
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli-responsive nanogel composites and their application in nanomedicine
Maria
Molina
a,
Mazdak
Asadian-Birjand
a,
Juan
Balach
b,
Julian
Bergueiro
a,
Enrico
Miceli
ac and
Marcelo
Calderón
*ac
aFreie Universität Berlin, Institute of Chemistry and Biochemistry, Takustr. 3, 14195 Berlin, Germany. E-mail: marcelo.calderon@fu-berlin.de; Fax: +49-30-838-459368; Tel: +49-30-83859368
bInstitute for Complex Materials, Leibniz-Institute for Solid State and Materials Research Dresden, Helmholtzstr. 20, D-01069 Dresden, Germany
cHelmholtz Virtual Institute “Multifunctional Biomaterials for Medicine”, Kantstr. 55, 14513 Teltow, Germany
First published on 1st June 2015
Abstract
Nanogels are nanosized crosslinked polymer networks capable of absorbing large quantities of water. Specifically, smart nanogels are interesting because of their ability to respond to biomedically relevant changes like pH, temperature, etc. In the last few decades, hybrid nanogels or composites have been developed to overcome the ever increasing demand for new materials in this field. In this context, a hybrid refers to nanogels combined with different polymers and/or with nanoparticles such as plasmonic, magnetic, and carbonaceous nanoparticles, among others. Research activities are focused nowadays on using multifunctional hybrid nanogels in nanomedicine, not only as drug carriers but also as imaging and theranostic agents. In this review, we will describe nanogels, particularly in the form of composites or hybrids applied in nanomedicine.
 Maria Molina | Dr Maria Molina received her BS (Hons) in Chemistry from the National University of Rio Cuarto (Argentina) in 2007. In 2007 she obtained a CONICET Fellowship to undertake her PhD studies (2011) in the group of Prof. Cesar Barbero (National University of Rio Cuarto) working on the preparation of smart hydrogels as drug delivery systems. In 2012 she started her postdoctoral research at Freie Universität Berlin, under the supervision of Prof. M. Calderón and Prof. R. Haag. Her current research is focused on the preparation of responsive nanogels for anticancer therapy. In 2013 she was awarded an Alexander von Humboldt postdoctoral fellowship from the Georg Forster Program. |
 Mazdak Asadian-Birjand | Mazdak Asadian-Birjand received his master degree in chemistry at the Freie Universitaet Berlin (Germany) in 2011. In the same year he started his PhD studies under the supervision of Prof. M. Calderón and Prof. R. Haag. His research is focused on the synthesis and biochemical evaluation of environmentally responsive nanogels for biomedical applications. |
 Juan Balach | Dr Juan Balach received his PhD degree in Chemistry from National University of Rio Cuarto in 2011 supervised by Prof. César Barbero and was a postdoctoral fellow (2012) in the Department of Colloid Chemistry at the Max Planck Institute of Colloids and Interfaces, Germany. Since 2013, he has been a Research Associate at the Institute for Complex Materials, Leibniz Institute for Solid State and Materials Research (IFW) Dresden, Germany. His research interests mainly focus on the rational design of advanced carbon-based materials for electrochemical energy storage applications. |
 Julian Bergueiro | Dr Julian Bergueiro received his BS and MS in Chemistry from the University of Santiago de Compostela (Spain) in 2007 and 2008, respectively. In 2013 he received his PhD from USC under the supervision of Prof. S. Lopez. In 2012 he joined the group of Prof. R. Riguera to work on the synthesis and characterization of stimuli-response helical polymers and poly(phenylacetylene)s@gold nano-particle nanocomposites. He joined Prof. M. Calderón group in 2013 to carry out his postdoctoral research at Freie Universität Berlin. Recently he was awarded with a Dahlem International Net-work Postdocs Fellowship to develop gold based thermoresponsive nanogels as nanocarriers. |
 Enrico Miceli | Enrico Miceli received the BSc in Chemistry from the University of Padova (Italy) in 2007. In 2013 he received the MSc in Chemistry from the Freie Universität Berlin (Germany). In 2014 he started his PhD studies in the research group of Prof. Marcelo Calderón in collaboration with Prof. Joachim Dzubiella. He is involved in the investigation of the interaction between proteins and thermoresponsive nanogels. |
 Marcelo Calderón | Marcelo Calderón is an Assistant Professor for Macromolecular and Organic Chemistry at the Department of Biology, Chemistry, and Pharmacy from the Freie Universität Berlin, Germany. Dr Calderón received his PhD in organic chemistry in 2007 from the National University of Córdoba, Argentina, under the supervision of Prof. Miriam Strumia. In 2007 he joined the Research Group of Prof. Rainer Haag at the Freie Universitat Berlin for his Postdoctoral work, where he pursued the development of polymer-drug conjugates for the passive targeted delivery of drugs, genes, and imaging probes. Dr Calderón was the recipient of the Arthur K. Doolittle Award in 2010 (ACS, Polymer Materials), the Cesar Milstein Fellowship in 2011 (Ministry of Science, Technology and Productive Innovation, Argentina), and the NanoMatFutur Grant for Young Scientists from the German Ministry of Science in 2012 (BMBF). |
1. Introduction
Nanogels are nanosized hydrogel particles that combine the properties of both hydrogels and nanomaterials.1 Like hydrogels, they show high water content, tunable chemical and physical structures, good mechanical properties, and biocompatibility.2 The nanoscale mainly provides a large surface area for bioconjugation, long time of circulation in blood, and tunable size from nanometers to micrometers with the possibility of being actively or passively targeted to the desired site of action, e.g., tumor sites.3 The sum of these properties plus the presence of an interior network for the encapsulation of biomolecules makes nanogels ideal candidates for their application in nanomedicine.4,5 Nanomedicine is the application of nanotechnologies to the medical field as drug delivery systems, imaging and sensing agents, theranostic materials, among others. Smart nanogels have attracted much attention for their application in medicine due to their capacity to respond to diverse medically relevant stimuli6 like pH,7 temperature,8–11 ionic force,12 redox environment,13etc., by changing their volume, refractive index, and hydrophilicity–hydrophobicity.14In recent years, the development of advanced or hybrid nanogels with multifunctionality and novel properties has been of interest in many research fields ranging from materials science to nanomedicine.15,16 Hybrid nanogels can be classified based on their different properties. For the purpose of this review, we divided the hybrid nanogels into two categories: nanomaterial–nanogel and polymer–nanogel composites and we use @ in the nomenclature of the nanomaterial–nanogel composites meaning that the new component is hybridized with the nanogel. Nanomaterial–nanogel composites have also been synthesized by incorporation of nanosized materials such as plasmonic,17,18 magnetic,19–21 and carbonaceous nanoparticles,22 like carbon nanotubes,23,24 graphene,25 and fullerenes.26 These materials are of great interest in nanomedicine due to their applicability for imaging,27 guided therapy,19 triggered drug release,28 and hyperthermia,29 among others. Furthermore, polymer–nanogel composites include interpenetrated networks (IPNs) and core–shell particles. The advantage of hybridizing the nanogels with other polymers and not only copolymerizing them is that each component maintains its original properties. The core–shell strategy is especially useful for targeting therapy30 while the interpenetration allows the development of multiresponsive nanogels31 and the control of the release profile.32 Several reviews about synthesis and characterization of nanogels have been published.4,33–36 In this review, we will focus on the description of different hybrid nanogels and their application in the nanomedicine field (Fig. 1). In addition, it is hoped that this review will promote further design and development of new hybrid materials as well as deeper studies for shortening the path to their clinical application.
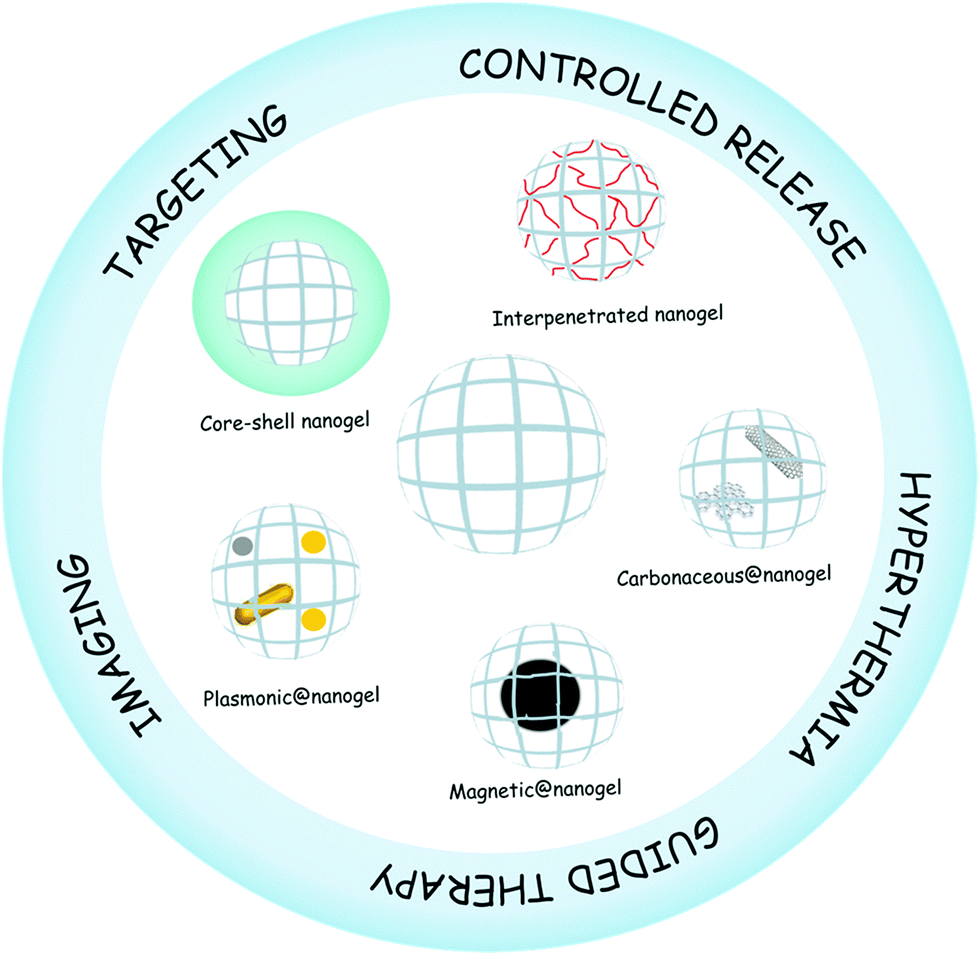 | ||
| Fig. 1 Hybrid nanogels and their application in nanomedicine. | ||
2. Nanomaterial–nanogel composites
The possibility of combining the properties of organic and inorganic components in one material widens their applications in different fields like materials science. However, the so-called hybrid inorganic–organic materials are not physical mixtures but intimately mixed systems. The resulting properties, such as nanostructure, degree of organization, etc., not only depend on their components' nature but also on the synergy between the components. These inorganic–organic composites can be classified according to the nature of the interactions between the components into (a) class I systems where no covalent bonds are formed between the organic and inorganic components and only weak interactions are present such as hydrogen bonding, van der Waals, or electrostatic forces and (b) class II where strong chemical bonds such as covalent or Lewis acid–base bonds are present.37,38 Without any doubt, these new generations of hybrid materials will open up new possibilities for application in different areas of nanomedicine such as, for example, optical sensing,39 on demand drug delivery,40 imaging,41 and hyperthermia therapy.19In this section different nanomaterial–nanogels and their application in nanomedicine will be described, including plasmonic@, magnetic@, and carbonaceous material@nanogels.
2.1. Plasmonic@nanogels
Photothermal therapy (PTT) and photodynamic therapy (PDT) are currently the most promising techniques for treating cancer.42 This is because of the possibility of engineering devices in the nanometric scale that present photothermal transductors (PTs) with absorption in the biological window. Near infrared irradiation (NIR) is known as a biological window since it is hardly absorbed by water and blood cells and can penetrate deeply into the body. PDT is based on PTs that can use NIR light to produce reactive oxygen species which induce tissue destruction. On the other hand, PTT makes use of specific PTs that can effectively transform NIR light into local heat, surpassing the traditional hyperthermia methods such as hot-water bath or heated blood perfusion.43 Nanogels composed of PTs and different polymers are ideal nanodevices for PTT because of their controlled size and architecture, biocompatibility, degradation ability, physical properties, accumulation in tumors, loading capacity, and postsynthetic modification.14 Moreover state-of-the-art drug delivery systems have emerged that make use of thermoresponsive polymers that can swell or shrink with the temperature in combination with PTs.44 The local heat produced by PTs upon exposure to specific radiation causes the transition of the thermoresponsive polymer and expels the drug retained in it.Gold nanoclusters have proven to be useful agents for PTT after they were shown to have an absorption in the NIR region four times higher than conventional photo-absorbing dyes. More specifically, gold nanorods (AuNRs) have been extensively explored due to their aspect ratio that enables efficient NIR light absorption. Photoexcitation of metal nanostructures causes local heating which is useful for PTT. Magnetic nanoparticles (Section 2.2) can also generate local heating when they are exposed to an alternating magnetic field (AMF) acting as magnetic transductors.45,46 Some of the most employed PTs like gold nanoparticles (AuNPs) as well as magnetic and silver nanoparticles (AgNPs) also have the advantage of acting as a contrast agent for bioimaging. All these properties favor the use of plasmonic hybrid nanogels as theranostic agents in chemo- and thermotherapy as well as in in vitro or in vivo imaging.47Table 1 summarizes recent examples that are discussed in the following sections and highlights the potential of the plasmonic NPs@nanogels as theranostic agents.
| Plasmonic NPs | Polymer | Stimuli | Application | Theranostic agent | Ref. |
|---|---|---|---|---|---|
| AuNPs | PEG | NIR light | Imaging (internalization) | AuNPs | 55 |
| AuNPs | PEAMA–PEG | Caspase-3 | Imaging (FRET, internalization) | FITC | 56 |
| AuNPs | PAA | NIR light | Imaging (biodistribution) | Au Nanoclusters | 57 |
| AuNPs | PEAMA-PEG | NIR light | Hyperthermia | AuNPs | 58 |
| AuNRs | Pluronic | NIR light | PTT and PDT | Ce6 | 59 |
| AuNPs | Polyamine | X-rays | RF hyperthermia | — | 60 |
| AuNPs-MnO2NRs | Chitin | Radiofrequency | RF hyperthermia | AuNPs–MnO2NRs | 61 |
| AuNPs | PNIPAm-IPN | Temperature | Hyperthermia – drug delivery | 5-FU | 62 |
| Au nanoclusters | P(NIPAm-co-AA) | Temperature | Hyperthermia – drug delivery | DOX | 63 |
| AuNRs | PNIPAm | NIR light-temperature | Hyperthermia – drug delivery | — | 64 |
| AuNRs | PNIPAm | NIR light-temperature | Hyperthermia – drug delivery | DOX | 65 |
| Au–AgNRs | PAA-aptamers | NIR light | Hyperthermia – drug delivery | DOX | 66 |
| Au–AgNPs | PS-PEG | NIR light | Hyperthermia – drug delivery | Curcumin | 67 |
| AgNPs | P(NIPAm-co-AA) | pH | Imaging – drug delivery | Dipyridamole | 69 |
| AgNPs | P(NIPAm-co-DMAEA) | pH | Imaging – drug delivery | Insulin | 70 |
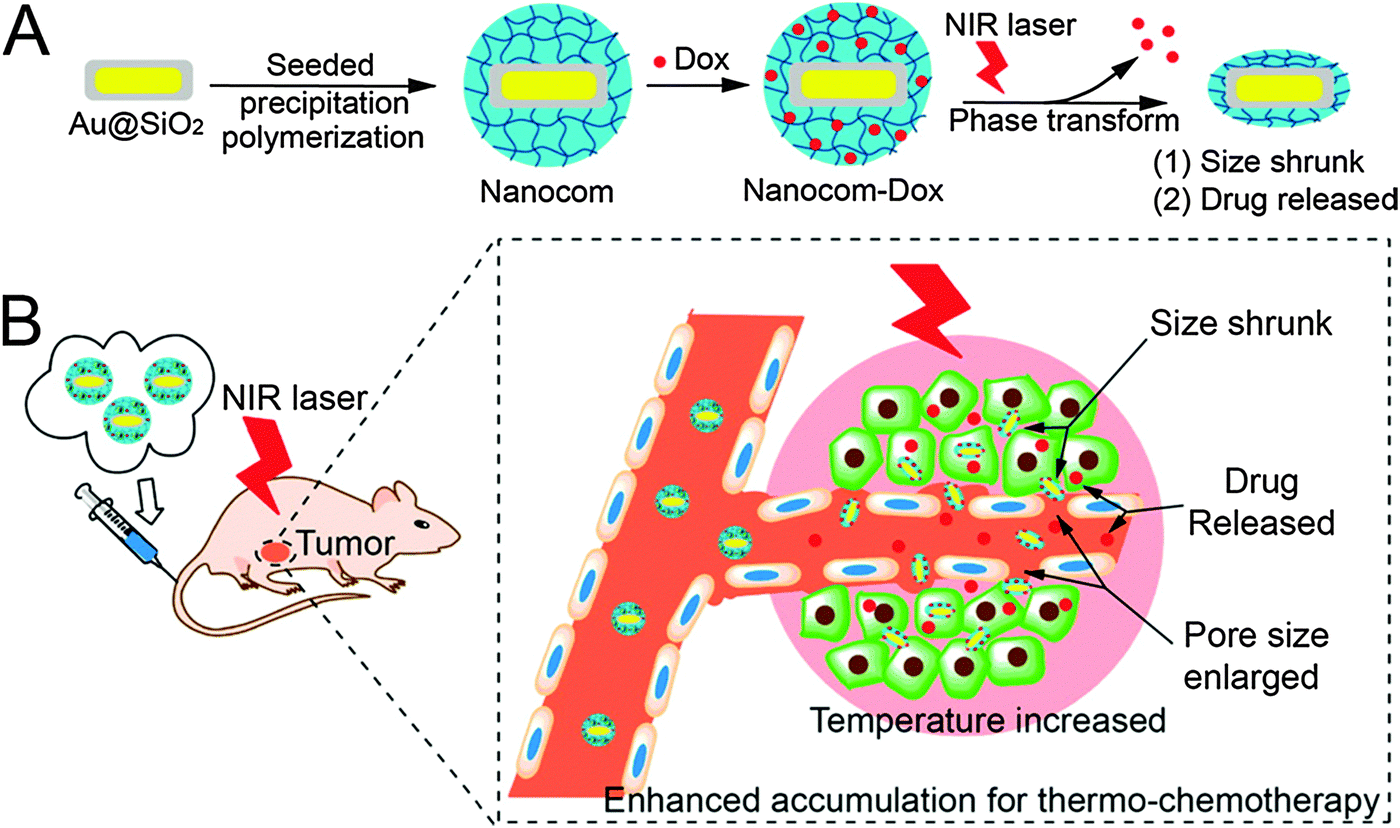 | ||
| Fig. 2 (A) Hybrid nanogel synthesis and (B) NIR laser induced targeted therapy using the hybrid nanogel. Reprinted with permission from ref. 65. Copyright 2014 American Chemical Society. | ||
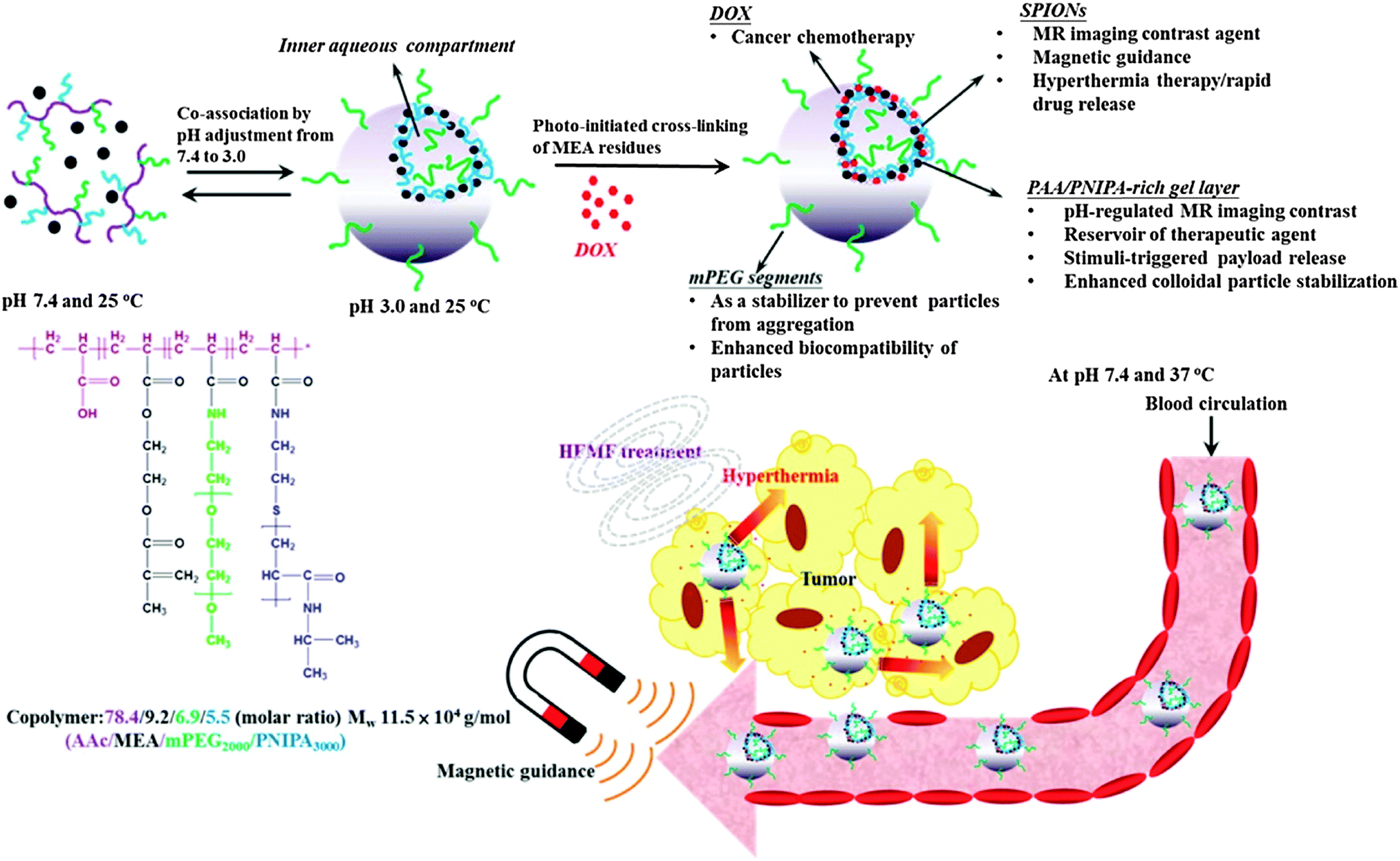 | ||
| Fig. 3 DOX-loaded hybrid nanogels as a multifunctional anticancer theranostic system. Reprinted with permission from ref. 95. Copyright 2013 American Chemical Society. | ||
| Compound | Magnetic material | Matrix/coating | Relaxivity@1.5T [mM−1 s−1] r1/r2 | Stimuli | Application | Ref. |
|---|---|---|---|---|---|---|
| Feridex® (ferumoxides) | Fe3O4 | Dextran | 24/98 | — | Commercial (T2) MRI contrast agent | 73 |
| Resovist® (Ferucarbotran) | Fe3O4 | Carboxydextran | 25/151 | — | Commercial (T1 and T2) MRI contrast agent | 73 and 75 |
| Ferumoxtran-10® (Combidex) | Fe3O4 | Carboxydextran | 10/60 | — | Commercial (T2) MRI contrast agent | 73 and 76 |
| γ-Fe2O3@P(VA-b-VCL) | γ-Fe2O3 | P(VA-b-VCL) | —/37 | Glucose, pH, temperature, AMF | Multiresponsive drug delivery system and T2 contrast agent | 94 |
| SPION@AA-NIPAm-MEA-PEG | Fe3O4 | AA-NIPAm-MEA-PEG | —/265.5 | Temperature, pH, AMF | Multiresponsive drug delivery system and T2 contrast agent | 95 |
| MnFe2O4@PGA-PLL(PEG) | MnFe2O4 | PGA–PLL(PEG) | —/382.6 | AMF | Intravenous (i.v.) injectable T2 contrast agent for dual bio-imaging | 92 |
| SPION@NIPAm-AA-Cy5.5-Lf | Fe3O4 | PNIPAm-co-AA | —/142.7 | Temperature, pH, AMF | i.v. injectable T2 contrast agent for dual bio-imaging | 90 |
| Magnevist® (Gd(III)–DTPA) | Gd(III) | DTPA chelate | 4.7/9.6 | — | Commercial (T1) MRI contrast agent | 99 |
| Gd@nanogel (surface coordinated) | Gd(III) | PEOMA, AEMA, EGDMA, DTPA | 17.5/— | AMF | i.v. injectable T1 contrast agent for bio-imaging | 91 |
| Gd@nanogel (incorporated) | Gd(III) | bPEI (core), mMePEG (shell), Cy5.5 | 2.1/82.6 | AMF | i.v. injectable T2 contrast agent for dual bio-imaging | 89 |
| Gd@nanogel (incorporated) | Gd(III) | PAA DOTA-chelate-crosslinker | 17.6/— | AMF | Potential T1 contrast agent | 101 |
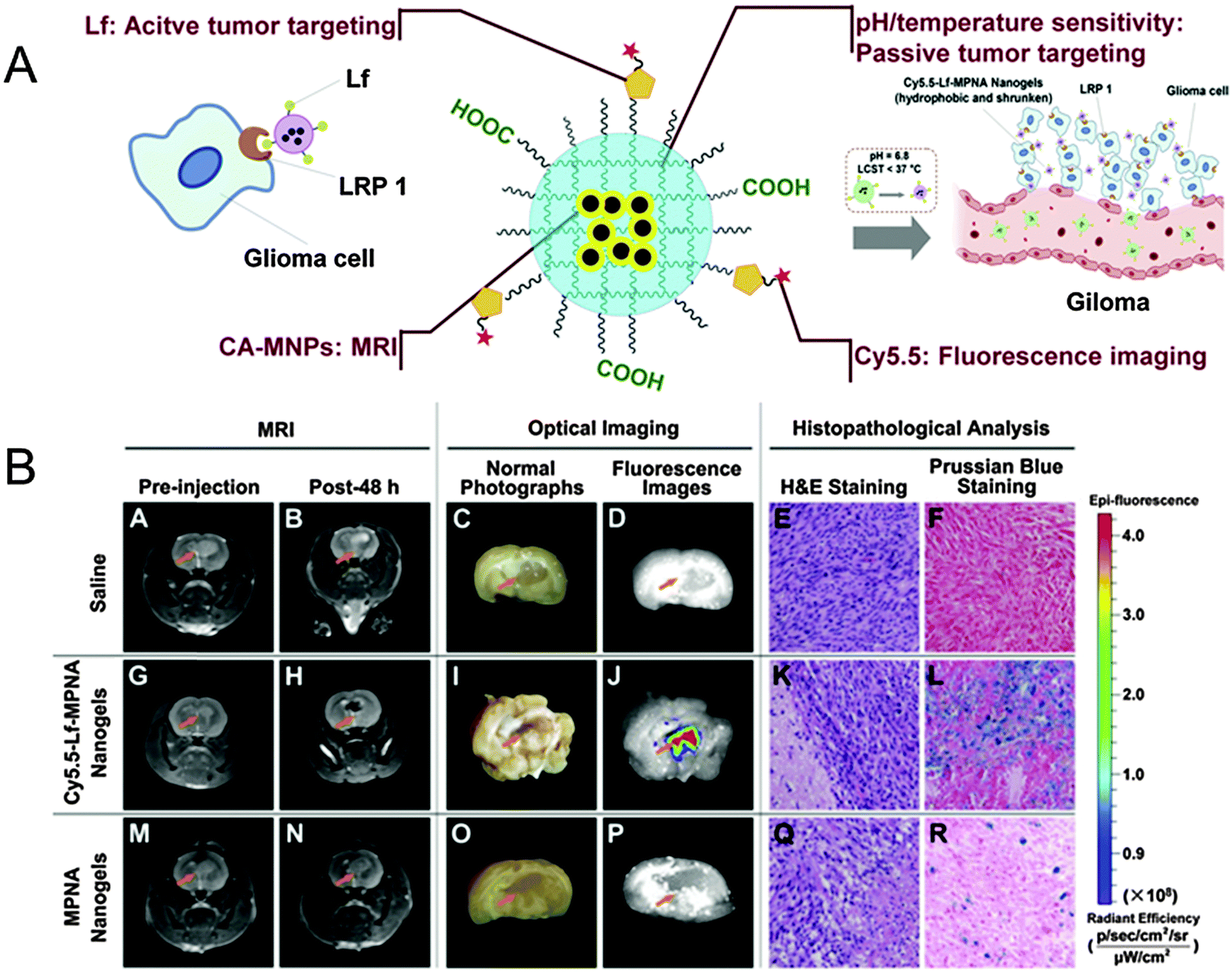 | ||
| Fig. 4 (A) Schematic representation of SPION@NIPAm-AA-Cy5.5-Lf. (B) In vivo studies: MRI, optical imaging, and histopathological analysis of rats bearing gliomas treated with saline as a control (upper row), SPION@NIPAm-AA-Cy5.5-Lf nanogels (middle row), and NIPAm-AA nanogels (lower row), respectively. Reprinted from ref. 90. Copyright (2013), with permission from Elsevier. | ||
| Nanomaterial: QDs, PSNPs, and nanostructured carbon materials | Polymer network | Preparation methods | Stimuli | Enhanced property and application | Ref. |
|---|---|---|---|---|---|
| Protein coated-QDs | CHPNH2 | Self-assembly of negatively charged QDs and positively charged nanogels | — | — | 113 |
| MPA-capped CdTe QDs | Chitin | Incorporation of QDs into preformed nanogels | pH | pH-sensitive drug delivery system with simultaneous biosensing | 114 |
| MPA-capped CdTe QDs | PNIPAm | Copolymerization of MPA-QDs during PNIPAm polymerization | Temperature | Temperature dependent on–off fluorescence properties | 115 |
| CdS QDs | p(NIPAm-AAm-PBA) | In situ immobilization of QDs within the microgels | Temperature and [glucose] | Non-invasive continuous optical detection of saccharides | 116 |
| CdSe-ZnS core–shell QDs | Polypeptide | Self-assembly of QDs and polypeptide complex | pH and temperature | Targeted nanogel for simultaneous cancer diagnosis, imaging, and therapy | 41 |
| Bi2O3 QDs | PVA | In situ immobilization of QDs within the nanogel-network | Temperature | Temperature-responsive anticancer drug release with dual-model imaging for theranostic actions | 122 |
| Hollow silica spheres | PNIPAm | Crosslinking polymerization | Temperature | Enhanced stability and well-controlled drug release | 134 |
| Silica nanoparticles | Poly(N-isopropylacrylamide-co-2-(dimethylamino) ethyl methacrylate, methyl-chloride) | Surfactant-free emulsion polymerization | Temperature | Controlled slow drug release of more than 24 h | 133 |
| PSNPs | P(VCL-s-s-MAA) | Precipitation polymerization in the presence of functionalized PSNPs | pH and temperature | Controlled pH- and temperature-triggered DOX release | 135 |
| PSNPs | PNIPAm | In situ radical polymerization in mesopores | Temperature | Temperature-responsive controlled ibuprofen delivery | 136 |
| PSNPs | Alginate/CHI | Layer-by-layer self-assembly | pH | pH-responsive DOX release | 137 |
| Fullerene (C60) | DMA-GC | Self-assembly of chitosan-based polymer and C60 | Light | Advanced endosomal pH targeting for photodynamic antitumor therapy | 26 |
| f-SWCNTs | PVI-co-AA | Crosslinking polymerization | pH | Improved thermal stability of the pH-sensitive nanogel composite | 23 |
| CHI-CNT | PNIPAm | Crosslinking polymerization | NIR light and temperature | Triggered DOX release upon NIR irradiation | 155 |
| rGO | PNIPAm/PEG-d/chitosan | Crosslinking polymerization | NIR light and temperature | Improved photothermal response resulting in a fast drug release | 163 |
| PEI-coated NDs | N-Acetylated chitosan | Crosslinking polymerization | Enzyme-responsive | Enhanced mechanical properties | 168 |
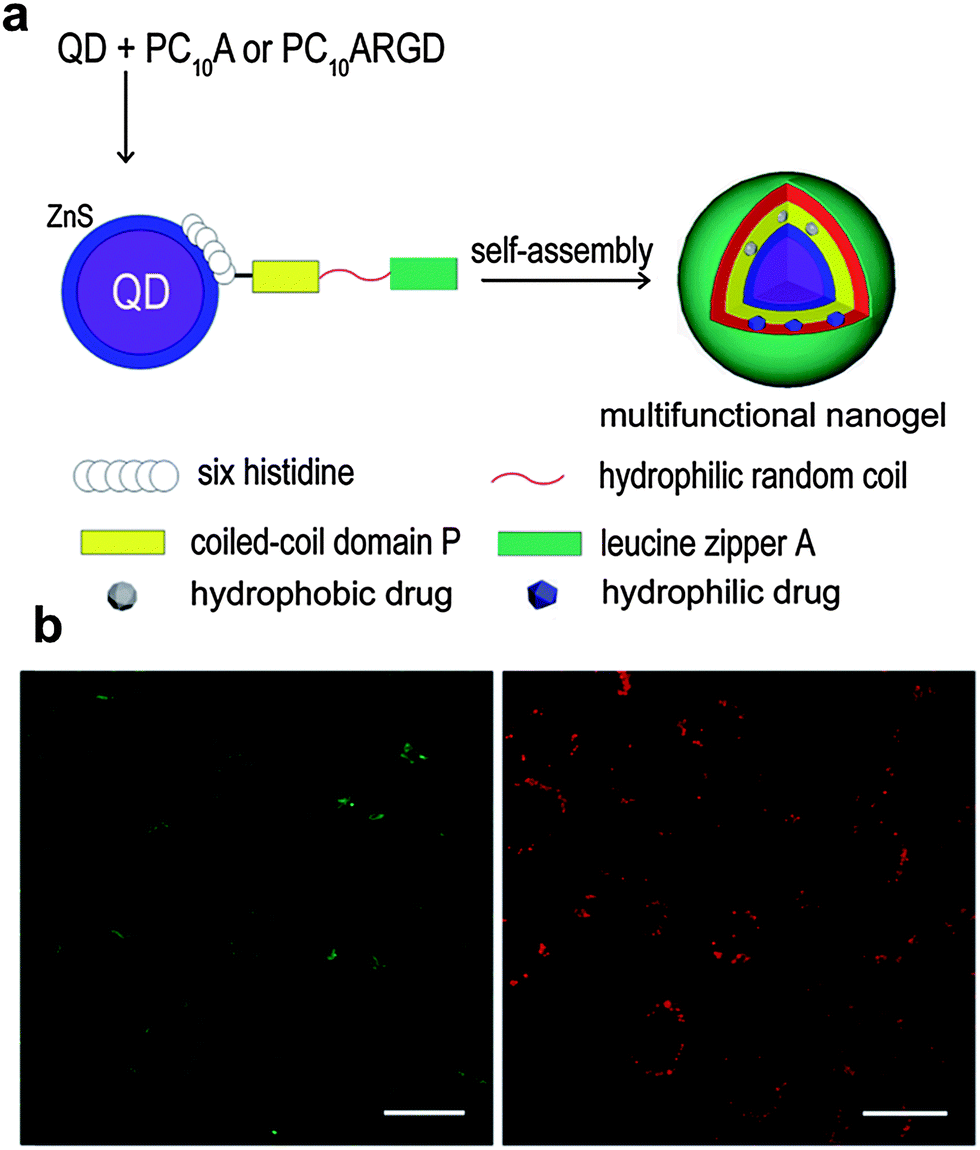 | ||
| Fig. 5 (a) Schematic representation of the QDs@polypeptide nanogel. (b) Confocal fluorescence images of HeLa cells incubated with the QDs@polypeptide nanogel; green and red channels show the green dye (fluorescein sodium) signal and red QDs signal. Reproduced from ref. 41 with permission from The Royal Society of Chemistry. | ||
 | ||
| Fig. 6 Schematic representation of DOX loaded PNIPAm@CHI-CNT and the triggered drug release by NIR irradiation. Reprinted from ref. 155, Copyright (2015), with permission from Elsevier. | ||
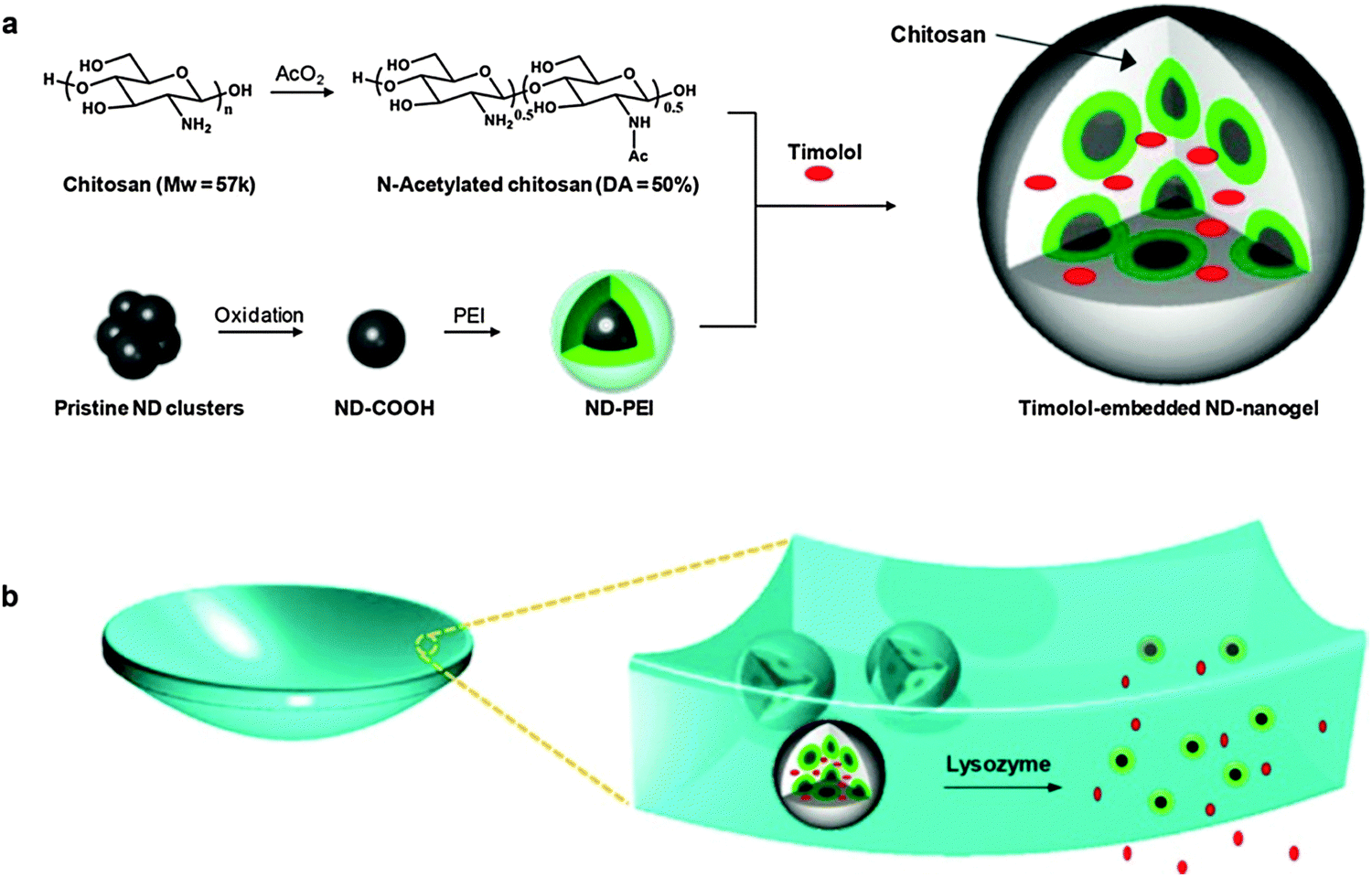 | ||
| Fig. 7 Schematic representation of (a) timolol-loaded ND@nanogel and (b) ND@nanogel embedded contact lenses. Reprinted with permission from ref. 168. | ||
Gold hybrid nanogels (Au@NGs) have been reported as relevant bioimaging devices. Siegwart et al.55 described uniformly crosslinked poly(ethylene glycol) (PEG) based nanogels synthesized by atom transfer radical polymerization (ATRP) in which AuNPs were entrapped. Cell uptake and internalization of these nanodevices have been studied in human umbilical vascular endothelial cells (HUVECs) and human mesenchymal stem cells (hMSCs) validating an endocytosis mechanism. A similar approach by Oishi et al.56 made use of the fluorescence resonance energy transfer (FRET) between AuNPs and fluorescein isothiocyanate (FITC) in a PEGylated Au@NG. A caspase-3-responsive system was designed to monitor the cancer response to therapy. A pronounced fluorescence was observed when the activated caspase-3 in apoptotic cells cleaved the Asp–Glu–Val–Asp peptide that resulted in the release of the FITC and dequenching of its fluorescence. The biodistribution of poly(acrylic acid) (PAA) nanogels with AuNPs formed in situ was recently studied in H22 tumor-bearing mice.57 Gels mainly accumulated in the liver and spleen because of their capture by phagocytic cells of the reticuloendothelial system (RES) of these organs.
Au@NGs are interesting not only for bioimaging by employing the optical properties of the gold, but also for other applications. In the last decade, the use of nonspherical AuNPs as PTs in Au@NGs enabled their advantageous application in PDT and PTT. PEGylated poly-[2-(N,N-diethylamino)ethyl methacrylate] (PEAMA) core nanogels with AuNPs post-synthesized by reduction of Au(III) ions reported by Nakamura et al.58 showed high biocompatibility and remarkable photothermal efficacy. PTT in response to 514.4 nm light was achieved in HeLa cells, killing only the cells in the laser area with low IC50 values depending on the gold concentration of the nanogel. One of the most relevant examples in this field is the PPT and PDT synergistic combination from Kim et al.59 with Pluronic based nanogels loaded with AuNRs as the PTT agent and chlorin e6 (Ce6) as the photosensitizer. In vitro SCC7 cell line and in vivo SCC7 tumor bearing mice remarkably enhanced the combination of PTT and PDT. But even more interestingly, the tumor decreased in size when the PDT was applied before PTT. When PTT was applied first the combined therapy was not so effective as with just PTT. This comparative study therefore revealed a more convenient way to the use of photo-therapies. The radiosensitizing potential of gold containing nanogels enhancing the biological effect of X-irradiation was studied with PEGylated polyamine nanogels containing AuNPs.60 This report proved that these devices enhanced the cell radiosensitivity because of the induction of apoptosis and the inhibition of DNA double-strand break repair by Au@NG mediated endoplasmic reticulum stress. A combination of chitin nanogels with AuNPs and MnO2 rods was also used for radiofrequency (RF) assisted hyperthermia.61 Au@NGs showed better results in the ablation of T47D cancer cells than the bare nanogels. Au@NGs formed by thermoresponsive polymers controlled by PTs were used as smart drug delivery systems in combination with hyperthermia. Thus, these nanogels are theranostic systems with drug delivery, hyperthermia, and bioimaging capabilities. Poly(N-isopropylacrylamide) (PNIPAm) is a thermoresponsive polymer with a lower critical solution temperature (LCST) of around 32 °C which is suitable for medical applications. PNIPAm based nanogels were reported in combination with AuNPs in a core–shell fashion.62 Au@PNIPAm nanogels were employed in cell imaging trespassing cellular barriers to enter the cytoplasm. 5-Fluorouracil (5-FU) served as a model anticancer drug to test the viability of HeLa cells upon exposure to 515 nm laser. Cell death increased significantly in the drug loaded systems in comparison to non-loaded ones, which demonstrated the higher therapeutic efficacy of the combined chemo-photothermal treatments. P(NIPAm-co-AA) nanogels with incorporated gold nanoclusters were employed as a doxorubicin (DOX) delivery system.63 In this case, the dual thermo- and pH-responsive systems were decorated with tumor targeting peptides like (CRGDRCPDC)-SH (iRGD) that showed good results in HUVECs and extravascular tumor cells (B16). More convenient PTs like gold nanorods (AuNRs) were studied as building blocks in nanogel based drug delivery systems. Kawano et al.64 proved that silica coated AuNR core–shell PNIPAm hybrid nanogels underwent phase transition and accumulated in NIR irradiated target sites. A similar system was used as a NIR guided DOX delivery system.65 After NIR irradiation the accumulation in the tumor following systemic administration was enhanced with almost a total tumor growth inhibition as well as lung metastasis (Fig. 2).
Gold–silver nanorods (Ag–AuNRs), which can absorb NIR photons more efficiently than spherical gold and silver nanoparticles but combining their basic properties, were used as PTs in polyacrylamide based nanogels, crosslinked by complementary DNA strands. Nanogels were functionalized with DNA aptamers for specific tumor cell targeting.66 DOX was loaded into DNA aptamers that responded upon local heating by NIR irradiation of the PT releasing the drug. Studies in CCRF-CEM cells and in the Ramos cell line as the control demonstrated low cytotoxicity of the nanogels as well as high specificity for tumor cells after NIR irradiation. Bimetallic gold–silver nanoparticles were also reported in core–shell polystyrene/PEG based nanogels.67 Au–Ag@NGs were able to encapsulate drugs like curcumin that has pro-apoptotic effects. Upon NIR light exposure, the release of drug could be triggered by thermoresponsivity of the outer PEG shell to local temperature increase. In vitro comparative study of these systems in B16F10 cells revealed an enhanced effect of chemo- and thermotherapies.
2.2. Magnetic@nanogels
Within nanomaterial–nanogel composites, magnetic nanoparticles have attracted interest because of their application as PTs and in imaging. In the past, the most common methodology for preparing superparamagnetic iron nanoparticles (SPION)71 consisted of reduction and coprecipitation of ferrous and ferric salts in aqueous media in the presence of stabilizers. This procedure has led to several commercial products like Feridex® (Ferumoxides),72,73 Resovist® (Ferucarbotran),73–75 and Ferumoxtran-10® (Combidex).73,74,76 As their biodistribution depended on their size and surface modification, much effort has been made in developing magnetic nanomaterials with increased colloidal stability under physiological conditions. Moreover, the research focus was on improving contrast properties while lowering cytotoxicity, maintaining longer blood half-life, improving biocompatibility, and enhancing tissue/organ targeting ability as well as pharmaceutical efficacy due to surface functionalization.77 Today, to accomplish the requirements for safer and more efficient nanostructure-based contrast agents, the developed materials have become more complex such as nanoclusters,78 polymersomes,79,80 nanocomposites,81–88 and nanogels.89–93 Considering the obtained impressive biodistributions and remarkable magnetic resonance imaging (MRI) properties, the following section presents recent developments of hybrid nanogels for magneto-transducers (MTs) and MRI.MTs like SPIONs were used as an alternative to PTs. As an example, γ-Fe2O3 magnetic NPs were internalized in poly(vinyl alcohol-b-N-vinylcaprolactam) [P(VA-b-VCL)] based nanogels for glucose-, pH-, and thermo-responsive release of DOX.94 AMF induced heating could be generated because of the superparamagnetic properties of the SPIONs that accelerated the drug release performance of the system. Chiang et al. also reported the use of AMFs to enhance drug release95 from hybrid hollow nanogels formed with PEG, AA, and PNIPAm, and crosslinked by photo-initiated polymerization of 2-methacryloylethyl acrylate (MEA). The DOX loaded nanogels exhibited accelerated drug release in response to pH reduction and hyperthermia. Magnetic properties of the nanogel were employed not only for AMF therapy but, moreover, for guided transport toward the target and MIR. Enhanced in vitro cytotoxicity against HeLa cells in comparison with free DOX demonstrated the great potential of the multimodal theranostic system capable of combining hyperthermia and chemotherapy (Fig. 3).
MRI is a widely known noninvasive imaging technique that achieves notable high spatial and temporal resolution. It displays contrast by tracking changes in water proton density and longitudinal (T1) or transversal (T2/T2*) relaxation times of protons in normal and diseased tissues.96 Since their first use as MRI contrast agents 30 years ago,97 iron oxide nanoparticles have gained much attention because of their ability to dramatically shorten T2/T2* relaxation times and therefore produce a decreased signal intensity in T2/T2* weighted MR images. With their specific accumulation in the liver, spleen, and bone marrow, their high magnetization, high contrast visualization compared to conventional paramagnetic T1 contrast agents, they have exerted a large impact on the field of molecular and bio-imaging.98
In spite of a multi-modal approach for magnetic nanogels, Kim et al.92 developed a self-fluorescent and, at the same time, high-relaxivity T2-weighted MRI contrast agent for both MRI and optical bio-imaging. The formulation was achieved by electrostatic assembly and crosslinking of poly(γ-glutamic acid) (γ-PGA) coated manganese iron oxide (MnFe2O4) nanoparticles with the positively charged polyelectrolyte poly(L-lysine) (PLL). During the ionic gelation process, glutaraldehyde was added to the mixture which provided structural integrity to the nanogel and involved the formation of a self-fluorescent chemical bond. These fluorescent bonds were generated by the crosslinking of PLL with the glutaraldehyde monomer. Finally, PEG was conjugated to the surfaces of the MnFe2O4@PGA–PLL(PEG) nanogels. The r2 value, which is known as relaxivity, is the reciprocal of the T2 relaxation time per unit concentration of metal ions. This value was 382.6 (Fe + Mn) mM−1 s−1 for the nanogel, two fold higher as compared to the r2 of conventional SPION contrast agents such as Feridex® and Resovist® (Table 2). The remarkable increase in r2 resulted from the synergistic magnetism of the multicore MnFe2O4 nanoparticle satellites that were embedded in the nanogels. Moreover, MnFe2O4@PGA–PLL(PEG) nanogels also generated a strong fluorescence signal indicating that these compounds can be used as an imaging technique in living tissues. In vivo NIR fluorescence and MRI of the lymph node in a mouse model highlighted the nanogel's passive targeting ability to successfully migrate from the injection site to the lymph node. The results suggest that after the incorporation of therapeutic moieties these systems could be a potent tool in theranostic technology.
A similar approach in multi-modality was followed by Jiang et al.90 when engineering pH and temperature sensitive magnetic nanogels conjugated with Cyanine 5.5-labeled lactoferrin (Cy5.5-Lf) for MRI and fluorescence imaging of glioma in rats (Fig. 4). In this study, citric acid coated SPIONs were incorporated in nanogels by free radical dispersion polymerization with NIPAm and AA as environmental sensitive monomers, and N,N′-methylenebisacrylamide (MBAAm) as the crosslinker. After polymerization, the surface coating was achieved by peptide coupling chemistry mediated conjugation of Cy5.5-Lf providing both a dual imaging modality and a targeting ligand for specific targeting of lipoprotein receptor-related protein 1 (LRP1) expressing cells. The obtained nanogels showed an r2 value of 142.7 mM−1 s−1, similar to that of Resovist®, and revealed pH and temperature sensitiveness for enhanced targeting modalities. In fact, SPION@NIPAm-AA-Cy5.5-Lf nanogels were hydrophilic and swollen under physiological conditions (pH 7.4, 37 °C), which could prolong the blood circulation time, but became hydrophobic and shrunken in the acidic environment of tumor tissues (pH 6.8, 37 °C). As a result, they could be more easily accumulated in tumor tissue and internalized by tumor cells. Cellular uptake studies on both rat C6 glioma cells (high LRP1 expression) and human umbilical vein endothelial (ECV 304) cells (no LRP1 expression) interestingly revealed that the cellular uptake of both SPION@NIPAm-AA-Cy5.5-Lf nanogels and NIPAm-AA nanogels at pH 6.8 was significantly higher than that at pH 7.4. These results suggested that the nanogels' hydrophilic/hydrophobic transition and their change in size would increase their cellular uptake, as well as improve their internalization in malignant tissues. SPION@NIPAm-AA-Cy5.5-Lf nanogels were applied in vivo and their imaging properties were evaluated in rats bearing C6 glioma with both MRI and fluorescence imaging technique. As a result, histopathological analyses obtained significant targeting ability for SPION@NIPAm-AA-Cy5.5-Lf nanogels on gliomas both in vitro and in vivo.
Even though such impressive investigations have been performed as mentioned above, SPIONs generally provide some conceptual disadvantages that may limit their advanced clinical use. On the one hand, T2 contrast agents are negative contrast agents that can lead to signal-decreasing effects. As a result, the obtained dark signals could be mistaken for other pathogenic conditions leading to images of lower contrast than T1 contrasted images. On the other hand, the high susceptibility of T2 contrast agents might induce distortion of the magnetic field in the neighboring tissues. This background distortion is the so-called susceptibility artifact, which can yield obscure images and ruin the background around the malignant tissue.71,96 Therefore, the most used clinical contrast agents are based on gadolinium complex T1 agents such as Magnevist (Table 2). Since these commercially available T1 agents provide a relatively short blood half-life and low contrast efficiency due to low relaxivity values,99 the scientific community aimed to improve their clinical impact through nanotechnology.77,96 As an example, Soleimani et al.91 recently developed small nanogels (∼10 nm diameter) based on poly(ethylene glycol) methyl ether methacrylate (PEOMA) and N-(2 aminoethyl) methacrylamide hydrochloride (AEMA) as monomers, and ethylene glycol dimethacrylate (EGDMA) as the crosslinker. The nanogel's surface was decorated with an iso-thiocyanate derivative of the clinically used chelate diethylenetriaminepentaacetic acid (DTPA) which was able to complex gadolinium (Gd(III)) ions out of an aqueous GdCl3–nanogel solution mixture. Compared with the clinical agent Magnevist® (Gd(III)–DTPA),99 the Gd(III) coated nanogels provided a 4 fold enhancement in r1 relaxivity (Table 2). The higher contrast at the same dose in T1 weighted MR images of C.B.-17 SCID mice bearing MDA-MB-231 tumors achieved for nanogels indicated an increased accumulation in the tumor due to enhanced circulation in the vasculature.
A similar observation has been made in another approach incorporating Gd(III) ions into a nanogel matrix by Lim et al.89 Here, GdCl3 was coordinated to branched polyethyleneimines (bPEI) through an inverse miniemulsion technique of Tween 80 stabilized water droplets in cyclohexane. The obtained physically crosslinked nanogels (160 nm in diameter) were subsequently modified with a Cy5.5 dye to yield nanogels with a dual-imaging modality. Moreover, the labeled nanogels were coated with linear poly(ethylene glycol) N-hydroxy succinimide (PEG-NHS) as a stealth agent in order to increase the blood circulation time. As expected, low cytotoxicity and high deformability in terms of Young's modulus were obtained, which subsequently led to minimal filtration by the RES and therefore increased passive accumulation in the tumor of a SCC7 tumor bearing mouse. Interestingly, no enhanced T1-weighted MRI contrast was observed (r1 = 2.1 mM−1 s−1), but the remarkable r2 value was 82.6 mM−1 s−1 for T2-weighted MRI. As T1 and T2 are known to rely on the number of Gd(III) ions or the size,100 the dense multi Gd(III) complex, represented as nanogel, resulted in a transverse relaxivity that lied in the range of SPIONs. Finally, these nanogels should be considered as potential candidates for theranostic purposes since the bPEI core surface could be easily modified with suitable drugs and bioactive molecules.
A study that tested different chelators for efficient Gd nanogel formation was recently performed by Lux et al.101 They designed polyacrylamide (PAAm) based nanogels with different acrylic DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) and DTPA based Gd(III) chelate complexes as crosslinkers. Similar to the work of Soleimani et al.,91r1 relaxivities in the range of 9.7–17.6 mM−1 s−1 were obtained which were 3–4 fold higher than the values of commercially available agents such as Magnevist® (Table 2). Since Gd(III) ions were embedded by crosslinking in the nanogel matrix, DTPA-based nanogels were more inert against transmetallation than Magnevist®, suggesting that crosslinker chelates may represent an important approach towards stable metal-chelating biomedical agents. As this methodology appeared in two steps, namely, the acrylic chelate Gd(III) complex formation followed by free radical emulsion polymerization with PAAm, incorporation of the crosslinkers into nanogels with a biocompatible and biodegradable polymer backbone might be a promising perspective towards clinical relevance.
As shown in this section, most of the presented nanogel based MRI contrast agents are still in the preliminary stage of in vitro and in vivo testing. Several key issues have to be addressed to provide nanogel based MRI contrast agents with clinical relevance. These issues mainly deal with pharmacokinetics, long-term stability, and toxicological effects. Therefore, intensive research is necessary to accomplish the ultimate task of using nanogel based MRI contrast agents for molecular and bio-imaging and thus achieve the transition of these concepts from bench to the bedside.
2.3. Other nanomaterials–nanogels: quantum dots, porous silica nanoparticles, and nanostructured carbon materials
As shown in Sections 2.1 and 2.2, plasmonic and magnetic nanoparticles have been playing a significant role in the development of novel stimuli-responsive nanogel composites for potential application in nanomedicine. In addition to these examples, a great deal of effort has been devoted to creating new types of nanomaterial–nanogel composites with enhanced functions, including the use of QDs, porous silica nanoparticles (PSNPs), and nanostructured carbon materials (Table 3). This section highlights the features of such nanomaterials as part of stimuli-responsive hybrid nanogel systems and their applications for delivery of therapeutic agents.In the last few years, different strategies have been used to obtain stimuli-sensitive QDs-based fluorescent probe nanogel composites as drug delivery systems with simultaneous imaging and biosensing. A simple approach is to incorporate surface-modified QDs into the polymer network of the preformed nanogels. For example, Hasegawa et al. prepared monodisperse hybrid nanoparticles of 38 nm by mixing protein-coated QDs with amino-modified cholesterol-bearing pullulan nanogels (CHPNH2-QD) in phosphate buffered saline (PBS) solution for 30 min at room temperature.113 Although the incorporation of QDs into unmodified, neutral cholesterol-bearing pullulan (CHP) nanogels was suppressed, electrostatic interaction between negatively charged QDs and positively charged amino-containing nanogels induced the successful formation of the hybrid nanogel complex. Furthermore, the QDs@CHPNH2 nanogel composite did not form aggregation after its internalization into various human cells. In a similar way, Rejinold et al. reported the preparation of multifunctionalized biodegradable nanogels by simply mixing mercaptopropionic acid-capped CdTe Ds and chitin nanogels in an aqueous solution.114 The unreacted –OH and –NH2 groups of the repeating N-acetylglucosamine units, that compose the chitin chains, acted as anchor points to sequester Cd2+ ions and further immobilize CdTe QDs in the nanogel matrix. The obtained CdTe QDs–chitin nanogel composite as a fluorescent probe and drug delivery system could be easily loaded with bovine serum albumin (BSA) showing promising applications for simultaneous bioimaging during drug release under local environmental conditions. Following the same trend, different nanogel networks have been used to seize QDs in nanomedicine as sensing and imaging materials.115–117 Nevertheless, the design of multitasking hybrid drug carriers capable of integrating various functionalities in one single nano-object is highly desirable.118–120 Recently, Yang et al. developed an interesting QDs@polypeptide nanogel composite with a dual hydrophilic/hydrophobic character (Fig. 5).41 The main formation of the novel hybrid nanogels can be summarized in two steps: (a) specific metal–affinity interaction between hydrophilic glutathione-coated CdSe–ZnS QDs and N-terminal polyhistidine sequences of PC10A or PC10ARGD coiled-coil polypeptides and (b) nanogel formation and final encapsulation of QDs via self-assembly. The thus obtained pH and temperature dual responsive QDs-polypeptide nanogels of 23 nm in diameter were further loaded simultaneously with hydrophobic and hydrophilic dyes (2-amino-4,6-bis-[(4-N,N′-diphenylamino)styryl]pyrimidine and fluorescein sodium, respectively) as model drugs. Their evaluation in HeLa cancer cells revealed remarkable overexpressed αvβ3-integrin receptors for simultaneous optical pH-sensing and targeted delivery of drugs.
Other strategy for the preparation of QDs@nanogel composites can be the in situ synthesis of QDs in the interior of the nanogel. This approach has been illustrated by Wu et al.121 by conducting a successful in situ synthesis of CdSe QDs inside of a temperature- and pH-sensitive polysaccharide-based nanogel network. In the construction of a hybrid QDs-nanogel complex, the synthesis of QDs within the gel matrix offered a confined growth process that could improve the chemical interaction of quantum nanocrystals with the polymeric gel to confer them optimal dimensions and stable optical signals. Following a similar strategy, Zhou's group moved forward in their research and developed pH sensitive CdSe@CHI–poly(methacrylic acid) and temperature responsive Bi2O3@PVA nanogels by in situ formation and immobilization of QDs.122,123 The main goal of such custom-designed engineering nanohybrid systems was to integrate multifunctionalities for simultaneous biosensing, bioimaging, and effective therapy.
Motivated by the potential application of spherical hollow silica matrices, Liu et al. constructed a thermoresponsive drug release system based on hollow silica nanospheres coated with a PNIPAm nanogel shell.134 The prepared hollow silica nanogels showed good biocompatibility under in vitro cytotoxicity evaluation as well as excellent thermoresponsive controlled-release behavior of rhodamine B (RHB) during release studies. This hollow silica PNIPAm-nanogels appear to be highly suitable for stimuli-responsive controlled-release drug delivery applications. Similar systems have been developed using nanostructured mesoporous silica covered with stimuli-responsive polymer shells in order to retard and control the release rate of the cargo under environmental stimulus control.135–137 The development of hollow or mesoporous silica materials offers new possibilities for incorporating specific drugs within silica cavities followed by controlled release of its cargo from the matrix due to its well-defined structure.
Despite the interesting properties of the fullerenes, the spotlight of scientific research has been cast on the utilization of other nanostructured carbon materials in recent years. In terms of mechanical, optical, conductive, and thermal properties, carbon nanotubes (CNTs) have received enormous attention in the field of bioapplications.151–153 Although many research groups have incorporated CNTs into hydrogel polymers154 in order to reinforce gel structure, increase electrical conductivity, and enhance NIR sensitivity properties, only a few studies have reported the preparation of the CNT–nanogel composite for drug delivery purposes.23,24,155 Nevertheless, this is a topic that will be much studied in the forthcoming years. Very recently, Qin et al.155 published the development of a NIR triggered DOX delivery system based on CHI-coated CNT encapsulated in a PNIPAm nanogel (PNIPAm@CHI–CNT, Fig. 6). In this work, they reported a high DOX loading capacity of ∼43% and a faster drug release at 40 °C and pH 5. Moreover, due to the CNT these nanogels showed a triggered DOX release upon NIR irradiation.
Although CNTs have been actively researched for their possible application in nanomedicine since the late 90s, graphene has garnered tremendous attention in biomedicine in the past few years because of its high mechanical strength, great optical absorbance, excellent thermal conductivity, and low toxicity.156–159 Graphene is a sheet-like two-dimensional sp2-bonded carbon structure and a single layer of graphene sheet has a thickness of 0.34 nm (one-atom thick). Particularly, reduced graphene oxide (rGO) is capable of adsorbing NIR light and converting such optical energy into thermal energy.160–162 Under this consideration, Wang et al. constructed a photothermal NIR light-responsive drug delivery platform based on CHI-modified rGO crosslinked with NIPAm and polyethylene glycol-diacrylate (PEG-d).163 The rGO hybrid nanogel was further loaded with DOX as an anticancer drug showing high loading capacity up to 48 wt%. In vivo studies showed that the photothermal response of the rGO to NIR irradiation led to a rapid rise in the temperature of the surrounding gel and resulted in a fast release of the loaded DOX with high local controllability. The weight composition of rGO/nanogel not only had a dramatic effect on the thermo- and photo-responsive properties of the hybrid nanogel composite, but also a defect or excess in rGO could switch-off or switch-on a specific skill of the rGO–nanogel complex.161 For example, at a rGO composition of ∼47.5 wt%, the nanogel composite showed both thermo- and photo-sensitivities, while at lower (≤32 wt%) or higher (≥64.5 wt%) rGO composition the hybrid nanogel only presented thermo- or photo-sensitive properties, respectively. Furthermore, an excess of rGO (≥78.5 wt%) caused the nanogel composite to lose its responsive properties.
Lately, a particular allotropic form of carbon has been considered for its use in bioimaging, biosensing, and therapeutic applications: diamond. Particularly, nanodiamonds (NDs) are three-dimensional sp3-bonded carbons with a distinctive faceted surface architecture where, depending on the size and morphology, the sp3-surface of the NDs is stabilized through either termination with functional groups or reconstruction of sp3 carbon into sp2 carbon. The presence of such surface functional groups on the NDs allows them (i) to coordinate water molecules around the surface and ensure well-dispersion in an aqueous medium/matrix and (ii) to interact chemically with specific molecules or drugs in a biocompatible environment.164,165 Furthermore, NDs have demonstrated excellent biocompatibility in many in vitro and in vivo studies.166,167 Very recently, the rational design of therapeutic contact lenses through the incorporation of NDs into nanogel matrices as effective ocular drug delivery platforms has been demonstrated by Kim et al.168 They have synthesized a drug-loaded ND@nanogel composite via crosslinking polyethyleneimine-coated NDs and partially N-acetylated CHI in the presence of timolol maleate. The timolol-containing NDs@nanogels were then embedded within a hydrogel matrix and cast into enzyme-responsive contact lenses (Fig. 7). The main role of the coated NDs is to retain the timolol by short distance chemical interactions and to avoid the premature elution of the drug, but keeping it physically active for its further release under degradation of the nanogel via lysozyme cleavage. In addition to the improved timolol-controlled release, the incorporation of a small amount of NDs into the lens matrix enhances tensile strength and boosts elastic modulus by the reinforcement of the polymer matrix. Overall, the obtained lysozyme degradable, timolol-loaded ND@nanogel embedded contact lenses not only had excellent water and oxygen permeability, optical clarity, and improved mechanical properties, but most importantly also displayed sustainable drug release under lysozyme activation. This novel ND-based system provided an interesting platform for the development of advanced enzyme-triggered drug release devices for sustained therapy applications.
Undoubtedly, the use of QDs, PSNPs, and nanostructured carbon materials to control the mechanical, optical, electrical, and thermal properties of smart nanogel composites for potential on-demand high-controlled drug delivery nanovehicles has made tremendous advances in nanomedicine. However, the capability of the nanomaterials, mentioned in this section, to enhance hybrid nanogel composite abilities is still not fully explored. We believe that forthcoming research on the topic and further full-understanding of biological systems will change dramatically today's concepts and a huge progress in biomedical technology will take place in a near future.
3. Polymer–nanogel composites
Composites of nanogels with other polymers not only extend the applicability of these systems due to the multisensitiveness,169 but moreover overcome some disadvantages of single networks as, for example, slow-rate response,170 the hysteresis during repeated swelling–shrinking cycles,171etc. Based on the nature of the composite polymer–nanogels can be classified into two categories: (a) IPNs and (b) core–shell particles. The utilization of these strategies for obtaining multiresponsive hybrid nanogels and their further application in nanomedicine will be described in Sections 3.1 and 3.2.3.1 Interpenetrated and semi-interpenetrated polymer networks
To enhance the mechanical strength, the swelling/deswelling response, and to add new sensitivities to a nanogel, multicomponent networks as IPNs have been designed (Table 4). IPNs are defined by IUPAC as “two or more networks that are at least partially interlaced on a molecular scale but not covalently bonded to each other and cannot be separated unless chemical bonds are broken”.172 The combination of the polymers results in a polymeric system with a new profile. IPNs can be classified into full IPNs (IPN)173 when the interpenetrated polymer is crosslinked inside the first network, or semi-IPNs (sIPNs)27 when a linear polymer is embedded within the first network.| Network | Polymers | Stimuli | Application | Ref. |
|---|---|---|---|---|
| IPN | PNIPAm/PAA | Temperature and pH | Stomach-specific drug delivery system | 31, 173 and 178 |
| sIPN | HPC/PAA | Temperature and pH | Oxaliplatin delivery | 180 |
| IPN | CHI/POEG | Temperature and pH | Chemo-cryo cancer therapy | 182 |
| IPN | P(NIPAm–Dex–PBA) | Temperature and [glucose] | Insulin delivery | 170 and 220 |
| IPN | [P(NIPAm-co-FPBA)]/PAAm | Temperature and [glucose] | Nanoglucometer | 171 |
| IPN | NaAlg/PAA | pH | Ib controlled release | 184 |
| sIPN | CHI/PMAA | pH | TMZ release | 123 |
| IPN | PNIPAM/PAA | Temperature and pH | Imaging by SERS | 185 |
There are two synthetic pathways for obtaining IPNs:174 (1) simultaneous synthesis, where both monomers are mixed and the polymers synthesized at the same time by noninterfering routes175 and (2) sequential synthesis, where the second monomer is polymerized in situ, inside the first single network.176
One of the major uses of IPNs is obtaining biomedically relevant dual responsive hybrid nanogels with little interference between stimuli.31 Several examples of IPNs for synthesizing dual pH/temperature responsive networks as drug delivery systems were published in the last decade.27,173,177–180 do Nascimento Marques et al.181 found that the introduction of pH-sensitive polymers to crosslinked PNIPAm particles not only produced dual-sensitive materials but also allowed particle stability to be adjusted depending on the desired application. In this context, Xing et al.173 reported temperature/pH dual stimuli responsive hollow nanogels with an IPN structure based on a PAA and PNIPAm (PNIPAm/PAA IPN hollow nanogels). These nanogels showed a high drug loading capacity of isoniazid (INH), an antitubercular drug, up to 668 mg INH per gram of the nanogel. In vitro drug release studies showed an acid triggered drug release behavior, thus making them a potential stomach-specific drug delivery system.
Besides PNIPAm, other thermoresponsive polymers were used in IPNs. Chen et al.180 reported the synthesis of a thermo and pH dual-responsive nanogel of hydroxypropylcellulose–poly(acrylic acid) (HPC–PAA) particles with sIPN polymer network structure. The novelty of this system was that depending on the chemical composition and the degree of crosslinking, the thermoresponsive behavior could be shifted from the upper critical solution temperature (UCST) to the LCST. Additionally, oxaliplatin was successfully loaded in the nanogel showing a high in vitro anticancer activity.
Another thermoresponsive system, based on poly(oligo ethylene glycol) (POEG), was used by Zhou et al.,182 who combined the advantages of linear PEG and temperature-responsive polymers in a single macromolecular structure. They have designed a new class of pH-responsive CHI-based nanogels by IPN of CHI chains into a POEG chain network (Fig. 8). The POEG–CHI nanogels responded to the changes in environmental pH and the cell internalization is facilitated by the positive surface charge of the nanogels in the extracellular pH conditions of the tumor (∼6.0–6.2). More interestingly, the IPN enabled a remote modulation of the pH response by external cooling/heating (cryo/thermo treatment). The nanogel was loaded with 5-FU, a model anticancer drug, which could be released from the drug carriers upon increased acidity in subcellular compartments (∼5.0). The in vitro studies in B16F10 melanoma cells showed reduced toxicity in combined chemo-thermo treatments but significantly enhanced therapeutic efficacy in combined chemo-cryo treatments.
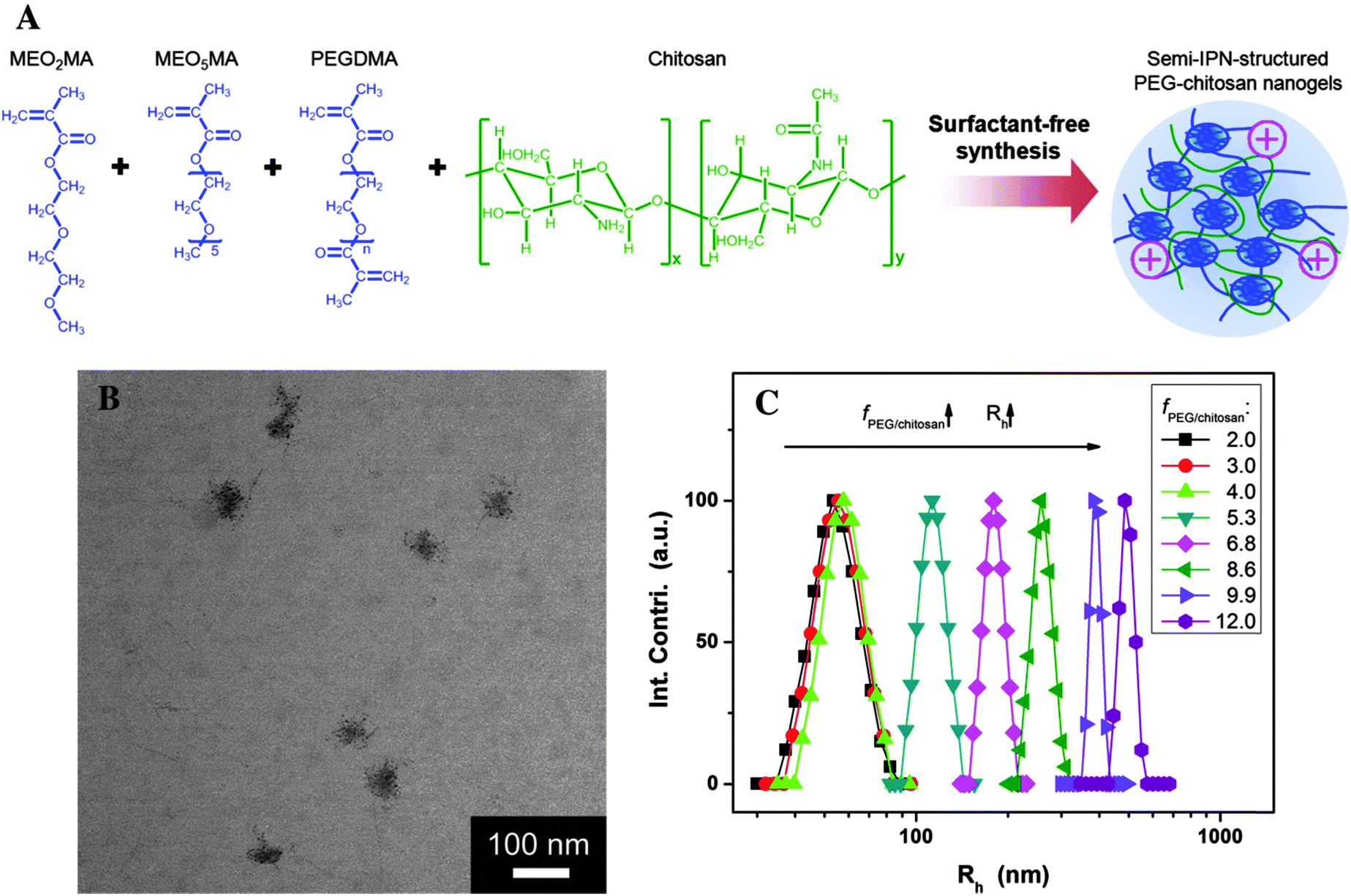 | ||
| Fig. 8 (A) Schematic illustration of POEG–chitosan nanogels with a semi-IPN structure. (B) TEM images of POEG–chitosan nanogels. (C) Effect of molar ratio of fPOEG/CHI on the average hydrodynamic radius (Rh) of POEG-CHI nanogel particles. Reprinted from ref. 182 with permission from John Wiley & Sons, Inc. | ||
Additionally, the IPN strategy was extensively studied for the production of glucose sensitive nanogels in order to address the slow-time-response issue of the other insulin delivery systems reported previously.183 Wu et al.170 reported the synthesis of a nanogel dispersion with three interpenetrating polymer networks consisting of PNIPAm, Dex, and poly(3-acrylamidophenylboronic acid) (P(NIPAm–Dex–APBA)). The particle size could be tuned with the Dex content. The swelling behavior of the nanogels at different glucose concentrations revealed their sensitivity to glucose. Furthermore, the nanogels had good biocompatibility with L-929 mouse fibroblast cells. The loading amount of insulin was 16.2%, whereas drug release was dependent on the composition of Dex and the glucose concentration in the release medium. In vivo experiments have shown the efficacy of the insulin-nanogels to decrease the glucose levels in the blood of diabetic rats and the hypoglycemic effect of the insulin-nanogels was similar to that of the free insulin. They have further studied diabetes and developed a nanoglucometer based on a fluorescent hybrid nanogel glucometer (FNG) for intracellular glucometry.171 This nanogel contained ZnO QDs covalently bonded onto a crosslinked network of PAAm, which was interpenetrated in a network of poly(N-isopropylacrylamide-co-2-acrylamidomethyl-5-fluorophenylboronic acid) [poly(NIPAm-co-FPBA)]. This double-network-structured FNG responded to the glucose levels of the surrounding media and converted the disruptions in the homeostasis of glucose level into fluorescence signals at a fast time response. The high sensitivity and selectivity together with the good penetration in model B16F10 cells showed the promise of the FNG for application as a nanoglucometer. In this example, the IPN strategy was used in order to overcome the problems during repeated swelling–shrinking cycles that are inherent to the simple network systems.
Another application of the IPN strategy was the modification of the release profile of different drugs.123,184 In this context, Babu et al.184 developed a pH-sensitive IPN of sodium alginate (NaAlg) and AA for controlled release of ibuprofen (IB). While NaAlg disintegrated in the intestinal fluid, PAA provided pH-sensitivity to the nanogel.
Wu et al.123 reported the synthesis of hybrid nanogels based on the immobilization of CdSe QDs in poly(methacrylic acid) (PMAA) networks sIPN with CHI. They have shown that the crosslinked structure conferred colloidal and structural stability compared with the non-crosslinked hybrid nanogels. The covalently crosslinked hybrid nanogels proved to have negligible cytotoxicity, could sense the environmental pH change, and regulated the release of anticancer drug, temozolomide (TMZ), in the typical abnormal pH range of 5–7.4.
Very recently, a new application of the IPN strategy was developed by Liu et al.185 They have synthesized an AgNP-loaded PNIPAM–PAA interpenetrated microgel for its use in surface-enhanced Raman scattering (SERS). SERS is an optical detection technique with higher sensitivity than traditional techniques and, more importantly, it can be used for detection in blood and other biological environments.186 In this work, they have proved that the SERS intensity can be reversibly tuned with temperature or pH, showing an increase when lowering the pH from 7 to 3 or rising the temperature from 20 to 45 °C. With this study, new opportunities in the field of theranostics are open.
3.2. Core–shell polymer networks
Nanogels with core–shell architectures were designed to yield separate but covalently bound compartments, which result in inhomogeneous but chemically stable single particles. This inhomogeneity can be exploited for the design of nanogels that respond to multiple signals. Typical examples describe thermoresponsive nanogels exhibiting more than one phase transition temperature,169,187 amphoteric nanogels that swell in both basic and acidic media but shrink near the isoelectric point,188 nanogels that are both thermo- and pH-responsive,133,169,188–192 core–shell nanogels with controlled drug delivery,193,194etc.The grafting of specific (macro-)molecules to the core of nanogels was used to yield star-like core–shell nanogels, which are active towards enhanced cell epitope recognition30,195,196 or increased protein resistance of the nanogels.197–201 PEGylated nanogels are the most common example of core–shell nanogels in which PEG acts as the shell. The grafting of linear PEG chains on the surface of the nanogels contributes to a drastic increase in the bioavailability of the particle due to the protein-resistant properties of PEG.202 Its use is widespread due to its approval by the Food and Drug Administration (FDA) for pharmacological use. The readers are referred to the review by Knop et al. for a more comprehensive discussion of PEG as the shell for drug delivery applications.202
A review by Richtering and Pich extensively discussed the peculiar swelling properties of stimuli-responsive core–shell nanogels.203 It was observed that the mechanical properties of such particles are determined by both core and shell, which dynamically influence each other. The architecture of core–shell particles confers physical constrictions to the swelling of the two single compartments. This leads to varied LCSTs as well as swelling degrees.
In the pioneering work of Jones and Lyon,189 which reported in 2000 the first example of a core–shell nanogel, it is observed that the shell swelling behavior plays a major role in the swelling of the whole particle. This effect is especially observed when a charged PNIPAm-co-AA polymer is in the shell rather than its uncharged PNIPAM counterpart. When added to the shell, the decreased interchain distance in the core polymer leads to decreased LCST as well as lower swelling degree.
Since then, a large number of studies have been published in this field describing the synthesis and characterization of core–shell nanogels. A review by Hendrickson et al.204 in 2010 already described multifunctional core–shell nanoparticles. We will therefore preferentially focus our attention on more recent papers regarding core–shell nanogels applied in nanomedicine. A number of studies reported the synthesis and characterization of novel core–shell nanogels, as listed in Table 5.
| Core | Shell | Responsiveness | Ref. |
|---|---|---|---|
| P(NIPAm-co-CMA) | P(DMA-co-CMA) | Temperature (LCST NIPAm + UCST nanogel) | 187 |
| P(MeO2MA) | P(MeO2MA-co-OEGMA) | Temperature (2 LCST) and pH | 169 |
| P(IADME-co-VIm) | P(VCL) | Temperature, pH (amphoteric) | 188 |
| OEG | OEG or PEG | Temperature | 200 |
| PEAMA | PEG | pH | 201 |
| Poly(DVB)-co-(PEG, NIPAm, MBA, MPA) | PNIPAm-co-(MBA, MPA) | Temperature | 207 |
| OEG | PEG | Temperature | 208 |
| BAP | PEG | Reducing conditions | 210 |
| PEAMA | PEG | pH | 211 |
The synthesis of core–shell nanogels was reported following different procedures. The most common technique is the seed precipitation polymerization, in which the already prepared thermoresponsive core is used in its collapsed state for the growth of the shell on its surface via a second step of radical polymerization.30,188,189,191,195,196,205 Another widespread synthetic technique is the crosslinking of amphiphilic micelles preformed by self-assembly133,187,192,197,206 or the reversible addition–fragmentation chain-transfer polymerization (RAFT).169,187,190,200,206–208 All these synthetic methodologies can give core–shell nanogels with facile scalability and low polydispersity.
The imaging of core–shell nanogels was typically performed via transmission and scanning electron microscopy (TEM, SEM)133,169,187–189,191,198,199,206,209,210 or atomic force microscopy (AFM).192,200,208,209 The visualization of the two distinct compartments in polymer-only core–shell nanoparticles is usually problematic due to the soft properties of these materials. Nevertheless, TEM and AFM microscopy provided some nice micrographs of core–shell nanogels displaying the distinct environments, as shown in Fig. 9.189,191,192 The data obtained by characterization via small-angle X-Ray scattering (SAXS) and small-angle neutron scattering (SANS) were used to retrieve radial structural information for the nanogels. The data were fitted to a concentric core–shell model function to give a more accurate description of the size of core and shell compartments.197,201,211
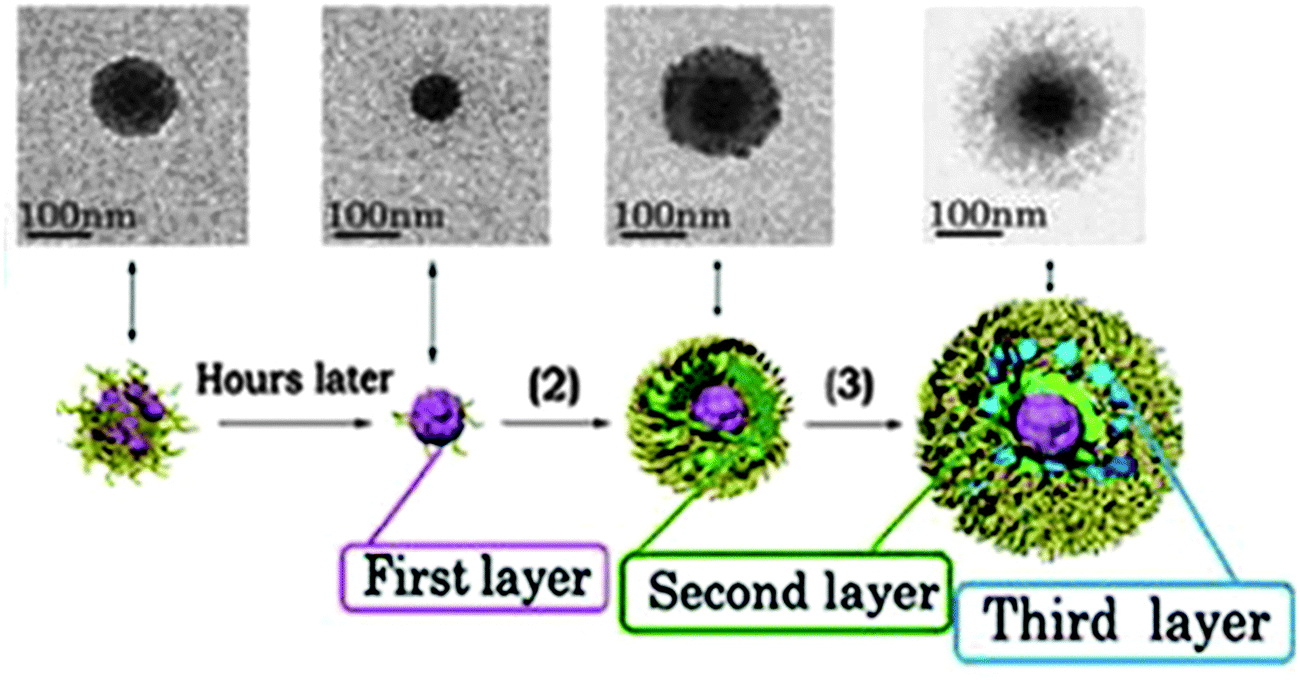 | ||
| Fig. 9 TEM image of the controlled formation of microgels/nanogels. Adapted with permission from ref. 210. Copyright (2012) American Chemical Society. | ||
Among other biomedical applications, several studies were conducted for the synthesis of core–shell nanogels as effective carriers for short interfering ribonucleic acid (siRNA) delivery. An interesting review about this subject was published in 2012 by Smith and Lyon.212
Glucose-responsive core–shell nanogels were developed for the treatment of diabetes mellitus. Lapeyre et al.205 and Zhao et al.198 developed systems that efficiently prolonged the release of insulin in the presence of elevated blood glucose levels. PNIPAm-based core–shell nanogels including a comonomerized glucose-responsive phenylboronic acid (PBA) derivative in the shell were synthesized.205 It was found that PBA forms stable complexes with glucose, and as a result the increased hydrophilicity of the complex leads ultimately to the swelling of the nanogels. The synthesis of the thermoresponsive PNIPAm core occurred via precipitation polymerization, and the collapsed cores were subsequently used as seeds for the second analogous polymerization step, which was performed to include the PBA derivatives APBA or 4-(1,6-dioxo)-2,5-diaza-7-oxamyl phenylboronic acid (DDOPBA) as the glucose-responsive comonomer together with NIPAm. Core–shell nanogels obtained with DDOPBA were used for biological studies because of their higher biocompatibility at physiological pH, the pKa of DDOPBA being 7.8. The swelling behavior of the PNIPAm–APBA core–shell nanogels was extensively investigated in relation with glucose binding, and a complex behavior was observed. At temperatures below the LCST of PNIPAm (T = 32 °C), binding of glucose to the nanogels led to swelling of the shell, which then allowed the relaxation of the otherwise constrained core. Moreover, the swelling degree of the shell was found to be a tuned response, directly proportional to the glucose concentration. The results for PNIPAm–DDOPBA nanogels were found to be very similar to those of PNIPAm–APBA nanogels, although a higher transition temperature was observed in this case. The breathing-in technique was used to encapsulate FITC-insulin in the nanogels, and a prolonged retention of the drug was observed for the loaded nanogels. The release of insulin from PNIPAm–DDOPBA nanogels proved to be dependent on the presence of glucose over a 9 h time span, as shown from fluorescence spectroscopy.
Park et al.197 reported the synthesis of pH-responsive, core–shell poly(aspartic acid)–PEG nanogels, which were also able to prolong the release of insulin. A linear block copolymer of PEG-co-poly(succinimide) (PEG-co-PSI) was synthesized, which formed micelles via self-assembly. After micellation the PSI residues were crosslinked with hexamethylenediamine to form the precursor nanogel. Subsequently, an alkaline hydrolysis of the PSI core led to the formation of the poly(aspartic acid)–PEG nanogel (PAsp–PEG). Thus, the nanogel core turned hydrophilic and pH-responsive. The PAsp–PEG nanogels were incubated in a solution of recombinant human insulin, and its release from the nanogels was investigated. The release curve was found to be dependent on the pH: the release was completed within 48 h at pH 7.4, while at pH 2 only 50% of the model drug was released in the same time span.
Analogous protein carriers were developed as core–shell nanogels. Chen et al. reported the synthesis of pH-responsive hollow core–porous shell PAA nanogels that exhibited surprisingly high loading capacities (800 wt% for BSA and 200 wt% for DOX).209 The synthesis proceeded via the semi-interpenetration of PAA with HPC, the latter being removed in a subsequent step by alkaline hydrolysis, to give a core–shell nanogel of PAA alone. The removal of HPC left a void in the nanogel core. It was assumed that this structural organization was responsible for the very high loading capacities obtained after incubation of BSA and DOX. For BSA, the loading capacity was 8.0 mg per mg nanogel. Interestingly, TEM images showed that the BSA loaded nanogels became solid nanoparticles, while before encapsulation the nanogels had been soft materials. For DOX, the loading capacity (2 mg per mg nanogel) was lower than that of BSA, but nevertheless very high if compared with the values obtained by other nanocarriers.213 The release behavior for BSA was shown to be almost independent of the pH, indicating that the driving force of encapsulation and release was not of electrostatic nature, but dependant on hydrophobic interactions. Circular dichroism spectra proved that the native structure of BSA was retained after encapsulation and release. On the other hand, the release profile of DOX was shown to be strongly dependent on the pH. In more acidic conditions (pH 4.0), the release of DOX was shown to be much faster than at a physiological pH of 7.4.
Another example of a protein carrier was developed by Bhuchar et al.206 Core–shell nanogels exhibiting a thermoresponsive poly(methoxydiethylene glycol methacrylate)-co-poly(2-amino-ethyl methacrylamide hydrochloride) (MeODEGM-co-AEMA) core and a shell of poly(2-methacryloyloxyethyl phosphorylcholine) were synthesized via a one-step RAFT. The resulting nanogels exhibited a thermoresponsive core with 2,2-dimethacroyloxy-1-ethoxypropane as the crosslinker, which degrades in acidic conditions. The nanogel loading occurred via incubation in a protein solution (BSA, insulin, or β-galactosidase). The cationic AEMA residues present in the core of the nanogels increased the binding affinity of the oppositely charged proteins. The release of proteins was investigated via BCA assay, and the observed rate increased with decreasing pH, as expected from pH-degradable nanogels. Moreover, the release rates were found to be dependent on the size of the proteins: the smaller the protein, the faster its release out of the nanogels. Table 6 recapitulates the most recent studies on core–shell nanogels with controlled drug release.
| Core | Shell | Responsiveness | Controlled release | Ref. |
|---|---|---|---|---|
| PNIPMAm | PNIPMAm-co-aminopropyl methacrylate-YSA peptide | Temperature | siRNA and chemosensitization to docetaxel | 30 and 192 |
| Poly(aspartic acid) | PEG | pH | Rh-insulin | 197 |
| PEG-co-poly(phenylboronic acid) | PEG | [Glucose] | Insulin | 198 |
| Poly(L-cystine-co-γ-benzyl-glutamate) | PEG | Reducing conditions | Indometacin | 199 |
| PNIPAm | PNIPAm-co-4-(1,6-dioxo-2,5-diaza-7-oxamyl)phenylboronic acid | [Glucose], temperature | Insulin | 205 |
| Poly(methoxydiethylene glycol)methacrylate and poly(2-aminoethyl methacrylamide hydrochloride) | Poly(2-methacryloyloxyethyl phosphorylcholine) | Temperature responsive and pH degradable | BSA, insulin, β-galactosidase | 206 |
| PAA (hollow) | PAA (porous) | pH | Very high loading (800% BSA, 200% DOX) but slow release | 209 |
Wu et al. reported the synthesis of an enzyme-degradable polyphosphoester (PPE) core–galactosylated (Gal) PEG shell nanogel for the treatment of hepatic carcinoma (Fig. 10).214 The synthesis of the nanogels proceeded via ring-opening polymerization of 3,6-dioxaoctan-1,8-diyl bis(ethylene phosphate) (TEGDP) together with monomethoxy-PEG (mMePEG) chains, initiated by stannous octoate in dimethylsulfoxide (DMSO) at 60 °C. The resulting nanogels were coupled with lactobionic acid in the presence of PEG–NH2 residues, assisted by sulfo-NHS and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) coupling agents, to give Gal-PEG chains. The galactosyl ligand on PEG was used to target the asialoglycoprotein receptor (ASGP-R), which is overexpressed in mammalian hepatocytes.215 The nanogels were loaded with DOX and incubated with HepG2 cells at 4 °C to investigate the receptor binding of Gal-nanogels, and the flow cytometry data showed a remarkable increase of fluorescence for Gal-nanogels compared to the non-galactosylated ones. Addition of lactobionic acid to the cell environment showed a competitive inhibition of the binding of Gal-nanogels on the cell receptors. Induced hepatocarcinoma in rats was treated with DOX-loaded Gal-nanogels, and a decrease in tumor mass was observed for DOX-loaded Gal-nanogels compared to all other samples and controls.
 | ||
| Fig. 10 (A) Schematic representation of DOX-loaded Gal-nanogels. (B and C) In vivo studies on HCC-bearing rats after treatment with PBS, free DOX, m-PPE–DOX, or Gal-PPE–DOX. Adapted from ref. 214 with permission from The Royal Society of Chemistry. | ||
Moreover, biodegradable core–shell nanogels were reported by Xing et al. with the synthesis of a PEGylated poly(L-cystine-co-γ-benzyl-glutamate) nanogel via ring-opening polymerization.199 The core was degradable under reducing conditions due to the presence of cystine residues. Interestingly, the reduction of the disulfide bonds of the cystine residues led to the formation of linear polymer chains rather than smaller products. This was due to the fact that the crosslinker was not present in the backbone of the nanogel but only on the single chains. The nanogels were loaded with the anti-inflammatory drug, indometacin, and a release study under both reducing (10−2 M dithiothreitol (DTT)) and non-reducing conditions was conducted. The release of indometacin was complete after 200 h under reducing conditions, while for non-reducing conditions a maximum release of 35% was observed after 80 h.
4. Preclinical and clinical development/application
Nanogel-based formulations have proven to be useful scaffolds in nanomedicine including drug delivery, anticancer therapy, imaging, etc., in in vivo animal models, but still many safety issues have to be overcome before the results can be applied to clinical practice. Without doubt, cancer is one of the most challenging and studied applications for hybrid nanogels, as was discussed in the previous sections and in numerous reviews.216–219Nevertheless, hybrid nanogels were applied not only in anticancer therapies but also in different medical applications like diabetes,220 skin treatment,221 cosmetics,222 lenses,168 vaccines,223 neurosciences,224 lupus,25etc.Table 7 summarizes the hybrid nanogels in preclinical and clinical trials.
| Phase | Hybrid nanogel | Application | Ref. |
|---|---|---|---|
| Preclinical | Plasmonic@NG | Cancer therapy | 69 |
| Core–shell nanogels | Neurodegenerative disorders | 224–227 | |
| Core–shell nanogels | Treatment of acute pulmonary inflammation | 228 | |
| CHP-W9-peptide | Bone loss disorder | 237 | |
| CHP | Vaccines | 233, 238 and 239 | |
| Clinical | CHP | Vaccines | 234, 235, 240 and 241 |
Kavanov and Vinogradov225 have extensively studied the use of nanogels for the treatment of neurodegenerative disorders in order to overcome the rapid clearance of the macromolecules injected in blood. They have developed cationic nanogels based on PEG and polyethylenimine for the systemic delivery of oligonucleotides (ODN) to the central nervous system.226 Further studies proved that by using the core–shell strategy for modifying the surface of those nanogels with polypeptides (transferrin or insulin) the permeability of the ODN increased two-fold.227
So far, the results of using the core–shell strategy in order to obtain a targeted therapy are promising for further clinical trials.118 For example, Coll Ferrer et al.228 designed a biocompatible nanogel composed of a Dex shell and a lysozyme core conjugated with an antibody (AntiICAM-1) directed to the pulmonary endothelium as a dexamethasone carrier to treat acute pulmonary inflammation in an animal model of LPS-induced lung injury. In vivo studies demonstrated the therapeutic efficiency of ICAM-NG-DEX in mice, while when a control nanogel was used it did not have an anti-inflammatory effect of the encapsulated drug.
Despite great advances in the development and biological evaluation of nanogels, only a few examples of clinical trials have been reported. The most advanced studies are for CHP nanogels. In 1991 Akiyoshi et al. showed that these hydrophobized polysaccharides form monodisperse and colloidally stable nanogels in water upon self-aggregation229,230 and that they have a strong binding for hydrophobic guest molecules,231 proteins, and enzymes.232 Since then, CHP nanogels have been used as drug delivery systems in vivo and in clinical trials, not only for cancer therapy233–235 but also in treating Alzheimer's disorder,236 for bone loss disorder,237 and vaccines.238,239
As an example of CHP preclinical studies, Nochi et al.239 have used CHP nanogels as an intranasal vaccine-delivery system. In this work, they synthesized a non-ionic CHP nanogel and a cationic, amino modified, CHP nanogel (cCHP) (Fig. 11), thereby demonstrating that a neurotoxin BoHc/A administered intranasally with the cCHP nanogel is effectively taken up by mucosal dendritic cells after its release (Fig. 11b). Moreover, the hybrid nanogels did not accumulate in the olfactory bulbs or brain and induced tetanus-toxoid-specific systemic and mucosal immune responses.
 | ||
| Fig. 11 (a) Schematic representation of the hybrid nanogels. (b) Computed tomography images of the drug delivered to the nasal mucosa. (c) Retention of BoHc/A in the nasal tissues after intranasal immunization. Reprinted with permission from Macmillan Publishers Ltd: Nature Materials from ref. 239, copyright (2010). | ||
Clinical trials of CHP nanogels yielded promising results. In a phase I clinical trial, a CHP–HER2 complex vaccine was administrated to HER2-expressing cancer patients to evaluate the safety and immune response to 146HER2.240 The CHP-HER2 vaccine proved to be safe, well tolerated, and to induce HER2-specific CD8+ and/or CD4+ T-cell immune response. In a phase II clinical trial 146HER2-specific antibody responses following CHP-HER2 vaccination were analyzed.235 The results indicated that CHP-HER2 induced HER2-specific humoral responses. Moreover, the antibody titers increased over repeated vaccinations, and all reached the plateau levels within nine vaccinations.
Similar results were obtained by Kawabata et al.234,241 when they evaluated the safety and the humoral immune responses of CHP-HER2 and CHP-NY-ESO-1 combination vaccines with the immuno-adjuvant OK-432 administered to therapy-refractory esophageal cancer patients.
CHP nanogels have shown encouraging results in clinical trials. However, additional optimization will be necessary for clinical approval.242
5. Future perspectives and conclusions
We have discussed here various aspects of hybrid nanogels and their applicability in nanomedicine. In recent years, it has been demonstrated that nanotechnology when applied to medicine can provide superior therapeutics and ways to diagnose different diseases. This research area is of such interest that further developments are occurring almost on a daily basis. The research on multifunctional hybrid nanogels is mostly focused on the synthesis of drug delivery systems due to their ability to efficiently encapsulate therapeutics and release them upon an environmental stimulus. However, the capacity of the hybrid materials to convert external signals to heat, generate highly oxidative species, etc., has increased the potential of hybrid nanogels for combinatorial therapies and theranostics.Furthermore, due to their size and softness, nanogels are easily internalized by the cells and show less accumulation in non-target tissues. Besides these inherent properties of smart nanogels, hybrid nanogels are very attractive because they let new sensitivities to be added to the system via a simple hybridization of the components which allows multiresponsive materials to be obtained. As we have shown in this review, the incorporation of new responsivities to smart nanogels widens the utilization of these systems in several applications such as imaging, guided therapy, and hyperthermia. In this way, it is possible to envision the development of novel materials for nanomedicine that rely not only on the biological endogenous signals like acidic pH, reductive potential, enzyme overexpression, etc., but also on external stimulus for the triggering of activity. This will enable the preparation of smart nanomaterials with superior targetability and minimized side effects that exert their therapeutic or diagnostic function only at the site where the external trigger was applied.
The hybridization of different kinds of materials will need to follow a rational screening that ensures the synergistic combination of the best properties of both components toward applications in nanomedicine. This represents a great opportunity for chemists, material scientists, and biologists to develop new methodologies that allow one to quickly access the properties and application potential of newly synthesized hybrid nanogels. In particular, novel biological techniques that provide information about biodegradability, biological fate, and in vitro and in vivo therapeutic or diagnostic effects upon irradiation are of great need.
Nevertheless, the application of these new materials in the industry is still in its first steps and there is an urgent need for relevant clinical data relating to safety and efficacy of hybrid nanogels in vivo. In particular, a substantial number of unmet issues of the plasmonic, magnetic, and carbonaceous nanoparticles, regarding the pharmacodynamics, metabolism, and pharmacokinetics still need to be assessed before hybrid nanogels can fully make the transition from bench to bedside. We hope that by highlighting the latest and more significant studies in the field, we will inspire the community to develop and perform studies toward the clinical use of such promising systems.
Abbreviations
| AA | Acrylic acid |
| AEMA | 2-Amino-ethyl methacrylamide hydrochloride |
| AFM | Atomic force microscopy |
| AgNPs | Silver nanoparticles |
| AMF | Alternating magnetic field |
| APBA | 3-Acrylamidophenylboronic acid |
| ARS | Alizarin red S |
| ASGP-R | Asialoglycoprotein receptor |
| ATRP | Atom-transfer radical polymerization |
| Au@NGs | Gold hybrid nanogels |
| AuNPs | Gold nanoparticles |
| BAP | Poly(1-(2-aminoethyl)piperazine-co-N,N′-bis(acryloyl)cystamine) |
| BCA | Bicinchoninic acid |
| bPEI | Branched polyethyleneimines |
| BSA | Bovine serum albumin |
| Ce6 | Chlorin e6 |
| CHI | Chitosan |
| CHP | Cholesterol-bearing pullulan |
| CHPNH2 | Amino-modified CHP |
| CNTs | Carbon nanotubes |
| Cy5.5 | Cyanine5.5 |
| DDOPBA | 4-(1,6-Dioxo-2,5-diaza-7-oxamyl)phenylboronic acid |
| Dex | Dextran |
| DMAP | 4-Dimethylaminopyridine |
| DMSO | Dimethylsulfoxide |
| DOTA | 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid |
| DOX | Doxorubicin |
| DTPA | Diethylenetriaminepentaacetic acid |
| DTT | Dithiothreitol |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| EGDMA | Ethylene glycol dimethacrylate |
| FDA | Food and Drug Administration |
| FITC | Fluorescein isothiocyanate |
| FPBA | 2-Acrylamidomethyl-5-fluorophenylboronic acid |
| FRET | Fluorescence resonance energy transfer |
| 5-FU | 5-Fluorouracil |
| Gal | Galactosylated |
| Gd | Gadolinium |
| HPC | Hydroxypropylcellulose |
| hMSCs | Human mesenchymal stem cells |
| HUVECs | Human umbilical vascular endothelial cells |
| IB | ibuprofen |
| INH | isoniazid |
| IPN | Interpenetrating polymer network |
| iRGD | Tumor targeting peptides (CRGDRCPDC)-SH |
| i.v. | Intravenous |
| LCST | Lower critical solution temperature |
| LRP1 | Lipoprotein receptor-related protein 1 |
| MBAAm | N,N′-Methylenebisacrylamide |
| MEA | Methacryloylethyl acrylate |
| MeODEGM | Methoxydiethylene glycol methacrylate |
| mMePEG | Monomethoxy-PEG |
| MePEGA | Methoxyl-PEG methacrylate |
| MnFe2O4 | Manganese iron oxide |
| MRI | Magnetic resonance imaging |
| MTs | Magneto-transducers |
| NaAlg | Sodium alginate |
| NDs | Nanodiamonds |
| NHS | N-Hydroxysulfosuccinimide |
| NIPAm | N-Isopropylacrylamide |
| NIPMAm | N-Isopropylmethacrylamide |
| NIR | Near infrared |
| ODN | Oligonucleotides |
| PAA | Polyacrylic acid |
| PAAm | Polyacrylamide |
| Pasp | Poly(aspartic acid) |
| PBA | Phenylboronic acid |
| PBS | Phosphate buffered saline |
| PDT | Photodynamic therapy |
| PEAMA | poly-[2-(N,N-diethylamino)ethyl methacrylate] |
| PEG | Polyethyleneglycol |
| PEG-d | Polyethylene glycol-diacrylate |
| PEOMA | Poly(ethylene glycol) methyl ether methacrylate |
| γ-PGA | Poly(γ-glutamic acid) |
| PLL | Poly(L-lysine) |
| PMAA | Poly(methacrylic acid) |
| PNIPAm | Poly(N-isopropylacrylamide) |
| PNIPMAm | Poly(N-isopropylmethacrylamide) |
| POEG | Poly(oligo ethylene glycol) |
| PPE | Polyphosphoester |
| PSI | Poly(succinimide) |
| PSNPs | Porous silica nanoparticles |
| PT | Photothermal transductor |
| PTT | Photothermal therapy |
| PVA | Polyvinyl alcohol |
| P(VA-b-VCL) | poly(vinyl alcohol-b-N-vinylcaprolactam) |
| p(VPBA-co-DMAEA) | Poly(4-vinylphenylboronic acid-co-2-(dimethylamino)ethyl acrylate) |
| QDs | Quantum dots |
| QD@NGs | Quantum dot hybrid nanogels |
| QT | Pentaerythritol tetra(3-mercaptopropionate) |
| RAFT | Reversible addition–fragmentation chain-transfer polymerization |
| RES | Reticuloendothelial system |
| RF | Radiofrequency |
| rGO | Reduced graphene oxide |
| RHB | Rhodamine B |
| SANS | Small-angle neutron scattering |
| SAXS | Small-angle X-ray scattering |
| SEM | Scanning electron microscopy |
| SERS | Surface-enhanced raman scattering |
| sIPN | Semi interpenetrating polymer network |
| siRNA | Short interfering RNA (ribonucleic acid) |
| SPIONs | Superparamagnetic iron nanoparticles |
| TEGDP | 3,6-Dioxaoctan-1,8-diyl bis(ethylene phosphate) |
| TEM | Transmission electron microscopy |
| TMZ | Temozolomide |
| UCST | Upper critical solution temperature |
| YSA | 12 amino acid peptide (YSAYPDSVPMMS) |
Acknowledgements
We gratefully acknowledge financial support from the Bundesministerium für Bildung und Forschung (BMBF) through the NanoMatFutur award (Thermonanogele, 13N12561), the Helmholtz Virtual Institute, Multifunctional Biomaterials for Medicine, the Freie Universität Focus Area Nanoscale, and the Deutsche Forschungsgemeinschaft (DFG)/German Research Foundation via SFB1112, Project A04. Dr Maria Molina acknowledges financial support from the Alexander von Humboldt Foundation. Dr Julian Bergueiro acknowledges financial support from Dahlem Research Center through the Dahlem International Network Postdocs program. We gratefully acknowledge Dr Pamela Winchester for proofreading the manuscript.References
- M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS PubMed.
- E. M. Ahmed, J. Adv. Res., 2013, 6, 105–121 CrossRef PubMed.
- Y. Gao, J. Xie, H. Chen, S. Gu, R. Zhao, J. Shao and L. Jia, Biotechnol. Adv., 2014, 32, 761–777 CrossRef CAS PubMed.
- M. Asadian-Birjand, A. Sousa-Herves, D. Steinhilber, J. C. Cuggino and M. Calderon, Curr. Med. Chem., 2012, 19, 5029–5043 CrossRef CAS PubMed.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS PubMed.
- R. Tong, L. Tang, L. Ma, C. Tu, R. Baumgartner and J. Cheng, Chem. Soc. Rev., 2014, 43, 6982–7012 RSC.
- D. Steinhilber, T. Rossow, S. Wedepohl, F. Paulus, S. Seiffert and R. Haag, Angew. Chem., Int. Ed., 2013, 52, 13538–13543 CrossRef CAS PubMed.
- J. C. Cuggino, C. I. Alvarez I, M. C. Strumia, P. Welker, K. Licha, D. Steinhilber, R.-C. Mutihac and M. Calderon, Soft Matter, 2011, 7, 11259–11266 RSC.
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1–33 CrossRef CAS PubMed.
- M. Molina, M. Giulbudagian and M. Calderón, Macromol. Chem. Phys., 2014, 215, 2414–2419 CrossRef CAS.
- M. Giulbudagian, M. Asadian-Birjand, D. Steinhilber, K. Achazi, M. Molina and M. Calderon, Polym. Chem., 2014, 5, 6909–6913 RSC.
- S. Kazakov, M. Kaholek, I. Gazaryan, B. Krasnikov, K. Miller and K. Levon, J. Phys. Chem. B, 2006, 110, 15107–15116 CrossRef CAS PubMed.
- X. Zhang, K. Achazi, D. Steinhilber, F. Kratz, J. Dernedde and R. Haag, J. Controlled Release, 2014, 174, 209–216 CrossRef CAS PubMed.
- L. Zha, B. Banik and F. Alexis, Soft Matter, 2011, 7, 5908–5916 RSC.
- S. Nayak and L. A. Lyon, Angew. Chem., Int. Ed., 2005, 44, 7686–7708 CrossRef CAS PubMed.
- J. Khandare, M. Calderon, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- Y. Chen, X. Zheng, X. Wang, C. Wang, Y. Ding and X. Jiang, ACS Macro Lett., 2013, 3, 74–76 CrossRef.
- M. C. CollFerrer, R. Ferrier Jr., D. Eckmann and R. Composto, J. Nanopart. Res., 2012, 15, 1–7 Search PubMed.
- W.-H. Chiang, V. T. Ho, H.-H. Chen, W.-C. Huang, Y.-F. Huang, S.-C. Lin, C.-S. Chern and H.-C. Chiu, Langmuir, 2013, 29, 6434–6443 CrossRef CAS PubMed.
- T. M. Ruhland, P. M. Reichstein, A. P. Majewski, A. Walther and A. H. E. Müller, J. Colloid Interface Sci., 2012, 374, 45–53 CrossRef CAS PubMed.
- J.-M. Shen, X.-M. Guan, X.-Y. Liu, J.-F. Lan, T. Cheng and H.-X. Zhang, Bioconjugate Chem., 2012, 23, 1010–1021 CrossRef CAS PubMed.
- H. Wang, F. Ke, A. Mararenko, Z. Wei, P. Banerjee and S. Zhou, Nanoscale, 2014, 6, 7443–7452 RSC.
- R. M. Sankar, K. M. Seeni Meera, D. Samanta, P. Jithendra, A. B. Mandal and S. N. Jaisankar, Colloids Surf., B, 2013, 112, 120–127 CrossRef CAS PubMed.
- R. M. Sankar, K. M. S. Meera, D. Samanta, A. Murali, P. Jithendra, A. Baran Mandal and S. N. Jaisankar, RSC Adv., 2012, 2, 12424–12430 RSC.
- M. Look, E. Stern, Q. A. Wang, L. D. DiPlacido, M. Kashgarian, J. Craft and T. M. Fahmy, J. Clin. Invest., 2013, 123, 1741–1749 CAS.
- S. Kim, D. J. Lee, D. S. Kwag, U. Y. Lee, Y. S. Youn and E. S. Lee, Carbohydr. Polym., 2014, 101, 692–698 CrossRef CAS PubMed.
- W. Wu and S. Zhou, Nanomaterials in Drug Delivery, Imaging, and Tissue Engineering, John Wiley & Sons, Inc., 2013, pp. 269–319 Search PubMed.
- J. Ge, E. Neofytou, T. J. Cahill, R. E. Beygui and R. N. Zare, ACS Nano, 2011, 6, 227–233 CrossRef PubMed.
- T. Kawano, Y. Niidome, T. Mori, Y. Katayama and T. Niidome, Bioconjugate Chem., 2009, 20, 209–212 CrossRef CAS PubMed.
- W. H. Blackburn, E. B. Dickerson, M. H. Smith, J. F. McDonald and L. A. Lyon, Bioconjugate Chem., 2009, 20, 960–968 CrossRef CAS PubMed.
- X. Liu, H. Guo and L. Zha, Polym. Int., 2012, 61, 1144–1150 CrossRef CAS.
- A. Lohani, G. Singh, S. S. Bhattacharya and A. Verma, J. Drug Delivery, 2014, 2014, 11 Search PubMed.
- J. Bergueiro and M. Calderón, Macromol. Biosci., 2014, 15, 183–199 CrossRef PubMed.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- G. Liu and Z. An, Polym. Chem., 2014, 5, 1559–1565 RSC.
- D. M. Eckmann, R. J. Composto, A. Tsourkas and V. R. Muzykantov, J. Mater. Chem. B, 2014, 2, 8085–8097 RSC.
- C. Sanchez, G. J. d. A. A. Soler-Illia, F. Ribot, T. Lalot, C. R. Mayer and V. Cabuil, Chem. Mater., 2001, 13, 3061–3083 CrossRef CAS.
- C. Sanchez, B. Julian, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559–3592 RSC.
- W. Wu and S. Zhou, Nano Rev., 2010, 1, 5730 Search PubMed.
- T. Hoare, J. Santamaria, G. F. Goya, S. Irusta, D. Lin, S. Lau, R. Padera, R. Langer and D. S. Kohane, Nano Lett., 2009, 9, 3651–3657 CrossRef CAS PubMed.
- J. Yang, M.-H. Yao, L. Wen, J.-T. Song, M.-Z. Zhang, Y.-D. Zhao and B. Liu, Nanoscale, 2014, 6, 11282–11292 RSC.
- D. Jaque, L. Martinez Maestro, B. Del Rosal, P. Haro-Gonzalez, A. Benayas, J. L. Plaza, E. Martin Rodriguez and J. Garcia Sole, Nanoscale, 2014, 6, 9494–9530 RSC.
- V. Shanmugam, S. Selvakumar and C. S. Yeh, Chem. Soc. Rev., 2014, 43, 6254–6287 RSC.
- N. Fomina, J. Sankaranarayanan and A. Almutairi, Adv. Drug Delivery Rev., 2012, 64, 1005–1020 CrossRef CAS PubMed.
- J. Thevenot, H. Oliveira, O. Sandre and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 7099–7116 RSC.
- H. Wang, J. Yi, S. Mukherjee, P. Banerjee and S. Zhou, Nanoscale, 2014, 6, 13001–13011 RSC.
- R. Liang, M. Wei, D. G. Evans and X. Duan, Chem. Commun., 2014, 50, 14071–14081 RSC.
- X. Lian, J. Jin, J. Tian and H. Zhao, ACS Appl. Mater. Interfaces, 2010, 2, 2261–2268 CAS.
- T. Niidome, J. Phys.: Conf. Ser., 2010, 232, 012011 CrossRef.
- M. Oishi, H. Hayashi, T. Uno, T. Ishii, M. Iijima and Y. Nagasaki, Macromol. Chem. Phys., 2007, 208, 1176–1182 CrossRef CAS.
- N. Singh and L. A. Lyon, Chem. Mater., 2007, 19, 719–726 CrossRef CAS.
- M. Oishi, T. Nakamura, Y. Jinji, K. Matsuishi and Y. Nagasaki, J. Mater. Chem., 2009, 19, 5909–5912 RSC.
- S. Shi, Q. Wang, T. Wang, S. Ren, Y. Gao and N. Wang, J. Phys. Chem. B, 2014, 118, 7177–7186 CrossRef CAS PubMed.
- C. Xiao, S. Chen, L. Zhang, S. Zhou and W. Wu, Chem. Commun., 2012, 48, 11751–11753 RSC.
- D. J. Siegwart, A. Srinivasan, S. A. Bencherif, A. Karunanidhi, J. K. Oh, S. Vaidya, R. Jin, J. O. Hollinger and K. Matyjaszewski, Biomacromolecules, 2009, 10, 2300–2309 CrossRef CAS PubMed.
- M. Oishi, A. Tamura, T. Nakamura and Y. Nagasaki, Adv. Funct. Mater., 2009, 19, 827–834 CrossRef CAS.
- Y. Chen, X. Zheng, X. Wang, C. Wang, Y. Ding and X. Jiang, ACS Macro Lett., 2014, 3, 74–76 CrossRef CAS.
- T. Nakamura, A. Tamura, H. Murotani, M. Oishi, Y. Jinji, K. Matsuishi and Y. Nagasaki, Nanoscale, 2010, 2, 739–746 RSC.
- J. Y. Kim, W. I. Choi, M. Kim and G. Tae, J. Controlled Release, 2013, 171, 113–121 CrossRef CAS PubMed.
- H. Yasui, R. Takeuchi, M. Nagane, S. Meike, Y. Nakamura, T. Yamamori, Y. Ikenaka, Y. Kon, H. Murotani, M. Oishi, Y. Nagasaki and O. Inanami, Cancer Lett., 2014, 347, 151–158 CrossRef CAS PubMed.
- N. S. Rejinold, R. Ranjusha, A. Balakrishnan, N. Mohammed and R. Jayakumar, RSC. Adv., 2014, 4, 5819–5825 RSC.
- X.-Q. Zhao, T.-X. Wang, W. Liu, C.-D. Wang, D. Wang, T. Shang, L.-H. Shen and L. Ren, J. Mater. Chem., 2011, 21, 7240–7247 RSC.
- S. Su, H. Wang, X. Liu, Y. Wu and G. Nie, Biomaterials, 2013, 34, 3523–3533 CrossRef CAS PubMed.
- T. Kawano, Y. Niidome, T. Mori, Y. Katayama and T. Niidome, Bioconjugate Chem., 2009, 20, 209–212 CrossRef CAS PubMed.
- Z. Zhang, J. Wang, X. Nie, T. Wen, Y. Ji, X. Wu, Y. Zhao and C. Chen, J. Am. Chem. Soc., 2014, 136, 7317–7326 CrossRef CAS PubMed.
- H. Kang, A. C. Trondoli, G. Zhu, Y. Chen, Y.-J. Chang, H. Liu, Y.-F. Huang, X. Zhang and W. Tan, ACS Nano, 2011, 5, 5094–5099 CrossRef CAS PubMed.
- W. Wu, J. Shen, P. Banerjee and S. Zhou, Biomaterials, 2011, 32, 598–609 CrossRef CAS PubMed.
- E. V. Panfilova, B. N. Khlebtsov and N. G. Khlebtsov, Colloid J., 2013, 75, 333–338 CrossRef CAS.
- W. Wu, T. Zhou, A. Berliner, P. Banerjee and S. Zhou, Chem. Mater., 2010, 22, 1966–1976 CrossRef CAS.
- W. Wu, N. Mitra, E. C. Y. Yan and S. Zhou, ACS Nano, 2010, 4, 4831–4839 CrossRef CAS PubMed.
- J. W. M. Bulte and D. L. Kraitchman, NMR Biomed., 2004, 17, 484–499 CrossRef CAS PubMed.
- C. W. Jung and P. Jacobs, Magn. Reson. Imaging, 1995, 13, 661–674 CrossRef CAS PubMed.
- Y.-X. J. Wang, Quant. Imaging Med. Surg., 2011, 1, 35–40 Search PubMed.
- Y.-X. Wang, S. Hussain and G. Krestin, Eur. Radiol., 2001, 11, 2319–2331 CrossRef CAS PubMed.
- G. Shahnaz, C. Kremser, A. Reinisch, A. Vetter, F. Laffleur, D. Rahmat, J. Iqbal, S. Dünnhaupt, W. Salvenmoser, R. Tessadri, U. Griesser and A. Bernkop-Schnürch, Eur. J. Pharm. Biopharm., 2013, 85, 346–355 CrossRef CAS PubMed.
- R. A. M. Heesakkers, G. J. Jager, A. M. Hövels, B. de Hoop, H. C. M. van den Bosch, F. Raat, J. A. Witjes, P. F. A. Mulders, C. H. van der Kaa and J. O. Barentsz, Radiology, 2009, 251, 408–414 CrossRef PubMed.
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- X. Wang, Z. Zhou, Z. Wang, Y. Xue, Y. Zeng, J. Gao, L. Zhu, X. Zhang, G. Liu and X. Chen, Nanoscale, 2013, 5, 8098–8104 RSC.
- W.-H. Chiang, W.-C. Huang, C.-W. Chang, M.-Y. Shen, Z.-F. Shih, Y.-F. Huang, S.-C. Lin and H.-C. Chiu, J. Controlled Release, 2013, 168, 280–288 CrossRef CAS PubMed.
- H. K. Patra, N. U. Khaliq, T. Romu, E. Wiechec, M. Borga, A. P. F. Turner and A. Tiwari, Adv. Healthcare Mater., 2014, 3, 526–535 CrossRef CAS PubMed.
- C. Niu, Z. Wang, G. Lu, T. M. Krupka, Y. Sun, Y. You, W. Song, H. Ran, P. Li and Y. Zheng, Biomaterials, 2013, 34, 2307–2317 CrossRef CAS PubMed.
- W. Dong, Y. Li, D. Niu, Z. Ma, J. Gu, Y. Chen, W. Zhao, X. Liu, C. Liu and J. Shi, Adv. Mater., 2011, 23, 5392–5397 CrossRef CAS PubMed.
- Y. Wang, F. Xu, C. Zhang, D. Lei, Y. Tang, H. Xu, Z. Zhang, H. Lu, X. Du and G.-Y. Yang, Nanomedicine, 2011, 7, 1009–1019 CrossRef CAS PubMed.
- J. H. Hwang, Y.-W. Noh, J.-H. Choi, J.-R. Noh, Y.-H. Kim, G.-T. Gang, K.-S. Kim, H. S. Park, Y. T. Lim, H. Moon, K. S. Hong, H. G. Lee, B. H. Chung and C.-H. Lee, Magn. Reson. Med., 2014, 71, 1054–1063 CrossRef CAS PubMed.
- J. Lu, S. Ma, J. Sun, C. Xia, C. Liu, Z. Wang, X. Zhao, F. Gao, Q. Gong, B. Song, X. Shuai, H. Ai and Z. Gu, Biomaterials, 2009, 30, 2919–2928 CrossRef CAS PubMed.
- S. Srivastava, R. Awasthi, D. Tripathi, M. K. Rai, V. Agarwal, V. Agrawal, N. S. Gajbhiye and R. K. Gupta, Small, 2012, 8, 1099–1109 CrossRef CAS PubMed.
- L. Zhu, D. Wang, X. Wei, X. Zhu, J. Li, C. Tu, Y. Su, J. Wu, B. Zhu and D. Yan, J. Controlled Release, 2013, 169, 228–238 CrossRef CAS PubMed.
- H. Wang, J. Shen, G. Cao, Z. Gai, K. Hong, P. R. Debata, P. Banerjee and S. Zhou, J. Mater. Chem. B, 2013, 1, 6225–6234 RSC.
- C.-K. Lim, A. Singh, J. Heo, D. Kim, K. E. Lee, H. Jeon, J. Koh, I.-C. Kwon and S. Kim, Biomaterials, 2013, 34, 6846–6852 CrossRef CAS PubMed.
- L. Jiang, Q. Zhou, K. Mu, H. Xie, Y. Zhu, W. Zhu, Y. Zhao, H. Xu and X. Yang, Biomaterials, 2013, 34, 7418–7428 CrossRef CAS PubMed.
- A. Soleimani, F. Martinez, V. Economopoulos, P. J. Foster, T. J. Scholl and E. R. Gillies, J. Mater. Chem. B, 2013, 1, 1027–1034 RSC.
- H. M. Kim, Y.-W. Noh, H. S. Park, M. Y. Cho, K. S. Hong, H. Lee, D. H. Shin, J. Kang, M.-H. Sung, H. Poo and Y. T. Lim, Small, 2012, 8, 666–670 CrossRef CAS PubMed.
- C. Prashant, M. Dipak, C.-T. Yang, K.-H. Chuang, D. Jun and S.-S. Feng, Biomaterials, 2010, 31, 5588–5597 CrossRef CAS PubMed.
- J. Liu, C. Detrembleur, A. Debuigne, M.-C. De Pauw-Gillet, S. Mornet, L. Vander Elst, S. Laurent, E. Duguet and C. Jérôme, J. Mater. Chem. B, 2014, 2, 1009–1023 RSC.
- W. H. Chiang, V. T. Ho, H. H. Chen, W. C. Huang, Y. F. Huang, S. C. Lin, C. S. Chern and H. C. Chiu, Langmuir, 2013, 29, 6434–6443 CrossRef CAS PubMed.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- M. H. M. Dias and P. C. Lauterbur, Magn. Reson. Med., 1986, 3, 328–330 CrossRef CAS.
- C. Corot, P. Robert, J.-M. Idée and M. Port, Adv. Drug Delivery Rev., 2006, 58, 1471–1504 CrossRef CAS PubMed.
- J. Pintaske, P. Martirosian, H. Graf, G. Erb, K. P. Lodemann, C. D. Claussen and F. Schick, Invest. Radiol., 2006, 41, 213–221 CrossRef PubMed.
- K. M. L. Taylor, A. Jin and W. Lin, Angew. Chem., Int. Ed., 2008, 47, 7722–7725 CrossRef CAS PubMed.
- J. Lux, M. Chan, L. Vander Elst, E. Schopf, E. Mahmoud, S. Laurent and A. Almutairi, J. Mater. Chem. B, 2013, 1, 6359–6364 RSC.
- M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS PubMed.
- P. Mitchell, Nat. Biotechnol., 2001, 19, 1013–1017 CrossRef CAS PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- H. Mattoussi, G. Palui and H. B. Na, Adv. Drug Delivery Rev., 2012, 64, 138–166 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- V. Biju, S. Mundayoor, R. V. Omkumar, A. Anas and M. Ishikawa, Biotechnol. Adv., 2010, 28, 199–213 CrossRef CAS PubMed.
- G. O. Menendez, M. Eva Pichel, C. C. Spagnuolo and E. A. Jares-Erijman, Photochem. Photobiol. Sci., 2013, 12, 236–240 CAS.
- Z. Li, W. Xu, Y. Wang, B. R. Shah, C. Zhang, Y. Chen, Y. Li and B. Li, Carbohydr. Polym., 2015, 121, 477–485 CrossRef CAS PubMed.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CAS.
- W. C. W. Chan, D. J. Maxwell, X. Gao, R. E. Bailey, M. Han and S. Nie, Curr. Opin. Biotechnol., 2002, 13, 40–46 CrossRef CAS PubMed.
- U. Hasegawa, S.-I. M. Nomura, S. C. Kaul, T. Hirano and K. Akiyoshi, Biochem. Biophys. Res. Commun., 2005, 331, 917–921 CrossRef CAS PubMed.
- N. S. Rejinold, K. P. Chennazhi, H. Tamura, S. V. Nair and J. Rangasamy, ACS Appl. Mater. Interfaces, 2011, 3, 3654–3665 Search PubMed.
- Y.-Q. Wang, Y.-Y. Zhang, F. Zhang and W.-Y. Li, J. Mater. Chem., 2011, 21, 6556–6562 RSC.
- W. Wu, T. Zhou, J. Shen and S. Zhou, Chem. Commun., 2009, 4390–4392 RSC.
- D. Jańczewski, N. Tomczak, M.-Y. Han and G. J. Vancso, Macromolecules, 2009, 42, 1801–1804 CrossRef.
- Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- A. M. Smith, H. Duan, A. M. Mohs and S. Nie, Adv. Drug Delivery Rev., 2008, 60, 1226–1240 CrossRef CAS PubMed.
- K. Park, S. Lee, E. Kang, K. Kim, K. Choi and I. C. Kwon, Adv. Funct. Mater., 2009, 19, 1553–1566 CrossRef CAS.
- W. Wu, M. Aiello, T. Zhou, A. Berliner, P. Banerjee and S. Zhou, Biomaterials, 2010, 31, 3023–3031 CrossRef CAS PubMed.
- H. Zhu, Y. Li, R. Qiu, L. Shi, W. Wu and S. Zhou, Biomaterials, 2012, 33, 3058–3069 CrossRef CAS PubMed.
- W. Wu, J. Shen, P. Banerjee and S. Zhou, Biomaterials, 2010, 31, 8371–8381 CrossRef CAS PubMed.
- P. Yang, S. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- I. I. Slowing, J. L. Vivero-Escoto, B. G. Trewyn and V. S. Y. Lin, J. Mater. Chem., 2010, 20, 7924–7937 RSC.
- Z. Tao, RSC Adv., 2014, 4, 18961–18980 RSC.
- X. Hu, X. Hao, Y. Wu, J. Zhang, X. Zhang, P. C. Wang, G. Zou and X.-J. Liang, J. Mater. Chem. B, 2013, 1, 1109–1118 RSC.
- A. Agostini, L. Mondragón, A. Bernardos, R. Martínez-Máñez, M. D. Marcos, F. Sancenón, J. Soto, A. Costero, C. Manguan-García, R. Perona, M. Moreno-Torres, R. Aparicio-Sanchis and J. R. Murguía, Angew. Chem., Int. Ed. Engl., 2012, 51, 10556–10560 CrossRef CAS PubMed.
- H. Meng, M. Xue, T. Xia, Y.-L. Zhao, F. Tamanoi, J. F. Stoddart, J. I. Zink and A. E. Nel, J. Am. Chem. Soc., 2010, 132, 12690–12697 CrossRef CAS PubMed.
- I. I. Slowing, J. L. Vivero-Escoto, C.-W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- I. I. Slowing, B. G. Trewyn, S. Giri and V. S. Y. Lin, Adv. Funct. Mater., 2007, 17, 1225–1236 CrossRef CAS.
- J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S. Y. Lin, Small, 2010, 6, 1952–1967 CrossRef CAS PubMed.
- S. Chai, J. Zhang, T. Yang, J. Yuan and S. Cheng, Colloids Surf., A, 2010, 356, 32–39 CrossRef CAS.
- G. Liu, C. Zhu, J. Xu, Y. Xin, T. Yang, J. Li, L. Shi, Z. Guo and W. Liu, Colloids Surf., B, 2013, 111, 7–14 CrossRef CAS PubMed.
- B. Chang, D. Chen, Y. Wang, Y. Chen, Y. Jiao, X. Sha and W. Yang, Chem. Mater., 2013, 25, 574–585 CrossRef CAS.
- B.-S. Tian and C. Yang, J. Nanosci. Nanotechnol., 2011, 11, 1871–1879 CrossRef CAS PubMed.
- W. Feng, W. Nie, C. He, X. Zhou, L. Chen, K. Qiu, W. Wang and Z. Yin, ACS Appl. Mater. Interfaces, 2014, 6, 8447–8460 CAS.
- D. F. Acevedo, J. Balach, C. R. Rivarola, M. C. Miras and C. A. Barbero, Faraday Discuss., 2006, 131, 235–252 RSC.
- J. Balach, H. Wu, F. Polzer, H. Kirmse, Q. Zhao, Z. Wei and J. Yuan, RSC Adv., 2013, 3, 7979–7986 RSC.
- S. Jokar, A. Pourjavadi and M. Adeli, RSC Adv., 2014, 4, 33001–33006 RSC.
- A.-H. Lu, G.-P. Hao, Q. Sun, X.-Q. Zhang and W.-C. Li, Macromol. Chem. Phys., 2012, 213, 1107–1131 CrossRef CAS.
- M. Boot-Handford, J. C. Abanades, E. Anthony, M. Blunt, S. Brandani, N. Mac Dowell, J. Fernandez, M.-C. Ferrari, R. Gross, J. Hallett, S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, M. Pourkashanian, G. Rochelle, N. Shah, J. Yao and P. Fennell, Energy Environ. Sci., 2014, 7, 130–189 CAS.
- Y. Gogotsi and V. Presser, Carbon nanomaterials, CRC Press, 2013 Search PubMed.
- J. Gu, S. Su, Y. Li, Q. He and J. Shi, Chem. Commun., 2011, 47, 2101–2103 RSC.
- M. Adeli, R. Soleyman, Z. Beiranvand and F. Madani, Chem. Soc. Rev., 2013, 42, 5231–5256 RSC.
- E. Mehdipoor, M. Adeli, M. Bavadi, P. Sasanpour and B. Rashidian, J. Mater. Chem., 2011, 21, 15456–15463 RSC.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- S. Bosi, T. Da Ros, G. Spalluto and M. Prato, Eur. J. Med. Chem., 2003, 38, 913–923 CrossRef CAS PubMed.
- Z. Chen, L. Ma, Y. Liu and C. Chen, Theranostics, 2012, 2, 238–250 CrossRef CAS PubMed.
- P. Mroz, Y. Xia, D. Asanuma, A. Konopko, T. Zhiyentayev, Y.-Y. Huang, S. K. Sharma, T. Dai, U. J. Khan, T. Wharton and M. R. Hamblin, Nanomedicine, 2011, 7, 965–974 CrossRef CAS PubMed.
- N. Saito, H. Haniu, Y. Usui, K. Aoki, K. Hara, S. Takanashi, M. Shimizu, N. Narita, M. Okamoto, S. Kobayashi, H. Nomura, H. Kato, N. Nishimura, S. Taruta and M. Endo, Chem. Rev., 2014, 114, 6040–6079 CrossRef CAS PubMed.
- E. L. Hopley, S. Salmasi, D. M. Kalaskar and A. M. Seifalian, Biotechnol. Adv., 2014, 32, 1000–1014 CrossRef CAS PubMed.
- C. Barbero, R. Coneo Rodriguez, R. Rivero, M. Martinez, M. Molina, J. Balach, M. Bruno, G. Planes, D. Acevedo, C. Rivarola and M. Miras, Aquananotechnology, CRC Press, 2014, pp. 15–54 Search PubMed.
- R. Bellingeri, F. Alustiza, N. Picco, D. Acevedo, M. A. Molina, R. Rivero, C. Grosso, C. Motta, C. Barbero and A. Vivas, J. Appl. Polym. Sci., 2014, 132 DOI:10.1002/app.41370.
- Y. Qin, J. Chen, Y. Bi, X. Xu, H. Zhou, J. Gao, Y. Hu, Y. Zhao and Z. Chai, Acta Biomater., 2015, 17, 201–204 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- H. Y. Mao, S. Laurent, W. Chen, O. Akhavan, M. Imani, A. A. Ashkarran and M. Mahmoudi, Chem. Rev., 2013, 113, 3407–3424 CrossRef CAS PubMed.
- K. Yang, L. Feng, X. Shi and Z. Liu, Chem. Soc. Rev., 2013, 42, 530–547 RSC.
- R. Mo, T. Jiang, W. Sun and Z. Gu, Biomaterials, 2015, 50, 67–74 CrossRef CAS PubMed.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. Sanchez Casalongue, D. Vinh and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS PubMed.
- N. Lu, J. Liu, J. Li, Z. Zhang, Y. Weng, B. Yuan, K. Yang and Y. Ma, J. Mater. Chem. B, 2014, 2, 3791–3798 RSC.
- Y. Zhang, T. R. Nayak, H. Hong and W. Cai, Nanoscale, 2012, 4, 3833–3842 RSC.
- C. Wang, J. Mallela, U. S. Garapati, S. Ravi, V. Chinnasamy, Y. Girard, M. Howell and S. Mohapatra, Nanomedicine, 2013, 9, 903–911 CrossRef CAS PubMed.
- A. M. Schrand, S. A. C. Hens and O. A. Shenderova, Crit. Rev. Solid State Mater. Sci., 2009, 34, 18–74 CrossRef CAS.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2012, 7, 11–23 CrossRef CAS PubMed.
- E. K. Chow, X.-Q. Zhang, M. Chen, R. Lam, E. Robinson, H. Huang, D. Schaffer, E. Osawa, A. Goga and D. Ho, Sci. Transl. Med., 2011, 3, 73ra21 Search PubMed.
- L. Moore, E. K.-H. Chow, E. Osawa, J. M. Bishop and D. Ho, Adv. Mater., 2013, 25, 3532–3541 CrossRef CAS PubMed.
- H.-J. Kim, K. Zhang, L. Moore and D. Ho, ACS Nano, 2014, 8, 2998–3005 CrossRef CAS PubMed.
- Y. Kotsuchibashi and R. Narain, Polym. Chem., 2014, 5, 3061 RSC.
- Z. Wu, X. Zhang, H. Guo, C. Li and D. Yu, J. Mater. Chem., 2012, 22, 22788–22796 RSC.
- J. Fan, X. Jiang, Y. Hu, Y. Si, L. Ding and W. Wu, Biomater. Sci., 2013, 1, 421–433 RSC.
- M. Nic, J. Jirat and B. Kosata, IUPAC Compendium of Chemical Terminology Gold Book, 2014 Search PubMed.
- Z. Xing, C. Wang, J. Yan, L. Zhang, L. Li and L. Zha, Soft Matter, 2011, 7, 7992–7997 RSC.
- E. S. Dragan, Chem. Eng. J., 2014, 243, 572–590 CrossRef CAS.
- V. Koul, R. Mohamed, D. Kuckling, H.-J. P. Adler and V. Choudhary, Colloids Surf., B, 2011, 83, 204–213 CrossRef CAS PubMed.
- R. C. Mundargi, S. A. Patil, P. V. Kulkarni, N. N. Mallikarjuna and T. M. Aminabhavi, J. Microencapsulation, 2008, 25, 228–240 CrossRef CAS PubMed.
- H. Jiang, C. W. Lo and D. Zhu, Google Pat., US 20130137054 A1, 2013 Search PubMed.
- Z. Li, J. Shen, H. Ma, X. Lu, M. Shi, N. Li and M. Ye, Soft Matter, 2012, 8, 3139–3145 RSC.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- Y. Chen, D. Ding, Z. Mao, Y. He, Y. Hu, W. Wu and X. Jiang, Biomacromolecules, 2008, 9, 2609–2614 CrossRef CAS PubMed.
- N. do Nascimento Marques, P. S. Curti, A. M. da Silva Maia and R. d. C. Balaban, J. Appl. Polym. Sci., 2013, 129, 334–345 CrossRef CAS.
- T. Zhou, C. Xiao, J. Fan, S. Chen, J. Shen, W. Wu and S. Zhou, Acta Biomater., 2013, 9, 4546–4557 CrossRef CAS PubMed.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS PubMed.
- V. Ramesh Babu, K. S. V. Krishna Rao, M. Sairam, B. V. K. Naidu, K. M. Hosamani and T. M. Aminabhavi, J. Appl. Polym. Sci., 2006, 99, 2671–2678 CrossRef.
- X. Liu, X. Wang, L. Zha, D. Lin, J. Yang, J. Zhou and L. Zhang, J. Mater. Chem. C, 2014, 2, 7326–7335 RSC.
- M. Y. Sha, H. Xu, S. G. Penn and R. Cromer, Nanomedicine, 2007, 2, 725–734 CrossRef CAS PubMed.
- J. He, B. Yan, L. Tremblay and Y. Zhao, Langmuir, 2011, 27, 436–444 CrossRef CAS PubMed.
- S. Schachschal, A. Balaceanu, C. Melian, D. E. Demco, T. Eckert, W. Richtering and A. Pich, Macromolecules, 2010, 43, 4331–4339 CrossRef CAS.
- C. D. Jones and L. A. Lyon, Macromolecules, 2000, 33, 8301–8306 CrossRef CAS.
- X.-B. Liu, J.-F. Zhou and X.-D. Ye, Chin. J. Chem. Phys., 2012, 25, 463–468 CrossRef CAS.
- W. Zhang, R. Yao, W. Tao, H. He and S. Shui, Colloid Polym. Sci., 2013, 292, 317–324 Search PubMed.
- S. Zschoche, J. C. Rueda, M. Binner, H. Komber, A. Janke, K.-F. Arndt, S. Lehmann and B. Voit, Macromol. Chem. Phys., 2012, 213, 215–226 CrossRef CAS.
- W. He, Y. Lv, Y. Zhao, C. Xu, Z. Jin, C. Qin and L. Yin, Int. J. Pharm., 2015, 484, 163–171 CrossRef CAS PubMed.
- X. Li, P. Du and P. Liu, RSC Adv., 2014, 4, 56323–56331 RSC.
- S. Nayak, H. Lee, J. Chmielewski and L. A. Lyon, J. Am. Chem. Soc., 2004, 126, 10258–10259 CrossRef CAS PubMed.
- E. Dickerson, W. Blackburn, M. Smith, L. Kapa, L. A. Lyon and J. McDonald, BMC Cancer, 2010, 10, 10 CrossRef PubMed.
- C. W. Park, H.-M. Yang, H. J. Lee and J.-D. Kim, Soft Matter, 2013, 9, 1781–1788 RSC.
- L. Zhao, C. Xiao, J. Ding, P. He, Z. Tang, X. Pang, X. Zhuang and X. Chen, Acta Biomater., 2013, 9, 6535–6543 CrossRef CAS PubMed.
- T. Xing, B. Lai, X. Ye and L. Yan, Macromol. Biosci., 2011, 11, 962–969 CrossRef CAS PubMed.
- W. Shen, Y. Chang, G. Liu, H. Wang, A. Cao and Z. An, Macromolecules, 2011, 44, 2524–2530 CrossRef CAS.
- G. Tamura, Y. Shinohara, A. Tamura, Y. Sanada, M. Oishi, I. Akiba, Y. Nagasaki, K. Sakurai and Y. Amemiya, Polym. J., 2011, 44, 240–244 CrossRef.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- W. Richtering and A. Pich, Soft Matter, 2012, 8, 11423 RSC.
- G. R. Hendrickson, M. H. Smith, A. B. South and L. A. Lyon, Adv. Funct. Mater., 2010, 20, 1697–1712 CrossRef CAS.
- V. Lapeyre, C. Ancla, B. Catargi and V. Ravaine, J. Colloid Interface Sci., 2008, 327, 316–323 CrossRef CAS PubMed.
- N. Bhuchar, R. Sunasee, K. Ishihara, T. Thundat and R. Narain, Bioconjugate Chem., 2012, 23, 75–83 CrossRef CAS PubMed.
- L. A. Picos-Corrales, A. Licea-Claveríe and K.-F. Arndt, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4277–4287 CrossRef CAS.
- Y. Chang, W. Shen, H. Wang, G. Liu, Z. An and A. Cao, J. Controlled Release, 2011, 152, e75–e76 CrossRef PubMed.
- Y. Chen, X. Zheng, H. Qian, Z. Mao, D. Ding and X. Jiang, ACS Appl. Mater. Interfaces, 2010, 2, 3532–3538 CAS.
- J. Zhang, F. Yang, H. Shen and D. Wu, ACS Macro Lett., 2012, 1, 1295–1299 CrossRef CAS.
- Y. Shinohara, G. Tamura, I. Akiba, A. Tamura, M. Oishi, Y. Nagasaki, K. Sakurai and Y. Amemiya, J. Phys.: Conf. Ser., 2011, 272, 012018 CrossRef.
- M. H. Smith and L. A. Lyon, Acc. Chem. Res., 2012, 45, 985–993 CrossRef CAS PubMed.
- Y. Zhu, J. Shi, W. Shen, X. Dong, J. Feng, M. Ruan and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 5083–5087 CrossRef CAS PubMed.
- J. Wu, T.-M. Sun, X.-Z. Yang, J. Zhu, X.-J. Du, Y.-D. Yao, M.-H. Xiong, H.-X. Wang, Y.-C. Wang and J. Wang, Biomater. Sci., 2013, 1, 1143–1150 RSC.
- G. Ashwell and J. Harford, Annu. Rev. Biochem., 1982, 51, 531–554 CrossRef CAS PubMed.
- D. Dorwal, Int. J. Pharm. Pharm. Sci., 2012, 4, 67–74 CAS.
- S. Maya, B. Sarmento, A. Nair, N. S. Rejinold, S. V. Nair and R. Jayakumar, Curr. Pharm. Des., 2013, 19, 7203–7218 CrossRef CAS PubMed.
- G. Soni and K. S. Yadav, Saudi Pharm. J. DOI:10.1016/j.jsps.2014.1004.1001.
- M. M. Yallapu, M. Jaggi and S. C. Chauhan, Drug Discovery Today, 2011, 16, 457–463 CrossRef CAS PubMed.
- W. Wu and S. Zhou, Macromol. Biosci., 2013, 13, 1464–1477 CrossRef CAS PubMed.
- S. Dasgupta, S. K. Ghosh, S. Ray, S. Singh Kaurav and B. Mazumder, Curr. Drug Delivery, 2014, 11, 132–138 CrossRef CAS.
- P. Somasundaran, S. C. Mehta, L. Rhein and S. Chakraborty, MRS Bull., 2007, 32, 779–786 CrossRef CAS.
- S. A. Ferreira, F. M. Gama and M. Vilanova, Nanomedicine, 2013, 9, 159–173 CrossRef CAS PubMed.
- J. Gilmore, X. Yi, L. Quan and A. Kabanov, J. Neuroimmune Pharmacol., 2008, 3, 83–94 CrossRef PubMed.
- S. V. Vinogradov, Expert Opin. Drug Delivery, 2007, 4, 5–17 CrossRef CAS PubMed.
- S. Vinogradov, E. Batrakova and A. Kabanov, Colloids Surf., B, 1999, 16, 291–304 CrossRef CAS.
- S. V. Vinogradov, E. V. Batrakova and A. V. Kabanov, Bioconjugate Chem., 2003, 15, 50–60 CrossRef PubMed.
- M. C. Coll Ferrer, V. V. Shuvaev, B. J. Zern, R. J. Composto, V. R. Muzykantov and D. M. Eckmann, PLoS One, 2014, 9, e102329 Search PubMed.
- K. Akiyoshi, S. Yamaguchi and J. Sunamoto, Chem. Lett., 1991, 1263–1266 CrossRef CAS.
- K. Akiyoshi, S. Deguchi, N. Moriguchi, S. Yamaguchi and J. Sunamoto, Macromolecules, 1993, 26, 3062–3068 CrossRef CAS.
- K. Akiyoshi, I. Taniguchi, H. Fukui and J. Sunamoto, Eur. J. Pharm. Biopharm., 1996, 42, 286–290 CAS.
- K. Akiyoshi, S. Kobayashi, S. Shichibe, D. Mix, M. Baudys, S. Wan Kim and J. Sunamoto, J. Controlled Release, 1998, 54, 313–320 CrossRef CAS PubMed.
- T. Shimizu, T. Kishida, U. Hasegawa, Y. Ueda, J. Imanishi, H. Yamagishi, K. Akiyoshi, E. Otsuji and O. Mazda, Biochem. Biophys. Res. Commun., 2008, 367, 330–335 CrossRef CAS PubMed.
- A. Uenaka, H. Wada, M. Isobe, T. Saika, K. Tsuji, E. Sato, S. Sato, Y. Noguchi, R. Kawabata, T. Yasuda, Y. Doki, H. Kumon, K. Iwatsuki, H. Shiku, M. Monden, A. A. Jungbluth, G. Ritter, R. Murphy, E. Hoffman, L. J. Old and E. Nakayama, Cancer Immun., 2007, 7, 9–20 Search PubMed.
- S. Kageyama, S. Kitano, M. Hirayama, Y. Nagata, H. Imai, T. Shiraishi, K. Akiyoshi, A. M. Scott, R. Murphy, E. W. Hoffman, L. J. Old, N. Katayama and H. Shiku, Cancer Sci., 2008, 99, 601–607 CrossRef CAS PubMed.
- Y. Lee, S. Y. Park, C. Kim and T. G. Park, J. Controlled Release, 2009, 135, 89–95 CrossRef CAS PubMed.
- N. Alles, N. S. Soysa, M. D. A. Hussain, N. Tomomatsu, H. Saito, R. Baron, N. Morimoto, K. Aoki, K. Akiyoshi and K. Ohya, Eur. J. Pharm. Sci., 2009, 37, 83–88 CrossRef CAS PubMed.
- H. F. Staats and K. W. Leong, Nat. Mater., 2010, 9, 537–538 CrossRef CAS PubMed.
- T. Nochi, Y. Yuki, H. Takahashi, S.-I. Sawada, M. Mejima, T. Kohda, N. Harada, I. G. Kong, A. Sato, N. Kataoka, D. Tokuhara, S. Kurokawa, Y. Takahashi, H. Tsukada, S. Kozaki, K. Akiyoshi and H. Kiyono, Nat. Mater., 2010, 9, 572–578 CrossRef CAS PubMed.
- S. Kitano, S. Kageyama, Y. Nagata, Y. Miyahara, A. Hiasa, H. Naota, S. Okumura, H. Imai, T. Shiraishi, M. Masuya, M. Nishikawa, J. Sunamoto, K. Akiyoshi, T. Kanematsu, A. M. Scott, R. Murphy, E. W. Hoffman, L. J. Old and H. Shiku, Clin. Cancer Res., 2006, 12, 7397–7405 CrossRef CAS PubMed.
- R. Kawabata, H. Wada, M. Isobe, T. Saika, S. Sato, A. Uenaka, H. Miyata, T. Yasuda, Y. Doki, Y. Noguchi, H. Kumon, K. Tsuji, K. Iwatsuki, H. Shiku, G. Ritter, R. Murphy, E. Hoffman, L. J. Old, M. Monden and E. Nakayama, Int. J. Cancer, 2007, 120, 2178–2184 CrossRef CAS PubMed.
- M. Amidi, E. Mastrobattista, W. Jiskoot and W. E. Hennink, Adv. Drug Delivery Rev., 2010, 62, 59–82 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |