
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe selective addition of water
Verena
Resch
ab and
Ulf
Hanefeld
*a
aGebouw voor Scheikunde, Biokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Julianalaan 136, 2628BL Delft, The Netherlands. E-mail: u.hanefeld@tudelft.nl
bOrganische und Bioorganische Chemie, Institut für Chemie, Karl-Franzens-Universität Graz, Heinrichstrasse 28, 8010 Graz, Austria
First published on 14th August 2014
Abstract
Water is omnipresent and essential. Yet at the same time it is a rather unreactive molecule. The direct addition of water to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds is therefore a challenge not answered convincingly. In this perspective we critically evaluate the selectivity and the applicability of the different catalytic approaches for water addition reactions: homogeneous, heterogeneous and bio-catalytic. Here we would like to discuss how to speed up water addition and even make it selective.
C double bonds is therefore a challenge not answered convincingly. In this perspective we critically evaluate the selectivity and the applicability of the different catalytic approaches for water addition reactions: homogeneous, heterogeneous and bio-catalytic. Here we would like to discuss how to speed up water addition and even make it selective.
 Verena Resch and Ulf Hanefeld | Verena Resch was born in Austria in 1984. She studied biochemistry and molecular biology at the University of Graz and graduated with an MSc in 2008 under the supervision of Prof. Wolfgang Kroutil, establishing multi-enzyme cascades. Staying in the same group, she received her PhD in organic chemistry in 2011 working on the use of alkaloid pathway enzymes in organic synthesis. In 2012 she started as a postdoctoral fellow for two years with an Erwin-Schrödinger Fellowship from the Austrian Science Fund at the University of Technology in Delft in the group of Prof. Ulf Hanefeld working on hydratases and chemo-enzymatic cascades. She has just returned to Graz, where she continues her work as an Erwin Schrödinger fellow. |
Ulf Hanefeld was born in 1966 in Cologne, Germany and grew up in then (West) Berlin and London. In 1993 he received his PhD from the Georg-August-Universität Göttingen, having performed research both in Göttingen (Prof. H. Laatsch) and in Seattle (Prof. H. G. Floss). After postdoctoral years with Prof. C. W. Rees (Imperial College London), Prof. J. Staunton (Cambridge) and Prof. J. J. Heijnen and Dr. A. J. J. Straathof (TU Delft), he received a fellowship from the Royal Netherlands Academy of Arts and Sciences (KNAW). He rose through the ranks at the Technische Universiteit Delft and his research in Delft focuses on enzymes, their immobilisation and application in organic synthesis. |
1. Introduction
Water is often seen as something that disturbs a reaction. Indeed, it is commonly used to “quench a reaction”. Yet at the same time it is actually rather unreactive, being both a poor nucleophile and a poor electrophile. Consequently, the selective addition of water to carbon–carbon double bonds is known to be a chemically very challenging reaction. This, even though it is taught in every undergraduate course.1–4 How then to take up this gauntlet and bring undergraduate chemistry to real life? How to realise short and efficient routes for the synthesis of alcohols by straightforward water addition to double bonds?Even though water – in terms of sustainability and abundance – is an attractive reagent, it is rarely applied for the addition to double bonds in chemical processes. Indeed, only a few methods for the hydration of alkenes are reported,5–9 of which only one is applied as a direct process for the synthesis of ethanol and similar alcohols.10
Difficulties lie mainly in the activation of water as a nucleophile. Compared to carbon or nitrogen nucleophiles, oxygen-containing nucleophiles, such as water or hydrogen peroxide, are known to be bad nucleophiles. In general, nucleophilicity can be increased when charged species of the nucleophile are employed. This is of course also true for carbon- and nitrogen-containing nucleophiles (nucleophilicity in decreasing order: CH3− > NH2− > OH− > F−). To improve the reactivity of water for its addition to carbon–carbon double bonds, a strong activation is necessary. In the textbook, in electrophilic addition reactions to carbon–carbon double bonds, the activation of the double bond is acid induced (Scheme 1). Consequently, water is protonated and loses any nucleophilic character, the opposite of activation. Indeed, under these conditions almost any other nucleophile will add to the activated double bond, outcompeting water even if it is the solvent and thus present in excess. This is also shown in a few published procedures for the addition of water to double bonds; the presence of other nucleophiles is carefully avoided.11
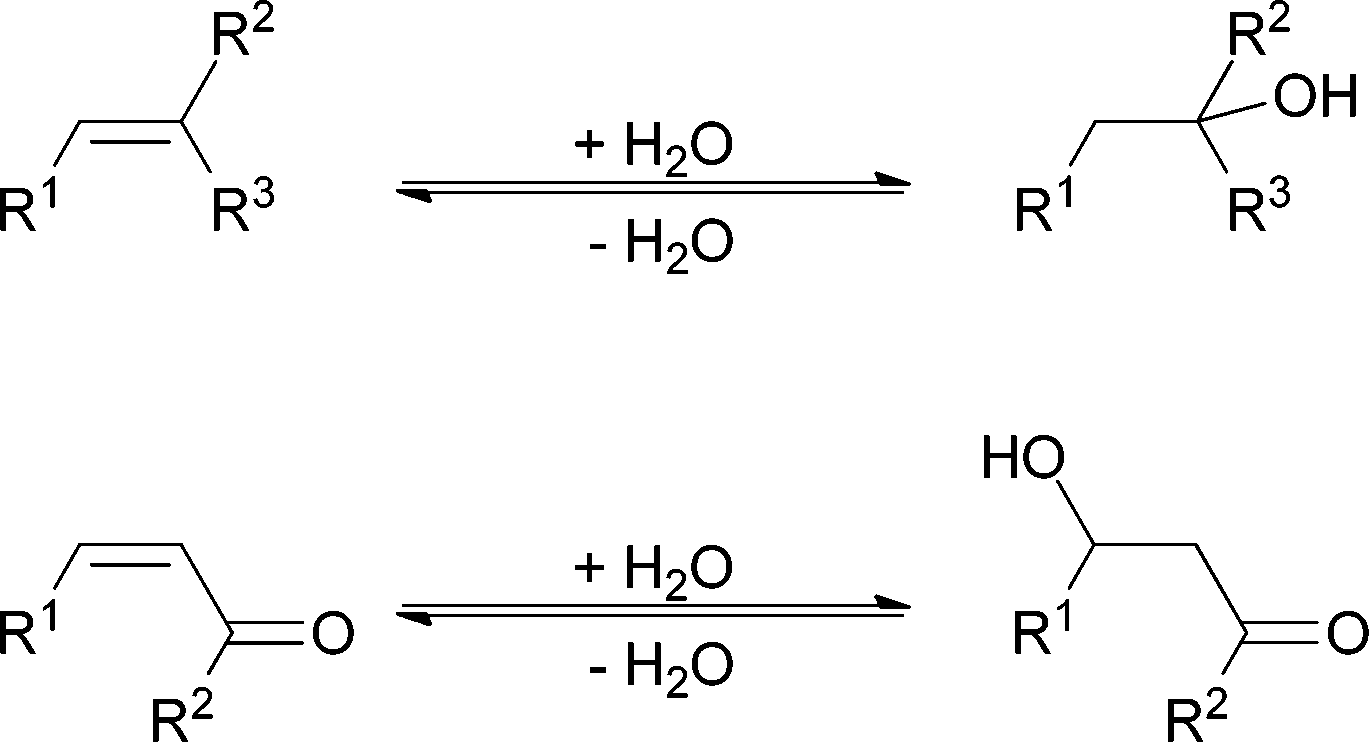 | ||
| Scheme 1 Water addition to isolated CC bonds, i.e. an electrophilic addition, follows Markovnikov's rule. Nucleophilic addition to a polarised double bond, a Michael addition, is observed for α,β-unsaturated carbonyl compounds. | ||
Nucleophilic addition to carbon–carbon double bonds proceeds more readily in polarised, electron-deficient double bonds. These are the conditions of the Michael reaction (Scheme 1). But even here water addition is the exception and many successful Michael reactions with water as inert solvent have been described as part of efforts to make the procedure more environmentally benign.12–14
The use of water as a benign and unreactive solvent has a second reason besides its poor nucleophilicity. In many addition reactions to carbon–carbon double bonds the equilibrium of the water addition reaction is unfavourable, impeding it (see also Table 2). Both for electron-rich and isolated double bonds and for conjugated, electron-poor double bonds the equilibrium can be on the side of the starting material, even if the reaction is performed in water. Thus poor nucleophilicity slows down a reaction with an unfavourable equilibrium, so that it is often not even noticed and other nucleophiles can be used in water. Here we would like to discuss how to speed up water addition and even make it selective.
2. Chemical catalysts
The textbook addition of water to carbon–carbon double bonds displays a very poor selectivity.2–4 This is due to the fact that both addition to electron-rich and addition to polarised double bonds (Scheme 1) is normally performed under acid catalysis. This does, however, induce a vast range of undesired side reactions such as isomerisations, polymerisations and rearrangements.2.1. Acid-catalysed addition of water to electron-rich C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C bonds
C bonds
The large-scale synthesis of “simple” alcohols is based on small alkenes normally derived from fossil fuels. One of the first bulk petrochemical processes from the 1920s was the hydration of propene.15,16 This is, however, an indirect process, in which propene is first treated with 60% sulfuric acid. In the second step the formed sulfate is steam-treated in a stripper, and subsequently, isopropanol is removed at the top while the acid is collected at the bottom and recycled (Fig. 1). The process has to be run carefully to avoid high temperatures that would cause ether formation. This indirect hydration process is actually a prime example of how difficult it is to add water to an electron-rich CC bond. The process with concentrated sulfuric acid is not commonly used anymore.
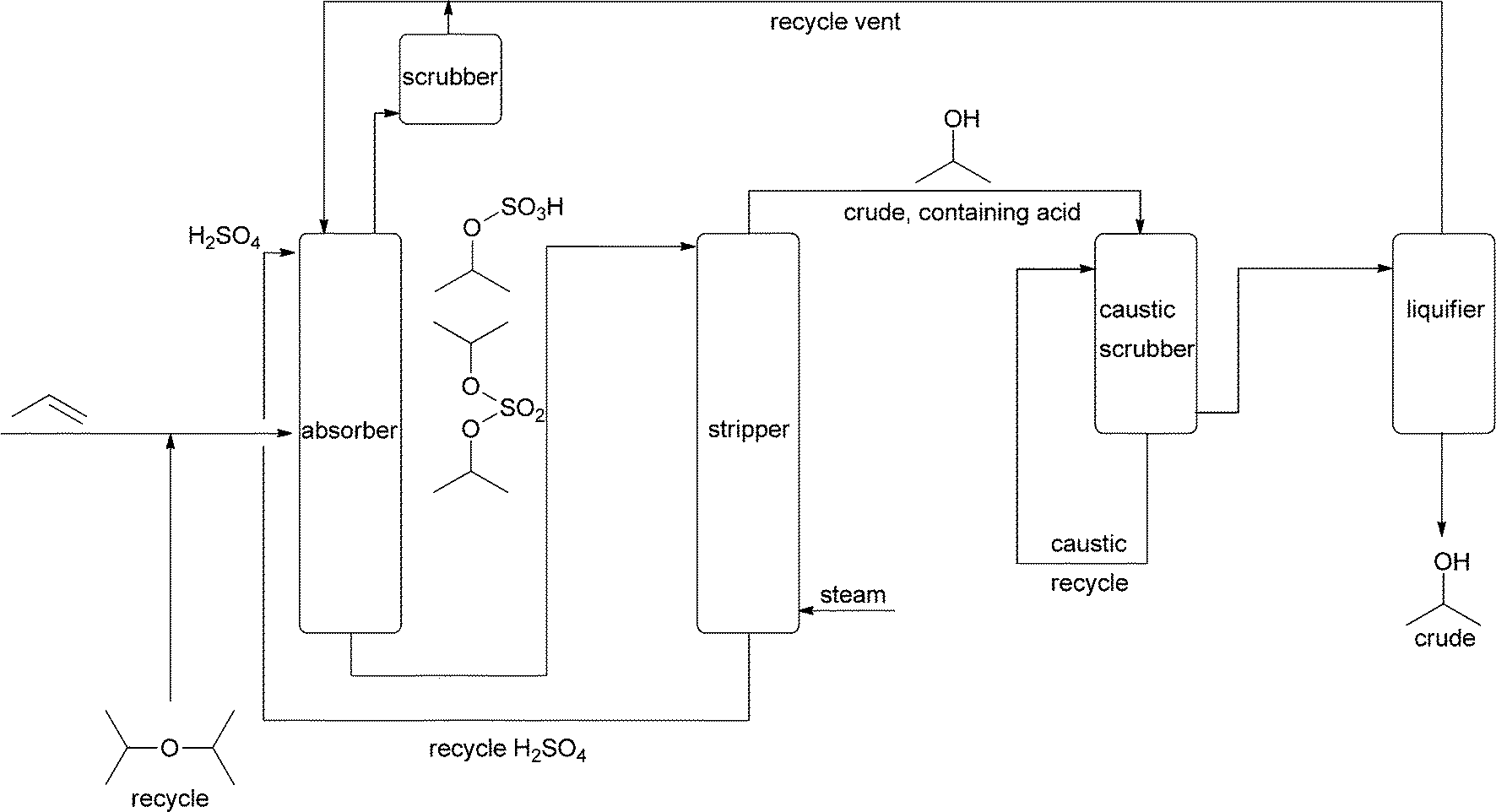 | ||
| Fig. 1 Indirect hydration of propene. This process demonstrates many of the problems that water addition reactions have - low reactivity (therefore sulfuric acid reacts first, leading to multiple steps) and side product formation (in particular ether). | ||
In addition to the indirect process, a direct process was developed that utilised heterogeneous acids. This came on stream commercially in 1951.16 Processes based on vapour-liquid phase reactions with either sulfonated polystyrene ion-exchangers or tungsten oxide catalysts utilise high pressure. This calls for expensive equipment but has the advantage that the reaction equilibrium is forced towards the product side, as two molecules combine to form one product. With both catalysts, high conversions per pass through the reactor were achieved (>60%). Vapour-phase hydration at high pressure also utilising tungsten oxide, now immobilised on silica, yields even 95%. When low pressure and phosphoric acid on silica are used, conversions are only 10% per pass and large recycling streams have to be handled. Nonetheless, this has also been commercialised. Detailed investigations of the equilibrium and the kinetics under various reaction conditions are available.15
For ethanol, fermentation was replaced by indirect hydration via the sulfuric acid method in 1930, and in 1948 this started to be superseded by direct hydration.10 Although countless different acidic materials have been suggested as catalysts, phosphoric acid supported on celite (a natural silicate, the skeletons of diatoms), montmorillonite or similar carriers is used. The reaction is plagued by two side reactions, ether formation and polymerisation. At low temperatures ether synthesis becomes dominant, and at high pressures, polymerisation. Therefore an equimolar feed at 250–300 °C is used with 5–8 MPa. Under these conditions, the equilibrium is then on the ethylene side with conversions below 25%. This causes large recycle streams (Fig. 2), again demonstrating how difficult water addition reactions are.
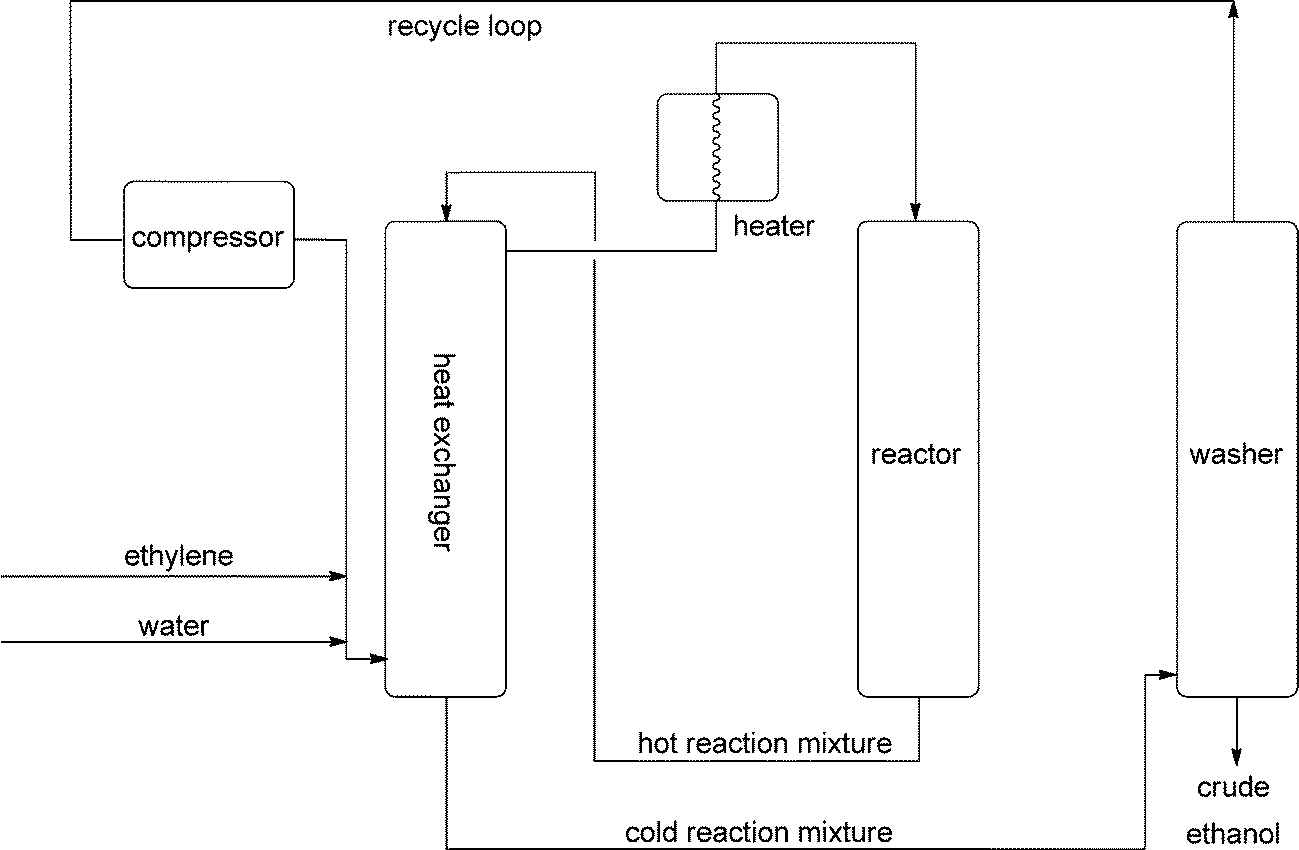 | ||
| Fig. 2 Direct hydration of ethene. This process demonstrates one of the problems that water addition reactions have - side product formation - here ether and polymer. Therefore the process cannot be run under optimum conditions for ethanol formation and a large recycle stream has to be taken care of. | ||
Water addition to isobutene is industrially less important since the production of tert-butanol is often coupled with propene oxide production, starting from isobutane.17 However, both the indirect hydration and the direct hydration of isobutene are employed.18,19 In particular, indirect hydration is utilised with the technical C4 feeds containing isobutene and n-butene. Again detailed studies on the kinetics and the equilibrium have been performed, revealing a situation less favourable than that in the propene case.20
While “simple” alcohols can be produced from an alkene that cannot isomerise and where Markovnikov's rule ensures that only one regioisomer can be formed, problems become even more pronounced when terpenes are the starting materials.21 Only two terpenes have been investigated more thoroughly for selective water addition reactions. Dihydromyrcene can be converted into dihydromyrcenol with relatively high selectivity due to the difference in the stability of intermediate carbenium ions (Scheme 2). Biphasic systems with zeolites as catalysts as well as triflic acid in ionic liquids have been studied for this.5,22,23 Similar studies for the structurally more demanding α-pinene gave product mixtures. The alcohols dominated but no single alcohol could be obtained as the only product.24–27
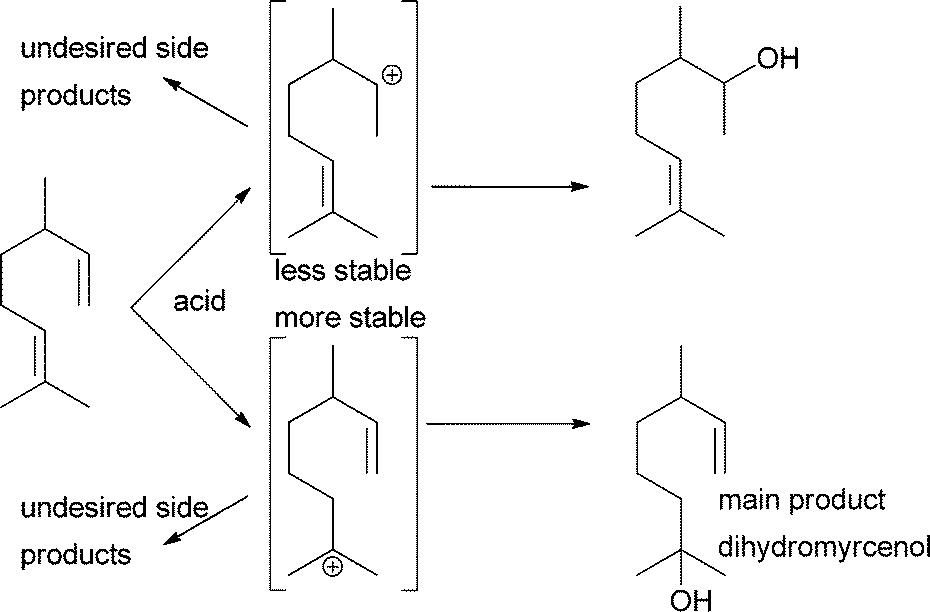 | ||
| Scheme 2 Dihydromyrcene can be converted into dihydromyrcenol with good selectivity. | ||
2.2. Chemocatalytic addition of water to electron-deficient CC bonds
The Michael addition of water can be either acid or base catalysed, activating either the α,β-unsaturated carbonyl compound or the nucleophile, water. However, it needs to be emphasised that almost every other nucleophile reacts more readily in Michael additions, making water addition an exception. One recent example for a base-catalysed addition of water was the use of amines to catalyse the addition of water to α,β-unsaturated carbonyl compounds.6 In this study, proteinogenic α-amino acids were tested as catalysts to convert, for example, cyclohex-2-enone to 3-hydroxycyclohexanone. The best results were obtained using L-lysine as the catalyst. However, this reaction is also limited by its equilibrium, which allows a maximum conversion of approx. 25%. Nevertheless, the conditions – in comparison to some of the examples mentioned above – are very mild, and α-amino acids are non-toxic and sustainable catalysts. No stereo-induction was observed, although chiral α-amino acids were used as catalysts. Other recent approaches based on mimicking hydratases will be discussed in section 3.3.
The only process that was run on an industrial scale is the addition of water to acrolein,28 yielding 3-hydroxypropanal. Starting with approx. 20% acrolein in water, it was possible to reach greater than 50% conversion with a selectivity of greater than 80% for the product. Addition of acids to the acidic ion exchange catalyst (pH ~4) suppressed the polymerisation of acrolein that commonly decreased catalyst activity.29,30 3-Hydroxypropanal was exclusively used to produce 1,3-propanediol (Scheme 3).31 However, this process has been replaced with a biological route to this diol. Today it is produced via fermentation, starting either from glycerol or from sugar. A key step in the bio-process is a dehydratase-catalysed elimination of water from glycerol.32–35
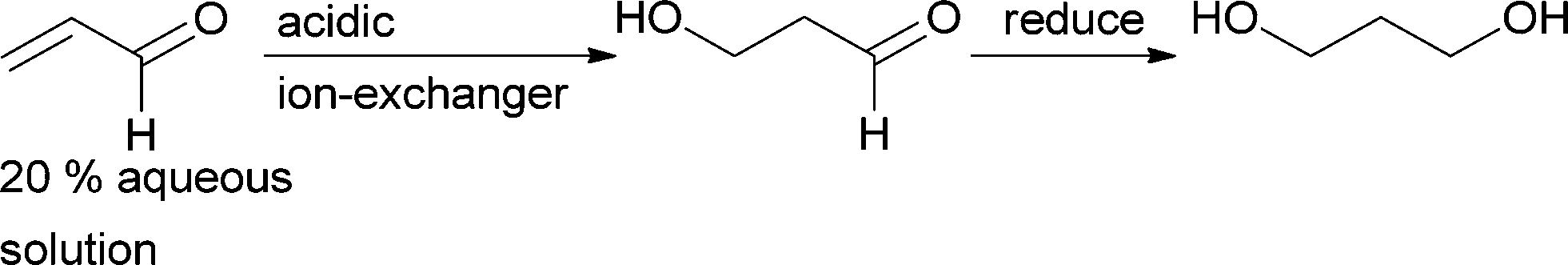 | ||
| Scheme 3 Michael addition of water to acrolein. 3-Hydroxypropanal is then reduced to 1,3-propanediol. | ||
3. Enzymes as catalysts
In contrast to chemical catalysts, nature is well capable of providing the right activation to use water as a nucleophile. Enzymes are able to use water as a substrate and also provide the right environment for asymmetric transformations allowing the synthesis of enantiomerically pure alcohols.36 Given the small size of water, asymmetric addition is even more remarkable. The active-site geometry and the potential cofactors involved in enzyme-catalysed water addition reactions are essential for the reaction to proceed. Furthermore, their ability to bind both the nucleophile and the electrophile – which leads to the stabilisation of the transition state – enhances the reaction. Basically an intramolecular reaction takes place.37,38Enzymes that catalyse the addition of water to carbon–carbon double bonds are called hydratases or hydro-lyases (E.C. 4.2.1.-). Living organisms harbour a vast variety of hydratases, which are involved in primary metabolism such as the citric acid cycle. Apart from enzymes of primary metabolism, some are also employed in the energy storage and release system of living organisms, where they are, for example, in charge of degrading fatty acids. The hydratases involved in metabolic pathways display high selectivities. In primary metabolism perfect selectivities are indispensable to life. However, from a chemist's point of view, enantioselectivity is highly desired, but in contrast, substrate acceptance should be as broad as possible. This is unfortunately not always the case for hydratases. Even though this limits their application, there are several examples that prove their potential, and the application of hydratases on the industrial scale is well established.39
3.1. Mechanistic aspects of enzymatic water addition
As for the chemical catalysts, the addition of water catalysed by enzymes can be grouped into two different types depending on the substrate. The double bond can be either an isolated (electron-rich) double bond or conjugated to a carbonyl functionality (polarised and electron-poor), representing a Michael-type addition (Scheme 1).40Hydratases have different features that allow the activation of water. In some cases, activation is performed with the help of a metal ion, which is located in the active site; in others the reaction is catalysed without cofactors. This of course also leads to a different mechanism. Aconitase (an iron–sulfur cluster containing enzyme) and fumarase C (which requires no cofactor) serve as two distinguished examples. Both enzymes catalyse the addition of water to similar substrates (see Scheme 4).
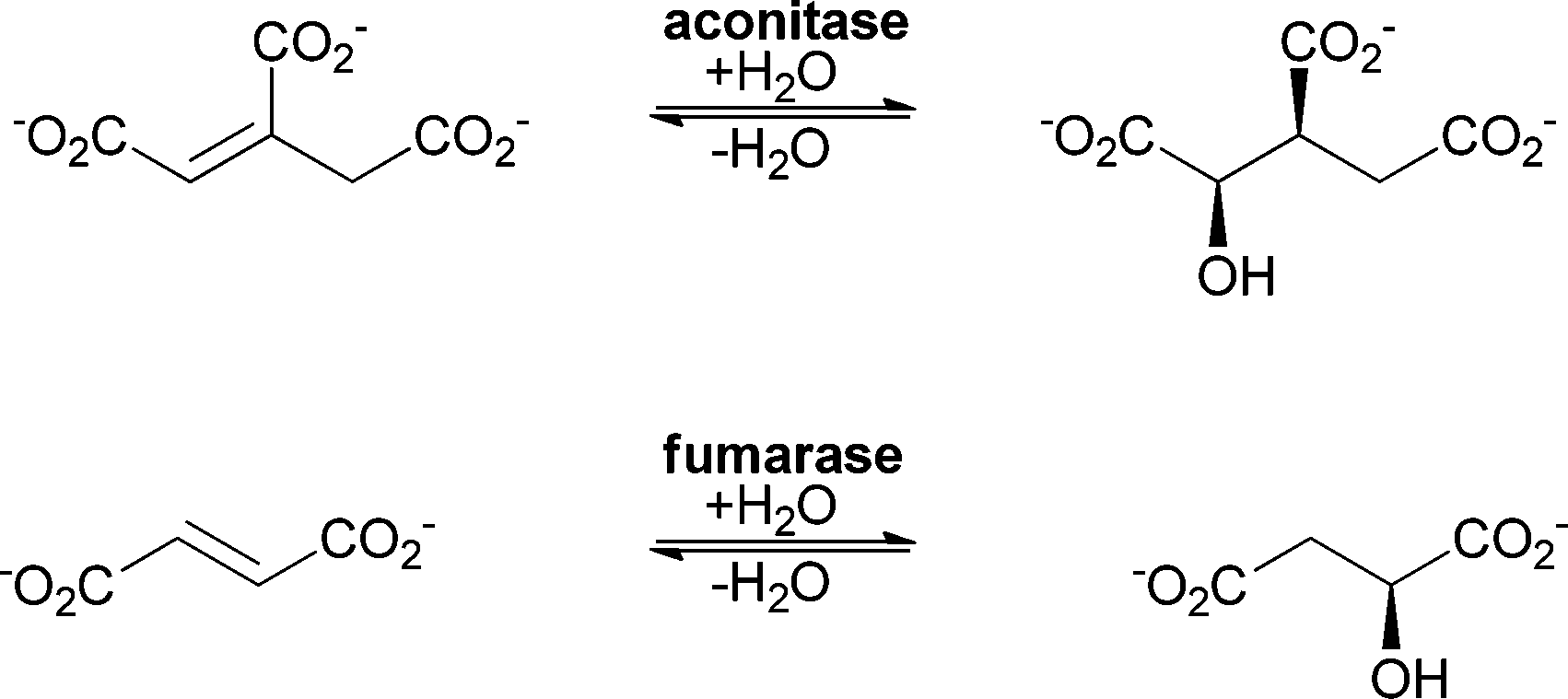 | ||
| Scheme 4 Aconitase and fumarase catalyse the hydration of similar substrates but the cofactor chemistry and the mechanistic aspects differ. | ||
Class I fumarases (fum A and B) harbour an iron–sulfur cluster that is involved in catalysis. A second class of fumarases (class II, fum C) performs the same reaction without the help of any cofactor. The iron–sulfur cluster acts as a Lewis acid and is involved in the activation of the water molecule.41 Aconitase is also an iron–sulfur cluster containing enzyme. Since detailed mechanistic studies on aconitase exist,42 we will take aconitase as an example to illustrate the different activation mechanisms of water in comparison with fumarase C, a cofactor-independent fumarase.
The iron–sulfur cluster of aconitase consists of four iron and four sulfur atoms [4Fe–4S] forming a cube-like structure that is bound to the protein backbone by three cysteine residues (see Fig. 3). This allows one iron to remain without a binding partner and it can therefore act as a Lewis acid. It contributes to the reaction in two ways: first it helps to orient the substrate in the active site by forming a coordination bond to the hydroxyl group of the carboxylate, and second it binds water which serves as the second substrate. The binding of both substrates allows a close interaction and an intramolecular reaction is possible. The formed product – which is isocitrate in the case of aconitase – is released from the active site and the iron–sulfur cluster can be employed in another catalytic cycle (see Scheme 5).
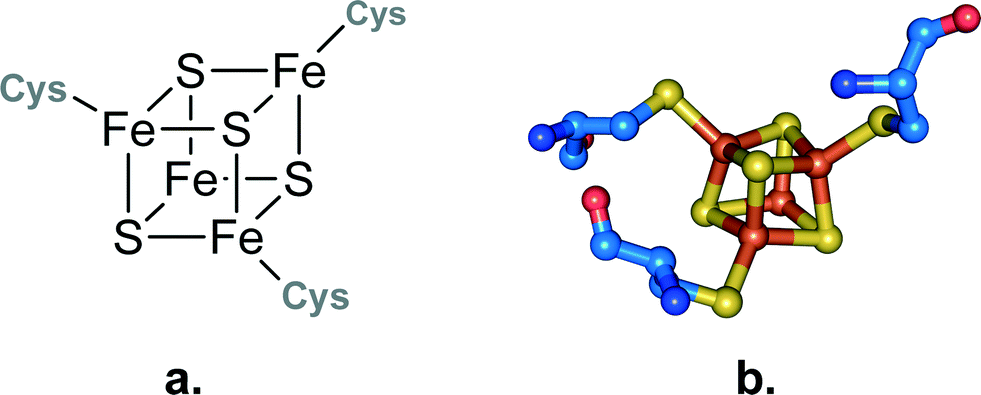 | ||
| Fig. 3 a. Schematic representation of an iron–sulphur cluster with three irons bound to cysteine residues. b. Iron–sulphur cluster as found in the crystal structure of aconitase from Bos taurus.42 | ||
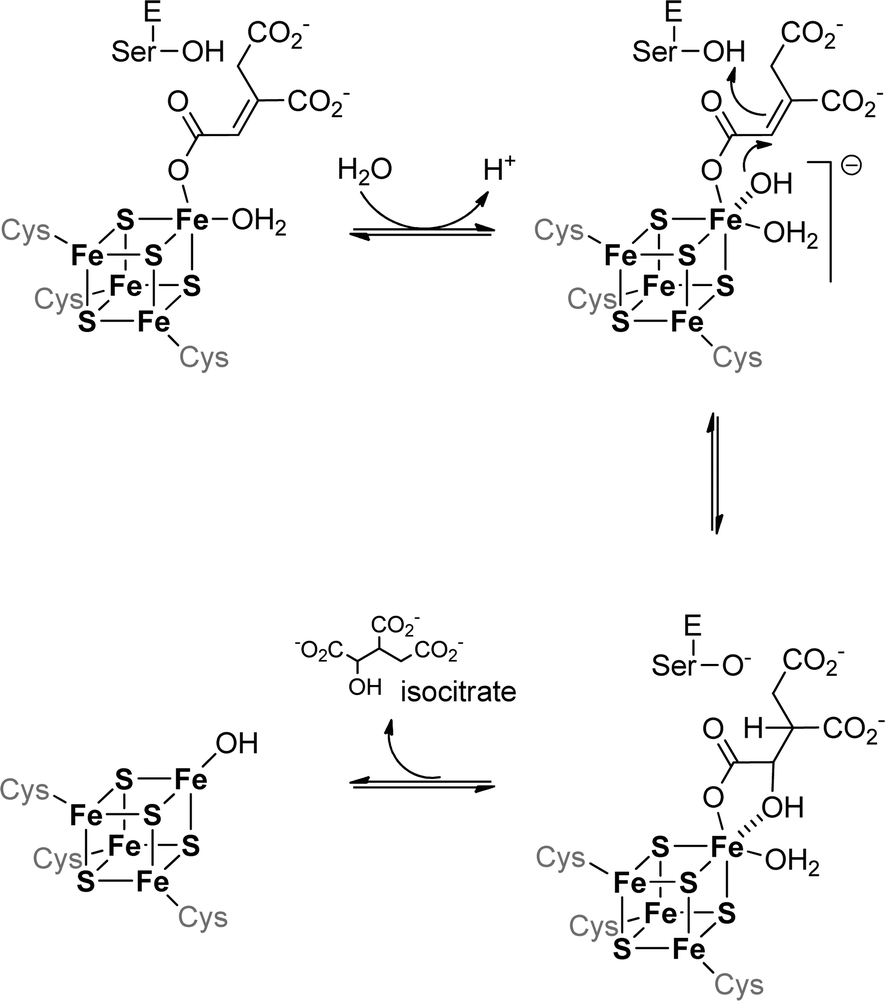 | ||
| Scheme 5 Hydration part of the catalytic cycle of the iron–sulfur cluster containing aconitase. | ||
In the case of metal-independent fumarase, the mechanism for water addition differs strongly due to the absence of a cofactor. Here proton transfer is performed by two acid–base α-amino acid residues. The mechanism involves two states that are defined by the ionisation state of these two residues. They can be in the form of either a protonated acid and a deprotonated base (state E1) or a deprotonated acid and a protonated base (state E2). In the E1 state, the enzyme is able to bind fumarate as the substrate and water addition to malate occurs. For the dehydration of malate, the enzyme needs to be in the E2 state. In general, the basic residue is involved in the deprotonation of the water molecule that is added to the double bond. The primary function of the acidic residue is to donate a proton to the substrate (see Scheme 6).43 The two very different catalytic mechanisms for aconitase and fumarase show how diverse the mechanisms of activation for both water and substrate can be.
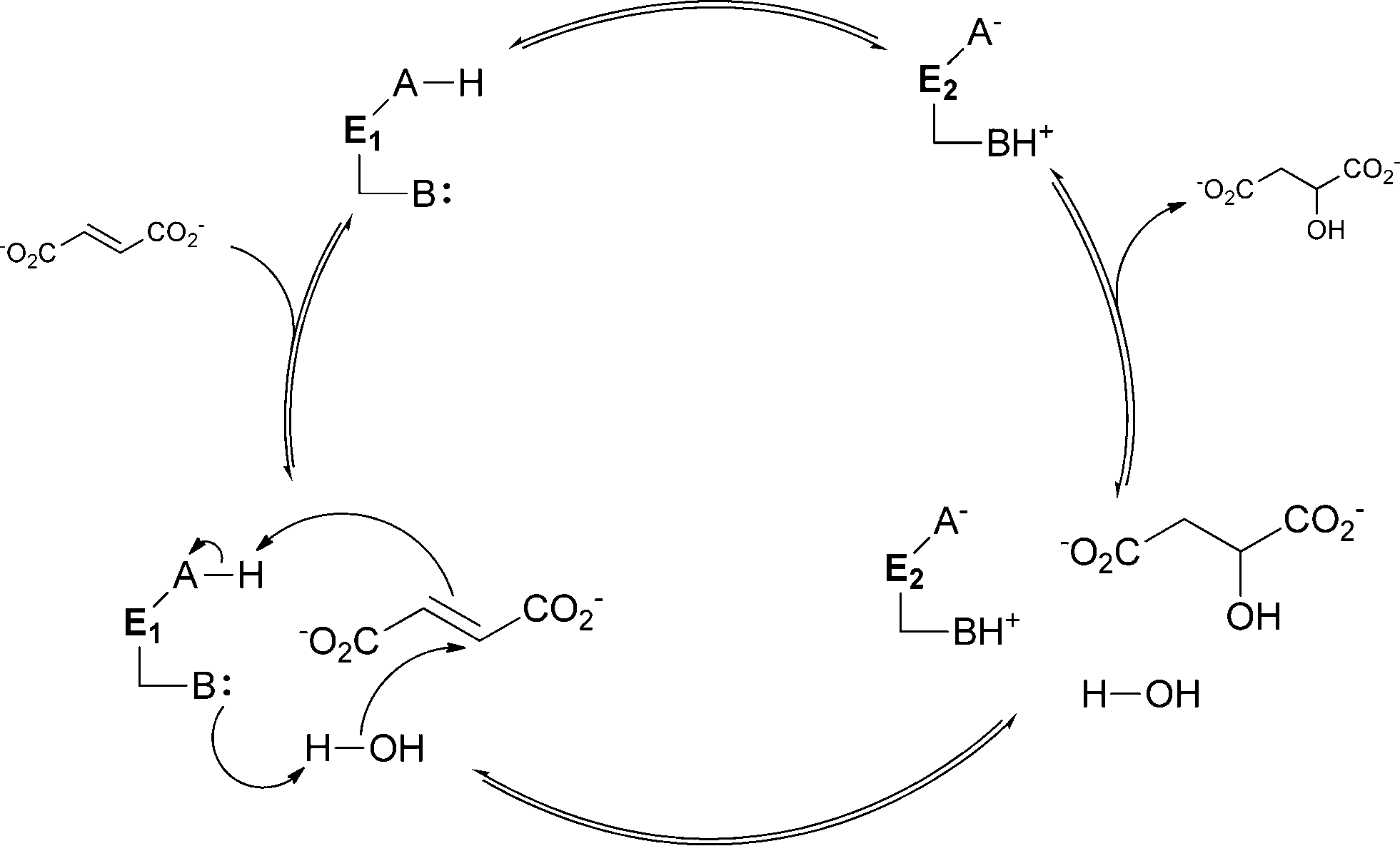 | ||
| Scheme 6 Catalytic mechanism of metal-independent fumarase. The two acid/base residues (A and B) are essential. Depending on their ionisation state, either the E1 or the E2 form is present. | ||
Most hydration reactions are equilibrium reactions and hydratases are also able to perform both addition and elimination of water. Depending on the substrate, the equilibrium can either lie on the substrate or product side (for equilibrium yields of different industrially employed hydratases see Table 2).
In general, both addition and elimination of water can occur either in syn or in anti fashion (see Scheme 7). Depending on their mechanism, chemically (acid/base) catalysed addition and elimination reactions can show selectivity towards the anti-product or no selectivity is observed. In the case of an E2 mechanism (concerted) anti-stereoselectivity is again observed. If the reaction proceeds via an E1 mechanism (stepwise), no selectivity is observed. In contrast to chemical methods, enzymes are also able to perform syn-addition/elimination. Depending on the enzyme, biocatalytic hydration and elimination reactions can show either syn or anti preference.44
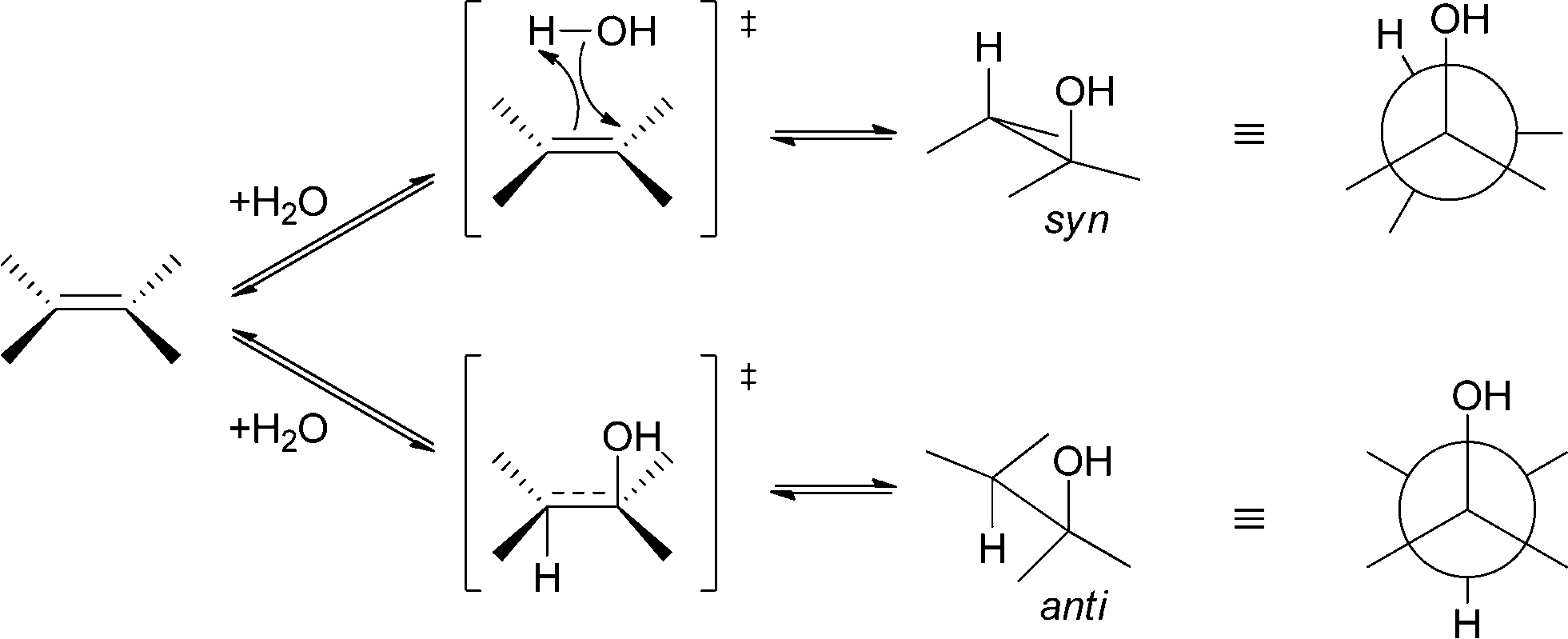 | ||
| Scheme 7 Depending on the enzyme, either syn- or anti-addition and elimination reactions are observed. | ||
Studies showed that the preference depends, for example, on the position of the abstracted proton. If the proton is in the α-position to the carboxylate group, anti-selectivity is observed. Abstracted protons that are in the α-position to the carbonyl group of a thioester lead to syn-selectivity.41 Enzymes that catalyse anti-addition/elimination belong to the aspartase/fumarase superfamily, for example, fumarase, aconitase and enolase. Enzymes belonging to the enoyl-CoA hydratase superfamily such as enoyl-CoA hydratase can catalyse syn-addition/elimination (see Scheme 8).45
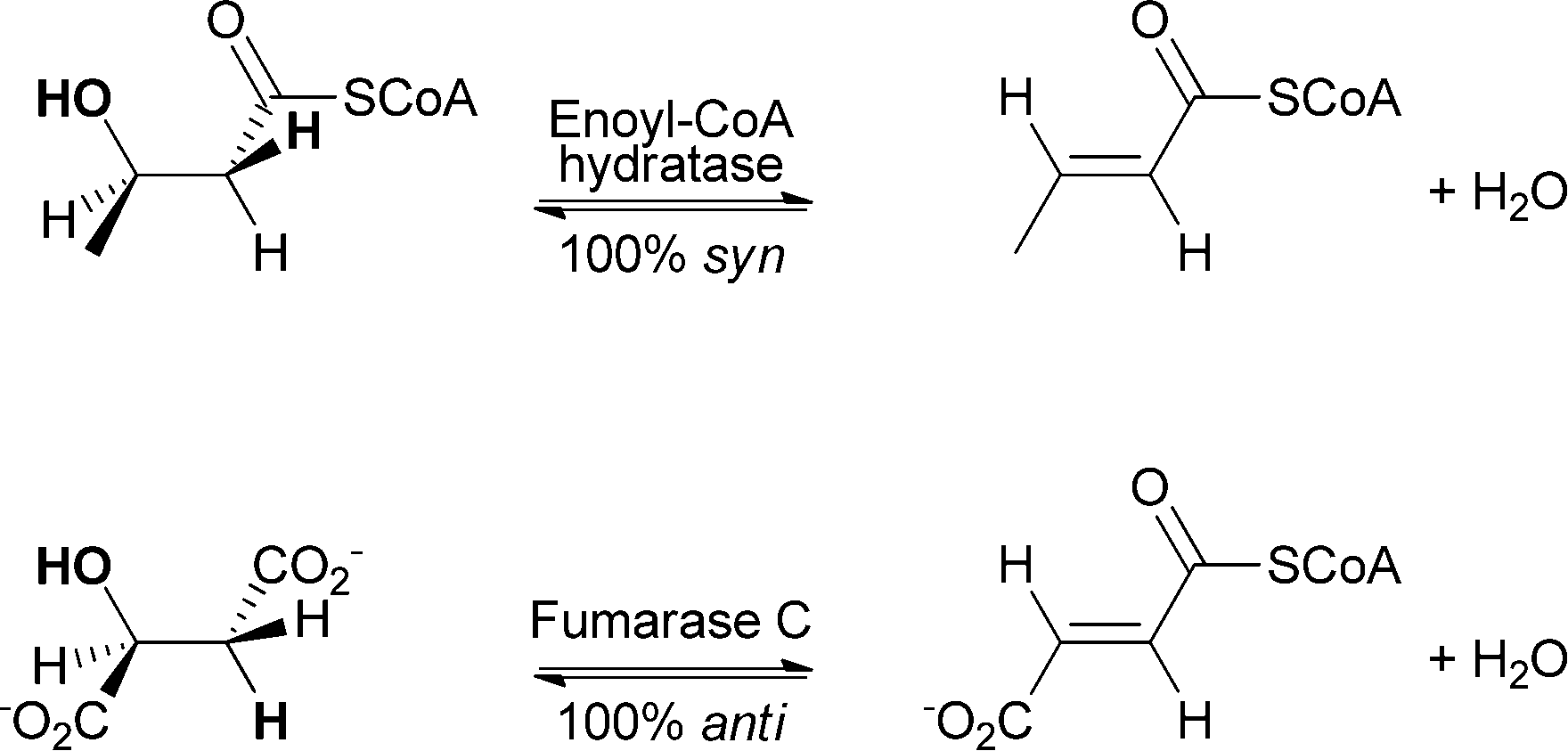 | ||
| Scheme 8 Enoyl-CoA hydratase and fumarase C catalyse the addition and elimination of water with different stereo-preferences. In the case of enoyl-CoA, hydratase selectivity towards syn-addition and elimination is observed, while fumarase C shows anti-preference. | ||
Theories to explain the difference in selectivity suggest taking the nature of the substrate and the structural features of the enzyme into account. It is assumed that the acidity of the proton attached to the α-carbon plays a crucial role.46,47 In terms of enzyme structure, two important features were recognised as important. For enzymes belonging to the enoyl-CoA hydratase superfamily performing exclusively the syn-addition and elimination of water a conserved oxyanion hole involved in the stabilisation of the enolate anion is key. It is assumed that the reaction follows an E1cB-elimination mechanism, where first the elimination of a proton takes place, leading to the formation of an enolate. In the final step, the hydroxide serves as a leaving group.44 If the addition or elimination is taking place in an anti fashion – as is the case with enzymes from the aspartase/fumarase superfamily – the reaction follows a concerted E2 mechanism. Enzymes from this group share active-site residues that allow the stabilisation of aci-carboxylate intermediates (Scheme 9).44,48
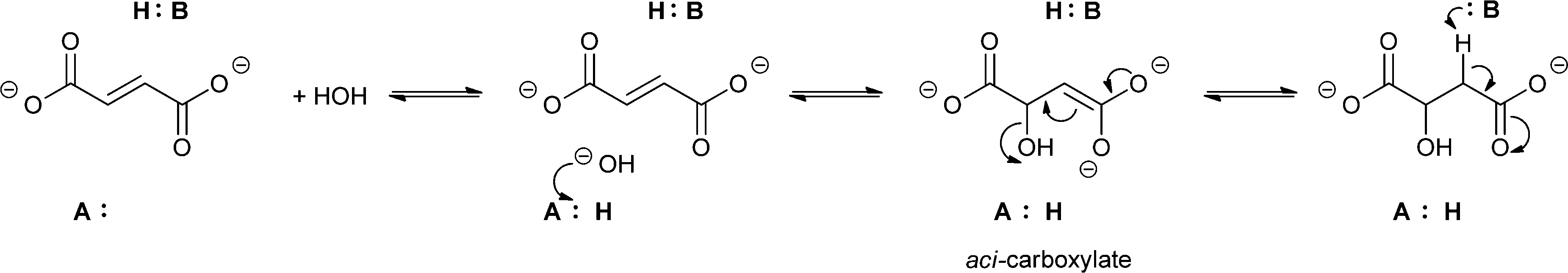 | ||
| Scheme 9 Mechanism of the acid–base catalysed anti-addition/elimination reaction of enzymes from the aspartase/fumarase superfamily involving aci-carboxylate as an intermediate. | ||
While syn/anti-selectivity differs with the type of enzyme used for catalysis, the chemically catalysed reaction shows only preference towards anti-addition. Therefore, in chemically acid-catalysed reactions regioselectivity but not stereoselectivity can be observed. In contrast to these findings, both chemical and biocatalytic methods for the addition of water to electron-rich double bonds follow Markovnikov's rule.
3.2. Enzyme-catalysed addition of water to electron-rich CC bonds
The addition of water to electron-rich double bonds is catalysed by a variety of different hydratases such as oleate hydratase, carotenoid hydratases, linalool dehydratase-isomerase, kievitone hydratase, phaseollidin hydratase, limonene hydratase and acetylene hydratase, to name just a few.1 The focus of this review lies on hydratases that are employed either in industrial processes or on the laboratory scale; therefore we would like to focus on oleate hydratase and limonene hydratase in particular.
In general, many hydratases from different organisms are described. However, in most cases detailed characterisation is still missing. Oleate hydratases are nonetheless successfully applied in larger-scale biotransformations. One example is an oleate hydratase from a bacterial strain used in the production of γ-dodecalactone, which is known as an essential flavour compound in whiskey. In this process, (R)-10-hydroxystearic acid is produced in a fermentative approach and is then further converted to γ-dodecalactone by baker's yeast, giving an ee of 87% (see Scheme 10).56,57
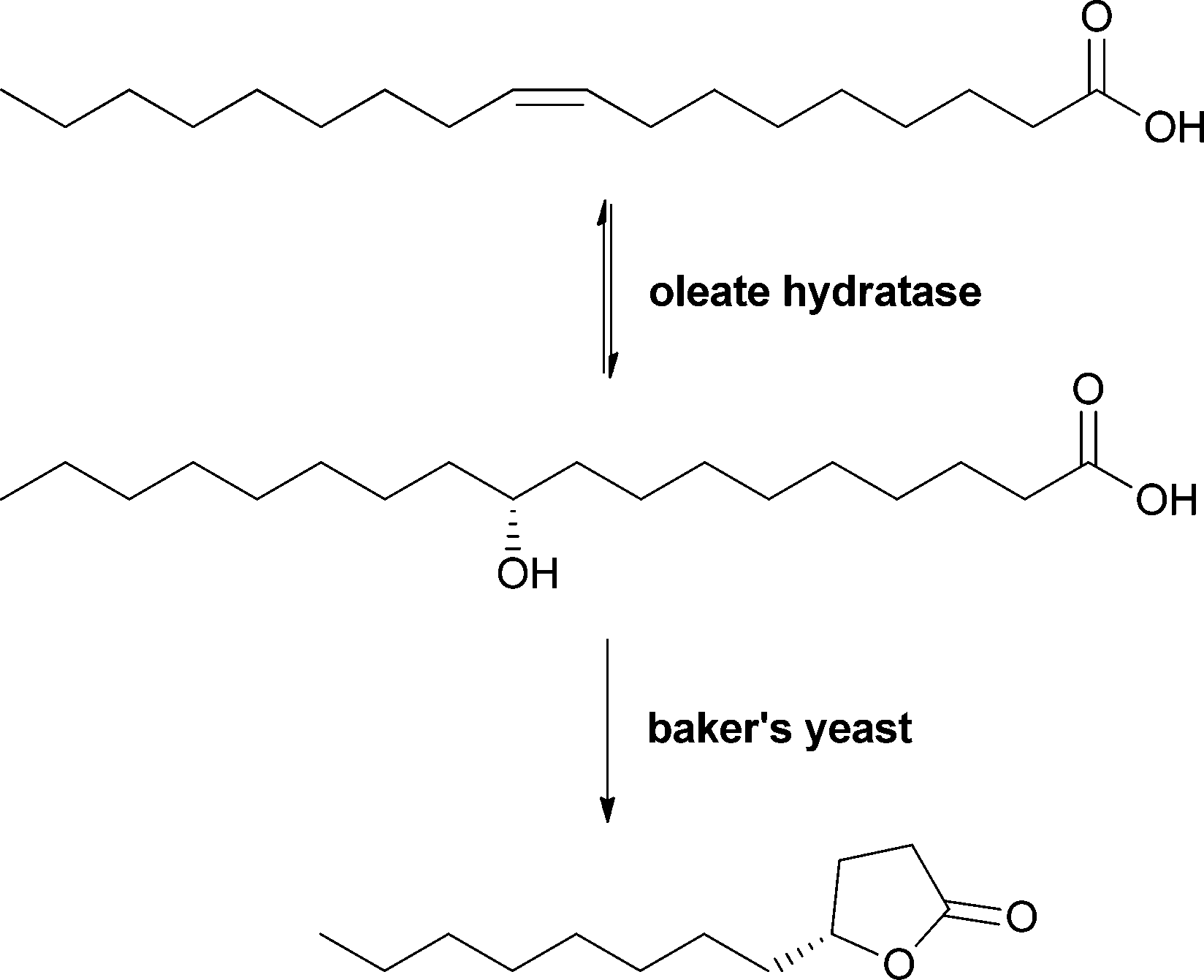 | ||
| Scheme 10 Oleate hydratase in combination with baker's yeast for the enantioselective synthesis of the whiskey flavour compound γ-dodecalactone. | ||
Oleate hydratase from different sources is also employed for the large-scale production of 10-hydroxystearic acid starting from oleic acid (Table 1). For example an oleate hydratase from Stenotrophomonas nitritireducens was employed in this bioprocess and a productivity of 7.9 g L−1 h−1 of 10-hydroxystearic acid was achieved.58 In another process the use of oleate hydratase from Stenotrophomonas maltophilia (overexpressed in E. coli) is reported, producing 10-hydroxystearic acid in 98% yield (w/w) which corresponds to a volumetric productivity of 12.3 g L−1 h−1 or 49 g L−1 after 4 h.59 A third bioprocess again uses heterologously expressed oleate hydratase from S. maltophilia. Using a whole-cell approach, a productivity of 8.2 g L−1 h−1 (46 g L−1) of 10-hydroxystearic acid was reached. Furthermore, the isolation of 10-hydroxystearic acid gave rise to a yield of 70.9% and after recrystallisation a purity of 99.7% was achieved.60
Oleate hydratase showed also great potential as an enzyme embedded in cascade reactions. A multistep enzyme-catalysed reaction sequence shows the combination of oleate hydratase, alcohol dehydrogenase, two different Baeyer–Villiger monooxygenases and an esterase. The enzymes were combined for the synthesis of long-chain α,ω-dicarboxylic and ω-hydroxycarboxylic acids using renewable fatty acids and plant oils.61 The reaction started with oleic acid, which was converted into either n-nonanoic acid and ω-hydroxynonanoic acid or n-octanol and 1,10-decanedioic acid. In the sequence, oleate hydratase catalysed the addition of water, which was followed by the oxidation of the hydroxyl group by alcohol dehydrogenase. Baeyer–Villiger monooxygenase-catalysed oxidation leads to the formation of an ester, which is further hydrolysed by the esterase (see Scheme 11). Very recently also the conversion of the hydroxyacid by an alcohol dehydrogenase into an aldehyde and subsequently transaminase catalysed into the amino acid has been described. Thus monomers for nylon production are accessible from unsaturated fatty acids.62
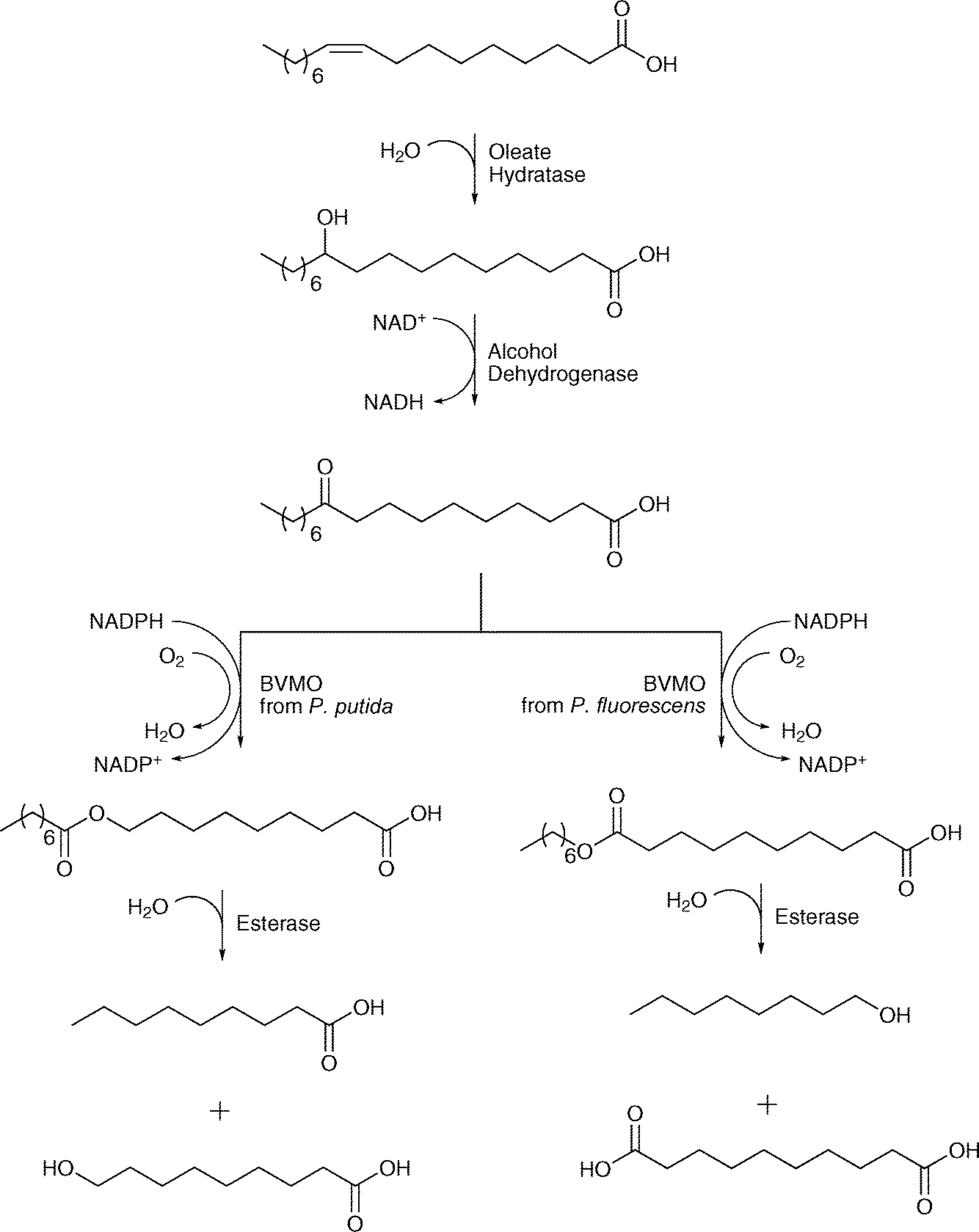 | ||
| Scheme 11 A multi-enzymatic cascade employing an oleate hydratase, an alcohol dehydrogenase, two different Baeyer–Villiger monooxygenases and an esterase producing α,ω-dicarboxylic and ω-hydroxycarboxylic acids. | ||
Especially during processing of citrus fruits, limonene is produced as a waste product and is a perfect precursor for a variety of different flavour and fragrance compounds such as menthol and carvone.63 (R)-(+)-α-Terpineol for example is a common fragrance in the perfume industry, since it is known for its strong lilac-like smell. In contrast, its (S)-enantiomer displays a strong conifer-like odour. Since the olfactory properties strongly depend on the enantiomer, the enantioselective addition of water to the double bond is highly important for the production of pure fragrance. All studies on the enantioselective addition of water showed that limonene hydratase is very specific and only (R)-(+)-limonene was converted to (R)-(+)-α-terpineol (see Scheme 12).
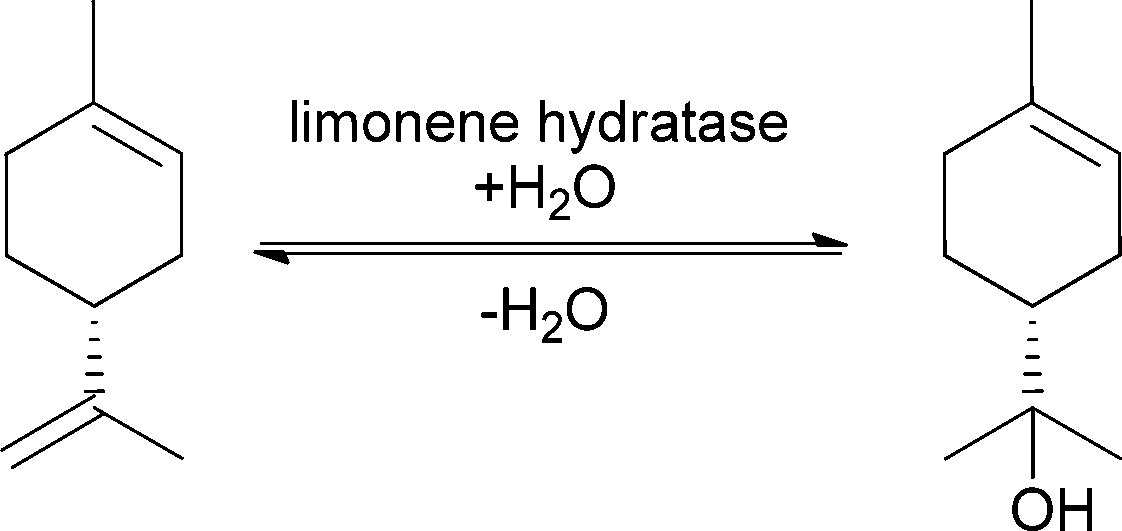 | ||
| Scheme 12 Addition of water to (R)-(+)-limonene catalysed by limonene hydratase to form (R)-(+)-α-terpineol. | ||
Limonene hydratases are often found not only in fungi (especially in those that grow on rotting citrus peel) such as Fusarium oxysporum 152B,64–66Pleurotus sapidus,67Aspergillus niger (ATCC 16404, ATCC 9642 and ATCC 1004 strains)68 and Penicillium spp.69–71 but also in bacterial sources such as Pseudomonas gladioli63,72Escherichia coli,73 and Sphingobium spp.74 are known to harbour limonene hydratases. Because (R)-(+)-limonene is a cheap starting material, the chemical industry has also turned its interest to limonene hydratase. Several small-scale processes are running, producing 0.1 to 15.5 g of (R)-(+)-α-terpineol per litre of fermentation medium.65,68,71,75,76 The most profitable process uses resting cells of Sphingobium sp., allowing the production of 130 g of (R)-(+)-α-terpineol per litre of medium within 96 h.74
3.3. Enzyme-catalysed addition of water to electron-deficient CC bonds
The addition of water to electron-deficient double bonds opens up a completely different class of substrates. Many enzymes that can perform this addition are known and well investigated. The most famous one is fumarase, but also malease, citraconase, aconitase, urocanase, enzymes with hydratase-tautomerase bi-functionality, enoyl-CoA hydratase, carnitine dehydratase, hydroxycinnamoyl-CoA hydratase lyase, Michael hydratase,77 phenolic acid decarboxylases78 and an artificial hydratase are described.1,79
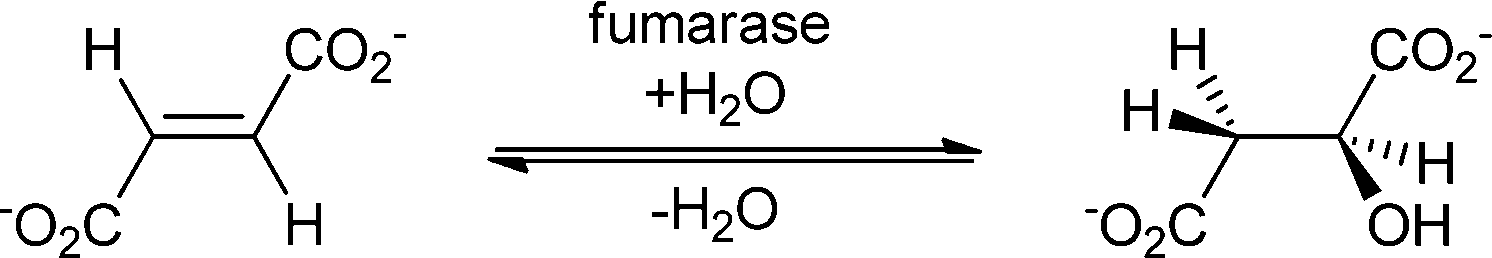 | ||
| Scheme 13 Fumarase catalyses the addition of water to fumarate, forming (S)-malate. | ||
Due to their important role in primary metabolism, fumarases are ubiquitous in nature. Three different types of fumarases (fum A, fum B and fum C) are found in E. coli and are categorised in two different classes. Class I fumarases are dependent on Fe2+ and sensitive to heat. E. coli fumarase A and B are grouped in this class.81,82 In contrast, class II fumarases – fum C belongs to this group – are independent of Fe2+; these enzymes are also not sensitive towards elevated temperatures and maintain activity at 50 °C.83–86 Investigations on the structure of fum C also allowed detailed insight into the mechanism behind the hydration reaction (see Scheme 6).43,87–89 Due to their high stability only class II fumarases are used for industrial applications.
Malate is a very important compound in the food industry. It is the second-most widely used acidulant holding approx. 10% of this market and can be an alternative to citric acid.90 It is furthermore a potential monomer for biodegradable polymers. In the traditional process for the production of (S)-malate, apple juice was used as a source for isolation of the compound. Since apple juice contains only 0.4–0.7% (S)-malate, this method was soon proven to be inefficient. Chemical hydration processes for the production of malate often require harsh conditions. For example, in the largest chemical process, maleic anhydride is converted to racemic malate by hydration. To hydrate maleic anhydride the reaction needs to be carried out at 180 °C and 1 MPa.91
Already in the late 1970s, fermentative processes using fumarase were invented to replace the traditional isolation method. Whole-cell approaches using immobilised cells of Brevibacterium ammoniagenes,92Brevibacterium flavum93,94 (representing two industrially established processes) or Saccharomyces cerevisiae90 to develop continuous production systems were investigated. Using Brevibacterium flavum cells immobilised on κ-carrageenan gel gave a conversion of 80%, which represents approximately the equilibrium conversion, and a production capacity of 468 t a−1.95,96 In 1984, an enzyme-membrane-reactor-based production system was started by the former Degussa company.97 This system allowed the recycling of the enzyme while still performing homogenous enzyme catalysis. The same system was applied for the production of not only (S)-malate but also natural α-amino acids.
Suspended whole cells from Corynebacterium glutamicum were used by AMINO GmbH in 1988 in a process that allowed the production of approx. 2000 t a−1.95,98,99 The main limitation in water addition to fumarate is the equilibrium that governs the reaction. Even though the product side is favoured, full conversion cannot be achieved. To overcome this limitation, a precipitation strategy was used in some industrial processes. Calcium carbonate is used in an elegant way to precipitate both the fumarate and the malate and the reaction takes place in a slurry of the salts and the biocatalyst, where only approx. 1% of the calcium salt is in solution (see Fig. 4).95
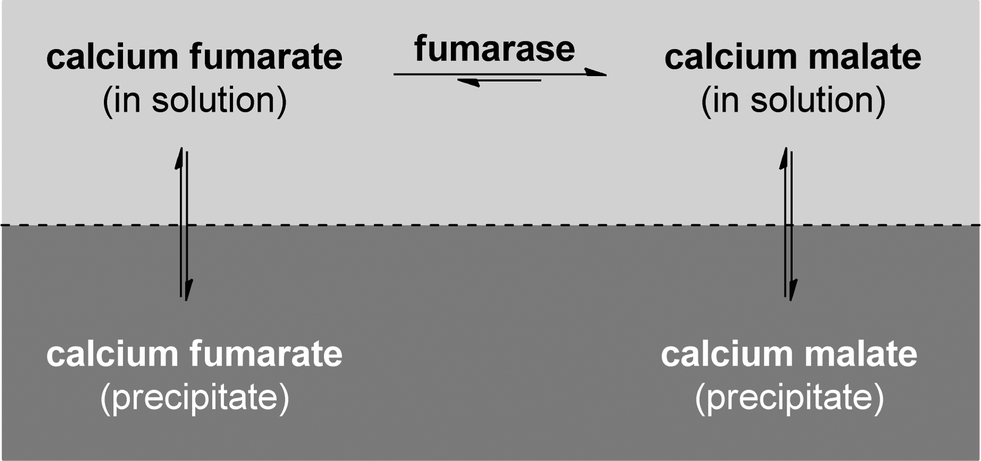 | ||
| Fig. 4 The calcium carbonate method allows shifting the equilibrium of the reaction by precipitating both fumarate and malate. The reaction mixture is a slurry of the salts and the biocatalyst. | ||
The industrial production of (S)-malate by, for example, AMINO GmbH comprises the following steps. The reaction mixture contains imidazole buffer, fumarase (whole cells of Corynebacterium glutamicum), fumarate and calcium carbonate to achieve the shift of equilibrium. The biocatalyst can be removed from the reaction mixture by a simple filtration step. The calcium salt is removed by the addition of sulphuric acid leading to the formation of insoluble calcium sulfate – removed by filtration – and the soluble free (S)-malate. After evaporation of the solvent, (S)-malate is purified by crystallisation and isolated after a final centrifugation step (see Fig. 5).
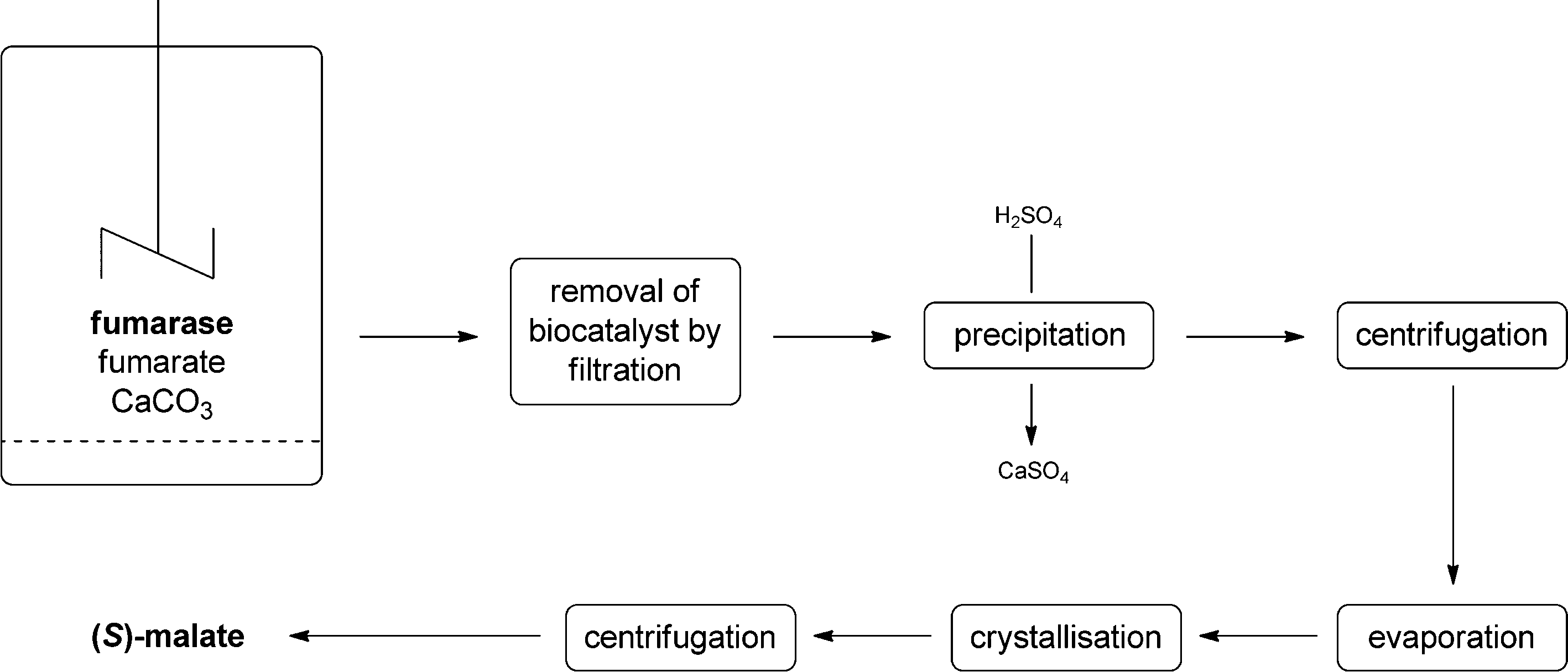 | ||
| Fig. 5 Flow scheme of the industrial process for the production of (S)-malate using whole cells of Corynebacterium glutamicum. | ||
As an enzyme that is involved in primary metabolism, fumarase shows strict substrate specificity. What is desired and highly important for living organisms is a drawback when it comes to biocatalysis, where a broader substrate spectrum is appreciated. Experiments to elucidate the substrate scope were already carried out in 1968 using fumarase from pig heart. The enzyme showed activity towards the following substrates: fluorofumarate, fumarate, chlorofumarate, bromofumarate, acetylenedicarboxylate, iodofumarate and mesaconate (in decreasing order of activity). For nearly all tested substrates, water addition took place in a trans fashion. The cis product was found for α-fluoromalate, where spontaneous decomposition to oxaloacetate was also observed.100 Later tests on substrate specificity showed that chloro-, fluoro- and difluoro-fumarate are also accepted as substrates. Chlorofumarate, for example, was converted to L-threo-chloromalate, which was chemically transformed further to trans-D-erythro-sphingosine and 2-deoxy-D-ribose (see Scheme 14).101
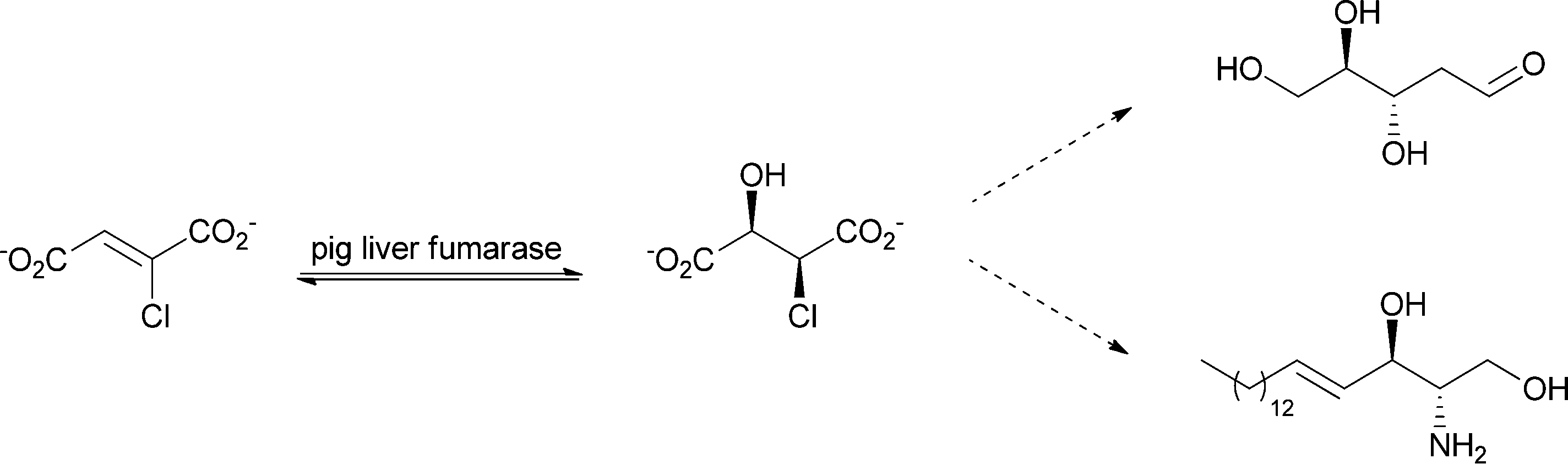 | ||
| Scheme 14 Pig liver fumarase was used for the conversion of chlorofumarate to L-threo-chloromalate, which was further converted to trans-D-erythro-sphingosine and 2-deoxy-D-ribose. | ||
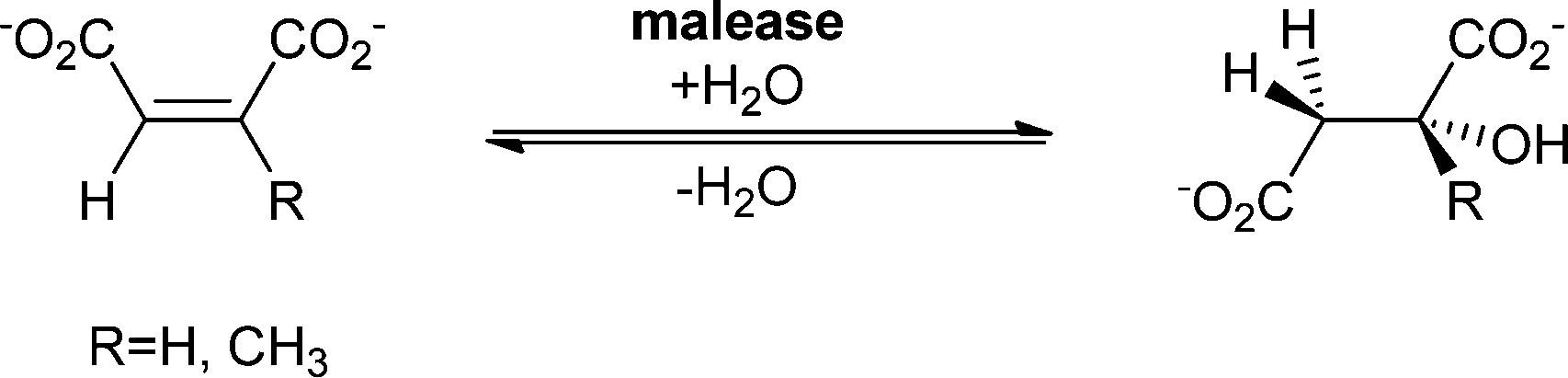 | ||
| Scheme 15 Malease catalyses the addition of water to maleic acid or citraconic acid. | ||
The substrate spectrum of malease is – as is the case for fumarase – rather narrow, and maleic acid and citraconic acid are accepted best, but small changes in the functional group pattern of these substrates are allowed.104–106 For example chloromaleate and bromomaleate are hydrated to give α-substituted malates (2S,3S)-3-chloromalate and (2S,3S)-3-bromomalate.107
In industry, Pseudomonas pseudoalcaligenes containing malease is used for the large-scale production of malic acid. Starting this production process from maleic anhydride, which undergoes spontaneous hydrolysis under aqueous conditions, allows a more cost-efficient process.95 In more recent studies, the use of permeabilised P. pseudoalcaligenes cells in a continuous process is reported.108
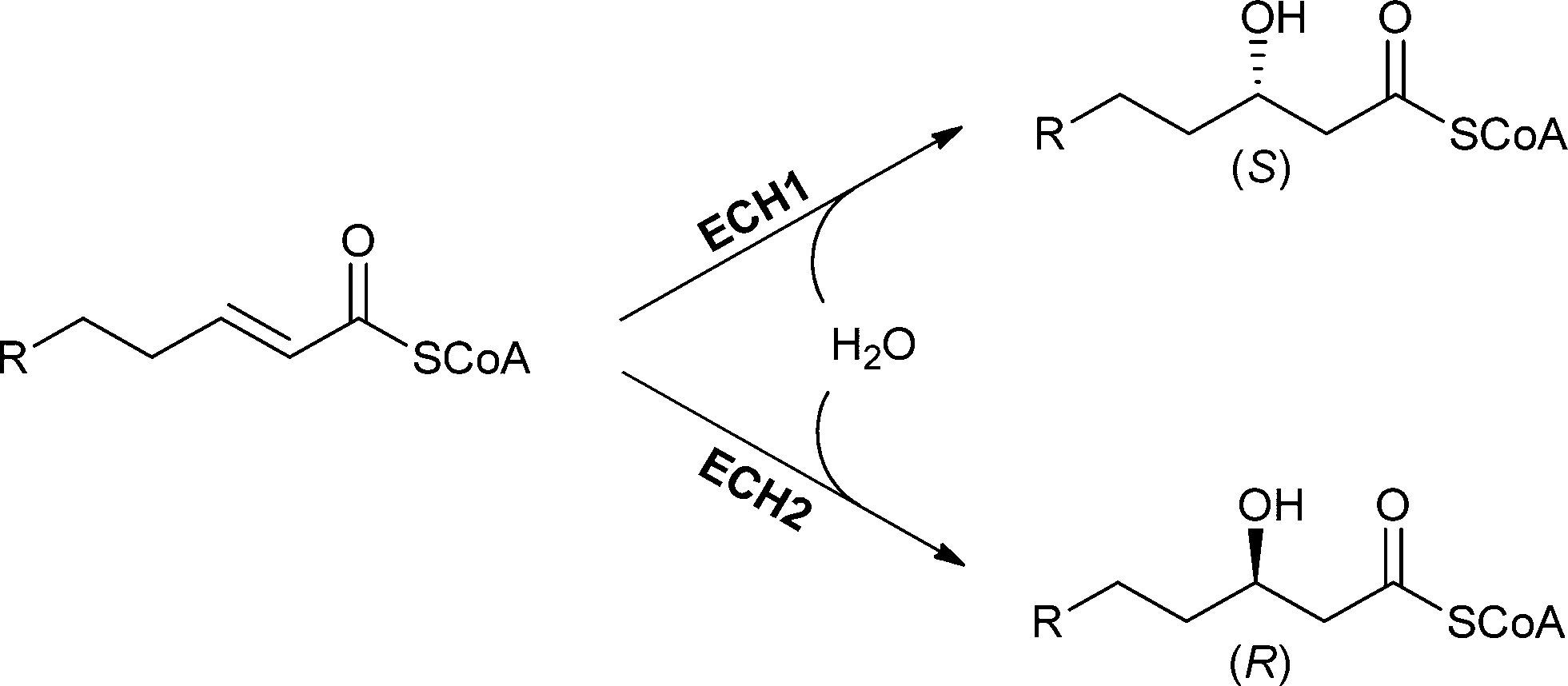 | ||
| Scheme 16 The addition of water catalysed by ECH1 and ECH2 proceeds in a syn fashion but displays a different enantioselectivity depending on the enzyme used. | ||
One example of an (S)-selective enoyl-CoA hydratase is ECH1. This enzyme is also known as crotonase and is able to perform the addition of water to substrates with a chain length between 4 and 20 carbon atoms.45,109 This rather broad substrate spectrum is achieved by a flexible loop in the active site, which allows the increase of hydrophobic binding pockets to bind larger substrates.112 Water addition to a trans-enoyl-CoA-thioester takes place in a syn fashion and (S)-3-hydroxyacyl-CoA is formed.45,112–115 ECH1 from bovine liver can be heterologously expressed in E. coli, which allows the production of larger amounts of enzyme.
