
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DYNAMERS: dynamic polymers as self-healing materials
Nabarun
Roy†
*a,
Bernd
Bruchmann
b and
Jean-Marie
Lehn
*a
aLaboratoire de Chimie Supramoléculaire, ISIS, Université de Strasbourg, 8, allée Gaspard Monge, 67000, Strasbourg, France. E-mail: lehn@unistra.fr; Fax: +33 368 855140
bBASF SE, Joint Research Network on Advanced Materials and Systems (JONAS), Carl-Bosch Str. 38, D-67056, Ludwigshafen, Germany. E-mail: bernd.bruchmann@basf.com
First published on 5th May 2015
Abstract
Importing self-repair or self-healing features into inert materials is of great relevance to material scientists, since it is expected to eliminate the necessity of replenishing a damaged material. Be it material chemistry or more specifically polymer chemistry, such materials have attracted the imagination of both material scientists and chemists. A stroll down the memory lane 70 years back, this might have sounded utopian. However with the current progress in supramolecular chemistry and the emergence of dynamic covalent and non-covalent chemistries, novel perspectives have been opened up to materials science towards the development of dynamic materials (DYNAMATS) and in particular dynamic polymers (DYNAMERS), with the ability to produce such species by custom made designs. Chemistry took giant strides to gain control over the structure and features of materials and, besides basic progress, to apply it for tailor-making matter for applications in our daily life. In that applied perspective, materials science plays a paramount role in shaping our present and in contributing to a sustainable future. The goal is to develop materials, which would be dynamic enough to carry out certain functions as effectively as in biological systems with, however, the freedom to recruit the powers of chemistry on a wider scale, without the limitation imposed by life. Material scientists and in particular polymer chemists may build on chemistry, physics and biology for bridging the gap to develop dynamic materials presenting a wide range of novel functionalities and to convert dreams into reality. In this current review we will focus on developments in the area of dynamic polymers, as a class of dynamic materials presenting self-healing features and, more generally, the ability to undergo adaptation under the effect of physical and/or chemical agents, and thus function as adaptive polymers or ADAPTAMERS.
 Nabarun Roy | Nabarun Roy completed his Masters (MSc Specialisation in Organic Chemistry) in Chemistry from IIT Bombay in 2005. Following his Masters, he pursued his doctoral research in Bio-inorganic chemistry from Max Planck Institute for Chemical Energy Conversion (formerly MPI for Bioinorganic chemistry), Mülheim a.d. Ruhr, Germany under the guidance of Prof. Karl Wieghardt. In 2010, he began his postdoctoral sojourn at Institut de Science et d'Ingénierie Supramoléculaires, at the University of Strasbourg, under the supervision of Prof. Jean-Marie Lehn working on self-healing supramolecular polymers. In 2013, he joined BASF Polyurethanes GmbH, Germany and presently works in the field of polyurethane research. |
 Bernd Bruchmann | Bernd Bruchmann received his PhD degree in Chemistry from the Technical University of Clausthal, Germany, in 1988. In the same year he joined BASF, working on polymers for foams, thermoplastic and coatings applications. From 2000 to 2010 he was heading a research group focused on the development of branched and highly branched polymeric structures. Since 2010 he has been responsible for BASF's European Post Doc Initiative “Joint Research Network on Advanced Materials and Systems” (“JONAS” for short), which is a scientific collaboration of BASF with the I.S.I.S.-Institute at the University of Strasbourg, the University of Freiburg (Germany) and ETH Zurich. |
 Jean-Marie Lehn | Jean-Marie Lehn became a Professor of Chemistry at the Université Louis Pasteur in Strasbourg in 1970 and from 1979 to 2010 he was a Professor at the Collège de France in Paris. He is presently a Professor at the University of Strasbourg Institute for Advanced Study (USIAS). He shared the Nobel Prize in Chemistry in 1987 for his studies on the chemical basis of “molecular recognition”. His work led him to define “supramolecular chemistry” which concerns the chemical species held together by non-covalent intermolecular forces. It developed into the chemistry of “self-organization” processes and more recently towards “adaptive chemistry” and complex systems. |
1. Introduction
Extending the dynamic features inherent to supramolecular chemistry to molecular chemistry has led to the emergence of a constitutional dynamic chemistry based on either covalent bonds or non-covalent interactions that may form or break reversibly, thus allowing for a continuous modification in constitution by reorganization and exchange of building blocks. On the molecular level, considerable research efforts have been vested in developing dynamic materials, which could undergo component exchange or reorganization via reversible chemical reactions under the influence of external agents, thus being able to respond to physical stimuli and/or chemical effectors and behaving as adaptive materials. Dynamicity is imparted into the material owing to the presence of covalent chemical bonds capable of undergoing reversible formation and cleavage, such as for instance the various imine type bonds (imines, acylhydrazones, hydrazones, etc.), disulfides and reversible 4+2 cyclo-additions or Diels–Alder (DA) reactions. Dynamicity is intrinsic on the supramolecular level due to the lability and reversibility of non-covalent interactions, such as H-bonding. As materials of particular significance from both the basic and applied points of view, such dynamic features have been introduced into macromolecules, resulting in the generation of constitutional dynamic polymers or Dynamers.1–4 By virtue of reversible chemical connections (both reversible covalent bonds and non-covalent interactions) between the interconnecting monomeric units, DYNAMERs are able to perform dynamic functions in response to physical stimuli and chemical effectors, by establishing adaptable networks for instance of covalent type.5 They thus represent an intriguing class of adaptive polymers, which one may term Adaptamers. The dynamism within such materials allows for reversible exchange and reorganization of components in response to multiple physical and/or chemical agents (Fig. 1).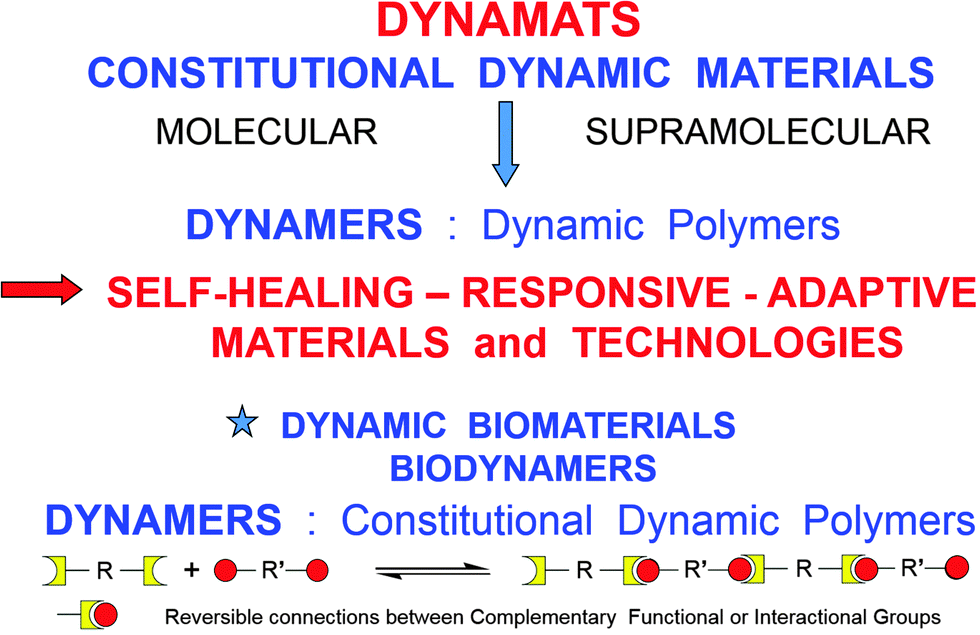 | ||
| Fig. 1 DYNAMATS and DYNAMERS: constitutional dynamic materials and polymers formed through covalent and non-covalent dynamic connections. | ||
In the present article, we would like to focus on two specific types of dynamic materials, namely: dynamic polymers constituted through dynamic covalent and non-covalent connections between building blocks exhibiting self-healing features6–11 and biodynamers obtained by introducing such dynamic features into analogs of biomacromolecules.12–17 We primarily focus on recently reported dynamers, exhibiting dynamism through certain selected covalent reactions such as reversible DA reactions, and imine-type functional groups as well as on dynamic supramolecular polymers.
Various pathways have been implemented to get access to self-healing materials. An exhaustive and comprehensive overview of self-healing polymers through other routes (e.g. crack healing,18 optically healable polymers,19,20 healing through encapsulated healing agents,21 self-healing gels22,23) will not be within the scope of this article, since it has been presented in a number of review articles and books published in the recent past.7–11,24–27
2. DYNAMERS based on reversible Diels–Alder reactions
2.1 DYNAMERS based on reversible DA reactions at elevated temperatures
In the last decade various strategies have been adopted to develop smart materials implementing indigenous methodologies. One of the pathways to achieve this, is to incorporate dynamic covalent bonds, which may form and reversibly cleave so as to allow an intrinsically dynamic exchange of molecular components through reversible chemical reactions. One of such dynamic covalent reactions, which has been extensively studied to develop self-healing materials, is the reversible Diels–Alder (DA) reaction. The DA reaction is a self-contained reversible dynamic reaction, owing to the fact that all atoms of the starting components are also present within the product. There has been a considerable research focus in this direction endeavored to develop self-healing materials based on the DA reaction.24–27 One of the best known DA reactions is that of furan based moieties as dienes with maleimide based motifs as dienophiles. Exploration of the retro-DA reaction based on such [4+2] cyclo-addition is also not a recent discovery. This technology was reported as early as in 196928 and subsequently in the late 1970's, a report described a thermally-crosslinked polymer network incorporating a reversible DA reaction at elevated temperature.29 Following a decade later, a number of publications came to light using furan based components as dienes and maleimide based ones as dienophiles to get access to the reversible DA reaction.30–33In a study of the thermal reversibility of furan-maleimide based benzyl aryl ether dendrons and dendrimers, a DA dendrimer exhibited around 40% dissociation of the DA links after 1 h at 110 °C, and reassembly to the extent of 82% of the original dendron structure was achieved upon cooling the material to 65 °C in a course of 5 days.34 This work was probably the first example of a thermally labile remendable dendrimer making use of the DA/retro-DA (rDA) approach. Subsequently, furan-maleimide based reversible DA chemistry was developed for the design of remendable epoxy resins.35–38 Reversibility of the material could be achieved at temperatures above 90 °C. However, most of these systems are only reversible at elevated temperatures and multiple cycles of fracture and repair were hard to achieve.
A thermally remendable cross-linked polymeric material was described based on a thermally reversible DA cycloaddition of a tetra-furan based multi diene and a tri-maleimide based multi dienophile (Fig. 2).39
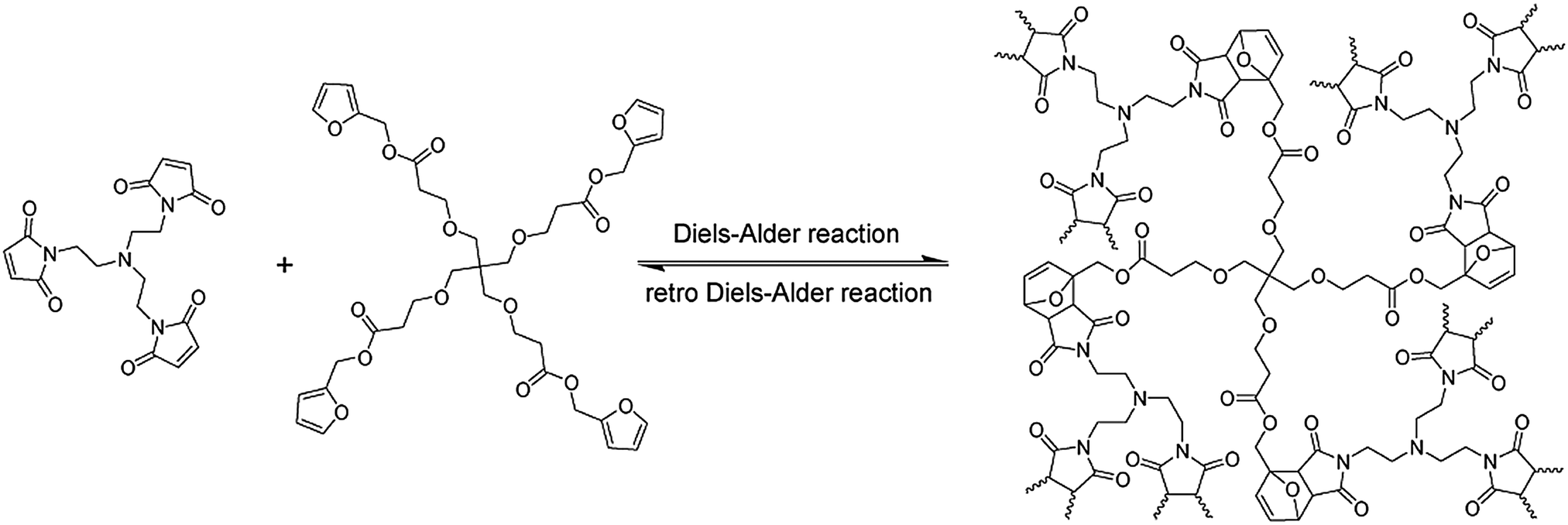 | ||
| Fig. 2 Furan and maleimide based cross-linked healable polymer networks making use of the thermally reversible Diels–Alder reaction reported by Wudl.39 Reproduced by permission26 of The Royal Society of Chemistry. | ||
At a temperature above 120 °C around 30% (determined from the solid-state NMR) of inter-monomer linkages disconnect, however upon cooling the system, they reconnect back generating a reversible mending process. Subsequently another highly cross-linked polymer was reported, based on bis-maleimide and furan moieties, which could remend fractured surfaces through a reversible DA reaction.40,41
A thermally reversible crosslinked polyamide system making use of a reversible DA reaction, based on maleimide and furan functionalized aromatic polyamides, was shown to exhibit self-repairing behavior in maleimide and polyamide furan cross-linked gels42,43 and films44 presenting adduct formation at 50 °C and rDA reaction at 150 °C (Fig. 3).
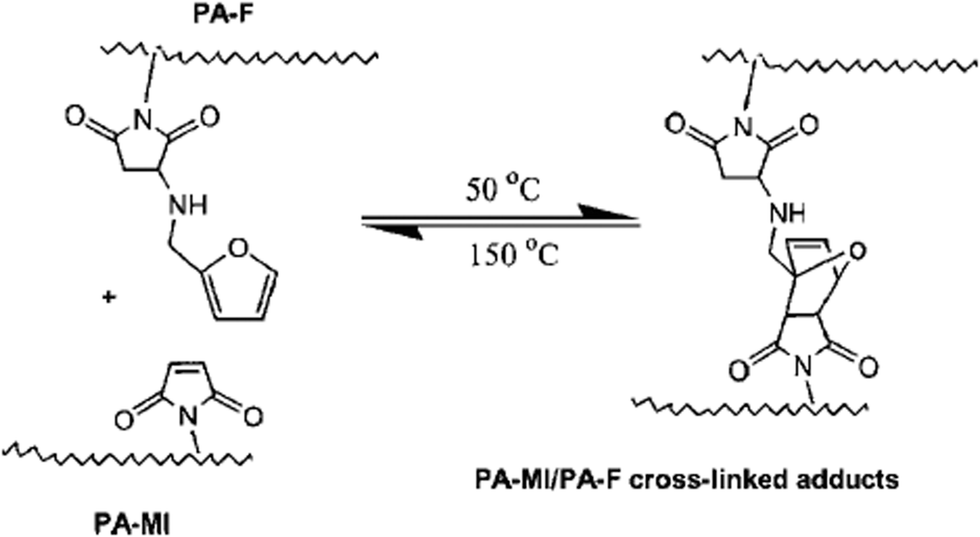 | ||
| Fig. 3 Thermally reversible cross-linking reactions between PA-MI (polyamide-maleimide) and PA-F (polyamide furan). Reproduced with permission.44 Copyright 2007, Wiley-VCH. | ||
There have been a number of publications utilizing the DA reaction at elevated temperatures to get access to thermally or photochemically cross-linkable polyamides with pendant maleimide or phthalimide groups.45–47 Recently, an elegant one pot synthesis of thermo-remendable dynamic polymer system was described, that made use of reversible DA chemistry and click chemistry.48,49
2.2 DYNAMERS based on reversible DA reactions at room temperature
Most of the reactions reported in the literature required elevated temperature beyond 100 °C to initiate the rDA reaction. It was thus of interest to develop DA reactions, which would be reversible under ambient conditions. Such systems would facilitate the generation of self-healing dynamers at a lower temperature or even at room temperature. In search of a room temperature dynamic reversible system in our group, we had uncovered a couple of reversible DA reactions.50–53 One of them, which is of particular interest for the development of self-healing dynamic polymer system, is based on the reaction of 6,6′-substituted fulvenes with dicyano- and tricyano-ethylene carboxyesters in an organic solvent under ambient conditions (Fig. 4).50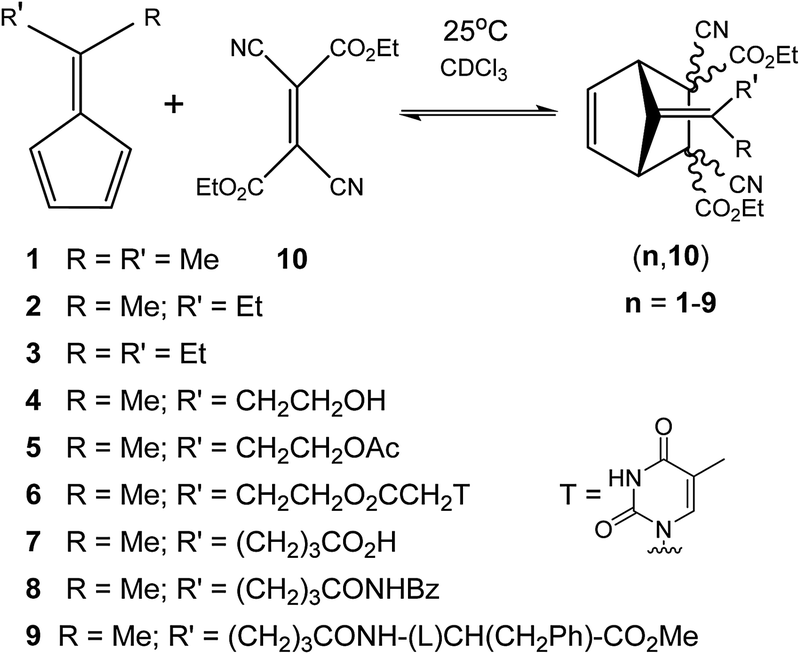 | ||
| Fig. 4 Ambient temperature reversible Diels–Alder reactions between fulvenes (1–9) and diethyldicyanofumarate 10.50 | ||
This series of fulvene based dienes and diethyldicyanofumarate based dienophiles underwent a reversible Diels–Alder reaction at 25 °C and their reversibility has been validated by a component exchange reaction. The dynamics of these DA reactions were studied by performing diene exchange experiments monitoring the reaction by 1H NMR spectroscopy.
Upon addition of 5 equivalents of 1 to an equilibrating mixture of (3,10) and components 3 and 10, within a time span of less than 1 minute, almost the entire (3,10) adduct was displaced in favor of the newly formed adduct (1,10) at 25 °C. This phenomenon was monitored by 1H NMR and the signal at 1.05 ppm corresponding to the terminal methyl group protons of the bridged ethyl group of adduct (3,10) completely disappeared upon addition of 1 (Fig. 5).50
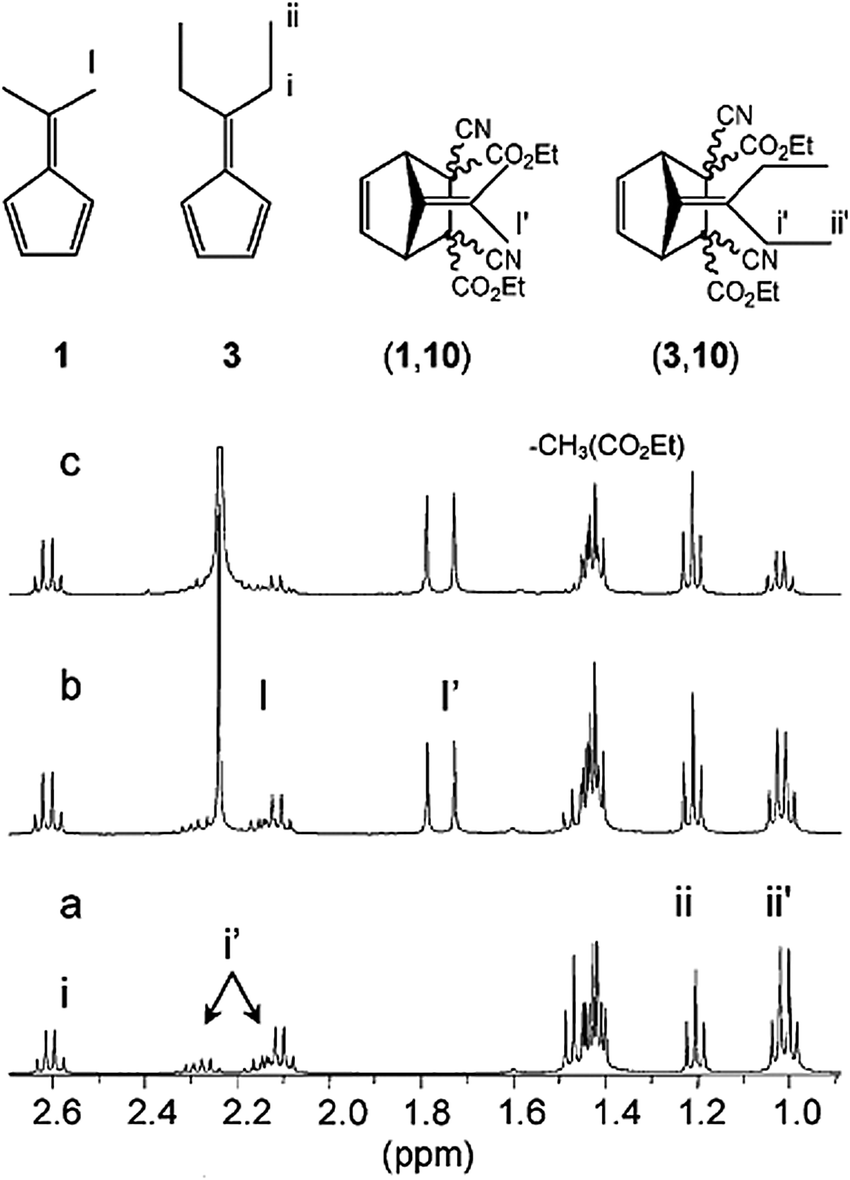 | ||
| Fig. 5 1H NMR spectra indicating the dynamic component exchange between fulvenes and dicyanofumarate esters: (a) 1/1 mixture of diethyl dicyanofumarate 10 (100 mM) and diethylfulvene 3 (100 mM) in CDCl3. (b) Mixture obtained after addition of 1 eq. 1 to solution (a), (c) mixture obtained after addition of 5 eq. of dimethyl fulvene, 1, to solution (a).50 | ||
Along these lines two other room temperature reversible DA reactions were developed using anthracene derivatives as dienes together with cyanofumarates and N-phenyltriazolinedione as dienophiles.51,52 Implementation of the basic process incorporating fulvenes as dienes and cyanofumarates as dienophiles, a polymeric material was developed, which by virtue of the room temperature reversible DA reaction gave access to a self-healing polymeric material. For this purpose bis(dicyanofumarate) (11) and bis(fulvene) terminated polydispersed polyethylene substituted (12) diene were synthesized (Fig. 6).53
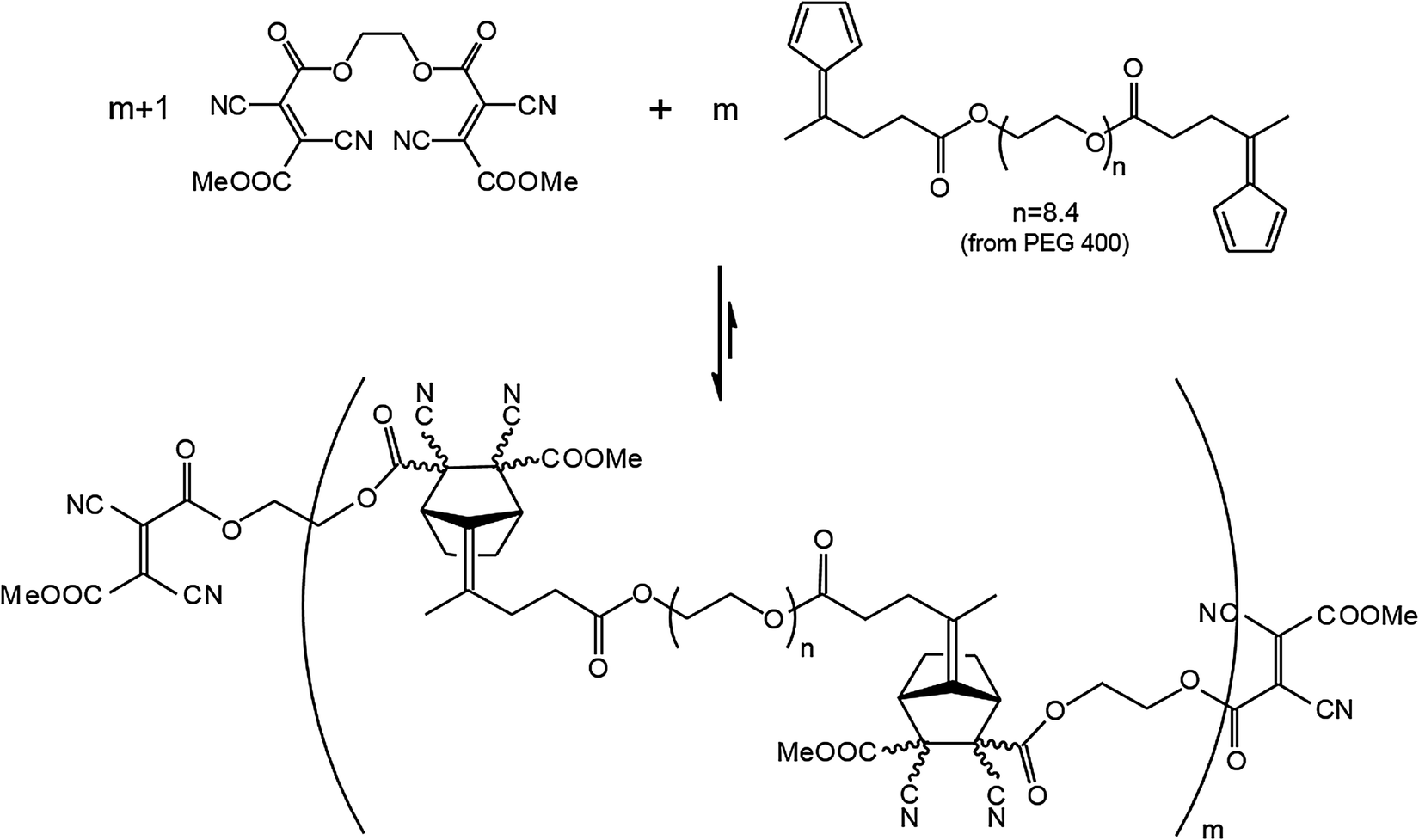 | ||
| Fig. 6 Dynamer (11,12) formed through reversible [4+2] cyclo-addition reaction between dienophile 11 and diene 12.53 | ||
Reaction of 11 and 12 at room temperature yielded a soft stretchy polymer film. This film was cut and the two pieces were overlapped and gently pressed to ensure contact. Within about ten seconds the two separated films could be stretched without tear, demonstrating the self-healing behavior at room temperature (Fig. 7).
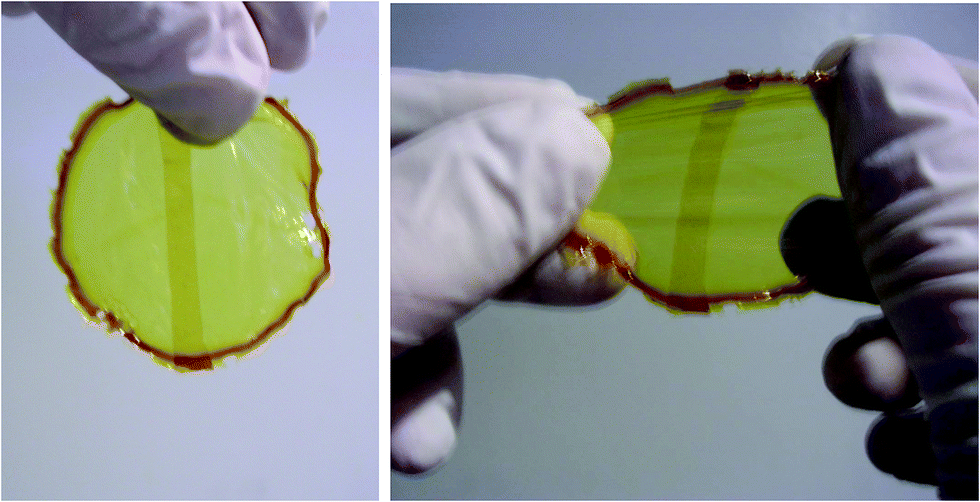 | ||
| Fig. 7 Self-healing of the DYNAMER film (11,12) formed via a room temperature reversible dynamic Diels–Alder reaction between two superimposed halves of the initial film (left) and under elongation strain (right). The central rectangular darker area is the region of overlap of the superimposed two pieces of cut films.53 | ||
It should be noted that in this study, self-healing took place through the overlap of the cut films and not between the edges of freshly cut pieces of material.53
2.3 Materials presenting self-healing features imparted by DA reactions
Contrary to self-healing routes incorporating reversible DA systems, alternative pathways to achieve healing phenomena have been reported. A solvent based system involved self-healing composites based on the functionalization of an epoxy amine network and a reactive healing agent. The healing was achieved through a reversible covalent linkage across the cracked composite surface through a DA reaction: a furan functionalized epoxy amine thermoset was cured by making use of the DA adduct formation with a bis-maleimide DMF solution injected under low pressure (Fig. 8).54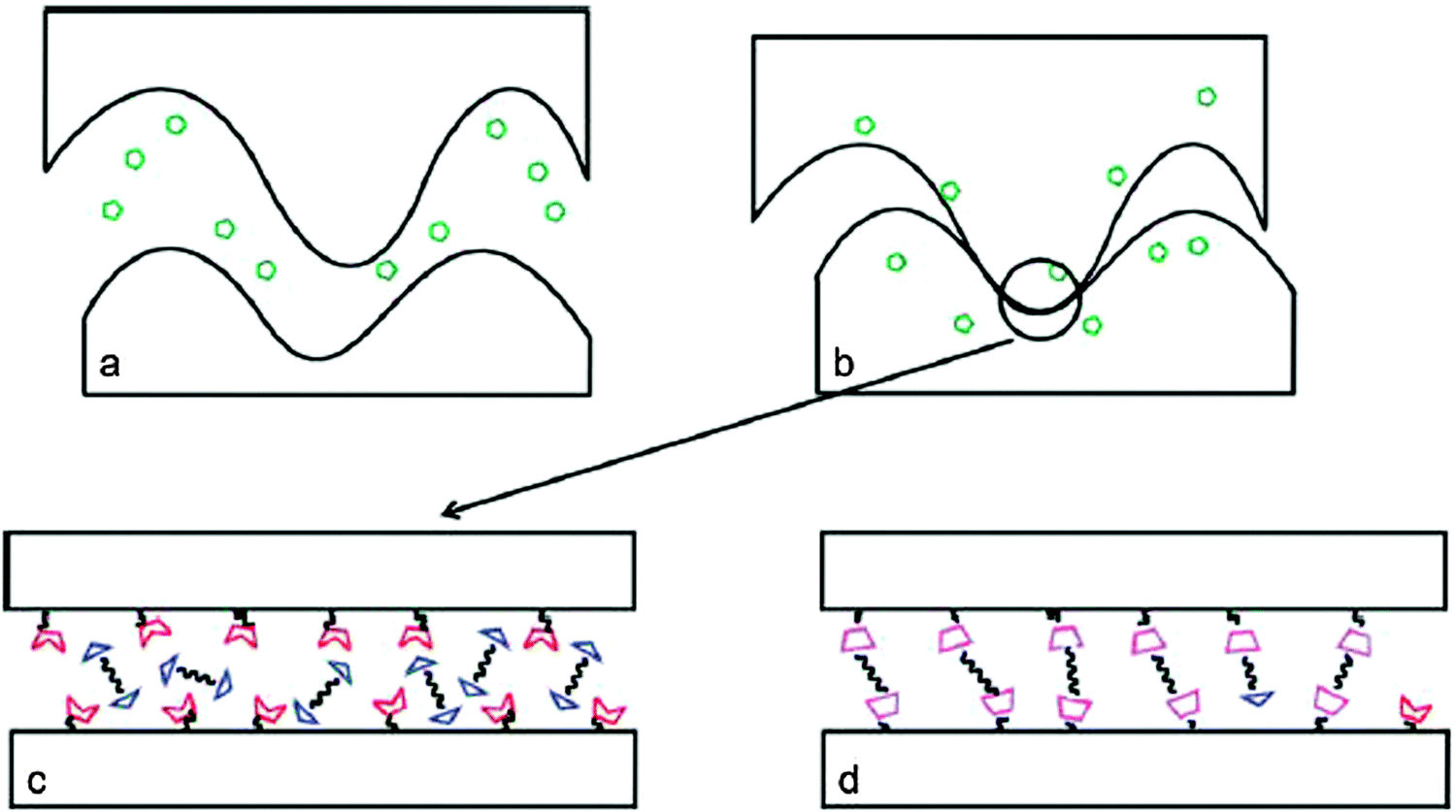 | ||
| Fig. 8 Hypothetical healing mechanism of solvent based DA reaction between a furan functionalized epoxy amine resin and a bis-maleimide (BMI). (a) Injection of the healing agent into crack (green symbols: BMI solution; red notched trapezoids: furan moieties; blue triangles: bis-maleimide; magenta trapezoids: DA adducts). (b) Solvent swelling of the polymer network. (c) Furan motifs coming into contact with BMI along the crack surface. (d) BMIs upon reaction with furan functionalized motifs generated DA adducts to heal the cracked surface. Reproduced with permission54 Copyright 2010, ACS Publications. | ||
This procedure could be carried out multiple times without any substantial reduction in its healing efficiency. In this case, DMF solvent induced swelling and softening of the crack surfaces resulted in the mechanical interlocking giving access to physical linkages as well as covalent chemical linkages, which both contributed to healing efficiency. The other such solvent system based delivery of healing agents at the cracked surface has been described.55,56
The same group also reported a self-healing epoxy-amine based thermoset, where the healing was achieved through the reaction of furans in the thermoset and the multi-maleimides encapsulated as healing agents via a urea-formaldehyde encapsulation technique. The healing agent was also solvent based, consisting of a solution of the multi-maleimides in phenyl acetate. The self-healing efficiency was found to be around 70% after first fracture.57 One may note that self-healing materials based on integrated microencapsulated or vascular systems containing healing agents do not possess self-sustainability – once the material runs out of the incorporated healing agent, it will no longer be able to repair itself.
Hetero-DA reaction based motifs have also been employed to get access to self-healing systems. Thus, a reversibly cross-linked polymer was generated via a hetero-DA reaction of biscyclopentadiene end functionalized PMMA with a trifunctional pyridinyl dithioformate cross-linker.58
3. Supramolecular DYNAMERS implementing non-covalent interactions
3.1 Self-healing systems based on non-covalent interactions
Self-healing systems described above in Section 2.3 are not self-sustainable and more often than not self-healing polymers based on such systems would require the intervention of an additional external physical or chemical initiator to achieve the desired self-healing action. The use of healing agents renders the material extrinsically self-healing, thus there has been a sustained effort to develop intrinsically self-healing systems based on non-covalent interactions, in particular by exploiting hydrogen bonding. However, self-healing systems based on non-covalent interaction of bulk polymeric materials, other interactions such as stacking,8,10,59,60 fiber formation,61–65 cluster formation65,66 also play a role in the healing process.The first step towards developing self-healing materials by exploitation of H bonding is achieved through the generation of supramolecular polymers. The area of supramolecular polymers has been developing rapidly and has been subject to many extensive reviews.2,3,67–71,85
Main-chain supramolecular polymers based on H-bonding are formed by polycondensation of molecular monomers bearing terminal complementary motifs of H bonding donor (D) and acceptor (A) sites such as DAD and ADA arrays.74 Similarly, supramolecular dynamers were obtained via extensive sextuple H bonding with a high association constant through the use of a Janus type cyanuric wedge (ADA–ADA array) (A) and a complementary diaminopyridine substituted isophthalamide receptor unit (DAD–DAD array) (B) (Fig. 9).72
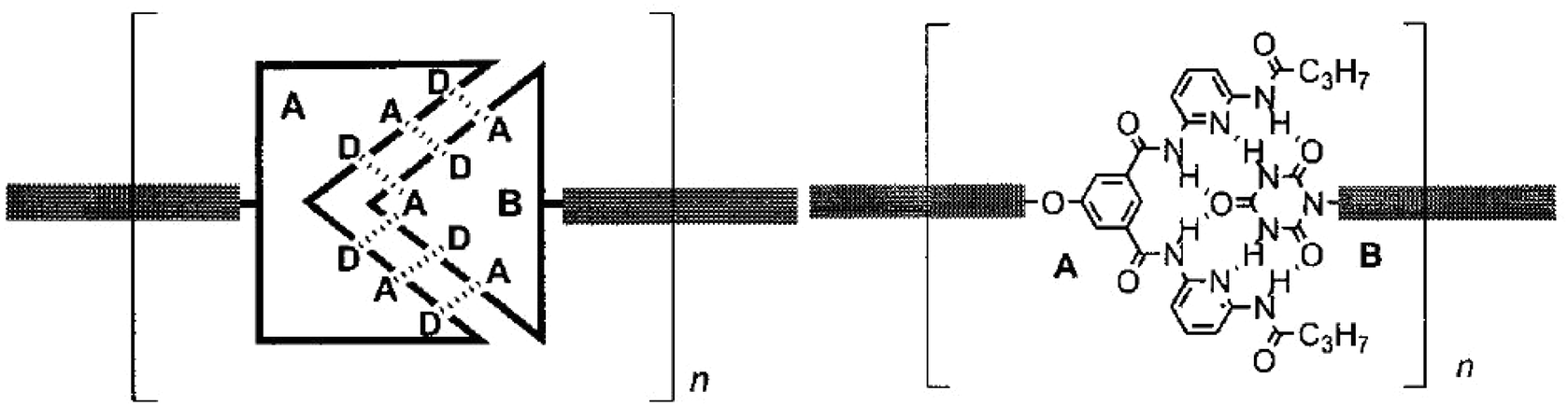 | ||
| Fig. 9 Generation of main-chain supramolecular polymers through polyassociation of molecular monomers bearing complementary H-bonding double-faced, Janus type molecular recognition units. | ||
H bond mediated molecular recognition between such hetero complementary binding sites led to polyassociation with the formation of linear chain supramolecular polymeric helical fibers (Fig. 10).73,74
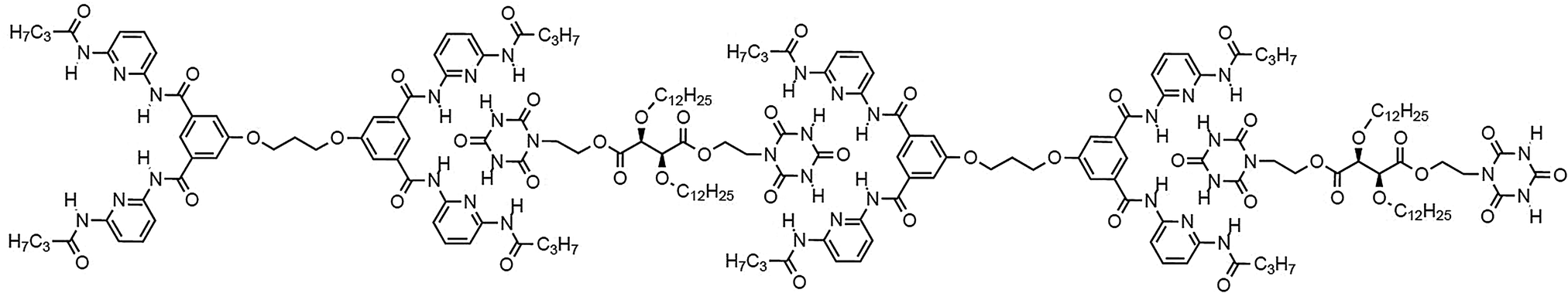 | ||
| Fig. 10 Generation of supramolecular polymers formed via sextuple H-bonding based molecular recognition of hetero complementary units.72 | ||
Supramolecular polymers exploiting quadruple H bonding motifs have been extensively studied.75–86 Specifically, using the 2 ureido-4-pyrimidone moiety (UPy) self-complementary end groups, which form dimer units of a high association constant (>106 M−1 in H-bond assisting solvents, such as chloroform), supramolecular polymers with a high degree of polymerization were obtained.76,86
Recent studies exploit H bonding to develop soft rubbery polymeric materials.87,88 These materials, owing to their low Tg and flexible backbone, presented rubber like features with an elongated plastic regime. Spontaneous self-healing making use of H bonding has been described for poly(isobutylene)s of varying molecular weights bearing barbituric acid based end groups.88,89 DMA (Dynamic Mechanical Analysis) of those materials indicated an extended rubbery plateau. These materials are joined by supramolecular cross points formed via the barbituric acid end groups.
The self-healing behaviour was demonstrated by cutting small round shaped films prepared from these barbituric acid terminated poly(isobutylene), and bringing the cut edges into contact with each other. The material underwent autonomous healing in the course of several hours (Fig. 11). The healing mechanism was explained by complex aggregation in the molten state of the polymer.89
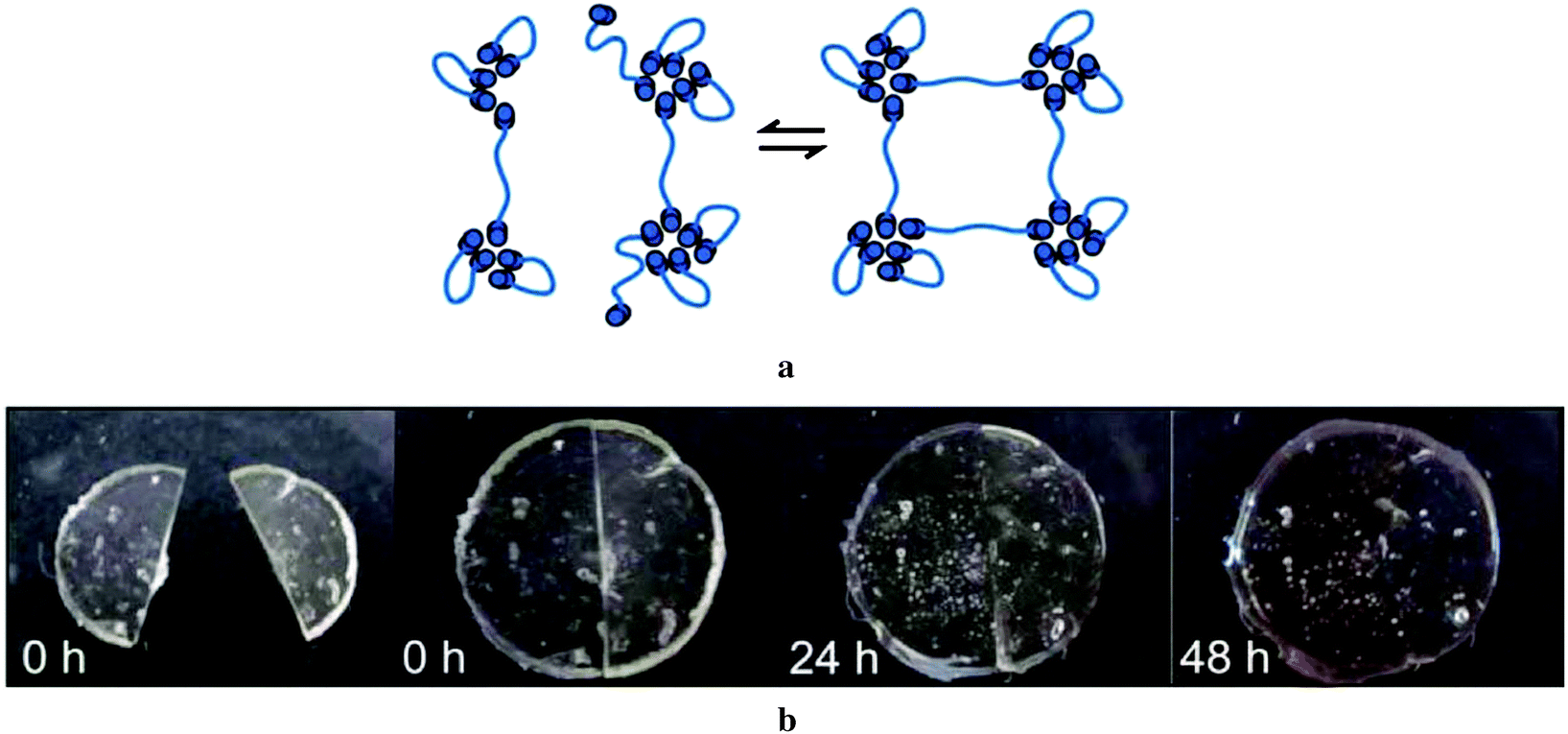 | ||
| Fig. 11 (a) Reversible supramolecular network formation through barbituric acid functionalized poly(isobutylene) (PIB). (b) Illustration of self-healing experiment at different time intervals. Reproduced from ref. 89 with permission from The Royal Society of Chemistry. | ||
The first reported examples of self-healing polymers based on extensive H bonding90 were achieved by making use of extensive H bonding involving three motifs of urea-derived groups (see Fig. 12). The system was obtained by a simple one step synthesis from a mixture of fatty di-acid and triacid condensed with diethyl triamine and subsequently treated with urea. The polymer thus generated was plasticized with 11 wt% of dodecane to optimize Tg of the material from 28 to 8 °C. The soft rubbery nature of the polymer was observed at large deformations.
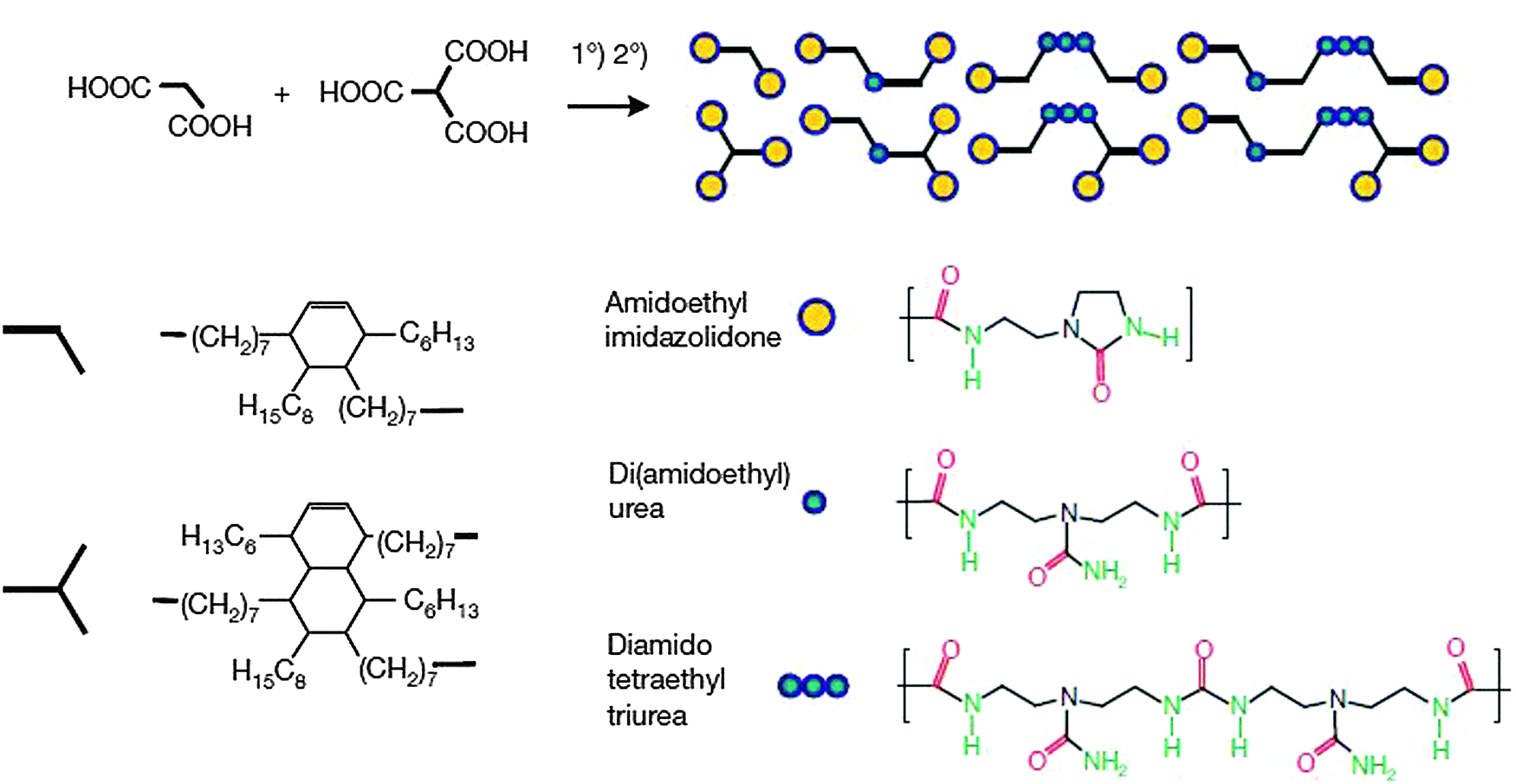 | ||
| Fig. 12 Synthesis of a self-healing supramolecular polymer equipped with complementary H bonding units. Reproduced with permission.90 Copyright 2008, Nature Publishing Group. | ||
The stress–strain curve exhibited similar behavior to that of soft rubbers exceeding 500% strain at break. Upon releasing the stress, the residual strain was found to be less than 5% (Fig. 13).
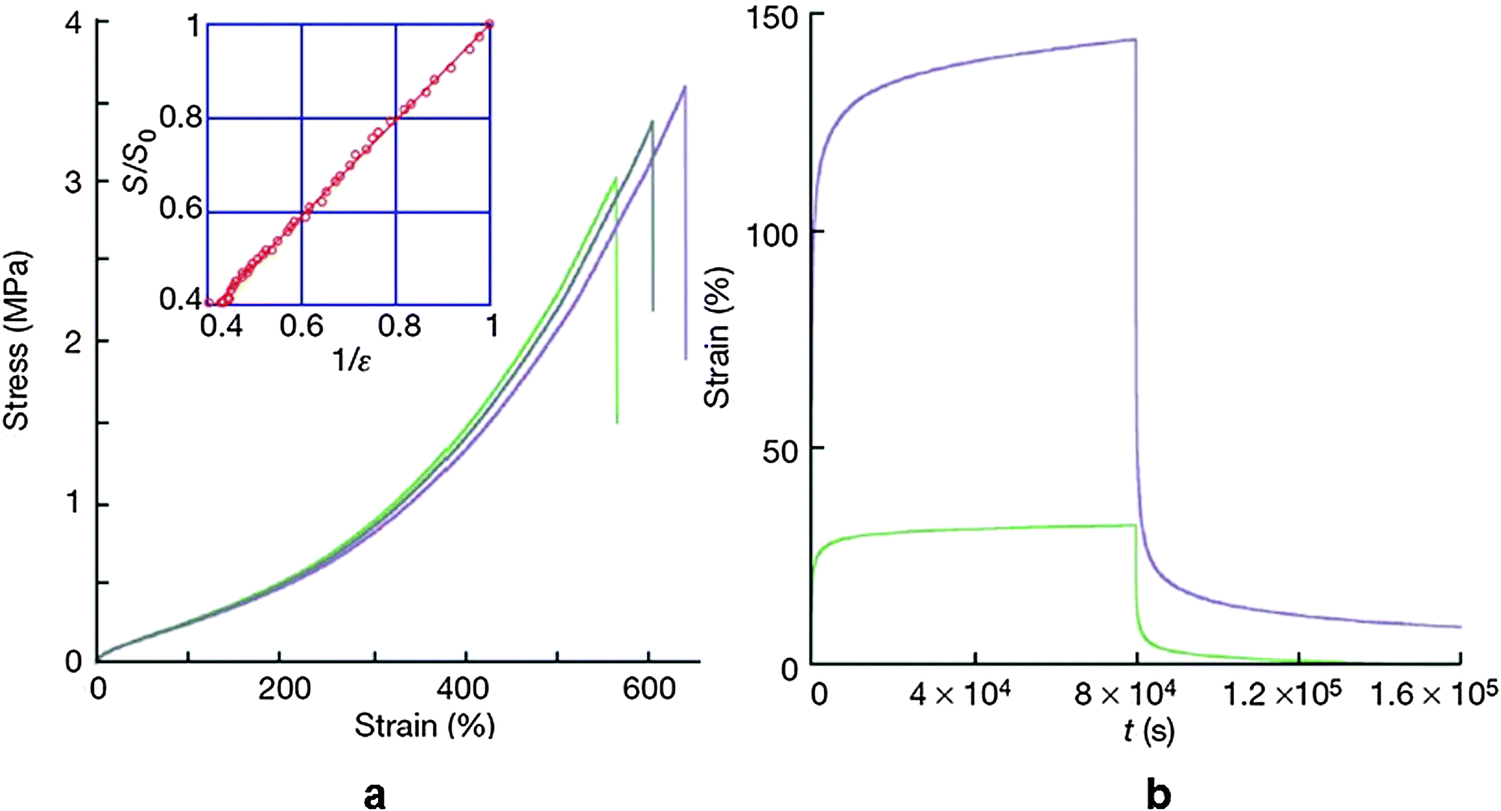 | ||
| Fig. 13 (a) Stress–strain curve of the self-healing supramolecular rubber (3 data set points were shown to exhibit reproducibility). The inset shows that the cross-sectional area is inversely proportional to tensile deformation. (b) Creep recovery experiment for an applied stress of 5 MPa (green) and 20 MPa (purple). Reproduced with permission.90 Copyright 2008, Nature Publishing Group. | ||
Rheological experiments indicated that an applied stress of 5 MPa for 8 × 104 s resulted in 32% strain and it increased at a rate of 0.04% per hour. When the stress was relieved the material recovered its initial dimension with negligible residual strain. Even though the material behaved like a soft rubber, it had certain differences with conventional rubbers. In contrast to classical rubbers, this supramolecular soft rubber-like material underwent self-healing under ambient conditions. When the sample was broken up or cut into fragments and pieces brought into contact again with each other, the cut surfaces or the breaking edges immediately underwent self-healing. The healing efficiency was found to be adversely affected by increasing the waiting time of bringing the surfaces in contact with each other. This was indirect evidence that healing was achieved through the freely available H bonding sites. A longer waiting time resulted in a decreasing number of available H bonding sites, which in turn affected the H bonding efficiency. The self-healing mechanism through H bonding was confirmed by the slow dynamics of H bond re-association and illustrated by time dependent infra-red spectroscopy.90
Subsequently another supramolecular self-healing rubber like material was obtained through the oligocondensation of fatty mono, di and triacids with diethylenetriamine (DETA). The extent of branching and molecular weight was controlled by selection of the ratio of di- and triacids and through end capping before the condensation reaction with 2-aminoethylimidazolidine (UDETA). These moieties could also act as complementary H bonding units. Additional complementary units could also be introduced through grafting of urea onto all the secondary amines of DETA.91 This synthetic strategy was proposed to give access to a vast variety of supramolecular soft rubbers.
Another approach that has also been exploited in the recent past is the formation of thermo-reversible elastomers by making use of cross-linkable motifs. A recent illustration concerns the formation of a self-healing nano-composite elastomer using functionalised graphene oxide nanoparticles as the macro cross-linker. It involves a matrix of randomly amide-terminated oligomer assembled through H bonding interactions and cross linked via suitably functionalised graphene nanoparticles.92
A temperature induced self-healing system was obtained and commercialized under the brand name SupraB (Suprapolix BV).93 Exploiting the thermo-reversible behavior of quadruply H bonded UPy groups, this material behaves as a soft melt at elevated temperature owing to the breakup of H bonds and as an elastic plastic at room temperature generated by the re-association of H bonds of UPy motifs. Thus, SupraB could be dissolved in a suitable solvent and spray casted into flexible films of different thicknesses with an excellent mechanical performance. Moreover, when the surface was damaged through scratching it underwent spontaneous healing by briefly elevating the temperature to 140 °C. This healing was found to be efficient for multiple cycles. A telechelic polymer making use of the same quadruple H bonding UPy units has also been described (Fig. 14).80
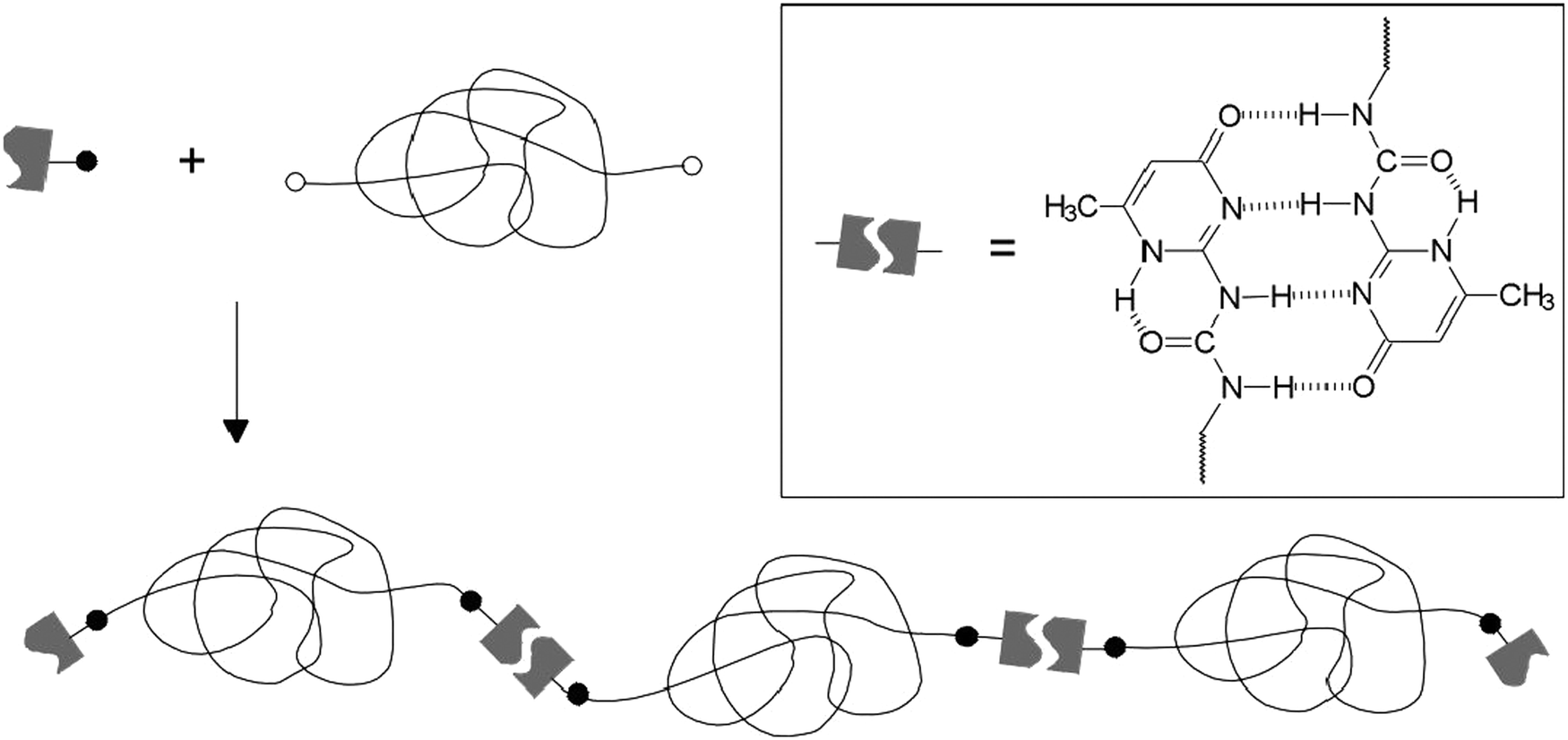 | ||
| Fig. 14 Synthesis of telechelic polymers with quadruple H bonded ureidopyrimidinone (UPy) units. Reproduced with permission.80 Copyright 2000, Wiley-VCH. | ||
The OH terminated poly(ethylene/butylene) diol is a viscous liquid, however functionalization with UPy moieties resulted in the formation of an elastic solid material owing to the H bonding imparted by the UPy motifs.80 A SupraB polymer is a spin-off product of this concept (Fig. 15).93
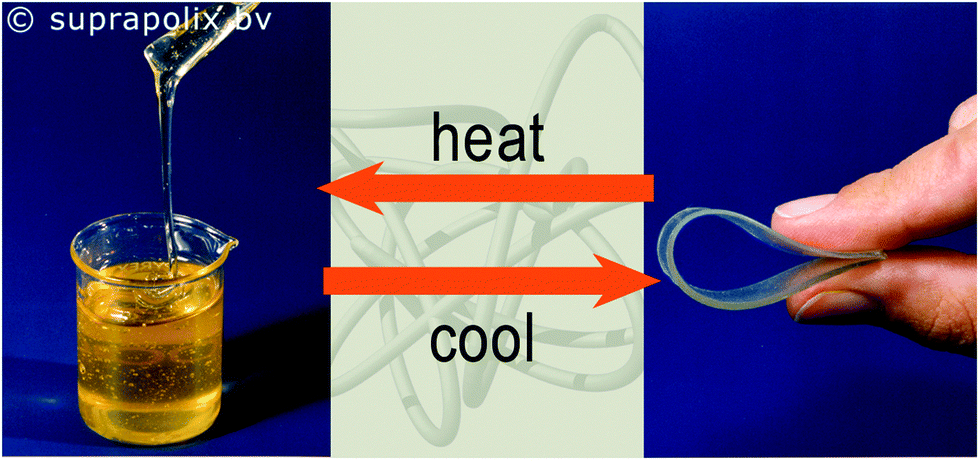 | ||
| Fig. 15 The OH terminated poly(ethylene/butylene) diol is a viscous liquid (left); same diol functionalized with UPy moieties (right).93 Reproduced with permission.93 Suprapolix BV. | ||
In our group we explored the formation of a tris-urea synthon, which would give access to sextuple H bonding.94 Bis-urea motifs have been extensively studied.95–101 Extension to a triple urea motif was achieved by reacting an isocyanate with carbodihydrazide. X-ray diffractable single crystals could be obtained from a DMSO solution of the product of the reaction of di-tert-butyl phenylisocyanate with carbohydrazide to generate the tris-urea motif (Scheme 1).
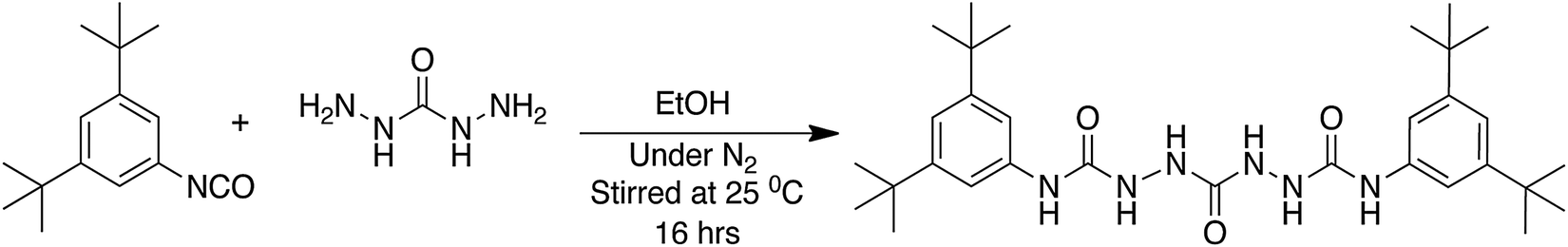 | ||
| Scheme 1 Synthesis of tris-urea compound 2,2′-carbonyl-bis-N-(3,5-di-tert-butylphenyl)-hydrazine-carboximide.94 | ||
The tris-urea motif in principle would give rise to a perpendicular and a horizontal array of H bonding with a twisted orientation around the N–N bonds (Fig. 16).
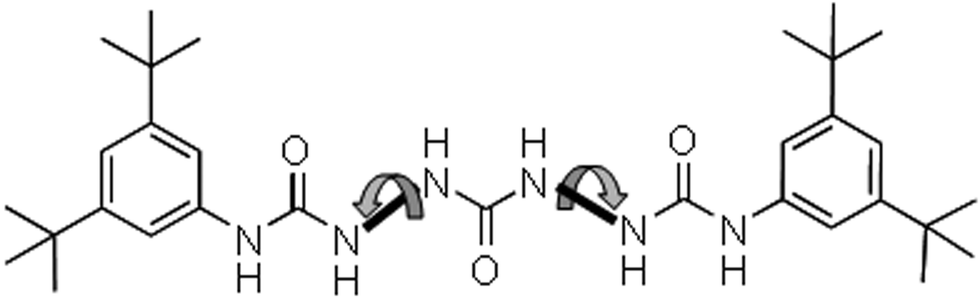 | ||
| Fig. 16 Schematic representation of twisted orientation around the N–N bond of the tris urea motif. | ||
The solid state molecular structure showed the parallel and perpendicular arrays of H bonding giving rise to sextuple H bonding. However, owing to the presence of DMSO, the perpendicular H bonding sites were occupied by solvent molecules (shown in Fig. 17).94
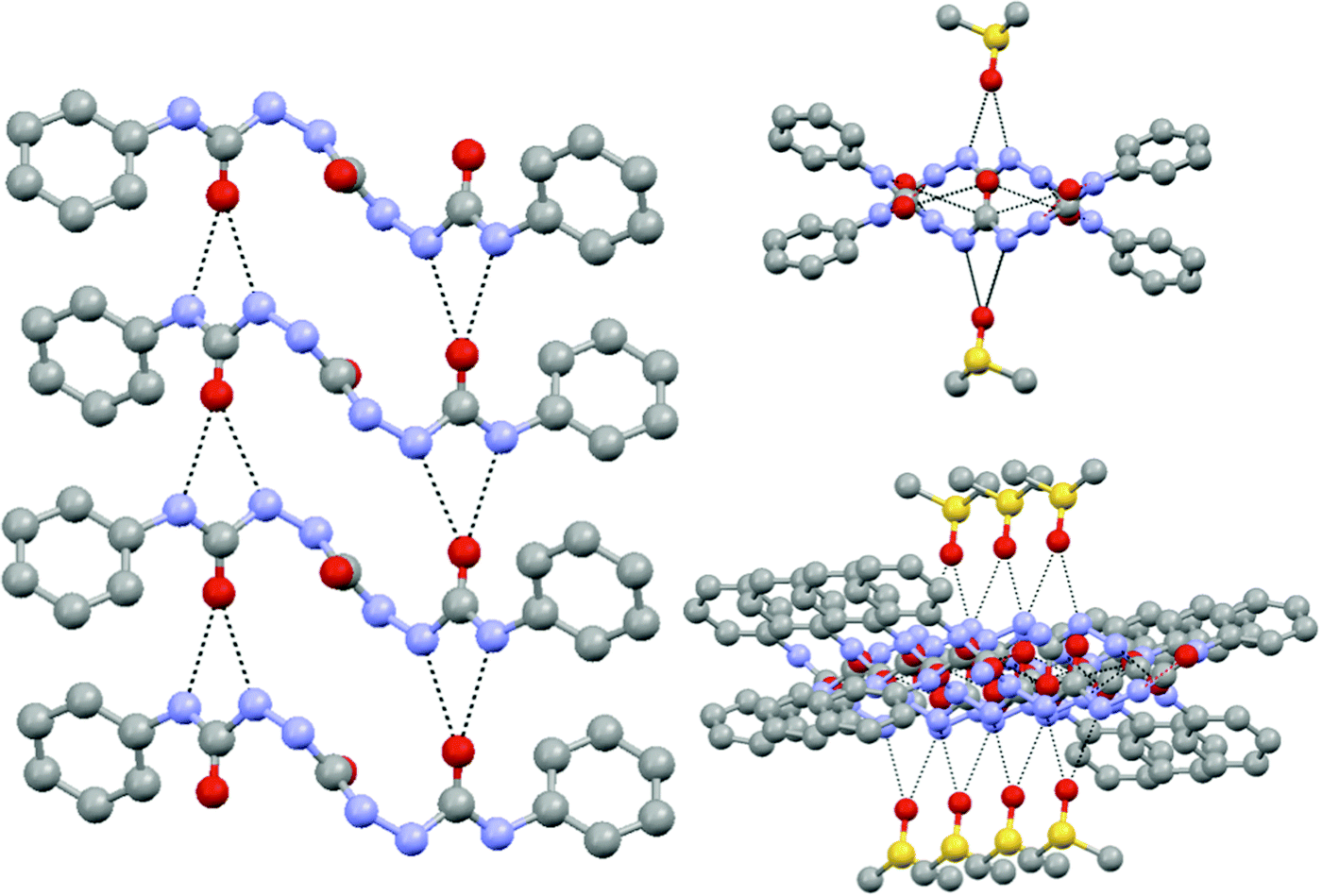 | ||
Fig. 17 Intermolecular hydrogen-bonding arrays displayed by the tris-urea compound within the unit cell: (left) H-bonds between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H sites of the terminal urea groups in adjacent molecules; (right) H bonding between the N–H sites of the central urea group and the O site of the solvent of crystallization DMSO.94 O and N–H sites of the terminal urea groups in adjacent molecules; (right) H bonding between the N–H sites of the central urea group and the O site of the solvent of crystallization DMSO.94 | ||
For developing such an extensive H bonding into a soft polymeric material that could present self-healing properties, NCO terminated PDMS compounds were reacted with carbodihydrazide (Scheme 2).
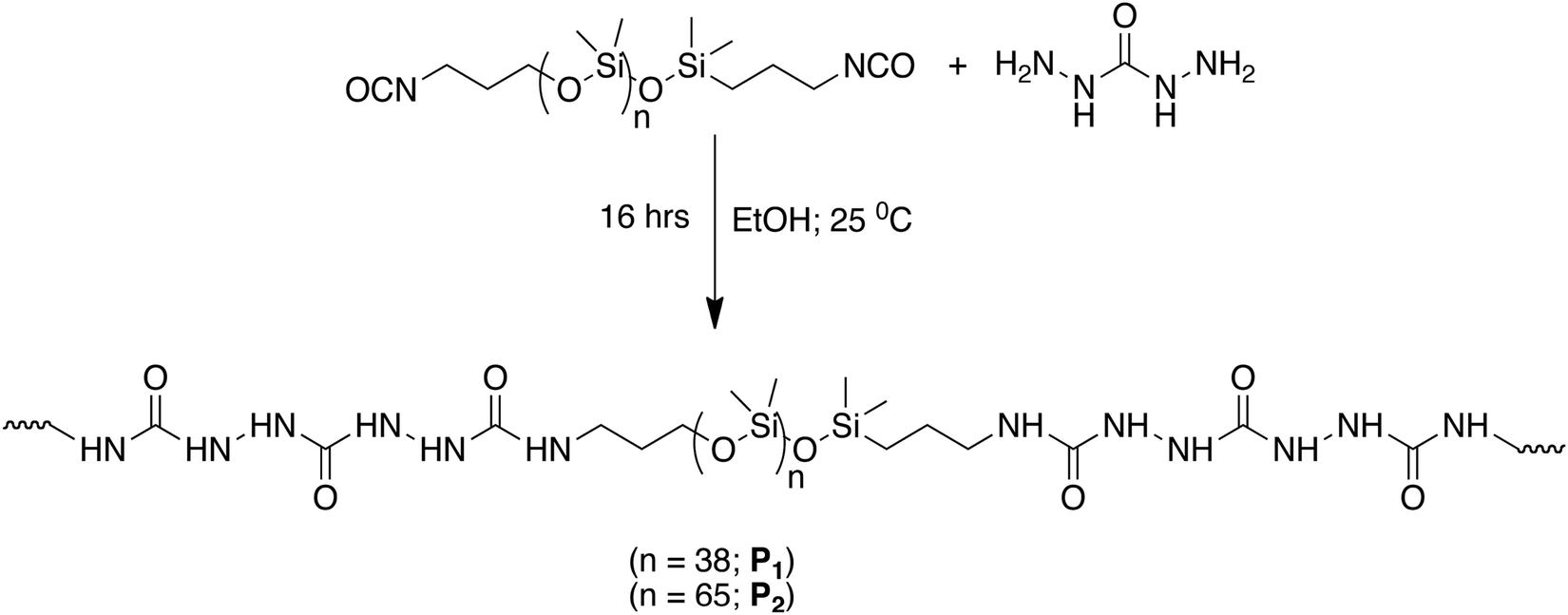 | ||
| Scheme 2 Synthesis of two different polymers P1 and P2 containing tris-urea motifs.94 | ||
The resulting two polymers P1 and P2 were characterized by SANS and then casted into films by removal of the solvent from a solution in chloroform. In the case of P1 a brittle material was formed and when broken fragments were brought into contact with each other, the material self-repaired to form a bulk healed polymer block in the course of an hour. In contrast, in the case of P2, a more elastic polymer was generated and when a polymer block of about 1 mm thickness was cut into pieces the fragments brought into contact with each other underwent self-healing at the interface (Fig. 18). However, in accordance with what had also been previously observed with other self-healing H bonding supramolecular polymers, this healing process was time dependent. Keeping the broken fragments apart for a longer duration (more than 10 minutes) did not result in self-healing any more.
 | ||
| Fig. 18 (left) (a) Native damaged polymer P1; (b) self-repaired polymer P1; (center) (c): mechanically cut polymer P2; (right) (d) self-healed polymer P2.94 | ||
This behaviour may be explained by noting that the free hydrogen bonding groups exposed at the freshly cut surface will in course of time undergo non-covalent interaction with available partners on the same surface or in the interior of the polymer sample, thus leading to a progressive reduction in self-healing capability between pieces. The H bonding patterns in polymers P1 and P2 were substantiated by detailed ATR FT-IR spectroscopy.94
Hindered urea bonds give access to catalyst free dynamic material properties.102 The dynamicity of the hindered urea bond (HUB) was demonstrated and this concept was then further extrapolated towards self-healing HUB containing polymers. The dynamicity of the hindered urea bond was shown by an exchange experiment, involving the exchange of 1-(tert-butyl)-1-ethylurea (TBEU) by mixing TBEU with t-butylmethyl amine and a bulkier N-substituted secondary amine similar to t-butylmethyl amine. The exchange reaction was monitored by 1H NMR (Fig. 19).102
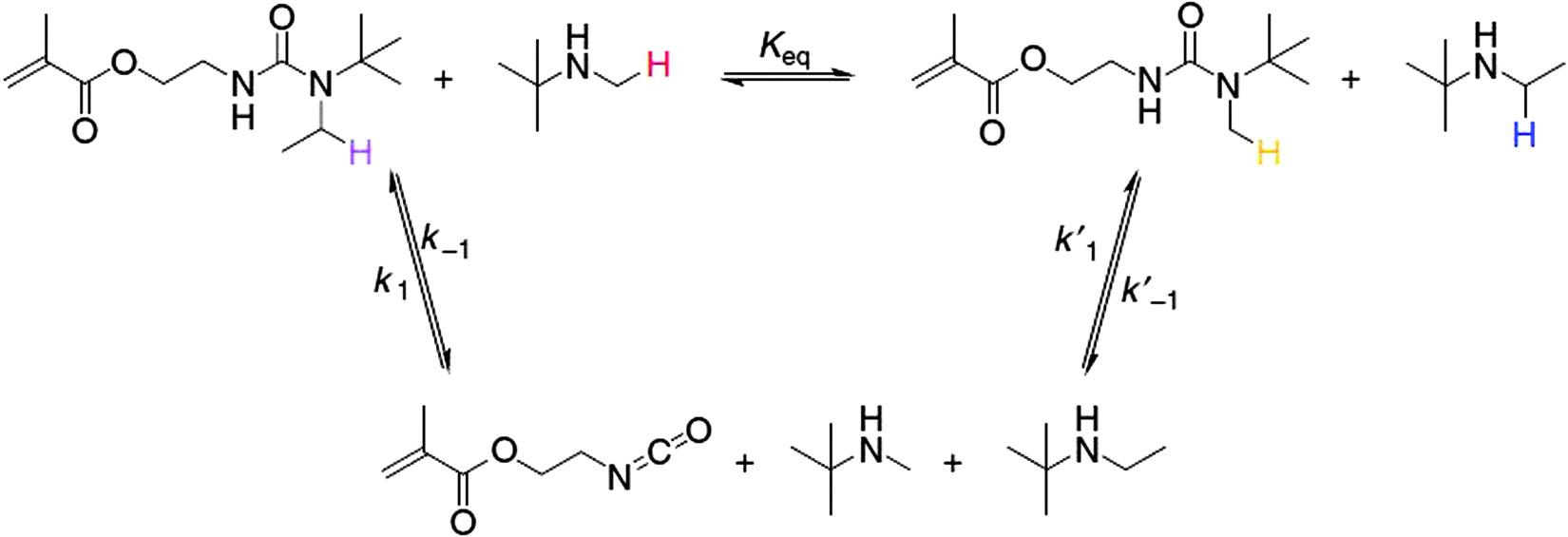 | ||
| Fig. 19 Dynamic exchange reaction of TBEU with hindered primary and secondary amines. Reproduced with permission.102 Copyright 2014, Nature Publishing Group. | ||
The postulated isocyanate intermediate was beyond the detection limit of NMR, however the presence of t-butylmethyl urea and t-butylethyl amine could be detected. The reversible exchange was substantiated by the linear regression of the reaction kinetics. After demonstrating the dynamic exchange in small molecules, HUB was incorporated into the backbone of polyureas and poly(urethane-urea) polymers, which subsequently exhibited self-healing behavior (Scheme 3).
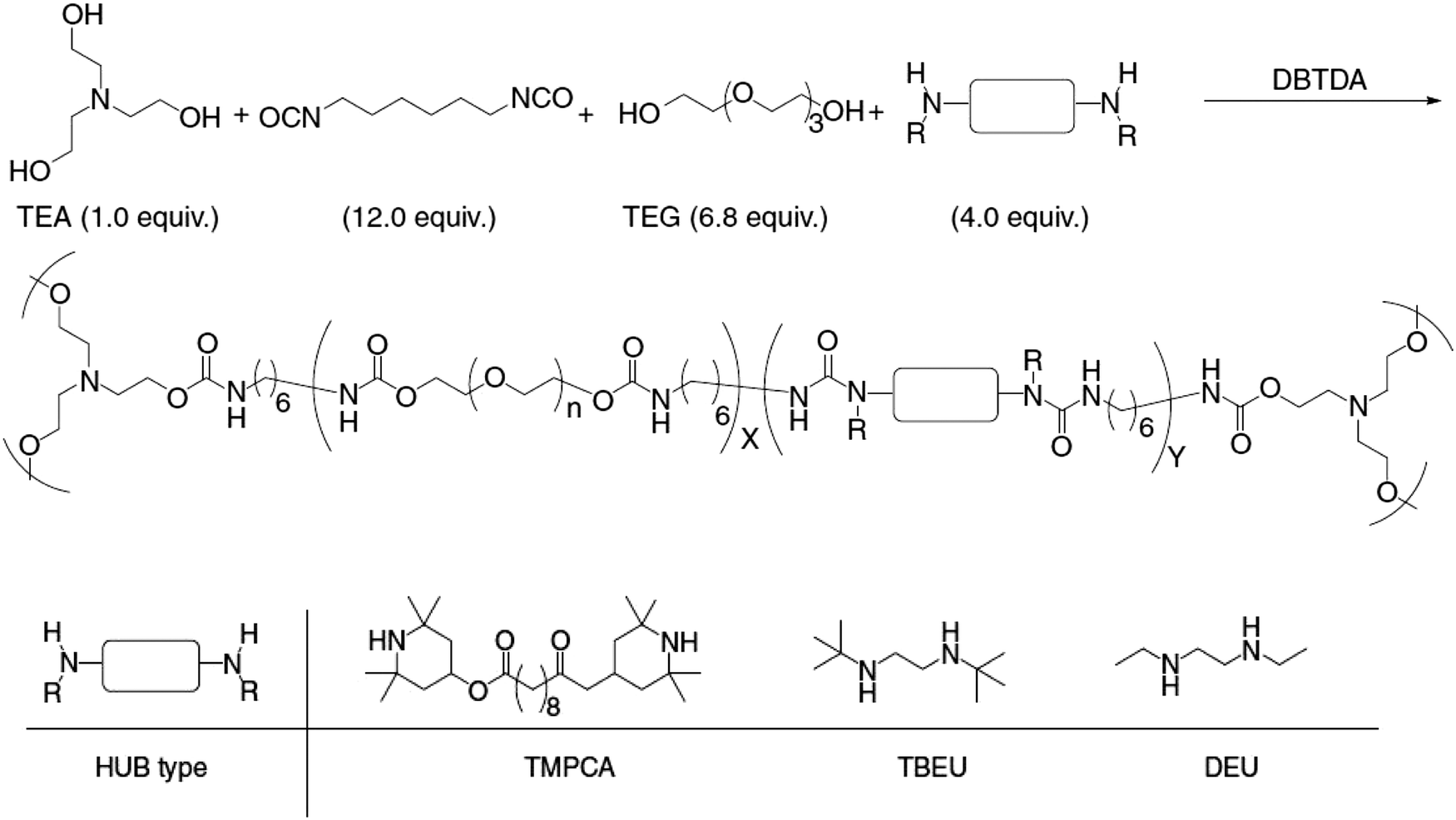 | ||
| Scheme 3 Synthetic scheme of the generation of hindered urea bonds based cross-linked poly(urethane-urea). Reproduced with permission.102 Copyright 2014, Nature Publishing Group. | ||
The self-healing experiment was carried out by cutting a polymer sample into pieces, bringing them in contact with each other with gentle pressing and leaving to heal for 12 h at 37 °C. The gel was subsequently stretched and no fracture was observed in the cut region, depicting the efficiency of the healing process. (Fig. 20).102
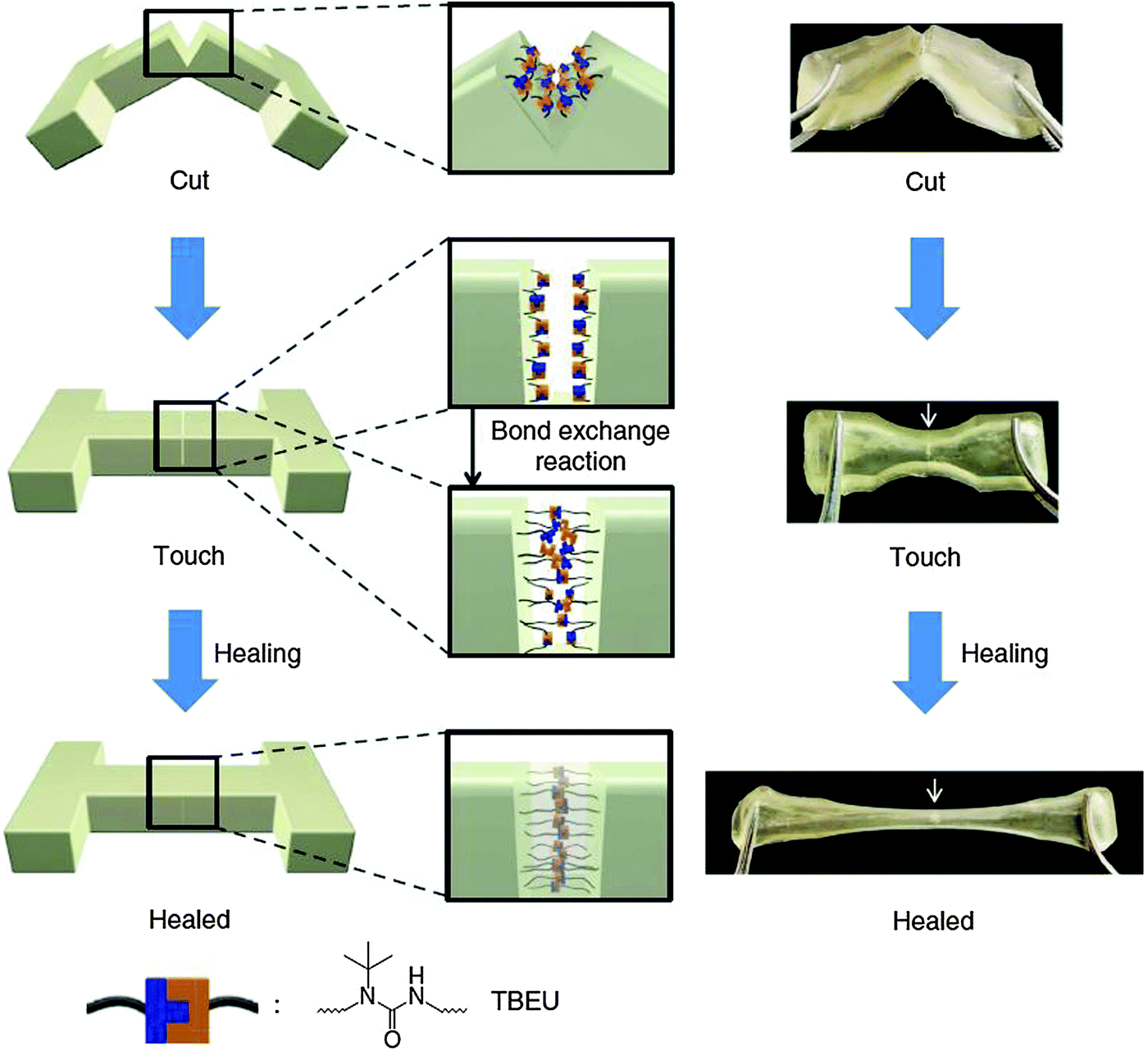 | ||
| Fig. 20 Schematic and pictorial illustration of the self-healing process based on the materials of Scheme 3. Reproduced with permission.102 Copyright 2014, Nature Publishing Group. | ||
A recent review stresses the importance of non-covalent interactions in conferring reversibility and adaptive features to functional supramolecular materials.103 Along similar lines a tunable self-assembled polyurethane type elastomer has been reported, where the self-assembly is driven by the triple H bonding between amidoisocytosine and ureidoimidazole motifs.104
3.2 Self-healing systems based on non-conventional dynamic routes
Along with the implementation of covalent and non-covalent dynamic chemistry for the development of functional materials, there have also been reports from different groups, where certain other reversible connections were used to generate self-healing systems. Thus, a self-healing material making use of the covalent dynamicity of diarylbibenzofuranone (DABBF) was described.105 DABBF readily undergoes cleavage into the corresponding monomer units under ambient conditions and the generated radical species are also stable under aerobic conditions. The introduction of this motif within the polymer backbone gave a self-healing gel like material. In order to generate three-dimensional motifs, a custom made cross-linked polymeric material was synthesized using polyaddition chemistry via urethane formation (Fig. 21).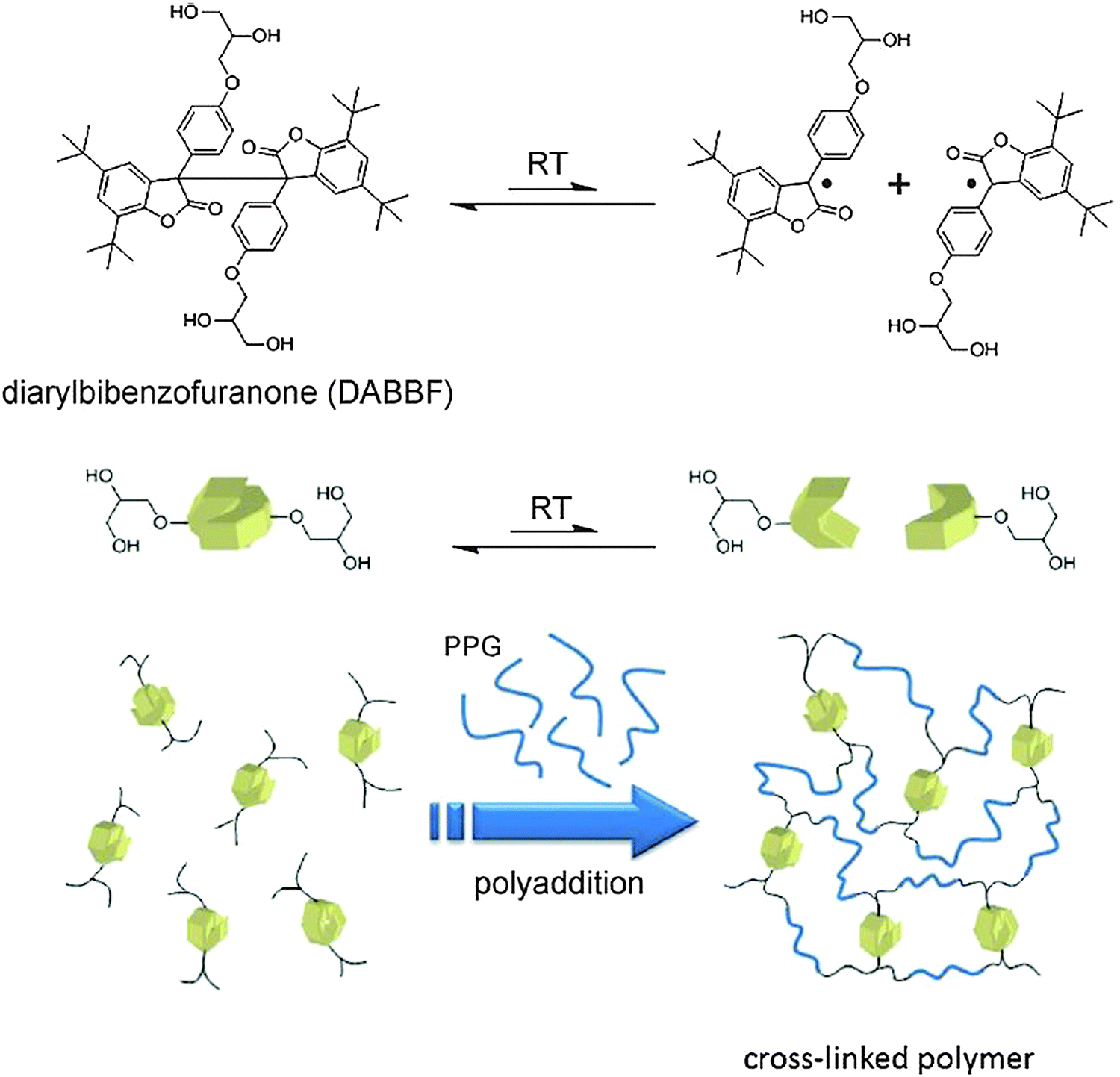 | ||
| Fig. 21 Synthesis of a DABBF incorporated cross-linked polymer. Reproduced with permission.105 Copyright 2012, Wiley-VCH. | ||
The self-healing ability of the cross-linked polymer was investigated under air at room temperature. After cutting the sample into two pieces, the interface of the cut surfaces was saturated with DMF to prevent any H bonding from urethane bonds. Then they were brought into contact with each other without any pressing. In the course of 24 hours, the sample underwent complete healing and the polymer could not be cleaved through manual stretching (Fig. 22).105
 | ||
| Fig. 22 Depiction of self-healing of DABBF incorporated in a cross-linked polyurethane–polymer. Reproduced with permission.105 Copyright 2012, Wiley-VCH. | ||
The self-healing polymers described so far are generally soft in nature and based on supramolecular non-covalent interactions or covalent dynamic bonds resulting in soft flexible materials with low glass transition temperatures. The concept of self-healing was extended into comparatively harder grade materials. In a classical thermoplastic elastomer (TPE), there are phase segregated soft and hard domains, and self-healing in the hard domain would generally lead to sticky material, which is undesirable. The gap between classical TPE and supramolecular interactions involving H bonds was bridged by developing a self-healing system based on polyacrylate-amide (PA-amide) brushes constituting the soft segment, which was grafted onto a polystyrene backbone, forming the hard block. These secondary amide functionalities could be modified in a synthetically simple route, giving access to both H bond donor and acceptor capabilities generating a dynamic network of supramolecular interactions (Fig. 23).106
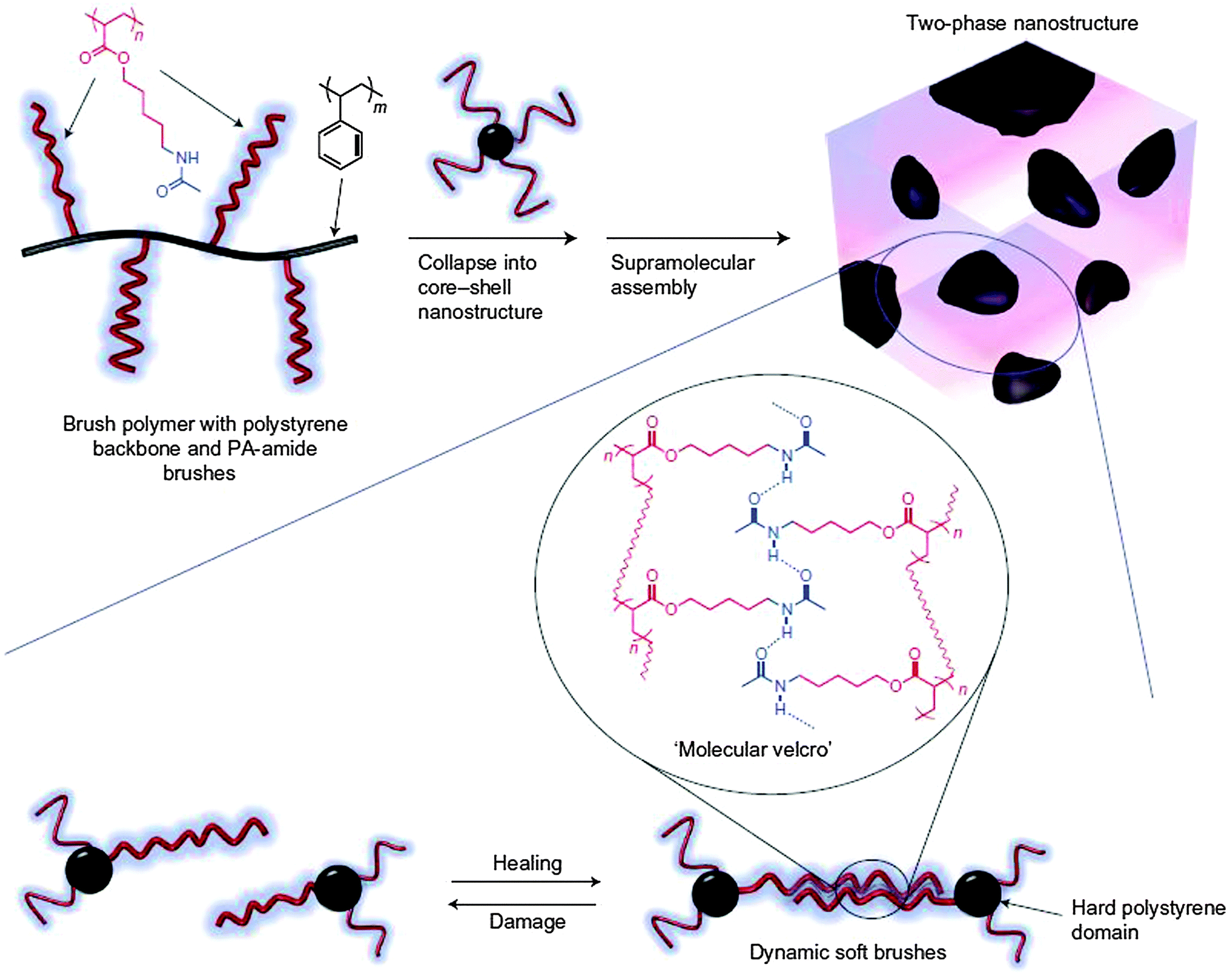 | ||
| Fig. 23 (top) Schematic representation of the H bonding brush polymer self-assembly process into two-phase nano-structure morphology during processing. (bottom) Pictorial illustration of reversible cleavage of supramolecular interconnectivities through mechanical stress and subsequent healing owing to the presence of H bonding interactions through soft brush polymers. Reproduced with permission.106 Copyright 2012, Nature Publishing Group. | ||
This example demonstrated the self-healing process operating only through the H bonding interactions between soft polymer brushes containing H bond donor and acceptor sites. Thus, cutting a sample of material into two separate parts, bringing them together again and leaving it to heal for several hours, resulted in an optimal healing efficiency of 92% relatively compared to the undamaged starting sample. The healing efficiency was found to increase with additional healing times.106 It could be inferred here that upon application of mechanical stress, selective damage of only soft segments is not achievable. The decrease in healing efficiency could also be attributed to irreversible damage that might have also taken place in the hard domain. However, this example is a step forward towards developing hard self-healing TPE materials.
Certain other harder grade self-repairing materials were also reported based on a polyimide copolymer bearing naphthalene-diimide units in combination with pyrenyl end capped polyamide motifs. These materials were solution casted into flexible homogenous films, which underwent self-repairing upon cutting the films into two halves and then subsequently overlapping them with gentle pressing at 80 °C.107,108 In this case, the self-healing was achieved through the π–π stacking of adjacent naphthalene units and H bonding via the vicinal pyrene groups.
4. Double DYNAMERS: dynamic polymers based on two-fold dynamism
In the previous section, we principally focused on dynamic features originating from a single type of dynamism, working either through dynamic covalent reactions or through dynamic non-covalent interactions. The combination of these two orthogonal dynamic behaviours may be expected to provide materials exhibiting a two-fold dynamism. The formation of double dynamic polymeric materials or double dynamers was reported earlier by our group by taking advantage of dual dynamicity originating from (i) sextuple H bonding between a DAD–DAD (D = donor, A = acceptor) receptor and an ADA–ADA cyanuric acid wedge (see also above, Fig. 9 and 10) and (ii) reversible acylhydrazone bond formation through condensation of hydrazides and aldehydes (Fig. 24).109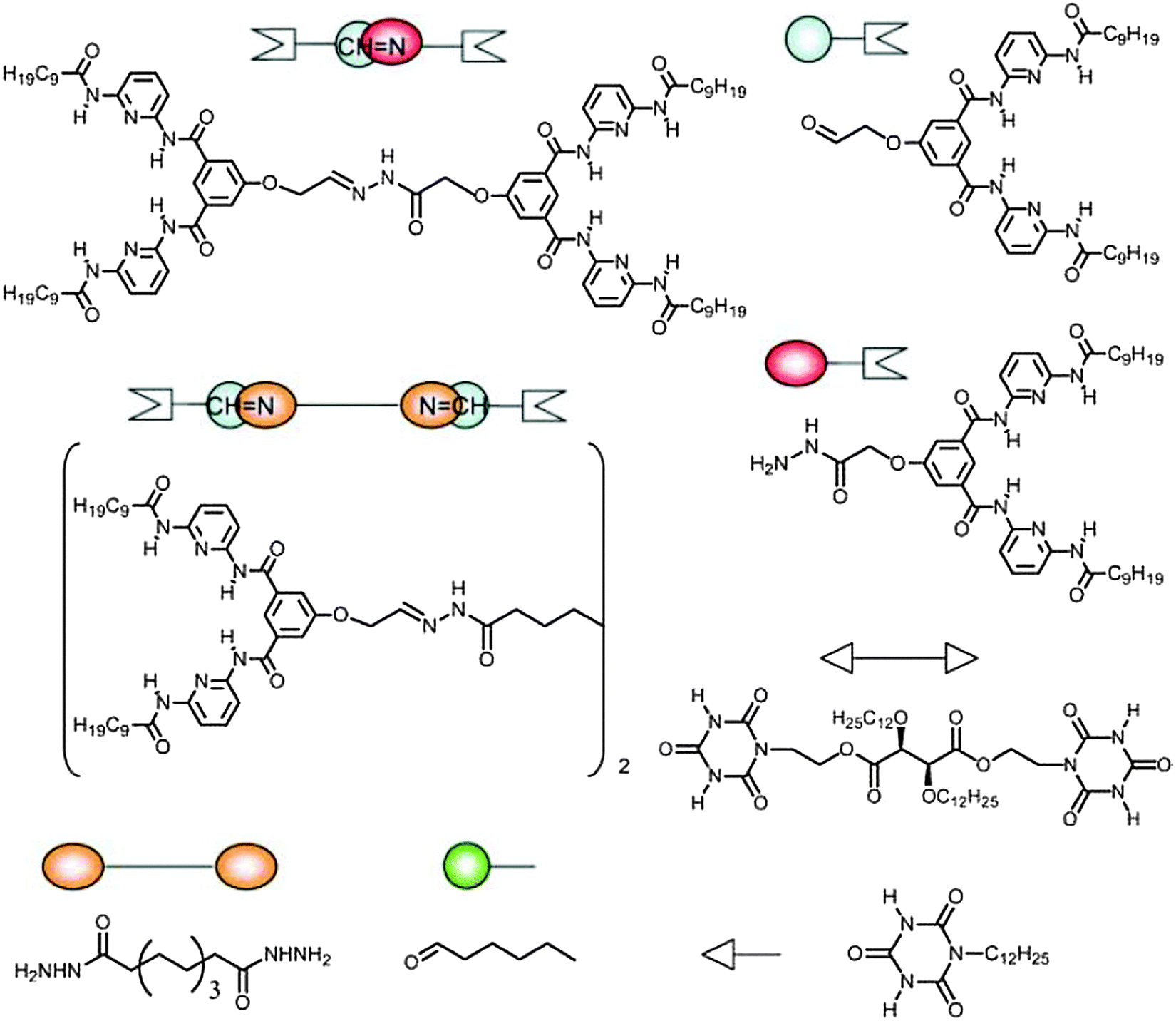 | ||
| Fig. 24 Molecular components and building blocks for the formation of double dynamic polymeric material incorporating both non-covalent and covalent dynamics.109 | ||
Two types of supramolecular polymers were essentially generated along this design, depicted by SPI and SPII as illustrated in Scheme 4.
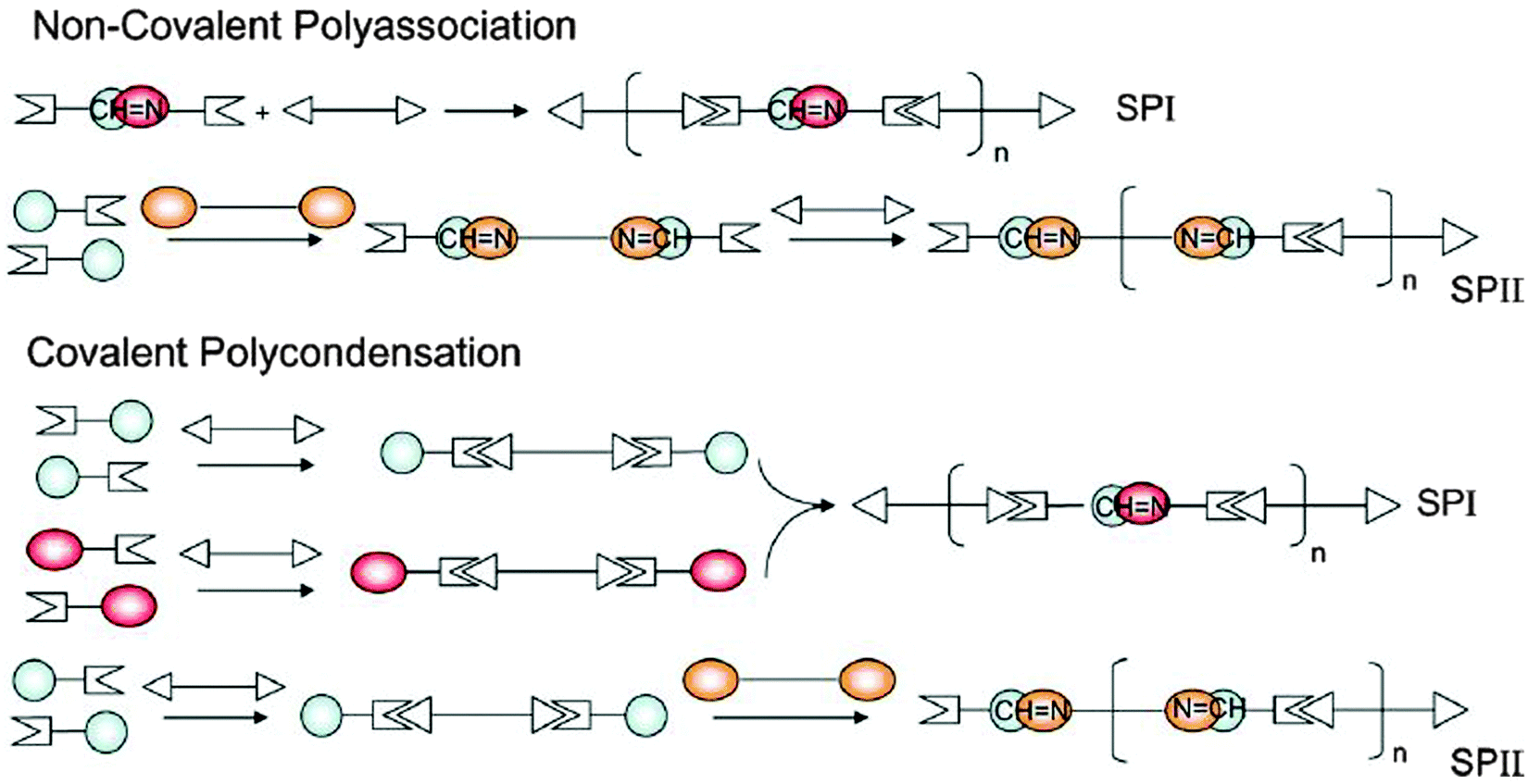 | ||
| Scheme 4 Methods for the generation of the double dynamic polymers SPI and SPII combining imine type covalent dynamics and supramolecular dynamics via complementary H bonding head groups.109 | ||
The double dynamicity was exhibited by covalent and non-covalent end capping experiments.109 Recently, along similar lines, a supramolecular polymer was generated by polyassociation of an acylhydrazone-based covalent dynamic building block through a complementary hydrogen bonding component, following a molecular dynamic/supramolecular non-covalent sequence.110
This concept of double dynamicity was further exploited to get access to a self-repairing material. A PDMS functionalized dialdehyde was condensed with carbodihydrazide and the polymer was subsequently solvent casted into a film (Scheme 5).111
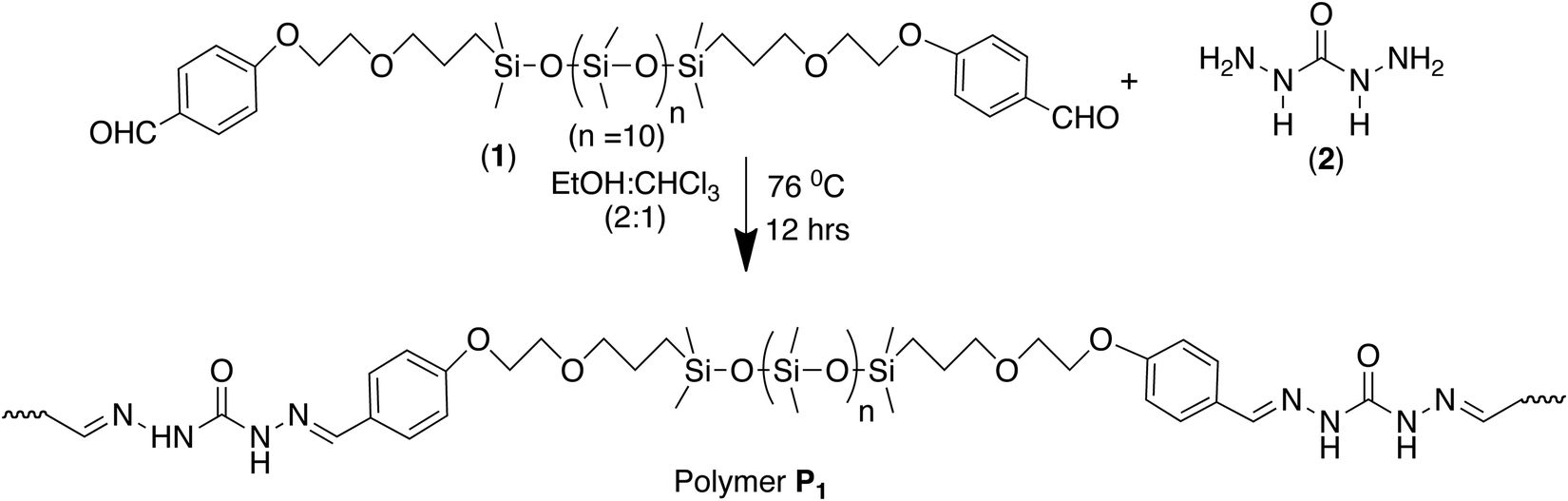 | ||
| Scheme 5 Synthesis of dynamic polymer P1 by polycondensation of the monomeric components, bis-aldehyde and carbodihydrazide.111 | ||
Cutting the film with scissors and overlapping the two cut surfaces within a few minutes, followed by gentle pressing, yielded a completely healed polymer film in the course of a few hours (Fig. 25). This self-healing experiment was carried out under ambient conditions without application of any additional pressure or load. Varying the duration of healing time showed that below 4 hours the healing process was not efficient.
 | ||
| Fig. 25 (left) (a) Uncut soft-stretchy polymer film; (b) soft-polymer film after healing and illustration of progressive stretching of the self-healed polymer film (the blue arrow indicates the healed overlapping domain). (right) True stress vs. true strain curve of the native and self-healed polymer film.111 | ||
The elasticity of the self-healed film was found to be partially retained and the self-healed polymer could withstand 90% of the original strain, indicating the efficiency of the healing process.
The reversibility of the acylhydrazone bonds within the simple monomeric motifs was verified by a component exchange experiment using an acid catalyst. However, in order to validate the covalent dynamicity of the healing process, the same healing experiment was also carried out with and without the acid catalyst (pentadecafluoro octanoic acid). It was found out that the time scale of healing did not improve significantly with the addition of an acid catalyst, suggesting that the healing process in this particular case involved predominantly the hydrogen bonding motif, as was also the case in another system based on urea groups, reported earlier by our group.94
5. Self-healing in the context of degradable dynamic polymers
Another aspect of DYNAMATs and DYNAMERs that might in particular be of great interest for future applications would be to develop materials that could undergo dynamic formation and degradation. Subsequently, upon removal of the source of degradation, it could resort to self-healing, reverting back into the original polymer. Such dynamers owing to their subsequent biodegradation may also be ascribed as Green DYNAMERs (GD). Recently, films of such a type of green dynamer were developed by covalently linking biodegradable oligomeric units (polybutylene adipate (PBA) and polybutylene succinate (PBS)) connected through hydrolytically degradable reversible imine bonds.112 These GD's can also be envisaged as possessing self-healing properties because the imine bonds may be restored by evaporation of water and drying the sample.From the perspective of material design, such GDs should satisfy the following three requirements: (i) thermoplastic nature exhibiting good processibility/mechanical properties; (ii) a controllable disintegration rate in water (no water solubility) (iii) conversion into CO2 and H2O upon degradation (Fig. 26).
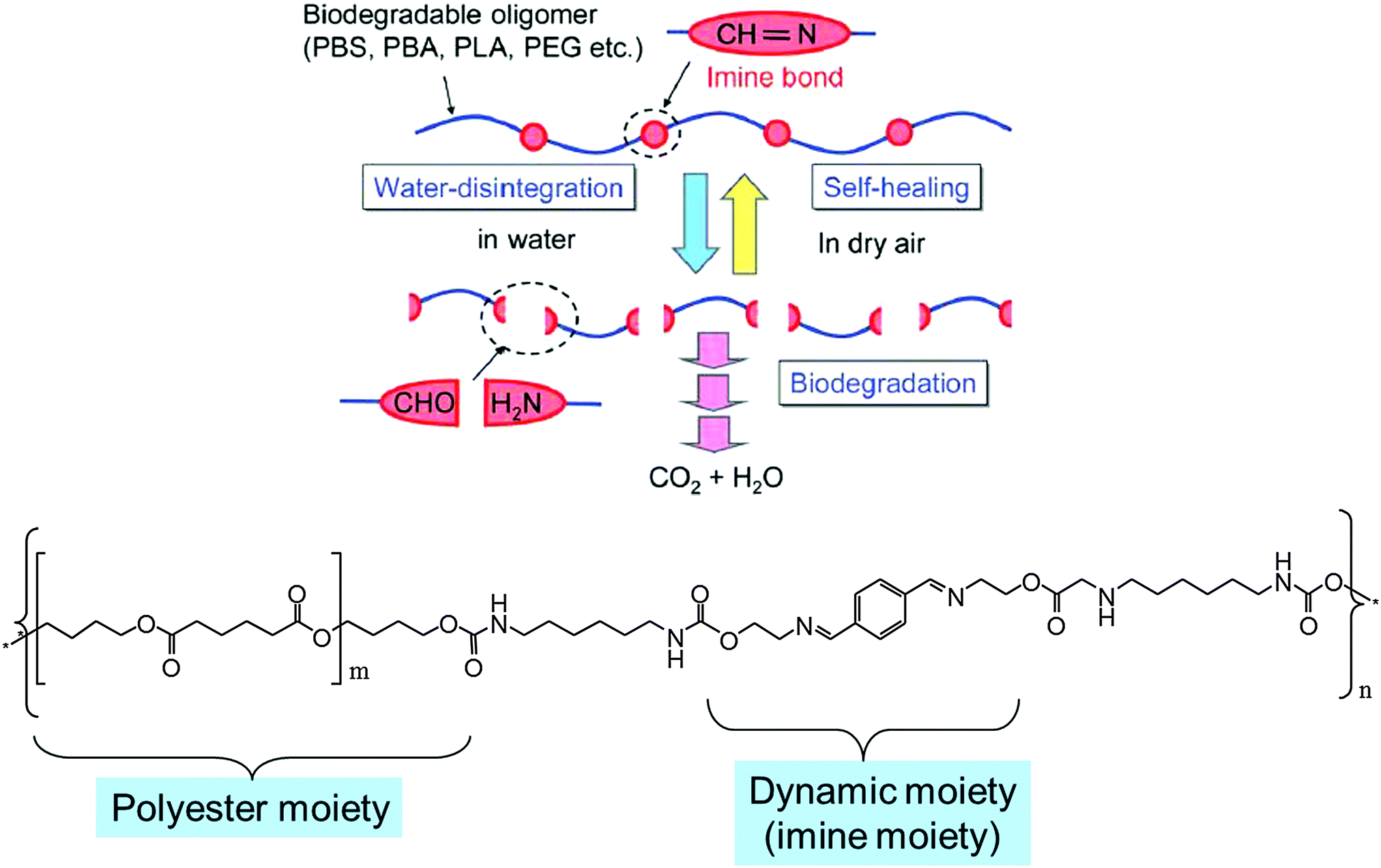 | ||
| Fig. 26 (top) Schematic depiction of the molecular design for the synthesis of a Green DYNAMER; (bottom) representative example of molecular design of a Green DYNAMER.112 | ||
Two different types of GD's were built upon the connection of biodegradable PBS segments through environmentally/hydrolytically degradable imine bonds. In order to demonstrate that these GD's were not water soluble, a control experiment was performed by immersing prepared films into distilled water at 35 °C for 24 h. After drying the films, the weight retention was found to be 98.8% indicating their water insolubility.
The samples were then subjected to water disintegration tests and it was observed that an increasing proportion of imine bonds within the polymer backbone promoted the extent of water disintegration (Fig. 27). Dynamer 2 with a number average Mn of 2500 Da disintegrated faster compared to dynamer 1 with Mn 6300 Da with a lesser number of imine bonds. Thus, through modulation of imine bonds within the polymer backbone it is possible to regulate the extent of water disintegration and in turn its green dynamism.
 | ||
| Fig. 27 Water disintegration test for PBS films (a) and green dynamer (imine functionalized) films of 15 mm thickness based on PBS of Mn = 6300 (b) and Mn = 2500 (c). | ||
In order to investigate the self-healing properties of these green dynamers, NMR measurements were performed on freshly prepared films after immersion in water at 50 °C for 15 h and subsequent drying at 80 °C for 240 h. The Mn values were calculated from NMR data and were found to exhibit a reduction from 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 7000 Da. However, after subsequent drying of the film, the Mn recovered its initial value of 13000, demonstrating that these green dynamers could also exhibit self-healing features upon removal of the disintegrating agent, in this case water.112
000 to 7000 Da. However, after subsequent drying of the film, the Mn recovered its initial value of 13000, demonstrating that these green dynamers could also exhibit self-healing features upon removal of the disintegrating agent, in this case water.112
6. Biodynamers
Extending the concept of dynamic polymers into the domain of biopolymers gives rise to a new area of biologically relevant dynamic polymeric materials namely biodynamers. In this last section, we will give a brief overview of such dynamic biopolymers. Biologically inspired supramolecular polymers have also received considerable research focus in the recent past.12–17,113–117 The features of dynamers may also be applied to biologically active moieties, such as nucleic acid, carbohydrate and amino acid derived units, yielding in principle dynamic analogues of the corresponding biological macromolecules (Scheme 6) capable of undergoing component exchange through reversible chemical reaction, with component reorganization and exchange under the influence of external physical and chemical effectors, such as pH and temperature.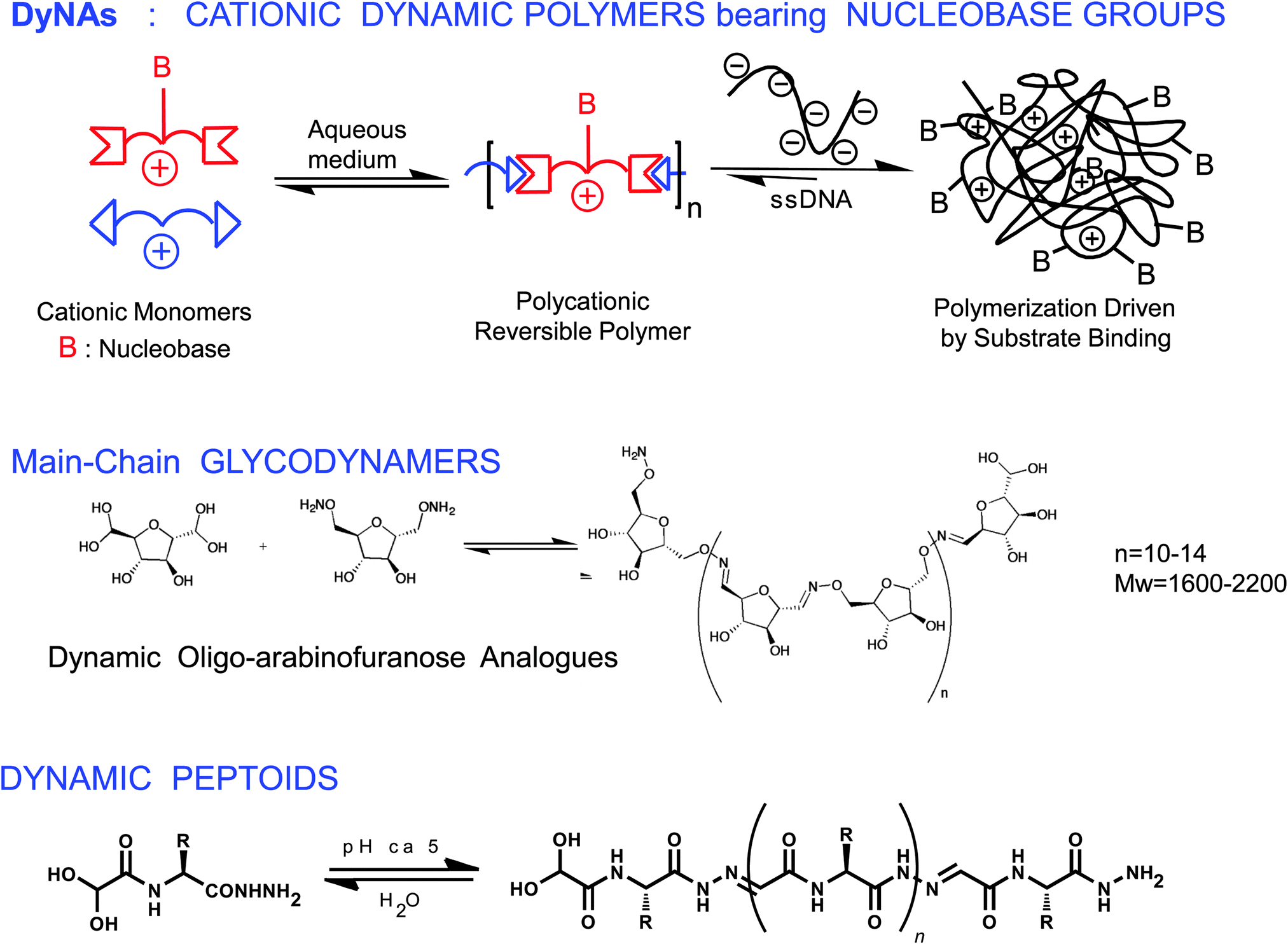 | ||
| Scheme 6 Biodynamers. Generation of dynamic analogues of nucleic acids, DyNAs (top),17 of polysaccharides (middle)122 and of proteins (bottom).123 | ||
Dynamic analogues of nucleic acids, DyNAs, may be formed through polycondensation of suitably functionalized monomers bearing nucleic acid residues, via reversible covalent reaction (Scheme 6, top).17,118–121 Such DyNAs were generated through polycondensation of dialdehydes with nucleobase appended dihydrazides. The monomeric components were chosen to be cationic, protonated amines, so as to interact with various polyanionic entities. It was observed that the DPn of the polymer increased with an increase in the negative charge of the target species, driven by electrostatic interactions, corroborating the adaptability of the biodynamer towards a target entity.
Dynamic analogues of glycopolymers, oligo(poly)saccharides, glycodynamers, could present adaptive character, being in principle able to reorganize their sequence or constitution for preferential selection of bioactive units in response to interaction with target entities. One may envisage to generate three types of glycodynamers via three methodologies, namely (a) main chain glycodynamers via reversible polycondensation of saccharide residues. (b) Side chain appended or grafted saccharide residues on a dynamic main chain and (c) double dynamic glycodynamers through simultaneous incorporation of main chain and side chain dynamics. Main chain dynamic analogues of oligofuranosides were prepared through oxime polycondensation (Scheme 6, middle).15 On the other hand, covalent dynamers bearing glycosidic side-chains were synthesized by reversible acylhydrazone formation. The dynamism in these materials was verified through component exchange experiments and monitoring through 1H NMR spectroscopy as well as by opto-dynamic features, whereby dynamic incorporation of different monomeric components led to marked changes in fluorescence.122 The target-binding ability of these biodynamers was substantiated by surface plasmon resonance (SPR).122
Dynamic analogues of polypeptides/proteins, proteoid biodynamers were formed by polycondensation of water-soluble amino acid derived constituents, via acyl hydrazone and imine formation (Scheme 6, bottom). The process was driven through self-organization enforced primarily by hydrophobic effects, which are also a major factor in protein folding. It involved the polycondensation of an amphiphilic dialdehyde, presenting a hydrophobic aromatic core and a hydrophilic hexaglyme side-chain, with hydrazides of different amino acids, generating the hybrid biodynamers shown in Scheme 7.123
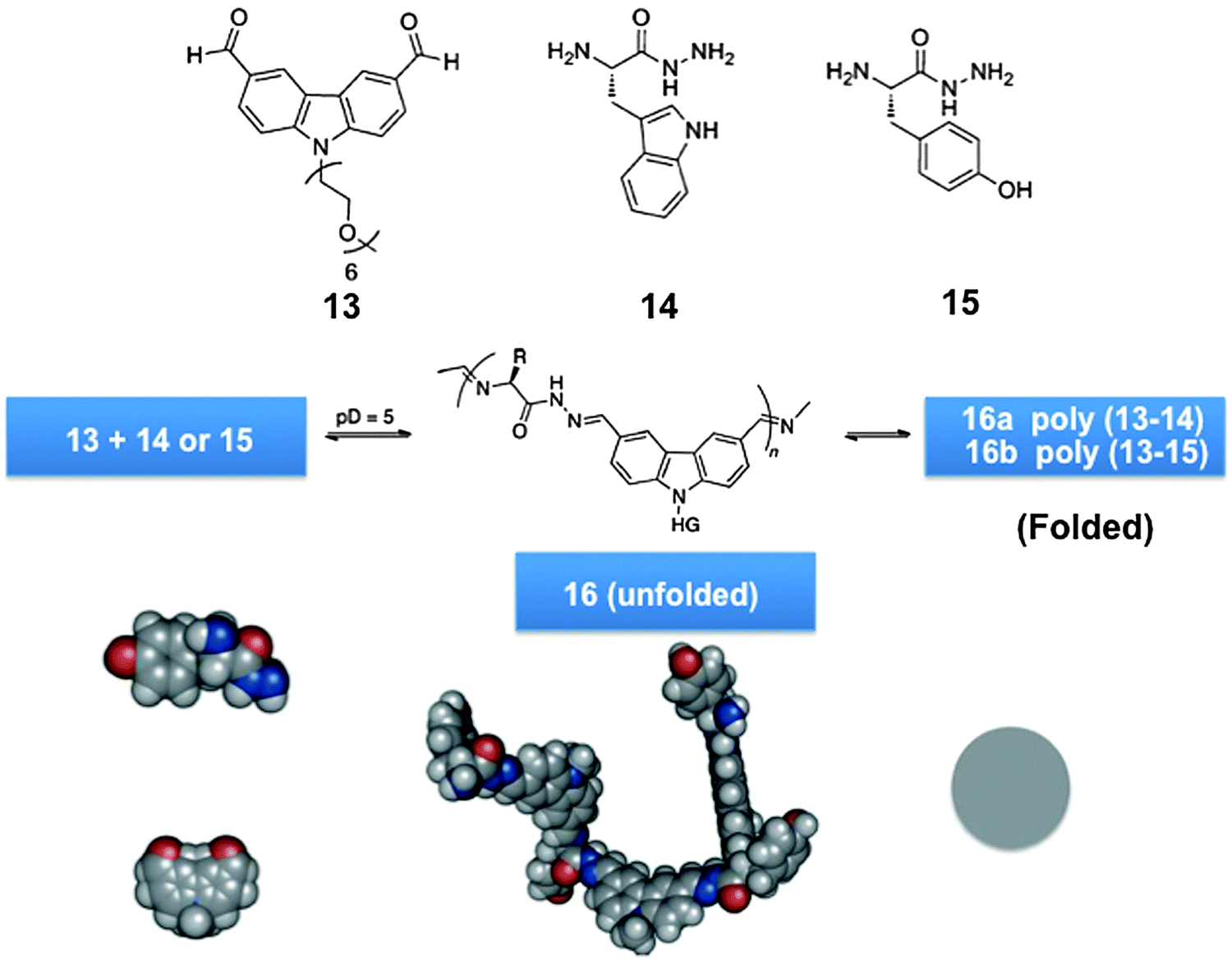 | ||
| Scheme 7 Generation of hybrid proteoidic globular biodynamers of type 16via dynamic polycondensation of monomers.123 | ||
The reversible polycondensation of the monomers yielded a dynamic copolymer with a globular structure with remarkable monodispersity. It featured double covalent dynamics allowing for fine-tuning of both exchange and incorporation processes through pH control. It displayed self-organization driven by hydrophobic effects with component selection, whereby in the course of build-up, selection of the most suitable amino acid building block occurred, as indicated by the preferential incorporation of the more hydrophobic amino-acid component with an increased rate and higher molecular weight of the polymer formed.
Although biodynamic materials of the types discussed above were not yet investigated with respect to potential self-healing behaviour, we included them in the present review in order to stress that they provide strong incentives to further explore this field towards the development of self-healing biodynamers that would be compatible with biological tissues and present medical applications, for instance in reparative medicine (see below).
7. Conclusions and outlook
Dynamic materials and in particular dynamic polymers developed in the last decade or so are bridging the gap between materials science, chemistry and biology.124 Dynamic covalent and non-covalent chemistries represent a significant step along the development of more complex forms of materials. The notion and implementation of self-healing behaviour are especially intriguing as well as promising immediate outcomes of dynamic constitutional chemistry that forms the conceptual background. In the past ten years, more than a thousand scientific reports have been published on the topic of dynamic materials and specifically on self-healing systems. Self-healing materials could be rightly recognized as materials of the future.125 However, out of the different approaches that have been evaluated to synthesize them, the systems, which are self-contained or self-sustainable, may be considered as the most important ones. These materials are principally based on dynamic covalent chemistry such as the DA reaction or on non-covalent interactions, like H bonding. Such self-healing materials are truly self-sustainable as the connections between the components can be reformed even after multiple cycles without compromise to the material performance. As a recent illustration, supramolecular polymers have been used as healable coating materials. Thus, DYNAMERs may also be envisaged as potential materials for self-healing coatings and paints for scratch free surface functionalization.126On another line, one may note that in a recent development supramolecular polymers have been put to use as biomaterials for the fabrication of heart implants, which have indeed been surgically applied with success in several children presenting severe congenital cardiac malformation.127 These achievements are particularly satisfying and rewarding with regard to the transformation, more than twenty years later, of an initial concept of basic research into a major medical/surgical advance.74,75,128 One may expect that numerous future advances will be made in many areas of materials science and beyond through the implementation of constitutional dynamic materials and polymers of both molecular and supramolecular nature.
Acknowledgements
This work was supported by the ERC Advanced Grant SUPRADAPT 290585.References
- W. G. Skene and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8270–8275 CrossRef CAS PubMed.
- J.-M. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS PubMed.
- J.-M. Lehn, Adv. Polym. Sci., 2013, 261, 155–172 CrossRef CAS.
- J.-M. Lehn, Top. Curr. Chem., 2012, 322, 1–32 CrossRef CAS.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS PubMed.
- S. K. Ghosh, Self-Healing Materials: Fundamental, Design Strategies and Applications, Wiley-VCH, Weinheim, 2009 Search PubMed.
- S. van der Zwaag, Self-Healing Materials: An Alternative Approach to 20 Centuries of Materials Science, Springer-Verlag, Dordrecht, 2007 Search PubMed.
- W. Binder, Self-Healing Polymers: From Principles to Applications, Wiley-VCH, Weinheim, 2013, ISBN:978-3-527-33439-1 Search PubMed.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- N. K. Guimard, K. Oehlenschlaeger, J. Zhou, S. Hilf, F. G. Schmidt and C. Barner-Kowollik, Macromol. Chem. Phys., 2012, 213, 131–143 CrossRef CAS PubMed.
- A. B. W. Brochu, S. L. Craig and W. M. Reichert, J. Biomed. Mater. Res., Part A, 2011, 96, 492–506 CrossRef PubMed.
- L. Marin, B. C. Simionescu and M. Barboiu, Chem. Commun., 2012, 48, 8778–8780 RSC.
- Y. Ruff, E. Buhler, S. Candau, E. Kesselman, Y. Talmon and J.-M. Lehn, J. Am. Chem. Soc., 2010, 132, 2573–2584 CrossRef CAS PubMed.
- Y. Ruff and J.-M. Lehn, Biopolymers, 2008, 89, 486–496 CrossRef CAS PubMed.
- Y. Ruff and J.-M. Lehn, Angew. Chem., Int. Ed., 2008, 47, 3556–3559 CrossRef CAS PubMed.
- N. Sreenivasachary, D. T. Hickman, D. Sarazin and J.-M. Lehn, Chem. – Eur. J., 2006, 12, 8581–8588 CrossRef CAS PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- G. L. Fiore, S. J. Rowan and C. Weder, Chem. Soc. Rev., 2013, 42, 7278–7288 RSC.
- A. C. Jackson, J. A. Bartelt and P. V. Braun, Adv. Funct. Mater., 2011, 21, 4705–4711 CrossRef CAS PubMed.
- M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., Int. Ed., 2012, 51, 7011–7015 CrossRef CAS PubMed.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrinyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- S. D. Bergmann and F. Wudl, J. Mater. Chem., 2008, 18, 41–62 RSC.
- J. A. Syrett, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 978–987 RSC.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- A. Gandini, Polím.: Ciênc. Tecnol., 2005, 15, 95–101 CrossRef CAS PubMed.
- J. M. Craven, US Pat., 3435003, 1969 Search PubMed.
- M. Stevens and A. Jenkins, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 3675–3685 CrossRef CAS PubMed.
- J. S. Park, T. Darlinton, A. F. Starr, K. Takahashi, J. Riendeau and H. T. Hahn, Compos. Sci. Technol., 2010, 70, 2154–2159 CrossRef CAS PubMed.
- A. A. Kavitha and N. K. Singha, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4441–4449 CrossRef CAS PubMed.
- M. Watanabe and N. Yoshie, Polymer, 2006, 47, 4946–4952 CrossRef CAS PubMed.
- A. Sanyal, Macromol. Chem. Phys., 2010, 211, 1417–1425 CrossRef CAS PubMed.
- J. R. McElhanon and D. R. Wheeler, Org. Lett., 2001, 3, 2681–2683 CrossRef CAS PubMed.
- J. R. McElhanon, E. M. Russick, D. R. Wheeler, D. A. Loy and J. H. Aubert, J. Appl. Polym. Sci., 2002, 85, 1496–1502 CrossRef CAS PubMed.
- D. A. Loy, D. R. Wheeler, J. R. McElhanon and M. L. Durbin-Voss, US Pat., 6 403 753, 2002 Search PubMed.
- D. A. Loy, D. R. Wheeler, E. M. Russick, J. R. McElhanon and R. S. Sanders, US Pat., 6 337 384, 2002 Search PubMed.
- J. H. Small, D. A. Loy, D. R. Wheeler, J. R. McElhanon and R. S. Saunders, US Pat., 6 271 335, 2001 Search PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- X. Chen, F. Wudl, A. K. Mal, H. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802–1807 CrossRef CAS.
- F. Wudl and X. Chen, US Pat., 6 933 361, 2005 Search PubMed.
- Y.-L. Liu, C. Y. Hsieh and Y. W. Chen, Polymer, 2006, 47, 2581–2586 CrossRef CAS PubMed.
- Y.-L. Liu and C. Y. Hsieh, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 905–913 CrossRef CAS PubMed.
- Y.-L. Liu and Y. W. Chen, Macromol. Chem. Phys., 2007, 208, 224–232 CrossRef CAS PubMed.
- J. S. Felipe, J. De Abajo and J. G. De La Campa, J. Appl. Polym. Sci., 1985, 30, 61–69 CrossRef PubMed.
- M. K. Kim and S. Y. Kim, Macromolecules, 2002, 35, 4553–4555 CrossRef CAS.
- Y. J. Kim, I. S. Chung, I. In and S. Y. Kim, Polymer, 2005, 46, 3992–4004 CrossRef CAS PubMed.
- G. Rivero, L. T. Nguyen, X. K. D. Hillewaere and F. E. Du Prez, Macromolecules, 2014, 47, 2010–2018 CrossRef CAS.
- S. Billiet, K. D. Bruycker, F. Driessen, H. Goossens, V. V. Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS PubMed.
- P. J. Boul, P. Reutenauer and J.-M. Lehn, Org. Lett., 2005, 7, 15–18 CrossRef CAS PubMed.
- P. Reutenauer, P. J. Boul and J.-M. Lehn, Eur. J. Org. Chem., 2009, 1691–1697 CrossRef CAS PubMed.
- N. Roy and J.-M. Lehn, Chem. – Asian J., 2011, 6, 2419–2425 CrossRef CAS PubMed.
- P. Reutenauer, E. Buhler, P. J. Boul, S. J. Candau and J.-M. Lehn, Chem. – Eur. J., 2009, 15, 1893–1900 CrossRef CAS PubMed.
- A. M. Peterson, R. E. Jensen and G. R. Palmese, ACS Appl. Mater. Interfaces, 2010, 2, 1141–1149 CAS.
- M. M. Caruso, D. A. Delafuente, V. Ho, N. R. Sottos, J. Moore and S. R. White, Macromol. Rapid Commun., 2007, 40, 8830–8832 CrossRef CAS.
- B. J. Blaiszik, M. M. Caruso, D. A. McIlroy, J. S. Moore, S. R. White and N. R. Sottos, Polymer, 2009, 50, 990–997 CrossRef CAS PubMed.
- P. A. Pratama, M. Sharifi, A. M. Peterson and G. R. Palmese, ACS Appl. Mater. Interfaces, 2013, 5, 12425–12431 CAS.
- A. J. Inglis, L. Nebhani, O. Altintas and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515–5520 CrossRef CAS.
- S. Sivakova, D. A. Bohnsack, M. E. Mackay, P. Suwanmala and S. J. Rowan, J. Am. Chem. Soc., 2005, 127, 18202–18211 CrossRef CAS PubMed.
- J.-L. Wietor and R. P. Sijbesma, Angew. Chem., Int. Ed., 2008, 47, 8161–8163 CrossRef CAS PubMed.
- D. J. M. van Beek, A. J. H. Spiering, G. W. M. Peters, K. te Nijenhuis and R. P. Sijbesma, Macromolecules, 2007, 40, 8464–8475 CrossRef CAS.
- N. E. Botterhuis, D. J. M. van Beek, G. M. L. van Gemert, A. W. Bosman and R. P. Sijbesma, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3877–3885 CrossRef CAS PubMed.
- E. Kolomiets, E. Buhler, S. J. Candau and J. M. Lehn, Macromolecules, 2006, 39, 1173–1181 CrossRef CAS.
- M. T. Hunley, A. S. Karikari, M. G. McKee, B. D. Mather, J. M. Layman, A. R. Fornof and T. E. Long, Macromol. Symp., 2008, 270, 1–7 CrossRef CAS PubMed.
- W. H. Binder, S. Bernstorff, C. Kluger, L. Petraru and M. Kunz, Adv. Mater., 2005, 24, 2824–2828 CrossRef PubMed.
- K. E. Feldman, M. J. Kade, E. W. Meijer, C. J. Hawker and E. J. Kramer, Macromolecules, 2010, 43, 5121–5127 CrossRef CAS.
- J.-M. Lehn, in Supramolecular Polymers, ed. A. Ciferri, Dekker, New York, 2000, pp. 615–641 Search PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS PubMed.
- A. Ciferri, Supramolecular Polymers, New York, 2nd edn, 2005 Search PubMed.
- Themed issue: Supramolecular Polymers, Chem. Soc. Rev., 2012, 18, 5869–6216.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- V. Berl, M. Schmutz, M. J. Krische, R. G. Khoury and J.-M. Lehn, Chem. – Eur. J., 2002, 8, 1227–1244 CrossRef CAS.
- J.-M. Lehn, Polym. Int., 2002, 51, 825–839 CrossRef CAS PubMed.
- C. Fouquey, J.-M. Lehn and A. M. Levelut, Adv. Mater., 1990, 2, 254–257 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. Ky Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- J. H. K. Ky Hirschberg, F. H. Beijer, H. A. van Aert, P. C. M. M. Magusin, R. P. Sijbesma and E. W. Meijer, Macromolecules, 1999, 32, 2696–2705 CrossRef CAS.
- R. F. M. Lange, M. van Gurp and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3657–3670 CrossRef CAS.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- A. El-Ghayoury, E. Peeters, A. P. H. J. Schenning and E. W. Meijer, Chem. Commun., 2000, 1969–1970 RSC.
- J. H. K. Ky Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167–170 CrossRef PubMed.
- A. P. H. J. Schenning, P. Jonkheijm, E. Peeters and E. W. Meijer, J. Am. Chem. Soc., 2001, 123, 409–416 CrossRef CAS.
- A. El-Ghayoury, A. P. H. J. Schenning, P. A. van Hal, J. K. J. van Duren, R. A. J. Janssen and E. W. Meijer, Angew. Chem., Int. Ed., 2001, 40, 3660–3663 CrossRef CAS.
- H. M. Keizer, R. P. Sijbesma, J. F. G. A. Jansen, G. Pasternack and E. W. Meijer, Macromolecules, 2003, 36, 5602–5606 CrossRef CAS.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- F. Herbst, K. Schröter, I. Gunkel, S. Gröger, T. Thurn-Albrecht, J. Balbach and W. H. Binder, Macromolecules, 2010, 43, 10006–10016 CrossRef CAS.
- F. Herbst, D. Döhler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 CrossRef CAS PubMed.
- F. Herbst, S. Seiffert and W. H. Binder, Polym. Chem., 2012, 3, 3084–3092 RSC.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- D. Montarnal, F. Tournilhac, M. Hidalgo, J.-C. Couturier and L. Leibler, J. Am. Chem. Soc., 2009, 131, 7966–7967 CrossRef CAS PubMed.
- C. Wang, N. Liu, R. Allen, J. B.-H. Tok, Y. Wu, F. Zhang, Y. Chen and Z. Bao, Adv. Mater., 2013, 25, 5785–5790 CrossRef CAS PubMed.
- SupraPolix BV, http://www.suprapolix.com/.
- N. Roy, E. Buhler and J.-M. Lehn, Chem. – Eur. J., 2013, 19, 8814–8820 CrossRef CAS PubMed.
- L. Bouteiller, Adv. Polym. Sci., 2007, 207, 79–112 CrossRef CAS.
- S. Boileau, L. Bouteiller, F. Lauprêtre and F. Lortie, New J. Chem., 2000, 24, 845–848 RSC.
- V. Simic, L. Bouteiller and M. Jalabert, J. Am. Chem. Soc., 2003, 125, 13148–13154 CrossRef CAS PubMed.
- O. Colombani and L. Bouteiller, New J. Chem., 2004, 28, 1373–1382 RSC.
- M. Bellot and L. Bouteiller, Langmuir, 2008, 24, 14176–14182 CrossRef CAS PubMed.
- J. Courtois, I. Baroudi, N. Nouvel, E. Degrandi, S. Pensec, G. Ducouret, C. Chanéac, L. Bouteiller and C. Creton, Adv. Funct. Mater., 2010, 20, 1803–1811 CrossRef CAS PubMed.
- E. Sabadini, K. R. Francisco and L. Bouteiller, Langmuir, 2010, 26, 1482–1486 CrossRef CAS PubMed.
- H. Yang, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218–3227 Search PubMed.
- K. Liu, Y. Kang, Z. Wang and X. Zhang, Adv. Mater., 2013, 25, 5530–5548 CrossRef CAS PubMed.
- A. Gooch, C. Nedolisa, K. A. Houton, C. I. Lindsay, A. Saiani and A. J. Wilson, Macromolecules, 2012, 45, 4723–4729 CrossRef CAS.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 124, 1164–1168 CrossRef PubMed.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- E. Kolomiets and J.-M. Lehn, Chem. Commun., 2005, 1519–1521 RSC.
- G. Schaeffer, E. Buhler, S. J. Candau and J.-M. Lehn, Macromolecules, 2013, 46, 5664–5671 CrossRef CAS.
- N. Roy, E. Buhler and J.-M. Lehn, Polym. Int., 2014, 63, 1400–1405 CrossRef CAS PubMed.
- K. Fukuda, M. Shimoda, M. Sukegawa, T. Nobori and J.-M. Lehn, Green Chem., 2012, 14, 2907–2911 RSC.
- I. Nakazawa, S. Suda, M. Masuda, M. Asai and T. Shimitu, Chem. Commun., 2000, 881–882 RSC.
- D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477–488 CrossRef CAS PubMed.
- Z. Shriver, S. Raguram and R. Sasisekharan, Nat. Rev. Drug Discovery, 2004, 3, 863–873 CrossRef CAS PubMed.
- L. Marin, I. Stoica, M. Mares, V. Dinu, B. C. Simionescu and M. Barboiu, J. Mater. Chem., 2013, 1, 3353–3358 RSC.
- Y. Lu and J. Liu, Acc. Chem. Res., 2007, 40, 315–323 CrossRef CAS PubMed.
- J. T. Goodwin and D. G. Lynn, J. Am. Chem. Soc., 1992, 114, 9197–9198 CrossRef CAS.
- A. Bugaut, J.-J. Toulmé and B. Rayner, Angew. Chem., Int. Ed., 2004, 43, 3144–3147 CrossRef CAS PubMed.
- B. Klekota and B. L. Miller, Tetrahedron, 1999, 55, 11687–11697 CrossRef CAS.
- C. Karan and B. Miller, J. Am. Chem. Soc., 2001, 123, 7455–7456 CrossRef CAS.
- Y. Ruff, E. Buhler, S.-J. Candau, E. Kesselman, Y. Talmon and J.-M. Lehn, J. Am. Chem. Soc., 2010, 132, 2573–2584 CrossRef CAS PubMed.
- A. K. H. Hirsch, E. Buhler and J.-M. Lehn, J. Am. Chem. Soc., 2012, 134, 4177–4183 CrossRef CAS PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2832–2850 CrossRef PubMed.
- N. Notman, Chem. World, 2012, 50–53 CAS.
- Q. Wei, C. Schlaich, S. Prévost, A. Schulz, C. Böttcher, M. Gradzielski, Z. Qi, R. Haag and C. A. Schalley, Adv. Mater., 2014, 26, 7358–7364 CrossRef CAS PubMed.
- The supramolecular biomaterial and the implants have been developed by the company XELTIS. The surgical implantations have been realized by Professor Leo Bokeria at the Bakoulev Scientific Center of Cardiovascular Surgery in Moscow. For brief reports, see: L. A. Bokeria, Cardiology today, 2014; F. J. Schoen, Cardiology today, 2014.
- J.-M. Lehn, Makromol. Chem., Macromol. Symp., 1993, 69, 1–17 CrossRef CAS PubMed.
Footnote |
| † Current address: BASF Polyurethanes GmbH, Elastogranstr. 60, D-49448, Lemfoerde, Germany. E-mail: E-mail: nabarun.roy@basf.com. |
| This journal is © The Royal Society of Chemistry 2015 |