
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversibility of asymmetric catalyzed C–C bond formation by benzoylformate decarboxylase†
Marco
Berheide
a,
Selin
Kara
*ab and
Andreas
Liese
*a
aInstitute of Technical Biocatalysis, Hamburg University of Technology, Denickestrasse 15, 21073, Hamburg, Germany. E-mail: liese@tuhh.de; Fax: (+) 49 40 42878 2127; Tel: (+) 49 40 42878 3018
bInstitute of Microbiology, Chair of Molecular Biotechnology, Technische Universität Dresden, 01062 Dresden, Germany. E-mail: selin.kara@tu-dresden.de; Fax: (+) 49 351 463 39520; Tel: (+) 49 351 463 39517
First published on 3rd February 2015
Abstract
Benzoylformate decarboxylase (BFD) from Pseudomonas putida catalyzed the formation of 2-hydroxy-1-phenylpropanone (2-HPP), a 2-hydroxy ketone, from the kinetic resolution of rac-benzoin in the presence of acetaldehyde. The formation rate of 2-HPP via kinetic resolution of benzoin was 700-fold lower compared to the formation via direct carboligation of benzaldehyde and acetaldehyde. Further investigations revealed that BFD not only accepts (R)-benzoin but also 2-HPP as the substrate. A typical Michaelis–Menten type kinetics was observed starting from enantiopure (S)- or (R)-2-HPP. The formation of racemic 2-HPP while using benzoin as the donor in the presence of acetaldehyde and the racemization of (R/S)-2-HPP were detected. The equilibrium constant determined, showed favoured conditions towards the product side i.e. (R)-benzoin and 2-HPP. In the end, an extended reaction mechanism was proposed by supplementing the already known mechanism with the C–C bond cleavage activity of BFD towards 2-hydroxy ketones.
Introduction
Synthesis of chiral 2-hydroxy ketones has attracted great attention as these compounds are applied broadly for the synthesis of biologically active compounds such as pharmaceuticals, agrochemicals, and pheromones.1 Thiamine diphosphate (ThDP) dependent enzymes have been widely applied for the synthesis of these crucial compounds and among them, benzoylformate decarboxylase (BFD)2 and benzaldehyde lyase (BAL)3 have been investigated in detail.It was reported that BFD naturally catalyzes the non-oxidative decarboxylation of benzoylformate to form benzaldehyde and carbon dioxide.4 Later, in the beginning of the 1990s, (S)-2-hydroxy-1-phenylpropanone ((S)-2-HPP) was described as the product of benzoylformate decarboxylation in the presence of acetaldehyde.5 Since the 1990s, enantioselective C–C bond formation, termed carboligation, has gained increased interest in the research community. After the crystal structure of Pseudomonas putida BFD (EC 4.1.1.7)6 was known, stereoselectivity of BFD-catalyzed carboligations was investigated by molecular modelling studies revealing an ‘S-pocket’ responsible for the (S)-selectivity of BFD.7 The size of the ‘S-pocket’ is found to be large enough for the binding of acetaldehyde but not for benzaldehyde hence yielding (S)-2-HPP and (R)-benzoin, respectively.
Another ThDP-dependent enzyme is BAL from Pseudomonas fluorescens Biovar I (EC 4.1.2.38), reported on for the first time in 1989.8 It was shown that Pseudomonas fluorescens Biovar I can grow on benzoin as sole carbon source owing to the BAL-catalyzed cleavage of acyloin linkage of (R)-benzoin. It took a decade until the potential activity of BAL for the reverse acyloin condensation was reported.3 Since then, BAL has been commonly applied for the synthesis of chiral 2-hydroxy ketones, the catalytic mechanism of which involves two steps: (1) the nucleophilic attack of the ylide form of ThDP on the carbonyl C-atom of (R)-benzoin (or an araliphatic compound) yielding an enamine–carbanion intermediate after the release of the aromatic aldehyde and (2) the attack of the enamine–carbanion intermediate on an acceptor aldehyde yielding a 2-hydroxy ketone.3a,b,h
The C–C bond cleavage of benzoin catalyzed by BFD has been already a topic discussed in the literature.7a,9 As (R)-benzoin is formed from two molecules of benzaldehyde catalyzed by BFD, it is clear that the active site of BFD can accommodate benzoin. In fact, the reversible benzoin synthesis by BFD was reported to be possible but disfavored due to the low solubility of benzoin in the aqueous medium (i.e. ~1.4 mM at 25 °C), which hinders the binding of BFD to benzoin.9 On the other hand, BFD-catalyzed cleavage of benzoin was reported to be impossible from the mechanistic point of view as benzoin cannot sterically fit into the active site of BFD.7a The present study was based on initial evidences that hint to a carbolyase activity also present in BFD. Therefore, in this work the catalytic activity of BFD on the cleavage of C–C bond of 2-hydroxy ketones was investigated assuming a similar mechanism as for BAL.
An experimental evidence for the BFD-catalyzed cleavage of (R)-benzoin is however difficult to obtain, due to the low solubility of benzoin in aqueous buffer and the thermodynamic equilibrium, which strongly favors the formation of benzoin and the presumable low activity of BFD for the cleavage of (R)-benzoin. However, the equilibrium can be shifted towards benzaldehyde formation by in situ removal of the benzaldehyde formed. Therefore, acetaldehyde was added to the reaction medium to form 2-HPP (4) (Scheme 1).
 | ||
| Scheme 1 Postulated reaction sequence for the cleavage of benzoin (1) to benzaldehyde (2) and further carboligation with acetaldehyde (3) yielding 2-HPP (4) catalyzed by BFD. | ||
Results and discussion
Earlier, BFD-mediated C–C bond formations were conducted in phosphate buffer, whereby no C–C bond cleavage activity of BFD could be detected under these conditions so far. Alternatively, triethanolamine (TEA) was also applied for ThDP-dependent enzymes, which is in fact the buffer of choice for BAL-catalyzed reactions.3 Therefore, we became interested in using TEA buffer to evaluate the C–C bond cleavage activity of BFD. In addition, due to the limited solubility of benzoin, DMSO, a common cosolvent used for BFD and BAL,2,3 was applied.Kinetic resolution of rac-benzoin catalyzed by BFD
Firstly, the kinetic resolution of rac-benzoin was investigated, whereby the enantiomeric excess (ee) of benzoin was monitored during the course of reaction. Since BFD catalyzes the formation of (R)-benzoin,2a only (R)-benzoin can be accepted as substrate in a kinetic resolution. In an ideal situation the ee of (S)-benzoin would continuously increase as the cleavage of (R)-benzoin proceeds, reaching 100% at 50% of conversion. Indeed, our results revealed an ideal kinetic resolution catalyzed by BFD as shown in Fig. 1.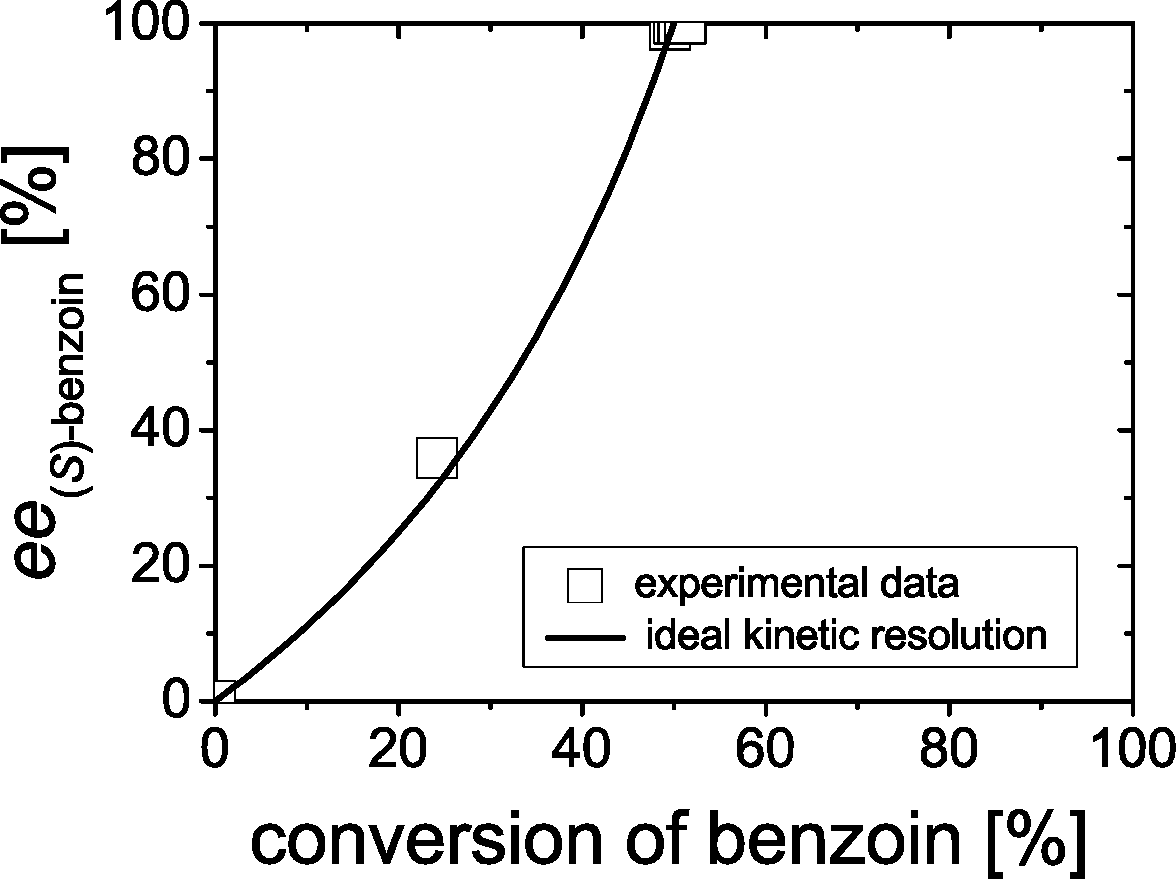 | ||
| Fig. 1 The ee value of (S)-benzoin as a function of the conversion of benzoin in the kinetic resolution of rac-benzoin (1) (with 50 mM acetaldehyde). Reaction conditions: 1.6 mM rac-benzoin, 50 mM acetaldehyde 50 mM TEA, 30% (v/v) DMSO, 0.5 mM ThDP, 2 mM Mg2+, wtBFD (81 U2-HPP mL−1, 9.8 mg mL−1 wtBFD) at pH 7.5 and 25 °C. Lines indicate visual aids. Data points are average values of duplicates. The curve represents an ideal kinetic resolution (i.e. 50% conversion and 100% ee (S)-benzoin). | ||
Starting from 1.6 mM rac-benzoin the concentrations as well as the ee values of benzoin (1) and 2-HPP (4) were monitored during the course of the reaction, showing a perfect kinetic resolution. As shown in Fig. 2A, the concentration of benzoin reached 0.8 mM after 2 h. Only (S)-benzoin was detectable in the reaction mixtures. Simultaneously, 1.6 mM of rac-2-HPP (Fig. 2A) were formed in the presence of 50 mM acetaldehyde.
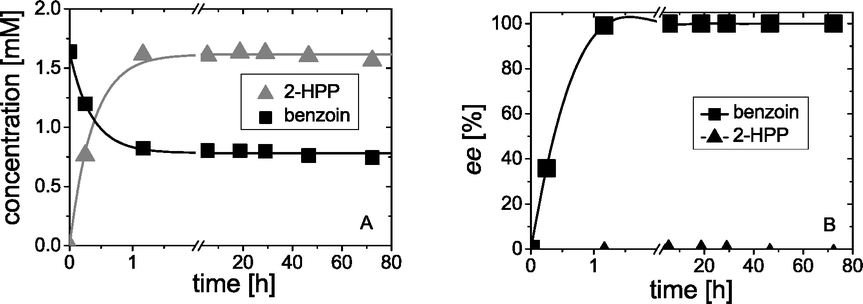 | ||
| Fig. 2 Kinetic resolution of rac-benzoin (1.6 mM) catalyzed by wtBFD in the presence of 50 mM acetaldehyde. Plots show the concentrations of benzoin and 2-HPP (left) and the respective ee-values for (S)-benzoin and 2-HPP (right). Reaction conditions: 50 mM TEA, 30% (v/v) DMSO, 0.5 mM ThDP, 2 mM Mg2+, wtBFD (81 U2-HPP mL−1, 9.8 mg mL−1 wtBFD) at pH 7.5 and 25 °C. Lines indicate visual aids. Data points are average values of duplicates. | ||
The BFD-catalyzed kinetic resolution of benzoin shown here is very similar to that catalyzed by BAL. When (R)-benzoin is incubated with BAL in the presence of acetaldehyde enantiopure (R)-4 is formed.3a,10 Regarding the kinetic parameters and microscopic rate constants, it was shown that BFD and BAL exhibit differences.11 Nevertheless, in addition to their catalytic activities, BFD and BAL are structurally similar, since (i) both enzymes are homotetrameric and (ii) both bind to the cofactor ThDP in the active site.6,12 In contrast to BFD, no ‘S-pocket’ was observed in the structure BAL and hence the strict (R)-enantioselectivity (>99%) of BAL for the formation of (R)-2-HPP is attributed to only one possible arrangement of acetaldehyde in the active site prior to C–C bond formation with ThDP-bound benzaldehyde.7a In this study, formation of benzaldehyde from benzoin catalyzed by BFD could not be verified, as benzaldehyde immediately reacts with the added acetaldehyde to afford 4. For BAL it was postulated that there is a direct reaction involving an enzyme–benzoin complex with acetaldehyde and benzaldehyde to form 4,13,14 which might also be valid for BFD.
Next, we determined the formation rate of 4 from 1.6 mM rac-benzoin and 50 mM acetaldehyde by linear regression. Table 1 illustrates the formation rate of 4via kinetic resolution of benzoin compared with that of via direct carboligation. Here, it was clearly seen that the formation of 4 starting from benzoin and acetaldehyde was ~700-fold slower than the direct carboligation of benzaldehyde and acetaldehyde.
| V Direct carboligation [U2-HPP mg−1]a | V Kinetic resolution [U2-HPP mg−1]b |
|---|---|
| a Conditions for direct carboligation: 40 mM benzaldehyde, 400 mM acetaldehyde, 50 mM phosphate buffer, 0.5 mM ThDP, 2 mM Mg2+ at pH 7.5 and 30 °C. b Conditions for kinetic resolution of benzoin: 1.6 mM rac-benzoin, 50 mM acetaldehyde, 50 mM TEA buffer, 0.5 mM ThDP, 2 mM Mg2+, 30% (v/v) DMSO at pH 7.5 and 25 °C. Results are average values of duplicates. | |
| 8.3 | 11.3 × 10−3 |
Cleavage of 2-HPP catalyzed by BFD
Encouraged by the results obtained for the kinetic resolution of rac-benzoin, we evaluated the cleavage of 4 as it might also be accepted as the substrate by BFD (Scheme 2). | ||
| Scheme 2 Postulated reaction scheme for the cleavage of (S)- or (R)-4 to benzaldehyde and acetaldehyde, with subsequent reformation of rac-4. | ||
To evaluate 4 as a substrate for BFD and to investigate the formation of surprising rac-4 in detail, we applied enantiopure (>99.9% ee) (S)-4 or (R)-4 and monitored the ee values. Our results showed that low concentrations of acetaldehyde are required to accelerate the racemization of 4 (data not shown); hence, we applied 25 mM acetaldehyde. The formation of the enantiocomplementary form of 4 was analyzed starting from different concentrations of 4 (≤170 mM) (Fig. 3). The maximum solubility of 4 in aqueous medium was determined to be 180 mM at 25 °C (measured for both enantiomers). The BFD-mediated cleavage of 4 showed a ‘classic’ Michaelis–Menten activity depending on the substrate concentration. The KM values were determined as 62 ± 9 mM for (R)-4 and 62 ± 4 mM for (S)-4 and the Vmax values were found as 1.0 mU mg−1 for the formation of (R)-4 and 2.1 mU mg−1 for the formation of (S)-4. Despite of the high enantioselectivity of BFD for the formation of (S)-4 in the direct carboligation of benzaldehyde and acetaldehyde (ee = 92% (S)),2a we observed a similar affinity of BFD for (R)- and (S)-4 in the cleavage reaction. Presently, there is no fully satisfactory explanation for this observation and further kinetic and molecular modelling investigations are required to explain this observation.
 | ||
| Fig. 3 Formation rates of (R)-4 from enantiopure (S)-4 (A) and formation rates of (S)-4 from enantiopure (R)-4 (B). Reaction conditions: (R)- or (S)-4 (0–170 mM), 25 mM acetaldehyde, 50 mM TEA buffer, 0.5 mM ThDP, 2 mM Mg2+, wtBFD (51 U2-HPP mL−1, 6 mg mL−1 wtBFD) at pH 7.5 and 25 °C. Data points are average values of duplicates. | ||
Further on, we investigated the formation rates of (R)- or (S)-4 at different acetaldehyde concentrations. As shown in Fig. 4, maximum rates for the formation of (R)-4 or (S)-4 were detected at low acetaldehyde concentrations (<50 mM). Here, of particular importance is the course of the respective formation rates of 4 at higher acetaldehyde concentrations. An exponential decay in the formation rates of (R)- or (S)-4 from the cleavage of their enantiocomplementray forms was observed when higher acetaldehyde concentrations (>50 mM) were applied. This observed decrease in the enzyme activity with increasing acetaldehyde concentrations was not in agreement with the data reported for the formation of 4 for the direct carboligation of benzaldehyde and acetaldehyde by Wilcocks and Ward (1992).5a Iding et al. (2000)2a also reported a decreased activity of BFD at higher acetaldehyde concentrations for the direct carboligation of benzaldehyde and acetaldehyde to afford 4, however, first when acetaldehyde concentrations exceeded ~500 mM.
 | ||
| Fig. 4 Formation rates of (R)-4 from 30 mM of enantiopure (S)-4 (A) and formation rates of (S)-4 from 30 mM of enantiopure (R)-4 (B) as a function of the acetaldehyde concentration (0–700 mM). Reactions were carried out in 50 mM TEA buffer, 0.5 mM ThDP, 2 mM Mg2+, wtBFD (21 U2-HPP mL−1 (2.5 mg mL−1 wtBFD) for (S)-4 and 33 U2-HPP mL−1 (4 mg mL−1 wtBFD) for (R)-4), at pH 7.5 and 25 °C. Data points are average values of duplicates. | ||
Next, we investigated the formation of benzaldehyde as a cleavage product of (S)- or (R)-4 in the presence of different acetaldehyde concentrations (Fig. 5). Similar to the behavior shown in Fig. 4, formation rates of benzaldehyde also decreased with increasing acetaldehyde concentrations. For concentration values of acetaldehyde higher than 400 mM, benzaldehyde formation rates were ~0.2 mU mg−1. It is important to mention that benzaldehyde formation rates presented here are only apparent values, since only free benzaldehyde in the reaction solution could be detected.
 | ||
| Fig. 5 Formation of benzaldehyde from 30 mM (S)-4 (A) or (R)-4 (B) in the presence of varying acetaldehyde concentrations catalyzed by wtBFD. Reaction conditions: 30 mM (R)- or (S)-4, 0–700 mM acetaldehyde, 50 mM TEA buffer, 0.5 mM ThDP, 2 mM Mg2+, wtBFD (21 U2-HPP mL−1 (2.5 mg mL−1 wtBFD) for (S)-4 and 33 U2-HPP mL−1 (4 mg mL−1 wtBFD) for (R)-4), at pH 7.5 and 25 °C. Data points are average values of duplicates. | ||
Overall, the here presented BFD-catalyzed formation of (R)-4 from (S)-4, and vice versa, follows a two-step process: (1) C–C bond cleavage: formation of benzaldehyde and acetaldehyde from 4 and (2) C–C bond formation: reformation of (R)- or (S)-4 from benzaldehyde and acetaldehyde (Scheme 2).
Negative control for the kinetic resolution of rac-benzoin
To exclude a non-enzymatic cleavage of benzoin, three negative control experiments were performed: (1) without BFD to examine a possible cleavage of benzoin catalyzed by buffer components, (2) using heat-inactivated BFD (a variant with similar activity) (30 min incubation at 80 °C) and lastly (3) using BFD (a variant with similar activity) with additional 20 mM of 3-chloromethylbenzoyl-phosphonate (3-Cl-MBP) added as inhibitor.15 The amount of enzyme used in negative controls (2 and 3) was 30 U2-HPP mL−1 (3.6 mg mL−1) BFD defined under standard synthesis conditions (see experimental section). In all negative controls 1.6 mM of rac-benzoin and 50 mM of acetaldehyde were used and no formation of 4 was detected over 72 h.Negative control for the racemization of 4
In order to exclude a potential auto-catalytical racemization of 4 in the absence of enzyme two negative controls (starting from 30 mM (R)-4 (>99% ee) and 25 mM acetaldehyde) were performed: (1) with heat-inactivated 33 U2-HPP mL−1 (4 mg mL−1) of wtBFD (30 min incubation at 80 °C) and (2) with 33 U2-HPP mL−1 (4 mg mL−1) of wtBFD in the presence of 20 mM of 3-Cl-MBP used as inhibitor.15 In the positive control, the ee dropped to 78% (R)-4 after 24 h, whereas no change was observed in the negative controls. Even no formation of benzaldehyde was detected in the negative controls. Whereas, 0.17 mM benzaldehyde were found after 24 h in the positive control.Equilibrium conditions for 2-HPP, benzoin, benzaldehyde and acetaldehyde
In order to determine the equilibrium concentrations of the reaction components, a racemic mixture of 4 (at 140 mM) was incubated in the presence of acetaldehyde (at 50 mM). Although equimolar amounts of acetaldehyde are formed due to the cleavage of 4, the supply of 50 mM acetaldehyde was necessary since formed acetaldehyde may evaporate due to its low boiling point (20.4 °C). Therefore, it is highly recommended to run the reactions as well as to perform sampling under slightly pressurized conditions to prevent evaporation of acetaldehyde. As shown in Fig. 6, benzaldehyde and (R)-benzoin concentrations increased during the course of reaction. After 90 h, benzoin and benzaldehyde reached 0.4 mM and 1.2 mM, respectively, whereby the concentration of 4 was almost constant at 140 mM during the course of reaction. | ||
| Fig. 6 Concentrations of 2-HPP, benzaldehyde and (R)-benzoin at approached equilibrium. The reaction was catalyzed by wtBFD. Reaction conditions: 140 mM rac-4, 50 mM acetaldehyde, 50 mM TEA buffer, 0.5 mM ThDP, 2 mM Mg2+, BFD (204 U2-HPP mL−1, 25 mg mL−1 wtBFD), at pH 7.5 and 25 °C. Data points are average values of duplicates. | ||
Based on the aforementioned results showing: (i) the formation of 4 from (R)-benzoin and acetaldehyde and (ii) the racemization of 4, we postulate a mechanism shown in Scheme 3, whereby the substrates (e.g. benzaldehyde and acetaldehyde) and the carboligation products (e.g.4 and (R)-benzoin) are in equilibrium.
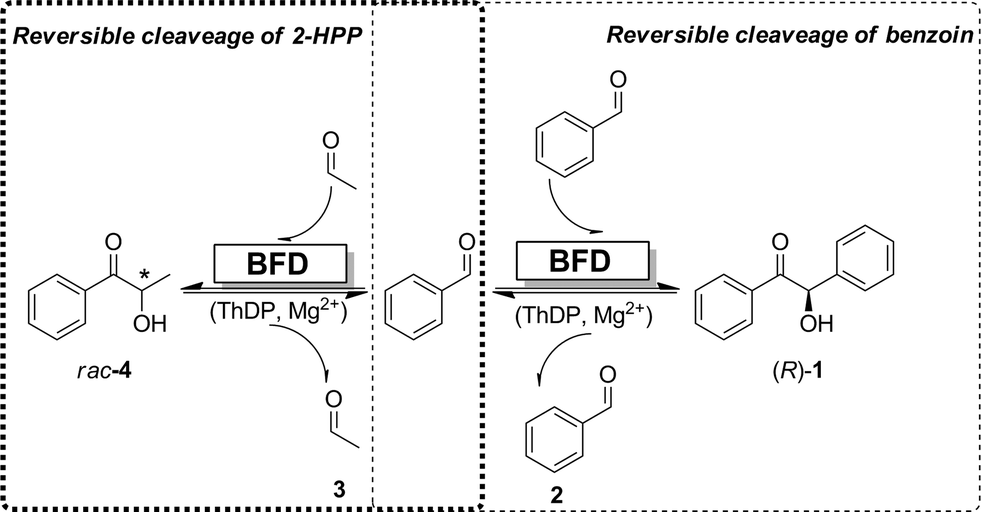 | ||
| Scheme 3 Schematic representation of equilibrium between benzaldehyde (2), acetaldehyde (3), 2-HPP (4) and (R)-benzoin (1). | ||
To describe the whole reaction (Scheme 3), as a reversible-, coupled- and isolated system, the following equation was used:
| 3 benzaldehyde + acetaldehyde ⇌ (R)-benzoin + 2-HPP |
Thus, the equilibrium constant “K” can be calculated as:

When the equilibrium concentrations of all reaction components are known, the equilibrium constant can be determined based on the below given assumptions:
1. The equilibrium concentration of 4 is the average value of the data measured during the reaction course, as no significant change in the concentrations of 4 was detectable (Fig. 6).
2. The concentration of acetaldehyde at equilibrium equals to its initial concentration since the formation of acetoin and/or evaporation of acetaldehyde from the reaction medium is neglected.
3. The equilibrium concentrations of (R)-benzoin and benzaldehyde are calculated from exponential regression for t → ∞.
The concentrations of reaction components at equilibrium and the equilibrium constant are illustrated in Table 2. A very high equilibrium constant of 6.3 × 105 L2 mol−2 clearly indicates that the reaction is favored to the side of 2-hydroxy ketones (e.g.4 and (R)-benzoin), whereby 4 is the main product. The very low concentration of benzaldehyde at equilibrium shows also that the formation of 2-HPP proceeds very slowly due to the high KM value for benzaldehyde (~80 mM ref. 2a, 9 and 16). The same is also true for benzoin formation, since for (R)-benzoin formation no substrate saturation was achieved up to ~40 mM of benzaldehyde.16 As mentioned above, the synthesis of acetoin by BFD catalysis was neglected due to the previously reported low activity of BFD for this reaction.2a,16,17
| 2-HPP [mM] | AA [mM] | BA [mM] | (R)-Benzoin [mM] | K Eq. [L2 mol−2] |
|---|---|---|---|---|
| 141.3 ± 4.2 | 50 | 1.37 ± 0.04 | 0.58 ± 0.04 | 6.3 × 105 |
Based on our results, the existing reaction mechanism for decarboxylation of benzoylformate6b and for the formation of (S)-4ref. 2a can be extended as shown in Scheme 4. In the proposed enhanced reaction mechanism the following principles and reaction steps are considered:
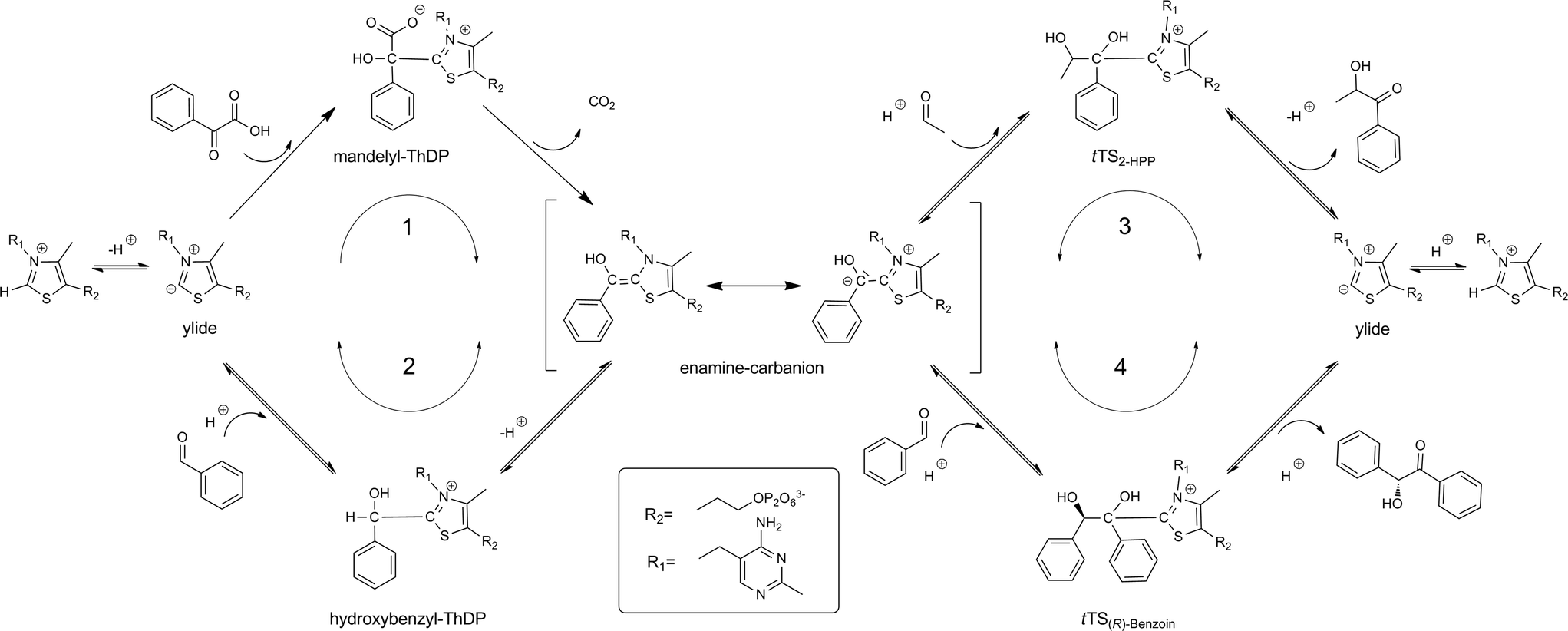 | ||
| Scheme 4 Extended reaction mechanism of BFD based on the reaction mechanisms given in literature.2a,5b In addition to the irreversible decarboxylation of benzoylformate (route 1), the binding of benzaldehyde (route 2), 2-HPP (4) (route 3) and (R)-benzoin (route 4) to the ylide-form of the cofactor ThDP is possible. Starting from a respective tetrahedral transition state (tTS) and followed elimination of acceptor electrophile (e.g. proton (route 2), acetaldehyde (route 3) and benzaldehyde (route 4); irreversible elimination of CO2 (route 1)), the enamine–carbanion intermediate is formed. As the tetrahedral transition states: mandelyl-ThDP (route 1), hydroxybenzyl-ThDP (route 2), tTS2-HPP (route 3) and tTS(R)-benzoin (route 4) are formed. | ||
1. The cofactor ThDP is in equilibrium with its reactive, deprotonated ylide-form.
2. The mandelyl-ThDP complex15 is formed from benzoylformate and ThDP-ylide, followed by an irreversible formation of enamine–carbanion intermediate via elimination of CO2 (route 1).
3. Benzaldehyde can reversibly bind to ThDP-ylide and thus forms hydroxybenzyl-ThDP15 and the enamine–carbanion intermediate is formed by reversible deprotonation (route 2).
4. The binding of acetaldehyde as an acceptor substrate at C2-atom of the enamine–carbanion forms a new tetrahedral transition state, 1-phenyl-1,2-dihydroxypropyl-ThDP (tTS2-HPP), which reversibly yields the ThDP-ylide by elimination of 4 (route 3).
5. The binding of benzaldehyde as an acceptor at C2-atom of the enamine–carbanion complex provides another tetrahedral complex, 1,2-dihydroxy-1,2-diphenylethyl-ThDP (tTS(R)-benzoin), which reacts to the ThDP-ylide by reversible elimination of (R)-benzoin (route 4).
6. The chiral information of the carboligation products 4 and (R)-benzoin at the tetrahedral transition states is included, since these complexes already have the chirality of the products (route 3 and 4).
7. All reaction steps are in principle reversible, only the decarboxylation activity of BFD can be regarded as quasi-irreversible.
The BFD-mediated racemization of (S)-4 can be explained by the above postulated reaction mechanism (Scheme 4). Based on the considerations given above, (S)-4 binds first to the ThDP-ylide forming the tTS2-HPP adduct (route 3) which is followed by the elimination of acetaldehyde to afford the enamine–carbanion intermediate. However, acetaldehyde can rebind to the enamine–carbanion and thus reforms the tTS2-HPP adduct (route 3) which predominantly yields (S)-4 (e.g. 92% (S)5a and 90% (S)17) but tiny amounts of (R)-4 are also formed. While the reaction proceeds these ‘selectivity mistakes’ accumulate and hence give a racemic mixture at the end, since the formation of (S)-4 is highly preferred over (R)-4 whereas the kinetic parameters for the BFD-catalyzed cleavage reaction of both enantiomers are similar. In principle, the same might also be true for (R)-benzoin; however, since only (R)-benzoin is formed via BFD catalysis, racemization of (R)-benzoin is not possible. This is due to the fact that benzaldehyde cannot fit into the ‘S-pocket’7 found in BFD due to steric hindrance and hence only (R)-benzoin is formed.
Herein, the orientation of the acceptor aldehyde to the enamine–carbanion has to be justified since binding of the acceptor aldehyde to the prochiral enamine–carbanion intermediate is the crucial step. Iding et al. (2000) analyzed the ee of the carboligation products (e.g.4 or benzoin) based on Re- or Si-attack on the acceptor aldehyde (e.g. acetaldehyde in the synthesis of 4 or benzaldehyde in the synthesis of benzoin).2a
The above described extended reaction model can also explain the observed BFD-catalyzed racemization of 4. Racemization takes place since the tetrahedral transition state (tTS2-HPP) is cleaved into the enamine–carbanion (EC) and acetaldehyde (AA). Subsequently, 4 is formed from the cleavage products. Therefore, this elementary step can be expressed as:

At equilibrium if c(acetaldehyde) is increased then c(EC) should decrease or c(tTS2-HPP) should increase. However, a decrease of the average concentration of c(EC) leads to decreased reaction rates from EC to tTS2-HPP, which means decreased racemization rates. Furthermore, reduced racemization rates of 4 in the presence of low acetaldehyde concentrations (0–25 mM) (Fig. 4) can result from a substrate-limited reaction of the EC and acetaldehyde towards tTS2-HPP. In addition, increased reaction rates by increasing the concentrations of 4 (Fig. 3) can be simply explained by the increased amounts of 4 reacting with the ThDP-ylide to form tTS2HPP.
Further on, the formation of 4 from (R)-benzoin and acetaldehyde can be explained through our postulated mechanism. Here, (R)-benzoin binds first to ThDP-ylide to form the enamine–carbanion (route 4). Subsequently, the enamine–carbanion can further react with acetaldehyde yielding 4 (route 3). Starting from rac-benzoin, whereby only (R)-benzoin is accepted as the substrate, a classical resolution yields in 4 and (S)-benzoin (Fig. 2). Consequently, tTS(R)-benzoin can only be formed in the (R)-form since only (R)-benzoin is synthesized from benzaldehyde. As aforementioned, due to steric hindrance benzaldehyde cannot fit into the previously defined S-pocket7a and thus no proper alignment of enamine–carbanion and benzaldehyde is possible to form tTS(S)-benzoin.
Conclusion
This work represents a first time report of the C–C bond cleavage activity of benzoylformate decarboxylase from Pseudomonas putida towards 2-hydroxy ketones. This has been demonstrated by the formation of rac-2-HPP from rac-benzoin via the in situ removal of benzaldehyde in the presence of acetaldehyde. However, the carboligation reaction to form (R)-benzoin and 2-HPP is favored by BFD as also indicated by the very high equilibrium constant. A racemic mixture of 2-HPP was detected while using benzoin as the donor in the presence of acetaldehyde. Not only (R)-benzoin but also both enantiomers of 2-HPP are accepted by BFD as a substrate for the cleavage reaction. The similar affinity of BFD observed for the cleavage of (R)- and (S)-2-HPP necessitates further kinetic investigations. Herein, in silico substrate docking for both enantiomers to investigate the orientation of phenyl- and methyl groups of 2-HPP in the ‘S-pocket’ might be useful.The formation of (R)-2-HPP from (S)-2-HPP, and vice versa, was demonstrated and the formation rates were shown to be dependent of the acetaldehyde concentration. The formation of benzaldehyde as the cleavage product of 2-HPP was successfully monitored. Lastly, the reaction mechanism reported previously was supplemented with the observed C–C bond cleavage activity of BFD, which explains the ‘reversibility’ of the BFD-catalyzed C–C bond formations.
The practical usefulness of the BFD-catalyzed C–C cleavage in the kinetic resolution of benzoin can be improved by increasing the solubility of benzoin. To overcome this challenge the reaction can be performed under low water activity conditions (e.g. neat substrate, organic media etc.), where BFD is applied in an immobilized form or as in the whole cells.
Experimental
The chemicals used in this study were commercially obtained in analytical-grade quality and were used as received. Aldehydes were distilled prior to their use.Preparation of wtBFD
Detailed description of cell cultivation, gene expression, and enzyme purification are found in the PhD thesis of M. Berheide18 and Berheide et al. (2010).19Determination of the activity of BFD for direct carboligation of benzaldehyde and acetaldehyde
Activity analysis was performed in 50 mM phosphate buffer, 0.5 mM ThDP, 2 mM MgCl2 at pH 7.5 and 30 °C. The substrate stock of benzaldehyde and acetaldehyde was prepared in the above given buffer, respectively. The substrate solution (4.9 mL) was incubated at 30 °C for 3 min in a glass vessel with a volume of 10 mL. The reaction was started by the addition of wtBFD stock solution (0.1 mL) which was prepared in 50 mM phosphate buffer, 0.5 mM ThDP, 2 mM MgCl2 at pH 7.5. Final concentrations of benzaldehyde and acetaldehyde were 40 mM and 400 mM, respectively. Samples were taken at definite time intervals and quenched with stop-solution (acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H3PO4 (95:5), v/v) at a 2:1 ratio (sample:stop-solution, v/v). After centrifugation of the precipitate (13000 rpm, 3.5 min), samples were analyzed by HPLC.
:H3PO4 (95:5), v/v) at a 2:1 ratio (sample:stop-solution, v/v). After centrifugation of the precipitate (13000 rpm, 3.5 min), samples were analyzed by HPLC.
Here, one unit of activity (U2-HPP) is defined as the amount of enzyme which catalyzes the formation of 1 μmol of 2-HPP in 1 min at 30 °C under the conditions given above. Protein amounts were determined by the standard Bradford method20 using bovine serum albumin (BSA) as a standard.
Analysis of kinetic resolution of rac-benzoin
The experiments for kinetic resolution of benzoin were performed in 1.93 mL of total volume using 30% (v/v) DMSO. The stock solution of rac-benzoin was prepared in DMSO at a concentration of 5.2 mM in the presence of 50 mM acetaldehyde. The enzyme solution was prepared by dissolving lyophilized wtBFD (157 U2-HPP, 19 mg wtBFD) in 1.35 mL of 50 mM TEA buffer, 0.5 mM ThDP, 2 mM MgCl2 at pH 7.5 and 25 °C, which also contained 50 mM of acetaldehyde. The enzyme solution was cooled on ice and the reactions were started by addition of 0.58 mL of rac-benzoin stock solution (also containing acetaldehyde) into the enzyme solutions. Therefore, the final concentrations were 1.6 mM of rac-benzoin and 50 mM of acetaldehyde. Samples were taken at definite time intervals over a period of 312 h and quenched with stop-solution (acetonitrile:H3PO4 (95:5), v/v) at a 2:1 ratio (sample:stop-solution, v/v). This was followed by pelleting of the precipitate (13,000 rpm, 3.5 min) and subsequent HPLC analysis.
Analysis of cleavage of 2-HPP (4)
All experiments were performed in 50 mM TEA buffer, 0.5 mM ThDP, 2 mM MgCl2 at pH 7.5 and 25 °C. The stock solutions of (S)- and (R)-4 were prepared at a concentration of 180 mM which also contained 25 mM of acetaldehyde and the final concentrations were adjusted to be 5, 50, 80, 110, 140 and 170 mM of (S)- or (R)-4. To dilute the stock solutions of 4 TEA buffer in the presence of 25 mM acetaldehyde was used. The enzyme solutions were prepared by dissolving lyophilized wtBFD in TEA buffer in the presence of 25 mM acetaldehyde. Reactions were started by addition of the enzyme solution (51 U2-HPP wtBFD) to the substrate solution. Samples were taken over a period of 30 h, quenched with stop-solution (as above described) and subsequently analyzed by HPLC. Total reaction volume was 1 mL. The Michaelis–Menten kinetic parameters were determined using Origin 9.1 (Origin Lab Corporation, Northampton, MA, USA).To analyze the reactions using varying acetaldehyde concentrations, a stock solution of 2 M acetaldehyde in 50 mM TEA buffer, 0.5 mM ThDP, 2 mM MgCl2 at pH 7.5 and 25 °C was prepared. The concentration of (S)- or (R)-4 was 30 mM. The final concentrations of acetaldehyde were adjusted to 0, 25, 50, 200, 400 and 700 mM. The concentration of enzyme used per reaction was 21 U2-HPP mL−1. Only in the case of 30 mM (R)-4 as substrate, 33 U2-HPP mL−1 of wtBFD was applied (Fig. 4(B)). Samples were taken over a period of 30 h and handled as described before. Total reaction volume was 1 mL.
Analysis of reaction equilibrium starting from rac-4 and acetaldehyde
A stock solution containing 180 mM of (S)-and (R)-4 and 50 mM of acetaldehyde was prepared in TEA buffer. The lyophilized enzyme was dissolved in 222 μL TEA buffer which also contained 50 mM of acetaldehyde. Reactions were started by addition of 778 μL of rac-4 solution (containing 50 mM acetaldehyde) so that the final concentrations were 140 mM rac-4 and 50 mM acetaldehyde. Reactions contained 204 U2-HPP of wtBFD. Samples were taken over a period of 92 h, processed as previously described and analyzed by HPLC. Total reaction volume was 1 mL.Standard deviations observed in duplicated experiments
In case of determination of conversion values (e.g.Fig. 2A, 5 and 6) the deviation between duplicated experiments was 3–15%, whereas in case of the determination of ee values (Fig. 1, 2B, 3, and 4) the standard deviation between experiments was 3–5%.Negative control for kinetic resolution of rac-benzoin
Information on the BFD variant: the used BFD variant (BFD A460I) showed cleavage activity like wild type BFD under the same reaction conditions.19Synthesis of enantiopure (S)-4
Substrate solution containing benzaldehyde (47.25 g, 445 mmol) and acetaldehyde (195 g, 4.42 mol) was prepared in 10.5 L potassium phosphate buffer (20 mM), ThDP (0.2 mM) and MgCl2 × 6H2O (2 mM). The pH was adjusted to 7.5 using concentrated H3PO4 and NaOH. Reaction was started by the addition of 1400 U2-HPP of freshly purified wtBFD to the substrate solution and run at room temperature. After 36 h the total reaction mixture was extracted with dichloromethane (5 × 200 mL). The organic phase was washed with water and brine then dried over using magnesium sulfate and lastly the solvent was removed with a rotary evaporator. The crude product (68.3 g, ee(S)-2-HPP = 85%) was obtained as a yellow viscous oil. After recrystallization using isohexane, (S)-4 (ee > 99.9%, yield = 44.85 g, 67.2%) was isolated as needle-shaped crystals (~3 cm long).1H-NMR (400 MHz, CDCl3) δ (ppm): 1,47 (d, 3H, ,3J = 7,1 Hz; CH3); 3,82 (br, 1H; OH); 5,19 (q, 1H, 3J = 7,1 Hz; CHOH); 7,52 (“t”, 2H, 3J = 7,47 Hz; Ar–H); 7,64 (tt, 1H, 3J = 7,47 Hz, 4J = 1,3 Hz; Ar–H); 7,95 (dd, 2H, 3J = 7,07 Hz, 4J = 1,3 Hz; Ar–H).
13C-NMR (100 MHz, CDCl3) δ (ppm): 22,32 (CH3); 69,32 (CHOH); 128,67/128,89 (CH); 133,31 (Cq); 134,01 (CH); 202,4 (CO).
Synthesis of enantiopure (R)-4
Benzaldehyde (4.6 g, 43 mmol) and acetaldehyde (17.64 g, 0.4 mol) were dissolved in 1 L of potassium phosphate buffer (50 mM), ThDP (0.5 mM) and MgCl2 × 6H2O (2 mM) and 5% MTBE (v/v). The pH was adjusted to 8.0 using concentrated H3PO4 and NaOH and the reaction was started by addition of 40 mg of BAL (provided by Dr. Nils Kurlemann). Reaction was run at room temperature and after 130 h the reaction mixture was extracted with dichloromethane (5 × 100 mL). The combined organic phase was washed with water and brine, dried over magnesium sulfate and lastly the solvent was removed with a rotary evaporator. The crude product (9.69 g) was obtained in pale yellow, viscous form and recrystallized using isohexane. In the end, (R)-4 (ee > 99.9%, yield = 6.3 g, 97%) was isolated as needle-shaped crystals (~4 cm long).1H-NMR (400 MHz, CDCl3) δ (ppm): 1,47 (d, 3H, 3J = 7,05 Hz; CH3); 3,82 (br, 1H; OH); 5,19 (q, 1H, 3J = 7,05 Hz; CHOH); 7,52 (“t”, 2H, 3J = 7,4 Hz; Ar–H); 7,64 (tt, 1H, 3J = 7,4 Hz, 4J = 1,3 Hz; Ar–H); 7,95 (dd, 2H, 3J = 7,0 Hz, 4J = 1,3 Hz; Ar–H).
13C-NMR (100 MHz, CDCl3) δ (ppm): 22,32 (CH3); 69,32 (CHOH); 128,67/128,89 (CH); 133,31 (Cq); 134,01 (CH); 202,4 (CO).
Analytics
Reactions were analyzed by HPLC (Agilent 1100, Hewlett Packard) equipped with a LiChrosphere RP-8 column (Hypersil, 250 × 4 mm, Merck) and detections were at 254 nm. Triethanolamine (0.2%, pH 3.0):(60:40, v/v) was used as an eluent at a flow rate of 1.0 mL min−1 at 30 °C. Retention times were t2-HPP = 5.0 min, tbenzaldehyde = 8.1 min and tbenzoin = 11.9 min. For ee determination, samples were extracted with isohexane and analyzed by HPLC using a Daicel Chiralcel OD-H column (5 μm). Isohexane:isopropanol (98:2, v/v) was used as a mobile phase at a flow rate of 0.75 mL min−1 at 20 °C and detections were at 254 nm. Retention times were t(S)-2-HPP = 17.8 min, t(R)-2-HPP = 21.8 min, t(S)-benzoin = 30.0 min and t(R)-benzoin = 48.0 min.
Acknowledgements
Special thanks to Prof. Dr. Martina Pohl (Institute of Molecular Enzyme Technology, Heinrich-Heine University of Düsseldorf, Research Center Juelich) for generously providing the wild type BFD. Dr. A. Bornadel is acknowledged for proofreading parts of the manuscript. We thank anonymous referees for critically reading our manuscript and for providing valuable inputs and comments.Notes and references
- (a) M. Pohl and A. Liese, ed. R. N. Patel, in Biocatalysis in the Pharmaceutical and Biotechnology Industries., CRC Press Taylor & Francis Group, Florida, 2007, pp. 661–676 Search PubMed; (b) S. Kara and A. Liese, Enzymatic C–C bond formation, asymmetric, in, Encyclopedia of Industrial Biotechnology. Bioprocess, Bioseparation, and Cell Technology, ed. M. C. Flickinger, Wiley-VCH, 2010, pp. 2034–2049 Search PubMed; (c) L. Hilterhaus and A. Liese, Industrial Application and Processes Using Carbon–Carbon Lyases, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH, 3rd edn, 2012, pp. 991–998 Search PubMed; (d) M. Müller, G. A. Sprenger and M. Pohl, Curr. Opin. Chem. Biol., 2013, 17, 261–270 CrossRef PubMed.
- (a) H. Iding, T. Dünnwald, L. Greiner, A. Liese, M. Müller, P. Siegert, J. Grötzinger, A. S. Demir and M. Pohl, Chem. – Eur. J., 2000, 6, 1483–1495 CrossRef CAS; (b) A. S. Demir, T. Dünnwald, H. Iding, M. Pohl and M. Müller, Tetrahedron: Asymmetry, 1999, 10, 4769–4774 CrossRef CAS; (c) T. Dünnwald, A. S. Demir, P. Siegert, M. Pohl and M. Müller, Eur. J. Org. Chem., 2000, 2161–2170 CrossRef; (d) S. Kara, W. S. Long, M. Berheide, S. Peper, B. Niemeyer and A. Liese, J. Biotechnol., 2011, 152, 87–92 CrossRef CAS PubMed; (e) T. Gerhards, U. Mackfeld, M. Bocola, E. von Lieres, W. Wiechert, M. Pohl and D. Rother, Adv. Synth. Catal., 2012, 354, 2805–2820 CrossRef CAS PubMed.
- (a) A. S. Demir, M. Pohl, E. Janzen and M. Müller, J. Chem. Soc., Perkin Trans. 1, 2001, 633–635 RSC; (b) M. Pohl, B. Lingen and M. Müller, Chem. – Eur. J., 2002, 8, 5288–5295 CrossRef CAS; (c) A. S. Demir, O. Şeşenoğlu, E. Eren, B. Hosrik, M. Pohl, E. Janzen, D. Kolter, R. Feldmann, P. Dünkelmann and M. Müller, Adv. Synth. Catal., 2002, 344, 96–103 CrossRef CAS; (d) A. S. Demir, O. Şeşenoğlu, P. Dünkelmann and M. Müller, Org. Lett., 2003, 5, 2047–2050 CrossRef CAS PubMed; (e) P. Domínguez de María, T. Stillger, M. Pohl, S. Wallert, K.-H. Drauz, H. Gröger, H. Trauthwein and A. Liese, J. Mol. Catal. B: Enzym., 2005, 38, 43–47 CrossRef PubMed; (f) N. Kurlemann and A. Liese, Tetrahedron: Asymmetry, 2004, 15, 2955–2958 CrossRef CAS PubMed; (g) S. Shanmuganathan, D. Natalia, A. van den Wittenboer, C. Kohlmann, L. Greiner and P. Domínguez de María, Green Chem., 2010, 12, 2240–2245 RSC; (h) P. Ayhan and A. S. Demir, Adv. Synth. Catal., 2011, 353, 624–629 CrossRef CAS; (i) C. R. Müller, M. Pérez-Sánchez and P. Domínguez de María, Org. Biomol. Chem., 2013, 11, 2000–2004 RSC.
- (a) I. C. Gunsalus, F. C. Gunsalus and R. Y. Stanier, J. Bacteriol., 1953, 66, 538–542 CAS; (b) G. D. Hegeman, J. Bacteriol., 1966, 91, 1155–1160 CAS; (c) G. D. Hegeman, Methods Enzymol., 1970, 17, 674–678 Search PubMed; (d) A. Y. Tsou, S. C. Ransom, J. A. Gerlt, D. D. Buechter, P. C. Babbitt and G. L. Kenyon, Biochemistry, 1990, 29, 9856–9862 CrossRef CAS.
- (a) R. Wilcocks and O. P. Ward, Biotechnol. Bioeng., 1991, 39, 1058–1062 CrossRef PubMed; (b) R. Wilcocks, O. P. Ward, S. Collins, N. J. Dewdney, Y. Hong and E. Prosen, Appl. Environ. Microbiol., 1992, 58, 1699–1704 CAS.
- (a) M. S. Hasson, A. Muscate, M. J. McLeish, L. S. Polovnikova, J. A. Gerlt, G. L. Kenyon, G. A. Petsko and D. Ringe, Biochemistry, 1998, 37, 9918–9930 CrossRef CAS PubMed; (b) E. S. Polovnikova, M. J. McLeish, E. A. Sergienko, J. T. Burgner, N. L. Anderson, A. K. Bera, F. Jordan, G. L. Kenyon and M. S. Hasson, Biochemistry, 2003, 42, 1820–1830 CrossRef CAS PubMed.
- (a) M. Knoll, M. Müller, J. Pleiss and M. Pohl, ChemBioChem, 2006, 7, 1928–1934 CrossRef CAS PubMed; (b) D. Gocke, D. L. Walter, E. Gauchenova, G. Kolter, M. Knoll, C. L. Berthold, G. Schneider, J. Pleiss, M. Müller and M. Pohl, ChemBioChem, 2008, 9, 406–412 CrossRef CAS PubMed.
- B. Gonzalez and R. Vicuna, J. Bacteriol., 1989, 171, 2401–2405 CAS.
- M. M. Kneen, I. D. Pogozheva, G. L. Kenyon and M. J. McLeish, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1753, 263–271 CrossRef CAS PubMed.
- P. Dominguez de Maria, T. Stillger, M. Pohl, S. Wallert, K. Drauz and H. Gröger, J. Mol. Catal. B: Enzym., 2006, 38, 43–47 CrossRef CAS PubMed.
- M. Kokova, M. Zavrel, K. Tittmann, A. C. Spiess and M. Pohl, J. Mol. Catal. B: Enzym., 2009, 61, 73–79 CrossRef CAS PubMed.
- (a) T. G. Mosbacher, M. Müller and G. E. Schulz, FEBS J., 2005, 272, 6067–6076 CrossRef CAS PubMed; (b) H. C. Hailes, D. Rother, M. Müller, R. Westphal, J. M. Ward, J. Pleiss, C. Vogel and M. Pohl, FEBS J., 2013, 280, 6374–6394 CrossRef CAS PubMed.
- T. Stillger, M. Pohl, C. Wandrey and A. Liese, Org. Process Res. Dev., 2006, 10, 1172–1177 CrossRef CAS.
- F. Hildebrand, S. Kuhl, M. Pohl, D. Vasic-Racki, M. Müller, C. Wandrey and S. Lütz, Biotechnol. Bioeng., 2007, 96, 835–843 CrossRef CAS PubMed.
- M. Bruning, M. Berheide, D. Meyer, R. Golbik, H. Bartunik, A. Liese and K. Tittmann, Biochemistry, 2009, 48, 3258–3268 CrossRef CAS PubMed.
- S. Kara, Online Monitoring of Biocatalytic 2-Hydroxy Ketone Synthesis under Ambient and High Pressure, PhD Thesis, Hamburg University of Technology, Hamburg, 2011 Search PubMed.
- (a) P. Siegert, M. J. McLeish, M. Baumann, H. Iding, M. M. Kneen, G. L. Kenyon and M. Pohl, Protein Eng., Des. Sel., 2005, 18, 345–357 CrossRef CAS PubMed; (b) P. Siegert, Vergleichende Charakterisierung der Decarboxylase- und Carboligasereaktion der Benzoylformiatdecarboxylase aus Pseudomonas putida und der Pyruvatdecarboxylase aus Zymomonas mobilis mittels gerichteter Mutagenese, PhD Thesis, Heinrich-Heine University of Düsseldorf, Düsseldorf, 2000 Search PubMed.
- M. Berheide, Untersuchungen zur Enantioselektivität Thiamindiphosphat abhängiger Enzyme: Reaktionstechnische Optimierung der C-C Bindungsbildung, PhD Thesis, Hamburg University of Technology, Hamburg, 2010 Search PubMed.
- M. Berheide, S. Peper, S. Kara, W. S. Long, S. Schenkel, M. Pohl, B. Niemeyer and A. Liese, Biotechnol. Bioeng., 2010, 106, 18–26 CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
Footnote |
| † In memory of Ayhan S. Demir (1950–2012). |
| This journal is © The Royal Society of Chemistry 2015 |