The (R)-selective enoyl-CoA hydratase ECH2 also catalysed syn-addition with opposite enantioselectivity. Trans-2-enoyl CoA thioesters are accepted as substrates and (R)-3-hydroxyacyl-CoA is formed as the product.116 The substrate specificity of ECH2 depends on the source. For example, ECH2s from bacterial sources show preference towards short-chain substrates, while ECH2s from eukaryotic sources prefer long-chain substrates.114 The difference in enantioselectivity between ECH1 and ECH2 can easily be explained by the geometry of the active site. The active sites of the ECH1 and ECH2 behave like mirror images of each other.115
In industry, enoyl-CoA hydratase is employed for the production of (R)-3-hydroxybutyric acid and (R)-3-hydroxyisobutyric acid from butyric and isobutyric acid respectively. In whole-cell processes run by Kanegafuchi Japan, cells from Candida rugosa IFO 0750M are used.95,117 This process consists of three steps starting from butyric acid, which is converted into the corresponding CoA-thioester. This thioester is then enzymatically dehydrated to form the α,β-unsaturated compound, which serves as the substrate for the hydratase. In additional steps the hydrated thioester can then be converted to (R)-3-hydroxybutanoic acid (>98% ee). The efficiency of this process is represented by a space-time yield of 5–10 g L−1 d−1.95 (R)-3-Hydroxybutanoic acid is an important building block for the synthesis of a carbapenem intermediate (see Scheme 17a).95,118 This process can also be employed for the production of (R)-3-hydroxyisobutyric acid, which is a precursor for captopril (see Scheme 17b). For this process a space-time yield of 5–10 g L−1 d−1 and a yield of 98% are reported.95,117,118
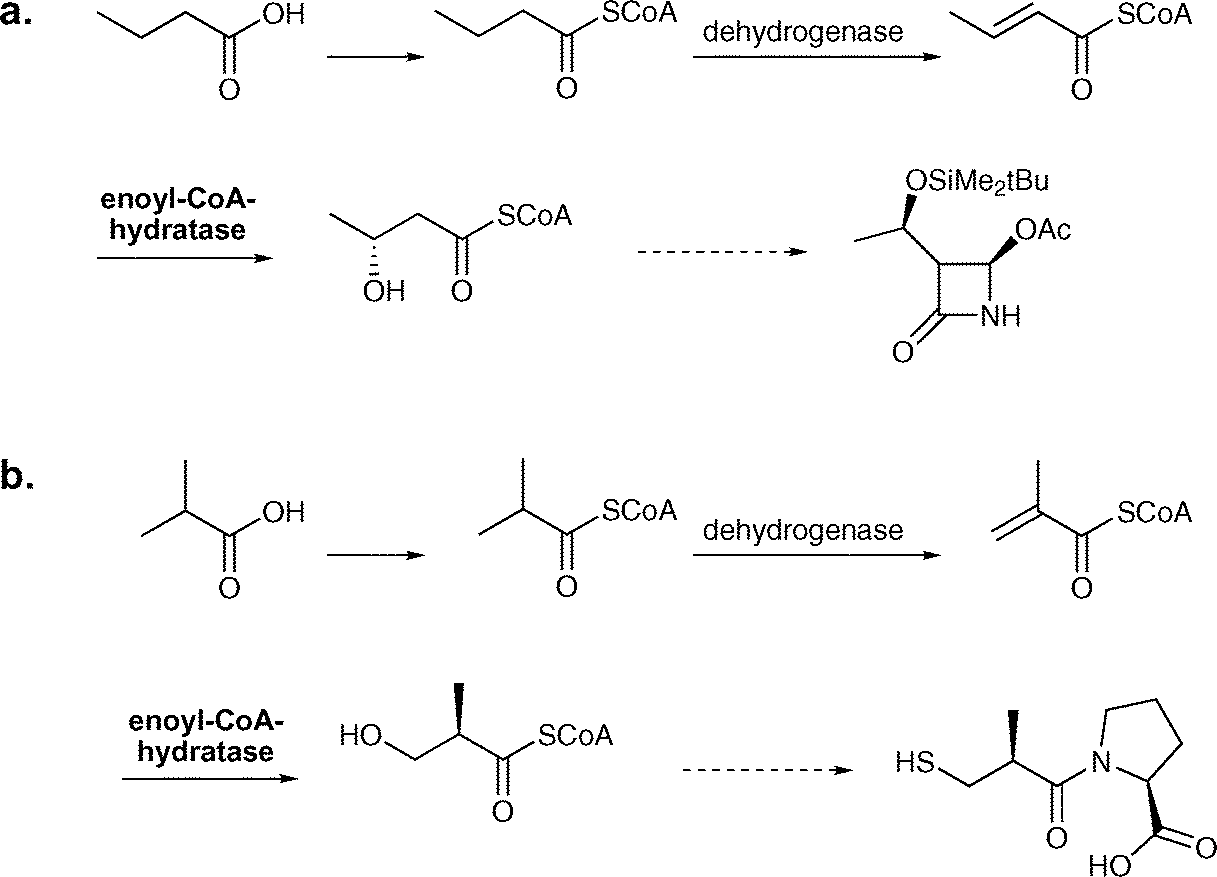 | ||
| Scheme 17 Industrial processes employing enoyl-CoA hydratase from Candida rugosa IFO 0750M. a. Route starting from butyric acid for the synthesis of (R)-3-hydroxybutyric acid, which serves as a precursor for a carbapenem intermediate. b. Route starting from isobutyric acid for the synthesis of (R)-3-hydroxyisobutyric acid, a building block in captopril synthesis. | ||
Recently the concept of designing new enzymes with new activities has become more popular. Two examples with pyridine-based ligands of Cu catalysts were described. In one case, DNA was utilised as the chiral backbone, while in the other case the complex was embedded into a homodimeric protein. Remarkably, the DNA-based catalyst catalysed the syn-addition of water.119 When alcohol was present in the reaction mixture this was used as a nucleophile, rather than water, again demonstrating the difficulty in utilising water.120 In the other example the design of an artificial metalloenzyme with hydratase activity was achieved by employing the homodimeric protein LmrR, a transcriptional regulator, as the second coordination sphere for a Cu(II) phenanthroline complex that is responsible for the activation of water.79 Even though this method is not yet applicable on the large scale, it represents a different approach for the design of novel enzymes. Again other nucleophiles were preferred over water.121
4. Conclusion and outlook
Water is and remains both a poor electrophile and a poor nucleophile that is difficult to activate. Its direct application as a reagent in addition reactions to CC double bonds therefore remains challenging. Chemical catalysts to date are not very successful in the activation of water (section 2), yet there is hope. Building on the knowledge of biological systems the first artificial hydratases were developed, opening up new avenues for chemo-catalysis (section 3.3).79,119 In stark contrast to chemical approaches, hydratases are the backbone of life. They may serve as models for better chemical catalysts, but of course they are catalysts in their own right. Indeed they have proven their value in several industrial processes (section 3).
In general, the asymmetric addition of water to double bonds is a very efficient and green method for the production of secondary and tertiary alcohols.122 Water is an abundant and safe nucleophile, being both the solvent and the substrate, and hydratases are the perfect tools to perform this reaction. Selected hydratases such as fumarase, malease and enoyl-CoA hydratase have already proven to be versatile tools for the biocatalytic synthesis of fine chemicals. They are employed on the industrial scale in well-established processes still running today on large annual production scales (see Table 2).
| Product | Enzyme | Organism | Company | Yield [%] | Equilibrium yield [%] | Annual production [t] |
|---|---|---|---|---|---|---|
| a No numbers available. b Equilibrium yield for (3R)-3-hydroxybutanoyl-CoA, which serves as the precursor for β-hydroxy-n-butyric acid. | ||||||
| (S)-Malate | Fumarase | Corynebacterium glutamicum (suspended whole cells) | AMINO GmbH | 85 | 82 (ref. 123, 124) | 2000 |
| (S)-Malate | Fumarase | Brevibacterium flavum (immobilised whole cells) | Tanabe Seiyaku Co., Ltd. | >70 | 82 (ref. 123, 124) | 468 |
| (R)-Malate | Malease | Pseudomonas pseudoalcaligenes (immobilised whole cells) | DSM | >99 | 100 (ref. 105) | —a |
| β-Hydroxy-n-butyric acid | Enoyl-CoA hydratase | Candida rugosa IFO 0750M (suspended whole cells) | Kanegafuchi Chemical Industries Co., Ltd | —a | 85b,125 | —a |
| β-Hydroxyisobutyric acid | Enoyl-CoA hydratase | Candida rugosa IFO 0750M (suspended whole cells) | Kanegafuchi Chemical Industries Co., Ltd | 98 | —a | —a |
These hydratases are employed for very specific reactions, and due to their narrow substrate spectrum, broad applicability is not possible. However, in recent years methods for the discovery of novel enzymes as well as the techniques to engineer known biocatalysts have significantly improved. Structural investigations allow deeper insight into the mechanisms behind hydratase-catalysed reactions and catalytically essential residues can be identified. This might allow the expansion of the range of hydratases. Furthermore, promiscuous hydratase activity for different decarboxylases was discovered. In this study, seven phenolic decarboxylases from different sources were used to hydrate five different hydroxystyrenes as substrates.78
Promisingly there are also organisms with reported hydratase activity that were not yet investigated in detail. For example, the use of resting cells from Rhodococcus rhodochrous for the hydration of the lactones 3-methyl-2-butenolide and 3-ethyl-2-butenolide to give the corresponding (R)-3-hydroxy-3-alkylbutanolides is reported, but detailed investigations on the enzyme performing this interesting reaction are still lacking.77,126,127 A hydratase activity from Alicycliphilus denitrificans was, however, recently shown to be an artefact. This was due to a coupled assay used, indicating that great care has to be taken when identifying these enzyme activities.128,129
To summarise, the addition of water is a demanding task. Chemically only the most simple molecules with a limited number of functional groups can be converted reasonably well. But even in these cases activity and in particular selectivity are insufficient. Hydratases, on the other hand, can be efficient and highly selective catalysts for the addition of water, a reaction that is still underrepresented and difficult to achieve in organic chemistry. Using water as the nucleophile and substrate at the same time is not only an elegant route but also a very green route to the production of a variety of different alcohols. The current limitations arising from the substrate scope of the known enzymes can be challenged by modern protein engineering techniques and new enzyme discovery to broaden the toolbox of hydratases available for industrial applications.130
Acknowledgements
An “Erwin Schrödinger” Fellowship (J3292) is generously provided by the Austrian Science Fund (FWF) for V.R. The authors thank all of their colleagues and group members for the fruitful discussions and the joint work that helped in forming the ideas formulated in this perspective.References
- J. Jin and U. Hanefeld, Chem. Commun., 2011, 47, 2502–2510 RSC.
- J. E. McMurry, Organic Chemistry, Cengage Learning, 8th edn, 2012 Search PubMed.
- K. Schwetlick, Organikum, Wiley, 23 edn, 2009 Search PubMed.
- J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, 2nd edn., 2012 Search PubMed.
- P. N. Davey, M. J. Earle, J. T. Hamill, S. P. Katdare, D. W. Rooney and K. R. Seddon, Green Chem., 2010, 12, 628–631 RSC.
- V. Resch, C. Seidler, B.-S. Chen, I. Degeling and U. Hanefeld, Eur. J. Org. Chem., 2013, 7697–7704 CrossRef CAS PubMed.
- S. Wang, Z. Zhang, C. Chi, G. Wu, J. Ren, Z. Wang, M. Huang and Y. Jiang, React. Funct. Polym., 2008, 68, 424–430 CrossRef CAS PubMed.
- X. Wang, D. Sui, M. Huang and Y. Jiang, Polym. Adv. Technol., 2006, 17, 163–167 CrossRef CAS PubMed.
- L. Xue, B. Jia, L. Tang, X. F. Ji, M. Y. Huang and Y. Y. Jiang, Polym. Adv. Technol., 2004, 15, 346–349 CrossRef CAS PubMed.
- N. Kosaric, Z. Duvnjak, A. Farkas, H. Sahm, S. Bringer-Meyer, O. Goebel and D. Mayer, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, DOI:10.1002/14356007.a09_587.pub2.
- V. J. Nowlan and T. T. Tidwell, Acc. Chem. Res., 1977, 10, 252–258 CrossRef CAS.
- Y. Li, C. Wang, G. Jia, S. Lu and C. Li, Tetrahedron, 2013, 69, 6585–6590 CrossRef CAS PubMed.
- H. M. Meshram, N. Satish Kumar, J. B. Nanubolu, L. Chandrasekhara Rao and N. Nageswara Rao, Tetrahedron Lett., 2013, 54, 5941–5944 CrossRef CAS PubMed.
- C. Pan and Z. Wang, Coord. Chem. Rev., 2008, 252, 736–750 CrossRef CAS PubMed.
- L. Petrus, R. W. De Roo, E. J. Stamhuis and G. E. H. Joosten, Chem. Eng. Sci., 1984, 39, 433–446 CrossRef CAS.
- A. J. Papa, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, DOI:10.1002/14356007.a22_173.pub2.
- D. A. O'Sullivan, Chem. Eng. News, 1985, 63, 10–11 Search PubMed.
- H.-D. Hahn, G. Dämbkes, N. Rupprich, H. Bahl and G. D. Frey, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2013, DOI:10.1002/14356007.a04_463.pub3.
- Z. Lei, Y. Yang, Q. Li and B. Chen, Catal. Today, 2009, 147S, S352–S356 CrossRef PubMed.
- L. Petrus, R. W. De Roo, E. J. Stamhuis and G. E. H. Joosten, Chem. Eng. Sci., 1986, 41, 217–226 CrossRef CAS.
- J. L. F. Monteiro and C. Veloso, Top. Catal., 2004, 27, 169–180 CrossRef CAS.
- P. Botella, A. Corma, J. M. L. Nieto, S. Valencia, M. E. Lucas and M. Sergio, Appl. Catal., A, 2000, 203, 251–258 CrossRef CAS.
- Y. Liu, Z. Zhou, G. Yang, Y. Wu and Z. Zhang, Int. J. Chem. React. Eng., 2010, 8, 1542–6580 Search PubMed.
- N. Comelli, M. C. Avila, C. Volzone and M. Ponzi, Cent. Eur. J. Chem., 2013, 11, 689–697 CrossRef CAS PubMed.
- A. M. Chibiryaev, A. Yermakova and I. V. Kozhevnikov, J. Supercrit. Fluids, 2010, 51, 295–305 CrossRef CAS PubMed.
- M. C. Ávila, N. A. Comelli, E. Rodríguez-Castellón, A. Jiménez-López, R. Carrizo Flores, E. N. Ponzi and M. I. Ponzi, J. Mol. Catal. A: Chem., 2010, 322, 106–112 CrossRef PubMed.
- T. Mochida, R. Ohnishi, N. Horita, Y. Kamiya and T. Okuhara, Microporous Mesoporous Mater., 2007, 101, 176–183 CrossRef CAS PubMed.
- D. Arntz, A. Fischer, M. Höpp, S. Jacobi, J. Sauer, T. Ohara, T. Sato, N. Shimizu and H. Schwind, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, DOI:10.1002/14356007.a01_149.pub2.
- C. Kohlpaintner, M. Schulte, J. Falbe, P. Lappe, J. Weber and G. D. Frey, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2013, DOI:10.1002/14356007.a01_321.pub3.
- T. Haas, T. Hahm, R. Vanheertum, W. Hofen and L. Deusser, WO 01/09073 A1, 2001.
- C. J. Sullivan, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, DOI:10.1002/14356007.a22_163.
- E. Celinska, Biotechnol. Adv., 2010, 28, 519–530 CrossRef CAS PubMed.
- D. M. Adkesson, A. W. Alsop, T. T. Ames, L. A. Chu, J. M. Disney, B. C. Dravis, P. Fitzgibbon, J. M. Gaddy, F. G. Gallagher, W. F. Lehnhardt, J. C. Lievense, M. L. Luyben, M. Seapan, R. E. Trotter, G. M. Wenndt and E. K. Yu, WO 2004/101479 A2, 2004.
- V. E. T. Maervoet, M. De Mey, J. Beauprez, S. De Maeseneire and W. K. Soetaert, Org. Process Res. Dev., 2011, 15, 189–202 CrossRef CAS.
- A. J. Mattam, J. M. Clomburg, R. Gonzalez and S. S. Yazdani, Biotechnol. Lett., 2013, 35, 831–842 CrossRef CAS PubMed.
- H. Gröger, Angew. Chem., Int. Ed., 2014, 53, 3067–3069 CrossRef PubMed.
- V. E. Anderson, M. W. Ruszczycky and M. E. Harris, Chem. Rev., 2006, 106, 3236–3251 CrossRef CAS PubMed.
- X. Zhang and K. N. Houk, Acc. Chem. Res., 2005, 38, 379–385 CrossRef CAS PubMed.
- M. J. van der Werf, W. J. J. van den Tweel, J. Kamphuis, S. Hartmans and J. A. M. de Bont, Trends Biotechnol., 1994, 12, 95–103 CrossRef CAS.
- T. Tokoroyama, Eur. J. Org. Chem., 2010, 2009–2016 CrossRef CAS PubMed.
- J. R. Mohrig, K. Moerke, D. Cloutier, B. Lane, E. Person and T. Onasch, Science, 1995, 269, 527–529 CAS.
- H. Lauble, M. C. Kennedy, H. Beinert and C. D. Stout, J. Mol. Biol., 1994, 237, 437–451 CrossRef CAS PubMed.
- I. A. Rose and T. M. Weaver, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 3393–3397 CrossRef CAS PubMed.
- J. R. Mohrig, Acc. Chem. Res., 2013, 46, 1407–1416 CrossRef CAS PubMed.
- H. M. Holden, M. M. Benning, T. Haller and J. A. Gerlt, Acc. Chem. Res., 2001, 34, 145–157 CrossRef CAS PubMed.
- K. R. Hanson and I. A. Rose, Acc. Chem. Res., 1975, 8, 1–10 CrossRef CAS.
- J. A. Gerlt and P. G. Gassman, J. Am. Chem. Soc., 1992, 114, 5928–5934 CrossRef CAS.
- V. Puthan Veetil, G. Fibriansah, H. Raj, A.-M. W. H. Thunnissen and G. J. Poelarends, Biochemistry, 2012, 51, 4237–4243 CrossRef CAS PubMed.
- E. N. Davis, L. L. Wallen, J. C. Goodwin, W. K. Rohwedder and R. A. Rhodes, Lipids, 1969, 4, 356–362 CrossRef CAS.
- W. G. Niehaus, A. Kisic, A. Torkelson, D. J. Bednarczyk and G. J. Schroepfer, J. Biol. Chem., 1970, 245, 3790–3797 CAS.
- L. L. Wallen, R. G. Benedict and R. W. Jackson, Arch. Biochem. Biophys., 1962, 99, 249–253 CrossRef CAS.
- A. Kisic, Y. Miura and G. J. Schroepfer Jr., Lipids, 1971, 6, 541–545 CrossRef CAS.
- L. E. Bevers, M. W. H. Pinkse, P. D. E. M. Verhaert and W. R. Hagen, J. Bacteriol., 2009, 191, 5010–5012 CrossRef CAS PubMed.
- Y.-C. Joo, K.-W. Jeong, S.-J. Yeom, Y.-S. Kim, Y. Kim and D.-K. Oh, Biochimie, 2012, 94, 907–915 CrossRef CAS PubMed.
- B.-N. Kim, Y.-C. Joo, Y.-S. Kim, K.-R. Kim and D.-K. Oh, Appl. Microbiol. Biotechnol., 2012, 95, 929–937 CrossRef CAS PubMed.
- S. Gocho, N. Tabogami, M. Inagaki, C. Kawabata and T. Komai, Biosci., Biotechnol., Biochem., 1995, 59, 1571–1572 CrossRef CAS.
- A. Wanikawa, K. Hosoi, I. Takise and T. Kato, J. Inst. Brew., 2000, 106, 39–44 CrossRef CAS PubMed.
- B.-N. Kim, S.-J. Yeom and D.-K. Oh, Biotechnol. Lett., 2011, 33, 993–997 CrossRef CAS PubMed.
- Y.-C. Joo, E.-S. Seo, Y.-S. Kim, K.-R. Kim, J.-B. Park and D.-K. Oh, J. Biotechnol., 2012, 158, 17–23 CrossRef CAS PubMed.
- E.-Y. Jeon, J.-H. Lee, K.-M. Yang, Y.-C. Joo, D.-K. Oh and J.-B. Park, Process Biochem., 2012, 47, 941–947 CrossRef CAS PubMed.
- J.-W. Song, E.-Y. Jeon, D.-H. Song, H.-Y. Jang, U. T. Bornscheuer, D.-K. Oh and J.-B. Park, Angew. Chem., Int. Ed., 2013, 52, 2534–2537 CrossRef CAS PubMed.
- J.-W. Song, J.-H. Lee, U. T. Bornscheuer and J.-B. Park, Adv. Synth. Catal., 2014, 356, 1782–1788 CrossRef CAS PubMed.
- K. R. Cadwallader, R. J. Braddock, M. E. Parish and D. P. Higgins, J.Food Sci., 1989, 54, 1241–1245 CrossRef CAS PubMed.
- M. R. Marostica and G. M. Pastore, Food Chem., 2006, 101, 345–350 CrossRef PubMed.
- J. Bicas, F. Barros, R. Wagner, H. Godoy and G. Pastore, J. Ind. Microbiol. Biotechnol., 2008, 35, 1061–1070 CrossRef CAS PubMed.
- J. L. Bicas, C. P. de Quadros, I. A. Néri-Numa and G. M. Pastore, Food Chem., 2010, 120, 452–456 CrossRef CAS PubMed.
- J. Onken and R. G. Berger, J. Biotechnol., 1999, 69, 163–168 CrossRef CAS.
- I. Rottava, G. Toniazzo, P. F. Cortina, E. Martello, C. E. Grando, L. A. Lerin, H. Treichel, A. J. Mossi, D. de Oliveira, R. L. Cansian, O. A. C. Antunes and E. G. Oestreicher, LWT--Food Sci. Technol., 2010, 43, 1128–1131 CrossRef CAS PubMed.
- A. Adams, J. C. R. Demyttenaere and K. N. De, Food Chem., 2002, 80, 525–534 CrossRef.
- A. Z. M. Badee, S. A. Helmy and N. F. S. Morsy, Food Chem., 2011, 126, 849–854 CrossRef CAS PubMed.
- Q. Tan and D. F. Day, Appl. Microbiol. Biotechnol., 1998, 49, 96–101 CrossRef CAS.
- K. R. Cadwallader, R. J. Braddock and M. E. Parish, J. Food Sci., 1992, 57, 241–244 CrossRef CAS PubMed + 248.
- N. Savithiry, T. Cheong and P. Oriel, in Biotechnology for Fuels and Chemicals, ed. B. Davison, C. Wyman and M. Finkelstein, Humana Press, 1997, ch. 20, vol. 63–65, pp. 213–220 Search PubMed.
- J. L. Bicas, P. Fontanille, G. M. Pastore and C. Larroche, Process Biochem., 2010, 45, 481–486 CrossRef CAS PubMed.
- M. Pescheck, M. Mirata, B. Brauer, U. Krings, R. Berger and J. Schrader, J. Ind. Microbiol. Biotechnol., 2009, 36, 827–836 CrossRef CAS PubMed.
- S. G. A. Prieto, V. J. A. Perea and L. C. C. Ortiz, Vitae, 2011, 18, 163–172 Search PubMed.
- H. L. Holland and J.-X. Gu, Biotechnol. Lett., 1998, 20, 1125–1126 CrossRef CAS.
- C. Wuensch, J. Gross, G. Steinkellner, K. Gruber, S. M. Glueck and K. Faber, Angew. Chem., Int. Ed., 2013, 52, 2293–2297 CrossRef CAS PubMed.
- J. Bos, A. Garcia-Herraiz and G. Roelfes, Chem. Sci., 2013, 4, 3578–3582 RSC.
- C. A. Lamartiniere, H. D. Braymer and A. D. Larson, Arch. Biochem. Biophys., 1970, 141, 293–302 CrossRef CAS.
- D. H. Flint, Arch. Biochem. Biophys., 1994, 311, 509–516 CrossRef CAS PubMed.
- Y. Ueda, N. Yumoto, M. Tokushige, K. Fukui and H. Ohya-Nishiguchi, J. Biochem., 1991, 109, 728–733 CAS.
- S. A. Woods, S. D. Schwartzbach and J. R. Guest, Biochim. Biophys. Acta, 1988, 954, 14–26 CrossRef CAS.
- D. H. Flint, M. H. Emptage and J. R. Guest, Biochemistry, 1992, 31, 10331–10337 CrossRef CAS.
- T. Weaver, M. Lees, V. Zaitsev, I. Zaitseva, E. Duke, P. Lindley, S. McSweeny, A. Svensson, J. Keruchenko, I. Keruchenko, K. Gladilin and L. Banaszak, J. Mol. Biol., 1998, 280, 431–442 CrossRef CAS PubMed.
- J. Yang, Y. Wang, E. M. Woolridge, V. Arora, G. A. Petsko, J. W. Kozarich and D. Ringe, Biochemistry, 2004, 43, 10424–10434 CrossRef CAS PubMed.
- T. Weaver, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 1395–1401 CrossRef PubMed.
- T. Weaver and L. Banaszak, Biochemistry, 1996, 35, 13955–13965 CrossRef CAS PubMed.
- T. Weaver, M. Lees and L. Banaszak, Protein Sci., 1997, 6, 834–842 CrossRef CAS PubMed.
- E. Bressler, O. Pines, I. Goldberg and S. Braun, Biotechnol. Prog., 2002, 18, 445–450 CrossRef CAS PubMed.
- K. Lohbeck, H. Haferkorn, W. Fuhrmann and N. Fedtke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI:10.1002/14356007.a16_053.
- K. Yamamoto, T. Tosa, K. Yamashita and I. Chibata, Eur. J. Appl. Microbiol., 1976, 3, 169–183 CrossRef CAS.
- I. Takata, K. Yamamoto, T. Tosa and I. Chibata, Enzyme Microb. Technol., 1980, 2, 30–36 CrossRef CAS.
- I. Chibata, T. Tosa and I. Takata, Trends Biotechnol., 1983, 1, 9–11 CrossRef CAS.
- A. Liese, K. Seelbach and C. Wandrey, Industrial Biotransformations; Second, Completely Revised and Extended Edition, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006 Search PubMed.
- T. Tosa and T. Shibatani, Ann. N. Y. Acad. Sci., 1995, 750, 364–375 CrossRef CAS PubMed.
- W. Leuchtenberger, M. Karrenbauer and U. Plöcker, Ann. N. Y. Acad. Sci., 1984, 434, 078–086 CrossRef CAS PubMed.
- H. J. Danneel and R. Geiger, DE 4430010 C1, 1996.
- H. J. Danneel and R. Geiger, DE 4424664 C1, 1995.
- J. W. Teipel, G. M. Hass and R. L. Hill, J. Biol. Chem., 1968, 243, 5684–5694 CAS.
- M. A. Findeis and G. M. Whitesides, J. Org. Chem., 1987, 52, 2838–2848 CrossRef CAS.
- J.-L. Dreyer, Eur. J. Biochem., 1985, 150, 145–154 CrossRef CAS PubMed.
- B.-F. He, T. Nakajima-Kambe, T. Ozawa and T. Nakahara, Process Biochem., 2000, 36, 407–414 CrossRef CAS.
- M. J. van der Werf, W. J. van den Tweel and S. Hartmans, Appl. Environ. Microbiol., 1992, 58, 2854–2860 CAS.
- M. J. van der Werf, W. J. J. van den Tweel and S. Hartmans, Appl. Environ. Microbiol., 1993, 59, 2823–2829 CAS.
- M. J. van der Werf, W. J. J. van den Tweel and S. Hartmans, Eur. J. Biochem., 1993, 217, 1011–1017 CrossRef CAS PubMed.
- M. Ueda, H. Yamada and Y. Asano, Appl. Microbiol. Biotechnol., 1994, 41, 215–218 CrossRef CAS.
- M. J. F. Michielsen, C. Frielink, R. H. Wijffels, J. Tramper and H. H. Beeftink, J. Biotechnol., 2000, 79, 13–26 CrossRef CAS.
- J. K. Hiltunen and Y.-M. Qin, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2000, 1484, 117–128 CrossRef CAS.
- G. Agnihotri and H.-W. Liu, Bioorg. Med. Chem., 2003, 11, 9–20 CrossRef CAS.
- P. Bhaumik, M. K. Koski, T. Glumoff, J. K. Hiltunen and R. K. Wierenga, Curr. Opin. Struct. Biol., 2005, 15, 621–628 CrossRef CAS PubMed.
- B. J. Bahnson, V. E. Anderson and G. A. Petsko, Biochemistry, 2002, 41, 2621–2629 CrossRef CAS PubMed.
- A. F. Bell, Y. Feng, H. A. Hofstein, S. Parikh, J. Wu, M. J. Rudolph, C. Kisker, A. Whitty and P. J. Tonge, Chem. Biol., 2002, 9, 1247–1255 CrossRef CAS.
- M. K. Koski, A. M. Haapalainen, J. K. Hiltunen and T. Glumoff, J. Biol. Chem., 2004, 279, 24666–24672 CrossRef CAS PubMed.
- M. K. Koski, A. M. Haapalainen, J. K. Hiltunen and T. Glumoff, J. Mol. Biol., 2005, 345, 1157–1169 CrossRef PubMed.
- T. Hisano, T. Tsuge, T. Fukui, T. Iwata, K. Miki and Y. Doi, J. Biol. Chem., 2003, 278, 617–624 CrossRef CAS PubMed.
- J. Hasegawa, M. Ogura, H. Kanema, N. Noda, H. Kawaharada and K. Watanabe, J. Ferment. Technol., 1982, 60, 501–508 CAS.
- J. Crosby, Tetrahedron, 1991, 47, 4789–4846 CrossRef CAS.
- A. J. Boersma, D. Coquière, D. Geerdink, F. Rosati, B. L. Feringa and G. Roelfes, Nat. Chem., 2010, 2, 991–995 CrossRef CAS PubMed.
- R. P. Megens and G. Roelfes, Chem. Commun., 2012, 48, 6366–6368 RSC.
- J. Bos, F. Fusetti, A. J. Driessen and G. Roelfes, Angew. Chem., Int. Ed., 2012, 51, 7472–7475 CrossRef CAS PubMed.
- R. Kourist and U. Bornscheuer, Appl. Microbiol. Biotechnol., 2011, 91, 505–517 CrossRef CAS PubMed.
- R. M. Bock and R. A. Alberty, J. Am. Chem. Soc., 1953, 75, 1921–1925 CrossRef CAS.
- H. A. Krebs, Biochem. J., 1953, 54, 78–82 CAS.
- J. R. Stern and A. del Campillo, J. Biol. Chem., 1956, 218, 985–1002 CAS.
- B.-S. Chen, L. G. Otten, V. Resch, G. Muyzer and U. Hanefeld, Stand. Genomic Sci., 2013, 9, 175 CrossRef PubMed.
- M. Müller, ChemBioEng Rev., 2014, 1, 14–26 CrossRef PubMed.
- J. Jin, A. J. Straathof, M. H. Pinkse and U. Hanefeld, Appl. Microbiol. Biotechnol., 2011, 89, 1831–1840 CrossRef CAS PubMed.
- V. Resch, J. Jin, B.-S. Chen and U. Hanefeld, AMB Express, 2014, 4, 30 CrossRef PubMed.
- T. Davids, M. Schmidt, D. Böttcher and U. T. Bornscheuer, Curr. Opin. Chem. Biol., 2013, 17, 215–220 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |