
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding and exploiting nanoparticles' intimacy with the blood vessel and blood
Magdiel Inggrid
Setyawati
a,
Chor Yong
Tay
a,
Dominic
Docter
b,
Roland H.
Stauber
b and
David Tai
Leong
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: cheltwd@nus.edu.sg
bMolecular and Cellular Oncology, ENT/University Medical Center of Mainz, Langenbeckstr. 1, 55101 Mainz, Germany
First published on 4th August 2015
Abstract
While the blood vessel is seldom the target tissue, almost all nanomedicine will interact with blood vessels and blood at some point of time along its life cycle in the human body regardless of their intended destination. Despite its importance, many bionanotechnologists do not feature endothelial cells (ECs), the blood vessel cells, or consider blood effects in their studies. Including blood vessel cells in the study can greatly increase our understanding of the behavior of any given nanomedicine at the tissue of interest or to understand side effects that may occur in vivo. In this review, we will first describe the diversity of EC types found in the human body and their unique behaviors and possibly how these important differences can implicate nanomedicine behavior. Subsequently, we will discuss about the protein corona derived from blood with foci on the physiochemical aspects of nanoparticles (NPs) that dictate the protein corona characteristics. We would also discuss about how NPs characteristics can affect uptake by the endothelium. Subsequently, mechanisms of how NPs could cross the endothelium to access the tissue of interest. Throughout the paper, we will share some novel nanomedicine related ideas and insights that were derived from the understanding of the NPs' interaction with the ECs. This review will inspire more exciting nanotechnologies that had accounted for the complexities of the real human body.
 Magdiel Inggrid Setyawati | Dr Magdiel Inggrid Setyawati is currently a postdoctoral fellow at the Department of Chemical and Biomolecular Engineering, National University of Singapore (NUS). She obtained her MS degree from National Taiwan University of Science and Technology and her PhD from NUS. Her research interests include nanomaterials-cell interactions, biomedical applications and potential toxicity of nanomaterials. |
 Chor Yong Tay | Dr Chor Yong Tay is currently a Lee Kuan Yew Postdoctoral Fellow at the Department of Chemical and Biomolecular Engineering, National University of Singapore (NUS). He obtained his PhD from the Department of Materials Science and Engineering, Nanyang Technological University (Singapore) in 2012. His main research interests include cell–material interactions, nanomaterials for biomedical applications, cell mechanobiology, and regenerative medicine. |
 Dominic Docter | Dr Dominic Docter is currently a junior group leader in the Department of Nanobiomedicine at the ENT University Medical Center of Mainz and is supported by the Peter and Traudl Engelhorn Foundation. He received his diploma degree in biology in 2008 and his PhD degree at the Johannes Gutenberg University Mainz in 2014 after participating in the “Schwerpunktprogramm: Biological response to Nanoscale Particles” of the German Research Foundation. He has received the NMFZ-research award from the University Medical Center of Mainz. His research is focused on understanding events at the host-nano–bio interface to optimize and explore possible nano-diagnostic and nano-therapeutic applications. |
 Roland H. Stauber | Dr Roland H. Stauber is currently a professor at the Molecular and Cellular Oncology/Nanobio-medicine department at the ENT Medical University Mainz. His research interests focus on the understanding of molecular mechanisms at the nano–bio interface, the development of nano- and microtechnology for cancer treatment and diagnosis, as well as drug development. He has received the Alexander Karl Award for Cancer Research and a President's Fellowship of the Chinese Academy of Sciences. Dr Stauber received his PhD degree from Würzburg University in 1994 and did his post-doctoral training at the National Cancer Institute (USA). |
 David Tai Leong | Dr David Tai Leong is currently an assistant professor at the Department of Chemical and Biomolecular Engineering, National University of Singapore (NUS). He obtained his PhD in Biology and Bachelor in Chemical Engineering from NUS and received his post-doctoral training at the Howard Hughes Medical Institute at the University of Pennsylvania. His research interests span across fundamental understanding of biological effects of nanomaterials to their applications. He has received the Lee Kuan Yew Postdoctoral Fellowship. |
1. Introduction
The applications of engineered nanomaterials (NMs) are not only increasing in technical products but also more and more in biotechnology and biomedicine.1–5 Thus, the ‘marriage’ of nanotechnology with biomedicine defines one of the most exciting and cross-disciplinary developments over the last decade.1–4 In the field of nanobiomedicine, NMs and nanoparticles (NPs) have exhibited promise as tools with improved efficacy, biodistribution, and pharmacokinetics.6–10 Recent advancements in synthesis and the ability to rationally manipulate NMs'/NPs' features, such as their physical, chemical, and biological properties, open up new horizons to rationally design a variety of clinically relevant applications, like drug delivery, in vitro diagnostics, imaging nanoprobes, contrast agents and photodynamic therapy agents.11–20 Moreover, with the advent of the concept of so called ‘personalized medicine’, the field has started to grow producing a huge variety of different (multi-) functional NMs.21–24 However, despite the increasing production of new nano-tools, still few of them reached the clinics. One of the most challenging hurdles that NMs are facing in biomedicine, is to successfully crossing biological barriers and still are able to specifically recognize the target. Moreover, besides the current enthusiasm for nanotechnology, the use of nanomaterials may pose unknown risks to patients and thus, ‘safety comes first’ particularly in the field of nanobiomedicine.25–32While the vasculature is not the common target of interest by these NPs, due to the intravenous injection as a popular route of entry for this diagnostic imaging and therapeutic drug delivery systems, has inevitably made the vasculature as the main organ of tissue where there are unintended effects of those nanomedicine formulations (Fig. 1). Despite its obvious importance for any intravenously introduced nanomedicine, testing for side effects or toxicity of nanomedicine on these cell types are often overlooked. Instead, sometimes irrelevant cell types such as NIH-3T3 mouse fibroblasts were used to test for toxicity towards non-targeted cell types.33,34 With this review, we can educate the field on the diversity of endothelial cells (ECs) found in the human body so that they can better appreciate and thereby include more appropriate EC model in accordance to their target tissue. These would further enhance overall nanosafety and better align animal and in vitro studies with real anatomical relevance.
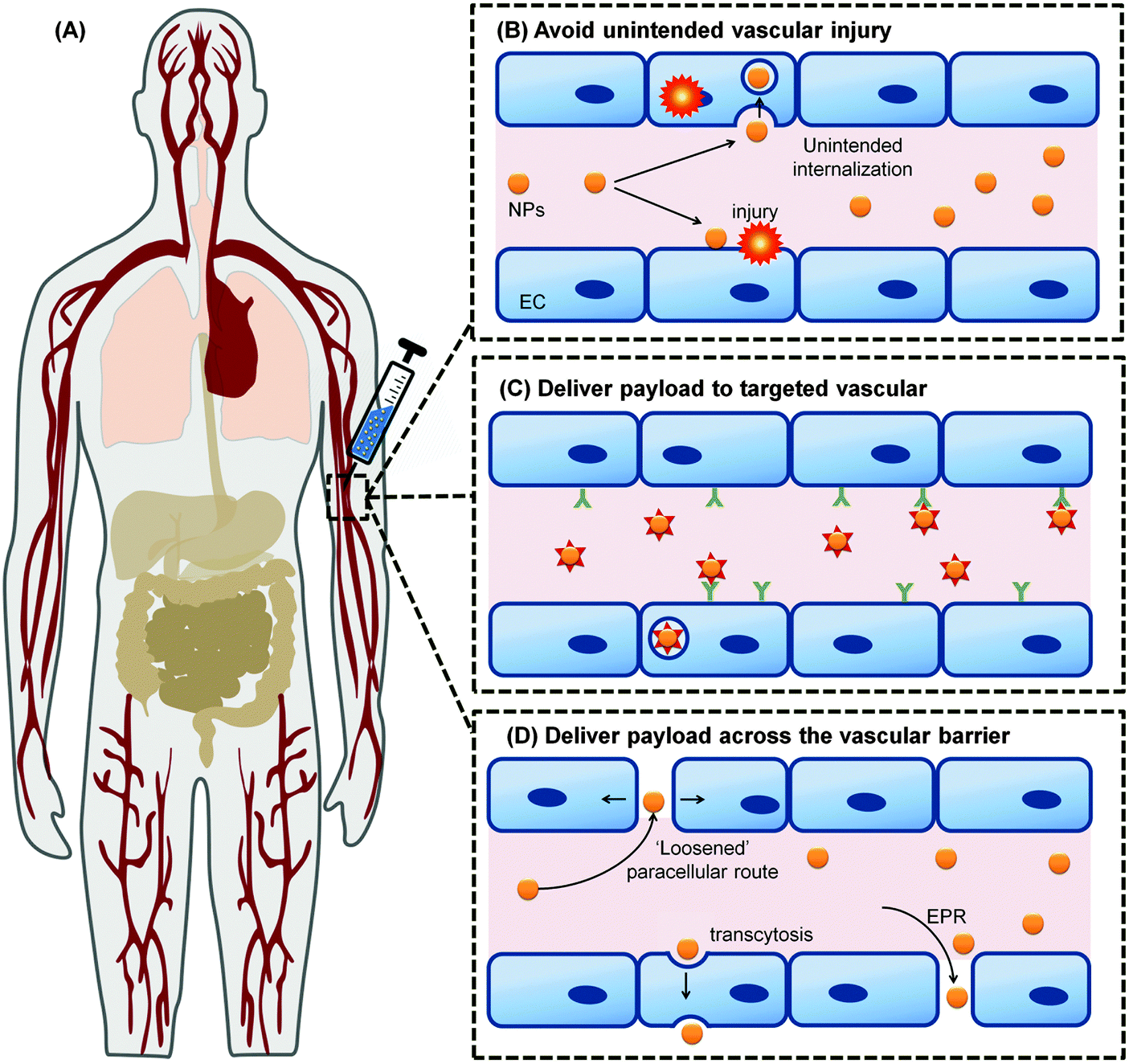 | ||
| Fig. 1 The vascular system permeates through all tissues in the body; (A) making the vasculature as the main route of intentional introduction of nanomedicine. Understanding the interaction between these NMs with the blood vessel and blood that it carries will allow the nanomedicine to (B) avoid unintended interaction with the ECs and causing vascular injury. It also allows optimum nanomedicine design that could engage the vascular to deliver their payload (C) following their internalization into the vascular target, or (D) across the vascular barrier. Panel (A) is adapted from ref. 35 with the permission of The Royal Society of Chemistry. Panel (B)–(D) adapted from ref. 36 under the license of ACS AuthorChoice. This is an unofficial adaptation of an article that appeared in an ACS publication. ACS has not endorsed the content of this adaptation or the context of its use. | ||
Among the multiple components of the blood system, ECs could be considered as a main target tissue due to its dual role as a regulated barrier and ‘unintended victim’ of nanobiomedical approaches (Fig. 1). Hence, emphasis will be given to the role of ECs as an important target tissue and interfering barrier to the nanomaterial-based therapeutics. Besides the relatively static nature of blood vessels, NMs will also interact with the more ‘fluid’ tissue of the vascular system; the blood itself. Blood contains an abundance of diversity in type and number of cells mixed in with protein rich plasma. Upon entry into the blood environment, it is thought that the NPs would acquire an interfacial layer of proteins which is in dynamic equilibrium and highly varied and aptly defined as the protein corona. Since this protein corona is what separates the underlying material of the NP from the bulk plasma, any eventual outcome would be highly dependent on this interfacial layer. Understanding the formation, kinetics and final interaction of this quintessential protein corona with blood cells, with ECs and subsequently the target cells would therefore bring about alterations in our current emphasis on the NMs itself.
In this review, we have delved deep into the exposing the field to the plethora of EC types and certain important nuances in their morphological differences in normality and in pathological states and linking these differences with their functions with the intent of exploiting this knowledge in nanomedicine design. Coupling to this theme, we systematically categorized the various parameters of the protein corona that should be considered when designing nanomaterials that may have interactions with blood and ECs. Finally, we have embedded our insights and presented interesting new ideas that synergize protein corona presentations with EC biology to perhaps stimulate other new studies and even more exciting novel strategies.
2. Endothelial cell heterogeneity in health and disease
The ECs in human body possess a coverage equal to a tennis court (nearly 270 m2 in surface area).37 They share a common characteristic of forming the inner lining of the vast extensive blood vessel network in human body. It is estimated that an average adult human (male, 70 kg) possess a 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kilometer-long of blood vessel network, requiring approximately 1–6 × 1013 ECs to form its interior surface.38 All ECs share a common characteristic that they are tethered on the luminal surface of blood vessels, bringing them into intimate contact with blood and non-blood components of the blood vessel. They also exert a potent anti-coagulant activity39 and express common biomarkers.40 While all ECs are similar in makeup, they are not called to function in exactly the same manner. Different ECs in different tissues have different structural adaptions to fulfil the diversity of functions. In addition, they express protein biomarkers that are tissue specific. This ECs heterogeneity exists not only between the different sizes of the vascular conduits but also between different organs and even within the same vascular loop of the same organ (e.g. kidney). Moreover, notable fundamental differences are observed between the normal and the diseased ECs. Thus, an understanding on the underlining ECs heterogeneity, both in normal and pathological conditions, is important in formulating better nanomedicine strategies to treat the disease without inducing adverse effect on the normal vasculature.
000 kilometer-long of blood vessel network, requiring approximately 1–6 × 1013 ECs to form its interior surface.38 All ECs share a common characteristic that they are tethered on the luminal surface of blood vessels, bringing them into intimate contact with blood and non-blood components of the blood vessel. They also exert a potent anti-coagulant activity39 and express common biomarkers.40 While all ECs are similar in makeup, they are not called to function in exactly the same manner. Different ECs in different tissues have different structural adaptions to fulfil the diversity of functions. In addition, they express protein biomarkers that are tissue specific. This ECs heterogeneity exists not only between the different sizes of the vascular conduits but also between different organs and even within the same vascular loop of the same organ (e.g. kidney). Moreover, notable fundamental differences are observed between the normal and the diseased ECs. Thus, an understanding on the underlining ECs heterogeneity, both in normal and pathological conditions, is important in formulating better nanomedicine strategies to treat the disease without inducing adverse effect on the normal vasculature.
ECs heterogeneity in healthy vascular beds
ECs are diverse. They adopt various size and shape, resulting in their structural heterogeneity across the blood vessel network. The ECs of the microvessels are characteristically flat but adopt a cuboidal shape in the high endothelial venules.41 ECs thickness start from 1 μm in the aorta to less than 0.1 μm in the veins and capillaries.42 Rabbit inferior vena cava is lined by larger ECs (average length and width of 108 and 14 μm, respectively) when compared to those that line the aorta (average length and width of 96 and 11 μm, respectively).43 Aortic blood vessels in rat were reported to be covered by long and narrow ECs (55 × 10 μm) while the ECs found in the pulmonary artery, which forms in rectangular configuration, were broader and shorter (30 × 14 μm) and the inferior vena cava was paved with ECs with long, narrow and rectangular phenotype.44 Rat tracheal microcirculation is populated by elongated and spindle-like ECs in the arterioles, irregularly shaped ECs of the capillaries, large elliptically shaped ECs in the postcapillary venules, and rounded ECs in the collecting venules.45 Even at the subcellular level, there are differences. The ECs nuclei position from the midpoint of the longitudinal axis of the cells. An aortic EC positions its nucleus downstream from the midpoint of its longitudinal axis while an EC of inferior vena cava positions its nucleus upstream of the axis.46However, endothelial heterogeneity is most notable at the morphological level (Fig. 2). In continuous capillaries, continuous non-fenestrated ECs pave their wall from side to side. These continuous ECs are joined to each other by the tight junction (e.g. claudins) and adherens junction (e.g. VE-cadherins) proteins.47 The arrangement is typically found in endothelium bed such as the brain, skin, lung and heart,40,48 where the restrictive nature of endothelium bed is required to maintain the luminal and abluminal fluids (e.g. blood and cerebrospinal fluid (CSF)) separated and the solute transfer between the two kept in a highly selective nature.48 In contrast, the endothelium beds of exocrine and endocrine glands, gastric and intestinal mucosa are lined by ECs with small cytoplasmic openings called fenestrae.40 The fenestrae are 60–80 nm in size and extend through the full thickness of the cells, compartmentalizing the cells cytoplasm into small bodies of plaques.49 The fenestrae openings typically are sealed by a thin non-membranous diaphragm (5–6 nm),40,49 allowing increased filtration and solute transport across the endothelium beds while maintaining certain degree of size selectivity. The sinusoidal endothelium is similar to the fenestrated endothelium to a certain extent. For example, the ECs of the liver, the most notable discontinuous vascular bed, possess cytoplasmic pores (0.1–1 μm in diameter) with no diaphragm sealing those gaps.50,51 The sinusoidal vascular bed typically displays disorganized basal membrane. This, in addition to the larger cytoplasmic pores and the absence of the diaphragm, forms a much more permissive endothelium bed where much larger particles transfer to and from the blood circulation could occur. For instance, the liver endothelium permits the transport of small to medium sized chylomicrons (∼75–200 nm) through its pores.52
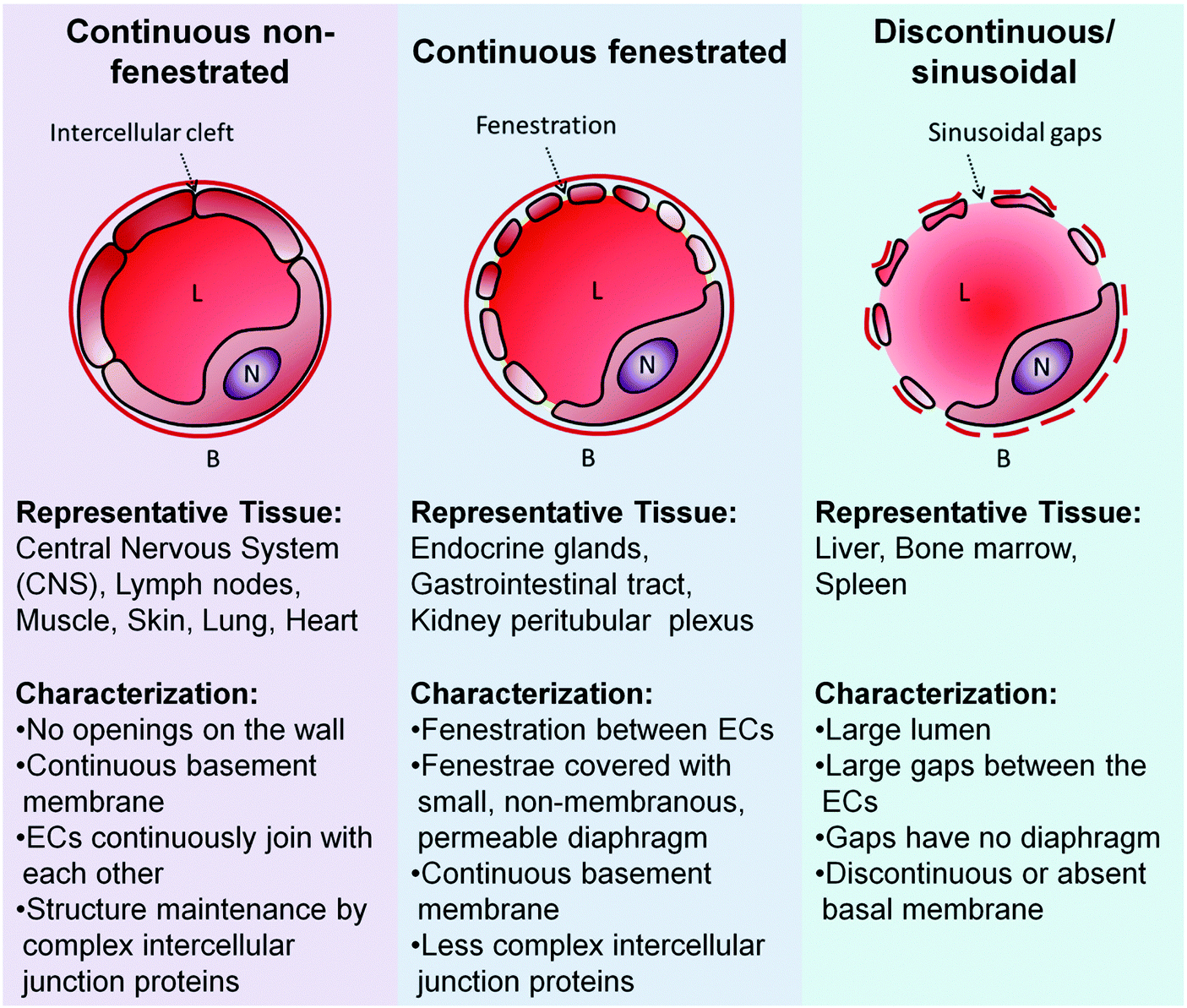 | ||
| Fig. 2 ECs morphological heterogeneity translates to diversity in the capillaries. B: Basal membrane; L: lumen; N: Nucleus of ECs. Adapted from ref. 54. Copyright 2003 by Nature Publishing Group. Adapted with permission. | ||
ECs structural heterogeneity is not only observed in within different organ but also evident within the individual organs. An extreme case is the kidney which comprises of discontinuous ECs in the glomerulus, while its peritubular region contains both highly fenestrated ECs as well as the continuous non-fenestrated ECs.53
In addition to being morphologically different, their gene and protein are as diverse as their different localities (Table 1).46,55–57 Lung ECs, for instance, express specific adhesion molecule (LuECAM-1) on their cell surface that are not found in ECs of other tissues.58,59 P-glycoproteins which remove foreign substances from the brain back to the blood circulation are expressed exclusively by the ECs of the blood brain barrier (BBB).60 Anti-coagulant protein, tissue-type plasminogen activator (t-PA) expression is reported to be expressed strictly in arteries of the pulmonary system and central nervous system.40,61 In addition to the exclusive expression of these specific biomarkers, the endothelium bed heterogeneity is defined by differential or uneven expression pattern of the commonly shared ECs biomarkers. Anti-coagulant, tissue factor pathway inhibitor (TFPI), is unevenly distributed with the highest expression in the placenta and lung and lowest in the brain.62 Similarly, the pro-coagulant protein, von Willebrand factor (vWF), though almost ubiquitously present in most type of endothelium bed, yet is absent in the sinusoidal ECs. There is higher expression of vWF expression on the ECs of the venous circulation than on the arterial portion.63,64
| Biomarkers | Vascular localizations | Functions | Antigen | Regulation | Ref. |
|---|---|---|---|---|---|
| PECAM-1, platelet endothelial cell adhesion molecule-1; ACE, angiotensin converting enzyme; vWF, von Willebrand factor; TFPI, tissue factor pathway inhibitor; TM, thrombodulin; VE-cadherin, vascular endothelial cadherin; t-PA, tissue plasminogen activator; LuECAM-1, lung endothelial cell adhesion-1 molecule; DANCE, developmental arteries and neural crest EGF-like protein; BBB, blood brain barrier. | |||||
| General ECs biomarker | |||||
| PECAM-1 (CD31) | Ubiquitous | Facilitate leukocyte transendothelial migration | Leukocytes | Expressed consecutively | 68 and 69 |
| ACE | Ubiquitous, enriched in the lung capillaries | Converts Angiotensin I to Angiotensin II, degrades the bradykinin, regulates blood pressure | Angiotensin | Expressed consecutively | 70 |
| vWF | Ubiquitous but absent in sinusoidal ECs. Expressed more in veins than in arteries | Mediate platelet adhesion and blood coagulation process (pro-coagulant) | Factor VIII antigen | Expressed consecutively | 63 and 64 |
| TFPI | Ubiquitous, highest in placenta and lung, lowest in brain | Anti-coagulant | TFPI antigen | Expressed consecutively | 62 |
| TM (CD141) | Ubiquitous but absent in the brain | Anti-coagulant | Thrombin | Expressed consecutively | 71 and 72 |
| VE-cadherin (CD144) | Ubiquitous | Maintain vascular integrity. Control the paracellular transport | VE-cadherin | Expressed consecutively | 73 and 74 |
| Organ specific ECs biomarker | |||||
| t-PA | heart and brain | Anti-coagulant | t-PA antigen | Expressed consecutively | 61 |
| LuECAM-1 | Lung | Promotes cell adhesion and trafficking | Melanoma cells | Expressed consecutively | 58 and 75 |
| DANCE | Heart, ovary, and colon | Play a role in vascular development and remodelling | Integrin and the RGD motif peptide | Diminished expression in adult vasculature. Expressed on developing, atherosclerotic, or injured vasculature | 76 |
| P-glycoprotein | BBB | Responsible for drug transport from brain to blood circulation | Amphipathic drugs | Expressed consecutively | 60 |
| Transferrin receptor | BBB | Mediating iron binding transferrin transport | Transferrin | Expressed consecutively | 77 |
Moreover, high heterogeneity is observed on the some of the interendothelial junction proteins that facilitate the endothelial cell-to-cell junction and regulate the paracellular route of solute transport. Junctional adhesion molecule (JAM)-2 is highly expressed in the intercellular cleft of high endothelial venules (HEV), JAM-1 is highly expressed in the EC of the brain, while the bulk of the vascular beds broadly express the JAM-3.65 Occludin, one of the tight junction proteins is highly expressed in the EC of the brain, yet its presence is barely detectable in other endothelium beds.66 Moreover, the occludin expression pattern differs within the brain itself. The ECs in the nerve fiber-rich regions express occludin in their interendothelial junction. Conversely, occludin expression is absent on those which are in the cell body-rich area.67 In the kidney, the tight junction claudin-5 is exclusively expressed by the ECs in the arterial circulation, and undetected on the ECs of the veins and capillaries.65
The diverse structure and protein expression combinations forms the canvas for this common cell type to paint a landscape of sharp as well as gradual differences in functionality. A pertinent example could be observed on how the endothelium beds perform their common function to keep the blood in a fluid state and to manage any breach in the blood vessel which accounts for hemostasis in blood circulation. The hemostasis state is maintained by the ECs via pro-coagulants and anti-coagulants expressions which are heterogeneous in nature across different endothelium beds. Repertoire of EC-derived hemostatic factors in the arterial circulation comprised of TM, t-PA and endothelial cell protein C receptor (EPCR); the venal circulation concoction includes TM, EPCR and vWF while the TM and TFPI mixture is normally found in the capillaries.40 In animal study involving mice with deficiency of functional anti-coagulant TM, blood clots formation were observed in major organs (e.g. lung, heart, spleen and liver) with exception of brain which is known for not expressing TM.78
A more obvious example on the endothelium's functional heterogeneity could be glimpsed by way of vascular bed regulates their permeability to accommodate the solute transport from blood circulation to the tissue. The sinusoidal beds found in the liver control overall blood lipid level by filtering out the larger chylomicrons while permitting smaller lipoprotein macromolecules (75–200 nm) through the sinusoidal gaps.52,79 These lipoproteins finally reach the abluminal side of the endothelium beds where they meet hepatocytes and get cleared from the body.79,80 The sinusoidal gaps of the liver are also the reason for rapid drug clearance from the body.80
In contrast, other endothelium beds that are continuous are highly selectively and do not permit macromolecules movement through their fenestrae and/or intercellular gaps. In order to cross the vascular beds, the macromolecules have to be taken up by the EC, transported across the EC, and released to the surrounding tissue at the other end.40 These fenestrae and gaps on continuous endothelium, however, allow small solutes to passively move between and through them, in effect creating a more selective type of endothelial barrier than its sinusoidal counterpart. Moreover, within the group of continuous endothelium beds itself, there exists a spectrum of barrier permeability control. Barrier derived from the pulmonary microvascular beds showed 5-fold more restrictive albumin permeability than that of barrier derived from pulmonary arterial beds (1.1 × 10−6 cm s−1 < 5.1 × 10−6 cm s−1).70,81 This differential permeability control is attributable to the tight junction protein complex between the neighboring ECs that make up the barrier in the pulmonary microvascular context. The vascular beds permeability control is inversely proportional to the number and the complexity of its tight junction protein complexes. Among all the vascular beds in the body, the BBB is built with the most number of tight junction components and with the highest complexity. The albumin permeability across the BBB was found to be 25-fold lower than that of the pulmonary microvascular beds (0.043 × 10−6 cm s−1),82 aptly illustrates the restrictive nature of this understandably restrictive bed to protect a privileged organ, the brain. This makes the BBB a highly impenetrable barrier to most of the solutes in the blood circulation and presents one of the toughest challenges to nanomedicine drug delivery.
ECs heterogeneity in diseased vascular beds
Many pathological conditions can dramatically change the status quo endothelium condition, giving raise to another set of diversity amongst these diseased vascular beds. One very common medical condition of notable significance in vascular beds is atherosclerosis; a disease where plaque build-up along the blood vessel. ECs isolated from this atherosclerotic vascular beds showed a distinctly different morphological appearance, in which the cells are transformed into multinucleated giant cells.83 In addition, the atherosclerosis ECs showed a drastically reduced expression of anti-coagulant TM on their cell surface.84 Compared to the normal ECs, the atherosclerotic ECs produced higher amount thrombin and promoted higher occurrence of thrombosis (blood clot formation inside the blood vessel), which appeared to be closely associated with the atherosclerotic plaque.ECs derived from inflamed blood vessel show significant increase expression of adhesion molecules which are involved in the leukocyte trafficking (Table 2). E-selectin and P-selectin slow down the leukocyte rolling along the blood vessel, whereas intracellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) participate in the leukocyte adhesion on the inflamed EC.85 Pathological cues such as cytokines, reactive oxidants, and abnormal blood flow mechanics can heighten the expression of ICAM-1 by 50 to 100-fold compared to healthy endothelium.86 In addition, the pathological cues activate the mobilization of the P-selectin from its intracellular storage (Weibel–Palade bodies) to the cellular surface.87 This mobilization of P-selectin is then followed by de novo synthesis process of E-selectin88 and VCAM-1, which are absent in healthy endothelium.89
| Biomarkers | Vascular localizations | Functions | Antigen | Regulation | Ref. |
|---|---|---|---|---|---|
| ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1; VEGFR-2, vascular endothelial growth factor receptor-2. | |||||
| ICAM-1 (CD54) | Ubiquitous | Facilitate leukocyte firm arrest and transendothelial migration | Leukocyte | Upregulated by inflammatory cytokines | 99 |
| E-selectin (CD62E) | Absent in normal vasculature | Facilitate leukocyte rolling | Leukocyte | Upregulated by inflammatory cytokines | 100 and 101 |
| P-selectin (CD62P) | Expression in normal vascular bed is highest in lung and lowest in muscle and brain | Facilitate leukocyte rolling | Leukocyte | Upregulated by inflammatory cytokines | 100 and 101 |
| VCAM-1 (CD106) | High expression in heart, brain, small intestine | Facilitate leukocyte firm arrest and transendothelial migration | Leukocyte | Upregulated by inflammatory cytokines | 99 and 102 |
| αvβ3 Integrin | Ubiquitous | Modulate the angiogenesis process | RGD motif peptide | Expressed consecutively in normal ECs, overexpressed in the ECs of tumor vessel. Modulated by b-FGF, TNF-α | 103 and 104 |
| VEGFR-2 | Ubiquitous |
Modulate the paracellular transport
Involve in the angiogenesis |
VEGF | Overexpressed in the ECs of tumor vessel | 105 |
| Tie-2 | Ubiquitous | Involve in the angiogenesis | Angiopoietins | Overexpressed in the ECs of tumor vessel | 106 |
| Endosialin (CD248) | ECs of the tumor vessel | Tumor neoangiogenesis | Mac-2BP/90K | Overexpressed in the ECs of tumor vessel | 107 |
In addition, the increased expression of ICAM-1 and VCAM-1 is also observed on tumor derived ECs possibly due to the deluge of growth factors excreted by the tumor.90 Other highly expressed biomarkers on the tumor ECs surface are integrin, endosialin, VEGFR-2, Tie-1 and Tie-2 protein.90 Tumor ECs next to malignant colorectal tumor tissue show a total of 46 expressed transcripts that were expressed at least 10-fold higher than their normal ECs counterpart.91 In contrast, PECAM-1 and the tight junction proteins which are required to maintain normal vascular barrier function were notably absent in tumor ECs.92 This abnormality resulted to an immature leaky tumor vasculature with large gaps (∼1.5 μm)92,93 in which the outcome is aptly termed as ‘enhanced permeability and retention (EPR) effect’.94,95 This effect is exploited by many anti-cancer nanomedicine strategies. The tumor ECs are also marked with the presence of transcellular pores and fenestrae (0.5 μm)92,96 and the absence of basal membrane.97 All these structural abnormalities in the tumor ECs is a double edged sword in that they are the gateway for the tumor cells to escape into the blood circulation and as easy influx of precious growth nutrients but provide the same ease in drug and nanomedicine delivery to the tumor. Sometimes, hemorrhages often observed in tumors can reverse the situation with a high interstitial pressure in the blood circulation which compromises the blood flow and thwart many drug carrier delivery strategies.96,98 Compounding the problem is that in pathological vasculature (atherosclerosis, aneurism, etc.), the increased turbulent blood flow also increased the collision frequency of the NPs with diseased ECs and possibly heightened interactions between particles and ECs and likely increased uptake.36
The heterogeneity of vascular beds found in the various organs is part of the very fabric of endothelium that governs its versatility as a supportive tissue and could be observed both in health and disease states of vascular beds. In the context of nanomedicine, the vascular bed can take on multiple roles; (i) recipient of collateral damage, (ii) be the target tissue, and (iii) presents a barrier for the nanomedicine therapeutic effort. Understanding the heterogeneity inherent to the endothelium beds is critically important for rational design of safe, effective and specific therapies. For instance, understanding the vascular beds structural heterogeneity gives the knowledge which endothelium bed in a certain system could pose the biggest obstacle for nanomedicine therapy. The knowledge also offers the ability to choose alternative delivery route of nanomedicine. Instead of designing the delivery via paracellular route that is extremely restrictive in nature, the nanomedicine could be devised to be trafficked to the target tissue via a transendothelial manner (see Section 5 for more detailed discussion).
In addition, the understanding of biomarker expression heterogeneity (Tables 1 and 2) between vascular beds is analogous to knowing the precise ‘zip code’ for a specific vascular bed to deliver the nanomedicine to the right address, the basis of specific targeting vascular nanomedicine rational design (Table 3). This specific delivery to the vascular target as contrasted to the many current design which is addressed to the tumors offers substantial improvement on therapeutic index of the nanomedicine payload as well as reduces the systemic toxicity (in the context of drug delivery platform) and undesired off-target effect giving false-positive (in the context of imaging and detection platforms). The rationale is very straightforward because the vascular bed is the first location encountered by the nanomedicine instead of the tumor tissues. For this to work, the targeted molecule on the ECs of the target vascular bed needs to be first accessible. This allows the initial engagement of nanomedicine with the ECs and subsequent interaction. This would mean that the target molecules have to be extracellular, where it is fully exposed on the EC surface. Most of intracellular molecules are unsuitable as the possible target, unless due to certain pathological condition (e.g. P-selectin) they are exposed on the surface. In order to maximize any possibility contact with the nanomedicine, the extracellular domain of these molecules need to project beyond the glycocalyx layer.108
| Target | Ligand | Nanoparticle carrier | Model | Application | Ref. |
|---|---|---|---|---|---|
| DOTA, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid; HAEC, human aortic endothelial cell; Ldlr, low density lipoprotein receptor; MBEC, mouse brain endothelioma cells; MCEC, murine cardiac endothelial cell; MRI, magnetic resonance imaging; PEG, poly ethylene glycol; PLGA, poly(lactic-co-glycolic acid). | |||||
| ICAM-1 | Anti-ICAM-1 antibody | Liposome | In vitro (HUVEC) | Stem cell delivery | 122 |
| ICAM-1 | Anti-ICAM-1 antibody | Liposome | In vitro (MBEC bEnd.5) | Imaging | 123 |
| ICAM-1 | Anti-ICAM-1 antibody | PLGA | In vitro (HUVEC) In vivo (C57BL/6 mouse) | Proof of concept | 124 |
| ICAM-1 | Anti-ICAM-1 antibody | Polystyrene | In vitro (HUVEC) In vivo (C57BL/6 mouse) | Size and shape dependent delivery of catalase | 125 |
| ICAM-1 | Peptide (cLBAL) | PLGA | In vitro (HUVEC) | Proof of concept | 126 |
| ICAM-1 | Peptide NNQKIVNLKEKVAQLEA (fibrinogen binding sequence) | Polystyrene | In vitro (HUVEC) In vivo (C57BL/6 mouse) | Proof of concept | 127 |
| VCAM-1 | Cyclic peptide VSHPNKK | Iron oxide | In vitro (MCEC) In vivo (C57BL/6 mouse) | MRI imaging | 128 |
| VCAM-1 | Peptide VHPKQHR | Iron oxide | In vitro (human carotid artery) In vivo (C57BL/6 mouse) | MRI imaging | 129 |
| VCAM-1 | Anti-VCAM-1 antibody | Liposome | In vivo (Ldlr−/− mouse) | Drug delivery | 130 |
| VCAM-1 | Anti-VCAM-1 antibody | Liposome | In vitro (HUVEC, HAEC) | siRNA delivery | 131 |
| PECAM | Anti-PECAM antibody | PEG-PLGA | In vitro (HUVEC) In vivo (C57BL/6 mouse) | Antioxidant (catalase, peroxidase, xanthine oxidase) delivery | 132 |
| E-selectin | Anti-E-selectin antibody | Liposome | In vitro (HUVEC, HAEC) | siRNA delivery | 131 |
| P-selectin | Anti P-selectin antibody | Cu-DOTA | In vivo (Ldlr−/− mouse) | MRI imaging | 133 |
For systemic delivery to treat generalized vascular conditions, including sepsis, intravascular coagulation and hypertension, ubiquitously expressed moieties (e.g. PECAM-1) are excellent candidates.109 For localized delivery, moieties that are highly expressed in certain vascular beds or in sites of diseased vascular bed provide the suitable means for local delivery. ACE that is highly expressed in the lung is utilized to deliver antioxidant and gene therapies to the injured lung endothelium.110–113 Endosialin targeting nanomedicine could deliver anti-angiogenesis treatment to the tumor vasculature and starve the malignant tumor cells. Consecutively expressed surface moieties (e.g. PECAM-1 and ACE) are potentially useful as nanomedicine target for prophylactic type of therapy.109 Inducible moieties (e.g. VCAM-1, ICAM-1, E-selectin, and P-selectin) that are present due to vasculature pathological alteration could be used as a good addressing beacon for the much needed detection and therapeutic intervention.90,114–116
An important consideration is that the vascular bed targeting strategy should not drastically affect the normal functioning of the target tissue (with the exception of targeting cancer cells). Targeting TM results in its binding inhibition to thrombin and reduces the anti-coagulant in the blood which may lead to thrombosis. Consideration should be made whether there is a change in the target functionality upon being targeted by the nanomedicine. Targeting the selectins and the cell adhesion molecules (CAMs) could potentially block the leukocyte binding process, and thus suppress any inflammation, an added advantage for nanomedicine delivering anti-oxidant. Targeting ACE could inhibit the conversion of angiotensin I to angiotensin II, which might benefit the nanomedicine for hypertension management.
Lastly, the heterogeneity in the endothelium highlights the importance of choosing the correct representative of endothelium barrier in designing and testing the efficacy of nanomedicine strategies. For instance, the bulk of nanomedicine strategies for cancer treatment117–120 were tested against ECs derived from human umbilical vein endothelium (HUVEC), which possesses no structural and biomarker expression resemblance to the tumor ECs nor any close relevance to adult ECs since umbilical endothelium is only present at a specific stage of an adult life; during pregnancy.121 In fact, any introduced nanomedicine would tend to target the placenta in a pregnant female and not target any vascular bed in a male or non-pregnant female. Therefore, it comes as no surprise that many nanomedicine studies fail to perform as their intended design when tested in a more complex model such as in animal model. Clearly, an appreciation of the endothelium beds heterogeneous nature, either when they are in the healthy or disease states, is essential for successful nanomedicine formulation.
3. Engineered versus ‘natural’ surface coatings – the relevance of the blood derived protein corona for cross-talk with the blood system
Engineered surface coatings of NPs are not only important for obtaining stable colloidal systems and may increase the NPs' water-dispersibility, but also allow functionalization via conjugation with targeting ligands and/or bioactive molecules for obtaining multifunctional ‘intelligent’ NPs that could recognize ECs (Table 3).134–137 NP stabilization can be achieved via engineering of various surface coatings, such as the use of polymeric stabilizers/surfactants, by deposition of layers of inorganic metals, non-metals or oxide surfaces, by generating polymeric shells or by the formation of lipid-like coatings.137–139 Besides affecting the NPs' ‘technical identity’, such distinct surface coatings were shown to have also a profound impact on the NPs' biocompatibility, including cell vitality, cell adhesion, and the NPs' cellular uptake and biodistribution in the blood system as well as in tissues belonging to the reticuloendothelial system (RES).139–142However, it is often neglected that in complex physiological vascular system, a certain degree of in situ biotransformation will most likely occur for all NMs. For the majority of all current nanobiomedical applications, such devices will be intravenously injected, at which point they are immediately exposed to a highly complex biological milieu. Here, a plethora of biomolecules such as lipids, metabolites, sugars, and especially proteins will adsorb onto the surface of NMs', mediated by van der Waals, electrostatic, hydrogen bonding, and hydrophilic/hydrophobic interactions.143–147 The sum of all adsorption processes will results in the formation of the so-called ‘biomolecule corona’, of which the protein corona has mostly been studied so far. It is now accepted but far from being understood in detail that the formation of a protein/biomolecule corona seems capable to critically affect not only the physicochemical characteristics of the NMs but also the (patho)physiological and biomedical identity of NMs' in general.148,149 Hence, it becomes obvious that the bio-physical properties of formulated NPs will differ (significantly in most cases) from the contextual corona-covered NMs.25,144,148,150,151 Strictly speaking (chemically), no NPs will be ‘naked’ or ‘pristine’.
In the area of biomolecule corona research, the term ‘hard corona’ was coined as a protein adsorption signature of a NM, but is sometimes also used to describe a NM's ‘long-lived’ equilibrium protein signature, e.g. a plasma protein signature of a NM in the blood.144,148,152,153 On top of this ‘hard corona’ some models also suggested the existence of a ‘soft corona’, which can be conceptualized as a tentative, low adherence and dynamically transient layer of biomolecules (Fig. 3).148,153–157 However, since such a ‘soft corona’ seems to be lost during typical purification steps, its existence, let alone its (patho)biological and medical relevance is still unclear.35 The terminologies used in current literature further accentuated the confusing situation. ‘Soft’ (versus ‘hard’) corona was used as a ‘scapegoat excuse’ in explaining off unexplainable observations. Hence, we strongly suggest a standardized definition of the ‘protein corona’ (PC) as that of analytically accessible NM-protein complexes.
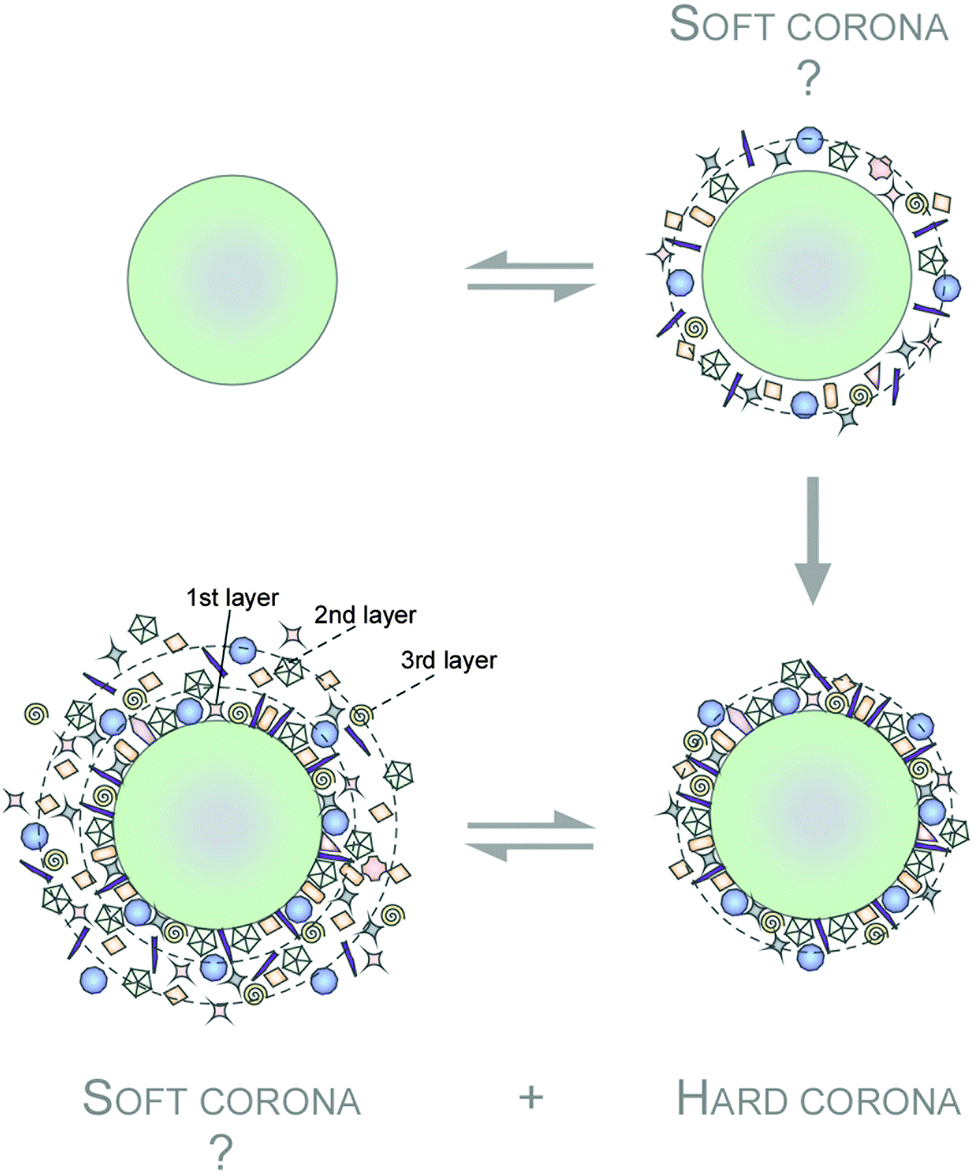 | ||
| Fig. 3 Hypothetical model of protein corona formation and terminology. A highly complex protein corona is established on NPs in the blood system. On top of this ‘hard corona’ the existence of a dynamic ‘soft corona’ of loosely associated biomolecules is also suggested. Adsorbed proteins are indicated. | ||
Notably, such protein corona forms the interface between NPs and biological systems and can likely initiate the transformation of the NPs' through alterations of colloidal stability. Their colloidal stability can either be further enhanced through induction of steric stabilization or lowered due to protein-mediated bridging, charge compensation or simply charge inhomogeneity on the NPs' surface.145,158–161 When the NPs aggregate, these multiple interactions of proteins with NPs could further strengthened beyond what would otherwise be when compared to the same proteins bound to discrete NPs. Being in the aggregated state, might even encapsulate weak or non-binding proteins within the aggregate. To complicate matters further, such aggregations have kinetic and thermodynamic components which overall determine the transient and steady state condition of the protein corona.158–161 Accessing the protein corona, characterizing it at the molecular resolution at the historical pace of the protein corona evolution is very important. Ex situ, the sub-fractionation of such aggregates by centrifugation techniques is possible during NPs and proteins mixing. But this aggregation status is almost impossible to obtain in vivo, let alone predict. In summary, the aggregation of NPs certainly adds an additional level of complexity to the already complicated system, but nonetheless should be taken into account in the design and application context of NPs within physiological systems.
Clearly, adsorbed proteins also define the surface of NMs and have been shown to significantly affect the interactions between the NMs and the biological environment, including cellular components found within the blood system.148,153,156,159,162–166 Notably, recent studies demonstrated that the plasma PC is highly complex.144,166 Quantitative snapshot proteomics identified over 200 different corona proteins.167,168 The PC was found to be established in less than one minute, and was rather stable, changing almost exclusively quantitatively but not qualitatively over time.144 In contrast, previous reports suggested that the plasma derived PC consisted of less than one hundred proteins. The PC although evolved at an overall slow rate, is highly dynamic at any given time due to continuous protein association and dissociation events.145,153,166,169 However, such knowledge is the key for understanding and predicting the relevance of the PC for biomedical applications and its impact on ECs in the blood system. Thus, PC profiling needs to be performed on a high technological level and with consistent protocols to ensure data quality and meaningful inter-laboratory data comparison. Particularly, for the plasma PC quantification, quantitative high-resolution LC-MS/MS is capable to reduce PC characterization time and in principle can provide qualitative and quantitative data even from large libraries of NPs.167,170
Physicochemical properties of NMs and factors affecting PC formation in the blood
Several studies have shown that the physicochemical properties of the pristine NMs, such as size, shape and surface chemistry (collectively termed the 3 ‘S’) can influence the amount, composition and in situ evolution of the PC, which in turn can (co)determine the NMs' bioactivity in the blood system.25,144,145,168,171,172 For example, there is evidence that the PC is capable of regulating various cell-NM interactions,144,163,165,166,173 blood residence time,174,175 (tumor)cell targeting activity and pharmacokinetic profiles,174 albeit the underlying molecular mechanisms are not yet fully resolved. Consequently, the nanoscience community has recognized the need to better understand the NMs' parameters controlling the formation of the PC, which is important within the framework of both, nanosafety and nanomedicine. As such, numerous studies have been conducted to systemically dissect and mechanistically understand the PC and its quintessential dependence on the NPs' innate physico-chemical properties and then linking the PC to possible (patho)biological implication and rational biomedical exploits.25,144,145,168,171,172 Typically, PC profiles are significantly different from the protein composition of the biological fluid investigated suggesting that different proteins in the fluid have different propensities to bind to the NP.144–146,153,158,166 Distinct proteins will be either enriched out or rejected by the NP surface. The determination of the corona by the protein source is also discussed as an important factor within the context of the so called ‘personalized protein corona’ (PPC)176 From the biological standpoint, it is envisaged that humans with a certain disease may have specific NP coronas, which could be an important factor in nano-biomedical science.176 At the other end of the spectrum, deciphering the main determinants of the NMs properties that governs the formation (qualitative and quantitative) of the PC is imperative to realize the notion of the PPC (Fig. 4).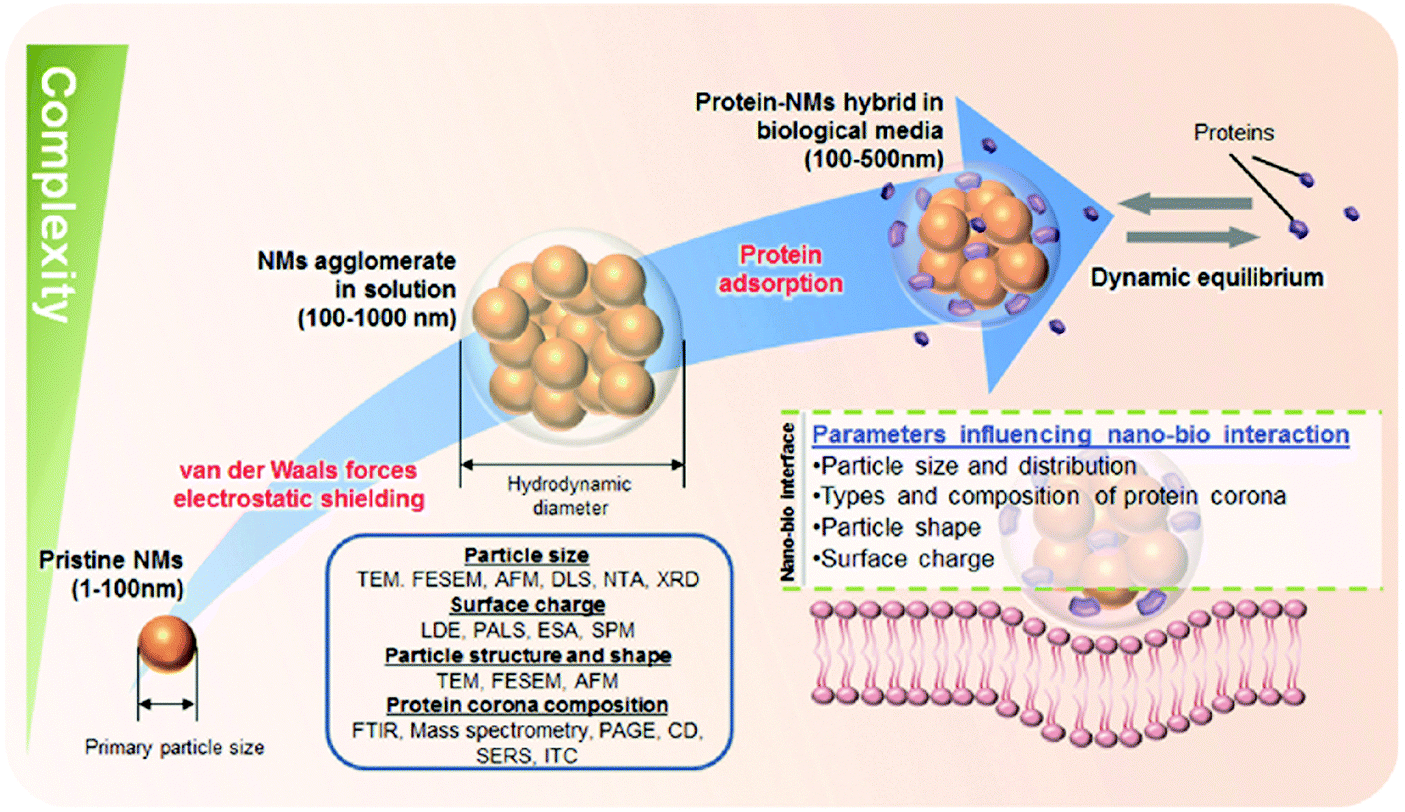 | ||
| Fig. 4 Schematics to illustrate the level of complexity to characterize NPs in biological relevant fluids. A wide spectrum of characterization techniques should be employed to characterize NP size, surface charge, and particle structure and shape which are main determinants in the formation of protein corona. TEM: Transmission Electron Microscopy; FESEM: Field Emission Scanning Electron Microscopy; AFM: Atomic Force Microscopy; DLS: Dynamic Light Scattering; NTA: Nanoparticle Tracking Analysis; XRD: X-Ray Diffraction; LDE: Laser-Doppler Electrophoresis; PALS: Phase Analysis Light Scattering; ESA: Electronic Sonic Amplitude; SPM: Scanning Probe Miscroscopy; FTIR: Fourier Transform Infrared Spectroscopy; PAGE: Polyacrylamide Gel Electrophoresis; CD: Circular Dichroism; SERS: Surface-Enhanced Raman Sepectroscopy, ITC: Isothermal Titration Calorimetry. Reproduced from ref. 163. Copyright 2014 by Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Reproduced with permission. | ||
The size of the NMs is an important intrinsic physicochemical parameter that is linked to its bioactivity. There are several manners which NP size can impact formation of protein. Firstly, NM size will directly determine the available surface area for protein adsorption.169 Using a panel of mono-dispersed gold NPs ranging from 4–40 nm, it was shown that the amount and composition of the surface bound protein corona is heavily dependent on the NMs size and adsorption time. Particles with size smaller than the size of a typical protein (i.e. ∼10 nm) displayed little adsorption of surface proteins. In contrast, Au NPs with intermediate size (10, 13, and 16 nm) favored the formation of a dense hard protein corona layer, while a less dense protein layer was formed on Au NPs with bigger size (24 and 40 nm).169 These empirical findings are consistent with the classical Langmuir adsorption model in which the adsorbate (proteins) is generally considered to be much smaller than the adsorbent (NPs). However, when both the adsorbent and adsorbate are of comparable size, the effect of shear forces due to Brownian motion may play a more dominant role, leading to the weak or unfavorable binding events.
A slight change in NPs size in the tens of nanometers is sufficient to induce a protein compositional change in the hard protein corona layer.167 As the surface curvature (κ) of a sphere can be defined simply as a reciprocal to the radius (R), κ = 1/R,177 a decrease in particle size would therefore translate to a significant increase in the local curvature. This brings about the notion that changes to the nanoscale size-related surface curvature could also impact the specific surface energetics and therefore modulate the process of self-assembly of small molecules on the surface of the NPs.178 When a protein approaches a NM with a diameter that is so much bigger than the size of the protein, what the protein ‘perceives’ is essentially a flat surface. As the size of the NM becomes smaller, the effects of surface curvature become more pronounced and significant.179 Along the same line, the manner in which the proteins adsorb, arrange and pack on the nanomaterials surface will therefore be highly dependent on the nanomaterials surface curvature.180 Despite significant work, the relation between the original surface functionality of the NPs and the nature of the corona is far from being trivial and currently still remains impossible to predict or to simulate in complex physiological environments.144–146,153,158,166,168 Though, some studies still suggest of having identified ‘the major NM factor’ controlling protein/biomolecule corona formation. However, as convincingly shown by recent comprehensive studies, none of the above-mentioned factors, such as the NPs' physicochemical properties or exposure time, alone is able to determine formation and composition of the PC.144,166 Not only the 3 ‘S’, but also the relative ratio of the physiological fluid to the NP dispersion seems to play a role affecting the composition and evolution of the PC.144–146,148,153,158,166,168,181 However, for most biomedical applications in the blood there will be an excess of plasma proteins versus NMs. Despite the complexity and analytical challenges of the PC already during its ex situ characterization, researchers are facing additional challenges during its in situ analysis. Particularly, when NPs move from one physiological (micro)environment to another, e.g. from the circulation via different cellular uptake mechanisms into cells (e.g. EC, monocytes or macrophages), a key question is whether the original corona remains stable or is subjected to substantial changes, which again adds an additional level of complexity.144–146,148,153,166,168,181 So far, it is assumed that even after passing through several ‘physiological (micro)environments’, the final corona would still contain a fingerprint of its history and keeps a memory of its prior journey through the body,145,182 which is in line with recent reports showing the stability of PC signatures ex situ.144
Among the many types of possible protein-nanomaterials interactions, Coulomb forces between two charged entities has been extensively studied and has been viewed as playing a vital role in the formation of protein corona. While it was generally thought that proteins that are predominantly negatively charged under physiological conditions will display a high affinity to cationic (positively charged) NPs, there are also numerous studies that have shown that plasma proteins could also bind to a wide range of anionic and neutral NPs.183,184 Although proteins possesses an overall net negative charge in physiological pH, there are regions of positively charged domains which may function as potential binding sites to the anionic NPs.185 Regardless of the initial surface charge of the pristine NPs, all of the nanomaterials will become anionic following the formation of the protein corona186–189 and still be internalized into the cells (Fig. 5). In this regard, the conventional wisdom that cationic NPs can promote binding to the negatively charged cell membrane and thus improve cellular internalization may appear to be overly simplified. So how can one account for the enhanced cellular binding of cationic NPs commonly observed by different labs? It is possible that NPs bearing different net surface charge may recruit different types of proteins onto their surface which may either promote of hinder binding of the NP–protein complex to the cell receptors. NP–protein complex formed by anionic or cationic particles may also engage different cell receptors, leading to differential binding and uptake profile. For example, NP–protein complex formed from cationic polystyrene NP was found to bind directly with scavenger receptors while NP–protein complex derived from the anionic polystyrene NPs have to compete with the FBS proteins to bind to the protein receptors.190 Further study revealed that is the NP charge mediated changes to the surface bound protein conformation, epitope presentation and denaturation that lead to this difference.191 Collectively, these studies demonstrated the importance of NPs surface charge in the protein adsorption process which has deep implications in the biological outcomes. In summary, as we are still at the beginning of understanding the role of the protein corona for biomedical applications, the topic of a hypothetical ‘PPC’ adds an additional level of complexity to the field, which certainly has to be addressed and most importantly be confirmed by future studies.
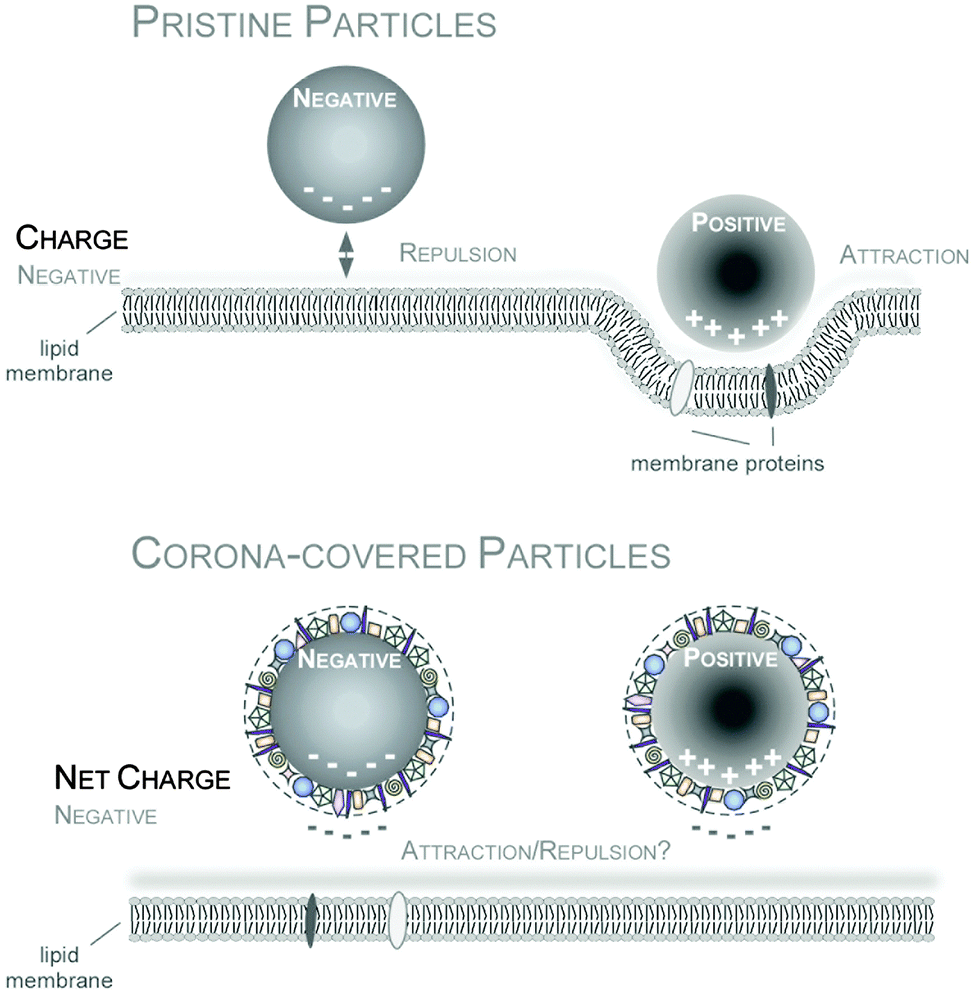 | ||
| Fig. 5 Impact of NP charge on cellular uptake in the absence or presence of the PC. Improved cellular uptake of positively charged NPs can be mediated by enhanced interaction with the negatively charged cell membrane for pristine NPs (upper panel). In contrast, PC covered NPs are overall negatively charged, that might hinder NP-charge driven cell membrane interaction. Reproduced from ref. 35 with the permission of The Royal Society of Chemistry. | ||
Nanoparticles in blood: from cloak-and-dagger operations to known biological effectiveness
Once the nanomaterials gain entry into the vascular milieu, the nanomaterials will interact with a plethora of blood constituents such as red blood cells, dissolved nutrients, bioactive factors and plasma proteins. Protein/biomolecule adsorption onto NMs in the blood system will clearly generate new/modified contact interfaces between NMs and the cellular components of the environment. Thus, protein corona formation and evolution may confer the NMs with a new ‘biological identity’, potentially also having opposing effects. Proteins that are adsorbed onto the NMs' surfaces can vary greatly in terms of their amount, densities, conformation and orientation. In the human blood plasma coronas of various NPs, proteins involved in physiological, pathological and nanotoxicological relevant activities were identified.144–146,168,192–194 Such PC components have dynamic range that spans about three to four orders of magnitude.144From these data it is almost certain that the PC immediately changes the facial identity of the NPs and may trigger responses not only from blood cells but also from the endothelium. Albeit mostly based on data of ex situ plasma studies, it is expected that upon intravenous administration, blood complement factors bind rapidly to NPs' surfaces and mark them as ‘foreign entities’.144,168,192 It was shown in animal studies that within minutes following intravenous administration, erythrocytes, and resident phagocytes such as macrophages, monocytes, granulocytes and dendritic cells begin to engulf and phagocytose majority of the injected NPs.168,194 Moreover, binding of opsonins is expected to promote rapid clearance of NPs, including NPs, from the vasculature through capture by the RES, i.e. the spleen itself, Kupffer cells of the liver and monocytes of the bone marrow.144,149,194 All of which screens through large volume of blood and thus any introduced NPs component. Accumulation of opsonised NPs into some of the RES organs may be favorable side effects if these organs are the intended target sites, but not so if the NPs are intended for example to target breast cancer cells. Hence, to deliver the NPs to tissues other than the RES organs, minimizing the rapid systemic clearance of the NPs becomes essential. In most cases, albumin, which is a protein that is found in abundance (∼55%)195,196 in the serum was found associated with various NPs at relatively high levels. Dysopsonins, such as albumin may antagonize the biological reactions triggered by NPs bound opsonins,144,149,168 might prolong NP circulation times in the blood. Although detailed in vivo studies for the many possible types of NPs are yet to be performed, we can surmise logically that an increase in the circulation time by bound dysopsonins as PC on the NPs should also increase the interaction time with components of the blood clotting system. While some reports found that NMs can induce platelet aggregation, the underlying molecular and nanoscale mechanisms are not well understood.144,168 Employing human primary cells from the blood system, it was demonstrated that PC formation affected processes at the nano-bio interface.144 Despite the short-lived presence of pristine NPs in the blood system, the NPs caused EC death, triggered thrombocyte activation and induced hemolysis (Fig. 6).35,144
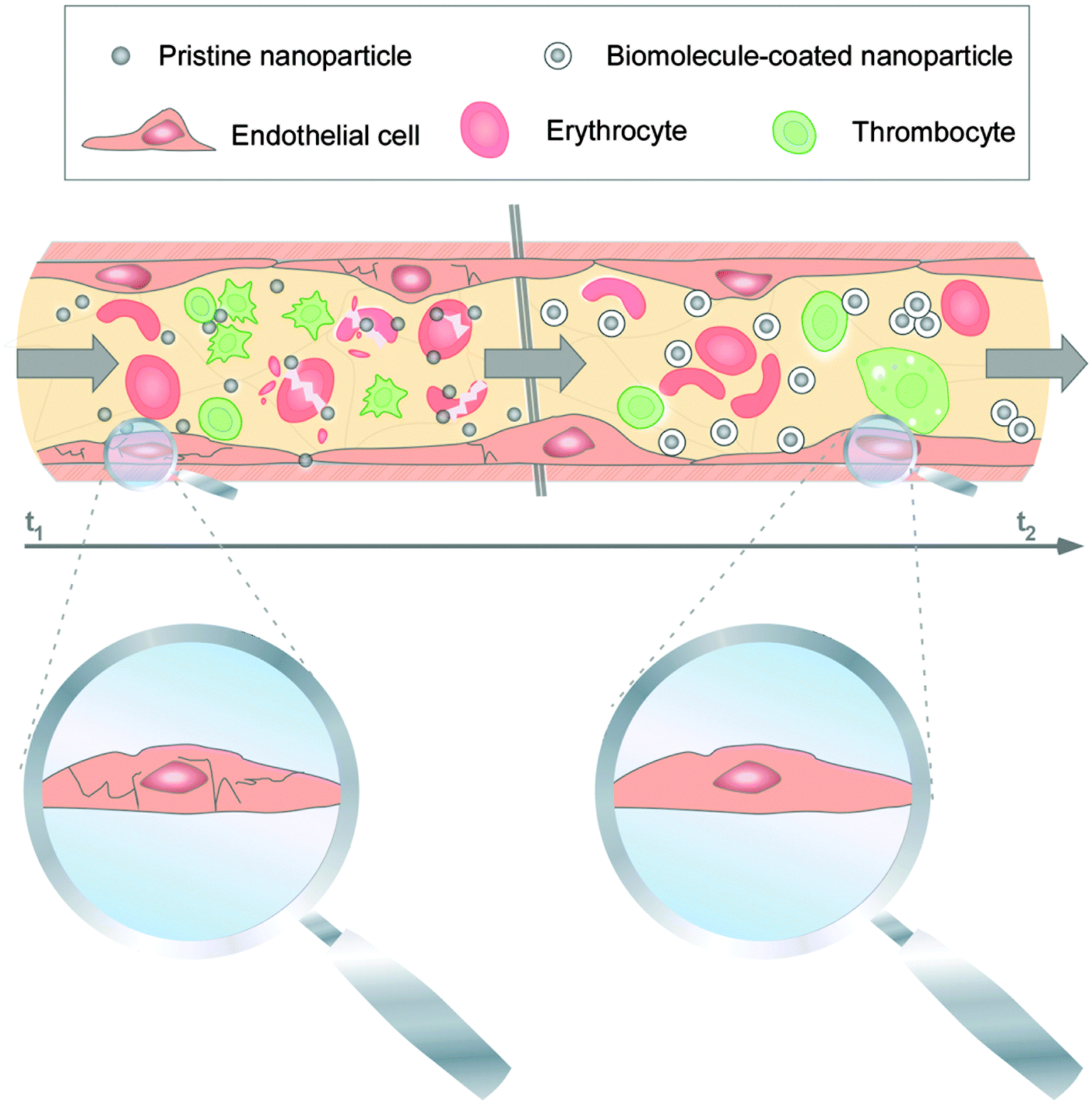 | ||
| Fig. 6 Impact of PC formation on cellular components of the human blood system. Upon biomedical application, pristine NPs seem to exist for a short period of time, but are capable to affect vitality of ECs (magnified), may induce hemolysis, and activate thrombocytes. PC development modulates the NPs' decoration with bioactive proteins is protecting cells against NP-induced pathobiological processes, and can also affect the NPs' cellular uptake. Adapted from ref. 35 with the permission of The Royal Society of Chemistry. | ||
A functionally diverse group of high abundance plasma proteins also detectable in the PC of various NMs are apolipoproteins. These plasma proteins are involved in important lipid and cholesterol transport and metabolism.144,149 As apolipoproteins are able to bind to specific receptors of various organs, the apolipoproteins in the NMs' PC will affect their biodistribution.144,149,168 Albeit, the functionalization of NPs with certain apolipoproteins was reported to improve the NPs' ability to overcome biological barriers, such as the BBB.197 It is logical to exploit the PC-mediated ‘natural functionalization’ of NMs occurring upon injection into the blood system to increase their nanotherapeutic activity. In contrast to antibodies or peptides, natural proteins are less immunogenic, and have usually a higher association constant than peptides for their cognate receptors. Such in situ PC-engineering, would also decrease the cost for the production of nanotherapeutics. However these ‘designer coronas’ need to be well controlled and fulfil the desired biological requirements, such as high affinity receptor binding and/or the ability to trigger endocytosis, which is still difficult to predict in situ.
Notably, even complete profiling of a NM's PC may not always allow direct extrapolation how the bound proteins may affect the homeostasis of the blood system. This is primarily due to the fact that the composition of the protein corona is far from being static and was shown to be a dynamic entity that evolves with time that is dependent on a multitude of biological parameters such as the physiological environment, cell type and culture condition.153,198 For instance, simply by transferring a 15 nm citrated capped Au NPs from a 10% FBS solution to a conditioned medium enriched with the cells secretome is sufficient to bring about a drastic compositional change in the protein corona.198 Furthermore, the identity of the adsorbed proteins on silicon dioxide (SiO2) NPs was shown to change as a function of plasma concentration.199 These results suggest that blood-borne NPs that are constantly subjected to different types of physiological transport profile, local plasma concentration fluctuation and microenvironment will possess different biological identity at different time. As such, this makes accurate characterization of the protein corona at any one point in time an insurmountable task. We anticipate that development of technological platforms capable of characterizing the protein corona in real time will be an area of intensive research in the future. A possible approach is to employ the concept of ‘organ on chip’ to mimic certain aspects of physiological conditions that the NPs may be exposed to and sample it as a function of time, locality and different fluid flow rate.
During protein adsorption on the NPs surfaces, drastic conformational changes may be induced in proteins, particularly when hydrophobic or charged protein domains interact with hydrophobic or charged surfaces. Clearly, the degree of this conformational change depends on several aspects; the structure and chemistry of the protein in question as well as the physicochemical characteristics of the NM. For instance, dimerization of β-lactoglobulin (β-LG) decreases with decreasing polystyrene NPs size (increase in localized surface curvature) as less space is available for adjacent protein–protein interaction.179 Conformational changes may even denature the protein activity directly via loss of active site configuration and even indirectly by altering the exposed facet of the adsorbed protein to the ligand. In this case, exposing normally non-accessible domains or by hiding critical binding or catalytic domains.155,191,200 For example, it was observed for polyacrylamide coated Au NPs, to be able to unfold the fibrinogen in the corona and activate the Mac-1 receptor and the downstream NF-κB inflammatory signalling pathway, resulting in the release of inflammatory cytokines.201 Collectively, the PCs' ‘dagger function’ may not only be responsible for off target biodistribution, inflammation, complement activation and/or NM clearance but may also result in the activation of undesired cell pathways due to unpredictable unfolding of adsorbed proteins.
The PC remains as a major unpredictable complexity for nanomedical applications. Therefore to reduce the uncertainties, there are currently numerous attempts to chemically prevent and/or modulate protein adsorption.145,153,174,202–204 Such chemical strategies aiming to increase plasma half-life have been proposed as possibilities to functionalize the surface of NPs with a variety of different molecules. As examples, hydrophilic oligomeric or polymeric ethylene glycol units (PEGylation), zwitterionic low molecular weight and polymeric coatings have been used as potential stealth materials.145,153,174,202–204 However, despite the reduction of protein adsorption by surface functionalization with these stealth materials, design of PC-free NMs remains a challenge for the field.145,153,174,202–204
4. Nanomaterials properties that dictate their intracellular delivery to vascular target
There are four distinct endocytosis pathways that could be used by nanomedicine to gain entry to the ECs (Fig. 7). Caveolin-mediated endocytosis involves invagination of the solute by lipid raft and mediated by the caveolin protein that is present in the caveolar coat.205 The caveolin mediated pathway, which is preferentially inhibited by lipid chelators (e.g. cyclodextrin, and filipin), is the predominant endocytosis pathway used by ECs to take in solutes from the blood circulation, including nanomedicine particles.205,206 Ligands on nanomedicine bind to the cellular surface receptors and enter into cells via the clathrin-mediated pathway. The interaction between the nanomedicine and the surface receptors trigger the formation of a clathrin coated pits that transport the nanomedicine into the cells.205 Although an EC is not as phagocytic as a macrophage, they can still phagocytose large (>1 μm) particle (e.g. bacteria)207 through actin remodeling to form the large invagination necessary to bring the particle into the cell.205 Reminiscent to phagocytosis process, the micropinocytosis process requires cytoskeleton remodeling to form the membrane ruffles and protrusion of plasma membrane in order to engulf the cargo into the cells along with substantial amount of extracellular fluid into the cell.115,205 Particles like human immunodeficiency virus (HIV)208 reportedly enter the brain ECs through micropinocytosis.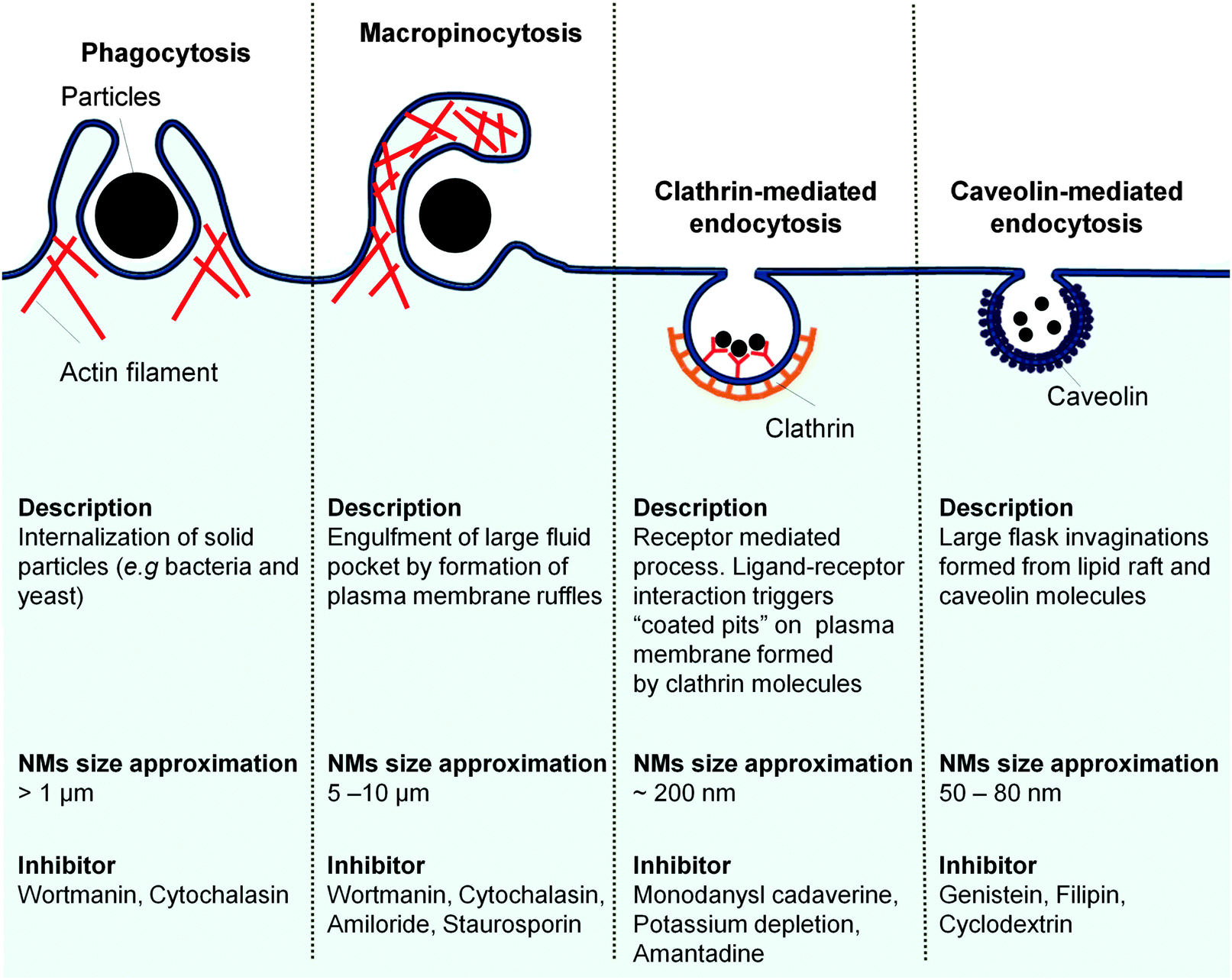 | ||
| Fig. 7 ECs endocytic pathways, the route of entry for nanomedicine intracellular delivery to target endothelium. Adapted from ref. 209. Copyright 2007 Nature Publishing Group. Adapted with permission. | ||
The existing endocytosis pathways vary not only in the protein signaling involved but also, most notably, on their size of the vesicle employed to internalize the particle. It is tempting to claim that if one took the inherent size limitation of each endocytosis vesicle (Fig. 7) into design consideration, one could deliver nanomedicine to the target ECs via certain endocytosis pathway. However, the complex nature of nanomaterials interaction with the cells makes it difficult for the identification of the exact entry pathway involved based solely on the nanomaterial size. Macropinocytosis is traditionally conserved for the large solid particle (5–10 μm) uptake.209,210 It was reported to be activated in HUVEC to bring in DNA coated single-walled carbon nanotubes (SWCNT, major axis length 100–500 nm)211 and polystyrene (PS) nanosphere (100, 200, and 500 nm).212 Clathrin coated vesicles with upper size limit of 200 nm,213 are shown to facilitate the entry of 500 nm PS NPs.212 The flask-shaped caveolae pit involved in the uptake of PS nanosphere which were much larger (500 nm)212 than the pit neck size limit of 50–80 nm.116,214 In addition to nanomaterials size indiscriminating uptake tendency, activation of multiple entry pathways in the ECs is also noted. Silica (15 nm)215 and PS (200 nm)212 NPs enter ECs via three different pathways, micropinocytosis, caveolin-mediated and clathrin-mediated pathways.
Both factors, i.e. size indiscriminate uptake and concurrent activation of multiple pathways, have made the uptake of non-targeting NP too technically challenging to be ascribed to one distinct pathway. Nevertheless, a number of studies demonstrate that overall internalization process of nanomedicine into the EC has a strong correlation with the physiochemical properties of the nanomaterial carrier.147,163,216–218 The strong correlation is expected because NP physicochemical properties mediate the initial engagement, through specific or non-specific interaction with cellular components (Fig. 8). This initial interaction then has to overcome the restrictive forces in order for the internalization process to occur. As such, the time required for the internalization as well as the efficiency of the internalization process itself could be expressed through the balance of various factors, namely size, shape, and surface chemistry of the NP in addition to the receptor distribution, elasticity of the cell membrane, and energy required to engulf the NP and transport it into the cell (Fig. 8).147,163,216–218
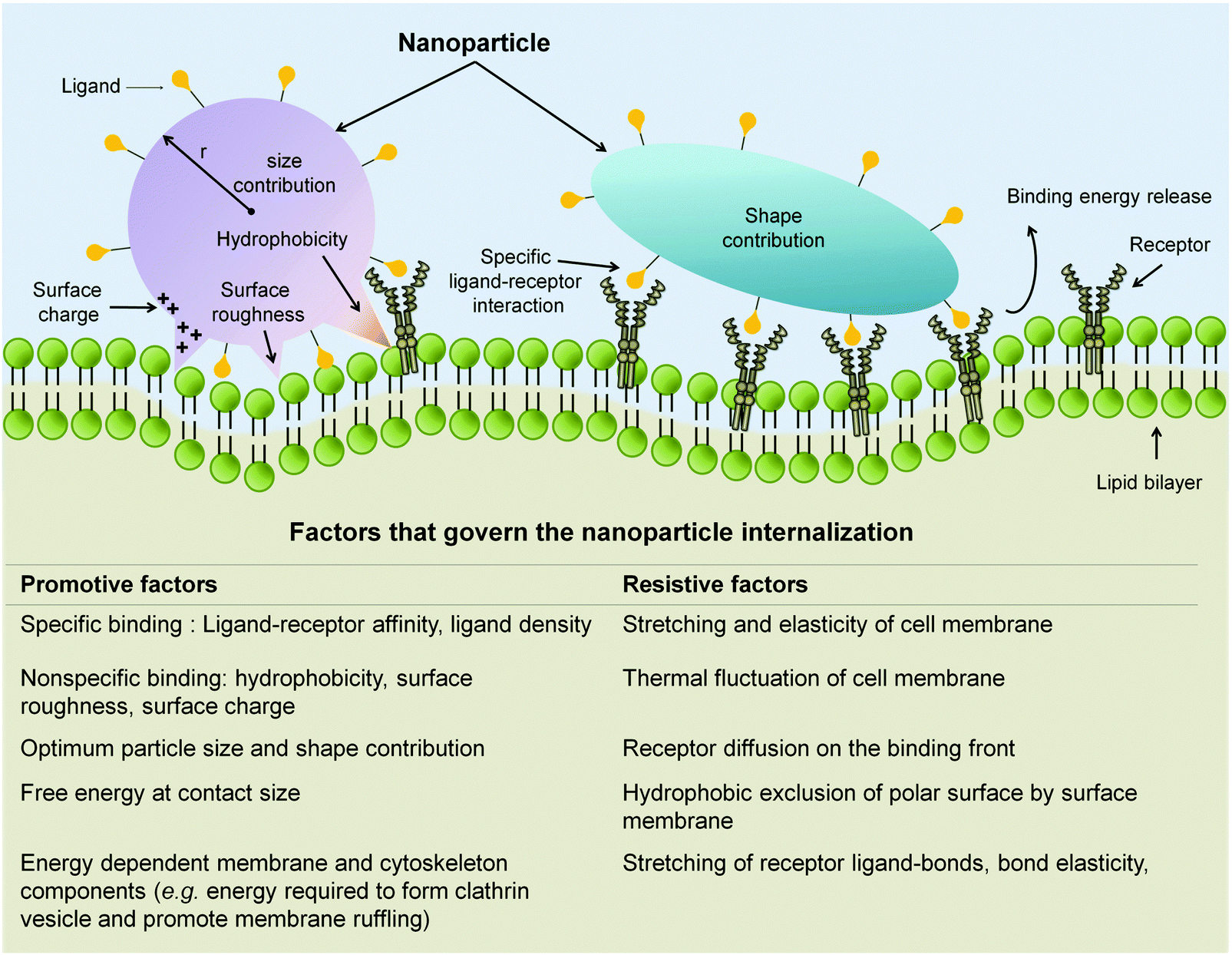 | ||
| Fig. 8 Attributed factors that dictate the NP internalization process. Through specific ligand-receptor interaction in addition to those non-specific ones (i.e. hydrophobic interaction, electrostatic pull, and surface roughness), NP promote their attachment and engulfment. NP size and shape also contribute to the promotive force required to overcome the resistive factors of internalization. Adapted from ref. 147. Copyright 2009 by Nature Publishing Group. Adapted with permission. | ||
Surface characteristic matters
As the initiation of internalization process occurs at the interface between the NP and the cells, NP surface characteristic is one factor that dictates the internalization process effectiveness. One component of the NP surface characteristic that could be tuned to facilitate nanomedicine delivery is the targeting ligand. There is a whole host of moieties (Table 2) that could be used as target for nanomedicine formulation (Table 3). Anti-E-selectin targeted liposomes and PS NPs are reported to be successfully delivered to the activated ECs.219,220 Targeting E-selectin with its ligand in delivering the porous silica NPs to metastatic breast cancer treatment to the bone marrow endothelium.221,222 In a similar manner, targeting VCAM-1 moiety on the cell surface ensured the intracellular delivery of the nanomedicine.223 This has benefited the imaging224 as well as drug delivery effort to inflamed ECs.225 The strategy also enhanced in vivo detection and therapeutic intervention of pathological conditions (i.e. atherosclerosis and thrombosis).220,226The perceivable benefit of attaching the targeting ligand is the ligand-receptor specific interaction which activates a clathrin mediated endocytosis pathway. However, targeting capability is affected by a number of factors. To name a few of them: (1) distribution profile of the targeted surface receptors on the cell attachment front;36,147 (2) the stability of the bond used to attach the ligand on the NP surface;36,227 (3) the ligand change of binding affinity due to surface attachment, presumably due to the orientation of the attached ligand that block the recognition site.36,227 Thus, optimization on the ligand density is required and the possibility of multiple ligands attachment should be considered (reviewed here36,115,228).
In addition to those of biological origin, internalization could be mediated by groups that are derived from chemical technique (e.g. DNA, peptide, and chemical group). DNA conjugation on SWCNT was reported to facilitate the cell internalization and imaging of HUVEC.211 Likewise, the cell penetrating peptides attachment offers a versatile method for gene delivery to EC.229 Alternatively, surface functionalization with chemical moieties such as amine groups allow initial attachment of NP on the cell surface to be mediated by the electrostatic interaction.
Indeed, surface charge of the NP is one major determiner in the non-specific binding between NP and cell surface. Iron oxide (Fe3O4), SiO2 and titanium dioxide (TiO2) NPs of the similar size (117–127 nm) but different surface charge were internalized by the ECs with different efficiency.230 TEM images show positively charged Fe3O4 NP to be taken in the most, followed by moderate internalization of the neutral charge of TiO2 NP. In contrast, the cells were shown to take negatively charged SiO2 NP the least.230 The effect of surface charge is also recapitulated in a more controlled study utilizing polymer modified gold NPs (average particle diameter of ∼75 nm). Dermal EC showcases extremely high up take (100% uptake) of positively-charged ethanediamine-modified gold NP following 24 h exposure. Moderate uptake was observed for neutral charged NP modelled by the hydroxypropylamine-coated gold NP while the negative-charged (taurine coated gold NP) was observed with almost no internalization.231 The importance of nanomaterials surface charge in regulating the ECs internalization is well documented by other studies.232–234 The surface charge effect is postulated to occur due to the electrostatic interaction (i.e. electrostatic attraction/repulsion) between the NP and the negatively charged phospholipid layer of the cell membrane232,235 or with protein domain on the cell surface.147 In addition, NPs' surface charge could affect the types and orientations of the proteins that were acquired during circulation from the blood flow. As previously discussed (see Section 3 for more details), it is possible that the positively charged NP recruited proteins that could promote their binding and uptake profile.190
Though there is no direct comparative study for EC, extrapolation from other mammalian cell model and simulation studies suggest that hydrophobicity nature of the NP surface effect its internalization profile. Profound internalization was observed for natural organic matter coated fullerene (C70-NOM), while its counterpart, the hydrophilic fullerol (C60(OH)20) was excluded from the cells.236 This hydrophobicity effect is also recapitulated in a molecular dynamic simulation where the hydrophobic fullerene (C60) was described to ‘jump into’ the model lipid bilayer, dipalmitoylphosphatidylcholine (DPPC). This results in the C60 instantaneous internalization (t = 4.09 ns). The more hydrophilic fullerol, C60(OH)20, requires 48 ns to get internalized by the cells.218 More recent studies identify that the this spontaneous internalization is accommodated by the hydrophobic interaction between the NMs (e.g. fullerene, graphene) and the lipid tail found in the cell membrane,216,217,236,237 which concomitant with the lipid peroxidation prominently found following their exposure to cells.
Size matters
In addition to the surface characteristics of the NP, several other factors (e.g. NP size and shape) also promote the internalization process (Fig. 8). Indeed, the bulk of evidence highlights the size dependent NP uptake to ECs. HUVEC internalized higher amount of 100 nm nanosphere PS when compared to their 200 nm counterpart.212 Conjugation of targeting moiety on the nanomaterials does not to abolish the size dependence effect of nanomaterials uptake. A study utilizing anti-ICAM-1 conjugated PS nanosphere (0.1 and 1 μm) showed more rapid internalization of smaller size nanosphere by ECs.125 In contrast, citrate-capped gold NPs (18, 35, and 65 nm) internalization increased with an increase of NPs diameter, in which the gold NP with diameter of 65 nm showing the highest uptake.231 A more recent study that investigated a series of gold NP sizes (20, 50, 70, and 100 nm), reported highest uptake of gold NPs with diameter of 70 nm.238 This suggests that 70 nm to be a certain critical NP size that ensures nanoparticles optimum uptake into ECs. This value is skewed from other reported critical diameter of 50 nm for optimum NP uptake but from other mammalian cell models.163,239,240 Differences in the composition of the plasma membrane between the cell types in addition to the inherent differences in the preferential of endocytic pathway could contribute to the difference in determining the critical NP diameter. Conversely, critical dimension required for optimum EC uptake is observed for other shapes like nanodiscs. HUVEC showed the highest uptake of medium size nanodisc (d × h = 220 × 100 nm) as compared to its other counterparts (small, d × h = 80 × 70 nm; large, d × h = 325 × 100 nm).212In a controlled system, all variability from the cells could be considered as negligible as only one type of cell is used to make the comparison. NP uptake to the cells is then determined by two factors: (1) adhesion force between the NP and the cell surface and (2) the Gibbs free energy required for the membrane deformation.212,241 Within a given geometric shape, both factors strongly depend on the dimensions of the NP. Compared to small particles, large particles are postulated to have a larger contact area per particle, enabling them to have more contact with the cell surface and to initiate the uptake process. In a recent study, Demokritou and coworkers utilized NPs-coated AFM tip to measure the real-time interaction force between the NPs and cell membrane.242 It was found that the large NPs create more bonds with the cell membrane and thus require larger amount of force to detach them after the interaction with the cell membrane was formed. This supports the notion that the large NPs have larger contact area and thus form more interaction bonds with the cell membrane, resulting with their higher internalization. If this is the case, it would be logical to surmise that smaller NPs of the same geometrical shape will also be internalized by the cells; and indeed so but to a lesser degree. Smaller NP required a smaller Gibbs free energy cost to form the vesicle,243 and for those NP that entered the cell via non-vesicle formation; an even smaller adhesion force requirement.244 The critical dimension for NP internalization is created in a ‘sweet spot’ where NP could engage sufficient interaction with the cell and require a minimum Gibbs free energy for vesicle formation or via a penetration force through the lipid bilayer.
Shape matters
Beside the size aspect, nanomaterials internalization to ECs is heavily influenced by their shape. Shape influences increase with increase in size of the NP.244 Higher efficiency of siRNA delivery was observed for PS, PLGA, and PEG nanoneedle (aspect ratio, AR = 9) compared to their spherical counterparts (AR = 1) of similar volume and material composition, suggesting a more efficient uptake of the nanoneedle structure.245 ICAM-1 conjugated nanorods are taken into the cells in a greater amount than the nanopsheres of identical volume, resulting in the enhanced brain and lung targeting in the in vivo setting by the nanorods.246 In a more comprehensive study utilizing PS nanomaterials of similar volume comprising of nanospheres (d = 200 nm), nanorods (w × l × h = 400 × 100 × 100 nm) and nanodiscs (d × h = 220 × 100 nm), Agarwal et al. observed different uptake of these geometrically different NP. Nanodiscs was taken in most efficiently by ECs, followed by the nanorods, and lastly nanospheres.212 This different internalization profile further exemplify that the same controlling factors, i.e. the adhesion force between NP and cells in addition to the Gibbs free energy requirement, profoundly influenced the internalization process of geometrically different NP. The disc, the rod, and the needle shaped NP offers larger surface area per unit volume compared to spherical shaped NP. This facilitates more interaction with the cells and leads to higher internalization. Moreover, the fact that the EC favors the nanodisc over nanorod of equal volume, when the surface area difference between the two NP are less than 5%, highlight the important role of Gibbs free energy requirement.212,243 Indeed, Agarwal et al. reported that discoidal NP required less energy to fold the membrane around the NP when compared to the rod-shaped NP of equal volume.212However, the comparative study between different shapes of gold NP resulted in a similar amount of nanorods and nanospheres being internalized by the ECs.247 Similarly, PLGA elliptical nanodiscs and nanospheres of the same material and volume were taken in the same amount by the HUVEC.248 Interestingly, the elliptical nanodisc uptake rate was much slower than the spherical particle. Overall, these studies suggested that the interpretation of geometrical dependency on EC uptake in some cases is not as straightforward as it seems. One also has to consider the intertwined role of geometrical orientation in the internalization process.244,249 In comparison to the spherical shaped NP, rod and disc shaped will only provide larger surface area for interaction when their long axis is in the parallel plane with the cell membrane. In contrast, when their long axis is perpendicular with the cell membrane plane, they will offer no appreciable advantage over the sphere with regard to the surface area required for the interaction.250 This is postulated to cause the negation of shape dependency effect.
Not limited to those that have been discussed previously, NP properties such as rigidity,36,246 curvature,239,251 surface roughness252,253 could also help in promoting the internalization to the EC. Surface roughness, for example, becomes relevant at nanoscale. The small radii protrusion and depression was simulated to greatly reduce the repulsive force (e.g. electrostatic, hydrophilic), cementing stronger adhesion compared to the smooth NP.252,253
Interestingly, physiochemical properties of the nanocarrier, especially its surface chemistry, also play a major role to determine the subcellular addressing of the nanomedicine once it gets internalized.254 Cationic liposomes are known for its ability to escape the endosome compartments. It is postulated that due to its charge the cationic liposome interact with the anionic phospholipid layer of the endosomal compartment, resulting in the destabilization of the endosome and the nanomedicine release to the cytoplasm.255 NP with tertiary amine groups on its surface (e.g. polyethylenimine and polyamidoamine) is also reported to escape the endosomal and lysosomal compartment by acting as proton sponges. The presence of the NP inside the endosome/lysosome prevented acidification of these compartments. As such, counter-ions were pumped into the compartments to reach the desired pH. Nevertheless, the counter ions influx resulted in the disruption of the osmotic balance in the compartment, leading to the rupture of the compartments and the release of the NPs into the cytoplasm.256 Negatively charged polymer NP was observed to polymerize actin filament and mediate the DNA payload delivery to nucleus, while the positively charged NP did not.257 Governance over the internalization process is important to determine the successful delivery of nanomedicine. The effect of NP physicochemical properties has been shown to promote initial engagement of cell component that lead to its internalization and subcellular addressing.
From another perspective, it is also important to note that in situations where the ECs are not the target tissue, the internalization of the NPs in the ECs should therefore be avoided. The common strategy is to PEGylate the NPs, however, that same strategy is not suitable as PEGylation would also decrease internalization of the NPs into the target cells due to their crowding effect of the PEG over the targeting moieties.36,258 Finding the optimum ratio between the targeting moieties to PEG density conjugated on the surface allows a compromise that minimizes the off-target issue (reviewed extensively by Jokerst et al.258). Alternatively, one could factor in the fundamental environment differences between the targeted and non-targeted vasculature into the PEGylation design. For instance, the immediate vasculature surrounding the tumor is usually more acidic (pH 6.5–6.8) than the physiological pH (7.4).259 Taking this fact to consideration, one could incorporate pH sensitive linkers in designing their PEG layer, as was reported by Torchilin et al. The authors utilized hydrazone, a low pH sensitive linker, to link the targeting ligands to the PEG chain. At low pH, the hydrazone bond degrades, exposing the targeting ligand from the otherwise crowding PEG chain; this exposure then facilitated the NP initial interaction with the cells.260 Similarly, poly histidine chain was reported to successfully mediate the pH induced biotin (targeting moiety) reposition process from the PEG shield on the polymer NP surface.261 Another possible strategy is to forgo the need of minimizing the interaction of the nanoparticles with the luminal cell membrane of the ECs altogether. This could be easily done if the NPs have a path of escape between the ECs through the relaxation of the rather exclusive paracellular route (described in more detail in Section 5).
5. Nanomaterials properties that dictates their transport across the vascular barrier
In most pathological conditions, the actual target for the nanomedicine is beyond the endothelium, necessitating the nanomedicine to traverse across the endothelium barrier. For instance, nanomedicine intended for cancer treatment has to traverse across the tumor vasculature prior to find its way to the target tumor cells, though the delivery is greatly helped by the leakiness of tumor vasculature (known as EPR effect94,95). Nevertheless, this becomes a major hurdle for numerous therapeutic strategies intended to treat those pathological conditions that are barred by the presence of intact continuous vascular barrier, such for the case intended for intracerebral drug.Following traditional solute transport paradigm (Fig. 9), nanomedicine could cross over the vascular barrier via two routes: the transcellular (i.e. it gets internalized at the apical (luminal) side, transported across the cell body and exited at the basolateral side of the EC) and paracellular where it diffuses through the EC intercellular junction space.
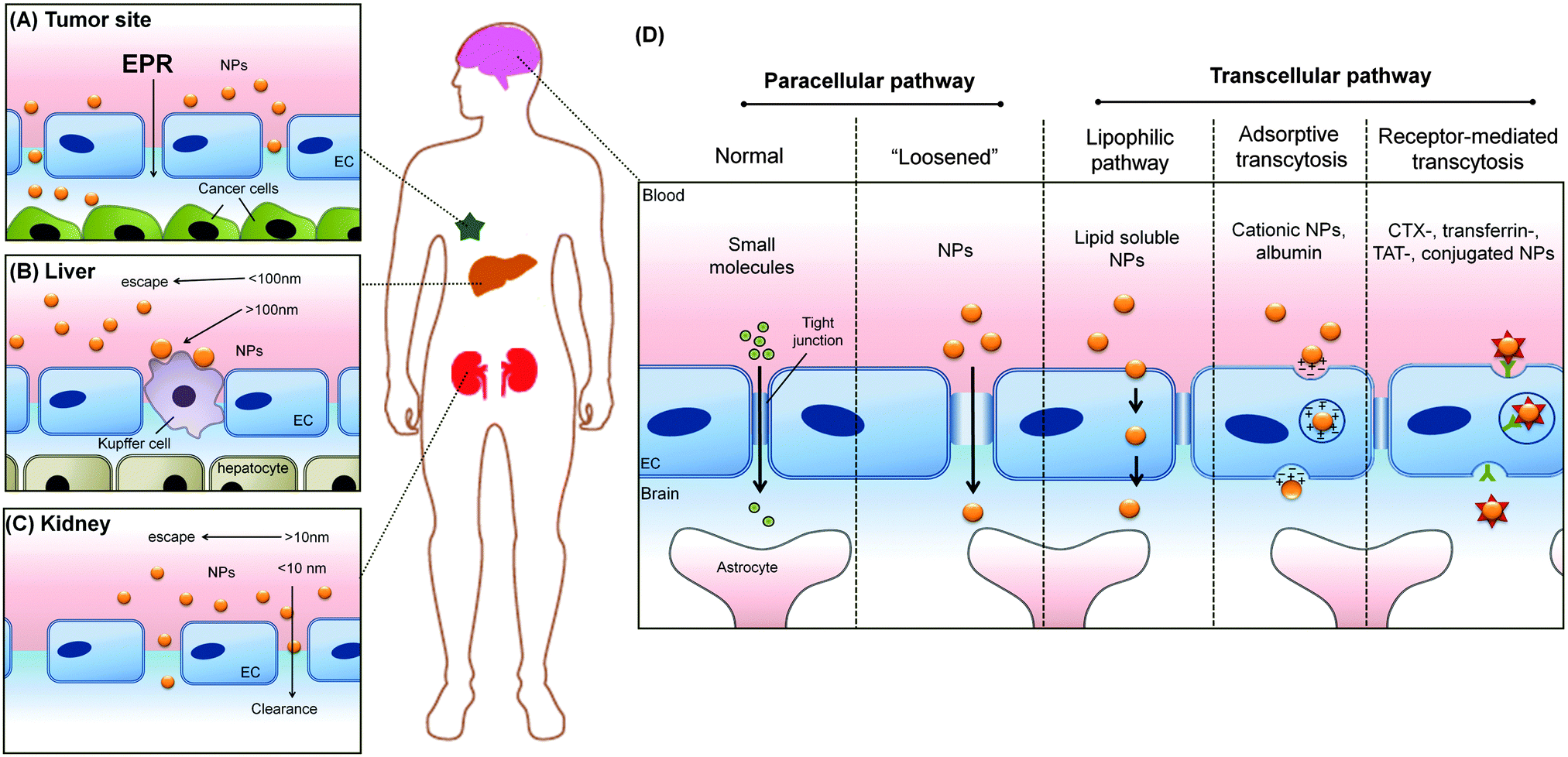 | ||
| Fig. 9 Physiological barriers encountered by circulating nanomedicine particles. (A) NPs with the size of 30–200 nm could passively reach the tumor site due to the EPR effect. (B) The NPs with the size more than 100 nm triggers the Kupffer cells to remove them from the blood circulation. (C) NPs (<10 nm) that reach the kidney are cleared out from by way of size exclusion through the glomerulus fenestration. (D) The tight junction structure in the BBB prevents the NPs passive penetration to the brain. Thus, nanomedicine formulation is required to actively engage the transcellular pathway in addition to approach the delivery by ‘loosening’ the interendothelial gaps. Adapted from ref. 262. Copyright 2011 by Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Adapted with permission. | ||
Transcellular route
As transcellular transport (Fig. 9) is initiated with the internalization of nanomedicine, it is then anticipated that those physicochemical properties that affect its internalization will also affect its transcellular process. Transferrin-conjugated gold NP (20, 40, and 80 nm) shows size dependent transcytosis capability, with the highest delivery across the BBB observed for NP with size of 80 nm. Optimum association of NP and transferrin receptors was thought to be the reason that the NP with largest diameter is found to most efficient in facilitating delivery into the brain.263Surface identity is shown to be most prominent aspect of the NP that influences it transport via the transcellular route. Electrostatic interaction mediated the enhanced transcytosis of cationized albumins over the negatively charged BBB.264 Similarly, positively charged tripalmitin NP attained the highest etoposide delivery to the brain as compared to its negatively charged counterpart.265 Positively charged maltodextrin NPs (60 nm) showed close to 20-fold improvement of BBB penetration when compared to the uncoated.266 Coating or conjugating nanomedicine with albumin could enhance the transendothelial transport of the nanomedicine.267 The albumin on the NP surface acts on as ligand that mediates the interaction between the nanomedicine with the albumin binding proteins (gp60 and gp90) located at the caveolae.36,267 Upon interaction with gp60 and gp90 proteins, the nanomedicine is brought into the cell via the caveolin-mediated pathway, where the NP is exempted from the endosomal and lysosomal processing, resulting in the overall enhancement of transendothelial transport.115,268,269 Incorporation of prion proteins,270 GM-1 binding peptide,271 and rabies virus glycoprotein272 on the NP surface targeted the caveolin mediated pathway internalization and improved the nanomedicine transcytosis over BBB. A number of studies showed the crossing of even the most impenetrable barrier, BBB, was possible by targeting receptors with transcytotic capacities like transferrin receptor,273,274 leptin receptor,275,276 and insulin receptor.277,278
Paracellular route
Due to the size constraint of the intercellular gap, the paracellular pathway traditionally is conserved for the route small solutes (Fig. 9). Early effort in nanomedicine design strategy showed that poly-amindoamine (PAMAM) dendrimers (15–45 Å) utilized this paracellular route to gain entrance to the brain.279 In a more recent study, Leong et al. observed that TiO2 NP (23 nm) by virtue of its small size could squeeze into the intercellular junction of the dermal ECs.280 In addition to size, surface charge of the nanomedicine also influenced the transport route to cross the vascular barrier. Cationic glucose NP was primarily found in the paracellular area, while its neutral NP counterpart was found mostly at the surface of the EC. Further investigation revealed that the neutral NP entered the brain through caveolae pathway, while the cationic NP entered via the paracellular route.281Paracellular route that is highly restrictive in nature has deterred many nanomedicine strategies to make this route as the main route of delivery. Nevertheless, recent developments show possible strategy to ‘loosened’ the paracellular route, making this pathway a viable option for nanomedicine. Leong et al. observed that the TiO2 NP found in the paracellular route could interact with the VE-cadherin junction protein. This interaction resulted in the disruption of the VE-cadherin homophilic pairing, triggered intracellular signaling and cytoskeleton remodeling in the EC, resulting in a gap formation in the range of microns (Fig. 10). At this gap scale, many drugs and NPs can easily cross the endothelial barrier in effect causing the opening of paracellular route. This effect was coined to be ‘nanoparticle induced endothelial leakiness’ (NanoEL).280
 | ||
| Fig. 10 Opening the paracellular route could potentially assist the delivery of drug payload to the restricted disease site. (A) The opening of the paracellular route is obvious from the interendothelial cells gaps formed. (B) The opening of the paracellular route is initiated by (1) physical interaction of the NPs with adherens junction component, VE-cadherin. This (2) triggers intracellular signal transduction which leads to (3) cytoskeleton modulation, (4) cell shape contraction. Adapted from ref. 280 with the permission of The Nature Publishing Group. | ||
Aluminium oxide NP was reported to reduce the expression of junctional protein in brain EC, claudin-5, providing less restrictive paracellular route.282 Most recently, targeting the adenosine receptor with nanoagonists has been shown to induce the brain ECs contraction that leads to the loss of junctional protein integrity and result in the temporary increased paracellular route permeability for approximately 30 min. Upon the increased paracellular route permeability mediated by the nanoantagonist, the therapeutic load entered the brain more easily, as evidenced by the enhanced delivery rate to the mouse brain (Fig. 11).283
 | ||
| Fig. 11 Schematic overview of the brain drug delivery strategy achieved by up-regulating the paracellular pathway via nanoagonist mediated A2A adenosine receptor signalling. Enhanced delivery of drug payload is observed with the increase of BBB permeability. Reproduced from ref. 283. Copyright 2014 by American Chemical Society. Reproduced with permission. | ||
6. Concluding remarks
In this review, we have discussed extensively on how ECs are not made the same. Even within the same tissue, there are diverse EC types. Even for the same segment of blood vessel in the same tissue, the target's diseased state would already create deviations from normal behavior. These diversity and deviations are very important considerations to be factored in. In most nanomedicine strategies, there is a need to overcome the endothelial barrier. The diseased state of the vasculature would either increase or lower the endothelial barrier. This minimally would affect the diffusion and pharmacokinetics of the nanomedicine at the site of interest. Thus, it is advisable to investigate through an in vitro co-culture system with the normal or diseased EC type with the cell type of interest. Many reported studies only have a direct exposure of the nanomedicine to the cell type of interest. An additional advantage is that the co-culture model can mimic to a certain extent, spatial, cell-endothelial interactions and paracrine exchanges between both cell layers. This could recapitulate the in vivo conditions, in vitro and reduces any artefactual outcomes of simply adding the nanomedicine to the target cell. If the endothelial barrier is taken into account in future nanomedicine design and investigation, one can also consider breaking down the overall nanomedicine strategy in such a way that there is one nanomaterial system that overcomes the barrier, possibly through the NanoEL effect, and another system that actually delivers the drug payload. One could design NPs that can elicit the NanoEL effect as needed at the site of interest with the sole intent of crossing the barrier transiently and with little collateral damage to the endothelium. That would then require a better understanding of the intrinsic physiocochemical properties of NP that induces (or not induce) the NanoEL effect. All the existing auxiliary technologies like bioimaging and targeting can also be deployed to supplement the ‘overcoming endothelial barrier’ system. Understanding the diversity of the ECs can certainly bring about new paradigms in targeting. Currently, many nanomedicine strategies target the cell type of interest. However, that address is hidden behind the endothelial barrier. Therefore, if there is a discernible marker difference between the proximal ECs to the target tissue compared to other tissues, then it would therefore be easier to target those proximal ECs instead of just the target tissue. This would certainly reduce the often undesirable side effects and reduce the overall dose received by the patient. Finally, by also taking into account how the intrinsic NP properties like size, shape, surface charge and density affect physiological and pathological ECs both proximal to or away from the target site will certainly build a holistic approach towards tackling the many complex interlocking challenges of negotiating or working cooperatively with the vasculature.Acknowledgements
This work was funded by Ministry of Education, Singapore (R-279-000-418-112 to DTL), Peter und Traudl Engelhorn Foundation, Zeiss ChemBioMed, BMBF-NanoBEL and Fonds der Chemischen Industrie.References
- T. J. Webster, Nanomedicine, 2013, 8, 525–529 CrossRef CAS PubMed.
- M. Reese, Health matrix, 2013, 23, 537–572 Search PubMed.
- K. Zheng, X. Yuan, N. Goswami, Q. Zhang and J. Xie, RSC Adv., 2014, 4, 60581–60596 RSC.
- Z. Luo, K. Zheng and J. Xie, Chem. Commun., 2014, 50, 5143–5155 RSC.
- G. Pyrgiotakis, J. McDevitt, Y. Gao, A. Branco, M. Eleftheriadou, B. Lemos, E. Nardell and P. Demokritou, Nanomedicine, 2014, 10, 1175–1183 CrossRef CAS PubMed.
- M. Pautler and S. Brenner, Int. J. Nanomed., 2012, 5, 803–809 Search PubMed.
- S. E. McNeil, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 264–271 CrossRef CAS PubMed.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872–897 CrossRef CAS PubMed.
- Wahajuddin and S. Arora, Int. J. Nanomed., 2012, 7, 3445–3471 CrossRef CAS PubMed.
- X.-D. Zhang, Z. Luo, J. Chen, H. Wang, S.-S. Song, X. Shen, W. Long, Y.-M. Sun, S. Fan, K. Zheng, D. T. Leong and J. Xie, Small, 2015, 11, 1683–1690 CrossRef CAS PubMed.
- X. Qian, X. H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS PubMed.
- M. Ferrari, Nat. Nanotechnol., 2008, 3, 131–132 CrossRef CAS PubMed.
- X. Yuan, M. Setyawati, D. Leong and J. Xie, Nano Res., 2014, 7, 301–307 CrossRef CAS.
- X.-D. Zhang, Z. Luo, J. Chen, X. Shen, S. Song, Y. Sun, S. Fan, F. Fan, D. T. Leong and J. Xie, Adv. Mater., 2014, 26, 4565–4568 CrossRef CAS PubMed.
- M. Helou, M. Reisbeck, S. F. Tedde, L. Richter, L. Bar, J. J. Bosch, R. H. Stauber, E. Quandt and O. Hayden, Lab Chip, 2013, 13, 1035–1038 RSC.
- M. I. Setyawati, R. V. Kutty, C. Y. Tay, X. Yuan, J. Xie and D. T. Leong, ACS Appl. Mater. Interfaces, 2014, 6, 21822–21831 CAS.
- C. Y. Tay, L. Yuan and D. T. Leong, ACS Nano, 2015, 9, 5609–5617 CrossRef CAS PubMed.
- X. Liang, X. Li, L. Jing, X. Yue and Z. Dai, Biomaterials, 2014, 35, 6379–6388 CrossRef CAS PubMed.
- A. Rajala, Y. Wang, Y. Zhu, M. Ranjo-Bishop, J. X. Ma, C. Mao and R. V. Rajala, Nano Lett., 2014, 14, 5257–5263 CrossRef CAS PubMed.
- C. Mi, J. Zhang, H. Gao, X. Wu, M. Wang, Y. Wu, Y. Di, Z. Xu, C. Mao and S. Xu, Nanoscale, 2010, 2, 1141–1148 RSC.
- D. E. Lee, H. Koo, I. C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656–2672 RSC.
- M. S. Muthu, D. T. Leong, L. Mei and S. S. Feng, Theranostics, 2014, 4, 660–677 CrossRef CAS PubMed.
- R. V. Kutty, S. L. Chia, M. I. Setyawati, M. S. Muthu, S.-S. Feng and D. T. Leong, Biomaterials, 2015, 63, 58–69 CrossRef CAS PubMed.
- X. Liang, Y. Li, X. Li, L. Jing, Z. Deng, X. Yue, C. Li and Z. Dai, Adv. Funct. Mater., 2015, 25, 1451–1462 CrossRef CAS PubMed.
- A. M. Nystrom and B. Fadeel, J. Controlled Release, 2012, 161, 403–408 CrossRef PubMed.
- R. Bawa, Curr. Drug Delivery, 2011, 8, 227–234 CrossRef CAS.
- A. Baun and S. F. Hansen, Nanomedicine, 2008, 3, 605–608 CrossRef PubMed.
- M. Rahman, M. Z. Ahmad, I. Kazmi, S. Akhter, M. Afzal, G. Gupta and V. R. Sinha, Curr. Drug Discovery Technol., 2012, 9, 319–329 CrossRef CAS.
- C. L. Ventola, P. T., 2012, 37, 631–639 Search PubMed.
- M. I. Setyawati, X. Yuan, J. Xie and D. T. Leong, Biomaterials, 2014, 35, 6707–6715 CrossRef CAS PubMed.
- A. C. Jachak, M. Creighton, Y. Qiu, A. B. Kane and R. H. Hurt, MRS Bull., 2012, 37, 1307–1313 CrossRef CAS PubMed.
- R. Foldbjerg, X. Jiang, T. Miclaus, C. Chen, H. Autrup and C. Beer, Toxicol. Res., 2015, 4, 563–575 RSC.
- K. Y. Choi, H. Y. Yoon, J.-H. Kim, S. M. Bae, R.-W. Park, Y. M. Kang, I.-S. Kim, I. C. Kwon, K. Choi, S. Y. Jeong, K. Kim and J. H. Park, ACS Nano, 2011, 5, 8591–8599 CrossRef CAS PubMed.
- M. Ding, N. Song, X. He, J. Li, L. Zhou, H. Tan, Q. Fu and Q. Gu, ACS Nano, 2013, 7, 1918–1928 CrossRef CAS PubMed.
- D. Docter, D. Westmeier, M. Markiewicz, S. Stolte, S. K. Knauer and R. H. Stauber, Chem. Soc. Rev., 2015 10.1039/C5CS00217F.
- M. Howard, B. J. Zern, A. C. Anselmo, V. V. Shuvaev, S. Mitragotri and V. Muzykantov, ACS Nano, 2014, 8, 4100–4132 CrossRef CAS PubMed.
- H. Wolinsky, Circ. Res., 1980, 47, 301–311 CrossRef CAS.
- H. G. Augustin, D. H. Kozian and R. C. Johnson, BioEssays, 1994, 16, 901–906 CrossRef CAS PubMed.
- E. Langenkamp and G. Molema, Cell Tissue Res., 2009, 335, 205–222 CrossRef CAS PubMed.
- W. C. Aird, Circ. Res., 2007, 100, 158–173 CrossRef CAS PubMed.
- M. Miyasaka and T. Tanaka, Nat. Rev. Immunol., 2004, 4, 360–370 CrossRef CAS PubMed.
- L. Florey, Br. Med. J., 1966, 2, 487–490 CrossRef.
- J. B. Silkworth, W. E. Stehbens and D. Phil, Angiology, 1975, 26, 474–487 CrossRef PubMed.
- G. Kibria, D. Heath, P. Smith and R. Biggar, Thorax, 1980, 35, 186–191 CrossRef CAS.
- A. Hirata, P. Baluk, T. Fujiwara and D. M. McDonald, Am. J. Physiol., 1995, 269, L403–L418 CAS.
- W. C. Aird, Crit. Care Med., 2003, 31, S221–S230 CrossRef PubMed.
- A. Hartsock and W. J. Nelson, Biochim. Biophys. Acta, 2008, 1778, 660–669 CrossRef CAS PubMed.
- C. Weidenfeller, E. V. Shusta, C. Weidenfeller and E. V. Shusta, in Endothelial Biomedicine, ed. W. C. Aird, Cambridge University Press, Cambridge, UK, 2007, pp. 1124–1139 Search PubMed.
- M. Simionescu and N. Simonescu, Handbook of Physiology: The Cardiovascular System, American Physiological Society, Bathesda, USA, 1984, vol. 4, pp. 44–164 Search PubMed.
- E. Wisse, J. Ultrastruct. Res., 1970, 31, 125–150 CrossRef CAS.
- A. R. Pries and W. M. Kuebler, Handb. Exp. Pharmacol., 2006, 1–40 CAS.
- W. C. Aird, Circ. Res., 2007, 100, 174–190 CrossRef CAS PubMed.
- B. Molitoris, in Endothelial Biomedicine, ed. W. C. Aird, Cambridge University Press, Cambridge, UK, 2007, pp. 1271–1277 Search PubMed.
- O. Cleaver and D. A. Melton, Nat. Med., 2003, 9, 661–668 CrossRef CAS PubMed.
- J. T. Chi, H. Y. Chang, G. Haraldsen, F. L. Jahnsen, O. G. Troyanskaya, D. S. Chang, Z. Wang, S. G. Rockson, M. van de Rijn, D. Botstein and P. O. Brown, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10623–10628 CrossRef CAS PubMed.
- P. N. Belloni and G. L. Nicolson, J. Cell. Physiol., 1988, 136, 398–410 CrossRef CAS PubMed.
- M. I. Fatehi, D. Z. Gerhart, T. G. Myers and L. R. Drewes, Brain Res., 1987, 415, 30–39 CrossRef CAS.
- D. J. Goetz, M. E. el-Sabban, D. A. Hammer and B. U. Pauli, Int. J. Cancer, 1996, 65, 192–199 CrossRef CAS.
- D. B. Cines, E. S. Pollak, C. A. Buck, J. Loscalzo, G. A. Zimmerman, R. P. McEver, J. S. Pober, T. M. Wick, B. A. Konkle, B. S. Schwartz, E. S. Barnathan, K. R. McCrae, B. A. Hug, A. M. Schmidt and D. M. Stern, Blood, 1998, 91, 3527–3561 CAS.
- A. H. Schinkel, Adv. Drug Delivery Rev., 1999, 36, 179–194 CrossRef CAS.
- E. G. Levin, C. L. Banka and G. C. Parry, Arterioscler., Thromb., Vasc. Biol., 2000, 20, 1668–1674 CrossRef CAS.
- M. S. Bajaj, M. N. Kuppuswamy, A. N. Manepalli and S. P. Bajaj, Thromb. Haemostasis, 1999, 82, 1047–1052 CAS.
- R. R. Turner, J. H. Beckstead, R. A. Warnke and G. S. Wood, Am. J. Clin. Pathol., 1987, 87, 569–575 CAS.
- K. Yamamoto, V. de Waard, C. Fearns and D. J. Loskutoff, Blood, 1998, 92, 2791–2801 CAS.
- M. E. Gerristen, Microvascular Research: Biology and Pathology, Academic Press, Oxford, UK, 1st edn, 2005 Search PubMed.
- T. Hirase, J. M. Staddon, M. Saitou, Y. Ando-Akatsuka, M. Itoh, M. Furuse, K. Fujimoto, S. Tsukita and L. L. Rubin, J. Cell Sci., 1997, 110, 1603–1613 CAS.
- H. Hirakawa, S. Okajima, T. Nagaoka, T. Kubo, T. Takamatsu and M. Oyamada, NeuroReport, 2004, 15, 405–408 CrossRef CAS PubMed.
- H. M. DeLisser, P. J. Newman and S. M. Albelda, Immunol. Today, 1994, 15, 490–495 CrossRef CAS.
- A. Vecchi, C. Garlanda, M. G. Lampugnani, M. Resnati, C. Matteucci, A. Stoppacciaro, H. Schnurch, W. Risau, L. Ruco and A. Mantovani, et al. , Eur. J. Cell Biol., 1994, 63, 247–254 CAS.
- P. N. Belloni and R. J. Tressler, Cancer Metastasis Rev., 1990, 8, 353–389 CrossRef CAS.
- H. Ishii, H. H. Salem, C. E. Bell, E. A. Laposata and P. W. Majerus, Blood, 1986, 67, 362–365 CAS.
- M. C. Boffa, B. Burke and C. C. Haudenschild, J. Histochem. Cytochem., 1987, 35, 1267–1276 CrossRef CAS PubMed.
- E. Dejana, Nat. Rev. Mol. Cell Biol., 2004, 5, 261–270 CrossRef CAS PubMed.
- M. Giannotta, M. Trani and E. Dejana, Dev. Cell, 2013, 26, 441–454 CrossRef CAS PubMed.
- D. B. Cines, E. S. Pollak, C. A. Buck, J. Loscalzo, G. A. Zimmerman, R. P. McEver, J. S. Pober, T. M. Wick, B. A. Konkle, B. S. Schwartz, E. S. Barnathan, K. R. McCrae, B. A. Hug, A.-M. Schmidt and D. M. Stern, Endothelial Cells in Physiology and in the Pathophysiology of Vascular Disorders, 1998 Search PubMed.
- T. Nakamura, P. Ruiz-Lozano, V. Lindner, D. Yabe, M. Taniwaki, Y. Furukawa, K. Kobuke, K. Tashiro, Z. Lu, N. L. Andon, R. Schaub, A. Matsumori, S. Sasayama, K. R. Chien and T. Honjo, J. Biol. Chem., 1999, 274, 22476–22483 CrossRef CAS PubMed.
- W. A. Jefferies, M. R. Brandon, S. V. Hunt, A. F. Williams, K. C. Gatter and D. Y. Mason, Nature, 1984, 312, 162–163 CrossRef CAS PubMed.
- H. Weiler-Guettler, P. D. Christie, D. L. Beeler, A. M. Healy, W. W. Hancock, H. Rayburn, J. M. Edelberg and R. D. Rosenberg, J. Clin. Invest., 1998, 101, 1983–1991 CrossRef CAS PubMed.
- Y. Fujioka and Y. Ishikawa, J. Atheroscler. Thromb., 2009, 16, 145–154 CrossRef CAS.
- F. Braet and E. Wisse, Comp. Hepatol., 2002, 1, 1 CrossRef.
- P. del Vecchio, A. Siflinger-Birnboim, P. Belloni, L. Holleran, H. Lum and A. Malik, In Vitro Cell. Dev. Biol., 1992, 28, 711–715 Search PubMed.
- N. Sándor, F. R. Walter, A. Bocsik, P. Sántha, B. Schilling-Tóth, V. Léner, Z. Varga, Z. Kahán, M. A. Deli, G. Sáfrány and H. Hegyesi, PLoS One, 2014, 9, e112397 Search PubMed.
- A. S. Antonov, M. A. Nikolaeva, T. S. Klueva, Y. A. Romanov, V. R. Babaev, V. B. Bystrevskaya, N. A. Perov, V. S. Repin and V. N. Smirnov, Atherosclerosis, 1986, 59, 1–19 CrossRef CAS.
- A. S. Antonov, N. S. Key, M. D. Smirnov, H. S. Jacob, G. M. Vercellotti and V. N. Smirnov, Thromb. Res., 1992, 67, 135–145 CrossRef CAS.
- J. S. Pober and W. C. Sessa, Nat. Rev. Immunol., 2007, 7, 803–815 CrossRef CAS PubMed.
- S. Muro, X. Cui, C. Gajewski, J. C. Murciano, V. R. Muzykantov and M. Koval, Am. J. Physiol.: Cell Physiol., 2003, 285, C1339–C1347 CrossRef CAS PubMed.
- T. Kumasaka, W. M. Quinlan, N. A. Doyle, T. P. Condon, J. Sligh, F. Takei, A. Beaudet, C. F. Bennett and C. M. Doerschuk, J. Clin. Invest., 1996, 97, 2362–2369 CrossRef CAS PubMed.
- T. K. Kishimoto and R. Rothlein, Adv. Pharmacol., 1994, 25, 117–169 CAS.
- S. M. Albelda, Am. J. Respir. Cell Mol. Biol., 1991, 4, 195–203 CrossRef CAS PubMed.
- C. Garlanda and E. Dejana, Arterioscler., Thromb., Vasc. Biol., 1997, 17, 1193–1202 CrossRef CAS.
- D. Ribatti, B. Nico, A. Vacca, L. Roncali and F. Dammacco, J. Hematother. Stem Cell Res., 2002, 11, 81–90 CrossRef PubMed.
- A. C. Dudley, Cold Spring Harbor Perspect. Med., 2012, 2, a006536 Search PubMed.
- H. Hashizume, P. Baluk, S. Morikawa, J. W. McLean, G. Thurston, S. Roberge, R. K. Jain and D. M. McDonald, Am. J. Pathol., 2000, 156, 1363–1380 CrossRef CAS.
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- K. Greish, Methods Mol. Biol., 2010, 624, 25–37 CAS.
- P. Baluk, H. Hashizume and D. M. McDonald, Curr. Opin. Genet. Dev., 2005, 15, 102–111 CrossRef CAS PubMed.
- R. Kalluri, Nat. Rev. Cancer, 2003, 3, 422–433 CrossRef CAS PubMed.
- R. K. Jain, L. L. Munn and D. Fukumura, Nat. Rev. Cancer, 2002, 2, 266–276 CrossRef CAS PubMed.
- T. A. Springer, Nature, 1990, 346, 425–434 CrossRef CAS PubMed.
- M. J. Eppihimer, B. Wolitzky, D. C. Anderson, M. A. Labow and D. N. Granger, Circ. Res., 1996, 79, 560–569 CrossRef CAS.
- G. S. Kansas, Blood, 1996, 88, 3259–3287 CAS.
- D. D. Henninger, J. Panes, M. Eppihimer, J. Russell, M. Gerritsen, D. C. Anderson and D. N. Granger, J. Immunol., 1997, 158, 1825–1832 CAS.
- P. C. Brooks, A. M. Montgomery, M. Rosenfeld, R. A. Reisfeld, T. Hu, G. Klier and D. A. Cheresh, Cell, 1994, 79, 1157–1164 CrossRef CAS.
- S. Kim, K. Bell, S. A. Mousa and J. A. Varner, Am. J. Pathol., 2000, 156, 1345–1362 CrossRef CAS.
- N. R. Smith, D. Baker, N. H. James, K. Ratcliffe, M. Jenkins, S. E. Ashton, G. Sproat, R. Swann, N. Gray, A. Ryan, J. M. Jürgensmeier and C. Womack, Clin. Cancer Res., 2010, 16, 3548–3561 CrossRef CAS PubMed.
- V. Martin, D. Liu, J. Fueyo and C. Gomez-Manzano, Histol. Histopathol., 2008, 23, 773–780 CAS.
- W. J. Rettig, P. Garin-Chesa, J. H. Healey, S. L. Su, E. A. Jaffe and L. J. Old, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 10832–10836 CrossRef CAS.
- C. Garnacho, S. M. Albelda, V. R. Muzykantov and S. Muro, J. Controlled Release, 2008, 130, 226–233 CrossRef CAS PubMed.
- V. R. Muzykantov, ISRN Vasc. Med., 2013, 2013, 27 Search PubMed.
- K. Nowak, S. Weih, R. Metzger, R. F. Albrecht, 2nd, S. Post, P. Hohenberger, M. M. Gebhard and S. M. Danilov, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2007, 293, L162–L169 CrossRef CAS PubMed.
- P. N. Reynolds, K. R. Zinn, V. D. Gavrilyuk, I. V. Balyasnikova, B. E. Rogers, D. J. Buchsbaum, M. H. Wang, D. J. Miletich, W. E. Grizzle, J. T. Douglas, S. M. Danilov and D. T. Curiel, Mol. Ther., 2000, 2, 562–578 CrossRef CAS PubMed.
- W. H. Miller, M. J. Brosnan, D. Graham, C. G. Nicol, I. Morecroft, K. M. Channon, S. M. Danilov, P. N. Reynolds, A. H. Baker and A. F. Dominiczak, Mol. Ther., 2005, 12, 321–327 CrossRef CAS PubMed.
- P. N. Reynolds, S. A. Nicklin, L. Kaliberova, B. G. Boatman, W. E. Grizzle, I. V. Balyasnikova, A. H. Baker, S. M. Danilov and D. T. Curiel, Nat. Biotechnol., 2001, 19, 838–842 CrossRef CAS PubMed.
- E. T. Keelan, A. A. Harrison, P. T. Chapman, R. M. Binns, A. M. Peters and D. O. Haskard, J. Nucl. Med., 1994, 35, 276–281 CAS.
- S. Muro, M. Koval and V. Muzykantov, Curr. Vasc. Pharmacol., 2004, 2, 281–299 CrossRef CAS.
- J. E. Schnitzer, N. Engl. J. Med., 1998, 339, 472–474 CrossRef CAS PubMed.
- A. R. Patel, M. B. Chougule, E. Lim, K. P. Francis, S. Safe and M. Singh, Nanomedicine, 2014, 10, 1053–1063 CrossRef CAS PubMed.
- B. Liu, S. M. Han, X. Y. Tang, L. Han and C. Z. Li, Asian Pac. J. Cancer Prev., 2014, 15, 4915–4918 CrossRef.
- F. Bai, C. Wang, Q. Lu, M. Zhao, F. Q. Ban, D. H. Yu, Y. Y. Guan, X. Luan, Y. R. Liu, H. Z. Chen and C. Fang, Biomaterials, 2013, 34, 6163–6174 CrossRef CAS PubMed.
- J.-H. Kim, Y.-S. Kim, K. Park, E. Kang, S. Lee, H. Y. Nam, K. Kim, J. H. Park, D. Y. Chi, R.-W. Park, I.-S. Kim, K. Choi and I. Chan Kwon, Biomaterials, 2008, 29, 1920–1930 CrossRef CAS PubMed.
- R. G. Bagley, J. Walter-Yohrling, X. Cao, W. Weber, B. Simons, B. P. Cook, S. D. Chartrand, C. Wang, S. L. Madden and B. A. Teicher, Cancer Res., 2003, 63, 5866–5873 CAS.
- S. M. Herbst, M. E. Klegerman, H. Kim, J. Qi, H. Shelat, M. Wassler, M. R. Moody, C.-M. Yang, X. Ge, Y. Zou, J. A. Kopechek, F. J. Clubb, D. C. Kraemer, S. Huang, C. K. Holland, D. D. McPherson and Y.-J. Geng, Mol. Pharmaceutics, 2010, 7, 3–11 CrossRef CAS PubMed.
- L. Paulis, I. Jacobs, N. van den Akker, T. Geelen, D. Molin, L. Starmans, K. Nicolay and G. Strijkers, J. Nanobiotechnol., 2012, 10, 25 CrossRef CAS PubMed.
- S. Muro, T. Dziubla, W. Qiu, J. Leferovich, X. Cui, E. Berk and V. R. Muzykantov, J. Pharmacol. Exp. Ther., 2006, 317, 1161–1169 CrossRef CAS PubMed.
- S. Muro, C. Garnacho, J. A. Champion, J. Leferovich, C. Gajewski, E. H. Schuchman, S. Mitragotri and V. R. Muzykantov, Mol. Ther., 2008, 16, 1450–1458 CrossRef CAS PubMed.
- N. Zhang, C. Chittasupho, C. Duangrat, T. J. Siahaan and C. Berkland, Bioconjugate Chem., 2008, 19, 145–152 CrossRef CAS PubMed.
- C. Garnacho, D. Serrano and S. Muro, J. Pharmacol. Exp. Ther., 2012, 340, 638–647 CrossRef CAS PubMed.
- M. Nahrendorf, F. A. Jaffer, K. A. Kelly, D. E. Sosnovik, E. Aikawa, P. Libby and R. Weissleder, Circulation, 2006, 114, 1504–1511 CrossRef CAS PubMed.
- K. A. Kelly, J. R. Allport, A. Tsourkas, V. R. Shinde-Patil, L. Josephson and R. Weissleder, Circ. Res., 2005, 96, 327–336 CrossRef CAS PubMed.
- P. I. Homem de Bittencourt Jr, D. J. Lagranha, A. Maslinkiewicz, S. M. Senna, A. M. V. Tavares, L. P. Baldissera, D. R. Janner, J. S. Peralta, P. M. Bock, L. L. P. Gutierrez, G. Scola, T. G. Heck, M. S. Krause, L. A. Cruz, D. S. P. Abdalla, C. J. Lagranha, T. Lima and R. Curi, Atherosclerosis, 2007, 193, 245–258 CrossRef PubMed.
- P. S. Kowalski, L. L. Lintermans, H. W. M. Morselt, N. G. J. Leus, M. H. J. Ruiters, G. Molema and J. A. A. M. Kamps, Mol. Pharmaceutics, 2013, 10, 3033–3044 CrossRef CAS PubMed.
- T. D. Dziubla, V. V. Shuvaev, N. K. Hong, B. J. Hawkins, M. Madesh, H. Takano, E. Simone, M. T. Nakada, A. Fisher, S. M. Albelda and V. R. Muzykantov, Biomaterials, 2008, 29, 215–227 CrossRef CAS PubMed.
- I. Nakamura, K. Hasegawa, Y. Wada, T. Hirase, K. Node and Y. Watanabe, Biochem. Biophys. Res. Commun., 2013, 433, 47–51 CrossRef CAS PubMed.
- V. V. Shuvaev, M. A. Ilies, E. Simone, S. Zaitsev, Y. Kim, S. Cai, A. Mahmud, T. Dziubla, S. Muro, D. E. Discher and V. R. Muzykantov, ACS Nano, 2011, 5, 6991–6999 CrossRef CAS PubMed.
- K. Pollinger, R. Hennig, A. Ohlmann, R. Fuchshofer, R. Wenzel, M. Breunig, J. Tessmar, E. R. Tamm and A. Goepferich, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6115–6120 CrossRef CAS PubMed.
- J. E. Dahlman, C. Barnes, O. F. Khan, A. Thiriot, S. Jhunjunwala, T. E. Shaw, Y. Xing, H. B. Sager, G. Sahay, L. Speciner, A. Bader, R. L. Bogorad, H. Yin, T. Racie, Y. Dong, S. Jiang, D. Seedorf, A. Dave, K. Singh Sandhu, M. J. Webber, T. Novobrantseva, V. M. Ruda, K. R. Lytton-JeanAbigail, C. G. Levins, B. Kalish, D. K. Mudge, M. Perez, L. Abezgauz, P. Dutta, L. Smith, K. Charisse, M. W. Kieran, K. Fitzgerald, M. Nahrendorf, D. Danino, R. M. Tuder, U. H. von Andrian, A. Akinc, D. Panigrahy, A. Schroeder, V. Koteliansky, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2014, 9, 648–655 CrossRef CAS PubMed.
- T. Gillich, C. Acikgoz, L. Isa, A. D. Schluter, N. D. Spencer and M. Textor, ACS Nano, 2013, 7, 316–329 CrossRef CAS PubMed.
- S. D. Kong, J. Lee, S. Ramachandran, B. P. Eliceiri, V. I. Shubayev, R. Lal and S. Jin, J. Controlled Release, 2012, 164, 49–57 CrossRef CAS PubMed.
- S.-D. Li and L. Huang, J. Controlled Release, 2010, 145, 178–181 CrossRef CAS PubMed.
- X.-M. Zhu, Y.-X. J. Wang, K. C.-F. Leung, S.-F. Lee, F. Zhao, D.-W. Wang, J. M. Y. Lai, C. Wan, C. H. K. Cheng and A. T. Ahuja, Int. J. Nanomed., 2012, 7, 953–964 CrossRef CAS PubMed.
- C. Tay, Y. Yu, M. Setyawati, J. Xie and D. Leong, Nano Res., 2014, 7, 805–815 CrossRef CAS.
- L. Wang, J. Li, J. Pan, X. Jiang, Y. Ji, Y. Li, Y. Qu, Y. Zhao, X. Wu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17359–17368 CrossRef CAS PubMed.
- X.-R. Xia, N. A. Monteiro-Riviere and J. E. Riviere, Nat. Nanotechnol., 2010, 5, 671–675 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- M. P. Monopoli, F. B. Bombelli and K. A. Dawson, Nat. Nanotechnol., 2011, 6, 11–12 CrossRef CAS PubMed.
- A. Jedlovszky-Hajdu, F. B. Bombelli, M. P. Monopoli, E. Tombacz and K. A. Dawson, Langmuir, 2012, 28, 14983–14991 CrossRef CAS PubMed.
- E. R. Toke, O. Lorincz, E. Somogyi and J. Lisziewicz, Int. J. Pharm., 2010, 392, 261–267 CrossRef CAS PubMed.
- J. Wolfram, Y. Yang, J. Shen, A. Moten, C. Chen, H. Shen, M. Ferrari and Y. Zhao, Colloids Surf., B, 2014, 124, 17–24 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- C. D. Walkey and W. C. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- I. Lynch, T. Cedervall, M. Lundqvist, C. Cabaleiro-Lago, S. Linse and K. A. Dawson, Adv. Colloid Interface Sci., 2007, 134–135, 167–174 CrossRef CAS PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- S. Milani, F. B. Bombelli, A. S. Pitek, K. A. Dawson and J. Radler, ACS Nano, 2012, 6, 2532–2541 CrossRef CAS PubMed.
- J. S. Gebauer, M. Malissek, S. Simon, S. K. Knauer, M. Maskos, R. H. Stauber, W. Peukert and L. Treuel, Langmuir, 2012, 28, 9673–9679 CrossRef CAS PubMed.
- L. Treuel, D. Docter, M. Maskos and R. H. Stauber, Beilstein J. Nanotechnol., 2015, 6, 857–873 CrossRef CAS PubMed.
- D. Bargheer, J. Nielsen, G. Gebel, M. Heine, S. C. Salmen, R. Stauber, H. Weller, J. Heeren and P. Nielsen, Beilstein J. Nanotechnol., 2015, 6, 36–46 CrossRef CAS PubMed.
- C. Bantz, O. Koshkina, T. Lang, H. J. Galla, C. J. Kirkpatrick, R. H. Stauber and M. Maskos, Beilstein J. Nanotechnol., 2014, 5, 1774–1786 CrossRef CAS PubMed.
- L. Treuel and M. Malissek, Methods Mol. Biol., 2013, 991, 225–235 CAS.
- C. Y. Tay, M. I. Setyawati, J. Xie, W. J. Parak and D. T. Leong, Adv. Funct. Mater., 2014, 24, 5936–5955 CrossRef CAS PubMed.
- S. H. Lacerda, J. J. Park, C. Meuse, D. Pristinski, M. L. Becker, A. Karim and J. F. Douglas, ACS Nano, 2010, 4, 365–379 CrossRef PubMed.
- C. C. Fleischer and C. K. Payne, Acc. Chem. Res., 2014, 47, 2651–2659 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, F. Song, R. Liu, H. Guo, D. W. Olsen, Y. Cohen, A. Emili and W. C. Chan, ACS Nano, 2014, 8, 2439–2455 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, B. W. Neun, S. Man, X. Ye, M. Hansen, A. K. Patri, R. M. Crist and S. E. McNeil, Nanomedicine, 2014, 10, 1453–1463 CrossRef CAS PubMed.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. Puntes, ACS Nano, 2010, 4, 3623–3632 CrossRef CAS PubMed.
- D. Docter, U. Distler, W. Storck, J. Kuharev, D. Wunsch, A. Hahlbrock, S. K. Knauer, S. Tenzer and R. H. Stauber, Nat. Protoc., 2014, 9, 2030–2044 CrossRef CAS PubMed.
- P. P. Adiseshaiah, J. B. Hall and S. E. McNeil, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 99–112 CrossRef CAS PubMed.
- J. Leszczynski, Nat. Nanotechnol., 2010, 5, 633–634 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- C. Sacchetti, K. Motamedchaboki, A. Magrini, G. Palmieri, M. Mattei, S. Bernardini, N. Rosato, N. Bottini and M. Bottini, ACS Nano, 2013, 7, 1974–1989 CrossRef CAS PubMed.
- U. Sakulkhu, M. Mahmoudi, L. Maurizi, J. Salaklang and H. Hofmann, Sci. Rep., 2014, 4, 5020 CAS.
- M. J. Hajipour, S. Laurent, A. Aghaie, F. Rezaee and M. Mahmoudi, Biomater. Sci., 2014, 2, 1210–1221 RSC.
- R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203–2237 RSC.
- D. A. Walker, E. K. Leitsch, R. J. Nap, I. Szleifer and B. A. Grzybowski, Nat. Nanotechnol., 2013, 8, 676–681 CrossRef CAS PubMed.
- M. Kurylowicz, H. Paulin, J. Mogyoros, M. Giuliani and J. R. Dutcher, J. R. Soc., Interface, 2014, 11, 20130818 CrossRef CAS PubMed.
- M. Kurylowicz, M. Giuliani and J. R. Dutcher, ACS Nano, 2012, 6, 10571–10580 CAS.
- G. Caracciolo, D. Pozzi, A. L. Capriotti, C. Cavaliere, P. Foglia, H. Amenitsch and A. Lagana, Langmuir, 2011, 27, 15048–15053 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, T. Cedervall, T. Berggard, M. B. Flanagan, I. Lynch, G. Elia and K. Dawson, ACS Nano, 2011, 5, 7503–7509 CrossRef CAS PubMed.
- Z. J. Deng, M. Liang, I. Toth, M. Monteiro and R. F. Minchin, Nanotoxicology, 2013, 7, 314–322 CrossRef CAS PubMed.
- Z. J. Deng, G. Mortimer, T. Schiller, A. Musumeci, D. Martin and R. F. Minchin, Nanotechnology, 2009, 20, 455101 CrossRef PubMed.
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153–203 CrossRef CAS.
- M. I. Setyawati, C. Y. Tay and D. T. Leong, Biomaterials, 2013, 34, 10133–10142 CrossRef CAS PubMed.
- M. Giovanni, C. Y. Tay, M. I. Setyawati, J. Xie, C. N. Ong, R. Fan, J. Yue, L. Zhang and D. T. Leong, Environ. Toxicol., 2014 DOI:10.1002/tox.22015.
- C. Y. Tay, W. Fang, M. I. Setyawati, S. L. Chia, K. S. Tan, C. H. L. Hong and D. T. Leong, ACS Appl. Mater. Interfaces, 2014, 6, 6248–6256 CAS.
- C. Y. Tay, P. Cai, M. I. Setyawati, W. Fang, L. P. Tan, C. H. L. Hong, X. Chen and D. T. Leong, Nano Lett., 2014, 14, 83–88 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, J. Phys. Chem. B, 2012, 116, 8901–8907 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, J. Phys. Chem. B, 2014, 118, 14017–14026 CrossRef CAS PubMed.
- S. Barkam, S. Saraf and S. Seal, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 544–568 CrossRef CAS PubMed.
- S. M. Moghimi and Z. S. Farhangrazi, Nanomedicine, 2013, 9, 458–460 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, A. K. Patri, J. Zheng, J. D. Clogston, N. Ayub, P. Aggarwal, B. W. Neun, J. B. Hall and S. E. McNeil, Nanomedicine, 2009, 5, 106–117 CrossRef CAS PubMed.
- N. L. Anderson and N. G. Anderson, Mol. Cell. Proteomics, 2002, 1, 845–867 CAS.
- R. Pieper, C. L. Gatlin, A. J. Makusky, P. S. Russo, C. R. Schatz, S. S. Miller, Q. Su, A. M. McGrath, M. A. Estock, P. P. Parmar, M. Zhao, S. T. Huang, J. Zhou, F. Wang, R. Esquer-Blasco, N. L. Anderson, J. Taylor and S. Steiner, Proteomics, 2003, 3, 1345–1364 CrossRef CAS PubMed.
- S. Meister, I. Zlatev, J. Stab, D. Docter, S. Baches, R. H. Stauber, M. Deutsch, R. Schmidt, S. Ropele, M. Windisch, K. Langer, S. Wagner, H. von Briesen, S. Weggen and C. U. Pietrzik, Alzheimer's Res. Ther., 2013, 5, 51 CrossRef PubMed.
- A. Albanese, C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. Chan, ACS Nano, 2014, 8, 5515–5526 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- Y. Zhang, Y. Bai, J. Jia, N. Gao, Y. Li, R. Zhang, G. Jiang and B. Yan, Chem. Soc. Rev., 2014, 43, 3762–3809 RSC.
- Z. J. Deng, M. Liang, M. Monteiro, I. Toth and R. F. Minchin, Nat. Nanotechnol., 2011, 6, 39–44 CrossRef CAS PubMed.
- K. Natte, J. F. Friedrich, S. Wohlrab, J. Lutzki, R. von Klitzing, W. Osterle and G. Orts-Gil, Colloids Surf., B, 2013, 104, 213–220 CrossRef CAS PubMed.
- A. K. Murthy, R. J. Stover, A. U. Borwankar, G. D. Nie, S. Gourisankar, T. M. Truskett, K. V. Sokolov and K. P. Johnston, ACS Nano, 2013, 7, 239–251 CrossRef CAS PubMed.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti, S. Palchetti, S. De Grossi, A. Riccioli, H. Amenitsch and A. Lagana, Nanoscale, 2014, 6, 2782–2792 RSC.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718–2739 RSC.
- J. E. Schnitzer, Adv. Drug Delivery Rev., 2001, 49, 265–280 CrossRef CAS.
- E. Eugene, I. Hoffmann, C. Pujol, P. O. Couraud, S. Bourdoulous and X. Nassif, J. Cell Sci., 2002, 115, 1231–1241 CAS.
- N. Q. Liu, A. S. Lossinsky, W. Popik, X. Li, C. Gujuluva, B. Kriederman, J. Roberts, T. Pushkarsky, M. Bukrinsky, M. Witte, M. Weinand and M. Fiala, J. Virol., 2002, 76, 6689–6700 CrossRef CAS.
- S. Mayor and R. E. Pagano, Nat. Rev. Mol. Cell Biol., 2007, 8, 603–612 CrossRef CAS PubMed.
- J. A. Swanson and C. Watts, Trends Cell Biol., 1995, 5, 424–428 CrossRef CAS.
- S. Bhattacharya, D. Roxbury, X. Gong, D. Mukhopadhyay and A. Jagota, Nano Lett., 2012, 12, 1826–1830 CrossRef CAS PubMed.
- R. Agarwal, V. Singh, P. Jurney, L. Shi, S. V. Sreenivasan and K. Roy, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17247–17252 CrossRef CAS PubMed.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- L. Pelkmans and A. Helenius, Traffic, 2002, 3, 311–320 CrossRef CAS.
- G. Naudin, S. Poussard, M. Decossas, S. Mornet and O. Lambert, Condensed Matter, Paris, France, 2014 Search PubMed.
- D. Bedrov, G. D. Smith, H. Davande and L. Li, J. Phys. Chem. B, 2008, 112, 2078–2084 CrossRef CAS PubMed.
- J. Wong-Ekkabut, S. Baoukina, W. Triampo, I. M. Tang, D. P. Tieleman and L. Monticelli, Nat. Nanotechnol., 2008, 3, 363–368 CrossRef CAS PubMed.
- R. Qiao, A. P. Roberts, A. S. Mount, S. J. Klaine and P. C. Ke, Nano Lett., 2007, 7, 614–619 CrossRef CAS PubMed.
- S. Kessner, A. Krause, U. Rothe and G. Bendas, Biochim. Biophys. Acta, 2001, 1514, 177–190 CrossRef CAS.
- K. Namdee, A. J. Thompson, A. Golinski, S. Mocherla, D. Bouis and O. Eniola-Adefeso, Atherosclerosis, 2014, 237, 279–286 CrossRef CAS PubMed.
- J. Mai, Y. Huang, C. Mu, G. Zhang, R. Xu, X. Guo, X. Xia, D. E. Volk, G. L. Lokesh, V. Thiviyanathan, D. G. Gorenstein, X. Liu, M. Ferrari and H. Shen, J. Controlled Release, 2014, 187, 22–29 CrossRef CAS PubMed.
- A. P. Mann, T. Tanaka, A. Somasunderam, X. Liu, D. G. Gorenstein and M. Ferrari, Adv. Mater., 2011, 23, H278–H282 CrossRef CAS PubMed.
- I. Ricard, M. D. Payet and G. Dupuis, Eur. J. Immunol., 1998, 28, 1708–1718 CrossRef CAS.
- H. Yang, F. Zhao, Y. Li, M. Xu, L. Li, C. Wu, H. Miyoshi and Y. Liu, Int. J. Nanomed., 2013, 8, 1897–1906 Search PubMed.
- G. Morral-Ruiz, P. Melgar-Lesmes, C. Solans and M. J. Garcia-Celma, J. Controlled Release, 2013, 171, 163–171 CrossRef CAS PubMed.
- M. A. McAteer, A. M. Akhtar, C. von Zur Muhlen and R. P. Choudhury, Atherosclerosis, 2010, 209, 18–27 CrossRef CAS PubMed.
- V. S. Trubetskoy, J. Narula, B. A. Khaw and V. P. Torchilin, Bioconjugate Chem., 1993, 4, 251–255 CrossRef CAS.
- B. S. Ding, T. Dziubla, V. V. Shuvaev, S. Muro and V. R. Muzykantov, Mol. Interventions, 2006, 6, 98–112 CrossRef CAS PubMed.
- H. Margus, K. Padari and M. Pooga, Mol. Ther., 2012, 20, 525–533 CrossRef CAS PubMed.
- Y. Hou, M. Lai, X. Chen, J. Li, Y. Hu, Z. Luo, X. Ding and K. Cai, J. Biomed. Mater. Res., Part A, 2014, 102, 1726–1736 CrossRef PubMed.
- C. Freese, M. I. Gibson, H.-A. Klok, R. E. Unger and C. J. Kirkpatrick, Biomacromolecules, 2012, 13, 1533–1543 CrossRef CAS PubMed.
- D. Huhn, K. Kantner, C. Geidel, S. Brandholt, I. De Cock, S. J. Soenen, P. Rivera Gil, J. M. Montenegro, K. Braeckmans, K. Mullen, G. U. Nienhaus, M. Klapper and W. J. Parak, ACS Nano, 2013, 7, 3253–3263 CrossRef PubMed.
- D. Yu, Y. Zhang, X. Zhou, Z. Mao and C. Gao, Biomacromolecules, 2012, 13, 3272–3282 CrossRef CAS PubMed.
- S. Krasnici, A. Werner, M. E. Eichhorn, M. Schmitt-Sody, S. A. Pahernik, B. Sauer, B. Schulze, M. Teifel, U. Michaelis, K. Naujoks and M. Dellian, Int. J. Cancer, 2003, 105, 561–567 CrossRef CAS PubMed.
- T. Jung, W. Kamm, A. Breitenbach, E. Kaiserling, J. X. Xiao and T. Kissel, Eur. J. Pharm. Biopharm., 2000, 50, 147–160 CrossRef CAS.
- R. Chen, T. A. Ratnikova, M. B. Stone, S. Lin, M. Lard, G. Huang, J. S. Hudson and P. C. Ke, Small, 2010, 6, 612–617 CrossRef CAS PubMed.
- Y. Li, H. Yuan, A. von dem Bussche, M. Creighton, R. H. Hurt, A. B. Kane and H. Gao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12295–12300 CrossRef CAS PubMed.
- M. Shilo, A. Sharon, K. Baranes, M. Motiei, J.-P. M. Lellouche and R. Popovtzer, J. Nanobiotechnol., 2015, 13, 19 CrossRef PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- W. Jiang, Y. S. KimBetty, J. T. Rutka and C. W. ChanWarren, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- H. Gao, W. Shi and L. B. Freund, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9469–9474 CrossRef CAS PubMed.
- G. Pyrgiotakis, C. O. Blattmann and P. Demokritou, ACS Sustainable Chem. Eng., 2014, 2, 1681–1690 CrossRef CAS PubMed.
- S. Dasgupta, T. Auth and G. Gompper, Nano Lett., 2014, 14, 687–693 CrossRef CAS PubMed.
- K. Yang and Y.-Q. Ma, Nat. Nanotechnol., 2010, 5, 579–583 CrossRef CAS PubMed.
- P. Kolhar, N. Doshi and S. Mitragotri, Small, 2011, 7, 2094–2100 CrossRef CAS PubMed.
- P. Kolhar, A. C. Anselmo, V. Gupta, K. Pant, B. Prabhakarpandian, E. Ruoslahti and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10753–10758 CrossRef CAS PubMed.
- D. Bartczak, O. L. Muskens, S. Nitti, T. Sanchez-Elsner, T. M. Millar and A. G. Kanaras, Small, 2012, 8, 122–130 CrossRef CAS PubMed.
- J.-W. Yoo, N. Doshi and S. Mitragotri, Macromol. Rapid Commun., 2010, 31, 142–148 CrossRef CAS PubMed.
- H. Herd, N. Daum, A. T. Jones, H. Huwer, H. Ghandehari and C.-M. Lehr, ACS Nano, 2013, 7, 1961–1973 CrossRef CAS PubMed.
- B. D. Chithrani and W. C. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef CAS PubMed.
- P. Decuzzi and M. Ferrari, Biomaterials, 2007, 28, 2915–2922 CrossRef CAS PubMed.
- E. M. Hoek and G. K. Agarwal, J. Colloid Interface Sci., 2006, 298, 50–58 CrossRef CAS PubMed.
- X. Huang, S. Bhattacharjee and E. M. Hoek, Langmuir, 2010, 26, 2528–2537 CrossRef CAS PubMed.
- L. Rajendran, H.-J. Knolker and K. Simons, Nat. Rev. Drug Discovery, 2010, 9, 29–42 CrossRef CAS PubMed.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- C. P. Ng, T. T. Goodman, I. K. Park and S. H. Pun, Biomaterials, 2009, 30, 951–958 CrossRef CAS PubMed.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- Y. Wang, K. Zhou, G. Huang, C. Hensley, X. Huang, X. Ma, T. Zhao, B. D. Sumer, R. J. DeBerardinis and J. Gao, Nat. Mater., 2014, 13, 204–212 CrossRef CAS PubMed.
- S. Biswas, N. S. Dodwadkar, R. R. Sawant and V. P. Torchilin, Bioconjugate Chem., 2011, 22, 2005–2013 CrossRef CAS PubMed.
- E. S. Lee, K. Na and Y. H. Bae, Nano Lett., 2005, 5, 325–329 CrossRef CAS PubMed.
- F. M. Kievit and M. Zhang, Adv. Mater., 2011, 23, H217–H247 CrossRef CAS PubMed.
- D. T. Wiley, P. Webster, A. Gale and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8662–8667 CrossRef CAS PubMed.
- D. Triguero, J. B. Buciak, J. Yang and W. M. Pardridge, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 4761–4765 CrossRef CAS.
- L. H. Reddy, R. K. Sharma, K. Chuttani, A. K. Mishra and R. R. Murthy, AAPS J., 2004, 6, e23 Search PubMed.
- L. Fenart, A. Casanova, B. Dehouck, C. Duhem, S. Slupek, R. Cecchelli and D. Betbeder, J. Pharmacol. Exp. Ther., 1999, 291, 1017–1022 CAS.
- C. Chen, H. Hu, M. Qiao, X. Zhao, Y. Wang, K. Chen and D. Chen, J. Drug Targeting, 2015, 23, 311–322 CrossRef CAS PubMed.
- P. Oh, P. Borgström, H. Witkiewicz, Y. Li, B. J. Borgström, A. Chrastina, K. Iwata, K. R. Zinn, R. Baldwin, J. E. Testa and J. E. Schnitzer, Nat. Biotechnol., 2007, 25, 327–337 CrossRef CAS PubMed.
- J. Harris, D. Werling, J. C. Hope, G. Taylor and C. J. Howard, Trends Immunol., 2002, 23, 158–164 CrossRef CAS.
- J. V. Georgieva, D. Kalicharan, P. O. Couraud, I. A. Romero, B. Weksler, D. Hoekstra and I. S. Zuhorn, Mol. Ther., 2011, 19, 318–325 CrossRef CAS PubMed.
- J. V. Georgieva, R. P. Brinkhuis, K. Stojanov, C. A. G. M. Weijers, H. Zuilhof, F. P. J. T. Rutjes, D. Hoekstra, J. C. M. van Hest and I. S. Zuhorn, Angew. Chem., Int. Ed., 2012, 51, 8339–8342 CrossRef CAS PubMed.
- T. E. Park, B. Singh, H. Li, J. Y. Lee, S. K. Kang, Y. J. Choi and C. S. Cho, Biomaterials, 2015, 38, 61–71 CrossRef CAS PubMed.
- H. Ding, V. Sagar, M. Agudelo, S. Pilakka-Kanthikeel, V. S. Atluri, A. Raymond, T. Samikkannu and M. P. Nair, Nanotechnology, 2014, 25, 055101 CrossRef PubMed.
- P. Zhang, L. Hu, Q. Yin, L. Feng and Y. Li, Mol. Pharmaceutics, 2012, 9, 1590–1598 CrossRef CAS PubMed.
- M. Tamaru, H. Akita, T. Fujiwara, K. Kajimoto and H. Harashima, Biochem. Biophys. Res. Commun., 2010, 394, 587–592 CrossRef CAS PubMed.
- Y. Liu, J. Li, K. Shao, R. Huang, L. Ye, J. Lou and C. Jiang, Biomaterials, 2010, 31, 5246–5257 CrossRef CAS PubMed.
- S. V. Vinogradov, E. V. Batrakova and A. V. Kabanov, Bioconjugate Chem., 2004, 15, 50–60 CrossRef CAS PubMed.
- K. Ulbrich, T. Knobloch and J. Kreuter, J. Drug Targeting, 2011, 19, 125–132 CrossRef CAS PubMed.
- M. El-Sayed, M. F. Kiani, M. D. Naimark, A. H. Hikal and H. Ghandehari, Pharm. Res., 2001, 18, 23–28 CrossRef CAS.
- M. I. Setyawati, C. Y. Tay, S. L. Chia, S. L. Goh, W. Fang, M. J. Neo, H. C. Chong, S. M. Tan, S. C. J. Loo, K. W. Ng, J. P. Xie, C. N. Ong, N. S. Tan and D. T. Leong, Nat. Commun., 2013, 4, 1673 CrossRef CAS PubMed.
- Y. Jallouli, A. Paillard, J. Chang, E. Sevin and D. Betbeder, Int. J. Pharm., 2007, 344, 103–109 CrossRef CAS PubMed.
- L. Chen, R. A. Yokel, B. Hennig and M. Toborek, J. Neuroimmune Pharmacol., 2008, 3, 286–295 CrossRef PubMed.
- X. Gao, J. Qian, S. Zheng, Y. Changyi, J. Zhang, S. Ju, J. Zhu and C. Li, ACS Nano, 2014, 8, 3678–3689 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |