
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crucial aspects in the design of chirally modified noble metal catalysts for asymmetric hydrogenation of activated ketones
Alfons
Baiker
Department of Chemistry and Applied Biosciences, Institute of Chemical and Biochemical Engineering, ETH-Zurich, Hönggerberg, CH-8093 Zurich, Switzerland. E-mail: baiker@chem.ethz.ch
First published on 17th July 2015
Abstract
In view of the importance of optically pure chiral products there is ample reason to develop methods that facilitate their efficient production. Compared to the mostly applied homogeneous catalysts based on transition metals coordinated to suitable chiral ligands, heterogeneous chiral catalysts could offer several features that are beneficial for practical application such as stability, ease of catalyst separation and regeneration as well as straightforward access to continuous process operation. Various strategies have been developed for imparting chirality to catalytic active surfaces, among which the chiral modification of active metal surfaces by adsorption of suitable chiral organic compounds has so far been among the most successful. In this tutorial review lessons learned from research on asymmetric hydrogenation on chirally modified noble metals will be presented. Key aspects for the design of such catalysts will be elucidated using chirally modified platinum catalysts for the asymmetric hydrogenation of α-activated ketones as an example.
 Alfons Baiker | Alfons Baiker, studied chemical engineering at ETH Zurich and earned his PhD degree in 1974. After several postdoctoral stays at various universities he finished his habilitation at Stanford University (California) and returned to ETH in 1980, where he started his own research group focusing on heterogeneous catalysis and reaction engineering. He moved up to the ranks to become Full Professor in 1990. His research interests, documented in over 900 publications in refereed journals and numerous patents, are centered on catalyst design and novel catalytic materials, mechanisms and kinetics of catalytic surface processes, asymmetric hydrogenation, selective oxidation, environmental catalysis, chiral surfaces, in situ spectroscopy, and the application of supercritical fluids and ionic liquids in catalysis. |
Key learning points(1) Understanding the behavior of catalytic systems based on chirally modified metals requires that all (n − 1)n/2 interactions between (n) reaction components are considered.(2) Basic requirements for an efficient chiral modifier are proper anchoring on the noble metal surface, efficient enantiodifferentiation and structural stability under reaction conditions. (3) Subtle changes of the structure of the anchoring moiety as well as of the stereogenic center(s) of the modifier strongly influence enantioselection. (4) A transient surface complex formed via hydrogen bonding(s) between chiral modifier and prochiral substrate is at the origin of enantiodifferentiation. (5) Interaction of solvent with chiral modifier or substrate can alter the sense of enantiodifferentiation. |
1. Introduction
The importance of obtaining optically pure products can hardly be overstated in view of their broad application as pharmaceuticals, agrochemicals, flavors and fragrances. Among the various methods applied for the production of optically pure compounds asymmetric catalysis is unique in the sense that with a small amount of a chiral catalyst large quantities of a chiral product can be produced. Homogeneous asymmetric hydrogenation catalyzed by transition metals coordinated to suitable chiral ligands has advanced to an important enantioselective methodology in synthetic organic chemistry.1 A similar development has not been witnessed in heterogeneous asymmetric hydrogenation catalysis. A major reason for the slower development of chiral heterogeneous catalysis is the intrinsic heterogeneity of solid surfaces and the geometric constraints imposed by them, which leads to structurally less well-defined active sites. However, heterogeneous chiral catalysis could offer several beneficial features for practical application, among them catalyst separation, stability and regeneration as well as ready access to continuous processes are considered the most important. A prerequisite for efficient chiral catalysis is high activity combined with enantioselectivity.The crystal structure of catalytically active metals is highly symmetrical and thus achiral. Therefore, various strategies have been developed for imparting chirality to catalytic active metal surfaces.2 Among these approaches the chiral modification by adsorption of chiral organic compounds, which is the focus of this tutorial review, belongs to the most successful. The concept of chiral modification has been applied primarily to platinum group metals and Ni used for the asymmetric hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC bonds. The knowledge accumulated in these fields has been covered in various comprehensive reviews.3–5
O and CC bonds. The knowledge accumulated in these fields has been covered in various comprehensive reviews.3–5
The aim of this tutorial review is to provide a guide for the development of catalytic systems for asymmetric hydrogenation based on chirally modified metals. For illustrating the crucial aspects that have to be considered in the design of this kind of catalysts we will chose the best understood catalytic system, namely the platinum-catalyzed asymmetric hydrogenation of activated ketones (Scheme 1).
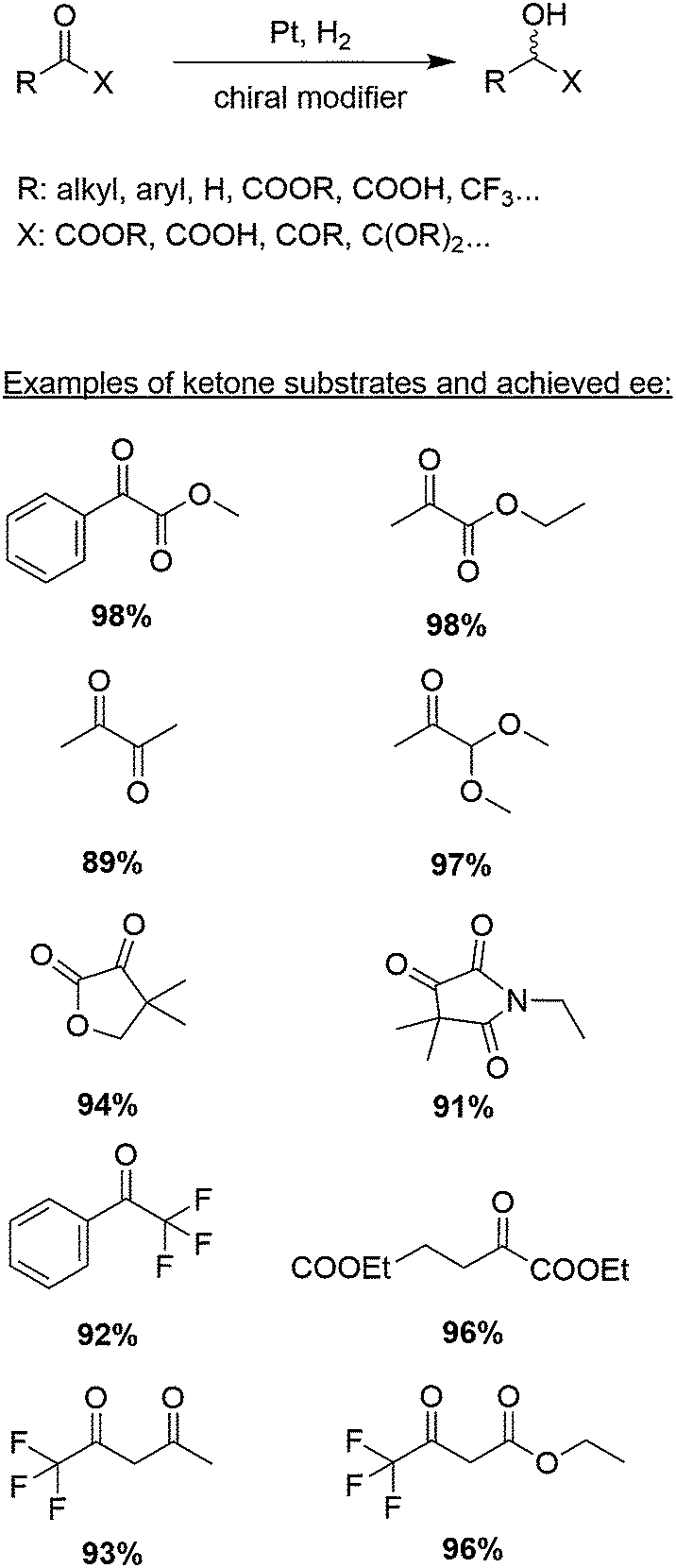 | ||
| Scheme 1 Asymmetric hydrogenation of α-activated ketones over chirally modified Pt-catalysts. Percentages given for different ketone substrates indicate highest achieved enantiomeric excesses ee. | ||
2. Important features of chirally modified Pt systems
2.1 Reaction pathway, thermodynamic and kinetic factors
Fig. 1 shows a simplified version of the reaction pathway as it has first been proposed for the asymmetric hydrogenation of ethyl pyruvate over 10,11-dihydrocinchonidine modified platinum.4 We distinguish between two enantioselective cycles leading to (R)- and (S)-products, respectively, and a racemic cycle showing no enantiodifferentiation. Parallel to the enantioselective hydrogenations on chirally modified sites racemic hydrogenation occurs on non-modified achiral Pt surface sites. According to this scheme desirable 100% enantiomeric excess, ee = 100[(R) − (S)]/[(R) + (S)], can be theoretically achieved if only one of the enantioselective cycles is active and no racemic hydrogenation occurs. This prerequisite imposes rigorous requirements on the catalyst design in the sense that the catalyst should not expose non-chiral active sites and the chiral sites should control the enantioface (Re or syn) with which the prochiral substrate is adsorbed on the modified metal surface. The requirement that no active achiral sites should be exposed is less stringent if the reaction rate of the enantioselective cycle is much higher than that of the racemic cycle. In fact a rate enhancement (ligand acceleration) for the enantioselective cycle has been observed for the hydrogenation of several substrates over cinchona-modified platinum.3–5 Crucial steps in the mechanism are the adsorption of the modifier, which bestows the chiral information to the active metal surface, the adsorption of the ketone and hydrogen on the active Pt surface, the stepwise addition of two hydrogen atoms to the CO group via an enantiodifferentiating half-hydrogenated intermediate modifier–ketone surface complex, and finally the desorption of the product alcohol. There is wide agreement that the reaction occurs on the platinum surface and thus the surface coverages of both modifier–ketone complexes (θ(CD–ketone)R, θ(CD–ketone)S) and hydrogen (θH) are decisive for the reaction kinetics. The product formation rate of the three different cycles can formally be expressed as:| rR = kRθ(CD–ketone)RθH; rS = kSθ(CD–ketone)SθH; rrac = kracθketoneθH |
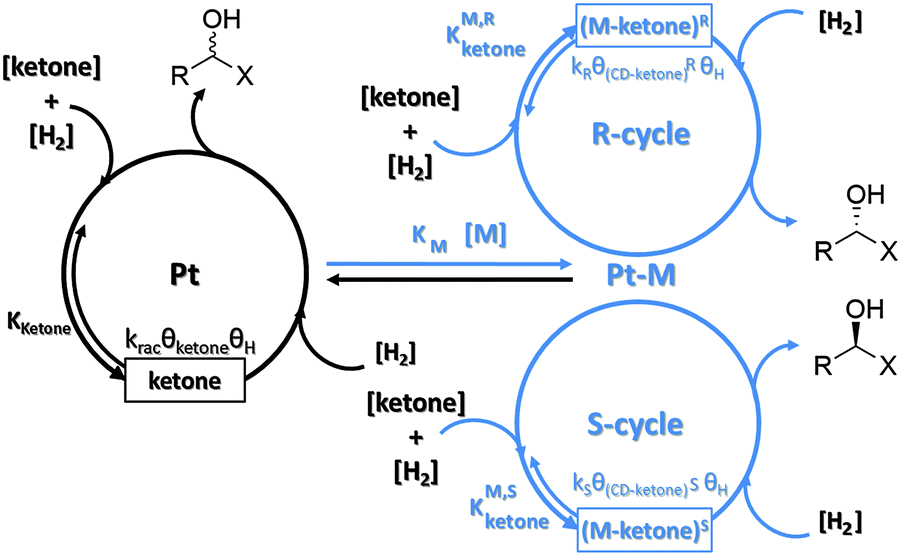 | ||
| Fig. 1 Simplified scheme of catalytic cycles occurring in the hydrogenation of activated ketones on chirally modified Pt catalysts. (M-ketone)R and (M-ketone)S represent intermediate enantiodifferentiating pro-(R) and pro-(S) modifier–ketone surface complexes, respectively. | ||
This indicates that the ratio of the coverages of pro-(R) and pro-(S) surface complexes, (θ(CD–ketone)R/θ(CD–ketone)S), is a very influential factor determining the sense of enantiodifferentiation. This ratio can be roughly estimated by theoretical calculations of the structure and stability of the intermediate surface complexes using the relation:
| θ(CD–ketone)R/θ(CD-ketone)S = exp[−ΔΔG0/RT] |
As an example the complex formed in the hydrogenation of ketopantolactone (4,4-dimethyldihydrofuran-2,3-dione) on cinchonidine-modified Pt/Al2O3 is shown in Fig. 2. The theoretical calculations revealed some important features of the interaction of the ketone substrate with the cinchonidine-modified platinum. A surface complex is formed between the cinchona modifier and the reactant ketone via hydrogen bonding (N–H⋯O) between the quinuclidine N atom of the modifier and the oxygen atom of the keto carbonyl group. As a rough guide the structures of these complexes could be considered to resemble the structure of corresponding enantiodifferentiating transition states and thereby provide a helpful criterion for judging the potential of a chiral compound to act as a suitable modifier.6–9 Steric constraints can easily be inspected and energetic considerations allow identification of the more stable diastereomeric complex favoring one of the enantiomers. Limitations of this approach arise mainly from the fact that enantioselection is accompanied by small energetic differences of a few kJ mol−1 and accurate quantum chemical calculations of binding energies and structures of large complexes adsorbed on metal surfaces are still challenging. Furthermore, the assumption that enantiodifferentiation can be traced to different stability of the adsorbed diastereomeric surface complexes (thermodynamic control) needs to be proven and possible solvent influences complicate the theoretical prediction. Thus predictions for such complex systems are only reliable in conjunction with experimental verification of important boundary conditions (adsorption modes, conformations, solvent interaction etc.). Furthermore, note that minimum energy structures are compared whereas at the transition state, at least one of the degrees of freedom will be at a maximum and a choice has to be made as to the likely reaction pathway. Enantioselectivity is considered to come about by the different stability of the adsorbed diastereomeric modifier–substrate complexes, i.e. predictions of the sense of enantiodifferentiation will fail if kinetics of hydrogen addition are different for the diastereomeric pro-(R) and pro-(S) complexes affording the (R)- and (S)-products. Nevertheless, it is noteworthy that in spite of these critical points, the predictability of the sense of enantiodifferentiation by this approach seems to work reasonably in several cases.6–9
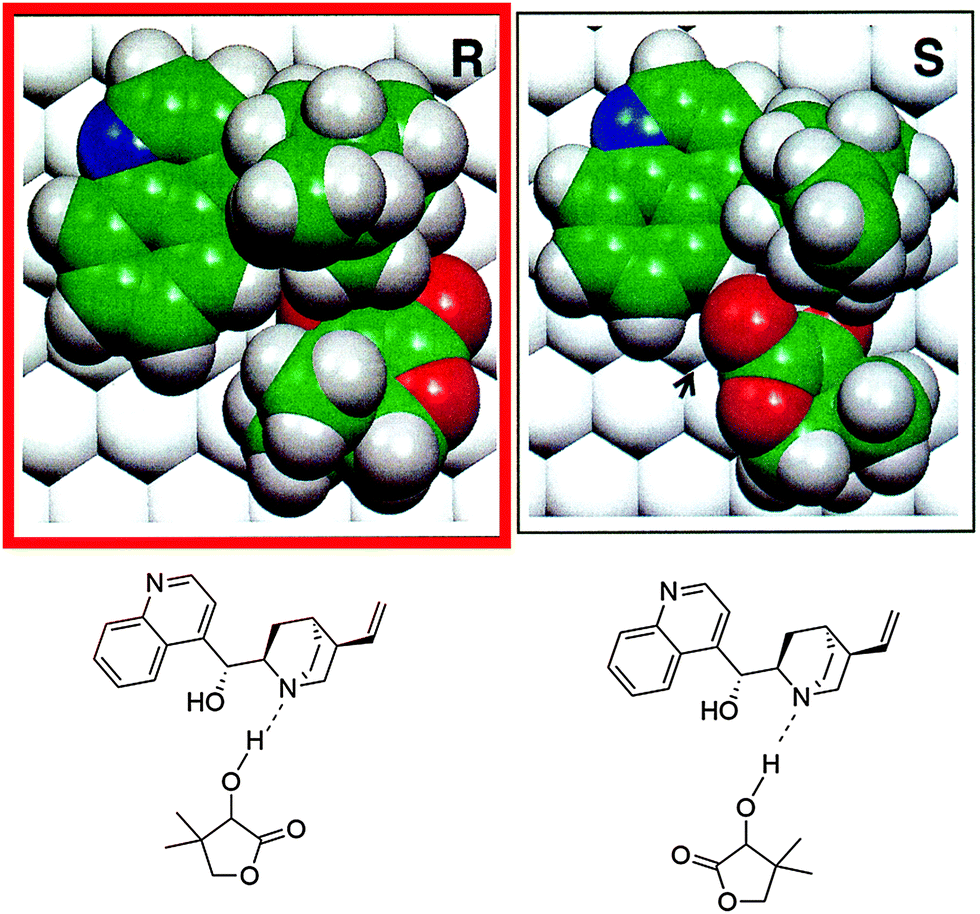 | ||
| Fig. 2 Theoretically calculated intermediate enantiodifferentiating surface complexes (CD–ketopantolactone) of pro-(R) and pro-(S) cycles in the asymmetric hydrogenation of ketopantolactone over cinchonidine-modified Pt. The pro-(R) complex is favored by 2.2 kcal mol−1 which indicates an achievable ee > 90% to (R)-pantolactone if the reaction is thermodynamically controlled. Experimentally 94% ee to (R)-pantolactone are achieved (cf.Scheme 1). Adapted with permission from ref. 6. Copyright © 1997 Academic Press. | ||
2.2 Choice of catalyst
The basic requirement of the catalyst is that it is active for the desired hydrogenation and allows anchoring (adsorbing) the chiral modifier in a way that it induces enantioselection. For the asymmetric hydrogenation of activated ketones supported platinum chirally modified by cinchona alkaloids originally developed by Orito and coworkers is still one of the most efficient systems.10 Similarly modified supported palladium first reported by Nitta and coworkers11 proved to be most suitable for the asymmetric hydrogenation of α-functionalized olefins, but this catalytic system has not been investigated in the same depth. These two noble metals clearly outperform others, such as Rh, Ru and Ir.3,5 The reasons for this specificity is rather complex and not completely understood as yet. Clearly it is not a sufficient prerequisite if a metal shows hydrogenation activity; there are other important requirements such as the appropriate stable adsorptive anchoring of the chiral modifier and the suppression of detrimental side reaction. Besides the nature of the metal, its dispersion (particle size) and shape are important factors. This structure sensitivity is evident by the fact that catalysts with Pt particles of about 3–4 nm size perform best,12 and the finding that higher ee is achieved on Pt particles exposing predominantly the (111) faces (Fig. 3).13 There are several steps in the reaction pathway, which can be affected by the surface structure (exposed faces) of the supported platinum particles. Likely candidates are the adsorption of the modifier and substrate as well as the hydrogenation and product desorption. | ||
| Fig. 3 Structure sensitivity of the asymmetric hydrogenation of ketopantolactone over Pt nanoparticles of different shapes. Adapted with permission from ref. 13. Copyright, 2009 © American Chemical Society. | ||
Special attention has to be given to the pretreatment of the catalyst. Thermal pretreatment in hydrogen at ca. 400 °C (prereduction) proved to be essential with Pt-based catalysts to achieve optimal catalytic performance.12 The striking effect of reductive catalyst pretreatment was proposed to be due to cleaning of the surface from adsorbed impurities and/or change of size and shape of Pt particles. Another pretreatment, which enhances the catalytic performance, is ultrasonification of the prereduced catalyst in the presence of the chiral modifier, as demonstrated for the hydrogenation of ethyl pyruvate on cinchonidine-modified Pt/Al2O3 affording 97% ee.14
Beside the active noble metal, the support material has a crucial influence on the catalytic behavior. While originally almost exclusively carbon and sometimes alumina supports were used, later a variety of other supports, such as silica, titania, and zeolites were employed.3–5 Recently, the scope of supports has been extended by the application of carbon nanotubes (CNTs). The encapsulation of chirally modified Pt nanoparticles in CNT channels showed significantly enhanced activity and enantioselectivity in the asymmetric hydrogenation of α-ketoesters compared to similar catalysts where the Pt nanoparticles were deposited at the outside of the CNTs.15 The enhanced catalytic performance of the encapsulated Pt nanoparticles was attributed to enrichment of the chiral modifier and reactants inside the channels of the CNTs.
Studies on the effect of acidic and basic supports indicated that both chemo- as well as enantioselectivity could strongly be influenced by tuning the acid–base properties of the support. This was demonstrated by comparing the catalytic behavior of Pt/Al2O3, Pt/Al2O3–SiO2, and Pt/Al2O3–Cs2O in the enantioselective hydrogenations of ketopantolactone (Fig. 4) and methyl benzoylformate over cinchonidine-modified catalysts.16 The acidic or basic properties of the Pt/Al2O3 catalyst were varied by gradual introduction of SiO2 and Cs2O, respectively. The gradual change of the acid–base properties of the support allowed to vary systematically the metal–support interaction and thus the electronic properties of the Pt particles, which were characterized by the bridged to linear (B/L) ratio of chemisorbed CO. The enantioselectivity in the hydrogenation of the above substrates was found to be correlated to the acid–base properties of the catalyst reflected by the B/L ratio of chemisorbed CO. Highest enantioselectivity was observed with acidic catalysts showing low B/L ratio. This behavior indicates that the acid–base properties of the support are crucial for optimizing the enantio-selectivity of these catalytic systems.
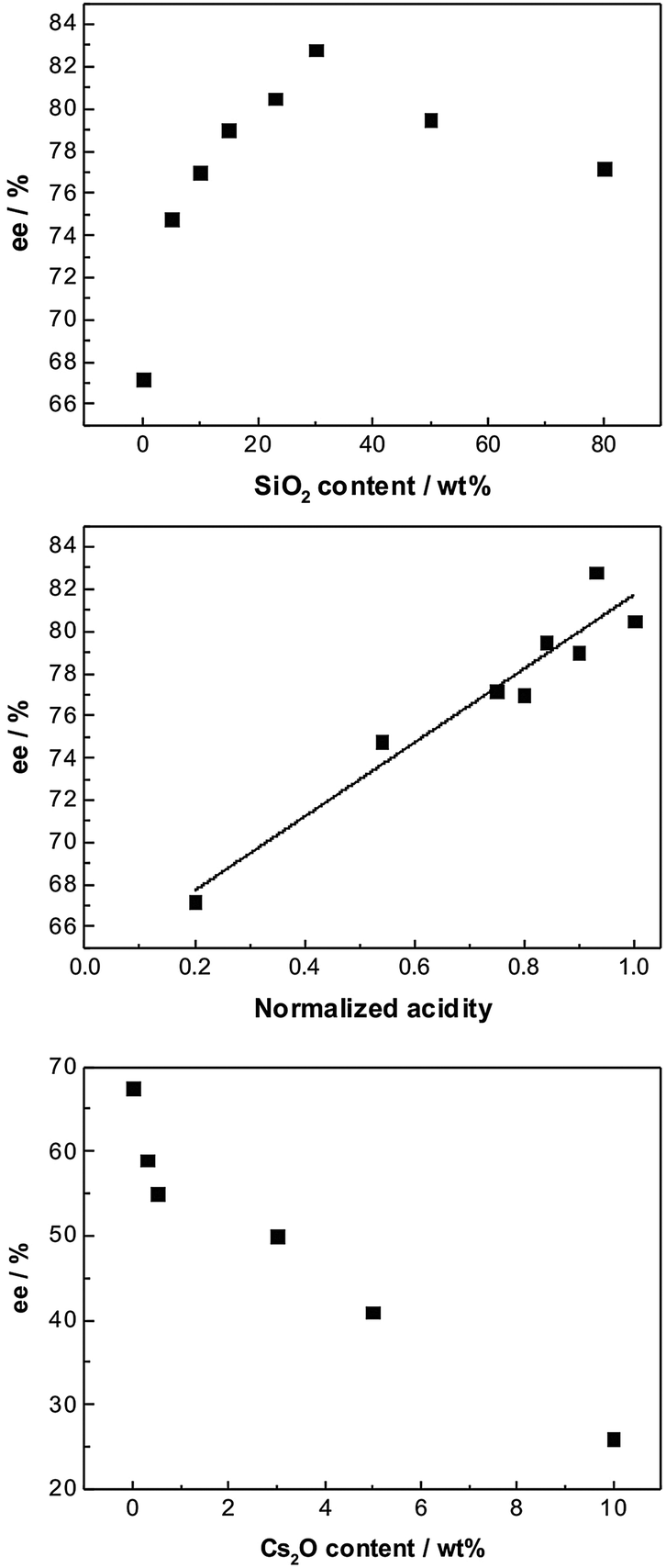 | ||
| Fig. 4 Effect of the acidity and basicity of the support on the asymmetric hydrogenation of ketopantolactone over cinchonidine-modified Pt/Al2O3 doped with SiO2 (top and middle) and Cs2O (bottom), respectively. Adapted with permission from ref. 16. Copyright © 2010 Elsevier Inc. | ||
Another property of the support affecting the catalytic performance is its pore structure, it influences the intraparticle diffusion of reactants, modifier and product.17 The pore size has to be adapted to the size of the chiral modifier otherwise noble metal particles in pores are not accessible to chiral modification and racemic hydrogenation may prevail. For the most frequently used chiral modifiers, cinchona alkaloids, this means that the pore size should be bigger than about 1–2 nm.
2.3 Chiral modification
Metal surfaces are normally achiral, albeit they may contain defects forming chiral sites such as kinks and steps. However, the probability of left-handed and right-handed chiral sites is equal rendering the overall behavior of a polycrystalline metal particle achiral. While on a single crystal surface an imbalance of such chiral sites can be achieved by cutting the crystal along specific planes,18 this procedure is not feasible for a technical catalyst since the number of chiral sites, which can be generated this way is much too small for efficient catalysis.2 The so far most effective procedure for the creation of chiral sites on the surface of polycrystalline metal particles is the adsorption of a suitable chiral modifier,2 the latter is thus a key component of these enantioselective catalytic systems. Basic requirements the modifier has to fulfill are proper anchoring on the metal surface, efficient enantiodifferentiation and structural stability under reaction conditions. The modifier is either brought onto the metal surface in a special pretreatment step or simply added to the reaction solution. Due to its simplicity, the latter method is mostly applied.3–5Originally almost exclusively natural cinchona alkaloids, cinchonidine (CD), cinchonine (CN) as well as quinidine (QD) and quinine (QN) (Scheme 2) were applied as chiral modifiers for the Pt-catalyzed asymmetric hydrogenation of activated ketones and these modifiers are still most frequently used. Cinchona alkaloids show considerable conformational flexibility due to their structure comprising two rigid moieties (quinoline and quinuclidine), which are separated by single carbon–carbon bonds. Several conformations have been identified by NMR and theoretical calculations,19 which can be classified in “open” and “closed” conformation, that means conformations where the nitrogen atom of the quinuclidine points away from or towards the aromatic quinoline ring, respectively. Fig. 5 shows the three most stable conformations of cinchonidine. The population density of these conformers is influenced by the surrounding medium (solvent), temperature and possible interaction with co-adsorbed species. The open conformation of cinchonidine has been proposed to be most suitable for the interaction with the ketone in the intermediate enantiodifferentiating modifier–ketone complex and the population of this conformation is favored in apolar and protic solvents.19
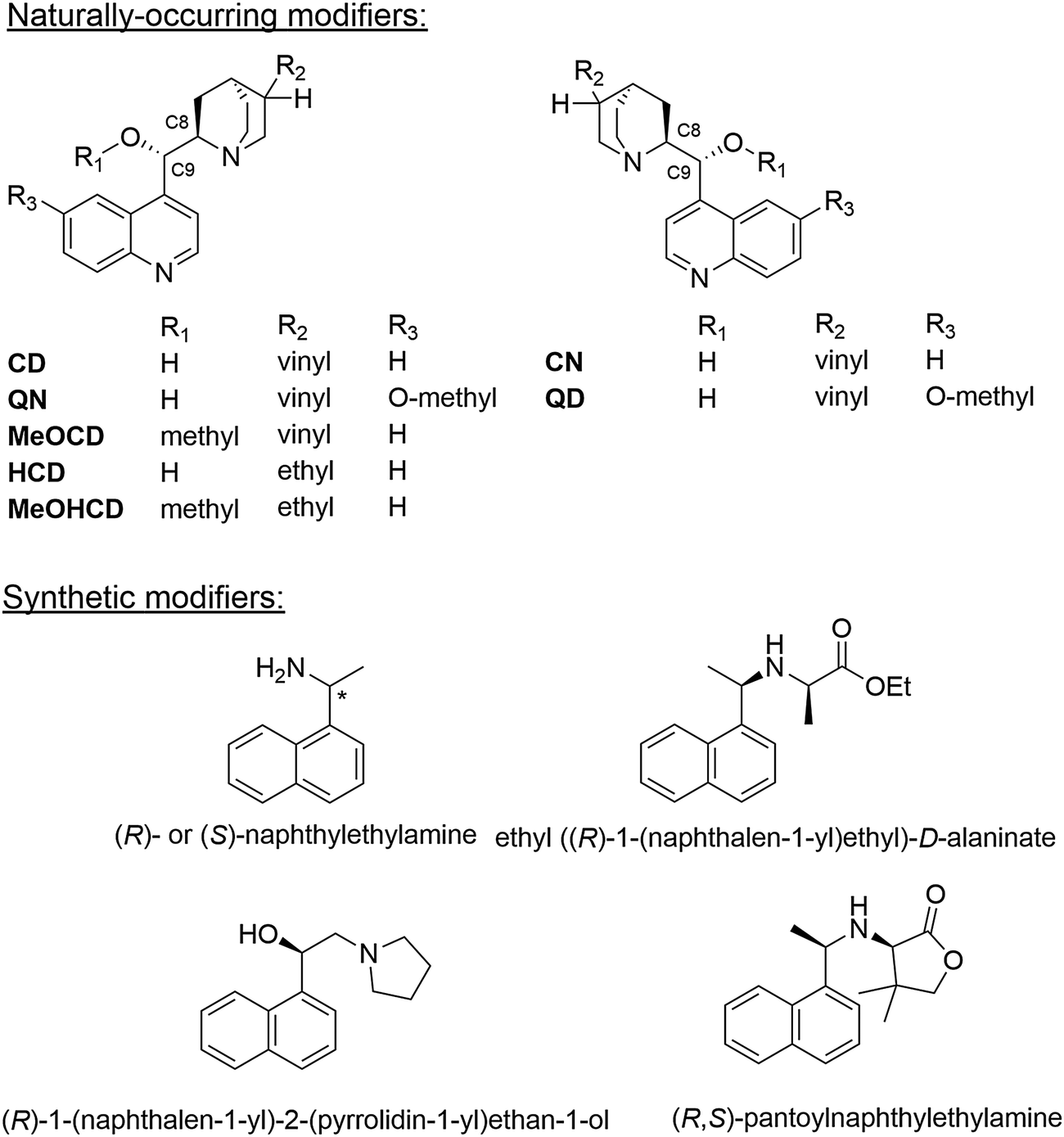 | ||
| Scheme 2 Some efficient natural and synthetic chiral modifiers. | ||
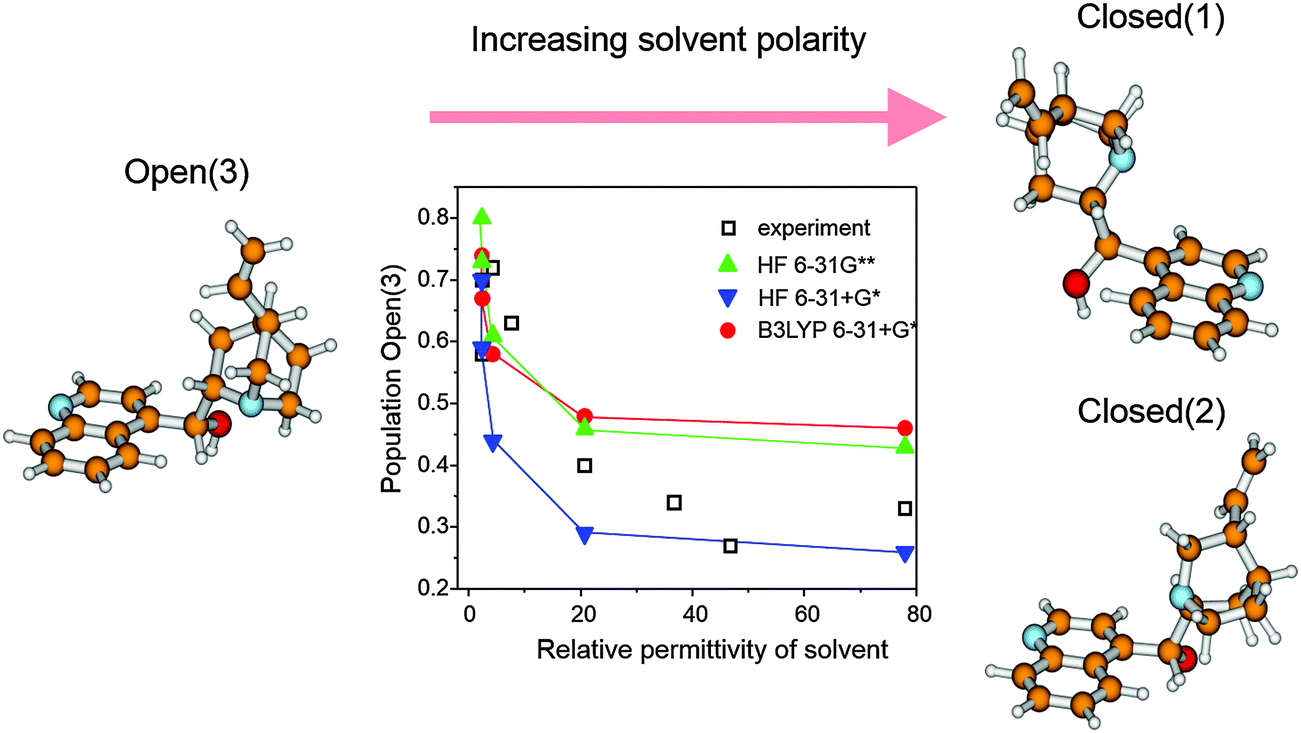 | ||
| Fig. 5 Most stable conformers of cinchonidine and their experimental and theoretically calculated populations in solvents of different permittivity. Adapted with permission from ref. 19. Copyright, 1998 © American Chemical Society. | ||
Systematic studies of the influence of structural changes of cinchona alkaloids on their enantiodifferentiating behavior and the hydrogenation reaction rate revealed some important structural features of the cinchona alkaloids, which are responsible for their unique behavior as chiral modifiers in Pt- and Pd-catalyzed asymmetric hydrogenations.3–5,20 With few exceptions, modifiers derived from cinchonidine lead to an excess of (R)-products, whereas cinchonine derivatives preferentially lead to the (S)-enantiomer. Important structural elements in the cinchona molecule, which affect ee and hydrogenation rate of activated ketones are:20 (i) the aromatic quinoline moiety; (ii) the substitution pattern of the quinuclidine; and, (iii) the substituents at C9. Strongest effects on ee have changes in the O–C9–C8–N part of the cinchona alkaloid and the partial or total hydrogenation of the quinoline moiety. The absolute configuration at C8 seems to be most influential in controlling the absolute configuration of the major product enantiomer (exceptions are large substituents at C9). The nature of substituents in the quinuclidine part has comparatively minor influence, except alkylation of the quinuclidine nitrogen, which leads to complete loss of ee.17,20 The amine function of the quinuclidine plays a crucial role in the formation of the transient enantio-differentiating surface complex between chiral modifier and reactant ketone. This 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex is formed via hydrogen bonding between the quinuclidine N atom and the O atom of the carbonyl group of the ketone substrate (N–H⋯O), as previously proposed based on theoretical calculations6–8 and later evidenced by ATR-IR spectroscopy studies.21 Recent operando ATR-IR studies corroborated the importance of this transient surface complex for the mechanism of the asymmetric hydrogenation.22 Based on all these studies,3–5 the following conclusions can be drawn about the effect of the various structural parts of cinchona alkaloids on their enantiodifferentiating ability: the aromatic ring system (quinoline) is important for adsorption (anchoring) of the modifier on the noble metal surface, the stereogenic center comprising C8 and C9, and the amine function adjacent to the stereogenic center are essential for enantiodifferentiation.
:1 complex is formed via hydrogen bonding between the quinuclidine N atom and the O atom of the carbonyl group of the ketone substrate (N–H⋯O), as previously proposed based on theoretical calculations6–8 and later evidenced by ATR-IR spectroscopy studies.21 Recent operando ATR-IR studies corroborated the importance of this transient surface complex for the mechanism of the asymmetric hydrogenation.22 Based on all these studies,3–5 the following conclusions can be drawn about the effect of the various structural parts of cinchona alkaloids on their enantiodifferentiating ability: the aromatic ring system (quinoline) is important for adsorption (anchoring) of the modifier on the noble metal surface, the stereogenic center comprising C8 and C9, and the amine function adjacent to the stereogenic center are essential for enantiodifferentiation.
Various models have been proposed for the 1:1 interaction between the cinchona modifier and α-ketoester.3–5 A common feature is the crucial role of the N–H⋯O bond. Based on STM studies under UHV conditions Mc Breen and coworkers proposed that additionally to the N–H⋯O interaction, two aromatic H bonds of the quinoline may interact with the ester carbonyl O atom, however, whether this additional binding modes are relevant under reaction conditions has not been verified yet.23 Until recently, it was assumed that the hydroxyl group at C9 of the cinchona alkaloid is not involved in the modifier–substrate interactions. However, a comparison of the efficiency of cinchonidine, cinchonine, and 9-epi-cinchonidine (or 8-epi-cinchonine) revealed that the configuration at C9 does affect ee and rate, indicating that in some cases it may be involved in the substrate–modifier interaction.24 The involvement of the C9–OH group in the diastereomeric complex between CD and ketopantolactone has recently been evidenced by ATR-IR combined with modulation excitation spectroscopy performed with C9–OH (CD) and C9–OCH3 (O-methyl-CD).25 Whether the C9–OH is also involved in the asymmetric hydrogenation of other activated ketones can hitherto not be answered.
The lessons learned from the studies of the influence of structural modification of cinchona alkaloids on their enantiodifferentiating ability have triggered the development of new synthetic modifiers. The knowledge about the structural requirements of the chiral modifier gained by the experiments with cinchona alkaloids and their derivatives has been used for a modular built-up of simpler modifiers based on enantiomerically pure 2-hydroxy-2-arylethylamines,26 2-(1-pyrolidinyl)-1-(1-naphthyl)ethanol,27,28 and 1-(1-naphtyl)ethylamine.29,30 Various derivatives of these compounds (Scheme 2) showed good enantiodifferentiation in the hydrogenation of some ketones, but generally not exceeding that of cinchona alkaloids. A common feature of all efficient synthetic modifiers is the aromatic moiety acting as anchoring unit. Recently the potential of chiral imidazolidinone and proline-derived surface modifiers has been explored, however these synthetic modifiers showed inferior performance compared to cinchona alkaloids.31
Finally it should be stressed that the optimal choice of the modifier is strongly dependent on the structure of the substrate and the solvent used. This specificity renders it difficult to provide a general rule for the choice of the most suitable modifier for a particular ketone.
2.4 Choice of solvent
The solvent plays a crucial role, not only for the dissolution of solid substrates and hydrogen. In the hydrogenation of α-ketoesters both conversion as well as ee decrease with higher solvent polarity.32 Depending on its chemical properties the solvent can interact with several components of the reaction system, such as modifier, substrate and products. It also affects the conformational behavior of the cinchona alkaloid modifiers19 and substrates33 and thus can influence the stereochemical outcome of the asymmetric hydrogenation. Among various solvents, toluene and acetic acid were found most suitable. Ethanol, which has often been applied in earlier investigations, is less suitable due to side reactions it can undergo with the reactant ketone (see Section 2.6 and Scheme 3). Protic solvents (e.g. acetic acid) lead to protonation of the quinuclidine nitrogen of the cinchona alkaloid. Apolar as well as protic solvents favor the “open conformation” (Fig. 5) of the cinchona modifier, which is considered to be involved in the crucial modifier–ketone interaction.2,3 The nature of the solvent also affects the conformational behavior of the substrate, which in turn can influence the specific enantiodifferentiating interaction between chiral modifier and substrate. The conformational behavior of various α-ketoesters, including the model substrate, ethyl pyruvate, has been investigated using solution FTIR in combination with ab initio calculations.33 α-Ketoesters can adopt s-trans and s-cis conformations, which interconvert by changing the dihedral angle OC–CO. Fig. 6 shows the dependence of the population of the rotational isomers s-trans and s-cis of ethyl pyruvate at room temperature. The population of the s-cis isomer increases with solvent polarity due to its higher dipol moment. Hydrogen bonding with alcoholic solvents also can lead to stabilization of the s-cis isomer, as demonstrated with p-fluorophenol, which is a strong hydrogen bond donor due to the acidity of the proton (Fig. 6).
 | ||
| Scheme 3 Possible side reactions of activated ketones over cinchona-modified platinum catalysts. | ||
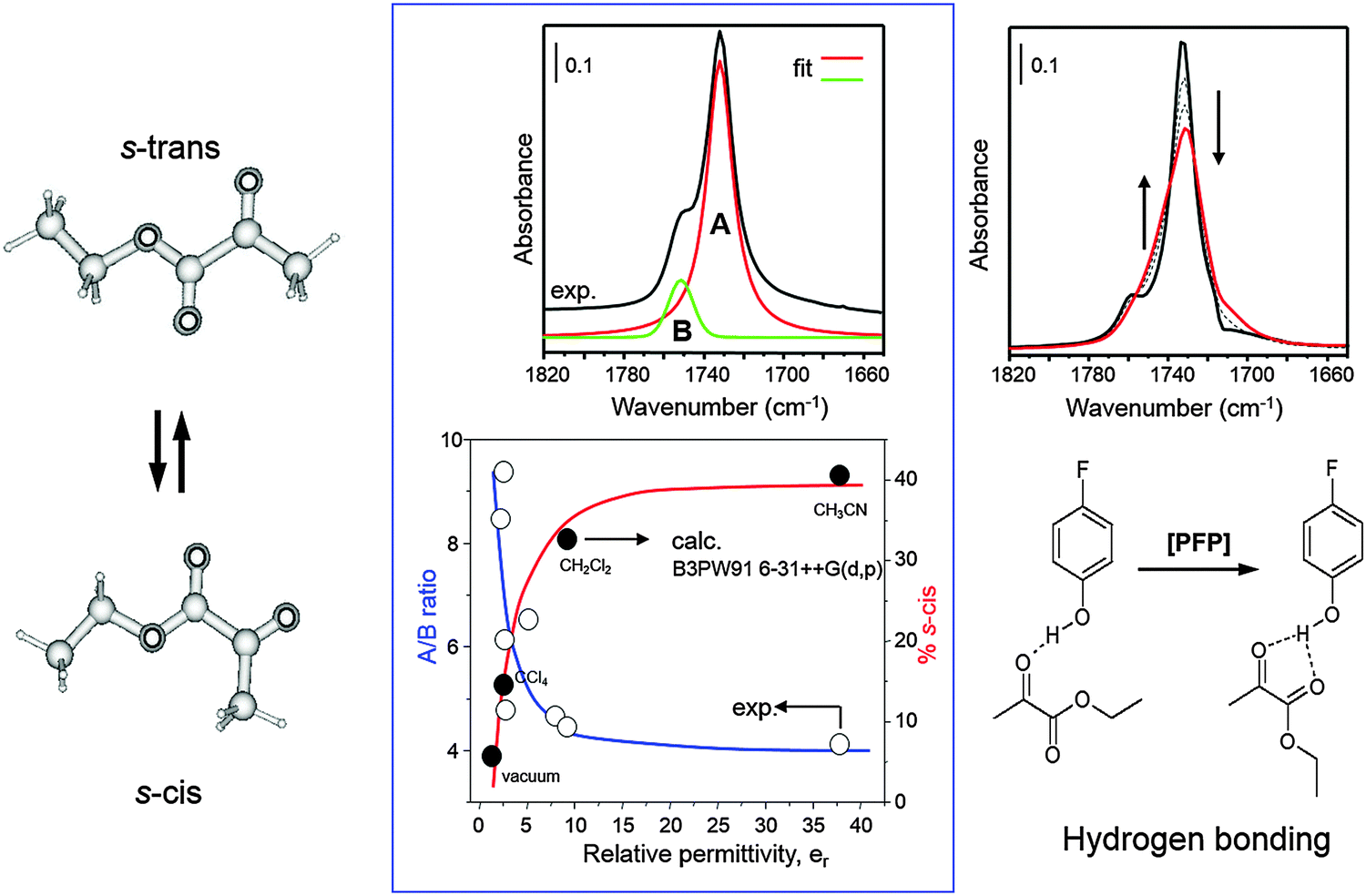 | ||
| Fig. 6 Conformational behavior of ethyl pyruvate in media of different permittivity and in the presence of a hydrogen donator. Adapted with permission from ref. 33. Copyright 2000 © Royal Society of Chemistry. | ||
Whether the conformational changes of the substrate induced by the solvent have a bearing on the structure of the enantiodifferentiating modifier–ketone complex is not clear yet and needs to be clarified.
At this point it should be stressed that there is a fundamental difference between the effect of acidic supports (see Section 2.2) and acid solvents. The support acidity affects the electronic state of Pt (metal–support interaction), which in turn influences the enantioselectivity and reaction rate. In contrast acid solvents, such as acetic acid and trifluoroacetic acid, protonate the basic N atoms of the cinchona alkaloid modifier and can form various linear or cyclic H-bonded complexes with the alkaloid and the ketone and these chemical changes in turn can influence the catalytic performance.34 An example, illustrating this difference is the enantioselective hydrogenation of ketopantolactone, where the acidic support improves the enantioselectivity, whereas the use of acetic acid as solvent diminishes it.35 The strong effect the solvent can have is particularly evident by examples of hydrogenations where the sense of enantiodifferentiation is switched if the solvent is changed. A striking example is the enantioselective hydrogenation of ethyl pyruvate on Pt modified with β-isocinchonine, an ether derivative with C8–C9 configuration identical to cinchonine. In acetic acid the expected (S)-ethyl lactate is the major enantiomer produced, whereas in toluene the sense of enantiodifferentiation changes to (R)-ethyl lactate.36
Finally, the solvent can also coadsorb on the surface and affect the surface coverages of reactants and modifier and thus the reaction kinetics.
2.5 Reaction conditions and catalyst reuse
Hydrogenations are normally performed at or slightly below room temperature in a stirred batch reactor (autoclave). Higher reaction temperatures are detrimental for achieving high ee due to the subtle energetic differences (few kJ mol−1) connected with the enantiodifferentiating process. Beside the reaction temperature, the amount of catalyst, the substrate/modifier ratio as well as the hydrogen pressure are crucial for optimal rate and enantioselectivity. Depending on the substrate–modifier pair the optimal molar substrate/modifier ratio can vary in a broad range (296000 to 130, Table 1 in ref. 37). While a too high substrate/modifier ratio leads to insufficient chiral modification and thus low ee, a too low ratio can also result in poor performance due to an imbalance of substrate and modifier coverage on the metal surface, which affects the surface reaction kinetics. The surface concentrations (coverages) of substrate and hydrogen are crucial for the reaction kinetics. The surface concentration of hydrogen is controlled by the rate of hydrogen supply through the liquid phase and the rate of its consumption by the substrate.
Normally reactions are performed in the pressure range 0.1–2 MPa, but pressures up to 10 MPa have also been used. While for most substrates (e.g. α-ketoesters, α-ketolactons, cyclic imidoketones) operation of the reactor in the kinetic regime at high hydrogen partial pressure (resulting in higher surface coverage of hydrogen) proved to be favorable, low pressure and mass-transfer limited conditions were found advantageous for some trifluoromethyl ketones and α-ketoacetals.37
Catalyst reuse is an important issue particularly for industrial production in a batch reactor. Beside the common reasons for catalyst deactivation (poisoning by impurities, structural and chemical changes) there is an additional point, which has to be considered for keeping the original catalytic performance of chirally modified noble metals. In spite of strong adsorption, some loss by desorption of the chiral modifier during multiple recycles can occur which may lead to some decrease of enantiodifferentiation and/or activity. This loss of modifier and the accompanying decrease in catalytic performance can easily be compensated by addition of some additional modifier to the reaction solution. Immobilization of the modifier by grafting, tethering or encapsulation are possible strategies to avoid loss of modifier upon reuse of the catalyst. However, so far only encapsulation15 afforded catalysts with promising catalytic performance.
2.6 Side reactions
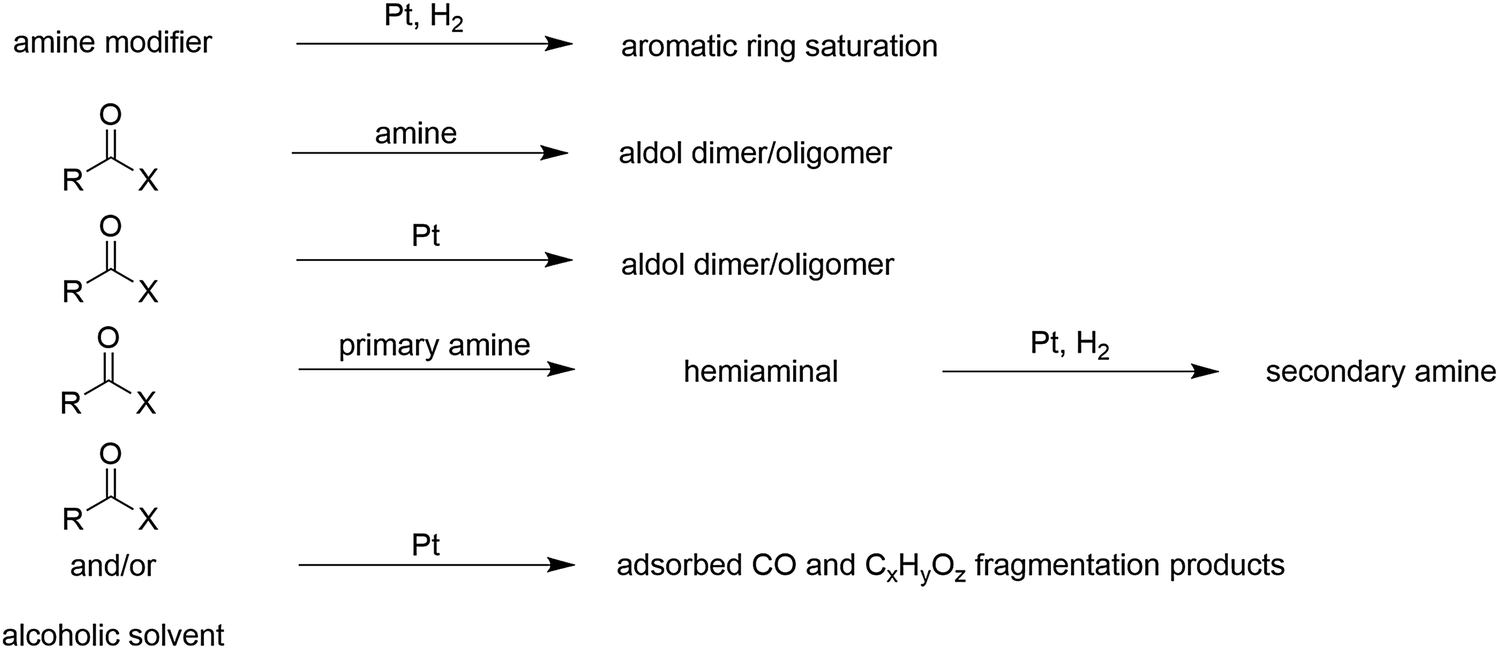
In some cases, the substrate and the chiral amine modifier can undergo undesired reactions either in solution or on the Pt surface. These side reactions can have striking effects on the rate and enantioselectivity. Scheme 3 summarizes the main side reactions, which have been observed.37 A detrimental side reaction, which leads to instable adsorption of modifiers, is the saturation of the aromatic ring system of the modifier resulting in partial loss of enantioselectivity. This loss of enantioselectivity due to partial desorption of the chiral modifier from the catalyst surface can be avoided by employing suitable modifier/catalyst ratios and is usually not observed under normal reaction conditions. The propensity for hydrogenation of the aromatic anchoring group increases at high hydrogen pressure and temperature. The resistance of the aromatic anchoring group against saturation decreases in the sequence: quinolyl > anthracenyl > naphthyl.Other relevant side reactions arise from interactions of amino- and hydroxyl groups of the modifier with some commonly used solvents. The most prominent are base-catalyzed aldol type reactions of ketones with enhanced reactivity (acidity of α-hydrogen), as first observed in the hydrogenation of ethyl pyruvate over CD-modified Pt/alumina.
Depending on the solvent (aprotic or protic) the quinuclidine N atom can either act as a nucleophile or an electrophile. Modifiers with a primary amino group such as naphthylethylamine can react with the ketone substrate providing the corresponding hemiaminal. After elimination of water and subsequent hydrogenation of the intermediate imine a secondary amine can be formed, which then acts as the actual modifier (cf.Scheme 3). This reaction can be utilized for the modular built-up of a suitable chiral modifier starting from commercially available simple napthylethylamine as mentioned in Section 2.3. If the diastereoselectivity of the reduction of the imine intermediate is high, as in the case of the hydrogenation of ethyl pyruvate and other α-activated ketones over napthylethylamine-modified Pt, high enantioselectivity can be achieved in spite of the transformation of the original modifier. Tertiary amine modifiers such as the cinchona alkaloids cannot undergo similar reductive alkylation reactions. The intermediate possessing the quaternary N atom is unstable and the reaction reverts, except for some amino-aldehydes where the intramolecular nucleophilic attack leads to a cyclic ionic adduct.
A side reaction, which can bias the proper functioning of the noble metal catalyst is the destructive adsorption of alcohols (solvent) as well as reactant ketones. This degradation can lead to the formation of CO and strongly adsorbed CxHyOz fragments, which poison the catalyst surface. Destructive adsorption is suppressed in the presence of hydrogen, which implies that these compounds should only be brought in contact with the noble metal catalysts in the presence of hydrogen. Disregard of this rule can lead to poor catalytic performance and false interpretation of mechanistic studies.
2.7 Inversion of enantioselectivity
Inversion of enantioselectivity can be induced by structural changes of the chiral modifier3–5 or by changing the solvent.36 A striking example for the effect of subtle changes of the modifier is provided by comparing the enantioselectivities achieved in the asymmetric hydrogenation over Pt/Al2O3 modified by cinchonidine, O-phenylcinchonidine and ether derivatives of cinchonidine bearing differently substituted phenyl rings, as shown in Fig. 7.38O-Phenylcinchonidine (PhOCD) is known to efficiently induce inversion of enantioselectivity with respect to cinchonidine (CD) in the enantioselective hydrogenation of various activated ketones on Pt/Al2O3.39 This inversion is rather surprising in light of the fact that the absolute configurations at the stereogenic centers C8 and C9 are the same in CD and PhOCD (Fig. 8A). ATR-IR spectroscopy under reaction conditions and theoretical calculations indicate that both modifiers are adsorbed via the quinoline rings but with different strength and that the spatial arrangement of the quinuclidine skeleton is critical for the chiral recognition.40 This arrangement is sensitive to different substituents at the phenyl ring leading to different sense of enantiodifferentiation (Fig. 7). Apparently, the presence of some types of ether substituents on CD, such as the O-phenyl moiety, closes the chiral space to the pro-(R) face of the substrate and opens it to the pro-(S) face. In contrast, the presence of trifluoromethyl groups on the O-phenyl moiety causes a conformational rearrangement of the carbon backbone of the modifier, which reshapes the chiral space and restores the pro-(R) selectivity.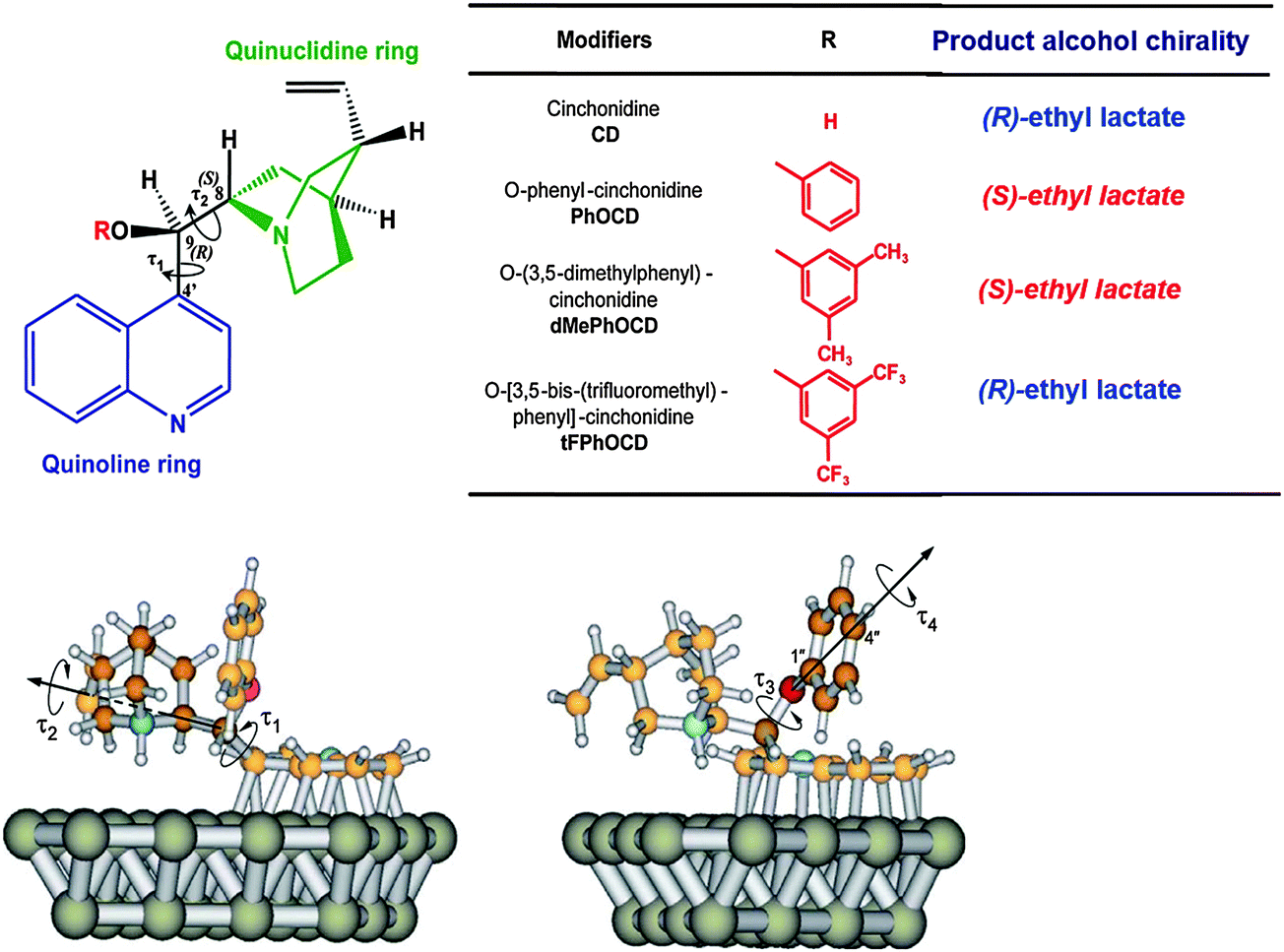 | ||
| Fig. 7 Top: Chemical structures of the cinchona-alkaloid modifiers; the main submolecular moieties, the absolute configurations at C8 and C9, and the torsional angles τ1 and τ2 are indicated. Bottom: Two views of a stable conformer of PhOCD adsorbed on platinum; in addition to τ and τ2, the two degrees of freedom associated with the phenyl ring, torsional angles τ3 and τ4, are also indicated; Pt gray, C orange, H white, N blue, O red; the carbon atoms of the quinuclidine (left) and of the phenyl moieties (right) have been darkened. Adapted with permission from ref. 38. Copyright 2007 © John Wiley and Sons. | ||
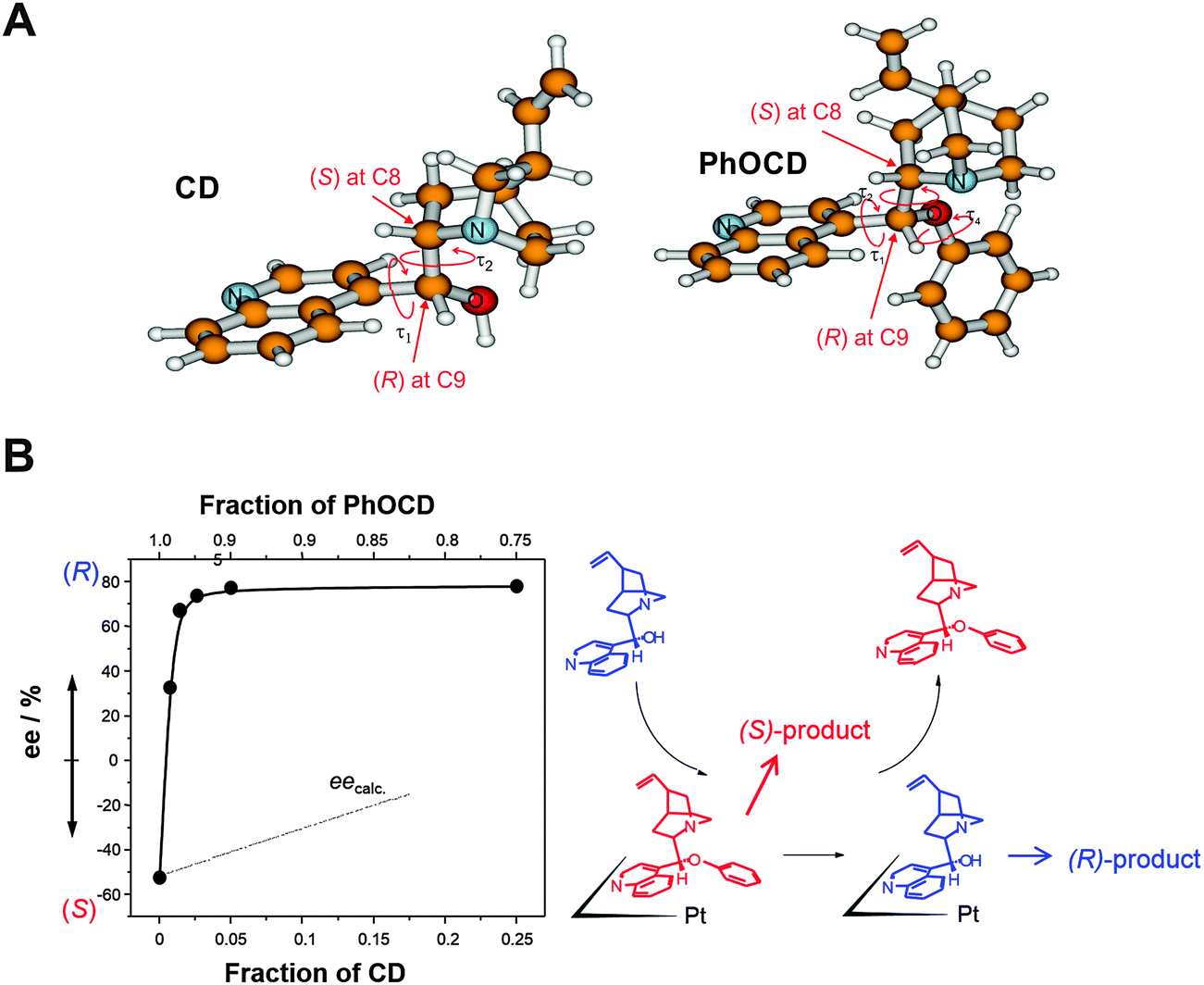 | ||
| Fig. 8 Inversion of enantioselectivity through competitive adsorption of chiral modifiers. (A) Structure of cinchonidine (CD) and O-phenyl-cinchonidine (PhOCD) show similar absolute configuration at the stereogenic centers C8 and C9 but induce different enantiodifferentiation. (B) Nonlinear behavior of CD–PhOCD mixture due to higher strength of adsorption of CD. Adapted with permission from ref. 39. Copyright 2003 © Elsevier Ltd. | ||
An interesting aspect, which has received considerable attention is the use of modifier mixtures whose individual components give rise to opposite enantio-differentiation, that is opposite product enantiomers. Such mixtures can show significant nonlinearity in their enantiodifferentiating behavior, that means the stereochemical outcome of the reaction is not predictable based on the expected individual contributions of the modifiers and their concentrations. A representative example of such a nonlinear behavior is shown in Fig. 8B. The enantioselectivity in the hydrogenation of ketopantolactone on Pt/Al2O3 originally modified with PhOCD is switched by simply adding CD to the reaction solution. This is explained by the higher adsorption strength of CD compared to PhOCD, leading to successive replacement of the latter on the surface.40 In most cases the main reason for the nonlinear behavior is the different adsorption strength of the modifiers resulting in a significant enrichment of the modifier with higher adsorption strength on the surface and thus higher number of the correspondingly modified surface sites. However, a mutual interaction of the co-adsorbed modifiers and influence on their adsorption modes cannot be ruled out and may in some cases also contribute to the nonlinearity of the behavior of modifier mixtures. Note that this type of nonlinear effect distinguishes considerably from the nonlinear behavior observed in homogeneous catalysis, where it is traced to molecular interactions (associations) between two enantiomers of the auxiliary or ligand.
2.8 Reactor operation mode
Typically, heterogeneous asymmetric hydrogenations are carried out in batch or semi-batch reactors (continuous hydrogen supply). An interesting option is the use of a continuous fixed-bed reactor41 (Fig. 9) instead of these commonly used reactors. The continuous operation mode leads to considerable process intensification. The time consuming steps of batch operation, including filling and emptying of the reactor content, are avoided and the thermal pretreatment (reduction) of the noble metal catalyst can be performed easily in the same reactor. However, continuous operation is only feasible if the reaction rate is sufficiently high and if the reaction can be carried out at high substrate/modifier ratio. In this case, the addition of a very small amount of the modifier to the reactor feed is sufficient to maintain stable chiral modification of the catalyst. If these conditions are fulfilled it is a very attractive option, as e.g. demonstrated for the Pt-catalyzed asymmetric hydrogenation of ketopantolactone41 and several other substrates.2 | ||
| Fig. 9 Process intensification by continuous reactor operation mode. | ||
3. Important interactions between reaction components
Catalytic processes are the result of the interplay of various components such as catalyst, reactant(s), product(s), solvent and additives such as a chiral modifier. In such multicomponent catalytic systems a variety of interactions have to be considered in order to attain a thorough understanding of their functioning. For a system containing (n) components the number of resulting interactions between the different components is n(n − 1)/2. Omitting a careful analysis of these interactions can lead to wrong interpretation of catalytic results and is thus misleading in catalyst design. This is particularly the case if the macroscopic behavior of a reaction system is interpreted based on studies of a single parameter, which is influenced by other parameters. The asymmetric hydrogenation of ketones on cinchona-modified platinum typically involves at least five different components (catalyst, substrate, solvent, chiral modifier, product) resulting in at least 10 different interactions. Rigorously there are even more interactions because the catalyst may contain several components which differently interact (e.g. active metal(s), support, and promotors). Fig. 10 illustrates the interactions, which generally have to be considered for the case of the asymmetric hydrogenation on chirally-modified noble metals.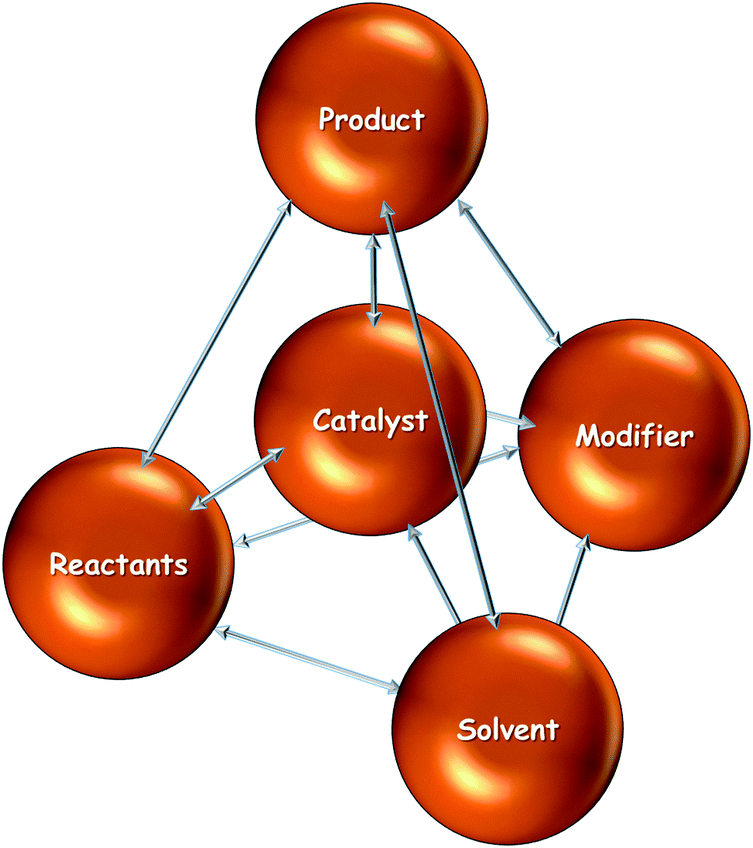 | ||
| Fig. 10 Interactions which have to be considered in the design of catalytic systems based on modified noble metals. | ||
For a deeper understanding that helps in the design of these catalytic systems all interactions need to be considered. Depending on the complexity of the catalyst system, this task can be very demanding. In a first step we may therefore sort out interactions that are likely to be of little significance such as e.g. the adsorption of solvent on metal particles and support. For hydrogenation reactions the catalyst is typically a supported metal that means one has to consider the interaction of the different reaction components with the active metal as well as with the support material, except the latter is inert. For the type of reactions considered here the catalyst is typically Pt/Al2O3. In the following we focus on some important interactions occurring in this catalytic system.
3.1 Adsorption of chiral modifier
Stable adsorption and proper adsorption geometry of the modifier on the noble metal surface is crucial for efficient enantiodifferentiation. Considerable effort has been expended in the spectroscopic analysis of the adsorption of cinchona alkaloids on platinum group metals. ATR-IR and theoretical investigations of the adsorption of cinchona alkaloids on Pt showed that depending on the surface coverage at least three different adsorption modes can be distinguished as shown for cinchonidine in Fig. 11: (1) CD adsorbed nearly parallel to the surface via the aromatic quinoline moiety, (2) tilted CD with hydrogen abstracted in α-position, and (3) CD bound to the surface by the nitrogen lone pair of the quinuclidine part.42,43 The aromatic quinoline moiety of these modifiers (CD and CN) acts as anchoring unit on the Pt surface.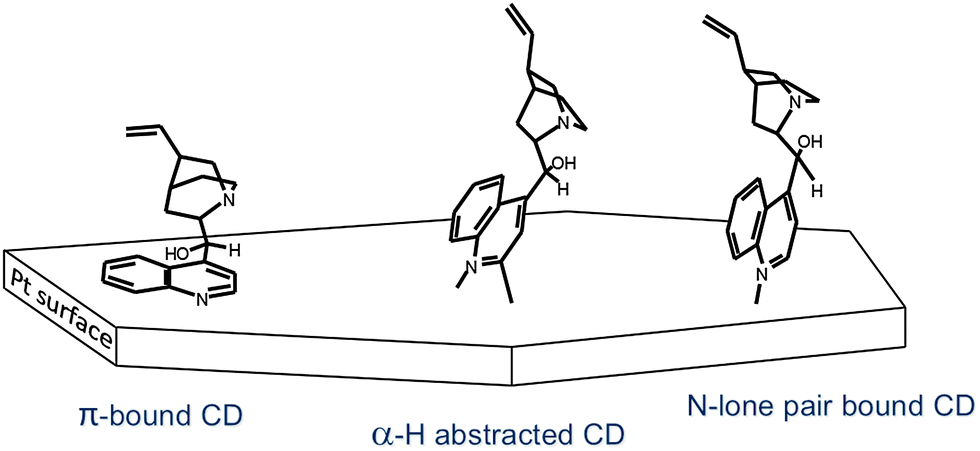 | ||
| Fig. 11 Adsorption modes of cinchonidine determined by ATR-IR spectroscopy. Adapted with permission from ref. 42. Copyright 2001© American Chemical Society. | ||
Time-lapsed scanning tunneling microscopy on Pt(111) surfaces at room temperature (normal reaction temperature) revealed that the adsorbed cinchona alkaloids possess considerable mobility, which increases with hydrogen pressure.44 Furthermore, interconversion between parallel and tilted adsorbed cinchona alkaloids was observed. The high mobility of adsorbed cinchona alkaloids may somewhat be suppressed in the liquid phase, where the asymmetric hydrogenations are normally carried out. Studies with modifiers where the anchoring group was varied indicated that extended aromatic ring systems such as quinoline, naphthalene or anthracene are suitable anchoring groups for chiral modifiers.20,26–30
3.2 Adsorption of substrate and products
Experimental and theoretical studies of the adsorption mode of ketone substrates and corresponding enantiomeric hydrogenation products are scarce, though this could be valuable mechanistic information. In situ X-ray absorption near edge structure (XANES),45 X-ray (XPS) and ultraviolet photoelectron spectroscopy (UPS),46 and reflection-absorption infrared spectroscopy (RAIRS)47 of the model substrates, ethyl- and methyl pyruvate, indicate that at least two important adsorption modes can be distinguished, namely bonding by the oxygen lone pairs leading to an adsorption where the molecule is oriented in upright or tilted position and bonding of the π-system of the CO groups resulting in an orientation nearly parallel to the surface (parallel adsorption mode). The ketone CO group was found to be more strongly involved in the chemisorption bond than the carboxyl CO group of ethyl pyruvate. In the absence of hydrogen the perpendicular O–lone pair bonded species seem to prevail, whereas in the presence of hydrogen the mean orientation is more inclined towards the surface. In this case, ethyl pyruvate also assumes nearly parallel adsorption interacting with Pt via its π-system. Parallel adsorption via π-bonding may also be favored under reaction conditions due to the hydrogen bonding between the oxygen of the α-carbonyl moiety and the quinuclidine N atom of the co-adsorbed cinchona alkaloid. This behavior illustrates the importance of the in situ approach for unraveling the adsorption modes of substrates and modifiers.
The adsorption modes observed for a single component (substrate, modifier, product) may alter significantly depending on the conditions applied and information collected at conditions far from those governing during reaction, though valuable on its own, has to be interpreted with caution for understanding the surface processes occurring under reaction conditions. Consequently, adsorption studies of substrates or modifiers alone are not very conclusive for helping in the evaluation of optimal modifier–substrate pairs for enantioselective hydrogenation. What is needed are spectroscopic investigations of co-adsorbed modifier–substrate pairs under reaction conditions, which is however a demanding task and still barely found in the literature.
The adsorption of the alcohol products can also significantly affect the surface processes and thus the overall kinetics of the hydrogenation because it may block active sites or interact with the modifier in a way that its interaction with the reactant ketone is disturbed.48
3.3 Modifier–substrate interaction
There is a general agreement that the 1:1 modifier–substrate surface complex plays a decisive role for the enantiodifferentiation.3–5 Nevertheless, there has been some debate whether this interaction already occurs in the bulk liquid phase or whether it takes place exclusively on the platinum surface. Most evidences collected so far indicate that the latter scenario holds true, as discussed in detail elsewhere.3,8 Although the structure of the enantiodiscriminating diastereomeric complex formed upon interaction of the reacting prochiral ketone and the chiral modifier depends on the structure of both interacting species, there are some common features. Operando ATR-IR studies of the coadsorption of cinchonidine and ketopantolactone confirmed the presence of the theoretically predicted N–H⋯O bond interaction between the quinuclidine nitrogen atom and the oxygen atom of the α-carbonyl bond of the ketone substrate.22 The involved hydrogen atom has been proposed to originate either from protonation (in protic solvent) or from dissociatively adsorbed dihydrogen (aprotic solvent),8 but experimental evidence for the latter was only achieved very recently by means of in situ ATR-IR combined with modulation excitation spectroscopy (Fig. 12).21 Beside this crucial hydrogen bond interaction also other hydrogen bonds between the aromatic H of the quinuclidine and the carbonyl O atom of the ester group have been proposed by Mc Breen and coworkers.23 It appears that depending on the chiral modifier and the substrate other bonding interactions become feasible, however, the proof that these additional hydrogen bonds are relevant under reaction conditions (presence of solvent) is lacking.
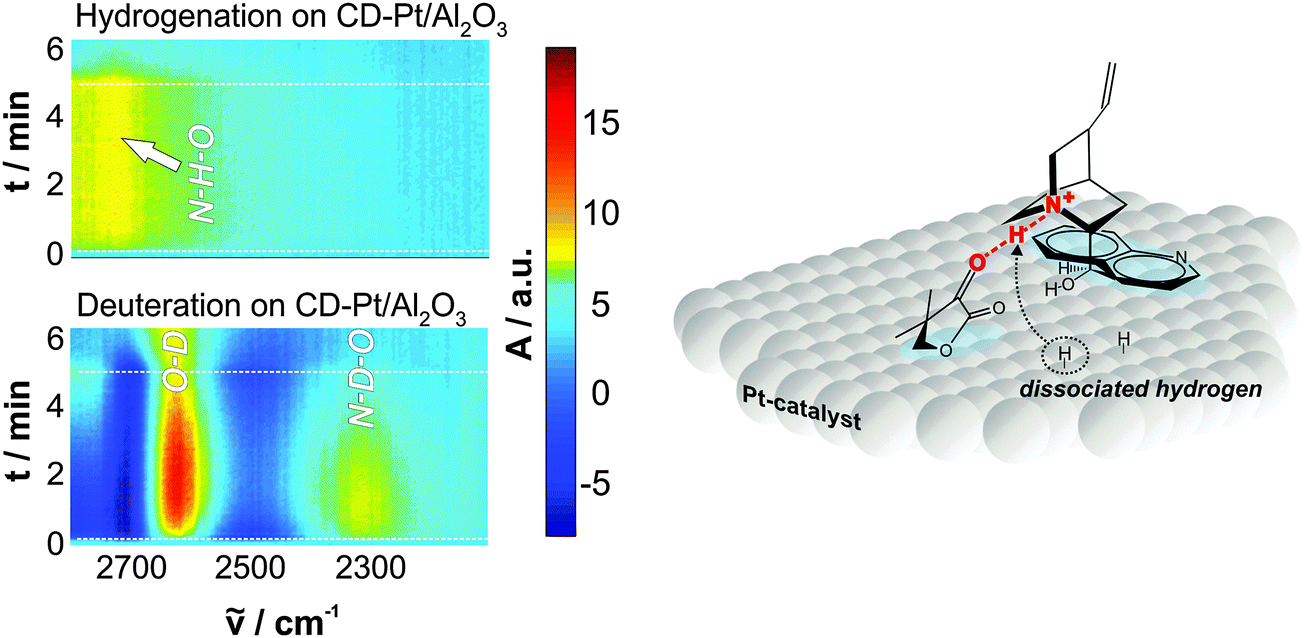 | ||
| Fig. 12 Origin of hydrogen in the N–H⋯O interaction between cinchonidine and ketone. Time domain ATR-IR spectra during hydrogenation and deuteration over cinchonidine-modified Pt and proposed H addition mechanism. Adapted with permission from ref. 22. Copyright 2014 © John Wiley and Sons. | ||
The influence of the configuration at the stereogenic centers at C8 and C9 positions of cinchona alkaloids on the stereochemical outcome of the asymmetric hydrogenations has been a matter of debate for a long time. The traditional, widely held notion was that the configuration at C8 is determining the stereochemical outcome, while that at C9 is less influential. However, this scenario has recently been questioned by comparative catalytic and theoretical studies of the action of cinchonidine (CD), cinchonine (CN), and 9-epi-cinchonidine (ECD) as chiral modifiers of supported Pt and Pd catalysts.24 The study revealed that in the hydrogenation of various activated ketones on Pt, a change in the absolute configuration at C9 did not affect the absolute configuration of the main product, however, the enantiomeric excesses were lowered by up to 30% and the reaction rate dropped by about an order of magnitude. In the Pd-catalyzed hydrogenation of different α-functionalized olefins, application of ECD – the C9 epimer of CD and C8 epimer of CN – led to a dramatic drop in enantioselectivity and reaction rate.24 This observation indicates that a subtle combination of both the C8 and C9 configurations is responsible for enantioselection on Pd. Furthermore calculations of the adsorption strength confirmed that the very low activity of ECD-modified noble metals cannot be related to the considerably different adsorption strength of ECD relative to CD or CN. The study indicated that for the hydrogenation of ketones on Pt both stereogenic centers are involved in the enantioselection. The Pd-catalyzed hydrogenation of olefins is even more demanding in the sense that only the right combination of the C8 and C9 configurations leads to effective chiral modifiers.24
The knowledge of the stability and structure of the adsorbed diastereomeric complex formed between substrate and modifier can give valuable information about the possible structure of the transition state and thus pave the way to a rationally guided design of new efficient chiral modifiers.8
3.4 Modifier–product interaction
Beside the modifier–substrate interaction, in specific cases the modifier–product interaction becomes relevant for explaining the observed reaction behavior. Such a system is the asymmetric hydrogenation of the trifluoroactivated ketone, 2,2,2-trifluoroacetophenone (TFAP), to (R)-1-phenyl-2,2,2-trifluoroethanol over cinchonidine-modified Pt. Catalytic hydrogenations in which enantiopure product alcohol was added to the reaction solution together with diastereoselective hydrogenation of cinchonidine and NMR analysis of the modifier–substrate–product interactions, revealed the key role of the minor product (S)-1-phenyl-2,2,2-trifluoroethanol in enantioselection. A multiple cycle mechanism including a racemic route on the unmodified sites and two different enantioselective routes was proposed. The enantioselective cycles involve a N–H–O type interaction between the quinuclidine N and the carbonyl O-atom of the substrate. At low conversion, the cinchona alkaloid alone is the source of chiral information. With increasing conversion, the weakly acidic minor (S)-product forms an adduct with the alkaloid and this complex starts to control the enantioselection affording lower ee. More recently this reaction network has been further evidenced by means of in situ ATR-IR spectroscopy.49 The three cycles: asymmetric hydrogenation of TFAP on CD-modified sites (Pt–CD), asymmetric hydrogenation on Pt–CD sites interfering with the acidic product alcohol (Pt–CD–P), and the racemic hydrogenation occurring on unmodified Pt sites are schematically presented in Fig. 13.49 The contributions of these reaction cycles to the overall reaction are different and change with progress of TFAP conversion. In the initial period the Pt–CD cycles (for the sake of clarity only the cycle leading to the major enantiomer is shown) and the racemic cycle determine the reaction behavior, while the Pt–CD–P cycles becomes relevant as soon as the concentration of the acidic product alcohol (P) is high enough. Whether this scenario is also typical for other α,α,α-trifluoro-activated ketones on CD-modified platinum needs to be verified.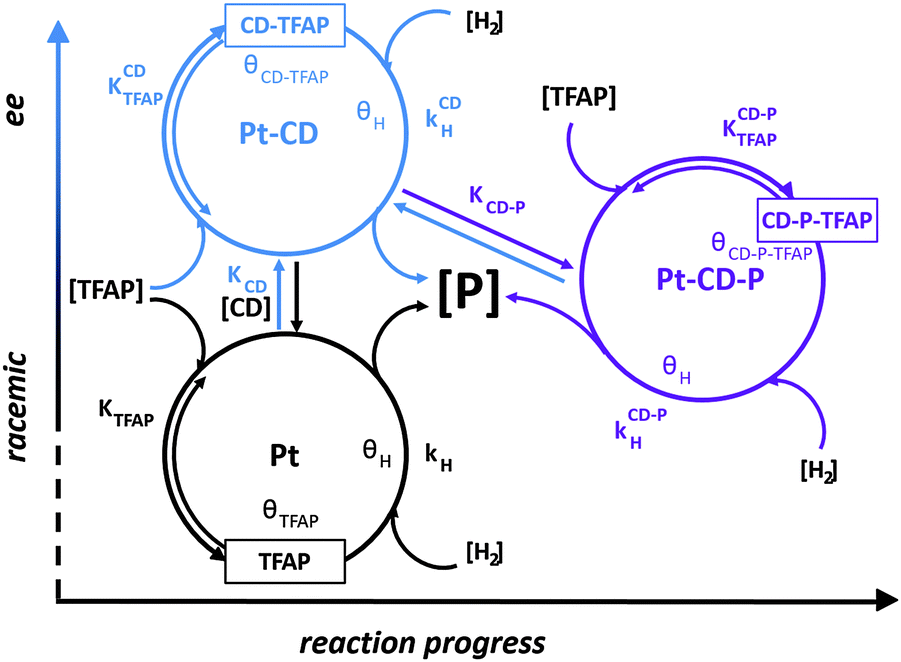 | ||
| Fig. 13 Schematic representation of three parallel reaction cycles occurring in the hydrogenation of trifluoroacetophenone (TFAP) in H2-saturated toluene on unmodified Pt Sites (red), CD-modified Pt sites (blue) and CD-modified Pt sites interfering with the product alcohol PTFE (violet) highlighting their dependence on liquid-phase concentrations and their contribution to enantioselection. Liquid-phase concentration of TFAP, product alcohol (P), CD, and H2 are indicated in square brackets and adsorbed species by θ. Equilibrium adsorption constants are indicated by K and hydrogenation rate constants by k. For the sake of simplicity only one of the enantioselective cycles is shown and the addition of hydrogenation on the surface is presented as one step. Adapted with permission from ref. 49. Copyright 2014 © American Chemical Society. | ||
3.5 Interactions with solvents or additives
Depending on its nature the solvent can interact with the reaction components (substrate, modifier, product, catalyst) in various ways. Some of these interactions are crucial to understand the reaction behavior. Protic solvents lead to the protonation of the nucleophilic quinuclidine N atom of cinchona alkaloids, which is involved in the N–H⋯O binding of the modifier–substrate complex. The relative permittivity of the solvent affects the conformational behavior of cinchona alkaloids19 and α-ketoesters,33 (cf.Fig. 5 and 6), which can have a bearing on the structure of the transient enantiodifferentiating modifier–substrate complex and thus affect reaction rate and enantiodifferentiation.In some cases the solvent or additive can even be part of the modifier–substrate complex forming a modifier–acid–ketone complex as demonstrated for hydrogenation of methyl-, ethyl- and isopropyl-4,4,4-trifluoroacetoacetate in the presence and absence of carboxylic acids (acetic acid, trifluoroacetic acid (TFA)).34 The involvement of the carboxylic acids in the enantiodifferentiating surface complex strongly altered the enantioselectivity and rate of the hydrogenations. More recently, the addition of small amounts of TFA in the asymmetric hydrogenation of trifluoroacetophenone over cinchonidine-modified Pt/Al2O3 was shown to strongly enhance the enantioselectivity. In order to elucidate the reason for this beneficial effect of TFA, the reaction system has been investigated by in situ ATR-IR combined with modulation excitation spectroscopy.50 Crucial molecular interactions between the chiral modifier (CD), acid additive (TFA) and the reactant trifluoroacetophenone at the catalyst surface were elucidated under reaction conditions. Evidence was provided that it is a monodentate acid–base adduct in which the carboxylate of TFA resides at the quinuclidine N-atom of CD. Two possible molecular structures of the enantiodifferentiating surface complex were proposed, which can explain the beneficial effect of TFA addition on enantioselectivity. These complexes are shown in Fig. 14. Note that the role of the acid in these complexes is different. In the complex “CD–TFA–ketone 1” TFA serves as a linker between the basic amine function of CD and the oxygen of the keto-carbonyl group, which provides a better fixation of the substrate on the surface and also facilitates a second interaction between the OH and the CF3 functions. In the complex “CD–TFA–ketone 2” the role of TFA is mainly the stabilization of the open conformation of the CD modifier thereby facilitating improved access to the C9–OH of CD.
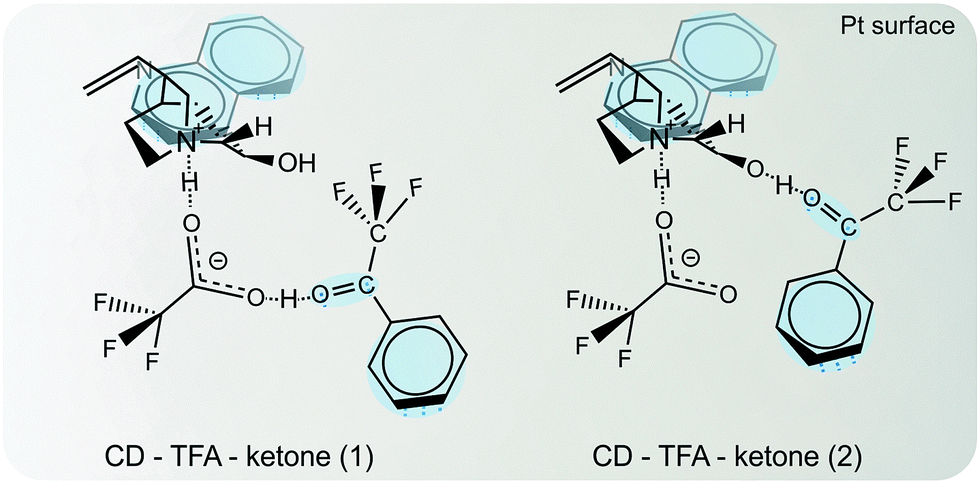 | ||
| Fig. 14 Enantiodifferentiating intermediate surface complexes between co-adsorbed ketone (TFAP) and chiral modifier CD in presence of a carboxylic acid (TFA). Conformation of adsorbed CD and N–H⋯O interaction between the quinuclidine N atom and the carbonyl group of the ketone adsorbed with its Re face allow for a second attractive interaction between OH and CF3. Adapted with permission from ref. 50. Copyright 2014 © John Wiley and Sons. | ||
Finally, it should be stressed that in the presence of carboxylic acids at least two different types of catalytic cycles may be operative: a cycle involving simply the modifier–ketone complex and cycles involving modifier–carboxylic acid–ketone complexes. Depending on the concentration of the carboxylic acid additive, the former or the later type of cycle may be predominant and control the enantioselectivity.
4. Conclusion and future challenges
Chiral modification by suitable chiral organic compounds is one of the most efficient methods to bestow chirality to active noble metal catalysts for the asymmetric hydrogenation of CO and CC bonds. While the application of this method is fairly simple, understanding of its functioning is demanding. The interactions between all components (noble metal, support, modifier, substrate, solvent, and product) have to be taken into account to explain the macroscopic behavior of these enantiodifferentiating catalytic systems. The principle aim of achieving a uniform chiral modification of the surface of polycrystalline noble metal particles is biased by their inherent heterogeneity. Surface metal atoms with different coordination and the coverage dependent geometry of adsorbed modifier render the modification of the surface with uniform enantiodifferentiating and active centers difficult. Furthermore, a significant amount of active metal sites may remain unmodified and contribute to racemic hydrogenation. In view of this difficulty the enantioselectivities achieved with some of these systems, well exceeding 90%, is striking. It is explainable by the fact that the enantioselective catalytic cycle often occurs with faster rate than the racemic cycle. While supported Pt is most efficient for CO bond hydrogenation of activated ketones, supported Pd is the choice for the hydrogenation of CC bonds. The reasons for this specificity are complex and not clearly understood yet.
A key component of these catalytic systems is the chiral modifier. Cinchona alkaloids originally applied by Orito and coworkers, still are the most versatile chiral modifiers. Systematic studies of structural changes of cinchona alkaloids and their effect on rate and enantioselectivity have revealed important structural requirements of efficient modifiers. These are an aromatic moiety that anchors the modifier on the noble metal surface and a stereogenic region adjacent to an amine function, which forms a N–H⋯O hydrogen bond with the O atom of the keto carbonyl group of the substrate. The latter is crucial for the formation of an enantiodifferentiating surface complex between the substrate and chiral modifier. The knowledge of the structure–enantiodifferentiation relationship has triggered the search of various new synthetic modifiers that are more suitable for a synthetic modular built-up than cinchona alkaloids. However, the experience gathered so far indicates that it is difficult to compete with the efficiency and versatility of the classical cinchona alkaloids. Nevertheless, some of the synthetic modifiers indicate that a rational structural tailoring of chiral modifiers for a specific substrate may be possible if the molecular interactions of the different components of the catalytic system are better understood. An important step towards this aim is the application of in situ and operando spectroscopic techniques, which allow characterizing the catalytic system under working conditions.
An inherent problem in the design of catalytic systems based on chirally modified noble metals is their high specificity. Depending on the structure of the substrate and modifier as well as on the solvent used different interactions seem to control their functioning rendering it difficult to give general guidelines for the design of chiral modifiers for a specific substrate. Theoretical studies together with experimental validation tests may help in finding suitable modifier–substrate combinations. However, their rational design needs a broader research effort, which embraces fundamental studies on the reaction mechanism and knowledge of the various interactions occurring in these systems. Without this deeper fundamental understanding the search for new catalytic system will remain in the realm of empiricism. Another future challenge is the broadening of the scope of reactions, where the concept of chirally modified active surfaces can be applied. Efforts towards these aims could be rewarding considering the inherent technical advantages such heterogeneous catalytic systems could offer.
Finally, improvement of the stability of the chiral modification could lead to catalysts, which do not need any further modification when used in many repetitive applications. Recently, encapsulation of the modifier in carbon nanotubes15 has been demonstrated to have some potential to achieve this goal.
Acknowledgements
Financial support by the Swiss National Science Foundation and the Foundation Claude & Giuliana is kindly acknowledged. The author thanks former and present coworkers for their invaluable contributions to this research.Notes and references
- D. A. Ager, A. H. M. de Vries and J. G. de Vries, Chem. Soc. Rev., 2012, 41, 3340–3380 RSC and references cited therein.
- A. Baiker, Catal. Today, 2005, 100, 159–170 CrossRef CAS PubMed.
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed.
- H. U. Blaser and M. Studer, Acc. Chem. Res., 2007, 40, 1348–1356 CrossRef CAS PubMed.
- M. Bartok, Curr. Org. Chem., 2006, 10, 1533–1567 CrossRef CAS.
- M. Schürch, O. Schwalm, T. Mallat, J. Weber and A. Baiker, J. Catal., 1997, 169, 275–286 CrossRef.
- T. Bürgi and A. Baiker, J. Catal., 2000, 194, 445–451 CrossRef.
- A. Baiker, J. Mol. Catal. A: Chem., 2000, 163, 205–220 CrossRef CAS.
- Y. Wang, N. Su, L. Ye, Y. Ren, X. Chen, Y. Du, Z. Li, B. Yue, S. Chi, E. Tsang, Q. Chen and H. He, J. Catal., 2014, 313, 113–126 CrossRef CAS PubMed.
- Y. Orito, S. Imai, S. Niwa and G.-H. Nguyen, J. Synth. Org. Chem., Jpn., 1979, 37, 137–138 CrossRef.
- Y. Nitta, Y. Ueda and T. Imanaka, Chem Lett., 1994, 6, 1095–1098 CrossRef.
- J. T. Wehrli, A. Baiker, D. M. Monti and H. U. Blaser, J. Mol. Catal., 1990, 61, 207–226 CrossRef CAS.
- E. Schmidt, A. Vargas, T. Mallat and A. Baiker, J. Am. Chem. Soc., 2009, 131(34), 12358–12367 CrossRef CAS PubMed.
- B. Török, K. Felföldi, G. Szakonyi, K. Balazsik and M. Bartok, Catal. Lett., 1998, 52, 81–84 CrossRef.
- Z. Chen, Z. Guan, M. Li, Q. Yang and C. Li, Angew. Chem., Int. Ed., 2011, 50, 4913–4917 CrossRef CAS PubMed.
- F. Hoxha, B. Schimmoeller, Z. Cakl, A. Urakawa, T. Mallat, S. E. Pratsinis and A. Baiker, J. Catal., 2010, 271, 115–124 CrossRef CAS PubMed.
- H. U. Blaser, H. P. Jalett, D. M. Monti, A. Baiker and J. T. Wehrli, Stud. Surf. Sci. Catal., 1991, 67, 147–155 CrossRef CAS.
- C. F. Mc Fadden, P. S. Cremer and A. J. Gellman, Langmuir, 1996, 12, 2483–2487 CrossRef CAS.
- T. Bürgi and A. Baiker, J. Am. Chem. Soc., 1998, 120, 12920–12926 CrossRef.
- H. U. Blaser, H. P. Jalett, W. Lottenbach and M. Studer, J. Am. Chem. Soc., 2000, 122, 12675–12682 CrossRef CAS.
- N. Maeda, K. Hungerbühler and A. Baiker, J. Am. Chem. Soc., 2011, 133, 19567–19569 CrossRef CAS PubMed.
- F. Meemken, K. Hungerbühler and A. Baiker, Angew. Chem., Int. Ed., 2014, 53, 8640–8644 CrossRef CAS PubMed.
- S. Lavoie, M. A. Laliberte, I. Temprano and P. H. McBreen, J. Am. Chem. Soc., 2006, 128, 7588–7593 CrossRef CAS PubMed.
- E. Schmidt, C. Bucher, G. Santarossa, T. Mallat, R. Gilmour and A. Baiker, J. Catal., 2012, 289, 238–248 CrossRef CAS PubMed.
- F. Meemken, N. Maeda, K. Hungerbühler and A. Baiker, Angew. Chem., Int. Ed., 2012, 51, 8212–8216 CrossRef CAS PubMed.
- K. E. Simons, G. Wang, T. Heinz, T. Giger, T. Mallat, A. Pfaltz and A. Baiker, Tetrahedron: Asymmetry, 1995, 505–518 CrossRef CAS.
- B. Minder, M. Schürch, T. Mallat, A. Baiker, T. Heinz and A. Pfaltz, J. Catal., 1996, 160, 261–268 CrossRef CAS.
- M. Schürch, T. Heinz, R. Aeschimann, T. Mallat, A. Pfaltz and A. Baiker, J. Catal., 1998, 173, 187–195 CrossRef.
- G. Z. Wang, T. Heinz, A. Pfaltz, B. Minder, T. Mallat and A. Baiker, J. Chem. Soc., Chem. Commun., 1994, 12, 2047–2048 RSC.
- E. Orglmeister, T. Mallat and A. Baiker, Adv. Synth. Catal., 2005, 347, 78–86 CrossRef CAS PubMed.
- M. Holland, F. Meemken, A. Baiker and R. Gilmour, J. Mol. Catal. A: Chem., 2015, 396, 335–345 CrossRef CAS PubMed.
- J. T. Wehrli, A. Baiker, D. M. Monti, H. U. Blaser and H. P. Jalett, J. Mol. Catal., 1989, 57, 245–257 CrossRef CAS.
- D. Ferri, T. Bürgi and A. Baiker, J. Chem. Soc., Perkin Trans. 2, 2000, 221–227 RSC.
- M. von Arx, T. Bürgi, T. Mallat and A. Baiker, Chem. – Eur. J., 2002, 8, 1430–1437 CrossRef CAS.
- M. Schürch, N. Künzle, T. Mallat and A. Baiker, J. Catal., 1997, 169, 275–286 CrossRef.
- M. Bartok, M. Sutyinski, K. Felföldi and G. Szöllösi, Chem. Commun., 2002, 1130–1131 RSC.
- M. von Arx, T. Mallat and A. Baiker, Top. Catal., 2002, 75–87 CrossRef CAS.
- A. Vargas, D. Ferri, N. Bonalumi, T. Mallat and A. Baiker, Angew. Chem., Int. Ed., 2007, 46, 3905–3908 CrossRef CAS PubMed.
- S. Diezi, A. Szabo, T. Mallat and A. Baiker, Tetrahedron: Asymmetry, 2003, 14, 2573–2577 CrossRef CAS.
- N. Bonalumi, A. Vargas, D. Ferri and A. Baiker, J. Phys. Chem. C, 2007, 111, 9349–9358 CAS.
- N. Künzle, R. Hess, T. Mallat and A. Baiker, J. Catal., 1999, 186, 239–241 CrossRef.
- D. Ferri and T. Bürgi, J. Am. Chem. Soc., 2001, 123, 12074–12084 CrossRef CAS PubMed.
- F. Zaera and J. Kubota, J. Am. Chem. Soc., 2001, 123, 11115–11116 CrossRef.
- M. Wahl, M. von Arx, T. A. Jung and A. Baiker, J. Phys. Chem. B, 2006, 110, 21777–21782 CrossRef CAS PubMed.
- T. Bürgi, F. Atamny, A. Knop-Gericke, M. Hävecker, T. Schedel-Niedrig, R. Schlögl and A. Baiker, Catal. Lett., 2000, 66, 109–1012 CrossRef.
- T. Bürgi, F. Atamny, R. Schlögl and A. Baiker, J. Phys. Chem. B, 2000, 104, 5953–5960 CrossRef.
- S. Lavoie, M. A. Laliberte and P. H. McBreen, J. Am. Chem. Soc., 2003, 125, 15756–15757 CrossRef CAS PubMed.
- Z. Cakl, S. Reimann, E. Schmidt, A. Moreno, T. Mallat and A. Baiker, J. Catal., 2011, 280, 104–115 CrossRef CAS PubMed.
- F. Meemken, A. Baiker, J. Dupre and K. Hungerbühler, ACS Catal., 2014, 4, 344–354 CrossRef CAS.
- F. Meemken, A. Baiker, S. Schenker and K. Hungerbühler, Chem. – Eur. J., 2014, 20, 1298–1309 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |