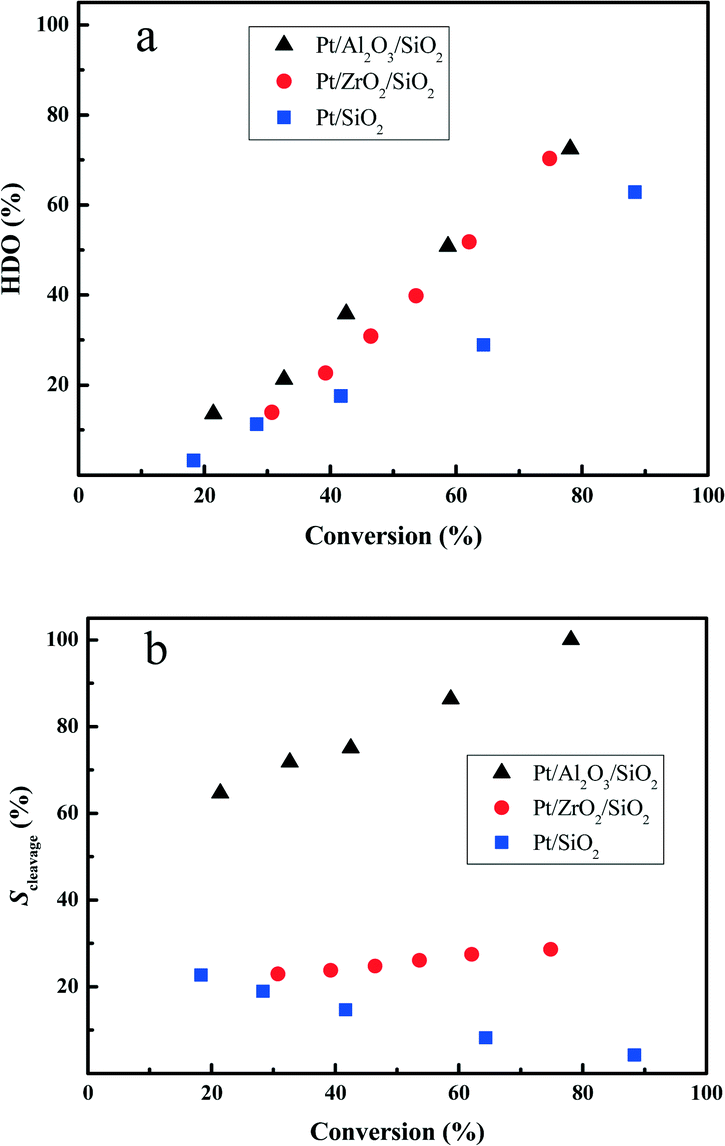 | ||
| Fig. 6 The deoxygenation degree (a) and selectivity for the C–O bond cleavage (b) as a function of the HDO conversion at 280 °C and 3.0 MPa total pressure. | ||
Guided by the proposed HDO reaction network in Scheme 1, the HHDBF is either hydrogenated to DHDBF or transformed to CHPOH via hydrogenolysis of the C–O bond. Hence, we calculate the selectivity for the C–O bond cleavage (Scleavage) with the following formula to reveal the effect of the supports on the reaction pathway.
| Scleavage = nCHPOH/(nCHPOH + nDHDBF) × 100% | (4) |
The Scleavage for the Pt/Al2O3/SiO2 catalyst with a value greater than 60% largely exceeds that for Pt/ZrO2/SiO2 or Pt/SiO2 with less than 30% selectivity from Fig. 6b. With increasing the HDO conversion, the Scleavage enlarges remarkably, which indicates that the HHDBF mainly transforms to CHPOH with cleavage of the saturated C–O bond over the Pt/Al2O3/SiO2 catalyst. For the Pt/ZrO2/SiO2 catalyst, the calculated Scleavage almost remains the same under the varied HDO conversions. However, the gradually reduced Scleavage for the Pt/SiO2 catalyst suggests that the deoxygenation of DHDBF is a major pathway in the hydrogenation reaction route. Comparing the Scleavage at a given HDO conversion of 40%, it is quite apparent that more scission of the saturated C–O bond takes place over the Pt/Al2O3/SiO2 catalyst with a value close to 75%. However, only 25% is obtained over the Pt/ZrO2/SiO2, and then 16% over the Pt/SiO2. The obvious difference in Scleavage for the three studied catalysts can be mostly attributed to the varied acidic properties of the supports. The modification by Al2O3 or ZrO2 changes the acidity of the supports and shows a positive effect on the hydrogenolysis of the sp3 C–O bond. In the conversion of anisole and guaiacol over Pt/Al2O3, the hydrogenolysis of the sp3 C–O bond to produce phenols and methane is kinetically significant.47,48 The Ru supported on Al2O3 catalysts show better performance of hydrogenolysis of phenol with a preferable yield of benzene than over silica supports.49 Over the alumina supported Pt catalysts, the vapor-phase HDO of meta-cresol produces a high amount of toluene with direct hydrogenolysis of the C–O bond.50 Zhang et al. prepared a series of ZrO2-SiO2 supported Ni and NiCu catalysts for the hydrodeoxygenation of guaiacol.51 The hydrogenolysis of the phenolic hydroxyl group with selective yield of benzene is attributed to the stronger acidity of the ZrO2–SiO2. These results are consistent with this work. Regarding the mechanism of the sp3 C–O bond cleavage involved in the hydrodeoxygenation of benzofuran over the sulfide catalysts, Romero et al. proposed that the cleavage occurs through nucleophilic substitution by SH− after protonation of the oxygen atom of 2,3-dihydrobenzofuran by a Brønsted acid site.13 The Al2O3 support contains mostly Lewis and a few Brønsted acid sites as proven by experimental and theoretical methods.52,53 The structure of the C–O bond in HHDBF is similar to that in 2,3-dihydrobenzofuran. It is reasonable to speculate that the cleavage of the C–O bond of HHDBF goes through a similar pathway in the HDO of DBF over Pt/Al2O3/SiO2. Consequently, the selective hydrogenolysis of C–O is preferable over Al2O3 modified SiO2 supported Pt catalysts, while the transformation is suppressed over Pt/ZrO2/SiO2 and Pt/SiO2. Besides, it is generally reported that the acidic sites of the catalysts promote the deoxygenation of those oxygen-containing intermediates in the HDO of phenolic molecules.19 The supported Pt catalysts over the acidic zeolite HBeta show superior deoxygenation performance than Pt/SiO2 in the HDO of anisole.20 It is speculated that an acid site adjacent to a Pt particle could facilitate the deoxygenation reaction by protonating the oxygen in phenol in a bifunctional reaction. Thus, the deoxygenation of those oxygen-containing intermediates could also facilitate the hydrogenolysis of the C–O bond in the consecutive reaction of HDO, which leads to the shift of Scleavage in this work.
Comparison of the HDO reaction results at 30% conversion in Table 3 further illustrates the influence of the supports on the HDO of DBF. It is noted that the further hydrocracking of BCHs to ring-opening products and CH scarcely occurs under the studied HDO conditions. The apparent order of TOFs for the tested catalysts is as follows: Pt/Al2O3/SiO2 > Pt/ZrO2/SiO2 > Pt/SiO2. From the comparison of product selectivity, it is clear that the total selectivity toward THDBF and HHDBF is 48% and only 7% selectivity toward DHDBF is obtained over the Pt/Al2O3/SiO2 catalyst. It is particularly worth mentioning that a high amount of oxygen-containing intermediates OCH (abbreviated as the sum of CHO and CHPOH) and totally deoxygenated products BCHs are yielded with 24% and 21% selectivity, respectively. This confirms that more partially hydrogenated DBFs (THDBF and HHDBF) transform to OCH via C–O bond cleavage instead of further hydrogenation of the aromatic ring in the HDO reaction, which is consistent with the results showing the selectivity of C–O bond cleavage over the Pt/Al2O3/SiO2 catalyst under varied conversions. The acidic properties of deposited Al2O3 oxides promote the cleavage of the saturated C–O bond with enhanced deoxygenation performance. The Pt/ZrO2/SiO2 and Pt/SiO2 catalysts showed better hydrogenation performance in the HDO of DBF with up to 70% selectivity to hydrogenated DBFs. Similar abilities of C–O bond scission are evidenced from the closed selectivity to OCH and BCHs.
| Sample | TOF (h−1) | Selectivity (%) | |||
|---|---|---|---|---|---|
| TH + HHDBFa | DHDBF | OCHb | BCHsc | ||
| a The total selectivity to THDBF and HHDBFF. b The total selectivity to CHO and CHPOH. c The hydrocracking of BCHs scarcely occurs and neglect the trace CHB. | |||||
| Pt/Al2O3/SiO2 | 2412 | 48 | 7 | 24 | 21 |
| Pt/ZrO2/SiO2 | 1784 | 56 | 18 | 12 | 14 |
| Pt/SiO2 | 1678 | 53 | 24 | 11 | 12 |
The kinetic analysis of the HDO reaction was conducted at 533–573 K with varied conversions of DBF by changing the weight time. A pseudo-first-order model for the HDO of DBF was represented by the following equation.
| −ln(1 − X) = kHDO × τ | (5) |
The essential linear relationship of −ln(1 − X) and τ is shown in Fig. 7 for the data obtained from the HDO reaction. The linear relationships illustrate that the overall reaction is pseudo-first-order. All of the fitting parameters are summarized in Table 4. For instance, the HDO rate constant (kHDO) over the Pt/Al2O3/SiO2 catalyst increases from 0.59 to 2.03 mol g−1 min−1 in the temperature range of 533 to 573 K. It is obvious that the increase of temperature promotes the transformation of the reactant DBF, which is quite similar to previous work on the HDO of biomass-derived molecules.3 At a constant temperature, the kHDO gradually decreases following the order: Pt/Al2O3/SiO2 > Pt/ZrO2/SiO2 > Pt/SiO2. This phenomenon could be ascribed to the more dispersed Pt nanoparticles in the prepared catalysts with superior activity in the HDO of DBF. The acidic properties of supports with promotion to the deoxygenation also accelerate the transformation of DBF. Analysis of the HDO rate constant-temperature dependence as shown in Fig. 8 yields the HDO activation energy (Ea) with values of 79, 72, 70 kJ mol−1 for the Pt/Al2O3/SiO2, Pt/ZrO2/SiO2, and Pt/SiO2 catalysts, respectively. Some previous works have pointed out that the catalytic HDO performance for phenol conversion was obtained over smaller Pd nanoparticles in the study of the hydrogenation of phenol in water.54 In the hydrogenation of furan, small particles preferentially yielded dihydrofuran by partial hydrogenation of the aromatic ring.55 It is concluded that the size of the supported metal nanoparticle affects the catalytic performance in the HDO of DBF. Pt nanoparticles with small size facilitate the hydrogenation of aromatic rings of DBF. As discussed from the NH3-TPD profiles in Fig. 3, the introduction of Al2O3 or ZrO2 to the surface of the SiO2 supports causes the formation of stronger acid sites. Mortensen et al. reported some similar results where the total acidity of the supports decreased in the following order: Ni/Al2O3 > NiZrO2 > Ni/SiO2.56 The decrease of the acidity is attributed to the fact that the low metal–oxygen bond energy in the order of E(Al–O) > E(Zr–O) > E(Si–O) can be linked to the formation of more oxygen vacancy sites in the oxides which function as Lewis acid sites.57,58 These Lewis acid sites are beneficial to the activation of the intermediate phenols via formation of phenoxide on Al2O3 as studied by IR spectroscopy.30 The cooperative effect of the support acidic sites and metal active sites has also been found to be favorable for the phenol hydrogenation on palladium catalysts, where the hydrogenation activity can be significantly increased by the introduction of a Lewis acid.22,59 Lee et al. reported the HDO of guaiacol over a series of Al2O3 based bifunctional catalysts. It is explained that the metal is responsible for the hydrogenation of aromatic rings and acidity appears to determine the degree of deoxygenation.60 The ZrO2/SiO2 supports also have some acidic characters, but significantly less than Al2O3/SiO2 supports from the NH3-TPD results. Thus, it is still thought to have the potential to activate the oxygen-containing intermediates on its surface. Besides, the deposited Al2O3 oxides have a few Brønsted acid sites, which are claimed to promote the protonation of the oxygen in the HDO process. This function could further facilitate the hydrogenolysis and deoxygenation, and thus accelerate the whole HDO reaction with increasing transformation of the reactant DBF. These explanations account for the gradual change of selectivity toward the deoxygenation and the C–O bond cleavage in the HDO reaction over Pt/Al2O3/SiO2, Pt/ZrO2/SiO2, and Pt/SiO2 catalysts. The better dispersed metal particles and acidic properties of the supports function cooperatively to promote the whole HDO reaction of DBF with apparently reduced HDO rate constant kHDO.61
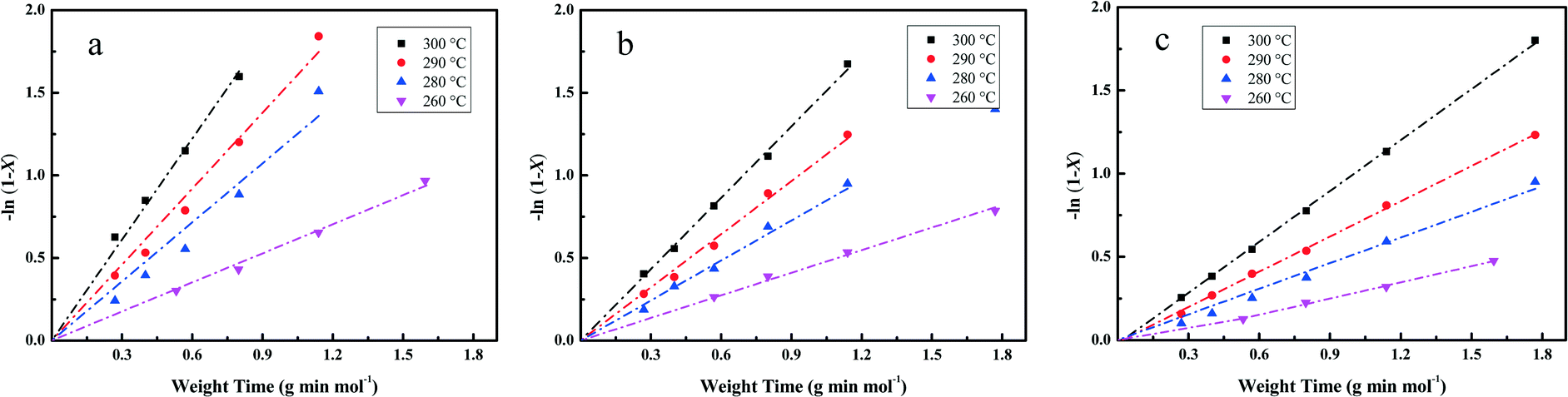 | ||
| Fig. 7 Fitting of the pseudo-first-order kinetic model to the HDO reaction experimental data collected over Pt/Al2O3/SiO2 (a), Pt/ZrO2/SiO2 (b), Pt/SiO2 (c) catalysts at a temperature range of 533 to 573 K. | ||
| Sample | HDO rate constant kHDO mol g−1 min−1 | r 1 | E a kJ mol−1 | r 2 | |||
|---|---|---|---|---|---|---|---|
| 573 K | 563 K | 553 K | 533 K | ||||
| a The coefficient of determination in the fitting of the pseudo-first-order kinetic model to the HDO reaction experimental data. b The coefficient of determination in the linear fitting of HDO rate constant. | |||||||
| Pt/Al2O3/SiO2 | 2.03 | 1.53 | 1.19 | 0.59 | >0.98 | 79 | >0.99 |
| Pt/ZrO2/SiO2 | 1.44 | 1.08 | 0.81 | 0.46 | >0.99 | 72 | >0.99 |
| Pt/SiO2 | 1.01 | 0.71 | 0.51 | 0.32 | >0.98 | 70 | >0.98 |
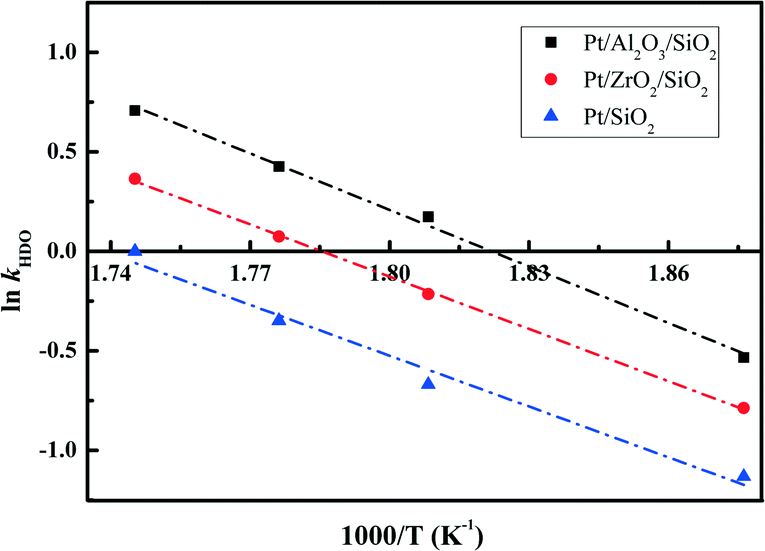 | ||
| Fig. 8 HDO rate constants kHDO as a function of inverse temperature. | ||
Conclusions
The deposited Al2O3 or ZrO2 oxides on the surface of silica with mesopores largely help to disperse Pt nanoparticles with smaller particle size. The HDO of DBF over the as-prepared Pt catalysts mainly goes through the hydrogenation reaction route. Pt/Al2O3/SiO2 and Pt/ZrO2/SiO2 catalysts with highly dispersed Pt nanoparticles show better HDO performance with superior hydrogenation activity of the aromatic rings. Al2O3/SiO2 and ZrO2/SiO2 supports with relatively strong acidic sites promote the cleavage of the saturated C–O bond with enhanced deoxygenation activity in the HDO reaction. From the varied values of Scleavage, it is obvious that the relatively strong acidic sites of the Al2O3/SiO2 and ZrO2/SiO2 supports selectively alter the reaction pathways to CHPOH through hydrogenolysis of the saturated C–O bond. However, further hydrogenation of HHDBF to DHDBF is favored over the Pt/SiO2 catalyst with less acidic properties. A detailed analysis based on the pseudo-first-order kinetics suggests that the activity of the as-prepared Pt catalysts in the HDO reaction of DBF declines in the following order: Pt/Al2O3/SiO2 > Pt/ZrO2/SiO2 > Pt/SiO2. It is concluded that the smaller sized Pt nanoparticles with better hydrogenation activity and the acidic properties of deposited Al2O3 or ZrO2 oxides function cooperatively in the HDO of DBF with an enhanced HDO performance being observed.Acknowledgements
We gratefully acknowledge the financial support provided by the National Natural Science Foundation of China (21373038) and the Fundamental Research Funds for the Central Universities (DUT12YQ03 and DUT13RC(3)41).Notes and references
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- M. Stocker, Angew. Chem., Int. Ed., 2008, 47, 9200–9211 CrossRef CAS PubMed.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493 RSC.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184–7201 CrossRef CAS PubMed.
- A. D. Sutton, F. D. Waldie, R. L. Wu, M. Schlaf, L. A. Silks and J. C. Gordon, Nat. Chem., 2013, 5, 428–432 CrossRef CAS PubMed.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS PubMed.
- J. S. Yoon, Y. Lee, J. Ryu, Y. A. Kim, E. D. Park, J. W. Choi, J. M. Ha, D. J. Suh and H. Lee, Appl. Catal., B, 2013, 142, 668–676 CrossRef PubMed.
- B. Dhandapani, T. St. Clair and S. T. Oyama, Appl. Catal., A, 1998, 168, 219–228 CrossRef CAS.
- S. Sitthisa and D. E. Resasco, Catal. Lett., 2011, 141, 784–791 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- C. M. Huelsman and P. E. Savage, Phys. Chem. Chem. Phys., 2012, 14, 2900–2910 RSC.
- Y. Romero, F. Richard, Y. Renème and S. Brunet, Appl. Catal., A, 2009, 353, 46–53 CrossRef CAS PubMed.
- A. Y. Bunch and U. S. Ozkan, J. Catal., 2002, 206, 177–187 CrossRef CAS.
- Y. X. Wang, Y. M. Fang, T. He, H. Q. Hu and J. H. Wu, Catal. Commun., 2011, 12, 1201–1205 CrossRef CAS PubMed.
- J. A. Cecilia, A. Infantes-Molina, E. Rodriguez-Castellon, A. Jimenez-Lopez and S. T. Oyama, Appl. Catal., B, 2013, 136, 140–149 CrossRef PubMed.
- L. Wang, M. M. Zhang, M. Zhang, G. Y. Sha and C. H. Liang, Energy Fuels, 2013, 27, 2209–2217 CrossRef CAS.
- V. Lavopa and C. N. Satterfield, Energy Fuels, 1987, 1, 323–331 CrossRef CAS.
- D. Y. Hong, S. J. Miller, P. K. Agrawal and C. W. Jones, Chem. Commun., 2010, 46, 1038–1040 RSC.
- X. L. Zhu, L. L. Lobban, R. G. Mallinson and D. E. Resasco, J. Catal., 2011, 281, 21–29 CrossRef CAS PubMed.
- R. Nava, B. Pawelec, P. Castano, M. C. Alvarez-Galvan, C. V. Loricera and J. L. G. Fierro, Appl. Catal., B, 2009, 92, 154–167 CrossRef CAS PubMed.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. B. Li and J. A. Lercher, Chem. Commun., 2010, 46, 412–414 RSC.
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250–1252 CrossRef CAS PubMed.
- Y. Yang, C. Ochoa-Hernández, V. A. de la Peña O'Shea, P. Pizarro, J. M. Coronado and D. P. Serrano, Appl. Catal., B, 2014, 145, 91–100 CrossRef CAS PubMed.
- S. K. Maity, M. S. Rana, B. N. Srinivas, S. K. Bej, G. M. Dhar and T. S. R. P. Rao, J. Mol. Catal. A: Chem., 2000, 153, 121–127 CrossRef CAS.
- T. Nimmanwudipong, C. Aydin, J. Lu, R. C. Runnebaum, K. C. Brodwater, N. D. Browning, D. E. Block and B. C. Gates, Catal. Lett., 2012, 142, 1190–1196 CrossRef CAS PubMed.
- A. L. Jongerius, J. R. Copeland, G. S. Foo, J. P. Hofmann, P. C. A. Bruijnincx, C. Sievers and B. M. Weckhuysen, ACS Catal., 2013, 3, 464–473 CrossRef CAS.
- A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, Green Chem., 2013, 15, 3049 RSC.
- A. Villa, M. Schiavoni and L. Prati, Catal. Sci. Technol., 2012, 2, 673–682 CAS.
- A. Popov, E. Kondratieva, J. M. Goupil, L. Mariey, P. Bazin, J. P. Gilson, A. Travert and F. Mauge, J. Phys. Chem. C, 2010, 114, 15661–15670 CAS.
- V. A. Yakovlev, S. A. Khromova, O. V. Sherstyuk, V. O. Dundich, D. Y. Ermakov, V. M. Novopashina, M. Y. Lebedev, O. Bulavchenko and V. N. Parmon, Catal. Today, 2009, 144, 362–366 CrossRef CAS PubMed.
- H. Gies, S. Grabowski, M. Bandyopadhyay, W. Grunert, O. P. Tkachenko, K. V. Klementiev and A. Birkner, Microporous Mesoporous Mater., 2003, 60, 31–42 CrossRef CAS.
- J. Strunk, W. C. Vining and A. T. Bell, J. Phys. Chem. C, 2011, 115, 4114–4126 CAS.
- P. Kustrowski, L. Chmielarz, R. Dziembaj, P. Cool and E. F. Vansant, J. Phys. Chem. B, 2005, 109, 11552–11558 CrossRef CAS PubMed.
- T. Klimova, L. Pena, L. Lizama, C. Salcedo and O. Y. Gutierrez, Ind. Eng. Chem. Res., 2009, 48, 1126–1133 CrossRef CAS.
- O. Y. Gutierrez, D. Valencia, G. A. Fuentes and T. Klimova, J. Catal., 2007, 249, 140–153 CrossRef CAS PubMed.
- H. Wan, J. Yan, L. Yu, X. Zhang, X. Xue, X. Li and X. Liang, Talanta, 2010, 82, 1701–1707 CrossRef CAS PubMed.
- C. K. Krishnan, T. Hayashi and M. Ogura, Adv. Mater., 2008, 20, 2131–2136 CrossRef.
- C. K. Krishnan, T. Hayashi and M. Ogura, Microporous Mesoporous Mater., 2008, 116, 406–414 CrossRef PubMed.
- X. T. Gao and I. E. Wachs, J. Catal., 2000, 192, 18–28 CrossRef CAS.
- X. T. Gao, J. L. G. Fierro and I. E. Wachs, Langmuir, 1999, 15, 3169–3178 CrossRef CAS.
- Y. Nagai, T. Hirabayashi, K. Dohmae, N. Takagi, T. Minami, H. Shinjoh and S. Matsumoto, J. Catal., 2006, 242, 103–109 CrossRef CAS PubMed.
- S. H. Jin, Z. H. Xiao, C. Li, X. Chen, L. Wang, J. C. Xing, W. Z. Li and C. H. Liang, Catal. Today, 2014, 234, 125–132 CrossRef CAS PubMed.
- S. J. Tauster and S. C. Fung, J. Catal., 1978, 55, 29–35 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- M. J. Girgis and B. C. Gates, Ind. Eng. Chem. Res., 1994, 33, 2301–2313 CrossRef CAS.
- R. C. Runnebaum, R. J. Lobo-Lapidus, T. Nimmanwudipong, D. E. Block and B. C. Gates, Energy Fuels, 2011, 25, 4776–4785 CrossRef CAS.
- T. Nimmanwudipong, R. C. Runnebaum, D. E. Block and B. C. Gates, Energy Fuels, 2011, 25, 3417–3427 CrossRef CAS.
- C. Newman, X. Zhou, B. Goundie, I. T. Ghampson, R. A. Pollock, Z. Ross, M. C. Wheeler, R. W. Meulenberg, R. N. Austin and B. G. Frederick, Appl. Catal., A, 2014, 477, 64–74 CrossRef CAS PubMed.
- P. T. M. Do, A. J. Foster, J. G. Chen and R. F. Lobo, Green Chem., 2012, 14, 1388–1397 RSC.
- X. Zhang, T. Wang, L. Ma, Q. Zhang, Y. Yu and Q. Liu, Catal. Commun., 2013, 33, 15–19 CrossRef CAS PubMed.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2002, 211, 1–5 CrossRef CAS.
- S. Jongpatiwut, Z. Li, D. E. Resasco, W. E. Alvarez, E. L. Sughrue and G. W. Dodwell, Appl. Catal., A, 2004, 262, 241–253 CrossRef CAS PubMed.
- Z. L. Li, J. H. Liu, C. G. Xia and F. W. Li, ACS Catal., 2013, 3, 2440–2448 CrossRef CAS.
- C. J. Kliewer, C. Aliaga, M. Bieri, W. Huang, C. K. Tsung, J. B. Wood, K. Komvopoulos and G. A. Somorjai, J. Am. Chem. Soc., 2010, 132, 13088–13095 CrossRef CAS PubMed.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen and A. D. Jensen, ACS Catal., 2013, 3, 1774–1785 CrossRef CAS.
- D. G. Rethwisch and J. A. Dumesic, Langmuir, 1986, 2, 73–79 CrossRef CAS.
- H. Idriss and M. A. Barteau, Adv. Catal., 2000, 45, 261–331 CAS.
- J. Matos and A. Corma, Appl. Catal., A, 2011, 404, 103–112 CrossRef CAS PubMed.
- C. R. Lee, J. S. Yoon, Y. W. Suh, J. W. Choi, J. M. Ha, D. J. Suh and Y. K. Park, Catal. Commun., 2012, 17, 54–58 CrossRef CAS PubMed.
- A. J. Foster, P. T. M. Do and R. F. Lobo, Top. Catal., 2012, 55, 118–128 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |