
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation
Oleksandr
Koniev
ab and
Alain
Wagner
*a
aLaboratory of Functional Chemo-Systems (UMR 7199), Labex Medalis, University of Strasbourg, 74 Route du Rhin, 67401 Illkirch-Graffenstaden, France. E-mail: alwag@unistra.fr
bSyndivia SAS, 4 rue Boussingault, 67000 Strasbourg, France
First published on 22nd May 2015
Abstract
Bioconjugation methodologies have proven to play a central enabling role in the recent development of biotherapeutics and chemical biology approaches. Recent endeavours in these fields shed light on unprecedented chemical challenges to attain bioselectivity, biocompatibility, and biostability required by modern applications. In this review the current developments in various techniques of selective bond forming reactions of proteins and peptides were highlighted. The utility of each endogenous amino acid-selective conjugation methodology in the fields of biology and protein science has been surveyed with emphasis on the most relevant among reported transformations; selectivity and practical use have been discussed.
 Oleksandr Koniev | Dr Oleksandr (Sasha) Koniev received an MSc in molecular chemistry from the University of Toulouse (France) in 2010. He subsequently joined the group of Dr Alain Wagner for his graduate studies, where he developed novel bioselective ligation reactions and obtained his PhD from the University of Strasbourg in 2014. The same year, Dr Koniev co-founded and became CEO of Syndivia, a biotechnology company that provides advanced bioconjugation techniques for the development of antibody–drug conjugates. His research interests focus on designing new bioconjugation approaches allowing preparation of biological materials with enhanced performance and their application in anticancer therapies. |
 Alain Wagner | Dr Alain Wagner received his PhD in organic chemistry from Université Louis Pasteur (1991, Strasbourg, France), followed by a post-doctoral fellowship with Prof. Peter Schultz at Affymax Research Institute (1991–1993, Palo Alto, USA). He joined CNRS in 1994 to develop projects involving chemistry and biology. Alain Wagner co-founded a number of successful start-up companies: Novalix Pharma (2002), Phytodia (2007), eNovalys (2009), and most recently Syndivia (2014). Returning to the CNRS (2008), he took the lead of the Laboratory of Functional Chemo-Systems. Dr Wagner has published more than 120 articles in peer-reviewed journals and he is an inventor of 17 patents. |
Introduction
Bioconjugation is a set of techniques allowing site-specific creation of a covalent link between a biomolecule and an exogenous moiety that endow it with desirable properties. This novel hybrid having the combined properties of its individual components can serve, for instance, as a more stable and efficient therapeutic,1–4 an assembly for studying proteins in the biological context,5–11 a new protein-based material,12–17 a microarray,18,19 a biological material,20–24 a tool for immobilisation,25 and for elucidation of the structure26,27 of proteins.A large number of reactions exist to modify proteins.28 However, site-specific conjugation continues to attract considerable research efforts to develop new methodologies that match continuously increasing requirements of modern applications in terms of selectivity, stability, mildness, and preserving biomolecule integrity. For the purpose of this overview, the focus will remain on recent developments in bond-forming approaches in bioconjugation of native amino acid residues. Among about 20 different amino acids involved in protein composition, only a smaller number comprises appropriate targets for practical bioconjugation methods. In fact, only one-third of all amino acid residues represent chemical targets for the vast majority of bond-forming approaches.
The bioconjugation methodology of choice is selected according to the intrinsic reactivity of the targeted amino acid residue (acidity/basicity, electrophilicity/nucleophilicity, oxido-reductive characteristics) and its specific special environment (in-chain, N-terminal, C-terminal, location in a specific sequence, accessibility, etc.). In this review we will thus present the known bioconjugation strategies in regard to these parameters ranked in a descending order of frequency they are reported in literature.
1. In-chain conjugation
1.1 Lysine
The use of chemical groups that react with primary amines is one of the oldest and most versatile techniques for protein conjugation. Virtually all proteins contain primary amino groups in their structure. They can be divided into two groups: the α-amino group situated in the N-terminus of most polypeptide chains and ε-amino groups of lysine residue (Lys, K). Because these amino groups possess pKa values of about 8 and 10 (for α- and ε-amines respectively), in a vast majority of cases they are protonated at physiological pH and, therefore, occur predominantly on the solvent-exposed outside surfaces of protein tertiary structures. As a result, they become easily accessible to conjugation reagents introduced into the aqueous media.Deprotonated primary amines are the most nucleophilic among the available functional groups present in a typical protein. However, protonation drastically decreases their reactivity. As a consequence, despite the generally higher intrinsic nucleophilicity of Lys ε-amino groups, they require higher pH values to be uncovered by deprotonation, which allows distinguishing α- and ε-amino groups by adjusting the pH. That is to say, at the higher pH level, when both types of primary amines are deprotonated, Lys side chain amino groups are generally more reactive towards electrophiles, while at the lower pH it is the opposite because of their prior protonation (Fig. 1). At the acidic pH all amines are protonated and possess no significant nucleophilicity compared to other side chains present in proteins. In particular, free (non-disulfide-bonded) Cys residues are much stronger nucleophiles and, if accessible, will readily be modified by most amine-reactive reagents.
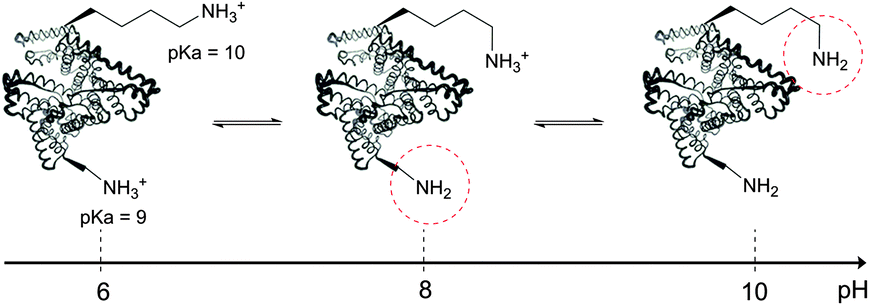 | ||
| Fig. 1 Deprotonation of different types of amino groups present in protein structure (more nucleophilic amine is encircled in red). Lysine ε-amino groups are more nucleophilic, but also more difficult to deprotonate. Generally, a pH of 8.5–9.5 is optimal for modifying lysine residues, while near neutral pH favours selective modification of N-termini. | ||
It is also to be mentioned that like any other parameters, nucleophilicity and basicity, as well as solvent exposure and accessibility of a particular amino group, are influenced by the microenvironment and can vary substantially, regarding the substrate. For instance, Westheimer and Schmidt have found the actual pKa of the amino group situated in the active site of acetoacetate decarboxylase to be 5.9, which is 4 pKa units less than that of an “ordinary” ε-amino group of lysine.29
Depending on reaction conditions, selective modification of either N-termini (see Section 2) or Lys ε-amino groups can be achieved by using various chemical reagents. They generally belong to one of the following classes (in the order of relevancy): activated esters (fluorophenyl esters, NHS (N-hydroxysuccinimides),30 sulfo-NHS, acyl azides), isothiocyanates, isocyanates,31 aldehydes, anhydrides, sulfonyl chlorides, carbonates, fluorobenzenes, epoxides and imidoesters. Among this vast variety of reactive functions, NHS esters (and their more soluble sulfo-NHS analogues) and imidoesters represent the most popular amine-specific functional groups that are incorporated into commercially available reagents for protein conjugation and labelling.28
Despite their name, amine-reactive reagents are not always entirely selective for amines. Firstly, as already mentioned before, they will react with any other stronger nucleophile, if the latter is present and accessible on a protein surface. Particularly, it concerns cysteine, tyrosine, serine and threonine side chains. Secondly, depletion of these highly activated reagents by hydrolysis is inevitable in aqueous solution. The rate of both side-reactions depends on the particular substrate, the conjugation partner, pH, temperature, and buffer composition. Evidently, buffers that contain free amines, such as TRIS (tris(hydroxymethyl)aminomethane), must be avoided when using any amine-reactive probes, since the rate of the reaction with buffer would greatly exceed that with protein amino groups.
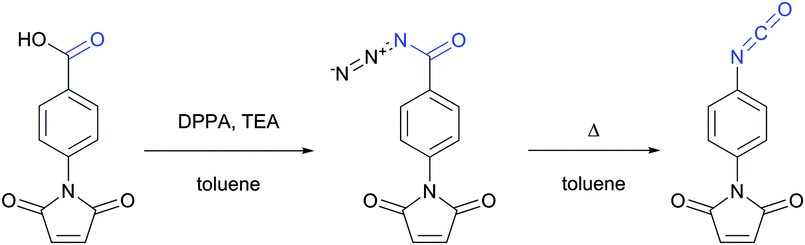 | ||
| Fig. 2 Synthesis of the p-maleimidophenyl isocyanate crosslinker via Curtius rearrangement proposed by Palumbo and associates.33 | ||
Several early studies were devoted to the elaboration of isocyanate conjugation methodology,34,35 but proven to be especially laborious and complicated mainly due to the high reactivity and low stability of isocyanates. Therefore they are of deferred interest today, being completely displaced by isothiocyanate-mediated approaches. Both isothiocyanates and isocyanates can be obtained from the corresponding aromatic amines upon reaction with thiophosgene and phosgene respectively (Fig. 3).36
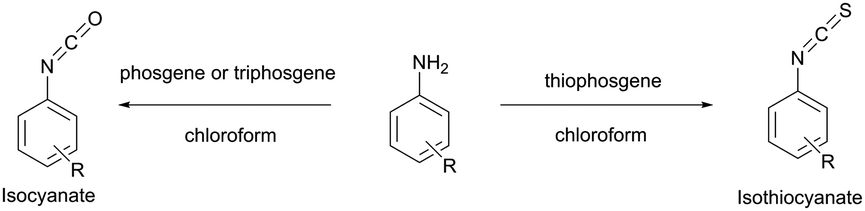 | ||
| Fig. 3 Synthesis of isothiocyanates and isocyanates from the corresponding aromatic amines.36 | ||
Isothiocyanate-based selective amino group modification was first reported in 1937 by Todrick and Walker,37 who found that the reaction of allyl isothiocyanate with cysteine in alkaline medium results selectively in thiourea – the product of amine addition to isothiocyanate. In 1950, exploiting the selectivity of amino-terminal labelling of the peptide with phenylisothiocyanate, Edman has developed a method for peptide sequencing that has changed cardinally the protein science and is known today as Edman degradation.38 Only 30 years later, Podhradský et al. have examined the reaction of isothiocyanates on complex substrates and demonstrated that the addition of the thiol and phenolate functions of cysteine and tyrosine residues is always prevalent, and that only at pH > 5 amino groups start to manifest themselves in the reaction.39 While thiol and alcohol additions result in reversible reactions to give dithiocarbamates and O-thiocarbamates respectively, amines add themselves irreversibly, thus shifting the reaction equilibrium towards thioureas (Fig. 4). One should however keep in mind that, despite the reversibility of the addition of thiols and alcohols to isocyanates, they can enhance the kinetics of their hydrolysis to unreactive amines or ureas and therefore significantly decrease the yield of the conjugation. Moderately reactive but quite stable in water and most solvents, isothiocyanates represent thus an appropriate alternative to the unstable isocyanates. As a consequence, they are much more popular in bioconjugation.
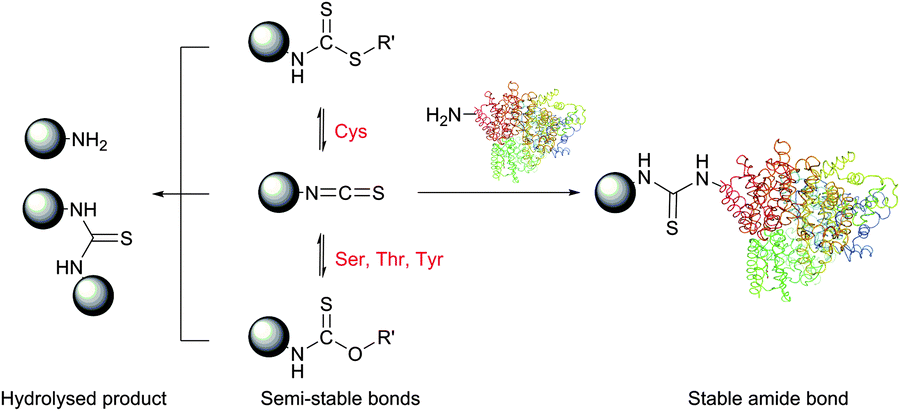 | ||
| Fig. 4 Reaction of isothiocyanates with nucleophilic amino acid residues present in proteins. Only the reaction of lysine and N-terminal residues (considerable at pH > 5) results irreversibly in obtaining the thiourea. Although the reactions of thiol and alcohol groups with isothiocyanates are reversible, they can largely accelerate the rate of isothiocyanate hydrolysis. | ||
Ever since the introduction of fluorescent isothiocyanate dyes as more stable analogues of corresponding isocyanates for fluorescent labelling of antibodies by Riggs et al.40 in 1958, they have found widespread use in research laboratories and proved to be an effective means for tagging proteins at specific sites.41
Fluorescein isothiocyanate (FITC) is arguably one of the most commonly used fluorescent derivatisation reagents for proteins. For instance, it was reported by Tuls et al.42 that cytochrome P-450 can be selectively labelled by FITC with 75% yield of a single-labelled LYS-338 conjugate in TRIS (particularly inappropriate buffer for amine-reactive reagents though) at pH 8.0 and 0 °C. Burtnick43 has described selective labelling of one out of 34 lysine residues of actin in borate buffer with 35-fold excess of the reagent at pH 8.5. Such a high level of selectivity towards the LYS-61 residue over other 33 lysine residues present in proteins (Fig. 5, shown in red) remains unclear, but it is hypothesised to be due to an anomalously low pKa value thereof. Following reports of Miki and collaborators44,45 further confirmed the selectivity of this labelling, yet without any explanation of such specificity. Bellelli et al.46 were able to covalently label ricin (pH 8.1, 6 °C for 4 h). In fact, the targets of the isothiocyanate-mediated labelling of proteins elaborated over the last 60 years are even difficult to enumerate. It was proven to be effective in diverse applications such as tagging of antibodies (usually in carbonate–bicarbonate buffer, pH 9),47–53 bleaching-based measurement of membrane protein diffusion of FITC-labelled cells (pH 9.5, 24 °C),54 surface topography of the Escherichia coli ribosomal subunit,55 α-actinin distribution in living and fixed fibroblasts,56 characterisation of a proton pump on lysosomes,57 and hematopoietic stem cells.58
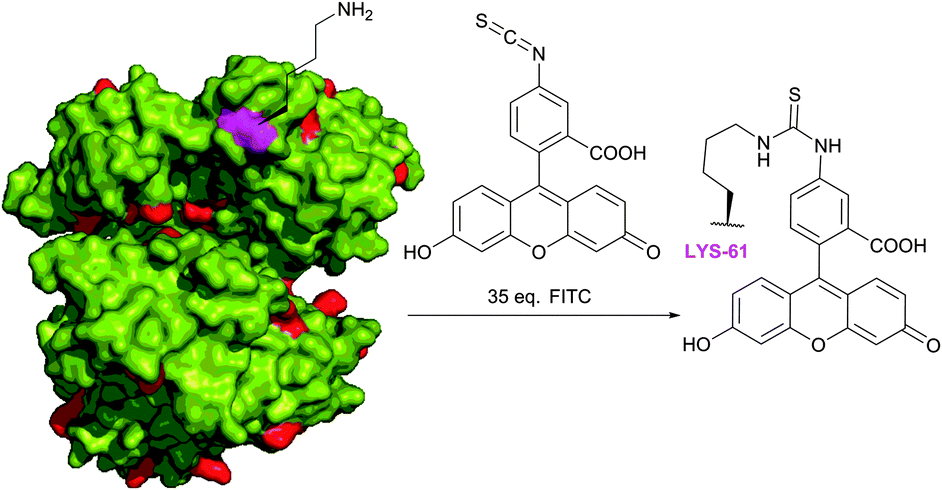 | ||
| Fig. 5 Selective fluorescent labelling of the Lys-61 residue (shown in magenta) of rabbit skeletal muscle G-actin (pdb: 2VYP) reported by Burtnick.43 | ||
The most stunning examples include 125I labelling by means of isothiocyanates, elaborated by Shapiro and colleagues59 for regional differentiation of the sperm surface (TRIS, pH 7.7, 12 °C for 30 min), and the application of a similar methodology by Schirrmacher et al.60 for 18F radioactive labelling of RSA, apotransferrin and bovine IgG (pH 9.0, room temperature for 10–20 min). Conjugation of antibodies with chelating agents for further radiometal labelling of antibodies has been described by several groups61–63 and is based on the use of phenylisothiocyanate-containing probes. Brechbiel et al.64 went even further by combining the chelating functionality with the biotin fragment in a scaffold of trifunctional conjugation reagents. The preparation of silica nanoparticles coated with isothiocyanate groups and their use in apoptosis detection has recently been elaborated.65
The classical protocol of isothiocyanate labelling involves the use of 5–10 equivalents at a slightly basic pH in the range of 9.0–9.5.66,67 Resulting thioureas are reasonably stable in aqueous medium and provide a suitable degree of conjugation.68 For example, Sandmaier and colleagues69 have recently demonstrated that radiolabelling of the anticanine CD45 antibody using isocyanate and isothiocyanate provides a more specific delivery to the targeted CD45-expressing cells than a method exploiting thiol–maleimide conjugation (see Section 1.3.2). However, it has been shown by Banks and Paquette70 that, compared to NHS ester based methodology, antibody conjugates prepared with isothiocyanates are less hydrolytically stable and deteriorate over time. Moreover, the reaction of NHS esters for amine labelling was found to be faster, to give more stable conjugates for both model amino acids and proteins, and to proceed readily at lower pH, compared to isothiocyanates. Consequently, NHS esters are preferable to isothiocyanates in many respects for synthesizing bioconjugates.
N-Hydroxysuccinimide (NHS) activated esters were introduced in 1963 by Anderson et al. as a better alternative to phenyl esters in forming the peptide bond.30,76,77 Possessing high selectivity towards aliphatic amines, NHS esters are today considered among the most powerful protein-modification reagents. Although several studies drew attention to a certain reactivity of NHS-activated esters with tyrosine,78–83 histidine,84 serine and threonine (especially when situated in certain locations, see Section 1.2),85–90 these side reactions possess largely decreased rates compared to the reaction with free amines and do not generally hinder the amine-selective derivatisation. High concentrations of nucleophilic thiols should however be avoided because, similarly to isothiocyanates, they may increase the rate of probe degradation by forming more easily hydrolysable intermediates (Fig. 6).
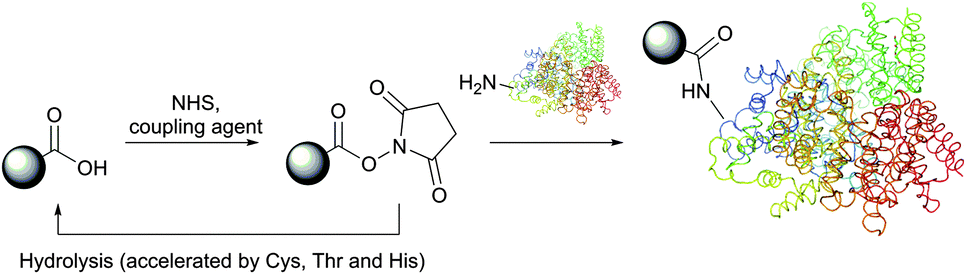 | ||
| Fig. 6 Preparation of activated NHS-esters and their reaction with nucleophilic amino acid residues present in proteins. Similarly to isothiocyanates, the reversible reactions of thiol and alcohol groups with NHS-esters largely increase the rate of hydrolysis thereof. | ||
The optimum pH for NHS-mediated labelling in aqueous systems was found to be lower than for other amine-selective reagents and ranges from 7 to 8 units (compared with 9–9.5 for isothiocyanates), which enlarges the prospect of its suitability for modifying alkaline-sensitive proteins. Several elaborated studies of the kinetics,72 the stoichiometry,91 and the selectivity (in-chain versus N-terminal modification)92 of NHS-mediated protein tagging have been recently reported.
Depending on the pH of the reaction solution and temperature, NHS esters are hydrolysed by water (possessing a half-life of 4–5 hours at pH 7, 1 hour at pH 8 and 10 minutes at pH 8.6),84,93 but are stable to storage if kept well desiccated. Virtually any molecule containing an acid functionality, or a moiety which can give an acid, can be transformed into its N-hydroxysuccinimide ester. While the activation with NHS generally decreases the water-solubility of the carboxylate molecule, the utilisation of sulfo-NHS94 preserves or even increases the water-solubility of the modified molecule by virtue of the charged sulfonate group. The development of new reagents based on NHS chemistry can be sometimes challenging,95 but the derivatives are frequently of very important use.96–100 Many NHS derivatives for the preparation of affinity reagents, fluorescent probes and cross-coupling reagents are now commercially available, enabling wide access to investigations.
The formed conjugates are linked by means of a very stable aliphatic amide bond with half-lives in the range of 7 years in water.101 This excellent stability and biocompatibility of the obtained bonds have provided an exceptional importance of NHS esters in the field of bioconjugation.
NHS ester-mediated covalent conjugation for protein modification has been first accomplished by Becker et al., who studied biotin transport first in yeast74 and then applied this technique to the covalent attachment of biotin to bacteriophage T4.102 Since then, the field of NHS-mediated conjugation of proteins has been unceasingly expanding its employability in countless applications.
Cross-linking of proteins often implies using NHS-containing homobifunctional or heterobifunctional cross-linking reagents. These were used for elucidation of protein–protein103–107 and protein–drug interactions,108 protein structural and subunit analysis,26,109 create protein complex models,110 and preparation of protein conjugates with enzymes, drugs or other macromolecules.111–113
Homobifunctional NHS cross-linkers are generally used in reaction procedures to randomly “fix” or polymerize peptides or proteins through their amino groups. Adding such crosslinkers to a cell lysate will result in the random conjugation of interacting proteins, protein subunits, and any other polypeptides whose Lys side chains happen to be in close proximity to each other. This represents a methodology for capturing a “snapshot” of all protein interactions at a certain instant of time. Using this approach, for instance, Sinz and collaborators were able to elucidate binding of calmodulin to mettilin, a polypeptide and principal component of honeybee venom, without chromatographic separation techniques.114 Cross-linking of the proteins with a number of different length NHS-homobifunctional cross-linkers, and the following digestion of obtained products with trypsin and analysis by HPLC enabled the possibility of three-dimensional structure modelling of the calmodulin–melittin complex (Fig. 7).
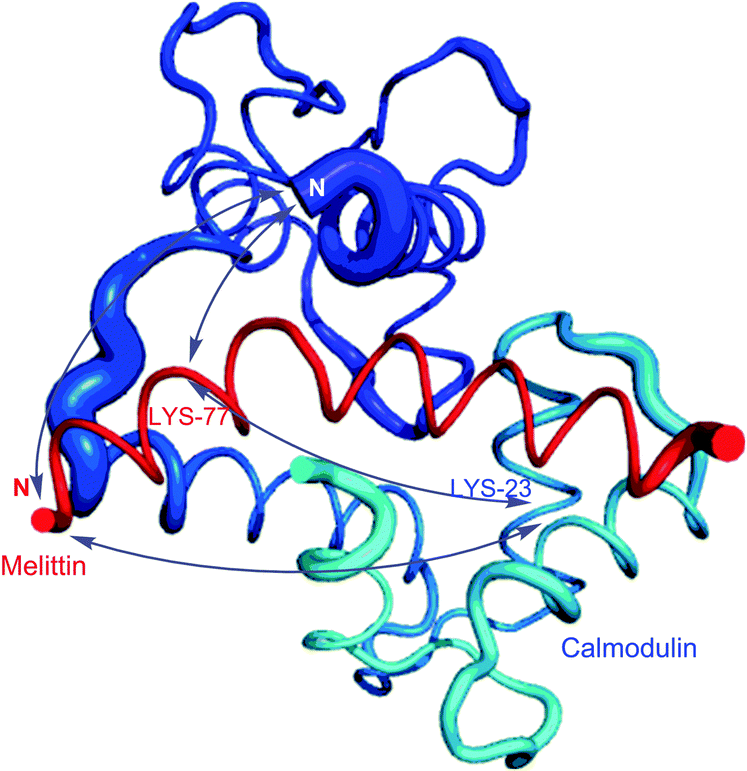 | ||
| Fig. 7 Mode of binding of melittin in the calmodulin–melittin complex (pdb: 2MLT and 1CDL) calculated from ambiguous distance restraints derived from the cross-linking data by Sinz and associates.114 | ||
Several applications however require the precision of crosslinking which cannot be provided by homobifunctional crosslinkers. For example, the preparation of an antibody–drug conjugate (ADC) implies selective linking of a cytotoxic payload to each molecule of the antibody without causing any antibody-to-antibody linkages to form. For such application the combination of different selective approaches in one linker is needed.
Therefore, heterobifunctional crosslinkers are designed to possess different reactive groups at either end. These reagents allow for sequential conjugations that diminish undesirable self-conjugation and polymerisation. Sequential procedures involve two-step processes, where heterobifunctional reagents (often in excess to ensure high conversion levels) are reacted with one protein using the most labile group of the crosslinker first. After eliminating the excess of the nonreacted crosslinker, the second protein is added to a solution containing modified first protein and another reaction occurs with the second reactive group of the crosslinker. According to the Pierce website (Rockford, IL, USA), the most popular heterobifunctional crosslinkers are those having amine-reactive NHS esters at one end and thiol-reactive maleimides (see Section 1.3.2) at the other end. Because of its less stability in aqueous solution compared to maleimide, the NHS-ester group should usually be reacted first. Takeda and co-workers115 used a bifunctional reagent that contained a NHS function and a benzylthioester function to prepare a DNA–protein hybrid. One of the fastest growing fields requiring heterobifunctional crosslinkers today is targeted drug delivery therapies – ADCs.22,116–120 They are constituted of three main components: one monoclonal antibody (mAb), targeting specific signs or markers of cancer cells, one cytotoxic agent, and one linker molecule that allows covalent drug binding to the mAb. The composition of trastuzumab emtansine (Kadcyla®, Genentech), an in clinic ADC for treatment of HER2-positive metastatic breast cancer, is depicted in Fig. 8.
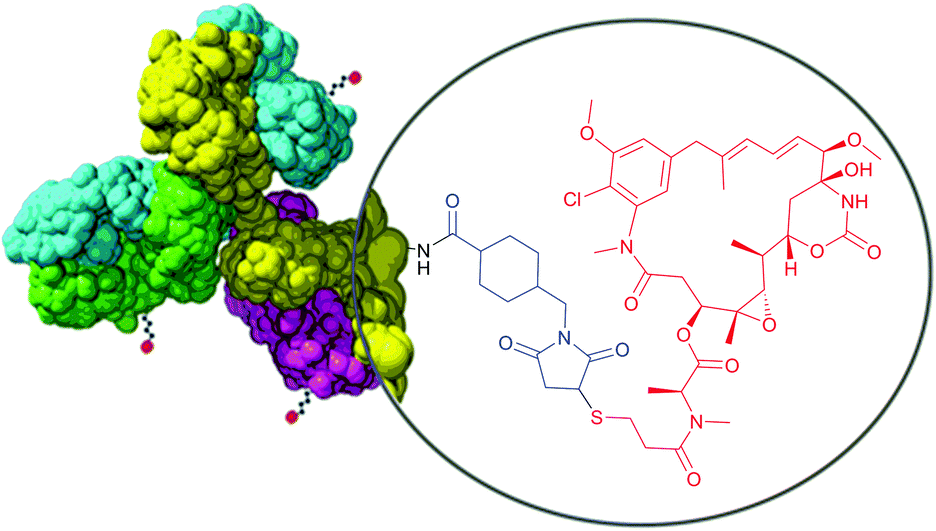 | ||
| Fig. 8 Structure of an antibody drug conjugate (ADC) Kadcyla®. SMCC linker (shown in blue) serves for conjugation of the antibody and cytotoxic payload (Mertansine, shown in red). | ||
The first example of a “cleavable” NHS cross-linking reagent, DSP, was reported by Lomant and Fairbanks93 and allowed effecting the reversal of the previously conjugated fragments under mild conditions of disulfide bond reduction. Further advances in the field have resulted in various types of linkers, cleavable under mild nucleophilic conditions (EGS),121 at basic pH (BSOCOES),122 in the presence of periodate (DST),123 or enzymatically.124 These found their applicability for studies in basic and applied research. The reader is directed to a recent review by Leriche et al.125 that provides an overview of chemical functions that can be used as cleavable agents and to a publication by Jin Lee126 for an overview of commercially available cross-linking reagents.
Other combinations of functionalities have been studied over the last 20 years and resulted in elaboration of heterotrifunctional127 linkers usually combining two bioselective reactive groups and a functionality for anchoring the obtained conjugate (e.g. the biotin moiety).
Many chemical probes widely used in bioconjugation contain the NHS-fragment in their structure and are designed to react with free amino groups of proteins. For example, biotinylation128–130 as well as PEGylation2 of proteins are most commonly achieved using NHS-activated probes today. It was recently reported by Anderson and collaborators that biotinylation of antibodies with NHS-biotin and their following adsorption on the surface of nanocrystal quantum-dots (QD) results in obtaining highly efficient QD-antibody conjugates for the detection of protein toxins.131 Other types of protein immobilisation on matrices have also been reported.84,132,133 The Bolton–Hunter reagent (SHPP),134 allowing the conjugation of tyrosine-like residues for increasing the yield of subsequent (radio)iodination, is also based on N-hydroxysuccinimide chemistry.135–140 Elaborated in 1982 by Ji et al.141 structurally similar SHPP photoactivable heterobifunctional probes for cross-linking experiments have been used in more than 100 studies ever since. The NHS ester-based strategy for isobaric, stable isotope labelling of peptides142–144 has recently found more widespread application in proteomic studies with simultaneous developments in enhancing peptide detection by electrospray ionisation mass spectrometry.78,145 This list can be continued and arguably utilisation of NHS-mediated techniques can be found in all major fields of protein conjugation and represents a gold standard in bioconjugation.
Historically, the conjugation of oligosaccharides to proteins has become the first target for this approach. In 1974, relying on the exceptional ability of the cyanoborohydride anion described three years earlier by Borch146 to reduce selectively Schiff bases generated in situ from an amine and an aldehyde, Gray has illustrated the possibility of mild synthesis of carbohydrate coated bovine serum albumin (BSA) and P150 protein (Fig. 9).147 However, because of low kinetics of conjugation, only 4 out of 59 BSA lysine residues (presumably those possessing the lowest pKa values) were derivatised after 300 hours of reaction.
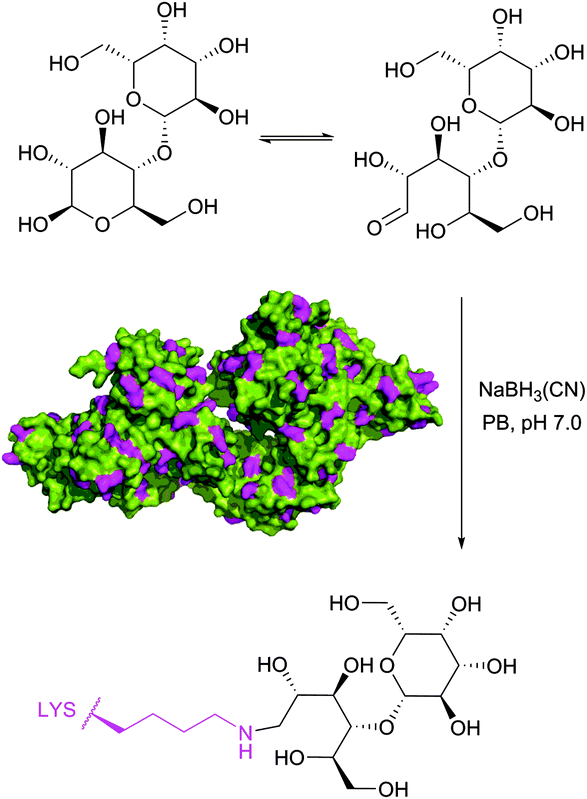 | ||
| Fig. 9 First example of reductive amination of lactose by bovine serum albumin (BSA, pdb: 3V03; lysine residues are shown in magenta) described by Gray.147 | ||
Reductive amination of proteins proceeds most readily at pH 6.5–8.5 where the reduction of aldehydes and ketones is negligible, and, if feasible, in an alcoholic solution under dehydrating conditions where the rate-limiting formation of the imine is favoured. According to Allred and colleagues,148 the addition of sodium sulfate (500 mM) may largely improve the coupling efficiency in aqueous media.
To date, reductive amination has played a central role in the synthesis of carbohydrate–protein conjugates,20,149–151 which have been used for years to study the molecular recognition of carbohydrates.152 Among these conjugates, polysaccharide–protein conjugate vaccines such as Menactra, HIBTiter, and Prevnar are FDA approved and used routinely for the prevention of invasive bacterial infections (Fig. 10),20,153 and potential anti-infective and anti-cancer agents are currently in clinical trials.23,151,154,155 The reader is directed to a recent comprehensive review of Adamo et al.24 covering the current status and future perspectives of carbohydrate–protein conjugates.
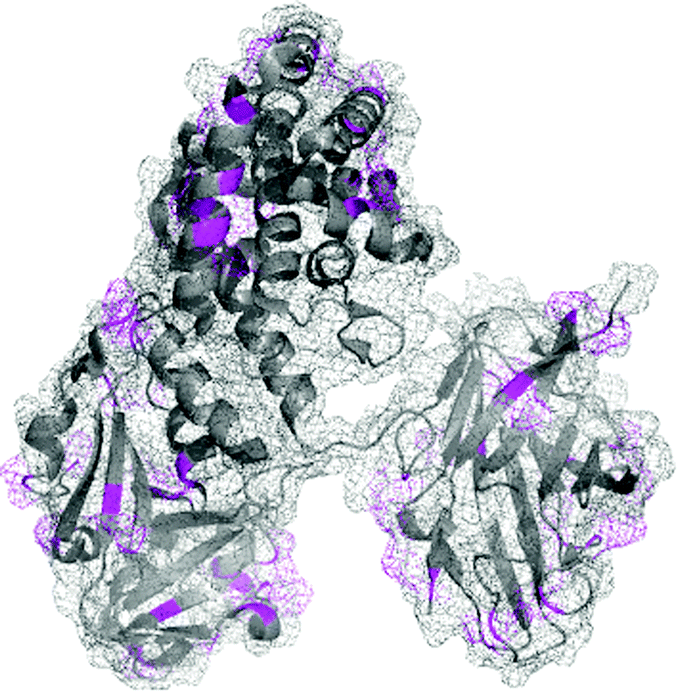 | ||
| Fig. 10 Structure of Prevnar 13 vaccine. The bacterial capsule sugars, a characteristic of the pathogens, are linked to CRM197, a nontoxic recombinant variant of diphtheria toxin (pdb: 4AE0), by reductive amination at lysine residues and N-terminus (shown in magenta). | ||
Another reported application of reductive amination includes the preparation of an organic trialdehyde to be used as a template for the synthesis of three-helix bundle proteins,156 protein PEGylation157,158 and immobilisation.159
Reductive amination however possesses several drawbacks preventing it from being generally applicable to protein conjugation.160 The most important is the necessity to use water-sensitive sodium cyanoborohydride, which has the potential for reducing disulfide bonds within proteins. As an alternative, McFarland and Francis161 have reported a water-stable iridium catalyst (Fig. 11). However, the efficiency of the method is lower than that of the classical reduction with cyanoborohydride.
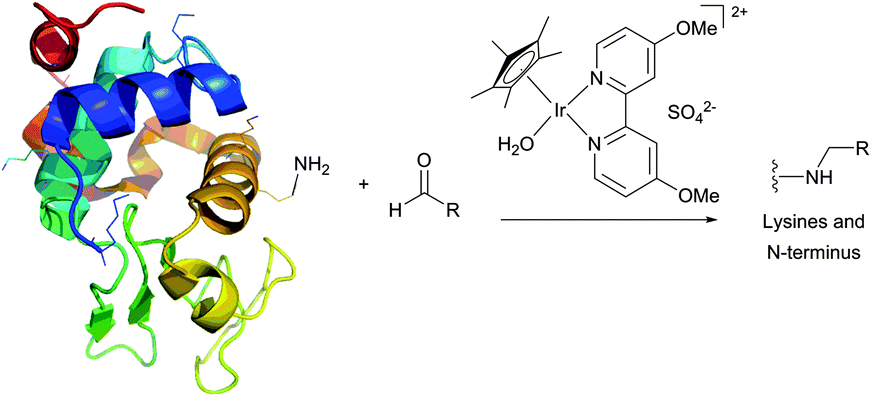
Sulfonyl halides are highly reactive but also very unstable, especially in aqueous media at the pH required for reaction with aliphatic amines. For example, Haugland and collaborators165 have demonstrated that the rate of hydrolysis of Texas Red (one of the most widely used long-wavelength fluorescent probes)166 and Lissamine rhodamine B sulfonyl chloride was much higher (complete hydrolysis within 5 minutes in pH 8.3 aqueous solution) than that of corresponding NHS esters (both retained most of their reactivity for more than an hour under the same conditions). Yet, the formed sulfonamide bonds are extremely stable and even survive amino acid hydrolysis,164,167 which makes sulfonamide conjugates useful for the applications where the stability of the conjugation bond is a crucial feature.
Optimal conditions of protein modification by sulfonyl chlorides are those under which free amino groups most effectively compete with water for a limited amount of the reagent. It is thus best done at low temperature at pH 8.5–9.5.168 At lower pH values, the unreactive protonated form of amines slows the labelling reaction compared to the hydrolysis by water, above this range the reagent is hydrolysed too rapidly.169,170 In practical experiments, a several-fold excess of the reagent is usually added, providing the unused probe is hydrolysed to the corresponding unreactive sulfonic acid after labelling. It must be borne in mind that unlike other amine-selective reagents, sulfonyl chlorides are unstable in dimethylsulfoxide, classically used for the preparation of stock solutions, and should never be used in this solvent (Fig. 12).171
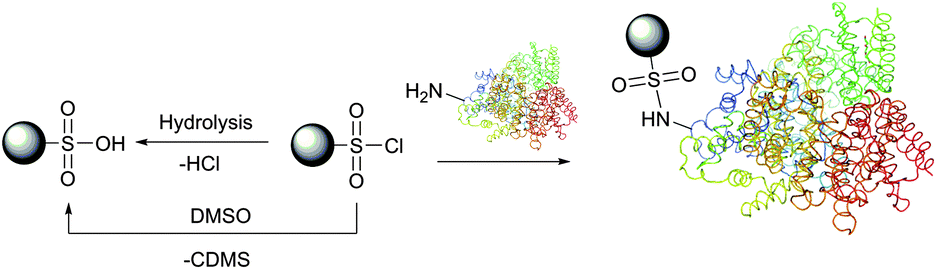 | ||
| Fig. 12 Reaction of sulfonyl chlorides with amino groups present in proteins. Hydrolysis of the starting material by water or dimethylsulfoxide171 (chlorodimethylsulfide, CDMS, is a leaving group) results in obtaining unreactive sulfonic acid. | ||
Apart from being reported for fluorescent labelling of proteins,172 sulfonyl chlorides were used to incorporate a chelate moiety into proteins,173 to study hydrodynamic properties or introduce long-lived fluorescence labels into macromolecules using tagging with pyrene derivatives174,175 or as cross-linking reagents.176
Because of their very high reactivity towards nucleophiles, sulfonyl halides also form conjugates with tyrosine, cysteine, serine, threonine, and imidazole residues of proteins;177 therefore, they are less selective than either NHS esters or isothiocyanates. These conjugates are however unstable and can be completely hydrolysed under basic conditions.
Covalent immobilisation of proteins on hydroxyl group containing carrying supports (such as agarose, cellulose, diol-silica, or polylactic acid films) is often accomplished by transforming the latter into corresponding sulfonates: tosylate, mesylate, or tresylate,178,179 serving as good leaving groups (Fig. 13).180–182 Albumin, cytokines and other therapeutic proteins and peptides were reported to undergo mild PEGylation by means of PEG tresylates.183–185 Although rather specific to amino groups, the chemistry of tresylate-mediated conjugation is not unique and well defined. For instance, Gais et al. have shown that PEG–tresylate conjugation can produce a product that contains a degradable sulfamate linkage resulting in heterodispersity of the reaction.186
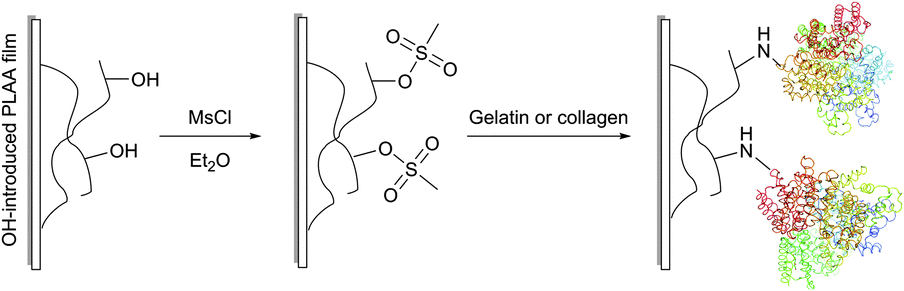 | ||
| Fig. 13 Schematic representation of the reaction protocol for immobilisation of protein on PLLA film surfaces described by Ma et al.181 | ||
Compared to other aryl halides, fluoro-substituted nitrobenzenes were found to be the most reactive in bimolecular nucleophilic substitution reactions.188 They are usually regarded as amino-selective reagents, despite their known reactivity towards thiolates, phenolates and imidazoles, as the products obtained in these reactions are either unstable at alkaline pH required for the reaction (Tyr and His) or can be thiolysed by excess β-mercaptoethanol (Cys).189
4-Fluoro-7-nitro-2,1,3-benzoxadiazole (NBD-F), which has been introduced as a fluorogenic reagent for more than 30 years ago by Imai and Watanabe,190 still remains important for several applications, mainly pre-column derivatisation and enrichment of peptides. The reader is referred to a recent review by Elbashir et al.191 providing an excellent overview of the NBD-F applicability to the analysis of peptides and to a complete overview of NBD-mediated methodologies for the fluorescent labelling of amino acid residues by Imai and associates.192,193
An elegant approach for improving protein crystallizability, still remaining a major challenge in protein structure research,194 was elaborated by Sutton and collaborators195 and consists in the introduction of a charged ammonium residue. It exploits the amine-selective derivatisation of protein by 1-fluoro-2-nitro-4-trimethylammoniobenzene iodide (Fig. 14) and results in increasing the hydrophilicity thereof. Using their approach, the authors were able to study the binding site196 and to obtain crystalline derivatives of modified bovine insulin,197 which is especially hard to crystallize without inducing structural changes.198 A similar protocol was used by Ladd et al.199 for chromophorical PEGylation of proteins with polyethylene glycol fluoronitrobenzene derivatives.
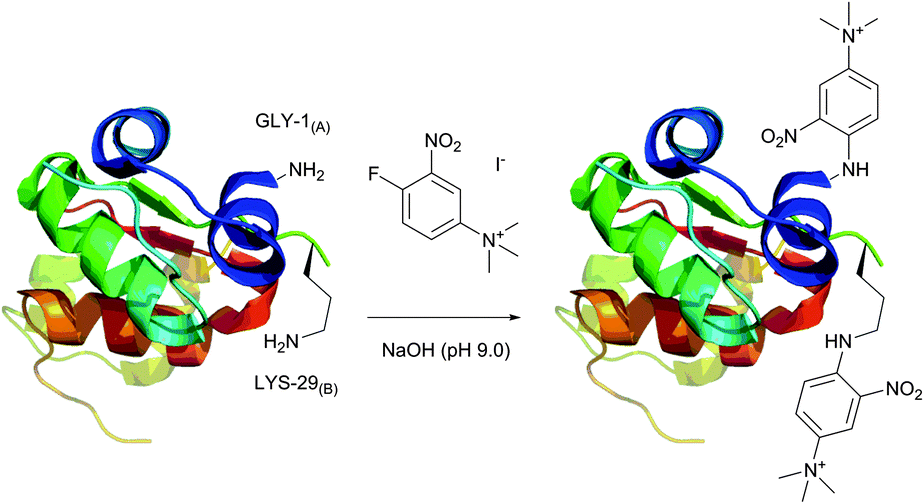 | ||
| Fig. 14 Derivatisation of bovine insulin with 1-fluoro-2-nitro-4-trimethylammoniobenzene iodide described by Sutton et al.195 Only amine-containing residues of A1 and B1 chains are shown. Two of the four tyrosine residues present in chains A1 and B1 (not shown) also react with the probe under described conditions. | ||
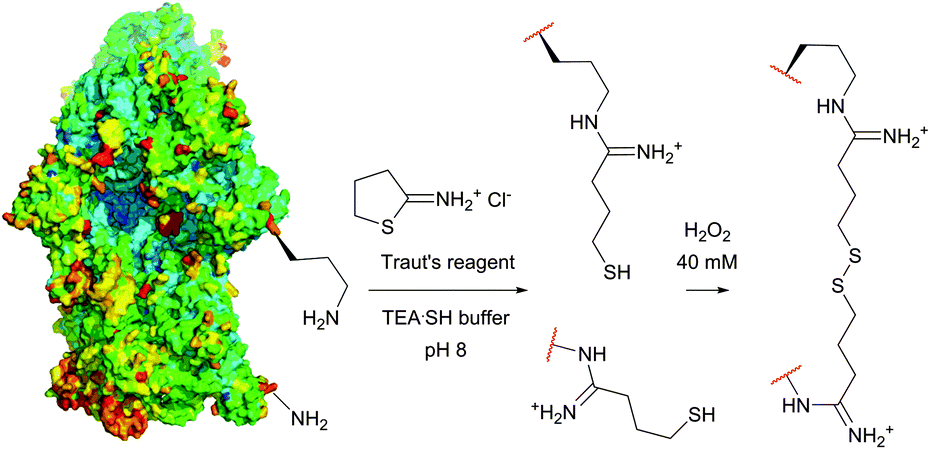 | ||
| Fig. 15 Procedure of the cleavable crosslinking of the intact 30s ribosomes (pdb: 1J5E) described by Traut et al.201 Lysine residues (Lys-72 and Lys-156) were chosen randomly for simplicity purposes (TEASH stands for triethanolamine buffer adjusted to 3% 3-mercaptoethanol). | ||
Imidoesters react with primary amines to form amidine bonds. A high specificity towards amines can be achieved when alkaline conditions (pH 10) and amine-free media, such as borate buffer, are used.202 This places imidoesters among the most specific agents for amine labeling. Because the resulting amidine bonds are protonated at physiological pH, positive charges near modified sites are preserved during the conjugation with lysines and N-termini. Consequently, as was first demonstrated by Wofsy et al.203 such modifications produce little or no significant changes in the conformational properties and biological activities of proteins.
Thiolates obtained after the ring opening of Traut's reagent by free amines enable a plentiful thiol-selective chemistry on modified α- or ε-amino groups of proteins (see Section 1.3). Although, many imidoesters other than Traut's reagent are today commercially available (for example, see DMA, DMP, or DTBP), the amount of described labelling imidoester probes is rather scarce.
Schramm et al.204 have described the synthesis of fluorescent imidoester dyes from corresponding nitriles; the approach was later used by Bozler et al.205 for the preparation of dansyl containing imidoester and selective modification of lysine residues in the active site of glucose dehydrogenase. New readily available reagents for the attachment of sugars to proteins via imidoester linkage,206 hydrophilic spin probes for determining membrane protein interaction using EPR,207 immunoreactive probes,208 tyrosine-like probes for radioactive labelling with 125I,209 protein PEGylation reagents,210 and the immobilisation of trypsin, yeast alcohol dehydrogenase, and E. coli asparaginase onto several types of organic polymer beads211 were achieved via imidoester conjugation and proven to have several advantages compared to other existing methodologies, namely, deprivation of solubility issues and retention of positive charge at the reaction site.
Azetidinone chemistry has recently been demonstrated by Barbas and collaborators212,213 to have potential for selective lysine labelling of a particular IgG framework, containing a very reactive lysine residue with an unusually low pKa of about 6. Some detailed procedures are described for a smooth opening of a β-lactam moiety resulting in a β-alanine peptide bond.213
Discovered by Tietze et al. as two-step sequential procedures for coupling of amines,214,215 squaric acid diester amine–amine conjugation is now actively developed by Wurm et al., who have recently reported their successful use for the one-pot preparation of poly(glycerol)–protein216 and glycol–protein conjugates217 in aqueous media (Fig. 16).
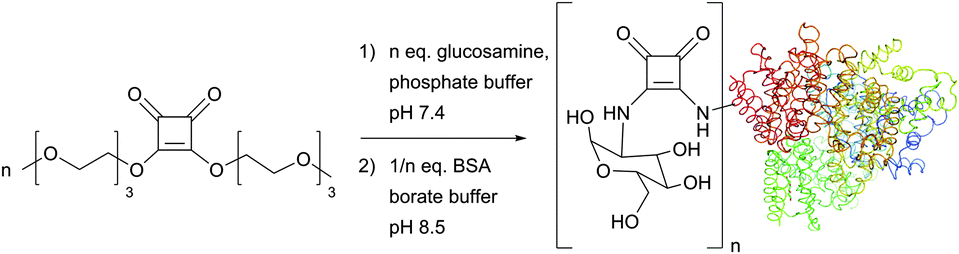 | ||
| Fig. 16 One-pot, two-step squaric acid diester mediated glycosylation of BSA (pdb: 3V03) described by Wurm et al.217 Up to 22 lysine residues of the 59 present in BSA (30–35 are available for post-modification) could be glycosylated with 25-fold excess of squaric diester and glucosamine in 12 hours. | ||
Dichlorotriazine derivatives were described for amine-selective conjugation mainly as fluorescent dyes218,219 and PEGylation probes.220–222 They were shown to possess high reactivity towards protein amines. However, as was demonstrated by Abuchowski et al.,220 because the hydrolysis of dichlorotriazine occurs readily under slightly basic conditions (pH 9.2) needed for reaction to take place with sufficient selectivity towards amines, a considerable excess of the probe must be used in the coupling reaction. Banks and Paquette70 have conducted a comparative study of three fluorescent probes, differing only in the moiety responsible for the reactivity with amines: CFSE (NHS ester), DTAF (dichlorotriazine) and FITC (isothiocyanate). It was found that the rate of conjugation is significantly faster for the NHS ester compared to the diclorotriazine probe, which, in turn, reacts faster than the isothiocyanate derivative. Each conjugate provided a satisfactory level of stability in solution over a period of 1 week at room temperature, although the hydrolysis of the remaining, relatively inert, chloro group of DTAF was observed (Fig. 17).
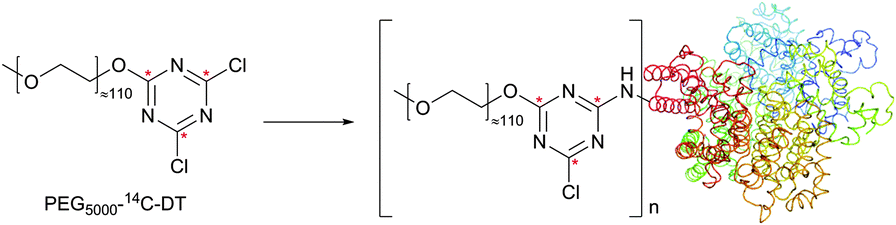 | ||
| Fig. 17 Modification of BSA (pdb: 3V03) by the PEG-[14C]dichlorotriazine probe reported by Abuchowski and colleagues (14C atoms are marked with red stars).220 | ||
Arpicco et al.223 have prepared thioimidoester activated PEG-containing derivatives and shown their superiority over the NHS-activated analogue for gelonin modification (the reaction was conducted in PBS at pH 7.4). In this particular case, PEGylation with a less active, compared to NHS ester, thioimidoester derivative resulted in the gelonin conjugate with higher inhibiting activity. Ikeda and associates224 have recently described a protocol for the preparation of the glutalaldehyde-functionalised PEG reagent, allowing for protein PEGylation under mild reaction conditions. Similarly, the modified protein exhibited higher biological activity than when reacted with a corresponding NHS-activated PEGylation reagent.
α-Halocarbonyls, such as iodoacetamides, can modify lysine residues at pH > 7.0,225 but the reaction rate is much slower than the reaction with cysteine residues. Another class of reagents usually used in cysteine-selective conjugation – vinyl-sulfones226 (see Section 1.3.3) – was recently reported to be applicable for lysine labelling at slightly basic pH.227,228
Modification of Lys residues with acid anhydrides, including succinic, citraconic, maleic, trimellitic, cis-aconitic, and various phthalic anhydride derivatives belongs to a pool of classically used protein modification methodologies229 and allows for transforming nucleophilic amines into acids and, as a result, enables carboxylate-selective chemistry thereof.
For more details of the practical aspects of using the above-described methodologies, the reader is referred to a recent review by Brun and Gauzy-Lazo230 on the preparation of antibody–drug conjugates by lysine conjugation.
1.2 Serine and threonine
With pKa values > 13, the hydroxyl groups of serine (Ser, S) and threonine (Thr, T) are rather poor nucleophiles close to physiological pH. No examples of direct conjugation of in-chain serine and threonine have been therefore reported to date.However, highly amine selective N-hydroxysuccinimide (NHS) esters have been documented to give occasional side reactions with hydroxyl side chains.83,90,92,231 In a series of experiments, Miller et al. have demonstrated that the presence of histidine in sequences of the type His-AA-Ser/Thr or His-AA-AA′-Ser/Thr (where AA and AA′ stand for any amino acid) can significantly increase the reactivity of hydroxyl groups toward classical amine labelling agents (Fig. 18).85,86,88,232
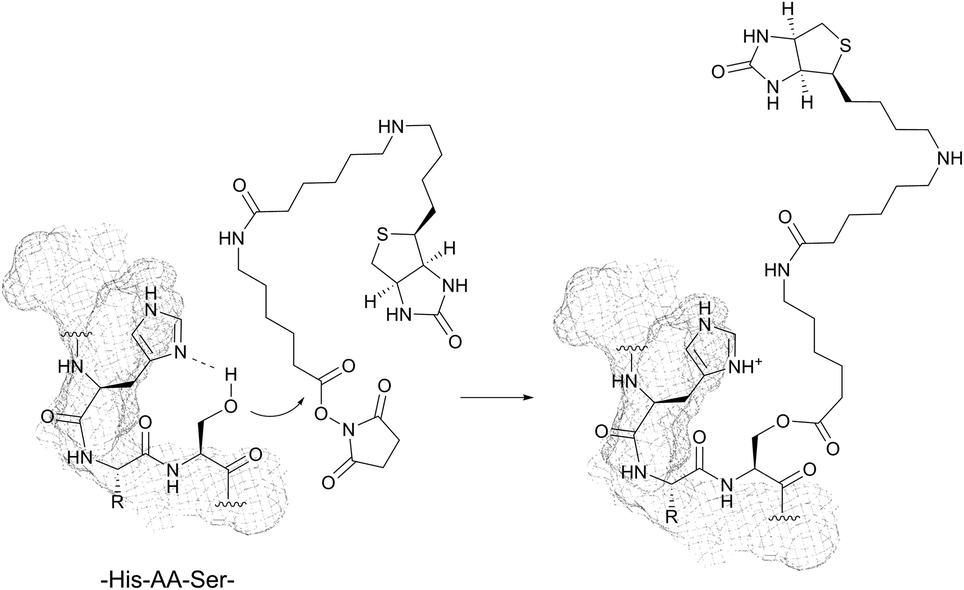 | ||
| Fig. 18 Nucleophilic attack of serine on the NHS-activated ester gives the stable O-acylated derivative. In addition to His-AA-Ser, this stable intermediate can be formed in the presence of linear sequences of His-AA-Thr and His-AA-Tyr, where AA refers to any amino acid. | ||
Similarly, Mädler and Zenobi have reported that the guanidinium group of arginine can contribute to the reactivity of hydroxyl groups toward NHS esters and catalyse the nucleophilic substitution.233 In both cases, it is hypothesised that the imidazolyl and guanidine moieties of histidine and arginine, respectively, catalyse the reaction by stabilizing the transition state by means of hydrogen bonds and electrostatic interactions. This promoting effect is thought to be responsible for side reactions on several substrates while using cross-linking reagents.92,233
Despite the fact that methodologies of selective in-chain serine and threonine labelling are rather scarce, these residues are of special interest for bioconjugation when located on the N-terminus (see Section 2.2).
1.3 Cysteine
Cysteine (Cys, C) is perhaps the most convenient target for bioconjugation owing to the exceptionally high nucleophilicity of its sulfhydryl (–SH) side chain which, and particularly in its deprotonated thiolate form (–S−), largely exceeds the reactivity of any other nucleophilic function in proteins.234 Furthermore, its relative rarity in proteins present in living organisms (1–2%)235,236 and the ease of its introduction into a specific site by site-directed mutagenesis allows access to protein assemblies with a single cysteine at a predetermined position.237 Even in proteins with multiple cysteines, the multiplicity is usually much smaller compared to lysines, which makes thiol-reactive labelling the preferred approach over amine-reactive methodologies.In proteins, thiols can also be generated by selectively reducing cystine disulfides with reagents such as dithiothreitol (DTT, D1532),238 2-mercaptoethanol (β-mercaptoethanol), or tris[2-carboxyethyl]phosphine (TCEP).239,240 Generally, all these reagents must be removed before conducting thiol-selective conjugation, as they will compete with target thiols in proteins otherwise.241 Unfortunately, removal of reducing agents is sometimes accompanied by air oxidation of thiols back to disulfides. Although, in contrast to the majority of thiol-reducing agents, TCEP does not contain the thiolate group, there have been several reports that it can react with α-halocarbonyls or maleimides and that labelling is inhibited when TCEP is present in the reaction medium.242,243
Direct labelling of the thiolate group is usually achieved by either a nucleophilic addition or displacement reaction with the thiolate anion as the nucleophile. The substantially less dissociation energy of sulfhydryl groups compared to the corresponding alcohols provides much higher acidity of the former and, as a consequence, a wider availability of its slightly nucleophilic anionic form at physiological pH.
Typically, the reaction of sulfhydryl groups with haloacetamides is conducted under physiological and alkaline conditions (pH 7.2–9.0). When iodoacetamides are used, the reaction is preferably carried out under subdued light in order to limit free iodine generation, which has the potential to react with Tyr, His and Trp residues. The reaction is most specific for sulfhydryl groups at pH 8.3. The iodoacetyl group is known to react with other amino acid side chains, especially when there is no cysteine present or if a gross excess of iodoacetyl is used. For instance, free amino groups, the thioester of methionine, and both imidazolyl side chain nitrogens will react with iodoacetyl groups above pH 7 and 5, although with much slower kinetics.246 This, however, can be resolved by the use of less reactive chloroacetamides247 or cautious control of pH and incubation time.
It is to be noted that the local environment has a profound effect on the reactivity of cysteine residues in proteins. If moderately reactive reagents such as iodoacetamide are used for bioconjugation, this difference in reactivities makes it possible to discern different types of Cys moieties present in the protein. Almost half a century ago, Gerwin248 reported dramatic differences in the reactivities of chloroacetic acid and chloroacetamide in the modification of the active-site cysteine of streptococcal proteinase, which was found to be due to the influence of the neighbouring histidine residue. As a general trend, cysteine residues possessing lower pKa values are more reactive when reaction is conducted under neutral or slightly acidic conditions, owing to their greater degree of dissociation and, as a consequence, higher concentration of the corresponding thiolate anions in the medium. For instance, Kim et al.249 have described a method for selective biotinylation of low-pKa cysteine residues in proteins simply by conducting the reaction at slightly acidic pH (Fig. 19).
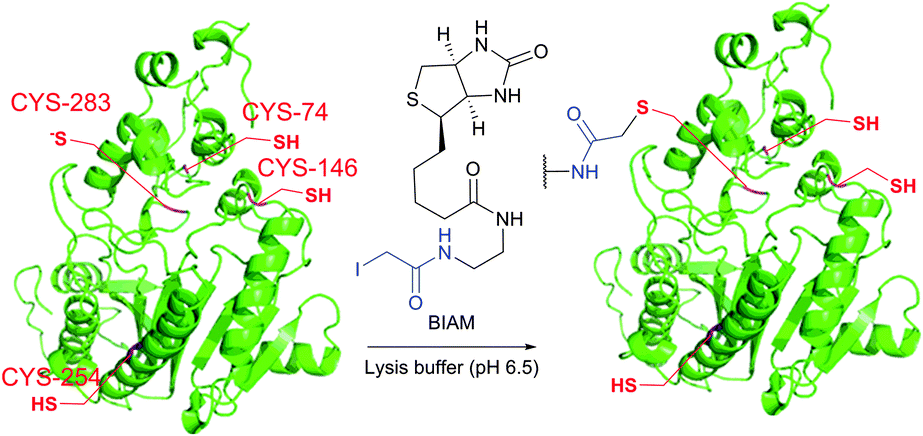 | ||
| Fig. 19 Biotinylation of the low-pKa cysteine residue of rabbit muscle creatine kinase (CK, pdb: 2CRK) by BIAM.249 The charge interaction between the negatively charged thiolate and the positively charged amino acid residues nearby results in a significantly lower pKa value of the CYS-283 residue (6.5). Consequently, selective alkylation thereof becomes possible in the presence of three other cysteine residues with higher pKa values (8.0–9.0). | ||
Davis and Flitsch250 described a procedure for the selective glycosylation of proteins at one or several sites by reacting the carbohydrate-tethered iodoacetamides with cysteine side chains, which allowed for preparing homogeneously glycosylated human erythropoietin251 and dihydrofolate reductase.252
In 1948, Mackworth253 published his study on the reactivity of the biochemical mechanism of the lachrymatory effect of certain war gases and first reported the reactivity of structurally relevant α-bromoacetophenones for the inhibition of several classes of thiol enzymes.
Despite advances made in the investigation of α-haloacetophenones and related ketoximes for the modification of the active sites of enzymes,256–259 their utility for the conjugation is very limited because of various side reactions.
An interesting approach that allows photochemical conversion of cysteine into corresponding thioaldehyde and then to aldehyde thought to be formed by Norrish type II cleavage was reported by Clark and Lowe.254,255 Photolysis of the enzyme, alkylated by a bromoacetophenone derivative, results in spontaneous loss of hydrogen sulfide from the generated thioaldehyde to give the corresponding aldehyde (Fig. 20), which can either be utilised as a locus for aldehyde-selective conjugation or be transformed into the corresponding serine or glycine residue by reduction or transamination respectively.
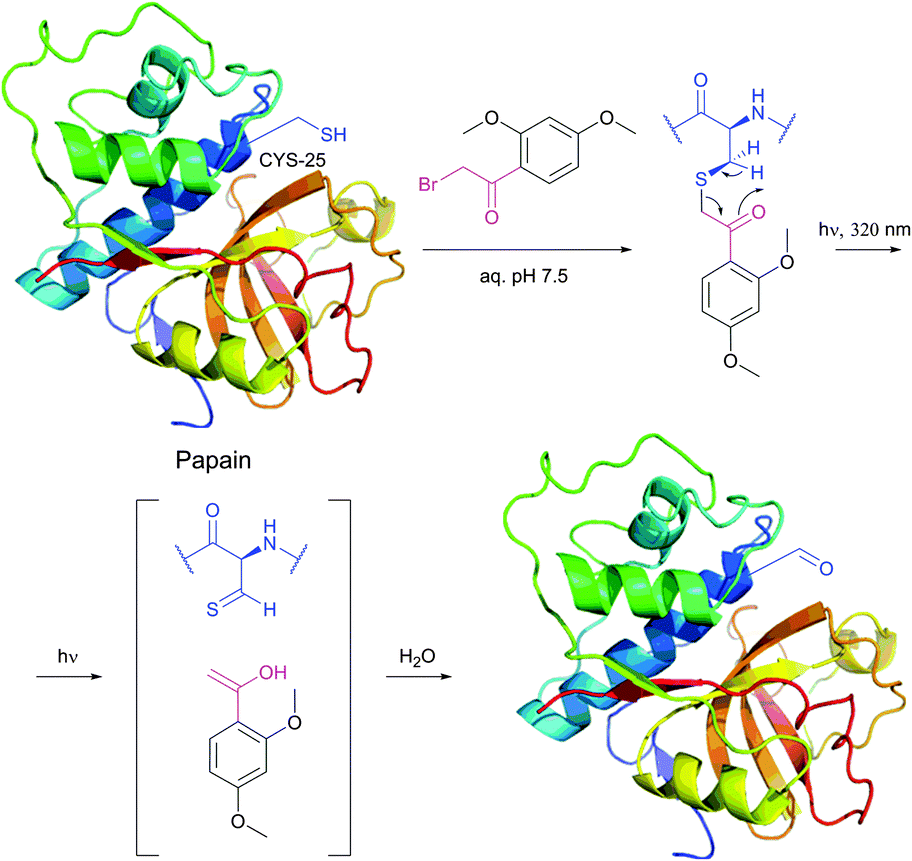
The reason for such remarkable reactivity of maleimide towards thiolates is worth being discussed. In general, the electrophilicity of alkenes is defined by their ability to serve as acceptors of nucleophile's electron density, and thus interrelated to the energy of electrophile's π* orbital (its lowest unoccupied molecular orbital, LUMO). Generally speaking, the rule is simple: the lower the energy of the alkene's π* orbital – the faster its reaction with nucleophiles. There exist two main approaches for decreasing alkene's LUMO energy: the direct attachment of an electron-withdrawing group (EWG) and the straining of the double bond. Although proceeding via two different mechanisms: by decreasing the energy of both orbitals or by diminishing the energetic gap between them, either approach results in lowering the LUMO energy of the alkene and, as a result, in the increase of its reactivity (Fig. 21). The unique reactivity of the maleimide moiety owes to the fact that it exploits these two mechanisms together.262,263
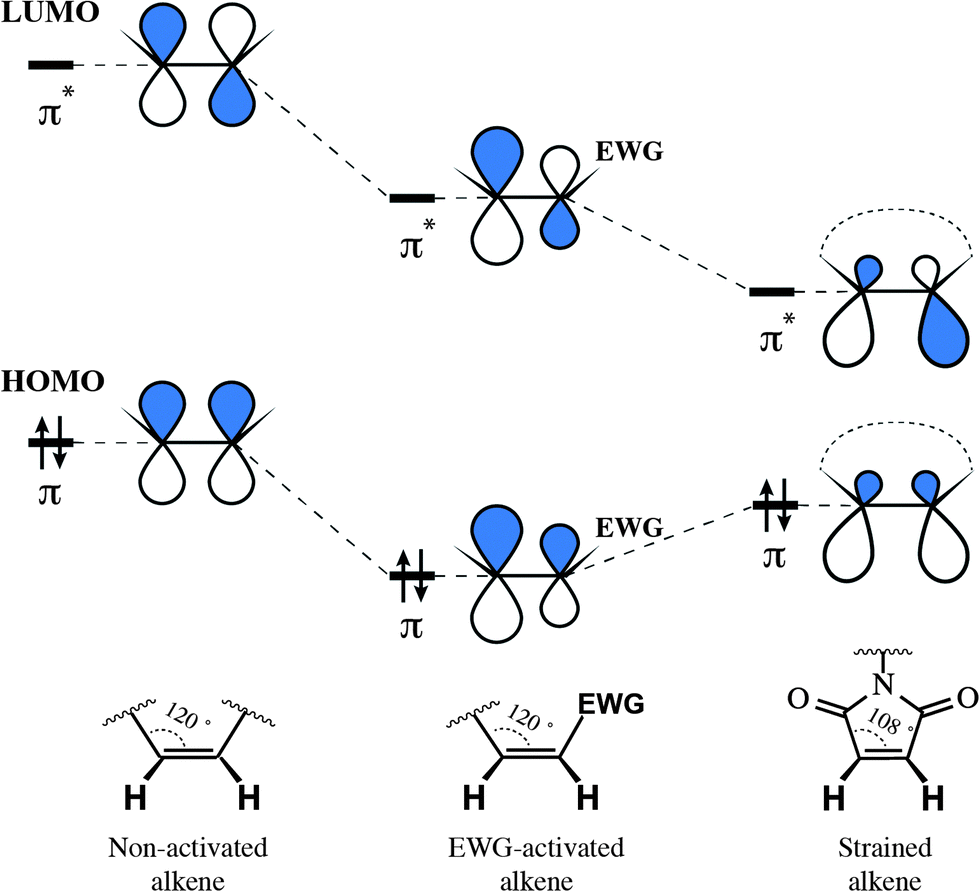 | ||
| Fig. 21 Influence of an electron-withdrawing group (EWG) and cycle strain on the frontier orbitals of alkenes, σ-orbitals omitted (the form of the orbital is presented approximately, based on the publication of Merchán et al.268). Decreasing the energy of the LUMO (lowest unoccupied molecular orbital) results in higher reactivity of the electrophile towards nucleophiles. Although via different mechanisms, both the EWG and strain of the cycle activate alkenes for the attack by nucleophiles. | ||
To date, a large variety of maleimide-based modifying reagents are available from a number of leading biochemical companies with even more being synthesised in laboratories around the world for specific applications. The applications of these reagents strongly overlap those of iodoacetamides, although maleimides apparently do not react with methionine, histidine or tyrosine.264,265
The optimum reaction conditions for maleimide-mediated conjugation, namely conducting the reaction under near neutral conditions (pH 6.5–7.5, Fig. 22), prevent the reaction of maleimide with amines, because the latter requires a higher pH to occur. At pH above 8 the hydrolysis of maleimide itself results in obtaining a mixture of isomeric non-strained maleamic acids unreactive toward sulfhydryls and can thus compete with thiol modification.28,266 Similarly, maleimide–thiol adducts hydrolyse, which either results in complete deconjugation or causes a significant change in the properties of the conjugate.266 Furthermore, especially at pH above 9, ring-opening by nucleophilic reaction with an adjacent amine may yield crosslinked products.267
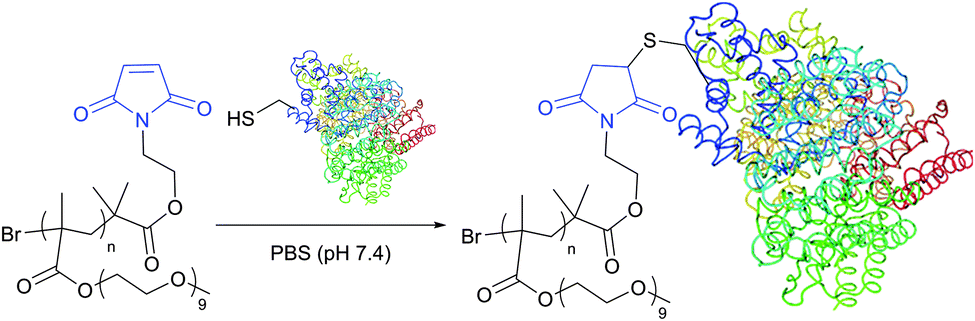 | ||
| Fig. 22 Conjugation of maleimide-functional poly(PEGMA) to the only free CYS-34 residue of BSA (pdb: 3V03).311 | ||
Schuber and co-workers269 have found that important kinetic discrimination can be achieved between the maleimide and bromoacetyl functions when the reaction with thiols is conducted at pH 6.5 and 9.0, respectively.
Maleimide–NHS heterobifunctional reagents are especially important for the formation of conjugates. Hydrolysis of both the maleimide moiety and the generated thioester linkage is considerably dependent on the type of chemical group adjacent to the maleimide. Interestingly, the cyclohexane ring was found to provide increased maleimide stability to hydrolysis due to its steric effects and its lack of aromatic character. For this reason, succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) and its water-soluble analogue (sulfo-SMCC) are today among the most popular crosslinkers in bioconjugation. They are often used in the synthesis of the protein–protein or protein–probe assemblies such as antibody–enzyme or antibody–drug conjugates respectively. These include enzyme immunoassays,270–274 carrier-protein conjugates,275–277 albumin-binding prodrugs,278,279 and even approved therapies.117,119,280,281
Short homobifunctional maleimides are commonly used to explore and characterize protein structure (i.e., oligomerisation) or protein interactions.282–288 Maleimide-mediated immobilisation of biomolecules is often achieved either by direct conjugation13,289,290 or by prior biotinylation of the molecule of interest.291 The latter approach has been used for protein enrichment,292 capture,293,294 and immobilisation on modified supports.295–298
Most of the optical thiol-selective fluorescent probes often used as sensors for monitoring biological processes are represented by maleimide-containing reagents.299–301 Another testament to maleimide utility is its use in glycosylation,302 radiolabelling,303,304 studying protein interactions,305–308 and quantitation of cysteine residues.309,310
Despite its successful application as a reagent for the chemical modification of proteins, the irreversibility of maleimide's addition makes it impossible to regenerate the unmodified protein by controlled disassembly of the conjugate. Such necessity is however often desirable for in vitro or in vivo applications. Several studies were devoted to a mild and specific hydrolysis of the imido group in maleimide conjugates.312,313 These approaches turned the originally irreversible maleimide-mediated thiol conjugation into the cleavable methodology. However, the harsh reaction conditions of this cleavage (strong basic conditions or the presence of a high amount of imidazole) make them incompatible with many fragile protein substrates.
Monobromomaleimide derivatives, introduced by Baker et al.314 in 2009, have expanded the class of reagents for the selective and reversible modification of cysteine (Fig. 23). In contrast to methanethiosulfonates (see Section 1.3.6),315 monobromomaleimides allow much more stable conjugation of thiolates, which are easily cleavable upon reaction with TCEP by the addition–elimination sequence (Fig. 23).
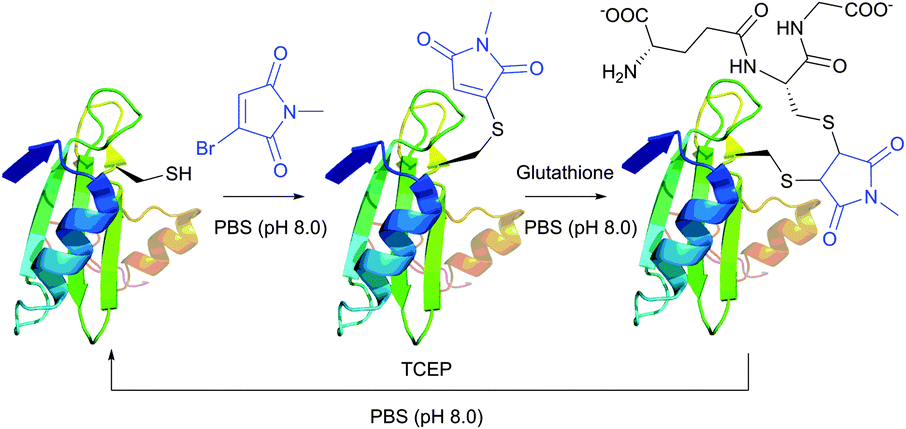 | ||
| Fig. 23 Conjugate addition of the L111C mutant of the SH2 domain of Grb2 (wild-type pdb: 1JYU) to bromomaleimide followed by second addition of glutathione resulting in the generation of the vicinal bis-cysteine adduct.316 | ||
Moreover, the initial modification of a protein resulted in obtaining a thiol–maleimide moiety, which was shown to be prone to a second thiol addition and resolved another recognised drawback of maleimide-based methodologies, namely the presence of only two points of attachment.316 Similar to a non-substituted maleimide, the hydrolysis of thiol–maleimide linkage results in a dramatic decrease in its reactivity towards thiolates, which can be used for “switching off” the linker after the first thiol addition (Fig. 24).317,318
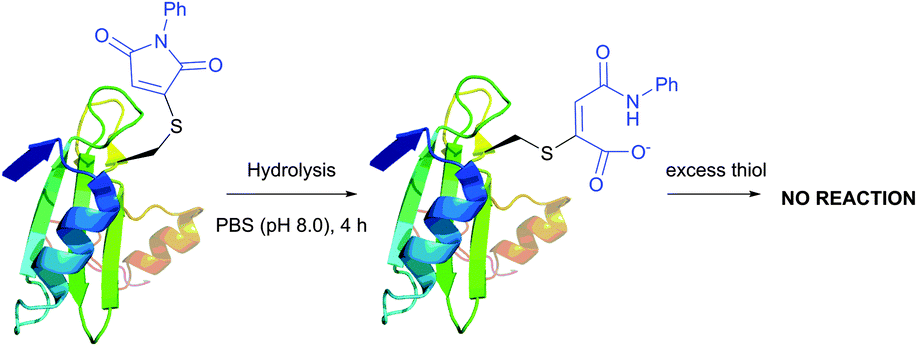
Initially, VS-mediated approaches have been used almost exclusively for PEGylation of proteins with end-functionalised PEG derivatives.322,324 Several studies on the immobilisation of macromolecules on solid supports using vinyl sulfones were reported, owing to the elaboration of new methods for the preparation of VS-modified surfaces.319,320,325–327 Versatile VS-containing probes, namely carbohydrates,227,328 chelating agents,329 fluorescent tags,330,331 and biotinylation reagents331 were recently developed and applied in the bioconjugation of proteins (Fig. 25). Ovaa and co-workers332 used vinyl sulfone handle to conjugate enzymes to a ubiquitin-like protein. The applications of VS-tags in proteomics have recently gained popularity and have been reviewed by Lopez-Jaramillo et al.331
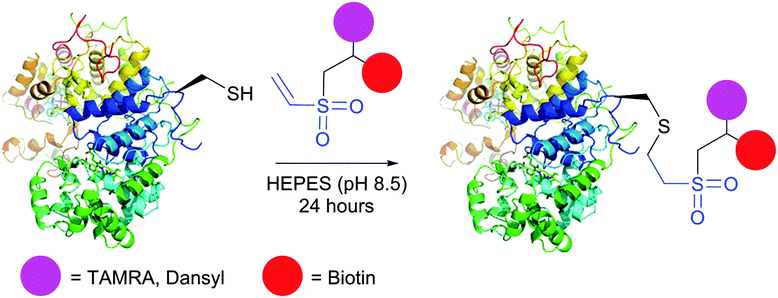 | ||
| Fig. 25 Bifunctional labelling of horseradish peroxidase (HRP, pdb: 2ATJ) with vinyl sulfone-functionalised tags described by Morales-Sanfrutos et al.330 | ||
Vinyl sulfones react with thiols to form a stable thioether linkage to the protein under slightly basic conditions (pH 7–8).228 The reaction may proceed faster if the pH is increased, but this usually also increases the amount of side-products (namely the modification of the Lys ε-amino groups and the His imidazole rings).320 The main advantage of VS-tags is their elevated stability in aqueous solutions, compared to more reactive thiols and maleimides, which can be subjected to ring opening or addition of water across the double bond.185
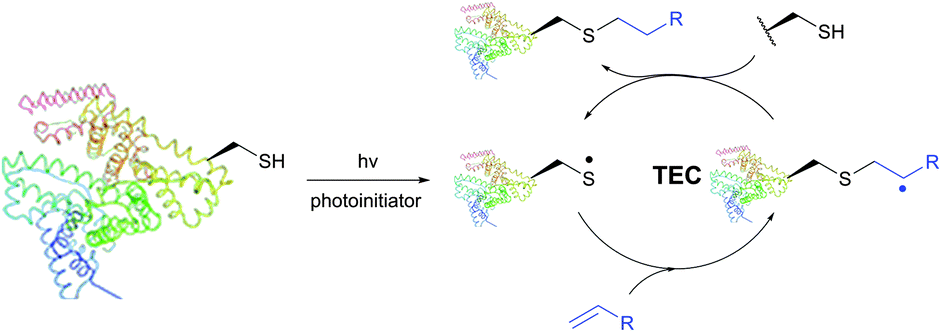 | ||
| Fig. 26 Mechanism of photoinduced free-radical thiol–ene coupling (TEC). | ||
The TEC conjugations (usually conducted in PBS-DMSO buffer at pH 7.0–7.5) are compatible with oxygen and aqueous media and are usually carried out upon irradiation at λmax 365 nm in the presence of Vazo44 (2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride) as an initiator. The resulting thioether linkage is biologically stable and robust.
The first approach to protein conjugation namely glycosylation via TEC was reported by Davis et al. in 2009.336 However, it consisted of photoinduced coupling of various glycosyl thiols with site-specifically introduced unnatural L-homoallylglycine. A complementary approach to peptide and protein glycoconjugation by photoinduced coupling on cysteines was first introduced almost at the same time by Dondoni and co-workers.337 The 66 kDa globular bovine serum albumin (BSA) possessing one free CYS-34 residue was selected as a model protein. Surprisingly, it revealed that not only the one CYS-34 SH group, as expected, but also two more SH groups arising from the 75 ↔ 91 disulfide bond were modified. It was suggested that such hyperglycosylation was due to well-documented disulfide bond degradation by UV-irradiation,338 namely to an electron transfer process from photoexcited tryptophan residues. Furthermore, prolonged irradiation of the reaction mixture up to 2 hours induced the introduction of seven glycoside residues into BSA. Despite the necessity for UV-irradiation, ensuing side-reactions, and often moderate yields,335 the fact that, in contrast to the majority of thiol-selective methodologies, TEC does not exploit elevated nucleophilicity of the thiolate but its readiness for the generation of radicals makes it especially tolerant to a wide range of functional groups. For instance, Garber and Carlson339 have used this feature of TEC for selective capping of thiols in the presence of thiophosphorylated groups, free alcohols and amines.
Several approaches involving the combination of cysteine-selective methodologies have been recently reported. Stolz and Northrop340 studied the reactivity of N-allyl maleimides and found this scaffold to be appropriate for consecutive two-step conjugation of thiols: via (1) base-initiated Michael-addition to maleimide moiety and (2) radical-mediated TEC of allyl-fragment. Scanlan and associates341 developed the sequential NCL-TEC approach (for more details on NCL, see Section 2.3.1) for the functionalisation of the cysteine thiolate generated at the ligation site during native chemical ligation.
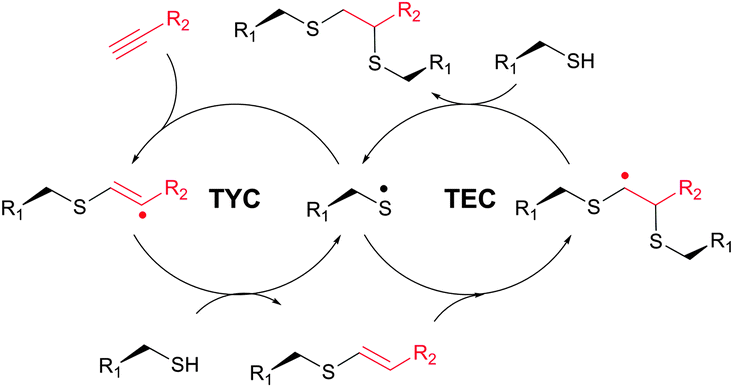 | ||
| Fig. 27 Mechanism of photoinduced free-radical thiol–yne coupling (TYC). | ||
TYC occurs under the same reaction conditions as TEC and smoothly proceeds at room temperature in aqueous solutions. First trials on the applicability of TYC on peptides were reported by Dondoni and collaborators in 2010.344 The authors have demonstrated the possibility of dual glycosylation of a series of peptides (up to 8 residues). Later on, Davis and Dondoni have expanded the dual conjugation strategy for achieving sequential glycosylation and fluorescent labelling of BSA (Fig. 28).345 Just as with TEC, the reaction also occurs at cysteine residues of the 75 ↔ 91 disulfide bridge.
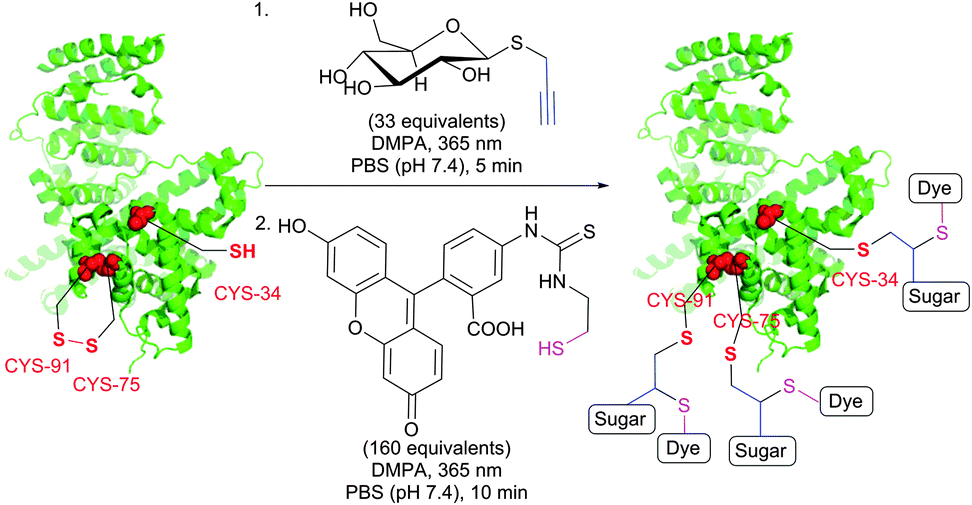 | ||
| Fig. 28 Glycosylation and fluorescent labelling of bovine serum albumin (BSA, pdb: 3V03) by TYC (first step) followed by TEC (second step) with 2,2-dimethoxy-2-phenylacetophenone (DMPA) as a photoinitiator.345 | ||
The necessity of using a photo- or a chemical initiator in both TEC and TYC conjugations represents the main drawback of these methodologies, as the presence of free radicals results in a series of side reactions, namely oxidation and crosslinking of proteins.
Diverse disulfides have been extensively used in the past decade for the modification of cysteine by disulfide exchange. This reversible reaction involves attack of cysteine thiolate at the disulfide, breaking the S–S bond, and subsequent formation of a new mixed disulfide. A well-known example of such reaction is colorimetric quantitation of free sulfhydryls with Ellman's Reagent.348 Several symmetric disulfide-containing fluorescent probes such as BODIPY L-cystine and fluorescein L-cystine are commercially available. However, because there is no thermodynamic preference for this disulfide exchange to pass one way or another, labelling with non-activated disulfides generally requires use of a large excess of the probe to achieve sufficient levels of tagging.349 In contrast, related activated thiols, namely thiosulfates (R–S–SO3−), thiosulfonates (R–S–SO2–R′, MTS), sulfenyl halides (R–S–X),350 pyridyl disulfides,351,352 and TNB-thiols (derivatives of 5-thio-2-nitrobenzoic acid) contain good leaving groups, which tautomerise to give unreactive forms thus shifting the reaction equilibrium (Fig. 29).
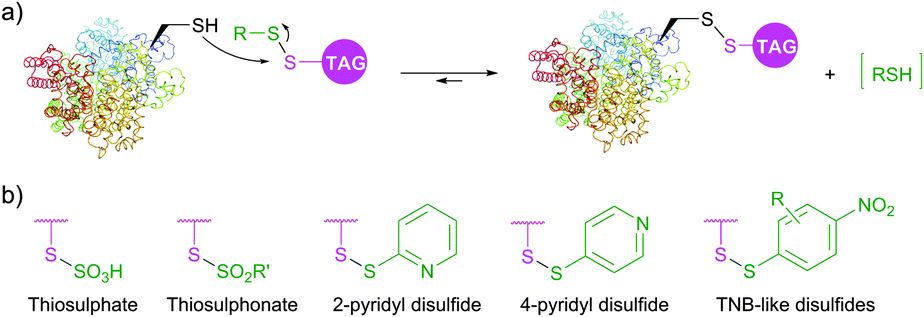 | ||
| Fig. 29 (a) General scheme of protein labelling via the formation of a disulfide bond. (b) Most relevant examples of activated thiols: thiosulfate, thiosulfonate, 2-pyridyl disulfide, 4-pyridyl disulfide, TNB-like thiol. | ||
PEGylation, fluorescent and biotinylation probes containing thiosulfate (commercialised as TS-link reagents) and pyridyl disulfide motifs are today widely commercially available. Thiosulfonates were first introduced for bioconjugation by Davis and co-workers353,354 in their work on the controlled glycosylation355 and further elaborated by Zhao et al.356 for site-selective PEGylation of proteins. Recent advances resulted in the further development of thiol activated methodologies towards selenenylsulfides357,358 and even methanedithiosulfonates, allowing for synthesizing trisulfide conjugates.359 Disulfide-based conjugation was recently reported for the preparation of antibody–drug conjugates and studying the influence of the spacer length on their stability.360 Diselene analogues of disulfide PEGylation reagents were proposed by Jevševar et al.361 as selective and fast alternatives for coupling. Although high conversion yield required the use of a large molar excess of the probe, this elegant approach represents an interesting technology which deserves further investigation.
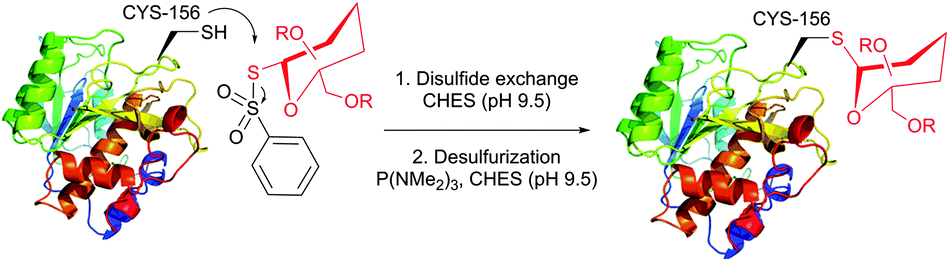
The main factor that has gained popularity to methodologies yielding disulfide and selenol-sulfide linkage is the reversibility they afford. However, the resulting conjugates are generally less stable than those obtained using bromomaleimides315 (Section 1.3.2) and can be readily cleaved with classical reducing agents such as DTT, β-mercaptoethanol or TCEP. Yet, in the case of disulfides, the modified protein can be made more stable and resistant to reduction by the corresponding thioether-linked conjugate by means of HMPT-mediated desulfurisation elaborated by Davis and associates (Fig. 30).362 The use of hindered disulfides represents another way to increase the resistance of generated conjugates to cleavage.363
In an attempt to resolve this problem, Brocchini et al.371 developed a clever methodology for the PEGylation of protein disulfide bonds with α,β-unsaturated bis-thiol alkylating reagents. Covalent rebridging of the two thiols derived from the disulfide after its mild reduction allowed obtaining the modified proteins with retained tertiary structure and biological activity. Interferon α-2b (IFN) was used in the initial studies, because it is representative of four-helical-bundle proteins with accessible disulfide bonds. Following reduction of the disulfide in IFN's, the two free cysteines were re-joined using a three-carbon linked functional PEG.368,371 The methodology was further expanded to PEGylation of therapeutic proteins,368,372,373 antigen-binding fragments of immunoglobulin G,374 and poly phosphocholine labelling of IFN.375 Simultaneously with the introduction of previously mentioned monobromomaleimides, Baker and co-workers have introduced a relevant class of reagents containing a highly reactive dibromomaleimide or dibromopyridazonedione scaffold, allowing rapid and efficient disulfide rebridging by installing a rigid two-carbon linker.316,376 This approach was first applied for equimolar PEGylation of 32-amino acid salmon calcitonin (sCT, Fig. 31)377 and very recently for the preparation of homogeneous antibody conjugates.378,379
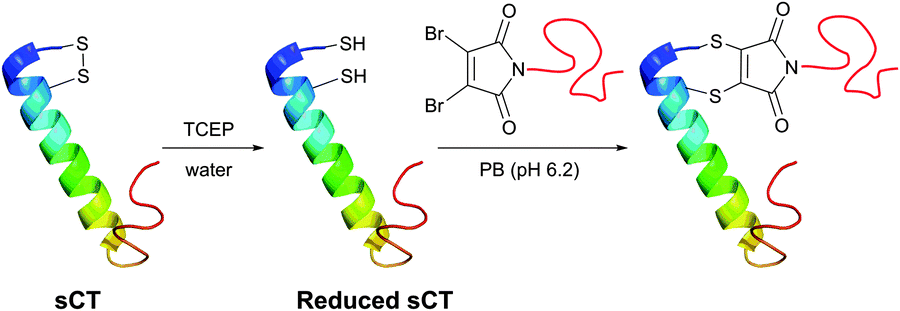 | ||
| Fig. 31 One-pot reduction-PEGylation of the disulfide bridge of sCT (pdb: 2GLH) followed by rebridging using dibromomaleimide.377 | ||
Although being very rapid (full conversion is achieved in less than 5 minutes), dibromomaleimide-based conjugation resulted in obtaining a small amount of the multimers while modifying complex polypeptides.378 Developed by Baker et al.380 and Haddleton et al.381 more stable and less reactive dithiophenolmaleimides allowed avoiding this apparent drawback of dibromomaleimide probes. In combination with benzeneselenols known for their efficiency in the catalysis of disulfide cleavage, the dithiophenolmaleimide approach allowed selective antibody fragment conjugation with no detectable formation of multimers and conserving a high reaction rate (Fig. 32).378 Haddleton and colleagues382 have recently reported an in situ one-pot preparation of oxytocin–polymer conjugates using dithiophenolmaleimide-containing probes.
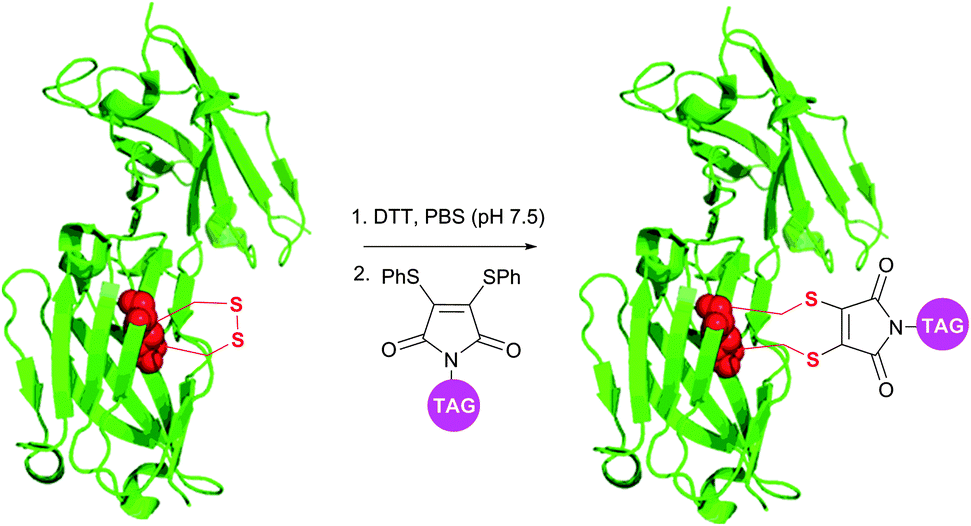 | ||
| Fig. 32 Dithiophenolmaleimide approach for in situ disulfide bridging of antibody fragments (pdb: 1QOK).378 | ||
Structurally similar to dibromomaleimides, but containing four attachment points instead of three, dibromopyridazinediones (PD) were recently described by Caddick et al.383,384 and provided a platform for IgG antibody dual labelling.384 Namely, the authors elaborated the preparation of a PD-linker containing two orthogonal reactive handles in its structure: (1) a strained alkyne, which readily reacts with azides in Cu-free strain-promoted azide–alkyne cycloaddition (SPAAC), and (2) a terminal alkyne, which reacts with azides in Cu-catalysed azide–alkyne cycloaddition (CuAAC). The construct obtained after rebridging a reduced antibody with the PD-linker was then used to selectively introduce two distinct functionalities (Fig. 33).
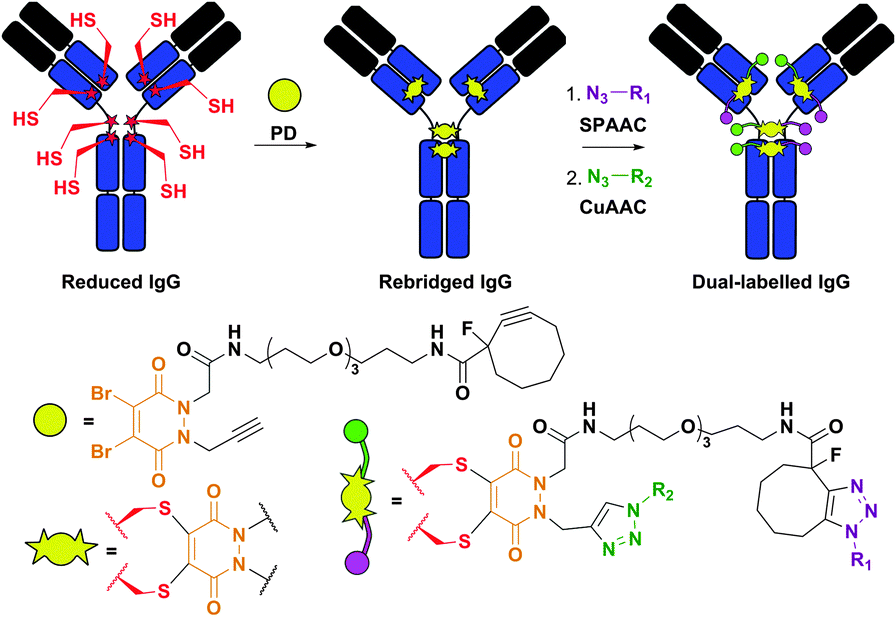 | ||
| Fig. 33 Dual labelling of IgG antibody using dibromopyridazinediones (PD, shown in orange).384 The construct obtained after rebridging of a reduced IgG with a PD-probe containing a strained alkyne (reactive in strain-promoted azide–alkyne cycloaddition, SPAAC) and a terminal alkyne (reactive in Cu-catalysed azide–alkyne cycloaddition, CuAAC) was subsequently labelled with two azide-containing probes (N3–R1 and N3–R2). | ||
An interesting disulfide stapling–unstapling strategy using dichloro-s-tetrazine was developed by Smith and collaborators (Fig. 34).385,386 In addition to their ability to be photochemically cleaved (i.e., unstaple thus regenerating reduced disulphide bonds; Fig. 34a), S,S-tetrazine macrocycles provide a possibility for labeling by exploiting the reactivity of the tetrazine in the inverse electron demand Diels–Alder reaction (Fig. 34b).
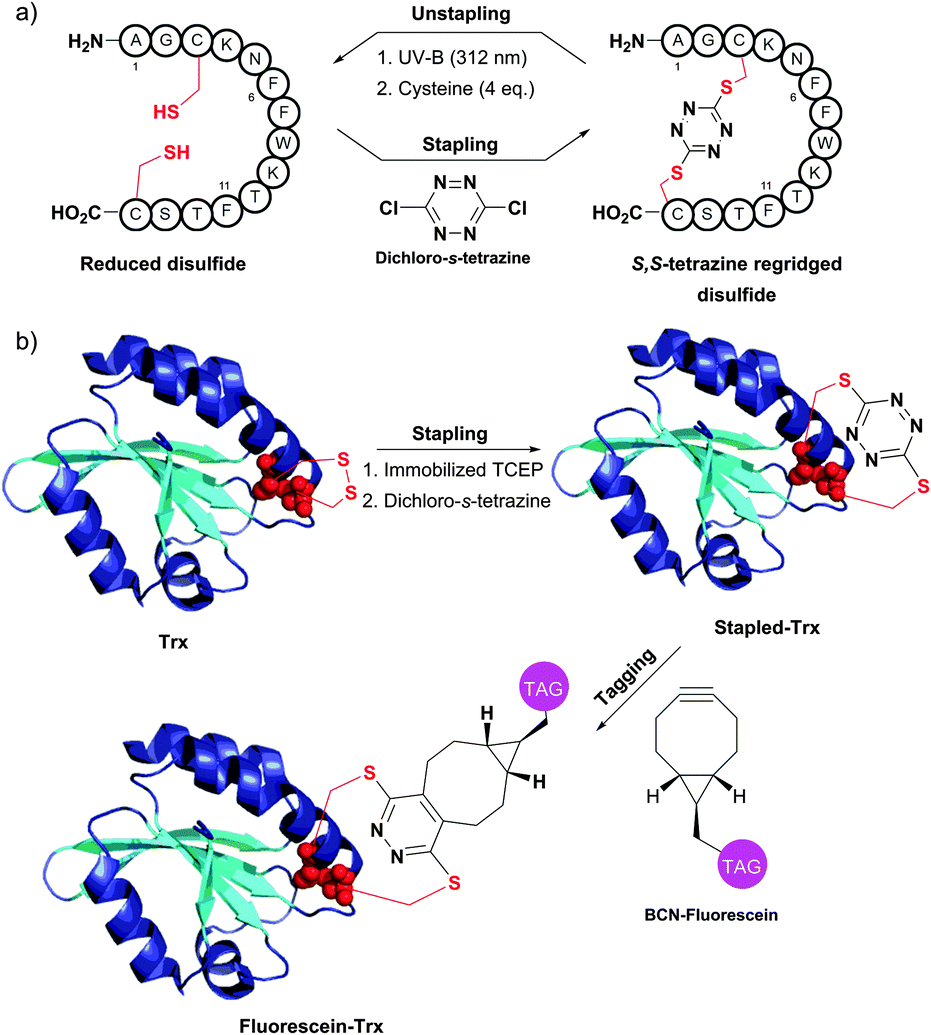 | ||
| Fig. 34 Protein stapling–unstapling using dichloro-s-tetrazine described by Smith et al. (a) Rebridging of a disulfide bond of a 14-mer peptide using dichloro-s-tetrazine and recovery of the starting product by photocleavage of the resulting S,S-tetrazine macrocycle (irradiation at 312 nm) followed by quenching of the thiocyanate intermediate with an excess of cysteine. (b) Rebridging of thioredoxin (Trx, pdb: 2F51) after the disulfide bond reduction followed by tagging of the resulting S,S-tetrazine with a strained alkyne containing probe (BCN-fluorescein). | ||
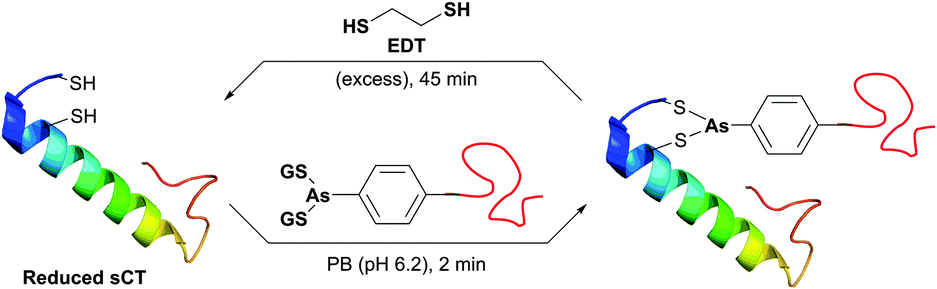
Organic arsenicals (similar to tetracysteine-selective biarsenical dyes initially developed by Tsien and co-workers, see Section 4) were recently exploited for efficient protein–polymer conjugation (Fig. 35).387 It is noteworthy that, in contrast to highly thiol-reactive dibromomaleimides, these reagents demonstrated enhanced selectivity for disulfide rebridging in the presence of free Cys residues. Namely, while dibromomaleimides reacted near quantitatively within 30 minutes with the free CYS-34 of native BSA, organic arsenicals exhibited limited reactivity and demonstrated only about 20% labeling over the same period of time.387 The authors hypothesised entropy-driven affinity of arsenicals for closely spaced dithiols to be the main reason of such specificity.
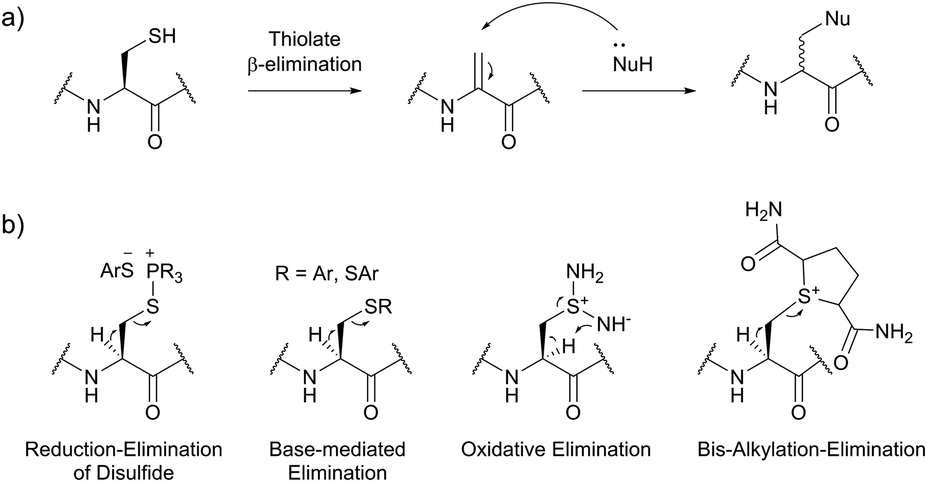 | ||
| Fig. 36 Utilisation of Cys-Dha transformation for bioconjugation. (a) General mechanism of the transformation. (b) Representative methods for the generation of dehydroalanine.395 | ||
Site-selective incorporation of Dha into proteins may be achieved by a considerable number of chemical transformations. Historically, the first example of such reaction was reported by Koshland and collaborators 50 years ago and consisted of transformation of nucleophilic serine of chymotrypsin to dehydroalanine via selective sulfonylation followed by base-mediated elimination.396–398 The method, however, exploited the particularly high nucleophilicity of the SER-195 residue located in the active site of chymotrypsin. Logically, more general methods based on the exceptional nucleophilic properties of cysteine received increased attention in the years to follow. These are represented in Fig. 36 and include: reduction–elimination, representing an often observed undesired side-reaction during reduction of disulfides; base-mediated elimination of activated thiolate, typically requiring temperatures incompatible with protein substrates and thus not being of synthetic interest; oxidative elimination, and bis-alkylation-elimination.395 Two last approaches seems to us the most promising among available Cys → Dha transformations and it is to these methodologies that we now turn.
1.3.8.1 Oxidative elimination. Oxidative elimination of thiolates is readily achievable, but often required high temperatures and severe reaction conditions yielded the methodology incompatible with fragile protein substances.399 However, recent efforts focused on finding milder conditions to carry out these desulfurisations, have resulted in developing two promising classes of chemical reagents: o-mesitylenesulfonylhydroxylamine (MSH, Fig. 37),392,400 and bromomaleimides (above-mentioned for the generation of bis-cysteine adducts).314
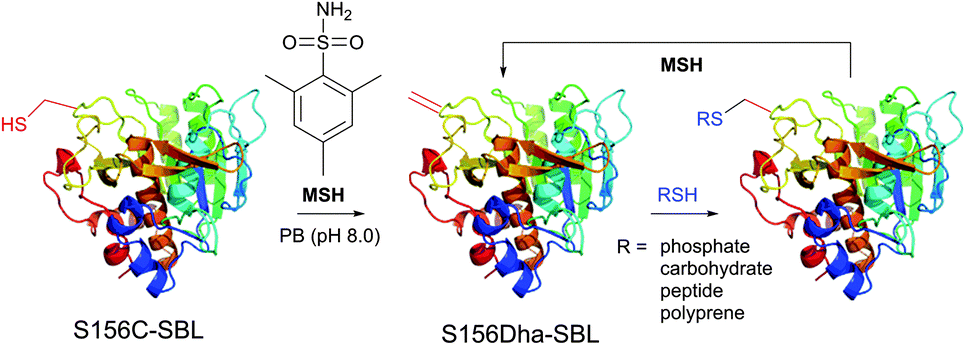 | ||
| Fig. 37 Conjugation of mutant S156C SBL (wild-type pdb: 1GCI) containing a single, surface-exposed cysteine residue CYS-156 by oxidative elimination followed by conjugation with thiol probes.392 | ||
Basic conditions are generally required for the reaction to achieve high conversion yields. Other amino acid residues may also react with MSH and bromomaleimide, but the reaction rates are largely inferior to those of thiolates and resulting products are generally unstable in basic conditions and decompose back to their starting unmodified forms.
1.3.8.2 Bis-alkylation–elimination. Conversion of cysteine to dehydroalanine by means of bis-alkylation–elimination approach was first introduced by Holmes and Lawton.401 Initially this transformation implied quite strenuous reaction conditions and was only compatible with a restricted number of protein substrates. Only recently have Davis and collaborators395 reported a more general method for Cys → Dha transformation by means of water-soluble α,α′-dibromoadipyl(bis)amide (DBAA) allowing generation of Dha moiety under mild conditions (37 °C, pH 7.0–8.0) at sufficiently high yields. This approach was evaluated on several model proteins, including SBL (see above),395 the single-domain antibody cAb-Lys3 A104C mutant,395 histone H3 (Fig. 38),393 AurA kinase domain,402 and GFP mutant.403
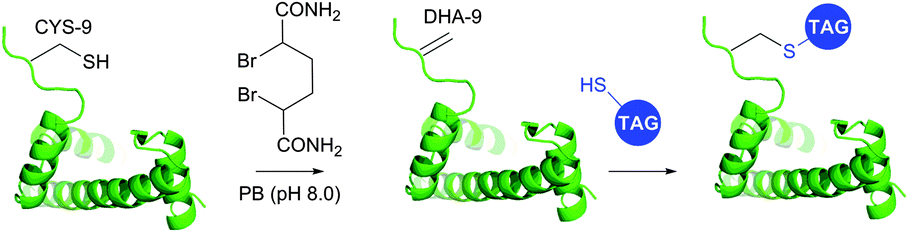 | ||
| Fig. 38 Dehydroalanine-mediated conjugation on histone 3 mutant H3.393 | ||
1.3.8.3 Other approaches. Despite recent advances in dehydroalanine-mediated conjugation methodologies, the inherent limitations of these methods preclude their general use for peptide and protein modification. None of these approaches enable general, chemo- and site-selective incorporation of dehydroalanine into proteins without the need for prior incorporation of an accessible Cys residue. Several other approaches continue to appear and are designed to overpass this problematic. These are oxidative elimination of aryl-selenocysteine,399,404–408 utilising of lacticin synthetase,409 transformation to selenocysteine thioethers.410
SNAr substitution chemistry approaches for cysteine modification in proteins were reported by several research groups.395,414,415 Davis et al.395 utilised Mukaiyama's reagent (2-chloro-1-methylpyridinium iodide) to generate an arylated cysteine as an intermediate for conversion thereof to dehydroalanine. Pentelute and co-workers have expanded the approach towards perfluoroaryls for protein stapling and conjugation.414,415 Finally, Barbas and associates416 have developed a class of Julia–Kocienski-like methylsulfonyl-functionalised reagents, that reacts rapidly and specifically with thiols under biologically relevant pH (5.8–8.0). Notably, the resulting conjugates possess superior hydrolytic stability compared to cysteine–maleimide, which makes this methodology appropriate for the preparation of stable protein conjugates and PEGylated proteins (Fig. 39).
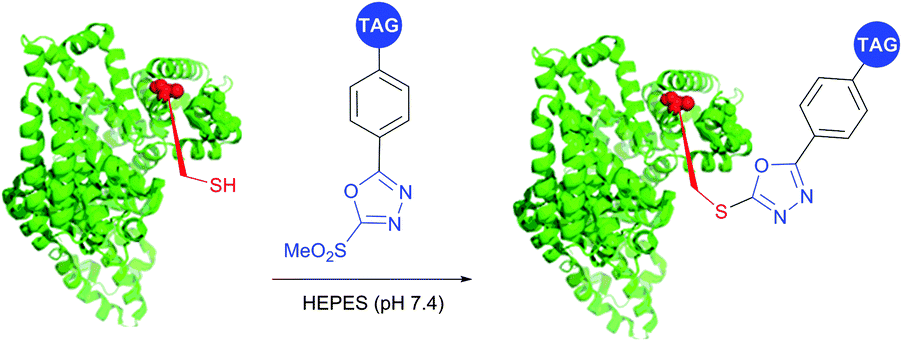 | ||
| Fig. 39 Labelling of the free cysteine residue of BSA (pdb: 3V03) with Julia–Kocienski-like reagents.417 | ||
A strategy exploiting selective cyclization of peptides containing three cysteines to generate combinatorial libraries of cysteine-rich bicyclic peptides was recently developed. This approach is based on utilisation of homotrifunctional linkers: TBMB (tribromomethyl-), TATA (triacryloyl-), or TBAB (tribromoacetamide-containing reagents).418,419
An efficient gold-catalysed allene-mediated coupling reaction has been recently developed by Che and colleagues.420 The method allowed direct thiol-selective functionalisation of model peptides and reduced RNase A (Fig. 40).
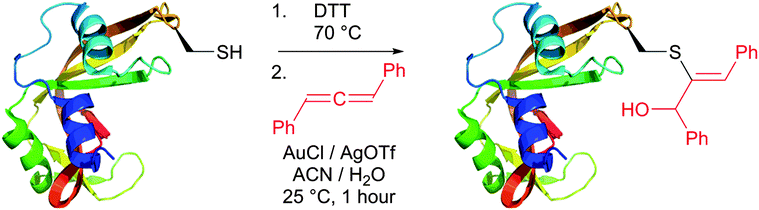
Reactions of thiols with electron deficient acetylenes have been known for decades, being, however, mostly conducted in organic solvents.421–425 Several examples of reactions in aqueous media have been recently reported.417,420,426,427 Che and co-workers417 have elaborated a versatile method for the selective cysteine labelling of unprotected peptides and proteins in aqueous media with arylalkynone reagents. Notably, modified peptides could be converted back into the unmodified peptides by treatment with thiols under mild reaction conditions (Fig. 41).
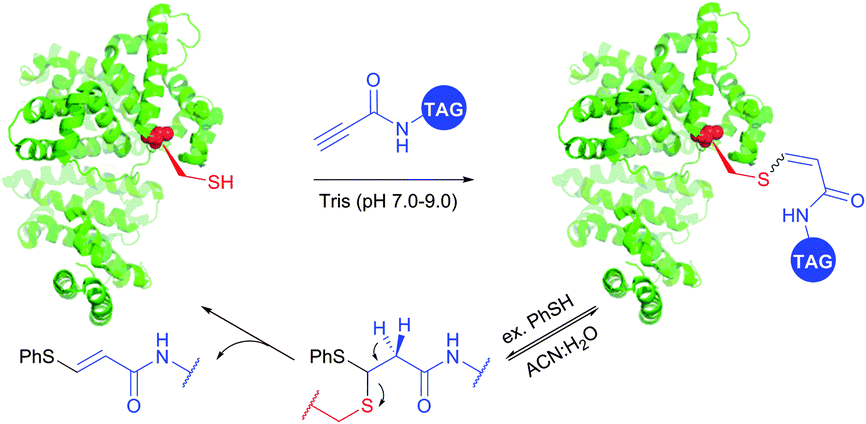 | ||
| Fig. 41 Cleavable labelling of BSA (pdb: 3V03) with arylalkynone reagents elaborated by Che and associates.417 | ||
Interestingly, in contrast to arylalkynones, structurally similar electron deficient acetylenes – 3-arylpropiolonitriles (APN) – were recently reported as a prominent class of reagents for irreversible tagging of cysteine.428,429 A superior stability of resulting conjugates in aqueous and biological media opens an interesting prospect in many fields where stability of obtained conjugates is crucial, e.g. for preparation of antibody conjugates possessing increased plasma stability (Fig. 42).429
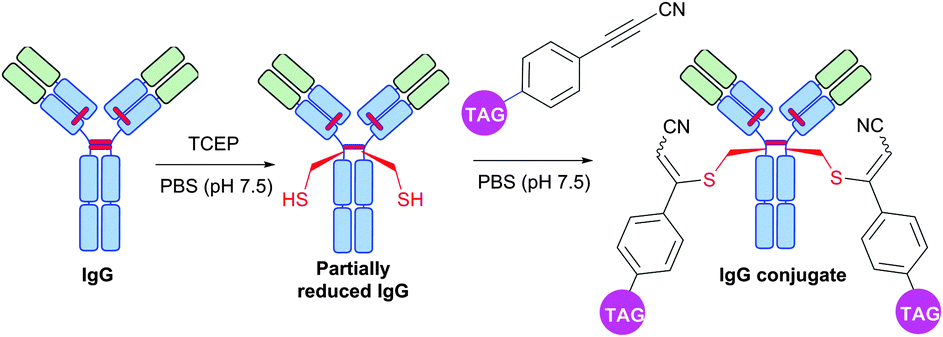 | ||
| Fig. 42 Preparation of IgG (pdb: 1IGY, red sticks represent four interchain disulfide bonds) conjugates with increased plasma stability using 3-arylpropiolonitriles (APN) described by Wagner and collaborators.429 | ||
Oxanorbornadienedicarboxylates (OND reagents), strained adducts of furans and electron-deficient alkynes, were found to provide better water stability while retaining selective, rapid, and fluorogenic reactivity towards cysteine compared to corresponding alkynes.430 α,β-Unsaturated ketones and amides (typically acrylamides) can undergo Michael-addition.431,432 However, the rate of addition is not generally high enough to provide it with competitive advantages compared with other approaches. Internal Cys residues were reported to accelerate native chemical ligation (see Section 2.3.1), an especially selective approach for N-terminal cysteine conjugation, via cyclic transition states.433–438
1.4 Tryptophan
Tryptophan (Trp, W) is the second (after cysteine) low abundance amino acid with about 1% frequency (depending on the living organism),235 but approximately 90% of proteins contain at least one Trp residue in their sequence.439 The specific reactivity of tryptophan in proteins is one of the most challenging problems in bioconjugation. In spite of the variety of reagents introduced over the years for selective modification of tryptophan, only a few can be used for conjugation. For instance, such classically used species as Koshland's reagent (2-hydroxy-5-nitrobenzylbromide)440 or chlorosulfonium ions441 present a high degree of cross reactivity with nucleophilic side chains, nonetheless still being used in numerous studies. These are, for example, investigation of the role of tryptophan in active sites of enzymes,442 estimation of its content in proteins,443 or determination of the surface accessibility of Trp residues in proteins.444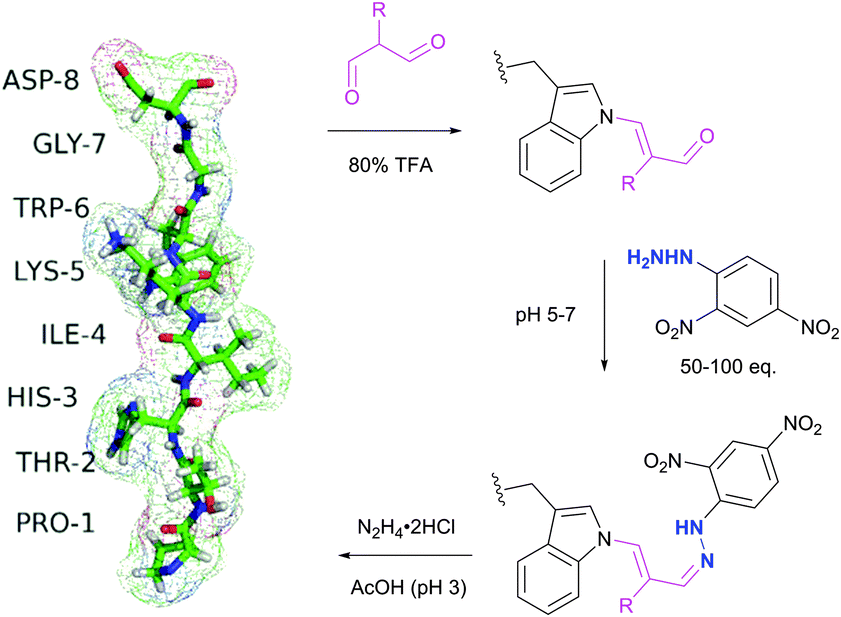 | ||
| Fig. 43 Selective labelling of Trp side-chain in a 8-mer peptide PTHIKWGD with malondialdehyde described by Foettinger et al.446 The peptide structure was simulated using RaptorX web server.447 | ||
To overpass selectivity issues, namely a known side reaction with Arg side chains,448 the conditions for the reactions with MDA, the hydrazone formation and the cleavage of the MDA derivative, had to be optimised concerning pH, buffer, temperature, and reagent. No side reactions of MDAs were observed only under strongly acidic conditions, such as aqueous TFA (80%). The following hydrazone formation requires approximately 50–100 fold molar reagent excess at a pH of 5–7 and sometimes increasing the temperature to 50 °C. Although unstable at acidic conditions and when the reagent excess is removed, the hydrazone bond remains firm in alkaline medium (pH > 9). The optimal conditions for the cleavage were found using hydrazine (applied as the dihydrochloride salt) in ammonium acetate solution at a pH ∼ 3. Demonstrably, these rather rough reaction conditions prevent this methodology from finding widespread use for sensitive protein targets, yet allowing its application in proteomics on peptide digests.449
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixtures of N-alkylated versus 2-alkylated products (Fig. 44) are generally obtained in moderate yields.450,451
:3 mixtures of N-alkylated versus 2-alkylated products (Fig. 44) are generally obtained in moderate yields.450,451
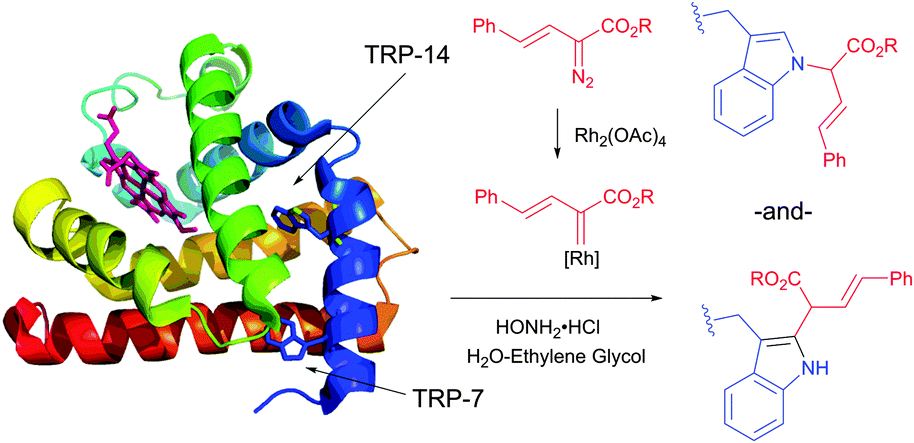 | ||
| Fig. 44 Modification of horse heart myoglobin (pdb: 1YMB) with rhodium carbenoids described by Francis et al.450 A 100 μM solution of myoglobin was exposed to stabilised vinyl diazo precursor (10 mM) and Rh2(OAc)4 (100 μM) for 7 h. N- and C-derivatisation of indole rings of both Trp residues – TRP-14 and TRP-7 – were identified by the mass reconstruction. An excess of hydroxylamine hydrochloride (75 mM) is crucial for the efficiency of the conjugation, although its mode of action was not elucidated. | ||
The initially reported reaction conditions tolerated several aqueous solvent systems and proceeded at room temperature. Yet, acidic conditions (pH 1.5–3.5) were still necessary for efficient protein labelling and stood out as the main drawback preventing this approach from being generally applicable. For instance, in the same work, authors have stated that myoglobin was denatured and the heme dissociated from the protein due to the high acidity of the medium.
To address these limitations, following efforts of the same group were to improve the pH range of the tryptophan modification methodology.452 For hydroxylamine was found to be ineffective at generating rhodium carbenoids at pH ≥ 6, a wide screening of commonly used buffers, as well as additives structurally similar to hydroxylamine H2NOH, was conducted in order to identify appropriate conditions. From these studies, tBuNHOH was found to be highly effective at promoting carbenoid addition. Despite the precise mode of action for tBuNHOH remains unclear, the authors attributed the substantial increase in catalytic activity to a specific interaction between this additive and Rh2(OAc)4. They speculated that, in contrast to hydroxylamine, tBuNHOH binds to Rh2(OAc)4 through the oxygen, rather than the nitrogen, the latter being disfavoured by the bulky tert-butyl substituent (Fig. 45a), and increases both the stability and the reactivity of the complex at neutral and slightly basic pH.
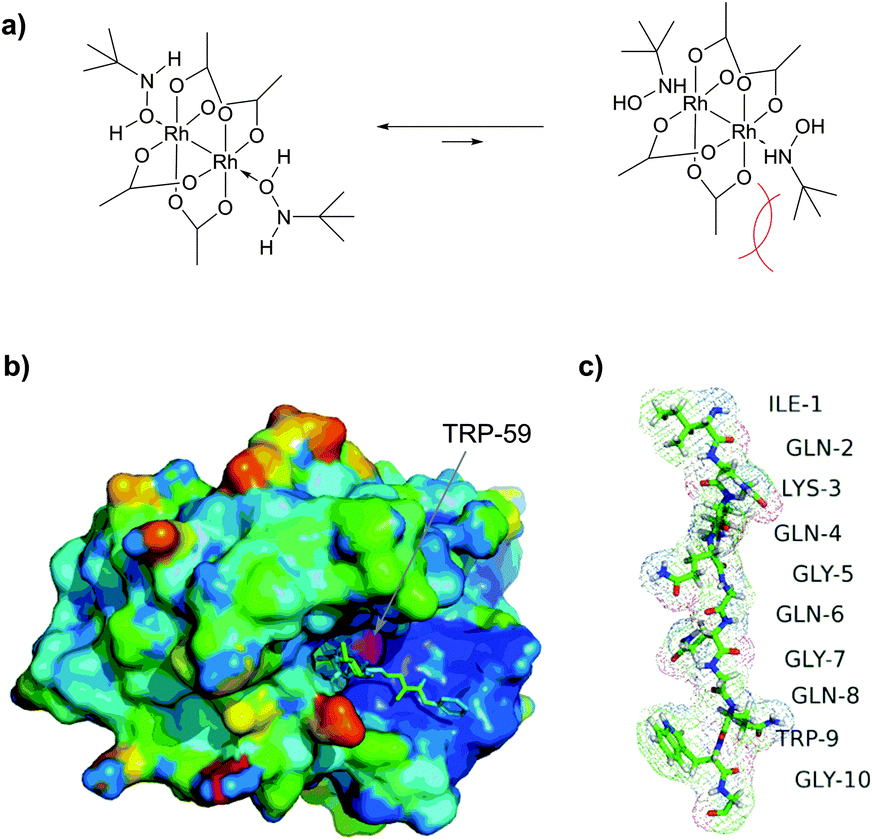 | ||
| Fig. 45 Optimised metallocarbenoids-based approach, described by Francis and collaborators.452 (a) Proposed binding of tBuNHOH with Rh2(OAc)4 at pH 6.0. O-Coordination is favoured due to a lower sterical hindrance of tert-butyl groups with acetate ligands. (b) Crystal structure of wild type FKBP (pdb: 1A7X), containing a single, buried tryptophan residue TRP-59 (shown in magenta), which is unavailable for modification under nondenaturating conditions. (c) A peptide tag (IQKQGQGQWG) incorporated into fusion FKBP protein expressed in E. coli as C-terminal intein fusion. The total level of modification was estimated to be in excess of 40%. | ||
Interestingly, in the same work, the authors have demonstrated the key role of solvent accessibility of residues in determining the outcome of conjugation on tyrosine using rhodium metallocarbenes. Human FK506 binding protein (FKBP) was identified as a suitable substrate for the study. The only Trp residue (TRP-59) of a wild type FDBP (containing an additional C-terminal threonine residue) is located at the base of the binding pocket, and therefore is unavailable for modification under nondenaturing conditions (Fig. 45b).
To overcome these difficulties, a labelling strategy based on tryptophan mutagenesis followed by chemoselective modification with rhodium carbenoids was utilised. Tryptophan-containing FKBP proteins were expressed in E. coli with C-terminal intein fusions containing a chitin binding domain for affinity purification and short tryptophan-containing peptides (Fig. 45c). Indeed, these newly obtained mutants with solvent accessible Trp residues showed significant level of conjugation (more than 40%) under optimised non-denaturating conditions at room temperature.
In 2010, further developing the rhodium-carbenoids methodology for selective tryptophan labelling described 6 years before by Francis et al.,453 Popp and Ball have reported structure-selective modification of aromatic side chains (expanding its scope to include Tyr and Phe residues) using proximity-driven approach (see Section 5 for details).454 Structurally similar to Rh2(OAc)4, metallopeptide complexes with a dirhodium center bounded with two glutamate residues were envisioned to provide delivering of the catalyst to a close proximity of the reactive side chains by exploiting the coiled coil matched peptides,455 for molecular peptide–peptide recognition (Fig. 46).
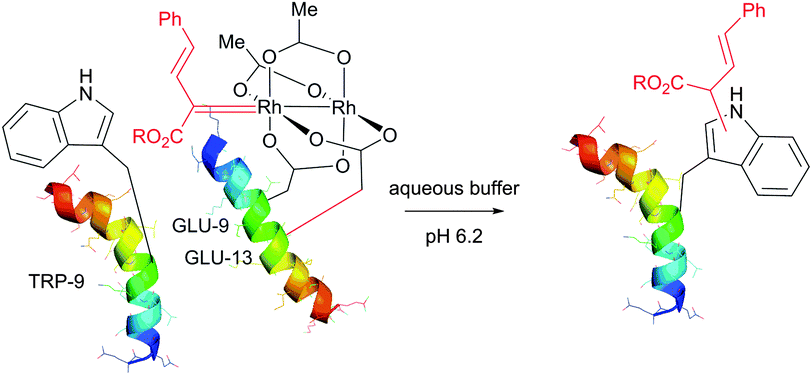 | ||
| Fig. 46 Selective covalent labelling of the TRP-9 residue of the peptide QEISALEKWISALEQEISALEK with its complementary dirhodium metallopeptide KISALQKQKESALEQKISALKQ described by Popp and Ball.454 The rhodium cluster, chelated with two glutamate residues, is brought closer to the reactive Trp residue by peptides coil self-assembling, resulting in selective peptide modification on TRP-9. Peptide structures were simulated using the RaptorX web server.447 | ||
By combining residue-selective chemistry with secondary-structure recognition, the authors have provided a strategy for selective covalent modification of biomolecules. However, only simple diazo reagents without functional handles were used in controlled environments on model peptide substrates.
In the following year, the same group has extended their initial studies to examine the reactivity of whole proteins in a complex, cell-like environment.456 For this, the proximity-driven catalysis approach was applied to a recombinant maltose binding protein (MBP), fused with the 21-amino-acid tryptophan-containing coil (almost identical to one used in the initial publication, Fig. 46). Directly after the expression, the lysate was subjected to metallopeptide-catalysed biotinylation. A single band in Western blot analysis indicated highly selective modification of the modified MBP protein with no nonselective modification to be observed.
1.5 Histidine
Histidine (His, H) is the only amino acid with a pKa in the physiological range, hence often found in active sites of many enzymes and is of crucial importance in mechanisms where abstraction or donation of a proton is needed. Because of its pKa value, both the acid and base forms are present at physiological pH (Fig. 47). Most of the studies on catalytic activity of enzymes and protein–protein interactions involving histidine-containing active centres were done by measuring the influence of site-specific modifications of His residues on the activity of the macromolecule. Main factors influencing histidine reactivity are (1) the pKa of the individual His residue, (2) solvent exposure of the residue, and (3) hydrogen bonding of the imidazolium ring. A vast number of studies have been conducted to date, nonetheless, exploiting a small number of chemical functions. | ||
| Fig. 47 Tautomerisation equilibrium of the neutral imidazole side chain (base forms A and C) occurring through the acid form B.457 Form A is somewhat favoured over C at neutral and acidic pH, while at basic pH the form C is preferred.458 | ||
 | ||
| Fig. 48 Fluorescent probes, described by Li et al. for selective ligation of histidine.459,460 | ||
The authors suggest that these probes can be used for specific labelling of His residues in proteins if a mild reaction condition (lower reaction temperature but longer reaction time) was used, but no example of such application was given. Moreover, considering known reactivity of epoxides with primary amines, thiolates, and hydroxyl groups, such selectivity towards histidine at physiological pH seems improbable.461
An affinity-based labelling approach (see Section 4) based on the epoxide opening was developed by Hamachi and collaborators for selective histidine labelling of bovine carbonic anhydrase II.462,463 Labelling reagents investigated by the authors must consist of at least three major fragments: (1) a benzenesulfonamide ligand directing specifically to bCA, (2) a reactive electrophilic epoxide for protein labelling, and (3) an exchangeable hydrazone bond between the ligand and the epoxide group for removing the ligand by hydrazone/oxime-exchange and restoring the enzymatic activity (Fig. 49a). Further developing their approach,463 the authors added an iodophenyl or acetylene handle on the epoxide-containing fragment to enable the possibility of further derivatisation of the obtained conjugate by Suzuki coupling464 or Huisgen cycloaddition465 either after or before removing the ligand from the active site of the enzyme (Fig. 49b).
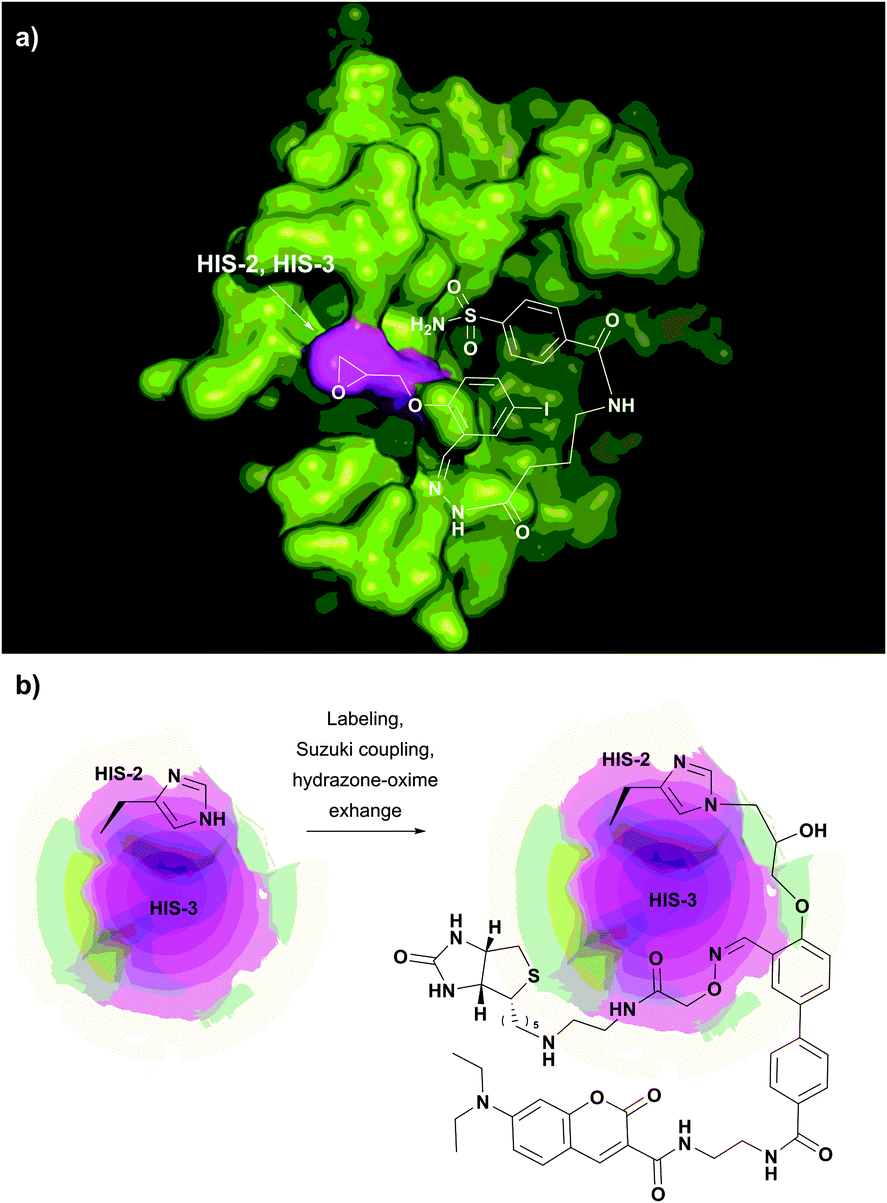 | ||
| Fig. 49 Affinity labelling, hydrazone–oxime-exchange reaction, and Suzuki coupling reaction on the surface of bCA (pdb: 1V9E) described by Hamachi and collaborators.463 (a) Principal structural fragments of the probe: a benzenesulfonamide ligand responsible for targeting bCA; an electrophilic epoxide responsible for reactivity towards vicinal His residues HIS-2 and HIS-3 (shown in magenta), cleavable hydrazone bond, responsible for the recovering of the enzymatic activity; an iodoaryl moiety, utilised for the further bioorthogonal transformation via Suzuki coupling. (b) Dual labelling of bCA in situ: fluorescent labelling of the HIS-2 or HIS-3 alkylated intermediate by Suzuki coupling with a coumarine derivative, followed by biotinylation by hydrazone/oxime-exchange. | ||
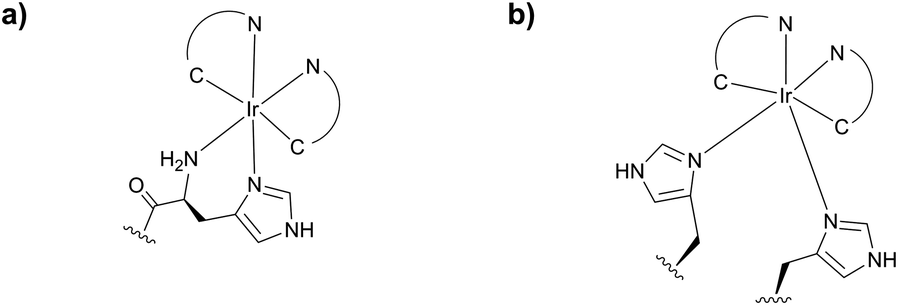 | ||
| Fig. 50 Luminescent histidine selective peptide tagging.467 (a) Labelling of a N-terminal histidine-containing HTat peptide (HRKKRRQRRR). (b) Labelling of a dihistidine motif placed in the middle of a P450dHTat peptide (MLAKGLPPKSVLVKGGHHGRKKRRQRRR). | ||
Although, reckoning obtaining of the coordination complexes to conjugation techniques would be stretching a point, they are however included in this survey because of histidine liability to complexation and increased stability of obtained complexes to decomposition.
Described by Pauly in the beginning of the 20th century,480,481 the reaction with diazonium salts has been only used for colorimetric determining of His residues482 and was not further elaborated for bioconjugation. Other classical examples of chemical modification of histidine are mainly reactions with pyrocarbonate, sulfonyl chlorides, sulfonic esters, phenacyl- and acylbromides and activated esters.229 These electrophiles react readily with other nucleophilic groups presented in proteins (thiols, amines, alcohols, or guanidino groups) and require a careful tuning of the reaction conditions to achieve sufficient selectivity. For instance, at low pH (generally < 6.0) these reactions are quite selective for histidine, as the main side reaction with the ε-amino group of lysine proceeds very slowly.483
Reader should, nonetheless, be aware, that these examples do not represent a general rule, but an exception from it. To avoid the reactions of more nucleophilic functions, the His residue must have been located in a unique microenvironment476,477 or have an enhanced nucleophilic character,478 but even in this case, prior modifications of highly reactive Cys residues are often inevitable.479
1.6 Tyrosine
Tyrosine (Tyr, Y) is one of the important amino acid residues which is known to be the active centre in many enzymes (notably tyrosine-specific protein kinases) and is used in signal transduction and cell signalling.484,485 Occurring with intermediate to low frequency in native proteins, tyrosine is often considered as an attractive target in bioconjugation, despite being often partially or completely buried due to the amphiphilic nature of the phenolic group.The reactivity of tyrosyl moiety is easily influenced by its deprotonation, which is a function of the microenvironment inside the protein. All described methodologies take advantage of either the peculiar chemical properties of the electron-rich aromatic ring, or the easiness of the tyrosine hydroxyl group to be transformed into highly reactive phenolate.
An elegant approach – the affinity labelling – allows surpassing the selectivity issues of tyrosine acylation by ligand-tethered directing of the reaction. In the case of tyrosine-selective modification, an acyl transfer catalyst is connected to a ligand with a high affinity to the target protein.462,486 The acyl group activated by the anchored catalyst is brought to the binging pocket of the protein and transfers an acyl moiety on the nucleophilic Tyr residue in close proximity. Utilizing this methodology, Hamachi et al.486 demonstrated selective tagging of Y51 residue of Congerin II (Fig. 52) using a suitable saccharide as a ligand to the target lectin (carbohydrate-binding protein) and DMAP (4-dimethylaminopyridine) as an acyl-transfer catalyst. In a similar way, Broo and collaborators487 have demonstrated the possibility of a site-specific acylation of a tyrosine residue situated in an active site of human glutathione transferase (hGST).
 | ||
| Fig. 52 Sugar-DMAP (4-dimethylaminopyridine) assisted Y51-specific acylation of Congerin II described by Hamachi et al.486 Schematic representation is made using lactose-ligated Congerin II crystal structure published by Muramoto et al.488 (pdb: 1IS4). | ||
Miller and collaborators have shown that biotinylation with NHS esters (see Section 1.1.2) may result in preferential O-acylation of hydroxyl-containing residues – serine, threonine and tyrosine (though to a greater extent of the first two) – when they are located two positions next to histidine (i.e. in sequences His-AA-Tyr, where AA refers to any amino acid).85,88
Several approaches for labelling involve the initial modification of tyrosine and successive conjugation of an obtained intermediate. For instance, an ortho-nitration of tyrosine with tetranitromethane (TNM)489 or peroxynitrite490 results in obtaining of o-nitrotyrosine that can be then reduced by sodium dithionite (Na2S2O4) to form an o-aminotyrosine.
Although much less reactive than aliphatic amines at neutral pH, the aromatic amine of o-aminotyrosine can selectively react with amine-reactive reagents at lower pH.491,492 Namely, Nikov et al.492 have demonstrated that selective labelling of aminotyrosines is achievable in the presence N-terminal and ε-amino groups of lysines by using NHS-activated ester at particular reaction conditions (acetate buffer, pH 5.0, 2 hours). Exploitation of the pKa difference between aminotyrosyl residues and other reactive groups in proteins (4.75 for aminotyrosine, whilst much higher values for N-terminal and side-chain amino groups, see Section 1.1) allows selective labelling thereof. The method was validated on model peptides and then applied to a human serum albumin modification (Fig. 53).
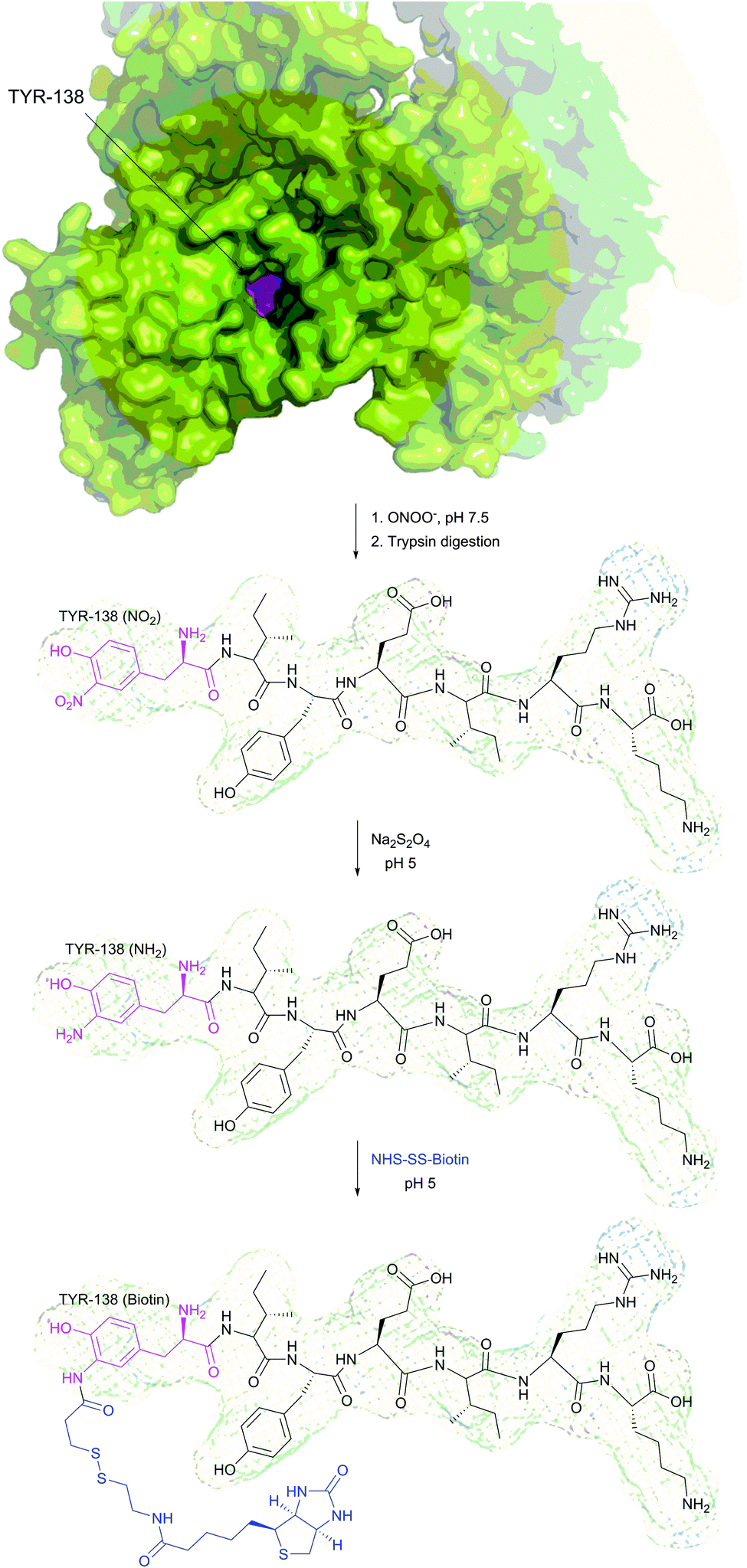 | ||
| Fig. 53 Two-step biotinylation of HSA (pdb: 1AO6) described by Nikov et al.492 Preferential nitration of TYR-138 (shown in magenta) and TYR-411 residues of HSA with peroxynitrite was achieved using protocol described by Jiao and colleagues.490 Aminotyrosine of the peptide 138YIYEIARK144, obtained after the step of reduction with sodium dithionite and following digestion of nitrated HSA with trypsin, were selectively modified with a cleavable biotin-containing reactant at pH 5.0. | ||
Despite the reaction of TNM and peroxynitrite with proteins being reasonably specific for tyrosine, side reactions with histidine, methionine and tryptophan have been reported, as has oxidation of sulfhydryl groups. The latter would seem to be the most common side reaction, as it can result in disulfide bond formation and the formation of oxidation products such as sulfone and sulfenic acid derivatives. As a general rule, it is normally assumed that the reaction of nitration reagents with Cys residues proceeds equally well at pH 6 and pH 8, while the reaction with tyrosine occurs at pH 8 and not at pH 6.
In a like manner, the phenol group in Tyr residues can be initially ortho-formylated with chloroform in an alkaline medium to a salicylaldehyde derivative, and then undergo a reaction with ortho-phenylenediamine derivatives to form fluorescent benzimidazoles as conjugation products (Fig. 54).493,494
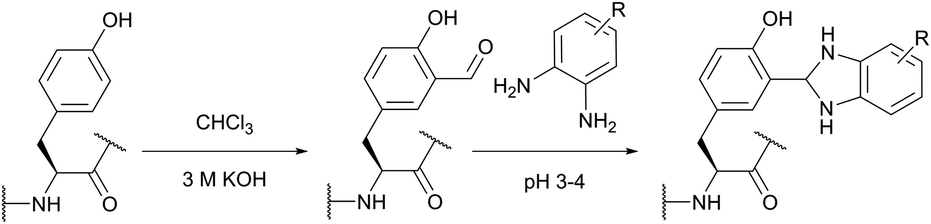 | ||
| Fig. 54 Two-step tyrosine modification by selective formylation of the tyrosyl residue at the ortho position of its phenolic moiety (reaction with chloroform in potassium hydroxide solution) and further derivatisation of the resulting aldehyde described by Kai et al. and Ishida et al.493,494 | ||
Further exploiting the methodology developed by Trost and Toste for selective O- and C-alkylation of phenols with π-allylpalladium complexes,495,496 Francis et al., have demonstrated the possibility of selective allylic alkylation of surface-exposed tyrosines of several full-size proteins.497
The photoactivable metal-catalysed version of tyrosine oxidation chemistry is significantly faster than the one achieved by Ni(II)–peptide complexes. It has been largely exploited by Kodadek and co-workers for cross-linking of closely associated proteins.503–505
These coupling reactions are hypothesised to occur through the addition of tyrosyl radicals to adjacent Tyr residues (Fig. 55). It is worth mentioning, that in some cases nearby tryptophan and other nucleophilic side chains can also participate in oxidation.503 For more details, the reader is directed to a review by Bonnafous and a publication of Francis that provide an excellent overview of oxidative cross-linking techniques.507,508
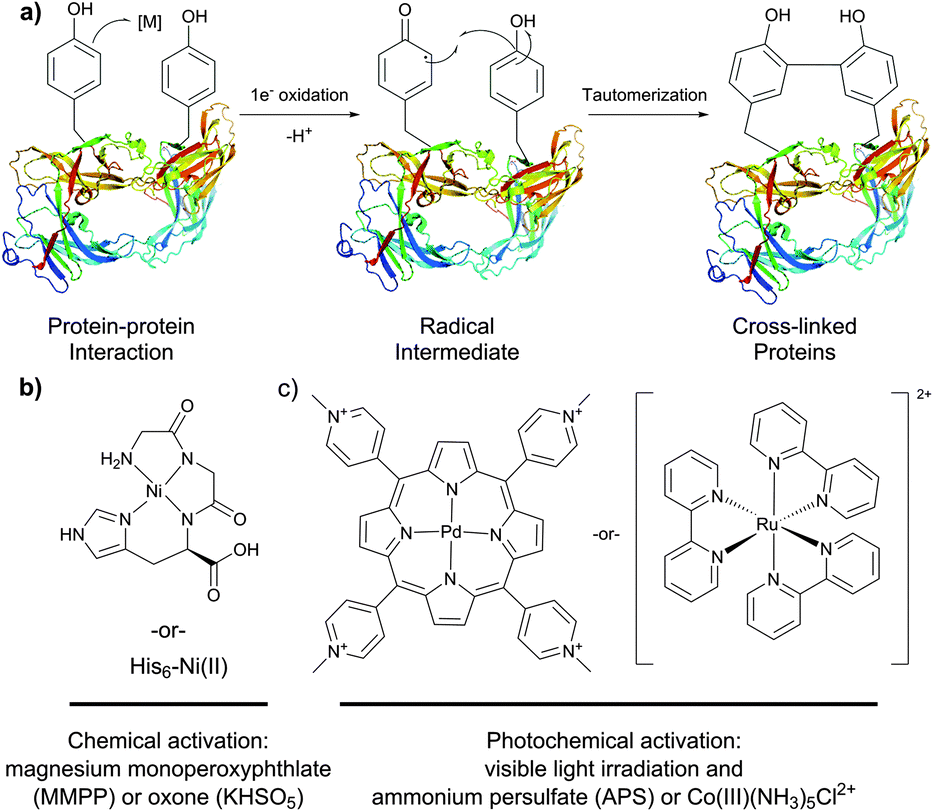 | ||
| Fig. 55 Transition metal catalysed oxidative cross-linking. (a) Schematic representation of the protein–protein cross-linking, described by Burlingame et al.506 The tyrosyl radical generated from ecotin (pdb: 1ECZ) D137H mutant after the abstraction of an electron with Ni(II)–GGH complex reacts with additional tyrosine residue on nearby protein that are in close proximity due to a significant protein–protein interaction to give a dimer. (b) Chemically activated by MMPP or oxone as stoichiometric oxidants, Ni(II)–peptide complexes can be used as efficient catalysts of cross-linking. (c) Photochemically activated by visible light in the presence of (NH4)2S2O8 or Co(III)(NH3)5Cl2+ as electron acceptors, transition metal complexes are used as catalysts of cross-linking. | ||
The use of cerium(IV) ammonium nitrate (CAN) – a classical one-electron oxidant – for chemoselective ligation on tyrosine was demonstrated by Francis et al. (Fig. 56).509 After the optimisation of reaction conditions, the authors could achieve modification of tyrosine-containing proteins with high yields at neutral pH and low substrate concentration and applied this strategy to modify both native and introduced residues on proteins with polyethylene glycol (PEG) and small peptides, although dealing with the concurrent reaction of Trp residues.509
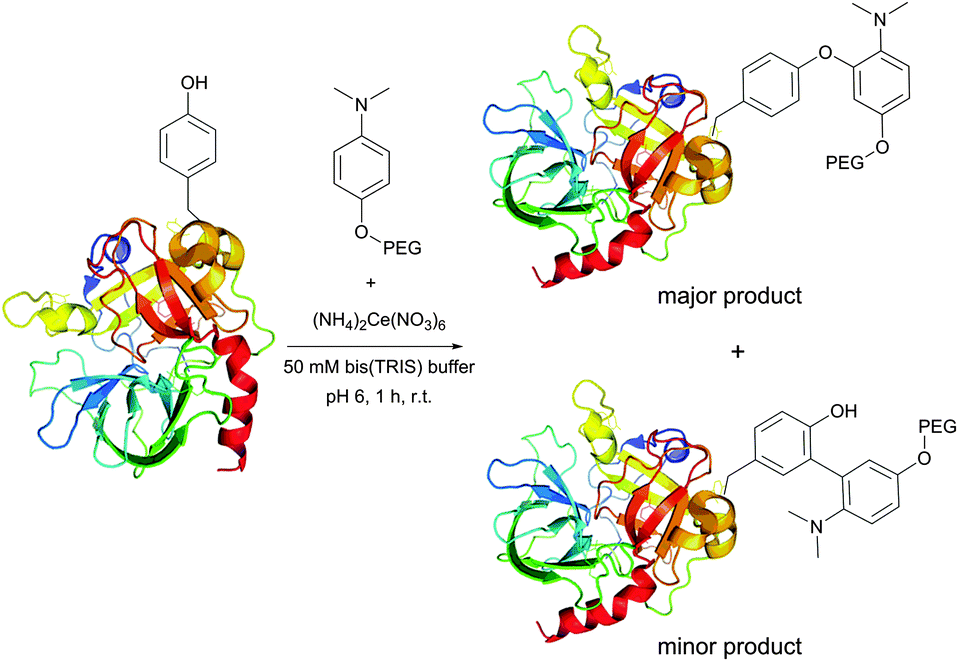 | ||
| Fig. 56 PEGylation of four solvent-accessible Tyr residues of chymotrypsinogen (pdb: 1EX3) reported by Francis et al.509 (only Y171 residue is shown for simplicity reasons). Intermediate tyrosyl radical, generated in the presence of cerium(IV) ammonium nitrate (CAN), gives two addition products with electron-rich aniline derivative (O- is preferred over C-arylation). | ||
Notwithstanding the issue of specificity, photo-oxidation and oxidation of techniques of tyrosine ligation continue to be of considerable interest for the study of protein–protein interactions,510 mapping multi-protein complexes,511 or assembling of macromolecules.512
The optimised conditions have nonetheless allowed its application for selective modification of tyrosines on the surface of bacteriophage MS2,515,516 the modification of the tobacco mosaic virus,517 and the direct conjugation on proteins.518 Francis and co-workers have demonstrated that highly reactive diazonium salts (i.e. containing electron withdrawing groups in their structure) should be utilised in order to achieve efficient Tyr targeting and avoid concurrent reaction with Lys and His residues (see Section 1.5.4).517 Recently described by Barbas et al., formylbenzene diazonium hexafluorophosphate reagent519 represents an elegant example of a stable ready-to-use reagent for tyrosine labelling and introduction of an aldehyde bioorthogonal tag, capable for future bioorthogonal modifications (Fig. 57).
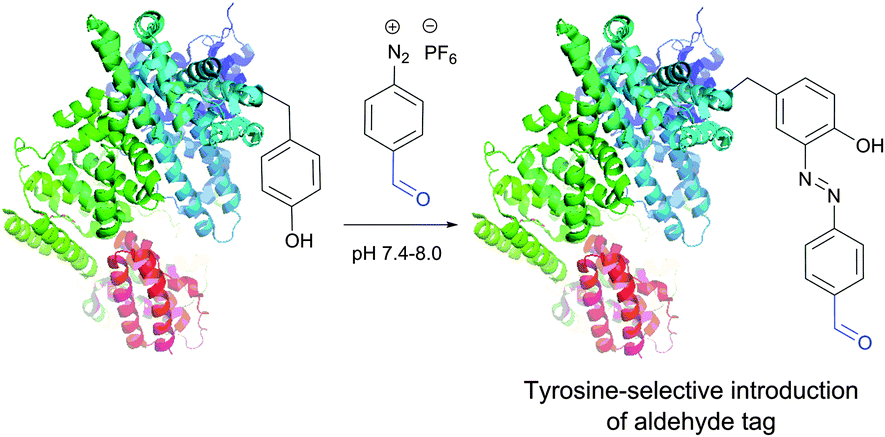
The three-component Mannich-type methodology – involving the in situ reaction between a Tyr residue, an amine and formaldehyde – was reincarnated more than 50 years later by Francis et al.521 The authors demonstrated the possibility of selective modification of tyrosine residues of α-chymotrypsinogen A under mild conditions (pH 6.5, 25–37 °C) and at low concentration of the protein (20–200 μM). However, 18 hours of incubation were needed to reach a reasonable level of tagging (66% in the case of a fluorescent labelling, Fig. 58). The same group then used this to incorporate synthetic peptides into full-sized proteins.522
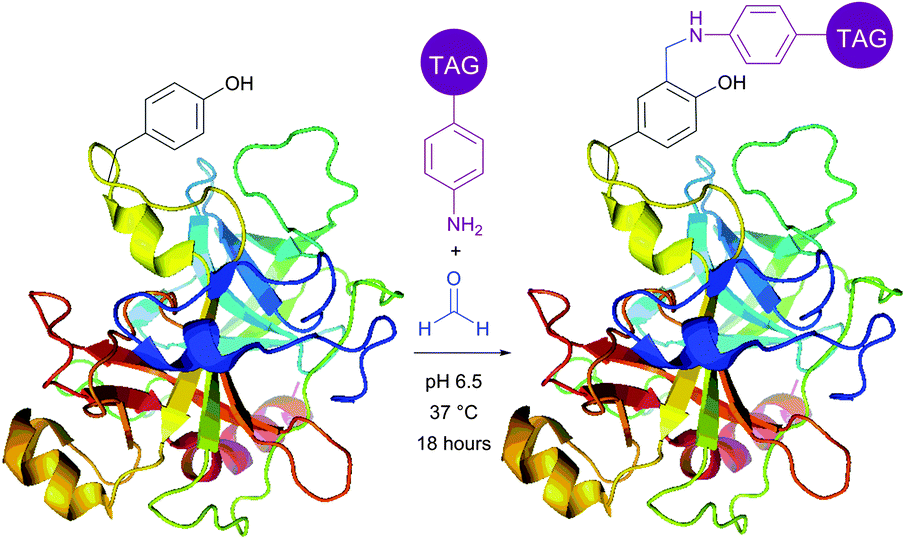 | ||
| Fig. 58 Three-component Mannich-type selective modification of the Y154 Tyr residue of α-chymotrypsinogen A (pdb: 1EX3) by a rhodamine dye, described by Francis et al.521 | ||
Despite recognised selectivity issues of a three-component Mannich-type approach for tyrosine labelling,523 its main advantage is the possibility to easily vary the participating partners: an aldehyde (Fig. 58, shown in blue) and an aniline residues (Fig. 58, shown in violet). In the following publication on the subject, Francis et al. have demonstrated the viability of NMR-based characterisation of the conjugate isotopically enriched by incorporation of 13C-formaldehyde into the coupling reaction.523 Interestingly, while a reaction by-product arising from tryptophan indole ring was revealed, Cys moiety was found to not participate in the reaction, except in the case of a reduced disulfide, which formed a dithioacetal.
Using similar precursors – electron-rich aniline derivatives –Tanaka et al.524 could demonstrate the potential of in situ obtained imines as fluorogenic probes for tyrosine labelling. While the educts, as well as the imine derivatives, exhibited weak or no fluorescence, the addition products had a significantly higher (more than 100-fold) level of fluorescence.
In the successive study, the same group has expanded this approach to presynthesised cyclic imines completely excluding the need for using an excess of highly reactive formaldehyde.525 Although, the authors have clearly demonstrated the applicability of their methodology in water at room temperature over a wide pH range (pH 2–10) on a set of model phenols, no example of peptide or protein conjugation has been given.
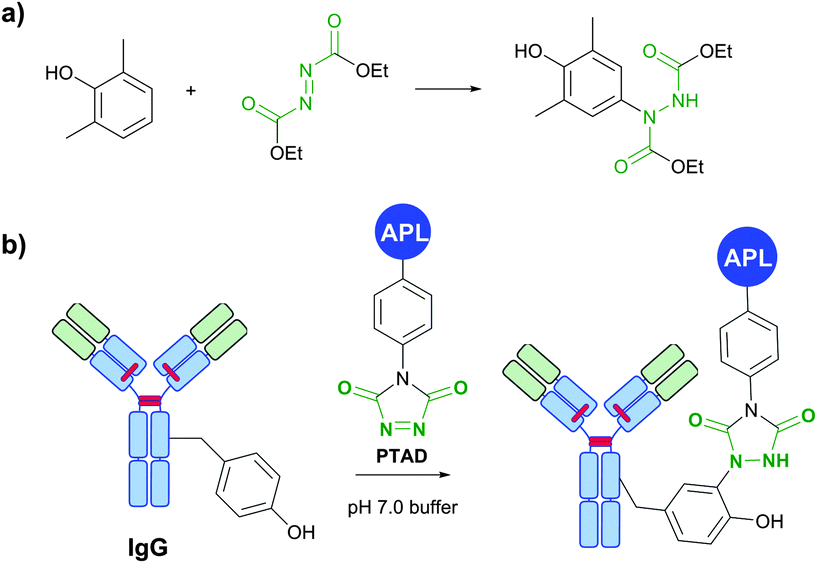 | ||
| Fig. 59 Reaction of electron-rich arenes with azodicarboxyl compounds (shown in green). (a) First example described by Schroeter in 1969. (b) Conjugation of an HIV entry inhibitor aplaviroc (APL) containing a cyclic diazodicarboxamide derivative – 4-phenyl-3H-1,2,4-triazoline-3,5(4H)-dione (PTAD) – with the IgG antibody demonstrated by Barbas.537 | ||
However, these highly reactive reagents decompose rapidly in aqueous media, which makes them not suitable for bioconjugation.535 On the other hand, corresponding diazodicarboxyamide reagents are too stabilised and do not react with phenols in aqueous media.535 Cyclic diazodicarboxyamides like 4-phenyl-3H-1,2,4-triazoline-3,5(4H)-dione (PTAD) were recently reported by Barbas and collaborators and represent a good compromise between reactivity and stability of diazodicarboxyl-containing reagents.536,537 Diazodicarboxylate-mediated tyrosine conjugation is applicable over a wide pH range, however the highest labeling efficiency was observed at pH 7–10.536 A versatile class of stable PTAD precursors, possessing different functional groups, was developed and applied for a selective tyrosine conjugation (Fig. 59b). Their utilisation implies prior to use oxidation with 1,3-dibromo-5,5-dimethylhydantoin and the addition of a small amount of TRIS (2-amino-2-hydroxymethyl-propane-1,3-diol) during the step of conjugation. The latter is of crucial importance for the coupling selectivity, for it is hypothesised to serve as a scavenger of a putative isocyanate by-product of the PTAD decomposition, which is promiscuous in labelling.
The non-selective labelling of other aromatic side chains of proteins is the Achilles' heel of the vast majority of approaches described for tyrosine labelling. Careful tuning of reaction conditions is important for achieving appropriate levels of selectivity. In some cases where purely chemical distinction of reactivity of amino acid moieties is not feasible, catalysis on the basis of molecular shape rather than local environment can be used to induce selectivity. This concept is routinely exploited by enzymes and allows enabling reactivity that would otherwise be kinetically impossible. In 2010, Popp and Ball used dirhodium metallopeptide catalysts for selective conjugation on tyrosine and tryptophan using the concept of the proximity-driven mechanism (see Section 5).454 In the following year, Silverman et al. have demonstrated a DNA-catalysed approach for selective labelling of tyrosine, although only on small peptide substrates.538
1.7 Arginine
With a pKa value above 12, arginine (Arg, R) is mainly presented in its protonated form in acidic, neutral, and even most basic environments. Effective delocalisation of a positive charge between nitrogen lone pairs and the double bond favours the formation of hydrogen bonds539,540 and makes the guanidinium side chain of arginine the least acidic cationic group among all 20 natural amino acids (Fig. 60).541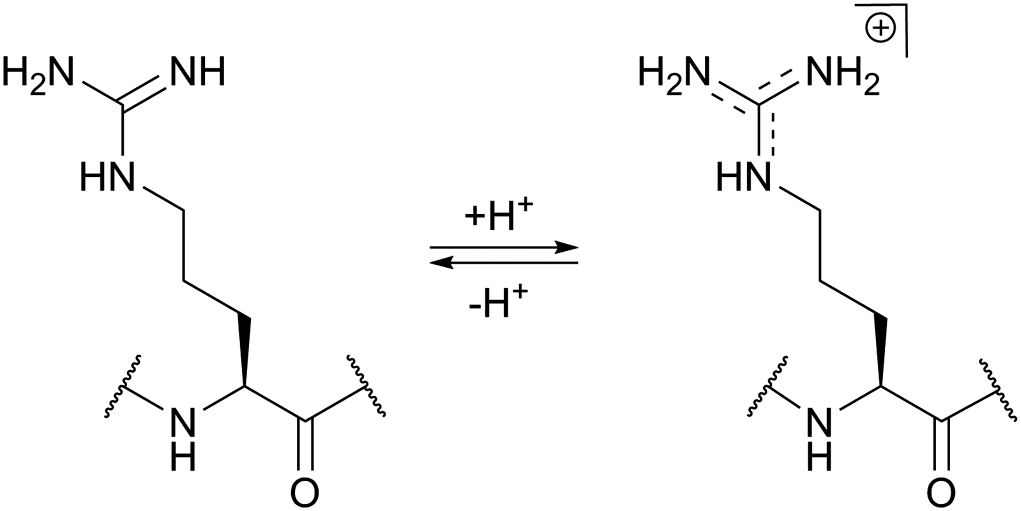 | ||
| Fig. 60 Protonation of the arginine side chain at neutral pH. | ||
However, the pKa value of arginine was found to vary significantly in the microenvironments within certain proteins,542,543 allowing, in terms of Leitner and Lindner,544 the grouping of arginines in “exposed” or “partially buried” residues, basing on the difference of their reactivities.
Most of the described approaches for arginine labelling and modification exploit the chemistry of α-dicarbonyl compounds. For instance, introduced by Takahashi545 as an arginyl reagent, phenylglyoxal has since been applied for the study of complex systems in the past decade.546–550 The reaction occurs under mild conditions and consists of two steps: first addition of phenylglyoxal resulting in the formation of hydrolytically instable imidazolidine diol, and the second step results in a relatively stable addition product (Fig. 61).
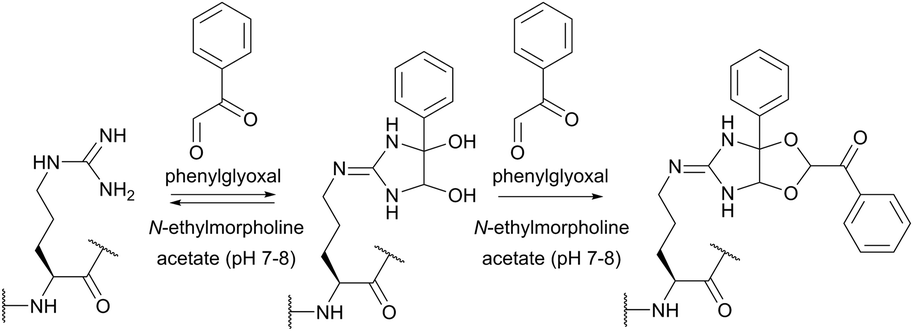 | ||
| Fig. 61 Reaction of phenylglyoxal with the arginine moiety (reaction condition: 0.2 M N-ethylmorpholine acetate buffer, pH 7–8). | ||
Substituted phenylglioxal analogs, such as p-hydroxyphenylglyoxal, p-nitrophenylglyoxal and 4-hydroxy-3-nitrophenylglyoxal, p-azidophenylglyoxal (APG) were reported for spectrophotometric and cross-linking study of the modification of arginine in proteins.551–555 None of these linkers have however been used in bond-forming conjugation. Because phenylglyoxal, like glyoxal, reacts with ε-amino groups at a significant rate,545 many efforts were made to increase its selectivity towards guanidinium residue. Cheung and Fonda have studied the effect of buffers and pH on the reaction rate556 and found that the reaction of arginine is greatly accelerated in bicarbonate–carbonate buffer systems, possibly due to the stabilisation of the obtained diol.
Geminal diones – namely 2,3-butanedione (introduced by Yankeelov)557,558 and 1,2-cyclohexanedione (introduced by Itano)559 – are another well-characterised reagents for the modification of Arg residues. The reaction progresses through the pathway that is similar to the phenylglyoxal addition. However, it was not until the observation that borate had a significant effect on the selectivity of the reaction that the use of this reagents became practical.560,561 The presence of borate in the solution allows shifting of the equilibrium during the addition to a guanidine moiety through the stabilisation of reversibly obtained diol (Fig. 62).
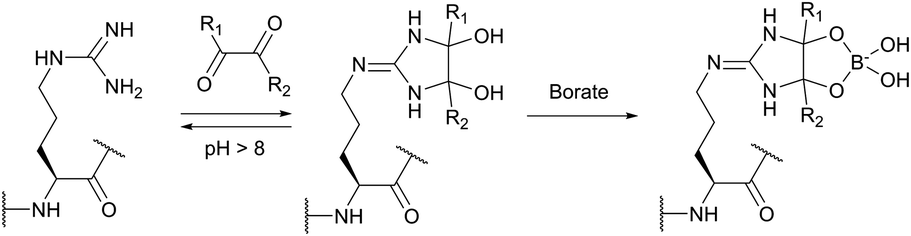 | ||
| Fig. 62 Reaction of 2,3-butanedione with arginine residues of Carboxypeptidaze A (pdb: 1HDU) in the presence of borate described by Riordan.560 | ||
In 2005, using this approach, Lindner and colleagues have described a method for the selection of arginine-containing peptides from a tryptic digest of the model proteins (BSA, lysozyme, ovalbumin) by a solid phase capture and release.562 First, arginine containing peptides presented in the digest were covalently modified on the guanidine moiety with 2,3-butanedione and phenylboronic acid under alkaline conditions. Polymeric materials allowing the immobilisation of phenylboronic acid were then used to capture the arginine-peptides on a solid support while washing away all not covalently bonded arginine-free peptides. Finally, the arginine-peptides were cleaved again from the boronic acid beads due to the reversibility of the reaction. Photoactivable bifunctional reagents for cross-linking of arginine moieties have been elaborated by Ngo et al. and Politz et al. to study enzymes with an arginine at their active sites.555,563
Arginine-specific PEGylation of lysozyme using polyethylene glycols containing an α-oxo-aldehyde motif in borate buffer was recently reported by Gauthier and Klok564 and represents mild and selective method for protein modification (Fig. 63). Other methods described to date565,566 possess selectivities, which are not sufficient (especially in the presence of Lys moieties) to consider them as suitable for bioconjugation.
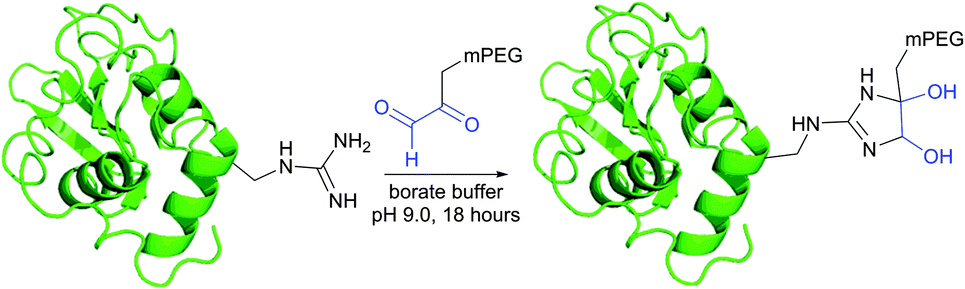 | ||
| Fig. 63 Selective PEGylation of lysozyme's arginine side chains (pdb: 2LYZ) described by Gauthier and Klok.564 | ||
1.8 Aspartic and glutamic acids
Carboxylic acid groups can be found in protein structure either on its C-terminus, or as side chains of Asp and Glu. Due to the low reactivity of carboxylate in water, it is usually difficult to selectively conjugate proteins at these moieties. Carboxylic acid should thus generally be converted to a more reactive ester by means of so called activating reagents.For more than a half century, carbodiimide-mediated activation is the most extensively used methodology for the modification of free-acids in protein.567,568 The reaction of carbodiimides with protonated carboxyl groups yields activated acylisoureas, which then react smoothly with a variety of nucleophiles, namely amines (Fig. 64).569 It is important to utilise weakly basic amines, that remain deprotonated and thus reactive at pH below 8.0, to avoid protein cross-linking occurring at higher pH values. For this reason, weakly basic hydrazides are often reagents of choice in coupling reactions with activated carboxylic acids.570 Although water-insoluble carbodiimides (DCC, DIC) still continue to be useful for acid-selective protein conjugation,571,572 most current reports exploit water-soluble carbodiimides such as 1-ethyl-3-(3-dimethyl-aminopropyl)carbodiimide (EDC). Developed by Sheehan and Hlavka573,574 these carbodiimides first proved their especial usefulness as zero-length cross-linking reagents to study proteins.574 Subsequent studies were devoted for the application of carbodiimides for quantitation of accessible carboxyl groups in proteins,567,568,575 preparation of antigenic conjugates,576 and protein immobilisation.577
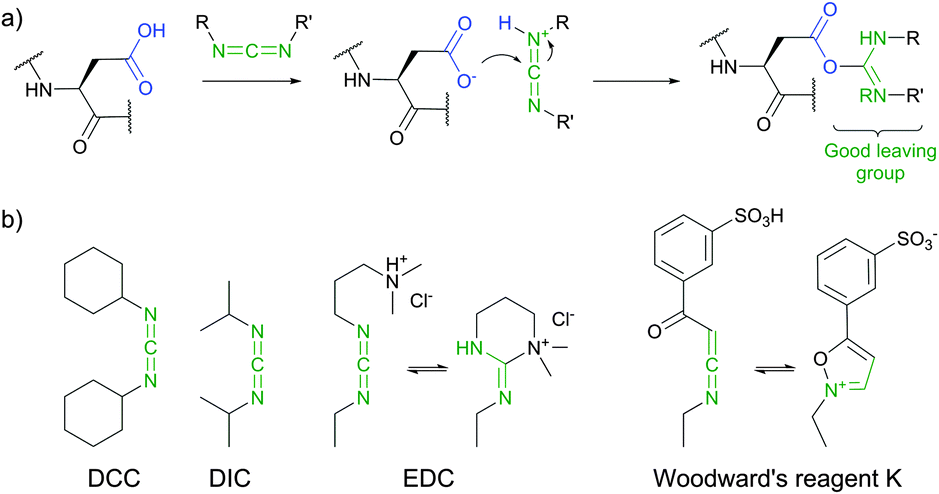 | ||
| Fig. 64 (a) Carbodiimide-mediated activation of the carboxylic acid side chain of glutamate. (b) Relevant examples of activating reagents. | ||
As mentioned previously, the upper limit for the optimal pH of carboxylate conjugation is defined by the reactivity of free amino groups present in protein. The lower limit is mainly determined by aqueous stability of the activating reagent. Borders and co-workers578 studied the stability of EDC in aqueous solution. It was found that EDC has a T12 of 37 hours (pH 7.0), 20 hours (pH 6.0), and 3.9 hours (pH 5.0) in 50 mM MES buffer at 25 °C; in the presence of 100 mM glycine, the T12 values were 15.8 hours, 6.7 hours, and 0.73 hours respectively. This supports the optimal pH for acid-selective conjugation to be in a range from 6.0 to 7.0. NHS (or its water-soluble analogue sulfo-NHS) is often included in coupling protocols to improve efficiency or to create a more stable intermediate. Possible side-reactions involving activating reagents were recently reviewed by Valeur and Bradley.579
Woodward's reagent K (N-ethyl-5-phenylisoxazolium-3′-sulfonate)580 and analogous substrates were used as activating reagents of carboxyl groups for synthetic purposes. Bodlaender et al.581 used N-ethyl-5-phenylisoxazolium-3-sulfonate, the N-alkyl derivatives of 5-phenylisoxazolium fluoroborate, to activate carboxyl groups on trypsin for subsequent modification with methylamine or ethylamine.
Lastly, several studies revealed unexpected examples of carboxyl group modification with reagents usually reacting far more effectively with other nucleophiles. For instance, p-bromophenacyls and iodoacetamides have been found to selectively alkylate carboxylic acid moieties of pepsin and ribonuclease T1 respectively.582–584 However, the applicability of these reagents is not general and is appropriate on specific substrates only.
1.9 Methionine
In spite being often considered a rather simple target for chemical modification (mainly through the oxidation and the reaction with α-halo acetic acids and their derivatives),585,586 only a handful of conjugation methodologies involving methionine (Met, M) were described up to the present.All approaches described in literature exploit alkylation of Met residues in acidic media. Although many nucleophilic functional groups present in proteins can react with alkylating reagents, at low pH all of them except methionine exist in protonated forms, which greatly decreases their reactivity.587 Consequently, alkylations of other nucleophilic functional groups, such as thiols, are commonly conducted at high pH,400 while methionine is the only functional group in proteins able to react with alkylating reagents at low pH.
Basing their research on pioneering studies being done by Toennies in the 1940s,589,590 Kramer and Deming have recently reported a reversible chemoselective labelling of methionine in peptides and polypeptides.588 Treatment of the model peptide (PHCRKM) with alkylating reagents of different structures in 0.2 M aqueous formic acid (pH 2.4) gave a single product, where only the Met residue was alkylated. This resulting sulfonium salts can readily be dealkylated by addition of pyridine-2(1H)-thione (PyS) to give the starting peptide as the sole product along with the alkylated PyS byproduct. The removal reaction was found to be selective and allows selective dealkylating using concentrations of PyS that do not react with the disulfide bond in cystine under identical conditions (Fig. 65).
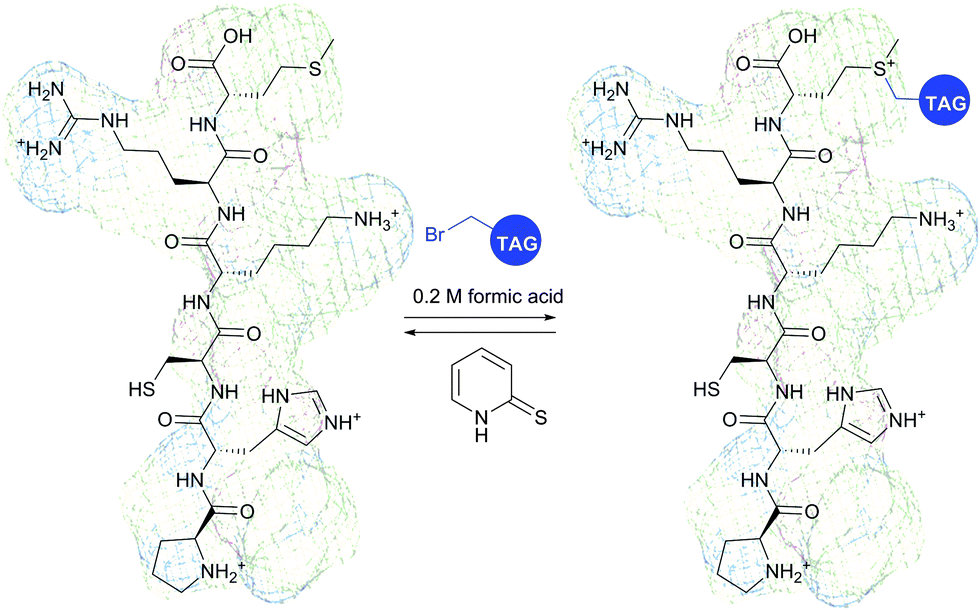 | ||
| Fig. 65 Selective reversible modification of a 6-amino-acid peptide (PHCRKM) via methionine alkylation reported by Kramer and Deming.588 No reactions with other amino acids were detected. | ||
2. N-terminal conjugation
2.1 α-Amino groups
The most important thing to note about N-terminal amino groups is that these are the only primary amines present in the protein structure that possess an adjacent amide bond in α-position, which slightly influences their reactivity. Consequently, the majority of classical methodologies exploit this peculiarity of the N-terminus.In the classical work on acetylation of the growth hormone, Reid592 has for the first time demonstrated the possibility of selective modification of N-termini, if acetylation is performed with a relatively small amount of acetic anhydride. Further development of this approach has in several protocols for selective labelling of α-amino groups of proteins,3,593 peptides,594,595 and proteomes.596–598 Under optimal reaction conditions, the use of a 5-fold excess of amine-reactive reagent in PBS (pH 6.5) at 4 °C, high levels of selectivity can be achieve after 2–24 hours of reaction. However, the preference for terminal amino groups achieved by control of pH is rather limited, mainly owing to the fickleness of the pKa of the amino group depending on the microenvironment and reaction conditions. Consequently, more proficient methods that rely upon increased chelating ability of N-termini,599 direct participation of the adjacent side chains or peptide bond,600,601 were developed and represent to date preferential approaches for bioconjugation.
 | ||
| Fig. 66 Selective labelling of lysozyme (pdb: 2LYZ) described by Che.603 Moderate-to-high level of selectivity towards N-terminal residue (K1) was achieved in the presence of 5 other in-chain lysines (LYS-13, LYS-33, LYS-96, LYS-97 and LYS-116). The ketene was synthesised from the corresponding acid by a two-step protocol: transformation into a mixed anhydride (oxalyl chloride, DCM) followed by the transformation thereof into ketene upon the reaction with a base (TEA, THF). | ||
Inspired by the pioneering works of Metzler and Snell,607 and Cennamo and collaborators608,609 on the transamination of simple amino acids and peptides under harsh conditions (heating at 100 °C and pH 5.0), Dixon and Moret610,611 developed a method for mild transamination in the presence of copper(II) salts, which allowed the reaction to pass at room temperature (Fig. 67). The isomerisation of the imine generated in situ by catalysed 1,3-proton shift transfer is the key step of the transformation which defines both its direction and the reaction rate. It is clearly the activation of α-protons of N-terminus by the adjacent peptide bond and the metal ions that makes this reaction specific to α-amino groups. Interestingly, Dixon reports that no traces of the reaction of lysine side chains were ever observed.600
 | ||
| Fig. 67 General scheme of the transamination reaction activated by metal ions (typically Cu2+ and Ni2+).610 Principal steps of the reaction mechanism: (a) generation of the imine; (b) isomerisation of the obtained imine by proton removal; (c) hydrolysis of isomeric imine to generate transaminated reaction partners. | ||
Quite a wide range of reaction conditions has been tested ever since. The discovery that pyridine612 and acetate613 greatly accelerated the transamination of amino acids led to a slightly milder reaction conditions, which however were still too harsh to maintain the folded structure of most proteins, and were therefore more appropriate for sequence-analysis applications.614
Only recently Francis and co-workers601 have re-examined Cennamo's approach608 of amino acid transamination in the presence of pyridoxal-5-phosphate (PLP, vitamin B6) at 100 °C and found much milder the reaction conditions when modifying the N-terminal residues of peptides (65 °C for reaction to be over in 2 hours; 25 °C to achieve a complete conversion in 24 hours). Screening experiments on different N-terminal amino acids of the peptides indicated that the aldehyde structure strongly influenced the reaction efficiency. Amusingly, known for more than 50 years PLP emerged as the most effective aldehyde among dozens being screened, affording the highest yields at milder reaction conditions.
The mechanism of PLP-catalysed transamination is depicted in Fig. 68. The reaction of PLP and N-terminal amine results in forming of a Schiff base aldimine (a). Then the α-proton the amino acid transferred to the 4′ position of the pyridoxal unit (b and c). Finally, hydrolysis of the obtained ketimine leads to the desired α-ketoacid and pyridoxamine phosphate (d). Quinoid is an important intermediate for the transformation of aldimine to ketimine and can be found in all transamination reagents described to date.
 | ||
| Fig. 68 Structure of PLP and mechanism of PLP-mediated transamination. Reaction pathway consists of (a) condensation reaction between pyridoxal and the amine; (b and c) tautomerisation of the obtained aldimine being favourable because of a much lower intrinsic pKa values of α-proton (shown in blue); (d) hydrolysis of the resulting ketimine, accompanied by decarboxylation in the case of aspartic acid (R = –CH2COOH).601 | ||
Under optimal reaction conditions: 10–50 μM protein and 10–50 mM PLP at 37 °C in PB (pH 6–7), a complete conversion is generally achieved after 2–24 hours. The resulting keto-proteins are generally rather stable and can be concentrated, stored, or lyophylised without any specific precautions.615 An example of transamination-conjugation methodology was demonstrated by Francis in the initial publication on selective labelling of an N-terminal glycine residue of horse heart myoglobin and enhanced green fluorescent protein (EGFP, Fig. 69).601
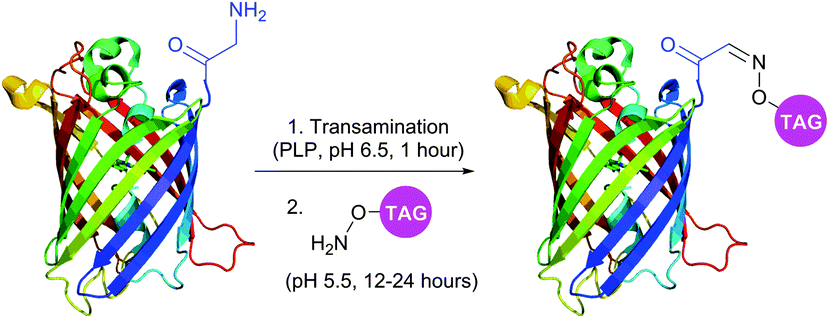 | ||
| Fig. 69 Site-specific N-terminal labelling of EGFP (pdb: 2Y0G). Proteins possessing N-terminal carbonyl groups obtained by in the first step PLP-mediated transamination were labelled with hydroxylamine probes in the second step.601 | ||
Although the side chain of the N-terminus does not participate directly in the transamination mechanism, the reaction rates were found to vary significantly depending on the amino acid in the N-terminal position.616 Generally, the majority N-terminal amino acids provide high yields of the desired transaminated products; however, some residues (His, Trp, Lys, and Pro) generate adducts with PLP itself, while other are incompatible with the technique because of known side reactions (Ser, Thr, Cys and Trp) or complete inertness (Pro).
In the attempts for the investigation of transamination reaction scope and limitations, Francis and collaborators have prepared an 8000-member one-bead-one-sequence combinatorial peptide library in which the three N-terminal residues were varied.617 Interestingly, the Ala-Lys (AK) motif was found to favour especially the transamination yields (the Lys residue is hypothesised to accelerate the isomerisation step of 1,3-proton shift acting as a general base). To demonstrate this, labelling of the Type III “Antifreeze” Protein and its mutant presenting the AKT sequence on the N-terminus were side-by-side benchmarked. At every time point analysed, the AKT terminus outperformed the wild-type one (GNQ) at different concentrations of PLP.
Although mild reaction conditions of PLP-mediated transamination render it amenable for the modification of intact proteins,618 the yields are generally not high and elevated temperatures are usually required, which largely limits the practical applicability of the approach. Given this situation, Francis and co-workers619 have utilised above-described combinatorial approach to identify another transaminative agents. As a result, N-methylpyridinium-4-carboxaldehyde benzenesulfonate salt (Rapoport's salt, RS, Fig. 70) was identified as a highly effective alternative to PLP. Furthermore, this was found to be particularly efficient for glutamate-rich sequences.619,620 The fact that several antibody isotypes dispose at least one glutamate-terminal chain makes RS particularly amenable for their selective conjugation. Remarkably, the difference of transamination reaction rates on Glu- and non-Glu polypeptides was significant enough for selective labelling of only the heavy chain of immunoglobulin G1 (containing the N-terminal Glu residue), while leaving unmodified the light chain. This was assigned to be due to the higher steric hindrance of the already less-reactive substrate (N-terminal Asp). To ensure these results an IgG1 mutant possessing Asp–Asp–Ser sequence on both chains was prepared. Indeed, this underwent the modification of both sites when exposed to RS. Facing another recognised drawback of Francis methodology, namely low efficiency for bulky amino acid termini (Leu, Ile, Val), Zhang et al.621 have elaborated an efficient PLP analogue, FHMDP (Fig. 70), that demonstrated much higher efficiency in transamination thereof.
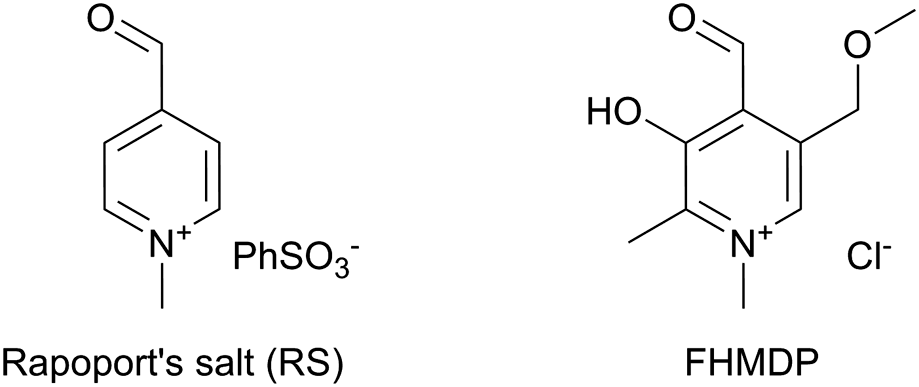
The above-mentioned transformations provide just a few examples of the rapidly growing field of transaminative modification of proteins. Recent advances have also resulted in elaboration of general approaches for protein immobilisation (Fig. 71),15,622 dual fluorescent modification of periplasmic solute binding proteins,623 protein PEGylation and PEG-like conjugation (e.g. OEGMAtion),624 preparation of phage conjugates,625,626 N-terminus proteomics,627,628 enabling Wittig629 and Pictet–Spengler ligation on transaminated proteins.630,631
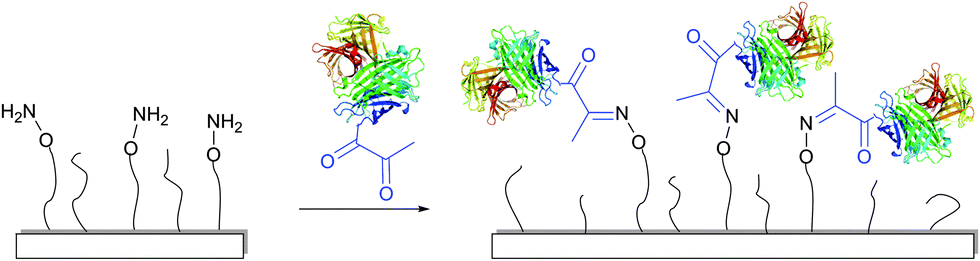
Rather than demonstrating any ability to transaminate, 2PCA exclusively showed conversion to a pair of cyclic imidazolidinone diastereomers upon reaction with model peptides. The key step of the reaction mechanism is a nucleophilic attack of the adjacent amide nitrogen on the electrophilic carbon of the initially formed N-terminal imine (Fig. 72a). The presence of nitrogen heterocycles (namely pyridines) was found to be crucial for efficiency of this stereoelectronically disfavored condensation. It is noteworthy that Lys residues are unreactive in such a pathway, because of the lack of a neighbouring amide group suitable for cyclisation and higher pKa values compared to α-amino groups. Furthermore, this methodology was also found to be compatible with the presence of free in-chain cysteines. This approach can therefore be considered as an orthogonal to other Lys-selective (see Section 1.1) and Cys-selective methodologies (see Section 1.3).
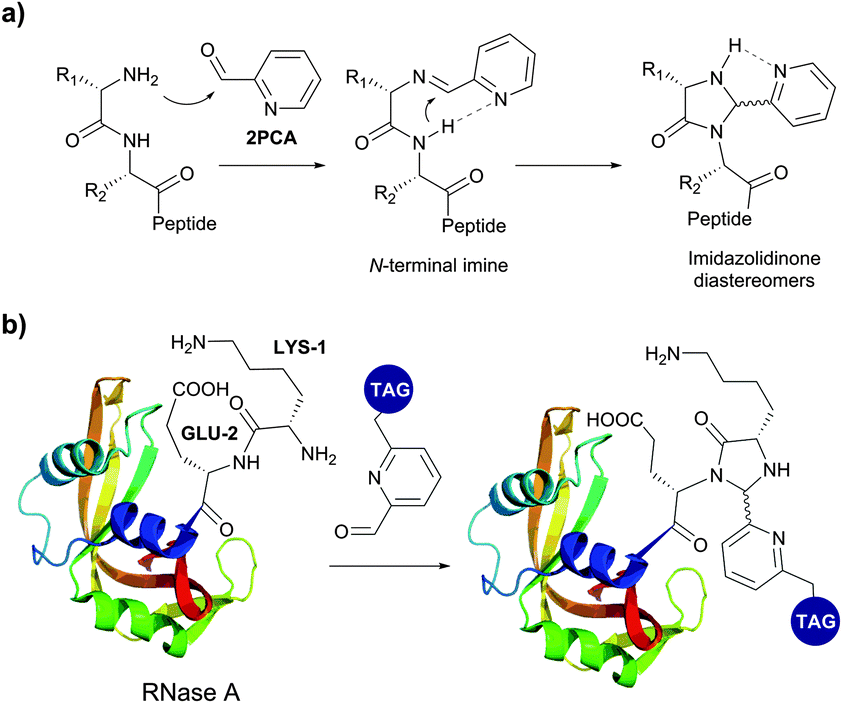 | ||
| Fig. 72 N-terminal protein modification using 2-pyridinecarboxyaldehydes (2PCT) reported by Francis et al.632 (a) Mechanism of the reaction of between N-terminal amino group and 2PCA. (b) Modification of native RNase A (pdb: 7RSA). | ||
2PCA-mediated conjugation was found to be generally applicable for protein labelling (the authors demonstrated its application on a broad set of 12 different proteins including RNase A; Fig. 72b) except for N-acylated proteins (no imine formation) and peptides containing proline in position 2 (no cyclisation of the formed N-terminal imine).632 The resulting imidazolidinone-containing conjugates are generally moderately stable and decompose to starting protein by about 20–30% after 12 hours of incubation at 37 °C (in case of RNase A). This may limit the suitability of this methodology for several applications where stability of generated conjugates is crucial; however, the substrate variation could resolve the issue and such efforts are underway.
2.2 Serine and threonine
In 1960 Waller and Dixon have described the first procedure for selective N-terminal modification of peptides. It consisted of preparation of corticotrophin selectively acetylated on its terminal serine.633 Although being possible only under highly denaturing conditions of alkali exposure, the approach allowed for a spontaneous intramolecular O → N acyl transfer634 on the N-terminal Ser residues, while O-acetyl groups of in-chain Ser, Thr and Tyr residues were hydrolysed. The general idea of overcoming the entropy barrier of an otherwise slow intermolecular process by bringing two reacting partners together through a covalent linkage to initialize an intramolecular reaction has been later transformed into a large variety of selective modification reactions.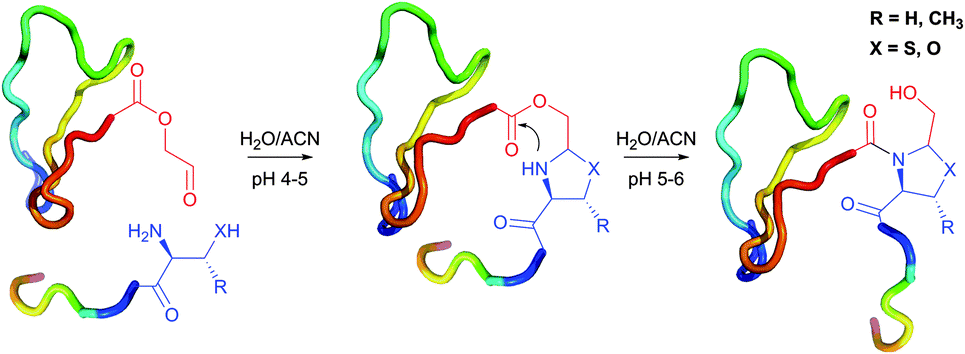 | ||
| Fig. 73 General scheme of the ligation strategy proposed by Liu and Tam.635 A model 50-residue peptide was obtained in good yield in ligation reaction between a 32-mer peptide VVSHFNDCPDSHTQFEFHGTCRFLVQEDKPAR containing C-terminal aldehyde function and a 17-mer peptide CHSGYVGARC(Ac-m)EHADLLA containing N-terminal cysteine; in the case of Thr- and Ser-peptides, the reaction was moderately efficient. The peptides structures were simulated using the RaptorX web server.447 | ||
Although this methodology has demonstrated its high potential for the ligation of unprotected peptides,635,638,639 the generation of a “non-native” heterocyclic fragment at the site of ligating the two peptides made it extruded almost completely by other “native ligation” approaches; notably, by a similar mechanistically native chemical ligation (NCL) (see Section 2.3.1).
Based on the periodic acid mediated oxidation,642 the reaction occurs only when there exists the target site for the periodate to form a cyclic intermediate, that is to say, when the N-terminal residue is represented by a serine or a threonine, or when hydroxylysine is present (rarely occurring in proteins). Possible side reactions include oxidation of the side chains of Met, Trp, and His. However, the potential for side reactions can be diminished by using very low periodate-to-protein molar ratios, as demonstrated by Geoghegan and Stroh in their experiments on two model peptides, SIGSLAK and SYSMEHFRWG, and with recombinant murine interleukin-1α (an 18 kDa cytokine with N-terminal Ser, Fig. 74),641 or by conducting the oxidation at neutral pH.643
 | ||
| Fig. 74 N-terminal serine labelling of recombinant murine interleukin-1α (pdb: 2KKI) with Lucifer yellow dye described by Geoghegan and Stroh.641 The method is also applicable if N-terminal threonine is present. | ||
As in the initial publication of Geoghegan and Stroh,641 obtained glyoxyloyl can serve as the locus for further chemical modification involving aldehyde-selective reactions (e.g. through the formation of stable oxime, hydrazone or previously described oxazolidine moieties).605,606 Robin and colleagues have demonstrated the possibility of using this two-step methodology for assembling two unprotected protein fragments: oxidised to glyoxyloyl-containing and hydrazide peptide derivatives.644 Rose and co-workers645 exploited the reactivity of generated glyoxyloyls towards O-alkyl hydroxylamine derivatives to synthesize a pentameric form of the cholera toxin subunit B.
Further investigation of periodate oxidation allowed its promoting for site-selective tagging, PEGylation, preparation of protein conjugates, protein capture and synthesis of large protein dendrimers.643,646–651 It is worth mentioning that periodate oxidation is incompatible with a number of protein classes. For instance, glycoproteins will undergo periodate-based cleavage of polysaccharide chains as a side reaction pathway.652
Lastly, glyoxyloyles can easily be transformed into corresponding amines via transamination reaction in the presence of copper(II) or nickel(II) salts.604,610,653 The reaction mechanism as well as need for both essential components of the system: the acceptor of the glyoxyloyl (usually aspartic acid or glycine) and the cation of a heavy metal are explained in Fig. 75. Despite being of moderate interest for bioconjugation by itself, this approach has initiated the development of a more general methodology for selective N-terminus modification – transaminative conjugation (Section 2.1.3). The reader is directed to a recent review by El Mahdi and Melnyk654 for a complete overview of the glyoxyloyl transformations in bioconjugation.
 | ||
| Fig. 75 Preparation of S1G mutant of corticotropin-releasing hormone (GRH) by regioselective transformation of N-terminal serine to glycine.604 Reaction steps: (a) selective serine oxidation by periodate; (b) formation of imine with amino group of glutamic acid precomplexated with Cu2+ (Cu-Glu); (c) base-assisted isomerisation of imine; (d) hydrolysis of imine, completed when aspartate and copper ions are removed. | ||
The inherent reactivity of an N-terminal phosphorylated Ser or Thr residues was demonstrated to significantly facilitate the amide bond formation with a range of C-terminal peptide thioesters. Although it is not yet clear what exact intermediate is formed during ligation, the authors have hypothesised that rapid acyl migration to the N-terminal amine of a peptide occurs through the formation of unstable acyl phosphate (Fig. 76).
 | ||
| Fig. 76 Hypothesised mechanism of the phosphate-assisted ligation reported by Thomas et al.655 | ||
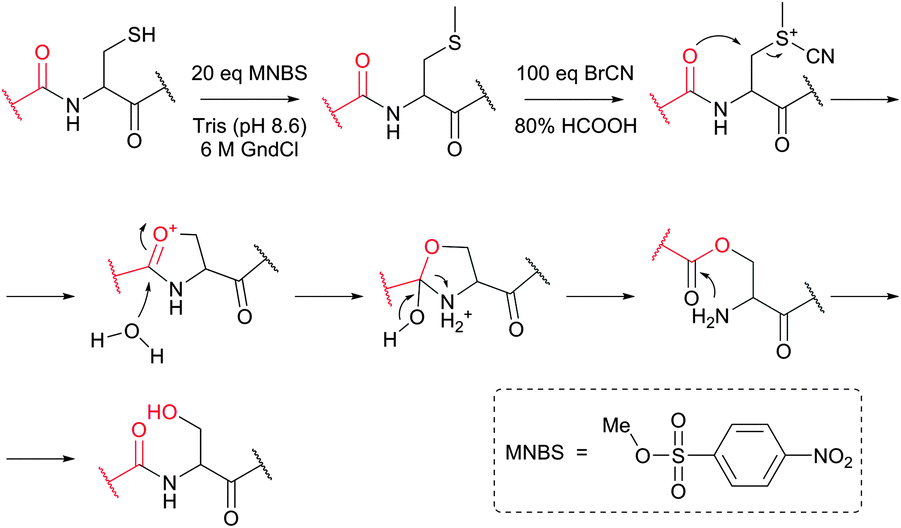
2.3 Cysteine
Generation of N-terminal Cys residue for native chemical ligation can be accomplished using solid-phase peptide synthesis,659 proteolytic processing,660 or by the spontaneous hydrolysis of intein fusion protein.661 Moreover, genetically directed, site-specific incorporation of 1,2-aminothiols handle into proteins has been recently reported by Chin and associates.662In 1994, confronted with this criticism, Kent and co-workers664 introduced a versatile approach to the linkage of peptide fragments using a native peptide bond – native chemical ligation (NCL). Based on the original principles of the chemical ligation methodology663 and the ability of thioesters to undergo S → N acyl shift discovered by Wieland et al.,665 NCL allowed to achieve chemoselective formation of the amide bond in the presence of unprotected nucleophilic amino acid side chains as alcohols (Ser/Thr), phenolates (Tyr), free amines (Lys), carboxylates (Glu/Asp), or other thiolates (Cys) presented in the macromolecule.
By analogy with previously developed O → N acyl shift, the reversibility of the thioester-thiol exchange in the presence of an exogenous thiol additive coupled with the capture of the acyl segment by S → N acyl shift, being possible only in the case when the latter is brought in the close proximity to an amine in a N-terminal cysteine thioester intermediate, result in an exquisite regioselectivity of this methodology. The product resulting from this S → N acyl shift represents a peptide, consisting of two fragments linked by a native peptide bond through a cysteine residue (Fig. 78).
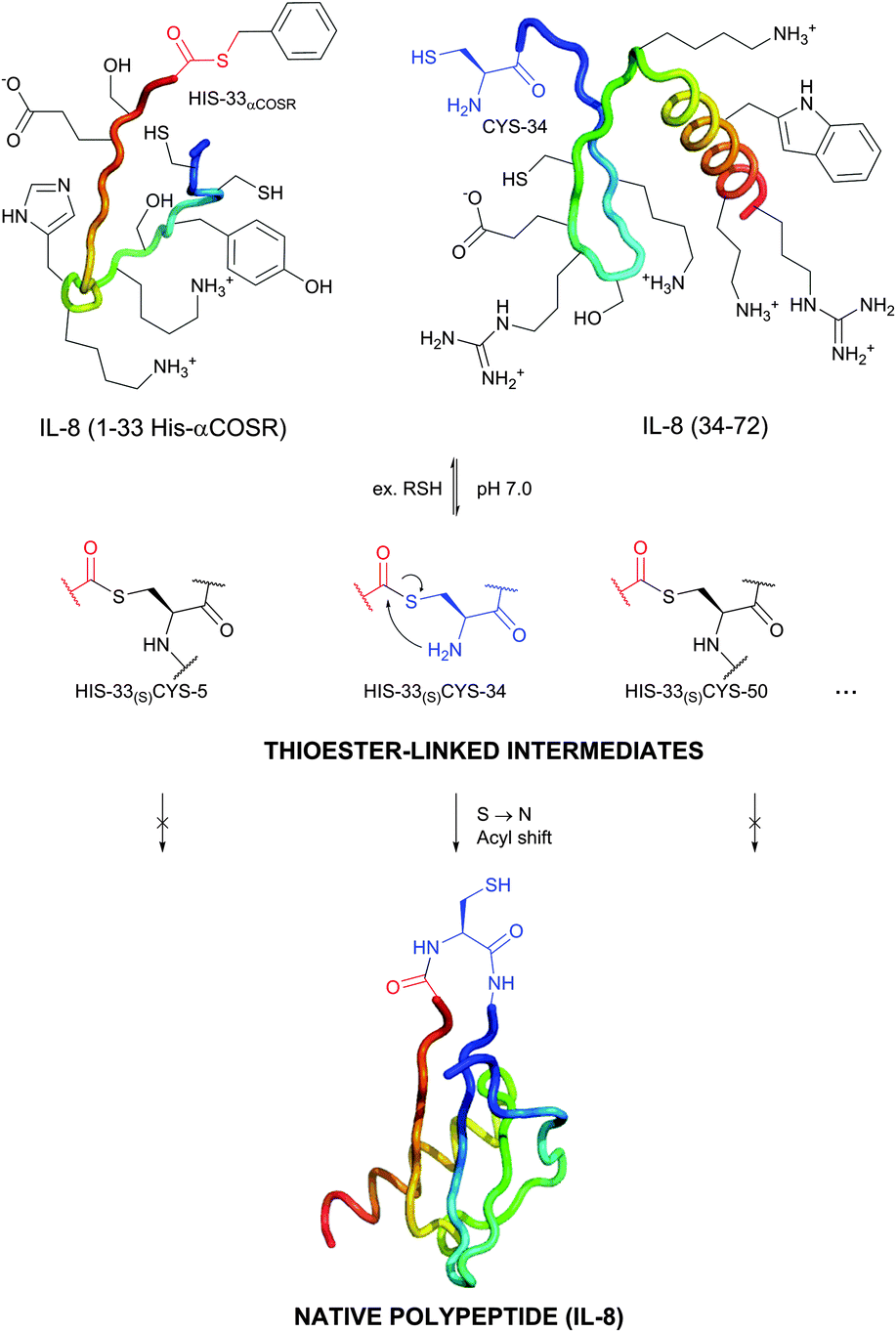 | ||
| Fig. 78 Synthesis of the human interleukin 8 by native chemical ligation elaborated by Kent et al.664 The ligation reaction occurs between an unprotected peptide thioester fragment – IL-8(1–33 His-αCOSR), and a second unprotected peptide possessing a N-terminal cysteine – IL-8(34–72). First step of thioester exchange results in different thioester-linked intermediates, among which only the peptide obtained from the corresponding N-terminal cysteine can undergo following irreversible step of an S–N acyl shift resulting in obtaining of a native amide bond at the linkage site. | ||
Typically, the reaction performed in PS or PBS buffer (pH 7.0–8.5) at 37 °C is complete in less than an hour and with high yields.666,667 Solubilizing agents such as guanidine hydrochloride or urea do not interfere with the ligation and are usually used to enhance the concentration of peptide segments, and thus increase reaction rate. It is important to prevent the thiolate of N-terminal cysteine from the oxidation resulting in a disulfide-linked dimer, which is unreactive in the ligation. A reductant (e.g. TCEP) or an excess of thiol corresponding to the thioester leaving group (4–5%, vol/vol) is generally added to keep the Cys residues in reduced form. Moreover, the latter largely increases the overall rate of NCL by reversing the first step of transesterification for in-chain intermediate adducts deprived from the possibility to undergo S → N acyl shift and to generate a stable amide bond.
The first step in synthesizing a protein by NCL generally consists in defining the fragments to be used in the ligation reactions. Preferentially, naturally occurring AA-Cys motifs in the native sequence should be chosen as the ligation sites (AA stands for any amino acid). Val, Ile, Asp, Asn, Glu, Gln and Pro represent less favourable choices, because of lower ligation rates and possible side reactions,666 which, however, can be accelerated either by transformation of the corresponding thioesters into selenoesters,668 or by tuning the reaction pH.669
Higher reaction rates were reported to be achievable while using good thiol-containing living groups, i.e. mildly acidic thiols such as thiophenol, 4-(carboxymethyl)thiophenol (MPAA), or 5-thio-2-nitrobenzoic acid (TNB, the reduced form of Elman's reagent). These are generally generated in situ by thiol–thioester exchange from the relatively unreactive peptide- (αCOSCH2CH2CO)-Leu alkylthioester by adding an excess thereof (1–5%, vol/vol).666
2.3.1.1 Sequential NCL. The complexity of proteins that can be obtained by NCL technique is limited by the maximum size of the accessible synthetic peptide segments. Two main approaches used today for the preparation thereof are: solid phase peptide synthesis pioneered by Merrifield (allowing the synthesis of proteins containing up to 50 residues),659 and recombinant DNA expression elaborated by Lobban et al.670 and Jackson et al.671 These allow for the preparation of peptide fragments containing up to 50, and up to 150 amino acid residues respectively.
Sequential native chemical ligation allows further extending of this limit by means of N-terminal cysteine protected peptides. Three polypeptide fragments: a peptide1-COSRAr (N-terminal fragment), a protected PG-Cys-peptide2-COSRAr (middle fragment), and a Cys-peptide3 (C-terminal fragment), are thus assembled in a one-pot three step synthesis. Firstly, the middle fragment and the C-terminal fragment are ligated under the classical reaction conditions of NCL. Then protecting group is removed, uncovering N-terminal cysteine of the obtained polypeptide (central plus C-terminal fragment), which undergoes the second NCL with the N-terminal fragment to give the target protein (Fig. 79).
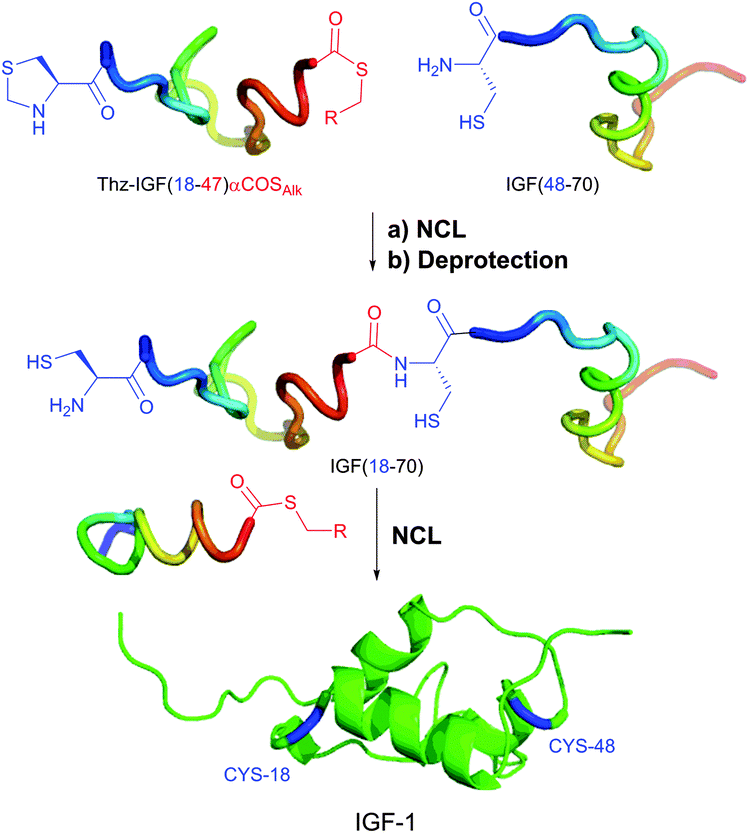 | ||
| Fig. 79 Synthesis of insulin-like growth factor 1 (IGF-1) via sequential native chemical ligation described by Sohma et al.672 The reversible protection of the α-amino group of the central peptide fragment IGF(18–47) prevents its self-reaction with the α-thioester moiety present in the same molecule. Thiazolidine protecting group can be easily removed by brief treatment with NH2OMe·HCl at pH 4. NCL reaction condition used: PB (pH 6.7), 6M GndCl, 10 mM MPPA, 20 mM TCEP. | ||
Since its introduction, sequential native chemical ligation has demonstrated its general applicability to the preparation of various complex assemblies. Consequently, several methodologies compatible with NCL for the protection of N-terminal Cys residues were elaborated. The most relevant among them are depicted in the Table 1.
| Protecting group | Deprotection | Examples | ||
|---|---|---|---|---|
| a Require a preparative-HPLC step before removal, gives lower overall yield compared with Thz.683 | ||||
| 1 | Thz |
|
NH2OMe pH 4 | IGF,672 HIV-1 Tat,674 crambin,675,676 EPO,677 PYP678 |
| 2 | Msc679 |
|
pH 12 | SOD,680 Abl-SH3681 |
| 3a | Acm682 |
|
AgOAc, DTT | Crambin,683 DAGK684 |
| 4 | Mapoc685 |
|
hν > 300 nm | hBNP-32685 |
The reader is referred to a recent review by Melnyk and collaborators673 for a more complete overview of the sequential ligation strategies on proteins.
2.3.1.2 Kinetically controlled ligation. The simple observation that the reaction rate of NCL largely depends on the thioester nature was exploited by Bang et al.675 to introduce a convergent strategy for the synthesis of native peptides – kinetically controlled ligation (KCL).
The fact that the kinetics of NCL with alkylthioesters is significantly inferior of those with arylthioester makes it possible to control the intrinsic dual reactivity of a bifunctional Cys-peptide2-COSRAlk so that it would selectively react with a peptide1-COSRAr and then undergo a classical NCL (i.e. in the presence of exogenous aryl thiol) with a third Cys-peptide3 to yield an assembled peptide1-Cys-peptide2-Cys-peptide3 with no need to use protecting groups. Bang and collaborators have applied this methodology to assemble a 46-residue protein crambin from six peptide fragments (Fig. 80).
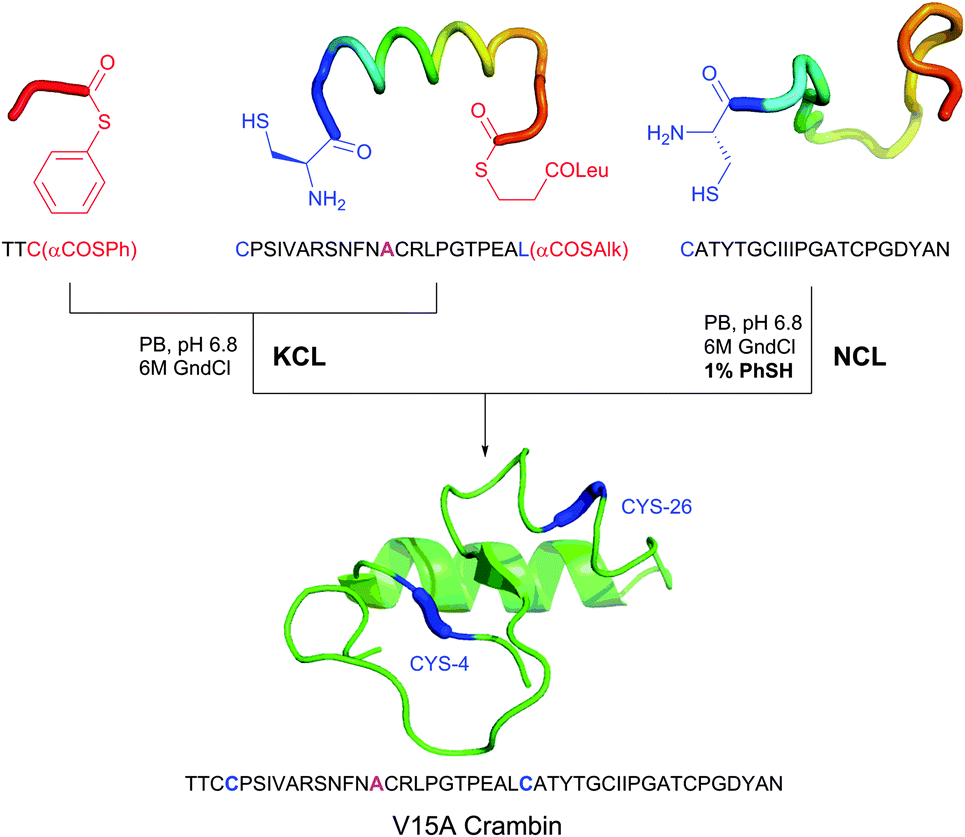 | ||
| Fig. 80 Two final steps of the synthesis of V15A crambin (pdb: 3NIR) described by Bang and associated.675 The mutation was introduced for simplifying the prior KCL step of assembling the second peptide. Kinetically controlled ligation of SAr thioester spontaneously occurs in aqueous media in the absence of exogenous thiophenol, while native chemical ligation of SAlk thioester must be accelerated by the addition of 1% PhSH. | ||
Further advancing pioneering works done by Botti et al.686 on in situ acyl migration, KCL methodology has been recently extended from alkylthioesters to a full class of O-esters undergoing a spontaneous transformation to produce a thioester when exposed to a reducing agent through disulfide bond reduction followed by O → S acyl shift (Fig. 81).687
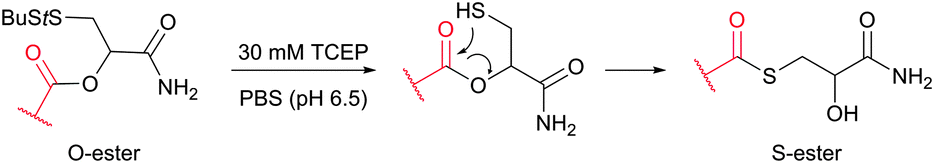 | ||
| Fig. 81 Schematic representation of the O → S acyl shift of O-esters containing a disulfide bond described by Zheng et al.687 | ||
Through a thorough investigation Zheng et al.687 have defined that structures of the O-esters have an important effect on their reactivity. The authors have side-by-side benchmarked their methodology with previously described KCL by synthesizing the same V15A crambin (Fig. 80) by a one-pot one-step condensation of peptide segments and found its applicability to this system. Readily available by Fmoc solid-phase synthesis, these O-ester scaffolds can expand the applicability of NCL to substrates with hardly accessible thioester peptide fragments.
2.3.1.3 Access to C-terminal peptide thioesters.
Classical approaches. The preparation of C-terminal α-thioesters involved in native chemical ligation is often associated with synthetic difficulties. Being especially reactive species, they either have to be introduced at the end of the synthetic pathway or be kept in a hidden form of thioester surrogates possessing higher stability.
Despite its recognised drawbacks due to hazardous acid treatment often leading to undesired side-reactions, the protocol of in situ neutralisation for Boc-based solid-phase peptide synthesis represents the most effective approach for the preparation of peptidyl thioesters.666,688–690 Alternatively, the Fmoc synthesis approach was investigated691–693 and found to be favoured when synthesizing phospho- and glycopeptides.
Expressed protein ligation (EPL). Introduced by Muir et al. in 1998,694 EPL represents another approach for the preparation of α-thioesters. It allows for obtaining the recombinant protein thioester by thiolysis of an intein fusion protein and thus enables a large pool of elaborated recombinant protein techniques for NCL. The reader is directed to several recent reviews of this area on chemical research for more details.695,696
X → S acyl-transfer. An elegant approach for keeping thioester in a relatively inert “hidden” form ready to be uncovered when required was first introduced by Danishefsky and colleagues in 2004 (Fig. 82).697 Several years later, the name “crypted thioesters” was coined by Otaka698 as a general term for such compounds. This approach is of especial interest, because it enables the assembly of peptide segments in N-to-C direction, which is rather rare and often difficult to achieve.673 Indeed, all above-described methodologies rely on the assembling of polypeptide chain in C-to-N direction: for instance, the sequential native chemical ligation per se consists of iterated cycle of ligation-deprotection-ligation…, i.e. adding new peptide fragments onto N-termini after deprotection thereof (see Section 2.3.1.1).
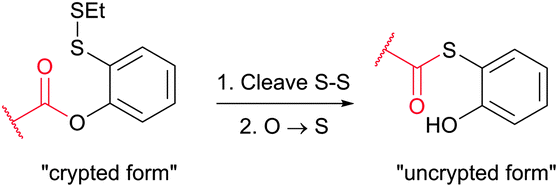 | ||
| Fig. 82 Principle of “crypted thioesters” demonstrated by Danishefsky and co-workers.697 Intramolecular O → S migration of acyl residue resulting in generation of highly active thioester (its “uncrypted form”) occurs only upon reductive cleavage of disulfide bond of reasonably stable “crypted form”. | ||
Synthesis of thioesters in situ from stable amides via N → S acyl-transfer was demonstrated by Ohta et al.699 who studied acylated oxazolidinones derived from S-protected cysteine. These possess a distorted amide planarity provoking so called ground-state destabilisation700 and, as a consequence, favour the acyl migration to a deprotected thiolate. Nakahara and collaborators have studied and elaborated two classes of secondary amides amenable to the N → S acyl shift at low pH values: 5-mercaptomethyl prolines701 and N-alkyl cysteamides (Fig. 83a).702 Oxazolidinones699 and N-sulfanylethylamilides (SEAlides)703 described by Otaka and collaborators were found to possess similar aptitude towards N → S acyl-transfer shift at low pH (for a complete overview of N → S acyl-transfer systems described before 2010 see review by Kang and Macmillan).704 Erlich et al.705 have recently applied N-alkyl cysteamide-based approach for the synthesis of 76-residue ubiquitin thioester, while Otaka and collaborators have demonstrated high potential of SEAlides by conducting the chemical synthesis of 162-redue active glycosylated GM2-activator protein.706
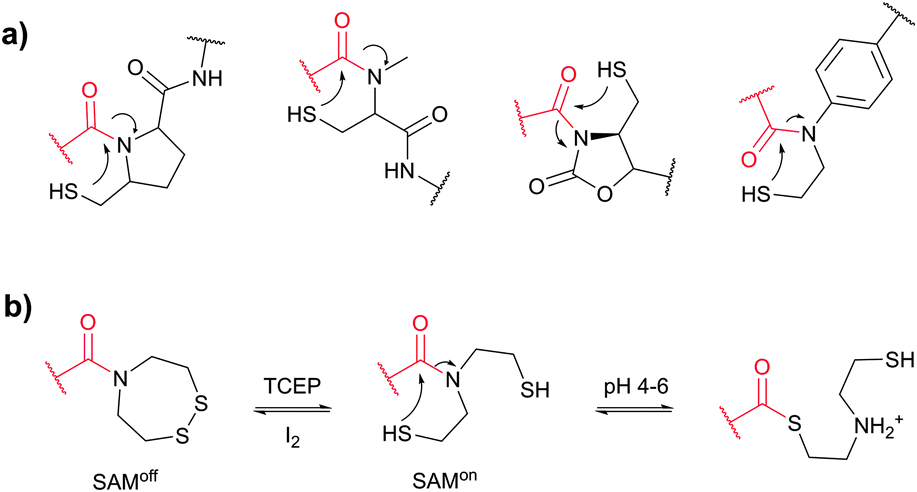 | ||
| Fig. 83 N → S migration of acyl residue. The thioester-amide equilibrium is shifted towards the thioester form at low pH due to the protonation of the secondary amides. In the presence of 3-mercaptopropionic acid (MPA) all intermediates produce MPA-thioester. (a) From left to right: reduced form of Danishefsky's ester,697 5-mercaptomethyl prolines,701N-alkyl cysteamides,702 oxazolidinones,699 and N-sulfanylethylamilides.703 (b) SAMon/off approach by Melnyk and collaborators.711 | ||
Almost simultaneously have two research groups reported a general approach, based on the application of bis(2-sulfanylethyl)amides (SAM) as precursors for NCL.707–710 An interesting extension of the methodology enabling the possibility of triggering the reactivity of SAM – so called SEAon/off system – has been further elaborated by Melnyk and collaborators.711 The transition between reactive (SEAon) and unreactive (SEAoff) states is simply triggered by mild oxidation/reduction procedures (Fig. 83b). SEAoff can be easily switched on via TCEP reduction, while the reverse switching off is achieved by mild oxidation with iodine. After few seconds, the excess of iodine is decomposed by the addition of dithiothreitol (DTT). Other amino acid residues susceptible to oxidation such as methionine or tryptophan are not affected because of this very short exposure to oxidant. However, cysteine residues must be protected with tert-butylsulfenyl groups to remain unaffected. At low pH values these are not reducible by DTT, allowing thus reliable protection of cysteines during the cycles of oxidative–reductive SAM triggering.
To demonstrate the potential of this newly established and optimised SAMon/off system, Melnyk and collaborators have synthesised 85-residue domain of the hepotocyte growth factor (HGF 125–209).711
2.3.1.4 Indirect ligation on other amino acid using NCL. In 2001, Yan and Dawson712 have introduced an approach that, for the first time, allowed virtually conducting NCL at alanine. While the ligation still occurred at N-terminal cysteine, its subsequent desulfurisation with freshly prepared RANEY® nickel produced the native target sequence containing alanine residue at the ligation site (Fig. 84). This strategy has inspired the development of a large pool of various ligation junctions that includes phenylalanine,713,714 glycine,715 valine,716,717 leucine,674,718 threonine,656 serine,657 proline,719–721 aspartate,722 glutamine,723 homocysteine,724,725 methionine (by subsequent S-methylation of the ligated homocysteinyl product by p-nitrobenzenesulfonate)724 arginine,726 and lysine (α- or ε-selective) ligations.727,728
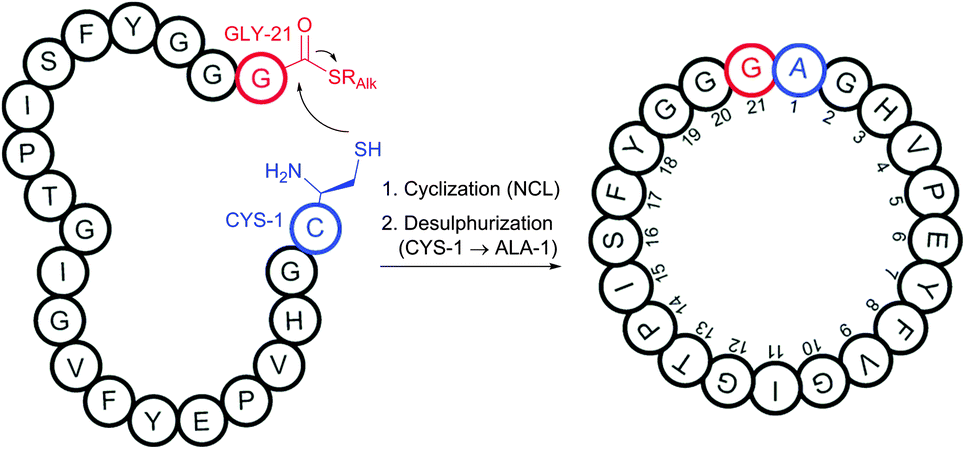 | ||
| Fig. 84 Synthesis of 21-residue cyclic antibiotic peptide Microcin J25 via NCL-desulfurisation strategy resulting in alanyl-linked polypeptide chain developed by Yan and Dawson.712 Reaction conditions: cyclisation – Tris-HCl, 6 M GndCl; desulfurisation – NaOAc, pH 4.5, 6 M GndCl, H2 (Pd/Al2O3). | ||
In order to extend native chemical ligation-desulfurisation approach to these amino acids, a SH-group should be attached to the carbon atom situated in the β-position to amino group (in some cases in the γ-position). The new building block containing a β-mercapto-α-amino or γ-mercapto-α-amino fragment is then introduced at the N-terminal position during solid-phase peptide synthesis or by means of DNA-recombinant technologies.714 Following ligation is conducted under classic NCL conditions: at pH 7.5–8.0 in the presence of TCEP as reducing agent and 1% of exogenous thiol additive. Finally, the desulfurisation of cysteine gives a nascent residue of interest (Table 2).
| Entry | AA | Intermediate | Product |
|---|---|---|---|
| 1 | Ala |
|
|
| 2 | Phe |
|
|
| 3 | Leu |
|
|
| 4 | Thr |
|
|
| 5 | Val |
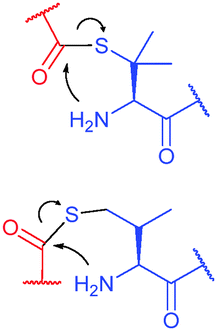
|
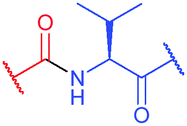
|
| 6 | Pro |
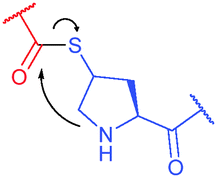
|
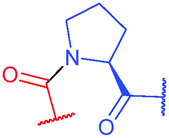
|
| 7 | Lysε |
|
|
| 8 | Lysα |
|
|
| 9 | Asp |
|
|
| 10 | Gln |
|
|
| 11 | Arg |
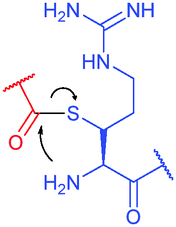
|
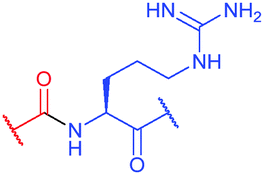
|
| 12 | Gly |
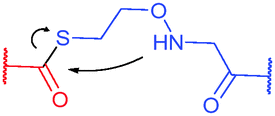
|
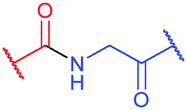
|
Besides aforementioned reduction on RANEY® nickel, various milder conditions such as nickel boride,712 Pd/Al2O3,729 or metal-free conditions730,731 were developed to achieve efficient desulfurisation. More recently, in situ ligation-desulfurisation approach was also reported.732,733
Desulfurisation-based methodology of the NCL expanding towards other amino acid junctions have contributed in many ways to prepare proteins and posttranslationally modified analogues for biochemical and structural analyses ever since its introduction. However, despite this broad utility, carrying out desulfurisation of the linkage site in the presence of other Cys residues in the protein sequence usually requires using of protecting groups.729,734 Not only does this necessity represent an undesirable step to protein synthesis, but it also implies some limitations on the applicability of the approach, mainly due to the solubility issues.
Developed by Dawson and collaborators735 the protocol for selective reduction of selenocysteines (Sec, U) in the presence of cysteine allowed overpassing the need for protection of thiolates and expanded the already established field of Sec-mediated native chemical ligations (see Section 2.6).736–739
The sensitivity of Sec peptides to reduction was noted in several works on selenocysteine ligations,737,738,740 During their preceding work on the synthesis of seleno-glutaredoxin 3 analogues (Se-Grx3), Dawson and associates741 have observed this incompatibility of Sec-containing proteins and peptides with TCEP-assisted native chemical ligation due to the generation of significant levels of a deselenised side product. In the publication to follow, the authors have successively applied the ligation-deselenisation strategy on a model peptide system. Accordingly, N-terminal Sec-peptide1 (UGLEFRSI-amide) prepared in the form of a diselenide dimer was ligated to the thioester peptide2 (Ac-LYRAG-SR) (Fig. 85) to produce the deselenised alanyl-peptide (Ac-LYRAGAGLEFRSI-amide) after the treatment with 50-fold excess TCEP at pH 5.5. Importantly, an excess of 200 mM 4-mercaptophenylacetic acid (MPAA) was needed for the ligation step. MPAA is both served as a catalyst to activate the alkyl thioester and as mild reducing agent to generate a small pool of free selenol to facilitate the ligation reaction.
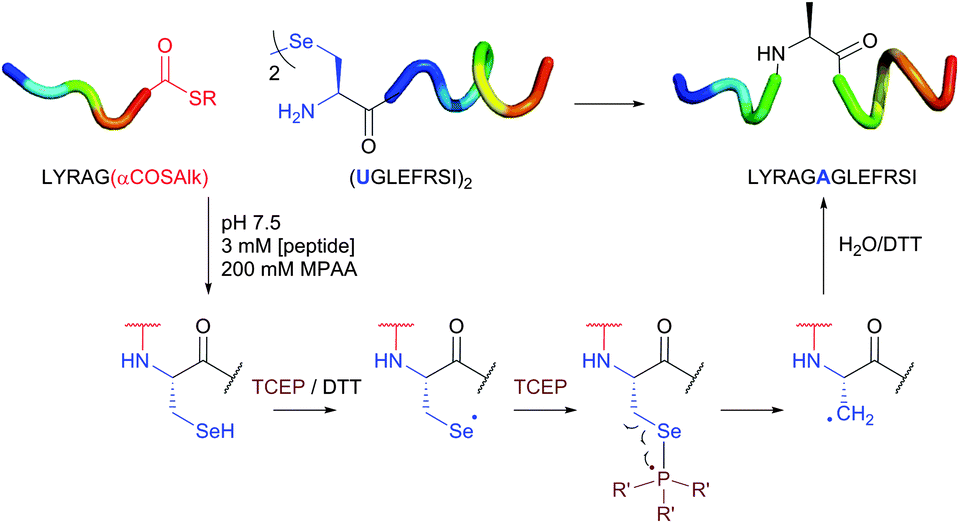 | ||
| Fig. 85 Traceless ligation of peptides using selective deselenisation described by Dawson and collaborators.735 | ||
This selectivity of reduction with TCEP is hypothesised to be due to the weaker Se–C bond compared with the S–C bond coupled with higher propensity of selenols to form radicals.742 It should be mention, however, that albeit upon heating, cysteine can be desulfurised in the absence of a radical initiator when treated with excess phosphine.743
Selenocysteine-mediated NCL deselenisation procedure have been recently exploited for selective ligation of selenolphenylalanine,744 and γ-selenolproline,720 easily transformable into peptides phenylalanine and proline respectively at the ligation site by treating Se-containing intermediates with TCEP or DTT. Analogously to N → S acyl transfer (Section 2.3.1.3), N → Se acyl shift was recently observed by Adams and Macmillan and allowed NCL to take place at lower temperatures and on shorter time scales.745 Corresponding selenoesters can be readily accessible by direct solid phase synthesis.746
NCL principles of S → N acyl transfer found their application in ligation assisted by proximity effect (see Section 5),747 allowing for conjugation of N-terminal residues other that cysteine by auxiliary-mediated acyl transfer. Furthermore, the Cys side chain thiolate introduced during NCL can also provide a synthetic handle for further functionalisation using cysteine-selective methodologies (see Section 1.2). It was recently demonstrated by Fang and co-workers that more accessible than thioesters simple phenyl esters could undergo native chemical ligation smoothly under the promotion of imidazole.748 Lastly, recently reviewed by Monbaliu and Katritzky749 Kemp's template-mediated thiol ligation,750–758 Tam's ligation by thiol/disulfide exchange,759–761 and other auxiliary-driven extensions of native chemical ligation,762,763 represent significant importance in the field of protein synthesis and can be considered as appropriate for bioconjugation.
The thiazolidine-mediated ligation was first applied by Tam and associates to the preparation of peptide dendrimers765,767 by attaching unprotected peptide dendrones containing Cys residues at their N-termini to a branched core matrix with aldehyde functions. Botti et al.766 have transformed this approach to a general method for the preparation of cyclic peptides. Villain et al. have demonstrated that obtained thiazolidines (sometimes referred to as pseudo-prolines)768 can be selectively cleaved by adding hydroxylamine derivatives, which react with aldehyde functions protected under the form of thiazolidine to form oximes. The authors applied this methodology for the covalent capture of proteins possessing N-terminal Cys or Thr residues (Fig. 86).769 Interestingly, under the same conditions N-terminal Ser residues reacted only poorly.
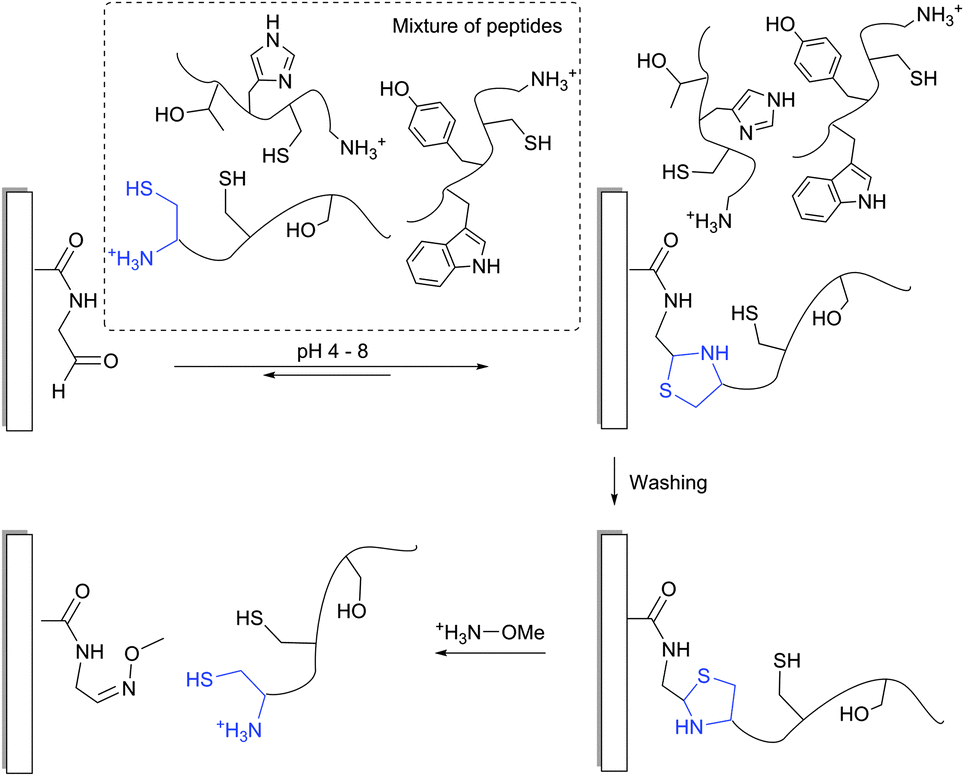 | ||
| Fig. 86 General scheme of covalent capture purification of N-terminal cysteine containing proteins developed by Villain et al.769 | ||
Because of the recent advances in the semisynthesis of proteins and the encoding of 1,2-aminothiols into recombinant proteins,662,770,771 thiazolidine-mediated conjugation is now experiencing a reappraisal of its potential for bioconjugation.772,773 For instance, Casi et al.772 have exploited thiazolidine formation for the preparation of antibody–drug conjugates by site-specific incorporation of a potent drug, containing an aldehyde moiety, to engineered recombinant antibodies displaying a Cys residue at their N-terminus, or a 1,2-aminothiol at their C-terminus.772 Lastly, thiazolidines represent one of the most often used N-terminal cysteine protecting groups for sequential NCL (see Table 1, Section 2.3.1.1).
000 firefly lanterns),775 luciferin enzymatic oxidation has been studied for the last 50 years.776
The regeneration pathway of luciferin in firefly was found to be consisted of the condensation of CBT with D-cysteine (Fig. 87).774 The reaction mechanism underlying this addition include first attack of cyano group of CBT by cysteine thiolate. This results in the formation of the electrophilic imidothiolate, subjected to the second attack by cysteine amino group to form thiazole structure after the yielding of ammonia gas.
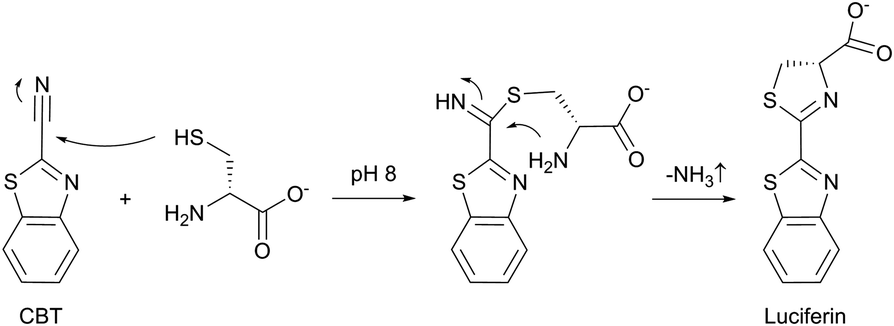 | ||
| Fig. 87 Synthesis of luciferin by the reaction between cyanobenzothiazole (CBT) and D-cysteine.774 | ||
Inspired by these early works, Rao and co-workers777 have further investigated the reaction of cyano-substituted aromatic compounds with amino-thiol substrates. They have demonstrated that benzotriazole motif plays an important role for the activation of the cysteine addition to a nitrile group. For instance, under optimal reaction conditions (PBS, pH 7.0–7.5) its replacement by other aromatic fragment such as picolinonitrile or benzonitrile largely decreases the reaction yield. All naturally occurring amino acids are unreactive towards CBT, except for cysteine owing the highest second-order rate constant among six other tested amino-thiol substrates (9.19 M−1 s−1, which is significantly higher than these of the majority of biocompatible click reactions).778 Finally, the efficiency and specificity of CBT-based labelling of terminal cysteine residues was demonstrated on proteins in vitro (Fig. 88) as well as on cell surfaces.
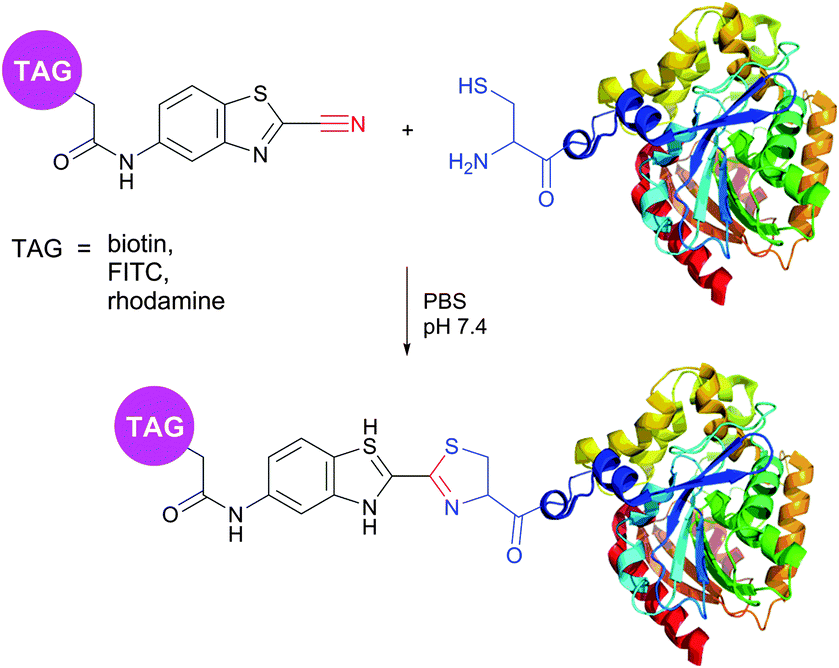 | ||
| Fig. 88 Labelling of Renilla luciferase (pdb: 2PSD) by CBT-based probes.777 N-terminal cysteine was generated by protease processing of the fusion protein. | ||
In the publications to follow, Rao and colleagues have extended the applicability of CBT towards biocompatible condensations to create polymer assemblies in vitro and in living cells under the control of either pH, disulfide reduction or enzymatic cleavage.779,780 Yuan et al. have taken advantage of this approach and developed a method for the determination of glutathione (GSH) concentration in vitro and in HepG2 human liver cancer cells.781 Jeon et al.782 have elaborated a CBT-based 18F-probe radiolabelling of N-terminal cysteine-bearing peptides and proteins. Two labelled substrates: a dimeric RGD-peptide – [18F]CBTRGD2, and Renilla lucifierase bearing a cysteine at N-terminus, have been synthesised with excellent radiochemical yields and shown good in vivo molecular PET imaging efficiency. Proceeding efficiently at physiological conditions, CBT-mediated N-terminal Cys conjugation represents a useful alternative to existing approaches for protein labelling.783
2.4 Tryptophan
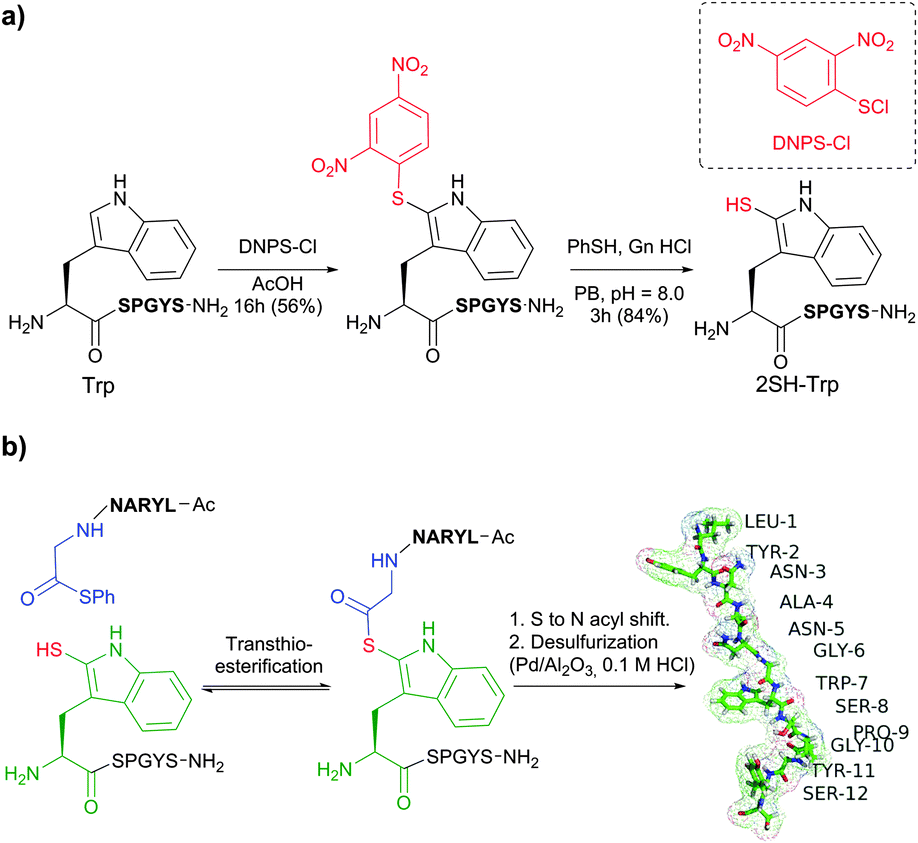 | ||
| Fig. 89 Ligation–desulfurisation at tryptophan, reported by Payne and collaborators.786 (a) Electrophilic sulfenylation of Trp in acidic conditions with DNPS-Cl and the obtention of the corresponding 2SH-Trp derivative. (b) Auxiliary-assisted native ligation of N-terminal 2SH-Trp peptide (2SH-WSPGYS-NH2) with a thioester of the peptide Ac-LYRANG-SPh resulting in 12-mer peptide Ac-LYRANGWSPGYS after the desulfurisation step. | ||
The proposed mechanism for this approach is mechanistically similar to NCL. It was hypothesised that the reaction would proceed via an initial step of the peptide thioester transthioesterification with an indole 2-thiol functionality followed by an S- to N-acyl shift through a 7-membered ring transition state to generate a native amide bond. The last step of 2-thiol Trp desulfurisation results in obtention of a ligated product with only naturally occurring amino acid residues (Fig. 89b). Although this methodology represents a clever chemoselective approach for the ligation of completely unprotected peptide fragments through Trp moiety, the harshness of the reaction conditions of sulfenylation and desulfurisation limit it only to peptide substrated and don't allow its application on complex biomolecules.
2.5 Histidine
Despite being known for its particular importance for the acyl transfer in many enzymatic processes,665 histidine has been rarely used for bioconjugation. The only study describing the ligation of N-terminal His peptides with activated thioester was reported by Zhang and Tam.791Ellman's regent348 was used to activate the C-terminal peptide thiocarboxylic acid by forming acyldisulfide derivative, which is then nucleophilically attacked by N-terminal histidine. Captured by the imidazole of the N-terminal histidine, the obtained Nim-acyl intermediate is hypothesised to undergo Nim → Nα shift to form histidine at the ligation site (Fig. 90). However, the Nim-acyl intermediate has not been isolated and it is quite possible that regioselectivity is obtained simply because of anchimeric assistance of the proximal imidazole moiety at the ligation site.
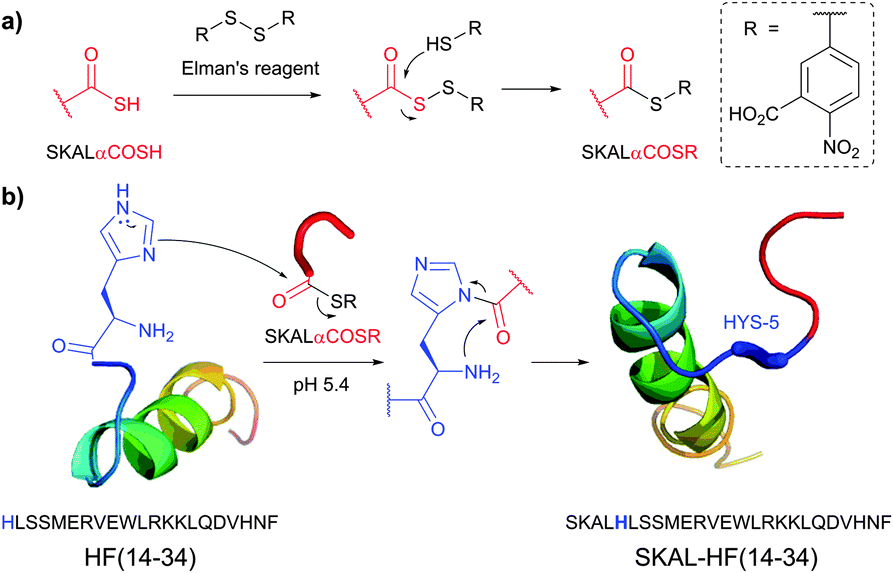 | ||
| Fig. 90 N-terminal histidine labelling described by Zhang and Tam.791 (a) Mechanism of thiocarboxylic acid activation by Ellman's reagent. (b) Labelling of bovine parathyroid hormone fragment (14–34, pdb: 1ZWC) with activated tetrapeptide thioacid. | ||
Interestingly, no sign of coupling reaction has occurred when a corresponding non-activated C-terminal thiocarboxylic acid is participating in the reaction instead of the acyldisulfide. The reaction pH plays an important role on the effectiveness of the reaction. Only when maintained at slightly acidic values (pH 5–6) and in the absence of the thiol nucleophiles, would the imidazolyl moiety of histidine be the sole nucleophile present in the polypeptide. This methodology has been applied to generate histidine-containing peptides with yields up to 75%.
2.6 Selenocysteine
Selenium and sulphur belong to the same main group of elements; therefore, 21st proteinogenic amino selenocysteine and Cys residue exhibit rather similar properties in terms of reactivity for bioconjugation.792–794 For instance, both Hilvert's738 and Raines's group736 demonstrated that C-terminal peptide thioesters react smoothly with peptide fragments containing a N-terminal selenocysteine in exactly the same manner as with corresponding cysteine analogues. Presumably proceeding through the same mechanism as NCL (Section 2.3.1), the first step of the ligation process consists in the nucleophilic attack on the thioester by selenolate to give a selenoester intermediate that subsequently rearranges to give a native chemical bond.Sec ligation can be chemoselective when conducted at slightly acidic pH. Low intrinsic pKa values of selenocysteine (5.2)795 and consequently its higher dissociation level at low pH, endows this amino acid with unique biochemical properties, allowing regiospecific covalent conjugation with electrophilic compounds in the presence of the side chains of all other natural amino acids including the thiol group of Cys (pKa 8.3). For instance, the reaction rate with selenocysteine was found to be 1000 fold faster than with cysteine at pH 5.0.736 Moreover, the lower pH generally suppresses β-elimination of the selenol group from selenocysteine resulting in the obtaining of unreactive dehydroalanine.392
Initially, considerable efforts were made to show the applicability of selenocysteine NCL for the preparation of selenium-containing derivatives of enzymes and benchmarking activities thereof. Hilvert and associates synthesised a C38U analogue of bovine pancreatic trypsin inhibitor (BPTI). Amusingly, the wild-type BPTI and its artificial analogue folded into alike conformations and demonstrated similar inhibiting affinity of trypsin and chymotrypsin.738 Raines et al. selected 124-residue ribonuclease A (RNase A) as a model protein for the investigations.736 DNA recombinant technology was utilised to prepare a C-terminal thioester fragment corresponding to residues 1–109, while standard solid phase peptide synthesis methodology was used to obtain a N-terminal Cys and Sec peptides corresponding to residues 110–124 (Fig. 91). Just as in the case of BPTI, the semisynthetic wild-type RNase A and C110U RNase A presented equivalent ribonucleolytic activities. Further advances in the field of Sec-NCL have resulted in synthesis and investigations of other different proteins such as seleno-glutaredoxin,796 azurin,797,798 and thioredoxine reductase.
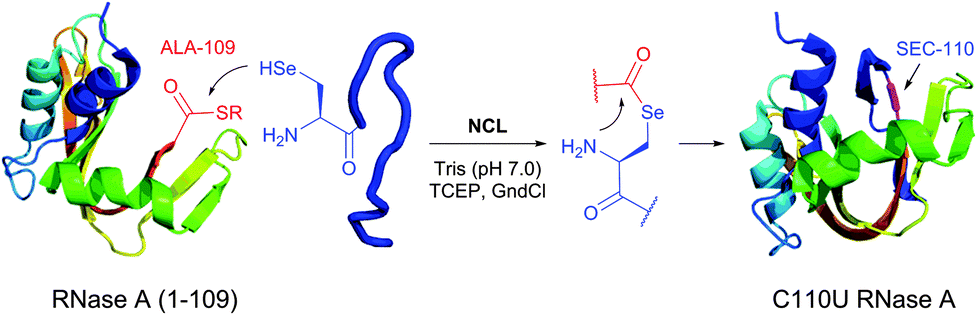 | ||
| Fig. 91 Selenocysteine native chemical ligation applied for the preparation of C110U mutant of RNase A (wild-type – pdb: 7RSA).736 | ||
Ease of the post-ligation transformation of selenocysteine to alanine (by deselenisation), dehydroalanine (by β-elimination) or non-natural amino acids (by addition reaction to dehydroalanine, see Section 1.3.8) became a spur to a further propagation of Sec-mediated methodologies as very effective tools in rational design of peptides and proteins. Quaderer and Hilvert799 exploited such transmutations of selenocysteine to access a series 16-residue cyclic peptides (Fig. 92).
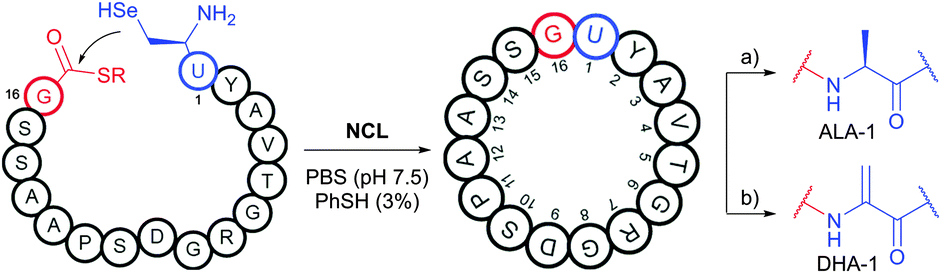 | ||
| Fig. 92 Synthesis of cyclic 16-resiudes selenopeptides by native chemical ligation and following transformation thereof to (a) alanine (RANEY® Ni, H2, TCEP, 20% AcOH); (b) dehydroalanines (H2O2, 10% ACN). | ||
In this initial report, the deselenisation step was conducted rather harshly (RANEY® Ni, H2), implying that all Cys residues (if there were any) would have been reduced as well. Recently, however, Dawson and collaborators735 have demonstrated that selenocysteine can be chemoselectively deselenated with TCEP in the presence of cysteines. This allowed overpassing the main limitation of the NCL-desulfurisation strategy (see Section 2.3.1.4), namely the inability to control regioselectivity of desulfurisations if several cysteines are present in the peptide or protein, and yielded in a pool of NCL-deselenisation strategies for mild incorporation of alanine, phenylalanine and proline into the ligation site by classic NCL approaches (see Table 2). Finally, selenocysteine peptides were found to efficiently undergo reverse NCL at acidic pH and thus to be of particular interest for the generation of thioesters by sequential N → Se acyl-transfer and substitution of the obtained selenoester by exogenous thiol (see Section 2.3.1.3).745
Because the incorporations of selenocysteine by the cell translational machinery are generally very laborious,800,801 selenopeptides are mainly obtained by SPPS.736,737,794
2.7 Proline
The oxidative coupling of o-aminophenols and o-catechols recently reported by Francis and collaborators802 represents an interesting approach for selective N-terminal conjugation on proteins. The strategy consists in prior in situ oxidation of o-aminophenols and o-catechols to active coupling species using potassium ferricyanide (Fig. 93a) followed by their reaction with a protein (Fig. 93a). This approach was shown to work particularly well with proline (due to increased nucleophilicity thereof) and can therefore be considered as N-terminal Pro selective methodology.802 The key advantage of the approach compared to the majority of N-terminal selective methodologies is its fast second-order kinetics in a single step transformation, which does not require prior oxidation of the N-terminus. Free cysteine residues, however, are also reactive during the oxidative coupling and must thus be protected, which represents the main drawback of the approach.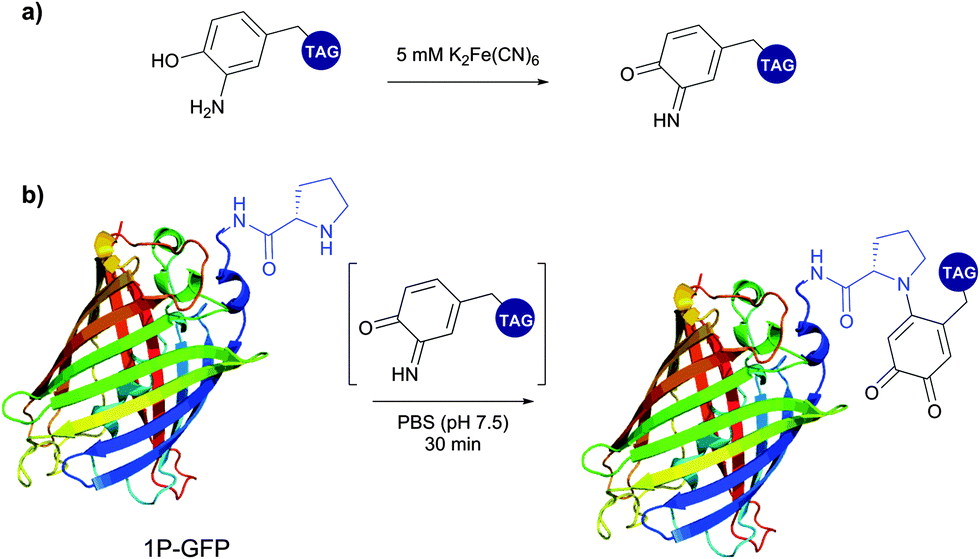 | ||
| Fig. 93 Selective modification of 1P-GFP (Pro residue is introduced on N-terminus) with o-aminophenol-PEG reagent (wild-type – pdb: 1GFL).802 (a) In situ oxidation of o-aminophenol derivatite. (b) Reaction of the active coupling species with N-terminus of 1P-GFP. | ||
3. C-terminal conjugation
Chemical methods for C-termini conjugation are rather scarce. Definitely, previously mentioned approaches for the conjugation of side-chain carboxylates (see Section 1.5) can also be applied effectively to modify the terminal carboxylates. The selectivity, however, would be the main issue, for up to date there is no described method for somewhat selective activation of N-terminal carboxylates by any activating reagent.Because of the impetuous development of NCL (see Section 2.3.1), requiring C-terminal thiolates as reacting partners for N-terminal Cys proteins, those became widely accessible, namely by means of SPPS. A perspective approach, exploiting these advances was proposed by Goody803 and collaborators, who developed a protocol for selective transformation of C-terminal thioesters to corresponding hydroxylamines, enabling thus the application of aldehyde- and ketone-selective methodologies on the C-terminus.
On the other hand, the unique position of protein C-termini has stimulated numerous efforts to target this location, which resulted in numerous enzymatic and intein-based approaches for C-terminal-selective protein modification.804–809 These methodologies are, however, not covered by this review devoted to chemical methods of bioconjugation.
4. Sequence-selective approaches
Several especially useful methodologies in bioconjugation exploit not a specific property of the residue, or its peculiar position in protein, but rather the synergetic effect of a batch of neighbouring amino acid residues. For instance, an example of such selectivity is the aforementioned selective modification of His-AA-Ser and His-AA-Thr peptide motifs (see Section 1.2) by usually promiscuous activated esters. In this case, the His imidazolyl side chain located in close proximity to Ser/Thr side chains increased the reactivity thereof towards electrophiles.Metal-chelation methodologies are perhaps the most elaborated among sequence-selective approaches (Fig. 94).810 The oligo-histidine sequence (usually H6), called His-tag, is known to interact robustly with transition-metal complexes, including a nitrilotriacetic acid (NTA) complex of Ni2+, therefore the sequence is widely used for protein purification by affinity chromatography. Similarly, oligo-aspartates (most often (D4)n, n = 1–3) were developed for selective labelling with multinuclear Zn2+ complexes.811,812 The tetracysteine motif (CCPGCC) was reported to especially effectively chelate biarsenical dyes such as 4′,5′-bis(1,3,2-dithioarsolan-2-yl)fluorescein (FlAsH),5,813 while a similar structural tetraserine sequence (SSPGSS) was found to be selective for diborate-scaffolds.
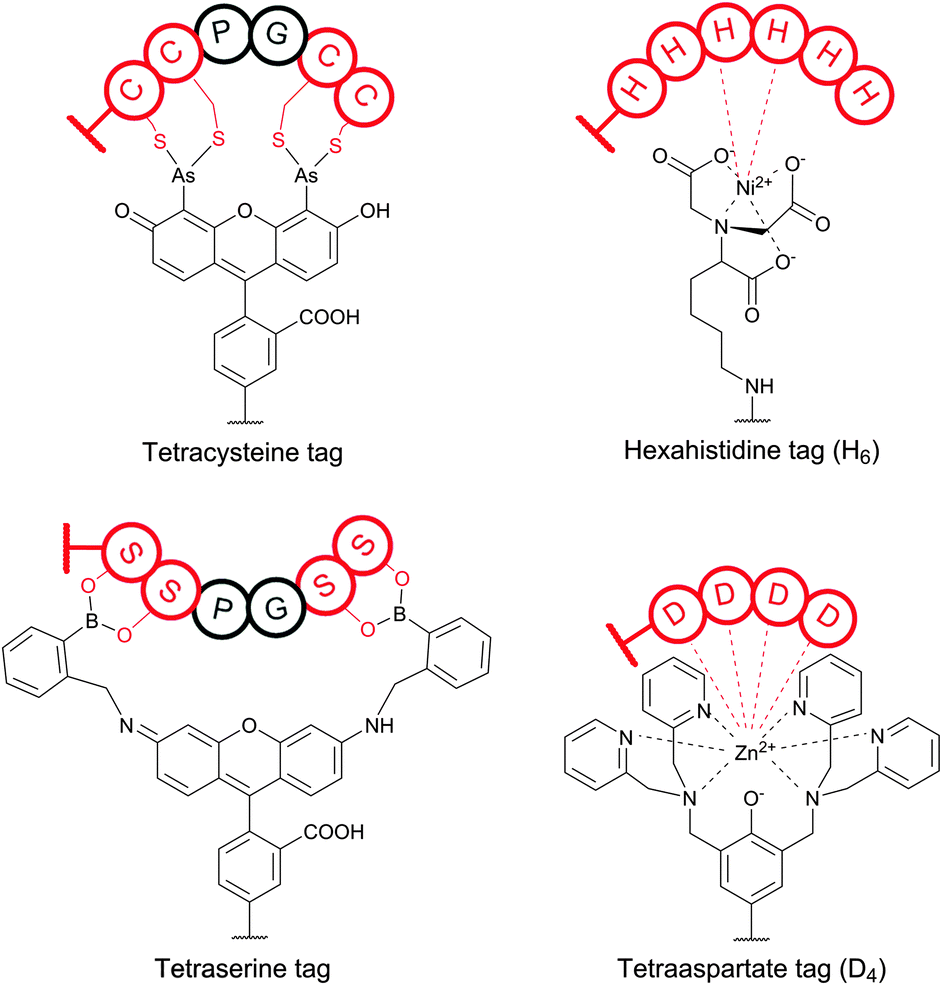 | ||
| Fig. 94 Strategies for the selective conjugation of proteins based on metal-chelation: tetracysteine/biarsenical system, oligohistidine/nickel-complex system, tetraserine–borate system, oligo-aspartate/zinc-complex system.27 | ||
5. Proximity-driven modifications
All the above-mentioned methodologies are mainly residue-specific. That is to say, they exploit specific reactivity of the functional group of interest or of an assembly of residues. As a result, bioconjugations of highly nucleophilic Cys, Lys, and Tyr residues with electrophilic reagents are definitely prevalent among described methodologies.The inherent reactivity is rare to be overcome, which largely limits of the scope of known methods for bioconjugation of the amino acids possessing low nucleophilicity. However, bringing the reaction partners into close proximity can accelerate a reaction thereof, which would not otherwise be possible because of the presence of other more reactive species. Routinely exploited by enzymes, this approach enables selectivity on the basis of the molecular shape rather than reactivity or the local environment (Fig. 95).
 | ||
| Fig. 95 Schematic representation of the proximity-driven approach for selective modification of macromolecules. Combining of promiscuous reactivity of the reactive group (RG, shown in red) with specific molecular recognition results in specific modification of the functional group of interest (FG, shown in blue). | ||
Developed by Hamachi and collaborators in 2006,462 the post-affinity-labelling approach can be used for the selective tethering a functional molecule at the proximity of the active site of enzymes. This method was then used on numerous substrates and enlarged the scope of selective conjugation especially on histidine and tyrosine (see Section 1.5.1).462,463,486,814 Despite these successes, a prevalent limitation of the applicability of the method due to the possible modification of the residues situated only in the vicinity of ligand-binding pocket of the target protein prevents this methodology from becoming a general approach for protein labelling.
In 2010, exploiting similar idea, Popp and Ball454 envisioned the combination of two previously described techniques: the coiled-coil based molecular recognition of complementary peptides455,815–817 and high catalytic activity of dirhodium complexes on carbene C–H insertion, previously reported by Francis and collaborators452 for the selective modification of tryptophan (see Section 1.4.2). Two complementary peptides: one, containing a dirhodium catalytic centre (precomplexated through two glutamate side chains), and another, containing a side chain to be modified are thus involved in this methodology (Fig. 96a). Because of the compelled proximity of the side chain of interest and the active catalytic centre in the obtained supramolecular assembly, the reaction of rhodium-catalysed C–H insertion is largely accelerated (more than 103 times).818
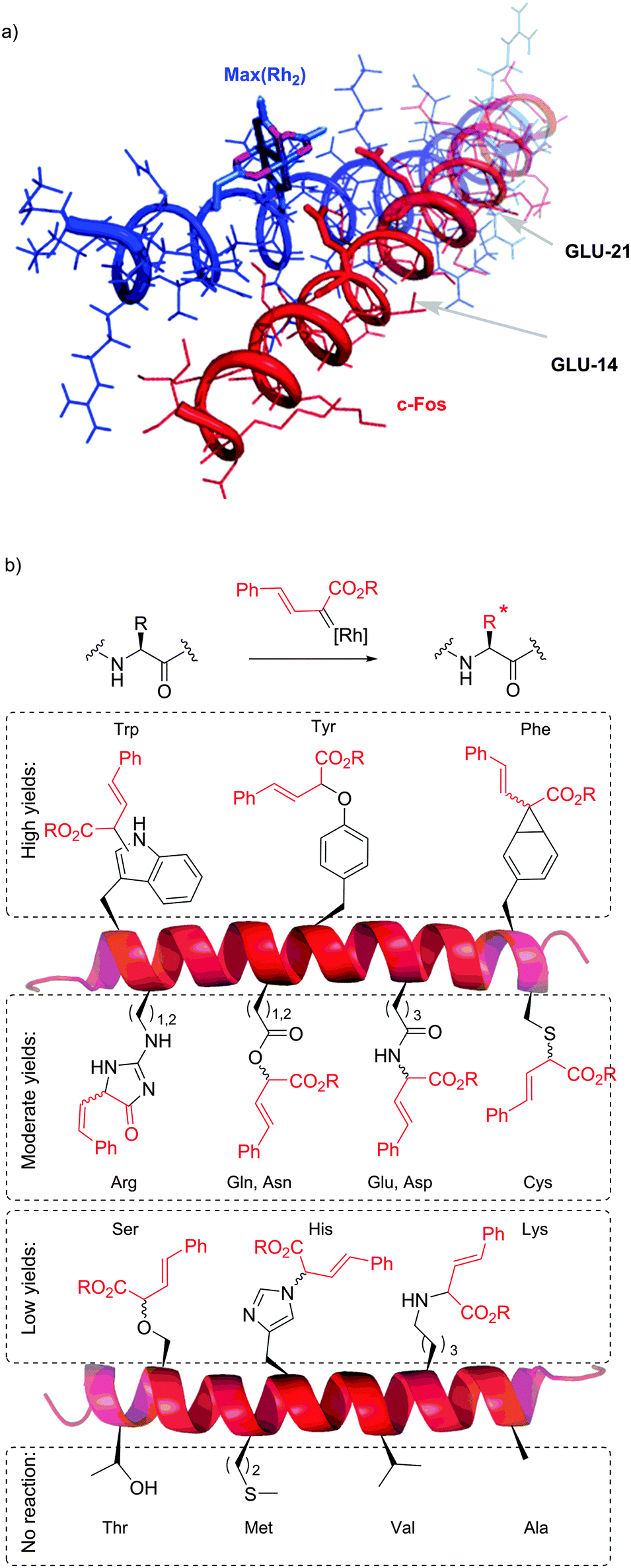 | ||
| Fig. 96 (a) Modification of c-Fos (shown in red) catalysed by Max(Rh2) (shown in blue) metallopeptide described by Popp and Ball.818 Two possible coiled-coil alignments result in modification of GLU-14 and GLU-21 residues, located in close vicinity to the dirhodium catalytic center. (b) Proposed product bond connectivity. | ||
As a result, the conjugation of amino acid residues with lower reactivities becomes possible. This allowed to expand the scope of originally tryptophan-selective dirhodium carbene methodology first to the other aromatic residues, phenylalanine and tyrosine,454 and then to over half of the naturally occurring amino acid residues (Fig. 96b).818 To date, dirhodium metallopeptide represent the only reported method for selective modification of Gln, Asn, and Phe side chains. The authors have also demonstrated the possibility to apply their methodology on chimeric proteins, containing fused coils,456 as well as on full-sised natural proteins possessing coiled-coils in their structures.818
Despite its important potential, the metallopeptides methodology is however not devoid of drawbacks. Because binding to dirhodium is nonselective and thus cannot be performed in the presence of other carboxylate-containing peptides, rhodium-peptide complexes must be synthesised beforehand, which is often challenging mainly due to their poor solubility.819 Moreover, the method is restricted to proteins containing coiled coil fragments in their structures, which in case of the vast majority of targets would mean the need for resource- and time-consuming expression of fused proteins.
Another approach, developed by Silverman and colleagues, exploits self-assembling of complementary DNA to bring into proximity two reacting fragments and allowed, although only on simple substrates, selective phosphorylation of tyrosine and serine, otherwise not feasible.538
Beyond coiled coils and DNA-based preorganisation, the principles of proximity-driven selectivity should be extended to other helix-binding protein domains and to biological molecular recognition generally. A significant broadening of the applicability of this elegant approach for protein modification, biochemistry and biomaterials engineering is anticipated in the nearest future.
Conclusions
The field of bioconjugation has expanded in the last 100 years and passed from a blind-eye modification of proteins one could found in nature to a well-established independent domain full of approaches allowing precise and reliable introduction of various tags into proteins' structure.Many, if not most of these methods, however, often possess drawbacks limiting their general applicability. This fact has become of special consideration with a rise of novel exigent applications for bioconjugation, namely the preparation of new therapeutic conjugates, vaccines, and biomaterials. Also tremendous progress in the sensitivity of analytical methodologies as well as the need to work with smaller and smaller amounts of sample, often-unstandardized patient samples, highlighted the need for more efficient, selective and reliable bioconjugation methods.
Moreover, some parameters of the mode of conjugation, previously completely neglected, were recently revealed to be of paramount importance. For instance, the stability of the generated linkage and the distribution of products generated upon conjugation can be determining for the overall efficiency of the conjugate.
Overall, we believe that intensive ongoing research in the field of bioconjugation will result in more efficient and selective methodologies allowing specific conjugation of native proteins in complex biological media, and ultimately in living organisms.
Acknowledgements
We thank Dr S. Kolodych (Syndivia SAS), Dr J.-Y. Bonnefoy (Syndivia SAS), Dr A. Osypenko (UDS), and Dr M. Recher (UDS) for fruitful discussions that led to the redaction of this manuscript. We also acknowledge the financial support from SATT Conectus “BioReLiable” project, CNRS, University of Strasbourg, and International Center for Frontier Research in Chemistry (icFRC).References
- S. Zalipsky, Bioconjugate Chem., 1995, 6, 150–165 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- D. P. Baker, E. Y. Lin, K. Lin, M. Pellegrini, R. C. Petter, L. L. Chen, R. M. Arduini, M. Brickelmaier, D. Wen, D. M. Hess, L. Chen, D. Grant, A. Whitty, A. Gill, D. J. Lindner and R. B. Pepinsky, Bioconjugate Chem., 2006, 17, 179–188 CrossRef CAS PubMed.
- S. J. Bell, C. M. Fam, E. A. Chlipala, S. J. Carlson, J. I. Lee, M. S. Rosendahl, D. H. Doherty and G. N. Cox, Bioconjugate Chem., 2008, 19, 299–305 CrossRef CAS PubMed.
- B. A. Griffin, Science, 1998, 281, 269–272 CrossRef CAS.
- H. M. O'Hare and K. Johnsson, Curr. Opin. Struct. Biol., 2007, 17, 488–494 CrossRef PubMed.
- X. Michalet, S. Weiss and M. Jäger, Macromolecules, 2006, 106, 1785–1813 CAS.
- C. A. Royer, Chem. Rev., 2006, 106, 1769–1784 CrossRef CAS PubMed.
- C. R. Bagshaw and D. Cherny, Biochem. Soc. Trans., 2006, 34, 979–982 CrossRef CAS PubMed.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- M. Fernández-Suárez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929–943 CrossRef PubMed.
- Q. Wang, T. Lin, L. Tang, J. E. Johnson and M. G. Finn, Angew. Chem., Int. Ed., 2002, 41, 459–462 CrossRef CAS.
- J. C. Smith, K.-B. Lee, Q. Wang, M. G. Finn, J. E. Johnson, M. Mrksich and C. A. Mirkin, Nano Lett., 2003, 3, 883–886 CrossRef CAS.
- D. W. P. M. Löwik, I. O. Shklyarevskiy, L. Ruizendaal, P. C. M. Christianen, J. C. Maan and J. C. M. van Hest, Adv. Mater., 2007, 19, 1191–1195 CrossRef PubMed.
- K. L. Christman, R. M. Broyer, Z. P. Tolstyka and H. D. Maynard, J. Mater. Chem., 2007, 17, 2021 RSC.
- M. J. Abedin, L. Liepold, P. Suci, M. Young and T. Douglas, J. Am. Chem. Soc., 2009, 131, 4346–4354 CrossRef CAS PubMed.
- L. S. Witus and M. B. Francis, Acc. Chem. Res., 2011, 44, 774–783 CrossRef CAS PubMed.
- M.-L. Chen, A. K. Adak, N.-C. Yeh, W.-B. Yang, Y.-J. Chuang, C.-H. Wong, K.-C. Hwang, J.-R. R. Hwu, S.-L. Hsieh and C.-C. Lin, Angew. Chem., Int. Ed., 2008, 47, 8627–8630 CrossRef CAS PubMed.
- A. Hucknall, D. H. Kim, S. Rangarajan, R. T. Hill, W. M. Reichert and A. Chilkoti, Adv. Mater., 2009, 21, 1968–1971 CrossRef CAS PubMed.
- Y.-L. Huang and C.-Y. Wu, Expert Rev. Vaccines, 2010, 9, 1257–1274 CrossRef CAS PubMed.
- S. Aggarwal, Nat. Biotechnol., 2010, 28, 1165–1171 CrossRef CAS PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2013, 12, 329–332 CrossRef CAS PubMed.
- Y.-L. Huang, J.-T. Hung, S. K. C. Cheung, H.-Y. Lee, K.-C. Chu, S.-T. Li, Y.-C. Lin, C.-T. Ren, T.-J. R. Cheng, T.-L. Hsu, A. L. Yu, C.-Y. Wu and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2517–2522 CrossRef CAS PubMed.
- R. Adamo, A. Nilo, B. Castagner, O. Boutureira, F. Berti and G. J. L. Bernardes, Chem. Sci., 2013, 4, 2995–3008 RSC.
- C. Gruttner, K. Muller and J. Teller, IEEE Trans. Magn., 2013, 49, 172–176 CrossRef.
- A. Sinz, J. Mass Spectrom., 2003, 38, 1225–1237 CrossRef CAS PubMed.
- M. J. Hinner and K. Johnsson, Curr. Opin. Chem. Biol., 2010, 21, 766–776 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, 2008 Search PubMed.
- F. H. Westheimer and D. E. Schmidt, Biochemistry, 1971, 10, 1249–1253 CrossRef CAS.
- G. W. Anderson, J. E. Zimmerman and F. M. Callahan, J. Am. Chem. Soc., 1964, 86, 1839–1842 CrossRef CAS.
- A. Lowe, Proc. R. Soc. Med., 1970, 63, 367–368 CAS.
- A. H. Coons, H. J. Creech, R. N. Jones and E. Berliner, J. Immunol., 1942, 45, 159–170 CAS.
- M. E. Annunziato, U. S. Patel, M. Ranade and P. S. Palumbo, Bioconjugate Chem., 1993, 4, 212–218 CrossRef CAS.
- H. J. Creech and R. N. Jones, J. Am. Chem. Soc., 1941, 63, 1670–1673 CrossRef CAS.
- L. Goodfriend and A. H. Sehon, Can. J. Biochem., 1958, 36, 1177–1184 CrossRef CAS.
- A. Rifai and S. S. Wong, J. Immunol. Methods, 1986, 94, 25–30 CrossRef CAS.
- A. Todrick and E. Walker, Biochem. J., 1937, 31, 297–298 CAS.
- P. Edman, Acta Chem. Scand., 1950, 4, 283–293 CrossRef CAS PubMed.
- D. Podhradský, Ĺ. Drobnica and P. Kristian, Experientia, 1979, 35, 154–155 CrossRef.
- R. J. Seiwald, J. L. Riggs, J. H. Burckhalter, C. M. Downs and T. G. Metcalf, Am. J. Pathol., 1958, 34, 1081–1097 Search PubMed.
- R. P. Haugland, Molecular probes, in Handbook of fluorescent probes and research chemicals, 1996 Search PubMed.
- J. Tuls, L. Geren and F. Millett, J. Biol. Chem., 1989, 264, 16421–16425 CAS.
- L. D. Burtnick, Biochim. Biophys. Acta, 1984, 791, 57–62 CrossRef CAS.
- M. Miki, Eur. J. Biochem., 1987, 164, 229–235 CrossRef CAS PubMed.
- M. Miki, J. Biochem., 1989, 106, 651–655 CAS.
- A. Bellelli, R. Ippoliti, M. Brunori, Z. Kam, M. Benveniste, F. Emmanuel, E. Turpin, A. Alfsen and J. P. Frénoy, Biochem. Biophys. Res. Commun., 1990, 169, 602–609 CrossRef CAS.
- R. E. Dedmon, A. W. Holmes and F. Deinhardt, J. Bacteriol., 1965, 89, 734–739 CAS.
- L. Amante, A. Ancona and L. Forni, J. Immunol. Methods, 1972, 1, 289–301 CrossRef CAS.
- A. F. Barbet and T. C. McGuire, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 1989–1993 CrossRef CAS.
- M. Schultzberg, J. M. Lundberg, T. Ho kfelt, L. Terenius, J. Brandt, R. P. Elde and M. Goldstein, Neuroscience, 1978, 3, 1169–1186 CrossRef CAS.
- W. P. Carney, R. H. Rubin, R. A. Hoffman, W. P. Hansen, K. Healey and M. S. Hirsch, J. Immunol., 1981, 126, 2114–2116 CAS.
- A. Varadarajan, R. M. Sharkey, D. M. Goldenberg and M. F. Hawthorne, Bioconjugate Chem., 1991, 2, 102–110 CrossRef CAS.
- R. Pei, M. Arjomand-Shamsai, C. T. Deng, A. Cesbron, J. D. Bignon and J. H. Lee, Tissue Antigens, 1993, 41, 200–203 CrossRef CAS PubMed.
- M. Edidin, Y. Zagyansky and T. Lardner, Science, 2003, 191, 466–468 Search PubMed.
- K. K. Huang and C. R. C. Cantor, J. Mol. Biol., 1972, 67, 265–275 CrossRef CAS.
- S. H. Blose, J. Cell Biol., 1980, 86, 608–615 CrossRef.
- S. Ohkuma, Y. Moriyama and T. Takano, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 2758–2762 CrossRef CAS.
- S. J. Morrison, H. D. Hemmati, A. M. Wandycz and I. L. Weissman, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 10302–10306 CrossRef CAS.
- C. A. Gabel, E. M. Eddy and B. M. Shapiro, J. Cell Biol., 1979, 82, 742–754 CrossRef CAS.
- P. Rosa-Neto, B. Wängler, L. Iovkova, G. Boening, A. Reader, K. Jurkschat and E. Schirrmacher, ChemBioChem, 2009, 10, 1321–1324 CrossRef CAS PubMed.
- K. E. Linder, M. D. Wen, D. P. Nowotnik, M. F. Malley, J. Z. Gougoutas, A. D. Nunn and W. C. Eckelman, Bioconjugate Chem., 1991, 2, 160–170 CrossRef CAS.
- C. F. Meares, M. J. McCall, D. T. Reardan, D. A. Goodwin, C. I. Diamanti and M. McTigue, Anal. Biochem., 1984, 142, 68–78 CrossRef CAS.
- S. S. Mirzadeh, M. W. M. Brechbiel, R. W. R. Atcher and O. A. O. Gansow, Bioconjugate Chem., 1989, 1, 59–65 CrossRef.
- D. S. Wilbur, M.-K. Chyan, D. K. Hamlin, B. B. Kegley, R. Nilsson, B. E. B. Sandberg and M. Brechbiel, Bioconjugate Chem., 2002, 13, 1079–1092 CrossRef CAS PubMed.
- H. Shi, X. He, K. Wang, Y. Yuan, K. Deng, J. Chen and W. Tan, J. Nanomed. Nanotechnol., 2007, 3, 266–272 CrossRef CAS PubMed.
- M. Roederer, Conjugation of monoclonal antibodies, 2004, http://drmr.com/abcon/index.html.
- E. C. E. Butcher and I. L. I. Weissman, J. Immunol. Methods, 1979, 37, 97–108 CrossRef.
- A. M. Landel, Anal. Biochem., 1976, 73, 280–289 CrossRef CAS.
- D. S. Wilbur, M.-K. Chyan, H. Nakamae, Y. Chen, D. K. Hamlin, E. B. Santos, B. T. Kornblit and B. M. Sandmaier, Bioconjugate Chem., 2012, 23, 409–420 CrossRef CAS PubMed.
- P. R. Banks and D. M. Paquette, Bioconjugate Chem., 1995, 6, 447–458 CrossRef CAS.
- M. B. Smith and J. March, March's Advanced Organic Chemistry, John Wiley & Sons, 2007 Search PubMed.
- G. P. Smith, Bioconjugate Chem., 2006, 17, 501–506 CrossRef CAS PubMed.
- O. Mäkelä, Immunology, 1966, 10, 81–86 Search PubMed.
- J. M. Becker, M. Wilchek and E. Katchalski, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 2604–2607 CrossRef CAS.
- M. Brinkley, Bioconjugate Chem., 1991, 3, 2–13 CrossRef.
- G. W. Anderson, J. E. Zimmerman and F. M. Callahan, J. Am. Chem. Soc., 1963, 85, 3039 CrossRef CAS.
- G. W. Anderson, F. M. Callahan and J. E. Zimmerman, J. Am. Chem. Soc., 1967, 89, 178 CrossRef CAS.
- W.-C. Yang, H. Mirzaei, X. Liu and F. E. Regnier, Anal. Chem., 2006, 78, 4702–4708 CrossRef CAS PubMed.
- F. E. Regnier and S. Julka, Proteomics, 2006, 6, 3968–3979 CrossRef CAS PubMed.
- Y. Ishikawa, Y. Yamamoto, M. Otsubo, S. M. Theg and N. Tamura, Biochemistry, 2002, 41, 1972–1980 CrossRef CAS PubMed.
- B. Holmquist, S. Blumberg and B. L. Vallee, Biochemistry, 1976, 15, 4675–4680 CrossRef CAS.
- S. Blumberg and B. L. Vallee, Biochemistry, 1975, 14, 2410–2419 CrossRef CAS.
- M. D. Leavell, P. Novak, C. R. Behrens, J. S. Schoeniger and G. H. Kruppa, J. Am. Soc. Mass Spectrom., 2004, 15, 1604–1611 CrossRef CAS PubMed.
- P. Cuatrecasas and I. Parikh, Biochemistry, 1972, 11, 2291–2299 CrossRef CAS.
- B. T. Miller, T. J. Collins, G. T. Nagle and A. Kurosky, J. Biol. Chem., 1992, 267, 5060–5069 CAS.
- B. T. Miller and A. Kurosky, Biochem. Biophys. Res. Commun., 1993, 196, 461–467 CrossRef CAS PubMed.
- B. T. Miller, Biochem. Biophys. Res. Commun., 1996, 218, 377–382 CrossRef CAS PubMed.
- B. T. Miller, T. J. Collins, M. E. Rogers and A. Kurosky, Peptides, 1997, 18, 1585–1595 CrossRef CAS.
- S. Mädler, C. Bich, D. Touboul and R. Zenobi, J. Mass Spectrom., 2009, 44, 694–706 CrossRef PubMed.
- C. L. Swaim, J. B. Smith and D. L. Smith, Mikrochim. Acta, 2004, 15, 736–749 CAS.
- J. S. Smith, B. T. Miller, S. L. Knock and A. Kurosky, Anal. Biochem., 1991, 197, 247–253 CrossRef CAS.
- S. Kalkhof and A. Sinz, Anal. Bioanal. Chem., 2008, 392, 305–312 CrossRef CAS.
- A. J. Lomant and G. Fairbanks, J. Mol. Biol., 1976, 104, 243–261 CrossRef CAS.
- J. V. Staros, R. W. Wright and D. M. Swingle, Anal. Biochem., 1986, 156, 220–222 CrossRef CAS.
- N. Abello, H. A. M. Kerstjens, D. S. Postma and R. Bischoff, J. Proteome Res., 2007, 6, 4770–4776 CrossRef CAS PubMed.
- A. W. Yem, H. A. Zurcher-Neely, K. A. Richard, N. D. Staite, R. L. Heinrikson and M. R. Deibel, J. Biol. Chem., 1989, 264, 17691–17697 CAS.
- V. C. Lombardi and D. A. Schooley, Anal. Biochem., 2004, 331, 40–45 CrossRef CAS PubMed.
- M. Morpurgo, E. A. Bayer and M. Wilchek, J. Biochem. Biophys. Methods, 1999, 38, 17–28 CrossRef CAS.
- M. Cooper, A. Ebner, M. Briggs, M. Burrows, N. Gardner, R. Richardson and R. West, J. Fluoresc., 2004, 14, 145–150 CrossRef CAS.
- Y. Deng, Z. Hou, L. Wang, C. Cherian, J. Wu, A. Gangjee and L. H. Matherly, Mol. Pharmacol., 2008, 73, 1274–1281 CrossRef CAS PubMed.
- D. Kahne and W. C. Still, J. Am. Chem. Soc., 1988, 110, 7529–7534 CrossRef CAS.
- J. M. Becker and M. Wilchek, Biochim. Biophys. Acta, 1972, 264, 165–170 CrossRef CAS.
- M. Trester-Zedlitz, K. Kamada, S. K. Burley, D. Fenyö, B. T. Chait and T. W. Muir, J. Am. Chem. Soc., 2003, 125, 2416–2425 CrossRef CAS PubMed.
- R. Kluger and A. Alagic, Bioinorg. Chem., 2004, 32, 451–472 CrossRef CAS PubMed.
- M. A. Trakselis, S. C. Alley and F. T. Ishmael, Bioconjugate Chem., 2005, 16, 741–750 CrossRef CAS PubMed.
- X. Tang, G. R. Munske, W. F. Siems and J. E. Bruce, Anal. Chem., 2005, 77, 311–318 CrossRef CAS PubMed.
- J. D. Chavez, N. L. Liu and J. E. Bruce, J. Proteome Res., 2011, 10, 1528–1537 CrossRef CAS PubMed.
- M. Q. Müller, F. Dreiocker, C. H. Ihling, M. Schäfer and A. Sinz, Anal. Chem., 2010, 82, 6958–6968 CrossRef PubMed.
- J. W. Back, L. de Jong, A. O. Muijsers and C. G. de Koster, J. Mol. Biol., 2003, 331, 303–313 CrossRef CAS.
- M. M. Young, N. Tang, J. C. Hempel, C. M. Oshiro, E. W. Taylor, I. D. Kuntz, B. W. Gibson and G. Dollinger, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5802–5806 CrossRef CAS PubMed.
- S. Avrameas, Immunochemistry, 1969, 6, 43–52 CrossRef CAS.
- B. F. Erlanger, Pharmacol. Rev., 1973, 25, 271–280 CAS.
- M. C. Garnett, Adv. Drug Delivery Rev., 2001, 53, 171–216 CrossRef CAS.
- D. M. Schulz, C. Ihling, G. M. Clore and A. Sinz, Biochemistry, 2004, 43, 4703–4715 CrossRef CAS PubMed.
- S. Takeda, S. Tsukiji, H. Ueda and T. Nagamune, Org. Biomol. Chem., 2008, 6, 2187–2194 CAS.
- N. K. Damle, Nat. Biotechnol., 2008, 26, 884–885 CrossRef CAS PubMed.
- S. C. Alley, N. M. Okeley and P. D. Senter, Curr. Opin. Chem. Biol., 2010, 14, 529–537 CrossRef CAS PubMed.
- S. Webb, Nat. Biotechnol., 2011, 29, 297–298 CrossRef CAS PubMed.
- W.-D. Janthur, N. Cantoni and C. Mamot, Int. J. Mol. Sci., 2012, 13, 16020–16045 CrossRef CAS PubMed.
- R. S. Zolot, S. Basu and R. P. Million, Nat. Rev. Drug Discovery, 2013, 12, 259–260 CrossRef CAS PubMed.
- P. M. Abdella, P. K. Smith and G. P. Royer, Biochem. Biophys. Res. Commun., 1979, 87, 734–742 CrossRef CAS.
- D. A. Zarling, A. Watson and F. H. Bach, J. Immunol., 1980, 124, 913–920 CAS.
- R. J. Smith, R. A. Capaldi, D. Muchmore and F. Dahlquist, Biochemistry, 1978, 17, 3719–3723 CrossRef CAS.
- R. Reents, D. A. Jeyaraj and H. Waldmann, Drug Discovery Today, 2002, 7, 71–76 CrossRef CAS.
- G. Leriche, L. Chisholm and A. Wagner, Bioorg. Med. Chem., 2012, 20, 571–582 CrossRef CAS PubMed.
- Y. Jin Lee, Mol. BioSyst., 2008, 4, 816–823 RSC.
- A. Sinz, S. Kalkhof and C. Ihling, J. Am. Soc. Mass Spectrom., 2005, 16, 1921–1931 CrossRef CAS PubMed.
- M. Wilchek and E. A. Bayer, Methods Enzymol., 1990, 184, 123–138 CAS.
- E. A. Bayer and M. Wilchek, Methods Enzymol., 1990, 184, 138–160 CAS.
- X. Chen, K. Muthoosamy, A. Pfisterer, B. Neumann and T. Weil, Bioconjugate Chem., 2012, 23, 500–508 CrossRef CAS PubMed.
- E. R. Goldman, E. D. Balighian, H. Mattoussi, M. K. Kuno, J. M. Mauro, P. T. Tran and G. P. Anderson, J. Am. Chem. Soc., 2002, 124, 6378–6382 CrossRef CAS PubMed.
- M. R. Lockett, M. F. Phillips, J. L. Jarecki, D. Peelen and L. M. Smith, Langmuir, 2008, 24, 69–75 CrossRef CAS PubMed.
- M. Yang, R. L. Teeuwen, M. Giesbers, J. Baggerman, A. Arafat, F. A. de Wolf, J. C. van Hest and H. Zuilhof, Langmuir, 2008, 24, 7931–7938 CrossRef CAS PubMed.
- A. E. Bolton and W. M. Hunter, Biochem. J., 1973, 133, 529–539 CAS.
- J. J. Langone, Methods Enzymol., 1980, 70, 221–247 CAS.
- P. Anderson, Methods Enzymol., 1986, 119, 281–283 CAS.
- M. Glaser, V. A. Carroll, D. R. Collingridge, E. O. Aboagye, P. Price, R. Bicknell, A. L. Harris, S. K. Luthra and F. Brady, J. Labelled Compd. Radiopharm., 2002, 45, 1077–1090 CrossRef CAS PubMed.
- J. Russell, J. A. O'Donoghue, R. Finn, J. Koziorowski, S. Ruan, J. L. Humm and C. C. Ling, J. Nucl. Med., 2002, 43, 671–677 CAS.
- D. R. Collingridge, V. A. Carroll, M. Glaser and E. O. Aboagye, Cancer Res., 2002, 62, 5912–5919 CAS.
- J. J. Park, T. S. Lee, J. H. Kang, R. Song and G. J. Cheon, Appl. Radiat. Isot., 2011, 69, 56–62 CrossRef CAS PubMed.
- T. H. Ji and I. Ji, Anal. Biochem., 1982, 121, 286–289 CrossRef CAS.
- P. L. Ross, Mol. Cell. Proteomics, 2004, 3, 1154–1169 CAS.
- L. R. Zieske, J. Exp. Bot., 2006, 57, 1501–1508 CrossRef CAS PubMed.
- K. Aggarwal, L. H. Choe and K. H. Lee, Briefings Funct. Genomics Proteomics, 2006, 5, 112–120 CrossRef CAS PubMed.
- H. Mirzaei and F. Regnier, Anal. Chem., 2006, 78, 4175–4183 CrossRef CAS PubMed.
- R. F. Borch, M. D. Bernstein and H. D. Durst, J. Am. Chem. Soc., 1971, 93, 2897–2904 CrossRef CAS.
- G. R. Gray, Arch. Biochem. Biophys., 1974, 163, 426–428 CrossRef CAS.
- J. C. Gildersleeve, O. Oyelaran, J. T. Simpson and B. Allred, Bioconjugate Chem., 2008, 19, 1485–1490 CrossRef CAS PubMed.
- B. A. Schwartz and G. R. Gray, Arch. Biochem. Biophys., 1977, 181, 542–549 CrossRef CAS.
- R. T. Lee and Y. C. Lee, Biochemistry, 1980, 19, 156–163 CrossRef CAS.
- T. Freire, S. Bay, S. Vichier-Guerre, R. Lo-Man and C. Leclerc, Mini-Rev. Med. Chem., 2006, 6, 1357–1373 CrossRef CAS.
- S. Y. Wong, Curr. Opin. Struct. Biol., 1995, 5, 599–604 CrossRef CAS.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS PubMed.
- L. Cipolla, F. Peri and C. Airoldi, Anti-Cancer Agents Med. Chem., 2008, 8, 92–121 CrossRef CAS.
- S. B. Oppenheimer, M. Alvarez and J. Nnoli, Acta Histochem., 2008, 110, 6–13 CrossRef CAS PubMed.
- D. C. Tahmassebi and T. Sasaki, J. Org. Chem., 1998, 63, 728–731 CrossRef CAS PubMed.
- M. D. Bentley, M. J. Roberts and J. M. Harris, J. Pharm. Sci., 1998, 87, 1446–1449 CrossRef CAS PubMed.
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220–13221 CrossRef CAS PubMed.
- R. Mallik, T. Jiang and D. S. Hage, Anal. Chem., 2004, 76, 7013–7022 CrossRef CAS PubMed.
- R. Tripathi, S. Verma, J. Pandey and V. Tiwari, Curr. Org. Chem., 2008, 12, 1093–1115 CrossRef CAS.
- J. McFarland and M. Francis, J. Am. Chem. Soc., 2005, 127, 13490–13491 CrossRef CAS PubMed.
- G. Weber, Biochem. J., 1952, 51, 155–167 CAS.
- B. S. Hartley and V. Massey, Biochim. Biophys. Acta, 1956, 21, 58–70 CrossRef CAS.
- W. R. Gray, Methods Enzymol., 1967, 11, 469–475 CAS.
- C. Lefevre, H. C. Kang, R. P. Haugland, N. Malekzadeh, S. Arttamangkul and R. P. Haugland, Bioconjugate Chem., 1996, 7, 482–489 CrossRef CAS PubMed.
- J. A. J. Titus, R. R. Haugland, S. O. S. Sharrow and D. M. D. Segal, J. Immunol. Methods, 1981, 50, 193–204 CrossRef.
- N. Seiler, Methods Biochem. Anal., 1970, 18, 259–337 CAS.
- Methods in Molecular Biology, Humana Press, Totowa, NJ, 2009, vol. 588, pp. 43–48 Search PubMed.
- W. R. Gray, Methods Enzymol., 1967, 11, 139–151 CAS.
- C. Gros and B. Labouesse, Eur. J. Biochem., 1969, 7, 463–470 CrossRef CAS PubMed.
- R. E. Boyle, J. Org. Chem., 1966, 31, 3880–3882 CrossRef CAS.
- J. K. Lin and J. Y. Chang, Anal. Chem., 1975, 47, 1634–1638 CrossRef CAS.
- R. A. Evangelista, A. Pollak, B. Allore, A. F. Templeton, R. C. Morton and E. P. Diamandis, Clin. Biochem., 1988, 21, 173–178 CrossRef CAS.
- A. Andreoni, G. Bottiroli, A. Colasanti, M. C. Giangarè, P. Riccio, G. Roberti and P. Vaghi, J. Biochem. Biophys. Methods, 1994, 29, 157–172 CrossRef CAS.
- B. M. Gorovits and P. M. Horowitz, J. Biol. Chem., 1995, 270, 13057–13062 CrossRef CAS PubMed.
- D. J. Herzig, A. W. Rees and R. A. Day, Biopolymers, 1964, 2, 349–360 CrossRef CAS PubMed.
- A. M. Gold, Biochemistry, 1965, 4, 897–901 CrossRef CAS.
- R. S. Tipson, J. Org. Chem., 1944, 9, 235–241 CrossRef CAS.
- R. K. Crossland, W. E. Wells and V. J. Shiner, J. Am. Chem. Soc., 1971, 93, 4217–4219 CrossRef CAS.
- K. Nilsson and K. Mosbach, Biochem. Biophys. Res. Commun., 1981, 102, 449–457 CrossRef CAS.
- Z. Ma, C. Gao, J. Ji and J. Shen, Eur. Polym. J., 2002, 38, 2279–2284 CrossRef CAS.
- K. Pugdee, Y. Shibata, N. Yamamichi, H. Tsutsumi, M. Yoshinari, Y. Abiko and T. Hayakawa, Dent. Mater. J., 2007, 26, 647–655 CrossRef CAS.
- C. Delgado, J. N. Patel, G. E. Francis and D. Fisher, Biotechnol. Bioprocess Eng., 1990, 12, 119–128 CAS.
- G. E. Francis, D. Fisher, C. Delgado, F. Malik, A. Gardiner and D. Neale, Int. J. Hematol., 1998, 68, 1–18 CrossRef CAS.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2012, 64, 116–127 CrossRef PubMed.
- H.-J. Gais and S. Ruppert, Tetrahedron Lett., 1995, 36, 3837–3838 CrossRef CAS.
- H. T. F. Sanger, Biochem. J., 1951, 49, 481–490 Search PubMed.
- H. N. Eisen, S. Belman and M. E. Carsten, J. Am. Chem. Soc., 1953, 75, 4583–4585 CrossRef CAS.
- S. Shaltiel, Biochem. Biophys. Res. Commun., 1967, 29, 178–183 CrossRef CAS.
- K. Imai and Y. Watanabe, Anal. Chim. Acta, 1981, 130, 377–383 CrossRef CAS.
- A. A. Elbashir and F. Suliman, Appl. Spectrosc., 2011, 46, 222–241 CrossRef PubMed.
- S. Uchiyama, T. Santa, N. Okiyama, T. Fukushima and K. Imai, Biomed. Chromatogr., 2001, 15, 295–318 CrossRef CAS PubMed.
- T. Santa, T. Fukushima, T. Ichibangase and K. Imai, Biomed. Chromatogr., 2008, 22, 343–353 CrossRef CAS PubMed.
- P. Smialowski and D. Frishman, in Data Mining Techniques for the Life Sciences, Humana Press, 2010, vol. 609, pp. 385–400, DOI:10.1007/978-1-60327-241-4_22.
- D. A. Sutton, S. E. Drewes and U. Welz, Biochem. J., 1972, 130, 589–595 CAS.
- M. Lesniak, J. Gliemann, J. Roth, B. R. Easter, D. A. Sutton and S. E. Drewes, Hoppe-Seyler's Z. Physiol. Chem., 1979, 360, 467–472 CrossRef CAS.
- D. A. Sutton, B. R. Easter and S. E. Drewes, Hoppe-Seyler's Z. Physiol. Chem., 1976, 357, 971–976 CrossRef CAS.
- G. Losse and G. Mützler, Z. Chem., 1981, 21, 394–403 CrossRef CAS PubMed.
- D. L. Ladd and R. A. Snow, Anal. Biochem., 1993, 210, 258–261 CrossRef CAS PubMed.
- M. J. Hunter and M. L. Ludwig, J. Am. Chem. Soc., 1962, 84, 3491–3504 CrossRef CAS.
- R. R. Traut, A. Bollen, T.-T. Sun, J. W. B. Hershey, J. Sundberg and L. R. Pierce, Biochemistry, 1973, 12, 3266–3273 CrossRef CAS.
- M. Nomura, Science, 1973, 179, 864–873 CAS.
- L. Wofsy and S. J. Singer, Biochemistry, 1963, 2, 104–116 CrossRef CAS.
- H. J. H. Schramm, Hoppe-Seyler's Z. Physiol. Chem., 1975, 356, 1375–1379 CrossRef CAS.
- H. Bozler, K. D. Jany and G. Pfleiderer, Biochim. Biophys. Acta, 1983, 749, 238–243 CrossRef CAS.
- Y. C. Lee, C. P. Stowell and M. J. Krantz, Biochemistry, 1976, 15, 3956–3963 CrossRef CAS.
- R. R. Mehlhorn, M. M. Swanson, L. L. Packer and P. P. Smith, Arch. Biochem. Biophys., 1980, 204, 471–476 CrossRef CAS.
- J. B. J. Denny and R. M. R. Roberts, J. Biol. Chem., 1982, 257, 2460–2468 CAS.
- F. T. Wood, M. M. Wu and J. C. Gerhart, Anal. Biochem., 1975, 69, 339–349 CrossRef CAS.
- D. E. Wright, EP0539167, 1993, http://patentscope.wipo.int/search/en/detail.jsf?docId=EP12397580.
- K. Ampon and G. E. Means, Biotechnol. Bioeng., 1988, 32, 689–697 CrossRef CAS PubMed.
- J. I. Gavrilyuk, U. Wuellner and C. F. Barbas, Bioorg. Med. Chem. Lett., 2009, 19, 1421–1424 CrossRef CAS PubMed.
- J. I. Gavrilyuk, U. Wuellner, S. Salahuddin, R. K. Goswami, S. C. Sinha and C. F. Barbas III, Bioorg. Med. Chem. Lett., 2009, 19, 3716–3720 CrossRef CAS PubMed.
- L. F. Tietze, M. Arlt, M. Beller, K.-H. Glüsenkamp, E. Jähde and M. F. Rajewsky, Chem. Ber., 1991, 124, 1215–1221 CrossRef CAS PubMed.
- L. F. Tietze, C. Schroeter, S. Gabius, U. Brinck, A. Goerlach-Graw and H. J. Gabius, Bioconjugate Chem., 1991, 2, 148–153 CrossRef CAS.
- F. Wurm, C. Dingels, H. Frey and H.-A. Klok, Biomacromolecules, 2012, 13, 1161–1171 CrossRef CAS PubMed.
- F. Wurm, T. Steinbach and H.-A. Klok, Chem. Commun., 2013, 49, 7815–7817 RSC.
- D. Blakeslee and M. G. Baines, J. Immunol. Methods, 1976, 13, 305–320 CrossRef CAS.
- D. Blakeslee, J. Immunol. Methods, 1977, 17, 361–364 CrossRef CAS.
- A. Abuchowski, T. Es, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578–3581 CAS.
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. Es and F. F. Davis, J. Biol. Chem., 1977, 252, 3582–3586 CAS.
- A. Matsushima, Chem. Lett., 1980, 773–776 CrossRef CAS.
- S. S. Arpicco, F. F. Dosio, A. A. Bolognesi, C. C. Lubelli, P. P. Brusa, B. B. Stella, M. M. Ceruti and L. L. Cattel, Bioconjugate Chem., 2002, 13, 757–765 CrossRef CAS PubMed.
- Y. Ikeda, J. Katamachi, H. Kawasaki and Y. Nagasaki, Bioconjugate Chem., 2013, 24, 1824–1827 CrossRef CAS PubMed.
- F. R. Gurd, Methods Enzymol., 1971, 25, 424–438 Search PubMed.
- I. Forristal, J. Sulfur Chem., 2005, 26, 163–185 CrossRef CAS PubMed.
- E. Valero, S. Tambalo, P. Marzola, M. Ortega-Muñoz, F. J. Lopez-Jaramillo, F. Santoyo-Gonzalez, J. de Dios López, J. J. Delgado, J. J. Calvino, R. Cuesta, J. M. Domínguez-Vera and N. Gálvez, J. Am. Chem. Soc., 2011, 133, 4889–4895 CrossRef CAS PubMed.
- A. Megia-Fernandez, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Org. Biomol. Chem., 2013, 11, 2586–2596 CAS.
- R. L. Lundblad, Chemical Reagents for Protein Modification, CRC Press, 3rd edn, 2010 Search PubMed.
- M.-P. Brun and L. Gauzy-Lazo, Antibody–Drug conjugates, 2013, 1045, 173–187 Search PubMed.
- S. Mädler, M. Seitz, J. Robinson and R. Zenobi, Mikrochim. Acta, 2010, 21, 1775–1783 Search PubMed.
- B. T. Miller, M. E. Rogers, J. S. Smith and A. Kurosky, Anal. Biochem., 1994, 219, 240–248 CrossRef CAS PubMed.
- S. Mädler, S. Gschwind and R. Zenobi, Role of arginine in chemical cross-linking with N-hydroxysuccinimide esters, 2010, vol. 398 Search PubMed.
- F. Brotzel and H. Mayr, Org. Biomol. Chem., 2007, 5, 3814–3820 CAS.
- D. Gilis, S. Massar, N. J. Cerf and M. Rooman, Genome Biol., 2001, 2, 1–12 CrossRef.
- M. N. Fodje and S. Al-Karadaghi, Protein Eng., 2002, 15, 353–358 CrossRef CAS PubMed.
- S. J. Sirk, T. Olafsen, B. Barat, K. B. Bauer and A. M. Wu, Bioconjugate Chem., 2008, 19, 2527–2534 CrossRef CAS PubMed.
- J. J. Gorman, G. L. Corino and S. J. Mitchell, Eur. J. Biochem., 1987, 168, 169–179 CrossRef CAS PubMed.
- J. A. Burns, J. C. Butler, J. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648–2650 CrossRef CAS.
- E. B. Getz, M. Xiao, T. Chakrabarty, R. Cooke and P. R. Selvin, Anal. Biochem., 1999, 273, 73–80 CrossRef CAS PubMed.
- R. E. Hansen and J. R. Winther, Anal. Biochem., 2009, 394, 147–158 CrossRef CAS PubMed.
- K. Tyagarajan, E. Pretzer and J. E. Wiktorowicz, Electrophoresis, 2003, 24, 2348–2358 CrossRef CAS PubMed.
- D. E. Shafer, J. K. Inman and A. Lees, Anal. Biochem., 2000, 282, 161–164 CrossRef CAS PubMed.
- D. R. Goddard and L. Michaelis, J. Biol. Chem., 1935, 112, 361–371 CAS.
- N. Lundell and T. Schreitmüller, Anal. Biochem., 1999, 266, 31–47 CrossRef CAS PubMed.
- F. R. Gurd, Methods Enzymol., 1972, 25, 424–438 CAS.
- M. L. Nielsen, M. Vermeulen, T. Bonaldi, J. Cox, L. Moroder and M. Mann, Nat. Methods, 2008, 5, 459–460 CrossRef CAS PubMed.
- B. I. Gerwin, J. Biol. Chem., 1967, 242, 451–456 CAS.
- J. R. Kim, H. W. Yoon, K. S. Kwon, S. R. Lee and S. G. Rhee, Anal. Biochem., 2000, 283, 214–221 CrossRef CAS PubMed.
- N. J. Davis and S. L. Flitsch, Tetrahedron Lett., 1991, 32, 6793–6796 CrossRef CAS.
- D. Macmillan, R. Bill, K. Sage, D. Fern and S. Flitsch, Chem. Biol., 2001, 8, 133–145 CrossRef CAS.
- R. S. Swanwick, A. M. Daines, L.-H. Tey, S. L. Flitsch and R. K. Allemann, ChemBioChem, 2005, 6, 1338–1340 CrossRef CAS PubMed.
- J. F. Mackworth, Biochem. J., 1948, 42, 82–90 CAS.
- P. I. Clark and G. Lowe, J. Am. Chem. Soc., 1977, 24, 923–924 Search PubMed.
- P. I. Clark and G. Lowe, Eur. J. Biochem., 1978, 84, 293–299 CrossRef CAS PubMed.
- R. W. Furlanetto and E. T. Kaiser, J. Am. Chem. Soc., 1970, 92, 6980–6982 CrossRef CAS.
- R. W. Furlanetto, M. Mochizuki and E. T. Kaiser, Biochem. Biophys. Res. Commun., 1974, 58, 192–196 CrossRef CAS.
- K. J. Schwartz, Y. Nakagawa and E. T. Kaiser, J. Am. Chem. Soc., 1976, 98, 6369–6378 CrossRef CAS.
- E. Kaiser and D. Lawrence, Science, 1984, 226, 505–511 CAS.
- E. Friedmann and D. H. Marrian, Br. J. Pharmacol., 1949, 4, 105–108 CrossRef CAS PubMed.
- E. Friedmann, Biochim. Biophys. Acta, 1952, 9, 65–75 CrossRef CAS.
- T. Kajfež, B. Kamenar, V. Piližota and D. Fleš, Croat. Chem. Acta, 2003, 76, 343–346 Search PubMed.
- S. G. Bodige, M. A. Méndez-Rojas and W. H. Watson, J. Chem. Crystallogr., 1999, 29, 57–66 CrossRef CAS.
- D. G. Smyth, O. O. Blumenfeld and W. Konigsberg, Biochem. J., 1964, 91, 589–595 CAS.
- R. D. Brown and K. S. Matthews, J. Biol. Chem., 1979, 254, 5128–5134 CAS.
- Y. Ishii and S. S. Lehrer, Biophys. J., 1986, 50, 75–80 CrossRef CAS.
- C.-W. Wu, L. R. Yarbrough and F. Y. Wu, Biochemistry, 1976, 15, 2863–2868 CrossRef CAS.
- T. Climent, R. González-Luque and M. Merchán, J. Phys. Chem. A, 2003, 107, 6995–7003 CrossRef CAS.
- P. Schelté, C. Boeckler, B. Frisch and F. Schuber, Bioconjugate Chem., 2000, 11, 118–123 CrossRef PubMed.
- S. Yoshitake, Y. Yamada, E. Ishikawa and R. Masseyeff, Eur. J. Biochem., 1979, 101, 395–399 CrossRef CAS PubMed.
- S. Yoshitake, M. Imagawa, E. Ishikawa, Y. Niitsu, I. Urushizaki, M. Nishiura, R. Kanazawa, H. Kurosaki, S. Tachibana and N. Nakazawa, J. Biochem., 1982, 92, 1413–1424 CAS.
- S. Hashida, M. Imagawa, S. Inoue, K. H. Ruan and E. Ishikawa, J. Appl. Biochem., 1984, 6, 56–63 CAS.
- S. S. Ghosh, P. M. Kao, A. W. McCue and H. L. Chappelle, Bioconjugate Chem., 1990, 1, 71–76 CrossRef CAS.
- N. Dickgreber, P. Stoitzner, Y. Bai, K. M. Price, K. J. Farrand, K. Manning, C. E. Angel, P. R. Dunbar, F. Ronchese, J. D. Fraser, B. T. Bäckström and I. F. Hermans, J. Immunol., 2009, 182, 1260–1269 CrossRef CAS.
- T. Hu and Z. Su, Biotechnol. Lett., 2002, 24, 275–278 CrossRef CAS.
- K. Kafi, D. J. Betting, R. E. Yamada, M. Bacica, K. K. Steward and J. M. Timmerman, Mol. Immunol., 2009, 46, 448–456 CrossRef CAS PubMed.
- G. K. Ehrlich, H. Michel, T. Truitt and W. Riboulet, Bioconjugate Chem., 2013, 24, 2015–2024 CrossRef CAS PubMed.
- F. Kratz, A. Warnecke, K. Scheuermann, C. Stockmar, J. Schwab, P. Lazar, P. Drückes, N. Esser, J. Drevs, D. Rognan, C. Bissantz, C. Hinderling, G. Folkers, I. Fichtner and C. Unger, J. Med. Chem., 2002, 45, 5523–5533 CrossRef CAS PubMed.
- A. Warnecke, I. Fichtner, G. Sass and F. Kratz, Arch. Pharm., 2007, 340, 389–395 CrossRef CAS PubMed.
- V. Kersemans, B. Cornelissen, M. D. Minden, J. Brandwein and R. M. Reilly, J. Nucl. Med., 2008, 49, 1546–1554 CrossRef CAS PubMed.
- D. J. Betting, K. Kafi, A. Abdollahi-Fard, S. A. Hurvitz and J. M. Timmerman, J. Immunol., 2008, 181, 4131–4140 CrossRef CAS.
- A. Alisio, J. Biol. Chem., 2004, 279, 26540–26545 CrossRef CAS PubMed.
- L. Giron-Monzon, J. Biol. Chem., 2004, 279, 49338–49345 CrossRef CAS PubMed.
- N. S. Green, E. Reisler and K. N. Houk, Protein Sci., 2008, 10, 1293–1304 CrossRef PubMed.
- F. T. Ishmael, M. A. Trakselis and S. J. Benkovic, J. Biol. Chem., 2003, 278, 3145–3152 CrossRef CAS PubMed.
- O. V. Kovalenko, X. Yang, T. V. Kolesnikova and M. E. Hemler, Biochem. J., 2004, 377, 407–417 CrossRef CAS PubMed.
- Y. K. Kwon, J. Cell Biol., 2003, 163, 375–384 CrossRef CAS PubMed.
- M. A. Stalteri and S. J. Mather, Bioconjugate Chem., 1995, 6, 179–186 CrossRef CAS.
- E. E. Leary Swan, K. C. Popat and T. A. Desai, Biomaterials, 2005, 26, 1969–1976 CrossRef CAS PubMed.
- A. Misra and P. Dwivedi, Anal. Biochem., 2007, 369, 248–255 CrossRef CAS PubMed.
- G. Elia, Curr. Protein Pept. Sci., 2010 Search PubMed , ch. 3. Unit 3.6.
- Y. Oda, T. Nagasu and B. T. Chait, Nat. Biotechnol., 2001, 19, 379–382 CrossRef CAS PubMed.
- S. Khurana, M. Kennedy, L. R. King and H. Golding, J. Virol., 2005, 79, 6791–6800 CrossRef CAS PubMed.
- H. Golding, J. Biol. Chem., 2005, 280, 29570–29577 CrossRef CAS PubMed.
- F.-U. Gast, I. Franke, G. Meiss and A. Pingoud, J. Biotechnol., 2001, 87, 131–141 CrossRef CAS.
- I. H. Cho, E. H. Paek, H. Lee, J. W. Choi and S. H. Paek, Biotechnol. Bioprocess Eng., 2004, 9, 112–117 CrossRef CAS.
- G. Y. Kang, G. Y. Han, J. Y. Kang, I.-H. Cho, H.-H. Park, S.-H. Paek and T. S. Kim, Sens. Actuators, B, 2006, 117, 332–338 CrossRef CAS PubMed.
- I.-H. Cho, E.-H. Paek, H. Lee, J. Y. Kang, T. S. Kim and S.-H. Paek, Anal. Biochem., 2007, 365, 14–23 CrossRef CAS PubMed.
- A. Pompella, S. Dominici, C. Cambiaggi, J. Frank and H. K. Biesalski, Methods Mol. Biol., 2002, 196, 305–312 CAS.
- J. M. Kuiper, R. Pluta, W. H. C. Huibers, F. Fusetti, E. R. Geertsma and B. Poolman, Protein Sci., 2009, 18, 1033–1041 CrossRef CAS PubMed.
- X. Chen, Y. Zhou, X. Peng and J. Yoon, Chem. Soc. Rev., 2010, 39, 2120 RSC.
- Y. Zhang, V. S. Bhatt, G. Sun, P. G. Wang and A. F. Palmer, Bioconjugate Chem., 2008, 19, 2221–2230 CrossRef CAS PubMed.
- S. Ahlgren, A. Orlova, D. Rosik, M. Sandström, A. Sjöberg, B. Baastrup, O. Widmark, G. Fant, J. Feldwisch and V. Tolmachev, Bioconjugate Chem., 2008, 19, 235–243 CrossRef CAS PubMed.
- C. A. Boswell, J. Marik, M. J. Elowson, N. A. Reyes, S. Ulufatu, D. Bumbaca, V. Yip, E. E. Mundo, N. Majidy, M. Van Hoy, S. N. Goriparthi, A. Trias, H. S. Gill, S. P. Williams, J. R. Junutula, P. J. Fielder and L. A. Khawli, J. Med. Chem., 2013, 56, 9418–9426 CrossRef CAS PubMed.
- M. K. Han, P. Lin, D. Paek, J. J. Harvey, E. Fuior and J. R. Knutson, Biochemistry, 2002, 41, 3468–3476 CrossRef CAS PubMed.
- J. M. West, J. Biol. Chem., 2003, 279, 945–951 CrossRef PubMed.
- F. Fernandes, L. M. S. Loura, R. Koehorst, R. B. Spruijt, M. A. Hemminga, A. Fedorov and M. Prieto, Biophys. J., 2004, 87, 344–352 CrossRef CAS PubMed.
- T. G. Nishino, K. Kitano, K. Kojima, T. Ogishima, A. Ito and S. Kitada, J. Biochem., 2007, 141, 889–895 CrossRef CAS PubMed.
- S. K. Wright and R. E. Viola, Anal. Biochem., 1998, 265, 8–14 CrossRef CAS PubMed.
- P. G. Mastroberardino, A. L. Orr, X. Hu, H. M. Na and J. T. Greenamyre, Free Radical Biol. Med., 2008, 45, 971–981 CrossRef CAS PubMed.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS PubMed.
- S. Matsui and H. Aida, J. Chem. Soc., Perkin Trans. 2, 1978, 1277–1280 RSC.
- J. Kalia and R. T. Raines, Bioorg. Med. Chem. Lett., 2007, 17, 6286–6289 CrossRef CAS PubMed.
- L. M. Tedaldi, M. E. B. Smith, R. I. Nathani and J. R. Baker, Chem. Commun., 2009, 6583–6585 RSC.
- D. D. Roberts, S. D. Lewis, D. P. Ballou, S. T. Olson and J. A. Shafer, Biochemistry, 1986, 25, 5595–5601 CrossRef CAS.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- C. P. Ryan, M. E. B. Smith, F. F. Schumacher, D. Grohmann, D. Papaioannou, G. Waksman, F. Werner, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 5452–5454 RSC.
- L. Castañeda, A. Maruani, F. F. Schumacher, E. Miranda, V. Chudasama, K. A. Chester, J. R. Baker, M. E. B. Smith and S. Caddick, Chem. Commun., 2013, 49, 8187–8189 RSC.
- J. Porath and R. Axén, Methods Enzymol., 1976, 44, 19–45 CAS.
- M. S. Masri and M. Friedman, J. Protein Chem., 1988, 7, 49–54 CrossRef CAS.
- G. Houen and O. M. Jensen, J. Immunol. Methods, 1995, 181, 187–200 CrossRef CAS.
- M. Morpurgo, F. M. Veronese, D. Kachensky and J. M. Harris, Bioconjugate Chem., 1996, 7, 363–368 CrossRef CAS PubMed.
- D. C. Meadows and J. Gervay-Hague, Med. Res. Rev., 2006, 26, 793–814 CrossRef CAS PubMed.
- P. Bailon and C.-Y. Won, Expert Opin. Drug Delivery, 2009, 6, 1–16 CrossRef CAS PubMed.
- F. Rusmini, Z. Zhong and J. Feijen, Biomacromolecules, 2007, 8, 1775–1789 CrossRef CAS PubMed.
- M. Ortega-Muñoz, J. Morales-Sanfrutos, A. Megia-Fernandez, F. J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, J. Mater. Chem., 2010, 20, 7189–7196 RSC.
- J. Morales-Sanfrutos, J. Lopez-Jaramillo, M. Ortega-Muñoz, A. Megia-Fernandez, F. Perez-Balderas, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Org. Biomol. Chem., 2010, 8, 667–675 CAS.
- F. J. Lopez-Jaramillo, M. Ortega-Muñoz, A. Megia-Fernandez, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Bioconjugate Chem., 2012, 23, 846–855 CrossRef CAS PubMed.
- L. Li, S.-W. Tsai, A.-L. Anderson, D. A. Keire, A. A. Raubitschek and J. E. Shively, Bioconjugate Chem., 2002, 13, 110–115 CrossRef CAS PubMed.
- J. Morales-Sanfrutos, F. J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, J. Org. Chem., 2010, 75, 4039–4047 CrossRef CAS PubMed.
- F. Lopez-Jaramillo, F. Hernandez Mateo, F. Santoyo Gonzalez and E. Leung, Integr. Proteom., 2012, 16, 301–326 Search PubMed.
- H. Ovaa, et al. , Angew. Chem., Int. Ed., 2003, 42, 3626–3629 CrossRef CAS PubMed.
- T. Posner, Chem. Ber., 1905, 38, 646–657 CrossRef PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- A. Dondoni and A. Marra, Chem. Soc. Rev., 2012, 41, 573–586 RSC.
- N. Floyd, B. Vijayakrishnan, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2009, 121, 7938–7942 CrossRef PubMed.
- A. Dondoni, A. Massi, P. Nanni and A. Roda, Chem. – Eur. J., 2009, 15, 11444–11449 CrossRef CAS PubMed.
- L.-Z. Wu, Y.-B. Sheng, J.-B. Xie and W. Wang, J. Mol. Struct., 2008, 882, 101–106 CrossRef CAS PubMed.
- K. C. A. Garber and E. E. Carlson, ACS Chem. Biol., 2013, 8, 1671–1676 CrossRef CAS PubMed.
- R. M. Stolz and B. H. Northrop, J. Org. Chem., 2013, 78, 8105–8116 CrossRef CAS PubMed.
- L. Markey, S. Giordani and E. M. Scanlan, J. Org. Chem., 2013, 78, 4270–4277 CrossRef CAS PubMed.
- A. Massi and D. Nanni, Org. Biomol. Chem., 2012, 10, 3791–3807 CAS.
- H. Bader, L. C. Cross, I. Heilbron and E. Jones, J. Chem. Soc., 1949, 619–623 RSC.
- M. Lo Conte, S. Pacifico, A. Chambery, A. Marra and A. Dondoni, J. Org. Chem., 2010, 75, 4644–4647 CrossRef CAS PubMed.
- M. L. Conte, S. Staderini, A. Marra, M. Sanchez-Navarro, B. G. Davis and A. Dondoni, Chem. Commun., 2011, 47, 11086–11088 RSC.
- M. W. Crankshaw and G. A. Grant, Curr. Protein Pept. Sci., 2001 Search PubMed , ch. 15. unit 15.1.
- M. L. Anson, J. Gen. Physiol., 1940, 23, 321–331 CrossRef CAS.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS.
- G. L. Kenyon and T. W. Bruice, Methods Enzymol., 1977, 47, 407–430 CAS.
- A. Fontana, E. Scoffone and C. A. Benassi, Biochemistry, 1968, 7, 980–986 CrossRef CAS.
- T. P. King, Y. Li and L. Kochoumian, Biochemistry, 1978, 17, 1499–1506 CrossRef CAS.
- W. M. Macindoe, A. H. van Oijen and G.-J. Boons, Chem. Commun., 1998, 847–848 RSC.
- B. G. Davis, M. A. Maughan, M. P. Green, A. Ullman and J. B. Jones, Tetrahedron: Asymmetry, 2000, 11, 245–262 CrossRef CAS.
- D. P. Gamblin, P. Garnier, S. J. Ward, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Org. Biomol. Chem., 2003, 1, 3642 CAS.
- B. G. Davis, Macromolecules, 2002, 102, 579–602 CAS.
- Y. J. Zhao, Y. Q. Zhai, Z. G. Su and G. H. Ma, Polym. Adv. Technol., 2010, 21, 867–873 CrossRef CAS PubMed.
- D. P. Gamblin, P. Garnier, S. van Kasteren, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Angew. Chem., Int. Ed., 2004, 116, 846–851 CrossRef PubMed.
- G. J. L. Bernardes, D. P. Gamblin and B. G. Davis, Angew. Chem., Int. Ed., 2006, 118, 4111–4115 CrossRef PubMed.
- G. J. L. Bernardes, J. P. Marston, A. S. Batsanov, J. A. K. Howard and B. G. Davis, Chem. Commun., 2007, 3145–3147 RSC.
- M. Steiner, I. Hartmann, E. Perrino, G. Casi, S. Brighton, I. Jelesarov, G. J. Bernardes and D. Neri, Chem. Sci., 2013, 4, 297–302 RSC.
- M. Kunstelj, K. Fidler, S. Skrajnar, M. Kenig, V. Smilović, M. Kusterle, S. Caserman, I. Zore, V. G. Porekar and S. Jevševar, Bioconjugate Chem., 2013, 24, 889–896 CrossRef CAS PubMed.
- G. J. L. Bernardes, E. J. Grayson, S. Thompson, J. M. Chalker, J. C. Errey, F. El Oualid, T. D. W. Claridge and B. G. Davis, Angew. Chem., Int. Ed., 2008, 47, 2244–2247 CrossRef CAS PubMed.
- K. Whiteman, C. Audette and S. D. Wilhelm, Bioconjugate Chem., 2009, 21, 84–92 Search PubMed.
- M. S. Rosendahl, D. H. Doherty, D. J. Smith, S. J. Carlson, E. A. Chlipala and G. N. Cox, Bioconjugate Chem., 2005, 16, 200–207 CrossRef CAS PubMed.
- M. T. Petersen, P. H. Jonson and S. B. Petersen, Protein Eng., 1999, 12, 535–548 CrossRef CAS PubMed.
- H. J. Leung, G. Xu, M. Narayan and H. A. Scheraga, J. Pept. Res., 2005, 65, 47–54 CrossRef CAS PubMed.
- T. Arakawa, S. J. Prestrelski, L. O. Narhi, T. C. Boone and W. C. Kenney, J. Protein Chem., 1993, 12, 525–531 CrossRef CAS.
- S. Brocchini, S. Balan, A. Godwin, J.-W. Choi, M. Zloh and S. Shaunak, Nat. Protoc., 2006, 1, 2241–2252 CrossRef CAS PubMed.
- J. M. Thornton, J. Mol. Biol., 1981, 151, 261–287 CrossRef CAS.
- R. Bhattacharyya, D. Pal and P. Chakrabarti, Protein Eng., 2004, 17, 795–808 CrossRef CAS PubMed.
- S. Shaunak, A. Godwin, J.-W. Choi, S. Balan, E. Pedone, D. Vijayarangam, S. Heidelberger, I. Teo, M. Zloh and S. Brocchini, Nat. Chem. Biol., 2006, 2, 312–313 CrossRef CAS PubMed.
- S. Balan, J.-W. Choi, A. Godwin, I. Teo, C. M. Laborde, S. Heidelberger, M. Zloh, S. Shaunak and S. Brocchini, Bioconjugate Chem., 2007, 18, 61–76 CrossRef CAS PubMed.
- S. Brocchini, A. Godwin, S. Balan, J.-W. Choi, M. Zloh and S. Shaunak, Adv. Drug Delivery Rev., 2008, 60, 3–12 CrossRef CAS PubMed.
- H. Khalili, A. Godwin, J.-W. Choi, R. Lever and S. Brocchini, Bioconjugate Chem., 2012, 23, 2262–2277 CrossRef CAS PubMed.
- A. Lewis, Y. Tang, S. Brocchini, J.-W. Choi and A. Godwin, Bioconjugate Chem., 2008, 19, 2144–2155 CrossRef CAS PubMed.
- L. Castañeda, Z. V. F. Wright, C. Marculescu, T. M. Tran, V. Chudasama, A. Maruani, E. A. Hull, J. P. M. Nunes, R. J. Fitzmaurice, M. E. B. Smith, L. H. Jones, S. Caddick and J. R. Baker, Tetrahedron Lett., 2013, 54, 3493–3495 CrossRef PubMed.
- M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 1847–1852 CrossRef CAS PubMed.
- F. F. Schumacher, V. A. Sanchania, B. Tolner, Z. V. F. Wright, C. P. Ryan, M. E. B. Smith, J. M. Ward, S. Caddick, C. W. M. Kay, G. Aeppli, K. A. Chester and J. R. Baker, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- M. E. B. Smith, J. Baker, F. F. Schumacher, S. Caddick, V. Chudasama and A. Maruani, WO2013132268, 2013, http://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013132268.
- F. F. Schumacher, M. Nobles, C. P. Ryan, M. E. Smith, A. Tinker, S. Caddick and J. R. Baker, Bioconjugate Chem., 2011, 22, 132–136 CrossRef CAS PubMed.
- M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, Chem. Commun., 2012, 48, 4064–4066 RSC.
- J. Collins, J. Tanaka, P. Wilson, K. Kempe, T. P. Davis, M. P. McIntosh, M. R. Whittaker and D. M. Haddleton, Bioconjugate Chem., 2015, 26, 633–638 CrossRef CAS PubMed.
- V. Chudasama, M. E. B. Smith, F. F. Schumacher, D. Papaioannou, G. Waksman, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 8781–8783 RSC.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
- M. J. Tucker, J. R. Courter, J. Chen, O. Atasoylu, A. B. Smith and R. M. Hochstrasser, Angew. Chem., Int. Ed., 2010, 49, 3612–3616 CrossRef CAS PubMed.
- S. P. Brown and A. B. Smith, J. Am. Chem. Soc., 2015, 137, 4034–4037 CrossRef CAS PubMed.
- P. Wilson, A. Anastasaki, M. R. Owen, K. Kempe, D. M. Haddleton, S. K. Mann, A. P. R. Johnston, J. F. Quinn, M. R. Whittaker, P. J. Hogg and T. P. Davis, J. Am. Chem. Soc., 2015, 137, 4215–4222 CrossRef CAS PubMed.
- C. J. Chapman, K. J. Wadsworth and C. G. Frost, J. Organomet. Chem., 2003, 680, 206–211 CrossRef CAS.
- C. J. Chapman, A. Matsuno, C. G. Frost and M. C. Willis, Chem. Commun., 2007, 3903–3905 RSC.
- C. J. Chapman, J. D. Hargrave, G. Bish and C. G. Frost, Tetrahedron, 2008, 64, 9528–9539 CrossRef CAS PubMed.
- L. Navarre, R. Martinez, J.-P. Genet and S. Darses, J. Am. Chem. Soc., 2008, 130, 6159–6169 CrossRef CAS PubMed.
- G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 5052–5053 CrossRef CAS PubMed.
- J. M. Chalker, L. Lercher, N. R. Rose, C. J. Schofield and B. G. Davis, Angew. Chem., Int. Ed., 2012, 124, 1871–1875 CrossRef PubMed.
- P. N. Collier, A. D. Campbell, I. Patel and R. J. K. Taylor, Tetrahedron Lett., 2000, 41, 7115–7119 CrossRef CAS.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- I. Photaki, J. Am. Chem. Soc., 1963, 85, 1123–1126 CrossRef CAS.
- D. H. Strumeyer, W. N. White and D. E. Koshland, Proc. Natl. Acad. Sci. U. S. A., 1963, 50, 931–935 CrossRef CAS.
- H. Weiner, W. N. White, D. G. Hoare and D. E. Koshland, J. Am. Chem. Soc., 1966, 88, 3851–3859 CrossRef CAS.
- D. H. Rich, J. Tam, P. Mathiaparanam, J. A. Grant and C. Mabuni, J. Chem. Soc., Chem. Commun., 1974, 897–898 RSC.
- J. Chalker, G. Bernardes, Y. Lin and B. Davis, Chem. – Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- T. J. Holmes and R. G. Lawton, J. Am. Chem. Soc., 1977, 99, 1984–1986 CrossRef CAS.
- F. C. Rowan, M. Richards, R. A. Bibby, A. Thompson, R. Bayliss and J. Blagg, ACS Chem. Biol., 2013, 8, 2184–2191 CrossRef CAS PubMed.
- R. I. Nathani, P. Moody, V. Chudasama, M. E. B. Smith, R. J. Fitzmaurice and S. Caddick, Chem. Sci., 2013, 4, 3455–3458 RSC.
- K. Hashimoto, M. Sakai, T. Okuno and H. Shirahama, Chem. Commun., 1996, 1139–1140 RSC.
- N. M. Okeley, Y. Zhu and W. A. van der Donk, Org. Lett., 2000, 2, 3603–3606 CrossRef CAS PubMed.
- F. P. Seebeck and J. W. Szostak, J. Am. Chem. Soc., 2006, 128, 7150–7151 CrossRef CAS PubMed.
- J. Wang, S. M. Schiller and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 119, 6973–6975 CrossRef PubMed.
- J. Guo, J. Wang, J. S. Lee and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 120, 6499–6501 CrossRef PubMed.
- Y. O. You, M. R. Levengood, L. A. F. Ihnken, A. K. Knowlton and W. A. van der Donk, ACS Chem. Biol., 2009, 4, 379–385 CrossRef CAS PubMed.
- Y. A. Lin, O. Boutureira and L. Lercher, J. Am. Chem. Soc., 2013, 135, 12156–12159 CrossRef CAS PubMed.
- H. Lindley, Nature, 1956, 178, 647–648 CrossRef CAS PubMed.
- M. A. Raftery and R. D. Cole, J. Biol. Chem., 1966, 241, 3457–3461 CAS.
- M. D. Simon, F. Chu, L. R. Racki, C. C. de la Cruz, A. L. Burlingame, B. Panning, G. J. Narlikar and K. M. Shokat, Cell, 2007, 128, 1003–1012 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- C. Zhang, A. M. Spokoyny, Y. Zou, M. D. Simon and B. L. Pentelute, Angew. Chem., Int. Ed., 2013, 52, 1–6 CrossRef PubMed.
- N. Toda, S. Asano and C. F. Barbas III, Angew. Chem., Int. Ed., 2013, 52, 12592–12596 CrossRef CAS PubMed.
- H.-Y. Shiu, T.-C. Chan, C.-M. Ho, Y. Liu, M.-K. Wong and C.-M. Che, Chem. – Eur. J., 2009, 15, 3839–3850 CrossRef CAS PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS PubMed.
- S. Chen, J. Touati and C. Heinis, Chem. Commun., 2014, 50, 5267–5269 RSC.
- A. O.-Y. Chan, J. L.-L. Tsai, V. K.-Y. Lo, G.-L. Li, M.-K. Wong and C.-M. Che, Chem. Commun., 2013, 49, 1428–1430 RSC.
- P. D. Halphen and T. C. Owen, J. Org. Chem., 1973, 38, 3507–3510 CrossRef CAS.
- W. E. Truce and G. J. W. Tichenor, J. Org. Chem., 1972, 37, 2391–2396 CrossRef CAS.
- O. De Lucchi, V. Lucchini, C. Marchioro, G. Valle and G. Modena, J. Org. Chem., 1986, 51, 1457–1466 CrossRef CAS.
- M. Journet, A. Rouillard, D. Cai and R. D. Larsen, J. Org. Chem., 1997, 62, 8630–8631 CrossRef CAS.
- O. Arjona, R. Medel, J. Rojas, A. M. Costa and J. Vilarrasa, Tetrahedron Lett., 2003, 44, 6369–6373 CrossRef CAS.
- B. A. Trofimov, A. G. Mal'kina, O. A. Shemyakina, V. V. Nosyreva, A. P. Borisova, S. S. Khutsishvili and L. B. Krivdin, Synthesis, 2009, 3136–3142 CrossRef CAS PubMed.
- H.-Y. Shiu, H.-C. Chong, Y.-C. Leung, M.-K. Wong and C.-M. Che, Chem. – Eur. J., 2010, 16, 3308–3313 CrossRef CAS PubMed.
- O. Koniev, G. Leriche, M. Nothisen, J.-S. Remy, J.-M. Strub, C. Schaeffer-Reiss, A. Van Dorsselaer, R. Baati and A. Wagner, Bioconjugate Chem., 2014, 25, 202–206 CrossRef CAS PubMed.
- S. Kolodych, O. Koniev, Z. Baatarkhuu, J.-Y. Bonnefoy, F. Debaene, S. Cianférani, A. Van Dorsselaer and A. Wagner, Bioconjugate Chem., 2015, 26, 197–200 CrossRef CAS PubMed.
- V. Hong, A. A. Kislukhin and M. G. Finn, J. Am. Chem. Soc., 2009, 131, 9986–9994 CrossRef CAS PubMed.
- S. Dziadek, S. Jacques and D. R. Bundle, Chemistry, 2008, 14, 5908–5917 CrossRef CAS PubMed.
- M. W. Jones, G. Mantovani, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, Chem. Commun., 2009, 5272–5274 RSC.
- C. Haase and O. Seitz, Justus Liebigs Ann. Chem., 2009, 2096–2101 CAS.
- F. K. Hansen, K. Ha, E. Todadze, A. Lillicotch, A. Frey and A. R. Katritzky, Org. Biomol. Chem., 2011, 9, 7162–7167 CAS.
- K. Ha, M. Chahar, J.-C. M. Monbaliu, E. Todadze, F. K. Hansen, A. A. Oliferenko, C. E. Ocampo, D. Leino, A. Lillicotch, C. V. Stevens and A. R. Katritzky, J. Org. Chem., 2012, 77, 2637–2648 CrossRef CAS PubMed.
- O. Bol'shakov, J. Kovacs, M. Chahar, K. Ha, L. Khelashvili and A. R. Katritzky, J. Pept. Sci., 2012, 18, 704–709 CrossRef PubMed.
- S. S. Panda, C. El-Nachef, K. Bajaj, A. O. Al-Youbi, A. Oliferenko and A. R. Katritzky, Chem. Biol. Drug Des., 2012, 80, 821–827 CAS.
- J.-C. M. Monbaliu, G. Dive, C. V. Stevens and A. R. Katritzky, J. Chem. Theory Comput., 2013, 9, 927–934 CrossRef CAS.
- K. Gevaert, P. Van Damme, L. Martens and J. Vandekerckhove, Anal. Biochem., 2005, 345, 18–29 CrossRef CAS PubMed.
- D. E. Koshland, Y. D. Karkhanis and H. G. Latham, J. Am. Chem. Soc., 1964, 86, 1448–1450 CrossRef CAS.
- W. E. Savige and A. Fontana, Methods Enzymol., 1977, 47, 442–453 CAS.
- J. E. Bell, F. J. Castellino, I. P. Trayer and R. L. Hill, J. Biol. Chem., 1975, 250, 7579–7585 CAS.
- H. R. Horton and D. E. Koshland, J. Am. Chem. Soc., 1965, 87, 1126–1132 CrossRef CAS.
- M. Strohalm, J. Šantrůček, R. Hynek and M. Kodíček, Biochem. Biophys. Res. Commun., 2004, 323, 1134–1138 CrossRef CAS PubMed.
- H. J. Teuber, O. Glosauer and U. Hochmuth, Eur. J. Inorg. Chem., 1964, 557–562 CAS.
- A. Foettinger, M. Melmer, A. Leitner and W. Lindner, Bioconjugate Chem., 2007, 18, 1678–1683 CrossRef CAS PubMed.
- M. Källberg, H. Wang, S. Wang, J. Peng, Z. Wang, H. Lu and J. Xu, Nat. Protoc., 2012, 7, 1511–1522 CrossRef PubMed.
- A. Foettinger, A. Leitner and W. Lindner, J. Mass Spectrom., 2006, 41, 623–632 CrossRef CAS PubMed.
- A. Foettinger, A. Leitner and W. Lindner, J. Proteome Res., 2007, 6, 3827–3834 CrossRef CAS PubMed.
- J. Antos and M. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS PubMed.
- T. Ye and M. A. McKervey, Macromolecules, 1994, 94, 1091–1160 CAS.
- J. M. Antos, J. M. McFarland, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 6301–6308 CrossRef CAS PubMed.
- J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS PubMed.
- B. V. Popp and Z. T. Ball, J. Am. Chem. Soc., 2010, 132, 6660–6662 CrossRef CAS PubMed.
- D. N. Woolfson, Adv. Protein Chem., 2005, 70, 79–112 CAS.
- Z. Chen, B. V. Popp, C. L. Bovet and Z. T. Ball, ACS Chem. Biol., 2011, 6, 920–925 CrossRef CAS PubMed.
- J. L. Howland, The Encyclopedia of Molecular Biology, Biochemical Education, 1995 Search PubMed.
- S. Li and M. Hong, J. Am. Chem. Soc., 2011, 133, 1534–1544 CrossRef CAS PubMed.
- X. Li, H. Ma, S. Dong, X. Duan and S. Liang, Talanta, 2004, 62, 367–371 CrossRef CAS PubMed.
- X. Li, H. Ma, L. Nie, M. Sun and S. Xiong, Anal. Chim. Acta, 2004, 515, 255–260 CrossRef CAS PubMed.
- R. E. Parker and N. S. Isaacs, Chem. Rev., 1959, 59, 737–799 CrossRef CAS.
- Y. Takaoka, H. Tsutsumi, N. Kasagi, E. Nakata and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 3273–3280 CrossRef CAS PubMed.
- H. Wakabayashi, M. Miyagawa, Y. Koshi, Y. Takaoka, S. Tsukiji and I. Hamachi, Chem. – Asian J., 2008, 3, 1134–1139 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Macromolecules, 1995, 95, 2457–2483 CAS.
- M. Meldal and C. W. Tornøe, Macromolecules, 2008, 108, 2952–3015 CAS.
- J. Z. Hearon, J. Natl. Cancer Inst., 1948, 9, 1–11 CAS.
- X. Wang, J. Jia, Z. Huang, M. Zhou and H. Fei, Chem. – Eur. J., 2011, 17, 8028–8032 CrossRef CAS PubMed.
- D.-L. Ma, W.-L. Wong, W.-H. Chung, F.-Y. Chan, P.-K. So, T.-S. Lai, Z.-Y. Zhou, Y.-C. Leung and K.-Y. Wong, Angew. Chem., Int. Ed., 2008, 47, 3735–3739 CrossRef CAS PubMed.
- K. Uchida and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 4544–4548 CrossRef CAS.
- R. Zamora, M. Alaiz and F. J. Hidalgo, Chem. Res. Toxicol., 1999, 12, 654–660 CrossRef CAS PubMed.
- F. J. Hidalgo and R. Zamora, J. Biol. Chem., 1993, 268, 16190–16197 CAS.
- R. Chand Bollineni, M. Fedorova and R. Hoffmann, Analyst, 2013, 138, 5081–5088 RSC.
- D. Wylie, M. Voloch, S. Lee, Y. H. Liu, S. Cannon Carlson, C. Cutler and B. Pramanik, Pharm. Res., 2001, 18, 1354–1360 CrossRef CAS.
- T. Nishino, V. Massey and C. H. Williams, J. Biol. Chem., 1980, 255, 3610–3616 CAS.
- E. B. Williams, S. Krishnaswamy and K. G. Mann, J. Biol. Chem., 1989, 264, 7536–7545 CAS.
- R. G. Fruchter and A. M. Crestfield, J. Biol. Chem., 1967, 242, 5807–5812 CAS.
- M. C. Lin, W. H. Stein and S. Moore, J. Biol. Chem., 1968, 243, 6167–6170 CAS.
- T. T. Inagami and H. H. Hatano, J. Biol. Chem., 1969, 244, 1176–1182 CAS.
- T. Y. Liu, J. Biol. Chem., 1967, 242, 4029–4032 CAS.
- H. Pauly, Hoppe-Seyler's Z. Physiol. Chem., 1905, 159–160 CrossRef CAS PubMed.
- H. Pauly, Hoppe-Seyler's Z. Physiol. Chem., 1915, 94, 284–290 CrossRef CAS PubMed.
- F. Zaheer and B. H. Nicholson, Biochim. Biophys. Acta, Protein Struct., 1971, 251, 38–43 CrossRef CAS.
- P. Dominici, B. Tancini and C. Borri Voltattorni, J. Biol. Chem., 1985, 260, 10583–10589 CAS.
- F. Marks, U. Klingmüller and K. Müller-Decker, Cellular signal processing: an introduction to the molecular mechanisms of signal transduction, lavoisier.eu, 2009 Search PubMed.
- M. A. Lemmon and J. Schlessinger, Cell, 2010, 141, 1117–1134 CrossRef CAS PubMed.
- Y. Koshi, E. Nakata, M. Miyagawa, S. Tsukiji, T. Ogawa and I. Hamachi, J. Am. Chem. Soc., 2008, 130, 245–251 CrossRef CAS PubMed.
- S. Håkansson, J. Viljanen and K. S. Broo, Biochemistry, 2003, 42, 10260–10268 CrossRef PubMed.
- T. Shirai, Y. Matsui, C. Shionyu-Mitsuyama, T. Yamane, H. Kamiya, C. Ishii, T. Ogawa and K. Muramoto, J. Mol. Biol., 2002, 321, 879–889 CrossRef CAS.
- J. F. Riordan, M. Sokolovsky and B. L. Vallee, J. Am. Chem. Soc., 1966, 88, 4104–4105 CrossRef CAS.
- K. Jiao, S. Mandapati, P. L. Skipper, S. R. Tannenbaum and J. S. Wishnok, Anal. Biochem., 2001, 293, 43–52 CrossRef CAS PubMed.
- H. Neurath and R. A. Kenner, Biochemistry, 1971, 10, 551–557 CrossRef CAS.
- G. Nikov, V. Bhat, J. S. Wishnok and S. R. Tannenbaum, Anal. Biochem., 2003, 320, 214–222 CrossRef CAS.
- M. Kai, J. Ishida and Y. Ohkura, J. Chromatogr. B, Biomed. Appl., 1988, 430, 271–278 CrossRef CAS.
- J. Ishida, M. Kai and Y. Ohkura, J. Chromatogr. A, 1986, 356, 171–177 CrossRef CAS.
- B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1998, 120, 9074–9075 CrossRef CAS.
- B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1998, 120, 815–816 CrossRef CAS.
- S. Tilley and M. Francis, J. Am. Chem. Soc., 2006, 128, 1080–1081 CrossRef CAS PubMed.
- K. C. Brown, S.-H. Yang and T. Kodadek, Biochemistry, 1995, 34, 4733–4739 CrossRef CAS.
- K. C. Brown, Z. Yu, A. L. Burlingame and C. S. Craik, Biochemistry, 1998, 37, 4397–4406 CrossRef CAS PubMed.
- D. A. Fancy, K. Melcher, S. A. Johnston and T. Kodadek, Chem. Biol., 1996, 3, 551–559 CrossRef CAS.
- D. A. Fancy and T. Kodadek, Tetrahedron, 1997, 53, 11953–11960 CrossRef CAS.
- G. Gill, A. A. Richter-Rusli, M. Ghosh, C. J. Burrows and S. E. Rokita, Chem. Res. Toxicol., 1997, 10, 302–309 CrossRef CAS PubMed.
- K. Kim, D. A. Fancy, D. Carney and T. Kodadek, J. Am. Chem. Soc., 1999, 121, 11896–11897 CrossRef CAS.
- D. A. Fancy and T. Kodadek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6020–6024 CrossRef CAS.
- D. A. Fancy, C. Denison, K. Kim, Y. Xie, T. Holdeman, F. Amini and T. Kodadek, Chem. Biol., 2000, 7, 697–708 CrossRef CAS.
- M. D. Person, K. C. Brown, S. Mahrus, C. S. Craik and A. L. Burlingame, Protein Sci., 2009, 10, 1549–1562 CrossRef PubMed.
- T. Kodadek, I. Duroux-Richard and J. Bonnafous, Trends Pharmacol. Sci., 2005, 26, 210–217 CrossRef CAS PubMed.
- J. Antos and M. Francis, Curr. Opin. Chem. Biol., 2006, 10, 253–262 CrossRef CAS PubMed.
- K. L. Seim, A. C. Obermeyer and M. B. Francis, J. Am. Chem. Soc., 2011, 133, 16970–16976 CrossRef CAS PubMed.
- I. Duroux-Richard, P. Vassault, G. Subra, J.-F. Guichou, E. Richard, B. Mouillac, C. Barberis, J. Marie and J.-C. Bonnafous, Chem. Biol., 2005, 12, 15–24 CrossRef CAS PubMed.
- C. Denison, J. Proteome Res., 2004, 3, 417–425 CrossRef CAS.
- S. Meunier, E. Strable and M. G. Finn, Chem. Biol., 2004, 11, 319–326 CrossRef CAS PubMed.
- H. G. Higgins and K. J. Harrington, Arch. Biochem. Biophys., 1959, 85, 409–425 CrossRef CAS.
- A. Roglans, A. Pla-Quintana and M. Moreno-Mañas, Chem. Rev., 2006, 106, 4622–4643 CrossRef CAS PubMed.
- J. M. Hooker, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 3718–3719 CrossRef CAS PubMed.
- J. M. Hooker, A. Datta, M. Botta, K. N. Raymond and M. B. Francis, Nano Lett., 2007, 7, 2207–2210 CrossRef CAS PubMed.
- T. L. Schlick, Z. Ding, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 3718–3723 CrossRef CAS PubMed.
- M. W. Jones, G. Mantovani, C. A. Blindauer, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 7406–7413 CrossRef CAS PubMed.
- J. Gavrilyuk, H. Ban, M. Nagano, W. Hakamata and C. F. Barbas, Bioconjugate Chem., 2012, 23, 2321–2328 CrossRef CAS PubMed.
- H. Fraenkel-Conrat and H. S. Olcott, J. Biol. Chem., 1948, 174, 827–843 CAS.
- N. S. Joshi, L. R. Whitaker and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 15942–15943 CrossRef CAS PubMed.
- D. Romanini and M. Francis, Bioconjugate Chem., 2008, 19, 153–157 CrossRef CAS PubMed.
- J. M. McFarland, N. S. Joshi and M. B. Francis, J. Am. Chem. Soc., 2008, 130, 7639–7644 CrossRef CAS PubMed.
- H.-M. Guo, M. Minakawa and F. Tanaka, Chem. Rev., 2008, 73, 3964–3966 CAS.
- M. Minakawa, H.-M. Guo and F. Tanaka, J. Org. Chem., 2008, 73, 8669–8672 CrossRef CAS PubMed.
- S. H. Schroeter, J. Org. Chem., 1969, 34, 4012–4015 CrossRef CAS.
- H. Mitchell and Y. Leblanc, J. Org. Chem., 1994, 59, 682–687 CrossRef CAS.
- Y. Leblanc and N. Boudreault, J. Org. Chem., 1995, 60, 4268–4271 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, G. M. Kumar and C. Madan, Synlett, 2001, 1781–1783 CrossRef CAS PubMed.
- J. S. Yadav, B. V. S. Reddy, G. Veerendhar, R. S. Rao and K. Nagaiah, Chem. Lett., 2002, 318–319 CrossRef CAS.
- S. Bombek, R. Lenaršič and M. Kočevar, Chem. Commun., 2002, 1494–1495 RSC.
- W. J. Kinart and C. M. Kinart, J. Organomet. Chem., 2003, 665, 233–236 CrossRef CAS.
- G. L. Chee, Synth. Commun., 2006, 36, 2151–2156 CrossRef CAS PubMed.
- S. Brandes, M. Bella, A. Kjærsgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2006, 45, 1147–1151 CrossRef CAS PubMed.
- G. Desimoni, G. Faita, P. P. Righetti, A. Sfulcini and D. Tsyganov, Tetrahedron, 1994, 50, 1821–1832 CrossRef CAS.
- H. Ban, J. Gavrilyuk and I. C. F. Barbas, J. Am. Chem. Soc., 2010, 132, 1523–1525 CrossRef CAS PubMed.
- H. Ban, M. Nagano, J. Gavrilyuk, W. Hakamata, T. Inokuma and C. F. Barbas, Bioconjugate Chem., 2013, 24, 520–532 CrossRef CAS PubMed.
- O. Y. Wong, P. I. Pradeepkumar and S. K. Silverman, Biochemistry, 2011, 50, 4741–4749 CrossRef CAS PubMed.
- C. L. Borders, J. A. Broadwater, P. A. Bekeny, J. E. Salmon, A. S. Lee, A. M. Eldridge and V. B. Pett, Protein Sci., 1994, 3, 541–548 CrossRef CAS PubMed.
- L. Shimoni and J. P. Glusker, Protein Sci., 1995, 4, 65–74 CrossRef CAS PubMed.
- R. M. C. Dawson, Data for Biochemical Research, Oxford University Press, USA, 3rd edn, 1989 Search PubMed.
- B. H. McMahon, M. Fabian, F. Tomson, T. P. Causgrove, J. A. Bailey, F. N. Rein, R. B. Dyer, G. Palmer, R. B. Gennis and W. H. Woodruff, Biochim. Biophys. Acta, 2004, 1655, 321–331 CrossRef CAS PubMed.
- Y. Xiao, M. S. Hutson, M. Belenky, J. Herzfeld and M. S. Braiman, Biochemistry, 2004, 43, 12809–12818 CrossRef CAS PubMed.
- A. Leitner and W. Lindner, Anal. Chem., 2005, 77, 4481–4488 CrossRef CAS PubMed.
- K. Takahashi, J. Biol. Chem., 1968, 243, 6171–6179 CAS.
- N. A. R. R. Manohar, Biochem. J., 1984, 224, 703–707 Search PubMed.
- C. L. Borders, J. E. Saunders, D. M. Blech and I. Fridovich, Biochem. J., 1985, 230, 771–776 CAS.
- G. Bailin, Biochem. Int., 1985, 11, 161–170 CAS.
- K. Varalakshmi, H. S. Savithri and N. A. Rao, Biochem. J., 1986, 236, 295–298 CAS.
- H. M. Eun, Biochem. Int., 1988, 6171–6179 Search PubMed.
- R. B. Yamasaki, A. Vega and R. E. Feeney, Anal. Biochem., 1980, 109, 32–40 CrossRef CAS.
- R. B. Yamasaki, D. A. Shimer and R. E. Feeney, Anal. Biochem., 1981, 111, 220–226 CrossRef CAS.
- J. Strévey, V. Vachon, B. Beaumier, S. Giroux and R. Béliveau, Biochim. Biophys. Acta, 1992, 1106, 110–116 CrossRef.
- E. B. Mukouyama, T. Hirose and H. Suzuki, J. Biochem., 1998, 123, 1097–1103 CrossRef CAS.
- T. T. Ngo, C. F. Yam, H. M. Lenhoff and J. Ivy, J. Biol. Chem., 1981, 256, 11313–11318 CAS.
- S.-T. Cheung and M. L. Fonda, Biochem. Biophys. Res. Commun., 1979, 90, 940–947 CrossRef CAS.
- J. A. Yankeelov Jr, Biochemistry, 1970, 9, 2433–2439 CrossRef CAS.
- J. A. Yankeelov Jr, Methods Enzymol., 1972, 25, 566–579 CAS.
- K. Toi, E. Bynum, E. Norris and H. A. Itano, J. Biol. Chem., 1967, 242, 1036–1043 CAS.
- J. F. Riordan, Biochemistry, 1973, 12, 3915–3923 CrossRef CAS.
- L. Patthy and E. L. Smith, J. Biol. Chem., 1975, 250, 557–564 CAS.
- A. Foettinger, A. Leitner and W. Lindner, J. Chromatogr. A, 2005, 1079, 187–196 CrossRef CAS PubMed.
- S. M. Politz, H. F. Noller and P. D. McWhirter, Biochemistry, 1981, 20, 372–378 CrossRef CAS.
- M. A. Gauthier and H.-A. Klok, Biomacromolecules, 2011, 12, 482–493 CrossRef CAS PubMed.
- K. Takahashi, J. Biochem., 1976, 80, 1173–1176 CAS.
- M. Pischetsrieder, J. Agric. Food Chem., 1996, 44, 2081–2085 CrossRef CAS.
- D. G. Hoare and D. E. Koshland Jr, J. Am. Chem. Soc., 1966, 88, 2057–2058 CrossRef CAS.
- D. G. Hoare and D. E. Koshland, J. Biol. Chem., 1967, 242, 2447–2453 CAS.
- H. G. Khorana, Chem. Rev., 1953, 53, 145–166 CrossRef CAS.
- R. Renthal, M. Cothran, N. Dawson and G. J. Harris, Biochim. Biophys. Acta, 1987, 897, 384–394 CrossRef CAS.
- A. Das and L. G. Ljungdahl, J. Bacteriol., 1997, 179, 3746–3755 CAS.
- G. M. Cook, S. Keis, H. W. Morgan, C. von Ballmoos, U. Matthey, G. Kaim and P. Dimroth, J. Bacteriol., 2003, 185, 4442–4449 CrossRef CAS.
- J. C. Sheehan and J. J. Hlavka, J. Org. Chem., 1956, 21, 439–441 CrossRef CAS.
- J. C. Sheehan and J. J. Hlavka, J. Am. Chem. Soc., 1957, 79, 4528–4529 CrossRef CAS.
- K. L. Carraway and D. E. Koshland Jr, Methods Enzymol., 1972, 25, 616–623 CAS.
- T. L. Goodfriend, L. Levine and G. D. Fasman, Science, 1964, 144, 1344–1346 CAS.
- N. Xia, Y. Xing, G. Wang, Q. Feng, Q. Chen, H. Feng, X. Sun and L. Liu, Int. J. Electrochem. Sci., 2013, 8, 2459–2467 CAS.
- M. A. Gilles, A. Q. Hudson and C. L. Borders, Anal. Biochem., 1990, 184, 244–248 CrossRef CAS.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- R. B. Woodward, R. A. Olofson and H. Mayer, J. Am. Chem. Soc., 1961, 83, 1010–1012 CrossRef CAS.
- P. Bodlaender, G. Feinstein and E. Shaw, Biochemistry, 1969, 8, 4941–4949 CrossRef CAS.
- B. F. Erlanger, S. M. Vratsanos, N. Wassermann and A. G. Cooper, J. Biol. Chem., 1965, 240, PC3447–PC3448 CAS.
- E. Gross and J. L. Morell, J. Biol. Chem., 1966, 241, 3638–3639 CAS.
- K. Takahashi, W. H. Stein and S. Moore, J. Biol. Chem., 1967, 242, 4682–4690 CAS.
- W. Vogt, Free Radical Biol. Med., 1995, 18, 93–105 CrossRef CAS.
- A. N. Glazer, Annu. Rev. Biochem., 1970, 39, 101–130 CrossRef CAS PubMed.
- K. Lindorff-Larsen and J. R. Winther, Anal. Biochem., 2000, 286, 308–310 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, Chem. Commun., 2013, 49, 5144–5146 RSC.
- G. Toennies, J. Biol. Chem., 1940, 132, 455–456 CAS.
- G. Toennies and J. J. Kolb, J. Am. Chem. Soc., 1945, 67, 849–851 CrossRef CAS.
- T.-L. Ho, Macromolecules, 1975, 75, 1–20 CAS.
- E. Reid, Nature, 1951, 1, 955 CrossRef PubMed.
- O. Kinstler, G. Molineux, M. Treuheit, D. Ladd and C. Gegg, Adv. Drug Delivery Rev., 2002, 54, 477–485 CrossRef CAS.
- G. Gaudriault and J. P. Vincent, Peptides, 1992, 13, 1187–1192 CrossRef CAS.
- I. Sélo, L. Négroni, C. Créminon, J. Grassi and J. M. Wal, J. Immunol. Methods, 1996, 199, 127–138 CrossRef.
- M. Yamaguchi, T. Nakazawa, H. Kuyama, T. Obama, E. Ando, T.-A. Okamura, N. Ueyama and S. Norioka, Anal. Chem., 2005, 77, 645–651 CrossRef CAS PubMed.
- G. Xu and S. R. Jaffrey, Cold. Spring. Harb. Protoc., 2010, 2010, 1–15 Search PubMed (pdb.prot5528).
- D. Bertaccini, S. Vaca, C. Carapito, F. Arsène-Ploetze, A. Van Dorsselaer and C. Schaeffer-Reiss, J. Proteome Res., 2013, 12, 3063–3070 CrossRef CAS PubMed.
- T. M. Rana and C. F. Meares, Bioconjugate Chem., 1990, 1, 357–362 CrossRef CAS.
- H. B. F. Dixon, J. Protein Chem., 1984, 3, 99–108 CrossRef CAS.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 CrossRef CAS PubMed.
- W.-K. Chan, C.-M. Ho, M.-K. Wong and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 14796–14797 CrossRef CAS PubMed.
- A. O.-Y. Chan, C.-M. Ho, H.-C. Chong, Y.-C. Leung, J.-S. Huang, M.-K. Wong and C.-M. Che, J. Am. Chem. Soc., 2012, 134, 2589–2598 CrossRef CAS PubMed.
- H. Dixon and L. R. Weitkamp, Biochem. J., 1962, 34, 462–468 Search PubMed.
- W. P. Jencks, J. Am. Chem. Soc., 1959, 81, 475–481 CrossRef CAS.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- D. E. Metzler and E. E. Snell, J. Am. Chem. Soc., 1952, 74, 979–983 CrossRef CAS.
- C. Cennamo, Naturwissenschaften, 1954, 41, 39 CrossRef CAS.
- C. Cennamo, B. Carafoli and E. P. Bonetti, J. Am. Chem. Soc., 1956, 78, 3523–3527 CrossRef CAS.
- H. Dixon, Biochem. J., 1964, 92, 661–666 CAS.
- H. Dixon and V. Moret, Biochem. J., 1965, 94, 463–469 CAS.
- H. Mix and F. W. Wilcke, Hoppe-Seyler's Z. Physiol. Chem., 1960, 318, 148–158 CrossRef CAS.
- S. Van Heyningen and H. B. Dixon, Biochem. J., 1969, 114, 70–71 Search PubMed.
- H. B. Dixon and R. Fields, Methods Enzymol., 1972, 25, 409–419 CAS.
- L. S. Witus and M. Francis, Curr. Protoc. Chem. Biol., 2010, 2, 125–134 Search PubMed.
- R. A. Scheck, M. T. Dedeo, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2008, 130, 11762–11770 CrossRef CAS PubMed.
- L. S. Witus, T. Moore, B. W. Thuronyi, A. P. Esser-Kahn, R. A. Scheck, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2010, 132, 16812–16817 CrossRef CAS PubMed.
- R. A. Scheck and M. B. Francis, ACS Chem. Biol., 2007, 2, 247–251 CrossRef CAS PubMed.
- L. S. Witus, C. Netirojjanakul, K. S. Palla, E. M. Muehl, C.-H. Weng, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17223–17229 CrossRef CAS PubMed.
- K. S. Palla, L. S. Witus, K. J. Mackenzie, C. Netirojjanakul and M. B. Francis, J. Am. Chem. Soc., 2015, 137, 1123–1129 CrossRef CAS PubMed.
- M. Zhang, X. Zhang, J. Li, Q. Guo and Q. Xiao, Chin. J. Chem., 2011, 29, 1715–1720 CrossRef CAS PubMed.
- M. Dettin, N. Muncan, A. Bugatti, F. Grezzo, R. Danesin and M. Rusnati, Bioconjugate Chem., 2011, 22, 1753–1757 CrossRef CAS PubMed.
- A. P. Crochet, M. M. Kabir, M. B. Francis and C. D. Paavola, Biosens. Bioelectron., 2010, 26, 55–61 CrossRef CAS PubMed.
- W. Gao, W. Liu, J. A. Mackay, M. R. Zalutsky, E. J. Toone and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15231–15236 CrossRef CAS PubMed.
- Z. M. Carrico, M. E. Farkas, Y. Zhou, S. C. Hsiao, J. D. Marks, H. Chokhawala, D. S. Clark and M. B. Francis, ACS Nano, 2012, 6, 6675–6680 CrossRef CAS PubMed.
- K. K. Palaniappan, R. M. Ramirez, V. S. Bajaj, D. E. Wemmer, A. Pines and M. B. Francis, Angew. Chem., Int. Ed., 2013, 125, 4949–4953 CrossRef PubMed.
- K. Sonomura, H. Kuyama, E.-I. Matsuo, S. Tsunasawa and O. Nishimura, Bioorg. Med. Chem. Lett., 2009, 19, 6544–6547 CrossRef CAS PubMed.
- K. Sonomura, H. Kuyama, E.-I. Matsuo, S. Tsunasawa, S. Futaki and O. Nishimura, Anal. Biochem., 2011, 410, 214–223 CrossRef CAS PubMed.
- M.-J. Han, D.-C. Xiong and X.-S. Ye, Chem. Commun., 2012, 48, 11079–11081 RSC.
- T. Sasaki, K. Kodama, H. Suzuki, S. Fukuzawa and K. Tachibana, Bioorg. Med. Chem. Lett., 2008, 18, 4550–4553 CrossRef CAS PubMed.
- P. Agarwal, J. van der Weijden, E. M. Sletten, D. Rabuka and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 46–51 CrossRef PubMed.
- J. I. MacDonald, H. K. Munch, T. Moore and M. B. Francis, Nat. Chem. Biol., 2015, 11, 326–331 CrossRef PubMed.
- J. P. Waller and H. B. Dixon, Biochem. J., 1960, 75, 320–328 CAS.
- M. Brenner, J. P. Zimmermann, J. Wehrmüller, P. Quitt and I. Photaki, Experientia, 1955, 11, 397–399 CrossRef CAS.
- C. F. C. Liu and J. P. J. Tam, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 6584–6588 CrossRef CAS.
- C.-F. Liu and J. P. Tam, J. Am. Chem. Soc., 1994, 116, 4149–4153 CrossRef CAS.
- J. P. Tam, C. Rao, C. F. Liu and J. Shao, Int. J. Pept. Protein Res., 1995, 45, 209–216 CrossRef CAS PubMed.
- Z. Miao and J. P. Tam, J. Am. Chem. Soc., 2000, 122, 4253–4260 CrossRef CAS.
- J. P. Tam, Q. Yu and J. L. Yang, J. Am. Chem. Soc., 2001, 123, 2487–2494 CrossRef CAS PubMed.
- R. Fields and H. Dixon, Biochem. J., 1968, 106, 883–887 Search PubMed.
- K. F. Geoghegan and J. G. Stroh, Bioconjugate Chem., 1992, 3, 138–146 CrossRef CAS.
- B. H. Nicolet and L. A. Shinn, J. Am. Chem. Soc., 1939, 61, 1615 CrossRef CAS.
- L. Zhang and J. P. Tam, Anal. Biochem., 1996, 233, 87–93 CrossRef CAS PubMed.
- H. F. Gaertner, K. Rose, R. Cotton, D. Timms, R. Camble and R. E. Offord, Bioconjugate Chem., 1992, 3, 262–268 CrossRef CAS.
- J. Chen, W. Zeng, R. Offord and K. Rose, Bioconjugate Chem., 2003, 14, 614–618 CrossRef CAS PubMed.
- K. F. Geoghegan, M. J. Emery, W. H. Martin, A. S. McColl and G. O. Daumy, Bioconjugate Chem., 1993, 4, 537–544 CrossRef CAS.
- S. D. Mikolajczyk, D. L. Meyer, J. J. Starling, K. L. Law, K. Rose, B. Dufour and R. E. Offord, Bioconjugate Chem., 1994, 5, 636–646 CrossRef CAS.
- K. Rose, W. Zeng, P.-O. Regamey, I. V. Chernushevich, K. G. Standing and H. F. Gaertner, Bioconjugate Chem., 1996, 7, 552–556 CrossRef CAS PubMed.
- H. F. Gaertner and R. E. Offord, Bioconjugate Chem., 1996, 7, 38–44 CrossRef CAS PubMed.
- E. H. Nardin, J. M. Calvo-Calle, G. A. Oliveira, P. Clavijo, R. Nussenzweig, R. Simon, W. Zeng and K. Rose, Vaccine, 1998, 16, 590–600 CrossRef CAS.
- D. Chelius and T. A. Shaler, Bioconjugate Chem., 2003, 14, 205–211 CrossRef CAS PubMed.
- Y. Zeng, T. N. C. Ramya, A. Dirksen, P. E. Dawson and J. C. Paulson, Nat. Methods, 2009, 6, 207–209 CrossRef CAS PubMed.
- H. B. Dixon, Biochem. J., 1968, 107, 124 CAS.
- O. El Mahdi and O. Melnyk, Bioconjugate Chem., 2013, 24, 735–765 CrossRef CAS PubMed.
- G. L. Thomas and R. J. Payne, Chem. Commun., 2009, 4260–4262 RSC.
- J. Chen, P. Wang, J. Zhu, Q. Wan and S. J. Danishefsky, Tetrahedron, 2010, 66, 2277–2283 CrossRef CAS PubMed.
- R. Okamoto and Y. Kajihara, Angew. Chem., Int. Ed., 2008, 47, 5402–5406 CrossRef CAS PubMed.
- R. Kaiser and L. Metzka, Anal. Biochem., 1999, 266, 1–8 CrossRef CAS PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- S. Chattopadhaya, R. Srinivasan, D. S. Y. Yeo, G. Y. J. Chen and S. Q. Yao, Bioorg. Med. Chem., 2009, 17, 981–989 CrossRef CAS PubMed.
- J. Xiao, A. Burn and T. J. Tolbert, Bioconjugate Chem., 2008, 19, 1113–1118 CrossRef CAS PubMed.
- D. P. Nguyen, T. Elliott, M. Holt, T. W. Muir and J. W. Chin, J. Am. Chem. Soc., 2011, 133, 11418–11421 CrossRef CAS PubMed.
- M. Schnolzer and S. Kent, Science, 1992, 256, 221 CAS.
- P. Dawson, T. Muir, I. Clark-Lewis and S. Kent, Science, 1994, 266, 776 CAS.
- T. Wieland, E. Bokelmann, L. Bauer, H. U. Lang and H. Lau, Justus Liebigs Ann. Chem., 1953, 583, 129–149 CrossRef CAS PubMed.
- T. M. Hackeng, J. H. Griffin and P. E. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10068–10073 CrossRef CAS.
- E. C. B. Johnson and S. B. H. Kent, J. Am. Chem. Soc., 2006, 128, 6640–6646 CrossRef CAS PubMed.
- T. Durek and P. F. Alewood, Angew. Chem., Int. Ed., 2011, 50, 12042–12045 CrossRef CAS PubMed.
- B. Dang, T. Kubota, K. Mandal, F. Bezanilla and S. B. H. Kent, J. Am. Chem. Soc., 2013, 135, 11911–11919 CrossRef CAS PubMed.
- P. E. Lobban and A. D. Kaiser, J. Mol. Biol., 1973, 78, 453–471 CrossRef CAS.
- D. A. Jackson, R. H. Symons and P. Berg, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 2904–2909 CrossRef CAS.
- Y. Sohma, B. L. Pentelute, J. Whittaker, Q.-X. Hua, L. J. Whittaker, M. A. Weiss and S. B. H. Kent, Angew. Chem., Int. Ed., 2008, 47, 1102–1106 CrossRef CAS PubMed.
- L. Raibaut, N. Ollivier and O. Melnyk, Chem. Soc. Rev., 2012, 41, 7001–7015 RSC.
- Z. Harpaz, P. Siman, K. S. A. Kumar and A. Brik, ChemBioChem, 2010, 11, 1232–1235 CrossRef CAS PubMed.
- D. Bang, B. L. Pentelute and S. B. H. Kent, Angew. Chem., Int. Ed., 2006, 45, 3985–3988 CrossRef CAS PubMed.
- D. Macmillan, Angew. Chem., Int. Ed., 2006, 45, 7668–7672 CrossRef CAS PubMed.
- Y. Yuan, J. Chen, Q. Wan, Z. Tan, G. Chen, C. Kan and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 5432–5437 CrossRef CAS PubMed.
- W. R. Gordon, D. Bang, W. D. Hoff and S. B. Kent, Bioorg. Med. Chem., 2013, 21, 3436–3442 CrossRef CAS PubMed.
- G. I. Tesser and I. C. Balvert-Geers, Int. J. Pept. Protein Res., 1975, 7, 295–305 CrossRef CAS PubMed.
- A. D. Gorse and J. E. Gready, Protein Eng., 1997, 10, 23–30 CrossRef CAS PubMed.
- J. A. Camarero, G. J. Cotton, A. Adeva and T. W. Muir, J. Pept. Res., 2009, 51, 303–316 CrossRef PubMed.
- D. Veber, J. Milkowski, S. Varga, R. Denkewalter and R. Hirschmann, J. Am. Chem. Soc., 1972, 94, 5456–5461 CrossRef CAS.
- D. Bang and S. Kent, Angew. Chem., Int. Ed., 2004, 43, 2534–2538 CrossRef CAS PubMed.
- S. Lahiri, M. Brehs, D. Olschewski and C. F. W. Becker, Angew. Chem., Int. Ed., 2011, 50, 3988–3992 CrossRef CAS PubMed.
- S. Ueda, M. Fujita, H. Tamamura, N. Fujii and A. Otaka, ChemBioChem, 2005, 6, 1983–1986 CrossRef CAS PubMed.
- P. Botti, M. Villain, S. Manganiello and H. Gaertner, Org. Lett., 2004, 6, 4861–4864 CrossRef CAS PubMed.
- J.-S. Zheng, H.-K. Cui, G.-M. Fang, W.-X. Xi and L. Liu, ChemBioChem, 2010, 11, 511–515 CrossRef CAS PubMed.
- R. Ingenito, E. Bianchi, D. Fattori and A. Pessi, J. Am. Chem. Soc., 1999, 121, 11369–11374 CrossRef CAS.
- S. Aimoto, Biopolymers, 1999, 51, 247–265 CrossRef CAS.
- M. Schnölzer, P. Alewood, A. Jones and D. Alewood, Int. J. Pept. Protein Res., 2007, 13, 31–44 Search PubMed.
- R. von Eggelkraut-Gottanka, A. Klose, A. G. Beck-Sickinger and M. Beyermann, Tetrahedron Lett., 2003, 44, 3551–3554 CrossRef CAS.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS PubMed.
- F. Thomas, J. Pept. Sci., 2013, 19, 141–147 CrossRef CAS PubMed.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS.
- R. David, M. P. O. Richter and A. G. Beck-Sickinger, Eur. J. Biochem., 2004, 271, 663–677 CrossRef CAS PubMed.
- R. R. Flavell and T. W. Muir, Acc. Chem. Res., 2009, 42, 107–116 CrossRef CAS PubMed.
- J. D. Warren, J. S. Miller, S. J. Keding and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 6576–6578 CrossRef CAS PubMed.
- K. Sato, A. Shigenaga, K. Tsuji, S. Tsuda, Y. Sumikawa, K. Sakamoto and A. Otaka, ChemBioChem, 2011, 12, 1840–1844 CrossRef CAS PubMed.
- Y. Ohta, S. Itoh, A. Shigenaga, S. Shintaku, N. Fujii and A. Otaka, Org. Lett., 2006, 8, 467–470 CrossRef CAS PubMed.
- A. Romanelli, A. Shekhtman, D. Cowburn and T. W. Muir, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6397–6402 CrossRef CAS PubMed.
- F. Nagaike, Y. Onuma, C. Kanazawa, H. Hojo, A. Ueki, Y. Nakahara and Y. Nakahara, Org. Lett., 2006, 8, 4465–4468 CrossRef CAS PubMed.
- H. Hojo, Y. Onuma, Y. Akimoto, Y. Nakahara and Y. Nakahara, Tetrahedron Lett., 2007, 48, 25–28 CrossRef CAS PubMed.
- A. Otaka, K. Sato, H. Ding and A. Shigenaga, Chem. Rec., 2012, 12, 479–490 CrossRef CAS PubMed.
- J. Kang and D. Macmillan, Org. Biomol. Chem., 2010, 8, 1993–2002 CAS.
- L. A. Erlich, K. S. A. Kumar, M. Haj-Yahya, P. E. Dawson and A. Brik, Org. Biomol. Chem., 2010, 8, 2392–2396 CAS.
- K. Sato, A. Shigenaga, K. Kitakaze, K. Sakamoto, D. Tsuji, K. Itoh and A. Otaka, Angew. Chem., Int. Ed., 2013, 52, 7855–7859 CrossRef CAS PubMed.
- N. Ollivier, J. Dheur, R. Mhidia, A. Blanpain and O. Melnyk, Org. Lett., 2010, 12, 5238–5241 CrossRef CAS PubMed.
- J. Dheur, N. Ollivier, A. Vallin and O. Melnyk, J. Org. Chem., 2011, 76, 3194–3202 CrossRef CAS PubMed.
- W. Hou, X. Zhang, F. Li and C.-F. Liu, Org. Lett., 2011, 13, 386–389 CrossRef CAS PubMed.
- N. Ollivier, L. Raibaut, A. Blanpain, R. Desmet, J. Dheur, R. Mhidia, E. Boll, H. Drobecq, S. L. Pira and O. Melnyk, J. Pept. Sci., 2013, 20, 92–97 CrossRef PubMed.
- N. Ollivier, J. Vicogne, A. Vallin, H. Drobecq, R. Desmet, O. El Mahdi, B. Leclercq, G. Goormachtigh, V. Fafeur and O. Melnyk, Angew. Chem., Int. Ed., 2011, 51, 209–213 CrossRef PubMed.
- L. Yan and P. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed.
- D. Crich and A. Banerjee, J. Am. Chem. Soc., 2007, 129, 10064–10065 CrossRef CAS PubMed.
- P. Botti and S. Tchertchian, WO2006133962, 2006, http://patentscope.wipo.int/search/en/detail.jsf?docId=WO2006133962.
- L. E. Canne, S. J. Bark and S. B. Kent, J. Am. Chem. Soc., 1996, 118, 5891–5896 CrossRef CAS.
- C. Haase, H. Rohde and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 6807–6810 CrossRef CAS PubMed.
- J. Chen, Q. Wan, Y. Yuan, J. Zhu and S. J. Danishefsky, Angew. Chem., Int. Ed., 2008, 47, 8521–8524 CrossRef CAS PubMed.
- Z. Tan, S. Shang and S. J. Danishefsky, Angew. Chem., Int. Ed., 2010, 49, 9500–9503 CrossRef CAS PubMed.
- S. Shang, Z. Tan, S. Dong and S. J. Danishefsky, J. Am. Chem. Soc., 2011, 133, 10784–10786 CrossRef CAS PubMed.
- S. D. Townsend, Z. Tan, S. Dong, S. Shang, J. A. Brailsford and S. J. Danishefsky, J. Am. Chem. Soc., 2012, 134, 3912–3916 CrossRef CAS PubMed.
- H. Ding, A. Shigenaga, K. Sato, K. Morishita and A. Otaka, Org. Lett., 2011, 13, 5588–5591 CrossRef CAS PubMed.
- R. E. Thompson, B. Chan, L. Radom, K. A. Jolliffe and R. J. Payne, Angew. Chem., Int. Ed., 2013, 52, 9723–9727 CrossRef CAS PubMed.
- P. Siman, S. V. Karthikeyan and A. Brik, Org. Lett., 2012, 14, 1520–1523 CrossRef CAS PubMed.
- J. P. Tam and Q. Yu, Biopolymers, 1998, 46, 319–327 CrossRef CAS.
- C. Dose and O. Seitz, Org. Biomol. Chem., 2004, 2, 59–65 CAS.
- L. R. Malins, K. M. Cergol and R. J. Payne, ChemBioChem, 2013, 14, 559–563 CrossRef CAS PubMed.
- K. K. Pasunooti, R. Yang, S. Vedachalam, B. K. Gorityala, C.-F. Liu and X.-W. Liu, Bioorg. Med. Chem. Lett., 2009, 19, 6268–6271 CrossRef CAS PubMed.
- R. Yang, K. K. Pasunooti, F. Li, X.-W. Liu and C.-F. Liu, J. Am. Chem. Soc., 2009, 131, 13592–13593 CrossRef CAS PubMed.
- Y.-Y. Yang, S. Ficht, A. Brik and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 7690–7701 CrossRef CAS PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 119, 9408–9412 CrossRef PubMed.
- C. Haase and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 1553–1556 CrossRef CAS PubMed.
- P. Siman, O. Blatt, T. Moyal, T. Danieli, M. Lebendiker, H. A. Lashuel, A. Friedler and A. Brik, ChemBioChem, 2011, 12, 1097–1104 CrossRef CAS PubMed.
- T. Moyal, H. P. Hemantha, P. Siman, M. Refua and A. Brik, Chem. Sci., 2013, 4, 2496–2501 RSC.
- B. L. Pentelute and S. B. Kent, Org. Lett., 2007, 9, 687–690 CrossRef CAS PubMed.
- N. Metanis, E. Keinan and P. E. Dawson, Angew. Chem., Int. Ed., 2010, 49, 7049–7053 CrossRef CAS PubMed.
- R. J. Hondal, B. L. Nilsson and R. T. Raines, J. Am. Chem. Soc., 2001, 123, 5140–5141 CrossRef CAS.
- M. D. Gieselman, L. Xie and W. A. van der Donk, Org. Lett., 2001, 3, 1331–1334 CrossRef CAS PubMed.
- R. Quaderer, A. Sewing and D. Hilvert, Helv. Chim. Acta., 2001, 84, 1197–1206 CrossRef CAS.
- N. A. McGrath and R. T. Raines, Acc. Chem. Res., 2011, 44, 752–761 CrossRef CAS PubMed.
- S. N. Gorlatov and T. C. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8520–8525 CrossRef CAS.
- N. Metanis, E. Keinan and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 16684–16691 CrossRef CAS PubMed.
- P. E. Dawson, Isr. J. Chem., 2011, 51, 862–867 CrossRef CAS PubMed.
- Z. Wang, T. Rejtar, Z. S. Zhou and B. L. Karger, Rapid Commun. Mass Spectrom., 2010, 24, 267–275 CrossRef CAS PubMed.
- L. R. Malins and R. J. Payne, Org. Lett., 2012, 14, 3142–3145 CrossRef CAS PubMed.
- A. L. Adams and D. Macmillan, J. Pept. Sci., 2013, 19, 65–73 CrossRef CAS PubMed.
- A. Ghassemian, X. Vila-Farrés, P. F. Alewood and T. Durek, Bioorg. Med. Chem., 2013, 21, 3473–3478 CrossRef CAS PubMed.
- D. M. Coltart, Tetrahedron, 2000, 56, 3449–3491 CrossRef CAS.
- G.-M. Fang, H.-K. Cui, J.-S. Zheng and L. Liu, ChemBioChem, 2010, 11, 1061–1065 CrossRef CAS PubMed.
- J.-C. M. Monbaliu and A. R. Katritzky, Chem. Commun., 2012, 48, 11601–11622 RSC.
- D. S. Kemp, S.-L. Leung and D. J. Kerkman, Tetrahedron Lett., 1981, 22, 181–184 CrossRef CAS.
- D. S. Kemp and D. J. Kerkman, Tetrahedron Lett., 1981, 22, 185–186 CrossRef CAS.
- D. S. Kemp, D. J. Kerkman, S.-L. Leung and G. Hanson, J. Org. Chem., 1981, 46, 490–498 CrossRef CAS.
- D. S. Kemp and N. G. Galakatos, J. Org. Chem., 1986, 51, 1821–1829 CrossRef CAS.
- D. S. Kemp, N. G. Galakatos, B. Bowen and K. Tan, J. Org. Chem., 1986, 51, 1829–1838 CrossRef CAS.
- D. S. Kemp, N. G. Galakatos, S. Dranginis, C. Ashton, N. Fotouhi and T. P. Curran, J. Org. Chem., 1986, 51, 3320–3324 CrossRef CAS.
- D. S. Kemp and R. I. Carey, J. Org. Chem., 1989, 54, 3640–3646 CrossRef CAS.
- D. S. Kemp and D. R. Buckler, Tetrahedron Lett., 1991, 32, 3009–3012 CrossRef CAS.
- D. S. Kemp and D. R. Buckler, Tetrahedron Lett., 1991, 32, 3013–3016 CrossRef CAS.
- C.-F. Liu, C. Rao and J. P. Tam, Tetrahedron Lett., 1996, 37, 933–936 CrossRef CAS.
- X.-H. Tan, X. Zhang, R. Yang and C.-F. Liu, ChemBioChem, 2008, 9, 1052–1056 CrossRef CAS PubMed.
- X. Zhang, F. Li and C.-F. Liu, Chem. Commun., 2011, 47, 1746–1748 RSC.
- A. Dirksen and P. E. Dawson, Curr. Opin. Chem. Biol., 2008, 12, 760–766 CrossRef CAS PubMed.
- G. Zou, H. Ochiai, W. Huang, Q. Yang, C. Li and L.-X. Wang, J. Am. Chem. Soc., 2011, 133, 18975–18991 CrossRef CAS PubMed.
- S. Ratner and H. T. Clarke, J. Am. Chem. Soc., 1937, 59, 200–206 CrossRef CAS.
- C. Rao and J. P. Tam, J. Am. Chem. Soc., 1994, 116, 6975–6976 CrossRef CAS.
- P. Botti, T. D. Pallin and J. P. Tam, J. Am. Chem. Soc., 1996, 118, 10018–10024 CrossRef CAS.
- J. Shao and J. P. Tam, J. Am. Chem. Soc., 1995, 117, 3893–3899 CrossRef CAS.
- T. Wöhr, F. Wahl, A. Nefzi, B. Rohwedder, T. Sato, X. Sun and M. Mutter, J. Am. Chem. Soc., 1996, 118, 9218–9227 CrossRef.
- M. Villain, J. Vizzavona and K. Rose, Chem. Biol., 2001, 8, 673–679 CrossRef CAS.
- T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249–289 CrossRef CAS PubMed.
- G. Casi, Curr. Opin. Struct. Biol., 2003, 13, 589–594 CrossRef CAS PubMed.
- G. Casi, N. Huguenin-Dezot, K. Zuberbühler, J. Scheuermann and D. Neri, J. Am. Chem. Soc., 2012, 134, 5887–5892 CrossRef CAS PubMed.
- G. J. L. Bernardes, M. Steiner, I. Hartmann, D. Neri and G. Casi, Nat. Protoc., 2013, 8, 2079–2089 CrossRef CAS PubMed.
- E. H. White, F. McCapra and G. F. Field, J. Am. Chem. Soc., 1963, 85, 337–343 CrossRef CAS.
- B. Bitler and W. D. McElroy, Arch. Biochem. Biophys., 1957, 72, 358–368 CrossRef CAS.
- J. Li, L. Chen, L. Du and M. Li, Chem. Soc. Rev., 2012, 42, 662–676 RSC.
- H. Ren, F. Xiao, K. Zhan, Y.-P. Kim, H. Xie, Z. Xia and J. Rao, Angew. Chem., Int. Ed., 2009, 48, 9658–9662 CrossRef CAS PubMed.
- C. P. Ramil and Q. Lin, Chem. Commun., 2013, 49, 11007–11022 RSC.
- G. Liang, H. Ren and J. Rao, Nat. Chem., 2010, 2, 54–60 CrossRef CAS PubMed.
- G. Liang, J. Ronald, Y. Chen, D. Ye, P. Pandit, M. L. Ma, B. Rutt and J. Rao, Angew. Chem., Int. Ed., 2011, 50, 6283–6286 CrossRef CAS PubMed.
- Y. Yuan, J. Zhang, M. Wang, B. Mei, Y. Guan and G. Liang, Anal. Chem., 2013, 85, 1280–1284 CrossRef CAS PubMed.
- J. Jeon, B. Shen, L. Xiong, Z. Miao, K. H. Lee, J. Rao and F. T. Chin, Bioconjugate Chem., 2012, 23, 1902–1908 CrossRef CAS PubMed.
- Y. Yuan and G. Liang, Org. Biomol. Chem., 2014, 12, 865–871 CAS.
- E. Scoffone, A. Fontana and R. Rocchi, Biochem. Biophys. Res. Commun., 1966, 25, 170–174 CrossRef CAS.
- E. Scoffone, A. Fontana and R. Rocchi, Biochemistry, 1968, 7, 971–979 CrossRef CAS.
- L. R. Malins, K. M. Cergol and R. J. Payne, Chem. Sci., 2014, 5, 260–266 RSC.
- J. Offer, Biopolymers, 2010, 94, 530–541 CrossRef CAS PubMed.
- X. Li, L. Zhang, S. E. Hall and J. P. Tam, Tetrahedron Lett., 2000, 41, 4069–4073 CrossRef CAS.
- A. Pictet and T. Spengler, Eur. J. Inorg. Chem., 1911, 2030–2036 CAS.
- V. Popov, S. S. Panda and A. R. Katritzky, Org. Biomol. Chem., 2013, 11, 1594 CAS.
- L. Zhang and J. Tam, Tetrahedron Lett., 1997, 1, 3–6 CrossRef.
- N. Metanis, J. Beld and D. Hilvert, The Chemistry of Selenocysteine, John Wiley & Sons, 2009, DOI:10.1002/9780470682531.pat0582.
- D. J. Craik and A. C. Conibear, J. Org. Chem., 2011, 76, 4805–4817 CrossRef CAS PubMed.
- L. R. Malins, N. J. Mitchell and R. J. Payne, J. Pept. Sci., 2013, 20, 64–77 CrossRef PubMed.
- L. A. Wessjohann, A. Schneider, M. Abbas and W. Brandt, Biol. Chem., 2007, 388, 997–1006 CrossRef CAS PubMed.
- G. Casi, G. Roelfes and D. Hilvert, ChemBioChem, 2008, 9, 1623–1631 CrossRef CAS PubMed.
- S. M. Berry, M. D. Gieselman, M. J. Nilges, W. A. van der Donk and Y. Lu, J. Am. Chem. Soc., 2002, 124, 2084–2085 CrossRef CAS PubMed.
- M. Ralle, S. M. Berry, M. J. Nilges, M. D. Gieselman, W. A. van der Donk, Y. Lu and N. J. Blackburn, J. Am. Chem. Soc., 2004, 126, 7244–7256 CrossRef CAS PubMed.
- R. Quaderer and D. Hilvert, Chem. Commun., 2002, 2620–2621 RSC.
- K. Forchhammer, J. Heider and C. Baron, Trends Biochem. Sci., 1991, 16, 463–467 CrossRef.
- M. Muttenthaler and P. F. Alewood, J. Pept. Sci., 2008, 14, 1223–1239 CrossRef CAS PubMed.
- A. C. Obermeyer, J. B. Jarman and M. B. Francis, J. Am. Chem. Soc., 2014, 136, 9572–9579 CrossRef CAS PubMed.
- L. Yi, H. Sun, Y.-W. Wu, G. Triola, H. Waldmann and R. S. Goody, Angew. Chem., Int. Ed., 2010, 49, 9417–9421 CrossRef CAS PubMed.
- A. Schwarz, C. Wandrey, E. A. Bayer and M. Wilchek, Methods Enzymol., 1990, 184, 160–162 CAS.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2002, 21, 86–89 CrossRef PubMed.
- I. Chen, M. Howarth, W. Lin and A. Y. Ting, Nat. Methods, 2005, 2, 99–104 CrossRef CAS PubMed.
- R. R. Flavell, P. Kothari, M. Bar-Dagan, M. Synan, S. Vallabhajosula, J. M. Friedman, T. W. Muir and G. Ceccarini, J. Am. Chem. Soc., 2008, 130, 9106–9112 CrossRef CAS PubMed.
- J. M. Antos, G.-L. Chew, C. P. Guimaraes, N. C. Yoder, G. M. Grotenbreg, M. W.-L. Popp and H. L. Ploegh, J. Am. Chem. Soc., 2009, 131, 10800–10801 CrossRef CAS PubMed.
- S. Tsukiji and T. Nagamune, ChemBioChem, 2009, 10, 787–798 CrossRef CAS PubMed.
- N. Soh, Sensors, 2008, 8, 1004–1024 CrossRef CAS PubMed.
- A. Ojida, K. Honda, D. Shinmi, S. Kiyonaka, Y. Mori and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 10452–10459 CrossRef CAS PubMed.
- C. T. Hauser and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3693–3697 CrossRef CAS PubMed.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063–6076 CrossRef CAS PubMed.
- T. Yamaguchi, M. Asanuma, S. Nakanishi, Y. Saito, M. Okazaki, K. Dodo and M. Sodeoka, Chem. Sci., 2014, 5, 1021–1029 RSC.
- P. Burkhard, J. Stetefeld and S. V. Strelkov, Trends Cell Biol., 2001, 11, 82–88 CrossRef CAS.
- G. Grigoryan and A. E. Keating, Curr. Opin. Struct. Biol., 2008, 18, 477–483 CrossRef CAS PubMed.
- D. A. D. Parry, R. D. B. Fraser and J. M. Squire, J. Struct. Biol., 2008, 163, 258–269 CrossRef CAS PubMed.
- B. V. Popp and Z. T. Ball, Chem. Sci., 2011, 2, 690 RSC.
- A. N. Zaykov, K. R. MacKenzie and Z. T. Ball, Chem. – Eur. J., 2009, 15, 8961–8965 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |