
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular polymer adhesives: advanced materials inspired by nature
Christian
Heinzmann
,
Christoph
Weder
* and
Lucas Montero
de Espinosa
*
Adolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, 1700 Fribourg, Switzerland. E-mail: christoph.weder@unifr.ch; lucas.montero@unifr.ch; Tel: +41 26 300 9465
First published on 23rd July 2015
Abstract
Due to their dynamic, stimuli-responsive nature, non-covalent interactions represent versatile design elements that can be found in nature in many molecular processes or materials, where adaptive behavior or reversible connectivity is required. Examples include molecular recognition processes, which trigger biological responses or cell-adhesion to surfaces, and a broad range of animal secreted adhesives with environment-dependent properties. Such advanced functionalities have inspired researchers to employ similar design approaches for the development of synthetic polymers with stimuli-responsive properties. The utilization of non-covalent interactions for the design of adhesives with advanced functionalities such as stimuli responsiveness, bonding and debonding on demand capability, surface selectivity or recyclability is a rapidly emerging subset of this field, which is summarized in this review.
 From left to right: Christian Heinzmann, Christoph Weder and Lucas Montero de Espinosa | Christian Heinzmann is currently a PhD student in Prof. Christoph Weder's research group at the Adolphe Merkle Institute of the University of Fribourg, Switzerland. He graduated from the Ruprecht-Karls-University of Heidelberg (Germany) in 2011, with the specialty in organic and inorganic synthesis. His ongoing research focuses on the synthesis of supramolecular polymers and their applications as stimuli-responsive, reversible adhesives with debond-on-demand features. |
Prof. Christoph (Chris) Weder is Director of the Adolphe Merkle Institute of the University of Fribourg, Switzerland, which was established in 2008 to conduct fundamental and application-oriented research in the domain of soft nanomaterials. He also leads the Swiss National Center of Competence in Research Bio-Inspired Materials. Weder was educated at ETH Zurich and the Massachusetts Institute of Technology, earned a Habilitation from ETH Zurich, and held a faculty position at Case Western Reserve University in Cleveland, before he joined the University of Fribourg. Chris' research interests are the design, synthesis, and investigation of bio-inspired and/or stimuli-responsive polymer-based nanomaterials. |
Lucas Montero de Espinosa is an Ambizione fellow of the Swiss National Science Foundation at the Adolphe Merkle Institute in Fribourg, Switzerland. He received his Chemistry degree from the Universidad Complutense de Madrid (Spain) in 2005 and his PhD from the Universitat Rovira i Virgili (Tarragona, Spain) in 2009. After a postdoctoral stay in the group of Prof. Michael A. R. Meier (University of Potsdam and Karlsruhe Institute of Technology, Germany), he joined the Adolphe Merkle Institute at the University of Fribourg in 2013 to build and lead a group that focuses on supramolecular polymers. |
Introduction
In principle, the phenomenon of adhesion is easily defined: if the work required to separate two dissimilar objects is higher than the work required to bring them into contact, then the objects will cling or stick to each other.1 However, if the objects are more complex than a pair of well-defined molecules in a well-defined environment, in which attractive forces outweigh repulsion, adhesion is often a highly complex process that involves several mechanisms spanning different length scales.1–3 Several theories have been developed over time to explain the chemical, physical and mechanical factors governing adhesion,4,5 but so far no unified theory has been developed. Adhesion phenomena are central to many biological processes involving both chemical and physical mechanisms, the latter often being emergent. In nature, adhesion is, on a molecular level, often driven by reversible interactions that are controlled by a wide range of stimuli that are imparted by either the environment or released by living organisms to satisfy a specific need. Non-covalent interactions are found in many natural adhesive systems such as those displayed by the gecko footpads (van der Waals forces)6 or the secretions of mussels and tubeworms (hydrogen bonds and/or metal–ligand complexes).7 Additionally, non-covalent interactions are involved in various biological processes including cell adhesion to extracellular biomolecules8 or bacterial adhesion.9,10 These examples illustrate that nature uses various types of non-covalent interactions to control adhesive forces and access complex functions. The possibility to exploit similar, albeit significantly simplified, mechanisms to design synthetic adhesives with advanced and stimuli-responsive properties has inspired a great deal of research activities in the last two decades. This contribution provides an overview of this exciting development, focusing on hydrogen bonding, host–guest interactions and metal–ligand complexation as the main non-covalent motifs that were utilized to design supramolecular adhesives. Mussel-inspired adhesives based on L-3,4-dihydroxyphenylalanine (DOPA) are intentionally excluded from this overview, since the related literature is vast and has been reviewed in several recent articles.11–13 Apart from one specific example, materials in which self-adhesion (cohesion) effects are emphasized, i.e. supramolecular polymers developed to be (self)healable, are also not covered, and the reader is referred to several recent reviews dedicated to this topic.14–17 Two general approaches to design reversible supramolecular adhesives can be distinguished, i.e. the modulation of the adhesive properties via the bulk material properties or, alternatively, the control of adhesion through a change of the surface interactions. While both approaches have been demonstrated to permit stimuli-responsive dynamic adhesion, the failure mechanisms are generally different; while a change of the bulk properties typically leads to cohesive failure, the modulation of surface interactions appears to cause adhesive failure.The first report of the adhesion of a supramolecular polymer on a substrate dates back to 1997,18–20 when Stupp and coworkers reported on a triblock oligomer that self-assembled into rod-like structures on account of long-range quadruple interactions (π–π stacking).18 When solution-cast onto a glass surface, the supramolecular material adhered tenaciously with a reported shear strength of 1.22 MPa, outperforming poly(vinyl phenol) (PVP) (0.53 MPa), whose side chains carry the same chemical functionality as the investigated supramolecular assemblies, and which is known for its hydrogen bonding ability21 and coating properties.22 The debonding on demand characteristics of the supramolecular polymer were not explored; however, its adhesion to the glass surface was shown to be resistant to washing with an aqueous HF solution, while PVP was washed from the glass surface within minutes under the same conditions.
Since the report of Stupp and coworkers, a broad range of supramolecular polymers have been explored as adhesives and numerous fundamental studies have been conducted, which are discussed in this review. The approach is also of interest in the context of technological exploitation, as evidenced by the number of patents filed in the field.23–33 Despite the variety of non-covalent interactions that could be used to synthesize supramolecular polymers,34,35 most of the examples found in the literature employ hydrogen bonding motifs to achieve reversible bonding and debonding. This may be explained by the relatively low synthetic efforts required to incorporate hydrogen bonding units into polymeric structures or polymerizable monomers.34,36,37 Additionally, hydrogen bonds are compatible with many other functional groups and can be comparatively strong.38 By contrast, the number of studies on supramolecular adhesives based on host–guest interactions, metal–ligand complexation, or other non-covalent interactions is much smaller. While this review is organized by the type of binding motif used, it appears that many of the concepts, functions and properties discussed could be realized by exploiting different types of non-covalent interactions.
Supramolecular adhesives based on hydrogen bonding
Early work by Long and coworkers focused on the synthesis of a methacrylic monomer (2-isocyanatoethyl methacrylate) functionalized with the self-complementary quadruple-hydrogen bond forming ureido-4-pyrimidinone (UPy) motif developed by Meijer, Sijbesma and coworkers,39 and its copolymerization with butyl acrylate to create polymers with different UPy content (Fig. 1a).36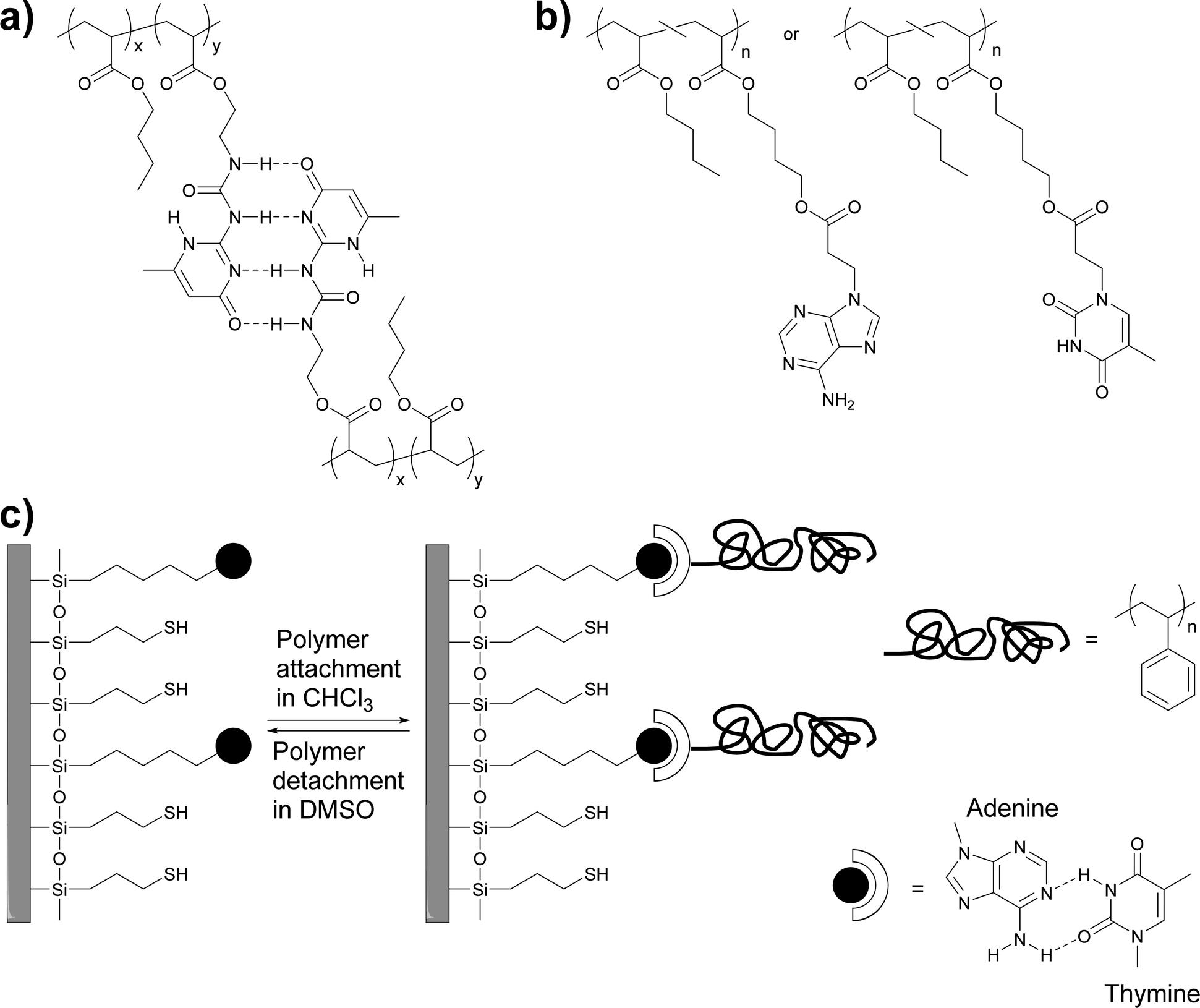 | ||
| Fig. 1 (a and b) Chemical structures of thermosensitive adhesives based on (self-)complementary hydrogen bonding motifs. (a) UPy-functionalized polyacrylates.36 (b) Adenine- and thymine-functionalized polyacrylates.40 (c) Schematic representation of the adhesion of thymine-functionalized polystyrene onto an adenine-functionalized surface.41 | ||
The incorporation of 2.5–10.4 mol% of the UPy-functionalized monomer caused an increase of the glass transition temperature (Tg) from −50 °C up to −23 °C, which is at least in part related to the formation of supramolecular cross-links via UPy dimerization. However, rheological measurements show clearly that the UPy dimers are completely dissociated at temperatures above 80 °C. The adhesion of these polymers to a glass surface was investigated by peel tests (Fig. 2),36 which showed an increase of the adhesion strength with increasing UPy content on account of strong UPy–glass interactions. Further evidence of such interactions was found in thin layer chromatography (TLC) analyses, where the UPy-containing copolymers did not ascend the TLC plate if a non-coordinating solvent such as chloroform was used as mobile phase. On the other hand, polar solvents such as THF or ethyl acetate moved all (co)polymers to the top of the TLC plate due to disruption of the UPy–silica interactions. Remarkably, the addition of only 3.3 mol% of the UPy–monomer led to a three-fold increase of the peel strength. Rheological measurements data suggested that the higher viscosity of the UPy-containing polymers could contribute to increase their peel strength, as is known that cross-linked materials have an increased creep resistance.42,43 The side-chain functionalization approach taken by Long and coworkers allows one to readily tune the supramolecular cross-link density as well as the glass transition temperature by simple variation of the polymer composition, in particular the content of the UPy–monomer and the choice of the comonomer.
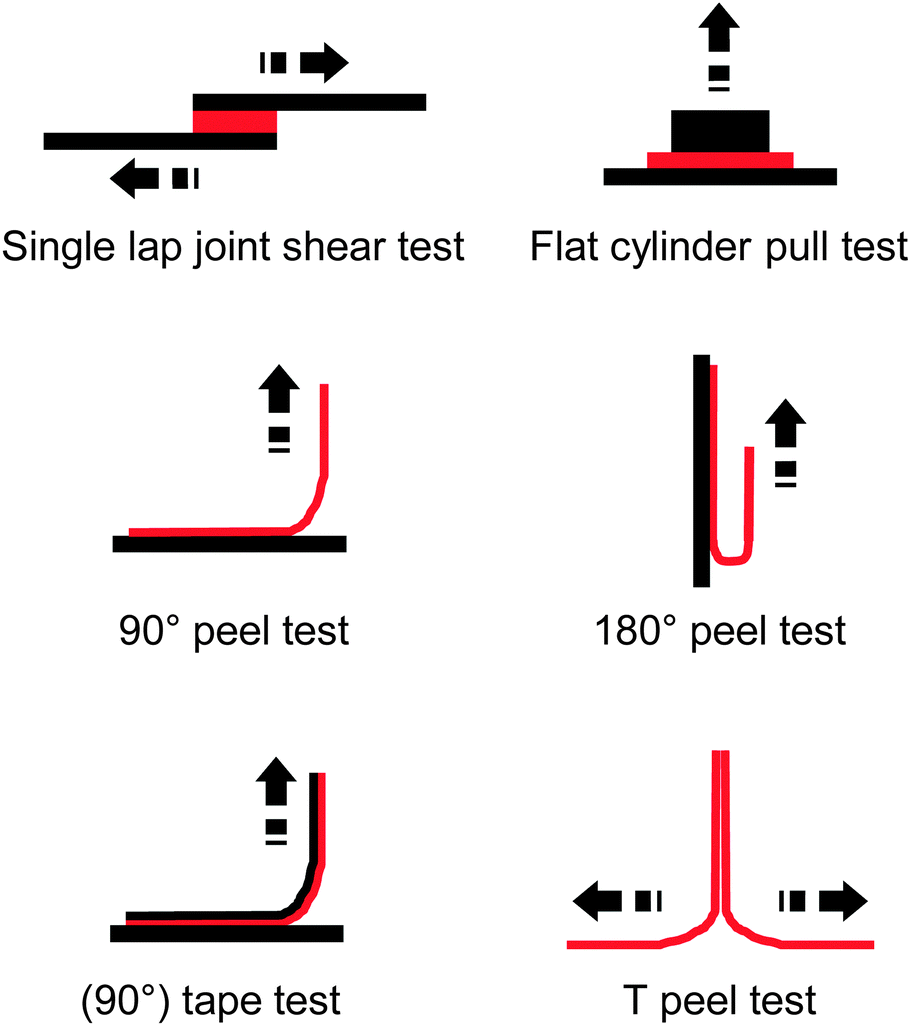 | ||
| Fig. 2 Overview of common test layouts to quantify the adhesive strength of an adhesive (red) to a substrate (black) or to itself. | ||
Long, Ward and coworkers later extended this approach to nucleobase-containing polyacrylates (Fig. 1b)40 and polystyrene (Fig. 1c).41 While nucleobases can self-dimerize via hydrogen bonding, the complementary binding of nucleobases, which plays a key role in the functions of biopolymers such as DNA or RNA, is of course much stronger and presents an intriguing design approach for molecular recognition-based selective adhesion. In the first example,41 thymine-terminated polystyrene was brought into contact with an adenine-functionalized silicon surface (Fig. 1c). The formation of adenine–thymine hydrogen bonds was confirmed with X-ray photoelectron spectroscopy (XPS) and through the increase of the water contact angle, which indicated the attachment of the hydrophobic polystyrene (PS) layer. When a hydroxyl-terminated polystyrene was used instead, only a very low adhesion to the adenine-functionalized surface was observed, due to the absence of specific interactions with the substrate. In a subsequent study, acrylate monomers carrying adenine (A) and thymine (T) motifs were synthesized and independently copolymerized with n-butyl acrylate (nBA) in different ratios.40 While the adenine containing polyacrylate was found to assemble in needle-like nanostructures (ca. 100 nm × 10 nm) at an adenine content of as low as 7 mol%, the thymine-carrying polyacrylates did not show any self-assembly, even at a thymine-monomer content of over 30 mol%. The differences observed were explained with the higher tendency of adenine dimers to form π–π stacks that can aggregate into larger crystalline domains. Temperature-dependent small-angle X-ray scattering (SAXS) and wide-angle X-ray diffraction (WAXD) measurements showed a decrease of the Bragg peak intensity as the temperature was increased until no structure could be observed over 80 °C. These observations were correlated with dynamic mechanical analyses (DMA), which showed that the materials lost their mechanical integrity at ca. 80 °C. The macroscopic adhesive properties of poly(T-co-nBA) (10 mol% thymine) and poly(A-co-nBA) (4 mol% adenine) (Fig. 1b) were characterized by a combination of tape peel and shear tests (Fig. 2) and compared to poly(acrylic acid-co-n-butyl acrylate) (poly(AA-co-nBA), 4 mol% AA) and poly(4-vinylpyridine-co-n-butyl acrylate) (poly(VP-co-nBA), 10 mol% VP), which were used as hydrogen bonding analogues to study the effect of hydrogen bonding strength on the peeling strength. The adenine-based polyacrylate displayed a higher shear strength but a lower peel strength than the thymine-based polyacrylate. Interestingly, the adhesive strength of a blend of the adenine and thymine containing polyacrylates was up to two times higher than that of the individual polymers, on account of the much stronger binding of the complementary base pair. The individual nucleobase-containing polyacrylates displayed a 3–4 times higher peel strength than the poly(AA-co-nBA) and poly(VP-co-nBA) reference polymers, on account of their cross-linked structure, which led to reduced creep and cohesive failure. The blend of the adenine and thymine containing polyacrylates, in turn, showed a 3–4 times higher peel strength than a reference blend of the AA- and VP-based analogues, which are cross-linked through weaker self-complimentary motifs. Overall, these early studies by Long, Ward and coworkers demonstrated the suitability of nucleobases as reversible cross-links in adhesive systems and underlined the advantage of complementary over self-binding.
Selective adhesion was also investigated by Zimmerman and coworkers, who employed a synthetic DNA base pair analog formed by 2,7-diamidonaphthyridine (DAN) and ureido-7-deazaguanine (DeUG).44 This quadruple hydrogen bonding pair was used to promote the adhesion of poly(styrene) (PS) bearing DAN as side chains (DAN–PS) onto a DeUG-functionalized glass surface (Fig. 3). The adhesion characteristics were investigated with a single lap joint layout (Fig. 2) and for comparison, three different control surfaces were tested, including the original primary amine-functionalized surface, an aminoalkyl-functionalized surface, and a surface displaying methylated DeUG units. It was shown that DAN–PS adhered better to the DeUG-functionalized surface (shear strength = 0.57 MPa) than to the control surfaces (shear strengths of below 0.30 MPa), reflecting a clear benefit of the complementary binding. When neat PS was used instead of DAN–PS, the shear strength decreased to 0.14 MPa for the DeUG-functionalized surface. Concerning the control surfaces, PS showed similar or lower adhesion than DAN–PS. Additional control surfaces were later prepared to rule out non-specific interactions by linking DeUG with a perfluorinated spacer.45 To explore the healability of the adhesive, which requires disassembly and reassembly of the supramolecular assemblies, the failed lap joints were re-attached via addition of a small portion of dichloromethane (disassembly) and subsequent solvent evaporation (re-assembly). A recovery of 55% of the initial shear strength was observed for DAN–PS after the first re-bonding (decrease from 0.70 MPa to 0.39 MPa), and the system could withstand two further debonding/re-bonding cycles before the adhesion strength was as low as that of neat PS (0.17 MPa). The degrading adhesive properties were attributed to contamination or damage of the sample after each shear test cycle, which may reduce the extent of formation of the DAN–DeUG complexes. The concept shown by Zimmerman and coworkers should be broadly applicable to other substrates and/or binding motifs and represents a universal platform that permits the selective and specific coating of surfaces with functional supramolecular polymers.
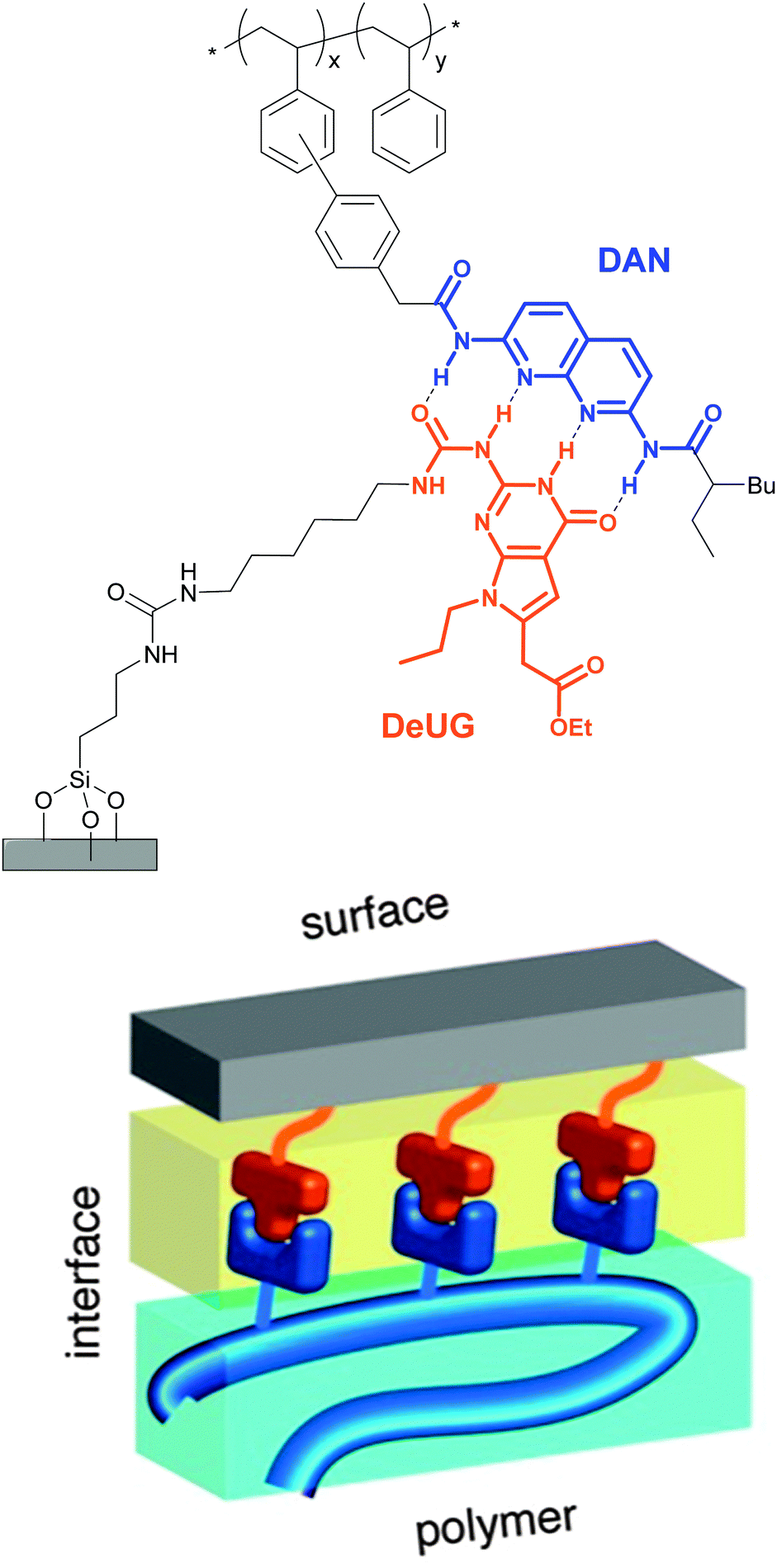 | ||
| Fig. 3 Schematic representation of polymer-surface selective adhesion based on DAN–DeUG recognition via hydrogen bonding. Adapted with permission from ref. 44 Copyright (2013) American Chemical Society. | ||
A common approach to synthesize supramolecular polymers involves the functionalization of a low-molecular weight telechelic polymer with supramolecular units as end-groups. The ditopic macromonomers thus obtained are sufficiently long to minimize cyclization and supramolecular polymerization thus leads to high apparent molecular weights through linear chain extension (as opposed to low-molecular weight cyclic structures, which are observed in the case of low-molecular weight ditopic monomers).34 In addition, if the polymer backbone and the supramolecular motifs are chemically dissimilar, the supramolecular polymer segregates into a biphasic morphology, much like a block-copolymer, and the mechanical properties of the material are enhanced if a physically cross-linking hard-phase is formed. Such reinforcement strategy is found in some of nature's strongest materials such as spider silk,46 where the segregation of protein's hydrophobic and hydrophilic domains contributes to their outstanding mechanical properties. Such well-defined structures are good models to study the effect that external factors such as the temperature have on the mechanical (and therefore adhesive) properties of supramolecular polymers. Hayes and coworkers highlighted the potential of urea- and urethane-terminated telechelic poly(isobutylene)s (PIBs) as thermosensitive adhesives.47 These macromonomers (number-average molecular weight, Mn ∼ 5500 g mol−1) form extended chains through simple hydrogen bonding, resulting in materials displaying a glass transition at ca. −65 °C and a second transition between −25 and −10 °C. On the basis of rheological measurements, the authors assigned this transition to the temperature at which the hydrogen bonds dissociate, leading to a decreased flow resistance. This is supported by the fact that the low-temperature Tg is almost independent of the hydrogen bonding end-group of the functionalized PIB, whereas the second transition strongly depended on the latter.
Using a similar approach, Bosman and coworkers tested the adhesive properties of a telechelic poly(dimethylsiloxane) (PDMS) functionalized with UPy end-groups (Fig. 4a).48 In this study, two glass slides were bonded with the supramolecular polymer by heating it to 120 °C, placing the melt between the two surfaces, and cooling the assembly to 20 °C. The joint thus formed (Fig. 2) was able to hold a 1 kg weight for 24 h, but failed after only 2 min when a 2 kg weight was mounted instead. The adhesive was shown to be fully reversible; this was shown in a qualitative experiment, in which the forcefully separated slides were brought again into contact and heated to 120 °C. After cooling, the original adhesive properties were restored. The pronounced temperature-response of these supramolecular polymers presents clear advantages with respect to their processability, and also promotes pronounced changes of the material's properties in a narrow temperature regime.
 | ||
| Fig. 4 (a) Chemical structure of UPy-functionalized telechelic PDMS, which forms a supramolecular adhesive upon linear chain extension via hydrogen bonding.48 (b) Schematic representation of the self-assembly of low-molecular weight PIB containing urea groups in the center of the chain molecules.49 | ||
The adhesive properties of assemblies of low-molecular weight polyisobutylene (PIB) containing two urea groups localized at the middle of the chain molecules (PIBUT) were investigated by Creton and coworkers (Fig. 4b).49 The authors demonstrated that upon introduction of a small fraction of hydrogen bonding units, this otherwise amorphous polymer with low glass transition temperature (−73 °C) self-assembled into an ordered structure with a periodicity of 4 nm as measured with SAXS. Although no further structure analyses were performed, the authors suggested that this supramolecular polymer may assemble into a comb-like structure on the basis of previous solution studies with similar polymers.50 Heating the polymer to temperatures above 100 °C caused dissociation of the hydrogen-bonded stacks (monitored by SAXS), which reformed after cooling to room temperature, restoring the material's properties. Interestingly, FTIR analysis suggests that the hydrogen bonds did not fully disassemble up to a temperature of 140 °C, which indicates that the observed order–disorder transition involves dissociation of van der Waals interactions rather than hydrogen bonds.49 The adhesive properties of PIBUT were investigated with a contact mechanics method, also known as Johnson–Kendall–Roberts (JKR) method, using a flat-ended cylinder as contacting probe (Fig. 2). Stainless steel and PDMS, the latter being amongst the most difficult surfaces to bind, were used as test surfaces and the performance of the modified PIB was compared to reference materials (neat PIB and PBA are common pressure sensitive adhesives (PSA)). First, the adhesion of PIBUT on a stainless steel surface was compared to that of three different molecular weight PIBs (Mn = 2800, 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, and 400000 g mol−1). While PIB2800 behaves like an unentangled Newtonian liquid and the molecular weight of PIB400000 is too high to form the fibrillary structures necessary for the tack test, the behavior of PIBUT is comparable to that of PIB85000, which has the closest behavior to that of a standard PSA such as linear high-Mn PBA. In a second adhesive test, PIBUT was compared with PIB85000 and PBA on stainless steel and PDMS. The modified PIB exhibited higher stress at debonding (0.85 MPa on steel and 0.6 MPa on PDMS) than the reference materials (neat PIB: 0.5 MPa and 0.1 MPa, respectively; PBA: 0.65 MPa and 0.2 MPa, respectively). According to the authors, the remarkably strong adhesion of PIBUT on PDMS, which is usually used as a release layer, make it potentially useful in applications involving difficult-to-bond surfaces.
000, and 400000 g mol−1). While PIB2800 behaves like an unentangled Newtonian liquid and the molecular weight of PIB400000 is too high to form the fibrillary structures necessary for the tack test, the behavior of PIBUT is comparable to that of PIB85000, which has the closest behavior to that of a standard PSA such as linear high-Mn PBA. In a second adhesive test, PIBUT was compared with PIB85000 and PBA on stainless steel and PDMS. The modified PIB exhibited higher stress at debonding (0.85 MPa on steel and 0.6 MPa on PDMS) than the reference materials (neat PIB: 0.5 MPa and 0.1 MPa, respectively; PBA: 0.65 MPa and 0.2 MPa, respectively). According to the authors, the remarkably strong adhesion of PIBUT on PDMS, which is usually used as a release layer, make it potentially useful in applications involving difficult-to-bond surfaces.
Weder and coworkers recently reported the use of UV light as a remote stimulus to induce the reversible debonding of supramolecular adhesives.51 A supramolecular system based on UPy-terminated telechelic poly(ethylene-co-butylene) (PEB) was synthesized and its adhesive properties were tested on steel and glass surfaces using a single lap joint shear test (Fig. 2 and 5a). As in the examples discussed above, UPy–PEB–UPy assembles into a supramolecular polymer via linear chain extension mediated by strong UPy dimerization.53 The resulting material displays polymer-like mechanical properties (tensile storage modulus E′ = 7.1 ± 1.8 MPa, Young's modulus E = 12 ± 1 MPa; measured at 25 °C, respectively) on account of its nanophase segregated lamellar morphology, in which the stacks formed by aggregating UPy dimers serve as the hard phase. In the adhesion tests, UPy–PEB–UPy displayed shear strengths of around 1 MPa on both steel and glass surfaces and, as in the previous examples, it underwent thermally induced reversible debonding; however, this material was also shown to debond on demand upon exposure to UV-light. Such functionality can be enabled through a light-heat conversion process whereby the UV-absorbing groups absorb UV light and produce a temperature increase on account of radiationless decay of the excited states. Since the UPy–PEB–UPy adhesive does not display a significant intrinsic absorption in the UV regime, the commercial UV absorber 2-(5-chloro-2H-benzotriazole-2-yl)-6-(1,1-dimethylethyl)-4-methyl-phenol was incorporated by simple blending to serve as light-heat converter and enable UV-light induced debonding, which took place after (optically) heating samples to ca. 70 °C. As a proof for reversibility/recyclability, the mechanically, thermally, or optically disassembled lap joints could be rebonded through exposure to light or heat, and the original adhesive properties were recovered. Further accounts of UPy–PEB–UPy as adhesive can be found in the patent literature. For example, an adhesive shear strength of 103.0 ± 23.7 MPa was reported for the adhesion to a zirconium ceramic substrate.23
 | ||
| Fig. 5 Chemical structures of the supramolecular polymers used by Weder and coworkers to achieve UV-light triggered debonding adhesives. (a) A telechelic poly(ethylene-co-butylene) (PEB) was functionalized with hydrogen bonding units (ureido-4-pyrimidinone; UPy), capable of intermolecular chain-extension via UPy–UPy-dimerization.51 (b) A side-chain functionalized methacrylate copolymer with UPy as the supramolecular cross-linking unit.52 | ||
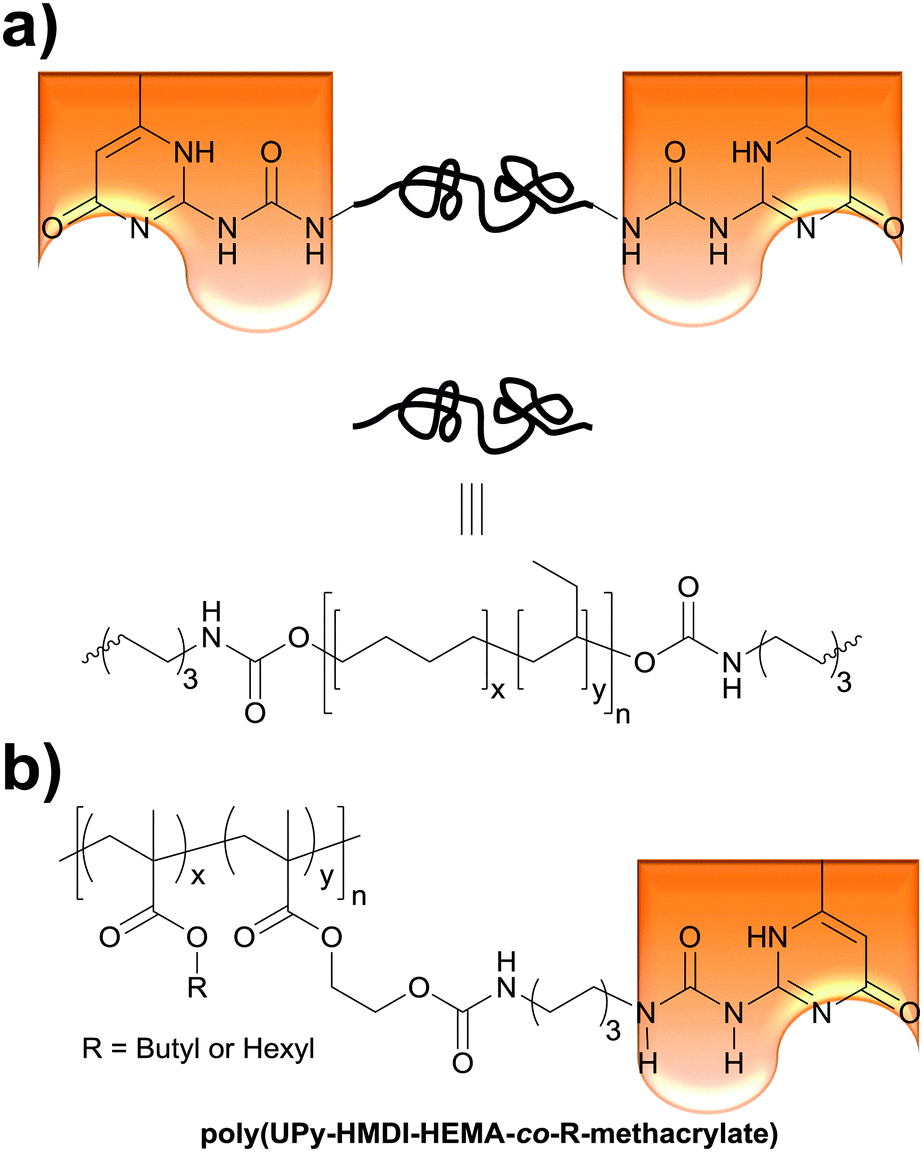
The Weder group recently extended the above concept of light- and temperature-responsive reversible adhesives to hydrogen-bonded, side-chain-functionalized supramolecular poly(alkyl methacrylate)s (Fig. 5b) that are reminiscent of the systems studied by Long (Fig. 1a),36 and demonstrated their bonding and debonding on demand capabilities.52 Thus, 2-hydroxyethyl methacrylate (HEMA) was functionalized with ureido-4-pyrimidinone (UPy) via a hexamethylenediisocyanate (HMDI) linker. The resulting (UPy–HMDI–HEMA) serves as a monomer that can, once polymerized, form supramolecular cross-links via hydrogen bonded dimers. Copolymers comprising 2.5, 5, or 10 mol% of the cross-linker were synthesized by copolymerizing UPy–HMDI–HEMA with hexyl methacrylate or butyl methacrylate. The adhesive properties of these copolymers were studied at temperatures between 20 and 60 °C by forming and testing single lap joints (Fig. 2) with stainless steel substrates. In a systematic study it was shown that increasing the concentration of UPy–HMDI–HEMA leads to improved mechanical and adhesive properties at elevated temperatures, on account of an increased concentration of supramolecular cross-links. The bond formation was reversible and debonded and subsequently rebonded lap joints displayed the same adhesive strength as pristine samples. The study also exemplarily confirmed the bonding and debonding on demand capabilities of one (co)polymer, which for light-induced debonding experiments was again blended with a UV-absorber. This work demonstrates the broad applicability of light-induced bonding and debonding on demand with supramolecular polymers and documents the ease with which the properties of such copolymers can be tailored.
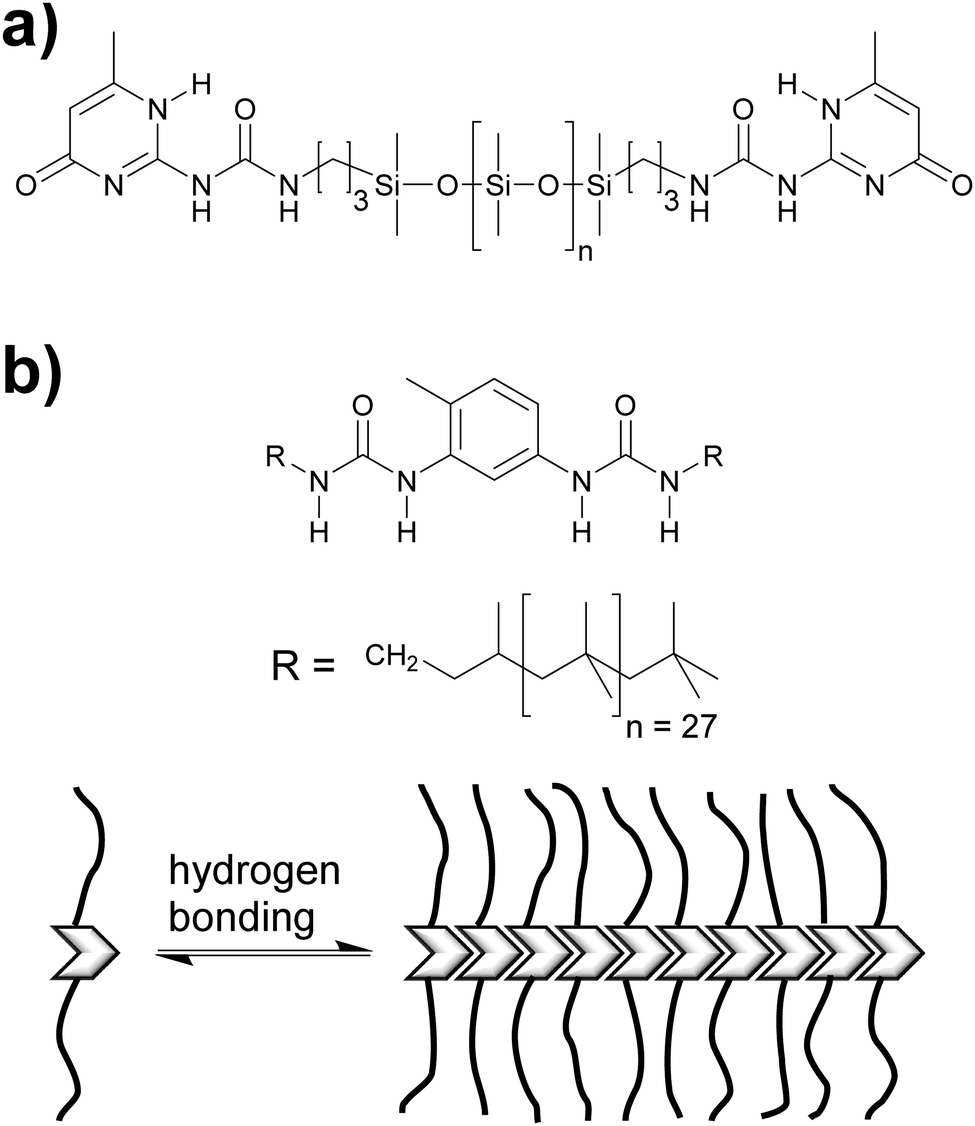
Thermoplastic polyamides are frequently used as hot-melt adhesives due to their high thermal and mechanical resistance. On the other hand, their use is limited to heat-resistant surfaces as they require high processing temperatures. Leibler and coworkers tackled this problem by blending a commercial polyamide (synthesized by the reaction of a mixture of fatty acid dimers and trimers with ethylenediamine and piperazine, Mn = 20000 g mol−1, Fig. 6) with a hydrogen bonding semicrystalline supramolecular polymer.54 The blend was designed to combine the good mechanical properties of polyamides with the highly temperature dependent properties of supramolecular polymers, yielding an easily processable material with low melt viscosity. The supramolecular polymer used consisted of a mixture of fatty acids dimers and trimers that had been modified by reaction with 1-(2-aminoethyl)-2-imidazolidinone (UDETA) in a 1:1 [COOH]/[NH2] ratio (Mn ∼ 800 g mol−1, Fig. 6). The adhesive properties of the neat polyamide and a blend containing 20 wt% of the supramolecular polymer were compared in T-peel tests (Fig. 2) using a non-woven polypropylene web as surface. Notably, the blend withstood a maximum peeling force of 3 N for an adhesive thickness of 5 μm, surpassing the neat polyamide by up to a factor of 6. The adhesive failure for the blend was always cohesive, indicating a strong adhesion at the interfaces through extensive hydrogen bonding. The improved mechanical strength of the blend was reported to be the result of the crystallization of the supramolecular polymer and of the presence of a higher number of hydrogen bonding units that attach to the surface. Additionally, the blend could be processed at a lower temperature than the neat polyamide, due to the plasticizing effect of the supramolecular polymer, which disconnected into low-viscosity low-molecular weight species at temperatures over 120 °C. Apart from their direct use as adhesives, these materials have been also proposed as additives in adhesive formulations such as hot-melt adhesives, pressure sensitive adhesives (PSA), and solvent-based adhesives.55,56
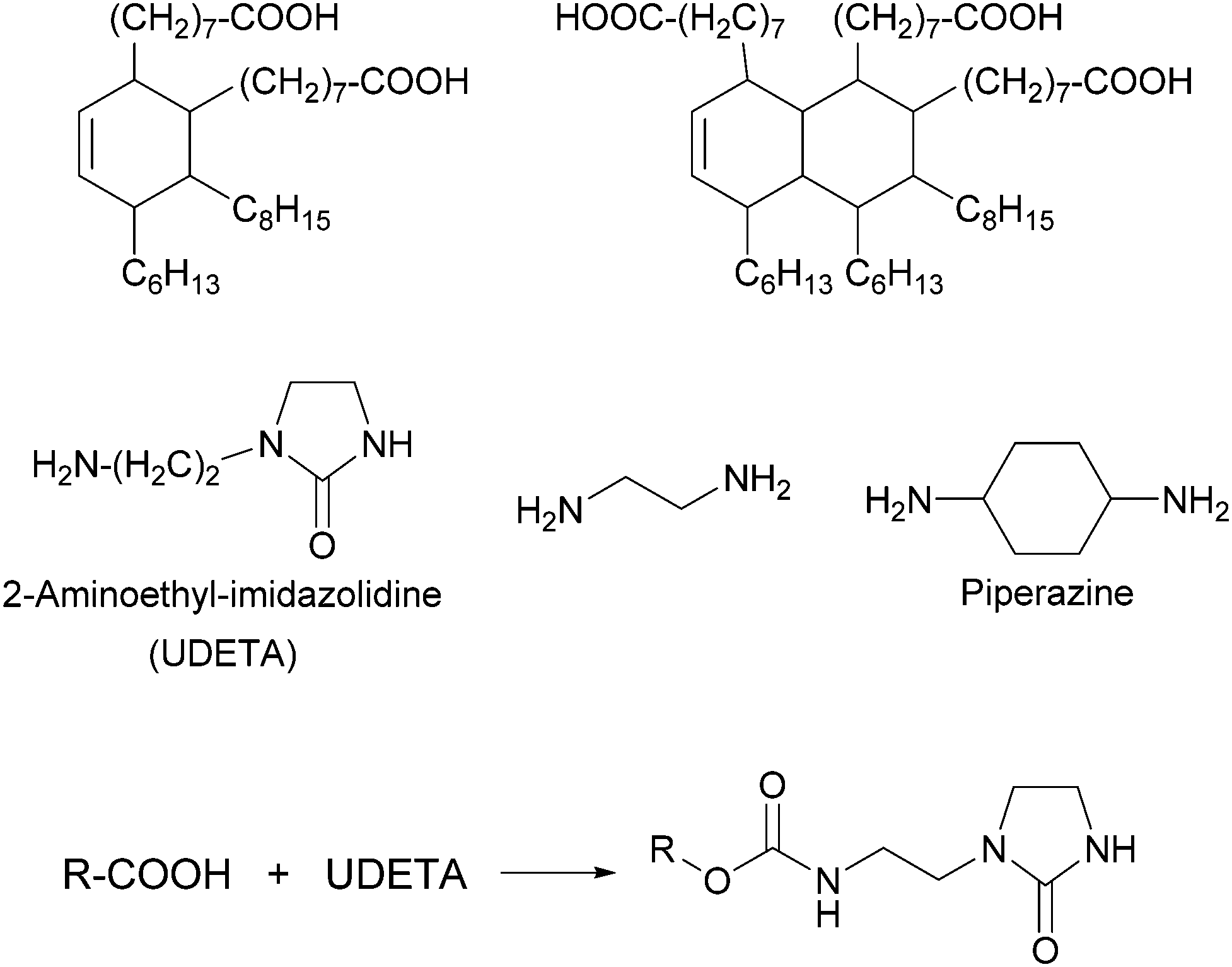 | ||
| Fig. 6 Chemical structures of fatty acid dimers and trimers and difunctional amines (top) that form the basis of the polyamides used by Leibler and coworkers in blends with supramolecular polymers formed by the reaction of fatty acids (and derivatives) with UDETA (bottom).54 | ||
The above discussed approach has also been used in various application-driven studies. Bostik, a company involved in the commercialization of adhesives, evaluated the adhesive properties of supramolecular polymers, based on carboxylic acids and amines as in Leibler's approach, using a 180° peel test layout (Fig. 2).24 Depending on the degree of amine functionalization, the studied supramolecular polymers exhibited peel strengths between 0.7 (10 wt% functionalization) and 2 N cm−1 (30 wt% functionalization), which were superior to that of the unfunctionalized reference (0.4 N cm−1). In another study, Bostik combined Leibler's supramolecular materials with styrene based-block copolymers such as poly(styrene-b-butadiene-b-styrene) (SBS), to obtain pressure-sensitive hot-melt adhesives.25 In 180° peel tests (Fig. 2), the supramolecular formulations achieved higher adhesive strengths (between 5.7 and 6.4 N cm−1) than a non-functionalized reference (4.0 N cm−1). The debond-on-demand ability of these adhesives was demonstrated using heat as stimulus, which caused debonding of the supramolecular adhesive within seconds, whereas the reference material required several minutes. The polymer company Arkema also used Leibler's approach to prepare various materials for bituminous compositions, to increase processability and to enhance cohesion and adhesion.26
The non-covalent adhesion of two rigid surfaces by means of supramolecular interactions is kinetically disfavored as some (surface) flexibility is required for non-covalent bonds to be formed. An approach to enhance adhesion between such surfaces involves the introduction of long and flexible spacers between the surface and the hydrogen bonding units. Xie and coworkers addressed this problem by chemically modifying the surface of an epoxy resin with hydrogen bonding polymers.57 Thus, the epoxy surface groups were functionalized with either low- or high-molecular-weight branched poly(ethyleneimine) (BPEI) (Fig. 7a). BPEI was chosen because of the high density of hydrogen bond donors and acceptors and on account of its branched architecture, which does not favor interfacial chain entanglements that would otherwise hinder reversible adhesion. In addition, a control sample was prepared by reacting the hydroxyl surface groups with acetic anhydride to investigate the role of hydroxyl hydrogen bonding in the adhesion, which was observed to be important. BPEI with Mn values of 1800, 10000, and 60000 g mol−1 were used, and the coverage of the epoxy polymer was dependent of the Mn, as demonstrated by AFM experiments. In the case of low Mn BPEI, AFM images showed isolated BPEI islands on the epoxy surface, whereas for the medium and high Mn BPEI, 2D network structures were observed. Additional XPS analysis confirmed the increase of the surface nitrogen content from 3% (non-functionalized epoxy) to 5, 10, and 12% (BPEI functionalized epoxies). In the adhesion experiments, all epoxy samples were bonded using the same procedure: heating to 90 °C (i.e. above Tg), adding a small amount of methanol (62 μL cm−2 per bonding area), pressing two surfaces together, and cooling under applied force. Methanol was shown to promote the dissociation of previously formed intra-surface hydrogen bonds, and this increases the formation of interfacial hydrogen bonds. As a result, the pull-off strength increased with increasing Mn from ∼ 450 N cm−2 (non-functionalized epoxy and Mn = 1800 g mol−1) to ∼ 640 N cm−2 (Mn = 10000 and 60000 g mol−1). Notably, these values are comparable to the adhesion achieved with a conventional superglue (500–900 N cm−2). When the methanol treatment was omitted, a substantially lower pull-off strength (132 N cm−2) was observed. In an additional experiment, the pH dependency of the pull-off strength was investigated with the BPEI of the highest Mn (60000 g mol−1) and was observed to slightly decrease with increasing pH from ∼640 N cm−2 (pH = 2.5) to ∼450 N cm−2 (pH = 11.5). This behavior was explained on the basis of the different BPEI ionization degrees at the different pH values. At low pH, the concentration of cationized nitrogen atoms (strong hydrogen bond acceptors) is higher than that of neutral nitrogen atoms (weaker acceptors), which leads to the observed stronger adhesion. Reversibility was investigated by heating the bonded samples to 90 °C, peeling them off, and reattaching them. The adhesive strength after two attach–detach cycles decreased to 67% of the original adhesive pull-off strength, which the authors related to minor changes in the surface morphology. This work documents a very useful approach to enhance adhesion to the surface of epoxy resins without changing their bulk properties.
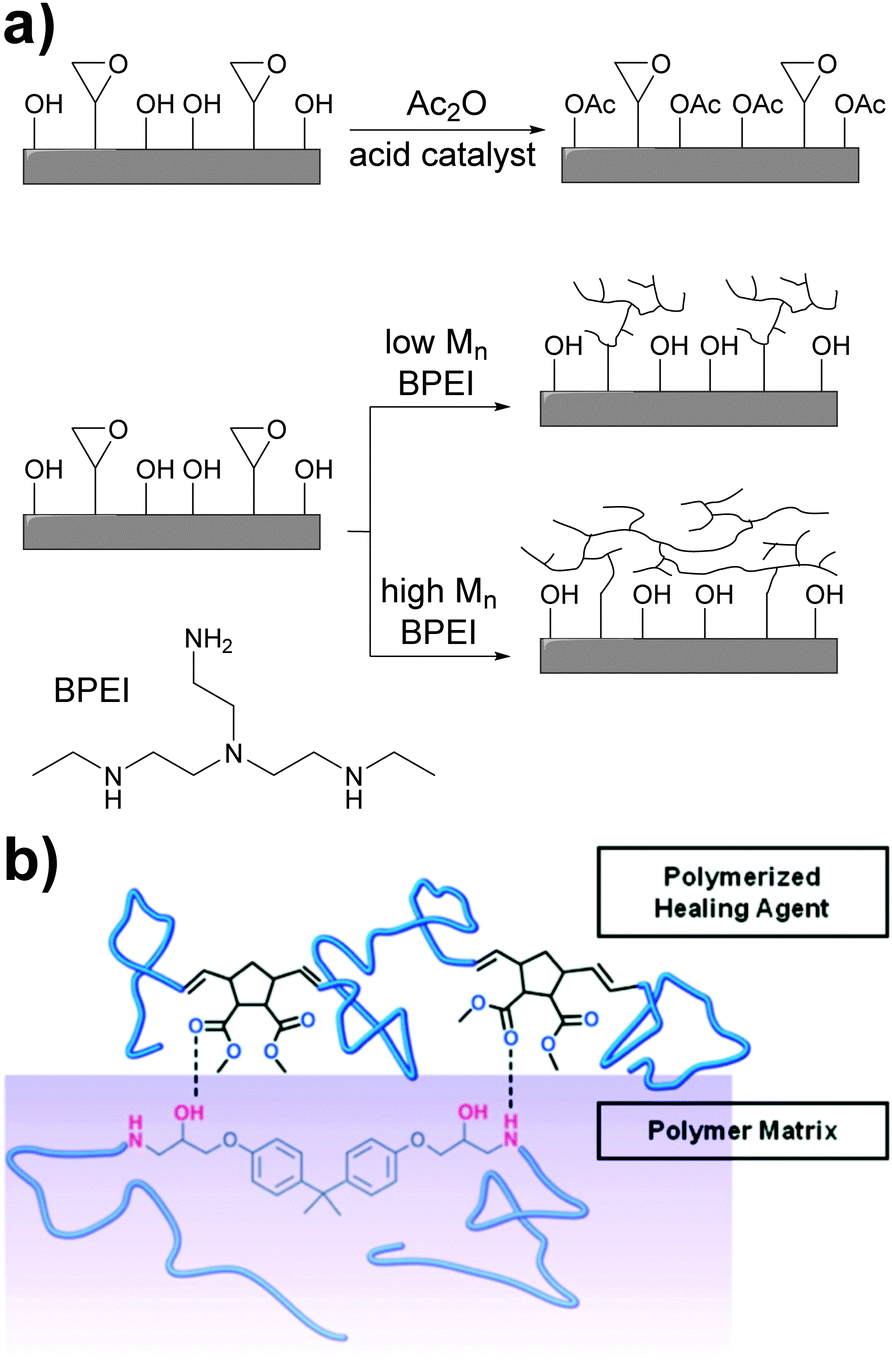 | ||
| Fig. 7 Approaches to achieve adhesion between rigid surfaces. (a) Surface functionalization of an epoxy resin with branched and hydrogen bonding BPEI to promote self-adhesion.57 (b) Enhancement of the adhesive properties of an in situ epoxy healing agent via hydrogen bonding.58 Part (b) was adapted with permission from ref. 58 Copyright (2011) American Chemical Society. | ||
A complementary study was reported by Moore and coworkers,58 who investigated the role of non-covalent interactions in the assisted surface adhesion of fractured epoxy resins. Taking the epoxy system EPON® 828/diethylenetriamine as model, the group optimized the composition of a healing agent mixture consisting of dimethylnorbornene ester (DNE), dicyclopentadiene (DCPD) and a ring-opening metathesis polymerization (ROMP) catalyst (Fig. 7b). DNE was selected to study the effect of hydrogen bonding between the ester groups and the amine functionalities of the epoxy resin, and DCPD was used to yield mechanical properties comparable of poly(DCPD) alone. A pre-crack was initiated in the epoxy resin with a razor blade and the specimens were pin loaded and shear tested. Once completely fractured, the healing agent mixture (DNE/DCPD/catalyst) was added inside capsules to the fractured surface at different concentrations and the adhesive strength after a curing/hardening step was measured. At 25 °C, the healing mixtures containing 5, 10 and 20 wt% of DNE displayed better adhesive properties than poly(DCPD) alone. In contrast, the mixture containing 2.5 wt% DNE showed limited healing, probably due the plasticizing effect of unreacted DNE counteracting any gain of adhesion promotion. At DNE contents of 10 and 20 wt%, the decrease of adhesive properties relative to 5 wt% can be explained by domination of non-covalent adhesion promotion over plasticization, yielding a slightly lower overall adhesion. The temperature at which curing/healing was carried out (25 and 50 °C) had also an effect of the adhesive properties, with higher adhesive properties at 50 °C (ca. 35 N vs. 10 N).
J. Zhang, X. Zhang and coworkers recently synthesized multi stimuli-responsive supramolecular hydrogels with reversible adhesive properties.59 These networks were prepared by addition of poly(ethylene polyamine) (PPA) to an aqueous dispersion of oxidized multi-walled carbon nanotubes (ox-MWCNTs). The resulting materials displayed a combination of weak (N–H⋯N) and strong (N–H⋯O from PPA–CNT interaction) hydrogen bonding motifs, and their ratio could be tuned to obtain different cross-linking degrees, which provided a means to control the gel–sol transition temperature of the supramolecular hydrogels between 23 °C and 48 °C as the CNT content was varied from 0.015–0.5 wt%. In addition to being highly thermosensitive, these materials were shown to be sensitive to pH and to NIR irradiation on account of the presence of ox-MWCNTs, which display photothermal energy conversion. Thus, the supramolecular hydrogels underwent a gel–sol transition in 2 min upon exposure to NIR light and returned to the gel state after 3 min at room temperature. The presence of ox-MWCNTs further contributed to an increase of the hydrogels' viscosity and enhanced their adhesive properties, which were investigated using partially dried gels on Teflon® and plasma-treated glass surfaces. In one experiment, a weight was glued with the dried gel onto a Teflon® plate. Once inverted, the device could hold 320 g (2.8 N cm−2) at 12 °C for 15 min. In a second experiment, the adhesion between two glass slides with a loading of 1.77 N cm−2 was tested and the temperature-dependent behavior explored. By varying the temperature between 5 and 40 °C, the authors observed adhesion failure times between ca. 40 s and ca. 20 min.
As mentioned before, non-covalent interactions play a key role in cell adhesion to biomaterials, which is a critical process for the formation of functional tissues. Cell adhesion is governed by a complex combination of adhesion mechanisms, which include the recognition of specific peptide sequences such as the well-studied RGD (Arg–Gly–Asp). Seeking to increase the versatility of current biomaterials, Meijer and coworkers reported a modular approach for the preparation of bioactive scaffolds using reversible supramolecular interactions.60 As building blocks, the authors synthesized a UPy-functionalized telechelic oligo(ε-caprolactone) (UPy–PCL–UPy, Mn = 2700 g mol−1) and two UPy-terminated peptides (GRGDS–UPy and PHSRN–UPy, each a sequence of 5 amino acids) (Fig. 8). PCL was chosen as it has been approved by the Food and Drug Administration (FDA) for specific applications in the human body due to its biocompatibility and biodegradability. The material properties of the neat PCL changed from a waxy, brittle solid (Tm = 43 and 49 °C) to a strong, elastic material (Tg = −59; Tm = 41 and 64 °C) upon functionalization with UPy units. Due to the reversibility of UPy dimers, UPy–PCL–UPy could be easily processed into several scaffolds (films, fibers, electrospun scaffold and meshes) and its biocompatibility was tested in vitro with two water-soluble model compounds, which indicated that the UPy-moiety is non-toxic (further verified by injection in rats). When PCLdiUPy was mixed with 4 mol% of GRGDS–UPy and/or PHSRN–UPy, a bioactive material was obtained on which mouse 3T3 fibroblast cells (5 × 104 cells cm−2) could be cultured. It was shown that the UPy-functionalized peptides are necessary to promote cell adhesion and spreading in vitro due to the dynamic binding of the peptides to the supramolecular material. The highest cell adhesion (lowest cell spreading) was observed when both UPy-functionalized peptides were mixed together (pointing to a synergistic effect). Different GRGDS–UPy concentrations (1, 2, or 4 mol%) were also tested; however, no clear correlation between peptide concentration and cell adhesion and spreading could be found. Control materials with UPy-free peptides were not able to promote cell adhesion and rapidly diffused out of the film. The strength of cell binding was determined by a wash-off test with or without prior incubation with fetal bovine serum (FBS), which acts as a detaching agent (competitive binding) of the cells to the bioactive material. While the cells attached to the blend containing both UPy-functionalized peptides could be washed off when incubated with FBS, the non-incubated cells were not washed off and started growing again after 1 day. These results reflect the significant potential of supramolecular polymers to reach new application fields if one fully exploits their dynamic character. The possibility to synthesize various bioactive materials in a modular approach is highly attractive, and the finding that the studied UPy-based building blocks are biocompatible, at least under a specific set of in vitro conditions, supports their usefulness in this context.
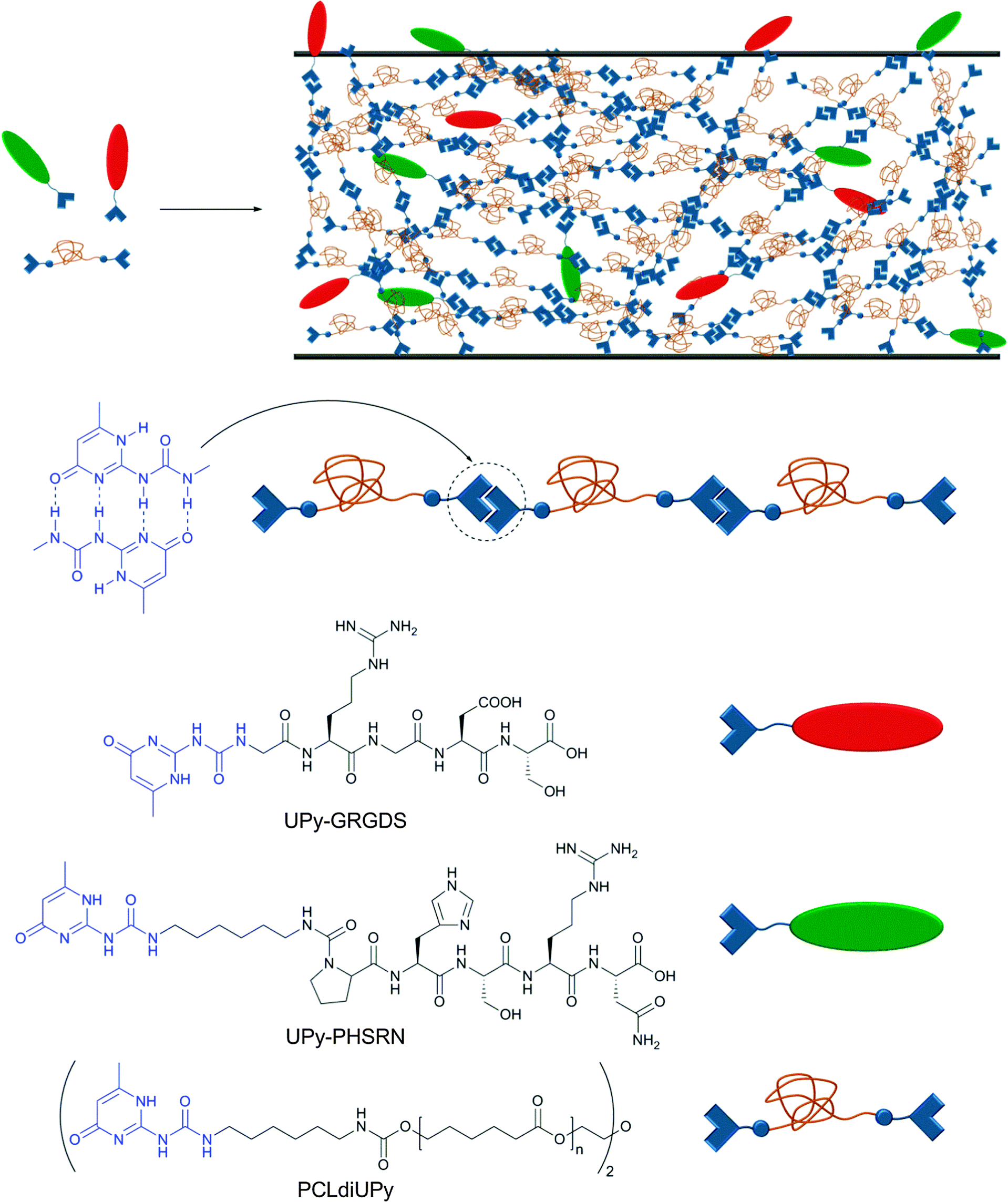 | ||
| Fig. 8 Modular approach for the preparation of bioactive surfaces based on supramolecular building blocks. Components and schematic representation of the supramolecular materials obtained via blending of the components.60 | ||
In anticipation of the possible technological exploitation of hydrogen-bonded polymers as adhesives, several patent applications have been filed in which the synthesis of such materials and the characterization of their adhesive properties is described. Apart from the examples mentioned throughout this section, of which some also formed the basis of pending patent applications or granted patents, the adhesive properties of several supramolecular polymers on steel by means of single lap joint tests (Fig. 2) were explored.27,28 Among them are materials based on poly(propylene oxide) (Mw = 2000 g mol−1) and 1,4-butanediol functionalized with various hydrogen bonding units (e.g. UPy, 2-aminopyrimidine, or isocytosine), which displayed adhesive strengths between 2.5 and 3.4 MPa.27 The failure mode was in most cases of adhesive nature as a result of limited interactions at the polymer–steel interface.
The patent literature also describes very general approaches, featuring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and/or CS acceptor groups and N–H, O–H, and/or S–H donor groups, to utilize supramolecular polymers as adhesives.29 Meijer and coworkers filed several patent applications in which the adhesive properties of UPy-based materials are at least mentioned, although only qualitative adhesive measurements were reported as these patents are mainly focussed on the synthesis and manufacturing/processing of the supramolecular polymers.30 With a different application focus, Smith and Nephew investigated the adhesive properties of dihydroxybenzoic acid-functionalized telechelic aliphatic oligomers (weight-average molecular weight, Mw, between 500 and 2000 g mol−1) in pull-tests (Fig. 2).31 The adhesive strengths varied between 1.8 and 15.1 MPa, while a reference cyanoacrylate adhesive reached 7.1 MPa.
O and/or CS acceptor groups and N–H, O–H, and/or S–H donor groups, to utilize supramolecular polymers as adhesives.29 Meijer and coworkers filed several patent applications in which the adhesive properties of UPy-based materials are at least mentioned, although only qualitative adhesive measurements were reported as these patents are mainly focussed on the synthesis and manufacturing/processing of the supramolecular polymers.30 With a different application focus, Smith and Nephew investigated the adhesive properties of dihydroxybenzoic acid-functionalized telechelic aliphatic oligomers (weight-average molecular weight, Mw, between 500 and 2000 g mol−1) in pull-tests (Fig. 2).31 The adhesive strengths varied between 1.8 and 15.1 MPa, while a reference cyanoacrylate adhesive reached 7.1 MPa.
Supramolecular adhesives based on host–guest interactions
While many supramolecular interactions display sufficiently high binding constants to serve as adhesives in a dry environment, most of them are disrupted in the presence of water, which acts as competitive binder. In nature, creatures such as mussels or sandcastle worms secrete adhesive coacervates that allow them to adhere to surfaces in aqueous environments.7 Kim and coworkers explored the potential of host–guest supramolecular interactions as a synthetic alternative to achieve underwater adhesion.61 In contrast to most macrocyclic hosts such as cyclodextrins, which exhibit relatively modest association constants in water (Ka ∼ 106 M−1), cucurbit[n]uril (CB[n], n = 5–8, 10) macrocycles containing a hydrophobic cavity and two carbonyl-fringed portals were demonstrated to be useful candidates for underwater adhesion. Among them, cucurbit[7]uril (CB[7]) forms very stable complexes with aminomethylferrocene (Fc, Ka ∼ 1012 M−1) and 1,1′-bis(trimethylammoniomethyl)ferrocene (BFc, Ka ∼ 1015 M−1) in water (Fig. 9). The experimental set-up used to explore the adhesive properties of surfaces modified with polymer brushes carrying these motifs consisted of two polymer-grafted silicon surfaces [Si] in which the free ends of the polymer chains were functionalized with either CB[7] (thickness of the brush layer 6.3 nm) or Fc (thickness 3.0 nm) moieties. Both surfaces were smooth as per AFM analysis and wettable as confirmed by contact angle measurements. Adhesion was promoted by manually pressing the surfaces together; they had previously been immersed in water (pH 7.4), which is required as the expulsion of water drives the inclusion of Fc into CB[7]. The bonded interface was shown to outperform bonds formed with commercial adhesives by withstanding a 2 kg weight in water and a 4 kg weight in the dry state (Fig. 9). The adhesive characteristics were further quantified in a lap shear adhesion test (Fig. 2), which resulted in a maximum lap shear strength of 1.12 ± 0.06 MPa. The so-called “supramolecular VELCRO®” maintained its adhesive properties through 5 debonding–rebonding cycles. While the maximum lap shear strength was reduced by ca. 30% after the first debonding–rebonding cycle, it remained constant afterwards. The stimuli responsive behavior of the assembled CB[7]–[Si]/Fc–[Si] system was investigated by exposing it to a NaClO (10 mM) solution while supporting a 0.5 kg weight. Under such conditions, Fc is oxidized to Fc+, which has a lower affinity to CB[7], and failure could be observed within minutes. The surfaces were then immersed in ascorbic acid to reduce Fc+ to Fc and the reassembled system could support a load of 1 kg for at least 1 h. With this contribution, Kim and coworkers provided a robust and stimuli-responsive underwater adhesion mechanism that should be applicable to a diverse range of surfaces.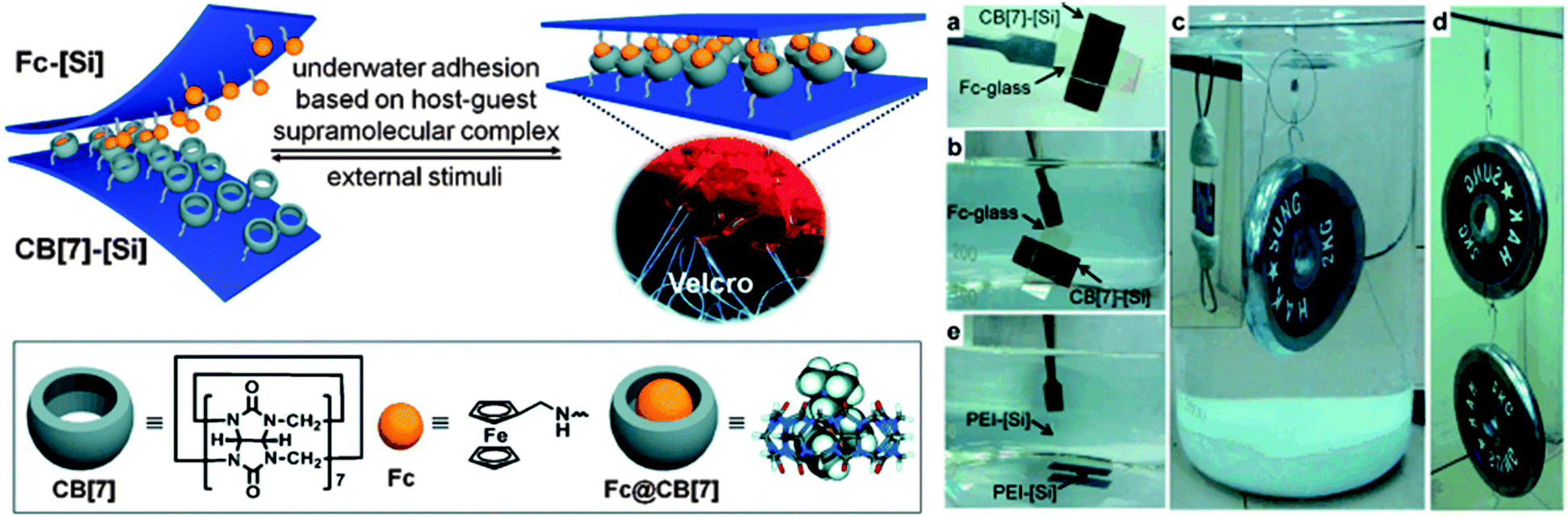 | ||
| Fig. 9 Redox-active adhesive based on CB[7]-Fc host–guest interactions and demonstration of underwater adhesion. Reprinted from ref. 61 with permission from John Wiley & Sons. | ||
In another example of host–guest-based supramolecular adhesion, Harada and coworkers reported a supramolecular hydrogel displaying selective adhesive properties on the basis of a molecular recognition process.62 The hydrogel studied was prepared by free-radical copolymerization of acrylamide with acrylamide derived monomers bearing β-cyclodextrin (βCD), bipyridine (bpy) and bisacrylamide (BAA) moieties. In water, the resulting copolymer (containing 2.2 mol% of βCD and 3.0 mol% of bpy moieties) forms a mixed network, where βCD–bpy inclusion complexes serve as reversible cross-linking points, whereas the bisacrylamide residues provide permanent cross-links. Upon addition of an appropriate metal salt, the bpy units bind to the metal ions to form M(bpy)n complexes and the degree of cross-linking decreases, although the mechanical integrity of the hydrogel is kept by the permanent cross-links. This allows modifying the properties of the hydrogel via the addition of metal salts. For instance, it was demonstrated that the adhesion of this hydrogel to another hydrogel containing tert-butyl units (which compete with bpy for the formation of inclusion complexes with βCD) could be switched on and off by addition of a metal salt (Fig. 10). The authors observed no adhesion between the βCD–bpy gel and the tert-butyl gel; however, both gels adhered (adhesion strength = 1000 ± 200 Pa) after the βCD–bpy gel was immersed for 1 min in a 100 mM CuCl2 aqueous solution, on account of the formation of inclusion complexes of the now available βCD with the tert-butyl groups. The addition of other transition metals such as Co2+, Ni2+ or Zn2+ in the form of MCl2 was also shown to promote gel adhesion using the same βCD- and tert-butyl-based hydrogel systems.
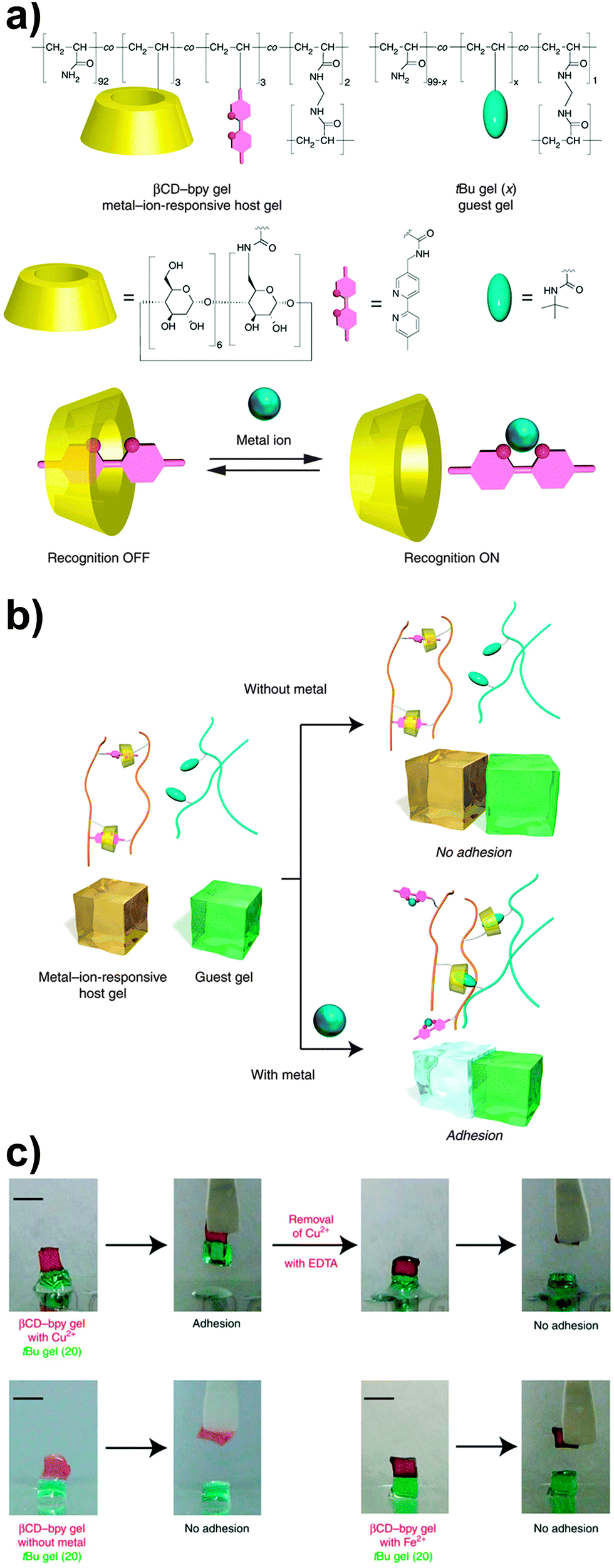 | ||
| Fig. 10 (a) Chemical structure of a βCD–bpy gel. (b) Schematic representation of its metal-triggered adhesion to a tert-butyl containing gel based on competing host–guest and metal–complex interactions. (c) Qualitative metal-triggered adhesion experiments. Figure reproduced from ref. 62 licensed under CC-BY 4.0. | ||
The recent development of synthetic mimics of reptile's adhesive setae63,64 motivated the same team to investigate the potential of host–guest interactions for hard surface adhesion.65 They synthesized acrylamide-based αCD- and βCD-containing hydrogels and studied their adhesion to hard surfaces functionalized with adamantane, ferrocene or azobenzene units. As control, they synthesized an acrylamide hydrogel, which showed very low adhesion to all surfaces. By contrast, friction tests revealed selective hydrogel-surface adhesion for the pairs αCD-hydrogel/azobenzene surface and βCD-hydrogel/ferrocene surface. Furthermore, the αCD-azobenzene and βCD-ferrocene interactions could be suppressed via irradiation with UV light, which causes trans/cis isomerization of the azobenzene motif, (Fig. 11a) and the addition of FeCl3, which causes oxidation of the ferrocene units to the weakly complexating ferrocenium species (Fig. 11b),66 respectively to promote partial debonding of the hydrogel from the surface. Although the debonding process was in both cases reversible upon irradiation with light of a wavelength of 430 nm and reduction with glutathione, respectively, some hysteresis was observed and the adhesion strength was somewhat lower than initially observed. While the adhesion of hydrogels to rigid surfaces is favored by their high flexibility, which facilitates the contact between complementary units, these results show the utility of host–guest interactions to achieve reversible adhesion in the presence of a hard surface and hints at the necessity of introducing certain mobility at the interface.
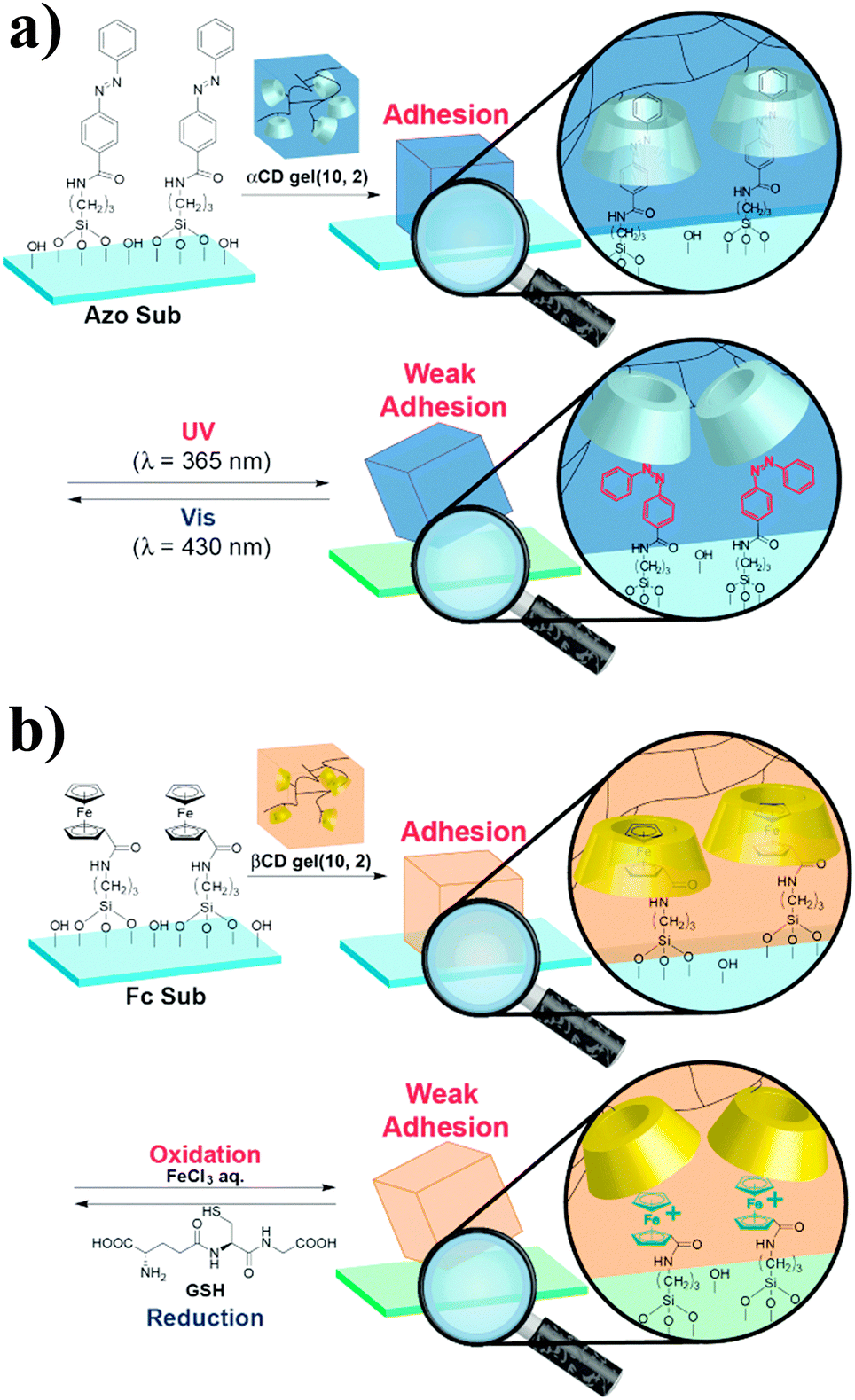 | ||
| Fig. 11 (a) Reversible adhesion of an αCD-hydrogel onto an azobenzene-functionalized surface via UV-promoted azobenzene isomerization. (b) Redox-responsive reversible adhesion of a βCD-hydrogel onto a ferrocene-functionalized surface. Reprinted from ref. 65 with permission from John Wiley & Sons. | ||
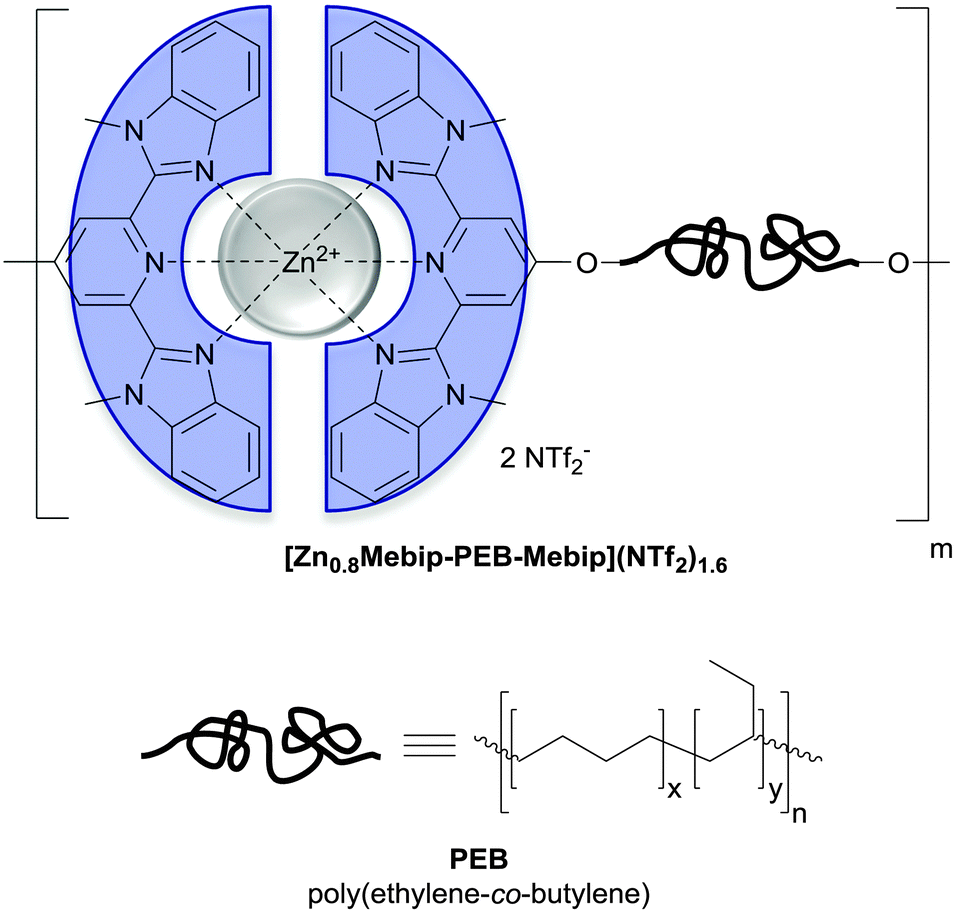 | ||
| Fig. 12 Chemical structure of the metallosupramolecular polymer used by Weder and coworkers to achieve UV-light triggered debonding adhesives. A telechelic poly(ethylene-co-butylene) (PEB) was functionalized with tridentate ligands (2,6-bis(1′-methylbenzimidazolyl)-pyridine; Mebip) capable of metal–ligand-coordination.51 | ||
In the examples discussed above, host–guest interactions were used to promote selective (reversible) adhesion between two gels or between a gel and a rigid surface. Burdick and coworkers provided an additional application of such interactions by studying the host–guest-driven adhesion of two βCD-functionalized rigid surfaces mediated by an adamantane-based (Ad) adhesive.67 The surfaces were created by functionalization of hyaluronic acid (HA) with βCD and methacrylate units, electrospinning of the functionalized HA into a fibrous mat-like structure and UV cross-linking of the methacrylate groups. On the other hand, the adhesive was prepared by functionalization of HA with Ad units. A first qualitative adhesion test was carried out by bonding two βCD-functionalized surfaces with the Ad-based adhesive, which was applied as a 0.5 wt% aqueous solution. After 15 min, the adhered surfaces were shown to remain bonded for up to seven weeks when kept in deionized water or phosphate-buffered saline (PBS) at 37 °C. A control experiment in which neat HA was used as adhesive resulted in immediate adhesive failure. The adhesive properties of this system were quantified by means of a single-lap joint test (Fig. 2), for which the βCD-methacrylate-functionalized HA was directly electrospun onto methacrylate-functionalized glass coverslips and then UV-cured. The coverslips were then bonded with the Ad-based adhesive (or neat HA as control) and a shear force was applied. The Ad-based adhesive showed a shear strength of 1 kPa; much higher than that of the control (ca. 20 Pa). According to the authors, this water-based adhesive system may be useful for in vivo applications such as the regeneration of structured tissues (cartilage, nervous tissue or cardiac muscle). The approach of Burdick and coworkers circumvents interface mobility issues with the aid of a supramolecular adhesive, offering an elegant alternative for reversible adhesion between hard surfaces.
The use of competitive interactions to achieve debonding on demand was also studied by Shi, Yang and coworkers.68 The authors prepared PDMS cubes (3 mm × 3 mm × 3 mm) that were surface-functionalized with either αCD or azobenzene units with the aim of promoting the assembly of macroscopic objects through molecular recognition. To overcome surface rigidity-derived problems that can lead to insufficient molecular binding (vide supra), the surface of the PDMS cubes was decorated with a flexible coating made by a layer-by-layer approach. The composition of the multilayer coating consisted of a flexible poly(ethyleneimine)/poly(acrylic acid) (PEI/PAA) layer, a barrier layer of poly(diallyldimethylammonium chloride)/poly(sodium-p-styrenesulfonate) (PDDA/PSS) to lower the stickiness of PEI/PAA and prevent nonselective association of the building blocks, and a final layer of either PDDA/PAA–αCD (αCD-grafted PAA) or PDDA/PAA–Azo (azobenzene-functionalized PAA). PDMS cubes functionalized with αCD units were placed in water together with PDMS cubes functionalized with Azo groups and the container containing the system was oscillated in a shaker. After 1 min of oscillation, the αCD-cubes were found to adhere to the Azo-cubes, as a result of a host–guest molecular recognition process. This was confirmed with control tests where either αCD-cubes or Azo-cubes were shaken independently resulting in no assembly. The intended role of the flexible PEI/PAA layer was also confirmed in an additional control experiment in which PDMS cubes were directly functionalized with either αCD or Azo units without intermediate PEI/PAA layer. The PDMS cubes thus prepared did not assemble, confirming that a certain degree of surface mobility is necessary for the host–guest interactions to take place. The stimuli-triggered debonding ability of this system was investigated through addition of the competitive host 1-adamantane carboxylic acid, which displays a higher binding constant to αCD (105 M−1) than the Azo units (103 M−1). The adamantane monomer was added as a 0.5 mM aqueous solution to the pre-assembled PDMS cubes and debonding took place after shaking the cubes as in the assembly process. The bond strength between the macroscopic cubes was determined using a microbalance-based method specially designed for the system studied,68 which showed that the adhesive force of the cubes having a flexible coating was 16 times higher than that between unmodified contact surfaces (3.05 × 10−3 N vs. 1.90 × 10−4 N). These results provide an interesting quantitative insight on the adhesion process that governs the oscillation-promoted self-assembly of complementary macroscopic objects.
Supramolecular adhesives based on metal–ligand complexation
Metallosupramolecular polymers are a well-studied class of stimuli-responsive materials,69–71 but their adhesive properties have been rarely reported. One prominent example is the above-discussed work by Harada and coworkers, in which selective adhesion of hybrid gels was achieved using a combination of host–guest and metal–ligand (bpy-M2+) interactions.62 In another example, Weder and coworkers studied the stimuli-responsive adhesion of a metallosupramolecular polymer based on the linear chain extension of Mebip-terminated telechelic PEB via metal–Mebip complex formation (Fig. 12).51 Chain extension of Mebip–PEB–Mebip takes place upon addition of a metal salt such as Zn2+, resulting in a metallosupramolecular polymer that exhibits a nanophase segregated lamellar morphology with the metal complexes forming the crystalline hard phase. The adhesive properties of this material were tested on steel and glass surfaces using a single lap joint shear test (Fig. 2 and 12) that showed strong adhesion to both surfaces with shear strengths of up to 2.5 MPa. As for the hydrogen bonded polymers discussed above, reversible debonding could be thermally triggered, but most interestingly, UV-light could also be used as stimulus to promote debonding in the absence of a UV-absorber additive. In contrast to UPy dimers,51 Mebip–metal complexes strongly absorb light in the UV regime and generate enough heat to cause complex dissociation upon radiationless decay of the excited states. A polymer prepared with a composition of [Zn0.8Mebip–PEB–Mebip](NTf2)1.6 was thus used as adhesive to bond two quartz glass slides and irradiated with light of a wavelength in the range of 320–390 nm and a power density of 900 mW cm−2. This caused an increase of the temperature up to ∼220 °C within 30 s and led to rapid debonding. The lap joints thus disassembled could be rebonded through exposure to light or heat to recover the original adhesive properties. This work thus expands the possibilities of UV-light-triggered debonding (vide supra) by using an intrinsically UV-active metal–complex as building block. The adhesion of [Zn0.8Mebip–PEB–Mebip](NTf2)1.6 to a zirconium ceramic substrate was also studied (shear strength = 33.0 ± 8.7 N),23 and the adhesive was shown to debond upon heating.In another example, Xie and coworkers explored metal cation-π interactions, which are amongst the strongest non-covalent interactions (up to 40 kcal mol−1), to bond two different rigid surfaces.72 The authors brought into contact an epoxy polymer with cross-linked poly(styrene) containing sodium sulfonate surface groups (Fig. 13) at 80 °C and performed tensile tests after cooling the bonded parts to room temperature. The adhesive strength was 721 N cm−2, which was almost double than that measured for a cross-linked poly(styrene) control (380 N cm−2), and superior to that observed with sulfonated cross-linked poly(styrene). The failure mode was cohesive as per XPS analysis and the nature of the interactions responsible for the enhanced adhesion was confirmed by Raman spectroscopy using model sulfonate and aromatic compounds. The adhesion mechanism reported by Xie and coworkers relies on a very simple synthetic scheme, which makes it interesting from the application point of view.
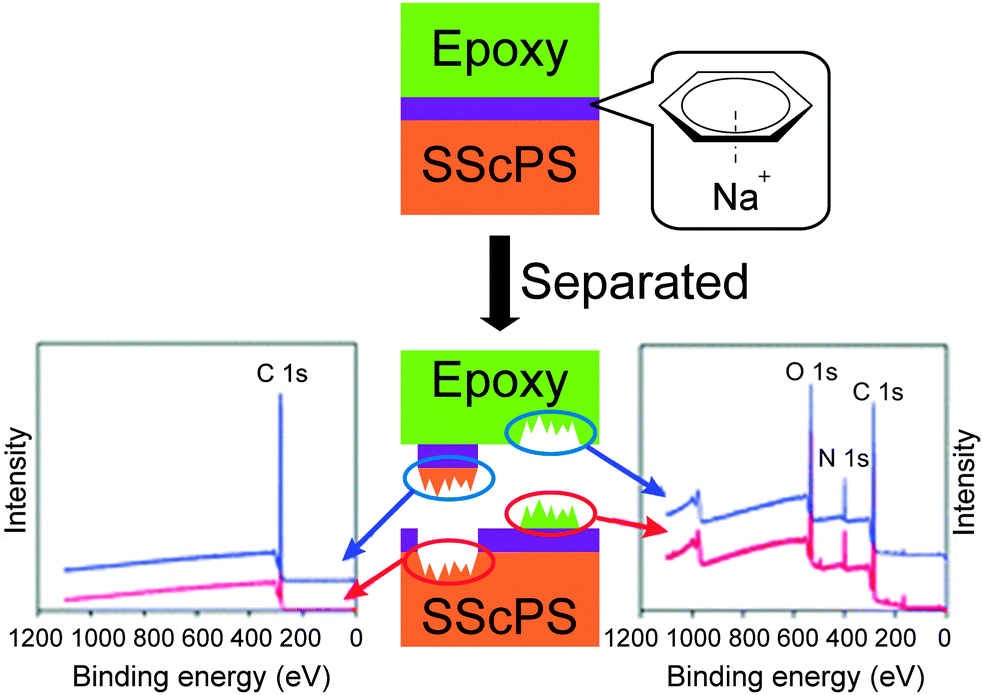 | ||
| Fig. 13 Approach to achieve adhesion between rigid surfaces with the use of metal cation-π interactions between epoxy resin and sulfonated polystyrene surfaces and determination of cohesive failure mechanism via XPS. Adapted from ref. 72 with permission from the Royal Society of Chemistry. | ||
Supramolecular adhesives based on other non-covalent interactions
This section discusses the use of non-covalent interactions such as hydrophobic/hydrophilic effects and ionic interactions to achieve adhesion, although it is noted that hydrogen bonds also play an important (complementary) role in many of these materials.An illustrative example of selective and reversible adhesion was reported by Creton and coworkers on the basis of a switchable surface.73 The authors grafted poly(styrene) (PS) and poly(2-vinyl pyridine) (P2VP) brushes and blends thereof onto a silicon surface and explored their adhesion to a blend of poly(vinyl pyrrolidone) and poly(ethylene glycol) (PVP–PEG) after different surface treatments. Upon exposing the surface to toluene, the PS chains swelled preferentially. After drying, the PS-enriched surface showed poor adhesion toward the PVP–PEG blend. However, when the surface was exposed to ethanol and subsequently to acidified water (pH 2) before drying, the protonated P2VP was brought to the surface and the PVP–PEG blend adhered well. This behavior was observed for surfaces with a P2VP brush content as low as ca. 2 mol% (relative to the content of PS) and the behavior was shown to be reversible. Thus, upon subsequent treatment of the protonated P2VP surface with aqueous ammonia, the adhesion of the PVP–PEG blend was significantly reduced.
Homogeneous hydrogels have been proposed as potential replacement for current two-phase dental adhesives.74–76 Prud'homme and coworkers77 reported hydrogels displaying tunable adhesion to both hydrophilic and hydrophobic surfaces relying on the coexistence of poly(methyl vinyl ether-co-maleic anhydride) (PMVE-MA) and poly(vinyl acetate) (PVAc) domains. Their synthetic approach involved the reaction of PVAc with hydrochloric acid to reach controlled hydrolysis degrees followed by cross-linking with PMVE-MA, which resulted in transparent gels when the reaction was carried out in a water/ethanol mixture. In order to elucidate the effect of molecular weight on the adhesive properties of these materials, 180° peel tests (Fig. 2) were performed with samples of different composition. The molecular weight of PMVE-MA and PVAc were shown to affect the adhesion in a non-linear fashion reaching a maximum peel strength of 15.4 N cm−2 with 500 kg mol−1 PVAc and 216 kg mol−1 PMVE-MA, which was ascribed to the optimum balance between entanglement and flow. The composition of the solvent mixture was shown to be critical for solubilizing the hydrophilic and hydrophobic polymeric components. While a 50:50 water/ethanol mixture provided homogeneous gels, dental applications require ethanol contents below 12% to avoid mucosal tissue irritation.77 Propylene glycol, glycerol and poly(ethylene glycol) (PEG-400) were thus investigated as amphiphilic alternatives to reduce the ethanol content and, although they all provided good miscibility at certain compositions, a 22.0:11.5:16.5 mixture of water/ethanol/PEG-400 was selected as the best alternative as propylene glycol and glycerol display a high number of hydroxyl groups that may interfere in the cross-linking reaction.
Pressure sensitive adhesives (PSAs) are a special class of polymers that adhere strongly to surfaces upon application of a slight pressure force.78 These materials have viscous liquid-like properties during adhesive bond formation but behave as slightly cross-linked rubbery polymers when a detaching force is applied. Thus, a complex combination of high fluidity in the bonding process and high cohesive strength to resist debonding stresses is required. At a molecular level, the strength of PSAs has been shown to depend on the balance between high cohesive strength and large free volume,79 which motivated Feldstein and coworkers80 to explore the adhesive properties of various mixtures of poly(N-vinyl pyrrolidone) (PVP) and PEG with an Mn of 400. In such mixtures, PEG-400 serves as a difunctional supramolecular cross-linker that bridges the carbonyl groups of PVP chains via hydrogen bonding (Fig. 14a). The resulting supramolecular network displays improved cohesive strength relative to PVP as a result of PVP/PEG-400 interactions, but also an increased free volume due to the introduction of spacers between PVP chains. The sub-nanoscale free-volume characteristics were studied with positron annihilation lifetime spectroscopy, which showed that an increase of the PEG-400 content from 0 to 43% led to an increase of ca. 40% in the free-volume fraction and of 15% in the mean hole radius. These observations indicate a reduction in number density of free-volume holes as the PEG-400 content is increased. Additionally, the effect of relative humidity (RH) on the free volume was found to be less pronounced. The adhesive properties of the blends were studied with peel and probe tack tests (Fig. 2). In order to study the effect that water has on the adhesive properties of these materials, the samples subjected to 180° peel tests were equilibrated at controlled vapor pressures and had a water content of 5–30%. On the other hand, the samples used in the probe tack tests were vacuum dried and contained ca. 3% water. In both tests, the maximum peel strength and probe tack adhesion were observed for a PVP–PEG-400 blend containing 36 wt% of PEG-400. Regarding the mechanical properties of these materials, tensile tests revealed an increasing linear correlation between the PEG-400 content and the elongation at break and, as expected, hydration caused a reduction of the tensile strength and an increase of the elongation at break on account of disrupting the hydrogen bonding between the PEG and the PVP. Overall, this study contributes to the understanding of the complex relationships between the nanoscopic free volume and the macroscopic adhesive and mechanical properties of PVP–PEG as PSAs models.
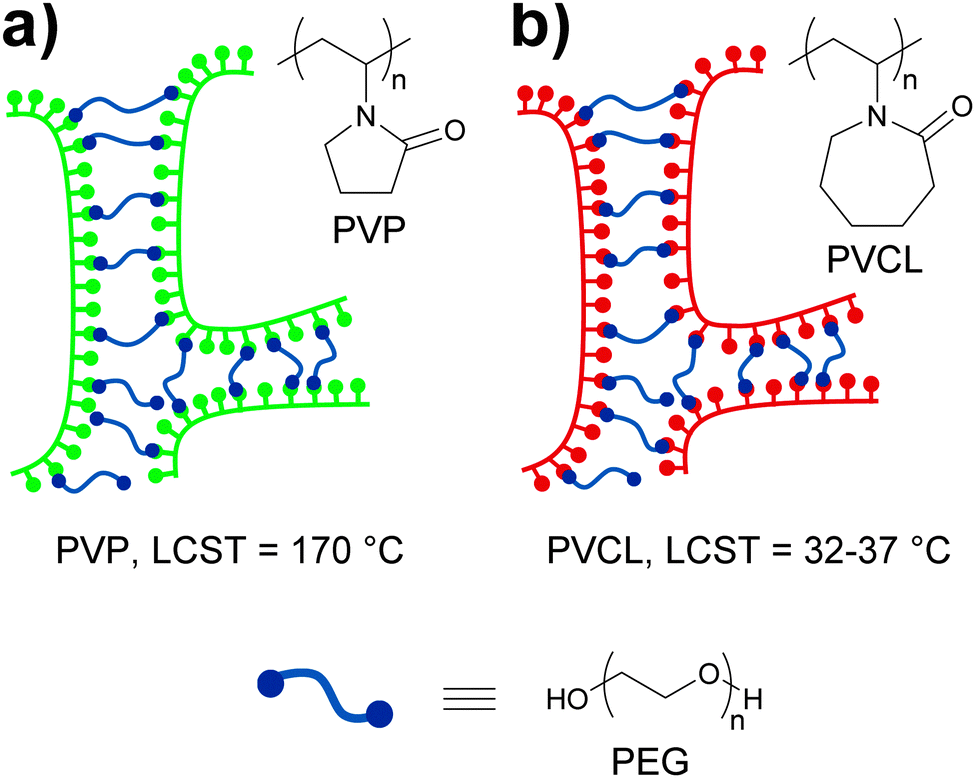 | ||
| Fig. 14 Temperature and water sensitive adhesives based on hydrogen bonded networks formed by blends of (a) PVP/PEG-40080 and (b) PVCL/PEG-400.81 | ||
In the context of stimuli-responsive adhesives for medical purposes, ideal PSAs should display strong adhesion to skin but could ideally be removed when no longer needed without skin damage and pain. Feldstein and coworkers81 continued their investigation of the blends discussed above by using poly(N-vinyl caprolactam) (PVCL) (Fig. 14b), a biocompatible polymer that shows a lower critical solution temperature (LCST) at 32–37 °C, i.e., near physiological temperature, instead of PVP. As in their previous study, the content of PEG-400 had a clear effect on the properties of the blends at ca. 22 °C and 50% relative humidity, which varied from a non-tacky solid (at 30 wt% of PEG-400) to a tacky liquid (at 60 wt% of PEG-400). Within this compositional range, a PEG-400 content of 45–50% (with a water content of 3–5 wt%) resulted in a PSA with well-balanced solid- and liquid-like properties, displaying a debonding stress of ∼0.67 MPa in a probe tack test at 22 °C. Interestingly, the same material shows a sharp decrease (∼0.35 MPa) of debonding stress at 35 °C and behaves as a tacky liquid at temperatures above 45 °C. In this case, the low water content of the sample did not allow to clearly establish the role of the LCST of PVCL on the adhesive properties of the blend. The effect of temperature and water content on the adhesive properties of the blend containing 45% of PEG-400 was therefore studied by means of 180° peel tests (Fig. 2). The blends containing 10 or 20 wt% of water experienced a gradual decrease of peel strength with increasing temperature with the 20% water blend displaying the highest values. On the other hand, at a water content of ca. 30%, a fast and clear loss of adhesion was observed at ca. 55 °C, and a further increase of the water content led to a gradual decrease of the detaching temperature. This behavior could be correlated to the variations on the cloud point of a diluted PVCL/PEG-400 with the temperature. Additionally, these results indicate that the PVCL/PEG-400 blends can accommodate more water than the PVP–PEG-400 blends before losing their adhesive properties. In other words, PVCL/PEG-400 blends form a denser supramolecular network that provides stronger adhesion. Overall, the mechanical properties of these materials can be (reversibly) switched by exposure to water at temperatures close to that of the body, which has clear implications in the development of adhesives for medical applications that would readily detach by simple exposure to water.
Conclusions
Despite many great advances made in the burgeoning field of supramolecular polymers, their use in the context of adhesive materials is a relatively recent development, although also this subdomain seems now to be rapidly growing. The studies reviewed here evidence the potential of supramolecular adhesives in many application areas and in many cases showcase the role of nature as a source of inspiration. Non-covalent interactions are central to most adhesion processes used by living organisms, which allow them to adhere to different surfaces in a variety of challenging environments and to detach in response to external stimuli. The work reviewed in this contribution suggests that some of such advanced functionalities can now, if only in crude ways, be emulated by synthetic supramolecular polymers based on hydrogen bonding, host–guest interactions, metal–ligand complexation and others, and combinations of such interactions. Thus, supramolecular adhesives displaying (multi)stimuli-triggered debonding-on-demand, substrate selectivity, underwater adhesion or bioactivity have been reported. Among them, two main approaches to switchable adhesion can be distinguished, namely the engineering of the adhesive forces at the interface of two substrates, and the design of the materials with stimuli-responsive bulk properties. While the first approach allows for the synthesis of adhesives for high-tech applications where selective/orthogonal adhesion is required, the second approach appears to offer a simpler and perhaps more cost-effective alternative that still allows for accessing appealing features such as remotely triggered debonding. Non-covalent interactions can thus provide the basis for the development of adhesives with the potential to impact different application fields; however, this is to date only reflected in a few emerging products including the customized materials developed by Suprapolix,82 Bostik,24,25 and Arkema.26 The main challenges in this field will therefore include the customer-driven design of supramolecular adhesives and the development of synthetic strategies leading to up-scalable productions. Nevertheless, considering the fast development of this research area, we expect an exciting future both in the fundamental and application arenas.Acknowledgements
The authors acknowledge funding from the Commission for Technology and Innovation (CTI, Grant No. 13746.1), the U.S. Army Research Office (W911NF-12-1-0339) and the Adolphe Merkle Foundation for support of research on supramolecular adhesives. LME is grateful for support through the Ambizione program of the Swiss National Science Foundation.References
- R. H. Lacombe, Adhesion Measurement Methods: Theory and Practice, CRC Press, Boca Raton, London, New York, 2005 Search PubMed.
- K. W. Allen, Int. J. Adhes. Adhes., 2003, 23, 87–93 CrossRef CAS.
- A. V. Pocius, Adhesion and Adhesives Technology, Carl Hanser Verlag GmbH & Co. KG, Munich, 3rd edn, 2012 Search PubMed.
- K. L. Mittal, Adhesion Aspects of Thin Films, VSP BV, Utrecht, 2001, vol. 1 Search PubMed.
- K. L. Mittal, Adhesion Aspects of Thin Films, VSP BV, Utrecht, 2005, vol. 2 Search PubMed.
- K. Autumn, Y. A. Liang, S. T. Hsieh, W. Zesch, W. P. Chan, T. W. Kenny, R. Fearing and R. J. Full, Nature, 2000, 405, 681–685 CrossRef CAS PubMed.
- E. Hennebert, B. Maldonado, P. Ladurner, P. Flammang and R. Santos, Interface focus, 2015, 5, 20140064 CrossRef PubMed.
- E. A. Dubiel, Y. Martin and P. Vermette, Chem. Rev., 2011, 111, 2900–2936 CrossRef CAS PubMed.
- C. Rodriguez-Emmenegger, S. Janel, A. de los Santos Pereira, M. Bruns and F. Lafont, Polym. Chem., 2015 10.1039/c1035py00197h.
- H. J. Busscher and H. C. van der Mei, PLoS Pathog., 2012, 8, e1002440 CAS.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS PubMed.
- S. C. Nicklisch and J. H. Waite, Biofouling, 2012, 28, 865–877 CrossRef CAS PubMed.
- L. Li, W. Smitthipong and H. Zeng, Polym. Chem., 2015, 6, 353–358 RSC.
- D. Habault, H. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244–7256 RSC.
- G. L. Fiore, S. J. Rowan and C. Weder, Chem. Soc. Rev., 2013, 42, 7278–7288 RSC.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- L. Montero de Espinosa, G. L. Fiore, C. Weder, E. J. Foster and Y. C. Simon, Prog. Polym. Sci., 2015 DOI:10.1016/j.progpolymsci.2015.1004.1003.
- S. I. Stupp, V. LeBonheur, K. Walker, L. S. Li, K. E. Huggins, M. Keser and A. Amstutz, Science, 1997, 276, 384–389 CrossRef CAS PubMed.
- A. E. Rowan, M. C. Feiters and R. J. M. Nolte, Proc. Annu. Meet. Adhes. Soc., 1997, 20, 271–273 Search PubMed.
- S. Abed, S. Boileau, L. Bouteiller, J. R. Caille, N. Lacoudre, D. Teyssie and J. M. Yu, Polym. Mater.: Sci. Eng., 1997, 76, 45–46 CAS.
- Q. Chuan, A. T. N. Pires and L. A. Belfiore, Polym. Commun., 1990, 31, 177–182 Search PubMed.
- S. Tanaka, T. Aoki and M. Yoshida, International Pat., WO9110756A1, 1991 Search PubMed.
- N. Moszner, T. Hirt, K. Rist, U. Salz, C. Weder, G. Fiore, C. Heinzmann and V. Rheinberger, International Pat., WO2013034778A2, 2013 Search PubMed.
- N. Sajot, C. Brunet and M. Lachhab, International Pat., WO2009150328A2, 2009 Search PubMed.
- D. Goubard and N. Sajot, Canada Pat., CA2740089A1, 2011 Search PubMed.
- J. A. Gonzalez Leon, J.-P. Gillet, G. Barreto, M. Hidalgo and V. Luca, International Pat., WO2011015773A2, 2011 Search PubMed.
- C. I. Lindsay and S. A. Woutters, US Pat., US20070149751A1, 2007 Search PubMed.
- M. K. C. Moens, EU Pat., EP1213309A1, 2002 Search PubMed.
- J. Zhan and R. Du, China Pat., CN201310168441A, 2013 Search PubMed.
- G. M. L. Hoorne-van Gemert, S. Chodorowski-Kimmès, H. M. Janssen, E. W. Meijer and A. W. Bosman, International Pat., WO2010002262A1, 2010 Search PubMed.
- B. Greener, International Pat., WO0107396A1, 2001 Search PubMed.
- G. Fichman, L. Adler-Abramovich, E. Gazit and P. B. Messersmith, International Pat., WO2014191997A1, 2014 Search PubMed.
- N. Boden, A. Agelli, E. Ingham and J. Kirkham, International Pat., WO2004007532A2, 2004 Search PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- K. Yamauchi, J. R. Lizotte and T. E. Long, Macromolecules, 2003, 36, 1083–1088 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. Ky Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- S. Cheng, M. Zhang, N. Dixit, R. B. Moore and T. E. Long, Macromolecules, 2012, 45, 805–812 CrossRef CAS.
- K. Viswanathan, H. Ozhalici, C. L. Elkins, C. Heisey, T. C. Ward and T. E. Long, Langmuir, 2006, 22, 1099–1105 CrossRef CAS PubMed.
- C. Creton, MRS Bull., 2003, 28, 434–439 CrossRef CAS.
- S. D. Tobing and A. Klein, J. Appl. Polym. Sci., 2001, 79, 2230–2244 CrossRef CAS.
- C. A. Anderson, A. R. Jones, E. M. Briggs, E. J. Novitsky, D. W. Kuykendall, N. R. Sottos and S. C. Zimmerman, J. Am. Chem. Soc., 2013, 135, 7288–7295 CrossRef CAS PubMed.
- Y. Zhang, C. A. Anderson and S. C. Zimmerman, Org. Lett., 2013, 15, 3506–3509 CrossRef CAS PubMed.
- L. Römer and T. Scheibel, Prion, 2014, 2, 154–161 CrossRef.
- P. Woodward, D. Hermida Merino, I. W. Hamley, A. T. Slark and W. Hayes, Aust. J. Chem., 2009, 62, 790–793 CrossRef CAS.
- G. M. L. van Gemert, J. W. Peeters, S. H. M. Söntjens, H. M. Janssen and A. W. Bosman, Macromol. Chem. Phys., 2012, 213, 234–242 CrossRef CAS.
- J. Courtois, I. Baroudi, N. Nouvel, E. Degrandi, S. Pensec, G. Ducouret, C. Chanéac, L. Bouteiller and C. Creton, Adv. Funct. Mater., 2010, 20, 1803–1811 CrossRef CAS.
- S. Pensec, N. Nouvel, A. Guilleman, C. Creton, F. Boue and L. Bouteiller, Macromolecules, 2010, 43, 2529–2534 CrossRef CAS.
- C. Heinzmann, S. Coulibaly, A. Roulin, G. L. Fiore and C. Weder, ACS Appl. Mater. Interfaces, 2014, 6, 4713–4719 CAS.
- C. Heinzmann, U. Salz, N. Moszner, G. L. Fiore and C. Weder, ACS Appl. Mater. Interfaces, 2015, 7, 13395–13404 CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. Ky Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- F. Chabert, F. Tournilhac, N. Sajot, S. Tencé-Girault and L. Leibler, Int. J. Adhes. Adhes., 2010, 30, 696–705 CrossRef CAS.
- S. Grimaldi, J.-P. Gillet, M. Hidalgo, F.-G. Tournilhac, P. Cordier and L. Leibler, International Pat., WO2008029065A2, 2008 Search PubMed.
- D. Montarnal, L. Leibler, F.-G. Tournilhac and M. Hidalgo, International Pat., WO2009141558A2, 2009 Search PubMed.
- R. Wang and T. Xie, Langmuir, 2010, 26, 2999–3002 CrossRef CAS PubMed.
- G. O. Wilson, M. M. Caruso, S. R. Schelkopf, N. R. Sottos, S. R. White and J. S. Moore, ACS Appl. Mater. Interfaces, 2011, 3, 3072–3077 CAS.
- R. Du, J. Wu, L. Chen, H. Huang, X. Zhang and J. Zhang, Small, 2014, 10, 1387–1393 CrossRef CAS PubMed.
- P. Y. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. van Luyn and E. W. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS PubMed.
- Y. Ahn, Y. Jang, N. Selvapalam, G. Yun and K. Kim, Angew. Chem., Int. Ed., 2013, 52, 3140–3144 CrossRef CAS PubMed.
- T. Nakamura, Y. Takashima, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2014, 5, 4622 CAS.
- A. K. Geim, S. V. Dubonos, I. V. Grigorieva, K. S. Novoselov, A. A. Zhukov and S. Y. Shapoval, Nat. Mater., 2003, 2, 461–463 CrossRef CAS PubMed.
- B. Yurdumakan, N. R. Raravikar, P. M. Ajayan and A. Dhinojwala, Chem. Commun., 2005, 3799–3801 RSC.
- Y. Takashima, T. Sahara, T. Sekine, T. Kakuta, M. Nakahata, M. Otsubo, Y. Kobayashi and A. Harada, Macromol. Rapid Commun., 2014, 35, 1646–1652 CrossRef CAS PubMed.
- T. Matsue, D. H. Evans, T. Osa and N. Kobayashi, J. Am. Chem. Soc., 1985, 107, 3411–3417 CrossRef CAS.
- C. B. Highley, C. B. Rodell, I. L. Kim, R. J. Wade and J. A. Burdick, J. Mater. Chem. B, 2014, 2, 8110–8115 RSC.
- M. Cheng, F. Shi, J. Li, Z. Lin, C. Jiang, M. Xiao, L. Zhang, W. Yang and T. Nishi, Adv. Mater., 2014, 26, 3009–3013 CrossRef CAS PubMed.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- R. Wang and T. Xie, Chem. Commun., 2010, 46, 1341–1343 RSC.
- H. Retsos, A. Kiriy, V. Senkovskyy, M. Stamm, M. M. Feldstein and C. Creton, Adv. Mater., 2006, 18, 2624–2628 CrossRef CAS.
- K. Zhao, X. R. Cheng, Y. L. Chao, Z. A. Li and G. L. Han, Dent. Mater., 2004, 20, 419–424 CrossRef CAS PubMed.
- M. Ozcan, Y. Kulak, C. de Baat, A. Arikan and M. Ucankale, J. Prosthodontics, 2005, 14, 122–126 CrossRef PubMed.
- R. Koppang, E. Berg, S. Dahm, C. Real and F. Floystrand, J. Prosthet. Dent., 1995, 73, 486–491 CrossRef CAS PubMed.
- X. Guo, F. Deng, L. Li and R. K. Prud'homme, Biomacromolecules, 2008, 9, 1637–1642 CrossRef CAS PubMed.
- M. M. Feldstein, Polym. Sci., Ser. A, 2009, 51, 1341–1354 CrossRef.
- M. M. Feldstein, Handbook of Pressure-Sensitive Adhesives and Products, CRC Press, Boca Raton, London, New York, 2009 Search PubMed.
- M. M. Feldstein, E. V. Bermesheva, Y. C. Jean, G. P. Misra and R. A. Siegel, J. Appl. Polym. Sci., 2011, 119, 2408–2421 CrossRef CAS.
- M. M. Feldstein, K. A. Bovaldinova, E. V. Bermesheva, A. P. Moscalets, E. E. Dormidontova, V. Y. Grinberg and A. R. Khokhlov, Macromolecules, 2014, 47, 5759–5767 CrossRef CAS.
- http://www.suprapolix.com/ .
| This journal is © The Royal Society of Chemistry 2016 |