
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Converting between the oxides of nitrogen using metal–ligand coordination complexes
Andrew J.
Timmons
and
Mark D.
Symes
*
WestCHEM, School of Chemistry, University of Glasgow, University Avenue, Glasgow, G12 8QQ, UK. E-mail: mark.symes@glasgow.ac.uk
First published on 9th July 2015
Abstract
The oxides of nitrogen (chiefly NO, NO3−, NO2− and N2O) are key components of the natural nitrogen cycle and are intermediates in a range of processes of enormous biological, environmental and industrial importance. Nature has evolved numerous enzymes which handle the conversion of these oxides to/from other small nitrogen-containing species and there also exist a number of heterogeneous catalysts that can mediate similar reactions. In the chemical space between these two extremes exist metal–ligand coordination complexes that are easier to interrogate than heterogeneous systems and simpler in structure than enzymes. In this Tutorial Review, we will examine catalysts for the inter-conversions of the various nitrogen oxides that are based on such complexes, looking in particular at more recent examples that take inspiration from the natural systems.
 Andrew J. Timmons | Andrew J. Timmons was born in Edinburgh in 1993 and attended George Watson's College where he first developed an interest in science. In 2011, he started a BSc (Hons) degree in Chemistry and Medicinal Chemistry at the University of Glasgow. His interests lie in renewable energy, small molecule activation and catalysis, which led him to undertake a final year project with Dr Mark Symes. At the time of writing, Andrew is focussed on his final year examinations and has aspirations to continue his education by pursuing a PhD after graduation. |
 Mark D. Symes | Mark D. Symes was born in London in 1982 and graduated from the University of Cambridge in 2005. After a PhD at the University of Edinburgh with David A. Leigh (FRS), he was a postdoctoral researcher in the group of Daniel G. Nocera at MIT. He returned to the UK as a postdoctoral research associate at the University of Glasgow in 2011, and he joined the faculty at Glasgow in 2013. His research interests span energy conversion, small molecule activations, electrochemistry and coordination chemistry. |
Key learning points(1) The key role of nitrogen oxides (especially nitrite) in the nitrogen cycle.(2) The coordination chemistry in the active sites of the various nitrogen oxide reductase enzymes. (3) How nitrite binds to, and is reduced at, metal ligand coordination complexes. (4) The various platforms that have been used for NO reduction. (5) Nitrate reduction with metal ligand coordination complexes. |
1. Introduction
Nitrogen is present in most of the building blocks of life and can be found in almost every major product of the chemical and pharmaceutical industries. It can exist in a number of oxidation states in various compounds (from +5 in nitrate to −3 in ammonia), but N2 itself is extremely inert on account of its strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond. It must therefore be “fixed” prior to incorporation into larger molecules. The most prevalent route of fixation is via conversion of N2 to ammonia, which is achieved industrially through the Haber Bosch process. The analogous transformation in natural systems (N2 reduction to ammonium using nitrogenase) is also a major contributor to total global nitrogen fixation.1
N triple bond. It must therefore be “fixed” prior to incorporation into larger molecules. The most prevalent route of fixation is via conversion of N2 to ammonia, which is achieved industrially through the Haber Bosch process. The analogous transformation in natural systems (N2 reduction to ammonium using nitrogenase) is also a major contributor to total global nitrogen fixation.1
Regardless of how N2 is fixed, there is a constant flux of nitrogen from one oxidation state to another mediated by a range of natural systems: this is the nitrogen cycle (Fig. 1). Inspection of Fig. 1 reveals the critical importance of the oxides of nitrogen and their inter-conversions in the nitrogen cycle. The delicate balance maintained by these competing processes is readily disturbed by anthropogenic nitrogen-cycle activity, such as excessive use of nitrate fertilisers. As shown in Fig. 1, nitrite stands out as the common intermediate in a number of nitrogen conversion reactions of major importance, and it can thus be viewed as the key to catalysis in the nitrogen cycle.1Fig. 1 also gives examples of some of the enzymes responsible for catalysing the transformation of nitrogen from one oxidation state to another. These enzymes contain transition metal coordination complexes in their active sites, the structures of many of which have been elucidated. In the following section, we will examine the structures of some of these enzymes and what is known of their mechanisms of action.
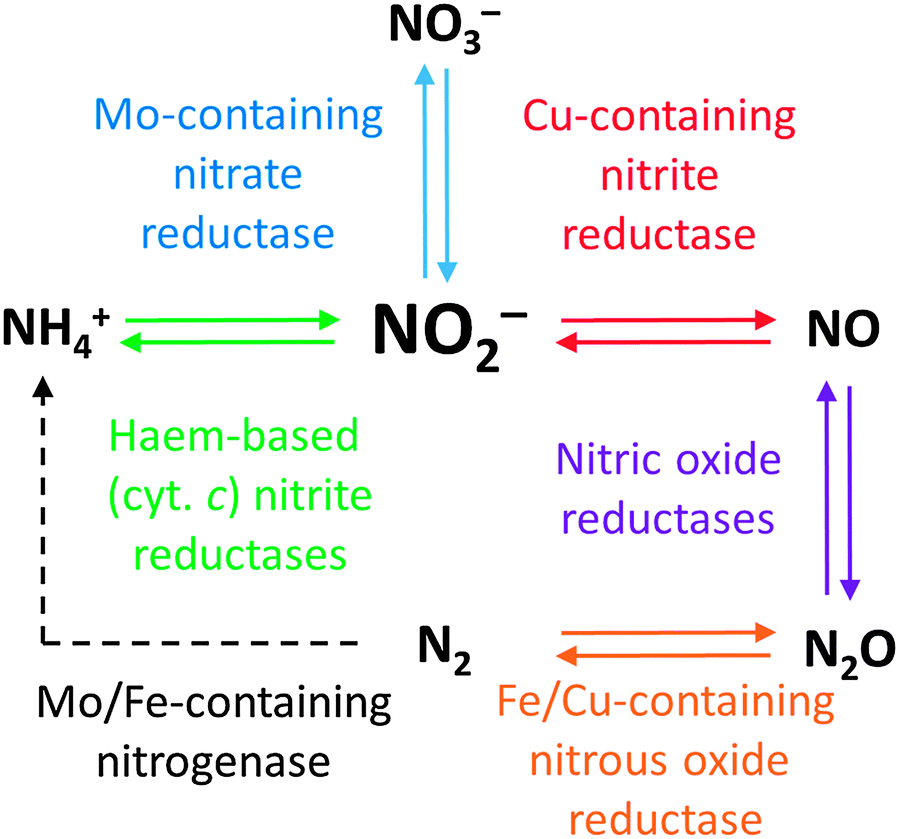 | ||
| Fig. 1 Overview of the nitrogen cycle, showing the prominent positions occupied by the nitrogen oxides (especially nitrite). Examples of some of the enzymes involved are given (more details on these can be found later in the text). All the reaction pathways shown with bold arrows will be addressed in this review. Processes shown by dashed arrows are not reviewed herein. | ||
To the best of our knowledge, there has been no comprehensive review of metal–ligand coordination complexes for the catalytic interconversion of the nitrogen oxides since Karlin's review in 2002.2 The aim of this review is therefore to provide an introduction to this fascinating field post-2002 that will give the interested reader an idea of the current state-of-the-art. Furthermore, we shall restrict ourselves as far as possible to a discussion of functional systems able to mediate conversions of the nitrogen oxides, rather than those systems that merely bind these oxides. Finally, this review will address only small, artificial metal–ligand coordination complexes for the nitrogen oxide transformations summarised by Fig. 1. Readers interested in heterogeneous catalysts and electrocatalysts are directed to an excellent review by Koper on this subject.3
2. Natural systems for the inter-conversions of the nitrogen oxides
N2 enters the nitrogen cycle not only through the fixation of N2 to ammonia but also in the form of nitrate and nitrite from mineral sources and physical events such as lightning strikes. Biological systems are then able to convert between the various oxidation states of nitrogen with high specificity using a handful of generic classes of enzymes, effectively allowing any oxidation state of nitrogen to be accessed. Where a single enzyme cannot carry out the desired transformation, sequences of enzymes are used.1 For example, nitrate conversion to ammonia can be achieved through a combination of molybdenum-dependent nitrate reductases (reducing nitrate to nitrite) and sirohaem-containing nitrite reductases (reducing nitrite to ammonia). All the enzymes featured in Fig. 1 contain metal–ligand coordination complexes in their active sites. In this section, we will examine the structures of these active sites, which lend such obvious inspiration to many of the metal–ligand complexes that we shall explore in this review.2.1 Nitrite reduction to ammonium with haem-based nitrite reductases
The 6-electron reduction of nitrite to ammonia is catalysed by a number of nitrite reductases that use haems as their active centres. The general equation for this reaction is:| NO2− + 6e− + 8H+ → NH4+ + 2H2O | (1) |
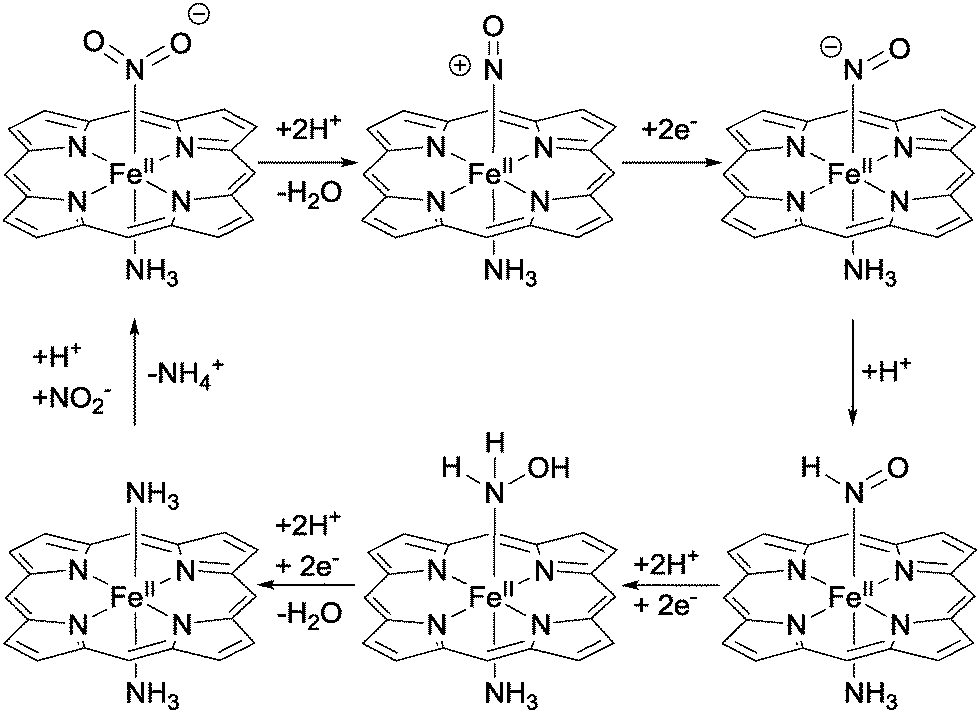 | ||
| Fig. 2 Mechanism of nitrite reduction by cytochrome c nitrite reductase according to Neese and co-workers.4 | ||
Upon binding of nitrite to the Fe(II) centre of the haem, N–O bond heterolysis occurs to generate a haem-nitrosyl ({Fe–NO}) complex. This is facilitated by a high degree of back-bonding from the filled dxz orbital of the Fe(II) to an empty orbital with π* character on the bound nitrite.4 This back-bonding serves to both strengthen the Fe–N bonding interaction and weaken the N–O bonds, promoting heterolysis. N–O bond heterolysis is also promoted by the formation of hydrogen bonds between protein residues (especially protonated histidine and arginine residues) and the nitrite oxygens. Indeed, the unique environment provided by the protein matrix may give a local pH below 3, aiding protonation of the oxygens bound to nitrite. Successive electron and proton transfers then result in the formation of the {Fe–NH(O)} adduct, where the nitrogen coordinated to iron is protonated in preference to the remaining oxygen. Further proton-coupled-electron transfers then lead to the formation of a putative iron–hydroxylamine complex ({Fe–NH2OH}), which is then itself protonated and reduced to break the final N–O bond, liberating water and giving the {Fe–NH3} intermediate. Further protonation then causes release of ammonium, allowing a second molecule of nitrite to bind and permitting the enzyme to enter the next catalytic cycle.1,4
2.2 Nitrite reduction to nitric oxide with copper-based nitrite reductases
Various bacteria remove nitrogen from their systems using the process of denitrification. This involves reduction of nitrite ions to NO and nitrous oxide (N2O) followed by a further, final reduction to dinitrogen gas. The first step in this denitrification pathway is the reduction of nitrite to NO:| NO2− + e− + 2H+ → NO + H2O | (2) |
| Cu(I) + NO2− + 2H+ → Cu(II) + NO + H2O | (3) |
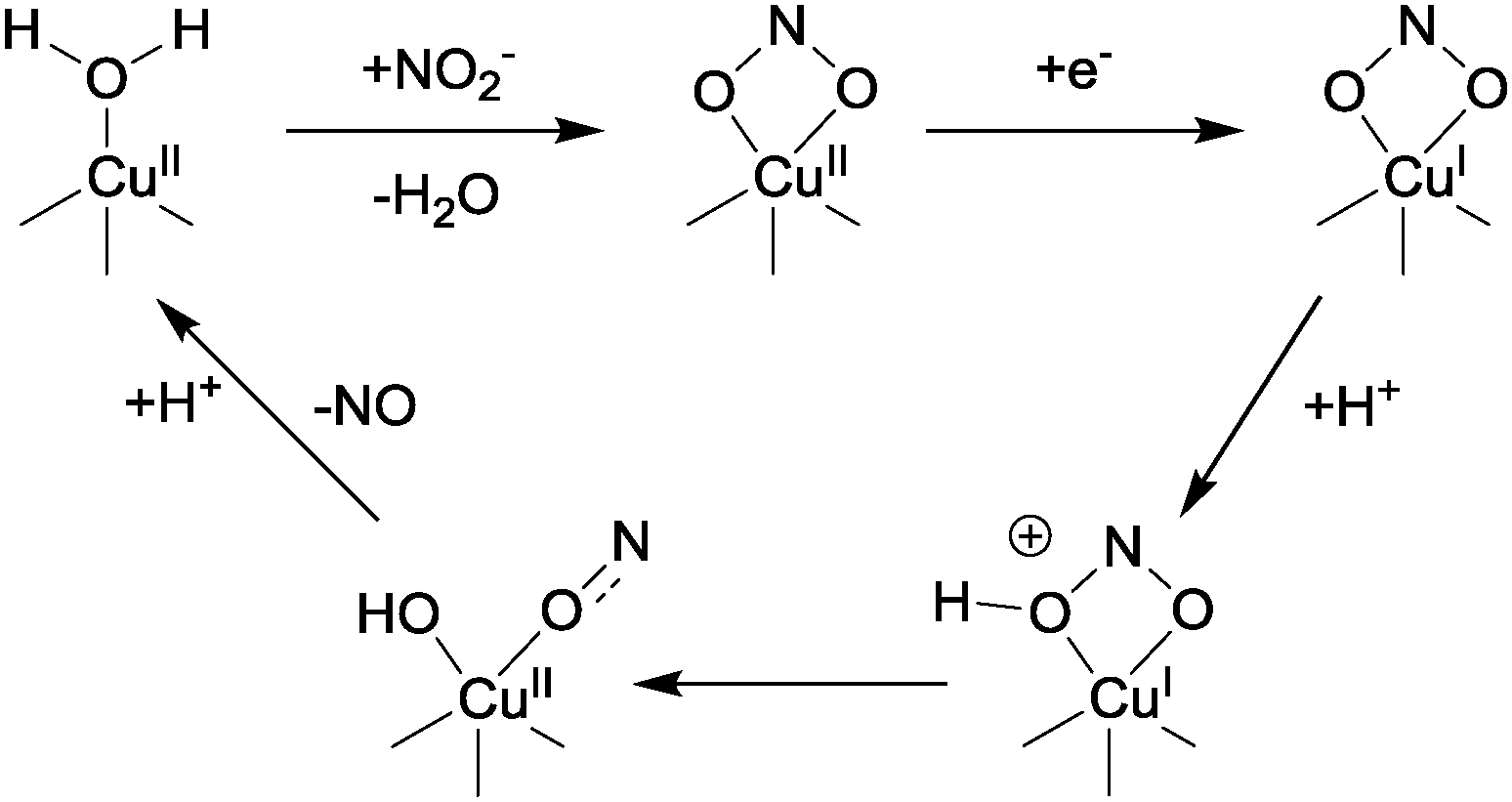 | ||
| Fig. 3 The proposed mechanism (simplified) of nitrite reduction to NO in CuNIR. | ||
In this proposed mechanism, nitrite displaces water and binds to the Cu(II) centre of CuNIR through its two oxygen atoms, producing a distorted square-based pyramidal geometry at the copper. Reduction of the Cu(II) centre then occurs as a result of electron transfer from a type-1 centre, and this electron transfer is itself probably triggered by protonation of a nearby conserved aspartate residue (pKa ∼ 5). There is currently some debate as to the ordering of the binding and reduction events, and it is possible that reduction precedes binding.1 In any case, the bound nitrite is then protonated at the oxygen that is to be abstracted (with this proton coming from the conserved aspartic acid previously mentioned), after which N–OH bond lysis occurs. This bond scission is facilitated by back-bonding from the copper dx2–y2 orbital into the N–OH σ* orbital. This interaction between the Cu and the oxygen atoms of the nitrite arises as a consequence of the bidentate binding mode of the NO2−.7 After the N–OH is broken, protonation of the {Cu–OH} species thus formed leads to NO liberation and the generation of a Cu(II)-aqua complex. This aqua complex can then re-enter the catalytic cycle by binding to nitrite followed by reduction (or vice-versa).
2.3 Nitrite oxidation to nitrate
The biological processes that underlie the interconversion of NO2− and NO3− are less well understood than the aforementioned reductions of nitrite. The dissimilatory pathway for conversion of nitrite to nitrate is performed using the molybdenum-containing nitrite oxidoreductase (MoNiOR) enzymes, in which two thiolate-bearing pyranopterin cofactors enforce a distorted trigonal prismatic geometry on the Mo centre, with the thiolates defining one rectangular face of the prism.8 The general reaction for nitrite oxidation is given in eqn (4):| NO2− + H2O → NO3− + 2H+ + 2e− | (4) |
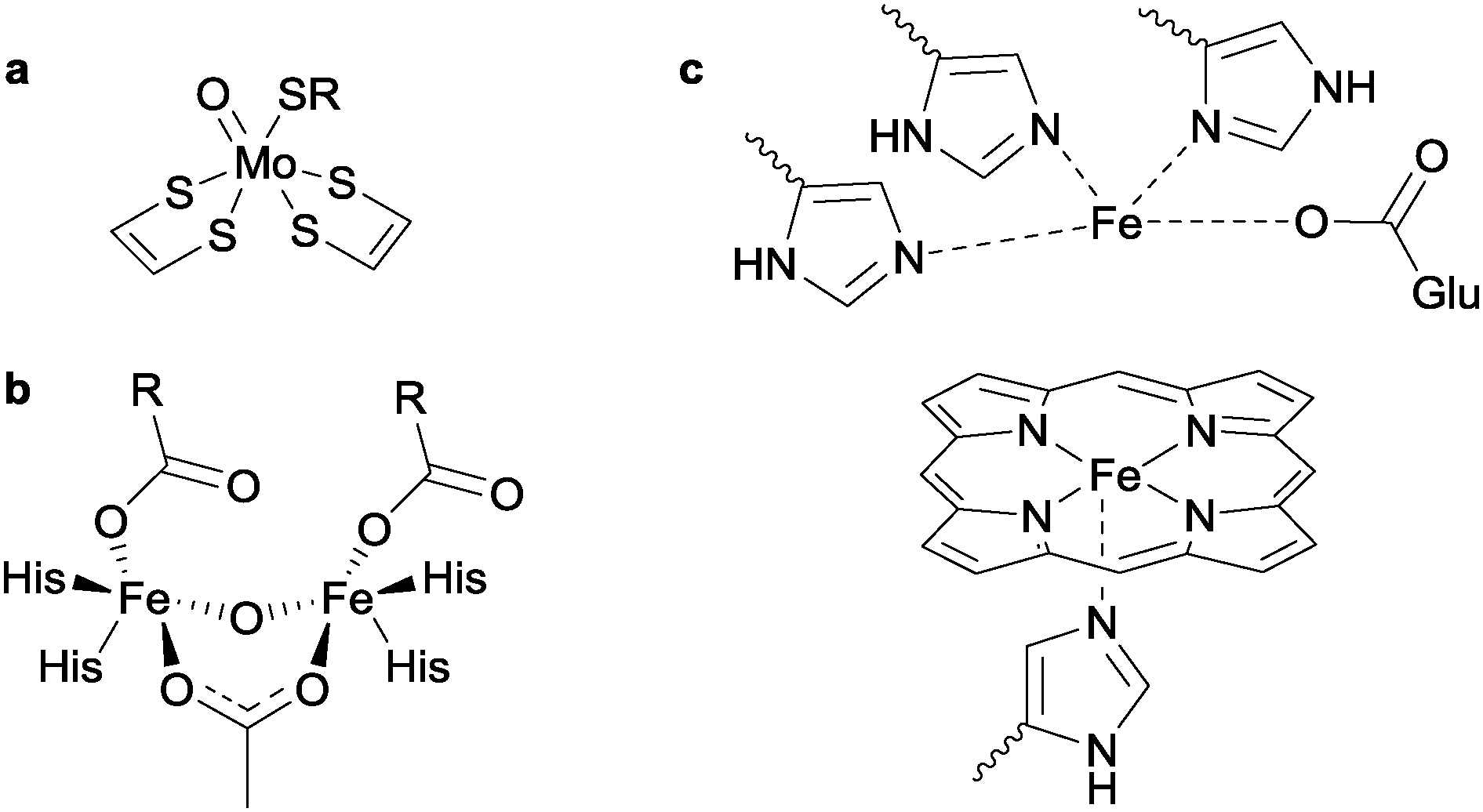 | ||
| Fig. 4 (a) Simplified active site structure of the dissimilatory nitrate reductases, showing the primary coordination environment around the Mo centre. SR is typically cysteine. Alternatively, this ligand could be present as –OR in the form of serine. (b) Active site structure of flavodiiron nitric oxide reductase, FNOR. The R-groups on FNOR are either aspartate or glutamate. His = histidine. (c) Simplified active site structure of nitric oxide reductase. Glu = glutamate. | ||
2.4 Nitric oxide reduction
Two types of enzymes exist for the conversion of NO to N2O: nitric oxide reductase (which contains two iron centres, one as haem and the other coordinated to a glutamate and three histidine ligands)9 and the non-haem flavodiiron nitric oxide reductase (FNOR, see structures b and c in Fig. 4).10 In both cases, the general reaction is given by:| 2NO + 2e− + 2H+ → N2O + H2O | (5) |
3. Artificial metal–ligand coordination complexes for nitrite reduction
3.1 Nitrite reduction with copper complexes
Copper coordination complexes have been developed as mimics of CuNIR in order to effect the reduction of nitrite to NO according to eqn (3) (see Section 2.2). The underlying mechanism for eqn (3) with reference to copper coordination complexes is generally thought to proceed as proposed by Averill,11 whereby nitrite binds to a reduced Cu(I) centre, with subsequent protonation yielding water and a Cu(I)-bound NO+ ligand. According to this postulated mechanism, binding of nitrite to a more oxidized Cu(II) centre would not lead to NO production, but might do so upon reduction of the metal centre.Various modes for the binding of nitrite to transition metal complexes have been observed (Fig. 5), including η1-NO2 (N-nitro), η1-ONO (O-nitrito), and η2-O–N–O (O,O-bidentate). In 1996, Tolman reported a functional, mononuclear model complex of CuNIR containing a Cu(I) centre supported by a sterically hindered triazacyclononane ligand that was somewhat reminiscent of the active site of enzyme.12 The nitrite was bound to the metal through its nitrogen atom (N-nitro), producing a distorted tetrahedral geometry at copper. Upon addition of acetic acid to this Cu(I)–nitrite adduct, NO was evolved according to eqn (3), with concomitant generation of the corresponding Cu(II)–di-acetate complex. Since this report, numerous other Cu complexes featuring tripodal ligands have shown activity for nitrite reduction to NO (see, for example, ref. 2 and 13). Here, we shall focus on a few of the more illustrative examples from the last decade.
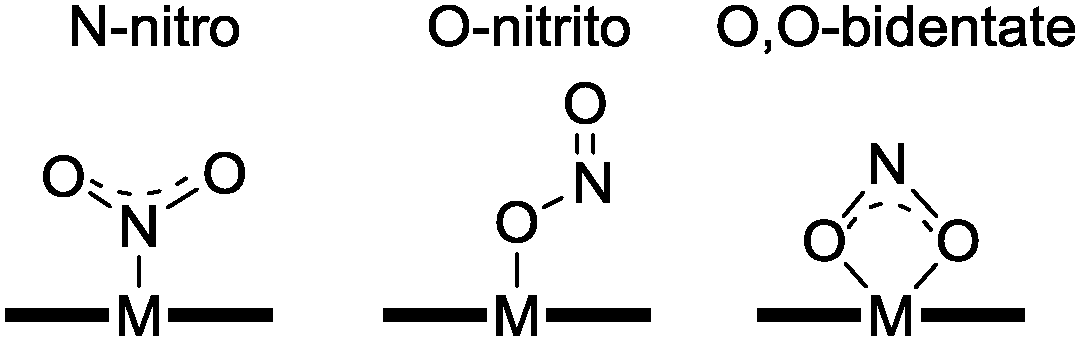 | ||
| Fig. 5 Common modes for binding of nitrite to metal centres. | ||
The first of these recent examples that we shall consider was reported by Kujime et al. in 2008,14 who studied several copper(I) nitrite complexes with various tridentate ligands (see Fig. 6) in order to determine the steric and electronic effects of the ligands on CuNIR-type activity. As with Tolman's CuNIR mimic, all five Cu(I)–nitrite complexes reacted with acetic acid to liberate NO and to form the corresponding Cu(II)–acetate complexes. However, there was a marked difference in the activity of the various complexes as a function of the tripodal ligand used, on account of both steric and electronic factors.
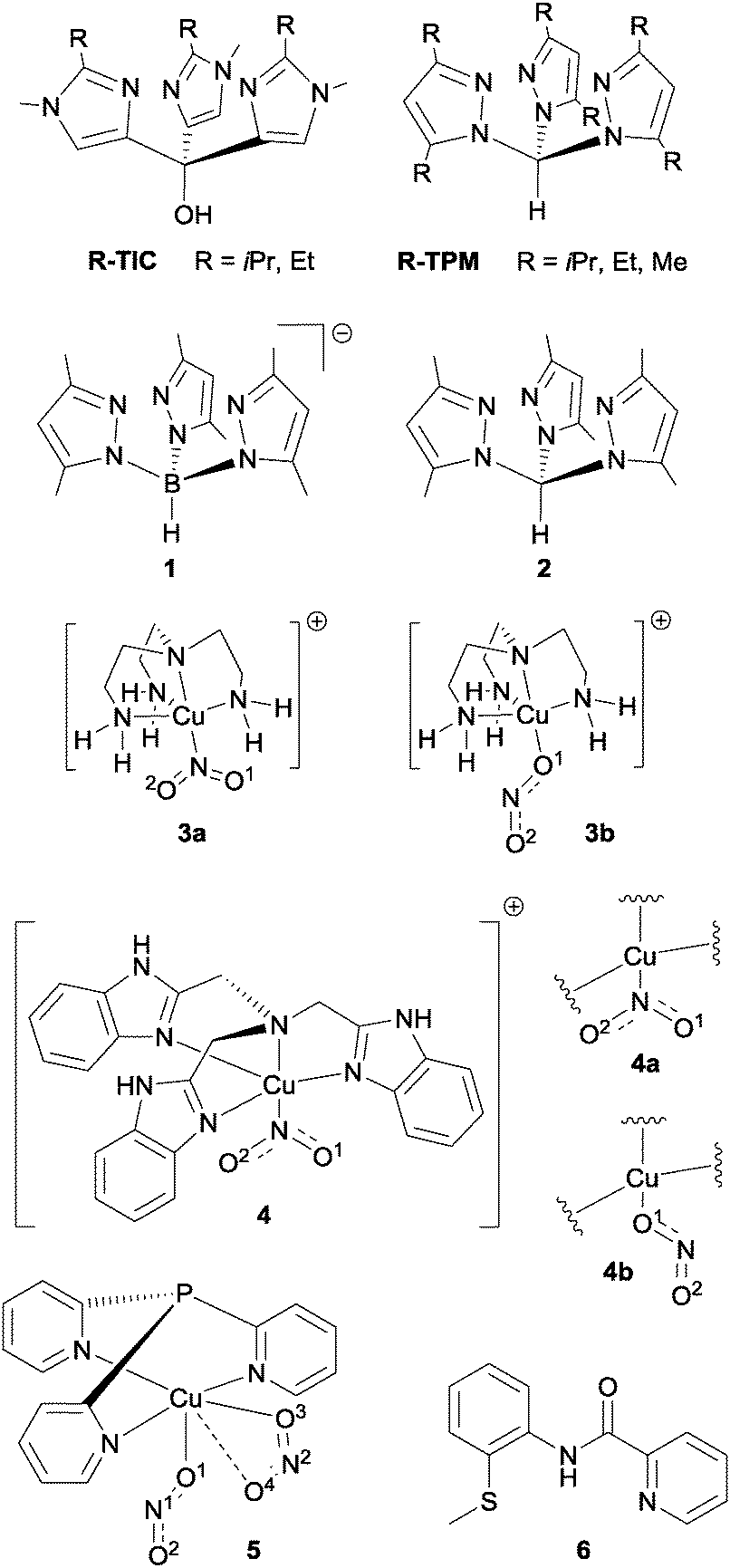 | ||
| Fig. 6 Chemical structures of the various ligands and complexes discussed in the text in relation to nitrite reduction with copper complexes. | ||
In terms of electronics, CuNIR-type activity was found to increase with increasing electron donation by the tridentate ligand. Upon coordination to the copper centre, the nitrite ion accepts electrons from the copper's occupied 3dπ-orbital into its pπ*-orbital (π-back-bonding), whilst donating electrons from its occupied pσ-orbital into the unoccupied 4s orbital of copper (σ-donation). As the π-back-bonding from the copper centre is less significant than the σ-donation from the nitrite ion, the electron density of nitrite when bound to Cu is smaller than in the free anion. The use of electron-donating ligands on the Cu centre should then act to push electron density onto the copper, increasing the π-back-bonding effect and therefore leading to a higher electron density on the nitrite when it is bound to Cu than might otherwise be expected. This means that the electron density on both the nitrite and copper can be controlled by careful choice of ligand.
For both (TIC)Cu(NO2) and (TPM)Cu(NO2)-type complexes (Fig. 6), the HOMO is formed by a combination of interactions between the unoccupied 4s-orbital of the Cu and the occupied pσ-orbital of NO2−. It is the HOMO of the copper complex which interacts with the protons during the reaction (allowing one oxygen of the bound nitrite to be lost as water, see eqn (3)). The protonation of bound nitrite is stepwise, with L–CuNO2H as the proposed intermediate (L = TIC or TPM) and the initial protonation of the copper-bound nitrite is the rate determining step.14 Hence the higher the energy of the HOMO, the faster initial protonation of the nitrite and hence the greater the reactivity observed. Kujime et al. found that the HOMO of the (TIC)Cu(NO2) complexes was about 0.5 eV higher than that of their (TPM)Cu(NO2) analogues, suggesting enhanced reactivity for the TIC–Cu complexes. The authors also calculated the stabilization energy for protonation of the Cu(I)–nitrite complexes and found that the stabilization energy for the (TIC)Cu(NO2) complexes was around 40 kJ mol−1 larger than for the (TPM)Cu(NO2) analogues (similar to the difference in energies between the HOMOs). This added further weight to the supposition that the enhanced Cu-NIR activity of the (TIC)Cu(NO2) complexes was due to the higher energy of the HOMOs in these compounds.
Kujime et al. also showed that steric effects have a large influence on reactivity. When more bulky ligand R-groups were used, X-ray crystallography revealed that the Cu–Nligand bond lengths (between the copper centre and the tripodal ligand) increased. This reduced electron donation from the tripodal ligand to the Cu, and hence decreased the overall electron density on the nitrite (due to decreased π-back-bonding from the Cu to the bound nitrite). This in turn led to slower nitrite reduction. Hence the authors concluded that less sterically encumbered complexes exhibited superior catalysis.
A distinct tripodal ligand for supporting Cu(I) was recently reported by Hsu and co-workers based on the scorpionate ligand hydrotris(3,5-dimethylpyrazolyl)borate (1, Fig. 6).15 The activity of the Cu(I)–NO2 complex of this ligand was compared to that of its C-bridged analogue (2), revealing that in both cases protonation was not the rate determining step during reaction with acetic acid to liberate NO (in contrast to the RDS observed by Kujime et al. with their complexes14). The more electron-rich B-bridged tripodal complex was found to be a more effective catalyst for nitrite reduction to NO than the C-bridged tripod complex, which the authors suggested could be due to greater electron density on the nitro ligand in the case of the B-bridged complex.
An alternative set of structurally-related copper–tripodal ligand complexes (based on Cu(II)) that are competent for nitrite reduction to NO and N2O was reported recently by Woollard-Shore et al.16 The structures of some of these complexes are shown in Fig. 6. The complexes of generic formula ([Cu(NO2)(Tren)][PF6]) (where Tren is tris-(2-aminoethyl)amine) and the complexes of formula ([Cu(NO2)(Tris-[(benzimidazol-2-yl)methyl]amine)][NO2]) were found to be a mixture of η1-NO2 and η1-ONO isomers. X-ray crystallography revealed that the η1-NO2 bound isomer of complex 3 (3a) displays two roughly equal nitrite N–O bond lengths (1.246 Å and 1.239 Å). In contrast, the η1-ONO bound isomer complex 3b showed unequal N–O bond lengths with r(N–O1) = 1.317 Å and r(N–O2) = 1.214 Å (where O1 is bound to the Cu centre). A disparity between N–O bond lengths may be expected for the η1-ONO bound isomers of each complex, but a significant difference in N–O bond lengths of the η1-NO2 bound isomer of complex 4 (4a) was also observed: r(N–O1) = 1.217 Å and r(N–O2) = 1.318 Å. This disparity in N–O bond lengths for complex 4a (which is not observed in the analogous isomer complex 3a) suggests that there is a difference in the distribution of electron density between the Cu2+ centre and the coordinated NO2− in complexes 3 and 4 brought about by the different ligand environments.16 In complex 4a, the dxz or dyz orbitals of the Cu2+ centre which are involved in bonding to the NO2− donate more strongly through back-bonding into the empty π*-orbitals of NO2− than in complex 3a, causing the overall electron density on the Cu2+ ion in 4a to be lower. This is reflected in a difference in the position of the Cu(I)/Cu(II) redox couples: 4a has a more positive reduction potential (and hence is easier to reduce) than is complex 3a.
The η1-ONO bound isomer complex 4b showed an elongation of the r(N–O1) bond (1.361 Å) (O1 is bound to Cu centre) with contraction of the r(N–O2) bond (1.210 Å) which is characteristic of the η1-ONO bound mode. Density functional theory calculations showed that the η1-NO2 isomers were lower in energy than the η1-ONO isomers by −5.2 kJ mol−1 and −3.8 kJ mol−1 for complexes 3 and 4 respectively in the gas-phase. This small difference in calculated energies helps to explain why both the η1-ONO and η1-NO2 linkage isomers were observed. Complex 4 demonstrated better catalytic properties on almost all fronts, including percentage of NO2− converted, reaction time, initial NO2− conversion rate and percentage of NO present in the final gas mixture.
Two different binding modes for nitrite were also observed for compound 5 ([Cu(NO2)(Tris-(4-phenyl-3-aza-3-butenyl)amine)][BF4]) with both an end-on η1-ONO ligand and a bidentate η2-ONO ligand. Comparison of the bond lengths in these complexes suggests that the bonding between the Cu2+ centre and the η1-ONO ligand is stronger than the interaction with the bidentate η2-ONO ligand. Complex 5 was found to have very selective catalytic properties – converting 88% of NO2− into NO with no trace of N2O. However, it remains unclear whether it is the η1-ONO, the η2-ONO, or possibly both nitrites, which are reduced and hence no correlation can be drawn between binding mode and reactivity with this complex.
CuNIR-type activity can also be observed in Cu–tripodal ligands that coordinate to the copper centre through heteroatoms other than solely nitrogen. A good example of this was published by Patra and co-workers in 2013, wherein the tridentate N,N,S ligand N-2-methylthiophenyl-2′-pyridinecarboxamide (6, Fig. 6) was used to synthesise both Cu(I) and Cu(II) complexes which were then coordinated to nitrite.17 The Cu(I)–nitrite adduct was found to evolve NO when exposed to acids (both acetic and perchloric acids were found to be effective), whilst the Cu(II)–nitrite adduct liberated only nitrite under the same conditions. The Cu(II)–nitrite adduct could, however, be electro-reduced to its Cu(I) form, from which NO could be evolved upon treatment with acid, in agreement with the mechanism of nitrite reduction proposed by Averill.11
Electrochemical methods for reducing nitrite using copper complexes attached to electrodes have also been explored. Yamaguchi and co-workers used the tripodal N,N,N ligand bis(6-methyl-2-pyridylmethyl)amine ethyl sulfide to coordinate to a Cu(I) centre and attached this to the surface of gold electrodes using the thiol tether.18 Under acidic conditions (pH 5.3), an electrocatalytic wave for the reduction of NO2− to NO was observed, but no such wave was observed in the presence of nitrite at higher pH. This underlines the fact that the reduction of nitrite as shown in eqn (3) requires protons, (whether delivered to the substrate in a stepwise fashion with regards to the electron, or in a concerted process) and that nitrite reduction to NO and water without proton transfer is not possible. In a similar fashion, Orain et al. have attached Cu(I) complexes based on the ligand tris(2-pyridylmethyl)amine to gold electrodes and have also observed a requirement for acidic conditions in order to evolve NO from nitrite solutions.19 In this case, no catalytic current for nitrite reduction was evident at pH 8.9 and only a reversible wave attributed to the Cu(II)/Cu(I) couple was observed. However, upon lowering the pH to 4.6, a dramatic increase in the current was detected and reversibility was lost as nitrite was catalytically reduced to NO.
3.2 Nitrite reduction with mono-nuclear iron complexes
An early example of nitrite reduction mediated by iron coordination complexes was supplied by Meyer and co-workers, who used the water-soluble porphyrin [FeIII(H2O)(TPPS)]3− (TPPS = (meso-tetrakis(p-sulfonatophenyl)porphyrin)) in mildly acidic aqueous solution to electro-reduce nitrite to ammonia, with significant amounts of N2O and hydroxylamine also being produced.20 The authors determined that the nitrosyl complex [FeII(NO+)(TPPS)]3− was an intermediate in this reaction pathway, a complex which itself formed via reaction of the reduced Fe(II) porphyrin complex with HONO:| [FeII(H2O)(TPPS)]4− + HONO + H+ → [FeII(NO+)(TPPS)]3− + 2H2O. |
| [FeII(NO+)(TPPS)]3− + e− → [FeII(NO˙)(TPPS)]4−. |
| [FeII(NO˙)(TPPS)]4− + e− → [FeII(NO−)(TPPS)]5−. |
| [FeII(NH3)(TPPS)]4− + 2H2O + H+ → [FeII(H2O)2(TPPS)]4− + NH4+. |
Iron porphyrins have also been employed for the generation of NO from nitrite. For example, Chi and co-workers used iron meso-tetrakis(4-N-methylpyridiniumyl)porphyrin ([FeIII(TMPyP)]5+) to generate NO from nitrite solutions electrochemically.22 In order to obtain free NO gas, both cathodic and anodic processes were required. In the first step, the Fe(III)–porphyrin complex was reduced in the presence of nitrite to give the bis-nitrito Fe(II)–porphyrin complex [FeII(NO2−)2(TMPyP)]2+ which subsequently disproportionated to give the [FeIII(TMPyP)]5+ starting material and the mono-nitrosyl complex [FeII(NO)(TMPyP)]4+. This mono-nitrosyl complex was then subjected to an electrochemical oxidation to re-generate [FeIII(TMPyP)]5+ and release NO gas. The authors combined these two electrochemical processes in a flow-cell apparatus in order to generate a constant stream of NO by this method.
Another example of nitrite reduction to NO with iron porphyrins that is of particular relevance to NO formation in biological systems was reported recently by Ford and co-workers.23 Using the water soluble Fe(III)(TPPS) complex, the authors were able to observe oxygen atom transfer from nitrite to species of biological relevance such as cysteine and glutathione, with simultaneous formation of the Fe(II)(TPPS)(NO) complex. However, despite the high stability commonly exhibited by such ferrous nitrosyl adducts (cf. Chi et al., who could liberate NO from their analogous Fe(II)–NO complex only by electrochemical oxidation of the iron centre22), Ford's team observed what they describe as “a matrix of redox transformations” that lead to spontaneous regeneration of the Fe(III)(TPPS) starting material with concomitant production of N2O and NO at pH 6. HNO (nitroxyl) was suggested as the most likely precursor for N2O formation on the basis of trapping experiments. Meanwhile, NO liberation was proposed to occur via coordination of a second equivalent of nitrite at the proximal site of Fe(II)(TPPS)(NO), giving first Fe(II)(TPPS)(NO)(NO2−) and then the oxidised complex Fe(III)(TPPS)(NO), from which NO can dissociate. Although the precise mechanism for NO liberation from Fe(II)(TPPS)(NO) remains somewhat unclear, what is obvious is that ferrous nitrosyls such Fe(II)(TPPS)(NO) may not be as inert as commonly assumed at pH values around 6.
Very recently, Harrop and co-workers have reported the first example of a non-heme complex ([Fe(LN4Im)(NO2)2], see Fig. 7) with trans-coordinated nitrite ligands that is capable of reducing nitrite to NO in a catalytic fashion.24 The reaction chemistry of this iron complex was investigated using thiols as these are able to function both as proton and electron sources for the reduction of the coordinated nitro ligands (cf. the use of thiols in ref. 23). Using p-chlorobenzenethiol (p-ClArSH) as a sacrificial reductant in conjunction with p-toluene sulfonic acid (p-TsOH) as a proton source, NO was produced selectively from this complex with no evidence for the formation of other nitrogen oxides such as N2O or NO2. In the presence of excess nitrite, catalytic turnover was demonstrated and the active complex could be isolated in 80% yield after 3 cycles (see Fig. 7). The selectivity of this catalyst is impressive, and bodes well for the development of systems that can generate NO to the exclusion of other nitrogen oxides.
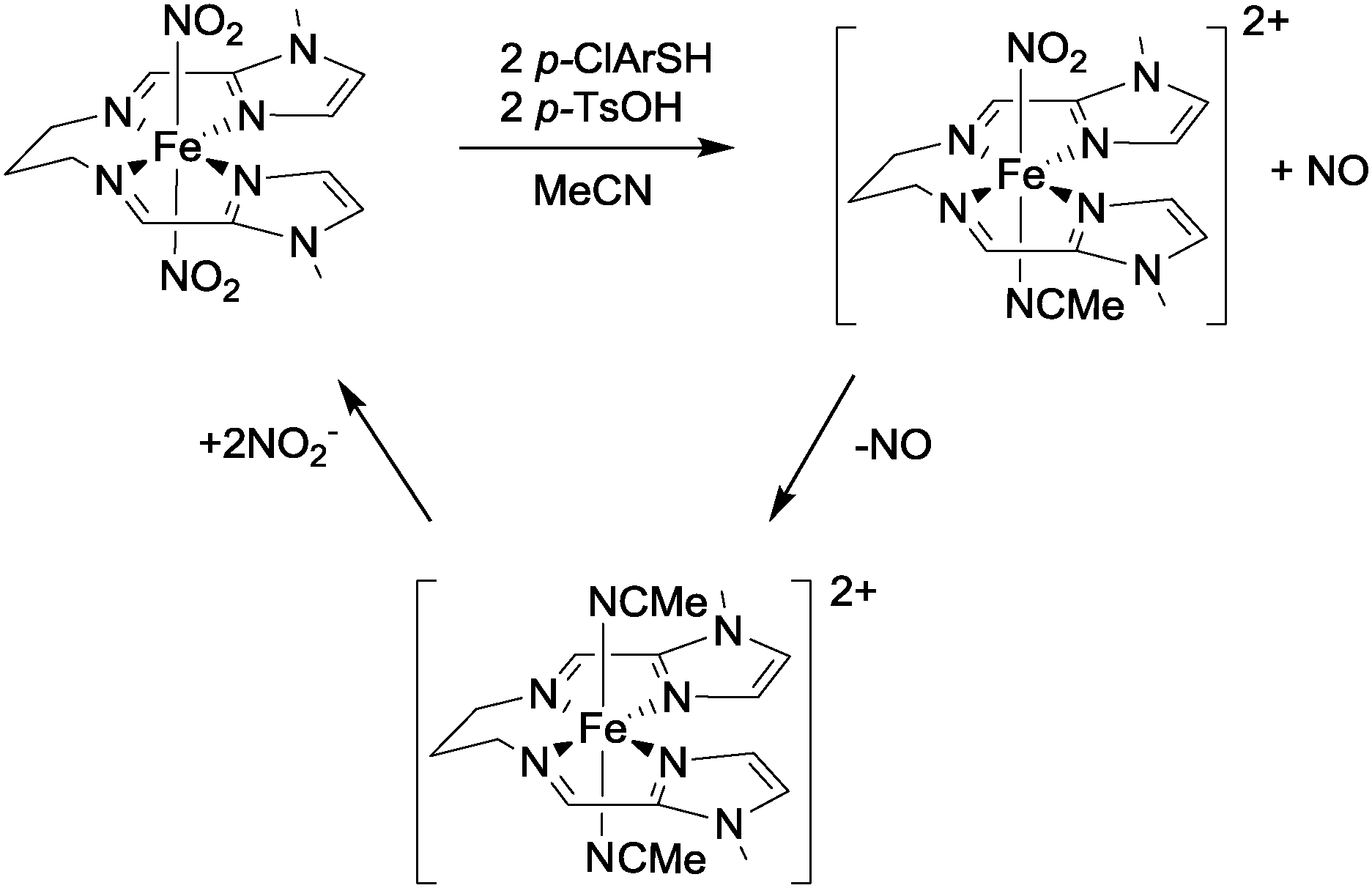 | ||
| Fig. 7 The structure of complex [Fe(LN4Im)(NO2)2] and details of its reaction with nitrite in the presence of p-chlorobenzenethiol and p-toluene sulfonic acid as reported by Harrop.24 | ||
Iron phthalocyanine complexes also display interesting activity with regard to nitrite reduction. Kudrik and van Eldik have studied the reactions of Fe(III) tetrasulfophthalocyanine with nitrite in the presence of both dithionite and sulfoxylate.25 With dithionite, NO was the dominant product observed (and the rate of reaction increased markedly when the pH was below 8), but when sulfoxylate was used as the reductant the main product was ammonia. This difference in reactivity was attributed to the different structures of the intermediate complexes formed during reduction by these two reagents: nitrite reduction by sulfoxylate (the stronger reducing agent) proceeds via an intermediate in which nitrite is coordinated to a highly reduced Fe(I) centre through nitrogen, whereas nitrite reduction by dithionite occurs through an intermediate in which an η1-ONO nitrite coordinates to an Fe(II) centre. As expected, the stronger reductant then leads to a greater degree of reduction of the nitrite – in this case the 6-electron reduction to NH3. Meanwhile, the less strong reducing agent is only able to effect the loss of one of the oxygen atoms of the nitrite (as water), leading to NO as the reduced product.
As a final example of the use of mono-nuclear iron complexes in the reduction of nitrite, we shall consider the system recently reported by Fout in which the role of hydrogen bonding in nitrite reduction is explicitly addressed.26 The Fe(II) complex [N(afaCy)3Fe(OTf)]+ (see Fig. 8) was observed to react cleanly with nitrite to yield the iron(III)-oxo species, [N(afaCy)3Fe(O)]+ and NO gas. The presence of a single hydrogen bond between the ligand and nitrite causes preferential binding of the nitrite to the metal centre in an η1-ONO fashion, which then in turn leads to the observed products of reduction: NO gas and an Fe(III)-oxo species that has retained the Fe–O bond from the η1-ONO-bound nitrite. Given the prominent role of hydrogen bonding in the natural nitrite reduction systems,1 investigation of the effects of hydrogen bonding in artificial systems is an area that offers great promise for altering binding orientations and hence product selectivity.
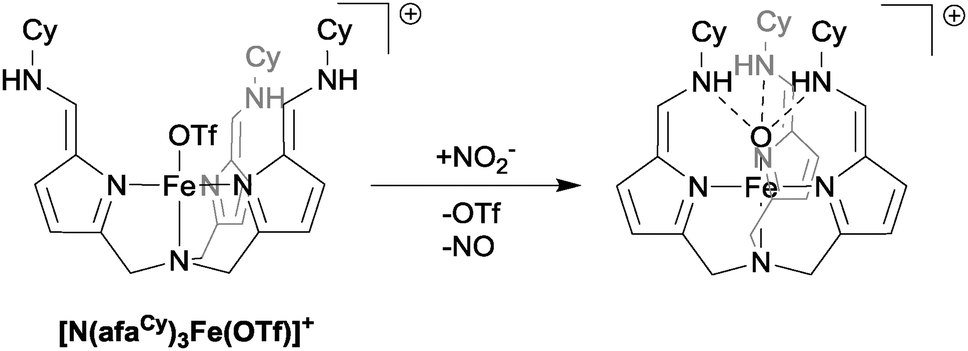 | ||
| Fig. 8 The structure of complex [N(afaCy)3Fe(OTf)]+ and its reaction with nitrite to generate NO. | ||
3.3 Nitrite reduction using other metal–ligand systems
There are a few other notable examples of metal–ligand coordination complexes that can reduce nitrite. Meyer has reported the reduction of nitrite and NO bound to metal centres to ammonia, using complexes of the form [M(tpy)(bipy)NOx]n+, where M = Ru or Os, tpy = terpyridine and bipy = bipyridine.27 For these conversions, a series of one-electron reductions were proposed, the first two of which were found to be pH-independent. The first of these processes was again postulated to be a one-electron transfer to a π* orbital of largely NO-character (see similar arguments advanced for the [FeIII(H2O)(TPPS)]3− system in Section 3.2 and ref. 20 and 21). A pH-dependency was seen for the third 1-electron reduction, and (as the terpyridine and bipyridine ligands should not undergo protonation when coordinated to the metal centres) protonation is likely to occur on the bound NO ligand. Hence a reaction scheme showing the intermediates of the first three reductions could be proposed, on the basis of the above and the fact that some related complexes containing a bound NHO ligand were known. Thus (taking the example of Ru):| [Ru(tpy)(bipy)NO]3+ + e− → [Ru(tpy)(bipy)NO]2+ |
| [Ru(tpy)(bipy)NO]2+ + e− → [Ru(tpy)(bipy)NO]+ |
| [Ru(tpy)(bipy)NO]+ + H+ → [Ru(tpy)(bipy)NHO]2+ |
| [Ru(tpy)(bipy)NHO]2+ + H+ + e− → [Ru(tpy)(bipy)N]2+ + H2O. |
Bimetallic systems have emerged as promising platforms for nitrite reduction. Uyeda and Peters have demonstrated selective conversion of NO2− to N2O using a heterobimetallic Co–Mg complex supported by diamine–dioxime ligands (Fig. 9).28 On the basis of data procured from cyclic voltammetry and electrolysis experiments, Uyeda and Peters devised a plausible electrocatalytic cycle for the reduction of nitrite based on the pre-catalyst depicted in Fig. 9.
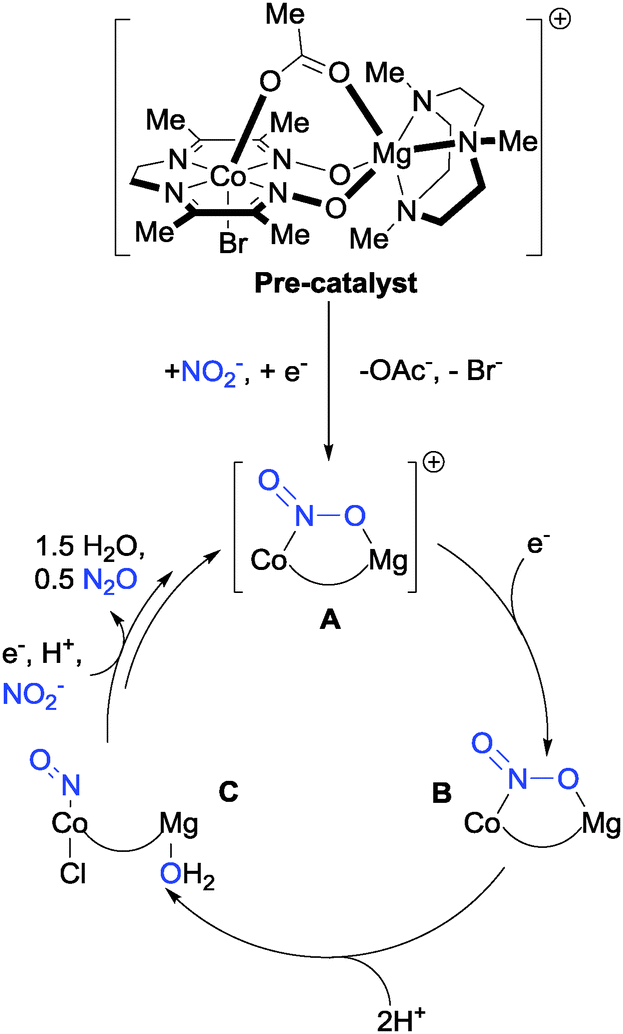 | ||
| Fig. 9 The mechanism of N2O production from nitrite with Uyeda and Peters' bimetallic Co–Mg catalyst.28 | ||
The proposed cycle begins with reduction of the pre-catalyst by 1 electron, which is followed by the loss of bromide and displacement of the OAc− ligand by nitrite to give the cationic complex A. Single electron reduction of complex A gives complex B, then subsequent protonation with two equivalents of acid yields nitrosyl complex C which, after further reduction and protonation steps, eliminates nitrous oxide and leaves a vacant site on the bimetallic catalyst to which NO2− can bind, regenerating complex A. Incorporation of the redox-innocent, Lewis-acidic Mg2+ centre into the framework was found to increase the stability of the Co–NO2− adduct sufficiently under acidic conditions that addition of the acid [Et3NH][BPh4] to complex B, did not produce hydrogen (as normally observed with such Co complexes in the absence of Mg), but instead led to N2O production via complex C. Although further mechanistic investigation is required to validate this proposed electrocatalytic cycle, it is consistent with the reactivity of the various intermediates isolated by the authors. The impressive selectivity for production of N2O over H2, NH2OH or NH3 makes this system attractive for further research and development.
Karlin has also reported a bimetallic catalyst for nitrite reduction, in this case a system that combines iron porphyrins and tripodal copper complexes to mediate the reversible interconversion of nitrite and NO.29 Upon addition of an Fe(II)–porphyrin to the Cu(II)–nitrite complex [(tmpa)CuII(NO2)]+, the (FeII)–haem-nitrosyl adduct was obtained, along with the oxygen-bridged [(porphyrin)FeIII–O–CuII(tmpa)]+ complex (Fig. 10). From this, the authors deduced that the haem played the role of the reductant whilst the Cu(II) ion provided a Lewis acid interaction (cf. Uyeda and Peters, ref. 28), facilitating N–O bond cleavage in nitrite and stabilising the oxo anion in the heterobimetallic oxygen-bridged product. Importantly, the authors were able to demonstrate that the reverse process (in this case the addition of two equivalents of NO to [(porphyrin)FeIII–O–CuII(tmpa)]+ to generate the (FeII)–haem-nitrosyl adduct and the [(tmpa)CuII(NO2)]+ starting material) was also possible, capturing some of the function of the haema3/CuB active site of cytochrome c oxidase, which is able to reduce nitrite to NO reversibly.
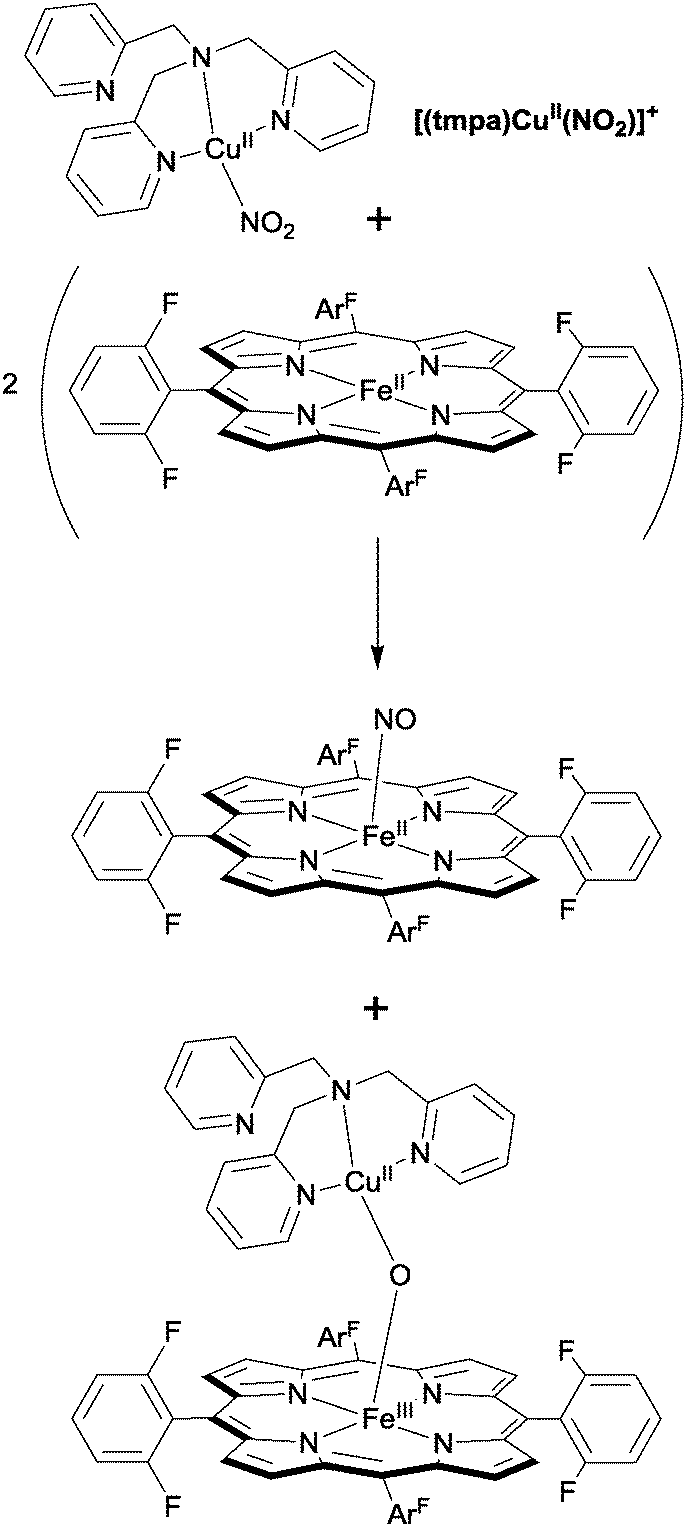 | ||
| Fig. 10 Nitrite reduction to NO using Karlin's bimetallic haem–Cu tripod system.29 | ||
4. Nitric oxide conversions with artificial metal–ligand coordination complexes
The reactions of nitric oxide with transition metal complexes were comprehensively reviewed by Ford in 2002.30 This section will look mostly at examples of NO reduction with metal–ligand coordination complexes since that time, concentrating largely on complexes with aromatic heterocyclic ligands. Readers interested in NO reduction to N2O using somewhat simpler metal–ligand complexes will find a useful overview of these in the first sections of ref. 31.Onishi and co-workers demonstrated N–N coupling of two nitrosyl ligands on bimetallic ruthenium clusters to produce N2O in 2007.32 Dimerisation of the ruthenium–nitrosyl complex TpRuCl2(NO) (complex 7, where Tp = hydrotris(pyrazolyl)borate; see Fig. 11) was achieved by refluxing complex 7 with stoichiometric amounts of pyrazole and excess Et3N in CH2Cl2 to give bimetallic complex 8 in 29% yield. Oxidation of 8 by AgBF4 in acetonitrile resulted in cleavage of the N–N bond to give dinitrosyl complex 9 ([{TpRu(NO)}2(μ-Cl)(μ-pyrazolyl)](BF4)2) in 56% yield. Cleavage of the N(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–N(O) bond was confirmed by X-ray crystallography of complex 9 which revealed a N⋯N distance of 3.006 Å. Comparison of the N–O bond distances in complex 9 (1.169 Å and 1.131 Å) shows significant shortening when compared to the N–O bond lengths of complex 8, indicating an increase in bond order as would be expected after N–N bond scission. Interestingly, when treated with zinc metal, complex 9 reductively reformed the N–N bond to afford the starting material in 70% yield. Treatment of complex 8 with HBF4·OEt2 in CH2Cl2 yielded bimetallic complex 10 ([(TpRu)2(μ-Cl)(μ-pyrazolyl)(μ-O)]; 21% yield), complex 9 (43% yield) and N2O gas (25% yield based on complex 8). Hence this system displays a range of interesting and reversible N–N bond formation and cleavage reactions.
O)–N(O) bond was confirmed by X-ray crystallography of complex 9 which revealed a N⋯N distance of 3.006 Å. Comparison of the N–O bond distances in complex 9 (1.169 Å and 1.131 Å) shows significant shortening when compared to the N–O bond lengths of complex 8, indicating an increase in bond order as would be expected after N–N bond scission. Interestingly, when treated with zinc metal, complex 9 reductively reformed the N–N bond to afford the starting material in 70% yield. Treatment of complex 8 with HBF4·OEt2 in CH2Cl2 yielded bimetallic complex 10 ([(TpRu)2(μ-Cl)(μ-pyrazolyl)(μ-O)]; 21% yield), complex 9 (43% yield) and N2O gas (25% yield based on complex 8). Hence this system displays a range of interesting and reversible N–N bond formation and cleavage reactions.
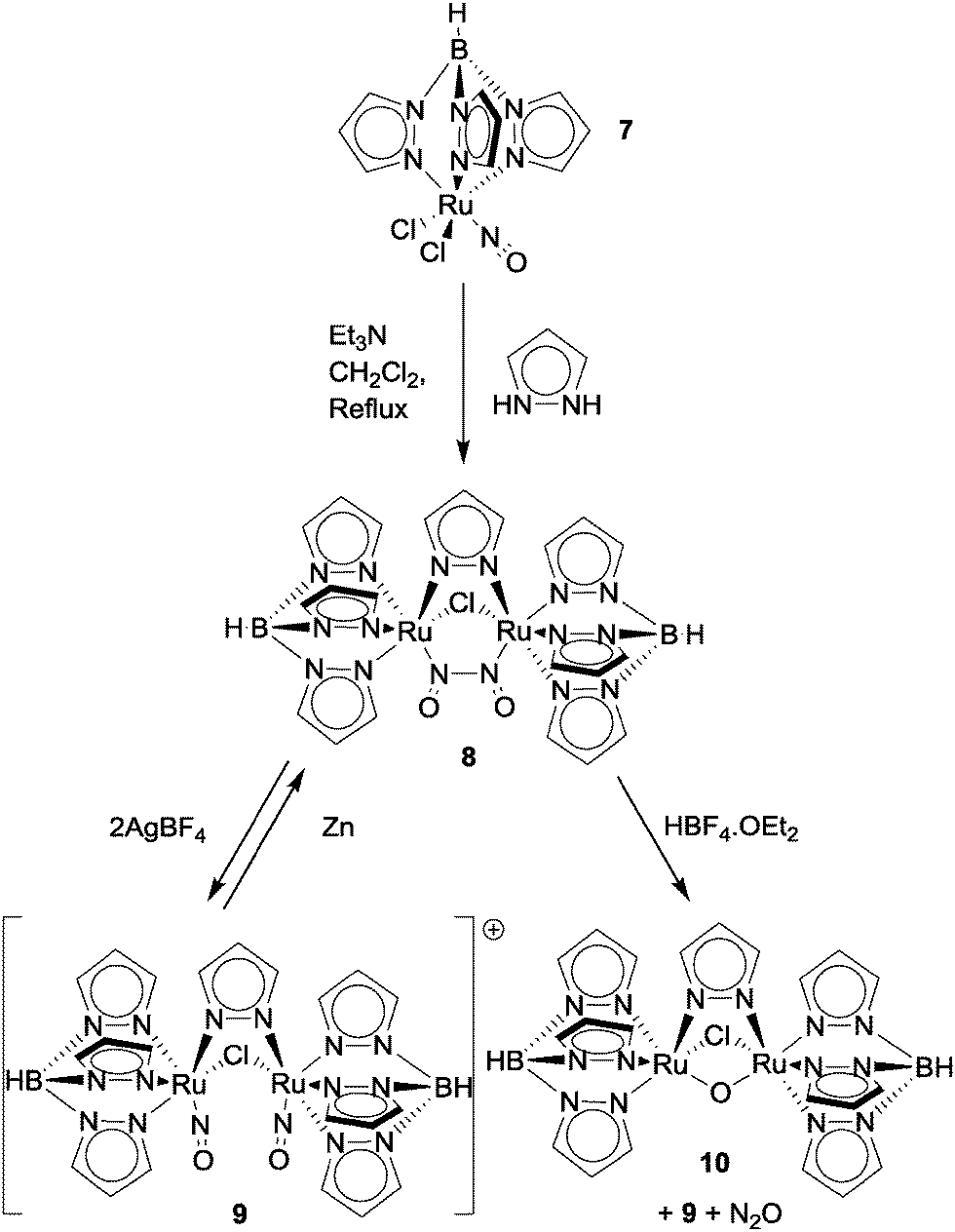 | ||
| Fig. 11 Onishi's di-ruthenium platform for NO reduction to N2O.32 | ||
Collman and co-workers reported the first functional synthetic nitric oxide reductase model complex in 2008, by decorating a porphyrin unit with imidazole appendages, such that metallation with iron gave rise to a complex containing both haem and non-haem iron centres, highly reminiscent of the active site of nitric oxide reductase (see Fig. 4c).33 When both irons were present as Fe(II), addition of NO gas led to the production of N2O. Detailed electronic, EPR and infra-red analysis allowed aspects of the mechanism of action to be determined. It was found that one molecule of NO binds to each Fe(II) centre (as opposed to both molecules binding to only one iron), and that production of N2O was achieved with simultaneous oxidation of both metal centres to the Fe(III) state. Mixed-valence complexes (where the haem iron was in the 3+ oxidation state and the non-haem iron in the 2+ state) were not competent for N2O production from NO.
A similar approach was taken by Karlin and co-workers, who reported NO reduction by a hetero-bimetallic complex containing tethered haem and copper centres analogous to those shown in Fig. 10.34 The activity of this system was investigated starting from the complex containing a haem iron, but no copper centre. Upon addition of excess NO to this mono-metallic complex, the dinitrosyl {Fe(NO)2} complex formed, wherein NO was bound to the iron centre both above and below the plane of the porphyrin ring, as confirmed by UV-vis and EPR spectroscopies. Addition of [CuI(MeCN)4]+ and 2 equivalents of acid to this dinitrosyl complex resulted in the production of nitrous oxide in >80% yield, with both metal centres undergoing oxidation in the process (Fe(II) → Fe(III) and Cu(I) → Cu(II)). In the absence of copper, addition of acid to the dinitrosyl complex yielded only the mono-nitrosyl adduct. On the other hand, addition of [CuI(MeCN)4]+ but no acid resulted in a haem–copper complex in which the haem iron remained bound to only one NO molecule (and also remained in the Fe(II) state), whilst the copper centre was oxidised to Cu(II) and found to be coordinated to nitrite. Meanwhile, N2O was evolved. The authors attributed the presence of both NO2 and N2O in this instance to disproportionation of the NO released from the {Fe(NO)2} complex via the process: 3NO → NO2 + N2O. The yield of N2O by this disproportionation reaction was significantly below that expected on the basis of eqn (5), which describes the selective conversion of NO to N2O as the sole nitrogen-containing product. Hence the authors reasoned that a haem centre, a copper ion and two equivalents of both acid and NO were all necessary in order to reduce NO to N2O in the same fashion as the cytochrome c oxidases (shown in eqn (5)).
Around the same time as this article by Karlin and co-workers, Xu et al. reported the production of N2O by protonation of a system comprising two haem units bridged by an N2O2 moiety (generated by the treatment of 2 equivalents of the haem precursor with hyponitrous acid, H2N2O2).35 Although the precise mechanism of protonation has yet to be determined, this study constituted the first iron–porphyrin hyponitrite complex ever isolated and established that the binding to iron in such complexes is through oxygen (as Fe–O–N–N–O–Fe), in contrast to the complexes described by Collman and Karlin, where NO binding to the metal centre was proposed to be through nitrogen. Thus, it seems that there may be more than one generic mechanism by which NO can be reduced to N2O using bimetallic haem-based complexes.
Entirely non-haem di-iron complexes have also been reported as being competent for NO reduction, in an analogous fashion to the flavodiiron nitric oxide reductases (FNORs). In 2013, Lehnert and co-workers described the rapid and quantitative production of N2O from NO and complex 11 (Fig. 12).36 NO was found to bind to both the iron centres in this complex to give a pseudo-octahedral coordination geometry at each iron. No N2O production was detected, but two-electron reduction of this complex by either chemical or electrochemical means resulted in fast and clean production of N2O in almost 100% yield. Monomeric complexes analogous to complex 11 but containing only a single iron centre did not mediate N2O production. Surprisingly (given eqn (5)), N2O formation by the di-iron complex was found not to be proton-dependent. The authors proposed that NO reduction in the absence of protons might proceed via the formation of a μ-oxo bridged di-iron unit, according to the equation: 2FeII–NO + 2e− → N2O + FeII–O–FeII and some spectroscopic evidence was found supporting this hypothesis.
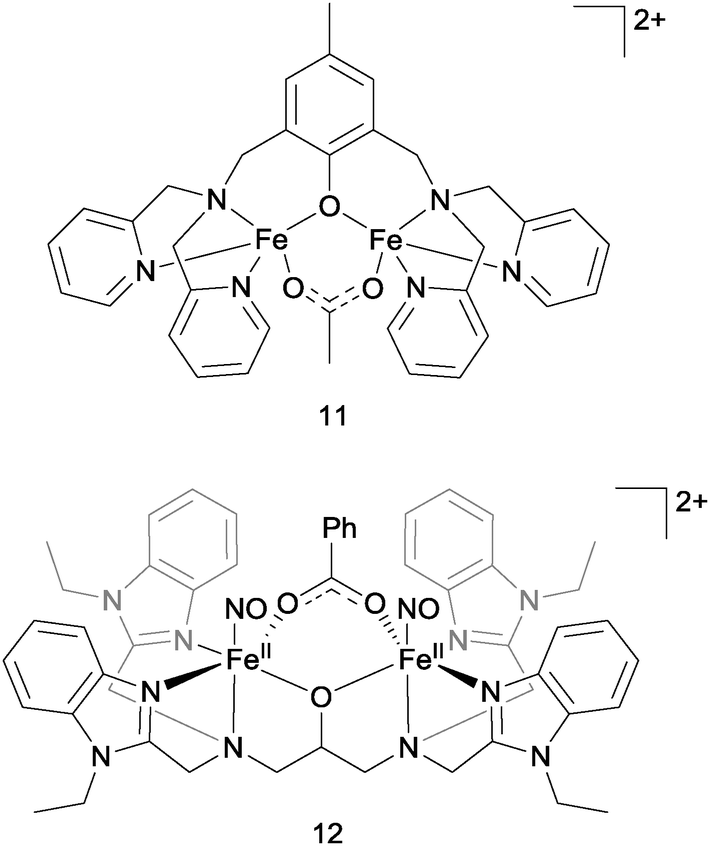 | ||
| Fig. 12 The structures of non-haem di-iron NO-reducing complexes 11 and 12. | ||
Jiang et al. recently reported the photo-driven production of N2O from a non-haem iron–nitrosyl dimer that also bears similarities to the flavodiiron nitric oxide reductases.37 In this case, irradiation of complex 12 (Fig. 12) with white light at both room temperature and 15 K produced N2O (up to 90% yield at 15 K). By detailed spectroscopic analysis of the low-temperature reaction, the authors established that irradiation of complex 12 leads to dissociation of at least one of the coordinated NO ligands. This dissociated NO might then bind in a side-on mode to a still coordinated NO ligand to produce a transient hyponitrite complex, which in turn then decomposes to liberate N2O upon further irradiation.
The final example of NO reduction that we shall consider concerns the production of N2O and water from the reaction of [Ni(NO)(bipy)2][PF6] (bipy = 2,2-bipyridine) with acetylacetone (H-acac).38 This reaction was observed to give rise to ligand re-arrangements at nickel, producing [Ni(bipy)3][PF6]2 and the complex [Ni(bipy)N2O2]. This latter complex (which could be isolated and characterised crystallographically) exhibited a square-planar Ni(II) centre coordinated to one bipy ligand and one cis-[N2O2]2− (hyponitrite) ligand. This hyponitrite was symmetrically coordinated to the Ni centre through its two oxygen atoms (both Ni–O bonds were found to be 1.800(1) Å), whilst the N–N bond length (1.240(3) Å) was consistent with a double bond. The hyponitrite complex then reacted further with H-acac, giving the complex [Ni(acac)2(bipy)] with concomitant N–O bond cleavage and N2O production. This work was the first well-characterised report of NO reduction mediated by nickel.
To conclude this section, we shall examine an otherwise unprecedented reaction pathway for the oxidation of NO to nitrite, as reported by Ibrahim and Pickett.39 Starting from the complex [Mo(NO)Cl(dttd)] (dttd = 1,2-bis(2-mercaptophenylthio)ethane, see Fig. 13), electroreduction by four electrons at a Hg-pool electrode in THF gave the highly reduced species [Mo(NO)(1,2-C6H4S2)2]3−, along with the liberation of chloride and ethylene (from the dttd ligand). Upon exposure to dioxygen, [Mo(NO)(1,2-C6H4S2)2]3− reacted rapidly to generate [MoO(1,2-C6H4S2)2]2− nearly quantitatively, with the production of free nitrite in 70% yield. A key step in this process is the first step of the reaction between [Mo(NO)(1,2-C6H4S2)2]3− and O2, which was suggested to be a single electron reduction of oxygen to give superoxide (O2˙−) and the paramagnetic species [Mo(NO)(1,2-C6H4S2)2]2−. These two species were then proposed to undergo radical–radical coupling to give nitrite and [MoO(1,2-C6H4S2)2]2−, possibly via the O-bound nitrato-complex [Mo(ONO2)(1,2-C6H4S2)2]3−. Intriguingly, six-electron reduction of [Mo(NO)Cl(dttd)] in the presence of phenol (as a proton source) leads directly to the production of [MoO(1,2-C6H4S2)2]2−, again with the liberation of chloride and ethylene but with ammonia as the main nitrogen-containing by-product (up to 55% yield).
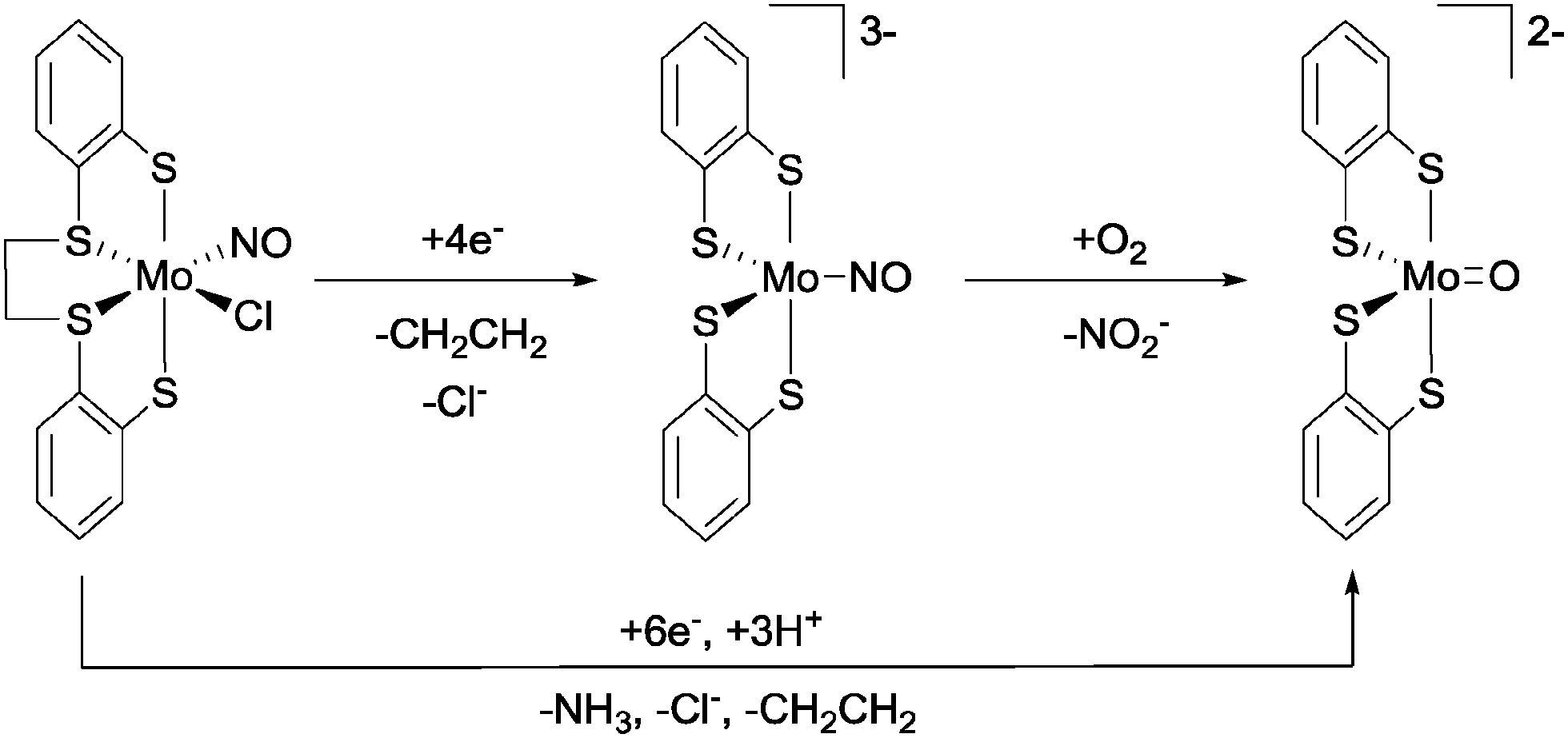 | ||
| Fig. 13 NO oxidation to nitrite and NO reduction to ammonia using the Mo platform described by Ibrahim and Pickett.39 | ||
5. Nitrous oxide reduction with artificial metal–ligand coordination complexes
The 2-electron, 2-proton reduction of nitrous oxide to nitrogen is mediated in nature by the copper-containing metallo-enzyme nitrous oxide reductase during the last step of bacterial denitrification according to the equation:1| N2O + 2H+ + 2e− → N2 + H2O | (6) |
| {Cu3S2}2+ → {Cu2S2}+ + {Cu}+ |
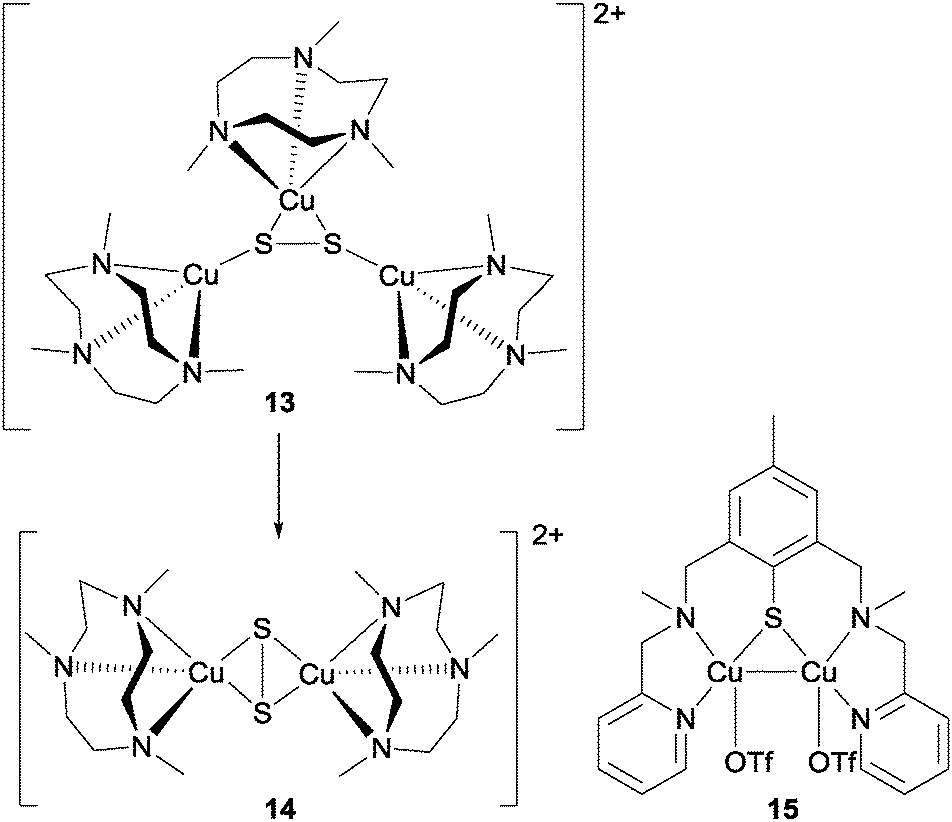 | ||
| Fig. 14 The structures of the N2O reduction mediators reported by Bar-Nahum et al.40 and Esmieu et al.41 | ||
This di-copper cluster was then suggested to reduce N2O via a transition state in which N2O bridges between the copper centres by way of its oxygen, although the authors noted that such a mechanism was not analogous to that believed to operate in the enzyme.
Very recently, Torelli and co-workers published an account of an alternative mixed-valence di-copper complex (15, Fig. 14) containing a {Cu2S} core that is competent for N2O reduction to N2 in acetone.41 The proposed mechanism calls for exchange of one of the labile trifluoromethanesulfonate ligands on the complex for N2O, leading to N2 evolution with the formation of a bridging Cu–O–Cu unit (where this oxygen is postulated to originate from N2O). Meanwhile, a second molecule of the di-copper complex that has undergone exchange of one of the labile trifluoromethanesulfonate ligands on the complex for N2O undergoes displacement of this N2O ligand by water, and then reacts with the Cu–O–Cu complex to generate the Cu–OH–Cu complex, which was found to be the final copper-containing product of the reaction.
Given nitrous oxide's potency as a greenhouse gas and ozone-depleter, catalysts able to mediate the selective removal of N2O from exhaust gas streams by conversion to N2 are attractive targets. Hence we can expect many more functional N2O-reducing platforms to be reported in the coming years.
6. Nitrate reduction with artificial metal–ligand coordination complexes
6.1 Nitrate reduction to nitrite and vice versa
Structural models of the oxotransferase enzymes (of which both the assimilatory and the dissimilatory enzymes are members) are numerous,42,43 but those competent for nitrate reduction are comparatively rare. In this subsection, we shall examine coordination complexes that are functional for nitrate reduction to nitrite or nitrite oxidation to nitrate (see eqn (4)).Seminal work in this area has been conducted by Holm and co-workers, who reported the first well-characterised reduction of NO3− to NO2− by molybdenum-mediated oxygen atom transfer in 1989.44 The metal–ligand complex in question was synthesised before the structure of any molybdoenzyme had been determined and was based on an S,N,S pincer. Hence it does not bear close structural analogy to the dithiolene-based active sites in the natural nitrate reductases. In later work, however, Holm modelled the active site structure of the dissimilatory nitrate reductases more closely and was able to synthesise a range of metal–ligand coordination complexes capable of oxygen atom transfer reactions (see Fig. 15).45
 | ||
| Fig. 15 Mechanism of nitrate reduction to nitrite with Holm's tungsten-centred catalysts as reported in ref. 45. | ||
On the basis of kinetic measurements, Holm proposed an associative pathway for nitrate reduction by the tungsten-based complexes reported in ref. 45 (the Mo-based complexes were insufficiently stable to be interrogated in such detail), as summarised in Fig. 15. The mechanism built on Holm's earlier work44 by suggesting that nitrate reduction by direct oxo transfer to the metal centre (with the metal undergoing WIV to WVI oxidation in the process) is a viable route to nitrate reduction in these systems.
A similar approach was taken by Sarkar and co-workers, who reported catalytic nitrate reduction to nitrite using Mo-centred catalysts bearing 1,2-dicyanoethylenedithiolate (mnt2−), SPh and PPh3 ligands.46,47 The first step in the proposed catalytic cycle (Fig. 16) is dissociation of the triphenylphosphine ligand, giving a penta-coordinated intermediate species able to undergo reaction with nitrate to generate nitrite, along with oxygen atom transfer to the molybdenum centre and concomitant MoIV to MoVI oxidation (cf. Holm's proposed mechanism for nitrate reduction at W centres in Fig. 15). The free PPh3 previously released then reacts with the molybdenum-oxo species to give OPPh3 and re-generate the active penta-coordinated MoIV complex. This complex can either react with more nitrate, or else re-combine with free PPh3 to form the catalytically-inert starting complex. Turn-over numbers of 50 mmol−1 s−1 (based on the amount of nitrite, OPPh3 and PPh3 recovered) were obtained.46
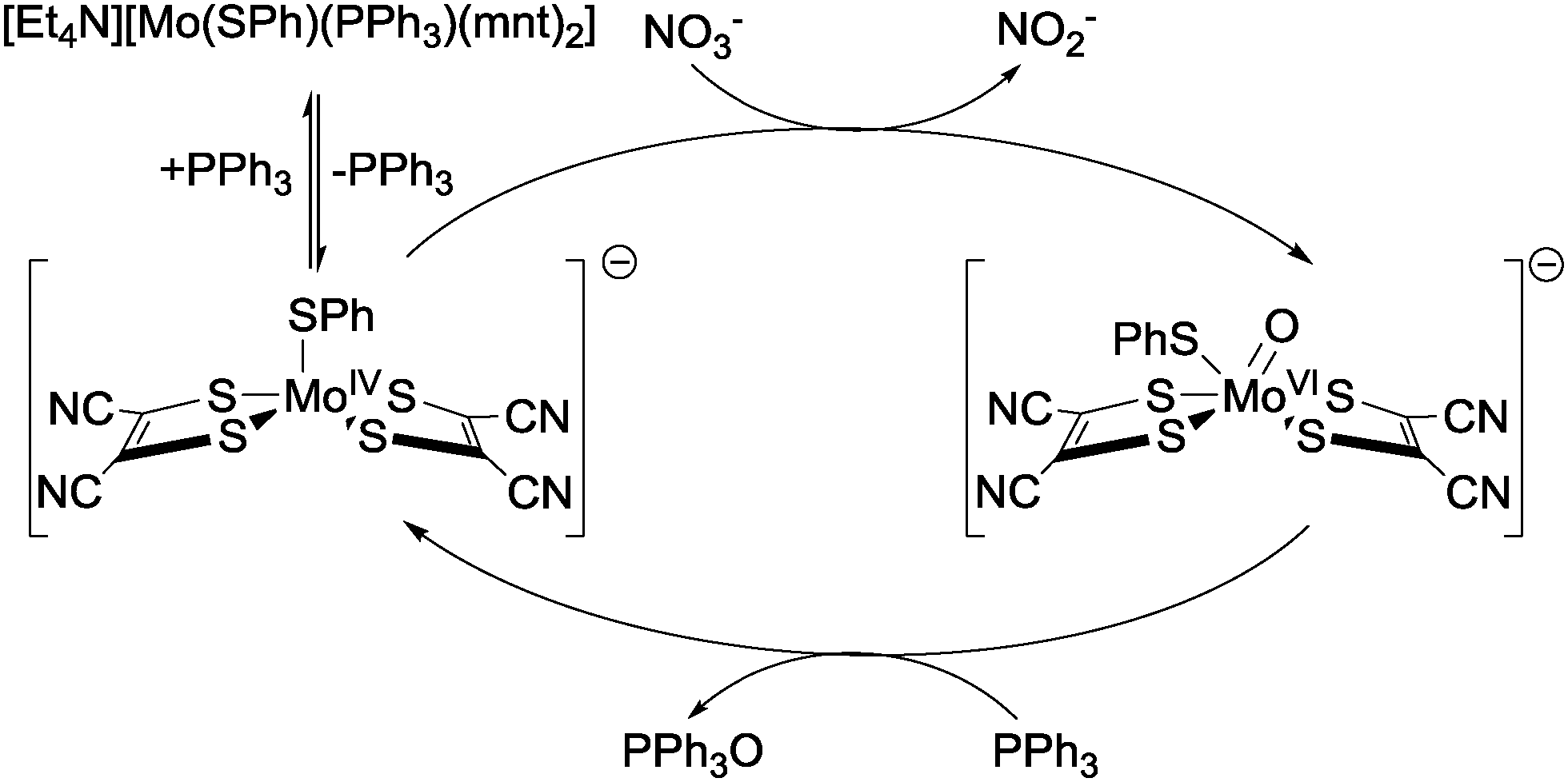 | ||
| Fig. 16 The catalytic cycle for the reduction of nitrate to nitrite using [Et4N][Mo(SPh)(mnt)2] and a triphenyl phosphine reducing agent as reported by Sarkar and co-workers.46,47 | ||
An example of the oxidation of nitrite to nitrate is provided by Man et al., who employed a trans-dioxoruthenium(VI) complex to mediate oxygen atom transfer from the metal-oxo to NO2−.48 Reaction of the trans-dioxoruthenium(VI) complex with nitrite was observed to lead to the reduced trans-{RuIV(ONO2)}+ species (where the nitrate ligand was bound to Ru through oxygen), as determined by spectrophotometric, mass spectroscopic and crystallographic analyses. Nitrate was then released by this RuIV complex upon reaction with water in stoichiometric yield after around 2 hours. 18O labelling studies showed that the oxygen that was transferred to the nitrite originated from the trans-dioxoruthenium(VI) complex via a reversible oxygen atom transfer reaction, rather than by an outer-sphere reaction involving solvent water.
6.2 Nitrate reduction to other nitrogen oxides
Nitrate reduction with metal–ligand coordination complexes to species other than nitrite has also been reported. An interesting dinitrosyl–iron complex which mediates the conversion of nitrate to first NO2 and then nitric oxide was reported in 2013 by Tsai et al.49 The thermally-unstable iron-centred complex 16 (Fig. 17) was formed by reaction between [(OAc)2Fe(NO)2]− and 2 equivalents of nitric acid and was found to react with bis-(diethylthiocarbamoyl)disulfide ((S2CNEt2)2) to form complex 18 (via intermediate 17) with release of N2O4. This N2O4 exists in equilibrium with NO2, and one equivalent of the NO2 thus formed then oxidises complex 18 to give the {Fe(NO2)(NO)} adduct 19 (Fig. 17). Reaction of complex 19 with dimethyl sulfide then re-generates complex 18, and also produces NO and dimethyl sulfoxide.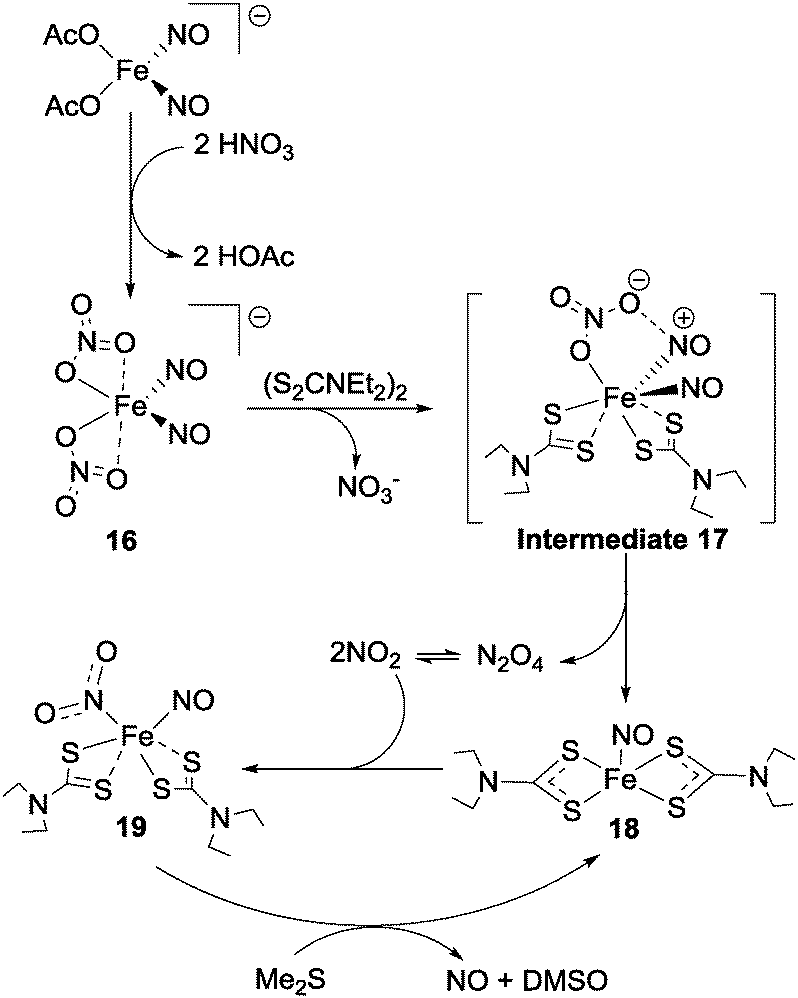 | ||
| Fig. 17 Nitrate reduction to NO2 and nitric oxide as reported by Tsai et al.49 | ||
The electrochemically-catalysed reduction of nitrate to hydroxylamine using metal–cyclams (cyclam = 1,4,8,11-tetra-azacyclotetradecane) was investigated by several groups in the 1980s and 1990s, as detailed in ref. 3. Whether the metal cyclam complexes were used directly in aqueous solution or absorbed on the electrode surface, the reduction reactions mediated by the metal–ligand complexes were six-electron processes: NO3− + 5H2O + 6e− → NH2OH + 7OH−. Depending on the underlying electrode material, hydroxylamine was sometimes the final product (e.g. when the cathode was Hg), whilst the use of other cathode materials (such as Ag, Cu or Pb) caused the hydroxylamine produced to be further reduced to ammonia at the potentials used.
As a final example, Kudrik et al. have used cobalt–phthalocyanines to mediate the conversion of nitrate to N2 and N2O by dithionite.50 Cobalt–phthalocyanine–nitrite complexes were identified as intermediates in these nitrate reduction reactions. However, when nitrite itself was used as the substrate, the final reaction product was found to be ammonia, and not N2 or N2O. These apparently contradictory results were rationalised by the authors by considering the different possible binding modes of nitrite to the cobalt centre during reaction (see Fig. 5 and Section 3.1). In all cases, the proposed mechanism of reaction called for the reduction of the initial Co(II)–phthalocyanine complex to the Co(I) oxidation state by dithionite, followed by coordination to and reaction with nitrate or nitrite. When nitrite was the substrate, the authors suggested that coordination was through nitrogen (η1-NO2) and that reduction and protonation then led to the Co(II)–NO adduct (bound through nitrogen), which then in turn gave ammonia upon further reduction and protonation. On the other hand, nitrate coordinated to the Co(I)–phthalocyanine complex was suggested to be through oxygen (η1-ONO), which upon reduction and protonation of the nitrogen oxide then gave the Co(II)–ON adduct (where the bond to cobalt was though oxygen). Further reduction of this intermediate was then postulated to give ON−, which dimerised to give N2O, which could in turn be further reduced to dinitrogen. Differences in the electrophilicity of nitrate and nitrite were held to account for the different binding preferences of these two substrates. This work has obvious parallels with the same authors' work on nitrite reduction with iron–phthalocyanine complexes (see Section 3.2)25 and serves to emphasise the importance that nitrite binding mode can have in determining the products of reduction.
7. Conclusions and outlook
In this Tutorial Review, we have presented an overview of the metal–ligand coordination complexes able to mediate the inter-conversions of the various nitrogen oxides. Many of these complexes bear similarities to the active sites of enzymes that perform the same transformations. Whilst intended in no way to be a comprehensive review of the topic, we believe that the examples presented herein represent a sufficiently broad cross-section of the recent research in this fascinating field to enable the reader to gain a ready understanding of the status of, and remaining challenges in, this area.The nitrogen oxides are a class of molecules central to biology, geochemistry and modern life. Nitrate fertilisers are essential for feeding the world's growing population, and their misuse constitutes a major environmental hazard. Species such as N2O and NO2 are produced by air-breathing internal combustion engines and are implicated in atmospheric pollution and the greenhouse effect. NO is an important signalling molecule in the body and a potent vasodilator. Converting between the oxides of nitrogen is thus an area of tremendous, and increasing, importance. The ability to selectively convert from one oxide to another would allow, for instance, the safe removal of excess nitrate from groundwater by conversion to nitrogen gas, the in situ generation of NO from nitrate salts in the bloodstream, or the efficient removal of N2O from exhaust gases. Nature possesses enzymes that can mediate these transformations with high specificity and high efficiency, but synthetic systems lag behind in both these respects. Some of the publications reviewed in this tutorial article perhaps suggest how synthetic catalysts could be improved in order to capture some of Nature's reactivity. For example, the explicit inclusion of hydrogen bonding by Fout26 shows how substrate binding and orientation can be tuned by weak non-covalent interactions, just as in the active sites of the corresponding enzymes.1 Kudrik and van Eldik's work shows how different binding modes can then lead to very different products during reduction.25,50 Meanwhile, Kujime et al. and Woollard-Shore et al. have shown elegantly how subtle variations in electronics and sterics can have big effects on catalysis.14,16 Hence by applying the principles of metal–ligand coordination chemistry, we can hope to produce more selective and efficient catalysts for the conversions of the nitrogen oxides. We hope that this review might in some small way inspire a new generation of scientists to strive to develop improved catalysts for these vital and challenging processes.
Acknowledgements
MDS acknowledges the University of Glasgow for a Kelvin Smith Research Fellowship. We thank Dr Stephen Goldup (University of Southampton) and Prof. Leroy Cronin (University of Glasgow) for useful discussions.References
- See: L. B. Maia and J. J. G. Moura, Chem. Rev., 2014, 114, 5273 CrossRef CAS PubMed and references therein.
- See: I. M. Wasser, S. de Vries, P. Moënne-Loccoz, I. Schröder and K. D. Karlin, Chem. Rev., 2002, 102, 1201 CrossRef CAS PubMed and references therein.
- See: V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209 CrossRef CAS PubMed and references therein.
- O. Einsle, A. Messerschmidt, R. Huber, P. M. H. Kroneck and F. Neese, J. Am. Chem. Soc., 2002, 124, 11737 CrossRef CAS PubMed.
- D. Bykov and F. Neese, J. Biol. Inorg. Chem., 2011, 16, 417 CrossRef CAS PubMed.
- See, for example: E. I. Tocheva, F. I. Rosell, A. G. Mauk and M. E. P. Murphy, Science, 2004, 304, 867 CrossRef CAS PubMed.
- S. Ghosh, A. Dey, Y. Sun, C. P. Scholes and E. I. Solomon, J. Am. Chem. Soc., 2009, 131, 277 CrossRef CAS PubMed.
- H.-K. Li, C. Temple, K. V. Rajagopalan and H. Schindelin, J. Am. Chem. Soc., 2000, 122, 7673 CrossRef CAS.
- T. Hino, Y. Matsumoto, S. Nagano, H. Sugimoto, Y. Fukumori, T. Murata, S. Iwata and Y. Shiro, Science, 2010, 330, 1666 CrossRef CAS PubMed.
- J. D. Caranto, A. Weitz, M. P. Hendrich and D. M. Kurtz, Jr., J. Am. Chem. Soc., 2014, 136, 7981 CrossRef CAS PubMed.
- B. A. Averill, Chem. Rev., 1996, 96, 2951 CrossRef CAS PubMed.
- J. A. Halfen, S. Mahapatra, E. C. Wilkinson, A. J. Gengenbach, V. G. Young, Jr., L. Que, Jr. and W. B. Tolman, J. Am. Chem. Soc., 1996, 118, 763 CrossRef CAS.
- A. C. Merkle and N. Lehnert, Dalton Trans., 2012, 41, 3355 RSC.
- M. Kujime, C. Izumi, M. Tomura, M. Hada and H. Fujii, J. Am. Chem. Soc., 2008, 130, 6088 CrossRef CAS PubMed.
- S. C. N. Hsu, Y.-L. Chang, W.-J. Chuang, H.-Y. Chen, I.-J. Lin, M. Y. Chiang, C.-L. Kao and H.-Y. Chen, Inorg. Chem., 2012, 51, 9297 CrossRef CAS PubMed.
- J. G. Woollard-Shore, J. P. Holland, M. W. Jones and J. R. Dilworth, Dalton Trans., 2010, 39, 1576 RSC.
- R. C. Maji, S. K. Barman, S. Roy, S. K. Chatterjee, F. L. Bowles, M. M. Olmstead and A. K. Patra, Inorg. Chem., 2013, 52, 11084 CrossRef CAS PubMed.
- T. Hiratsu, S. Suzuki and K. Yamaguchi, Chem. Commun., 2005, 4534 RSC.
- C. Orain, A. G. Porras-Gutiérrez, F. Evoung Evoung, C. Charles, N. Cosquer, A. Gomila, F. Conan, Y. Le Mest and N. Le Poul, Electrochem. Commun., 2013, 34, 204 CrossRef CAS PubMed.
- M. H. Barley, K. J. Takeuchi and T. J. Meyer, J. Am. Chem. Soc., 1986, 108, 5876 CrossRef CAS PubMed.
- For a discussion of the various electronic structure alternatives in nitrosyl complexes, see: G. K. Lahiri and W. Kaim, Dalton Trans., 2010, 39, 4471 RSC and references therein.
- Y. Chi, J. Chen and K. Aoki, Inorg. Chem., 2004, 43, 8437 CrossRef CAS PubMed.
- J. L. Heinecke, C. Khin, J. C. M. Pereira, S. A. Suárez, A. V. Iretskii, F. Doctorovich and P. C. Ford, J. Am. Chem. Soc., 2013, 135, 4007 CrossRef CAS PubMed.
- B. C. Sanders, S. M. Hassan and T. C. Harrop, J. Am. Chem. Soc., 2014, 136, 10230 CrossRef CAS PubMed.
- E. V. Kudrik, S. V. Makarov, A. Zahl and R. van Eldik, Inorg. Chem., 2005, 44, 6470 CrossRef CAS PubMed.
- E. M. Matson, Y. J. Park and A. R. Fout, J. Am. Chem. Soc., 2014, 136, 17398 CrossRef CAS PubMed.
- W. R. Murphy, Jr., K. J. Takeuchi and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 5817 CrossRef.
- C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 12023 CrossRef CAS PubMed.
- S. Hematian, M. A. Siegler and K. D. Karlin, J. Am. Chem. Soc., 2012, 134, 18912 CrossRef CAS PubMed.
- P. C. Ford and I. M. Lorkovic, Chem. Rev., 2002, 102, 993 CrossRef CAS PubMed.
- Y. Arikawa and M. Onishi, Coord. Chem. Rev., 2012, 256, 468 CrossRef CAS PubMed.
- Y. Arikawa, T. Asayama, Y. Moriguchi, S. Agari and M. Onishi, J. Am. Chem. Soc., 2007, 129, 14160 CrossRef CAS PubMed.
- J. P. Collman, Y. Yang, A. Dey, R. A. Decréau, S. Ghosh, T. Ohta and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15660 CrossRef CAS PubMed.
- J. Wang, M. P. Schopfer, A. A. N. Sarjeant and K. D. Karlin, J. Am. Chem. Soc., 2009, 131, 450 CrossRef CAS PubMed.
- N. Xu, A. L. O. Campbell, D. R. Powell, J. Khandogin and G. B. Richter-Addo, J. Am. Chem. Soc., 2009, 131, 2460 CrossRef CAS PubMed.
- S. Zheng, T. C. Berto, E. W. Dahl, M. B. Hoffman, A. L. Speelman and N. Lehnert, J. Am. Chem. Soc., 2013, 135, 4902 CrossRef CAS PubMed.
- Y. Jiang, T. Hayashi, H. Matsumura, L. H. Do, A. Majumdar, S. J. Lippard and P. Moënne-Loccoz, J. Am. Chem. Soc., 2014, 136, 12524 CrossRef CAS PubMed.
- A. M. Wright, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 9930 CrossRef CAS PubMed.
- S. K. Ibrahim and C. J. Pickett, J. Chem. Soc., Chem. Commun., 1991, 246 RSC.
- I. Bar-Nahum, A. K. Gupta, S. M. Huber, M. Z. Ertem, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2009, 131, 2812 CrossRef CAS PubMed.
- C. Esmieu, M. Orio, S. Torelli, L. Le Pape, J. Pécaut, C. Lebrun and S. Ménage, Chem. Sci., 2014, 5, 4774 RSC.
- J. H. Enemark, J. J. A. Cooney, J.-J. Wang and R. H. Holm, Chem. Rev., 2004, 104, 1175 CrossRef CAS PubMed.
- H. Sugimoto and H. Tsukube, Chem. Soc. Rev., 2008, 37, 2609 RSC.
- J. A. Craig and R. H. Holm, J. Am. Chem. Soc., 1989, 111, 2111 CrossRef CAS.
- J. Jiang and R. H. Holm, Inorg. Chem., 2005, 44, 1068 CrossRef CAS.
- A. Majumdar, K. Pal and S. Sarkar, J. Am. Chem. Soc., 2006, 128, 4196 CrossRef CAS PubMed.
- A. Majumdar, K. Pal and S. Sarkar, Inorg. Chem., 2008, 47, 3393 CrossRef CAS PubMed.
- W.-L. Man, W. W. Y. Lam, W.-Y. Wong and T.-C. Lau, J. Am. Chem. Soc., 2006, 128, 14669 CrossRef CAS PubMed.
- F.-T. Tsai, Y.-C. Lee, M.-H. Chiang and W.-F. Liaw, Inorg. Chem., 2013, 52, 464 CrossRef CAS PubMed.
- E. V. Kudrik, S. V. Makarov, A. Zahl and R. van Eldik, Inorg. Chem., 2003, 42, 618 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |