
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gas sensing using porous materials for automotive applications
Dominic J.
Wales
a,
Julien
Grand
b,
Valeska P.
Ting
a,
Richard D.
Burke
c,
Karen J.
Edler
d,
Chris R.
Bowen
c,
Svetlana
Mintova
b and
Andrew D.
Burrows
*d
aDepartment of Chemical Engineering, University of Bath, Claverton Down, Bath, BA2 7AY, UK
bLaboratoire Catalyse & Spectrochimie, ENSICAEN, 6 Boulevard Maréchal JUIN, 14000 Caen, France
cDepartment of Mechanical Engineering, University of Bath, Claverton Down, Bath, BA2 7AY, UK
dDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: a.d.burrows@bath.ac.uk
First published on 18th May 2015
Abstract
Improvements in the efficiency of combustion within a vehicle can lead to reductions in the emission of harmful pollutants and increased fuel efficiency. Gas sensors have a role to play in this process, since they can provide real time feedback to vehicular fuel and emissions management systems as well as reducing the discrepancy between emissions observed in factory tests and ‘real world’ scenarios. In this review we survey the current state-of-the-art in using porous materials for sensing the gases relevant to automotive emissions. Two broad classes of porous material – zeolites and metal–organic frameworks (MOFs) – are introduced, and their potential for gas sensing is discussed. The adsorptive, spectroscopic and electronic techniques for sensing gases using porous materials are summarised. Examples of the use of zeolites and MOFs in the sensing of water vapour, oxygen, NOx, carbon monoxide and carbon dioxide, hydrocarbons and volatile organic compounds, ammonia, hydrogen sulfide, sulfur dioxide and hydrogen are then detailed. Both types of porous material (zeolites and MOFs) reveal great promise for the fabrication of sensors for exhaust gases and vapours due to high selectivity and sensitivity. The size and shape selectivity of the zeolite and MOF materials are controlled by variation of pore dimensions, chemical composition (hydrophilicity/hydrophobicity), crystal size and orientation, thus enabling detection and differentiation between different gases and vapours.
 Dominic J. Wales | Dominic Wales is a Research Officer in the Department of Chemical Engineering at the University of Bath. His project involves the design and application of metal–organic frameworks and zeolites for exhaust gas sensing applications. |
 Julien Grand | Julien Grand is a Research Engineer in the Laboratoire Catalyse et Spectrochimie (LCS, CNRS), Caen. His project involves the synthesis of new types of zeolites and the development of selective zeolite sensors for automotive exhaust gases. |
 Valeska P. Ting | Valeska Ting is a Lecturer in the Department of Chemical Engineering and the University of Bath's Prize Fellow in Smart Nanomaterials. |
 Richard D. Burke | Richard Burke is a Prize Fellow in Automotive Powertrain Systems in the Department of Mechanical Engineering at Bath. |
 Karen J. Edler | Karen Edler is Professor of Soft Matter at the University of Bath. Her research group works on self-assembly in systems from inorganic surfactant-templated materials, to polyelectrolyte films, nanofibre gels and lipid-polymer particles. |
 Chris R. Bowen | Chris Bowen is Professor of Materials at the University of Bath and ERC Advanced Investigator on Novel Energy Materials, Engineering Science and Integrated Systems (NEMESIS). |
 Svetlana Mintova | Svetlana Mintova is Director of Research, 1st class at CNRS, Laboratoire de Catalyse et Spectrochimie, ENSICAEN and University of Caen, in Basse Normandy, France. |
 Andrew D. Burrows | Andrew Burrows is a Reader in Inorganic Chemistry at the University of Bath. His research interests lie in functionalised materials with extended structures such as metal–organic frameworks (MOFs). |
1. Introduction
Climate change and the need to combat its associated negative effects constitutes one of the key global challenges of the modern age. It is generally accepted that anthropogenic climate change is largely the result of the production and release of unprecedented levels of greenhouse gases, including carbon dioxide, since the industrial revolution as a result of the combustion of fossil-based fuels such as oil, coal and natural gas. Under conditions that allow for complete combustion, carbon dioxide is produced in large quantities. However, under conditions that do not allow for complete combustion, undesired and toxic by-products such as oxides of nitrogen (NOx), carbon monoxide, sulfur dioxide and various unburnt hydrocarbons are also emitted in smaller quantities, some from impurities present in the fuel.1 The release of these gases into the atmosphere has resulted in diminished air quality in major cities and built up areas.2–4 Consequently an increasing amount of research has been directed at the development of technological and materials-based mitigation strategies, for example, the capture and sequestration of CO2 and methane from the atmosphere. Porous materials, such as activated carbons, zeolites and most recently, metal–organic frameworks (MOFs) have been intensely studied for CO2 sequestration applications over the last two decades, as their high surface areas and large void volumes make them uniquely suited for adsorptive separation and storage of gases. However, rather than developing these materials for separating and capturing gases, such as CO2, from dilute mixtures in air (<1% CO2) or even from more concentrated flue gases (∼14% CO2), a more realistic approach could be to employ these materials to reduce the amount of gas emitted at the point of generation.One of the largest contributors to greenhouse gas emissions is the transportation sector, which currently accounts for over a quarter of the total global energy demand worldwide.5 Between 1990 and 2012, transportation was the second largest source of emitted greenhouse gases in the USA and accounted for 24% of CO2 emissions in the UK, according to an IMechE report from 2010, with the majority originating from road transport.3,6 While promising alternative energy technologies (e.g. battery and fuel-cell powered vehicles) are being developed in an effort to reduce future carbon emissions and remove the reliance on fossil fuels, there are many infrastructural and technological hurdles to overcome before these technologies become widespread. Similarly, while mid-term solutions such as the development of biofuels (including bioethanol and biodiesel) offer a lower-carbon alternative to traditional fossil fuels they can still lead to unwanted emissions. The outlook for the next 40 years is that the transportation industry will remain reliant on the internal combustion engine and hydrocarbon-based fuels.7,8 Hence there is an urgent need for concerted efforts to reduce emissions from transport sources, as well as ensuring that the most efficient use is made of the limited fossil fuel reserves remaining.9
1.1 Use of sensors in the automotive industry
The major motivations for the use of sensors to monitor automotive emissions are the legislative requirements to reduce harmful emissions that have negative effects on health and air quality and contribute to global climate change. However, the potential economic benefits from increasing fuel efficiency through reductions in unburnt fuels and increased fuel energy density is also a factor. The greatest gains in developing new multi-functional sensors based on porous materials for the automotive industry are in two main areas: for the design and manufacture of road vehicles, and for use in production vehicles for real-time fuel and emissions management.There is also a need to reduce the discrepancy between emission levels in diagnostic tests used to provide emissions certification in a factory setting (for example on rolling roads using the New European Drive Cycle (NEDC)) and real world emissions, which has become more significant over recent years. As an example, Carslaw et al. used road-side remote sensing equipment to measure real world vehicle NOx emissions from a large number of vehicles.12 The results suggested that whilst legislation imposed on factory-tested NOx emissions had achieved a reduction from 0.5 g km−1 to 0.2 g km−1 since the year 2000 (with all new vehicles sold meeting this standard), real world emissions had stagnated at around 1 g km−1.12 Another review of real world fuel consumption by Mock et al. summarising customer reviews of observed mileage, on-board emissions measurement and laboratory tests, showed that whilst in 2002 the average vehicle had 7% higher fuel consumption on the road compared to the NEDC, in 2012 the difference had increased to 25%.13 Such studies are motivating the automotive industry to move away from current legislative testing on idealised drive cycles and replace this with more dynamic and representative test cycles.14 This change presents significant opportunities for emissions sensing. Firstly, replacement drive cycles must be proven to be representative of real world driving and to establish the emissions associated with a real world drive cycle, real data must be captured under representative conditions. This is particularly challenging since current high-accuracy lab-based emissions sampling tends to be a large, intrusive mechanism which cannot be practically deployed on a significantly large scale. Secondly, engine emissions control is primarily achieved indirectly by modelling emissions empirically in order to determine optimum control of different engine features. Without a direct measure of emissions for use as feedback, less accurate indirect control with significant calibration and on-board diagnostics effort is required.15
(i) Catalytic converters:16 these reduce emissions of carbon monoxide, unburnt hydrocarbons and NOx gases. Optimal operation of catalytic converters requires precise control of the concentration of oxygen by the engine management system, in order to tailor the intake air![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :fuel ratio, known as λ.17 However, under cold start conditions, in order to raise the temperature of the catalytic convertor, the engine is run in a less fuel-efficient mode, involving higher speed idling, exhaust throttling and over-fuelling.
:fuel ratio, known as λ.17 However, under cold start conditions, in order to raise the temperature of the catalytic convertor, the engine is run in a less fuel-efficient mode, involving higher speed idling, exhaust throttling and over-fuelling.
(ii) Selective catalytic reduction (SCR) catalysts: these reduce emissions of NOx from diesel engines. Ammonia, as a urea–water solution, is injected into the exhaust stream for the reduction of NOx into nitrogen and water in the presence of oxygen on the catalyst. The amount of injected urea must be carefully controlled to match the NOx concentration to ensure full reduction and avoid NOx emission due to underdosing or ammonia emission from overdosing.
(iii) Particulate filters: exhaust particulates are captured in a ceramic filter, but must be routinely purged by raising the exhaust temperature either by overfuelling or throttling the intake air flow to reduce the particulates.
(iv) Lean NOx traps: NOx emissions bind onto barium nitrate coated surfaces when the engine is operating in a lean mode with excess oxygen in the exhaust. When the storage catalyst is full, the engine must induce an oxygen-deprived environment by throttling the engine intakes and operating with a close to stoichiometric air:fuel ratio, to reduce the NO2 into N2 and CO2.
Typically the use of the above technologies will have a detrimental effect on fuel economy either by requiring the injection of additional fuel or the throttling of the engine to control oxygen concentrations and exhaust temperatures. Many modern vehicles powered by an internal combustion engine are fitted with a solid state Zr and Pt-based oxygen sensor and/or a NOx sensor, which have response times in the order of a few tens of milliseconds.18,19 However, if the engine management system had access to sensors that could provide real-time information on the concentrations of a larger diversity of products in the exhaust stream, the engine management system could more finely tune the active emissions control, which would be greatly advantageous for increasing engine efficiency.
Thus, there is a need for emissions sensors across a range of applications from high cost, high accuracy research grade analysers for lab-based testing to robust, low cost production-level devices capable of differentiating between a large variety of gaseous species for on-board emissions monitoring. In both of these cases, the different applications will require sensors with different characteristics in terms of the intrusiveness of instrumentation (size, ability to install on vehicles), absolute measurement accuracy, sensor/measurement chain dynamics, feedback and cost.
2. Porous materials for sensing in the automotive industry
There is much potential for the use of porous materials as sensing elements for monitoring and feedback control of the wide variety of gases emitted in vehicle exhausts. The design and tailoring of porous materials for high selectivity allows more sensitive tracking and real-time response to emissions. The large-scale processing of porous materials is established and the production of miniaturised porous-materials sensors is eminently scalable. The vast array of different materials that could be incorporated lends itself to the optimisation of low cost and robust sensors that reduce the need for incorporation of expensive precious metals. In addition, the ability for porous materials to be made into layers of permeable thin films of differently selective materials can potentially be exploited to introduce additional multifunctionality to the final product, allowing a single sensor to detect a wide spectrum of sensing targets.This review will focus on two major classes of porous materials for sensing applications:
(i) Zeolites, which represent a well-known and industrially relevant class of porous aluminosilicate materials. Zeolites are thermally and chemically robust and made from readily-available and abundant starting materials, which make them attractive for low cost and robust sensors.
(ii) Metal–organic frameworks (MOFs), which constitute a relatively new class of hybrid framework material, consisting of metal nodes connected by organic linkers. These materials have incredible structural diversity and the combination of tailoring metal nodes and organic components to achieve desired interactions is an attractive possibility.
Other classes of porous materials have been used for gas sensing, such as mesoporous silica and metal oxides,20 carbon nanomaterials21 and porous silicon.22 These materials are, however, beyond the scope of this review.
There are several advantages to the use of porous materials in gas sensing applications. Porous materials offer flexibility and versatility in terms of structure and design. Size and shape selectivity for adsorbed species can be imparted to zeolites and MOFs through control of the dimensions of the pores enabling detection and differentiation between the multitude of combustion products in exhaust emissions.23 In addition the structural flexibility of some frameworks can add a further dynamic dimension to gas selectivity.24 Chemical selectivity and sensitivity can also be controlled by the incorporation of selected dopants into the zeolite and MOF frameworks in addition to the wide choice of organic linkers and metal nodes that can be incorporated into MOFs.25–28 Furthermore, MOFs and zeolites can exhibit high temperature stability, i.e. ∼500 °C for some MOF-related zeolitic imidazolate framework materials and ∼1200 °C for some zeolites.29,30 Indeed, MOFs and zeolites have been utilised as the active sensing element or as a gas selective filter/pre-concentrator in gas sensors which has been noted in several reviews on their applications.25,26,28,31–35 However, these reviews do not focus on the gases and vapours found in engine exhaust gases; some include examples of dissolved gases and none critically review zeolite- and MOF-based sensors in terms of thermal and selectivity suitability for sensing of gases in exhaust streams.
This review aims to provide an insight into the current state of the art of zeolites and MOFs for the sensing of gases found in vehicle exhausts along with a discussion of the associated sensor technologies allowing them to be employed. The review will also compare and contrast the performance of exemplar porous materials for such applications, to provide a perspective on the future prospects of zeolite- or MOF-based exhaust gas sensors in transportation. Examples of zeolite- and MOF-based sensors for the following important exhaust gases will be examined: water vapour (H2O), oxygen (O2), nitrogen oxides (NOx), carbon monoxide (CO) and carbon dioxide (CO2) together, hydrocarbons (in particular methanol, ethanol, aldehydes and alkanes), ammonia (NH3, the reagent in SCR catalysts), hydrogen sulfide (H2S), sulfur dioxide (SO2) and finally hydrogen (H2). Hydrogen is included due to the potential for it to be employed as an alternative low-carbon fuel for future vehicles. MOFs have been reported as sensing materials for nitroaromatic and nitroalkane vapours. Since these gases are not found in exhaust streams, sensors for these species are not included in this review. However, an excellent review by Banerjee et al. on the sensing of these species was published recently.36
2.1 Zeolites for sensing
Zeolites are defined as crystalline nano- and mesoporous materials with 3D framework structures that form regular and uniform pores and channels. They can be synthesised with different chemical compositions and frameworks which allows for a wide variety of materials; in fact more than 200 known synthetic zeolites have been reported, in addition to 40 natural ones.37,38 Zeolites consist of tetrahedral units of T (where T is typically Si, Al but can also be another element e.g. B, Ga, Ge, or P) bonded by oxygen atoms resulting in a framework containing cages and channels of distinct sizes and shapes. The internal microporosity of zeolites provides high surface areas and active sites for adsorption. In terms of designing zeolites for specific applications, zeolite crystallisation is an important process to control.39 The nature of precursors, the molar ratios, the type of solvents, the presence or absence of templates and additives or seeds all highly influence the final arrangement of the SiO4 and AlO4 tetrahedra. In addition, post-synthesis treatment can be used to tune the obtained properties. Some examples are ion-exchange reactions, surface modification reactions to increase or decrease the hydrophilic character of the zeolite, dealumination reactions to modify the Si/Al ratio of the zeolite framework, tailor the hydrophilic/hydrophobic character and to control the number of cations within the zeolite, and porosity alteration to obtain a hierarchical zeolite which exhibits additional porosity in the mesopore size region.40Molecular sieves are well known for application as adsorption media for various gases, and they can selectively adsorb molecules that are smaller than the pore sizes of the zeolites. Crystalline microporous materials, in particular zeolites, have significant impact in different applications ranging from petroleum refining and petrochemical processes to air separation and nuclear waste management. The completely controlled porous structure of zeolitic materials makes these materials true shape-selective molecular sieves. The presence of charge-compensating cations such as alkali metal, alkaline earth metal and protons within the inorganic frameworks allows fine-tuning of the ion exchange and catalytic properties. The hydrophobic nature of high-silica zeolites or the hydrophilic nature of aluminium-rich zeolites make these solids useful as specific adsorbents of organic molecules or water in the gas or liquid phase. Zeolites are therefore versatile materials in terms of potential applications, and are used as heterogeneous catalysts in industrial processes, ion-exchangers for water purification and water softening, as separating membranes and also as catalyst supports.41–44
Furthermore, a number of novel applications depend not only on the control of pore structure and intrazeolite chemistry, but also on the ability to control the morphology and pre-shaping, for example the growth of thin zeolite films for separation and sensing purposes. A number of novel applications of zeolites depend on the ability to create thin films. For numerous synthetic strategies, the main goal is to obtain adhesive layers of zeolites on various substrates such as noble and non-noble metals, glass, ceramics, silicon, and even three-dimensional substrates.45 The approach employed for a particular zeolite film preparation depends on the final application. The porous films can be deposited on the gas-sensitive element in a continuous and crack-free manner. The following three approaches are applied for deposition of zeolite crystals:
(a) spin (dip) coating (thickness in the range of 200–1000 nm)
(b) hydrothermal growth of seeded zeolite layers (thickness of 500–5000 nm)
(c) screen printing based on self-bonded zeolite crystals with/without additives (thickness in the range of 2000–10000 nm).
In sensing applications, zeolites are used in two main ways, as shown in Fig. 1. Firstly as a functional element with an active role (e.g. incorporating a physical response to adsorbed gases) or secondly as an auxiliary element with a supporting role, e.g. encapsulating the sensing agent, thereby increasing the concentration of an analyte via adsorption or molecular filtering of others gases to improve selectivity or sensitivity of the active sensing agent.
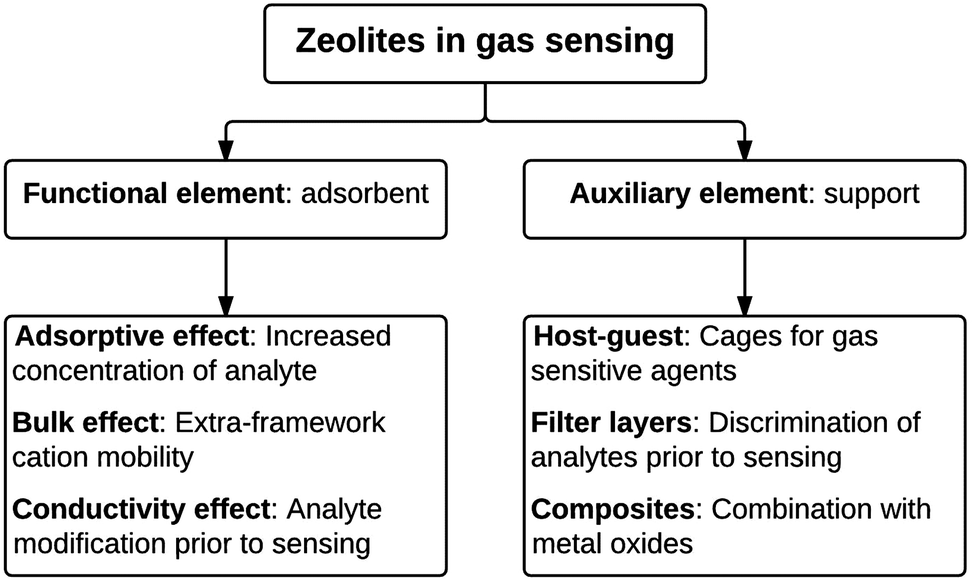 | ||
| Fig. 1 An overview of the different functions of zeolites in gas sensor applications. | ||
Nine types of zeolite have been reported for sensing in the literature (Table 1) and will be summarised in this review.
| Framework type | Zeolite | Pore openings (Å) | Analyte/s, sensor type (range) | Ref. |
|---|---|---|---|---|
| FER | Ferrierite | 4.2 × 5.4; 3.5 × 4.8 | Carbon monoxide (CO) – conductivity (ppm range) | 46 |
| MOR | Mordenite | 6.5 × 7.0; 2.6 × 5.7 | Water vapour (H2O) – UV-vis (9–92% RH), nitrogen dioxide (NO2) – conductivity (ppb range), ethanol vapour (C2H5OH) – conductivity (200 ppm) & ammonia (NH3) – conductivity (ppm range) | 47–54 |
| LTA | Zeolite A | 4.1 × 4.1 | Water vapour (H2O) – QCM (150 ppm), nitrogen dioxide (NO2) – QCM (50 ppm), nitric oxide (NO) – QCM (50 ppm), carbon monoxide (CO) conductivity (ppm range), ethanol vapour (C2H5OH) – cantilever (25 ppmv), hydrocarbons (HCs) – conductivity (1250 ppm), Metglas (6 ppm) & sulfur dioxide (SO2) – QCM (50 ppm) | 55–70 |
| MFI | ZSM-5 Silicalite-1 | 5.3 × 5.6; 5.1 × 5.5 | Water vapour (H2O) – cantilever (ng), QCM (250–750 ppm), UV vis (2.8% RH), conductivity (300 ppm), nitrogen dioxide (NO2) – QCM (200 ppm), carbon monoxide (CO) – impedance (350 ppm), carbon dioxide (CO2) – calorimetry (ppm range), ethanol vapour (C2H5OH) – QCM (50 ppm), cantilever (25 ppmv), IR (16.5 ppm), hydrocarbons (HCs) – conductivity (1250 ppm), IR (22 ppb), Metglas (180 ppm), ammonia (NH3) – conductivity (5 ppm; 612 ppmv) & hydrogen (H2) – fibre optic (0.14 nm kPa−1) | 50–52, 57–61, 66 and 71–93 |
| BEA | Beta | 6.6 × 6.7; 5.6 × 5.6 | Water vapour (H2O) – QCM (150 ppm), nitrogen dioxide (NO2) – conductivity (ppb range), carbon dioxide (CO) – IR (2–100 ppm), ethanol vapour (C2H5OH) – conductivity (200 ppm) & ammonia (NH3) – impedance (25 ppm) | 49, 51, 52, 55, 56, 58, 60, 61, 64 and 94–96 |
| FAU |
Zeolite X
Zeolite Y |
7.4 × 7.4
7.1 × 7.1 |
Water vapour (H2O) – UV vis (200 ppm), conductivity (40% RH), oxygen (O2) – UV vis, conductivity (100 ppm), nitrogen dioxide (NO2) – conductivity (204 ppb), potentiometry (200 ppb), amperometry (ppm range), carbon dioxide (CO) – impedance (1000 ppm), magnetic (0.33%), ethanol vapour (C2H5OH) – cantilever (1.4 ppmv), hydrocarbons (HCs) – UV vis (0.06–31.2 μg mL−1), impedance (10 ppm), cantilever (0.7 ppmv), ammonia (NH3) – conductivity (ppm range) & sulfur dioxide (SO2) – conductivity (100 ppm) | 49–52, 54, 60, 61, 66 and 97–127 |
| STI | Stilbite | 4.7 × 5.0; 2.7 × 5.6 | Water vapour (H2O) – conductivity (63% RH), methanol vapour (CH3OH) & hydrocarbons (HCs) – conductivity (ppm range) | 128 and 129 |
| LTL | Perlialite | 7.1 × 7.1 | Carbon monoxide (CO) – IR (2–100 ppm) | 94 |
| AFI | AlPO-5 | 7.3 × 7.3 | Carbon monoxide (CO) – microcalorimetry (ppm range) | 130 |
In summary, creation of materials with high surface area might allow the selective sensing of gases from exhausts. A porous structure with open accessible volume, high contact surface and strong interactions with only one or more desired analyte would be very important for the design of the required material.
2.2 Metal–organic frameworks for sensing
Metal–organic frameworks (MOFs) are nano- and mesoporous hybrid materials with open 2D or 3D framework structures that contain potential voids. MOFs are composed of metal cations or metal cation aggregates linked by multitopic organic linkers which ultimately assemble into crystalline networks. They can be synthesised from the combination of a variety of di-, tri- and tetravalent cations and many different linkers, and this allows for a huge variety of MOF structures.132 In addition this large degree of choice of the components in MOFs allows for the design of selective sensing interactions with analyte molecules, based on size or shape exclusion or relative strength of intermolecular interactions. Indeed many types of MOFs have been reported for sensing in the literature.28,32In a similar way to zeolite materials, the internal microporosity of MOFs provides high surface areas and active sites for adsorption in addition to selectivity provided by the dimensions of the pores. Thus, sensing elements comprised of MOFs can also be used for direct adsorption of and interaction with the analyte of interest or as a support to improve selectivity or sensitivity of the active sensing agent (Fig. 2).
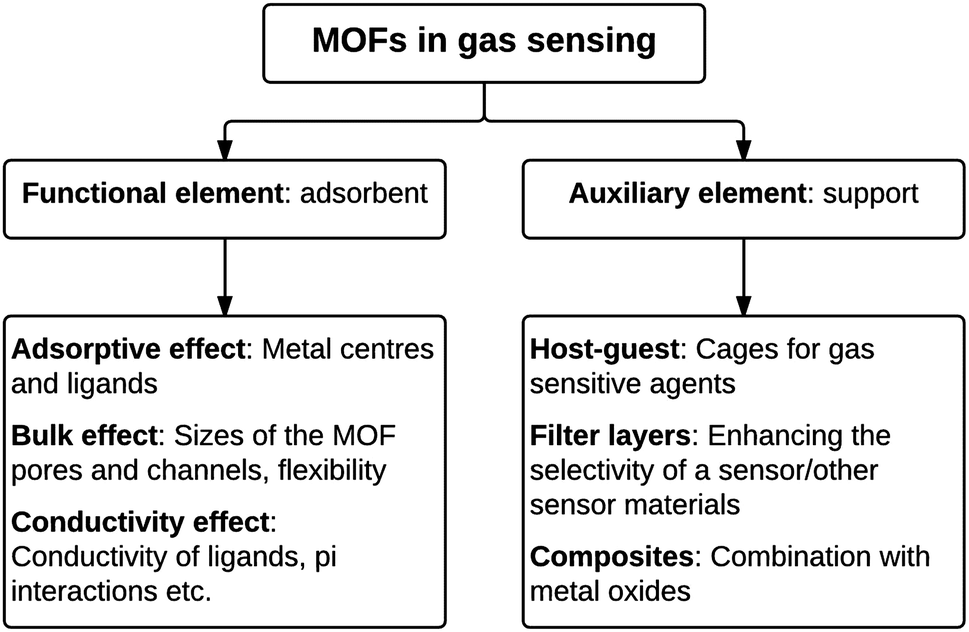 | ||
| Fig. 2 An overview of the different functions of metal–organic frameworks in gas sensor applications. | ||
Many different metal–organic frameworks have been reported for sensing in the literature (Table 2) and these will be summarised in this review.
| Metal–organic frameworka | Analyte/s | Detector/sensor type | Dynamic range (where stated) | Ref. |
|---|---|---|---|---|
| a L1 = pyridyl-4,5-imidazole dicarboxylate; tctpo = tris(p-carboxylate)triphenylphosphine oxide; dpe = 1,2-bis(4-pyridyl)ethane; bdc = 1,4-benzenedicarboxylate; btc = 1,3,5-benzenetricarboxylate; benztb = N,N,N′,N′-benzidinetetrabenzoate; abdc = 2-amino-1,4-benzenedicarboxylate; ad=adeninate; bpdc = 4,4′-biphenyldicarboxylate; L2 = [Ir(3-(2-pyridyl)benzoate)3]3−; L3 = [Ir(2-(4-carboxylato-2-pyridyl)pyridine-4-carboxylate)(2-phenylpyridine)2]−; L4 = [Ir(6-(4-carboxylatophenyl)pyridine-3-carboxylate)(2-phenylpyridine)2]2−; L5 = [Ir(6-(5-carboxylato-2-pyridyl)pyridine-3-carboxylate)(2-phenylpyridine)2]−; L6 = [Ru(6-(5-carboxylato-2-pyridyl)pyridine-3-carboxylate)(2,2′-bipyridine)2]; ip = imidazo[4,5-f][1,10]phenanthrolin-1-ide; bpz = 4-(3,5-dimethylpyrazol-1-id-4-yl)-3,5-dimethyl-pyrazol-1-ide; cbIM = 5-chlorobenzimidazol-1-ate; nIM = 2-nitroimidazolate; MV = methylviologen dication; mpba = N,N′-1,3-phenylenebis(oxamate); mIM = 2-methylimidazolate; 2,2′-bipy = 2,2′-bipyridine; 4,4′-bipy = 4,4′-bipyridine; m-bdc = 1,3-benzenedicarboxylate; ndc = 2,6-naphthalenedicarboxylate; bpee = 1,2-bis(4-pyridyl)ethylene; NO2-ip = 5-nitro-1,3-benzenedicarboxylate; tbtc = 4-[[3,5-bis[(4-carboxylatophenoxy)methyl]-2,4,6-trimethyl-phenyl]methoxy]benzoate; tpce = tetrakis(phenylcarboxylate)ethylene; dodbc = 2,5-dioxo-1,4-benzenedicarboxylate; att = 3-amino-1,2,4-triazole-5-thiol. | ||||
| [Eu(L1)(oxalate)0.5(H2O)]·2H2O | Water vapour (H2O) | Luminescence quenching | 75.8–85% RH | 133 |
| [Tb(L1)(oxalate)0.5(H2O)]·2H2O | Water vapour (H2O) | Luminescence quenching | 75.8–85% RH | 133 |
| [Tb(tcpto)(H2O)]·2DMF.H2O (PCM-15) | Water vapour (H2O) & ammonia (NH3) | Luminescence quenching | NH3: 4.5–13.4 μmol | 134 and 135 |
| [Zn(dpe)(bdc)]·4H2O (ZndB) | Water vapour (H2O) | Luminescence quenching | — | 136 |
| [In(OH)(bdc)] | Water vapour (H2O) & ethanol vapour (C2H5OH) | Luminescence spectroscopy | Saturated atmosphere | 137 |
| [Zn4O(bdc)3] (MOF-5) | Water vapour (H2O) & ethanol vapour (C2H5OH) | Luminescence spectroscopy | Saturated atmosphere | 137 |
| [Cu3(btc)2] (HKUST-1) | Water vapour (H2O), carbon dioxide (CO2), ethanol vapour (C2H5OH), methanol vapour (CH3OH), hydrocarbons (HCs), ammonia (NH3), hydrogen sulfide (H2S) & hydrogen (H2) | Piezoresistive microcantilever sensor (PRMS), colloidal crystal Bragg reflector (CCBR), microcantilever sensor (MCS), Kelvin probe (KP), quartz crystal microbalance (QCM), localised surface polarized resonance (LSPR), surface polarized resonance (SPR) & filter layer on optical sensor (FL) |
H2O: 277–1330 ppm (PRMS), 0–8000 ppm (CCBR), 0.2–1.6% (MCS)
CO2: measurement at 100 sccm (CCBR), 8–70% (MCS), measurement at 10% (in N2) (QCM), 0–100% (LSPR) C2H5OH: 0–6000 ppm (CCBR), 0.6–2.8% (MCS), 1–70% (SPR), 2–50 ppm (QCM), 2–50 ppm (KP) CH3OH: 1368–6580 ppm (PRMS), 1.2–6.0% (MCS), 1–70% SPR, 25–50 ppm (KP), 10–50 ppm (QCM) HCs: C2H6 = measurement at 100 sccm (CCBR), pentanal 1–15 ppm (KP) NH3: measurement at 5 ppm (KP) H2S: measurement at 2.5 ppm (KP) H2: (FL) |
138–147 |
| [Fe3(btc)2] (Basolite® F300) | Water vapour (H2O), ethanol vapour (C2H5OH) & methanol vapour (CH3OH) | Impedimetric gas sensors | H2O: 0–2.5% vol C2H5OH: 7–18% vol CH3OH: 18–35% vol | 148 |
| [Zn4O(benztb)(btc)2/3] (DUT-25) | Water vapour (H2O), ethanol vapour (C2H5OH) & methanol vapour (CH3OH) | Solvatochromism | — | 149 |
| [Fe3O(abdc)3X] (where X = Cl, OH) (NH2-MIL-88B) | Water vapour (H2O) & ethanol vapour (C2H5OH). | UV-visible reflection spectroscopy | — | 150 |
| [Ti8O8(OH)4(abdc)6] NH2-MIL-125(Ti) | Water vapour (H2O) | Impedimetric gas sensor | 11–95% RH | 151 |
| Yb3+@bio-MOF-1, where bio-MOF-1 = (Me2NH2)2[Zn8O(ad)4(bpdc)6]·8DMF·11H2O | Oxygen (O2) | Luminescence quenching | Measurements at 0% and 100% | 152 |
| [Zn4O(L2)2]·6DMF·H2O | Oxygen (O2) | Luminescence quenching | 0.05–1.00 atm PO2 | 153 |
| [Cd(L3)2(H2O)2]·3DMF·6H2O | Oxygen (O2) | Luminescence quenching | Lower limit 0.50% | 154 |
| [Zr6O4(OH)4(bpdc)5.971(L4)0.029] (derivative of UiO-67) | Oxygen (O2) | Luminescence quenching | 0.05–0.80 atm PO2 | 155 |
| [Zr6O4(OH)4(bpdc)5.942(L5)0.058] (derivative of UiO-67) | Oxygen (O2) | Luminescence quenching | 0.05–0.80 atm PO2 | 155 |
| [Zr6O4(OH)4(bpdc)5.821(L6)0.179] (derivative of UiO-67) | Oxygen (O2) | Luminescence quenching | 0.05–0.80 atm PO2 | 155 |
| [RuxZn7−x(ip)12](OH)2 (x = 0.10–0.16) (ruthenium doped MAF-34 MOFs) | Oxygen (O2) | Luminescence quenching | 0.03–1.013 bar PO2 | 156 |
| [Zn4O(bpz)2(abdc)] (MAF-X11) | Oxygen (O2) | Luminescence quenching | 0.05–1.00 bar PO2 | 157 |
| [(CH3)NH2][In3O(btc)2(H2O)3]2[In3(btc)4] (CPM-5) | Oxygen (O2) | Luminescence quenching | 0.0–1.0 atm PO2 | 158 |
| [In3O(OH)(H2O)2(btc)2] MIL-100(In) | Oxygen (O2) | Luminescence quenching | 0.0–1.0 atm PO2 | 158 |
| Cu-Y(btc), copper doped Y(btc) (JUC-32-Y) | Carbon monoxide (CO) | In situ IR spectroscopy | 0–100 ppm | 159 |
| [Zn(cbIM)(nIM)] (ZIF-69) | Carbon dioxide (CO2) | Microscale resonator | 1300–5200 ppm | 160 |
| MV[Mn2Cu3(mpba)3(H2O)3]·20H2O | Carbon dioxide (CO2) | Luminescence spectroscopy | Measurement at 1 bar | 161 |
| [Zn(mIM)2] (ZIF-8) | Carbon dioxide (CO2), ethanol vapour (C2H5OH), hydrocarbons (HCs) & hydrogen (H2) | Fabry–Pérot device (FPD), filter layer (FL) |
C2H5OH: 0–40% (FPD)
HCs: C3H8: 0–100% (FPD) H2: (FL) |
162 and 163 |
| [Co(mIM)2] (ZIF-67) | Formaldehyde (HCHO) & methanol vapour (CH3OH) | Interdigitated electrode resistance sensor (IERS) | HCHO: 5–500 ppm CH3OH: measurement at 100 ppm | 164 |
| [Zr6O4(OH)4(bdc)6] (UiO-66) | Methanol vapour (CH3OH) & ethanol vapour (C2H5OH) | UV-visible reflectance spectroscopy | CH3OH: 10 μL (vapour) C2H5OH: 10 μL (vapour) | 165 |
| [Cu2(OH)(2,2′-bipy)2(btc)]·2H2O | Methanol vapour (CH3OH) | Quartz crystal microbalance | 64–160 ppm | 166 |
| [Co(4,4′-bipy)(m-bdc)] | Methanol vapour (CH3OH) | Quartz crystal microbalance | 2–16 ppm | 166 |
| [Al12O(OH)18(H2O)3(Al2(OH)4)(btc)6]·24H2O | Methanol vapour (CH3OH) & ethanol vapour (C2H5OH) | Kelvin probe |
CH3OH: 100 ppm (lower limit)
C2H5OH: 10–50 ppm |
167, 168 |
| [Co3(btc)2]·H2O | Methanol vapour (CH3OH) & ethanol vapour (C2H5OH) | Kelvin probe |
CH3OH: 20–50 ppm
C2H5OH: 10–50 ppm |
167, 168 |
| [Ni3(btc)2]·H2O | Methanol vapour (CH3OH) & ethanol vapour (C2H5OH) | Kelvin probe |
CH3OH: 100 ppm (lower limit)
C2H5OH: 10–50 ppm |
167, 168 |
| [Cd3(btc)2]·H2O | Methanol vapour (CH3OH) & ethanol vapour (C2H5OH) | Kelvin probe |
CH3OH: 20–50 ppm
C2H5OH: 10–50 ppm |
167, 168 |
| [Zn2(ndc)2(dpe)]·2.5DMF·0.25H2O | Methanol vapour (CH3OH), ethanol vapour (C2H5OH) & hydrocarbons (HCs) | Luminescence quenching (LQ) | Concentration of all analytes was vapour pressure of each analytes at room temperature. | 169 |
| [Zn2(ndc)2(bpee)]·2.25DMF·0.5H2O | Methanol vapour (CH3OH), ethanol vapour (C2H5OH) & hydrocarbons (HCs) | Luminescence quenching (LQ) | Concentration of all analytes was vapour pressure of each analytes at room temperature. | 169 |
| [Zn2(bpdc)2(bpee)]·2DMF (RPM3) | Hydrocarbons (HCs) | Luminescence spectroscopy | — | 170 |
| [Zn(NO2-ip)(4-4′-bipy)] (Zn-CID-5) | Methanol vapour (CH3OH) | Quartz crystal microbalance | Measurement at 40% | 171 |
| [Co3(tbtc)2(DMF)2]·4DMF | Methanol vapour (CH3OH) | UV-visible spectroscopy and visible colour change | Vapour pressure of methanol at room temperature. | 172 |
| [Zn2(tpce)] | Ammonia (NH3) | Luminescence spectroscopy | 1% ammonia (NH3) in nitrogen carrier gas | 173 |
| [Mg2(dobdc)] | Ammonia (NH3) | Luminescence spectroscopy | 1% ammonia (NH3) in nitrogen carrier gas | 173 |
| [Cu(att)1.5] | Hydrogen sulfide (H2S) | Luminescence spectroscopy | 0–650 ppm | 174 |
2.3 Techniques for monitoring gas interactions
Chemical sensors are miniaturised devices that can deliver real-time and on-line information on the presence of specific compounds or ions in complex samples.175 In some cases MOFs and zeolites have been used to simply interact with gases and vapours in an irreversible manner and for the purpose of this review this will be referred to as gas detection rather than gas sensing.When a zeolite or a MOF is used as a functional sensing element with an active role, the real-time gas sensing response can be monitored through (i) mass/mechanical changes, for example via a quartz crystal microbalance (QCM), microcantilevers and/or microresonator, (ii) measuring optical property changes (e.g. IR spectroscopic measurements, refractive index measurements and/or luminescence spectroscopic measurements), (iii) variation in electric properties (e.g. impedance, capacitance and/or resistance), (iv) heat liberation (calorimetry) or even (v) by visible colour change.
Similarly, when the porous material is used as an auxiliary filtering element, the monitoring of the gas absorbance on corresponding sensing agents, such as metal oxides or polymers, can be achieved using the same techniques. The use of the porous materials as hosts for gasochromic sensing agents, such as reactive dyes, allows for visual monitoring in addition to conventional measurements. These monitoring techniques are briefly described below.
A piezoelectric microcantilever-based transducer uses a similar approach to a QCM transducer but consists of a cantilever paddle that is anchored at one end to a substrate with the other end modified with a sensing material and free to vibrate. When excited by an AC potential difference, the paddle vibrates at its resonance frequency. Upon adsorption of an analyte within or on the sensing layer this specific frequency changes due to mass gain and thus the frequency variation is related to the mass of analyte adsorbed. Further information is available in a review on the use of microcantilevers for chemical sensing.177 The closely related microresonator-based chemical sensors also operate by adsorption of an analyte leading to a change in mass which in turn changes the resonant frequency of the structure. An excellent paper describing microresonator theory was published by Schmidt and Howe.178
A magnetoelastic transducer usually consists of an amorphous ferromagnetic material, in a ribbon form, covered by a sensing film. When an alternating magnetic field is applied, the magnetoelastic material resonates at a specific frequency which changes due to adsorption of an analyte within or on the sensing film. Further information is available in a review on the use of magnetoelastic transducers for sensing.179
Optical fibre Bragg gratings can either be long-period gratings or short-period gratings. For both types of optical fibre Bragg grating, the period describes the distance between a fringe of low or high refractive index and the sequential fringe of the same refractive index. Long-period gratings typically have periods on the sub-millimeter length scale and work by coupling forward propagating light of a certain wavelength into forward coupling cladding modes. The coupled wavelength is dependent on the period of the Bragg grating. The power of the forward coupling modes is typically attenuated through absorption and scattering losses. If the cladding of the optical fibre is covered with a sensing material then interaction with analyte species will alter the absorbance, or change the magnitude of scattering losses within the cladding. This can result in a change in the magnitude of the received power at the coupled wavelength at the detector or even a wavelength shift of the transmission peak features. Further information is available in a review on the use of long period Bragg gratings for sensing.180
Luminescence-based detector or sensor systems measure changes in the fluorescence or phosphorescence emission spectrum of a luminescent material due to interaction with analyte molecules. Certain molecules, such as dioxygen O2, quench or “turn-off” the luminescence of the sensing material; the degree of change in luminescent intensity or wavelength shifting is related to the concentration of the quenching analyte.
Optical sensing can be employed if encapsulated reactive dyes are contained inside the zeolite cages. Colour changes caused by the presence of an analyte can be monitored with UV-visible spectroscopy and/or the naked eye.
The different types of sensing and detection techniques used in zeolite and MOF sensors are summarised in Fig. 3.
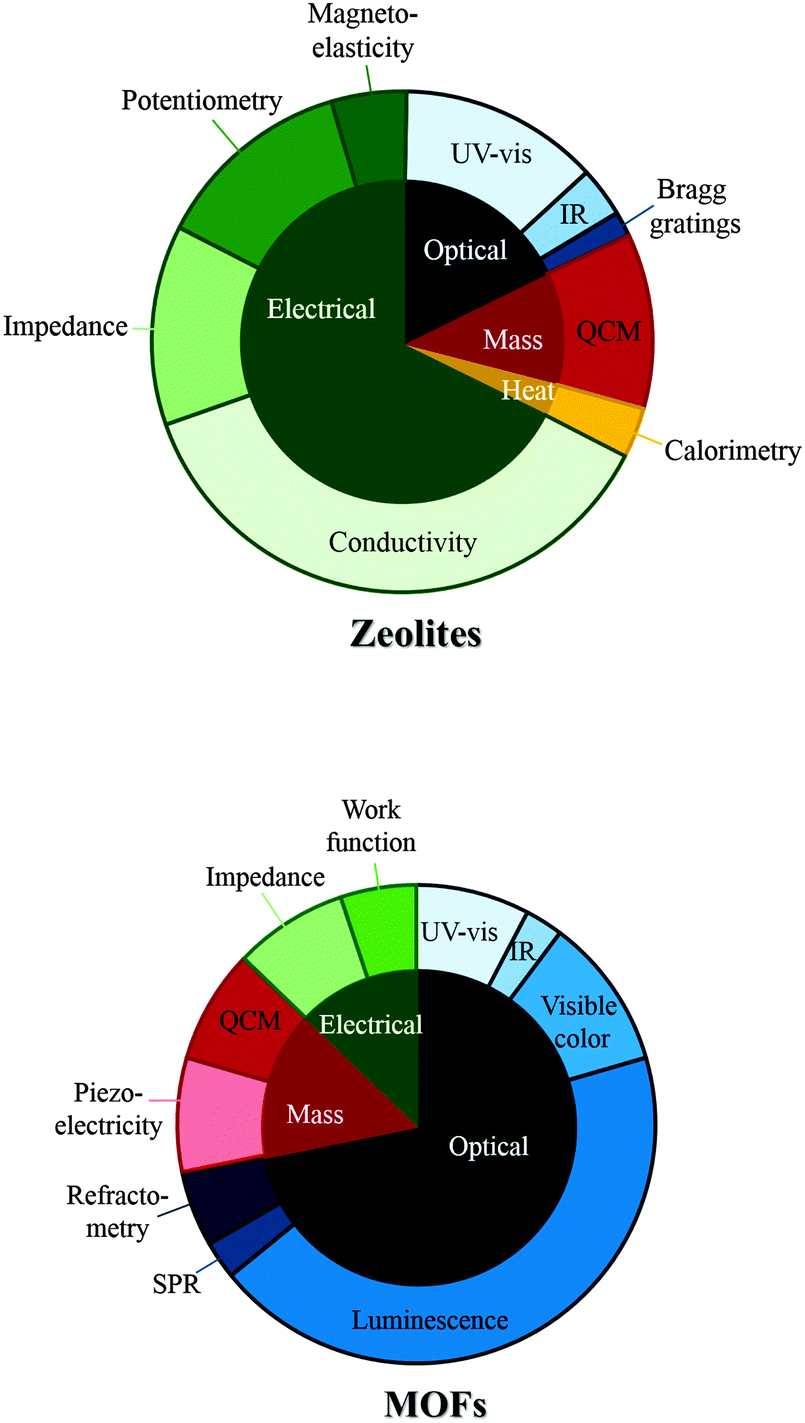 | ||
| Fig. 3 A comparison of the different types of sensing/detection techniques cited in this review. | ||
3. Exhaust gas sensing using porous materials
3.1 Humidity sensors
Host–guest chemistry using zeolites as hosts for a dye or conductive material has also been employed to construct optical or electrochemical detectors and sensors for humidity, respectively. Spectroscopic changes in the nature of the dye loaded into the zeolite cages indicated the presence or disappearance of water. Protonation/deprotonation of methylene blue held in mordenite-type zeolite showed spectral changes in a range of 9 to 92% of relative humidity with good reversibility and fast responses of approximately 2 min for the adsorption step and 4 min for the desorption step.47,48 Two absorption bands (the monomeric methylene blue and its protonated form at 650 and 745 nm, respectively) were monitored to obtain these results. Another example of an optical humidity detector utilised solvatochromic Nile Red dye encapsulated in a NaY zeolite.102 This device had a detection limit of 200 ppm humidity, a response time of around 4 min and discrimination against hexane was also demonstrated. One further example of an optical detector involved impregnation of silver on ZSM-5 type zeolite72 where the authors showed that Ag+ species in the Ag-ZSM-5 zeolite exhibited stable optical properties under high-temperature treatment. The behaviour of the 37700 cm−1 band was followed with in situ UV-vis spectroscopy. Above 150 °C, this band was found to be sensitive to the presence of water vapour in the gas stream (2.8% H2O in He), disappearing in the presence of water vapour while full intensity could be recovered under water-free conditions.
As an example of electrochemical sensing, Yu et al. employed a porous zeolite support to improve the stability and durability of LiCl at high humidity. Indeed, when the humidity exceeds 40% relative humidity (RH), the conductivity response of LiCl is no longer linear and thus it becomes difficult to use the material as a humidity sensor.103 The LiCl was dispersed into a NaY zeolite to obtain a linear response to up to 75% RH. The authors claimed that the well-defined cavities of NaY-type zeolites are more suitable as host materials for preparation of humidity-sensitive composite materials than those with channel structure, such as ZSM-5 or mordenite zeolites. Zou et al. similarly reported that the loading of LiCl into stilbite resulted in an electrically conductive material exhibiting very high humidity sensitivities with a linear response of 104 in conductivity with a humidity change from 0 to 63% and fast response times for adsorption and desorption.128
Changes in zeolite conductivity due to extra-framework cations contained in the framework have also been employed for the concept of humidity sensors with measurements based on changes in either impedance or capacitance. The impedance of the H-form of ZSM-5 was studied as a function of water concentration (0–300 ppm) in H2.73,74 ZSM-5 zeolite was chosen for its small pore size (5.5 Å), which improved water selectivity, and also for its acidic properties, due to the acid–base chemistry involved in impedance measurements. In addition, the high thermal stability of the zeolite in a reducing environment allowed sensing experiments in H2 up to 600 °C, measurements which are not possible with humidity sensors based on metal oxides or polymers.
To allow sensing under milder ambient experimental conditions, the capacitance of a zeolite can be measured instead of impedance. For this approach, electrodes coated with various zeolite films (A-, Y-, beta polymorph A- and silicalite-type) with different Si/Al ratios were studied.58 It was shown that the hydrophilic nature of zeolite A provided the most sensitive film, with a seven times higher response under a 350 ppm water concentrated atmosphere compared with the other films.
Metal oxide based sensors can be further modified with zeolite films to improve their response to gas analytes compared with uncoated metal oxides. A zeolite layer can be used as an adsorbent filter to enhance the selectivity of the metal oxide by retaining analytes and/or interfering species. An example was reported using SnO2 as the oxide and zeolite A and ZSM-5 zeolites as filters.59 Results showed that zeolite A improved the selectivity towards H2O by the elimination of the response to propane, hydrogen and methane while continuing to respond significantly to ethanol and CO. On the other hand, the ZSM-5-type silicalite-coated sensor gave enhanced response to both H2O and H2.
Humphrey et al. synthesised a terbium(III) MOF, ([Tb(tctpo)(H2O)]·2DMF·H2O) (DMF = N,N-dimethylformamide), known as PCM-15, with tris(p-carboxylate)triphenylphosphine oxide (tctpo) as the linker (Fig. 4).135 The resulting MOF is comprised of a 5,5-connected net structure, with each Tb3+ centre octa-coordinated, forming seven bonds to tctpo linkers and one to a water molecule. Upon activation of the MOF the intensity of the photoluminescence due to the terbium ion was found to double. Upon treatment with moisture-saturated N2, the photoluminescence intensity returned to that for the as-synthesised MOF. Thus, PCM-15 can act as a luminescence-based detector for humidity, though neither the response time nor the luminescence response at different % RH levels were reported. Another MOF which acted as a detector for water vapour was demonstrated by Chou et al.136 As with the examples above, changes in the luminescence intensity of the MOF upon dehydration and rehydration were used as the measurable output. However this MOF, [Zn(dpe)(bdc)]·4H2O (dpe = 1,2-bis(4-pyridyl)ethane, bdc = 1,4-benzenedicarboxylate), labelled as ZndB, utilises ligand-based luminescence rather than luminescent lanthanide metal ions. The MOF contains 2D hydrogen-bonded rings of sixteen water molecules and showed reversible on(dry)/off(hydrated) luminescent response to humidity upon cycling between room temperature and 85 °C. The mechanism behind the increase in fluorescence intensity upon heating at 85 °C was thought to be removal of the intra-channel water molecules resulting in a shortening of the inter-channel distance between two dpe moieties. This enhances the π–π stacking between the two interchain dpe moieties and thus increases dpe(π*)–dpe(π) excimer formation. An exposure time of >100 min of the MOF to the ambient % RH level at room temperature was required for the fluorescence intensity to return to as-prepared levels.
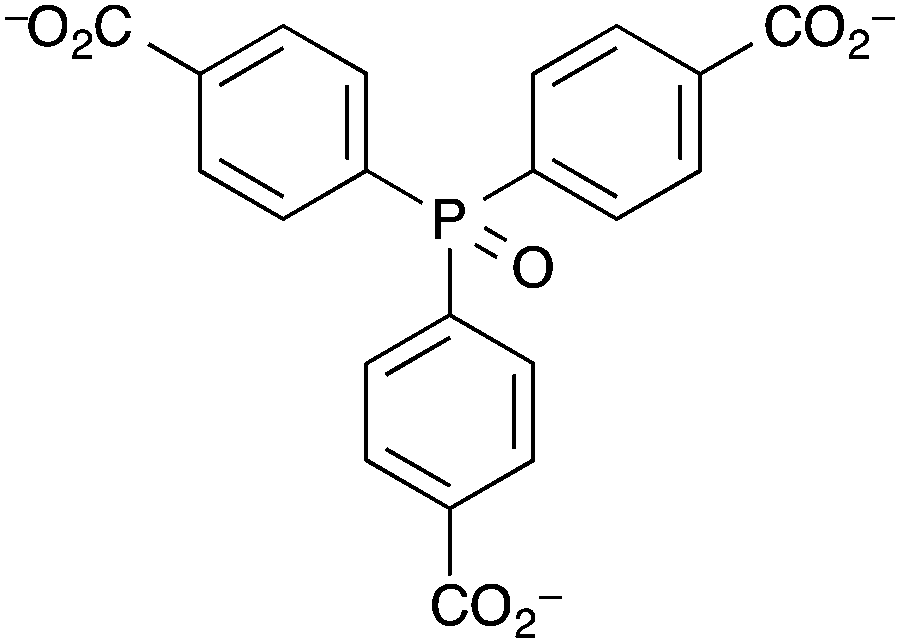 | ||
| Fig. 4 The tris(p-carboxylate)triphenylphosphine oxide (tctpo) ligand in the Tb3+ luminescent PCM-15 MOF prepared by Humphrey et al.135 | ||
Lee et al. also reported two MOFs that can be used to detect humidity by quantifying the differences in the luminescence emission spectra of a MOF when ‘wet’ and the same MOF when dry.137 Upon excitation at 270 nm, it was found that for dry [In(OH)(bdc)] emission λmax = 326 nm and for dry [Zn4O(bdc)3], MOF-5, emission λmax = 366 nm.183 Exposure to water vapour at 40 °C resulted in an emission wavelength redshift of 64 nm for [In(OH)(bdc)] (λmax water = 390 nm) and an emission wavelength blueshift of 27 nm (λmax water = 339 nm) for MOF-5. Therefore, both MOFs act as fluorescent probes for humidity. No reversibility experiments or experiments detailing a fluorescence change upon exposure to different concentrations of water vapour were reported for these MOFs, but they do show reasonable thermal stability, decomposing above 450 °C for [In(OH)(bdc)] and above 400 °C for MOF-5. Such thermal stability would be necessary for a MOF active sensing element for an exhaust gas sensor.184
The copper(II) MOF [Cu3(btc)2] (btc = 1,3,5-benzenetricarboxylate), HKUST-1, has also been used in the development of a piezoelectric cantilever sensor for both humidity and hydrocarbon vapours.138 Robinson et al. fabricated N-doped piezoelectric microcantilever sensors, ∼230 μm long and ∼80 μm wide, that were coated with a layer of HKUST-1. The microcantilever sensor device was then activated prior to the start of a sensing experiment by passage of dry nitrogen gas over the device for 15 min at 40 °C. Higher temperatures, required to completely remove adsorbed water, were not possible as the integrity of the device was found to be compromised at temperatures >50 °C at 1 atm pressure. Therefore, the authors waited until a steady state was achieved before sensing experiments were performed. The sensor device was exposed to different concentrations of water in nitrogen gas at 23 °C and at 1 atm pressure. Exposure to water vapour resulted in water being adsorbed on the HKUST-1 layer and a change in the resonant frequency of the microcantilever. The device demonstrated sensitivity between ∼300 ppm and ∼3000 ppm of water in nitrogen carrier gas and showed sensing reversibility. However, the magnitude of resonant frequency changes that were measured at equilibrium for different concentrations of water vapour in nitrogen carrier gas were identical to those measured upon exposure to different concentrations of methanol vapour. Therefore, based on measurable resonant frequency changes, the sensor device cannot differentiate between water and methanol in nitrogen. However, if the response times of the device are taken into account, i.e. the adsorption response time and the desorption response time, then analyte differentiation is possible. The adsorption and desorption response times for water were both <30 s, but this was only calculated at one water concentration. Robinson et al. defined the response time as the time taken to reach 63.2% of the equilibrium value either during desorption or adsorption as opposed to the definition of gas sensing response time t90, (the time taken to reach 90% of the equilibrium value during adsorption or desorption) that is used more routinely in the literature.185,186 No sensing results for mixed analyte gas streams were reported. The major disadvantage of this gas sensor in terms of application as a sensor for exhaust gases is the poor thermal instability of the microcantilever.
Impedance humidity sensors that utilise MOFs as the active sensing materials have been developed by Moos et al.148 The authors investigated two types of impedance humidity sensors; a planar interdigital electrode impedance sensor and a pellet-type metal-disc electrode impedance sensor, both of which are shown in Fig. 5.
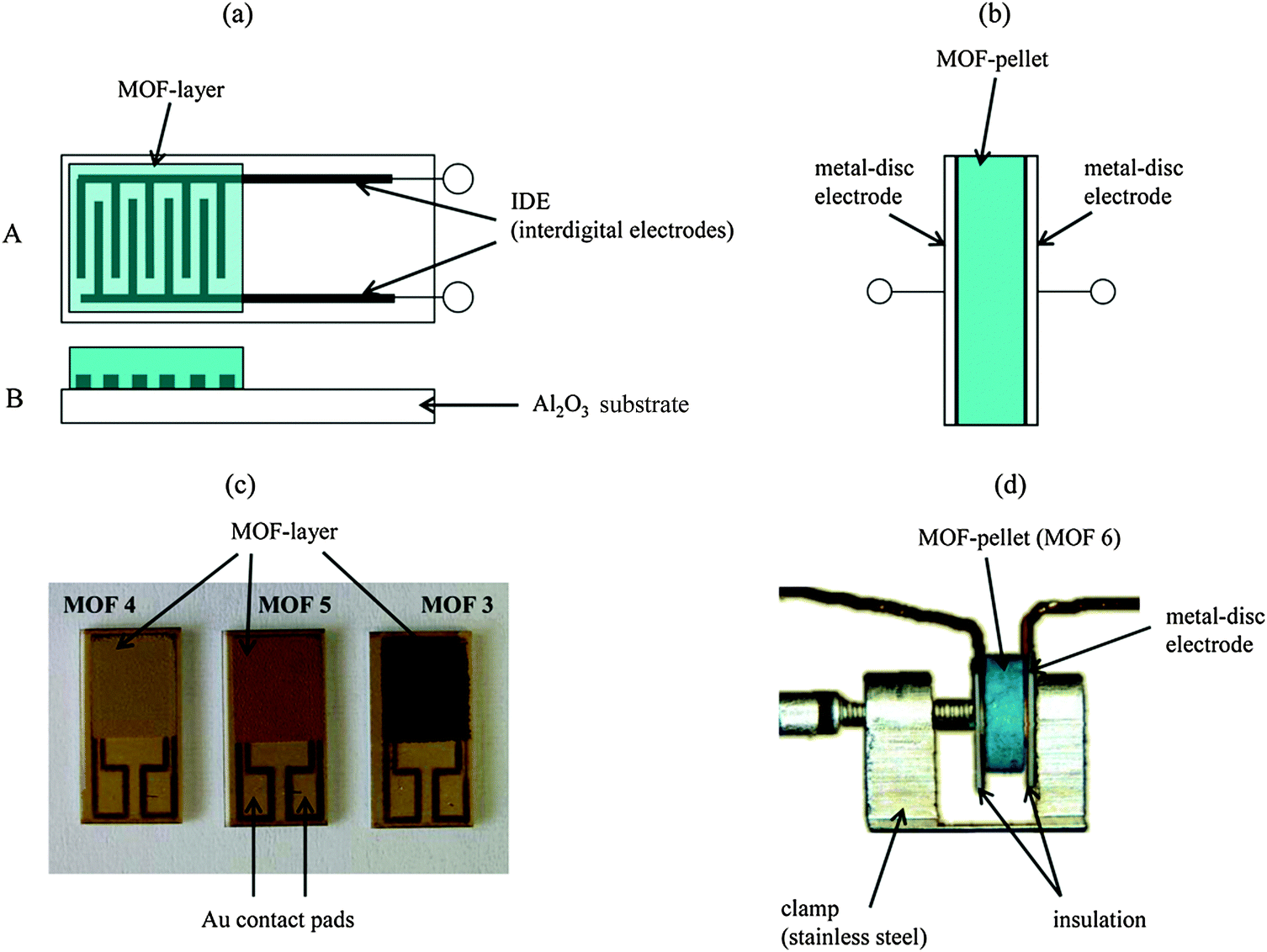 | ||
| Fig. 5 (a) Schematic of the planar interdigital electrode impedance sensor and (c) photographs of the actual planar interdigital electrode impedance sensors covered with thick films of different MOFs by Moos et al.148 (b) Schematic of the pellet-type metal-disc electrode impedance sensor and (d) photograph of the realised pellet-type metal-disc electrode impedance sensor, by Moos et al., containing a pellet of a MOF.148 Reproduced under the terms of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/) from ref. 148 with copyright belonging to the authors of ref. 148. | ||
For the pellet-type sensor, a pellet of a MOF was placed between the two disc electrodes. For the planar interdigital electrode sensor a thick film of a MOF was deposited on top of the interdigitated electrodes. The authors investigated several MOFs, but found that Fe-btc (Basolite® F300), the iron derivative of HKUST-1, afforded the highest magnitude measured changes in absolute impedance |![[Z with combining low line]](https://www.rsc.org/images/entities/i_char_005a_0332.gif) |, and capacitance C, upon exposure to humidity in a range of 0–3 vol% water in nitrogen carrier gas. Further experiments revealed that the best sensor response in terms of largest magnitude changes in measurable parameters and reversibility was achieved with the planar interdigital electrode impedance sensor format at the frequency of 1 Hz and with a thick film of Fe-btc MOF as the active sensing material.
|, and capacitance C, upon exposure to humidity in a range of 0–3 vol% water in nitrogen carrier gas. Further experiments revealed that the best sensor response in terms of largest magnitude changes in measurable parameters and reversibility was achieved with the planar interdigital electrode impedance sensor format at the frequency of 1 Hz and with a thick film of Fe-btc MOF as the active sensing material.
At an operating temperature of 120 °C, there was a linear response between || and water concentration (in vol%), an overall impedance range of 1.2 GΩ and a sensitivity of 590 MΩ/vol% water, in the range of 0–2.5 vol% water in nitrogen. In addition, the sensor response was reproducible with ∼100% recovery to pre-exposure impedance values after each exposure to water vapour. Therefore this sensor shows promise as a water sensor in exhaust emission sensing applications especially as Fe-btc is thermally stable up to ∼250 °C. However, a disadvantage of this sensor is that the sensing response becomes non-linear and tends towards exponential at temperatures higher than 120 °C; this limits potential use in hot exhaust gas sensing applications.
As stated above, the large surface areas and high porosity of MOFs allows for the uptake of gaseous analytes and thus favours MOFs as active sensing materials. In addition it is expected that the effective refractive index of the MOF will be sensitive to the uptake of analyte molecules in the pores. Therefore Hupp et al. fabricated a refractometric optical-based detector which utilised HKUST-1 as the detection material; adsorption of gaseous analytes in the pores within HKUST-1 changed the effective refractive index of the MOF.139 The refractometric optical detector consisted of an ordered hexagonal-packed array of sub-micrometer-sized silica microspheres coated in HKUST-1; this is known as a colloidal crystal. A colloidal crystal reflects light at particular wavelengths due to the periodic variation of refractive index within the structure. The reflected wavelength is known as the ‘stop band’. At normal incidence the ‘stop band’ λSB, in the [111] direction of a colloidal crystal is defined by:139
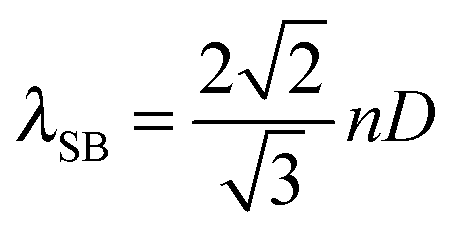 | (1) |
000 ppm in nitrogen carrier gas, ‘stop band’ redshifts were measured. The relationship between water concentration and measured ‘stop band’ wavelength shift was approximately linear between 100–1000 ppm and then plateaued after ∼4000 ppm. The authors estimated that the lowest change in concentration of water that could be measured with the device was 2.6 ppm, based on the wavelength resolution of 0.015 nm of the measuring equipment. The detection response was also reversible. However a major limitation of this proof-of-concept device was the MOF was not selective; many different vapours and gases could adsorb onto the MOF which resulted in a change in the effective refractive index of the colloidal crystal. For example, some concentrations of water, methanol and ethanol vapours in nitrogen carrier gas resulted in identical magnitude ‘stop band’ shifts. One advantage of this device is that it is potentially thermally robust for exhaust gas sensing applications; the silica microspheres were sintered at 600 °C during fabrication of the device and HKUST-1 is thermally stable up to ∼250 °C. However, a disadvantage of this current iteration of the detection device is that a light source and a high resolution optical spectrometer are needed to achieve high precision measurements of water vapour. The response of this device to other vapours and gases is discussed in the appropriate sections below.
HKUST-1 was also the chosen active sensing material for piezoelectric microcantilever humidity sensors demonstrated by Hesketh et al.140 The reason for their choice was that small distortions of the HKUST-1 framework occur upon adsorption of analytes in the pores. If the MOF is in the form of a thin film then the distortion within the framework creates a measurable stress on the piezoelectric microcantilever. The sensor device consisted of a 10-microcantilever array in which each microcantilever incorporated a piezoresistive element. This enabled measurement of changes in stress due to distortion of the framework of the MOF thin film caused by adsorption of analytes. For deposition of the HKUST-1 thin film (∼100 nm thick) some of the microcantilevers were coated with gold, which was then modified with a self-assembled monolayer of a thiol; the thiol facilitated the formation of the HKUST-1 thin film on those cantilevers when immersed in the reactant solutions. Some microcantilevers were masked to prevent gold deposition and thus were not subsequently coated with HKUST-1 in order to act as reference microcantilevers. The authors tested the response of the device to water vapour in nitrogen carrier gas with HKUST-1 in either the hydrated as-synthesised state or the ‘activated’ dehydrated state. The dehydrated state was achieved by passing current through the piezoresistive elements so that temperature reached 50 °C whilst under a flow of dry nitrogen gas for 2 h. It was found that the sensing response to water vapour did not significantly differ whether the HKUST-1 was hydrated or dehydrated. Measurements were performed by exposing the sensor to a flow of water vapour at various concentrations (0–1%) in nitrogen carrier gas at room temperature and pressure. The sensor response was non-linear within the range of water vapour concentrations investigated. The authors reported a measurable change in resistance for adsorption after 0.5 s upon exposure to moist nitrogen gas, though this was the shortest measurement interval used. The time constant for desorption was ∼10 s. Fitting of the water resistance curve to a Langmuir isotherm resulted in reasonable agreement with a 0–3 mbar isotherm of water adsorption on HKUST-1, which Hesketh et al. state corresponds to a resistance of 7.0 Ω for saturated HKUST-1.187 The main disadvantage of the sensor was that the HKUST-1 MOF layer did not demonstrate selectivity; methanol and ethanol vapour also adsorbed which resulted in a change in stress. Some concentrations of water, methanol and ethanol vapours in nitrogen carrier gas resulted in identical magnitude measured resistance values. The response of this device to other vapours and gases is discussed in the appropriate sections below.
Fleischer et al. also utilised HKUST-1 as the active sensing material for water and other analytes.141 The device was a Kelvin probe work function-based sensor that consisted of an alumina substrate coated on the top side with a ∼2 μm TiN back electrode and a screen printed platinum resistive heater element on the bottom side. On top of the TiN back electrode was a drop cast film of HKUST-1 as the sensing layer (Fig. 6).
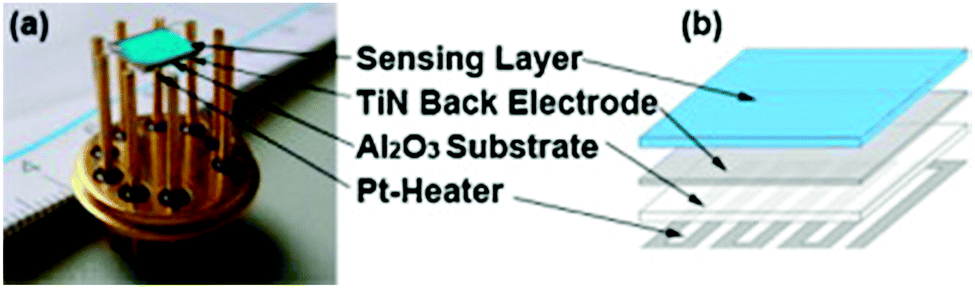 | ||
| Fig. 6 (a) Photograph of the Kelvin probe type sensor fabricated by Fleischer et al. and (b) schematic of the Kelvin probe highlighting the different layers that comprise the device.141 Reprinted from Sensors and Actuators B: Chemical, 187, Fleischer et al., Work function based gas sensing with Cu-BTC metal–organic framework for selective aldehyde detection, 142–146, Copyright 2012, with permission from Elsevier. | ||
Using this Kelvin probe setup, the measured signal represented the difference in the work function of the oscillating gold paddle and the sensing layer. As the gold paddle is not sensitive to the gases used, the signal represents the changes of the electronic structure of the sample under investigation. Humidity sensing measurements were performed in synthetic air (20% O2 and 80% N2) at a flow rate of 1 L min−1 at 25 °C, and the humidity was studied in the range 0–50%RH. It was found that the sensing response was reversible and stable with a decrease in work function (Δϕ) of 5.5 mV per percent increase in relative humidity. In addition it was found that there was only a small influence of variation in humidity on the work function response of the TiN sample; the HKUST-1 layer imparted sensitivity to water vapour. However, the authors also demonstrated the sensitivity of the device to aldehydes and thus the device suffers from cross sensitivity. No response time was given for sensing of water and the thermal stability of the sensor device was also not indicated.
Kaskel et al. reported a mesoporous MOF that acted as a humidity detector.149 The MOF was the first in a class of MOFs based on the N,N,N′,N′-benzidinetetrabenzoate ligand (benztb) (Fig. 7) with auxiliary 1,3,5-benzenetricarboxylate (btc) ligands to strengthen the mesoporous framework.
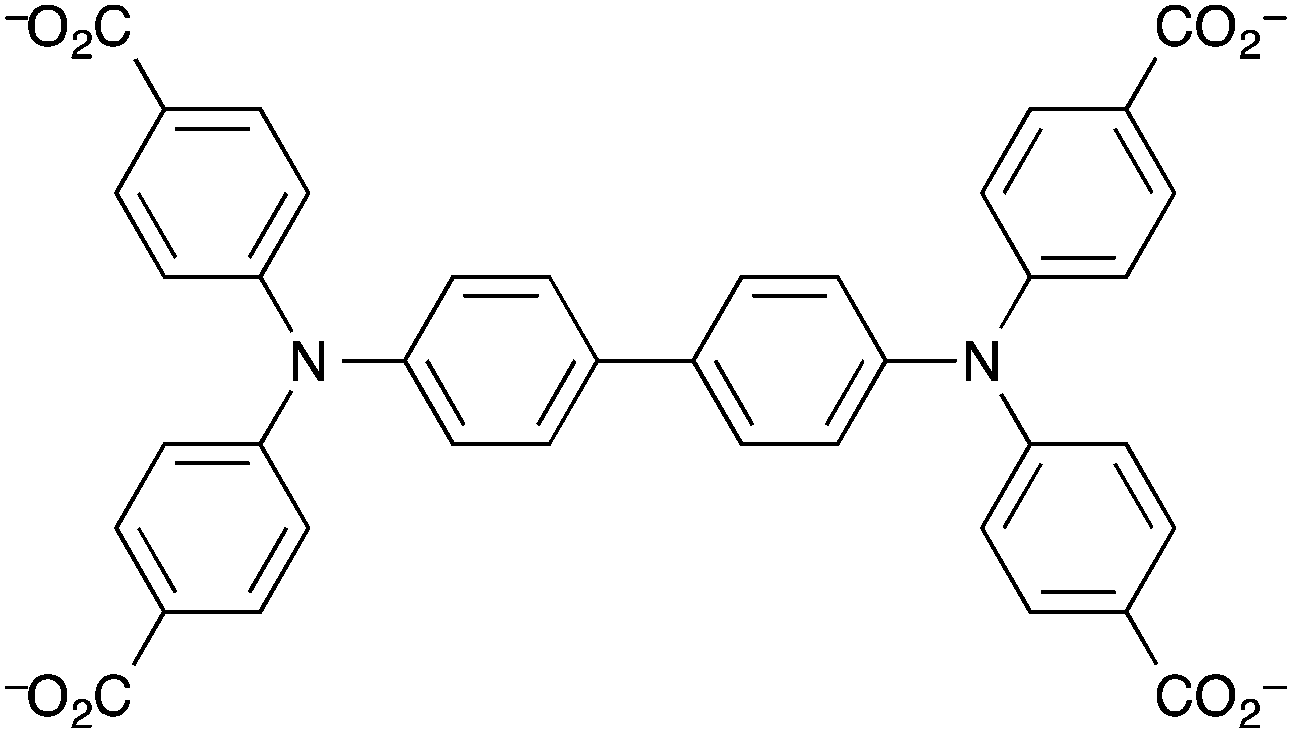 | ||
| Fig. 7 The N,N,N′,N′-benzidinetetrabenzoate (benztb) ligand. | ||
The particular MOF used in this work was [Zn4O(benztb)(btc)2/3] (DUT-25) in which two types of pore were found to exist: one mesopore with an internal diameter of 32 × 20 Å and the other with internal dimensions of 18 × 8 Å. The BET specific surface area of the MOF was found to be 4670 m2 g−1. This coupled with the large mesopore means that DUT-25 is suitable for doping with a gasochromic species to enable visual response detecting of gases and vapours. Kaskel et al. loaded the porous structure with the solvatochromic dye Nile Blue and demonstrated that the solvatochromism of the dye was intact despite encapsulation. Upon exposure to water vapour the colour changed from dark blue (due to the presence of Nile Blue in the ‘dry’ state) to light blue and thus the MOF is a visible indicator material for humidity. The highly porous material could be of interest to developers of exhaust gas sensors based on MOFs as DUT-25 was determined to be thermally stable up to ∼400 °C in air.
Wang et al. have reported a photonic film of a MOF which was used to detect water vapour.150 Nanoparticles of the iron-based MOF [Fe3O(abdc)3X], NH2-MIL-88B, (X = Cl, OH, abdc = 2-amino-1,4-benzenedicarboxylate) were deposited by spin deposition onto a silicon wafer. The coated silicon wafer was then placed into a quartz cuvette which contained a small volume of water, however the wafer did not come into contact with the water. The UV-visible reflection spectrum of the MOF film was then recorded every 2 min at 20 °C until the reflection spectra became stable. Adsorption of water into the pores of NH2-MIL-88B replaced air and thus the effective refractive index of the MOF increased. This increase in refractive index resulted in a redshift of the interference peaks in the reflection spectrum by 50 nm. However, greater redshifts were measured upon exposure to hydrocarbon species. These responses are described in Section 3.5.2.
Finally, Ruan et al. have reported an impedance-based sensor for humidity that utilises the MOF [Ti8O8(OH)4(abdc)6] (NH2-MIL-125(Ti)).151 A film of NH2-MIL-125(Ti) was deposited onto five pairs of Ag–Pd interdigitated electrodes on a ceramic substrate. Upon exposure to relative humidity ranging from 11–95%RH, the largest magnitude change in impedance was measured at 100 Hz. Within this humidity range the impedance decreased from 4.5 MΩ at 11% RH to ∼124 Ω at 95% RH in a non-linear manner. The t90 response time of the sensor, upon increasing concentration of water vapour, was 45 s and the t90 recovery time was ∼50 s.
3.2 Oxygen sensors
The supercages of a faujasite-type zeolite have been used to host the dye 2-hydroxymethylanthraquinone for oxygen detection.99 After photo-activation, the anthraquinone is transformed into oxygen-reactive dihydroxyanthracene, which in the presence of oxygen turns into a coloured anthraquinone allowing the reaction to be followed quantitatively using UV-visible spectroscopy. However, the utility of this detector is limited to one-off oxygen contamination detection by the fact that the reactive dihydroxyanthracene is irreversibly turned into the anthraquinone.
For potentiometric/amperometric O2 sensing, thin films of zeolites were used to cover conducting sensing electrodes to selectively limit CO2 exposure of the sensing surface.105 As the diffusion of oxygen through the zeolite film is preferential to that of CO2, the coated sensor showed a marked drop in response towards CO2 along with slightly diminished but still discernable response towards O2. A further amperometric and potentiometric bifunctional sensor was developed by Dutta et al. which could measure O2 and NO2 simultaneously in ppm concentrations.106 The O2 and NOx work by Dutta et al. is discussed further in Section 3.3.1.106
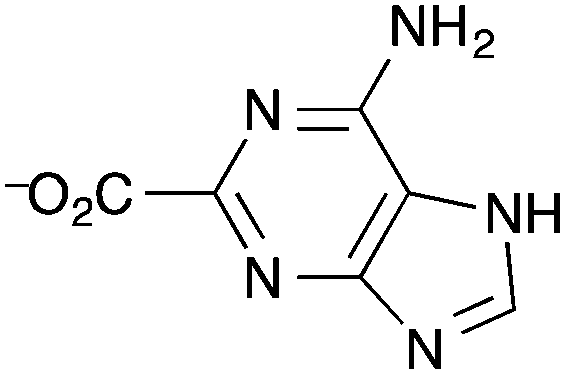 | ||
| Fig. 8 The adeninate ligand in the Yb3+@bio-MOF-1 reported by Rosi et al. as a potential near-infrared luminescent sensor for oxygen gas.152 | ||
Ir(ppy)3 complexes (ppy = 2-phenylpyridine) possess highly efficient phosphorescence owing to the facile intersystem crossing to populate the 3MLCT state. This 3MLCT phosphorescence can be readily quenched by molecules with a triplet ground state such as O2. Lin et al. devised a phosphorescent MOF that consisted of Zn-based secondary building units and ligands that are derivatives of Ir(ppy)3.153 The linker was a cyclometalated Ir complex based on the 2-phenylpyridine carboxylate derivative L2 (Fig. 9).
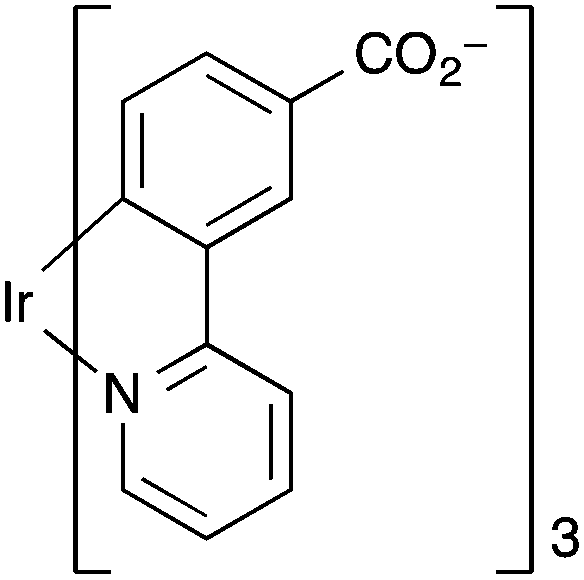 | ||
| Fig. 9 The iridium(III) metalloligand L2 used by Lin et al. to fabricate a luminescent MOF for oxygen gas detection.153 | ||
The porous 2D MOF [Zn4O(L2)2]·6DMF·H2O was ground and pressed onto the surface of a KBr pellet, which in turn was placed into a quartz cuvette that was equipped with an inlet/outlet port for O2 ingress or vacuum. Upon excitation at 385 nm, and after evacuation for 2 h under dynamic vacuum, the luminescence intensity centred at 538 nm decreased upon exposure to O2 with a sensitivity down to 0.05 atm and quenching efficiency of 59% at 1 atm of O2. The MOF also demonstrated reversible quenching by O2 with <5% of the original luminescence intensity lost after 8 cycles with an adsorption response time of ∼30 s and a desorption recovery time of ∼120 s. The MOF was thermally stable up to ∼300 °C.
Sheu et al. also used an Ir(III) complex as a ligand to create luminescent MOFs for which the luminescence was quenched in the presence of oxygen.154 The Ir(III) complex [Ir(ppy)2(dcbpy)]− (L3) contains two bidentate 2-phenylpyridine (ppy) ligands and 4,4′-dicarboxy-2,2′-bipyridine (dcbpy) as the third bidentate ligand (Fig. 10). Four MOFs were synthesised by stirring H2L3 with the perchlorate salts of zinc(II), cadmium(II), cobalt(II) and nickel(II) in aqueous DMF solution. Of these compounds, the cadmium MOF [Cd(L3)2(H2O)2]·3DMF·6H2O demonstrated the greatest oxygen detection characteristics with a quenching efficiency of 74% at 1 atm O2, a linear relationship on the Stern–Volmer plot (ratio of initial emission intensity Io, to measured emission intensity I, against mole fraction of oxygen χO2; Io/I vs. χO2) in the χO2 range 0–1, and the highest Stern–Volmer quenching constant, KSV, at both 1 atm O2 and at O2 concentration <0.2 mole fraction. In addition this material had favourable detection kinetics; a 95% response time of 70 s and a 95% recovery time of 30 s. The MOF also showed good reproducibility and the detection response was reversible. Despite the MOF being stable up to ∼300 °C, the detection response rapidly deteriorated at temperatures >50 °C. This was ascribed to the loss of co-ordinated guest/free solvent molecules that are involved in the oxygen-detection mechanism.
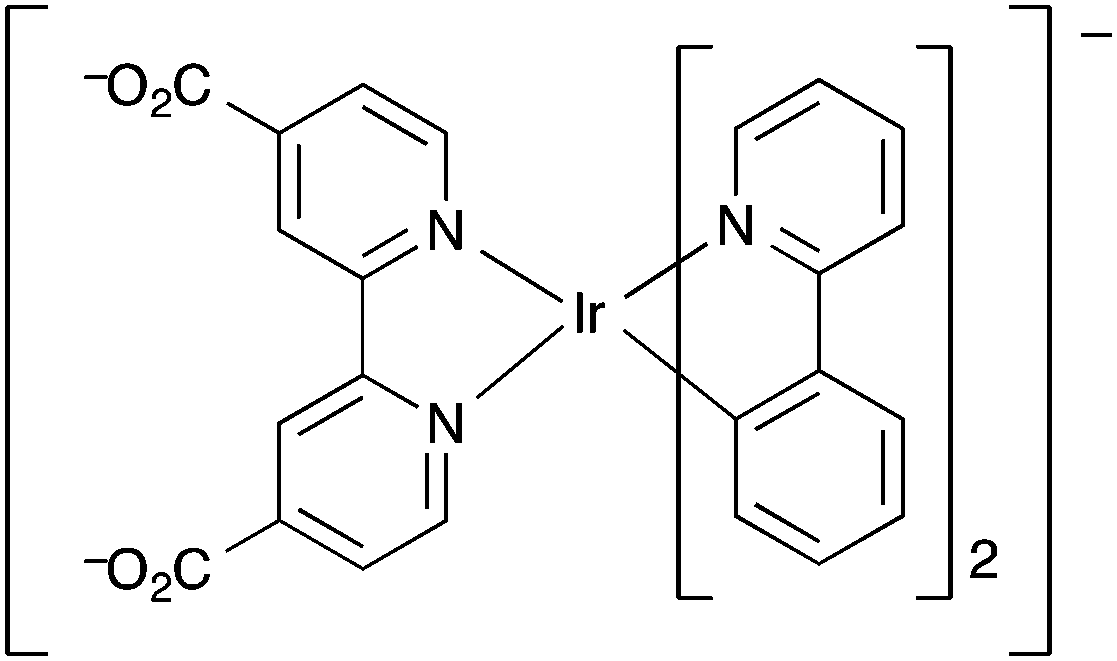 | ||
| Fig. 10 The iridium(III) metalloligand [Ir(ppy)2(dcbpy)]−, L3, used in the oxygen sensing MOFs synthesised by Sheu et al.154 | ||
Lin et al. further investigated the use of metal complexes as ligands in three-dimensional MOFs for luminescence-based detection of oxygen.155 By incorporating the iridium and ruthenium complex ligands L4, L5 and L6 (Fig. 11) into the zirconium-based MOF [Zr6O4(OH)4(bpdc)6], UiO-67, via a ligand substitution strategy, three phosphorescent MOFs were fabricated.
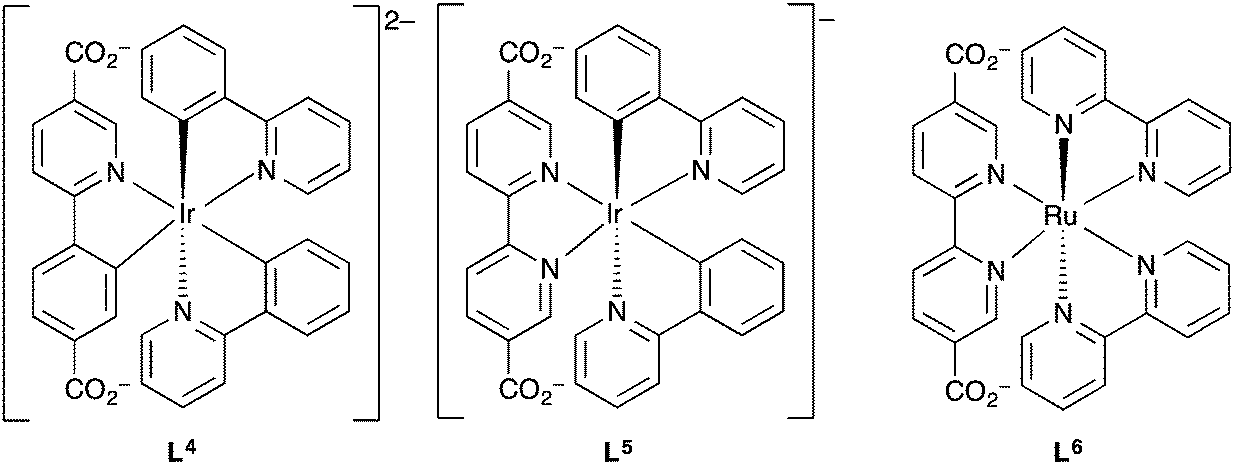 | ||
| Fig. 11 The phosphorescent metalloligands L4–L6 incorporated into UiO-67 by Lin et al. for oxygen sensing application.155 | ||
Nitrogen adsorption/desorption type 1 isotherms at 77 K were obtained for all three of the MOFs and [Zr6O4(OH)4(bpdc)5.971(L4)0.029], [Zr6O4(OH)4(bpdc)5.942(L5)0.058] and [Zr6O4(OH)4(bpdc)5.821(L6)0.179] all had BET surface areas >1000 m2 g−1 indicating microporosity. The MOFs were ground and pressed onto the surface of KBr pellets, which in turn were placed into quartz cuvettes that were equipped with an inlet/outlet port for O2 ingress or vacuum. Upon excitation at 420 nm, changes in the emission intensity at 600 nm were measured upon stepwise increase of O2 pressure starting from vacuum. Decreasing emission intensity with increasing oxygen concentration was observed for the MOFs between 0.001–0.839 atm O2. The phosphorescence emission of the L5-based MOF was most efficiently quenched by oxygen with a quenching efficiency of 65% at 0.8 atm O2. It was found for all three MOFs that the luminescence intensity decreased instantly after initial O2 concentrations and then much smaller extents of luminescence intensity reduction were observed for the subsequent stepwise O2 pressure increases. Thus all three MOFs displayed a non-linear response to increasing oxygen concentration which was further demonstrated by the non-linear Stern–Volmer curves. This suggested that the phosphorescent sites were distributed in an inhomogeneous manner within the MOF frameworks. The MOFs displayed reversible, reproducible sensing responses that returned to ∼1–3% of the initial intensities even after >5 oxygen sensing cycles.
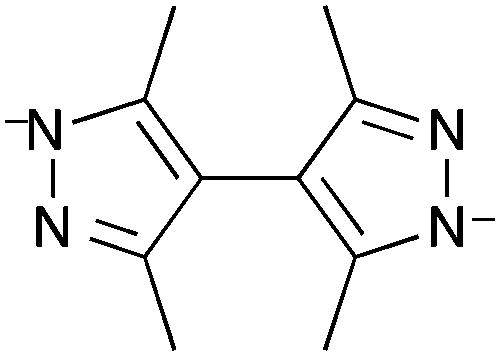
Chen et al. also used the approach of doping a parent MOF framework with phosphorescent ligands to afford a phosphorescent oxygen-detecting MOF.156 Chen et al. chose MAF-34 [Zn7(ip)12](OH)2 (ip = imidazo[4,5-f][1,10]phenanthrolin-1-ide, Fig. 12) as the parent MOF and doped this within varying amounts of ruthenium to form four phosphorescent MOFs of the formula [RuxZn7−x(ip)12](OH)2 where x = 0.10–0.16.
 | ||
| Fig. 12 The imidazo[4,5-f][1,10]phenanthrolin-1-ide ligand in the ruthenium-doped MAF-34 MOFs of Chen et al.156 | ||
The low concentration of the phosphorescent {Ru(ip)3}− units in the MOF meant that self-quenching was avoided. Upon excitation at 480 nm and exposure to increasing oxygen pressure the intensity of the phosphorescent emission at ∼600 nm decreased and blueshifted ∼5 nm with a very fast response. At 1 atm of O2 the emission intensities of the MOFs were quenched by between 75 and 88%, the authors attributing the high quenching efficiencies to the ultramicroporous channels within the MOFs and low ruthenium content which leads to more oxygen adsorbed per ruthenium phosphorescent centre. Indeed, high O2 uptake of 3.2 cm3 g−1 (0.20 mol L−1) was measured at 1 atm and 25 °C.
Time dependent luminescence emission profiles, in the absence of oxygen, show biexponential decay characteristics which suggests that the ruthenium phosphorescent centres are in two different environments within the MOFs. This was further confirmed by the non-linear Stern–Volmer plots recorded for all the MOFs, which indicate heterogeneous distribution of the luminescent species. However, a linear response to changing oxygen concentration is measured if a Freundlich adsorption isotherm treatment is applied to the data.
Chen et al. fabricated a composite thin film of [Ru0.16Zn6.84(ip)12](OH)2 in silicon rubber on a glass substrate. The thin film demonstrated very fast responses to changing oxygen concentration, good reversibility, reproducibility and stability. In addition, they demonstrated that this composite thin film could be applied to the outside of a commercial blue-light LED which enabled easy identification of changing oxygen concentration with a colour-changing device. The MOF-modified LED emitted magenta light in the presence of N2, due to the blue emission of the LED combined with the reddish-orange emission of the MOF. In the presence of O2 the intensity of the reddish-orange emission of the MOF decreased significantly so that the emitted light from the device was violet.
Chen et al. have also developed a fluorescent MOF which did not contain any precious metals.157 [Zn4O(bpz)2(abdc)] (bpz = 4-(3,5-dimethylpyrazol-1-id-4-yl)-3,5-dimethyl-pyrazol-1-ide, Fig. 13), MAF-X11, exhibited fluorescent emission at ∼470 nm upon excitation at 345 nm, the intensity of which decreased with increasing oxygen pressure. At 1 bar of O2, the fluorescence was quenched by 96.5% which is comparable to quenching intensities reported for luminescent hybrid materials containing precious metal complexes. The ratio of initial fluorescent intensity to measured luminescent intensity linearly related to changing pressure of O2 with a Stern–Volmer constant KSV = 27.1 bar−1. Upon repeated cycling between 1 bar O2 and vacuum the quenching response was found to be rapid, reversible and highly stable. Furthermore it was demonstrated that the quenching efficiency of the MOF in air was equivalent to the quenching efficiency at an O2 pressure of 0.21 bar, thus indicating that the MOF was not cross-sensitive to other gases in air. In addition the MOF was found to be thermally stable up to >450 °C.
 | ||
| Fig. 13 The 4-(3,5-dimethylpyrazol-1-id-4-yl)-3,5-dimethyl-pyrazol-1-ide ligand in the oxygen sensing fluorescent MOF MAF-X11, reported by Chen et al.157 | ||
Ligand-to-metal energy transfer (EnT) is responsible for the characteristic luminescence of lanthanide MOFs. It has been proposed that the quenching of the luminescence of lanthanide MOFs upon exposure to oxygen gas is due to oxygen deactivating the triplet-state of the organic ligands whilst having very little interaction with the lanthanide ions. For efficient oxygen sensing with lanthanide MOFs, the MOF needs to possess an effective EnT and an appropriate thermally activated energy back transfer (BenT) that will prolong the triplet-state lifetime of the ligands. With this in mind, Qian et al. proposed doping [(CH3)2NH2][In3O(btc)2(H2O)3]2[In3(btc)4], CPM-5, and [In3O(OH)(H2O)2(btc)2], MIL-100(In), with Tb3+ as they realised that the first triplet-state energy of the btc ligand is ∼23200 cm−1, which is close to the 5D4 excited state of Tb3+ (∼20400 cm−1).158 They proposed that this would result in an effective EnT and appropriate BenT, both of which would be beneficial to oxygen detection. Thin films (∼2.5 μm thick) of CPM-5 and MIL-100(In) were grown solvothermally on indium tin oxide glass and then doped with Tb3+ by a post-fabrication immersion treatment into an aqueous DMF solution of terbium(III) nitrate to form Tb3+-doped CPM-5 and MIL-100(In) known as CPM-5⊃Tb3+ and MIL-100(In)⊃Tb3+ respectively. When the luminescent properties of CPM-5⊃Tb3+ and MIL-100(In)⊃Tb3+ were investigated, it was found that the quantum yield of MIL-100(In)⊃Tb3+ was 16.8% which was much larger than the quantum yield of CPM-5⊃Tb3+ (1.1%).
Upon exposure to O2 in a N2 carrier gas the respective luminescent emissions, at λem = 544 nm for CPM-5⊃Tb3+ and λem = 546 nm for MIL-100(In)⊃Tb3+, were quenched with a quenching efficiency of 47% at 1 atm O2 for CPM-5⊃Tb3+ and 88% at 1 atm for MIL-100(In)⊃Tb3+. Stern–Volmer plots for the two MOFs showed good linearity suggesting the Tb3+ was distributed homogeneously within the MOFs. The Stern–Volmer quenching constant (KSV) for MIL-100(In)⊃Tb3+ was an order of magnitude greater than KSV for CPM-5⊃Tb3+. This was ascribed to the greater efficiency of the intramolecular energy transfer within MIL-100(In)⊃Tb3+ as opposed to the intermolecular energy transfer within CPM-5⊃Tb3+. Overall the MIL-100(In)⊃Tb3+ MOF possessed more desirable properties for O2 sensing as exemplified by a relatively faster sensing time. It took 6 s to achieve 95% of the maximum reduction in luminescent intensity upon changing from a 100% N2 atmosphere to a 100% O2 atmosphere for MIL-100(In)⊃Tb3+ and 90 s for CPM-5⊃Tb3+. Upon changing from a 100% O2 atmosphere to a 100% N2 it took 53 s for 95% of the maximum recovery in luminescent intensity to be achieved for MIL-100(In)⊃Tb3+ and 60 s for CPM-5⊃Tb3+. Furthermore, MIL-100(In)⊃Tb3+ exhibited good reversible oxygen quenching and nitrogen recovering properties.
3.3 NOx sensors
In the examples discussed below, the zeolite layer acts as a catalyst prior to sensing in order to maintain a constant composition of the NOx mixture thus enabling measurement of the equilibration between NO and NO2 at different temperatures by potentiometry between working and reference electrodes. A NOx sensor with minimal interferences from CO and O2 comprised yttria-stabilised zirconia (YSZ) pellets and a Pt-loaded zeolite Y layer.107 These sensors contain catalytic filters based on YSZ working at temperatures in the range of 300–700 °C. The development of a NO sensor capable of operating in automotive exhaust streams was reported from the same group based on a Pt-yttria-stabilised zirconia (Pt-YSZ) covered by zeolite. The sensors were tested at temperatures between 500 and 700 °C, and the zeolite was permeable to oxygen, thus, was minimising the interference from O2. The sensor shows interference from CO and NO2, however these tests indicate that the device is able to detect NOx in engine exhausts.109 Using Pt-doped zeolite Y on a yttria-stabilised zirconia electrolyte, the NOx concentration could be measured using a sensing electrode.108 This sensor device was then optimised to reduce interference from other gases. The addition of a WO3 electrode provided the highest reactivity difference with the Pt-doped zeolite Y and interference from 2000 ppm CO, 800 ppm propane, 10 ppm NH3, as well as the effects of 1–13% O2, CO2, and H2O were minimised.110,111 A human breath analysis was performed with 20 of those sensors connected in series for the diagnosis of asthma since NO in breath increases from 5–10 ppb (normal state) to 100 ppb in an asthmatic state.112
The imbalance in equilibration between NO2 and NO could also be managed by a voltage change, instead of temperature, to build an amperometric sensor. An example has been reported using the same material, a Pt-doped zeolite Y on a yttria-stabilised zirconia, connected to a potentiostat.113 It demonstrated a ppm detection of NOx with no interference from CO and hydrocarbons. Both amperometric and potentiometric monitoring were combined to build a bifunctional sensor which could measure NO2 and O2 simultaneously in ppm concentration with almost no interference between each other.106
A QCM oscillator-based sensor was also developed for NO sensing that took advantage of the adsorption of NO on zeolites. Zeolite A, silicalite-1 and sodalite were deposited on a QCM and NO adsorption was followed. The zeolite-A doped QCM was shown to be suitable for monitoring 50 ppm of NO in He at 443 K.57
Another approach involves preparation of sulfur-resistant bimetallic noble metal layers, where Cr and W improve the sulfur resistance of Pd–Y zeolite (FAU-type framework structure). On the other hand, La, Mn, Mo and Ag make the sulfur resistance worse and the second metal had no evident influence on product selectivity and the acidic properties of the catalysts. In all of these cases the metals were introduced by ion exchange, and the metal clusters tend to agglomerate with time and migrate to the surface of the sensing layers.188
Sensor protection in a destructive thermal and chemical environment was ensured by depositing a porous absorption material around the gas-sensitive section. A zeolite-type material or boehmite was introduced in the sensor substrate with additives and binders and subjected to thermal treatment up to 700 °C. The protective layers were stable at the described working conditions.189 Protective hydrophilic layers were prepared to prevent water from interfering with the operating sensors. These layers were based on highly hydrophilic materials such as zeolites A, X, and Y.68
It is notable that no examples of MOFs for NOx sensing could be found in the literature at the time of writing. However, NO adsorption in MOFs has been reported by Morris and co-workers for biological applications,190–193 suggesting MOFs that possess suitable stability for automotive exhaust gas sensing hold promise as potential sensing materials for NO.
3.4 CO and CO2 sensors
With respect to conductive polymer and zeolites for CO sensing, two composites have been investigated. Firstly, a polyaniline/zeolite A-type zeolite sensor has been developed which exhibited high selectivity towards CO, especially with the Ca2+ form of the zeolite.62 Indeed, Ca-zeolite A possesses larger pores than the K+ and Na+ analogues and thus does not greatly influence the diffusion of CO through the conductive polymer.69 The second example reported the utilisation of poly(3,4-ethylenedioxythiophene) doped with polystyrene sulfonic acid and ZSM-5.75 The sensitivity of the composite increased when the Si/Al mole ratio of the zeolite decreased. However, this composite only produced irreversible responses.
Under a CO reductive atmosphere, the reversible redox behaviour of TiO2 clusters within Y-type zeolite pores was followed by in situ UV-vis spectroscopy providing an efficient CO detector.114 The zeolite matrix permitted good stability of the clusters towards several oxidation–reduction cycles. In comparison with bulk TiO2, the TiO2–Y composite showed 10 times faster response times, of about 1 s at 800 °C.
As an alternative to conductivity measurements, a calorimetric sensor has been developed that measures the heat liberated during the catalytic reaction occurring in Pd-doped zeolites.63 Combined with a thin-film calorimeter, Pd-zeolite A exhibited a selective response towards CO over alkanes such as n- and i-butane, cyclo- and n-hexane.
Recently, Pd-containing perlialite and beta polymorph A zeolite films showed high molecular recognition towards CO through in situ IR spectroscopy.94 Fast detection of 2 to 100 ppm concentration of CO in the presence of highly concentrated vapours of methanol or pentane (400–4000 ppm) has been demonstrated. Similar results were obtained using a Pt-containing beta polymorph A zeolite film for CO detection in the presence of a highly concentrated methanol atmosphere.95
A convenient gas sensor combining a solid electrolyte NASICON, lithium carbonate and a NaY-type zeolite as filter has been reported for air quality control through CO2 sensing.115 The fabricated sensor was found to be stable over two years and could follow the CO2 concentration via impedance measurements. For example, when CO2 exposure increased from 350 to 1000 ppm, the response time was about 2 min. More recently, the NASICON was replaced with sodium conductive ZSM-5 zeolite which provided higher selectivity towards CO2.88 The zeolite layer works as both a catalyst and filter due to its sodium cation conductive activity and its well-defined small pore sizes. Conductive-based CO2 sensing has also been demonstrated using a polymer poly(aniline)–zeolite Y composite.194 This sensor showed good ability for CO2 sensing at room temperature.
A composite made of a zeolite Y film grown on a Metglas magnetoelastic strip has also been developed for CO2 sensing in a N2 atmosphere.195 Indeed, when an alternating current magnetic field is applied, the specific resonant frequency showed a discernable decrease down to a concentration of 0.33% CO2 in N2. However, a dry atmosphere is required to avoid interference with water as 2% relative humidity affected measurements. A similar device was subsequently reported using silicalite-1 thus permitting the sensing of CO2 in air instead of a N2 atmosphere.76
Microcalorimetric devices can now be made using lithographic techniques. One of the two sensitive areas of such a device (where evolved heat can be measured) was coated with a thin film of CoAlPO4-5, the other was kept open as a reference.130 The additional benefit of a zeolite with catalytic activity for such a device is the molecular sieving effect that can be combined in the response of the sensor (a molecule too big to enter the catalytically active interior of the zeolite should only show a weak response). The change in temperature was measured with a meandering Pt-wire resistor. This device was examined for the detection of CO and cyclohexane, and sensitivity and selectivity in the low ppm range was observed.
 | ||
| Fig. 14 Schematic of the layout of the in situ IR reactor cell and IR operando gas detection experimental set-up used by Mintova et al. for detection of carbon monoxide.159 Reprinted with permission from X. Zou, J.-M. Goupil, S. Thomas, F. Zhang, G. Zhu, V. Valtchev and S. Mintova, J. Phys. Chem. C, 2012, 116, 16593–16600. Copyright 2012 American Chemical Society. | ||
The IR spectral changes of the thin films of Cu–Y(btc) due to exposure to a range of concentrations of CO (1–100 ppm) were monitored in operando at room temperature. The adsorption of CO on the Cu0 metal clusters resulted in the appearance of an IR absorption peak centred at 2114 cm−1; the area of this peak could be quantified and related to the concentration of CO present in the Ar carrier gas. Remarkably a concentration as low as 1 ppm of CO in Ar can be detected by measuring the change in absorption at 2114 cm−1 and the detection was reversible, albeit requiring a purge of Ar gas for 4 h at room temperature. Additionally the MOF is thermally stable up to ∼500 °C and selectively detected CO over water, which was always present at a concentration of ∼100 ppm in the Ar carrier gas, and to vapours of chloroform and 2-ethylthiophene that were deliberately introduced. The MOF also showed reproducibility with no diminished response after 10 sensing cycles.
The piezoelectric microcantilever humidity sensor, modified with HKUST-1, demonstrated by Hesketh et al. (Section 3.1.2) also displayed sensitivity to carbon dioxide.140,142 The authors tested the response of the device to CO2 in nitrogen carrier gas at room temperature and pressure with HKUST-1 in the dehydrated state. The sensor response was non-linear within the range of water vapour concentrations investigated, ∼10%–∼70% concentration. However, when HKUST-1 was in the hydrated state no response to carbon dioxide was observed. The authors postulated that the removal of water molecules coordinated to the copper centres was required for carbon dioxide to bind.
The refractometric optical-based detector which utilised HKUST-1 as the sensing material, fabricated by Hupp et al. (Section 3.1.2), displayed limited sensitivity to carbon dioxide.139 Upon exposure to carbon dioxide the measured ‘stop band’ redshift was ∼3 nm relative to N2. The device also displayed sensitivity to other analytes other than water vapour, thus isolation of carbon dioxide from other exhaust gases and vapours would be required.
Candler et al. utilised zeolitic imidazolite framework (ZIF) nanoparticle-couple microresonators to sense carbon dioxide.160 The silicon microresonators were coated with particles of [Zn(cbIM)(nIM)] (cbIM = 5-chlorobenzimidazol-1-ate, nIM = 2-nitroimidazolate, Fig. 15), ZIF-69, by dielectrophoresis. The resonator structure was then exposed to various concentrations of CO2 at room temperature and the change in resonant frequency of the microresonator relative to the anchor was measured with a laser Doppler vibrometer set-up. A linear response was observed between the change in relative frequency and concentration of CO2 in the range 0–4000 ppm. At 5200 ppm of CO2 it was determined that 72 times more CO2 was adsorbed onto the ZIF-69 coated microresonator than on the bare silicon microresonator, thus highlighting the value of increasing the surface area for gas adsorption and thus enhancing the sensitivity of gas sensors. The sensor was cross-sensitive to isopropanol vapour, but differentiation was possible when the decay constant of the adsorption times were compared. The lowest mass change that was measureable and thus the lower detection limit of the sensor set-up was calculated to be ∼26 fg.
 | ||
| Fig. 15 The 5-chlorobenzimidazol-1-ate (cbIM) and 2-nitroimidazol-3-ate (nIM) ligands in ZIF-69. | ||
Duyne et al. utilised a localised surface plasmon resonance (LSPR) spectroscopic approach to detect the interaction of carbon dioxide with triangular silver nanoparticles that had been coated with HKUST-1.143 Upon exposure to CO2 a redshift in the localised surface plasmon resonance wavelength was measured due to adsorption of CO2 within the MOF and thus increase of the refractive index of the MOF-coated silver nanoparticles. The measured redshift in the LSPR wavelength for the device coated with HKUST-1 coated silver nanoparticles was 14 times greater than for the device coated with bare silver nanoparticles. The sensor device displayed a dynamic range in the concentration range 0–100% CO2 in N2 carrier gas and the sensing responses were reversible and reproducible over 5 cycles.
Pardo et al. demonstrated a detector for carbon dioxide based on the two-dimensional MOF MV[Mn2Cu3(mpba)3(H2O)3]·20H2O (MV = methylviologen dication and mpba = N,N′-1,3-phenylenebis(oxamate), Fig. 16), which is both magnetic and luminescent.161 Upon excitation at 400 nm, exposure to 1 bar carbon dioxide or a 1.5 bar 1:1 mixture of carbon dioxide and methane, a luminescent wavelength redshift of 17 nm was measured indicating an interaction of CO2 but not methane with the MOF. It was shown that the MOF was sensitive to both water and methanol in toluene. The authors predict that differentiation of carbon dioxide from both water and methanol would be possible as methanol and water exposure resulted in an emission wavelength blueshift whereas an emission wavelength redshift was measured upon exposure to carbon dioxide.
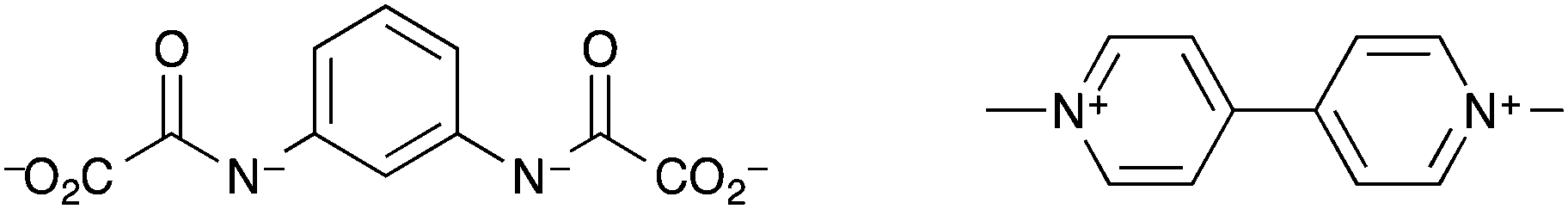 | ||
| Fig. 16 (left) N,N′-1,3-phenylenebis(oxamate) tetraanion and (right) methylviologen dication.161 | ||
3.5 Hydrocarbon and volatile organic compound sensors
As described in Section 3.1.1, sensors have been prepared using SnO2 with zeolite A- and ZSM-5-type zeolites as filters.59 Results showed that the SnO2/zeolite A sensor responded significantly to ethanol vapour along with improved selectivity towards H2O by the elimination of the response to hydrocarbons (propane and methane). Pd-doping of the SnO2/zeolite A sensor improved the selectivity against hydrocarbons as only 1250 ppm of propane provided any response.53,70 The water adsorbed by the hydrophilic zeolite A blocked the ingress of propane and any propane that did ingress was subsequently oxidised on Pd.
Another example reported the use of silicalite-1 on SnO2 for the sensing of ethylene in the presence of water, where the zeolite layer acted as a filter to improve selectivity towards hydrocarbons.89 The [010] oriented zeolite layer gave improved response and recovery time (14 s, 144 s) compared to a randomly oriented zeolite layer (25 s, 208 s). This was rationalised on the basis of vertical channels of the silicalite-1 permitting the concentration of ethylene faster than with randomly orientated silicalite-1.
Recently, a Si cantilever was used to detect toluene and ethanol vapours down to ∼25 ppmv in air.54 The adsorbent layer consisted of either organophilic silicalite or hydrophilic zeolites A and Y. To remove the adsorbed species from the nanoporous structure and thus improve performance, the hydrophilic zeolite-coated cantilevers were degassed at high temperature. Therefore, zeolite Y cantilevers with integrated heaters have been developed and could detect 1.4 ppmv of ethanol and 0.7 ppmv of toluene in air.
Semiconducting chromium titanium oxide covered with different types of zeolite (H-forms of zeolite A, ZSM-5, beta polymorph A and zeolite Y) have also been investigated as ethanol sensors.61 The highest sensitivity was obtained with the zeolite Y-modified sensor; the sensing response was doubled compared to a bare sensor. Computational studies showed that the improved sensitivity was due to a catalytic reaction and diffusion behaviour within the zeolite. Two semiconducting metal oxides (tungsten and zinc) covered with zeolite have also been described as ethanol sensors, but it has been reported that the cross-sensitivity to NO2 was significant (Section 3.3.1).49
Using two semiconducting tungsten and chromium titanium oxide sensors covered with the acidic form of zeolite A, it was reported that ethanol could be discriminated from isopropanol.64 Computational studies indicated that this discrimination was due to size and shape selectivity afforded by the zeolite A layer. Earlier, an electrical impedance study took advantage of the shape-selectivity of zeolite channels using natural stilbite zeolite for the discrimination of methanol, 2-propanol and 3-pentanol against water and 2,2-dimethylpropanol.129 Additionally, a study on the electrical conductivity response of three different zeolites (ZSM-5, mordenite and zeolite Y) towards ethanol was reported.50 The effects on sensing response of the framework structure, the nature of the charge balancing cation (NH4+ or H+ forms) and the hydrophilic/hydrophobic character due to Si/Al ratio were investigated. The sensitivity decreased with the increase of the Si/Al ratio and ammonium forms of zeolites gave negative responses whereas all H+ forms gave positive responses. The H+-zeolite Y with a Si/Al ratio of 30 provided the highest electrical conductivity sensitivity to ethanol vapour.
Another possibility for alcohol and hydrocarbon sensing is the use of a conductive polymer combined with zeolites. Two polymers combined with the K+ form of zeolite A were investigated. Both polypyrrole and polypyrrole–polyamide-6 composite sensors exhibited sensitivity towards acetone, methylether ketone, methanol and toluene.65 Interestingly, this study showed that the preparation of composites influenced the results, as pellets and films were found to be vapour sensitive whereas electrospun fibre bundles were insensitive. However, in the case of methanol sensing, the role of the zeolite remained unclear as the sensitivity could also be explained by the interaction between methanol and polyamide-6.
A silicalite-1–Metglas composite originally developed for CO2 sensing in air (see Section 3.4.1) was found to efficiently discriminate linear and branched hydrocarbons.76 It selectively detected n-butane while it did not respond to the presence of i-butane. Recently, a new study concerning zeolite–Metglas composites was reported for hydrocarbon sensing.66 It was found that a faujasite-based composite exhibited the lowest detection limits for o-xylene at just 6 ppm but suffered of a lack of selectivity with cross-sensitivity to p-xylene. On the other hand, a zeolite A-based sensor showed high selectivity between xylene isomers. Concerning n-hexane sensing, the best sensitivity (180 ppm) was obtained using the randomly-oriented ZSM-5-zeolite/Metglas sensor.
A composite comprised of zeolite Y cages as hosts for C60 has also been developed.116 Two types of composites, C60/zeolite Y and K-C60/zeolite Y, were deposited onto tantalum comb-type electrodes providing semiconductor sensors. In the presence of ethylene, the composites exhibited measurable changes in conductivity. Similarly, the zeolite ZSM-5 was used as a host for Pt clusters on two electrodes, Au and Cr, in order to build a propane-sensitive detector in the presence of O2/CO2/N2.77 The catalytic oxidation of propane in the presence of Pt and O2 resulted in a reversible drop in potential.
The conductivity induced by the mobility of extra-framework cations can also be measured by impedance or potentiometry methods. Impedance response towards hydrocarbons has been reported using a Cr2O3 layer on top of an ion-conducting Pt-doped ZSM-5.78 The device showed low interference to CO and H2, but no selectivity with NH3. Several studies by the same group were carried out to elucidate the mechanism of the sensing. It was found that the interfacial effect between Cr2O3 and Pt-doped ZSM-5 on the electrodes led to the selectivity of propane and propylene and that the impedance response was influenced by the presence of hydrocarbons at low frequencies.79 Two explanations were proposed to explain the impedance variation. On one hand, the presence of hydrocarbons might act as a charge density carrier with the Cr2O3 layer providing the impedance changes.197 On the other hand, the sorption of hydrocarbons could lead to the insertion of Na+ from the zeolite to the Cr2O3 layer which then led to the formation of sodium chromate at the interface providing the impedance changes.198
Potentiometric measurements between a reference Au/Na2CO3 electrode and a measuring electrode, comprised of BaCO3/ZSM-5/Au, have been studied for propane sensing at 400 °C.80,81 The differential between Na+ activity in the reference and Na+ activity in the ZSM-5 zeolite was monitored.82 The highest sensitivity was obtained using a high Si/Al ratio within the zeolite and time responses were improved by the use of films instead of pellets.67 More recently, the design of a planar potentiometric gas sensor based on Pt-doped zeolite films was envisaged by the same group.199
Catalytic reactions inside zeolite cages have also been developed for detection of hydrocarbons. As many zeolites possess acid sites, they can easily promote cracking reactions of C7–C10 linear alkanes. This property was exploited using Cr-zeolite Y and Mo-zeolite Y coated on semiconducting chromium titanium oxide.117,118 Using Cr-zeolite Y, nonane could be discriminated from heptane, octane and decane by producing a distinct response due to its catalytic transformation into methylpropanol and water. Using Mo-zeolite Y, linear alkanes provided mainly acetaldehyde along with water and ketones. Even if the discrimination and/or sensitivity was not high, an interesting point is that the sensing was monitored by the products of the catalytic reaction instead of by the analytes themselves.
A catalytic reaction was also used to increase selectivity to propane over propylene. A Pt-doped ZSM-5 layer covering semiconducting SrTi0.8Fe0.2O3 suppressed the propylene response while the propane was selectively detected.83,84 However, the mechanism of the suppression remains unclear. Pt was also used to dope zeolite Y which was then mixed with semiconducting TiO2.119 The obtained Pt-doped zeolite Y/TiO2 composite showed selectivity towards propane over CO. It was suggested that the propane oxidation catalysed by Pt-zeolite Y produced water which then interacted with TiO2 providing a resistance change which was monitored.
Optical measurements can also be performed. Adsorption within the zeolite increases the analyte concentration and thus enhances the sensitivity of the optical detector device. One example exploited the adsorption of n-hexane in silicalite-1 to provide a 180 times enhanced signal compared with an uncoated ATR element.90 The refractive index can also change due to the presence of the analyte. For example, when silicalite-1 was coated onto a long period fibre grafting detection limits of 16.5 ppm of isopropanol and 22 ppb of toluene were measured.91 However, the response time was faster for isopropanol than for toluene due to its smaller size; isopropanol undergoes a faster equilibration within the zeolite structure.
A different approach utilised the light emission obtained by the reaction of acetaldehyde with oxygen atoms in the zeolite cages.120 Followed by cataluminescence, the signal showed a linear response for 0.06–31.2 μg mL−1 acetaldehyde vapour. In addition, acetaldehyde could be discriminated from other aldehydes due to structural constraints within the zeolite; no response was measured with methanol, ethanol, isopropanol, toluene, chloroform, dichloromethane and acetonitrile.
An optical detector based on light emission upon detection of linear alkanes such as n-hexane which used a CsNaY zeolite has been developed.121 The catalytic activity has been explained by zeolite basicity which might enhance the formation of a four-membered ring transition state involving a carbonium ion and oxygen atom. The detection of linear alkanes was demonstrated down to a 0.55 μg mL−1 concentration limit with minor interference from alcohols and aromatics. In addition, linear alkanes could be discriminated from branched alkanes due to structural constraints within the zeolite under more concentrated atmospheres (3.88 μg mL−1).
000 ppm in nitrogen carrier gas, ‘stop band’ redshifts were measured. The relationship between ethanol vapour concentration and measured ‘stop band’ wavelength shift was approximately linear between 100–1000 ppm and then plateaued after ∼4000 ppm. It was estimated that the lowest change in concentration of ethanol vapour that could be measured with the device was 0.3 ppm. The sensitivity limit to ethanol vapour was lower than that for water vapour due to the higher relative refractive index of ethanol. However, a major limitation of this proof-of-concept device was the MOF was not selective; different concentrations of water and ethanol vapours in nitrogen carrier gas resulted in identical magnitude ‘stop band’ shifts. The device demonstrated limited sensitivity to ethane and ethylene gases in nitrogen carrier gas. The measured ‘stop band’ redshifts were ∼5 nm and ∼6 nm relative to the ‘stop band’ wavelength in N2 for ethane and ethylene respectively.
Hupp et al. also reported a ZIF-8 based Fabry–Pérot device as a selective sensor for propane and ethanol vapours.163 Adsorption of ethanol or propane into the pores of [Zn(mIM)2] (mIM = 2-methylimidazolate), ZIF-8, replaced nitrogen and thus the effective refractive index of the MOF increased. The increase in refractive index due to exposure to either ethanol or propane vapour resulted in a redshift of the interference fringes in the transmission spectrum. The response of the system to propane in nitrogen gas in a concentration range 0–100% was linear with a maximum interference fringe redshift of ∼50 nm measured in 100% propane. The device was unresponsive to sterically bulkier cyclohexane. In addition, the response of the system to ethanol vapour in nitrogen gas in a concentration range 0–100% was non-linear with a maximum interference fringe redshift of ∼60 nm measured in 40% ethanol with no further interference redshift occurring at concentration of ethanol vapour >40%.
The Kelvin probe sensor setup demonstrated by Fleischer et al. (Section 3.1.2) was also sensitive to aldehydes.141 Aldehyde vapour sensing measurements were performed in synthetic air (20% O2 and 80% N2) at a flow rate of 1 L min−1 at 25 °C, and at 40% RH. The sensing response was reversible and stable with changes in work function (Δϕ) measured upon exposure to the vapours of individual aldehydes. The change in work function upon exposure to 10 ppm of acetaldehyde and propanal were −1 mV and +1.4 mV respectively whereas the device was more sensitive to pentanal with a change in work function of −5 mV upon exposure to just 2 ppm. The device suffered from limited cross sensitivity with differences in the measured work function upon exposure to just pentanal, pentanal with acetaldehyde and pentanal with propanal.
Lee et al. reported two MOFs that can also be used to detect ethanol by quantifying the differences in the luminescence emission spectra before and after exposure to ethanol vapour.137 Upon excitation at 270 nm, it was found that for dry [In(OH)(bdc)] emission λmax = 326 nm and for dry MOF-5 emission λmax = 366 nm. Upon exposure to ethanol vapour the profile of the emission peak of [In(OH)(bdc)] changed from a single peak to a triple peak emission feature with λmax EtOH = 389 nm. However in the case of MOF-5 the emission wavelength blueshifted 27 nm to λmax EtOH = 339 nm, which was an identical to the λmax water value. Therefore MOF-5 was not able to differentiate between water and ethanol vapours.184
Robinson et al. fabricated N-doped piezoelectric microcantilever sensors, ∼230 μm long and ∼80 μm wide, that were coated with a layer of HKUST-1 (Section 3.1.2).138 The sensor device was exposed to different concentrations of methanol vapour in nitrogen gas at 23 °C and at 1 atm pressure. Exposure to methanol vapour resulted in methanol being adsorbed onto the HKUST-1 layer, changing the resonant frequency of the microcantilever. The device demonstrated sensitivity between ∼1000 ppm and ∼13000 ppm of methanol vapour in nitrogen carrier gas and demonstrated a reversible sensing response. However, the magnitude of changes in voltage that were measured at equilibrium for different concentrations of methanol vapour in nitrogen carrier gas were identical to those measured upon exposure to different concentrations of water vapour. The sensor device was however able to differentiate between water and methanol vapour by taking into account response times.
The piezoelectric microcantilever humidity sensor modified with HKUST-1, demonstrated by Hesketh et al. (Section 3.1.2), also displayed sensitivity to vapours of ethanol and methanol.140 The authors tested the response of the device to methanol and ethanol vapours in nitrogen carrier gas, at room temperature and pressure, with the HKUST-1 in both the dehydrated and hydrated state. The sensor response to both ethanol and methanol vapours was non-linear within the range of ethanol vapour concentrations (up to ∼2%) and methanol vapour concentrations (up to ∼8%) investigated. Whether the HKUST-1 was in the hydrated or dehydrated state made no discernible difference to the response for both alcohols.
The mesoporous MOF [Zn4O(benztb)(btc)2/3] loaded with Nile Blue, reported by Kaskel et al., also was a detector for vapours of ethanol and methanol.149 Upon exposure to ethanol and methanol vapours the colour changed from dark blue (due to the presence of Nile Blue in the ‘dry’ state) to light blue.
The planar interdigital electrode impedance sensor coated with a thick film of Fe-btc MOF and operating at a frequency of 1 Hz, reported by Moos et al. as a humidity sensor (Section 3.1.2), was also sensitive to methanol and ethanol vapours.148 At an operating temperature of 120 °C, there was a linear response between absolute impedance || and concentration of either methanol or ethanol in the range of 0–35 vol% methanol vapour in nitrogen and 0–18 vol% ethanol vapour in nitrogen. However, after exposure to either methanol or ethanol vapour, a longer desorption time was required for the sensor to return to the pre-exposure impedance value relative to water vapour. In addition, a drifting impedance value was observed upon repeated exposures to ethanol vapour.
The photonic film of NH2-MIL-88B MOF on a silicon wafer was also sensitive to ethanol vapour.150 Adsorption of ethanol into the pores of NH2-MIL-88B replaced air and thus the effective refractive index of the MOF increased. This resulted in a redshift of the interference peaks in the reflection spectrum by ∼300 nm.
Zhang et al. reported that the MOF [Co(mIM)2], ZIF-67, acted as a selective and highly sensitive sensing material for formaldehyde at 150 °C.164 ZIF-67 was coated onto silver–palladium interdigitated electrodes and the resistance of the MOF was measured during exposure to various analytes at 150 °C. The highest response was observed upon exposure to formaldehyde, but there was slight cross-sensitivity to methanol and acetone vapours. The response of the sensor to ammonia vapour and methane was negligible. The sensor was shown to be sensitive to formaldehyde in the concentration range 5–500 ppm with a linear sensing response between 5–50 ppm of formaldehyde. ZIF-67 is stable up to 250 °C in air.
Huo et al. reported that self-assembled crystals of a MOF can be used for optical detection of methanol and ethanol vapours.165 Particles of [Zr6O4(OH)4(bdc)6], UiO-66, were modified with polyvinylpyrrolidone and then films on the surface of a surfactant solution were formed using the Langmuir–Blodgett technique. These films were then transferred to a silica substrate using a dip coating technique. The thin films of self-assembled crystals of UiO-66 were then put into a quartz cuvette that was placed into a UV-visible spectrometer. Adsorption of ethanol or methanol into the pores of UiO-66 replaced nitrogen and the refractive index of the thin film changed. This caused the colour of the photonic crystal thin films to change. A redshift of the absorption peak by 30 nm occurred upon exposure to methanol vapour. This change in colour due to exposure to methanol vapour was also visible to the naked eye.
Zhao et al. reported two MOFs, [Cu2(OH)(2,2′-bipy)2(btc)]·2H2O and [Co(4,4′-bipy)(m-bdc)] (2,2′-bipy = 2,2′-bipyridine; 4,4′-bipy = 4,4′-bipyridine; m-bdc = 1,3-benzenedicarboxylate), which were highly sensitive to methanol vapour at room temperature when deposited as films on QCM sensors.166 When the QCM sensor coated with [Cu2(OH)(2,2′-bipy)2(btc)]·2H2O was exposed to methanol vapour it exhibited an approximately linear response with larger magnitude negative frequency shifts with higher concentrations of methanol vapour. The response time was approximately on the order of 1–2 min and the responses were reversible. The QCM sensor coated with [Co(4,4′-bipy)(m-bdc)] was also exposed to methanol vapour and it exhibited a non-linear response with larger magnitude negative frequency shifts with higher concentrations of methanol. Both MOFs had relatively poor thermal stability, degrading at 170 °C or below.
The techniques of surface plasmon resonance sensing and imaging, using HKUST-1 as the active sensing element, for the detection of straight chain alcohols was reported by Chen et al.144 Crystals of HKUST-1 were self-assembled from solutions of the reactants using a stepwise-immersion protocol onto a layer of gold, the surface of which had been modified so as to have terminal alcohol groups present. The polarisation of the surface plasmon mode was found to have a linear relationship with concentration of both ethanol and methanol vapour in the range 1% to 70% (v/v), although the sensor was overall more sensitive to methanol than ethanol. The response of the sensor was reversible and highly reproducible.
Urban et al. also used MOFs based on the btc ligand as the active sensing elements on a work function based sensor.167,168 The design and operation of the sensor was similar to the water vapour sensor reported by Hesketh et al. (Section 3.1.2) and the MOFs investigated were [Al12O(OH)18(H2O)3{Al2(OH)4}(btc)6]·24H2O, [Co3(btc)2]·H2O, [Ni3(btc)2]·H2O and [Cd3(btc)2]·H2O. Upon exposure to methanol or ethanol vapour at different concentrations in synthetic air carrier gas, at 25 °C and at 40% RH, the work function was found to decrease insignificantly for methanol and only marginally for ethanol vapour. The largest response was measured with the work function sensor modified with the layer of [Cd3(btc)2]·H2O. However, upon repeating the experiment in dry synthetic air (i.e. 0% RH), the work function decreased more significantly for both methanol and ethanol. In both cases the largest sensor response was measured upon exposure to ethanol vapour, however differentiation between the two alcohols in a mixed vapour stream would be difficult as the magnitude change in work function upon exposure to methanol at ∼50 ppm was identical to that upon exposure to ethanol at ∼10 ppm. Additionally, the sensor device showed cross sensitivity to other alcohols such as 2-propanol and n-butanol. Upon exposure to straight chain alkanes slight changes in measured work function were measured, but only in dry conditions. In all cases the sensing responses were reversible.
Work by Pohle et al. compared the response of this work function-based sensor to the response of a QCM sensor upon exposure to vapours of alcohols.145 Both sensors were modified with films of Cu-btc and exposed to different concentrations of methanol and ethanol vapours under different conditions. For both sensor types it was found that operation in dry conditions resulted in the largest measureable responses to methanol and ethanol vapour. In addition, it was found that the largest changes in frequency response from the QCM-based sensor were measured upon exposure to methanol vapour rather than ethanol vapour. In contrast, methanol vapour caused the smallest work function changes measured with the work function-based sensor compared to ethanol vapour. Therefore, if both sensors were operated at the same time, then two-dimensional discrimination of the nature of the analyte could be achieved.
Li et al. measured the change in fluorescence intensity and the wavelength shift of the fluorescence peak for two MOFs upon exposure to the vapours of many different organic compounds and then plotted these data onto a two-dimensional detection map which allowed differentiation between all of the analytes.169 The MOFs employed were [Zn2(ndc)2(dpe)]·2.5DMF·0.25H2O and [Zn2(ndc)2(bpee)]·2.25DMF·0.5H2O (ndc = 2,6-naphthalenedicarboxylate; bpee = 1,2-bis(4-pyridyl)ethylene). [Zn2(ndc)2(bpee)]·2.25DMF·0.5H2O displayed the larger responses to analytes in terms of fluorescence intensity change and fluorescence peak wavelength shift after exposure to the vapour of the analyte at room temperature for 5 min. The two-dimensional detection response map allowed for facile differentiation between methanol and ethanol. In addition it was found that both MOFs were stable up to ∼400 °C in a nitrogen atmosphere.
An orientated film of a fluorescent MOF on a glass substrate has been reported as a luminescence-based detector of alkenes by Balkus et al.170 [Zn2(bpdc)2(bpee)]·2DMF, RPM3, was grown solvothermally on a glass substrate, the surface of which had been seeded with zinc oxide particles. The resulting film of RPM3 was ∼3 μm thick and had grown preferentially in the crystallographic c-axis direction. Interestingly the thin film format of the MOF seemed to introduce stability to relative humidity compared to RPM3 powder. The MOF thin film was then doped with silver cations. The presence of Ag+ within the MOF partially quenched the fluorescence intensity at ∼422 nm due to the π–cation interaction with the bpee ligand. Upon exposure to propylene gas in dry nitrogen carrier gas the fluorescence intensity was enhanced by ∼43%, but partially quenched by 12% when exposed to propane gas in dry nitrogen. Thus this system can differentiate between alkanes and alkenes. The enhancement in fluorescence intensity upon exposure to propylene was proposed to be due to the formation of a π–cation interaction between an alkene analyte and the Ag+ cation thus weakening the π–cation interaction between the Ag+ cation and the bpee ligand. The sensing response to propylene was reversible, but heating at 60 °C under vacuum followed by nitrogen purging was required. The response to 1-hexene was also tested. Upon exposure to 1-hexene vapour a fluorescence intensity enhancement of ∼43% was measured which suggests that differentiation between different alkenes may not be possible with this MOF system.
Furukawa et al. investigated how the crystal orientation of a MOF impacted on its adsorption kinetics.171 The MOF used was [Zn(NO2-ip)(4,4′-bipy)] (NO2-ip = 5-nitroisophthalate), Zn-CID-5, and thin films of the MOF were grown solvothermally on QCM sensors, one of which had been modified with 16-mercaptohexadecanoic acid and the other remained bare. The presence of 16-mercaptohexadecanoic acid on the surface of the QCM favoured the orientation of the Zn-CID-5 crystals in the [1−1−1] direction whereas the preferred crystal direction of the Zn-CID-5 crystals on the bare QCM sensor was [100]. Zn-CID-5 crystals were also solvothermally grown on the surface of a bare QCM sensor in the presence of 4-phenylpyridine, which is a crystal size modulator. This led to a preferred orientation of the Zn-CID-5 crystals along the [010] direction. From adsorption kinetic studies it was found that Zn-CID-5 orientated along the [010] direction had the fastest adsorption kinetics and thus was the most suitable for sensing. The sensor showed the largest sensing responses to methanol compared to hexane as the methanol induced a ‘gate-opening’ phenomenon whereas hexane did to a far lesser extent. The sensing response to methanol was reversible, but the time response was over 100 s.
Finally Cao et al. reported a colorimetric detector for methanol vapour that was comprised of a solvothermally grown film of the MOF [Co3(tbtc)2(DMF)2]·4DMF (tbtc = 4-[[3,5-bis[(4-carboxylatophenoxy)methyl]-2,4,6-trimethyl-phenyl]methoxy]benzoate, Fig. 17) on an alumina support.172 Upon exposure to air saturated with methanol vapour the colour of the thin film changed from blue to pink over several hours of exposure. However, the colour could only be restored by exposing the MOF thin film to DMF vapour.
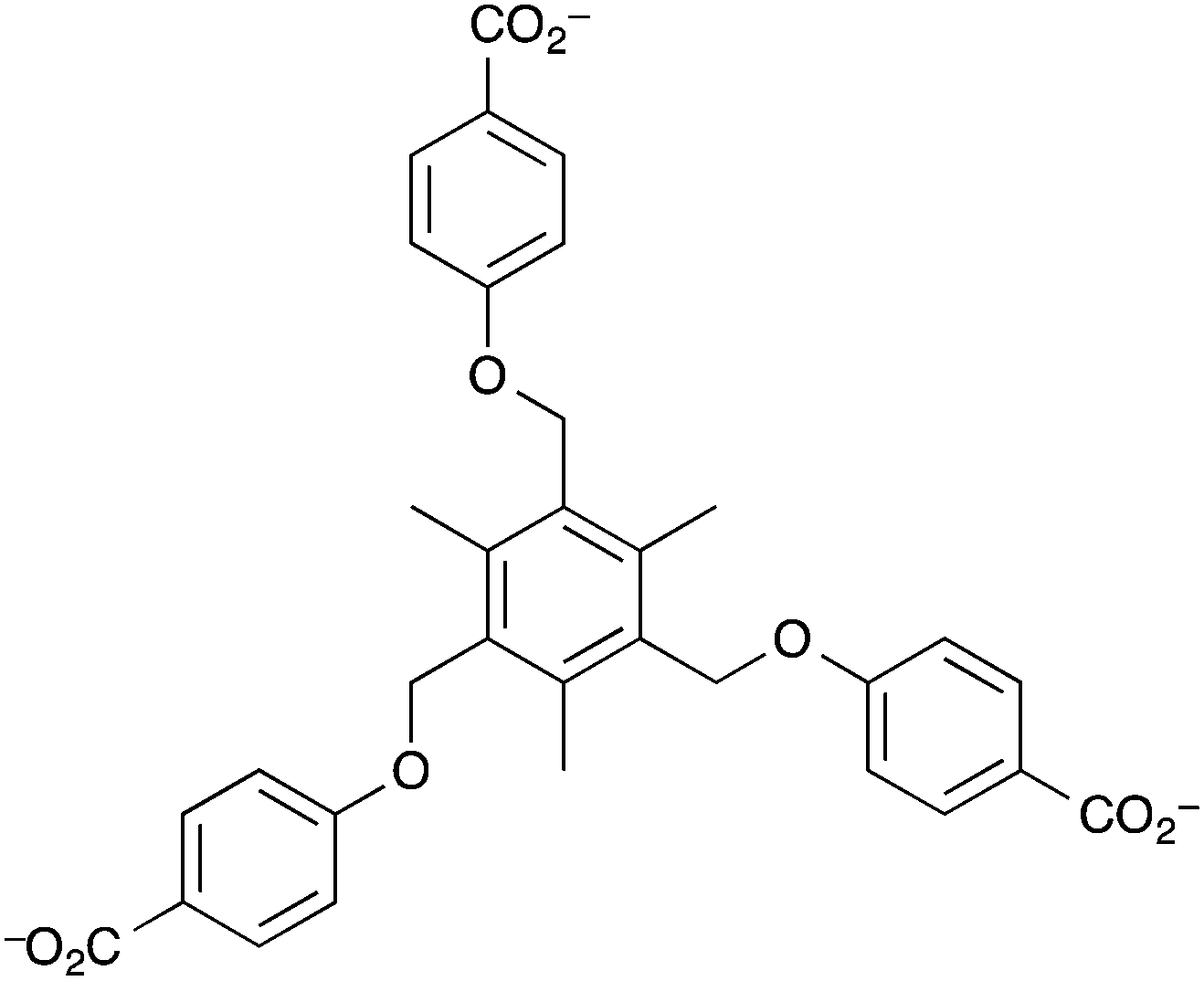 | ||
| Fig. 17 The 4-[[3,5-bis[(4-carboxylatophenoxy)methyl]-2,4,6-trimethyl-phenyl]methoxy]benzoate (tbtc) ligand.172 | ||
3.6 Ammonia sensors
Pt-doped zeolite Y assembled in films was promising, but showed a strong cross-sensitivity to NO.92 In contrast, the zeolite H-ZSM-5 had very small cross sensitivity to NO, CO, CO2 and hydrocarbons as no metal elements were incorporated in the zeolite structure. It provided a response time of seconds and a detection limit down to 5 ppm concentration of ammonia in synthetic air.85 To improve the sensitivity of a Fe-doped poly(p-phenylene) polymer (Fe-dPPP) sensor towards NH3, several forms of ZSM-5 zeolites (Na+, K+, NH4+, H+) were added into the conductive polymer matrix.86 The highest electrical conductivity sensitivity was obtained with the composite Fe-dPPP–H-ZSM-5. This was explained by the high acidity, pore volume and surface area of the H-form of ZSM-5 zeolite, as those properties favour both conductive polymer interaction and NH3 adsorption. In addition, the FTIR spectrum showed that the NH3–polymer interaction is irreversible while the NH3–zeolite interaction is reversible. The irreversibility of the NH3–polymer interaction was recently resolved by the same research group. Indeed, they reported that a composite comprised of HClO4-doped poly(3-thiopheneacetic acid) as polymer and a zeolite Y gave similar results with a fully reversible NH3–polymer interaction.122
The interaction of ammonia with Ag+-exchanged zeolite Y was recently exploited to demonstrate potential for NH3 detection.123 As no measurable self-reduction of Ag+, and therefore, no proton-mediated change of impedance could be recorded in the presence of NH3, the impedance measurements were based directly on the mobility change of the Ag+ in the presence of NH3. No cross-sensitivity was observed for O2, CO, CO2 and propane, though there was some interference from NO and water.
Sensors for ammonia based on non-proton conductive zeolite properties have also been developed. An H-ZSM-5 zeolite thin film was grown on a long-period fibre grating whereupon the adsorption-induced shift of the resonant wavelength of the fibre grating was monitored during exposure to ammonia.87 This sensor had a 2 min response time for adsorption–desorption of NH3 in 612 ppmv ammonia in nitrogen atmosphere. Recently, a sensor based on the conductivity of metal oxide was reported. Indeed, semiconducting zinc oxide covered with zeolite (Y, mordenite or beta polymorph A) showed sensitivity down to a few ppm concentration of NH3.49
Another gas detector containing a protective layer of a zeolite with a thickness of approximately 10 μm was applied as ammonia sensor in the control loop of a NOx storage catalyst for diesel engines.51 It was reported that ammonia could be detected in oxygen-rich gases containing nitrogen oxides such as exhaust gases or flue gases, using a condenser based on a hydrophobic, noble metal-free zeolite of low acidity having an ordered crystalline structure such as zeolite Y, mordenite or ZSM-5.52
Humphrey et al. have developed the PCM-15 MOF previously reported as a detector material for humidity (Section 3.1.2) for the detection of other gases and vapours.134,135 In particular, ammonia was found to be a more efficient quencher of the luminescence of the terbium(III) ion than water. Upon activation of the MOF at 150 °C under vacuum for 1 h, the intensity of the photoluminescence due to the terbium ion was found to double. Then upon treatment with 1 atm of ammonia gas, the photoluminescence intensity decreased 1.4 times relative to the photoluminescence intensity measured when the MOF was exposed to water vapour. PCM-15 exhibited selectivity to ammonia over hydrogen when exposed to low concentrations of ammonia gas in hydrogen carrier gas. This shows promise in detection of low concentrations of impurity gases or vapours in feedstock gases, but the major disadvantage with this system is that the detection of ammonia is irreversible.
Dincă et al. reported two MOFs that function as selective detectors for ammonia at 100 °C, but not at room temperature.173 [Zn2(tpce)] (tpce = tetrakis(phenylcarboxylate)ethylene, Fig. 18) and [Mg2(dobdc)] (dobdc = 2,5-dioxobenzene-1,4-dicarboxylate, Fig. 18), both displayed fluorescence “turn-on” upon exposure to ammonia as opposed to the typical fluorescence “turn-off” mechanism, otherwise known as quenching. Fluorescence “turn-on” describes the interaction between an analyte and a “dark” fluorophore resulting in the “switch on” of the “dark” species so that it luminesces. In addition the maximum emission wavelengths of both MOFs shifted upon interaction with ammonia at 100 °C. [Zn2(tpce)] exhibited a emission wavelength redshift from 487 nm to 511 nm when exposed to ammonia at 100 °C. In addition [Zn2(tpce)] was thermally stable up to ∼400 °C. Exposure to ammonia led to an irreversible phase change in [Zn2(tpce)] to an unidentified crystalline form, so the detection of ammonia is irreversible. However, [Mg2(dobdc)], in which Mg2+ ions are coordinated by dobdc ions, exhibited a reversible “turn-on” fluorescence response upon exposure to ammonia at 100 °C, after activation at 180 °C for 12 h. This MOF also exhibited selectivity over methanol and ethanol and was thermally stable up to ∼250 °C.
 | ||
| Fig. 18 The tetrakis(phenylcarboxylate)ethylene (tpce) ligand (left) and 2,5-dioxobenzene-1,4-dicarboxylate (dobdc) ligand (right) in the MOFs reported by Dinč et al. for detection of ammonia.173 | ||
3.7 Hydrogen sulfide sensors
 | ||
| Fig. 19 The 3-amino-1,2,4-triazole-5-thiol ligand used by Lu et al. to synthesise MOF nanoparticles which were fluorescently quenched in the presence of hydrogen sulfide gas.174 | ||
The material demonstrated a remarkable fluorescence Stokes shift of large magnitude upon excitation at 365 nm when dispersed in DMF. The resulting red fluorescence emission, λmax = 620 nm, was quenched by as little as 2 ppm of H2S in nitrogen carrier gas, but not by other gases or vapours such as CO2, ethanol or even air. In addition an accompanying colour change from yellow to brown could also act as an indicator of hydrogen sulfide exposure. The proposed mechanism for fluorescence quenching involves a reaction between the MOF and H2S to form a copper sulfide, Cu9S8 suggesting that the detection mechanism is irreversible.
The FGFET gas sensor modified with HKUST-1 reported by Fleischer et al., and described in Section 3.1.2, also demonstrated sensitivity to 3 ppm of hydrogen sulfide.146 The authors reasoned that the small kinetic diameter of hydrogen sulfide allowed for it to pass through the pores of the HKUST-1 overlayer and interact with the gold electrode. However, the magnitude of the change in the work function was similar to 5 ppm of ammonia.
3.8 Sulfur dioxide sensors
The history of sulfur dioxide removal from gas streams by using zeolite adsorbents is lengthy. A comparative study including natural and synthetic zeolites revealed that the SO2 capacity of the latter is higher, which is due to higher purity and the controlled characteristics of the synthetic materials.200 In the 1970s, synthetic mordenite- and faujasite (FAU)-type zeolites were employed for SO2 adsorption because of their stability under acidic conditions. A general understanding is that the sulfur dioxide is adsorbed by hydrogen bonding to one or two conveniently positioned surface hydroxyl groups of the zeolites. It was found that the stronger the acidity, the higher the electron deficiency, the better the sulfur resistance. It was also found that the adsorption of sulfur on Pd-containing FAU zeolite is reversible.124,125 The investigation of SO2 adsorption on silicalite-1 and de-aluminated zeolite Y showed the supremacy of all-silica pentasil-type molecular sieve towards SO2.
Faujasite-type zeolites were reported to be useful for sensing SO2 when combined with metal species. In a first example, a layer of faujasite-type zeolite deposited on QCM oscillators, with an electrode of gold, is active as the increased mass due to absorbed SO2 reduces the frequency of the QCM.101 In further work zeolite A, was deposited on the QCM. The zeolite-A doped QCM could monitor 50 ppm of SO2 in He at 170 °C.57
More recently, the effect of various transition metal cations (Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+) exchanged into hydrated Y-type zeolites for SO2 sensing were investigated by electrical conductivity.126 When exposed to a SO2 atmosphere, all poly(3,4-ethylenedioxythiophene)–poly(styrene sulfonic acid)/metal doped zeolite Y composites, and especially the Mn2+ doped zeolite, showed higher sensitivity than a pristine conductive polymer matrix used as reference though at the expense of longer response times. The authors attributed those results to the electronegativity of the metal cations which strongly improved the adsorption properties of the zeolite Y, and hence the electrical sensitivity of the composite.
Often sensing of different gases by zeolite molecular sieves faces problems due to catalytic side reactions. For instance, the sodium form of zeolite Y demonstrated impressive adsorptive and catalytic properties in NOx elimination from exhaust gases of diesel and lean burn engines. Physisorbed SO2, HSO3− and S2O52− species in the Na–Y adsorbent were observed, and the poisoning of N2O3 adsorption sites by SO2 was found to be a function of sodium hydrogen sulfite formed in the channels. A possibility of overcoming the poisoning of the N2O3 adsorption capacity of Na–Y zeolite by SO2 was based on short adsorption–desorption cycles, high temperature and low oxygen concentrations during the desorption process. Sultana et al.127 studied the adsorptive separation of NOx in the presence of SOx on Na-Y zeolite. They found that when Na-Y zeolite operates in a pressure swing process for NOx removal from engine exhausts, it is fairly resistant to SO2. However, poisoning by SO2 could be suppressed by using short adsorption–desorption cycles, by limiting the oxygen concentration in the regeneration gas and by working at temperatures higher than 255 °C.
The elimination of SO2 by microporous zeolite-type materials is based on either low or high silica zeolites. The SO2 molecule reacts with the metals situated in the pores of the zeolite structure leading to metal sulfate formation. This leads to channel blocking and a substantial decrease of transport is observed. Another disadvantage of low silica zeolite materials is their hydrophilic nature. Thus, the gas adsorption capacity is dramatically reduced by concurrent water adsorption. Furthermore, low-silica zeolites are not adapted for SO2 retention since their chemical stability in acidic media is limited. The increase of silicon content in zeolites leads to a decrease of their hydrophilicity, which can reach a level when the water molecules are rejected by the microporous structure. Such types of zeolitic materials are often employed in selective adsorption of SO2 and other sulfur compounds. The main advantage of these materials is their higher stability than other absorbers towards SO2 and possible regeneration for multiple usages due to their high thermal stability (900 °C) and high stability in an acidic media. In summary, the high silica zeolite films will allow discrimination of H2O molecules and selective adsorption of SO2. The latter is particularly important to retain the high capacity of the zeolite sorbent. SO2 detection on high silica zeolites is essentially unaffected by the presence of CO2 and other gases.
No examples of the use of MOFs for SO2 sensing were found in the literature. However, it should be noted that sulfur dioxide (SO2) adsorption in MOFs has been reported,201–205 thus MOFs that possess suitable stability for automotive exhaust gas sensing hold promise as potential sensing materials for SO2.
3.9 Hydrogen sensors
Silicalite zeolite can also be used as a filter. As described for humidity sensors, Santamaria et al. showed that the silicalite coating on SnO2 gave enhanced response to both H2O and H2.58 Recently, a fibre optic sensor consisting of a proton-conducting SrCe0.8Zr0.1Y0.1O2.95 thin film overcoated by silicalite zeolite layer was reported.93 Pores of the zeolitic filter were found to be small enough to permit H2 diffusion but restricted the diffusion of other gases and fine particles from dusty coal or biomass gasification processes. When the silicalite film was less than 2.5 μm thick, a high sensitivity of 0.14 nm kPa−1-H2 was observed.
Hupp et al. developed an optical sensor device that utilised Bragg grating stacks made from subsequential layers of ZIF-8 and palladium. A Bragg grating stack operates as a wavelength-selective mirror; some wavelengths are reflected whereas other wavelengths are transmitted. The wavelength reflected depends on the refractive index of the materials in the Bragg stack. If the refractive index of some of the layers within the Bragg grating stack changes then the wavelengths that are reflected also change. Hupp et al.'s Bragg grating stack comprised of two sets of a bilayer structure of Pd (∼10 nm)/ZIF-8 (∼300 nm) that operate at visible-light wavelengths, and the authors predicted, that upon exposure to certain analyte gases, the refractive index of the ZIF-8 layers would change due to adsorption of the analyte gases.162 Upon exposure to vapours of short chain length alkanes, which all have kinetic diameters >4 Å,208 the refractive index of the ZIF-8 layers increased due to adsorption of these compounds. However, the sensor device did not offer differentiation between the alkane analytes. In contrast, when the sensor was exposed to increasing concentrations of H2 in N2 carrier gas, from 0% up to 5%, the wavelengths reflected did not change but the transmission intensity increased. The authors therefore postulated that the H2 was insignificantly adsorbed within or on the ZIF-8 layers so as not to change the refractive index of the Bragg grating stack, but was reversibly adsorbed onto the palladium layer and forming a palladium hydride that is more transparent in the visible wavelengths than the ∼10 nm thick palladium layer. Therefore, by measuring the transmission intensity of the device during exposure to analytes, it was observed that the device possessed a selective affinity for hydrogen gas molecules over short alkyl chain vapour molecules which is due to difference in kinetic diameters; the kinetic diameter of H2 is 2.89 Å and the pore window size of ZIF-8 is 3.40 Å.209 Therefore the ZIF-8 layers acted as size selective filter layers. Sensing experiments with carbon dioxide, CO2, which has a kinetic diameter of 3.30 Å210 further confirmed that ZIF-8 acted as a size selective filter layer. Upon exposure to CO2 an increase in transmission intensity was measured; carbon dioxide is smaller than the pore size window of ZIF-8 and thus passes through the ZIF-8 pores and interacts with the palladium layer.
Dam et al. also utilised a MOF layer as a filter to achieve selective optical detection of hydrogen gas.147 The authors chose a copper btc material as the gas selective filter layer as this had been shown to possess increased selectivity towards H2 when in a thin film. Dam et al. deposited the MOF thin film through a layer-by-layer deposition process onto a palladium substrate that had been initially covered with a self-assembled monolayer of glycine. With this technique, the MOF thin film was found to be mostly amorphous though the authors argued that the chemical environment within this film was similar to that within HKUST-1 based on FTIR spectral data. The resulting sensor was interrogated with optical transmission spectroscopy whilst exposed to a H2 environment. Increases in the optical transmission were measured upon exposure to hydrogen gas at various concentrations at room temperature and 100 °C, but concentration differentiation was greatest when measurements were taken at 100 °C. The device showed stable repeatable and reversible responses, but the response time was on the order of 1000 s. Selectivity to hydrogen gas over O2, CO2 and CH4 in gas mixtures was demonstrated, but the kinetics of adsorption of hydrogen were significantly decreased when the device was exposed to H2–CO and H2–H2O mixtures. This could be due to the affinity of the copper MOF for H2O and CO; these gases preferentially adsorb and thus hinder the ingress of H2.211,212
4. Conclusions and future perspectives
It is clear that porous materials, exemplified by zeolites and MOFs, offer tremendous flexibility and versatility in terms of selectivity and sensitivity and thus demonstrate great promise for the fabrication of sensors for exhaust gases and vapours. Both size and shape selectivity have been demonstrated with zeolite- and MOF-based detectors via control of the dimensions of the pores, and in some cases the crystal orientation when in ordered films, thus enabling detection and differentiation between gases and vapours. Chemical selectivity and sensitivity have been controlled by the incorporation of select dopants into the frameworks so as to elicit desirable optical and or electrochemical responses upon interaction with analytes.In this review, we summarised 170 different published sensors including around a third based on MOFs and two-thirds based on zeolites (Fig. 20). Among those publications, hydrocarbon sensors are currently the most common for both zeolites and MOFs. Zeolite and MOF sensors were developed in similar numbers for automotive exhaust vapours, though no NOx and SO2 sensors based on MOFs were reported whereas only MOF-based sensors have been reported to detect H2S.
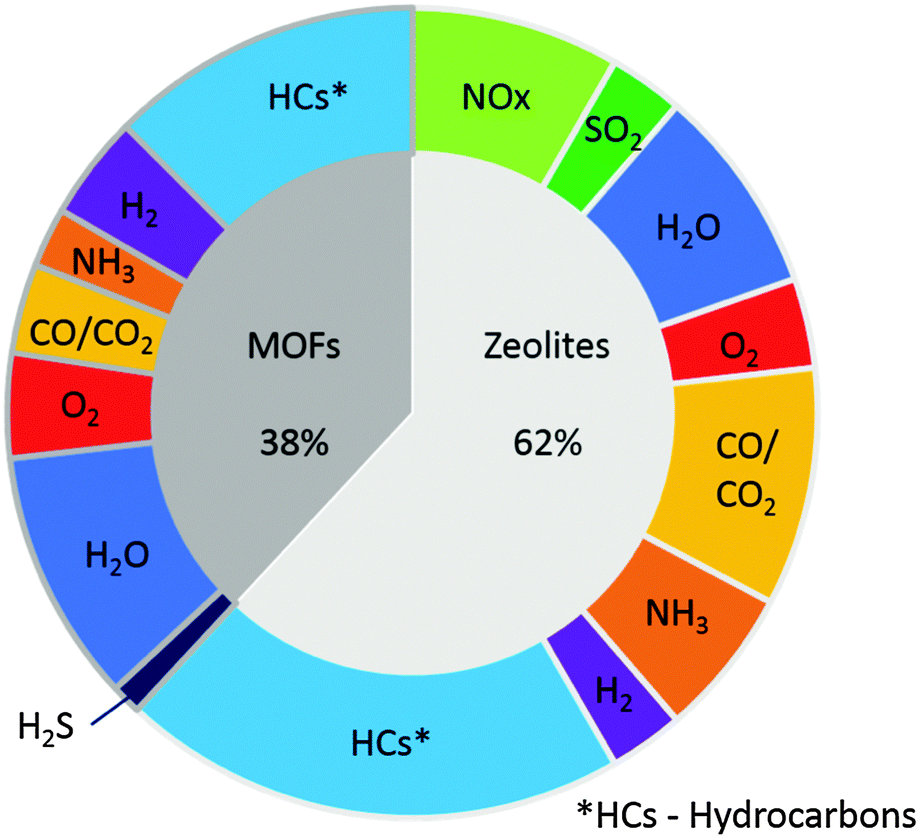 | ||
| Fig. 20 Reported sensors based on zeolites or MOFs along with the type of sensors. | ||
The greater chemical and thermal stability of zeolites, coupled with the ability to be more easily integrated into smaller electrical sensors, means zeolites may be the most suitable sensing materials for use in prospective on-board exhaust gas sensors. The lower chemical and thermal stability of MOFs suggests that these materials are more suited to fabricate sensors for exhaust gas emissions during commissioning of new vehicles in production; location in a production setting will allow for appropriate gas cooling and clean-up before sensing which would be impractical on-board vehicles.
The wide variety of organic linkers and metal nodes that can be incorporated into MOFs has allowed for increased sensitivity and selectivity based on control of the interactions between adsorbed analytes. Finally, it needs to be acknowledged that the field of MOFs for sensing is young in comparison to the research into the use of zeolites as sensor materials. While in most cases at the current time, zeolite-based sensors are more advanced than MOF-based analogues, it is expected that advances in discriminatory sensing, sensitivity and thermal stability will enable this gap to be closed in the future.
Despite their promise, few of these systems have to date been incorporated into devices that allow for real-time and on-line data collection and thus most only exist as detectors or probes under specific conditions. In addition, few examples of these materials have been shown to exhibit successful sensing of specific species when exposed to varying temperatures, pressures and in the presence of contaminants.
The sensitivity and selectivity that can be explicitly controlled by sensor designers when using MOFs and zeolites and the great need economically and environmentally for energy efficient transport indicates that the development of porous materials for automotive gas sensing will continue to be an active research field in the future.
Acknowledgements
Financial support from Materials for Energy Efficiency in Transport (MEET) funded by the European Regional Development Fund (ERDF) INTERREG IV programme is acknowledged.References
- M. A. Liberman, Introduction to Physics and Chemistry of Combustion: Explosion, Flame, Detonation, Springer, Berlin Heidelberg, 2008 Search PubMed.
- United Nations, Kyoto Protocol to the United Nations Framework Convention on Climate Change, 1998.
- United States Environmental Protection Agency, U.S. Greenhouse Gas Inventory Report, 2014.
- S. E. Manahan, Fundamentals of Environmental Chemistry, CRC Press, 2nd edn, 2000 Search PubMed.
- International Energy Agency (IEA), Key World Energy Statistics 2014, 2014.
- Institution of Mechanical Engineers, Low Carbon Vehicles: Driving the UK's Transport Revolution, 2010.
- Department for Business, Enterprise and Regulatory Reform, An Independent Report on the Future of the Automotive Industry in the UK, 2009.
- N. Jackson, Technology Roadmap, the R&D Agenda and UK Capabilities, Automotive Council UK, 2010.
- L. Chapman, J. Transp. Geogr., 2007, 15, 354 CrossRef PubMed.
- The European Parliament and Council of the European Union, Off. J. Eur. Union, 52, 1., 2009.
- A. M. K. P. Taylor, Energy Policy, 2008, 36, 4657 CrossRef PubMed.
- D. Carslaw, S. Beevers, E. Westmoreland, M. Williams, J. Tate, T. Murrells, J. Stedman, Y. Li, S. Grice, A. Kent and I. Tsagatakis, Trends in NOx and NO2 Emissions and Ambient Measurements in the UK, DEFRA, 2011.
- P. Mock, J. German, A. Bandivadekar, I. Riemersma, N. Ligterink and U. Lambrecht, From Laboratory to Road: A Comparison of Official and ‘Real-World’ Fuel Consumption and CO2 Values for Cars in Europe and the United States, The International Council on Clean Transportation, 2013.
- M. Tutuianu, A. Marotta, H. Steven, E. Ericsson, T. Haniu, N. Ichikawa and H. Ishii, in 68th Session of Working Party on Pollution and Energy, United Nations Economic Commission for Europe, 2013, 98.
- Diesel Engine Management: Systems and Components, ed. K. Reif, Springer Vieweg, Wiesbaden, 2014 Search PubMed.
- M. J. Nunney, Light and Heavy Vehicle Technology, Routledge, 4th edn, 2006 Search PubMed.
- N. Docquier and S. Candel, Prog. Energy Combust. Sci., 2002, 28, 107 CrossRef CAS.
- J. Riegel, H. Neumann and H.-M. Wiedenmann, Solid State Ionics, 2002, 152–153, 783 CrossRef CAS.
- U. Kiencke and L. Nielsen, Automotive Control Systems: For Engine, Driveline, and Vehicle, Springer-Verlag, Berlin, 2nd edn, 2005 Search PubMed.
- T. Wagner, S. Haffer, C. Weinberger, D. Klaus and M. Tiemann, Chem. Soc. Rev., 2013, 42, 4036 RSC.
- E. Llobet, Sens. Actuators, B, 2013, 179, 32 CrossRef CAS PubMed.
- S. Ozdemir and J. Gole, Curr. Opin. Solid State Mater. Sci., 2007, 11, 92 CrossRef CAS PubMed.
- E. L. First, C. E. Gounaris and C. A. Floudas, Langmuir, 2013, 29, 5599 CrossRef CAS PubMed.
- R. Matsuda, Nature, 2014, 509, 434 CrossRef CAS PubMed.
- Y. Zheng, X. Li and P. K. Dutta, Sensors, 2012, 12, 5170 CrossRef CAS PubMed.
- X. Xu, J. Wang and Y. Long, Sensors, 2006, 6, 1751 CrossRef CAS PubMed.
- X. Wan, H. Song, D. Zhao, L. Zhang and Y. Lv, Sens. Actuators, B, 2014, 201, 413 CrossRef CAS PubMed.
- J. Lei, R. Qian, P. Ling, L. Cui and H. Ju, TrAC, Trends Anal. Chem., 2014, 58, 71 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS PubMed.
- H. O. Pastore, E. C. Oliveira, A. Frache, S. R. Dutra, E. Boccaleri and L. Marchese, in Studies in Surface Science and Catalysis, ed. E. van Steen, M. Claeys and L. H. Callanan, Elsevier, 2004, vol. 154B, p. 1426 Search PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed.
- A. Bétard and R. A. Fischer, Chem. Rev., 2012, 112, 1055 CrossRef PubMed.
- O. Shekhah, J. Liu, R. A. Fischer and C. Wöll, Chem. Soc. Rev., 2011, 40, 1081 RSC.
- K. J. Mohan, K. Jagadeesan and A. Yadav, IOSR J. Environ. Sci., Toxicol. Food Technol., 2013, 6, 1 CrossRef PubMed.
- D. Banerjee, Z. Hu and J. Li, Dalton Trans., 2014, 43, 10668 RSC.
- S. M. Auerbach, K. A. Carrado and P. K. Dutta, Handbook of Zeolite Science and Technology, CRC Press, 2003 Search PubMed.
- Ordered Porous Solids: Recent Advances and Prospects, ed. V. Valtchev, S. Mintova and M. Tsapatsis, Elsevier, 2008 Search PubMed.
- M. Zaarour, B. Dong, I. Naydenova, R. Retoux and S. Mintova, Microporous Mesoporous Mater., 2014, 189, 11 CrossRef CAS PubMed.
- Post-Synthesis Modification I, ed. H. G. Karge and J. Weitkamp, Springer, Berlin Heidelberg, 2002 Search PubMed.
- Introduction to Zeolite Molecular Sieves, ed. J. Cejka, H. van Bekkum, A. Corma and F. Schueth, Elsevier, 3rd edn, 2007 Search PubMed.
- S. Babel and T. A. Kurniawan, J. Hazard. Mater., 2003, 97, 219 CrossRef CAS.
- M. M. Scherer, S. Richter, R. L. Valentine and P. J. J. Alvarez, Crit. Rev. Environ. Sci. Technol., 2000, 30, 363 CrossRef CAS PubMed.
- S. Mintova and E.-P. Ng, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, 2013, vol. 5, p. 285 Search PubMed.
- J.-P. Gilson, O. Marie, S. Mintova and V. Valtchev, Emerging Applications of Zeolites, 3rd FEZA School on Zeolites, Valencia, Spain, 2011, p. 245 Search PubMed.
- K. Fukui and S. Nishida, Sens. Actuators, B, 1997, 45, 101 CrossRef CAS.
- S. Sohrabnezhad, A. Pourahmad and M. A. Sadjadi, Mater. Lett., 2007, 61, 2311 CrossRef CAS PubMed.
- M. A. Zanjanchi and S. Sohrabnezhad, Sens. Actuators, B, 2005, 105, 502 CrossRef CAS PubMed.
- D. C. Pugh, E. J. Newton, A. J. T. Naik, S. M. V. Hailes and I. P. Parkin, J. Mater. Chem. A, 2014, 2, 4758 CAS.
- I. Yimlamai, S. Niamlang, P. Chanthaanont, R. Kunanuraksapong, S. Changkhamchom and A. Sirivat, Ionics, 2011, 17, 607 CrossRef CAS.
- E. Irion, A. Knezevic, H. Leye and M. Smuk, DE Pat., DE10133997, 2003 Search PubMed.
- U. Flesch, A. Kayser, W. Maunz, R. Moos, R. Muller, C. Plog, W. Schäfer and U. Simon, EP Pat., EP0871031A3, 1998 Search PubMed.
- M. Vilaseca, J. Coronas, A. Cirera, A. Cornet, J. R. Morante and J. Santamaria, Sens. Actuators, B, 2008, 133, 435 CrossRef CAS PubMed.
- M. A. Urbiztondo, A. Peralta, I. Pellejero, J. Sesé, M. P. Pina, I. Dufour and J. Santamaria, Sens. Actuators, B, 2012, 171–172, 822 CrossRef CAS PubMed.
- S. Mintova and T. Bein, Microporous Mesoporous Mater., 2001, 50, 159 CrossRef CAS.
- S. Mintova, S. Mo and T. Bein, Chem. Mater., 2001, 13, 901 CrossRef CAS.
- I. Sasaki, H. Tsuchiya, M. Nishioka, M. Sadakata and T. Okubo, Sens. Actuators, B, 2002, 86, 26 CrossRef CAS.
- M. Urbiztondo, I. Pellejero, A. Rodriguez, M. P. Pina and J. Santamaria, Sens. Actuators, B, 2011, 157, 450 CrossRef CAS PubMed.
- M. Vilaseca, J. Coronas, A. Cirera, A. Cornet, J. R. Morante and J. Santamaria, Sens. Actuators, B, 2007, 124, 99 CrossRef CAS PubMed.
- A. Afonja, S. Dungey, R. Binions, I. Parkin, D. Lewis and D. Williams, ECS Trans., 2009, 16, 77 CAS.
- R. Binions, H. Davies, A. Afonja, S. Dungey, D. Lewis, D. E. Williams and I. P. Parkin, J. Electrochem. Soc., 2009, 156, J46 CrossRef CAS PubMed.
- N. Densakulprasert, L. Wannatong, D. Chotpattananont, P. Hiamtup, A. Sirivat and J. Schwank, Mater. Sci. Eng., B, 2005, 117, 276 CrossRef PubMed.
- K. E. Yasuda, J. H. Visser and T. Bein, Microporous Mesoporous Mater., 2009, 119, 356 CrossRef CAS PubMed.
- R. Binions, A. Afonja, S. Dungey, D. W. Lewis, I. P. Parkin and D. E. Williams, IEEE Sens. J., 2011, 11, 1145 CrossRef CAS.
- L. Wannatong and A. Sirivat, React. Funct. Polym., 2008, 68, 1646 CrossRef CAS PubMed.
- T. Baimpos, L. Gora, V. Nikolakis and D. Kouzoudis, Sens. Actuators, A, 2012, 186, 21 CrossRef CAS PubMed.
- A. Zampieri, A. Dubbe, W. Schwieger, A. Avhale and R. Moos, Microporous Mesoporous Mater., 2008, 111, 530 CrossRef CAS PubMed.
- R. Bruck and K. Althoefer, DE Pat., DE102004053460, 2006 Search PubMed.
- C. Chuapradit, L. R. Wannatong, D. Chotpattananont, P. Hiamtup, A. Sirivat and J. Schwank, Polymer, 2005, 46, 947 CrossRef CAS PubMed.
- M. Vilaseca, J. Coronas, A. Cirera, A. Cornet, J. R. Morante and J. Santamaría, Catal. Today, 2003, 82, 179 CrossRef CAS.
- L. Scandella, G. Binder, T. Mezzacasa, J. Gobrecht, R. Berger, H. P. Lang, C. Gerber, J. K. Gimzewski, J. H. Koegler and J. C. Jansen, Microporous Mesoporous Mater., 1998, 21, 403 CrossRef CAS.
- P. Sazama, H. Jirglová and J. Dĕdeček, Mater. Lett., 2008, 62, 4239 CrossRef CAS PubMed.
- S. Neumeier, T. Echterhof, R. Bölling, H. Pfeifer and U. Simon, Sens. Actuators, B, 2008, 134, 171 CrossRef CAS PubMed.
- A. Satsuma, D. Yang and K. Shimizu, Microporous Mesoporous Mater., 2011, 141, 20 CrossRef CAS PubMed.
- P. Chanthaanont and A. Sirivat, e-Polym., 2012, 12, 106 Search PubMed.
- L. Gora, J. Kuhn, T. Baimpos, V. Nikolakis, F. Kapteijn and E. M. Serwicka, Analyst, 2009, 134, 2118 RSC.
- A. Dubbe, Sens. Actuators, B, 2009, 137, 205 CrossRef PubMed.
- G. Hagen, A. Dubbe, G. Fischerauer and R. Moos, Sens. Actuators, B, 2006, 118, 73 CrossRef CAS PubMed.
- S. Reiß, G. Hagen and R. Moos, Sensors, 2008, 8, 7904 CrossRef PubMed.
- A. Dubbe and R. Moos, Electrochem. Solid-State Lett., 2006, 9, H31 CrossRef CAS PubMed.
- A. Dubbe, Solid State Ionics, 2008, 179, 1645 CrossRef CAS PubMed.
- A. Dubbe and R. Moos, Sens. Actuators, B, 2008, 130, 546 CrossRef CAS PubMed.
- K. Sahner, D. Schönauer, R. Moos, M. Matam and M. L. Post, J. Mater. Sci., 2006, 41, 5828 CrossRef CAS.
- K. Sahner, R. Moos, M. Matam, J. J. Tunney and M. Post, Sens. Actuators, B, 2005, 108, 102 CrossRef CAS PubMed.
- M. E. Franke, U. Simon, R. Moos, A. Knezevic, R. Müller and C. Plog, Phys. Chem. Chem. Phys., 2003, 5, 5195 RSC.
- P. Phumman, S. Niamlang and A. Sirivat, Sensors, 2009, 9, 8031 CrossRef CAS PubMed.
- X. Tang, Z. Tang, S. J. Kim and J. Dong, Proc. SPIE, 2009, 7293, 72931E Search PubMed.
- G. Hagen, I. Marr and R. Moos, Sens. Lett., 2011, 9, 902 CrossRef CAS PubMed.
- D. Jadsadapattarakul, C. Thanachayanont, J. Nukeaw and T. Sooknoi, Sens. Actuators, B, 2010, 144, 73 CrossRef CAS PubMed.
- M. Grahn, Z. Wang, M. Lidström-Larsson, A. Holmgren, J. Hedlund and J. Sterte, Microporous Mesoporous Mater., 2005, 81, 357 CrossRef CAS PubMed.
- J. Zhang, X. Tang, J. Dong, T. Wei and H. Xiao, Sens. Actuators, B, 2009, 135, 420 CrossRef CAS PubMed.
- R. Moos, R. Müller, C. Plog, A. Knezevic, H. Leye, E. Irion, T. Braun, K.-J. Marquardt and K. Binder, Sens. Actuators, B, 2002, 83, 181 CrossRef CAS.
- H. Jiang, R. Yang, X. Tang, A. Burnett, X. Lan, H. Xiao and J. Dong, Sens. Actuators, B, 2013, 177, 205 CrossRef CAS PubMed.
- S. Thomas, P. Bazin, L. Lakiss, V. de Waele and S. Mintova, Langmuir, 2011, 27, 14689 CrossRef CAS PubMed.
- L. Lakiss, S. Thomas, P. Bazin, V. de Waele and S. Mintova, Microporous Mesoporous Mater., 2014, 200, 326 CrossRef CAS PubMed.
- U. Simon, U. Flesch, W. Maunz, R. Müller and C. Plog, Microporous Mesoporous Mater., 1998, 21, 111 CrossRef CAS.
- P. Payra and P. K. Dutta, Microporous Mesoporous Mater., 2003, 64, 109 CrossRef CAS PubMed.
- M. A. Coutant, P. Payra and P. K. Dutta, Microporous Mesoporous Mater., 2003, 60, 79 CrossRef CAS.
- K. L. McGilvray, M. N. Chrétien, M. Lukeman and J. C. Scaiano, Chem. Commun., 2006, 4401 RSC.
- C. Plog, W. Maunz, P. Kurzweil, E. Obermeier and C. Scheibe, Sens. Actuators, B, 1995, 25, 403 CrossRef CAS.
- M. Osada, I. Sasaki, M. Nishioka, M. Sadakata and T. Okubo, Microporous Mesoporous Mater., 1998, 23, 287 CrossRef CAS.
- I. Pellejero, M. Urbiztondo, D. Izquierdo, S. Irusta, I. Salinas and M. P. Pina, Ind. Eng. Chem. Res., 2007, 46, 2335 CrossRef CAS.
- N. Li, X. Li, T. Zhang, S. Qiu, G. Zhu, W. Zheng and W. Yu, Mater. Lett., 2004, 58, 1535 CrossRef CAS PubMed.
- T. A. Ruda-Eberenz, A. Nagy, W. J. Waldman and P. K. Dutta, Langmuir, 2008, 24, 9140 CrossRef CAS PubMed.
- W. L. Rauch and M. Liu, J. Mater. Sci., 2003, 38, 4307 CrossRef CAS.
- J.-C. Yang, J. V. Spirig, D. Karweik, J. L. Routbort, D. Singh and P. K. Dutta, Sens. Actuators, B, 2008, 131, 448 CrossRef CAS PubMed.
- P. Dutta and J. C. Yang, US Pat., US2007029210, 2007 Search PubMed.
- N. F. Szabo and P. K. Dutta, Sens. Actuators, B, 2003, 88, 168 CrossRef CAS.
- N. F. Szabo, H. Du, S. A. Akbar, A. Soliman and P. K. Dutta, Sens. Actuators, B, 2002, 82, 142 CrossRef CAS.
- J.-C. Yang and P. K. Dutta, Sens. Actuators, B, 2007, 125, 30 CrossRef CAS PubMed.
- J.-C. Yang and P. K. Dutta, J. Phys. Chem. C, 2007, 111, 8307 CAS.
- S. P. Mondal, P. K. Dutta, G. W. Hunter, B. J. Ward, D. Laskowski and R. A. Dweik, Sens. Actuators, B, 2011, 158, 292 CrossRef CAS PubMed.
- J.-C. Yang and P. K. Dutta, Sens. Actuators, B, 2007, 123, 929 CrossRef CAS PubMed.
- G. Grubert, M. Stockenhuber, O. P. Tkachenko and M. Wark, Chem. Mater., 2002, 14, 2458 CrossRef CAS.
- K. Kaneyasu, K. Otsuka, Y. Setoguchi, S. Sonoda, T. Nakahara, I. Aso and N. Nakagaichi, Sens. Actuators, B, 2000, 66, 56 CrossRef CAS.
- K. Tanaka, C.-K. Choo, S. Sumi, Y. Kamitani, T. Fujii, K. Satoh, K. Fukuda, R. Nakata, M. Yoshimune, Y. Yoshinaga and T. Okuhara, J. Phys. Chem. B, 2002, 106, 4155 CrossRef CAS.
- D. P. Mann, T. Paraskeva, K. F. E. Pratt, I. P. Parkin and D. E. Williams, Meas. Sci. Technol., 2005, 16, 1193 CrossRef CAS.
- D. P. Mann, K. F. E. Pratt, T. Paraskeva, I. P. Parkin and D. E. Williams, IEEE Sens. J., 2007, 7, 551 CrossRef CAS.
- J. Trimboli and P. K. Dutta, Sens. Actuators, B, 2004, 102, 132 CrossRef CAS PubMed.
- P. Yang, C. Lau, J.-Y. Liang, J.-Z. Lu and X. Liu, Luminescence, 2007, 22, 473 CrossRef CAS PubMed.
- P. Yang, X. Ye, C. Lau, Z. Li, X. Liu and J. Lu, Anal. Chem., 2007, 79, 1425 CrossRef CAS PubMed.
- S. Konkayan, P. Chanthaanont, W. Prissanaroon, P. Hormnirun and A. Sirivat, Mater. Technol., 2013, 28, 332 CrossRef CAS PubMed.
- Y. Zheng, M. R. Mullen, J. Wang and P. K. Dutta, Sens. Actuators, B, 2014, 193, 542 CrossRef CAS PubMed.
- L. Hu and L. Xuebao, Shiyou Jiagong/Petrochem. Technol., 1999, 15, 41 CAS.
- R. Moos, Int. J. Appl. Ceram. Technol., 2005, 2, 401 CrossRef CAS PubMed.
- P. Chanthaanont and A. Sirivat, Adv. Polym. Technol., 2013, 32, 21367 CrossRef PubMed.
- A. Sultana, D. D. Habermacher, C. E. A. Kirschhock and J. A. Martens, Appl. Catal., B, 2004, 48, 65 CrossRef CAS PubMed.
- J. Zou, H. He, J. Dong and Y. Long, J. Mater. Chem., 2004, 14, 2405 RSC.
- O. Schäf, V. Wernert, H. Ghobarkar and P. Knauth, J. Electroceram., 2006, 16, 93 CrossRef PubMed.
- S. Mintova, J. Visser and T. Bein, Stud. Surf. Sci. Catal., 2001, 135, 360 Search PubMed.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, http://www.iza-structure.org/databases/.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- Y. Yu, J.-P. Ma and Y.-B. Dong, CrystEngComm, 2012, 14, 7157 RSC.
- I. A. Ibarra, T. W. Hesterberg, J.-S. Chang, J. W. Yoon, B. J. Holliday and S. M. Humphrey, Chem. Commun., 2013, 49, 7156 RSC.
- I. A. Ibarra, T. W. Hesterberg, B. J. Holliday, V. M. Lynch and S. M. Humphrey, Dalton Trans., 2012, 41, 8003 RSC.
- C.-C. Wang, C.-C. Yang, W.-C. Chung, G.-H. Lee, M.-L. Ho, Y.-C. Yu, M.-W. Chung, H.-S. Sheu, C.-H. Shih, K.-Y. Cheng, P.-J. Chang and P.-T. Chou, Chem. – Eur. J., 2011, 17, 9232 CrossRef CAS PubMed.
- T. Lee, Z. X. Liu and H. L. Lee, Cryst. Growth Des., 2011, 11, 4146 CAS.
- I. Ellern, A. Venkatasubramanian, J. H. Lee, P. J. Hesketh, V. Stavilla, M. D. Allendorf and A. L. Robinson, ECS Trans., 2013, 50, 469 CrossRef PubMed.
- G. Lu, O. K. Farha, L. E. Kreno, P. M. Schoenecker, K. S. Walton, R. P. Van Duyne and J. T. Hupp, Adv. Mater., 2011, 23, 4449 CrossRef CAS PubMed.
- M. D. Allendorf, R. J. T. Houk, L. Andruszkiewicz, A. A. Talin, J. Pikarsky, A. Choudhury, K. A. Gall and P. J. Hesketh, J. Am. Chem. Soc., 2008, 130, 14404 CrossRef CAS PubMed.
- P. Davydovskaya, R. Pohle, A. Tawil and M. Fleischer, Sens. Actuators, B, 2013, 187, 142 CrossRef CAS PubMed.
- A. Venkatasubramanian, J.-H. Lee, R. J. Houk, M. D. Allendorf, S. Nair and P. J. Hesketh, ECS Trans., 2010, 33, 229 CAS.
- L. E. Kreno, J. T. Hupp and R. P. Van Duyne, Anal. Chem., 2010, 82, 8042 CrossRef CAS PubMed.
- J. Chen, J.-Y. Xu and Y. Chen, Chin. Chem. Lett., 2013, 24, 651 CrossRef CAS PubMed.
- P. Davydovskaya, A. Ranft, B. V. Lotsch and R. Pohle, Anal. Chem., 2014, 86, 6948 CrossRef CAS PubMed.
- R. Pohle, A. Tawil, P. Davydovskaya and M. Fleischer, Procedia Eng., 2011, 25, 108 CrossRef CAS PubMed.
- P. A. Szilágyi, R. J. Westerwaal, R. van de Krol, H. Geerlings and B. Dam, J. Mater. Chem. C, 2013, 1, 8146 RSC.
- S. Achmann, G. Hagen, J. Kita, I. M. Malkowsky, C. Kiener and R. Moos, Sensors, 2009, 9, 1574 CrossRef CAS PubMed.
- R. Grünker, V. Bon, A. Heerwig, N. Klein, P. Müller, U. Stoeck, I. A. Baburin, U. Mueller, I. Senkovska and S. Kaskel, Chem. – Eur. J., 2012, 18, 13299 CrossRef PubMed.
- Z. Hu, C. Tao, H. Liu, X. Zou, H. Zhu and J. Wang, J. Mater. Chem. A, 2014, 2, 14222 CAS.
- Y. Zhang, Y. Chen, Y. Zhang, H. Cong, B. Fu, S. Wen and S. Ruan, J. Nanopart. Res., 2013, 15, 2014 CrossRef.
- J. An, C. M. Shade, D. A. Chengelis-Czegan, S. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2011, 133, 1220 CrossRef CAS PubMed.
- Z. Xie, L. Ma, K. E. deKrafft, A. Jin and W. Lin, J. Am. Chem. Soc., 2010, 132, 922 CrossRef CAS PubMed.
- M.-L. Ho, Y.-A. Chen, T.-C. Chen, P.-J. Chang, Y.-P. Yu, K.-Y. Cheng, C.-H. Shih, G.-H. Lee and H.-S. Sheu, Dalton Trans., 2012, 41, 2592 RSC.
- S. M. Barrett, C. Wang and W. Lin, J. Mater. Chem., 2012, 22, 10329 RSC.
- X.-L. Qi, S.-Y. Liu, R.-B. Lin, P.-Q. Liao, J.-W. Ye, Z. Lai, Y. Guan, X.-N. Cheng, J.-P. Zhang and X.-M. Chen, Chem. Commun., 2013, 49, 6864 RSC.
- R.-B. Lin, F. Li, S.-Y. Liu, X.-L. Qi, J.-P. Zhang and X.-M. Chen, Angew. Chem., Int. Ed., 2013, 52, 13429 CrossRef CAS PubMed.
- Z. Dou, J. Yu, Y. Cui, Y. Yang, Z. Wang, D. Yang and G. Qian, J. Am. Chem. Soc., 2014, 136, 5527 CrossRef CAS PubMed.
- X. Zou, J.-M. Goupil, S. Thomas, F. Zhang, G. Zhu, V. Valtchev and S. Mintova, J. Phys. Chem. C, 2012, 116, 16593 CAS.
- Y. Hwang, H. Sohn, A. Phan, O. M. Yaghi and R. N. Candler, Nano Lett., 2013, 13, 5271 CrossRef CAS PubMed.
- J. Ferrando-Soria, H. Khajavi, P. Serra-Crespo, J. Gascon, F. Kapteijn, M. Julve, F. Lloret, J. Pasán, C. Ruiz-Pérez, Y. Journaux and E. Pardo, Adv. Mater., 2012, 24, 5625 CrossRef CAS PubMed.
- G. Lu, O. K. Farha, W. Zhang, F. Huo and J. T. Hupp, Adv. Mater., 2012, 24, 3970 CrossRef CAS PubMed.
- G. Lu and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 7832 CrossRef CAS PubMed.
- E.-X. Chen, H. Yang and J. Zhang, Inorg. Chem., 2014, 53, 5411 CrossRef CAS PubMed.
- C. Cui, Y. Liu, H. Xu, S. Li, W. Zhang, P. Cui and F. Huo, Small, 2014, 10, 3672 CrossRef CAS PubMed.
- L.-F. Song, C.-H. Jiang, C.-L. Jiao, J. Zhang, L.-X. Sun, F. Xu, W.-S. You, Z.-G. Wang and J.-J. Zhao, Cryst. Growth Des., 2010, 10, 5020 CAS.
- P. Davydovskaya, V. Pentyala, L. Hussein, R. Pohle and G. Urban, in 2013 Transducers & Eurosensors XXVII: The 17th International Conference on Solid-State Sensors, Actuators and Microsystems (Tranducers & Eurosensors XXVII), IEEE, 2013, 2067.
- P. Davydovskaya, V. Pentyala, O. Yurchenko, L. Hussein, R. Pohle and G. A. Urban, Sens. Actuators, B, 2014, 193, 911 CrossRef CAS PubMed.
- Z. Hu, S. Pramanik, K. Tan, C. Zheng, W. Liu, X. Zhang, Y. J. Chabal and J. Li, Cryst. Growth Des., 2013, 13, 4204 CAS.
- A. M. Marti, S. D. Perera, L. D. McBeath and K. J. Balkus, Langmuir, 2013, 29, 5927 CrossRef CAS PubMed.
- K. Hirai, K. Sumida, M. Meilikhov, N. Louvain, M. Nakahama, H. Uehara, S. Kitagawa and S. Furukawa, J. Mater. Chem. C, 2014, 2, 3336 RSC.
- W.-J. Li, S.-Y. Gao, T.-F. Liu, L.-W. Han, Z.-J. Lin and R. Cao, Langmuir, 2013, 29, 8657 CrossRef CAS PubMed.
- N. B. Shustova, A. F. Cozzolino, S. Reineke, M. Baldo and M. Dincă, J. Am. Chem. Soc., 2013, 135, 13326 CrossRef CAS PubMed.
- C. Zong, X. Liu, H. Sun, G. Zhang and L. Lu, J. Mater. Chem., 2012, 22, 18418 RSC.
- O. S. Wolfbeis, Anal. Chem., 2006, 78, 3859 CrossRef CAS PubMed.
- S. K. Vashist and P. Vashist, J. Sens., 2011, 2011, 571405 Search PubMed.
- K. M. Goeders, J. S. Colton and L. A. Bottomley, Chem. Rev., 2008, 108, 522 CrossRef CAS PubMed.
- M. A. Schmidt and R. T. Howe, Ceram. Eng. Sci. Proc., 1987, 8, 1019 CAS.
- C. A. Grimes, S. C. Roy, S. Rani and Q. Cai, Sensors, 2011, 11, 2809 CrossRef PubMed.
- S. W. James and R. P. Tatam, Meas. Sci. Technol., 2003, 14, R49 CrossRef CAS.
- F.-G. Banica, Chemical Sensors and Biosensors: Fundamentals and Applications, John Wiley & Sons, Chichester, 2012 Search PubMed.
- R. A. Potyrailo, C. Surman, N. Nagraj and A. Burns, Chem. Rev., 2011, 111, 7315 CrossRef CAS PubMed.
- M. Sabo, A. Henschel, H. Fröde, E. Klemm and S. Kaskel, J. Mater. Chem., 2007, 17, 3827 RSC.
- J.-S. Choi, W.-J. Son, J. Kim and W.-S. Ahn, Microporous Mesoporous Mater., 2008, 116, 727 CrossRef CAS PubMed.
- M. E. Azim-Araghi and A. Krier, in Selected Topics in Advanced Solid State and Fibre Optic Sensors, ed. S. M. Vaezi-Nejad, IET, 2000, p. 215 Search PubMed.
- D. J. Wales, R. M. Parker, J. C. Gates, M. C. Grossel and P. G. R. Smith, Sens. Actuators, B, 2013, 188, 857 CrossRef CAS PubMed.
- Q. M. Wang, D. Shen, M. Bülow, M. L. Lau, S. Deng, F. R. Fitch, N. O. Lemcoff and J. Semanscin, Microporous Mesoporous Mater., 2002, 55, 217 CrossRef CAS.
- D. Dou, M. Molinier and O. Bailey, WO Pat., WO2001056686, 2001 Search PubMed.
- J. Schneider, C. Schnabel and J. Waldrop, WO Pat., WO2005121763, 2005 Search PubMed.
- A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440 CrossRef CAS PubMed.
- N. J. Hinks, A. C. McKinlay, B. Xiao, P. S. Wheatley and R. E. Morris, Microporous Mesoporous Mater., 2010, 129, 330 CrossRef CAS PubMed.
- A. C. McKinlay, J. F. Eubank, S. Wuttke, B. Xiao, P. S. Wheatley, P. Bazin, J.-C. Lavalley, M. Daturi, A. Vimont, G. De Weireld, P. Horcajada, C. Serre and R. E. Morris, Chem. Mater., 2013, 25, 1592 CrossRef CAS.
- J. F. Eubank, P. S. Wheatley, G. Lebars, A. C. McKinlay, H. Leclerc, P. Horcajada, M. Daturi, A. Vimont, R. E. Morris and C. Serre, APL Mater., 2014, 2, 124112 CrossRef PubMed.
- Z. Safidine, Z. Ghebache and S. Lamouri, Polym. J., 2013, 45, 946 CrossRef CAS PubMed.
- I. G. Giannakopoulos, D. Kouzoudis, C. A. Grimes and V. Nikolakis, Adv. Funct. Mater., 2005, 15, 1165 CrossRef CAS PubMed.
- Y. Yan and T. Bein, Chem. Mater., 1992, 4, 975 CrossRef CAS.
- A. Fischerauer, G. Fischerauer, G. Hagen and R. Moos, Phys. Status Solidi A, 2011, 208, 404 CrossRef CAS PubMed.
- A. Dubbe, Phys. Status Solidi A, 2011, 208, 416 CrossRef CAS PubMed.
- G. Hagen and R. Moos, Sens. Lett., 2011, 9, 110 CrossRef CAS PubMed.
- I. Untea, D. Brasoveanu, M. Dancila and S. Bocskor, Environ. Eng. Manage. J., 2002, 1, 299 CAS.
- D. Britt, D. Tranchemontagne and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11623 CrossRef CAS PubMed.
- C. A. Fernandez, P. K. Thallapally, R. K. Motkuri, S. K. Nune, J. C. Sumrak, J. Tian and J. Liu, Cryst. Growth Des., 2010, 10, 1037 CAS.
- T. G. Glover, G. W. Peterson, B. J. Schindler, D. Britt and O. Yaghi, Chem. Eng. Sci., 2011, 66, 163 CrossRef PubMed.
- S. Yang, J. Sun, A. J. Ramirez-Cuesta, S. K. Callear, W. I. F. David, D. P. Anderson, R. Newby, A. J. Blake, J. E. Parker, C. C. Tang and M. Schröder, Nat. Chem., 2012, 4, 887 CrossRef CAS PubMed.
- K. Tan, P. Canepa, Q. Gong, J. Liu, D. H. Johnson, A. Dyevoich, P. K. Thallapally, T. Thonhauser, J. Li and Y. J. Chabal, Chem. Mater., 2013, 25, 4653 CrossRef CAS.
- X. Xu, J. Wang and Y. Long, Microporous Mesoporous Mater., 2005, 83, 60 CrossRef CAS PubMed.
- X.-W. Xu, J. Wang and Y.-C. Long, Chin. J. Chem., 2005, 23, 359 CrossRef CAS PubMed.
- S. J. Geier, J. A. Mason, E. D. Bloch, W. L. Queen, M. R. Hudson, C. M. Brown and J. R. Long, Chem. Sci., 2013, 4, 2054 RSC.
- J. Caro and M. Noack, in Advances in Nanoporous Materials, ed. S. Ernst, Elsevier, Oxford, 2010, vol. 1, p. 1 Search PubMed.
- N. Mehio, S. Dai and D. Jiang, J. Phys. Chem. A, 2014, 118, 1150 CrossRef CAS PubMed.
- P. Küsgens, M. Rose, I. Senkovska, H. Fröde, A. Henschel, S. Siegle and S. Kaskel, Microporous Mesoporous Mater., 2009, 120, 325 CrossRef PubMed.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308 RSC.
| This journal is © The Royal Society of Chemistry 2015 |