
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantum dots: bright and versatile in vitro and in vivo fluorescence imaging biosensors
K. David
Wegner
and
Niko
Hildebrandt
*
NanoBioPhotonics, Institut d'Electronique Fondamentale, Université Paris-Sud, 91405 Orsay Cedex, France. E-mail: niko.hildebrandt@u-psud.fr; Web: http://www.nanofret.com
First published on 17th March 2015
Abstract
Semiconductor quantum dots (QDs) have become important fluorescent probes for in vitro and in vivo bioimaging research. Their nanoparticle surfaces for versatile bioconjugation, their adaptable photophysical properties for multiplexed detection, and their superior stability for longer investigation times are the main advantages of QDs compared to other fluorescence imaging agents. Here, we review the recent literature dealing with the design and application of QD-bioconjugates for advanced in vitro and in vivo imaging. After a short summary of QD preparation and their most important properties, different QD-based imaging applications will be discussed from the technological and the biological point of view, ranging from super-resolution microscopy and single-particle tracking over in vitro cell and tissue imaging to in vivo investigations. A substantial part of the review will focus on multifunctional applications, in which the QD fluorescence is combined with drug or gene delivery towards theranostic approaches or with complementary technologies for multimodal imaging. We also briefly discuss QD toxicity issues and give a short outlook on future directions of QD-based bioimaging.
 K. David Wegner | K. David Wegner received his diploma in Chemistry in 2011 at the University of Potsdam (Germany). In the same year he joined the NanoBioPhotonics group of Prof. Niko Hildebrandt at University Paris-Sud (France) as graduate student. In his graduate studies he investigates multiplexed Förster resonance energy transfer between luminescent lanthanide complexes and quantum dots and their application in homogenous sandwich immunoassays for clinical diagnostics and as nanometric molecular rulers. |
 Niko Hildebrandt | Niko Hildebrandt (http://www.nanofret.com) holds a PhD in Physical Chemistry (2007) from the University of Potsdam (Germany), where he also carried out postdoctoral research until 2008. From 2008 to 2010 he was groupleader at the Fraunhofer Institute for Applied Polymer Research in Potsdam. Since 2010 he has been full professor at Université Paris-Sud in Orsay, France, where he is leading the group NanoBioPhotonics at the Institut d'Electronique Fondamentale with a research focus on time-resolved Förster resonance energy transfer (FRET) spectroscopy and imaging for multiplexed nanobioanalysis. Since 2014 he has been member of the Institut Universitaire de France. |
1. Introduction
Semiconductor nanocrystals (quantum dots, QDs) have arguably affected bioimaging research more than any other nanomaterial. The versatility of QD-based application was not imaginable when the relationship between size and band-gap of semiconductor materials was described in the early eighties.1 It took until 1998 when two Science articles pointed out the advantages of this new material for biosensing and established QDs as a new class of fluorophores in the toolkit of biological researchers.2,3 In the following years scientists used QDs for inorganic ion sensing, organic small molecule and biological macromolecule sensing, bioconjugation and cell staining, cellular effectors and reporters, animal imaging, and therapy.4–11 In particular, their unique photophysical properties were exploited for in vitro12–20 and in vivo21–24 imaging applications. The published conjugation strategies and commercial availability of QDs enlarged their application field and led to an exponential increase of QD related research articles, which were reviewed over the last few years in numerous articles.4,25–33In this review we summarize the developments of QD-based imaging methods in relevant biological materials with a focus on the recent advances for in vitro and in vivo biosensing. Although QDs can be prepared with atoms from groups II–VI, III–V, or IV–VI of the periodic table and in many different alloyed versions, we mainly discuss the most popular Cd-based QDs as a model for all other QDs. Information about the usage of other types of QDs (InP, GaAs, etc.), gold nanoparticles, iron particles, carbon dots, and further nanometric imaging agents can be found throughout this Chemical Society Reviews Themed Collection “Imaging Agents” and in the following ref. 34–38.
We begin with a short overview about the preparation of QDs, their optical properties, and advantages as imaging agents over conventional fluorophores and then discuss important contributions of QDs in super-resolution microscopy and single-particle tracking. Although QDs have been used for imaging applications in bacteria and yeast39–44 and for monitoring biological processes in leaf cells of plants,45 we focus our review on mammalian material and survey recent studies of in vitro, tissue, and in vivo imaging. After a brief journey into the controversial topic of QD toxicity we conclude our review and give a short outlook on the bright future of QD-based imaging.
2. Preparation and properties of QDs
A bottleneck for the use of the first inorganic QDs in biological applications was the reproducible synthesis of highly luminescent, water-soluble, and monodisperse QDs. The synthesis approach of Murray et al. in 1993 was a milestone for the preparation of uniform colloidal QDs.46 Their method was based on a high-temperature organometallic process, which resulted in colloidal QDs with a low polydispersity but unfortunately also a low photoluminescence (PL) quantum yield (QY). To improve the PL properties passivation of the QD core by a shell of a few atom layers of ZnS or CdS was found to be advantageous.47–49 An important property for the shell was that the material had a larger energy band-gap than the core, which led to a confinement of the excitons in the core and reduced the surface-related recombination in trap states. In addition to the enhanced PL properties, the core–shell QDs exhibited a better photochemical stability and a reduced Cd leaching from the core. Another important milestone, in particular for the later commercialization of QDs, was the improved synthesis protocol by Peng et al. The use of CdO as precursor instead of the sensitive and toxic Cd(CH3)2 was safer and allowed larger scale production.50 The use of toxic materials and the need for the transformation of QDs from the organic phase to the water phase, which comes at the cost of reduced QY, also led to the investigation of QD preparation in aqueous solution. Key features of the aqueous synthesis are direct water-solubility, facile preparation, good reproducibility, low costs, and improved biocompatibility.4,51 One of the first who demonstrated the aqueous synthesis of thiol-stabilized CdTe QDs were Rogach et al.,52 and various alternative approaches have been developed to produce QDs in the aqueous phase with good QY and small size.53,54 In a recent study Au et al. compared the conjugation efficacy of aqueous and organic synthesized QDs and their stability in biological media. The results showed both 10-fold increased conjugation efficacy and better stability for the aqueous synthesized QDs.55For all synthesis approaches the experimental parameters, such as temperature, growth time, etc., are used to control the shape and size of QDs. For in vitro and in vivo imaging applications the size plays a major role and smaller QDs are most often beneficial for such purposes. Small hydrodynamic radii of QDs are an important issue for avoiding perturbation of QD-functionalized biological molecules or to access biological interactions (e.g. by Förster resonance energy transfer (FRET) or other energy transfer processes) at distances of only a few nanometers. Diameters of water-soluble QDs are usually in the range of 5 to 10 nm but aqueous QDs (aqQDs) with average diameters down to 1.6 nm have also been produced.56 Apart from classical approaches starting with nucleation and growth of QDs in solution, it was shown that QDs can be grown on, for example, peptide templates.57 QDs were also “naturally” produced by exposing standard wild-type Lumbricus rubellus earthworms to soil spiked with CdCl2 and Na2TeO3 salts for 11 days. An intrinsic heavy-metal detoxification strategy was responsible for the intra-worm production of luminescent QDs, which could be successfully used in live-cell imaging after the isolation from the chloragogenous tissues.58 A similar effect could be obtained with rats after treatment with CdCl2. The detoxification of the heavy-metal salt produced QDs, whose luminescence could be divided in three colours (red, green, and yellow).59 The production of solid-state material inside living organisms is an emerging field in nanobiotechnology.58
On the interface between QD and the environment the surface molecules play an important role concerning water-solubility, biocompatibility, and bioconjugation. Different strategies of surface modification were used to render QDs biocompatible. These include surface cap exchange,2,60–62 amphiphilic surface ligands,63,64 and encapsulation in micelles23 or silica shells.65–69 Regarding fluorescence imaging specific binding of the final biosensor to the target is highly important and the large and charged surfaces of QDs were a leading cause of non-specific binding. An established strategy to avoid this effect is the use of poly(ethylene glycol) (PEG) as surface coating, which was shown to efficiently reduce non-specific binding by using at least 12 to 14 units of PEG.70 Hydroxyl coated QDs showed a 140-fold reduction in non-specific binding compared to carboxylate QDs and 10- to 20-fold reduction relative to that of PEG- and protein-coated QDs.71 To obtain water-soluble QDs with reduced non-specific binding comes at the cost of their hydrodynamic radius and surface charge. Because traditional transmission electron microscopy (TEM) can only visualize the inorganic core, methods like dynamic light scattering, liquid chromatography, and laser Doppler velocimetry are important tools to investigate the intrinsic properties of QDs in the water phase. For three-dimensional size and shape analysis of QD-bioconjugates under physiological conditions and at low concentrations FRET has been successfully employed.72,73 Too large QDs can be problematic in biological application because of reduced diffusion in tissues and tumours.74 In order to provide water-soluble QDs with small hydrodynamic size, low non-specific binding and high QY a cap exchange with PEGylated-dihydrolipoic acid (DHLA) ligands showed to be advantageous. Additionally, DHLA-QDs also provided good solution stability over a large pH range. By manipulating the terminal functional groups of the DHLA-PEG ligands with amino, carboxyl, or hydroxyl groups, the surface charge could be altered and QDs efficiently bioconjugated.75–77 Designing QDs with small and compact ligands that provide biocompatibility, stability, and significantly reduced overall hydrodynamic radii of the final QD-bioconjugates is highly important for successfull biosensing and cellular imaging.73,78 Another variant of surface coating is the use of soft-binding aminopropanol (APP) as cap exchange ligand for a rapid transfer of hydrophobic QDs into polar solvents. Due to the protonation of APP in aqueous solution, the ligand can be easily removed and the intermediate QD can be incorporated inside micelles under mild conditions.79
The large surface of QDs offers the possibility for the binding of multiple biomolecules of interest (BOI), such as antibodies or enzymes, that can enhance the sensitivity and activity in biological applications.80,81 Among the most prominent methods for QD bioconjugation are the aniline-catalyzed hydrazine bond formation for binding to amino groups and the use of maleimide groups for the conjugation of thiol groups present, for example, in the hinge regions of antibodies.82 For specific covalent binding Schieber et al. presented a method in which they used strain-promoted azide–alkyne cycloaddition reaction of azides with strained cyclooctynes. Therefore they conjugated azide-modified QDs with cyclooctyne-modified biomolecules for fluorescence imaging of tumour cells.83 For the preparation of a monovalent functionalized QD without chemical modification, Farlow et al. used a functionalized oligonucleotide, which wrapped around the QD monovalently due to steric exclusion.84 These are only a few impressions from the vast choice of QD preparation and bioconjugation. An overview of the different surface coating strategies and functionalization pathways are summarized in Fig. 1 and plenty of detailed additional information can be found in the following reviews.27,29,33,85,86 Although QD bioconjugation is very versatile, one should keep in mind that QDs are relatively large objects compared to small organic dyes and to the biomolecule they are conjugated to. This can lead to alterations in the biological function of the QD-bioconjugate. Moreover, the large and often charged surfaces of QDs provide a large space for non-specific interactions, which can interfere with the specific recognition of the QD-bioconjugates.
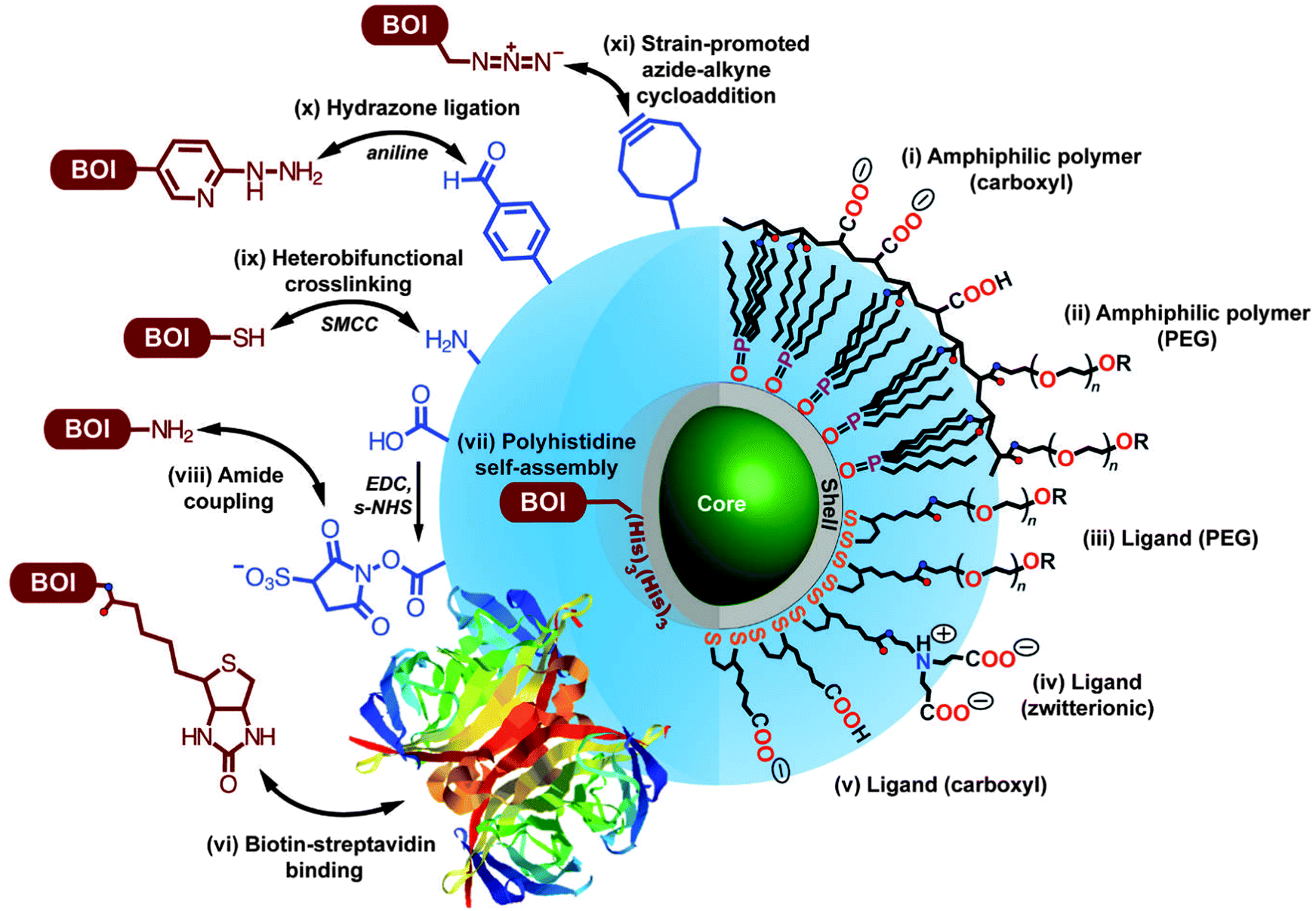 | ||
| Fig. 1 Overview of different bioconjugation (left side, BOI = biomolecule of interest) and surface coating (right side) strategies for QDs. Two surface coating strategies are presented: encapsulation with amphiphilic polymers (i, ii) and cap exchange with hydrophilic ligands exploiting the thiol-affinity of the ZnS shell of the QD (iii–v). Reprinted with permission from ref. 30. Copyright 2013 Society for Applied Spectroscopy. | ||
2.1 Photophysical properties and advantages of QD imaging agents
The unique photophysical properties of QDs are the main cause of their popularity and versatile usage in biosensing. QDs are semiconductor nanoparticles that have physical dimensions close to or smaller than the exciton Bohr radius.87 The spatial confinement of intrinsic electron and hole carriers leads to an increased band-gap energy and to a splitting of the continuous energy bands in discrete energy levels. This effect makes QDs to an intermediate between bulk materials and molecules. The absorbance of photons with energies higher than the band-gap leads to the creation of an electron–hole-pair, an exciton. With increasing excitation energy (shorter wavelength) there is also an increased absorption probability, which leads to a very broad absorption spectrum and large effective Stokes shifts (difference between excitation and emission wavelength) of more than 100 nm if necessary.88–92 Trap states caused by disturbed crystal structure (mainly on the QD surface) can lead to non-radiative deexcitation and is also one of the reasons for intermittent fluorescence (blinking) of QDs, which is visible on the single QD level.89 Another consequence of the quantum confinement effect is the size dependent emission, caused by an increased confinement of the excitons with smaller QD size and thus higher energy band-gap (Fig. 2B). This means that larger QDs of the same material exhibit a smaller energy band-gap and thus a PL emission in the red, whereas smaller QDs fluoresce in the blue (Fig. 2C). This effect allows to tune the PL colour of QDs by controlling their size and enables, in combination with different materials, to engineer QDs that cover the spectral range from the ultraviolet (UV) to the infrared (IR).93 The PL spectra of QDs have a nearly Gaussian shape with a narrow full-width-at-half-maximum (FWHM) of ca. 20 to 30 nm and show a negligible tendency for photobleaching.63,93 QD PL is typically circularly polarized or non-polarized for spherical QDs, whereas quantum rods show a polarized emission.94 The PL decay is most often multiexponential with average lifetimes ranging from ca. 10 to 100 ns. Important for imaging applications is the high brightness (product of QY and absorptivity), caused by high QYs and large molar extinction coefficients.95 Additionally, QDs provide large two-photon cross sections (ca. 103–104 Goeppert-Mayer units (GM)), which is advantageous for multiphoton excitation.96–98 Recently, CdSe/CdS-quantum-dots-quantum-rods with two-photon cross sections of ca. 105 GM and QYs of 78% have been produced.99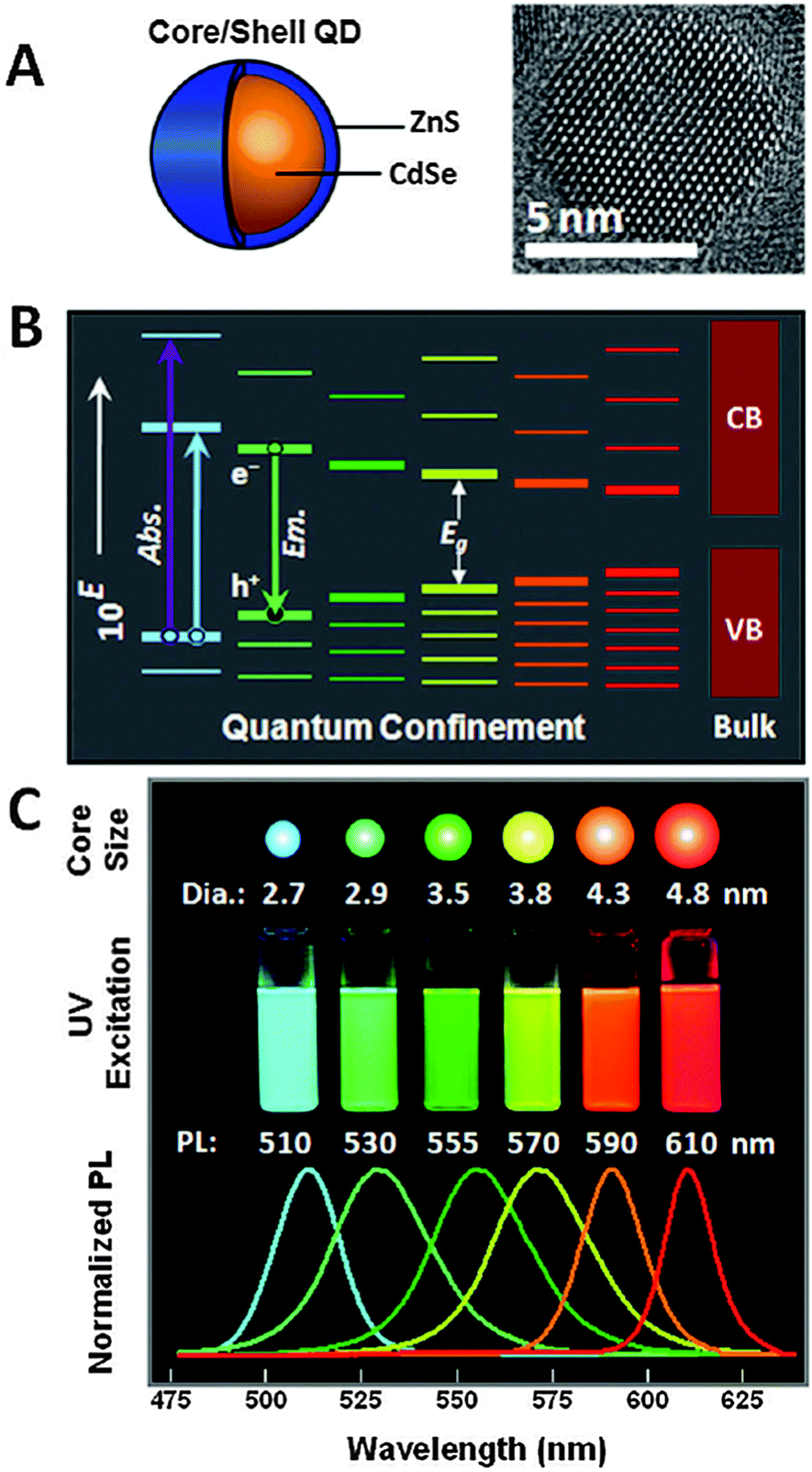 | ||
| Fig. 2 (A) Schematic drawing of a core–shell QD together with a TEM image illustrating the nanocrystalline structure of the QD core. (B) Quantum confinement of the exciton leads to an increasing band-gap energy with decreasing size as well as distinct energy levels in contrast to the continuous conduction band (CB) and valence band (VB) of the bulk semiconductor. (C) The larger band-gap energy of smaller QDs of the same material leads to lower PL wavelengths (large QDs are red and small QDs are blue). Reproduced with permission from ref. 85. Copyright 2011 American Chemical Society. | ||
One reason for the large interest in QDs for bioimaging is that fluorescent dyes and fluorescent proteins (FPs) have some shortcomings, such as narrow excitation spectra, small effective Stokes shifts, broad fluorescence bands, and susceptibility to photobleaching.33 Additionally, their PL lifetimes are in the same range (few nanoseconds) than biological autofluorescence, which does not allow time-gated detection for increasing the signal-to-noise ratio (SNR) and for better differentiation between different target molecules, as it was shown for QDs.13,100 The emission spectra of dyes show a pronounced shoulder in the red, which limits spectral multiplexing due to optical crosstalk. Moreover, the narrow absorption spectra make it hardly possible to efficiently excite several dyes at once. In contrast, multiple-QD excitation with a single excitation source (wavelength) and simultaneous multiple-QD PL detection offer the possibility of a multiplexing capability with almost negligible spectral crosstalk.18,63,101,102 In a direct comparison concerning photostability it could be shown that QDs are 100 times more stable and 20 times more bright than Rhodamine 6G.3 Further comparison with other dyes showed the superiority of QDs concerning their photostability3,12,16,63,103–105 also against one of the most stable organic dyes AlexaFluor 488 (Fig. 3).106 Another advantage of QDs over traditional fluorophores is their almost two orders of magnitude larger two-photon cross section,98,107 which is particularly interesting for in vivo applications with near infrared (NIR) excitation. A comparative study of QDs vs. organic dyes was performed by Resch-Genger et al.,108 and though QDs exhibit superior optical properties, they also bare some drawbacks such as blinking, nanocolloidal behaviour, and controversial toxicity issues. Despite their photophysical advantages QDs have not and will not replace organic dyes or fluorescent proteins for bioimaging. Each class of fluorophore has certain advantages and disadvantages and it is certainly a wise decision to exploit the entire fluorescence toolbox, which offers an almost unlimited choice of fluorescent probes for biosensing.5 In contrary to QDs most organic dyes are small and easy to conjugate to different biomolecules without altering the biological function of the dye-bioconjugate. Moreover, many dyes can be attached to larger biomolecules (e.g., antibodies), which increases the overall brightness of the bioconjugate PL. The commercial availability of a large variety (both biological and spectral) of dye-bioconjugates is another important advantage of organic dyes. Fluorescent proteins provide the unique possibility of expressing the fluorophore directly inside the biological system of interest, so that no chemical coupling becomes necessary, which can result in less disturbed biological functionality. More information about different types of fluorophores can be found in the literature,5,109–111 and one should always keep in mind that QDs are just one specific fluorescence tool among many others. For bioimaging applications it is important to evaluate the QD benefits against their drawbacks and to choose the right fluorophore (QD or not) for each specific biosensor. Tuneable absorption and emission spectra, spectrally broad and strong absorption, narrow emission bands, and high photostability are universal advantages of QDs for fluorescence imaging and therefore we do not explicitly discuss them for each application reviewed in the following sections.
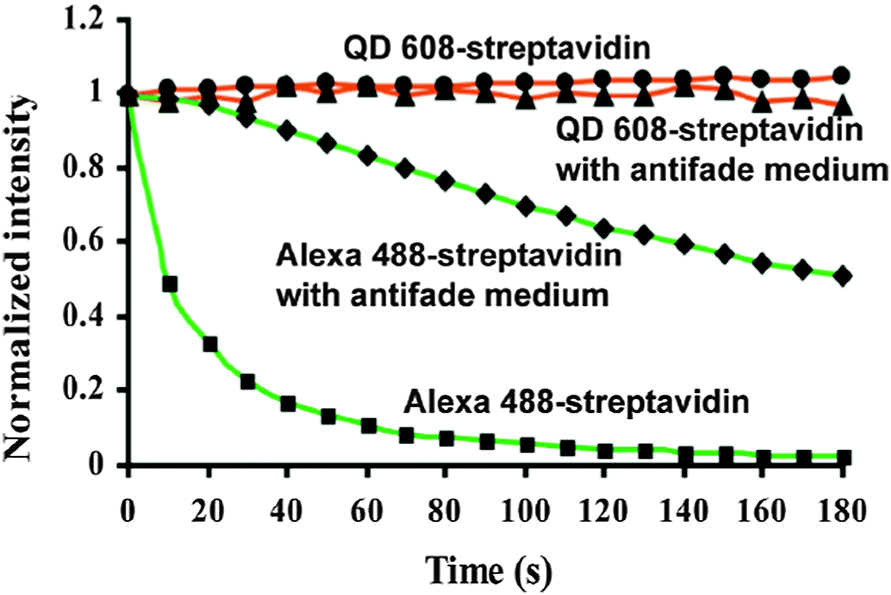 | ||
| Fig. 3 Comparison of the photostability of streptavidin conjugates with QD608 and AlexaFluor 488 over three minutes of continuous excitation demonstrated the superior photostability of QDs. Reprinted with permission from ref. 63. Copyright 2003 Nature Publishing group. | ||
3. Super-resolution microscopy and single-particle tracking
3.1 Super-resolution microscopy
In view of the 2014 Nobel Prize in Chemistry, which was awarded to Eric Betzig, Stefan W. Hell, and William E. Moerner for the development of super-resolved fluorescence microscopy,112 we will take a closer look at the impact of QDs on this particular field of imaging. Monitoring of biological processes at the subcellular level is important for the understanding of the relationship between cell compartment-triggered reactions. Super-resolution microscopy enables the localization of different proteins on the plasma membrane and in the cytoplasm and allows the investigation of how signals are transmitted within the cell or how molecules will be taken up and transported.28 The complexity of biochemical reactions in live cells cannot be fully resolved using standard wide-field ensemble measurements because many of these reactions are triggered by only a small number of molecules. For those reasons monitoring of nanometric subcellular structures or single-molecules is advantageous. The main challenges of measuring on the single-molecule level are the ability to detect a single-molecule in a dense medium and to distinguish two fluorophores that are separated by a distance below the diffraction limit of light (∼0.5 × the detection wavelength). For super-resolution images of fluorophore ensembles the point-spread-function (PSF) of a single emitter can be spatially reduced (by using, e.g., stimulated emission depletion microscopy – STED) or the centre of each separated PSF can be localized with high precision by collecting a large amount of photons per single emitter (by using, e.g., photoactivated localization microscopy – PALM, stochastic optical reconstruction microscopy – STORM, or points accumulation for imaging in nanoscale topography – PAINT). Using super-resolution techniques such as STED or saturated structural illumination microscopy (SSIM) requires high excitation powers and can cause phototoxicity in biological systems. STORM and PALM require long imaging times up to several minutes and much effort has been put into the development of faster super-resolution techniques to allow imaging of live biological systems.From the biological point of view three general key elements need to be considered for super-resolved microscopy. First, the probe must be transportable to the molecule or organelle of interest. This can be relatively easy when the target is a receptor expressed on the surface of the cell, but it can be more complicated when the probe has to be delivered inside the cytoplasm (cf. Section 4.1 for more details about cell delivery). Second, the fluorophore bioconjugate should be monovalent. Multivalency of the probe can be problematic because binding to more than one target can perturb the measurement. In one example of creating monovalent QDs Clarke et al. used peptide coating and applied the peptide–QD conjugates in single-molecule measurements.113 Third, the affinity between probe and target should be very high to allow a binding time that is longer than the required observation time. This is one of the reasons why the biotin–streptavidin recognition has been so popular inside super-resolution techniques.114 Streptavidin (sAv) is (in its native form) a tetramer and can bind four biotins. The reduction of the tetramer to a monomeric protein also leads to the reduction of the binding affinity. To overcome this problem Howarth et al. demonstrated the preparation of streptavidin with a controlled number of functional biotin binding subunits without decreased binding affinity.115–117 The three biological requirements mentioned above (biological functionality, monovalency, and high affinity) argue for the use of small fluorescent dyes, which are in fact the most often applied fluorophores for super-resolution microscopy.
From the photophysical point of view fluorophores should be bright and photostable to allow highly sensitive single-molecule detection, which places QDs in an advantageous position compared to organic dyes or FPs. Because their fluorescence intermittency, also known as blinking, limits the application possibilities, QDs are mainly used in super-resolution techniques that rely on spatial and temporal localization of single-molecules, such as PALM, fPALM, STORM, dSTORM, and GSD.118 In different studies using fluorescence fluctuation analysis it could be shown that the estimation of diffusional mobility can be highly influenced by blinking and that special analytical models are necessary to differentiate the contribution of photophysical fluctuations from those caused by transport.119–121 Improvements in synthesis and surface passivation of QDs could alter122 and even suppress blinking,123,124 whereas variation of the excitation wavelength had only minor influence on the blinking behaviour.125 Initial investigations assumed that blinking of QDs had a characteristic timescale.126 In contrast to these results, Bachir et al. concluded that QD blinking fluctuations obey a power law distribution and that there is no single characteristic fluctuation time for this phenomenon.120
In contrast to simultaneous ensemble measurements super-resolution imaging can take advantage of QD fluorescence blinking. Because complete on–off intermittency can be observed only for single QDs (and not for an ensemble of QDs because of a very low probability that the different QDs all blink simultaneously) it can be used to resolve closely spaced QDs.127 Dertinger et al. used a method, which relies on higher-order statistical analysis of temporal fluctuations (caused by blinking) recorded in a sequence of images. This method enabled a 5-fold improvement in spatial resolution and was demonstrated on QD-labelled microtubules of fibroblast cells. They coined their method super-resolution optical fluctuation imaging (SOFI) and achieved 55 nm spatial resolution (FWHM) in the x–y plane.128 With the synthesis of QDs with increased blinking rates also the temporal resolution of SOFI could be improved down to 80 ms.129 Chien and coworkers exploited the blinking phenomenon to map out the locations of individual QDs using total internal reflection microscopy (TIRF). The reconstructed image was in good correlation with the topographic image measured by atomic force microscopy (AFM).130 Hoyer et al. showed the possibility of using a simple webcam, QDs, and ground-state depletion microscopy to obtain fluorescence images with single-QD resolution.131 Wang et al. exploited QD blinking to achieve three-dimensional (3D) super-resolution imaging with 8 to 17 nm in the x–y plane and 58 to 81 nm in the z-direction. After successful simulation they applied their approach to resolve the 3D distribution of EGFR molecules on breast cancer cells. However, the technique also had limitations, such as limited resolution in the case of a too dense QD distribution. Moreover, the method was based on frame-subtraction, which requires extremely high spatial stability (no motion) from frame to frame.132
Most of the super-resolution techniques presented above used single-colour QD blinking. Shi et al. presented a spectral imaging nanoscopy approach for multicolour QD superlocalization. They used a transmission grating to disperse the emission light into zero and first order images. QDs in close proximity exhibited one overlapping zero-order spot but multiple (depending on the number of QDs) distinct first-order streaks (Fig. 4). The single QDs could be identified due to the disappearing of the first-order streak during the blinking off-state. The authors demonstrated their approach using up to three QDs connected with complementary oligonucleotides with different lengths and in human embryo kidney 239A cells.133
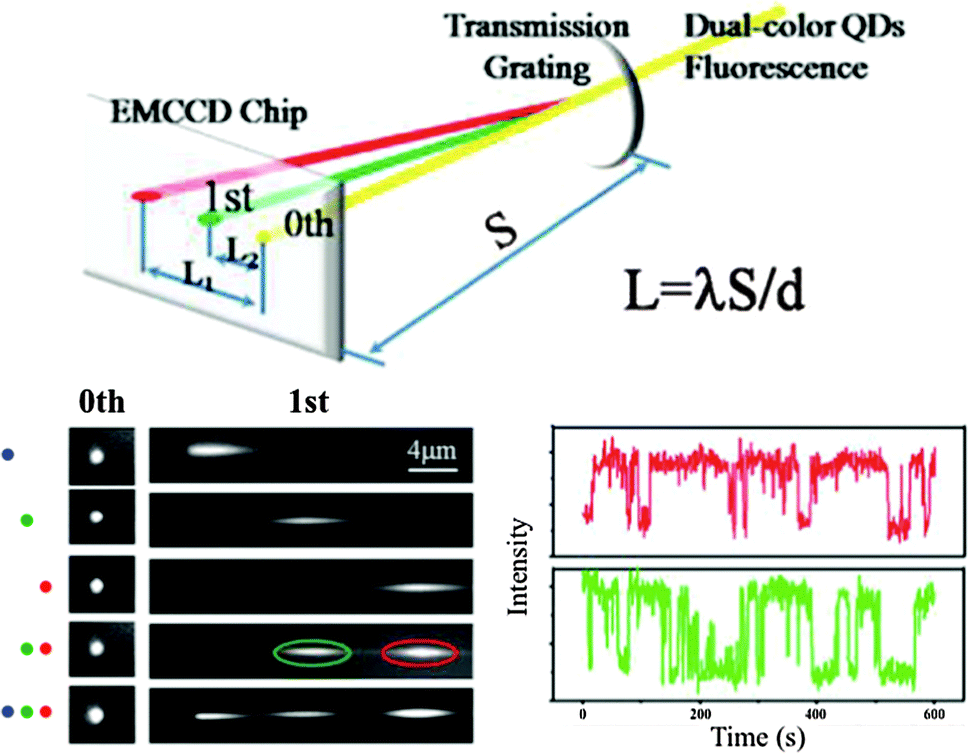 | ||
| Fig. 4 Multicolour super-resolution of three different QDs with PL maxima at 525 nm (blue), 585 nm (green), and 655 nm (red) separated by DNA at distances below 50 nm. Top: a transmission grating is used for colour separation (dual colour experiment is shown). Bottom left: zero and first order images of single QDs and combinations of two and three QDs. Bottom right: taking the first order images the blinking behaviour of the green and red QD can be distinguished for super-resolution imaging. Reproduced with permission from ref. 133. Copyright 2012 American Chemical Society. | ||
As demonstrated in the aforementioned study a high spatial density of QDs requires special techniques to distinguish the blinking of single QDs. Another possibility of imaging dense spatial QD distributions at super-resolution was proposed by Deng et al. using a reconstruction algorithm named spatial covariance reconstructive (SCORE) algorithm. The key point of this technique was statistic monitoring of the covariance between the intensities of pixels, which led to a preference of high labelling densities. SCORE does not take into account temporally non-drifting background and thus enables the analysis of fluctuating emitters in a background of constantly fluorescing molecules. The authors demonstrated the advantages of SCORE applied to images of QD-labelled microtubules in HeLa cells and could obtain a resolution below 90 nm within a few seconds of image acquisition (Fig. 5).134
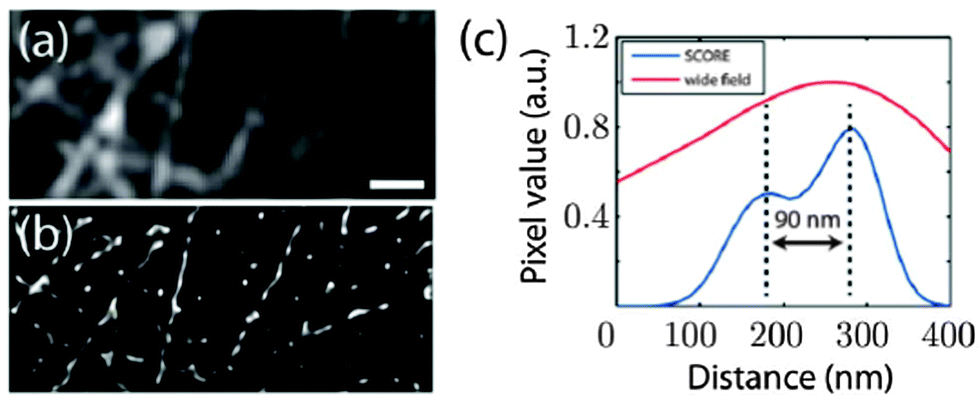 | ||
| Fig. 5 Super-resolution imaging of microtubules with dense QD–antibody labelling using the SCORE technique. Monitoring the intensity covariance between different image pixels allowed a significantly improved spatial resolution (ca. 90 nm) compared to standard wide field imaging: SCORE-image (b) and resolution (blue curve in (c)) versus wide flied image (a) and resolution (red curve in (c)). Reprinted with permission from ref. 134. Copyright 2014 Deng et al. | ||
Although blinking is one of the most often applied techniques for QD-based super-resolution, also other approaches, such as photon antibunching to narrow down the PSF135 or tri-exciton generation combined with spectral deconvolution and imaging,136 can create super-resolved images. Another alternative approach is near-field scanning optical microscopy (NSOM), which allows simultaneous acquisition of topographic and fluorescence images and was used to identify organized nanosized domains of molecules on the cell surface.137–139 The resolution for this imaging technique can reach ca. 50 nm.140 Zhong et al. used NSOM and two different emitting QDs to visualize the distribution and organization of T-cell receptors and gangliosides (GM1) as well as their nanospatial relationship and their correlation during T-cell activation. The results showed that a priori formation of GM1 nanodomains serves as a platform for the recruitment of T-cell receptor nanodomains.141 A very promising strategy for improving super-resolution microscopy is the use of controlled photoswitching of QDs. In a recent study Diaz and co-worker developed photoswitchable QDs based on FRET from QDs to photochromic acceptors that could be activated by UV radiation.142
3.2 Single-particle tracking
In addition to obtaining highly resolved images of cellular components, it is of large interest to study the dynamics of molecules with subcellular resolution. Single-molecule tracking (SMT) usually follows a single-molecule, whereas in single-particle tracking (SPT) the track of a mesoscopic particle bound to an ensemble of molecules is recorded.143 Their aforementioned unique photophysical properties have enabled frequent use of QD nanoparticles for SPT.144 Because in single-molecule imaging the localization of individual spots is not limited by diffraction but by signal-to-noise ratio (SNR), uncontrolled expression of genetically encoded FPs and low photostability of FPs and dyes do not allow long-term measurements with high SNR, which is essential for high resolution images.145 Other advantages of single QDs over dyes and FPs in single-molecule in vitro assays are for example their very narrow PL spectra with FWHM around 15 nm and a high SNR, which enable a significantly improved spatial resolution of 5 to 10 nm compared to 40 nm with organic dyes. The resistance to photodegradation allows SPT and SMT over several minutes (more than 20 minutes have been reported) compared to 5 to 10 s for standard organic dyes.146 Walter et al. reviewed different SPT microscopy techniques and proposed a guideline to choose the best technique for each target of interest.147 In general, a standard wide-field microscope can be used for SPT, but it may be very beneficial to use the auto-focus function to keep a constant distance of the sample in the z-position. The first step of identifying and selecting individual fluorescent particles is to reduce background via changing the intensity threshold. The x- and y-coordinates can be obtained by fitting a two-dimension (2D)-Gaussian function to the particle's intensity profile. This localization of the particles will be done for each frame and is called segmentation. In the next step an algorithm can be used for the calculation of the corresponding trajectories. In practice the algorithm links the centres of fluorescent spots across the adjacent frames in the image series. Further analysis using mean-square displacement as a function of time allows analysing the type of motion and provides information about different parameters, such as diffusion coefficient, transport velocity, and size of the confinement domain.115,148,149 For a continuous tracking of the target the intrinsic blinking of QDs is problematic and special algorithms that consider the disappearing of targets need to be used. Such tools can be freely downloaded.150,151 To cover the complexity and dynamics of biological processes it is of great interest to obtain the trajectories in three dimensions. Holtzer et al. enabled 3D-analysis of molecular mobility by introducing a cylindrical lens into the emission path and tracked passively diffused and actively transported QDs within life cells.152 In another study Ram and coworkers developed multifocal plane microscopy and used an algorithm allowing the 3D positioning of a point source. They demonstrated the strength of this technique by observing the complete endocytosis process of antibody-conjugated QDs to the sorting endosomes deep inside the cell.153 Wells et al. presented a new approach for 3D tracking of molecular motion using QDs and four overlapping confocal volume elements with an active feedback every 5 ms. Their approach showed several advantages compared to 3D tracking using charge-coupled device (CCD) cameras, namely lower excitation power, capability of time-resolved detection of the tracked molecules, and measurement of trajectories for minutes and over distances up to 10 μm in all spatial dimensions, which may enable tracking throughout the entire volume of many mammalian cells.154 The measurement of the angular components of a tracked molecule using polarization modulation techniques could provide information about conformational changes and add rotational orientation inside a living cell as another dimension to SPT.155,156 Similar to super-resolution one of the key factors for SPT measurements is the successful delivery of the conjugates into the cytoplasm, which can be problematic for QDs due to their large sizes or surface charges. Cellular delivery will be discussed in more detail in Section 4.1.Most probably because of complicated cell delivery initial applications of QD-based SPT targeted membrane proteins and studied entry/exit kinetics of receptors such as glycine,157,158 GABA,159 HER2,14,160 EGFR,14,161,162 and HIV receptor CD4,163,164 but also Interferon165 and NGF,166,167 various transmembrane proteins,168–170 and aquaporine171 were investigated by SPT. The high spatial resolution of QD-based SPT was used by Chung et al., who investigated the EGFR signalling processes with a novel time-dependent diffusivity analysis. Important steps within signal transduction are ligand binding and receptor dimerization. The authors were able to monitor single receptors in living cell and could observe that EGFR fluctuated continuously between monomer and dimer state and that preformed dimers were preferred for ligand binding.172
More recent studies were also able to track single QDs inside cells. Lowe et al. used QD-labelled cargos to study the entry to and exit from the nucleus by the nucleus pore complex. They were able to decipher characteristic steps for the import, namely cargo capture, filtering and translocation, and release into the nucleus.173 The investigation of altered expression or regulation of presynaptic serotonin (5-HT) transporter (SERT) is very important due to the involvement in multiple neuropsychiatric disorders. Chang and coworker used antagonist-conjugated QDs and monitored single SERT proteins on the surface of serotonergic cells and could identify two different pools defined by the lateral mobility of the proteins.174 In another study the authors investigated the diffusion kinetics of the lipid raft constituent ganglioside GM1, which acts as platform to facilitate neuronal signalling.175 Chen et al. developed an aptamer-based probe, which was capable to target membrane proteins (nucleolin and prion protein) and to provide a biotin functional group as versatile linker to different fluorescent probes including QDs. They investigated the endocytic pathway using SPT and trajectory analysis and observed three types of movements associated with distinct phases: membrane diffusion, vesicle transportation, and confined diffusion. Internalization into the cytoplasm was achieved through the clathrin-dependent/receptor-mediated pathway. Modification of the aptamer sequence could possibly allow to using this probe for the tracking of other markers.176 A further improvement for the investigation of the endocytic pathway was proposed by Ma et al., who were able to track the whole intracellular dynamic endocytosis process of phenylephrine conjugated to QDs via continuously filming fluorescent images in the same cell. The measured motion parameters and colocalization with specific fluorescent tags for different types of cell components enabled them to divide the endocytosis process into six stages after membrane passage: (i) directed movement along actin filaments and transportation to early endosomes; (ii) confined movement in early endosomes; (iii) directed movement along tubulin and transportation to late endosomes; (iv) confined movement in late endosomes; (v) directed movement along tubulin and transportation to lysosomes; (vi) confined movement in lysosomes.177
Ram et al. investigated the transferrin receptor pathway in a 10 μm thick epithelial cell monolayer using 3D single-molecule tracking with the aim to improve drug delivery across cellular barriers at specific body sites.178 The complexity of the neuronal structure offers a large playground for SPT to obtain information about molecular dynamics of lipids and transmembrane proteins in correlation to synaptic membrane compartments. Biermann et al. investigated the dynamics and organization of surface molecules in brain slices and were able to image QDs bound to cell surface molecules at depths of up to 60 μm.179 Penetration depths of QDs inside the organotypic brain slices and measured molecular dynamics of surface molecules are shown in Fig. 6. Dong and coworkers used a combined approach of fluorescence correlation spectroscopy and confocal imaging to investigate the binding of differently sized N-acetyl-L-cysteine capped QDs within live cells. With a single-molecule analysis procedure they were able to demonstrate that the binding efficiency and the targeted membrane sites in A-427 lung cancer model system were dependent on the QD size.180 QD-based SPT can also be used to increase the sensitivity of a biosensing platform as shown by Liu et al. They demonstrated a fluorescent colocalization assay based on single-molecule recognition for the detection of a single thrombin protein at a solid-phase surface using TIRF. Without target amplification or probe separation they obtained a limit of detection (LOD) of 0.8 pM.181
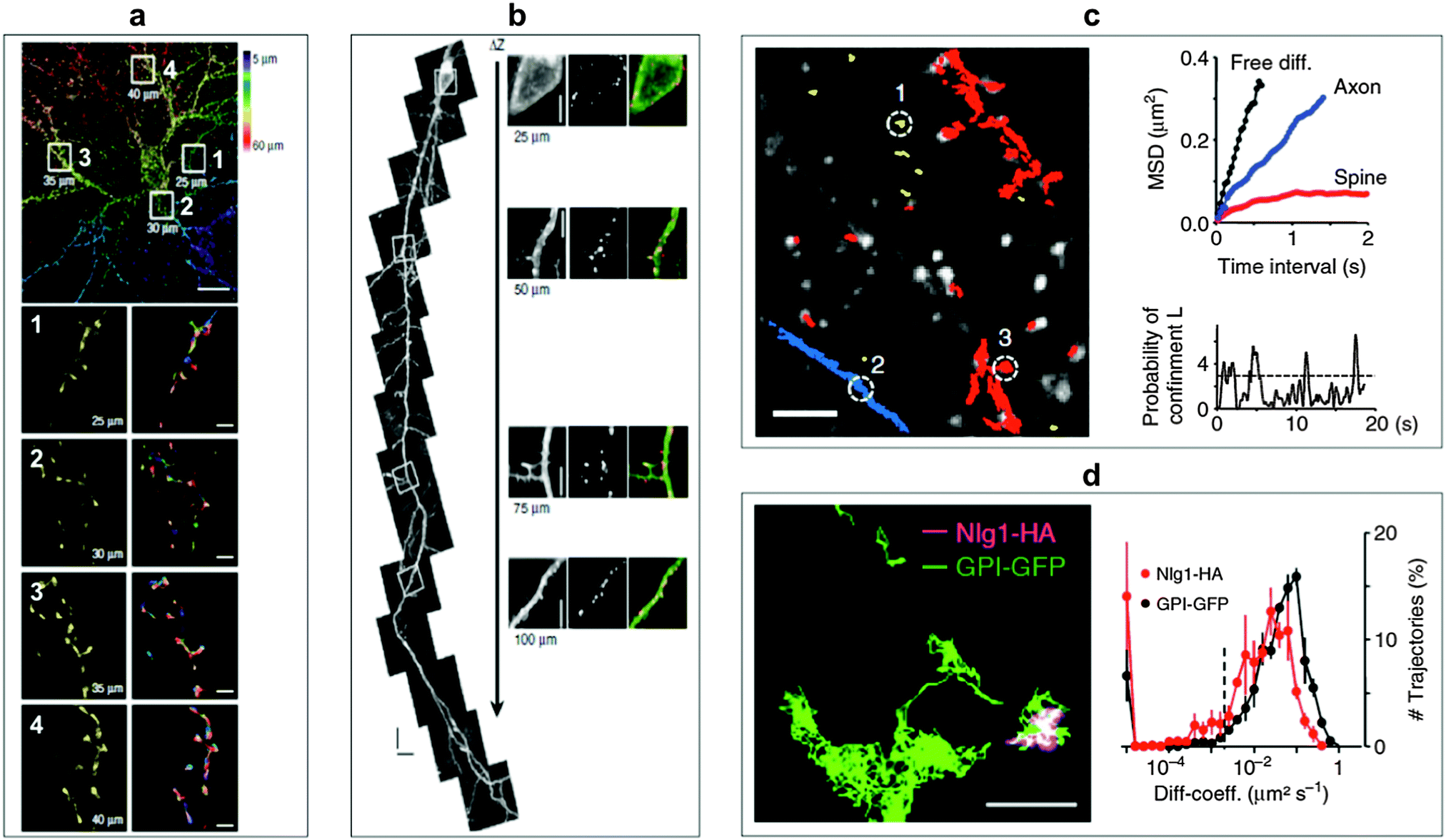 | ||
| Fig. 6 SPT for the monitoring of molecular surface dynamics of membrane-associated molecules in organotypic brain slices using QD–antibody conjugates. (a) Single-photon excitation allowed to extract different QD-trajectories (plotted in different colours in the bottom right images) on dendrites of a green fluorescent protein (GFP)-transfected neuron at penetration depths of up to 60 μm. Top scale bar, 10 μm; bottom scale bars, 1 μm. (b) Two-photon excitation permitted even deeper penetration of up to 150 μm. The right images show specific labelling with anti-GFP QDs at different depths without a loss of labelling density. Scale bar, 5 μm. (c) Individual QD trajectories (red: bound to dendrites; blue: bound to axons; yellow: extracellular; scale bar 10 μm) could be used to measure the mean square displacement (MSD) of extracellular free diffusion, in axons, and in dendritic spines (top right). The probability of confinement (defined as periods of time where a QD remains longer within a membrane area than predicted by assuming free Brownian diffusion; dotted line indicates level above which particles are considered to be confined) for the spine trajectory could also be investigated (bottom right). (d) Using two differently coloured QD-conjugates (trajectory images on the left, scale bar, 1 μm) against haemagglutinin (HA) and GFP allowed the simultaneous monitoring of molecular dynamics of the two surface molecules glycosylphosphatidylinositol (GPI) and neuroligin1 (Nlg1) and the extraction of distributions of diffusion coefficients (right). Reproduced with permission from ref. 179. Copyright 2014 Nature Publishing group. | ||
Myosin V was one of the first molecular motors to be investigated and it was demonstrated that the tagging of the head domains with two differently emitting QDs produced functional motors and that the position of the two heads during myosin stepping could be localized with a precision of ∼6 nm.183 Courty et al. demonstrated SMT measurements with QD-tagged kinesin-1 molecules to track the motion of intracellular proteins inside HeLa cells for several minutes. Using a recombinant biotinylated kinesin-1 conjugated to sAv–QD and pinocytotic influx allowed them to evaluate the velocity of the motion estimated to a value of 600 nm s−1. The unbinding–rebinding processes resulted in long directed trajectories, with an apparent processivity of several microns.184 By using QD–anti-phalloidin conjugates to target actin filaments and to image the motion of proteins Yoo and coworkers demonstrated the possibility of using antibodies for SMT.185 Watanabe et al. performed a systematic study of the molecular motors myosin, dynein, and kinesin in living cells. They monitored the movement of endocytosed QDs in vesicles along the microtubules towards the nucleus with a spatial precision of down to 1.9 nm and a temporal resolution of 330 μs.160 The influence of specific motor domains on their cellular routes and the visualization of the rotation of single microtubules by immobilized motor domains were also demonstrated.186,187 Using QD tagged myosin V in living cells internalized via pinocytotic influx revealed that 95% diffuse isotropically within the cytoplasm whereas 5% showed a directed movement, which was proposed to be “walking” on cortical actin filaments with step sizes of 74 nm.188,189 Zhang et al. reported a step size of 36 nm of individual myosin V motors with an accuracy of 2 to 3 nm in all three dimensions. The authors used 9 × 9 matrix excitation, an array detector, and QDs without intrinsic blinking. Their technique allowed an 80-fold increase of the imaging rate.190 Myosin V rotation around its own axis during the movement could be shown by Ohmachi et al., who developed a single-molecule fluorescence polarization technique for 3D orientation measurements. Their method allowed an orientation accuracy of 10° at 33 ms temporal resolution and revealed a myosin V rotation of 90° during each step.191 A single myosin step was characterized by Wang et al. by using a super-resolution approach for the investigation of actin gliding and immobilized myosin.192
4. In vitro applications
An enormous amount of research studies using QDs for in in vitro imaging applications have been published over the last decade and many of those have already been reviewed in focused articles.37,193 Here we try to give a useful and interesting overview about the impact of QDs in different imaging fields and to provide a good starting point for the interested reader to continue their research for accessing more specialized studies. We will first outline different techniques for delivering QDs to different subcellular compartments, followed by the description of some general QD-based in vitro imaging applications and then focus on investigations related to the characterization of the cellular environment, drug and gene delivery for theranostic purposes, and multimodal imaging.4.1 Cell delivery
There is no general strategy of QD cellular delivery and many different concepts have been proposed to deliver QDs to different places of interest inside various cells. An own comprehensive review could be written about this topic only. Here we limit ourselves to an overview of different concepts and strategies that were used for QD-uptake by cells. Because QDs are nanoparticles made of inorganic core materials the first challenge of using them for cellular imaging is the delivery into the cell. Cellular penetration of QDs can be achieved using different techniques divided in active and passive transportation. The major transport pathway of QDs into cells is endocytosis, which was illustrated by Osaki et al. as a drinking activity of eukaryotic cells, in which they ingest a part of their plasma membrane to swallow external objects.194 Passive transduction of QDs into the cell is mostly enabled via electrostatic interaction with the plasma membrane. For water-soluble QDs without functionalization with specific biomolecules different studies have shown the importance of the QD physical properties, such as type of surface groups, charge, and size, which determine uptake, transportation pathway, and accumulation inside the cell.195–198 Apart from the QD properties also cell type and cell incubation media and temperature can influence the uptake.199–201 In most cases amine surface groups, cationic charges, small sizes, normal cell culture incubation media, and human body temperatures are advantageous for cellular uptake. However, there is no general strategy of guaranteed efficient uptake and the experimental conditions need to be adapted to the cell type, the subcellular compartment of interest, and the imaging application itself.In one example for the importance of surface charge Lee et al. investigated the influence of the charge of small-sized QDs (hydrodynamic radii of 7 to 10 nm) on their ability to penetrate the nucleus of a cell. They observed successful staining of the nucleus of fixed and live HeLa cells only with positively charged QDs, whereas the negatively charged QDs remained in the cytoplasm. The authors exploited this charge-mediated placement of QDs to specific subcellular regions for two-colour QD imaging.202 Other possibilities to enhance cellular uptake are the encapsulation of QDs with saccharides,194 in virus like particles,203 in nanogels,204 or in carriers based on lipid bilayer vesicles.205–209
One of the main problems of passive QD transportation is entrapping inside the endocytic pathway and a minimal release into the cytoplasm. However, in order to allow bioactive QD-conjugates to target cell organelles, it is essential to release the QDs from endosomes or lysosomes. One approach is endosome disruption by using osmotic pressure.210,211 Bayles et al. used for the cytosolic delivery of QDs a cationic core–shell polymer, to whose surface anionic streptavidin–QDs were adsorbed via non-specific electrostatic interaction. The polymer colloids contained a pH-buffered proton sponge, which was able to increase its volume up to 50-fold at pH values below 6, due to the protonation of the tertiary amine groups. The volume increase but also the dramatic change of the zeta potential from +7 mV (pH 7.4) to +45 mV (pH 5.5) compromised the membrane integrity of late endosomes and led to their disruption.212 Kim et al. also exploited the local vesicular membrane destabilization for the escape of QDs into the cytosol. They used biodegradable polymeric nanospheres, which allowed endosomal to cytosolic translocation via pH-dependent reversal of the nanocomposite surface charge polarity. Once in the cytosol the polymer hydrolysed and released the QD-conjugates.213 In an effort to use the pH-responsive ability of polymers to enable target-specific delivery of QDs to tumour cells (solid tumours are known to have an acidic environment) Mok et al. modified the QD surface with grafted copolymers that exhibited charge reversal under acidic condition. They used poly(L-lysine) (PLL) whose backbones were post-modified using citraconic anhydride, which is a pH sensitive primary amine blocker. In acidic environment the initially negatively charged QDs cleaved off the citraconylated amide linkages, which resulted in positively charged QDs with enhanced passive cell delivery.214
Active transportation is characterized by ligand–receptor-mediated transportation using ligands, such as transferrin (Tf)215 or antibodies.66,216–219 A very popular approach is the use of cell-penetrating peptides (CPPs).220 The peptide sequences, generally referred to as protein transduction domains, often include short segments from the human immunodeficiency virus 1 transcriptional activator Tat protein (HIV-1 Tat) and homopolymers of arginine.221–227 Ruan et al. and Chen and coworkers investigated the mechanisms of Tat-peptide-mediated delivery. Their results demonstrated a delivery via macropinocytosis, a fluid-phase endocytosis process triggered by Tat–QD binding to the negatively charged cell membranes. The Tat–QDs were tethered to the inner vesicle surface and then trapped in cytoplasmic organelles. After transportation along the microtubule tracks, they ended up in the microtubule organization centre.228 Surprisingly, FITC–Tat conjugates could internalize into the cell via clathrin-dependent and caveolae-dependent endocytosis and lipid-raft-mediated macropinocytosis. These differences in the uptake (by replacing a QD with a dye on the same type of CPP) showed that both the CPP and the QD play a significant role in the cell delivery mechanism.229 A deeper insight into the transduction kinetics of Tat-peptide-labelled QDs was provided by Suzuki et al., who used confocal microscopy, TIRF, and four-dimensional microscopy. They studied the kinetics of Tat initially and immediately before, at the beginning of, and immediately after entry into living cells. Their results demonstrated an energy dependency of the Tat-mediated translocation and that triggering of heparan sulfate proteoglycans (HSPGs)-mediated events on the cell membrane was necessary. Furthermore, the delivery efficiency was correlated with the number of bound peptides per nanoparticle.230 Medintz et al. utilized CPP-functionalized QDs conjugated with differently sized fluorescent proteins to investigate the influence on endocytosis efficiency, endosomal escape, intracellular stability and intracellular fate. To bypass the endocytic pathway they also tested microinjection, which lead to a more homogeneous distribution of conjugates throughout the cytosol.231 Kim et al. developed thermo-sensitive QDs, whose cellular uptake was controlled via temperature-induced shielding/deshielding of CPPs. Temperature control was established by using poly(N-iso-propylacrylamide) PNIPAAm, which was co-attached with CPP via biotin–streptavidin recognition to the QD surface. Below the lower critical solution temperature (LCST) the PNIPAAm chains sterically hindered the cellular contact of CPPs. Above the LCST, the grafted PNIPAAm chains collapsed and the CPPs could induce cellular uptake.232 Boeneman Gemmill et al. studied different peptide motifs to enlarge the library of CPPs beyond the commonly used HIV-1 Tat-derived motif. Therefore they investigated four different peptides to deliver QDs into the cell. They found that the LAH4 motif, derived from a membrane-inserting antimicrobial peptide, and a chimeric sequence that combines a sweet arrow peptide with a portion originating from the superoxide dismutase enzyme, provided effective cellular delivery of QDs. They recognized that a small change within the peptide sequence can have a strong influence and can lead to inefficient cellular uptake. In comparison to the Tat-motif the new peptides have in common a strong positive charge at the N-termini, which leads to a “halo” of positive charges favourable for the interaction with the cell and for an efficient cellular uptake.233 In an additional study they used the structure–activity relationship analysis for the investigation of key elements within the peptide sequence of different CPPs. They were able to identify the responsible regions for efficient endocytosis and delivery of QDs and other material inside the cytosol of different cell lines.234 Delehanty et al. exploited peptide–endocytosis, peptide–membrane interaction, polymer-based transfection, and microinjection in live cells over a four-day staining period demonstrating simultaneous five-colour fluorescence imaging of a cell using QDs.235 This long-term staining procedure nicely illustrated the versatility and stability of QDs under different cellular delivery conditions. To enable the targeting of QDs to cell components inside the cytoplasm it is important to release the entrapped QD–CPPs conjugates from the endosomes or lysosomes. One option is photo-induced release as demonstrated by Yaghini et al. They used Tat–QDs conjugated to disulfonated aluminium phthalocyanine, which is an amphiphilic photosensitizer known to localize at phospholipid membranes. After the CPP-mediated internalization into the cell, the excitation of the photosensitizer via FRET with the QD as donor was leading to the photo-induced rupture of the endo/lysosomal membrane due to the creation of singlet oxygen. Successful release and efficient FRET was measured with steady-state confocal imaging and fluorescence lifetime imaging (FLIM).236 Liu et al. could circumvent the problem of QD–CPP conjugates entrapped in endosomes or lysosomes by using histidine- and arginine-rich peptides (HR9 peptides). Inhibition of typical energy-dependent endocytic pathways by CytD (an F-actin polymerization inhibitor) did not alter the transduction of the QD-conjugates and therefore the authors suggested a direct delivery through the cell membrane to be responsible for QD nanoparticle (NP) uptake.237 Another peptide used for cellular delivery is an insect neuropeptide, namely allatostin 1, from Drosophila.238,239
In contrast to the frequent use of polyarginine-based conjugates, Chakraborty et al. utilized cholera toxin B as QD delivery agent. The advantage of this toxin is the direct delivery of QDs inside the cytoplasm in small vesicles, which circumvents the endocytic pathway. Intracellular QDs could be imaged even after several days.240 Based on this investigation, Tekle et al. compared the plant toxin ricin and bacterial toxin Shiga with the ligand Tf for receptor-mediated uptake into cells. All three components led to cellular uptake but also tended to be retained in the endocytic structure without efficient exocytosis or route to the Golgi apparatus.241 This passive retention of QDs inside the cell can be advantageous for imaging applications but can also increase the risk of severe health problems caused by QD toxicity. Wu et al. developed a route for the active retention of QDs inside the cell. Their concept was based on intracellular recycling of ligand–receptor complexes. QDs were conjugated with transferrin (QD–Tf) targeting the transferrin receptor (TfR) on the cell surface. In the presence of two Fe3+ ions the QD–Tf was binding to the TfR and was internalized into early endosomes via clathrin-mediated endocytosis. Acidification inside the early endosomes led to the release of the Fe3+ and the QD–Tf returned to the cell surface, where the Tf–TfR complex dissociated upon exposure to the neutral pH. The binding of the Fe3+ ions to Tf was established by the binding to four amino sites and two carbonate synergistic anions. Transformation of the latter ones to oxalate strengthened the binding to iron ions and thus led to a slower release inside the endosomes. The adjustment of carbonate-/oxalate-Tf ratio was used to control the intracellular Tf retention time.242 The differences in the Tf cycle and imaging as well as flow cytometry results of different ligand-dependent retention times are presented in Fig. 7.
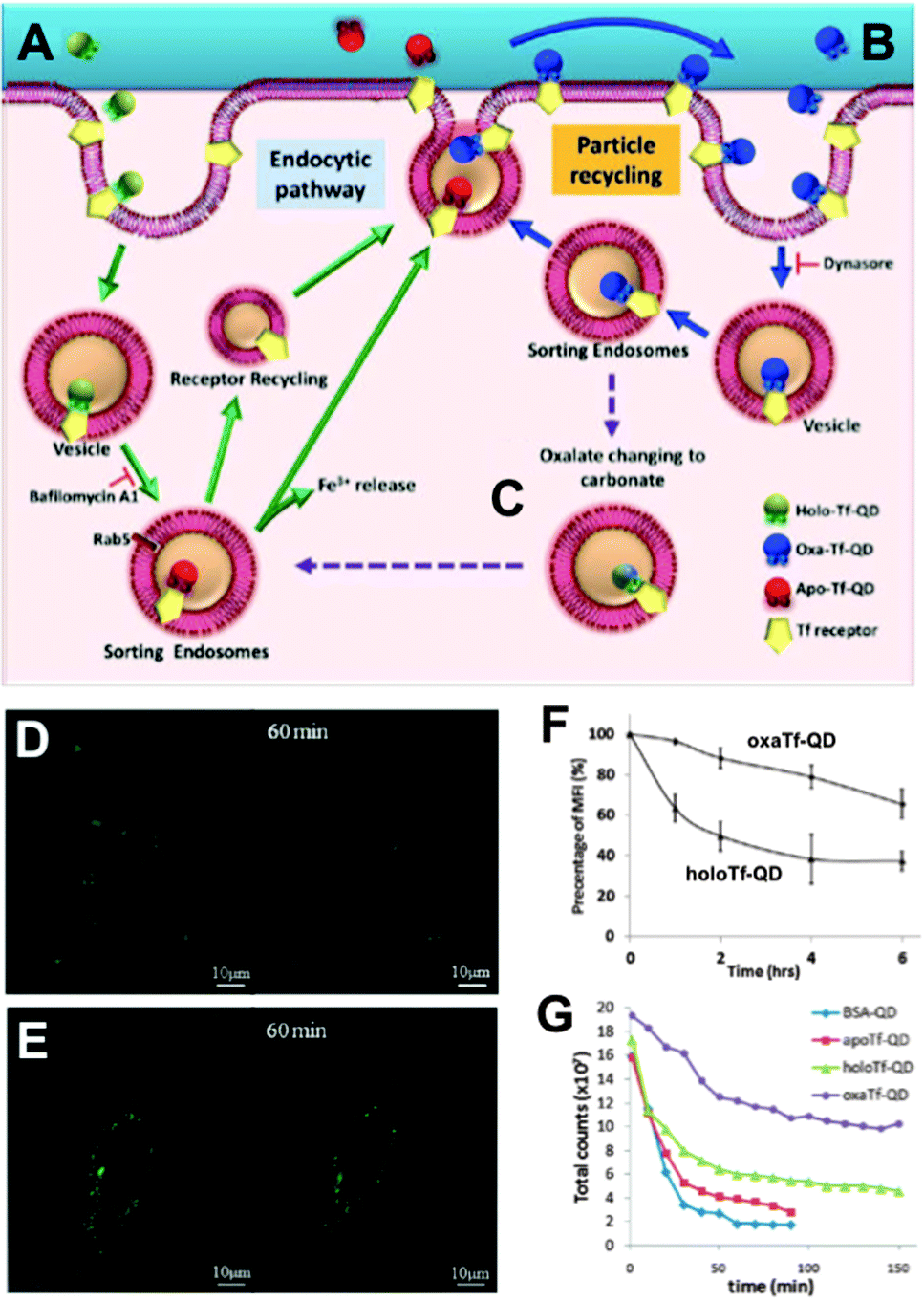 | ||
| Fig. 7 QD-conjugates for increased intracellular retention times. (A) Holo-Tf–QDs bind to TfR for cell internalization and transform into Apo-Tf–QD after pH-triggered Fe3+ release in endosomes. Apo-Tf–QD is removed from the cell and TfR is recycled. (B) Oxa-Tf–QDs bound to TfR recycle from the endosome to the cell surface and undergo reuptake. This recycling continues until the oxalate is replaced by carbonate (C), which leads to cellular removal as explained in A. Confocal images of HeLa cells incubated with Holo-Tf–QD (D) or Oxa-Tf–QD (E) and then washed and incubated with free Tf for TfR-binding competition illustrate the extended cellular retention of the Tf–QD conjugates, which is more efficient for Oxa-Tf–QDs as demonstrated by flow cytometry (F). Longer tumour retention times of Oxa-Tf–QDs compared to Holo-Tf, Apo-Tf, or BSA–QD conjugates were demonstrated in A549 xenograft tumours in mice (graph G shows the fluorescence intensity imaged at different time points after tumour injection). Reproduced with permission from ref. 242. Copyright 2013 American Chemical Society. | ||
Mechanical delivery techniques, such as electroporation and microinjection, can suffer from increased cell death and aggregation of QDs in the cytosol or a low throughput.243–245 Yum et al. presented a nanoscale mechanochemical method to deliver QDs into living cells using a membrane penetrating nanoneedle. This nanoneedle consisted of a chemically synthesized boron nitride nanotube, which was coated with a thin layer of Au. The Au layer facilitated the use of surface chemistry for attaching the QDs via disulfide bonds. Inside the cytoplasm these bonds were reduced into thiol-groups and led to the release of the QDs, which could also be delivered into the nucleus.246 Park and coworkers developed a novel platform for intracellular delivery of genetic material and nanoparticles, based on vertically aligned carbon nanosyringe arrays. Cargos like QDs could be loaded into hollow tubular compartments and simultaneously delivered to several cells without the need of external forces. The authors demonstrated this concept by QD delivery to the cytoplasm of cancer cells and stem cells.247
4.2 General applications
Successfully delivered QDs can be very useful fluorophores for investigating cellular structures and functions. One example is the measurement of cell motility by phagokinetic uptake of QDs and to correlate the migration of the cells to their metastatic potential.248 Also of great interest is stem cell tracking to investigate their fate after differentiation and the behaviour of their daughter cells.249,250 QDs bound to specific targets can be used to gain insight into endocytosis, distribution and shuttling of receptors on the membrane, or the nanoparticles themselves.251–253Many applications can avoid cellular delivery of QDs because several pathogen receptors are expressed on the outer membrane of cells. Those receptors can be targeted using ligands,254–256 aptamers,257,258 or antibodies.259–261 Because primary antibodies are relatively expensive compared to secondaries, most fluorescence imaging methods rely on a combination of primaries for receptor binding and fluorophore-conjugated secondaries (against the primaries) for fluorescence staining. Although very practical, this antibody combination causes larger sizes and lower specificity compared to direct staining with fluorophore-conjugated primaries, which can lead to limitations for nanometric distance measurements (e.g., with FRET) or multiplexing (where high specificity for multiple targets is required). To overcome the size limitation but still provide facile conjugation Howarth et al. used Escherichia coli biotin ligase for site-specific biotinylation of acceptor peptides that were genetically encoded at EGFR on HeLa cells and at AMPA receptors on neuron cells. This specific biotinylation allowed conjugation of QDs via biotin–streptavidin recognition instead of using the antibody approach.262 Chen et al. extended the application of biotin ligase using two orthogonal biotin ligases to enable duplex measurements with two QD colours. This allowed to simultaneously studying the trafficking and localization of two different cellular proteins for yeast-based applications.263 Le Gac et al. presented a study, in which they conjugated QDs with Annexin V to differentiate apoptotic cells due to the binding to phosphatidylserine (PS) moieties present on the outer membrane of apoptotic cells.264 Huang et al. developed QD-based quantification methodologies facilitating the analysis of subcellular distributions of multiple biomarkers, EGFR and E-cadherin (E-cad), which can help to predict the sensitivity to EGFR-targeted therapy.265 Zhang et al. presented a switchable probe to target folate receptors overexpressed on the membranes of different cancer cells. They used polyethyleneimine-coated CdS/ZnS QDs, whose fluorescence was turned off by adsorption of the folate receptor ligand folic acid. After ligand–receptor binding on the cell membrane the ligand was released from the QDs and the emission was switched on. This method aimed at improving imaging of folic receptors due to the avoidance of false/positive results by using the “off/on” switch principle.266 Another possibility to label QDs to cells is the genetic manipulation of proteins to express a histidine tag, which can be targeted, e.g., by Ni-nitrilotriacetic acid (Ni-NTA)-modified QDs165,267,268 or carboxylated QDs.269 Furthermore, the use of the HaloTag protein was demonstrated by So et al. to mediate specific labelling of living cells.270 Sunbul et al. developed an efficient method for one-step covalent labelling of cell surface proteins with QDs based on enzyme-catalysed site-specific modification of short peptide tags.271 In another example of colour-multiplexing with QDs Orndorff and coworkers used two different neurotoxins and conjugated them to differently coloured QDs, which bound to two targets on glioma cells.272 Kang et al. used three QD colours for the development of a multiplexed cellular imaging system, which was capable of targeting three different molecular markers in a single cell.273 In another study of multiplexed imaging with QDs multiple cancer markers were visualized using small molecules such as aptamers or peptides.274 Zrazhevskiy et al. combined the specificity of antibodies with the multiplexing capability of five different QDs for single-cell molecular profiling (Fig. 8).275,276 This allowed them to simultaneously stain Ki-67, HSP90, Lamin A, Cox-4, and b-tubulin in HeLa cells with QD–antibody probes emitting at 525, 545, 565, 585 and 605 nm, respectively.
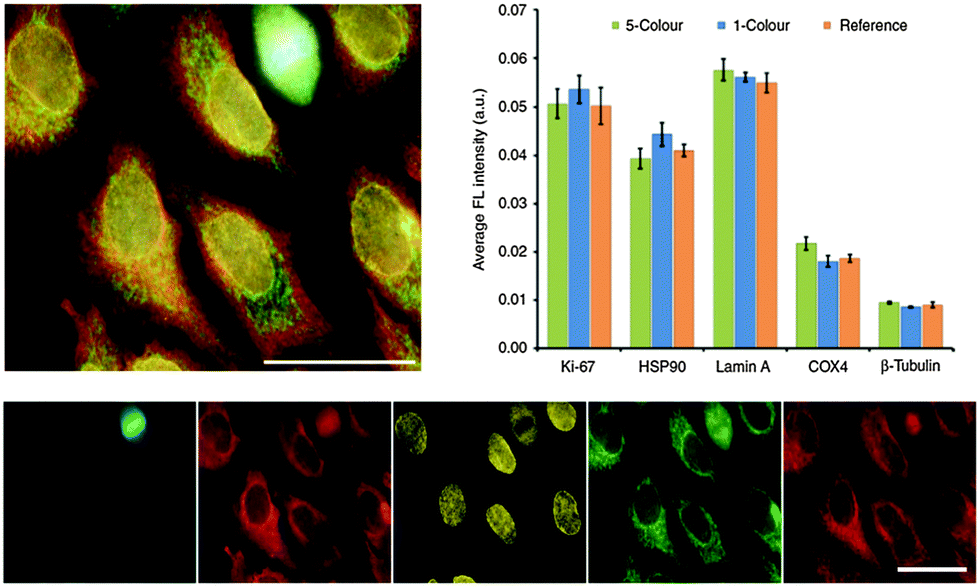 | ||
| Fig. 8 Five-fold multiplexed staining of Ki-67, HSP90, Lamin A, Cox-4, and b-tubulin in HeLa cells with QD–antibody conjugates (top left). Hyperspectral imaging and spectral unmixing were used to separate the individual QD colours (bottom). Comparison of fluorescence intensities from the 5-color multiplexed experiment with the ones from separate experiments with each individual colour (1-colour) and only a single colour (reference) demonstrated the reliability of the multiplexed method for detecting relative expression levels (top right). Reproduced with permission from ref. 275. Copyright 2013 Macmillan Publishers Ltd. | ||
4.3 Cell environment
Internalized QDs can be used to discover factors for cell integrity and structure or metabolic processes and related consequences in a subcellular manner. One example is the study of integrin dynamics of human bone marrow derived progenitor cells (BMPC) during differentiation. Chen et al. used laser optical tweezers to show that the cytoskeleton is weakly associated with its cell membrane by measuring the integrin diffusion on the surface of BMPC at successive stages of osteogenic differentiation.277 Helmick et al. investigated the spatial orientation of actin filaments during cell motility. The ability to monitor the spatial and temporal organization of such biopolymers within a cell is essential to enable an understanding of the complexity and dynamics existing in biological processes.278QD-based imaging nanosensors have been developed for the measurement of oxygen,279 the superoxide radical,280 chloride ion,281 calcium,282 and proteolytic activity283 within the cell. QDs and QD conjugates that showed temperature and pH dependent PL intensities and lifetimes were used as intracellular nanothermometers and pH-meters.284–287 Yang et al. developed a QD-based nanothermometer to characterize the response inside single living cells upon external chemical and physical stimuli.288 Haro-Gonzalez et al. studied laser-induced thermal effects with optical traps containing single trapped microspheres.289 The application of QD–dopamine bioconjugates as an intracellular pH sensor was demonstrated by Medintz and coworkers, who exploited PL quenching resulting from pH-dependent electron-transfer from QDs to oxidized dopamine–quinone.290 Gui et al. used a polydisperse QD solution, which showed a pH-dependent electrostatically tuned aggregation and disaggregation process with a simultaneous PL colour change.291 The combination of QDs and pH sensitive dyes in FRET was exploited by Snee et al. The authors observed a ratiometric response to pH owing to the modulation of the FRET efficiency. This approach can be a versatile method for chemical and biological sensing.292 Dennis et al. developed a QD-based sensor, comprising photostable QDs and pH-sensitive fluorescent proteins, which dramatically improved the sensitivity and photostability compared to common pH sensitive dyes. This FRET-based probe could be tailored by genetic engineering for different pH ranges.293
Charge transfer between dopamine and QDs is not limited to the measurement of intracellular pH. Khatchadourian et al. used QD–dopamine as a tool for indicating QD uptake, breakdown, and processing in living cells. Blinking was used to detect single QDs and the effects of dopamine on the blinking behaviour were investigated under different biochemical conditions.294 Clarke and coworkers presented a study, in which they observed a redox-sensitive pattern of cellular staining with QD–dopamine conjugates. Under reducing conditions fluorescence was only seen in the cell periphery and lysosomes. Increasing oxygen concentrations led to QD PL in the perinuclear region including mitochondria. Under full oxidization QD labelling was detected throughout the complete cell.295
QD-to-dye FRET can be a very useful technique to distinguish normal cells from cancer cells296 or to detect intracellular metabolic processes.297 Li et al. developed such a FRET biosensor for measuring the proteolytic activity of matrix metalloproteinase-2, which is heavily secreted by malignant tumour cells. The selective cleavage of the peptide resulted in the recovery of fluorescence from QDs. They developed a 535 nm emitting QD donor-based FRET probes (using Rhodamine B as acceptor) for in vitro and a 720 nm emitting QD donor-based FRET sensor (using an NIR emitting dye as acceptor) for in vivo use.298 Hu et al. developed a mercury biosensor based on FRET from N-acetyl-L-cysteine stabilized QDs to a Rhodamine 6G derivative–mercury conjugate. They could show intracellular imaging of mercury.299 QD/Au NP assemblies were used as fluorogenic substrate for BACE1 enzymatic assays by Choi and coworkers. BACE1 is a key enzyme for the production of amyloid-beta peptides, which are known to be related to Alzheimer's disease (AD). Classical FRET approaches can suffer from a low solubility of the peptide substrate and low SNR for the use in cellular BACE assays. The QD–Au system could overcome such problems and its relatively small size (∼12 to 15 nm) allowed efficient energy transfer from QDs to Au NPs. The latter are known as universal quenchers for diverse fluorophores due to long-range nanosurface energy transfer (NSET). NSET led to quenching of the luminescence of QDs that were assembled to the Au NPs via Ni–histidine interaction. Digestion of the BACE1 substrate cleaved the QDs off the Au NPs. This restored the QD PL and allowed the measurement of enzyme activity in vitro and in living cells.300
Wei et al. presented a DNA-templated heterobivalent QD FRET nanosensor. This probe exhibited dual motifs for extracellular and intracellular targeting and imaging of cancer cells. The first part of the DNA-based sensor (nucleolin-targeting motif – NTM) facilitated targeting of cancer cells and simultaneously stimulated macropinocytosis, which allowed the escape into the cytosol. Inside the cytosol the second part of the oligonucleotide (mRNA-targeting motif – MTM) targeted specific messenger RNA (mRNA), which was shown by switching off the FRET signal of a Cy5 dye.301 Probe preparation and the intracellular pathway together with fluorescence images demonstrating the specificity and versatility of the probe are shown in Fig. 9. This combination of specific targeting and cytosolic release of bioactive probes to measure tumour-associated mRNA is a promising tool for future imaging applications.
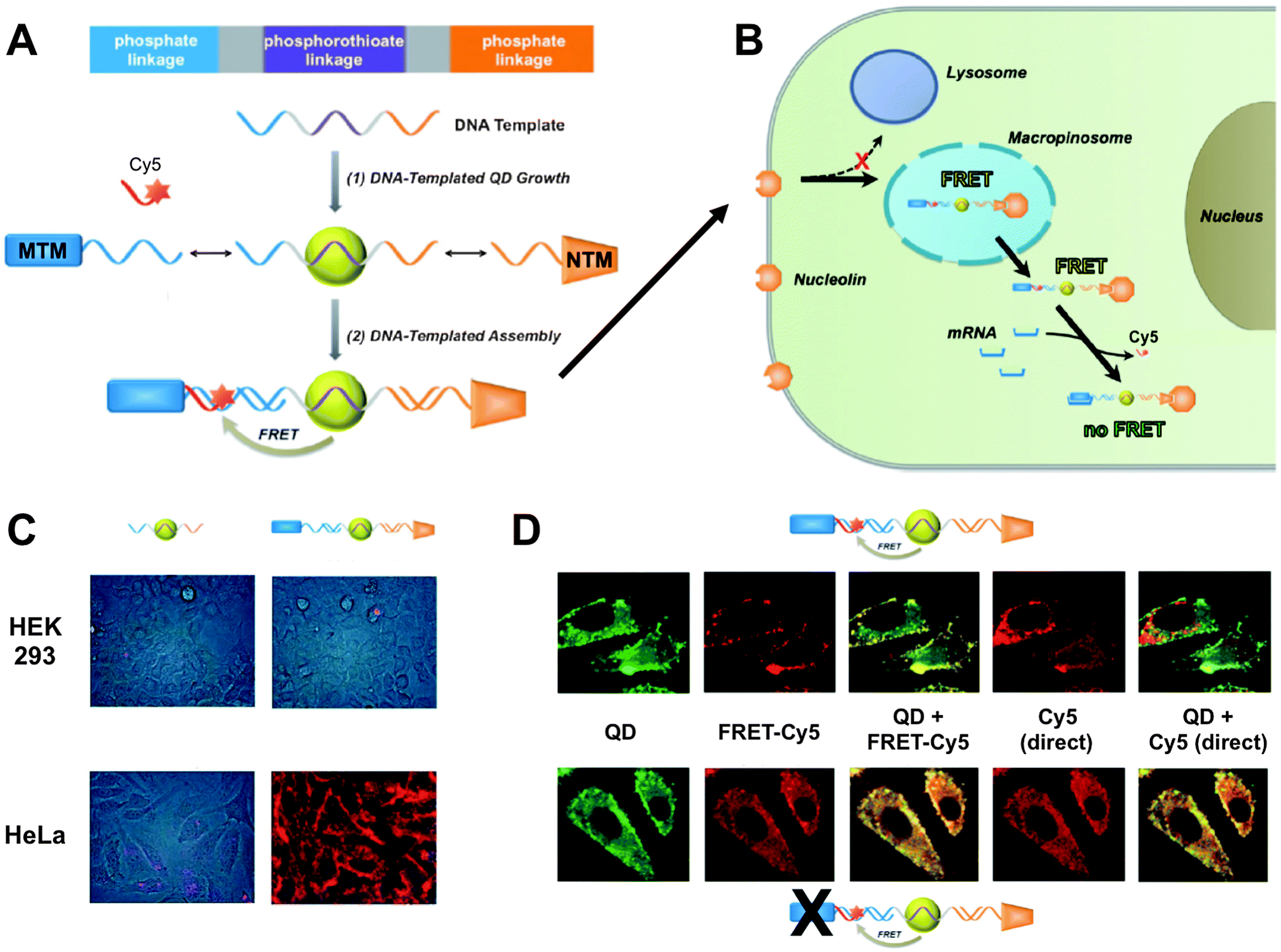 | ||
| Fig. 9 (A) DNA-templated heterobivalent QD-to-Cy5 FRET nanoprobe, which consists of a central QD with attached mRNA-targeting motif (MTM) and nucleolin-targeting motif (NTM). mRNA binding replaces the hybridized Cy5 dye and can therefore “switch-off” QD-to-Cy5 FRET. (B) Intracellular delivery route. NTM triggers cell targeting and macropinocytosis, whereas MTM targets intracellular mRNA, which is probed by reduced QD-to-Cy5 FRET. (C) Brightfield and QD fluorescence overlay images after 1 h nanoprobe incubation at 37 °C. In both HEK 293 and HeLa cells lacking targeting motifs led to only very faint QD background fluorescence and no cellular targeting (left images) whereas the complete MTM-QD-NTM probe led to cellular targeting (right images). (D) mRNA targeting in HeLa cells. After 4 h of incubation both the complete nanoprobes (top images) and MTM-deactivated nanoprobes (bottom images) have entered the cells. Only the complete nanoprobe can target mRNA, which leads to replacement of Cy5 and reduced FRET inside the cell. This can be seen in the QD (reduced FRET – green colour) + FRET-Cy5 (QD-sensitized Cy5 PL – yellow colour) overlay images, in which the complete probe contains Cy5 only on the cell surface, whereas the MTM-deactivated probe contains Cy5 also inside the cell (centre images). Overlay of QD and Cy5 PL from direct excitation of the two fluorophores shows a significant replacement of Cy5 by mRNA only in the case of the complete probes (non-colocalized QD and Cy5 PL – red colour in the right images). Reproduced with permission from ref. 301. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
4.4 Drug delivery
Although the relatively large sizes of QDs (compared to small molecules) may interfere with an efficient recognition of and delivery into cells, their physical and chemical stability and nanoparticle character can afford long systemic circulation times and possibly an enhanced permeability and retention (EPR) effect.302 Moreover, the large surfaces of QDs offer an ideal “playground” to attach several similar or different functionalities. This physical property can be exploited to combine diagnostics and therapy to so-called theranostics. For the treatment of diseases like cancer, inflammation, and pulmonary diseases it is of great interest to monitor the pathway, uptake inside targeted cells or organs, and the therapeutic efficiency of the applied drugs. In particular, for cancer treatment persists the problem of a potential development of multidrug resistance, due to overdosed anticancer-drugs. One of the first applications of QDs inside a drug delivery system was demonstrated by Lai et al., who used QDs to cap the mesoporous channels of a silica nanosphere with entrapped drugs. Only by reduction of the chemically labile disulfide linkage between nanosphere and the QDs the drug could be released.303 A relative young research field is the use of QDs inside photodynamic therapy, in which for example a non-covalently bound combination of QDs and chlorin e(6), a second generation photosensitizer, could be used for light induced damage to cancer cells.304 Other therapeutic agents are chimeric single chain antibodies (scFv-Fc), which can act as target biomolecules for diagnostics but also serve for treatment. One of them is the antibody against insulin-like growth factor receptor (IGF1R). This antibody was conjugated to QDs and used for the investigation of IGF1R, which is involved in the proliferation, survival, and metastasis of breast cancer. With a simultaneous targeting and down-regulation of IGF1R activity after endocytosis in the cell, such scFv-Fc antibodies have a great potential as theranostic tools.305A more common application is the combination of QDs with anti-cancer drugs and in some cases QDs can provide a synergistic effect for the uptake of drugs, even for cells that show drug resistance.306,307 Bagalkot et al. used the large surface area of QDs for the conjugation of an aptamer that targeted the extracellular domain of the prostate specific membrane antigen. The anticancer drug doxorubicin (Dox) was intercalated within the aptamer and thus in close proximity to the QD, which enabled FRET and quenched the PL of the QD. Successful binding of the aptamer to the cell membrane released Dox and re-activated the QD PL.308 The preparation of such nanoparticles with dual functionality of tracking and therapeutic aspect often includes a multistep procedure. A mild aqueous ‘one pot’ method to prepare a QD–Dox conjugate wrapped with a PEG shell was presented by Chen et al. The condensation reaction of glutathione (GSH)–QD with Dox established the drug carrier whereas the shell stabilized the NP and prevented a fast clearance and drug release during systemic circulation. To increase the specificity folic acid was used as ligand to target the folic acid receptor, which was demonstrated in vitro.309
In a recent study the advantageous properties of graphene (e.g., large surface area, low toxicity, and good stability) for improved delivery of Dox were combined with QDs as fluorescent reporters to monitor the drug delivery. A silica coating around the QDs suppressed potential toxicity and fluorescence quenching by graphene. One important advantage of using a graphene sheet was the large drug loading capacity of 1.4 mg mg−1. A simple electrostatic layer-by-layer approach allowed facile preparation and for specific targeting a transferrin ligand (Trf) was attached to the surface of the graphene sheet. Delivery of the nanoassembly inside the cell could be monitored using the QDs. Because Dox fluorescence was efficiently quenched by the graphene sheet, delivery of Dox in the nucleus (release from graphene) could be probed by restored Dox fluorescence (Fig. 10).310 The advantageous synergistic effects of QDs and carbon nanotubes for medical diagnostics and treatment have been reviewed by Madani et al.311
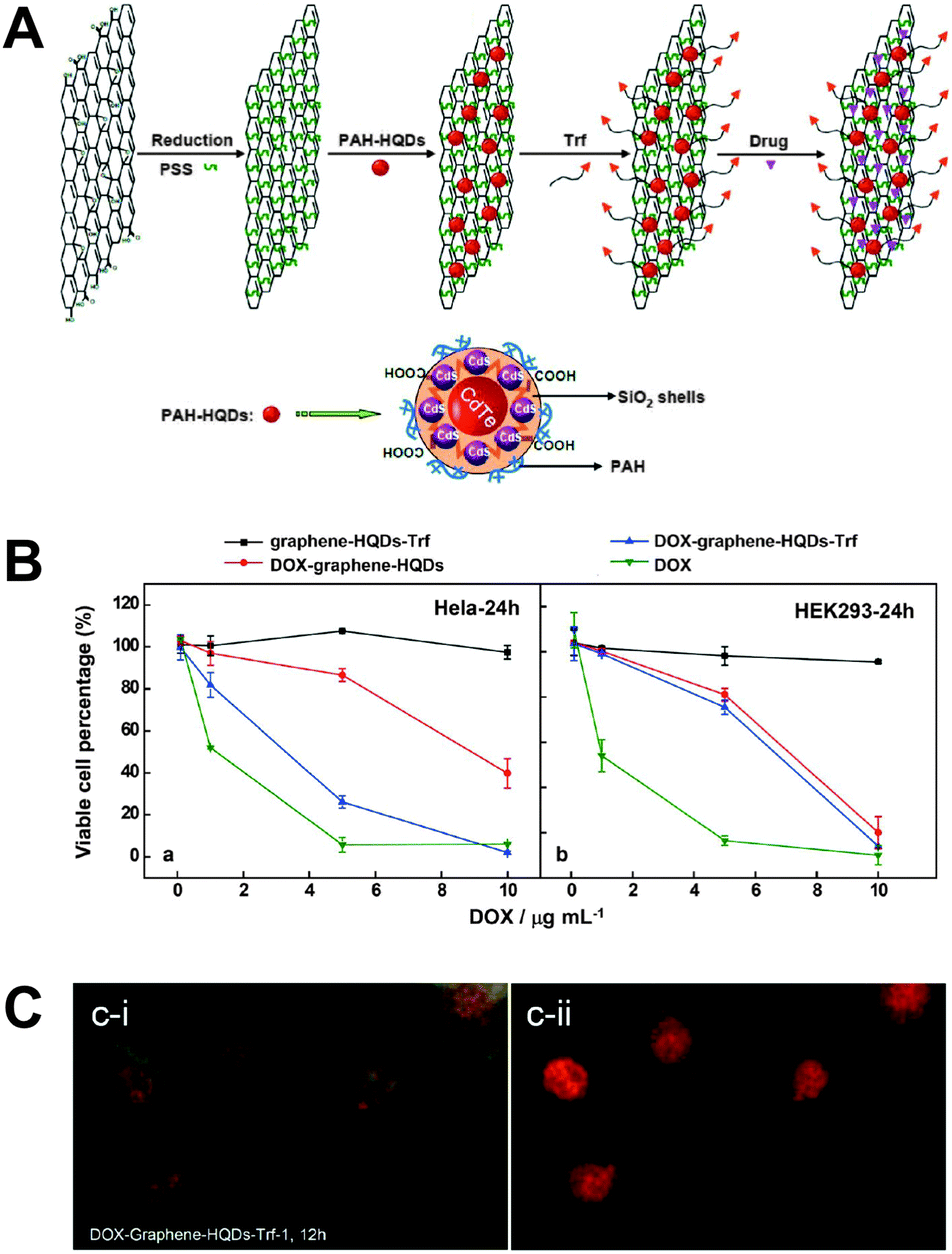 | ||
| Fig. 10 (A) Different preparation steps (electrostatic layer-by-layer approach) for a theranostic nanoassembly consisting of a graphene sheet, QDs, transferrin ligands (Trf), and doxorubicin (Dox). (B) After 24 h of incubation the nanoassembly without Dox does not show any significant cytotoxicity (black curves) for HeLa (left) and HEK293 (right) cells. The presence of Trf allows specific delivery only into HeLa cells (blue curves) whereas pure Dox (green curves) and the nanoassembly without Trf (red curves) do not show any specificity for a certain cell type. (C) Cellular delivery and release of Dox could be imaged by exciting the cells with blue (left) or green (right) light, which led to green QD and orange Dox PL and red Dox PL, respectively. After 24 h of incubation QDs show PL primarily in the cytosol (green PL in the left image), whereas free Dox can be found primarily in the nucleus (red PL in the right image) and partly in the cytosol (orange PL in the left image. Reproduced with permission from ref. 310. Copyright 2013 American Chemical Society. | ||
Liposomes and micelles are among the most frequently applied nanocarriers. Their hydrophilic heads make them water-soluble and biocompatible, whereas the hydrophobic tails can be used for drug delivery via encapsulation of hydrophobic drugs. Gopalakrishnan et al. used the hydrophobicity to incorporate hydrophobic QDs into the bilayer membrane of lipid vesicles. They showed that these vesicles were able to fuse inside the cell but depending on the surface functionalization they could also stain the plasma membrane. The attachment and partial fusion with the cell membrane could be used to release the vesicles' cargo into the cell.312 Another possibility is to use co-encapsulated hydrophobic QDs and Dox in a phospholipid-based polymeric micelle. PEG was used to increase the circulation time and allowed a very efficient passive accumulation in cancer cells, revealing a sustained release of the drug over seven days.313 Sigot et al. investigated the uptake and delivery capabilities of liposomes. Although PEG can prolong circulation times, the drawback is that the PEG coating can also prevent lipid–cell membrane association and the endosomal escape of the loaded cargo. The authors investigated the advantages of either fusogenic or pH-sensitive PEG-lipids inside the formulation of biotinylated lipid particles (BLPs) to promote the endosomal escape via PEG-lipid dissociation. Therefore they used a dual QD colour approach for targeting cancer cells and monitoring the release of the cargo. One type of QD was functionalized via biotin–streptavidin recognition with the epidermal growth factor (EGF), which targets the EGF receptor (EGFR) that is expressed on many cancer cells, whereas a different emitting QD was encapsulated inside the BLP and indicated the successful endosomal escape into the cytosol.314
Passive drug transportation does usually not allow a controlled release of the drug and can therefore lead to a distribution throughout the body and accumulation in undesired areas. Controlled drug release can dramatically increase the efficiency of a treatment. Biodegradable micelles were used for such a controlled delivery strategy but only provided limited application for stimulated drug release.315 A more promising approach used an in situ preparation of CdSe QDs inside dual-responsive (pH and temperature) hydroxypropylcellulose–poly(acryl acid) (HPC–PAA) hybrid nanogels. HPC was responsible for the sequestering and stabilization of the in situ formed QDs and PAA provided a pH responsive volume-phase transition. Using hydrogen bonding of the hybrid nanogel and the drug temozolomide (TMZ) was established at a low pH. With increasing pH the hydrogen bonds broke but this process was slowed down by a diffusion-controlled step provided by the mesh size of the nanogel. This approach avoided a random delivery of the drug. Additionally a change of the pH influenced the QD PL intensity, which could be used to gain information about the intracellular pH. The multifunctional hybrid nanogel offered optical pH sensing, cancer cell imaging, and controlled drug release within a single nanoparticle system.316 A more complex stimulated drug delivery system was proposed by Gui et al., who developed a thermo/pH-sensitive, magnetic/fluorescent, biocompatible, and low-toxic multifunctional microparticle. The magnetic core consisted of electrostatically adsorbed QDs on an iron oxide NP stabilized with thiodiglycolic acid and encapsulated in a silica sphere. A shell of a PNIPAM-g-chitosan copolymer provided temperature and pH regulated drug (adriamycin – ADM) release as a consequence of either an increased temperature or low pH, which led to the shrinking of the copolymer network (Fig. 11). These triggers could be controlled during the preparation of the probe. The authors demonstrated fluorescence imaging of in vitro cultured HepG2 cells and control experiments with free ADM and microparticles without ADM showed that the ADM anticancer drug retained its activity after release and that the microspheres did not show significant cytotoxicity.317
 | ||
| Fig. 11 (A) Schematic illustration of loading and pH and temperature controlled release of adriamycin (ADM) within hybrid microspheres containing magnetic quantum dots (black core with cyan shell) embedded in a copolymer network (magenta and orange parts). (B) Confocal fluorescence images of HepG2 cells after 24 h of incubation with 0.2 mg mL−1 of microspheres demonstrated successful cellular uptake. (C) Increasing temperatures led to microparticle size reduction and increased drug release. (D) Decreasing pH resulted in higher water content in the microparticle shell and a concomitant increase in drug release due to the higher solubility of ADM at lower pH. Reproduced with permission from ref. 317. Copyright 2014 Elsevier B.V. | ||
4.5 Gene delivery
RNA interference (RNAi) is a powerful tool to study gene functions and can be used for manipulating posttranscriptional inhibition of gene expression inside cells. Inhibition of protein translation, so called gene silencing, uses short, double-stranded, small-interfering (si) RNA and can be used as a therapeutic agent. A direct delivery of siRNA to the cell is not very efficient due to the negative charge of the nucleic acids, which prevents transfection to the cell. Nanocarriers that can be divided into viral and non-viral transporters, can be used to increase the transfection efficiency. Viral carriers offer precise and efficient exogenous nucleic acid transfection but they can also cause immune reactions by triggering the antiviral immune system.318 On the other hand non-viral carriers may suffer from low transfection efficiencies. One possibility of hiding the negative charge of nucleic acids is attachment to positively charged liposomes or polymers, which will retain their positive net charge after nucleic acid attachment. QD–liposome and QD–polymer conjugates have been successfully used for the imaging of siRNA transfection.319–321 A possibility to further improve cell transfection of QDs is surface-functionalization with peptides.318,322,323For optimized transfection efficiencies the delivery steps and the fate of siRNA and other nucleic acids need to be monitored. Because protein knockdown by RNAi can be delayed over 48 h after administration, the use of organic dyes or fluorescent proteins, which often suffer from photobleaching and do not allow efficient multiplexing, is limited.322 The unrivalled stability and colour-tuning capability of QDs are very advantageous for RNAi because they allow precise and multiplexed long-term tracking, while simultaneously using the QDs as nanocarriers. In an example of long-term imaging with QDs Ishihama et al. investigated the movement of mRNA within the nucleus. They could track mRNA–QD conjugates inside the nucleus for more than 60 s after microinjection with a temporal resolution of 30 ms.324 In another study Shao et al. studied the interplay and timing for suicide gene therapy, in which tumour cells are transfected with a gene that triggers the production of a specific enzyme that metabolizes an applied drug, which then triggers a cascade leading to tumour cell death. The authors used QDs labelled to plasmid DNA and tracked them at different time-intervals. It could be shown that the conjugates were shuttled to the nucleus after 24 h, which was identified as the perfect time point to administer the drug.325
Another crucial step in nanocarrier-based cell transfection is the dissociation of the nucleic acid from its carrier, also known as unpacking. Studying the dissociation kinetics is therefore highly important for designing efficient nanocarriers. Both a premature dissociation and an overly stable binding would be detrimental to the overall transfection efficiency. The binding of nucleic acids mainly occurs by electrostatic interactions with the carrier surface material or labile covalent binding. Cationic polymers can condense nucleic acids through electrostatic interactions to form polymer–nucleic acid nanocomplexes. Their dissociation could be studied by FRET from QD-donor-labelled nucleic acids to dye-acceptor-conjugated polymers. The dissociation led to reduced FRET, which was used to gain information about the unpacking and distribution of individual nanocomplexes.326 Chen et al. used the QD–FRET pair to compare unpacking of three different cationic polymers for gene transfer, namely condensed DNA in chitosan, polyethylenimine (PEI), and polyarginine (PA). The authors observed that the dissociation kinetics and the type of intracellular compartment where dissociation occurs have a significant impact on transfection efficiencies for the different gene carriers due to potential enzymatic barriers such as nuclease-mediated DNA degradation.327 A follow up study demonstrated simultaneous tracking of DNA dissociation from NPs and subsequent DNA degradation in a non-invasive manner. By using a QD-based FRET relay (consisting of QD, nuclear dye, and Cy5) three distinct states of DNA condensation and integrity could be observed. Inside this FRET relay the QD donor excited the nuclear dye that served as first acceptor and simultaneously as donor for the second acceptor Cy5. The nuclear dye acted as indicator for the DNA integrity, whereas the FRET to Cy5 displayed the dissociation of the DNA from the carrier. Condensed, free, and intact or partially degraded pDNA were identified in a single-particle manner within cells.328
One very promising application of RNAi is chemotherapy. siRNA can sequence-specifically inhibit the expression of targeted oncogenes, minimizing side-effects, such as immune suppression, caused by conventional chemotherapies. Jung et al. developed multifunctional siRNA–QD constructs, which fulfilled the challenging chemotherapeutic demand of target-oriented delivery. They demonstrated a selective and efficient inhibition of the expression of the epidermal growth factor receptor variant III (EGFRvIII) in target human U87 glioblastoma cells by monitoring the down-regulated signalling pathway. For the target of siRNA delivery, they used the arginine–glycine–aspartic acid (RGD) peptide, which is known to bind to the integrins overexpressed in various cancer cells. Additionally they increased the cellular uptake using the HIV-Tat derived peptide.329
Because nanocarriers are mainly endocytosed, the use of endosomal disrupting agents can improve endosomal release and enhance siRNA-mediated knockdown.322 Also QD-based siRNA nanocarriers that use the proton-sponge effect to deliver the carrier into the cytosol have been used. Yezhelyev et al. demonstrated 10- to 20-fold improved gene silencing activity and a 5-fold decrease of cytotoxicity compared to common delivery agents.330 Qi et al. used proton-sponge coated QDs to improve silencing of JAM-2, which belongs to a subfamily of junctional adhesion molecules and can be found around tumours and sites of inflammation. The proton-sponge coating consisted of amphiphol and PEI, which allowed (through electrostatic interaction) the transport of siRNA. By applying siRNA concentrations of 15 nM and 7.5 nM QDs in serum-containing medium the authors achieved a gene silencing efficiency of JAM-2 of more than 90% (Fig. 12).331 In an earlier publication Qi et al. used amphiphol to overcome possible limitations of conventional siRNA delivery agents, such as lipofectamine, for in vivo applications. Amphiphol is a linear polymer with alternating hydrophilic and hydrophobic side chains that are often used for solubilizing integral membrane proteins and delivering them into cell lipid bilayers, without disrupting the integrity of the cell membrane. In combination with NPs coated with hydrophobic surface ligands they can form stable complexes that are capable of carrying siRNA. At the same time they avoid enzymatic degradation, aid endosomal escape, and mediate siRNA delivery into the cytosol.332
 | ||
| Fig. 12 (A) Hydrophobic QDs were coated with the amphipol PMAL for solubility in aqueous media. These QDs were then functionalized (using EDC chemistry) with PEI to add a proton-sponge effect and decorated (via electrostatic interactions) with siRNA for silencing JAM-2. (B) siRNA release and accumulation in the cytoplasm of U251 cells was demonstrated by fluorescence imaging using AlexaFluor 488 (AF488) conjugated siRNA. When attached to the QDs AF488 PL was quenched and only QD PL (red) was visible. After ca. 30 min. siRNA started to be released from the QDs and green PL of AF488 appeared. After ca. 4 h siRNA was distributed over the cytoplasm. (C) Optimization of the concentrations and siRNA/QD ratios led to JAM-2 mRNA silencing efficiencies of more than 90% (red arrow on the right). Reproduced with permission from ref. 331. Copyright 2013 The Royal Society of Chemistry. | ||
4.6 Multimodal imaging in vitro
Similar to a double-functionality of theranostic QD-conjugates for therapy and diagnostics one can add multiple other physical, chemical, or biological functions to a QD for so-called multimodal imaging. This is a very challenging approach because one single nanoparticle may need to provide very different functions simultaneously. Research studies on QD-based multimodal imaging range from pure proofs of concept of double-functionality without any improvement of either function to actual biosensing applications that exploit multiple improved imaging modes or imaging and separation properties combined within one nanoparticle. Probably the most frequently investigated QD-based hybrid-NPs are magnetic-QDs. Various approaches have been used for the preparation of such particles. In one of the first studies Gaponik and coworkers encapsulated iron oxide NPs and QDs in a polymer microcapsule and demonstrated the possibility of the external manipulation of these capsules by a magnetic field.333 In other approaches, semiconductor material was grown on the surface of iron oxide NPs334 or on a cobalt core.335 Selvan et al. published a protocol for the preparation of nanocomposites with superparamagnetism and tuneable optical emission properties.336 In an extensive study Gao et al. investigated the factors, which lead to core–shell nanocrystals or to nanosponges in a one pot synthesis and they observed that altering the sequence of adding the different reagents is the most important factor.337 It was also shown that a too close proximity of QD and magnetic materials can influence the photophysical properties and lead to QD PL quenching.335 Silica-encapsulation of iron oxide NPs and QDs leads to larger particles but preserves the intrinsic properties of both materials. In addition, the silica shell provides a good biocompatibility and a large surface for further functionalization with biomolecules.338,339 A method which led to even higher QYs than those prepared with silica co-encapsulation was the covalent binding of silica-coated Fe3O4 and poly(styrene-co-maleic anhydride)-coated QDs via a ring-opening reaction. The functionalization of the resulting nanocomposite with the PEG derivate Jeffamine M-1000 polyetheramine resulted in a high colloidal stability over a wide pH range (pH 2–13).340 Cho et al. embedded iron oxide NPs in a spherical polystyrene matrix on which QDs were conjugated. They also showed the possibility to use the same system for drug storage. For this purpose the chemotherapeutic agent paclitaxel was loaded on the surface using a layer of biodegradable poly(lactic-co-glycolic acid) (PLGA) and antibodies were used for specific targeting and cancer treatment.341 To create bright probes a high concentration of the imaging agent is very beneficial. Shibu et al. presented a smart solution using photouncaging ligands for site-specific delivery of bimodal nanoparticles. Photoactivation of such ligands resulted in systematic degradation and led to a localized release of QDs and superparamagnetic iron oxide nanoparticles (SPIONs) for fluorescence and magnetic resonance imaging, respectively. The small contrast agents also facilitated removal after the imaging process.342 Another approach is layer-by-layer assembly using differently charged materials to form hybrid structures.343Because magnetic QDs provide advantages for imaging, sensing, and bioseparation they had a large impact on biomedical research. In particular, applications in labelling and sorting of cells demonstrated the benefits of such type of material.344,345 Zhang et al. developed a multi-shell structured multi-functional nanoprobe, composed of Fe3O4–SiO2–QD-SiO2–polydopamine core–triple-shell nanoarchitecture, which displayed high fluorescence intensity and rapid magnetic separation. They demonstrated the separation efficiency by sorting HL-60 cells from K562 cells on a microfluidic chip using a HL-60-specific aptamer (Fig. 13).346
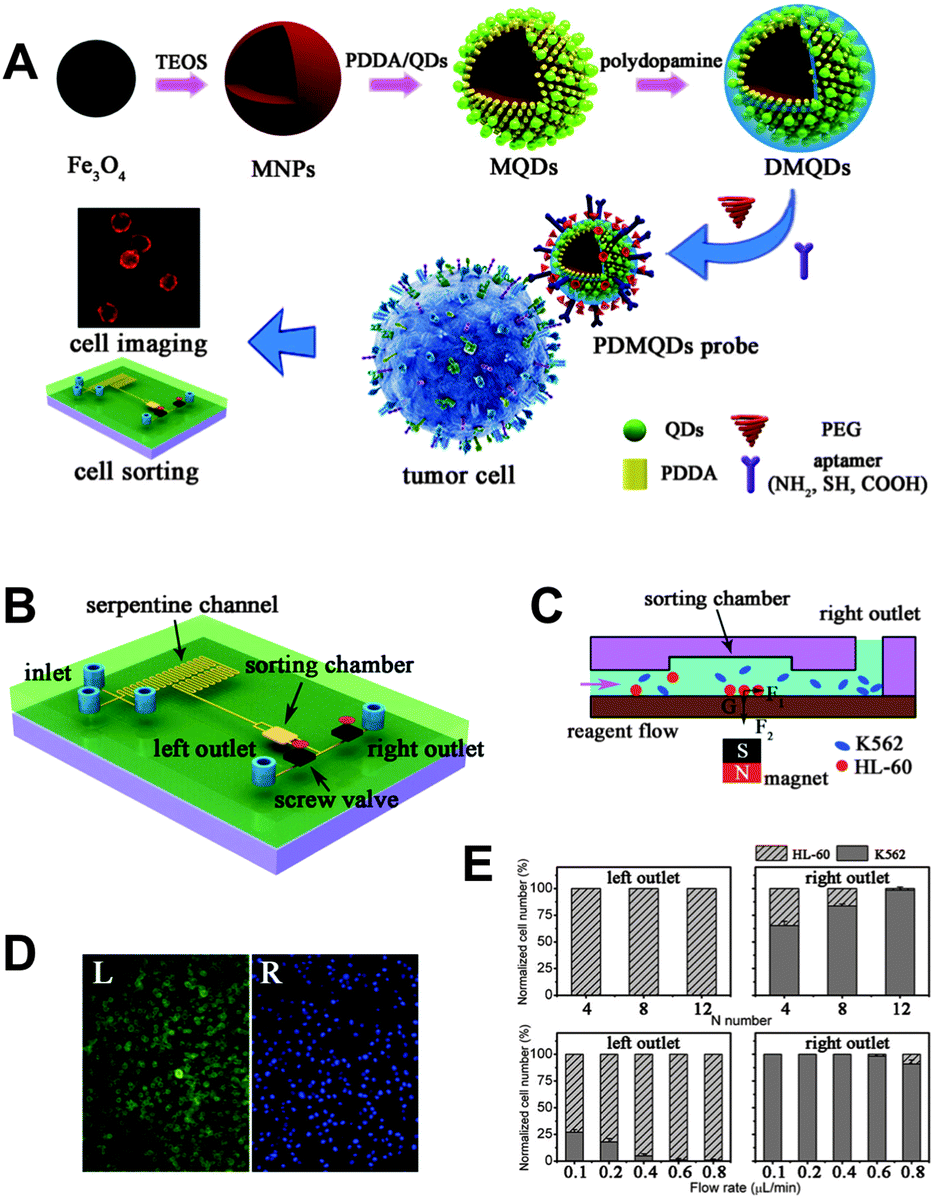 | ||
| Fig. 13 (A) Layer-by-layer assembly was used to produce PEGylated polydopamine coated magnetic QDs (PDMQDs) that could be conjugated with HL-60-specific aptamers for cell recognition to perform magnetic cell sorting and fluorescence imaging. (B) A microchip was used to mix and interact the aptamer-conjugated PDMQDs with HL-60 and K562 cells (serpentine channel). In the sorting chamber (C) a magnet is used to separate the two cell types. Fluorescence images (D) of cells from the two different outlets show the sorting efficiency that can be optimized by the number of repeated serpentine channel (E top) and the flow rate (E bottom). Reproduced with permission from ref. 346. Copyright 2013 American Chemical Society. | ||
Although in vitro measurements are most often an intermediate towards the application of multimodal probes in vivo, it is a crucial step for the acquisition of information concerning probe functionality, cell delivery, and cytotoxicity effects. Multimodal probes that combine fluorescence and magnetic resonance imaging (MRI) belong to the most important dual-imaging agents and SPIONs can be used as both a magnetic separation tool and a MRI contrast agent. Qiu et al. combined QDs, SPIONs, and Au NPs within a single PLGA-based NP. This particle was used to investigate the uptake of neutrophils, which are leukocytes and an integral part of the innate immune system. It is of great interest to understand the spatiotemporal aspects that relate neutrophils to the sites of infection and inflammation. QDs and SPIONs were embedded inside the PLGA core and Au NPs were assembled to the surface. As SPIONs can act as mediators in magnetic hyperthermia and as MRI contrast agents it is desirable to achieve a localized concentration for efficient cancer treatment and enhanced MRI contrast. Highly concentrated Au NPs are advantageous for the production of localized heat for photothermal therapies. Using the hybrid NPs the authors were able to image, track, and manipulate neutrophils.347
More common for MRI measurements is the use of Gd-complexes as contrast enhancers. One approach for the preparation of a bimodal probe was the encapsulation of QDs inside a paramagnetic micelle where Gd-complexes were embedded inside the micellular coating. These particles were functionalized with cyclic RGD to target αvβ3-integrins, which were overexpressed on the surface of both angiogenic endothelial and tumour cells.348 They were also used with annexin A5 to specifically target asymmetrically distributed PS, which is exposed on the cell surface of apoptotic cells. PS is one of the main targets for the investigations of programmed cell death (PCD). Acute myocardical infarction, heart failure, or unstable atherosclerotic plagues lead to increased PCD, whereas cancer results in suppressed PCD. The measurement of apoptotic activity may greatly enhance the capability for rating the status of a disease and can serve as an early indicator for the effect of therapeutic interventions.349 A relatively facile method for bimodal probe preparation was demonstrated by Prinzen et al., who used biotinylated Annexin A5 and biotinylated gadolinium–DTPA to bind to streptavidin-coated QDs for the visualization of cell death and activated blood platelets. Activated platelets also expose PS, which is necessary to trigger the blood coagulation process.350 Other approaches used silica-embedded QDs with paramagnetic molecules inside the silica sphere or immobilized on the surface.351,352 As an alternative of using Gd-complexes as paramagnetic molecules Wang et al. doped the shell of QDs with Mn. These particles showed a high QY and high MRI relaxivity values.353 An interesting in vitro study was performed by Xia et al., who developed a multifunctional NP for the combination of X-ray computed tomography (CT), fluorescence imaging, and photothermal therapy. The authors incorporated gold nanorods (GNRs) and QDs into silica spheres and used folate acid conjugated on the surface for targeting folate receptors expressed on cancer cells. The NPs exhibited strong X-ray attenuation for CT and strong fluorescence for optical imaging. They also showed an enhanced photothermal therapy effect for cancer cells due to the strong NIR absorption of GNRs and a better heat generation rate.354
5. Tissue
In an interlacing state between in vitro and in vivo applications the imaging of pathogens expressed in tissues is an important research field. The qualitative and quantitative measurement of biomarkers in tissue biopsy samples provides important information for the diagnosis, staging, and treatment efficiency of a disease. A sensitive and specific diagnosis is very hard to achieve by evaluating only a single biomarker and multiplexing is a very important issue, in particular for immunohistochemistry (IHC). The evaluation of a pool of biomarkers from a single sample affords multiple and therefore deeper disease information and takes into account the limited amount of biopsy material. Classical approaches of fluorescence IHC are based on organic dye antibody conjugates (e.g., FITC or rhodamine-based dyes), which often provide excellent biological and imaging properties and are readily available in a large spectral range. Drawbacks of such common IHC conjugates are photobleaching, spectral crosstalk, and low SNR caused by strong autofluorescence. Because efficient, sensitive, and selective fluorescence staining and imaging is more difficult for relatively complex tissue samples compared to cultured cells, the established IHC methods based on enzymes or dyes are still much more common than the use of QDs, which can suffer from more complicated bioconjugation and non-specific binding. Nevertheless, the photophysical benefits of QDs, in particular for multiplexing, have motivated a number of QD-based IHC research studies that showed the advantages of QD-based immunohistofluorometry (QD-IHF) compared to conventional detection methods.355–357 In particular the ability of exciting several QDs of different PL colours with the same excitation source (wavelength) for the detection of different biomarkers is extremely interesting for tissue imaging.358–361 Jennings et al. used five different emitting QDs conjugated with antibodies that targeted different components of the immune system present in a mouse spleen tissue. They performed a five-colour immunohistochemical staining and the specificity of each antibody was maintained under multiplexed conditions.82 Ferrara et al. demonstrated the unique possibilities of QD-based multiplexing for the investigation of coronary arteries. They combined the photostability and large two-photon absorption cross sections of QDs with the advantage of three-dimensional fluorescence imaging using two-photon excitation laser scanning microscopy. The results showed the feasibility of multicolour profiling and a 3D visualization of the vascular endothelium with outstanding resolution.362 Giepmanns and coworker exploited the tight packing of atoms in QDs for combining fluorescence microscopy with electron microscopy (EM). Due to different shapes of the QD cores they could differentiate three different QDs with EM and fluorescence microscopy in a single mouse tissue.363Tissue microarrays (TMA) enable fast and high throughput screening of biomarkers. Different studies showed the possibility of developing convenient data analysis software that can subtract autofluorescence, normalize tumour expression to cellular content, and produce a comprehensive profile of tumour derived antigens on a TMA.364 Another important aspect is the affinity of the antibody used for detection. Xiao et al. showed that a chicken-derived antibody, IgY, could be used for QD-IHF and provided high sensitivity and specificity for relative quantitation of cancer proteins. In direct comparison to conventional antibodies IgY showed an enhanced affinity to antigens measured by western blotting and IHC on normal and cancer cells. Further comparison in TMA for the quantitation of the human growth factor receptor HER2 using IgY and QDs showed a good agreement with chromogenic in situ hybridization.365
Yezhelyev and coworkers compared the use of multiplexed QD-IHF detection of hormone receptors and HER2 to traditional methods such as IHC, western blotting, and fluorescence in situ hybridization (FISH) in single paraffin embedded clinical tissue sections. Their results were in very good agreement with the already established clinical applications, which showed the translational potential of QDs for clinical tissue diagnostics. However, the authors also pointed out the need for more compact QD probes, which would allow deeper tissue penetration, controlled and site-specific conjugation, and an inclusion of housekeeping markers for standardization.366 A next step towards clinical applications was performed by Chen et al., who developed a QD-based HER2 kit for image acquisition in breast cancer tissue. The kit included an analysis software that allowed automated, sensitive, quantitative, and convenient detection and was successfully tested on 94 clinical samples.367 Li et al. used QD-IHF for image deconvolution and image analysis to improve Akt-1 quantification. The Akt/protein kinase B signalling pathway is implicated in tumour genesis and progression. The study of 840 TMAs of radical prostatectomy cases showed that high levels of Akt-1 could be associated with a higher risk of biochemical recurrence and prostate cancer-specific death.368 A general concern that could hamper the translation of QD-IHF in clinical practice is batch-to-batch variation of QD bioconjugate production. Xu et al. compared different batches of QDs concerning their consistency in tissue IHC using four-dimensional spectral imaging. The authors developed a quantitative method for evaluating the efficiency of QDs in staining tissues based on Hadamard transform spectral imaging. The Hadamard transform is a mathematical transform based on square waves and provides information about location and qualitative features. The four dimensions were location (the two coordinates x and y), fluorescence intensity (z) and wavelength (λ). For a proof-of-concept the authors used proliferating cell nuclear antigens (PCNA) in breast cancer tissues labelled with different batches of bioconjugated QDs. The results demonstrated that it is possible to maintain uniform bioconjugation efficiencies with different QD bioconjugation processes.369 Xu et al. investigated optimization of the labelling protocol for QD-IHF probes. In their study they pointed out the advantages of QDs over organic dyes and evaluated different staining methods as well as the variation of the number of antibodies per QD. The results demonstrated the multiplex detection and quantification of different biomarkers (which were validated by western blotting and conventional IHC) using QD-IHF.370 A direct comparison of conventional IHC, single-labelled, and double-labelled QD-IHF was demonstrated for a semi-quantitative in situ analysis of the nuclear markers estrogen receptor and progesterone receptor in ductal carcinoma in the breast (Fig. 14A). The results showed a concordance rate of 100% for the detection of both receptors between conventional IHC and both types of QD-based IHF using multispectral imaging (MSI).371
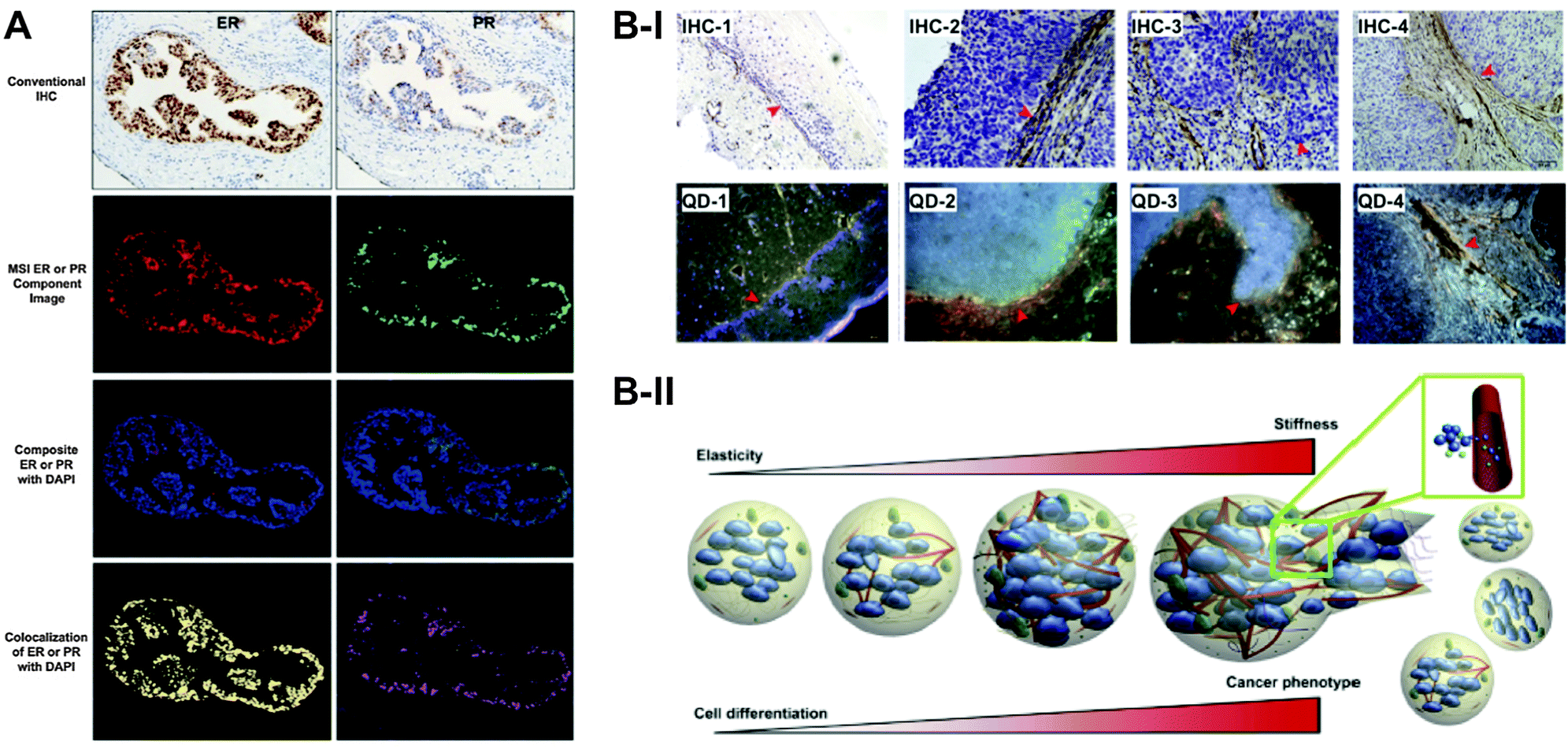 | ||
| Fig. 14 (A) Multispectral imaging (MSI) of double-labelled QD-IHF for the nuclear markers estrogen receptor (ER – red PL signals) and progesterone receptor (PR – green PL signals) showed excellent correlation with conventional IHC (top images). Co-staining with DAPI (blue) allowed colocalization studies to localize ER (yellow) and PR (magenta) in the cell nuclei. Reprinted with permission from ref. 371. Copyright 2013 Elsevier Inc. (B) Conventional IHC (B-I top) and QD fluorescence (B-I bottom) imaging to monitor stroma remodelling represented by collagen IV changes. Peri-tumour tissue (1) shows smooth, intact and linear collagen IV that was well-aligned with the basement membrane (BM). The smooth collagen line disappears in cervical carcinoma (CC) in situ, followed by collagen IV deposition to form dense layers (2). The collagen deposition becomes loose and fuzzy in micro-invasive CC (3) together with increased deposition in the extracellular matrix (ECM) leading to a stiff and rigid morphology of collagen IV (4). The results of this study proposed a pulse-mode of cancer invasion and metastasis with the process of tumour mass growth, prominent central hypoxia, collagen IV cross-linking and deposition, ECM remodelling, ECM stress build-up, and tumour nest burst leading to accelerated cancer progression (B-II). Reproduced with permission from ref. 377. Copyright 2013 Fang et al. | ||
Multiplexed QD-IHF and spectrally resolved fluorescence imaging can also be used for deciphering the molecular, cellular, and architectural heterogeneity of cancer specimens. In two studies Liu et al. showed the use of four protein biomarkers for the identification of single malignant tumour cells within complex tissue. This method provided new molecular and morphological information that was not accessible by traditional hematoxylin and eosin (H&E) staining and IHC.372,373 In contrast to conventional methods QD-IHF can provide both patterns and quantities, which are necessary for personalized diagnostics. Chen et al. used QD-based quantitative spectral analysis of hormone receptors and HER2 to establish a new molecular classification system of breast cancer. This classification could better reveal the breast cancer heterogeneity and identify five molecular subtypes with different five-year prognoses, which may help to formulate a more personalized and comprehensive therapy strategy as well as prognosis.374 QD-IHF also showed to be a useful tool to investigate the controversial question of the prognostic potential of the epidermal growth factor for breast cancer.375 Andrade et al. employed lectin to evaluate cell surface glycoconjugates, which play an important role in differentiation–dedifferentiation processes. They evaluated and compared the expression and distribution of glycoconjugates in normal human breast tissue and benign (fibroadenoma) and malignant transformed breast tissue. Fluorescence images showed that different regions of breast tissue expressed particular types of carbohydrates. This helped elucidating the glycoconjugate profile in biological processes and provided new insight into cellular and extracellular matrix structures, molecular content, and tumour environment for a better understanding of biological processes including the development and progression of cancer.376 Another interesting study was published by Fang et al., who investigated cancer invasion and metastasis. The mechanisms for these two properties of cancer have not yet been fully understood. In the focus of the investigation was the basement membrane (BM), which is a specialized form of the extracellular matrix (ECM) and has important cellular functions. The degradation of BM by metastasizing cancer cells leads to structural remodelling, which is characterized by increased deposition of, for example, collagen. Because of its role as a physical barrier to resist cancer invasion collagen, specifically collagen IV, is potentially of great interest. The authors used QD-based imaging to gain new insights in the dynamic changes of collagen IV during cancer invasion (Fig. 14B).377
In addition to provide complementary information to IHC, multiplexed QD-IHF was also used for the detection of mRNA using FISH assays. In a comparative study concerning the expression of specific mRNA in mouse brainstem sections, Chan et al. could measure a significantly increased sensitivity in FISH using QDs instead of organic dyes. Their approach provided ultrasensitive simultaneous detection of multiple mRNA and protein markers in tissues and histological sections.378 Tholouli et al. described a methodology for multiplexed QD-based in situ hybridization (QD-ISH) using QD-labelled oligonucleotides and spectral imaging for routinely processed, formalin-fixed paraffin embedded (FFPE) human biopsies. Single and multiplex QD-ISH was performed in samples with acute leukemia and follicular lymphoma. The advantage of multispectral imaging is the separation of spatially co-localized signals. The method relied on the generation of a complete optical profile for each pixel in the image field, from which multiple spectral distributions could be reconstructed via a least square fitting linear unmixing approach, enabling signal quantitation.379 mRNA displayed one cogwheel inside the whole protein production cycle. The understanding of the relationship between proteins and mRNA is an important aspect for different cellular functions and processes. Matsuno et al. tried to illustrate this relationship by visualizing the processes of transcription, translation, transport, and secretion of hormones. In situ hybridization and immunohistochemistry under electron microscopy provided sufficient ultrastructural resolution for two-dimensional images of subcellular localization of hormones and their mRNA. The combination with QDs and confocal laser scanning microscopy added the third spatial dimension.380,381
Tissue and collagen matrices, also called phantom tissue, can be used to optimize probes for later in vivo measurement. Investigations of the preferential uptake of QDs in tumour tissue,382 or the evaluation of the possible penetration depth of multiphoton excitation383 were performed to test the limits of the probes prior to in vivo use. Wong et al. presented a study in which they exploited the increased protease activity inside tumour microenvironment for enhanced intratumour delivery of a probe using a tissue model of collagen gel. They proposed a multistage system in which 100 nm NPs were degraded due to protease activity and led to the release of 10 nm QDs. They observed that the size change was efficient in the enhancement of diffuse transport.384
Conventional multiplexed tissue staining methods are based on fluorophores that are attached to secondary antibodies, which necessitates a sequential multicolour staining. Sweeney et al. demonstrated a more facile and less time-consuming staining method using primary antibodies labelled via biotin–streptavidin recognition to QDs. Although one should keep in mind the elevated costs of primary antibodies compared to secondaries, the direct use of labelled primaries enabled multiplexed staining of three antibodies for triplexed measurements in tonsillar tissue within eight hours.385
6. In vivo applications
Once an imaging agent has been successfully applied in vitro the transfer to in vivo systems is the consequent objective. First publications of QD applications in vivo appeared in the beginning of this millennium21,23 and since then the number of articles has increased tremendously as witnessed by many review articles.89,386–390 It should be noted that all in vivo applications of QDs have been limited to animals. However, this is a general limitation for fluorescence imaging and is mainly related to the limited tissue penetration depth of light and the very few imaging agents that have been approved for humans. Therefore only NIR-fluorescence based proof-of-concept studies, imaging of body parts, and endoscopic investigations have been performed so far.391,392 Other limiting factors for QD in vivo imaging in humans are their undefined biocompatibility and toxicity and a concomitant unavailability of QD probes that have been approved for applications in humans. Toxicity issues are reviewed in the last section of this article. In this chapter we give a short overview of QD properties and requirements for in vivo applications, discuss the use of QDs in studying the lymphatic system and for tumour localization, and report about QDs in multimodal imaging probes, a research field that has gained a lot of interest in the recent years due to the nanometric combination of fluorescence with other imaging techniques.6.1 Properties, requirements, and applications of QDs for in vivo imaging
The different available in vivo imaging technologies can be identified by mainly five properties: (i) resolution (spatial and temporal), (ii) penetration depth (in tissues), (iii) excitation energy (ionizing vs. non-ionizing), (iv) availability of injectable biocompatible molecular probes, and (v) the detection threshold of probes for a given technology.393 In contrast to other imaging techniques, such as CT or positron emission tomography (PET), fluorescence imaging uses non-ionizing radiation and is usually less hazardous. The unique photophysical and photochemical properties and large surfaces of QDs offer many in vivo imaging advantages that are hardly realizable with common fluorophores.394 The synthesis of QDs with emission in the NIR window from ca. 650 to 900 nm is an important aspect for non-invasive imaging methods. In this spectral range the absorption coefficients of water and haemoglobin are relatively low, which allows imaging in deeper tissue sections (ca. 0.5 to 10 mm). Jiang et al. compared NIR-QDs and their visible counterparts. They could observe a 10-fold increase of sensitivity for imaging deep tissues and organs using NIR-QDs.395 Although QDs may be used to stain whole animals in different colours, the major application aims at targeting specific parts of the body. A potential constraint of using fluorescent markers is that the SNR can be limited by receptor density, clearance kinetics, non-specific cellular uptake or adhesion, and the photophysical properties of the marker itself.393 High brightness and negligible photobleaching make QDs superior to other fluorophores such as organic dyes or fluorescent proteins.Although QDs have a broad absorbance spectrum, one-photon-excitation can be problematic due to the strong absorbance and scattering of tissue in the visible wavelength range, which limits the penetration depth and imaging possibilities. To avoid external light excitation, bioluminescence resonance energy transfer (BRET) can be used to promote QDs in an excited state, from which they can luminesce. Due to the oxidation of its substrate Renilla reniformis luciferase (Luc) can act as donor for a non-radiative BRET to QDs. So et al. demonstrated the advantages of that system using a covalent coupling between the amino groups of Luc and the carboxyl groups on the surface of QDs. The NIR-emitting QDs allowed minimal scattering after BRET for a sensitive QD detection inside mice. The results also showed that BRET is suitable for multiplexing using differently coloured QDs and that the sensitivity is strongly enhanced in small animal imaging. Although the excitation of the QD–Luc complex was possible via BRET, the QDs could also be excited directly, which made the complex compatible for both bioluminescence and fluorescence imaging.396 One of the drawbacks of the first BRET–QD complexes for improved deep tissue in vivo imaging was the lack of stability. Xing et al. resolved this problem by polyacrylamide gel encapsulation. The results demonstrated an improved stability in serum as well as for in vivo measurements.397 Instead of covalent binding of Luc and QD Hasegawa et al. used the interaction of glutathione-s-transferase (GST) tagged luciferase with glutathione coated QDs to enable BRET. This conjugate was successfully applied for whole body NIR imaging in mice.398 Although BRET avoids excitation by light, the “internal” excitation has limitations, such as the dependency on its substrate coelenterazine, which pose difficulties for a general QD-based in vivo imaging approach. The major excitation technique for in vivo tissue imaging is two-photon absorption because it allows QD excitation in the NIR window and therefore deeper tissue penetration depths. Larson et al. exploited the large two-photon absorption cross section of QDs for non-invasive multi-photon microscopy of the capillaries through the skin of living mice. The QD-containing vasculature was clearly visible and they were able to measure blood flow velocity and heart rates.107 In another study Lim et al. investigated the biological effects and tissue absorbance, scattering, and thickness related to the performance of two-photon-excited NIR-QDs.399 Apart from imaging QDs inside the tissue they can also be used for coating glass pipettes that are applied to target visually identified neurons for electrophysiological studies. Andrasfalvy et al. showed that such QD-coated glass pipettes can provide better two-photon excitation signals in deep brain tissues compared to other methods and they demonstrated the utility of their QD-pipettes in vitro and in vivo.400
In one of the early QD in vivo applications Akerman et al. faced some unexpected problems when using CdSe/ZnS QDs functionalized with lung targeting peptides. Although the QD bioconjugates showed excellent targeting specificity for the relevant vascular lung tissue, accumulation of QD fluorescence within the targeted tissue was not detectable. This was mainly attributed to a low pH in the microenvironment, to oxidation of the surface, or to adsorbed proteins.21 Dubertret and coworkers circumvented fluorescence quenching by encapsulating individual QDs in phospholipid block-copolymer micelles, which led to stable and non-toxic nanoparticles used for microinjection in early stage Xenopus embryos. Embryo development up to the tadpole stage could be followed by QD fluorescence imaging, which allowed lineage-tracing experiments in embryogenesis.23 These results demonstrated the importance of appropriate surface coatings and materials to produce efficient QD in vivo imaging agents. Many surface coating strategies have been developed, which used compact ligands,401,402 electrostatic interactions (e.g., with hyaluronic acid),403 immobilization on oligomeric nanoparticles,404 and encapsulation in micelles,405–407 to name only a few.
Advantages of PEG-containing QD surface coatings for in vivo imaging have been presented in a number of studies. Ballou et al. investigated the influence of different coatings on the distribution and circulating half-lives in vivo. Localization was determined by fluorescence imaging of living animals and by necropsy. Short circulating half-lives were found for amphiphilic poly(acryl acid), short chain (750 Da) methoxy-PEG, and long chain (3400 Da) carboxy-PEG QDs, whereas approximately 70 min were found for long-chain (5000 Da) methoxy-PEG QD.22 Supporting this study Daou et al. investigated the length of PEG chains on commercially available Qdot 705 ITK amino (PEG2000) QDs (Life Technologies) and studied their fate after tail injection in an anesthetized mouse by fluorescence imaging. They found that the speed of the first pass extraction towards the liver could be slowed down with increasing the hydrodynamic diameter (longer PEG chains) of the QDs.408 Al-Jamal et al. examined the modulation of surface and bilayer characteristics of functionalized QD–liposomes vesicles in terms of their blood circulation and retention time in tumour tissues. They could show that cationic hybrid vesicles have a rapid blood clearance, whereas PEG-incorporation on the QD surface dramatically prolonged the blood circulation half-life after systemic administration. The longer circulation time also led to a higher accumulation in the tumour.409 These results were supported by Papagiannaros and coworkers, who used QDs encapsulated in PEG-phospholipid micelles. Compared to commercially available QD-PEG they accumulated much faster (1 h instead of 4 h) and only half of the dosage was necessary to reach the same SNR. This QD complex presented a rapidly acting nanosized imaging agent that allowed an effective visualization of tumours using NIR imaging.410
Alternative synthesis approaches circumvent the organic phase by preparing QDs directly in water and sometimes using targeting molecules as stabilizers. The possibility to use such aqueous QDs for in vivo imaging has been shown in different studies.53,411–416 Also biocompatible alloyed QDs have been developed and CdTeSe–CdS core–shell QDs were successfully applied as NIR in vivo imaging probes. Injected to rodents the QDs did not show any apparent toxic effects over three months, which was confirmed by histological sectioning. Additionally, the number of QDs in the liver and spleen reduced over time, which suggested a clearance of the nanoparticles from the body.417 To introduce a paramagnetic effect in such alloyed QDs Yong et al. doped them with Mn, thus creating a magnetic semiconductor system. Based on the small size, NIR PL wavelengths, high QY, and paramagnetism such Mn doped QDs may be attractive multifunctional in vivo imaging probes.418 Apart from QDs also elongated Cd-based quantum rods (QR) have been used for in vivo imaging.419
The indefatigable amount of work in improving distribution, circulation time, and specific targeting of tissues and organs has led to a multiplicity of NIR-QD-based in vivo imaging applications. NIR-QDs have been used to track labelled stem cells,420 neutrophils,421 and viruses422 to improve the knowledge about their behaviour inside living animals by non-invasive imaging. In this context Pan et al. contributed an interesting study about monitoring of in vivo dynamics of avian influenza viruses, which are important for understanding viral pathogenesis and developing antiviral drugs. Bioorthogonal chemistry was used to label CdTeSe/ZnS alloyed QDs to avian influenza H5N1 pseudotype virus. This conjugated complex demonstrated bright and sustained PL signals in mouse lung tissue and allowed visualization of respiratory viral infection in a non-invasive real-time manner. The biodistribution in lungs and other organs could be quantified by measuring fluorescent signals and Cd concentrations of virus-conjugated QDs in tissue. This approach provided a simple, reliable, and quantitative strategy for tracking respiratory viral infections and antiviral drug screening.423
Due to the large surface area of QD nanoparticles they can also be used as drug delivery system to form a theranostic (drug therapy + fluorescence diagnostics) agent. Savla et al. demonstrated a QD-based drug delivery system for specific targeting of ovarian cancer cells and simultaneous delivery of an anti-cancer drug. The overexpression of mutated Mucin 1 was targeted with aptamer-conjugated QDs, to which Dox was bound via a pH-sensitive hydrazone. This bond is stable at neutral and slightly basic pH but undergoes a rapid hydrolysis in mildly acidic pH. Thus the theranostic QDs were stable during blood circulation but could efficiently release Dox inside the cancer cells. In vivo imaging studies showed a higher toxicity for cancer cells using the QD-conjugates compared to free Dox.424 The theranostic potential of liposomes for simultaneous bioimaging and drug delivery was investigated for the delivery of camptothecin for the treatment of melanoma. Cationic liposomes together with carboxylated QDs showed a high accumulation in solid tumours and the drug entrapment was nearly complete (>99%).425,426 Another cancer theranostic in vivo concept was developed by Shao et al., who used NIR-QDs conjugated with a DNA-encoded suicide gene and could monitor the therapeutic efficiency in mice. Transfection of tumour cells with suicide genes can lead to inhibition of the DNA polymerase and/or incorporation into DNA, which finally leads to DNA chain termination and tumour cell death. The authors could observe a tumour regression indicated by a significant decrease of the PL intensity at day 14 after administration of the QD-complexes.325
6.2 QD in the lymphatic system
The lymphatic system plays a crucial role as drainage system of the body. Its main functions are transportation of the lymph and filtration of prospective immunogens from the extracellular space. It consists of capillaries, vessels, and nodes, is spanned over the whole body, and is also correlating with the blood stream. If tumour cells invade a regional lymph node the large lymphatic network enables a fast spreading of the tumour cells over the whole body and is therefore playing an active role in metastasis. So-called sentinel lymph nodes (SLNs) are found in close distance to tumours and thus represent a gate for metastasis.427 Mapping of SLNs is important for assessing the stage of cancer and for planning an efficient treatment. Different studies showed the possibility of SLN mapping using NIR-QDs428–432 and demonstrated the possibility for an image-guided surgery for the resection of SLNs.433–435 Hama et al. demonstrated for the first time a simultaneous imaging of two different lymphatic drainages in vivo and their trafficking to lymph nodes.436 In the same year Kobayashi et al. used five different NIR-QDs for spectral fluorescence lymphangiography (Fig. 15). Their study demonstrated simultaneous visualization of five separate lymphatic flows in vivo and their trafficking to distinct lymph nodes, which had not been possible using other imaging techniques. The QDs were injected into five different sites and monitored the lymphatic drainages in the neck and the upper trunk. Ex vivo fluorescence imaging of the draining lymph nodes confirmed the in vivo results. This study evidenced the important advantages of QDs for multiplexed diagnostics by demonstrating the possibility of investigating separated drainage patterns and mixing of five adjacent lymphatic basins in vivo.437 A profitable application for this would be multiplexed in vivo monitoring of cellular trafficking inside the lymphatic system.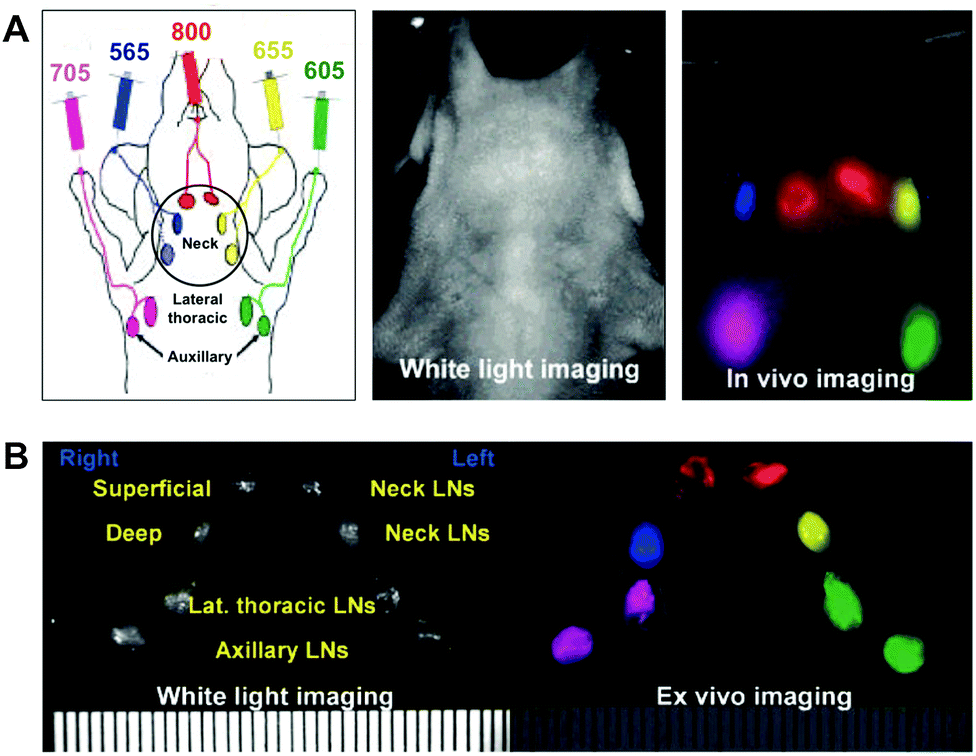 | ||
| Fig. 15 (A) Drainage into distinct lymph nodes of five different carboxylated QDs (central emission wavelengths in nm: 565, blue; 605, green; 655, yellow; 705, magenta; 800, red) intracutaneously injected into the middle digits of the bilateral upper extremities, the bilateral ears, and at the median chin of a mouse could be simultaneously imaged with different PL colours through the skin. (B) Ex vivo spectral fluorescence imaging of the eight draining lymph nodes after surgical resection confirmed the in vivo imaging results. Reproduced with permission from ref. 437. Copyright 2007 American Chemical Society. | ||
Noh et al. performed non-invasive tracking of dendritic cells via NIR-QDs during their migration into the lymph nodes. The monitoring of injected cells to their target tissue is very important for improving the performance of immunotherapeutic cells.438 Pic et al. studied the kinetics of NIR-QD accumulation in specific regional lymph nodes. In vivo fluorescence images could be confirmed by ex vivo elemental analysis (ICP-MS). QD accumulation was detectable 5 min after injection, reached a maximum after 4 h, and then decreased over a 10 days period. The overall level for the uptake of QDs by other organs remained rather lower.439 Kosaka and coworkers presented a method of real-time tracking of the lymph flow. The lymph flow is difficult to track in real-time because of the lack of appropriate imaging methods. The authors combined macro-zoom fluorescence microscopy with interstitial injected QDs that allowed tracking lymph through lymphatic channels into the lymph nodes. Further they used multiplexed two-colour real-time tracking with two QDs.440 Mathieu et al. used the combination of QDs and hyperspectral imaging to follow cerebrospinal fluid (CSF) drainage into the lymphatics. This is important to decipher central nervous system disorders, which are characterized by disturbances of CSF drainage. The authors could successfully monitor the QD signal in submandibular lymph nodes after 20 minutes. Compared to the monitoring using a surgically approach, this method allowed the imaging at multiple time points after injection in an intact organism without the need of surgery.441
6.3 QDs and tumours
The eminent relevance of cancer in the life sciences has made investigations of location and distribution of tumours and tumour cells to a high priority research field for in vivo imaging. To get an insight in the trafficking of cancer cells, it is possible to label such cells with QDs and to inject them subcutaneously or to use systemic application mostly in rodents.64,442 Antibodies are commonly applied to target tumours.216,443,444 Tada et al. used a dorsal skinfold chamber for single-particle tracking of QDs inside living mice. They were interested in the pathway of QDs labelled with antibodies and could decipher six steps of delivery, namely (i) circulation within a blood vessel, (ii) extravasation, (iii) access to the extracellular region, (iv) binding to a receptor on the cell membrane, (v) movement from the cell membrane to the perinuclear region, and (vi) access to the perinuclear region. Knowing antibody delivery pathways can provide helpful information for improving therapeutic efficacy.445 Diagaradjane and coworkers investigated the interaction of NIR-QD-labelled EGF with EGFR in xenografted tumours in mice. They were able to distinguish three phases of the nanoprobe: tumour influx within ca. 3 min, clearance within ca. 60 min, and enhanced accumulation within ca. 1–6 h. Although unconjugated QDs also showed tumour influx, the concentration gradually decreased over time. After 24 h the tumour fluorescence decreased to near-base-line levels for both pure QDs and EGF-QDs.446 Not only trafficking is important to understand the circulation of tumours cells. Also the interactions with membranes play a significant role in metastasis. Gonda et al. studied the membrane dynamics in metastatic tumour cells using QDs labelled with antibodies against a metastasis-promoting factor. Changes in membrane morphology and membrane protein dynamics based on its fluidity are critical for cancer metastasis. The authors were able to image the membrane dynamics with a spatial precision of 7 to 9 nm.447Yang et al. investigated the sensitivity of NIR-QDs for in vivo imaging. They used commercial Qdot 800 QDs (Life Technologies) conjugated with CPPs for endocytosis into oral carcinoma cells. The QD-labelled cells were inoculated in the dorsum, back muscle, and under the oral mucosa of nude mice in different amounts. The results revealed that at least 104 QD-labelled cells are necessary to obtain a sufficient signal. The highest cell amount used (106 cells) could be visually observed over 16 days.448 Similarly, Yukawa et al. investigated the intensity of NIR-QD-labelled Adipose tissue derived stem cells, which were transplanted subcutaneously into the backs of mice.449 Stroh et al. explored the multiplexing capability of QDs for in vivo imaging. They used multi-photon microscopy techniques and transgenic mice that expressed GFP and combined them with QDs. This allowed them to differentiate tumour vessels from both perivascular cells and the matrix.450 Lim et al. monitored natural killer cells that were used for immunotherapeutic cell-based cancer therapy via labelling with QDs. Qdot 705 QDs (Life Technologies) were attached to the killer cells using antibodies and a similar therapeutic effect was found compared to unlabelled killer cells. The conjugates were intratumourally injected and imaged using NIR fluorescence. Immunotherapeutic cells labelled with QDs can be a versatile platform for the effective tracking of injected therapeutic cell, which is very important in cell-based cancer therapies.451
Similar to the resection of lymph nodes Li et al. demonstrated a QD-based imaging-guided surgery (IGS) of U87 MG tumour xenografted mice (Fig. 16).452 NIR-QDs (728 nm PL maximum) were conjugated with tumour-specific peptides (cRGD) and injected into the tail veins of anesthetised mice. 48 h after injection the NIR-QDs were accumulated in the tumour, which could be used to distinguish it from other tissue and to resect it. Active targeting with cRGD–QD conjugates led to approximately five times stronger PL intensity in the tumour compared to passive targeting using QDs without cRGD.
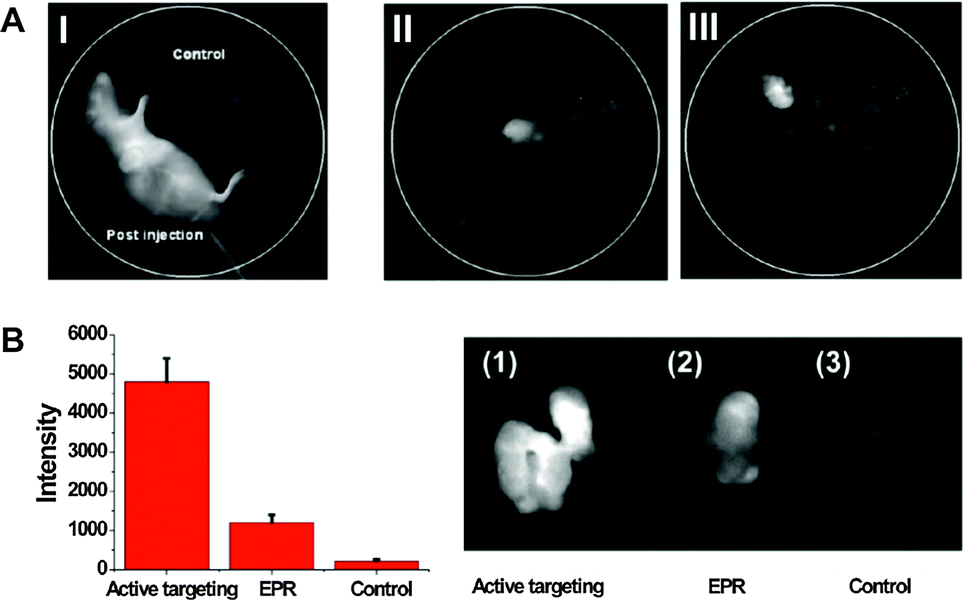 | ||
| Fig. 16 (A) NIR-PL image of a mouse directly after NIR-QD–cRGD conjugate injection (I) and imaging-guided surgery (II and III) of the QD-conjugate-accumulated tumour 48 h afterwards. The control mouse without QD-injection (I) does not show any significant NIR PL. (B) Active tumour targeting using NIR-QD–cRGD led to significantly higher PL intensity compared to passive targeting through the enhanced permeability and retention (EPR) effect (NIR-QDs without cRGD)) and the blank control (no QD injection). Reproduced with permission from ref. 452. Copyright 2012 Li et al. | ||
Apart from the tumour itself another important target is the vascular of the tumour. Angiogenesis is the process by which new vasculature establishes a blood supply to a growing tumour. Receptors, such as integrins, are highly expressed in tumour cells during angiogenesis and diagnostic targeting of such receptors can provide insight into the type and extent of diseases, including cancer.389 Integrins can be efficiently targeted by the peptide RGD.453–455 Chen and coworkers reported the development of vascular cell adhesion molecule-1 binding peptide (VCAM-1 binding peptide) functionalized QDs (VQDs). The authors observed an increase of the fluorescence intensity for in vivo and ex vivo experiments in inflamed endothelium as well as blue-shifted emission. Their investigations revealed that this shift was related to the binding of VQDs to VCAM-1, which could be a useful tool for VCAM-1 detection in vivo.456 Antibody conjugates can also be used to image the vasculature. Jayagopal et al. used QD–antibody conjugates for noninvasively assessing the retinal vasculature and to be able to monitor continuous vascular events using fluorescence angiography. They were able to image three cell adhesion molecules and one leukocyte subset within the same animal and without spectral overlap. This demonstrated the possibility of detailed multiplexed studies of diseases and biological processes.457 A study of Kwon et al. supported the usefulness of QD–antibody conjugates as a promising imaging tool for angiogenesis of cancer by labelling anti-vascular endothelial growth factor receptor 2 (VEGFR2) antibodies to QDs. For their xenograft models they injected human prostate cancer cells (PC3) to nude mice on hind limbs. The imaging results after QD-conjugate injection revealed an increased fluorescence at the tumour localization after 12 h.458
6.4 Multimodal imaging
For a more comprehensive view on a disease (or other biological systems of interest) it may be desirable to use a contrast agent that can be applied to several imaging techniques. Apart from the development and production of only one single imaging agent with equal biological properties for different imaging techniques, another advantage for in vivo use can be the avoidance of multiple administrations. Fluorescence imaging provides high spatial resolution and sensitivity but low tissue penetration depth. MRI, CT, and PET provide high penetration depths and are non-invasive. On the other hand they have a relatively low spatial resolution (for MRI and PET) and low target sensitivity (for MRI and CT). Multimodal probes allow the overall optimization of space, time, sensitivity, selectivity, and dose properties for gaining information about uptake efficiency, distribution in organs and tissue, and activities on the cellular and subcellular level.459 It should be noted that in proof-of-principle studies the demonstration of combining any multiple imaging techniques in a single nanoparticle could have higher priority than the usefulness of this combination for biosensing.Cai et al. developed a tumour targeting NIR-QD-based multimodal probe for PET and NIR imaging. They functionalized an NIR-QD with cyclic RGD-peptides for targeting the tumour vasculature and immobilized 64Cu–DOTA complexes on the QD surface to allow PET imaging. The combination of the mainly qualitative information of QD fluorescence imaging and highly quantitative tomographic imaging of PET permitted a sensitive and precise assessment of the pharmacokinetics and tumour-targeting efficacy. The authors could show an excellent linear correlation between in vivo PET and ex vivo NIR imaging results.460 Instead of a peptide Chen and coworkers used the vascular endothelial growth factor protein (VEGF) for their multimodal probe. VEGF and its receptor are major angiogenic regulators. The results revealed good correlation between ex vivo PET and NIR organ imaging and indicated that the assumed co-localization of 64Cu and QDs is true for different organs and tissue except for the liver. The 64Cu–DOTA complexes dissociated in the liver and led to an overestimation of QD-PET probe concentrations by PET.461 Duconge et al. were the first who reported of a fluorine-18-labeling and quantitative whole body imaging study of PEG-phospholipid QD micelles with PET. The long circulation in the blood stream (up to 2 h) and the slow uptake of the reticulo-endothelial system led to a distribution throughout the body. Using fibered confocal fluorescence imaging, they were also able to access the kinetics of cellular uptake.462
Oostendorp et al. circumvented the inherently low sensitivity of MRI using a commercially available PEGylated QD as scaffold for Gd-bearing complexes and tumour targeting Asn–Gly–Arg (cNGR)-peptides. This allowed non-invasive and selective in vivo detection of tumour neovascularization using quantitative molecular MRI. The MRI results were confirmed using two-photon laser scanning microscopy and it could be shown that the QDs were primarily located on the surface of tumour endothelial cells and to a lesser extent in the vessel lumen.463 Immobilization of Gd complexes on the QD surface can have significant effects, as was shown by Gerion and coworkers, who embedded QDs into a thin silica shell doped with paramagnetic Gd–DOTA complexes. Relaxivity measurements revealed a five to ten-fold enhancement depending on the type of relaxivity. The increase was not related to the nature of the core but rather to the immobilization on the silica shell, which reduced the rotational motion of the Gd complexes and to the hydrophilic nature of the silica surface, which allowed an efficient interaction with water molecules. Preliminary in vivo experiments indicated that these particles provided a MRI contrast enhancement.464
Ding et al. combined the high spatial resolution of CT with the high sensitivity and specificity of fluorescence imaging. Because CT contrast agents are non-specific and eliminate rapidly from blood vessels after intravenous injection, encapsulation via nanoemulsions or liposomes can be used to slow down the diffusion and to enable accumulation in lesions. Despite this increased blood circulation time, CT still suffers from low target sensitivity and large doses need to be applied, which can lead to adverse health effects. The authors developed a QD-iodinated oil nanoemulsion probe to target macrophages, which are key targets for atherosclerosis imaging. Different to other approaches they used hydrophobic QDs, embedded them in iodinated oil, and subsequently dispersed them in water to form an oil–water nanoemulsion. After thorough characterization of the samples, they extensively studied them in terms of cytotoxicity and affinity to three different cells in vitro. Micro-CT and in vitro cell confocal microscopy confirmed the staining of murine macrophages. The contrast agent was then used in atherosclerotic rabbits and showed in vivo accumulation in macrophages, which could be measured with clinical-CT and fluorescence imaging (Fig. 17). This investigation demonstrated the high potential of QD-based dual contrast agents also in larger animals.465
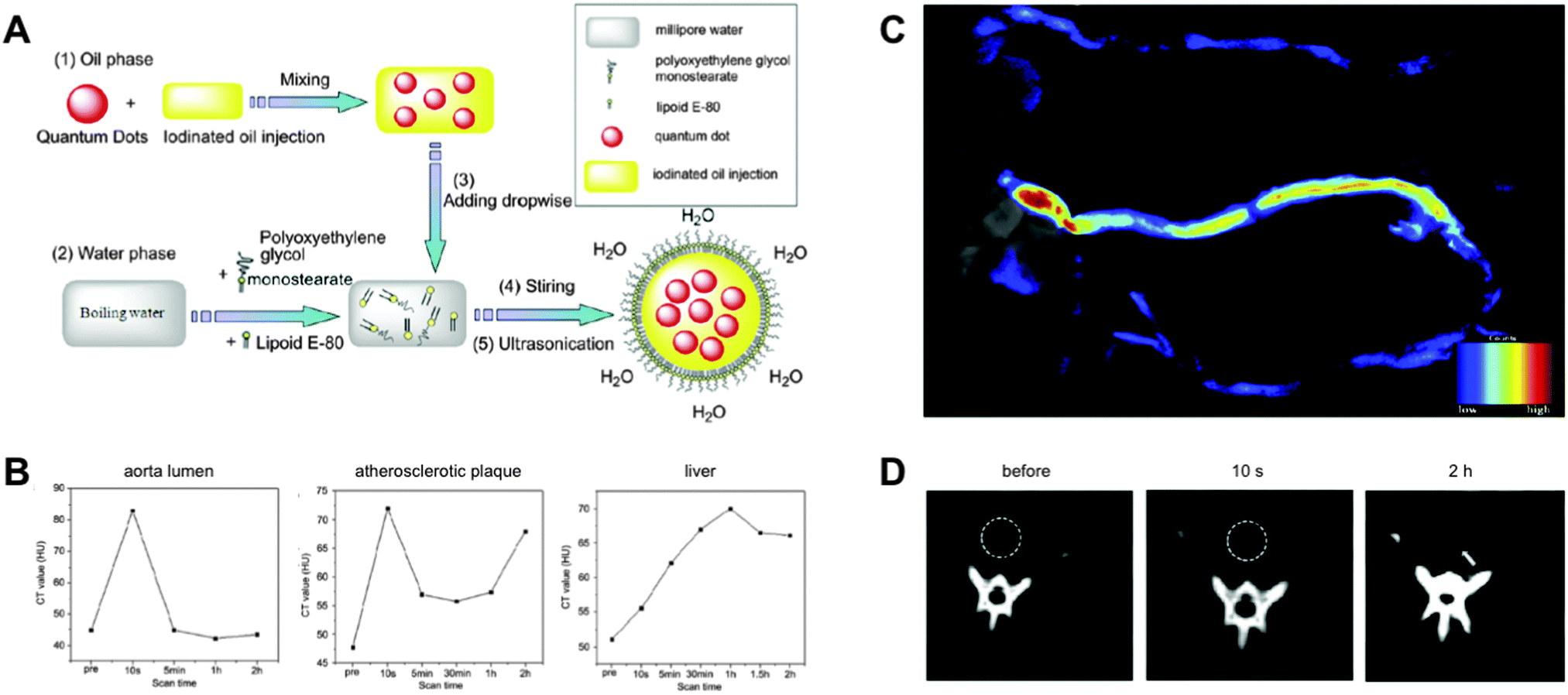 | ||
| Fig. 17 (A) Schematic of the preparation of QD-based nanoemulsions for CT/fluorescence imaging. (B) Kinetics of CT values of different organs within 2 h after injection of the nanoemulsion into rabbits. (C) Fluorescence images of excised aortas without (top) and 2 h after (bottom) injection of the QD-nanoemulsion. Due to accumulation over time QD-nanoemulsions led to a significantly higher PL intensity in the atherosclerotic plaques. (D) CT images of the abdominal aorta (circle) before and 10 s or 2 h after injection of the nanoemulsion. 2 h after injection atherosclerotic plaques (arrow) could be differentiated from the surrounding tissue. Reproduced with permission from ref. 465. Copyright 2012 Elsevier Ltd. | ||
7. Toxicology
Despite the superior photophysical properties of QDs compared to other fluorophores, there are certainly a couple of drawbacks, some of which we want to discuss in this section. One of the major reasons, why QDs are not implemented in clinical imaging and drug delivery applications on humans, is the concern about their toxic effects. The literature contains many reviews with a general overview about toxic effects of nanoparticles466–468 as well as reviews focused on QD toxicity.469–473 Because of the tremendous amount of related articles, we can only give an overview of the main reasons for toxic effects of QDs in vitro and in vivo as well as the related consequences. It should be mentioned that general toxicity properties for all QDs do not exist. Toxicity parameters must be determined for each QD–cell system in order to be sure about the toxic effects, which can be dependent on concentration, time, material, size, shape, coating, environment, and experimental conditions. In vitro cytotoxicity tests revealed that the release of Cd ions as well as the triggered formation of reactive oxygen intermediates (ROI) are responsible for induced toxic effects. Additionally, the intracellular distribution of QDs can enhance associated nanoscale effects.474 Cytotoxicity can be judged on many different parameters, such as cell number, cell growth, apoptosis, and cellular morphology of metabolic activity. The following examples from the literature give an outline of possible effects and their origins. We will start with in vitro experiments and then describe some in vivo systems.7.1 In vitro investigations
Shortly after the first applications of QDs in a biological context, first concerns were raised over the novel imaging agents. These concerns were mainly related to the chemical composition (heavy-metal ions), their size (nanomaterials), and the interference of these properties within cellular components and processes. In 2004 Derfus et al. investigated the effect of CdSe core materials on primary hepatocytes cells, which were used as a liver model.475 The toxic effect of CdSe was evaluated by measuring the mitochondrial activity using the colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) viability test. Exposure of the QDs with UV-light in the presence of air and subsequent incubation with cells led to a strong decrease of cell viability (97%). The authors suspected that the oxidation of the particles resulted in reduced Cd on the QD surface and that the release of Cd ions caused the cell damage. They also suggested that appropriate coating of the core would reduce the negative effects on cell viability.These early-observed toxicity results were supported in the following years by several studies, which showed that the surface modification had a tremendous influence on the cytotoxicity of QDs.198,476–484 Cell toxicity of QDs has been mainly attributed to reduction of the QD surfaces, leading to free Cd ions and generation of ROI. Because QDs are fluorophores that are usually excited by light, the photoactivated QDs can non-radiatively transfer their energy to proximal molecular oxygen to produce singlet oxygen, which will then produce ROI species like superoxide anions, hydroxyl radicals, and hydrogen peroxides.485 The molecular mechanisms of Cd ion-induced cell death were studied by many groups.486–490 Cd-ion leaching and ROI induce apoptotic biochemical changes, which create a loss of the mitochondrial membrane potential, mitochondrial release of cytochrome c, activation of caspases, and finally death.486 Brunetti et al. presented an alternative to the standard Cd based QDs. They compared the toxic effects of InP and CdSe QDs. Interestingly both materials were prone to ion leaching but In ions showed a lower intrinsic toxicity.491 Cho et al. measured the intracellular concentration of Cd when cells were exposed to CdCl2 and CdTe QDs. The Cd salt showed a linear behaviour of decreasing metabolic activity and intracellular Cd ion concentration. QDs also showed a dose-dependent correlation, but not linear as in the case of the Cd salt. The authors concluded that intracellular Cd ions and triggered ROI formation are not solely responsible for the toxic effect but that also lysosmal enlargement and intracellular distribution of QDs need to be taken into account.492 This was supported by a study of Stern et al., who investigated the toxic effects of CdSe and InGaP core materials. Both QDs led to toxic effects without a detectable elevation of the caspase activity or the upregulation of genes that were responsible for both the reaction with intracellular Cd ions and oxidative stress.493 This pointed out that the whole nanoparticle should be taken into account in terms of toxic behaviour. Domingos et al. investigated the consequences of QDs on the algae Chlamydomonas reinhardtii and they could show that free Cd ions and Cd-based QDs have distinctly different biological effects.494
Stern and coworkers also presented evidence for an induction of autophagy in QD-treated cells. Autophagy plays an important role in sensing oxidative stress, removing damaged proteins and organelles, and eliminating the damaged machinery that is responsible for excessive ROI production. This is an important defence/survival mechanism of the cell and can be activated by nanoparticles. In this regard Chen et al. investigated the effect of QDs on neurons. They found an enhanced autophagy formation due to an increased creation of ROI by QDs, which led to synaptic dysfunctions. By using an autophagy blocking agent the activity of the autophagosomes could be suppressed and the synaptic impairment restored.495 Luo et al. tried to decode the mechanism and functions of QD-induced autophagy in RAG (recombination-activation gene) cells. Experiments revealed that using an antioxidant to decrease QD-induced ROI creation led to a reduced autophagy activity and enhanced cell death. Similarly, the direct use of an autophagy inhibitor also led to increased cell mortality. These results supported the hypothesis that autophagy is a cellular protective mechanism in response to QD treatment.496
Cell compartments have different physiological properties that can have different impacts on QDs. For instance, the cytosol has a neutral pH whereas endosomes and lysosomes have an acidic pH, which will increase within the endocytosis process. The stability of QD coatings is a highly important property for imaging applications because degradation of the shell inside the cell would most probably lead to toxic effects. Corazzari et al. investigated ion release in artificial media representing the cytosolic and lysosomal cellular compartments.497 Their QDs showed a significant release of Cd and Zn ions in acidic pH. Because the endocytic pathway is one of the most common uptake mechanisms for QDs, pH-induced instabilities are a significant drawback and require profound investigation, which can allow to quantitatively and qualitatively assessing the toxic potential of QDs before their use in vitro or in vivo.
Apart from toxicity due to instable QDs, other aspects of QD–cell interactions are often equally important. A living organism provides many possible interactions with QDs (e.g., with proteins and ions) and therefore synergistic effects need to be scrutinized for future in vivo applications. For example, Zhao et al. could show the dramatic effect of QDs in presence of copper ions in human liver cells.490 Their investigations revealed a strong synergistic effect, leading to an 8-fold decrease of cell viability and a 3-fold increase of intracellular ROI levels. Nagy et al. reported about controversial behaviour of cytotoxicity and genotoxicity when using differently charged QDs. Negatively charged QDs with MPA (methoxy propyl acetate) capping ligands showed to be relatively noncytotoxic compared to positively charged, cystamine capped, QDs. In contrast, genetical analysis revealed that MPA-QDs induced a high number of DNA-strand breaks498 due to the upregulation of metallothioneins, which bind the Cd ions that leached from the core and thus reduce the ability for DNA repair. ROI can also induce nucleobase damage of DNA, which is attributed to the abstraction of hydrogen atoms from nucelobases and ribosyl groups.485 These results led to the conclusion that cytotoxicity assays must be complemented with genotoxic analysis in order to get the full picture of cellular damage.
Stem cell research is of high interest for clinical applications. Mesenchymal stem cells (MSC) are multipotent cells with the potential to differentiate into bone, cartilage, fat, and muscle cells. In clinical applications they are used in cell-based transplantation therapy499 and tracking of stem cells after implantation could be highly beneficial for evaluating the therapeutic outcome. Hsieh et al. investigated the effect of liposome transfected QDs into MSCs on cell proliferation for chondrogenic500 and osteogenic lineage.501 In both cases there was no effect on cell proliferation and the QDs were clearly visible after several cell passages. However, initiation of MSC differentiation was not successful as the internalized QDs prevented the full response to the induced differentiation. Muller-Borer et al. could show that cytotoxicity of QDs was dose-dependent and suggested to limit the QD exposure to MSC. An adjusted concentration of QDs led to minimal toxic effects and the QDs could be applied as useful fluorescent labels.499
Even if QDs induce a certain amount of toxicity, this toxicity needs to be compared to other existing and frequently used fluorescent imaging agents, such as organic fluorophores, under equal (or at least comparable) experimental conditions. Many imaging fluorophores, such as Hoechst dyes and BODIPY, are so commonly used that their cytotoxic effects are often completely disregarded. Bradburne et al. performed a direct cytotoxicity comparison of QDs versus organic dyes in cellular proliferation. The authors could identify small but significant effects on cellular viability when the dyes (acridine orange, Hoechst 33342) were used in a concentration range suggested by the suppliers. Using an iterative process to identify dosing conditions, surface ligands, and delivery modality, it was found that the applied type of QDs showed similar toxicity effects as the commonly used cell-labelling fluorophores.502 This study demonstrated the importance of the physical and chemical stability provided by appropriately selected surface ligands. Another comparative study using some of the most common imaging nanoparticles, namely CdTe QDs, Au NPs, and carbon dots, revealed that QDs had the strongest toxic effect on a variety of cell lines and plant growth. Uptake of QDs led to decreased cell metabolic activity, shrinkage of cells, breakage of chromatin, damage of cell membrane integrity, fragmentation of mitochondria, and finally cell death. The toxicity of QDs was time and dose dependent. In comparison, Au NPs led to decreased metabolic activity but no further effects of cellular/subcellular structures and carbon dots did not show any obvious toxic impact.503 It should be pointed out that the QDs used in the different studies discussed above were of different origins and that the results do not allow to draw a general conclusion about QD cytotoxicity.
7.2 In vivo investigations
In most cases in vitro cell culture investigations are not able to fully represent in vivo tissue systems, in which cell–cell and cell–matrix interactions as well as different diffusion/transport conditions play important roles. Lee et al. developed a 3D spheroid-interaction-based nanoparticle toxicology testing system. This consisted of hydrogel inverted colloidal crystal scaffolds that could be used to create a physiological relevant 3D-liver tissue. A comparison of toxicity of CdTe QDs in cell culture and in the 3D model revealed a lower toxicity in the latter, which correlated well with animal data. Although such model systems may be useful as intermediates between in vitro and in vivo measurements,504 it is still extremely difficult to make reliable predictions for in vivo effects based on results from in vitro investigations. The exact mechanisms of how QDs interact with the body and especially distribution, metabolism, and excretion are not well understood. In contrast to cell culture studies, the nanoparticles are not directly exposed to the target cell but have a rather long trip through different body transport systems. One of the major application methods for QD-based in vivo measurements is injection. The exogenous particles are then carried via the blood that contains various serum proteins. QD–protein interactions can lead to the formation of protein layers on the nanoparticle surface, known as “protein corona”. This protein corona influences the biodistribution and biocompatibility. With the bloodstream the nanoparticles will be delivered to the different organs and tissues, where their distribution is dependent on both the physio-anatomical features of the vasculature and the physicochemical properties of the nanoparticles. After successful distribution the QDs will undergo more or less metabolic processing and will be excreted from the body via the kidneys and/or in the feces. Retained nanoparticles can disturb over long-term periods the normal functions of organs or tissue and induce chronic organ toxicity, metabolic toxicity, immunotoxicity, or even genotoxicity.467 The immense surface of vascular endothelial cells provides plenty of space for interactions with QDs, in particular with negatively charged ones. Therefore, anionic QDs have a relatively low residence time in the blood stream and preferably accumulate in organs.505 On the other hand it was also shown that high doses of QDs could cause pulmonary vascular thrombosis by triggering the coagulation cascade via contact activation.506 Understanding the interaction of QDs with blood cells would help to improve QD design for theranostic drug delivery purposes. Fischer et al. characterized the in vivo kinetics, clearance, and metabolism of two different coated QDs in rodents. QDs coated with BSA accumulated to 99% in the liver and the authors were not able to detect them in the urine or feces for up two ten days after intravenous injection.507 Fitzpatrick et al. supported the hypothesis of a slow metabolic degradation of QDs. Their long-term study over two years using in vivo injected QDs in mice revealed a shift of the QD emission peak from the red to the blue with an increased spectral bandwidth, which they ascribed to QD degradation.508 Investigation of silica coated QDs showed that after five days only 8.6% of the injected QDs were not excreted and remained in hepatic tissue, whereas QDs with nearly no protein corona were cleared via urine. Larger aggregated particles were transported to the liver and cleared via bile excretion.509 Tang et al. examined the influence of QD surface charge and chemistry on biodistribution in mice. PEG-coated and negatively charged QDs accumulated primarily in the liver whereas positively charged QDs were found in the lung. Independent of their surface chemistry and charge, all QDs caused injuries in specific tissues, such as liver, lung, and kidney after acute and long-term exposure.510 While in general the PEG coating of QDs decrease the toxicity in cultured cells, PEGylated QDs show a different behaviour in vivo and can lead for example to lung tissue inflammation.511Apart from surface charge and chemical composition, the QD sizes play an important role for their distribution throughout the organism. Choi et al. could show that QDs with hydrodynamic radii of 5.5 nm and zwitterionic or neutral coatings led to a fast clearance through the kidneys.512 The fast clearance is important not only to avoid chronic long-term effects of the used imaging agent but also to minimize non-specific background for the imaging application. The authors confirmed their assumptions by receptor-specific imaging of prostate cancer and melanoma, where the fast clearance was very advantageous for the imaging procedure.513 A drawback of small-sized particles may be their ability to cross important barriers inside the human body, for example the blood–brain-barrier. Therefore it is important to investigate the effects of QDs on the central nervous system. Gao and coworkers investigated the consequences of chronic QD exposure in rats for their synaptic plasticity and spatial memory. The results show enhanced synaptic transmission and an impairment of the synaptic plasticity in the hippocampus that is important for learning and memory formation. Behavioural studies gave further evidence for impairment of the spatial memory. The authors concluded from the rat model that long-term exposure may induce impairments of hippocampal synaptic plasticity and spatial memory.514 King-Heiden et al. investigated QD toxicity on embryos of Zebrafish. They exposed the embryos to QDs and observed similar effects as in rodent models. In addition to the excellent transparency of Zebrafish embryos compared to rodents this animal model usually causes significantly lower costs for assessing structure–toxicity relationships of QDs.515
Similar to in vitro investigations also in vivo studies are controversial mainly due to the differences in QDs, their coatings, and their concentrations. Hauck et al. investigated biodistribution, animal survival, animal mass, haematology, clinical biochemistry, and organ histology for QDs injected in rats. They could not detect significant toxic effects in rats over four weeks of QD administration.516 Su et al. investigated aqQDs and their toxic effects.517 After short times (4 h) the aqQDs accumulated in the liver and after long-time blood circulation (80 days) in the kidneys. Larger molecules could be found in the spleen. Histological and biochemical analysis and body weight measurements demonstrated no evidence for overt toxicity even at long-time exposure. In another pilot study, rhesus macaques were injected with phospholipid micelle-encapsulated CdSe/CdS/ZnS QDs. The evaluation of blood and biochemical markers remained within the normal ranges. Also the histology of the major organs after 90 days of QD exposure showed no abnormalities. Using chemical analysis, traces of Cd accumulated in liver, spleen, and kidney were found. This was taken evidence for the slow breakdown and clearance of QDs from the body.518 These results were supported using similar QDs in mice. After 112 days of exposure there was no noticeable toxicity effect. The main accumulation areas were liver and spleen.519
To investigate an alternative body uptake mechanism, Hsieh et al. studied the toxic effects of QDs inhaled by mice. They measured the spatial distribution of QDs in lung tissue slices using laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS). With this method they were able to measure different elements and could show a localized distribution of Cd and Se ions in the bronchiolar area. The assumption that a close accumulation of both elements reflects the QDs were confirmed by fluorescence imaging. Inhaled QDs appeared in the same locations as the lymphocytes accumulated in the lungs. Over 17 days no degradation of the QDs was detected but they triggered inflammation. The results were supported by traditional H&E-staining.520 After extensive studies on cells via direct exposition of QDs, Zhang et al. investigated the skin permeability of QDs.521 QDs were topically applied to porcine skin via a flow-through diffusion cell system522 to assess the penetration effect. They found that QDs could penetrate through the uppermost stratum corneum layers of the epidermis and could be localized in the intracellular bilayer close to follicles. The penetration effect was enhanced when the skin was exposed to UV radiation.523
In summary, QDs have toxic effects that depend on many parameters, such as their material, composition, concentration, surface coating, and size, as well as the interacting system and the time of interaction. Due to these many factors it is unlikely that a general standard toxicity protocol for QDs will become available in the near future. Many QDs and many biological systems from in vitro cell cultures to several different in vivo animal models have been investigated. Many in vitro and in vivo studies discussed above have shown that the most important aspects of low toxicity are high stability and low concentrations and that advanced QD materials and preparation strategies can result in QDs with relatively low or negligible toxicity in some of the biological model systems. Regarding the almost infinite possibilities of combining different QDs with different biological systems as well as the long durations of representative toxicity studies it is doubtful that QD-based probes will be approved for application in humans in the near future. In cases where toxicity plays an important role for the application, careful and system-specific long-term investigations are necessary to justify the advantages QDs provide as imaging agents.
8. Conclusion and outlook
To end this review article we allow ourselves a few-sentences philosophic (or rather facetious) conclusion: QDs for imaging-based biosensing are like humans. Different sizes, shapes, compositions, concentrations, colours, time-scales, temperatures, and other properties of the QDs (or humans) and the surrounding lead to a large versatility of interactions with the biological system of interest (or the earth). There are strong (excellent photophysical properties) and weak (bad photophysical properties) QDs, good (efficient and harmless interaction with the biological system and other fluorophores) and evil (toxicity for the biological system and counterproductive interaction with other fluorophores) QDs, smart (multifunctional bioconjugates with highly preserved physicochemical and biological properties) and stupid (non-functional bioconjugates with inefficient photochemical and biological properties) QDs, rich (biosensing of large molecular ensembles) and poor (single-particle tracking) QDs, and maybe even beautiful and ugly QDs (which will strongly depend on the subjective point of view and the opinion of the society or community). However, each QD (even of very similar composition, size, and shape) can have very unique and distinct properties and a categorization is actually almost impossible although too often applied (as for humans).In a more scientific conclusion the combination of careful material composition and design with the versatile toolbox of biological recognition molecules can lead to very useful and multifunctional QD-based bioconjugates for advanced bioimaging. In this review we have summarized and discussed recent advances of QD-based fluorescence imaging for biosensing. QDs have influenced technologies (e.g., super-resolution microscopy and single-particle tracking, two-photon excitation imaging, multispectral/multicolour imaging, and multimodal imaging) as well as in vitro (e.g., cell delivery, cellular structures, functions, and environment, drug and gene delivery, and tissue diagnostics) and in vivo (e.g., investigations of the lymphatic system, tumours, and metastases) applications. On the other hand their materials (e.g., heavy metals) and their nanometric sizes have also opened an extensive discussion about their toxicity and many studies have reported different toxic effects ranging from completely harmless to very toxic. A careful analysis of QD–biosystem interactions becomes more necessary the more the bioimaging is involved in in vivo environments.
QD-based bioimaging is a relatively young research field and although applications as imaging agents in humans are a rather long-term perspective the future of QDs for more in vitro and in vivo investigations are brilliant just as the QD fluorescence. In particular, the well-studied spectroscopic advantages of using QDs within energy transfer processes such as FRET (QDs as donors and acceptors)524,525 for the detection of diffraction limited distances and the quantification of molecular interactions on the cellular and subcellular level at higher multiplexing dimensions (using PL intensity, lifetime, colour, and polarization) are very promising to further improve spatial and temporal resolution, sensitivity, and multiparameter detection. Advancement of bioimaging technologies and physicochemical and biocompatibility properties together with a more profound understanding of the biological interactions of QDs will pave the way for an even higher impact of QD-bioconjugates on bioimaging applications for the life sciences.
List of abbreviations
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| 4D | Four-dimensional |
| AD | Alzheimer's disease |
| ADM | Adriamycin |
| AFM | Atomic force microscopy |
| AMPA | Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| APP | Aminopropanol |
| aqQDs | Aqueous QDs |
| ATP | Adenosine triphosphate |
| BACE1 | Beta-site APP cleaving enzyme 1 |
| BLPs | Biotinylated lipid particles |
| BM | Basement membrane |
| BMPC | Human bone marrow derived progenitor cells |
| BOI | Biomolecules of interest |
| BRET | Bioluminescence resonance energy transfer |
| CC | Cervical carcinoma |
| CB | Conduction band |
| CCD | Charge-coupled device |
| CD4 | Cluster of differentiation 4 |
| CPPs | Cell-penetrating peptides |
| CSF | Cerebrospinal fluid |
| CT | X-ray computed tomography |
| DAPI | 4′,6-Diamidin-2-phenylindol |
| DHLA | Dihydrolipoic acid |
| DNA | Deoxyribonucleic acid |
| Dox | Doxorubicin |
| dSTORM | Direct stochastic optical reconstruction microscopy |
| E-cad | E-cadherin |
| ECM | Extracellular matrix |
| EDC | 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EGFRvIII | Epidermal growth factor receptor variant III |
| EM | Electron microscopy |
| EPR | Enhanced permeability and retention |
| ER | Estrogen receptor |
| FFPE | Formalin-fixed paraffin embedded |
| FISH | Fluorescence in situ hybridization |
| FITC | Fluoresceinisothiocyanat |
| FLIM | Fluorescence lifetime imaging |
| FPs | Fluorescent proteins |
| fPALM | Fluorescence photoactivation localization microscopy |
| FRET | Förster resonance energy transfer |
| FWHM | Full-width-at-half-maximum |
| GABA | Gamma-aminobutyric acid |
| GFP | Green fluorescent protein |
| GM1 | Monosialotetrahexosylganglioside |
| GM | Goeppert-Mayer |
| GNRs | Gold nanorods |
| GSD | Ground-state depletion |
| GSH | Glutathione |
| GST | Glutathione-s-transferase |
| GPI | Glycosylphosphatidylinositol |
| HA | Haemagglutinin |
| H&E | Hematoxylin and eosin |
| HER2 | Human epidermal growth factor receptor 2 |
| HIV | Human immunodeficiency virus |
| HIV-1 Tat | Human immunodeficiency virus 1 transcriptional activator Tat protein |
| HPC-PAA | Hydroxypropylcellulose-poly(acryl acid) |
| HSPGs | Heparin sulfate proteoglycans |
| IGF1R | Insulin-like growth factor receptor |
| IGS | Image-guided surgery |
| IHC | Immunohistochemistry |
| IR | Infrared |
| LCST | Lower critical solution temperature |
| LOD | Limit of detection |
| Luc | Luciferase |
| MRI | Magnetic resonance imaging |
| mRNA | Messenger ribonucleic acid |
| MPA | Methoxy propyl acetate |
| MSC | Mesenchymal stem cells |
| MSD | Mean square displacement |
| MSI | Multispectral imaging |
| MTM | mRNA-targeting motif |
| NGF | Nerve growth factor |
| NIg1 | Neuroligin1 |
| Ni-NTA | Ni-nitrilotriacetic acid |
| NP | Nanoparticle |
| NIR | Near infrared |
| NSET | Nanosurface energy transfer |
| NSOM | Near-field scanning optical microscopy |
| NTM | Nucleolin-targeting motif |
| PA | Polyarginine |
| PAINT | Points accumulation for imaging in nanoscale topography |
| PALM | Photoacitivated localization microscopy |
| PCD | Programmed cell death |
| PCNA | Proliferating cell nuclear antigens |
| PDMQDs | Polydopamine coated magnetic QDs |
| PEG | Poly(ethylene glycol) |
| PEI | Polyethylenimine |
| PET | Positron emission tomography |
| PL | Photoluminescence |
| PLGA | Poly(lactic-co-glycolic acid) |
| PLL | Poly(L-lysine) |
| PNIPAAm | Poly(N-iso-propylacrylamide) |
| PR | Progesterone receptor |
| PS | Phosphatidylserine |
| PSF | Point-spread-function |
| QDs | Quantum dots |
| QD-IHF | QD-based immunohistofluorometry |
| QD-ISH | QD-based in situ hybridization |
| QY | Quantum yield |
| RAG | Recombination activation gene |
| RGD | Arginylglycylaspartic acid |
| RNAi | RNA interference |
| ROI | Reactive oxygen intermediates |
| sAv | Streptavidin |
| scFv-Fc | Chimeric single chain antibodies |
| SCORE | Spatial covariance reconstructive |
| SERT | Serotonin (5-HT) transporter |
| siRNA | Small-interfering RNA |
| SMT | Single-molecule tracking |
| SNR | Signal-to-noise ratio |
| SLN | Sentinel lymph node |
| SOFI | Super-resolution optical fluctuation imaging |
| SPIONs | Superparamagnetic iron oxide nanoparticles |
| SPT | Single-particle tracking |
| SSIM | Saturated structural illumination microscopy |
| STED | Stimulated emission depletion microscopy |
| STORM | Stochastic optical reconstruction microscopy |
| TEM | Transmission electron microscopy |
| Tf/Trf | Transferrin |
| TfR | Transferrin receptor |
| TIRF | Total internal reflection microscopy |
| TMA | Tissue microarray |
| TMZ | Temozolomide |
| UV | Ultraviolet |
| VB | Valence band |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| VEGF | Vascular endothelial growth factor protein |
| VEGFR2 | Anti-vascular endothelial growth factor receptor 2 |
Acknowledgements
We thank the Agence National de la Recherche France (ANR project NanoFRET), the Innovative Medicines Initiative (IMI project OncoTrack), and the Institut Universitaire de France (IUF) for financial support.References
- L. E. Brus, J. Chem. Phys., 1984, 80, 4403–4409 CrossRef CAS PubMed.
- M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS.
- W. C. W. Chan and S. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS.
- C. Wang, X. Gao and X. Su, Anal. Bioanal. Chem., 2010, 397, 1397–1415 CrossRef CAS PubMed.
- B. Hötzer, I. L. Medintz and N. Hildebrandt, Small, 2012, 8, 2297–2326 CrossRef PubMed.
- Z. Jin and N. Hildebrandt, Trends Biotechnol., 2012, 30, 394–403 CrossRef CAS PubMed.
- I. L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11, 17–45 RSC.
- W. R. Algar, A. J. Tavares and U. J. Krull, Anal. Chim. Acta, 2010, 673, 1–25 CrossRef CAS PubMed.
- K.-T. Yong, Y. Wang, I. Roy, H. Rui, M. T. Swihart, W.-C. Law, S. K. Kwak, L. Ye, J. Liu, S. D. Mahajan and J. L. Reynolds, Theranostics, 2012, 2, 681–694 CrossRef CAS PubMed.
- M. F. Frasco and N. Chaniotakis, Anal. Bioanal. Chem., 2010, 396, 229–240 CrossRef CAS PubMed.
- Q. Ma and X. Su, Analyst, 2011, 136, 4883–4893 RSC.
- F. Chen and D. Gerion, Nano Lett., 2004, 4, 1827–1832 CrossRef CAS.
- M. Dahan, T. Laurence, F. Pinaud, D. S. Chemla, A. P. Alivisatos, M. Sauer and S. Weiss, Opt. Lett., 2001, 26, 825–827 CrossRef CAS.
- D. S. Lidke, P. Nagy, R. Heintzmann, D. J. Arndt-Jovin, J. N. Post, H. E. Grecco, E. A. Jares-Erijman and T. M. Jovin, Nat. Biotechnol., 2004, 22, 198–203 CrossRef CAS PubMed.
- F. Pinaud, D. King, H.-P. Moore and S. Weiss, J. Am. Chem. Soc., 2004, 126, 6115–6123 CrossRef CAS PubMed.
- A. Sukhanova, M. Devy, L. Venteo, H. Kaplan, M. Artemyev, V. Oleinikov, D. Klinov, M. Pluot, J. H. M. Cohen and I. Nabiev, Anal. Biochem., 2004, 324, 60–67 CrossRef CAS PubMed.
- J. O. Winter, T. Y. Liu, B. A. Korgel and C. E. Schmidt, Adv. Mater., 2001, 13, 1673–1677 CrossRef CAS.
- J. K. Jaiswal, H. Mattoussi, J. M. Mauro and S. M. Simon, Nat. Biotechnol., 2003, 21, 47–51 CrossRef CAS PubMed.
- J. K. Jaiswal, E. R. Goldman, H. Mattoussi and S. M. Simon, Nat. Methods, 2004, 1, 73–78 CrossRef PubMed.
- K. Hanaki, A. Momo, T. Oku, A. Komoto, S. Maenosono, Y. Yamaguchi and K. Yamamoto, Biochem. Biophys. Res. Commun., 2003, 302, 496–501 CrossRef CAS.
- M. E. Akerman, W. C. W. Chan, P. Laakkonen, S. N. Bhatia and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12617–12621 CrossRef CAS PubMed.
- B. Ballou, B. C. Lagerholm, L. A. Ernst, M. P. Bruchez and A. S. Waggoner, Bioconjugate Chem., 2004, 15, 79–86 CrossRef CAS PubMed.
- B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou and A. Libchaber, Science, 2002, 298, 1759–1762 CrossRef CAS PubMed.
- A. Hoshino, K. Hanaki, K. Suzuki and K. Yamamoto, Biochem. Biophys. Res. Commun., 2004, 314, 46–53 CrossRef CAS PubMed.
- R. J. Byers and E. R. Hitchman, Prog. Histochem. Cytochem., 2011, 45, 201–237 CrossRef PubMed.
- A. M. Smith, H. W. Duan, A. M. Mohs and S. Nie, Adv. Drug Delivery Rev., 2008, 60, 1226–1240 CrossRef CAS PubMed.
- P. Zrazhevskiy, M. Sena and X. Gao, Chem. Soc. Rev., 2010, 39, 4326–4354 RSC.
- M. M. Barroso, J. Histochem. Cytochem., 2011, 59, 237–251 CrossRef CAS PubMed.
- H. Mattoussi, G. Palui and H. B. Na, Adv. Drug Delivery Rev., 2012, 64, 138–166 CrossRef CAS PubMed.
- E. Petryayeva, W. R. Algar and I. L. Medintz, Appl. Spectrosc., 2013, 67, 215–252 CrossRef CAS PubMed.
- Y. Wang, R. Hu, G. Lin, I. Roy and K. T. Yong, ACS Appl. Mater. Interfaces, 2013, 5, 2786–2799 CAS.
- L. Liu, Q. Miao and G. Liang, Materials, 2013, 6, 483–499 CrossRef CAS PubMed.
- V. Biju, T. Itoh and M. Ishikawa, Chem. Soc. Rev., 2010, 39, 3031–3056 RSC.
- E. C. Wang and A. Z. Wang, Integr. Biol., 2014, 6, 9–26 RSC.
- J. Li and J. J. Zhu, Analyst, 2013, 138, 2506–2515 RSC.
- H. Xu, Q. Li, L. Wang, Y. He, J. Shi, B. Tang and C. Fan, Chem. Soc. Rev., 2014, 43, 2650–2661 RSC.
- Q. Li, L. Liu, J.-W. Liu, J.-H. Jiang, R.-Q. Yu and X. Chu, TrAC, Trends Anal. Chem., 2014, 58, 130–144 CrossRef CAS PubMed.
- V. Biju, Chem. Soc. Rev., 2014, 43, 744–764 RSC.
- N. I. Chalmers, R. J. Palmer, L. Du-Thumm, R. Sullivan, W. Shi and P. E. Kolenbrander, Appl. Environ. Microbiol., 2007, 73, 630–636 CrossRef CAS PubMed.
- P. M. A. De Farias, B. S. Santos, F. D. Menezes, A. G. Brasil, R. Ferreira, M. A. Motta, A. G. Castro-Neto, A. A. S. Vieira, D. C. N. Silva, A. Fontes and C. L. Cesar, Appl. Phys. A: Mater. Sci. Process., 2007, 89, 957–961 CrossRef CAS.
- S. Dwarakanath, J. G. Bruno, A. Shastry, T. Phillips, A. John, A. Kumar and L. D. Stephenson, Biochem. Biophys. Res. Commun., 2004, 325, 739–743 CrossRef CAS PubMed.
- J. A. Kloepfer, R. E. Mielke, M. S. Wong, K. H. Nealson, G. Stucky and J. L. Nadeau, Appl. Environ. Microbiol., 2003, 69, 4205–4213 CrossRef CAS.
- X. Xue, J. Pan, H. Xie, J. Wang and S. Zhang, Talanta, 2009, 77, 1808–1813 CrossRef CAS PubMed.
- S.-M. Wu, X. Zhao, Z.-L. Zhang, H.-Y. Xie, Z.-Q. Tian, J. Peng, Z.-X. Lu, D.-W. Pang and Z.-X. Xie, ChemPhysChem, 2006, 7, 1062–1067 CrossRef CAS PubMed.
- J. Xia, Y. Yu, Q. Liao, Y. Cao, B. Lin, X. Hu and J. Wu, J. Inorg. Biochem., 2013, 118, 39–47 CrossRef CAS PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS.
- M. A. Hines and P. Guyot-Sionnest, J. Phys. Chem., 1996, 100, 468–471 CrossRef CAS.
- Y. Tian, T. Newton, N. A. Kotov, D. M. Guldi and J. H. Fendler, J. Phys. Chem., 1996, 100, 8927–8939 CrossRef CAS.
- Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2001, 123, 183–184 CrossRef CAS.
- J. Weng, X. Song, L. Li, H. Qian, K. Chen, X. Xu, C. Cao and J. Ren, Talanta, 2006, 70, 397–402 CrossRef CAS PubMed.
- A. L. Rogach, L. Katsikas, A. Kornowski, D. Su, A. Eychmüller and H. Weller, Ber. Bunsenges. Phys. Chem., 1996, 100, 1772–1778 CrossRef CAS PubMed.
- W.-C. Law, K.-T. Yong, I. Roy, H. Ding, R. Hu, W. Zhao and P. N. Prasad, Small, 2009, 5, 1302–1310 CrossRef CAS PubMed.
- Y. Zheng, S. Gao and J. Y. Ying, Adv. Mater., 2007, 19, 376–380 CrossRef CAS PubMed.
- G. H. T. Au, W. Y. Shih and W. H. Shih, Analyst, 2013, 138, 7316–7325 RSC.
- A. C. A. Silva, S. L. V. de Deus, M. J. B. Silva and N. O. Dantas, Sens. Actuators, B, 2014, 191, 108–114 CrossRef CAS PubMed.
- J. Jia, P. Zhang, D. Gao, Z. Sheng, D. Hu, P. Gong, C. Wu, J. Chen and L. Cai, Chem. Commun., 2013, 49, 4492–4494 RSC.
- S. R. Stürzenbaum, M. Höckner, A. Panneerselvam, J. Levitt, J.-S. Bouillard, S. Taniguchi, L.-A. Dailey, R. A. Khanbeigi, E. V. Rosca, M. Thanou, K. Suhling, A. V. Zayats and M. Green, Nat. Nanotechnol., 2013, 8, 57–60 CrossRef PubMed.
- H. Trabelsi, I. Azzouz, M. Sakly and H. Abdelmelek, Int. J. Nanomed., 2013, 8, 1121–1128 CrossRef PubMed.
- D. Gerion, F. Pinaud, S. C. Williams, W. J. Parak, D. Zanchet, S. Weiss and A. P. Alivisatos, J. Phys. Chem. B, 2001, 105, 8861–8871 CrossRef CAS.
- S. Kim and M. G. Bawendi, J. Am. Chem. Soc., 2003, 125, 14652–14653 CrossRef CAS PubMed.
- S. Pathak, S.-K. Choi, N. Arnheim and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4103–4104 CrossRef CAS.
- X. Wu, H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41–46 CrossRef CAS PubMed.
- X. Gao, Y. Cui, R. M. Levenson, L. W. K. Chung and S. Nie, Nat. Biotechnol., 2004, 22, 969–976 CrossRef CAS PubMed.
- S. T. Selvan, T. T. Tan and J. Y. Ying, Adv. Mater., 2005, 17, 1620–1625 CrossRef CAS PubMed.
- M. Bottini, F. D'Annibale, A. Magrini, F. Cerignoli, Y. Arimura, M. I. Dawson, E. Bergamaschi, N. Rosato, A. Bergamaschi and T. Mustelin, Int. J. Nanomed., 2007, 2, 227–233 CAS.
- X. Wang, W. Li, B. Zhao, D. Zhang, K. Sun, X. An, Z. Zhang and Z. Shen, RSC Adv., 2013, 3, 3553–3556 RSC.
- Y. Zhu, Z. Li, M. Chen, H. M. Cooper and Z. P. Xu, J. Mater. Chem. B, 2013, 1, 2315–2323 RSC.
- Y. Zhang, M. Wang, Y.-G. Zheng, H. Tan, B. Y.-W. Hsu, Z.-C. Yang, S. Y. Wong, A. Y.-C. Chang, M. Choolani, X. Li and J. Wang, Chem. Mater., 2013, 25, 2976–2985 CrossRef CAS.
- E. L. Bentzen, I. D. Tomlinson, J. Mason, P. Gresch, M. R. Warnement, D. Wright, E. Sanders-Bush, R. Blakely and S. J. Rosenthal, Bioconjugate Chem., 2005, 16, 1488–1494 CrossRef CAS PubMed.
- B. A. Kairdolf, M. C. Mancini, A. M. Smith and S. Nie, Anal. Chem., 2008, 80, 3029–3034 CrossRef CAS PubMed.
- F. Morgner, D. Geißler, S. Stufler, N. G. Butlin, H. G. Löhmannsröben and N. Hildebrandt, Angew. Chem., Int. Ed., 2010, 49, 7570–7574 CrossRef CAS PubMed.
- K. D. Wegner, F. Morgner, E. Oh, R. Goswami, K. Susumu, M. H. Stewart, I. L. Medintz and N. Hildebrandt, Chem. Mater., 2014, 26, 4299–4312 CrossRef CAS.
- T. Pons, H. T. Uyeda, I. L. Medintz and H. Mattoussi, J. Phys. Chem. B, 2006, 110, 20308–20316 CrossRef CAS PubMed.
- W. Liu, M. Howarth, A. B. Greytak, Y. Zheng, D. G. Nocera, A. Y. Ting and M. G. Bawendi, J. Am. Chem. Soc., 2008, 130, 1274–1284 CrossRef CAS PubMed.
- B. C. Mei, K. Susumu, I. L. Medintz, J. B. Delehanty, T. J. Mountziaris and H. Mattoussi, J. Mater. Chem., 2008, 18, 4949–4958 RSC.
- K. Susumu, H. T. Uyeda, I. L. Medintz, T. Pons, J. B. Delehanty and H. Mattoussi, J. Am. Chem. Soc., 2007, 129, 13987–13996 CrossRef CAS PubMed.
- K. Susumu, E. Oh, J. B. Delehanty, J. B. Blanco-Canosa, B. J. Johnson, V. Jain, W. J. Hervey, W. R. Algar, K. Boeneman, P. E. Dawson and I. L. Medintz, J. Am. Chem. Soc., 2011, 133, 9480–9496 CrossRef CAS PubMed.
- M. Cao, L. Yu, P. Zhang, H. Xiong, Y. Jin, Y. Lu and L.-Q. Wang, Colloids Surf., B, 2013, 109, 154–160 CrossRef CAS PubMed.
- J. C. Claussen, A. Malanoski, J. C. Breger, E. Oh, S. A. Walper, K. Susumu, R. Goswami, J. R. Deschamps and I. L. Medintz, J. Phys. Chem. C, 2015, 119, 2208–2221 CAS.
- B. J. Johnson, W. R. Algar, A. P. Malanoski, M. G. Ancona and I. L. Medintz, Nano Today, 2014, 9, 102–131 CrossRef CAS PubMed.
- T. L. Jennings, S. G. Becker-Catania, R. C. Triulzi, G. Tao, B. Scott, K. E. Sapsford, S. Spindel, E. Oh, V. Jain, J. B. Delehanty, D. E. Prasuhn, K. Boeneman, W. R. Algar and I. L. Medintz, ACS Nano, 2011, 5, 5579–5593 CrossRef CAS PubMed.
- C. Schieber, A. Bestetti, J. P. Lim, A. D. Ryan, T.-L. Nguyen, R. Eldridge, A. R. White, P. A. Gleeson, P. S. Donnelly, S. J. Williams and P. Mulvaney, Angew. Chem., Int. Ed., 2012, 51, 10523–10527 CrossRef CAS PubMed.
- J. Farlow, D. Seo, K. E. Broaders, M. J. Taylor, Z. J. Gartner and Y.-W. Jun, Nat. Methods, 2013, 10, 1203–1205 CrossRef CAS PubMed.
- W. R. Algar, K. Susumu, J. B. Delehanty and I. L. Medintz, Anal. Chem., 2011, 83, 8826–8837 CrossRef CAS PubMed.
- V. Biju, T. Itoh, A. Anas, A. Sujith and M. Ishikawa, Anal. Bioanal. Chem., 2008, 391, 2469–2495 CrossRef CAS PubMed.
- W. C. W. Chan, D. J. Maxwell, X. Gao, R. E. Bailey, M. Han and S. Nie, Curr. Opin. Biotechnol., 2002, 13, 40–46 CrossRef CAS.
- A. P. Alivisatos, W. Gu and C. Larabell, Annu. Rev. Biomed. Eng., 2005, 7, 55–76 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- P. Alivisatos, Nat. Biotechnol., 2004, 22, 47–52 CrossRef CAS PubMed.
- J. M. Klostranec and W. C. W. Chan, Adv. Mater., 2006, 18, 1953–1964 CrossRef CAS PubMed.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CAS.
- J. Hu, L.-S. Li, W. Yang, L. Manna, L.-W. Wang and A. P. Alivisatos, Science, 2001, 292, 2060–2063 CrossRef CAS PubMed.
- C. A. Leatherdale, W.-K. Woo, F. V. Mikulec and M. G. Bawendi, J. Phys. Chem. B, 2002, 106, 7619–7622 CrossRef CAS.
- M. J. Levene, D. A. Dombeck, K. A. Kasischke, R. P. Molloy and W. W. Webb, J. Neurophysiol., 2004, 91, 1908–1912 CrossRef PubMed.
- T. Wang, J.-Y. Chen, S. Zhen, P.-N. Wang, C.-C. Wang, W.-L. Yang and Q. Peng, J. Fluoresc., 2009, 19, 615–621 CrossRef CAS PubMed.
- A. R. Clapp, T. Pons, I. L. Medintz, J. B. Delehanty, J. S. Melinger, T. Tiefenbrunn, P. E. Dawson, B. R. Fisher, B. O'Rourke and H. Mattoussi, Adv. Mater., 2007, 19, 1921–1926 CrossRef CAS PubMed.
- J. Dimitrijevic, L. Krapf, C. Wolter, C. Schmidtke, J.-P. Merkl, T. Jochum, A. Kornowski, A. Schüth, A. Gebert, G. Huttmann, T. Vossmeyer and H. Weller, Nanoscale, 2014, 6, 10413–10422 RSC.
- H. E. Grecco, K. A. Lidke, R. Heintzmann, D. S. Lidke, C. Spagnuolo, O. E. Martinez, E. A. Jares-Erijman and T. M. Jovin, Microsc. Res. Tech., 2004, 65, 169–179 CrossRef CAS PubMed.
- L. Zhu, S. Ang and W.-T. Liu, Appl. Environ. Microbiol., 2004, 70, 597–598 CrossRef CAS.
- Y. Choi, H. P. Kim, S. M. Hong, J. Y. Ryu, S. J. Han and R. Song, Small, 2009, 5, 2085–2091 CrossRef CAS PubMed.
- Z. Kaul, T. Yaguchi, S. C. Kaul, T. Hirano, R. Wadhwa and K. Taira, Cell Res., 2003, 13, 503–507 CrossRef PubMed.
- Y. Xiao and P. E. Barker, Nucleic Acids Res., 2004, 32, e28 CrossRef PubMed.
- L. Ma, S.-M. Wu, J. Huang, Y. Ding, D.-W. Pang and L. Li, Chromosoma, 2008, 117, 181–187 CrossRef CAS PubMed.
- N. Panchuk-Voloshina, R. P. Haugland, J. Bishop-Stewart, M. K. Bhalgat, P. J. Millard, F. Mao, W.-Y. Leung and R. P. Haugland, J. Histochem. Cytochem., 1999, 47, 1179–1188 CrossRef CAS PubMed.
- D. R. Larson, W. R. Zipfel, R. M. Williams, S. W. Clark, M. P. Bruchez, F. W. Wise and W. W. Webb, Science, 2003, 300, 1434–1436 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4588 CrossRef CAS PubMed.
- K. E. Sapsford, B. Wildt, A. Mariani, A. B. Yeatts and I. L. Medintz, in FRET-Förster Resonance Energy Transfer. From Theory to Applications, ed. I. L. Medintz and N. Hildebrandt, Wiley-VCH, Weinheim, 2013 Search PubMed.
- R. P. Haugland, The Molecular Probes® Handbook – A Guide to Fluorescent Probes and Labeling Technologies, Life Technologies Corporation, USA, 11th edn, 2010 Search PubMed.
- http://www.nobelprize.org/nobel_prizes/chemisty/laureates/2014/ .
- S. Clarke, F. Pinaud, O. Beutel, C. You, J. Piehler and M. Dahan, Nano Lett., 2010, 10, 2147–2154 CrossRef CAS PubMed.
- F. Pinaud, S. Clarke, A. Sittner and M. Dahan, Nat. Methods, 2010, 7, 275–285 CrossRef CAS PubMed.
- M. Howarth, D. J.-F. Chinnapen, K. Gerrow, P. C. Dorrestein, M. R. Grandy, N. L. Kelleher, A. El-Husseini and A. Y. Ting, Nat. Methods, 2006, 3, 267–273 CrossRef CAS PubMed.
- M. Howarth and A. Y. Ting, Nat. Protoc., 2008, 3, 534–545 CrossRef CAS PubMed.
- M. Howarth, W. Liu, S. Puthenveetil, Y. Zheng, L. F. Marshall, M. M. Schmidt, K. D. Wittrup, M. G. Bawendi and A. Y. Ting, Nat. Methods, 2008, 5, 397–399 CrossRef CAS PubMed.
- J. Xu, J. Chang, Q. Yan, T. Dertinger, M. P. Bruchez and S. Weiss, J. Phys. Chem. Lett., 2013, 4, 2138–2146 CrossRef CAS PubMed.
- N. Durisic, A. I. Bachir, D. L. Kolin, B. Hebert, B. C. Lagerholm, P. Grutter and P. W. Wiseman, Biophys. J., 2007, 93, 1338–1346 CrossRef CAS PubMed.
- A. I. Bachir, D. L. Kolin, K. G. Heinze, B. Hebert and P. W. Wiseman, J. Chem. Phys., 2008, 128, 225105 CrossRef PubMed.
- S. Boyle, D. L. Kolin, J. G. Bieler, J. P. Schneck, P. W. Wiseman and M. Edidin, Biophys. J., 2011, 101, L57–L59 CrossRef CAS PubMed.
- V. Biju, Y. Makita, T. Nagase, Y. Yamaoka, H. Yokoyama, Y. Baba and M. Ishikawa, J. Phys. Chem. B, 2005, 109, 14350–14355 CrossRef CAS PubMed.
- S. Hohng and T. Ha, J. Am. Chem. Soc., 2004, 126, 1324–1325 CrossRef CAS PubMed.
- O. Chen, J. Zhao, V. P. Chauhan, J. Cui, C. Wong, D. K. Harris, H. Wei, H.-S. Han, D. Fukumura, R. K. Jain and M. G. Bawendi, Nat. Mater., 2013, 12, 445–451 CrossRef CAS PubMed.
- M. H. W. Stopel, J. C. Prangsma, C. Blum and V. Subramaniam, RSC Adv., 2013, 3, 17440–17445 RSC.
- M. Nirmal, B. O. Dabbousi, M. G. Bawendi, J. J. Macklin, J. K. Trautman, T. D. Harris and L. E. Brus, Nature, 1996, 383, 802–804 CrossRef CAS.
- K. A. Lidke, B. Rieger, T. M. Jovin and R. Heintzmann, Opt. Express, 2005, 13, 7052–7062 CrossRef.
- T. Dertinger, R. Colyer, G. Iyer, S. Weiss and J. Enderlein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22287–22292 CrossRef CAS PubMed.
- T. M. Watanabe, S. Fukui, T. Jin, F. Fujii and T. Yanagida, Biophys. J., 2010, 99, L50–L52 CrossRef CAS PubMed.
- F. C. Chien, C. W. Kuo and P. Chen, Analyst, 2011, 136, 1608–1613 RSC.
- P. Hoyer, T. Staudt, J. Engelhardt and S. W. Hell, Nano Lett., 2011, 11, 245–250 CrossRef CAS PubMed.
- Y. Wang, G. Fruhwirth, E. Cai, T. Ng and P. R. Selvin, Nano Lett., 2013, 13, 5233–5241 CrossRef CAS PubMed.
- X. Shi, Z. Xie, Y. Song, Y. Tan, E. S. Yeung and H. Gai, Anal. Chem., 2012, 84, 1504–1509 CrossRef CAS PubMed.
- Y. Deng, M. Sun, P.-H. Lin, J. Ma and J. W. Shaevitz, PLoS One, 2014, 9, e94807 Search PubMed.
- O. Schwartz, J. M. Levitt, R. Tenne, S. Itzhakov, Z. Deutsch and D. Oron, Nano Lett., 2013, 13, 5832–5836 CrossRef CAS PubMed.
- A. Sporbert, Z. Cseresnyes, M. Heidbreder, P. Domaing, S. Hauser, B. Kaltschmidt, C. Kaltschmidt, M. Heilemann and D. Widera, PLoS One, 2013, 8, e64023 Search PubMed.
- J. Chen, Y. Wu, C. Wang and J. Cai, Scanning, 2008, 30, 448–451 CrossRef PubMed.
- L. Zhong, G. Zeng, X. Lu, R. C. Wang, G. Gong, L. Yan, D. Huang and Z. W. Chen, PLoS One, 2009, 4, e5945 Search PubMed.
- J. Chen, Y. Pei, Z. Chen and J. Cai, Micron, 2010, 41, 198–202 CrossRef CAS PubMed.
- Y. Chen, L. Shao, Z. Ali, J. Cai and Z. W. Chen, Blood, 2008, 111, 4220–4232 CrossRef CAS PubMed.
- L. Y. Zhong, Z. Zhang, X. X. Lu, S. P. Liu, C. Y. Chen and Z. W. Chen, BioMed Res. Int., 2013, 2013, 276498 Search PubMed.
- S. A. Diaz, F. Gillanders, E. A. Jares-Erijman and T. M. Jovin, Nat. Commun., 2015, 6, 6036 CrossRef PubMed.
- P. Pierobon and G. Cappello, Adv. Drug Delivery Rev., 2012, 64, 167–178 CrossRef CAS PubMed.
- M. J. Murcia, D. E. Minner, G.-M. Mustata, K. Ritchie and C. A. Naumann, J. Am. Chem. Soc., 2008, 130, 15054–15062 CrossRef CAS PubMed.
- N. P. Wells, G. A. Lessard and J. H. Werner, Anal. Chem., 2008, 80, 9830–9834 CrossRef CAS PubMed.
- T. Pons and H. Mattoussi, Ann. Biomed. Eng., 2009, 37, 1934–1959 CrossRef PubMed.
- N. G. Walter, C.-Y. Huang, A. J. Manzo and M. A. Sobhy, Nat. Methods, 2008, 5, 475–489 CrossRef CAS PubMed.
- N. Ruthardt, D. C. Lamb and C. Bräuchle, Mol. Ther., 2011, 19, 1199–1211 CrossRef CAS PubMed.
- M. P. Clausen and B. C. Lagerholm, Nano Lett., 2013, 13, 2332–2337 CrossRef CAS PubMed.
- A. Serge, N. Bertaux, H. Rigneault and D. Marguet, Nat. Methods, 2008, 5, 687–694 CrossRef CAS PubMed.
- K. Jaqaman, D. Loerke, M. Mettlen, H. Kuwata, S. Grinstein, S. L. Schmid and G. Danuser, Nat. Methods, 2008, 5, 695–702 CrossRef CAS PubMed.
- L. Holtzer, T. Meckel and T. Schmidt, Appl. Phys. Lett., 2007, 90, 053902 CrossRef PubMed.
- S. Ram, P. Prabhat, J. Chao, E. S. Ward and R. J. Ober, Biophys. J., 2008, 95, 6025–6043 CrossRef CAS PubMed.
- N. P. Wells, G. A. Lessard, P. M. Goodwin, M. E. Phipps, P. J. Cutler, D. S. Lidke, B. S. Wilson and J. H. Werner, Nano Lett., 2010, 10, 4732–4737 CrossRef CAS PubMed.
- T. M. Watanabe, F. Fujii, T. Jin, E. Umemoto, M. Miyasaka, H. Fujita and T. Yanagida, Biophys. J., 2013, 105, 555–564 CrossRef CAS PubMed.
- T. Tani, M. Oda, D. Araki, T. Miyashita, K. Nakajima, M. Arita and M. Yohda, J. Lumin., 2014, 152, 88–92 CrossRef CAS PubMed.
- M. Dahan, S. Levi, C. Luccardini, P. Rostaing, B. Riveau and A. Triller, Science, 2003, 302, 442–445 CrossRef CAS PubMed.
- M.-V. Ehrensperger, C. Hanus, C. Vannier, A. Triller and M. Dahan, Biophys. J., 2007, 92, 3706–3718 CrossRef CAS PubMed.
- C. Bouzigues, M. Morel, A. Triller and M. Dahan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11251–11256 CrossRef CAS PubMed.
- T. M. Watanabe and H. Higuchi, Biophys. J., 2007, 92, 4109–4120 CrossRef CAS PubMed.
- D. S. Lidke, K. A. Lidke, B. Rieger, T. M. Jovin and D. J. Arndt-Jovin, J. Cell Biol., 2005, 170, 619–626 CrossRef CAS PubMed.
- N. Kawashima, K. Nakayama, K. Itoh, T. Itoh, M. Ishikawa and V. Biju, Chem. – Eur. J., 2010, 16, 1186–1192 CrossRef CAS PubMed.
- P. Mascalchi, A. S. Lamort, L. Salome and F. Dumas, Biochem. Biophys. Res. Commun., 2012, 417, 409–413 CrossRef CAS PubMed.
- J. Huang, M. Brameshuber, X. Zeng, J. Xie, Q.-J. Li, Y.-H. Chien, S. Valitutti and M. M. Davis, Immunity, 2013, 39, 846–857 CrossRef CAS PubMed.
- V. Roullier, S. Clarke, C. You, F. Pinaud, G. Gouzer, D. Schaible, V. Marchi-Artzner, J. Piehler and M. Dahan, Nano Lett., 2009, 9, 1228–1234 CrossRef CAS PubMed.
- B. Cui, C. Wu, L. Chen, A. Ramirez, E. L. Bearer, W.-P. Li, W. C. Mobley and S. Chu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13666–13671 CrossRef CAS PubMed.
- S. S. Rajan, H. Y. Liu and T. Q. Vu, ACS Nano, 2008, 2, 1153–1166 CrossRef CAS PubMed.
- H. Chen, I. Titushkin, M. Stroscio and M. Cho, Biophys. J., 2007, 92, 1399–1408 CrossRef CAS PubMed.
- O. Lieleg, M. Lopez-Garcia, C. Semmrich, J. Auernheimer, H. Kessler and A. R. Bausch, Small, 2007, 3, 1560–1565 CrossRef CAS PubMed.
- K. M. Fichter, M. Flajolet, P. Greengard and T. Q. Vu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18658–18663 CrossRef CAS PubMed.
- J. M. Crane and A. S. Verkman, Biophys. J., 2008, 94, 702–713 CrossRef CAS PubMed.
- I. Chung, R. Akita, R. Vandlen, D. Toomre, J. Schlessinger and I. Mellman, Nature, 2010, 464, 783–788 CrossRef CAS PubMed.
- A. R. Lowe, J. J. Siegel, P. Kalab, M. Siu, K. Weis and J. T. Liphardt, Nature, 2010, 467, 600–604 CrossRef CAS PubMed.
- J. C. Chang, I. D. Tomlinson, M. R. Warnement, A. Ustione, A. M. D. Carneiro, D. W. Piston, R. D. Blakely and S. J. Rosenthal, J. Neurosci., 2012, 32, 8919–8929 CrossRef CAS PubMed.
- J. C. Chang and S. J. Rosenthal, ACS Chem. Neurosci., 2012, 3, 737–743 CrossRef CAS PubMed.
- L. Q. Chen, S. J. Xiao, P. P. Hu, L. Peng, J. Ma, L. F. Luo, Y. F. Li and C. Z. Huang, Anal. Chem., 2012, 84, 3099–3110 CrossRef CAS PubMed.
- J. Ma, L. Wu, Z. Hou, Y. Song, L. Wang and W. Jiang, Biomaterials, 2014, 35, 7042–7049 CrossRef CAS PubMed.
- S. Ram, D. Kim, R. J. Ober and E. S. Ward, Biophys. J., 2012, 103, 1594–1603 CrossRef CAS PubMed.
- B. Biermann, S. Sokoll, J. Klueva, M. Missler, J. S. Wiegert, J. B. Sibarita and M. Heine, Nat. Commun., 2014, 5, 3024 CAS.
- C. Dong, B. Chowdhury and J. Irudayaraj, Analyst, 2013, 138, 2871–2876 RSC.
- J. Liu, X. Yang, K. Wang, Q. Wang, W. Liu and D. Wang, Nanoscale, 2013, 5, 11257–11264 RSC.
- M. P. Bruchez, Curr. Opin. Chem. Biol., 2011, 15, 775–780 CrossRef CAS PubMed.
- D. M. Warshaw, G. G. Kennedy, S. S. Work, E. B. Krementsova, S. Beck and K. M. Trybus, Biophys. J., 2005, 88, L30–L32 CrossRef CAS PubMed.
- S. Courty, C. Luccardini, Y. Bellaiche, G. Cappello and M. Dahan, Nano Lett., 2006, 6, 1491–1495 CrossRef CAS PubMed.
- J. Yoo, T. Kambara, K. Gonda and H. Higuchi, Exp. Cell Res., 2008, 314, 3563–3569 CrossRef CAS PubMed.
- B. Nitzsche, F. Ruhnow and S. Diez, Nat. Nanotechnol., 2008, 3, 552–556 CrossRef CAS PubMed.
- J. Yajima, K. Mizutani and T. Nishizaka, Nat. Struct. Mol. Biol., 2008, 15, 1119–1121 CAS.
- P. Pierobon, S. Achouri, S. Courty, A. R. Dunn, J. A. Spudich, M. Dahan and G. Cappello, Biophys. J., 2009, 96, 4268–4275 CrossRef CAS PubMed.
- S. R. Nelson, M. Y. Ali, K. M. Trybus and D. M. Warshaw, Biophys. J., 2009, 97, 509–518 CrossRef CAS PubMed.
- R. Zhang, E. Rothenberg, G. Fruhwirth, P. D. Simonson, F. Ye, I. Golding, T. Ng, W. Lopes and P. R. Selvin, Nano Lett., 2011, 11, 4074–4078 CrossRef CAS PubMed.
- M. Ohmachi, Y. Komori, A. H. Iwane, F. Fujii, T. Jin and T. Yanagida, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5294–5298 CrossRef CAS PubMed.
- Y. Wang, K. Ajtai and T. P. Burghardt, Biochemistry, 2013, 52, 1611–1621 CrossRef CAS PubMed.
- M. Chen, X. He, K. Wang, D. He, X. Yang and H. Shi, TrAC, Trends Anal. Chem., 2014, 58, 120–129 CrossRef CAS PubMed.
- F. Osaki, T. Kanamori, S. Sando, T. Sera and Y. Aoyama, J. Am. Chem. Soc., 2004, 126, 6520–6521 CrossRef CAS PubMed.
- I. Nabiev, S. Mitchell, A. Davies, Y. Williams, D. Kelleher, R. Moore, Y. K. Gun'ko, S. Byrne, Y. P. Rakovich, J. F. Donegan, A. Sukhanova, J. Conroy, D. Cottell, N. Gaponik, A. Rogach and Y. Volkov, Nano Lett., 2007, 7, 3452–3461 CrossRef CAS PubMed.
- L. W. Zhang and N. A. Monteiro-Riviere, Toxicol. Sci., 2009, 110, 138–155 CrossRef CAS PubMed.
- X. Jiang, C. Röcker, M. Hafner, S. Brandholt, R. M. Dörlich and G. U. Nienhaus, ACS Nano, 2010, 4, 6787–6797 CrossRef CAS PubMed.
- S. J. Tan, N. R. Jana, S. Gao, P. K. Patra and J. Y. Ying, Chem. Mater., 2010, 22, 2239–2247 CrossRef CAS.
- J. P. Ryman-Rasmussen, J. E. Riviere and N. A. Monteiro-Riviere, Nano Lett., 2007, 7, 1344–1348 CrossRef CAS PubMed.
- S. Barua and K. Rege, Small, 2009, 5, 370–376 CrossRef CAS PubMed.
- T. Wang and X. Jiang, ACS Appl. Mater. Interfaces, 2013, 5, 1190–1196 CAS.
- J. Lee, Y. Choi, Y. Cho and R. Song, J. Nanosci. Nanotechnol., 2013, 13, 417–422 CrossRef CAS PubMed.
- F. Li, Z.-P. Zhang, J. Peng, Z.-Q. Cui, D.-W. Pang, K. Li, H.-P. Wei, Y.-F. Zhou, J.-K. Wen and X.-E. Zhang, Small, 2009, 5, 718–726 CrossRef CAS PubMed.
- U. Hasegawa, S.-I. M. Nomura, S. C. Kaul, T. Hirano and K. Akiyoshi, Biochem. Biophys. Res. Commun., 2005, 331, 917–921 CrossRef CAS PubMed.
- W. T. Al-Jamal, K. T. Al-Jamal, P. H. Bomans, P. M. Frederik and K. Kostarelos, Small, 2008, 4, 1406–1415 CrossRef CAS PubMed.
- W. T. Al-Jamal, K. T. Al-Jamal, B. Tian, L. Lacerda, P. H. Bomans, P. M. Frederik and K. Kostarelos, ACS Nano, 2008, 2, 408–418 CrossRef CAS PubMed.
- C. Ye, Y. Wang, C. Li, J. Yu and Y. Hu, Microchim. Acta, 2013, 180, 117–125 CrossRef CAS.
- M. Camblin, P. Detampel, H. Kettiger, D. Wu, V. Balasubramanian and J. Huwyler, Int. J. Nanomed., 2014, 9, 2287–2298 Search PubMed.
- K. Medepalli, B. W. Alphenaar, R. S. Keynton and P. Sethu, Nanotechnology, 2013, 24, 205101 CrossRef PubMed.
- H. Duan and S. Nie, J. Am. Chem. Soc., 2007, 129, 3333–3338 CrossRef CAS PubMed.
- H. N. Yang, J. S. Park, S. Y. Jeon, W. Park, K. Na and K.-H. Park, Biomaterials, 2014, 35, 8439–8449 CrossRef PubMed.
- A. R. Bayles, H. S. Chahal, D. S. Chahal, C. P. Goldbeck, B. E. Cohen and B. A. Helms, Nano Lett., 2010, 10, 4086–4092 CrossRef CAS PubMed.
- B. Y. Kim, W. Jiang, J. Oreopoulos, C. M. Yip, J. T. Rutka and W. C. Chan, Nano Lett., 2008, 8, 3887–3892 CrossRef CAS PubMed.
- H. Mok, J. W. Park and T. G. Park, Bioconjugate Chem., 2008, 19, 797–801 CrossRef CAS PubMed.
- Y. L. Pan, J. Y. Cai, L. Qin and H. Wang, Acta Biochim. Biophys. Sin., 2006, 38, 646–652 CrossRef CAS PubMed.
- K. C. Weng, C. O. Noble, B. Papahadjopoulos-Sternberg, F. F. Chen, D. C. Drummond, D. B. Kirpotin, D. Wang, Y. K. Hom, B. Hann and J. W. Park, Nano Lett., 2008, 8, 2851–2857 CrossRef CAS PubMed.
- G. Xu, K.-T. Yong, I. Roy, S. D. Mahajan, H. Ding, S. A. Schwartz and P. N. Prasad, Bioconjugate Chem., 2008, 19, 1179–1185 CrossRef CAS PubMed.
- Y. Xu, Q. Wang, P. He, Q. Dong, F. Liu, Y. Liu, L. Lin, H. Yan and X. Zhao, Adv. Mater., 2008, 20, 3468–3473 CrossRef CAS PubMed.
- J. Lee, Y. Choi, K. Kim, S. Hong, H.-Y. Park, T. Lee, G. J. Cheon and R. Song, Bioconjugate Chem., 2010, 21, 940–946 CrossRef CAS PubMed.
- F. Wang, Y. Wang, X. Zhang, W. Zhang, S. Guo and F. Jin, J. Controlled Release, 2014, 174, 126–136 CrossRef CAS PubMed.
- B. C. Lagerholm, M. Wang, L. A. Ernst, D. H. Ly, H. Liu, M. P. Bruchez and A. S. Waggoner, Nano Lett., 2004, 4, 2019–2022 CrossRef CAS.
- J. B. Delehanty, I. L. Medintz, T. Pons, F. M. Brunel, P. E. Dawson and H. Mattoussi, Bioconjugate Chem., 2006, 17, 920–927 CrossRef CAS PubMed.
- S. M. Rozenzhak, M. P. Kadakia, T. M. Caserta, T. R. Westbrook, M. O. Stone and R. R. Naik, Chem. Commun., 2005, 2217–2219 RSC.
- F. L. Xue, J. Y. Chen, J. Guo, C. C. Wang, W. L. Yang, P. N. Wang and D. R. Lu, J. Fluoresc., 2007, 17, 149–154 CrossRef CAS PubMed.
- Y. E. Koshman, S. B. Waters, L. A. Walker, T. Los, P. de Tombe, P. H. Goldspink and B. Russell, J. Mol. Cell. Cardiol., 2008, 45, 853–856 CrossRef CAS PubMed.
- Y. Lei, H. Tang, L. Yao, R. Yu, M. Feng and B. Zou, Bioconjugate Chem., 2008, 19, 421–427 CrossRef CAS PubMed.
- Y. Zhang, M. K. So and J. Rao, Nano Lett., 2006, 6, 1988–1992 CrossRef CAS PubMed.
- G. Ruan, A. Agrawal, A. I. Marcus and S. Nie, J. Am. Chem. Soc., 2007, 129, 14759–14766 CrossRef CAS PubMed.
- B. Chen, Q. Liu, Y. Zhang, L. Xu and X. Fang, Langmuir, 2008, 24, 11866–11871 CrossRef CAS PubMed.
- Y. Suzuki, C. N. Roy, W. Promjunyakul, H. Hatakeyama, K. Gonda, J. Imamura, B. Vasudevanpillai, N. Ohuchi, M. Kanzaki, H. Higuchi and M. Kaku, Mol. Cell. Biol., 2013, 33, 3036–3049 CrossRef CAS PubMed.
- I. L. Medintz, T. Pons, J. B. Delehanty, K. Susumu, F. M. Brunel, P. E. Dawson and H. Mattoussi, Bioconjugate Chem., 2008, 19, 1785–1795 CrossRef CAS PubMed.
- C. Kim, Y. Lee, J. S. Kim, J. H. Jeong and T. G. Park, Langmuir, 2010, 26, 14965–14969 CrossRef CAS PubMed.
- K. Boeneman Gemmill, M. Muttenthaler, J. B. Delehanty, M. H. Stewart, K. Susumu, P. E. Dawson and I. L. Medintz, Anal. Bioanal. Chem., 2013, 405, 6145–6154 CrossRef PubMed.
- K. Boeneman, J. B. Delehanty, J. B. Blanco-Canosa, K. Susumu, M. H. Stewart, E. Oh, A. L. Huston, G. Dawson, S. Ingale, R. Walters, M. Domowicz, J. R. Deschamps, W. R. Algar, S. DiMaggio, J. Manono, C. M. Spillmann, D. Thompson, T. L. Jennings, P. E. Dawson and I. L. Medintz, ACS Nano, 2013, 7, 3778–3796 CrossRef CAS PubMed.
- J. B. Delehanty, C. E. Bradburne, K. Susumu, K. Boeneman, B. C. Mei, D. Farrell, J. B. Blanco-Canosa, P. E. Dawson, H. Mattoussi and I. L. Medintz, J. Am. Chem. Soc., 2011, 133, 10482–10489 CrossRef CAS PubMed.
- E. Yaghini, F. Giuntini, I. M. Eggleston, K. Suhling, A. M. Seifalian and A. J. MacRobert, Small, 2014, 10, 782–792 CrossRef CAS PubMed.
- B. R. Liu, Y.-W. Huang, J. G. Winiarz, H.-J. Chiang and H.-J. Lee, Biomaterials, 2011, 32, 3520–3537 CrossRef CAS PubMed.
- V. Biju, D. Muraleedharan, K. Nakayama, Y. Shinohara, T. Itoh, Y. Baba and M. Ishikawa, Langmuir, 2007, 23, 10254–10261 CrossRef CAS PubMed.
- A. Anas, T. Okuda, N. Kawashima, K. Nakayama, T. Itoh, M. Ishikawa and V. Biju, ACS Nano, 2009, 3, 2419–2429 CrossRef CAS PubMed.
- S. K. Chakraborty, J. A. J. Fitzpatrick, J. A. Phillippi, S. Andreko, A. S. Waggoner, M. P. Bruchez and B. Ballou, Nano Lett., 2007, 7, 2618–2626 CrossRef CAS PubMed.
- C. Tekle, B. van Deurs, K. Sandvig and T.-G. Iversen, Nano Lett., 2008, 8, 1858–1865 CrossRef CAS PubMed.
- L. C. Wu, L. W. Chu, L. W. Lo, Y. C. Liao, Y. C. Wang and C. S. Yang, ACS Nano, 2013, 7, 365–375 CrossRef CAS PubMed.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Adv. Mater., 2004, 16, 961–966 CrossRef CAS PubMed.
- E. Muro, A. Fragola, T. Pons, N. Lequeux, A. Ioannou, P. Skourides and B. Dubertret, Small, 2012, 8, 1029–1037 CrossRef CAS PubMed.
- L. Damalakiene, V. Karabanovas, S. Bagdonas, M. Valius and R. Rotomskis, Int. J. Nanomed., 2013, 8, 555–568 CrossRef PubMed.
- K. Yum, S. Na, Y. Xiang, N. Wang and M.-F. Yu, Nano Lett., 2009, 9, 2193–2198 CrossRef CAS PubMed.
- S. Park, Y.-S. Kim, W. B. Kim and S. Jon, Nano Lett., 2009, 9, 1325–1329 CrossRef CAS PubMed.
- W. J. Parak, R. Boudreau, M. Le Gros, D. Gerion, D. Zanchet, C. M. Micheel, S. C. Williams, A. P. Alivisatos and C. Larabell, Adv. Mater., 2002, 14, 882–885 CrossRef CAS.
- S. Danner, H. Benzin, T. Vollbrandt, J. Oder, A. Richter and C. Kruse, Int. J. Cell Biol., 2013, 2013, 918242 CAS.
- B. S. Shah, P. A. Clark, E. K. Moioli, M. A. Stroscio and J. J. Mao, Nano Lett., 2007, 7, 3071–3079 CrossRef CAS PubMed.
- S. S. Rajan and T. Q. Vu, Nano Lett., 2006, 6, 2049–2059 CrossRef CAS PubMed.
- A. Cambi, D. S. Lidke, D. J. Arndt-Jovin, C. G. Figdor and T. M. Jovin, Nano Lett., 2007, 7, 970–977 CrossRef CAS PubMed.
- X. Gao, T. Wang, B. Wu, J. Chen, J. Chen, Y. Yue, N. Dai, H. Chen and X. Jiang, Biochem. Biophys. Res. Commun., 2008, 377, 35–40 CrossRef CAS PubMed.
- S. J. Rosenthal, I. Tomlinson, E. M. Adkins, S. Schroeter, S. Adams, L. Swafford, J. McBride, Y. Wang, L. J. DeFelice and R. D. Blakely, J. Am. Chem. Soc., 2002, 124, 4586–4594 CrossRef CAS PubMed.
- S. H. Young and E. Rozengurt, Am. J. Physiol.: Cell Physiol., 2006, 290, C728–C732 CrossRef CAS PubMed.
- J. Lee, Y.-J. Kwon, Y. Choi, H. C. Kim, K. Kim, J. Kim, S. Park and R. Song, ChemBioChem, 2012, 13, 1503–1508 CrossRef CAS PubMed.
- T. C. Chu, F. Shieh, L. A. Lavery, M. Levy, R. Richards-Kortum, B. A. Korgel and A. D. Ellington, Biosens. Bioelectron., 2006, 21, 1859–1866 CrossRef CAS PubMed.
- X.-C. Chen, Y.-L. Deng, Y. Lin, D.-W. Pang, H. Qing, F. Qu and H.-Y. Xie, Nanotechnology, 2008, 19, 235105 CrossRef PubMed.
- L. Y. Lee, S. L. Ong, J. Y. Hu, W. J. Ng, Y. Feng, X. Tan and S. W. Wong, Appl. Environ. Microbiol., 2004, 70, 5732–5736 CrossRef CAS PubMed.
- B. Barat, S. J. Sirk, K. E. McCabe, J. Li, E. J. Lepin, R. Remenyi, A. L. Koh, T. Olafsen, S. S. Gambhir, S. Weiss and A. M. Wu, Bioconjugate Chem., 2009, 20, 1474–1481 CrossRef CAS PubMed.
- J. Qian, K.-T. Yong, I. Roy, T. Y. Ohulchanskyy, E. J. Bergey, H. H. Lee, K. M. Tramposch, S. L. He, A. Maitra and P. N. Prasad, J. Phys. Chem. B, 2007, 111, 6969–6972 CrossRef CAS PubMed.
- M. Howarth, K. Takao, Y. Hayashi and A. Y. Ting, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7583–7588 CrossRef CAS PubMed.
- I. Chen, Y.-A. Choi and A. Y. Ting, J. Am. Chem. Soc., 2007, 129, 6619–6625 CrossRef CAS PubMed.
- S. Le Gac, I. Vermes and A. van den Berg, Nano Lett., 2006, 6, 1863–1869 CrossRef CAS PubMed.
- D.-H. Huang, L. Su, X.-H. Peng, H. Zhang, F. R. Khuri, D. M. Shin and Z. Chen, Nanotechnology, 2009, 20, 225102 CrossRef PubMed.
- Y. Zhang, J.-M. Liu and X.-P. Yan, Anal. Chem., 2013, 85, 228–234 CrossRef CAS PubMed.
- J. Kim, H.-Y. Park, J. Kim, J. Ryu, D. Y. Kwon, R. Grailhe and R. Song, Chem. Commun., 2008, 1910–1912 RSC.
- P. K. Bae, K. N. Kim, S. J. Lee, H. J. Chang, C. K. Lee and J. K. Park, Biomaterials, 2009, 30, 836–842 CrossRef CAS PubMed.
- K. Boeneman, J. B. Delehanty, K. Susumu, M. H. Stewart and I. L. Medintz, J. Am. Chem. Soc., 2010, 132, 5975–5977 CrossRef CAS PubMed.
- M.-K. So, H. Yao and J. Rao, Biochem. Biophys. Res. Commun., 2008, 374, 419–423 CrossRef CAS PubMed.
- M. Sunbul, M. Yen, Y. Zou and J. Yin, Chem. Commun., 2008, 5927–5929 RSC.
- R. L. Orndorff and S. J. Rosenthal, Nano Lett., 2009, 9, 2589–2599 CrossRef CAS PubMed.
- W. J. Kang, J. R. Chae, Y. L. Cho, J. D. Lee and S. Kim, Small, 2009, 5, 2519–2522 CrossRef CAS PubMed.
- M. H. Ko, S. Kim, W. J. Kang, J. H. Lee, H. Kang, S. H. Moon, D. W. Hwang, H. Y. Ko and D. S. Lee, Small, 2009, 5, 1207–1212 CAS.
- P. Zrazhevskiy and X. Gao, Nat. Commun., 2013, 4, 1619 CrossRef PubMed.
- P. Zrazhevskiy, L. D. True and X. Gao, Nat. Protoc., 2013, 8, 1852–1869 CrossRef CAS PubMed.
- A. K. Chen, M. A. Behlke and A. Tsourkas, Nucleic Acids Res., 2007, 35, e105 CrossRef PubMed.
- L. Helmick, A. A. de Mayolo, Y. Zhang, C. M. Cheng, S. C. Watkins, C. Y. Wu and P. R. Leduc, Nano Lett., 2008, 8, 1303–1308 CrossRef CAS PubMed.
- K. Higashi, T. Jin and E. Takahashi, Adv. Exp. Med. Biol., 2013, 789, 379–383 CrossRef CAS.
- D.-W. Li, L.-X. Qin, Y. Li, R. P. Nia, Y.-T. Long and H.-Y. Chen, Chem. Commun., 2011, 47, 8539–8541 RSC.
- M. J. Ruedas-Rama, A. Orte, E. A. H. Hall, J. M. Alvarez-Pez and E. M. Talavera, Analyst, 2012, 137, 1500–1508 RSC.
- A. I. Zamaleeva, M. Collot, E. Bahembera, C. Tisseyre, P. Rostaing, A. V. Yakovlev, M. Oheim, M. de Waard, J.-M. Mallet and A. Feltz, Nano Lett., 2014, 14, 2994–3001 CrossRef CAS PubMed.
- W. Zhang, P. Zhang, S. Zhang and C. Zhu, Anal. Methods, 2014, 6, 2499–2505 RSC.
- G. W. Walker, V. C. Sundar, C. M. Rudzinski, A. W. Wun, M. G. Bawendi and D. G. Nocera, Appl. Phys. Lett., 2003, 83, 3555–3557 CrossRef CAS PubMed.
- L. M. Maestro, C. Jacinto, U. R. Silva, F. Vetrone, J. A. Capobianco, D. Jaque and J. G. Sole, Small, 2011, 7, 1774–1778 CrossRef CAS PubMed.
- M. J. Ruedas-Rama, A. Orte, E. A. H. Hall, J. M. Alvarez-Pez and E. M. Talavera, Chem. Commun., 2011, 47, 2898–2900 RSC.
- A. Orte, J. M. Alvarez-Pez and M. J. Ruedas-Rama, ACS Nano, 2013, 7, 6387–6395 CrossRef CAS PubMed.
- J. M. Yang, H. Yang and L. Lin, ACS Nano, 2011, 5, 5067–5071 CrossRef CAS PubMed.
- P. Haro-Gonzalez, W. T. Ramsay, L. M. Maestro, B. del Rosal, K. Santacruz-Gomez, M. D. Iglesias-de la Cruz, F. Sanz-Rodriguez, J. Y. Chooi, P. R. Sevilla, M. Bettinelli, D. Choudhury, A. K. Kar, J. G. Sole, D. Jaque and L. Paterson, Small, 2013, 9, 2162–2170 CrossRef CAS PubMed.
- I. L. Medintz, M. H. Stewart, S. A. Trammell, K. Susumu, J. B. Delehanty, B. C. Mei, J. S. Melinger, J. B. Blanco-Canosa, P. E. Dawson and H. Mattoussi, Nat. Mater., 2010, 9, 676–684 CrossRef CAS PubMed.
- M. Gui, L. Bao, Y. Xia, C. Wei, S. Zhang and C. Zhu, Biosens. Bioelectron., 2011, 30, 324–327 CrossRef CAS PubMed.
- P. T. Snee, R. C. Somers, G. Nair, J. P. Zimmer, M. G. Bawendi and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 13320–13321 CrossRef CAS PubMed.
- A. M. Dennis, W. J. Rhee, D. Sotto, S. N. Dublin and G. Bao, ACS Nano, 2012, 6, 2917–2924 CrossRef CAS PubMed.
- R. Khatchadourian, A. Bachir, S. J. Clarke, C. D. Heyes, P. W. Wiseman and J. L. Nadeau, J. Biomed. Biotechnol., 2007, 2007, 70145 Search PubMed.
- S. J. Clarke, C. A. Hollmann, Z. Zhang, D. Suffern, S. E. Bradforth, N. M. Dimitrijevic, W. G. Minarik and J. L. Nadeau, Nat. Mater., 2006, 5, 409–417 CrossRef CAS PubMed.
- L. Shi, V. De Paoli, N. Rosenzweig and Z. Rosenzweig, J. Am. Chem. Soc., 2006, 128, 10378–10379 CrossRef CAS PubMed.
- R. Freeman, R. Gill, I. Shweky, M. Kotler, U. Banin and I. Willner, Angew. Chem., Int. Ed., 2009, 48, 309–313 CrossRef CAS PubMed.
- X. Li, D. Deng, J. Xue, L. Qu, S. Achilefu and Y. Gu, Biosens. Bioelectron., 2014, 61, 512–518 CrossRef CAS PubMed.
- B. Hu, L.-L. Hu, M.-L. Chen and J.-H. Wang, Biosens. Bioelectron., 2013, 49, 499–505 CrossRef CAS PubMed.
- Y. Choi, Y. Cho, M. Kim, R. Grailhe and R. Song, Anal. Chem., 2012, 84, 8595–8601 CrossRef CAS PubMed.
- W. Wei, X. He and N. Ma, Angew. Chem., Int. Ed., 2014, 53, 5573–5577 CrossRef CAS PubMed.
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS PubMed.
- S. Steponkiene, J. Valanciunaite, A. Skripka and R. Rotomskis, J. Biomed. Nanotechnol., 2014, 10, 679–686 CrossRef CAS PubMed.
- H. Zhang, D. Sachdev, C. Wang, A. Hubel, M. Gaillard-Kelly and D. Yee, Breast Cancer Res. Treat., 2009, 114, 277–285 CrossRef CAS PubMed.
- J. Li, C. Wu, F. Gao, R. Zhang, G. Lv, D. Fu, B. Chen and X. Wang, Bioorg. Med. Chem. Lett., 2006, 16, 4808–4812 CrossRef CAS PubMed.
- K. V. Chakravarthy, B. A. Davidson, J. D. Helinski, H. Ding, W.-C. Law, K.-T. Yong, P. N. Prasad and P. R. Knight, Nanomedicine, 2011, 7, 88–96 CrossRef CAS PubMed.
- V. Bagalkot, L. Zhang, E. Levy-Nissenbaum, S. Jon, P. W. Kantoff, R. Langer and O. C. Farokhzad, Nano Lett., 2007, 7, 3065–3070 CrossRef CAS PubMed.
- X. Chen, Y. Tang, B. Cai and H. Fan, Nanotechnology, 2014, 25, 235101 CrossRef PubMed.
- M.-L. Chen, Y.-J. He, X.-W. Chen and J.-H. Wang, Bioconjugate Chem., 2013, 24, 387–397 CrossRef CAS PubMed.
- S. Y. Madani, F. Shabani, M. V. Dwek and A. M. Seifalian, Int. J. Nanomed., 2013, 8, 941–950 Search PubMed.
- G. Gopalakrishnan, C. Danelon, P. Izewska, M. Prummer, P.-Y. Bolinger, I. Geissbühler, D. Demurtas, J. Dubochet and H. Vogel, Angew. Chem., Int. Ed., 2006, 45, 5478–5483 CrossRef CAS PubMed.
- R. Kumar, A. Kulkarni, D. K. Nagesha and S. Sridhar, Theranostics, 2012, 2, 714–722 CrossRef CAS PubMed.
- V. Sigot, D. J. Arndt-Jovin and T. M. Jovin, Bioconjugate Chem., 2010, 21, 1465–1472 CrossRef CAS PubMed.
- N. Abdullah Al, J. A. Nam, H. Mok, Y.-K. Lee and S. Y. Park, Macromol. Res., 2013, 21, 92–99 CrossRef.
- W. Wu, M. Aiello, T. Zhou, A. Berliner, P. Banerjee and S. Zhou, Biomaterials, 2010, 31, 3023–3031 CrossRef CAS PubMed.
- R. Gui, Y. Wang and J. Sun, Colloids Surf., B, 2014, 113, 1–9 CrossRef CAS PubMed.
- A. Hoshino, N. Manabe, K. Fujioka, S. Hanada, M. Yasuhara, A. Kondo and K. Yamamoto, Nanotechnology, 2008, 19, 495102 CrossRef PubMed.
- A. A. Chen, A. M. Derfus, S. R. Khetani and S. N. Bhatia, Nucleic Acids Res., 2005, 33, e190 CrossRef PubMed.
- W. B. Tan, S. Jiang and Y. Zhang, Biomaterials, 2007, 28, 1565–1571 CrossRef CAS PubMed.
- G. T. Hess, W. H. Humphries, N. C. Fay and C. K. Payne, Biochim. Biophys. Acta, Mol. Cell Res., 2007, 1773, 1583–1588 CrossRef CAS PubMed.
- A. M. Derfus, A. A. Chen, D.-H. Min, E. Ruoslahti and S. N. Bhatia, Bioconjugate Chem., 2007, 18, 1391–1396 CrossRef CAS PubMed.
- C. Walther, K. Meyer, R. Rennert and I. Neundorf, Bioconjugate Chem., 2008, 19, 2346–2356 CrossRef CAS PubMed.
- Y. Ishihama and T. Funatsu, Biochem. Biophys. Res. Commun., 2009, 381, 33–38 CrossRef CAS PubMed.
- D. Shao, J. Li, X. Xiao, M. Zhang, Y. Pan, S. Li, Z. Wang, X. Zhang, H. Zheng, X. Zhang and L. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 11082–11090 CAS.
- Y.-P. Ho, H. H. Chen, K. W. Leong and T.-H. Wang, J. Controlled Release, 2006, 116, 83–89 CrossRef CAS PubMed.
- H. H. Chen, Y.-P. Ho, X. Jiang, H.-Q. Mao, T.-H. Wang and K. W. Leong, Mol. Ther., 2008, 16, 324–332 CrossRef CAS PubMed.
- H. H. Chen, Y.-P. Ho, X. Jiang, H.-Q. Mao, T.-H. Wang and K. W. Leong, Nano Today, 2009, 4, 125–134 CrossRef CAS PubMed.
- J. Jung, A. Solanki, K. A. Memoli, K. Kamei, H. Kim, M. A. Drahl, L. J. Williams, H.-R. Tseng and K. Lee, Angew. Chem., Int. Ed., 2010, 49, 103–107 CrossRef CAS PubMed.
- M. V. Yezhelyev, L. Qi, R. M. O'Regan, S. Nie and X. Gao, J. Am. Chem. Soc., 2008, 130, 9006–9012 CrossRef CAS PubMed.
- L. Qi, W. Shao and D. Shi, J. Mater. Chem. B, 2013, 1, 654–660 RSC.
- L. Qi and X. Gao, ACS Nano, 2008, 2, 1403–1410 CrossRef CAS PubMed.
- N. Gaponik, I. L. Radtchenko, G. B. Sukhorukov and A. L. Rogach, Langmuir, 2004, 20, 1449–1452 CrossRef CAS.
- H. Gu, R. Zheng, X. Zhang and B. Xu, J. Am. Chem. Soc., 2004, 126, 5664–5665 CrossRef CAS PubMed.
- H. Kim, M. Achermann, L. P. Balet, J. A. Hollingsworth and V. I. Klimov, J. Am. Chem. Soc., 2005, 127, 544–546 CrossRef CAS PubMed.
- S. T. Selvan, P. K. Patra, C. Y. Ang and J. Y. Ying, Angew. Chem., Int. Ed., 2007, 46, 2448–2452 CrossRef CAS PubMed.
- J. Gao, B. Zhang, Y. Gao, Y. Pan, X. Zhang and B. Xu, J. Am. Chem. Soc., 2007, 129, 11928–11935 CrossRef CAS PubMed.
- D. K. Yi, S. T. Selvan, S. S. Lee, G. C. Papaefthymiou, D. Kundaliya and J. Y. Ying, J. Am. Chem. Soc., 2005, 127, 4990–4991 CrossRef CAS PubMed.
- V. Salgueiriño-Maceira, M. A. Correa-Duarte, M. Spasova, L. M. Liz-Marzan and M. Farle, Adv. Funct. Mater., 2006, 16, 509–514 CrossRef PubMed.
- X. Gong, Q. Zhang, Y. Cui, S. Zhu, W. Su, Q. Yang and J. Chang, J. Mater. Chem. B, 2013, 1, 2098–2106 RSC.
- H. S. Cho, Z. Dong, G. M. Pauletti, J. Zhang, H. Xu, H. Gu, L. Wang, R. C. Ewing, C. Huth, F. Wang and D. Shi, ACS Nano, 2010, 4, 5398–5404 CrossRef CAS PubMed.
- E. S. Shibu, K. Ono, S. Sugino, A. Nishioka, A. Yasuda, Y. Shigeri, S. Wakida, M. Sawada and V. Biju, ACS Nano, 2013, 7, 9851–9859 CrossRef CAS PubMed.
- S. R. Ahmed, J. Dong, M. Yui, T. Kato, J. Lee and E. Y. Park, J. Nanobiotechnol., 2013, 11, 28 CrossRef CAS PubMed.
- D. Wang, J. He, N. Rosenzweig and Z. Rosenzweig, Nano Lett., 2004, 4, 409–413 CrossRef CAS.
- H.-Y. Xie, C. Zuo, Y. Liu, Z.-L. Zhang, D.-W. Pang, X.-L. Li, J.-P. Gong, C. Dickinson and W. Zhou, Small, 2005, 1, 506–509 CrossRef CAS PubMed.
- P. H. Zhang, J. T. Cao, Q. H. Min and J. J. Zhu, ACS Appl. Mater. Interfaces, 2013, 5, 7417–7424 CAS.
- Y. Qiu, R. Palankar, M. Echeverria, N. Medvedev, S. E. Moya and M. Delcea, Nanoscale, 2013, 5, 12624–12632 RSC.
- W. J. M. Mulder, R. Koole, R. J. Brandwijk, G. Storm, P. T. K. Chin, G. J. Strijkers, C. D. Donega, K. Nicolay and A. W. Griffioen, Nano Lett., 2006, 6, 1–6 CrossRef CAS PubMed.
- G. A. F. van Tilborg, W. J. M. Mulder, P. T. K. Chin, G. Storm, C. P. Reutelingsperger, K. Nicolay and G. J. Strijkers, Bioconjugate Chem., 2006, 17, 865–868 CrossRef CAS PubMed.
- L. Prinzen, R.-J. J. H. M. Miserus, A. Dirksen, T. M. Hackeng, N. Deckers, N. J. Bitsch, R. T. A. Megens, K. Douma, J. W. Heemskerk, M. E. Kooi, P. M. Frederik, D. W. Slaaf, M. A. M. J. van Zandvoort and C. P. M. Reutelingsperger, Nano Lett., 2007, 7, 93–100 CrossRef CAS PubMed.
- R. Bakalova, Z. Zhelev, I. Aoki, K. Masamoto, M. Mileva, T. Obata, M. Higuchi, V. Gadjeva and I. Kanno, Bioconjugate Chem., 2008, 19, 1135–1142 CrossRef CAS PubMed.
- R. Koole, M. M. van Schooneveld, J. Hilhorst, K. Castermans, D. P. Cormode, G. J. Strijkers, C. D. Donega, D. Vanmaekelbergh, A. W. Griffioen, K. Nicolay, Z. A. Fayad, A. Meijerink and W. J. M. Mulder, Bioconjugate Chem., 2008, 19, 2471–2479 CrossRef CAS PubMed.
- S. Wang, B. R. Jarrett, S. M. Kauzlarich and A. Y. Louie, J. Am. Chem. Soc., 2007, 129, 3848–3856 CrossRef CAS PubMed.
- H.-X. Xia, X.-Q. Yang, J.-T. Song, J. Chen, M.-Z. Zhang, D.-M. Yan, L. Zhang, M.-Y. Qin, L.-Y. Bai, Y.-D. Zhao and Z.-Y. Ma, J. Mater. Chem. B, 2014, 2, 1945–1953 RSC.
- J. M. Ness, R. S. Akhtar, C. B. Latham and K. A. Roth, J. Histochem. Cytochem., 2003, 51, 981–987 CrossRef CAS PubMed.
- H. Chen, J. Xue, Y. Zhang, X. Zhu, J. Gao and B. Yu, J. Mol. Histol., 2009, 40, 261–268 CrossRef CAS PubMed.
- Y. Ruan, W. Yu, F. Cheng, X. Zhang and S. Larre, Sensors, 2012, 12, 5461–5470 CrossRef CAS PubMed.
- E. Zahavy, E. Freeman, S. Lustig, A. Keysary and S. Yitzhaki, J. Fluoresc., 2005, 15, 661–665 CrossRef CAS PubMed.
- T. J. Fountaine, S. M. Wincovitch, D. H. Geho, S. H. Garfield and S. Pittaluga, Mod. Pathol., 2006, 19, 1181–1191 CrossRef CAS PubMed.
- A. Karwa, E. Papazoglou, K. Pourrezaei, S. Tyagi and S. Murthy, Inflammation Res., 2007, 56, 502–510 CrossRef CAS PubMed.
- Y. Xing, Q. Chaudry, C. Shen, K. Y. Kong, H. E. Zhau, L. W. Chung, J. A. Petros, R. M. O'Regan, M. V. Yezhelyev, J. W. Simons, M. D. Wang and S. Nie, Nat. Protoc., 2007, 2, 1152–1165 CrossRef CAS PubMed.
- D. E. Ferrara, D. Weiss, P. H. Carnell, R. P. Vito, D. Vega, X. Gao, S. Nie and W. R. Taylor, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2006, 290, R114–R123 CrossRef CAS PubMed.
- B. N. G. Giepmans, T. J. Deerinck, B. L. Smarr, Y. Z. Jones and M. H. Ellisman, Nat. Methods, 2005, 2, 743–749 CrossRef CAS PubMed.
- A. A. Ghazani, J. A. Lee, J. Klostranec, Q. Xiang, R. S. Dacosta, B. C. Wilson, M. S. Tsao and W. C. W. Chan, Nano Lett., 2006, 6, 2881–2886 CrossRef CAS PubMed.
- Y. Xiao, X. Gao, G. Gannot, M. R. Emmert-Buck, S. Srivastava, P. D. Wagner, M. D. Amos and P. E. Barker, Int. J. Cancer, 2008, 122, 2178–2186 CrossRef CAS PubMed.
- M. V. Yezhelyev, A. Al-Hajj, C. Morris, A. I. Marcus, T. Liu, M. Lewis, C. Cohen, P. Zrazhevskiy, J. W. Simons, A. Rogatko, S. Nie, X. Gao and R. M. O'Regan, Adv. Mater., 2007, 19, 3146–3151 CrossRef CAS PubMed.
- C. Chen, J. Peng, H.-S. Xia, G.-F. Yang, Q.-S. Wu, L.-D. Chen, L.-B. Zeng, Z.-L. Zhang, D.-W. Pang and Y. Li, Biomaterials, 2009, 30, 2912–2918 CrossRef CAS PubMed.
- R. Li, H. Dai, T. M. Wheeler, M. Sayeeduddin, P. T. Scardino, A. Frolov and G. E. Ayala, Clin. Cancer Res., 2009, 15, 3568–3573 CrossRef CAS PubMed.
- H. Xu, C. Chen, J. Peng, H.-W. Tang, C.-M. Liu, Y. He, Z.-Z. Chen, Y. Li, Z.-L. Zhang and D.-W. Pang, Appl. Spectrosc., 2010, 64, 847–852 CrossRef CAS PubMed.
- H. Xu, J. Xu, X. Wang, D. Wu, Z. G. Chen and A. Y. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 2901–2907 CAS.
- J. Yu, S. E. Monaco, A. Onisko, R. Bhargava, D. J. Dabbs, K. M. Cieply and J. L. Fine, Hum. Pathol., 2013, 44, 394–401 CrossRef CAS PubMed.
- J. Liu, S. K. Lau, V. A. Varma, R. A. Moffitt, M. Caldwell, T. Liu, A. N. Young, J. A. Petros, A. O. Osunkoya, T. Krogstad, B. Leyland-Jones, M. D. Wang and S. Nie, ACS Nano, 2010, 4, 2755–2765 CrossRef CAS PubMed.
- J. Liu, S. K. Lau, V. A. Varma, B. A. Kairdolf and S. Nie, Anal. Chem., 2010, 82, 6237–6243 CrossRef CAS PubMed.
- C. Chen, S.-R. Sun, Y.-P. Gong, C.-B. Qi, C.-W. Peng, X.-Q. Yang, S.-P. Liu, J. Peng, S. Zhu, M.-B. Hu, D.-W. Pang and Y. Li, Biomaterials, 2011, 32, 7592–7599 CrossRef CAS PubMed.
- X.-Q. Yang, C. Chen, C.-W. Peng, J.-X. Hou, S.-P. Liu, C.-B. Qi, Y.-P. Gong, X.-B. Zhu, D.-W. Pang and Y. Li, Int. J. Nanomed., 2011, 6, 2265–2273 CAS.
- C. G. Andrade, P. E. C. Filho, D. P. L. Tenorio, B. S. Santos, E. I. C. Beltrao, A. Fontes and L. B. Carvalho, Int. J. Nanomed., 2013, 8, 4623–4629 Search PubMed.
- M. Fang, J.-P. Yuan, C.-W. Peng, D.-W. Pang and Y. Li, Biomaterials, 2013, 34, 8708–8717 CrossRef CAS PubMed.
- P. Chan, T. Yuen, F. Ruf, J. Gonzalez-Maeso and S. C. Sealfon, Nucleic Acids Res., 2005, 33, e161 CrossRef PubMed.
- E. Tholouli, J. A. Hoyland, D. Di Vizio, F. O'Connell, S. A. MacDermott, D. Twomey, R. Levenson, J. A. L. Yin, T. R. Golub, M. Loda and R. Byers, Biochem. Biophys. Res. Commun., 2006, 348, 628–636 CrossRef CAS PubMed.
- A. Matsuno, J. Itoh, S. Takekoshi, T. Nagashima and R. Y. Osamura, J. Histochem. Cytochem., 2005, 53, 833–838 CrossRef CAS PubMed.
- A. Matsuno, A. Mizutani, S. Takekoshi, J. Itoh, H. Okinaga, Y. Nishina, K. Takano, T. Nagashima, R. Y. Osamura and A. Teramoto, Brain Tumor Pathol., 2006, 23, 1–5 CrossRef CAS PubMed.
- A. Nair, J. Shen, P. Thevenot, L. Zou, T. Cai, Z. Hu and L. Tang, Nanotechnology, 2008, 19, 485102 CrossRef PubMed.
- L. M. Maestro, J. E. Ramirez-Hernandez, N. Bogdan, J. A. Capobianco, F. Vetrone, J. G. Sole and D. Jaque, Nanoscale, 2012, 4, 298–302 RSC.
- C. Wong, T. Stylianopoulos, J. Cui, J. Martin, V. P. Chauhan, W. Jiang, Z. Popovic, R. K. Jain, M. G. Bawendi and D. Fukumura, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2426–2431 CrossRef CAS PubMed.
- E. Sweeney, T. H. Ward, N. Gray, C. Womack, G. Jayson, A. Hughes, C. Dive and R. Byers, Biochem. Biophys. Res. Commun., 2008, 374, 181–186 CrossRef CAS PubMed.
- X. Gao, L. Yang, J. A. Petros, F. F. Marshal, J. W. Simons and S. Nie, Curr. Opin. Biotechnol., 2005, 16, 63–72 CrossRef CAS PubMed.
- W. Cai and X. Chen, Small, 2007, 3, 1840–1854 CrossRef CAS PubMed.
- L. A. Bentolila, Y. Ebenstein and S. Weiss, J. Nucl. Med., 2009, 50, 493–496 CrossRef CAS PubMed.
- M. A. Walling, J. A. Novak and J. R. E. Shepard, Int. J. Mol. Sci., 2009, 10, 441–491 CrossRef CAS PubMed.
- V. Biju, S. Mundayoor, R. V. Omkumar, A. Anas and M. Ishikawa, Biotechnol. Adv., 2010, 28, 199–213 CrossRef CAS PubMed.
- P. S. P. Thong, M. Olivo, W. W. L. Chin, R. Bhuvaneswari, K. Mancer and K. C. Soo, Br. J. Cancer, 2009, 101, 1580–1584 CrossRef CAS PubMed.
- S. K. Piper, C. Habermehl, C. H. Schmitz, W. M. Kuebler, H. Obrig, J. Steinbrink and J. Mehnert, PLoS One, 2013, 8, e83749 Search PubMed.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed.
- S. Rieger, R. P. Kulkarni, D. Darcy, S. E. Fraser and R. W. Köster, Dev. Dyn., 2005, 234, 670–681 CrossRef CAS PubMed.
- A. S. W. Jiang, B. Y. S. Kim, J. Zheng, J. T. Rutka, C. Wang and W. C. W. Chan, J. Lab. Autom., 2008, 13, 6–12 CrossRef PubMed.
- M.-K. So, C. Xu, A. M. Loening, S. S. Gambhir and J. Rao, Nat. Biotechnol., 2006, 24, 339–343 CrossRef CAS PubMed.
- Y. Xing, M.-K. So, A. L. Koh, R. Sinclair and J. Rao, Biochem. Biophys. Res. Commun., 2008, 372, 388–394 CrossRef CAS PubMed.
- M. Hasegawa, Y. Tsukasaki, T. Ohyanagi and T. Jin, Chem. Commun., 2013, 49, 228–230 RSC.
- Y. T. Lim, S. Kim, A. Nakayama, N. E. Stott, M. G. Bawendi and J. V. Frangioni, Mol. Imaging, 2003, 2, 50–64 CrossRef CAS.
- B. K. Andrasfalvy, G. L. Galinanes, D. Huber, M. Barbic, J. J. Macklin, K. Susumu, J. B. Delehanty, A. L. Huston, J. K. Makara and I. L. Medintz, Nat. Methods, 2014, 11, 1237–1241 CrossRef CAS PubMed.
- W. Liu, H. S. Choi, J. P. Zimmer, E. Tanaka, J. V. Frangioni and M. Bawendi, J. Am. Chem. Soc., 2007, 129, 14530–14531 CrossRef CAS PubMed.
- C. Li, Y. Ji, C. Wang, S. J. Liang, F. Pan, C. Zhang, F. Chen, H. Fu, K. Wang and D. Cui, Nanoscale Res. Lett., 2014, 9, 244 CrossRef PubMed.
- S. H. Bhang, N. Won, T. J. Lee, H. Jin, J. Nam, J. Park, H. Chung, H. S. Park, Y. E. Sung, S. K. Hahn, B. S. Kim and S. Kim, ACS Nano, 2009, 3, 1389–1398 CrossRef CAS PubMed.
- Y. Yuan, J. Zhang, L. An, Q. Cao, Y. Deng and G. Liang, Biomaterials, 2014, 35, 7881–7886 CrossRef CAS PubMed.
- L. Liu, K.-T. Yong, I. Roy, W.-C. Law, L. Ye, J. Liu, J. Liu, R. Kumar, X. Zhang and P. N. Prasad, Theranostics, 2012, 2, 705–713 CrossRef CAS PubMed.
- L.-W. Liu, S.-Y. Hu, Y. Pan, J.-Q. Zhang, Y.-S. Feng and X.-H. Zhang, Beilstein J. Nanotechnol., 2014, 5, 919–926 CrossRef CAS PubMed.
- O. Carion, B. Mahler, T. Pons and B. Dubertret, Nat. Protoc., 2007, 2, 2383–2390 CrossRef CAS PubMed.
- T. J. Daou, L. Li, P. Reiss, V. Josserand and I. Texier, Langmuir, 2009, 25, 3040–3044 CrossRef CAS PubMed.
- W. T. Al-Jamal, K. T. Al-Jamal, B. Tian, A. Cakebread, J. M. Halket and K. Kostarelos, Mol. Pharmaceutics, 2009, 6, 520–530 CrossRef CAS PubMed.
- A. Papagiannaros, T. Levchenko, W. Hartner, D. Mongayt and V. Torchilin, Nanomedicine, 2009, 5, 216–224 CrossRef CAS PubMed.
- Y. He, Y. Zhong, Y. Su, Y. Lu, Z. Jiang, F. Peng, T. Xu, S. Su, Q. Huang, C. Fan and S.-T. Lee, Angew. Chem., Int. Ed., 2011, 50, 5694–5697 Search PubMed.
- W.-C. Law, R. Hu, C.-K. Chen, G. Xu, X. Wang, I. Roy and K.-T. Yong, RSC Adv., 2013, 3, 11511–11514 RSC.
- Y. Lu, Y. Zhong, J. Wang, Y. Su, F. Peng, Y. Zhou, X. Jiang and Y. He, Nanotechnology, 2013, 24, 135101 CrossRef PubMed.
- J. Wang, Y. Lu, F. Peng, Y. Zhong, Y. Zhou, X. Jiang, Y. Su and Y. He, Biomaterials, 2013, 34, 9509–9518 CrossRef CAS PubMed.
- Y. Wang, R. Hu, G. Lin, W.-C. Law and K.-T. Yong, RSC Adv., 2013, 3, 8899–8908 RSC.
- C. Zhang, X. Ji, Y. Zhang, G. Zhou, X. Ke, H. Wang, P. Tinnefeld and Z. He, Anal. Chem., 2013, 85, 5843–5849 CrossRef CAS PubMed.
- K.-T. Yong, I. Roy, H. Ding, E. J. Bergey and P. N. Prasad, Small, 2009, 5, 1997–2004 CrossRef CAS PubMed.
- K.-T. Yong, Nanotechnology, 2009, 20, 015102 CrossRef PubMed.
- K. T. Yong, R. Hu, I. Roy, H. Ding, L. A. Vathy, E. J. Bergey, M. Mizuma, A. Maitra and P. N. Prasad, ACS Appl. Mater. Interfaces, 2009, 1, 710–719 CAS.
- A. B. Rosen, D. J. Kelly, A. J. T. Schuldt, J. Lu, I. A. Potapova, S. V. Doronin, K. J. Robichaud, R. B. Robinson, M. R. Rosen, P. R. Brink, G. R. Gaudette and I. S. Cohen, Stem Cells, 2007, 25, 2128–2138 CrossRef CAS PubMed.
- K. Kikushima, S. Kita and H. Higuchi, Sci. Rep., 2013, 3, 1913 Search PubMed.
- Y. Zhao, S. L. Lo, Y. Zheng, D. H. Lam, C. Wu, M. Y. Han and S. Wang, Biochem. Biophys. Res. Commun., 2013, 434, 110–116 CrossRef CAS PubMed.
- H. Pan, P. Zhang, D. Gao, Y. Zhang, P. Li, L. Liu, C. Wang, H. Wang, Y. Ma and L. Cai, ACS Nano, 2014, 8, 5468–5477 CrossRef CAS PubMed.
- R. Savla, O. Taratula, O. Garbuzenko and T. Minko, J. Controlled Release, 2011, 153, 16–22 CrossRef CAS PubMed.
- C.-J. Wen, C. T. Sung, I. A. Aljuffali, Y.-J. Huang and J.-Y. Fang, Nanotechnology, 2013, 24, 325101 CrossRef PubMed.
- S. H. Hsu, C. J. Wen, S. A. Al-Suwayeh, Y. J. Huang and J. Y. Fang, Nanomedicine, 2013, 8, 1253–1269 CrossRef CAS PubMed.
- G. Murtaza, K. Gao, T. Liu, I. Tariq, A. Sajjad, M. R. Akram, M. Niu, G. Liu, Z. Mehmood and G. Tian, BioMed Res. Int., 2014, 714674 Search PubMed.
- C. P. Parungo, Y. L. Colson, S.-W. Kim, S. Kim, L. H. Cohn, M. G. Bawendi and J. V. Frangioni, Chest, 2005, 127, 1799–1804 CrossRef PubMed.
- E. G. Soltesz, S. Kim, R. G. Laurence, A. M. DeGrand, C. P. Parungo, D. M. Dor, L. H. Cohn, M. G. Bawendi, J. V. Frangioni and T. Mihaljevic, Ann. Thorac. Surg., 2005, 79, 269–277 CrossRef PubMed.
- B. Ballou, L. A. Ernst, S. Andreko, T. Harper, J. A. J. Fitzpatrick, A. S. Waggoner and M. P. Bruchez, Bioconjugate Chem., 2007, 18, 389–396 CrossRef CAS PubMed.
- C. P. Parungo, D. I. Soybel, Y. L. Colson, S.-W. Kim, S. Ohnishi, A. M. DeGrand, R. G. Laurence, E. G. Soltesz, F. Y. Chen, L. H. Cohn, M. G. Bawendi and J. V. Frangioni, Ann. Surg. Oncol., 2007, 14, 286–298 CrossRef PubMed.
- A. Robe, E. Pic, H.-P. Lassalle, L. Bezdetnaya, F. Guillemin and F. Marchal, BMC Cancer, 2008, 8, 111 CrossRef PubMed.
- S. Kim, Y. T. Lim, E. G. Soltesz, A. M. De Grand, J. Lee, A. Nakayama, J. A. Parker, T. Mihaljevic, R. G. Laurence, D. M. Dor, L. H. Cohn, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2004, 22, 93–97 CrossRef CAS PubMed.
- C. P. Parungo, S. Ohnishi, S.-W. Kim, S. Kim, R. G. Laurence, E. G. Soltesz, F. Y. Chen, Y. L. Colson, L. H. Cohn, M. G. Bawendi and J. V. Frangioni, J. Thorac. Cardiovasc. Surg., 2005, 129, 844–850 CrossRef PubMed.
- E. Tanaka, H. S. Choi, H. Fujii, M. G. Bawendi and J. V. Frangioni, Ann. Surg. Oncol., 2006, 13, 1671–1681 CrossRef PubMed.
- Y. Hama, Y. Koyama, Y. Urano, P. L. Choyke and H. Kobayashi, Breast Cancer Res. Treat., 2007, 103, 23–28 CrossRef PubMed.
- H. Kobayashi, Y. Hama, Y. Koyama, T. Barrett, C. A. S. Regino, Y. Urano and P. L. Choyke, Nano Lett., 2007, 7, 1711–1716 CrossRef CAS PubMed.
- Y.-W. Noh, Y. T. Lim and B. H. Chung, FASEB J., 2008, 22, 3908–3918 CrossRef CAS PubMed.
- E. Pic, T. Pons, L. Bezdetnaya, A. Leroux, F. Guillemin, B. Dubertret and F. Marchal, Mol. Imaging Biol., 2010, 12, 394–405 CrossRef PubMed.
- N. Kosaka, M. Mitsunaga, P. L. Choyke and H. Kobayashi, Contrast Media Mol. Imaging, 2013, 8, 96–100 CAS.
- E. Mathieu, N. Gupta, R. L. Macdonald, J. Ai and Y. H. Yücel, Fluids Barriers CNS, 2013, 10, 35 CrossRef PubMed.
- E. B. Voura, J. K. Jaiswal, H. Mattoussi and S. M. Simon, Nat. Med., 2004, 10, 993–998 CrossRef CAS PubMed.
- L.-D. Chen, J. Liu, X.-F. Yu, M. He, X.-F. Pei, Z.-Y. Tang, Q.-Q. Wang, D.-W. Pang and Y. Li, Biomaterials, 2008, 29, 4170–4176 CrossRef CAS PubMed.
- H. Ding, K.-T. Yong, W.-C. Law, I. Roy, R. Hu, F. Wu, W. Zhao, K. Huang, F. Erogbogbo, E. J. Bergey and P. N. Prasad, Nanoscale, 2011, 3, 1813–1822 RSC.
- H. Tada, H. Higuchi, T. M. Wanatabe and N. Ohuchi, Cancer Res., 2007, 67, 1138–1144 CrossRef CAS PubMed.
- P. Diagaradjane, J. M. Orenstein-Cardona, N. E. Colon-Casasnovas, A. Deorukhkar, S. Shentu, N. Kuno, D. L. Schwartz, J. G. Gelovani and S. Krishnan, Clin. Cancer Res., 2008, 14, 731–741 CrossRef CAS PubMed.
- K. Gonda, T. M. Watanabe, N. Ohuchi and H. Higuchi, J. Biol. Chem., 2010, 285, 2750–2757 CrossRef CAS PubMed.
- K. Yang, Y.-A. Cao, C. Shi, Z.-G. Li, F.-J. Zhang, J. Yang and C. Zhao, Oral Oncol., 2010, 46, 864–868 CrossRef PubMed.
- H. Yukawa, Y. Kagami, M. Watanabe, K. Oishi, Y. Miyamoto, Y. Okamoto, M. Tokeshi, N. Kaji, H. Noguchi, K. Ono, M. Sawada, Y. Baba, N. Hamajima and S. Hayashi, Biomaterials, 2010, 31, 4094–4103 CrossRef CAS PubMed.
- M. Stroh, J. P. Zimmer, D. G. Duda, T. S. Levchenko, K. S. Cohen, E. B. Brown, D. T. Scadden, V. P. Torchilin, M. G. Bawendi, D. Fukumura and R. K. Jain, Nat. Med., 2005, 11, 678–682 CrossRef CAS PubMed.
- Y. T. Lim, M. Y. Cho, Y.-W. Noh, J. W. Chung and B. H. Chung, Nanotechnology, 2009, 20, 475102 CrossRef PubMed.
- Y. Li, Z. Li, X. Wang, F. Liu, Y. Cheng, B. Zhang and D. Shi, Theranostics, 2012, 2, 769–776 CrossRef CAS PubMed.
- W. Cai, D.-W. Shin, K. Chen, O. Gheysens, Q. Cao, S. X. Wang, S. S. Gambhir and X. Chen, Nano Lett., 2006, 6, 669–676 CrossRef CAS PubMed.
- W. Cai and X. Chen, Nat. Protoc., 2008, 3, 89–96 CrossRef CAS PubMed.
- B. R. Smith, Z. Cheng, A. De, A. L. Koh, R. Sinclair and S. S. Gambhir, Nano Lett., 2008, 8, 2599–2606 CrossRef CAS PubMed.
- Y. Chen, M. Molnar, L. Li, P. Friberg, L.-M. Gan, H. Brismar and Y. Fu, PLoS One, 2013, 8, e83805 Search PubMed.
- A. Jayagopal, P. K. Russ and F. R. Haselton, Bioconjugate Chem., 2007, 18, 1424–1433 CrossRef CAS PubMed.
- H. Kwon, J. Lee, R. Song, S. I. Hwang, J. Lee, Y. H. Kim and H. J. Lee, Korean J. Radiol., 2013, 14, 30–37 CrossRef PubMed.
- J. Cheon and J. H. Lee, Acc. Chem. Res., 2008, 41, 1630–1640 CrossRef CAS PubMed.
- W. Cai, K. Chen, Z.-B. Li, S. S. Gambhir and X. Chen, J. Nucl. Med., 2007, 48, 1862–1870 CrossRef CAS PubMed.
- K. Chen, Z.-B. Li, H. Wang, W. Cai and X. Chen, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35, 2235–2244 CrossRef CAS PubMed.
- F. Duconge, T. Pons, C. Pestourie, L. Herin, B. Theze, K. Gombert, B. Mahler, F. Hinnen, B. Kühnast, F. Dolle, B. Dubertret and B. Tavitian, Bioconjugate Chem., 2008, 19, 1921–1926 CrossRef CAS PubMed.
- M. Oostendorp, K. Douma, T. M. Hackeng, A. Dirksen, M. J. Post, M. A. M. J. van Zandvoort and W. H. Backes, Cancer Res., 2008, 68, 7676–7683 CrossRef CAS PubMed.
- D. Gerion, J. Herberg, R. Bok, E. Gjersing, E. Ramon, R. Maxwell, J. Kurhanewicz, T. F. Budinger, J. W. Gray, M. A. Shuman and F. F. Chen, J. Phys. Chem. C, 2007, 111, 12542–12551 CAS.
- J. Ding, Y. Wang, M. Ma, Y. Zhang, S. Lu, Y. Jiang, C. Qi, S. Luo, G. Dong, S. Wen, Y. An and N. Gu, Biomaterials, 2013, 34, 209–216 CrossRef CAS PubMed.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed.
- J. Li, X. Chang, X. Chen, Z. Gu, F. Zhao, Z. Chai and Y. Zhao, Biotechnol. Adv., 2014, 32, 727–743 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- M. Bottrill and M. Green, Chem. Commun., 2011, 47, 7039–7050 RSC.
- R. Hardman, Environ. Health Perspect., 2006, 114, 165–172 CrossRef.
- J. L. Pelley, A. S. Daar and M. A. Saner, Toxicol. Sci., 2009, 112, 276–296 CrossRef CAS PubMed.
- B. A. Rzigalinski and J. S. Strobl, Toxicol. Appl. Pharmacol., 2009, 238, 280–288 CrossRef CAS PubMed.
- F. M. Winnik and D. Maysinger, Acc. Chem. Res., 2013, 46, 672–680 CrossRef CAS PubMed.
- N. Chen, Y. He, Y. Su, X. Li, Q. Huang, H. Wang, X. Zhang, R. Tai and C. Fan, Biomaterials, 2012, 33, 1238–1244 CrossRef CAS PubMed.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS.
- E. Chang, N. Thekkek, W. W. Yu, V. L. Colvin and R. Drezek, Small, 2006, 2, 1412–1417 CrossRef CAS PubMed.
- A. R. Edmund, S. Kambalapally, T. A. Wilson and R. J. Nicolosi, Toxicol. In Vitro, 2011, 25, 185–190 CrossRef CAS PubMed.
- A. Hoshino, K. Fujioka, T. Oku, M. Suga, Y. F. Sasaki, T. Ohta, M. Yasuhara, K. Suzuki and K. Yamamoto, Nano Lett., 2004, 4, 2163–2169 CrossRef CAS.
- C. Kirchner, T. Liedl, S. Kudera, T. Pellegrino, A. M. Javier, H. E. Gaub, S. Stolzle, N. Fertig and W. J. Parak, Nano Lett., 2005, 5, 331–338 CrossRef CAS PubMed.
- J. Lovric, H. S. Bazzi, Y. Cuie, G. R. A. Fortin, F. M. Winnik and D. Maysinger, J. Mol. Med., 2005, 83, 377–385 CrossRef PubMed.
- J. Lovric, S. J. Cho, F. M. Winnik and D. Maysinger, Chem. Biol., 2005, 12, 1227–1234 CrossRef CAS PubMed.
- Y. Su, Y. He, H. Lu, L. Sai, Q. Li, W. Li, L. Wang, P. Shen, Q. Huang and C. Fan, Biomaterials, 2009, 30, 19–25 CrossRef CAS PubMed.
- M. Yu, Y. Yang, R. Han, Q. Zheng, L. Wang, Y. Hong, Z. Li and Y. Sha, Langmuir, 2010, 26, 8534–8539 CrossRef CAS PubMed.
- T. Zhang, J. L. Stilwell, D. Gerion, L. Ding, O. Elboudwarej, P. A. Cooke, J. W. Gray, A. P. Alivisatos and F. F. Chen, Nano Lett., 2006, 6, 800–808 CrossRef CAS PubMed.
- A. Anas, H. Akita, H. Harashima, T. Itoh, M. Ishikawa and V. Biju, J. Phys. Chem. B, 2008, 112, 10005–10011 CrossRef CAS PubMed.
- W.-H. Chan, N.-H. Shiao and P.-Z. Lu, Toxicol. Lett., 2006, 167, 191–200 CrossRef CAS PubMed.
- A. O. Choi, S. J. Cho, J. Desbarats, J. Lovric and D. Maysinger, J. Nanobiotechnol., 2007, 5, 1 CrossRef PubMed.
- K. G. Li, J. T. Chen, S. S. Bai, X. Wen, S. Y. Song, Q. Yu, J. Li and Y. Q. Wang, Toxicol. In Vitro, 2009, 23, 1007–1013 CrossRef CAS PubMed.
- S.-H. Oh and S.-C. Lim, Toxicol. Appl. Pharmacol., 2006, 212, 212–223 CrossRef CAS PubMed.
- Y. Zhao, K. Lin, W. Zhang and L. Liu, J. Environ. Sci., 2010, 22, 1987–1992 CrossRef CAS.
- V. Brunetti, H. Chibli, R. Fiammengo, A. Galeone, M. A. Malvindi, G. Vecchio, R. Cingolani, J. L. Nadeau and P. P. Pompa, Nanoscale, 2013, 5, 307–317 RSC.
- S. J. Cho, D. Maysinger, M. Jain, B. Röder, S. Hackbarth and F. M. Winnik, Langmuir, 2007, 23, 1974–1980 CrossRef CAS PubMed.
- S. T. Stern, B. S. Zolnik, C. B. McLeland, J. Clogston, J. Zheng and S. E. McNeil, Toxicol. Sci., 2008, 106, 140–152 CrossRef CAS PubMed.
- R. F. Domingos, D. F. Simon, C. Hauser and K. J. Wilkinson, Environ. Sci. Technol., 2011, 45, 7664–7669 CrossRef CAS PubMed.
- L. Chen, Y. Miao, L. Chen, P. Jin, Y. Zha, Y. Chai, F. Zheng, Y. Zhang, W. Zhou, J. Zhang, L. Wen and M. Wang, Biomaterials, 2013, 34, 10172–10181 CrossRef CAS PubMed.
- Y.-H. Luo, S.-B. Wu, Y.-H. Wei, Y.-C. Chen, M.-H. Tsai, C.-C. Ho, S.-Y. Lin, C.-S. Yang and P. Lin, Chem. Res. Toxicol., 2013, 26, 662–673 CrossRef CAS PubMed.
- I. Corazzari, A. Gilardino, S. Dalmazzo, B. Fubini and D. Lovisolo, Toxicol. In Vitro, 2013, 27, 752–759 CrossRef CAS PubMed.
- A. Nagy, J. A. Hollingsworth, B. Hu, A. Steinbrück, P. C. Stark, C. R. Valdez, M. Vuyisich, M. H. Stewart, D. H. Atha, B. C. Nelson and R. Iyer, ACS Nano, 2013, 7, 8397–8411 CrossRef CAS PubMed.
- B. J. Muller-Borer, M. C. Collins, P. R. Gunst, W. E. Cascio and A. P. Kypson, J. Nanobiotechnol., 2007, 5, 9 CrossRef PubMed.
- S.-C. Hsieh, F.-F. Wang, S.-C. Hung, Y.-J. Chen and Y. J. Wang, J. Biomed. Mater. Res., Part B, 2006, 79B, 95–101 CrossRef CAS PubMed.
- S.-C. Hsieh, F.-F. Wang, C.-S. Lin, Y.-J. Chen, S.-C. Hung and Y.-J. Wang, Biomaterials, 2006, 27, 1656–1664 CrossRef CAS PubMed.
- C. E. Bradburne, J. B. Delehanty, K. Boeneman Gemmill, B. C. Mei, H. Mattoussi, K. Susumu, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2013, 24, 1570–1583 CrossRef CAS PubMed.
- Y. Song, D. Feng, W. Shi, X. Li and H. Ma, Talanta, 2013, 116, 237–244 CrossRef CAS PubMed.
- J. Lee, G. D. Lilly, R. C. Doty, P. Podsiadlo and N. A. Kotov, Small, 2009, 5, 1213–1221 CAS.
- M. Praetner, M. Rehberg, P. Bihari, M. Lerchenberger, B. Uhl, M. Holzer, M. E. Eichhorn, R. Fürst, T. Perisic, C. A. Reichel, U. Welsch and F. Krombach, Biomaterials, 2010, 31, 6692–6700 CrossRef CAS PubMed.
- J. Geys, A. Nemmar, E. Verbeken, E. Smolders, M. Ratoi, M. F. Hoylaerts, B. Nemery and P. H. M. Hoet, Environ. Health Perspect., 2008, 116, 1607–1613 CrossRef CAS PubMed.
- H. C. Fischer, L. Liu, K. S. Pang and W. C. W. Chan, Adv. Funct. Mater., 2006, 16, 1299–1305 CrossRef CAS PubMed.
- J. A. J. Fitzpatrick, S. K. Andreko, L. A. Ernst, A. S. Waggoner, B. Ballou and M. P. Bruchez, Nano Lett., 2009, 9, 2736–2741 CrossRef CAS PubMed.
- Z. Chen, H. Chen, H. Meng, G. Xing, X. Gao, B. Sun, X. Shi, H. Yuan, C. Zhang, R. Liu, F. Zhao, Y. Zhao and X. Fang, Toxicol. Appl. Pharmacol., 2008, 230, 364–371 CrossRef CAS PubMed.
- Y. Tang, S. Han, H. Liu, X. Chen, L. Huang, X. Li and J. Zhang, Biomaterials, 2013, 34, 8741–8755 CrossRef CAS PubMed.
- C.-C. Ho, H. Chang, H.-T. Tsai, M.-H. Tsai, C.-S. Yang, Y.-C. Ling and P. Lin, Nanotoxicology, 2013, 7, 105–115 CrossRef CAS PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS PubMed.
- H. S. Choi, W. Liu, F. Liu, K. Nasr, P. Misra, M. G. Bawendi and J. V. Frangioni, Nat. Nanotechnol., 2010, 5, 42–47 CrossRef CAS PubMed.
- X. Gao, M. Tang, Z. Li, Y. Zha, G. Cheng, S. Yin, J. Chen, D.-Y. Ruan, L. Chen and M. Wang, J. Nanopart. Res., 2013, 15, 1575 CrossRef.
- T. C. King-Heiden, P. N. Wiecinski, A. N. Mangham, K. M. Metz, D. Nesbit, J. A. Pedersen, R. J. Hamers, W. Heideman and R. E. Peterson, Environ. Sci. Technol., 2009, 43, 1605–1611 CrossRef CAS.
- T. S. Hauck, R. E. Anderson, H. C. Fischer, S. Newbigging and W. C. W. Chan, Small, 2010, 6, 138–144 CrossRef CAS PubMed.
- Y. Su, F. Peng, Z. Jiang, Y. Zhong, Y. Lu, X. Jiang, Q. Huang, C. Fan, S.-T. Lee and Y. He, Biomaterials, 2011, 32, 5855–5862 CrossRef CAS PubMed.
- L. Ye, K.-T. Yong, L. Liu, I. Roy, R. Hu, J. Zhu, H. Cai, W.-C. Law, J. Liu, K. Wang, J. Liu, Y. Liu, Y. Hu, X. Zhang, M. T. Swihart and P. N. Prasad, Nat. Nanotechnol., 2012, 7, 453–458 CrossRef CAS PubMed.
- J. Liu, W.-C. Law, J. Liu, R. Hu, L. Liu, J. Zhu, H. Chen, J. Wang, Y. Hu, L. Ye and K.-T. Yong, RSC Adv., 2013, 3, 1768–1773 RSC.
- Y.-K. Hsieh, H.-A. Hsieh, H.-F. Hsieh, T.-H. Wang, C.-C. Ho, P.-P. Lin and C.-F. Wang, J. Anal. At. Spectrom., 2013, 28, 1396–1401 RSC.
- L. W. Zhang, W. W. Yu, V. L. Colvin and N. A. Monteiro-Riviere, Toxicol. Appl. Pharmacol., 2008, 228, 200–211 CrossRef CAS PubMed.
- R. L. Bronaugh and R. F. Stewart, J. Pharm. Sci., 1985, 74, 64–67 CrossRef CAS PubMed.
- L. J. Mortensen, G. Oberdörster, A. P. Pentland and L. A. DeLouise, Nano Lett., 2008, 8, 2779–2787 CrossRef CAS PubMed.
- N. Hildebrandt, K. D. Wegner and W. R. Algar, Coord. Chem. Rev., 2014, 273–274, 125–138 CrossRef CAS PubMed.
- W. R. Algar, H. Kim, I. L. Medintz and N. Hildebrandt, Coord. Chem. Rev., 2014, 263–264, 65–85 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |