
Tellurium: a maverick among the chalcogens†
Tristram
Chivers
*a and
Risto S.
Laitinen
*b
aDepartment of Chemistry, University of Calgary, Calgary, AB T2N 1N4, Canada. E-mail: chivers@ucalgary.ca
bLaboratory of Inorganic Chemistry, Center for Molecular Materials, University of Oulu, P. O. Box 3000, FI-90014, Finland. E-mail: risto.laitinen@oulu.fi
First published on 18th February 2015
Abstract
The scant attention paid to tellurium in both inorganic and organic chemistry textbooks may reflect, in part, the very low natural abundance of the element. Such treatments commonly imply that the structures and reactivities of tellurium compounds can be extrapolated from the behaviour of their lighter chalcogen analogues (sulfur and selenium). In fact, recent findings and well-established observations clearly illustrate that this assumption is not valid. The emerging importance of the unique properties of tellurium compounds is apparent from the variety of their known and potential applications in both inorganic and organic chemistry, as well as materials science. With reference to selected contemporary examples, this Tutorial Review examines the fundamental concepts that are essential for an understanding of the unique features of tellurium chemistry with an emphasis on hypervalency, three-centre bonding, secondary bonding interactions, σ and π-bond energies (multiply bonded compounds), and Lewis acid behaviour.
 Tristram Chivers | Tristram Chivers, a native of Bath, England, received his BSc, PhD and DSc degrees all from the University of Durham (UK). He is currently a Faculty Professor and Professor Emeritus of Chemistry at the University of Calgary. His primary research interests are in main group element chemistry with emphasis on the chalcogens. He is the author of two books: “A Guide to Chalcogen-Nitrogen Chemistry” and (with I. Manners) “Inorganic Rings and Polymers of the p-Block Elements: From Fundamentals to Applications”. He received the Alcan Lecture Award of the Canadian Society for Chemistry (CSC) in 1987, the E.W.R. Steacie Award from the CSC in 2001, and the Royal Society of Chemistry Award (UK) for Main-Group Element Chemistry in 1993. He was elected a Fellow of the Royal Society of Canada in 1991. In 2008 he was the recipient of the ASTech Outstanding Leadership in Alberta Science Award. |
 Risto S. Laitinen | Risto Laitinen received his MSc and PhD from Helsinki University of Technology (Finland). He joined University of Oulu (Finland) in 1988 as Professor of Inorganic and Analytical chemistry, and has served as Head of Chemistry Department in 1993–1999, 2003–2008, and 2010–2013. In 1984–1985 he was an Alexander von Humbold research fellow in Technische Universität Berlin (Germany). His research interests lie in synthetic, structural, and computational chemistry of sulfur, selenium, and tellurium. He has been an editor of the book: “Selenium and Tellurium Chemistry: From Small Molecules to Biomolecules and Materials” (with J. D. Woollins) He has long been involved in IUPAC (Member of Union Advisory Board 2004–2005, secretary, member, and national representative of Commission on Nomenclature of Inorganic Chemistry 1981–2001, and a titular member in Division of Chemical Nomenclature and Structure Representation 2015). |
Key learning points• Hypervalency and three-centre four-electron bonding account for the unusual structures of some inorganic and organic tellurium systems• Telluronium ions act as σ-acceptor ligands in anion detection • Secondary intra- and inter-molecular bonding interactions control structures and properties • Weak σ- and π-bond energies influence the structures and reactivities of tellurium compounds • Tellurium dihalides and tellurium mono- and di-cations are stabilised by coordination to electron-pair donors |
1 Introduction
Tellurium is the heaviest, non-radioactive member of the chalcogen family, which also includes oxygen, sulfur and selenium. The traditional major industrial use of tellurium has been in metallurgy, where it is used as an alloying agent, e.g. with steel or copper. In the 21st century, the emerging importance of the unique properties of tellurium compounds in both inorganic and organic chemistry, as well as in materials science, is apparent from the variety of their known and potential applications, notably in the electronics industry.1 For example, tellurium sub-oxides are used in phase-change memory chips as well as in re-writable CD, DVD and Blu-ray discs. Semi-conducting metal tellurides have a wide variety of both realised and potential uses, including as thermoelectric materials in cooling devices, in solar panels and, in the form of quantum dots, as biomarkers.Elemental tellurium has a lower abundance in the Earth's crust (ca. 1 ppb) than gold, platinum or the so-called “rare-earth” elements.1 In part, this may account for the scant attention paid to tellurium chemistry in most textbooks. Furthermore, it is commonly implied that the structures and reactivities of tellurium compounds can be inferred by extrapolation from the behaviour of their lighter chalcogen analogues. The purpose of this tutorial review is to point out with reference to contemporary tellurium chemistry, in addition to well-established observations, that this assumption is often not valid. The emphasis will be on examples that illustrate how the structures, properties and reactivities of tellurium compounds frequently differ from those of their lighter chalcogen analogues. An understanding of the fundamental chemistry of tellurium will be essential for the discovery of new functional materials and the advancement of novel applications in the coming decades. With that in mind, after a short general introduction, the following aspects of tellurium chemistry will be considered: (a) general bonding characteristics, (b) hypervalency, (c) secondary bonding interactions (SBIs), (d) multiple bonding, and (e) Lewis acid chemistry. For detailed accounts of specific topics the reader may consult the recent reviews and book chapters listed in ref. 2.
Naturally occurring tellurium is a mixture of several isotopes; those with natural abundance >1% are 122Te (2.5%), 124Te (4.6%) 125Te (7.0%), 126Te (18.7%), 128Te (31.8%) and 130Te (34.5%). This distribution gives rise to characteristic patterns in the mass spectra of compounds containing one (or more) tellurium atoms. Importantly, the nuclear spin I = 1/2 of the 125Te nucleus permits NMR spectroscopic studies of tellurium compounds both in solution and in the solid state; the low-abundant isotope 123Te (0.9%) also has I = 1/2. The scope of 125Te NMR chemical shifts relative to Me2Te (δ 0 ppm), which range from ca. −1800 ppm for the telluride ion (Te2−) to ca. +3100 ppm for tellurium cations, facilitates the identification of tellurium in varying chemical environments, e.g. different oxidation states. In addition, the magnitude of spin–spin coupling constants involving 125Te and other spin 1/2 nuclei, e.g.31P, provides valuable bonding information.
Tellurium, like sulfur and selenium, can adopt oxidation states ranging from −2 to +6 and polytellurium cations and anions both exhibit numerous fractional oxidation states (Section 3). However, as exemplified by the halides, higher oxidation states are more stable for selenium and, especially tellurium, in part as a result of lower ionization energies. For example, selenium and tellurium form stable solid tetrachlorides ECl4 (E = Se, Te), whereas SCl4 is thermally unstable. The stabilisation of higher oxidation states is also a feature of organotellurium compounds, as illustrated by the existence of air-stable hexaaryltellurium compounds Ar6Te (Ar = Ph, 4-CF3C6H4) and the related monoanions [Ar5Te]−, as well as Me6Te,3 for which there are no lighter chalcogen analogues. On the other hand, the Se and Te analogues of SCl2 are thermodynamically unstable towards disproportionation to ECl4, E2Cl2 and the element (Section 6). Tellurium subhalides e.g. Te3Cl2,4 for which there are no S or Se analogues provide a unique example of binary compounds in which the chalcogen exhibits two different oxidation states. In organotellurium chemistry the mixed-valent aryl tellurenyl halides RX2TeTeR (R = aryl; X = Cl, Br) are additional representatives of this phenomenon (Section 6).5
The homonuclear and heteronuclear single bond energies involving tellurium are significantly lower than those involving the sulfur or selenium congeners as exemplified by the following series:4
| Te–Te (149 kJ mol−1) < Se–Se (192 kJ mol−1) < S–S (266 kJ mol−1) |
| H–Te (ca. 238 kJ mol−1) < H–Se (276 kJ mol−1) < H–S (366 kJ mol−1) |
| C–Te (ca. 200 kJ mol−1) < C–Se (234 kJ mol−1) < C–S (272 kJ mol−1) |
This trend contributes to the higher lability, e.g. photochemical sensitivity, of tellurium compounds as manifested by the recent preparation of tellurium nanorods by photolysis of tBu2Te.6 Nevertheless, tellurium does show a tendency to catenate, albeit less pronounced than that for sulfur or selenium, e.g. in the formation of both inorganic and organic polytellurides (Section 3). By contrast, the propensity of tellurium to be involved in secondary bonding interactions (SBIs) is higher than those of selenium and sulfur owing to the decrease of the energy difference between the σ(E–X) and σ*(E–X) orbitals and stronger n2(X) → σ*(E–X) interactions generated by the higher polarisability of the heavier chalcogens (Section 4). Furthermore, the Pauling electronegativity of Te (2.1) is significantly lower than those of Se (2.6) or S (2.6).4 Consequently, E–X bonds (where X is more electronegative than the chalcogen) are more polar for E = Te leading to stronger SBIs, both intermolecular and intramolecular, in tellurium compounds.
The π-bond energies for Te–E (E = C, N, O, P) bonds, are significantly lower than those for selenium and, especially, sulfur counterparts. This trend has important consequences for the structures and reactivities of compounds containing these functionalities as discussed in Section 5. The Lewis acid behaviour of tellurium halides has been the subject of recent studies with an emphasis on stabilising the Te2+ and RTe+ cations (Section 6), which are potentially useful reagents in inorganic and organic tellurium chemistry, respectively.
2 General bonding features
The formation of chalcogen–chalcogen bonds, commonly referred to as catenation, is a characteristic trait in the chemistry of sulfur, selenium, and tellurium. The structural features involved in the bonding of chalcogen compounds are mainly due to two types of electronic interactions. The unstrained chalcogen–chalcogen bond adopts a torsional angle near 90° in order to minimise repulsion of the np lone pairs of the adjacent atoms (Fig. 1a). The np lone pairs may also be involved in hyperconjugative np2 → nσ* interactions leading to bond length alternations (Fig. 1b). The second lone pair occupies the valence ns orbital and has no stereochemical consequences. | ||
| Fig. 1 (a) The orthogonal lone pairs in RTe–TeR, (b) the hyperconjugative np2 → σ* interaction and (c) Te–Te bond lengths (Å) and C–Te–Te–C torsional angles (°) in acyclic organic ditellurides. | ||
The structural characteristics of the Te–Te bond are well exemplified by acyclic organic ditellurides RTe–TeR for which Te–Te distances fall within the range 2.66–2.78 Å (covalent radius of Te = 1.35 Å). As shown in Fig. 1c, the C–Te–Te–C torsional angle is generally close to 90°, but significant variations from that ideal value are observed. However, the Te–Te bond length does not seem not to be dependent on the torsional angle since, in addition to packing considerations, intermolecular contacts and steric effects may influence the Te–Te bond lengths. Interestingly, there are a few antiperiplanar ditellurides (<C–Te–Te–C = 180°), which either exhibit intramolecular heteroatom–tellurium SBIs, e.g. (2-MeOC6H4COTe–)2 or incorporate bulky substituents, e.g. TpsiTe–TeTpsi [Tpsi = tris(phenyldimethylsilyl)methyl].7
The trend to decreasing chalcogen–chalcogen bond strengths down the series S–S > Se–Se > Te–Te (Section 1) is amply illustrated from a consideration of catenated species. Whereas catenation is prevalent for sulfur compounds, this phenomenon becomes less important for selenium and tellurium, as illustrated by the number of known cyclic allotropes. While sulfur forms a large number of structurally characterised ring molecules Sn (n = 6–20), in the case of selenium, the crystal structures are known only for Se8 and Se6, although the structure of Se7 has been deduced by Raman spectroscopy combined with normal-coordinate analysis (ref. 2a and b, ESI†). The larger rings Se12 and Se19 have recently been characterized as ligands in silver(I) and copper(I) complexes.8 By contrast, homocyclic tellurium molecules are only stabilised in metal telluride or halide matrices (ref. 2a and b, ESI†). For instance, the Te8 ring is found in Cs3Te22,9a the Te6 ring is present in the complexes [(AgI)2Te6],9b [Rh(Te6)]Cl3 and [Re6Te8(TeCl3)2(Te6)],9c and the Te9 ring is incorporated in [Ru(Te9)][InCl4]2.9c The stabilisation of tellurium homocycles may be attributed to the stronger tellurium–halogen interactions compared to Te⋯Te SBIs (vide infra) in such matrices (Fig. 2).
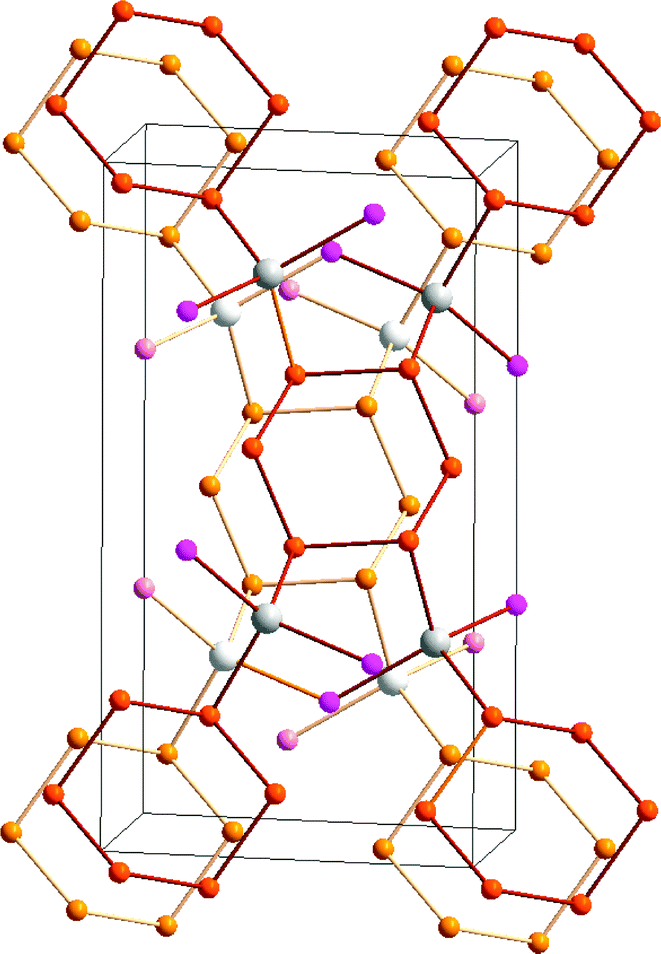 | ||
| Fig. 2 Stabilisation of cyclo-Te6 rings in a AgI matrix.9b In the front molecular layer, silver is displayed as grey, iodine as violet, and tellurium as orange. For clarity, the corresponding atoms in the second layer are shown in lighter colours. | ||
The relative stability of polymeric compared to cyclic allotropes is also a distinguishing feature of tellurium. Whereas cyclooctasulfur is the thermodynamically stable allotrope of the element at ambient conditions and the polymeric sulfur chain is unstable, polymeric tellurium is the only stable form of the element. As depicted in Fig. 3, secondary Te⋯Te interactions (Section 4), as well as weaker bond energies and the increasing importance of hypervalency (Section 3) contribute to these differences. Although it is highly insoluble in common solvents, microtubular tellurium crystals can be grown from ethylenediamine;10a hexagonal grey tellurium is also very soluble in a thiol–amine solvent mixture, presumably because the intermolecular contacts are disrupted in these solvents.10b Such solutions can be used for the facile preparation of binary tellurides, e.g. SnTe, which is used as an infrared detector.10b
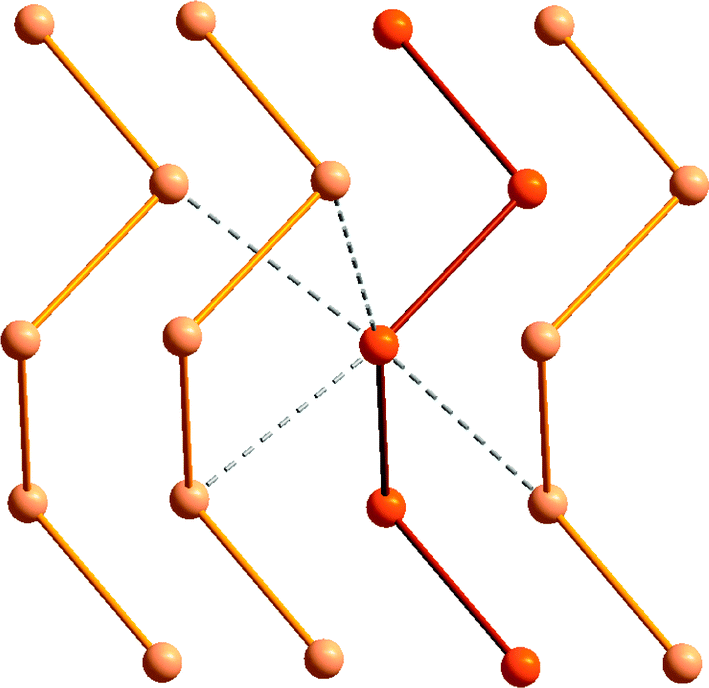 | ||
| Fig. 3 The helical structure of the hexagonal α-tellurium polymer showing the intermolecular Te⋯Te interactions. | ||
The decreasing propensity for catenation involving heavier chalcogens is also demonstrated by organic polychalcogenides. While there are numerous structurally characterised polysulfides up to RS11R and some polyselenides RSenR (n = 2–4) are known, examples of di- and tri-tellurides for tellurium are scarce and they incorporate either bulky substituents, e.g. (Me3Si)3CTeTeTeC(SiMe3)311 or intramolecular heteroatom coordination.7
3 Hypervalency
The terms hypervalency and hypervalent compounds were coined by Musher to describe a common situation for compounds of the heavier p-block elements, where the octet rule is apparently violated and the atom can formally accommodate more than eight valence electrons. The development of quantum chemistry and molecular orbital theory has resulted in the rationalization of hypervalency in terms of three centre-four electron (3c–4e) bonding, charge-transfer interactions, hyperconjugation, and SBIs. These effects become more prominent for heavier group 16 elements and explain the often unique structural characteristics and properties of tellurium compounds (ref. 2d, ESI†). At the same time, the tendency of tellurium to form π-bonds is much lower than that of sulfur and selenium. This section is concerned with σ-bonds; SBIs are discussed in Section 4 and π-bonding is covered in Section 5.The σ bonds in hypervalent species have been classified as m-E-n systems based on the formal number of electrons (m) around the chalcogen atom (E = S, Se, Te) together with the number of ligands (n). Scheme 1 shows illustrative examples of common geometries for tellurium compounds containing 3c–4e bonds. The 10-E-3 and 10-E-4 systems are based on trigonal bipyramidal geometry with two or one lone pairs of tellurium, respectively, lying on the equatorial positions; the 12-E-6 system is octahedral. The 3c–4e X–E–X fragments are approximately linear with bond orders significantly smaller than 1.
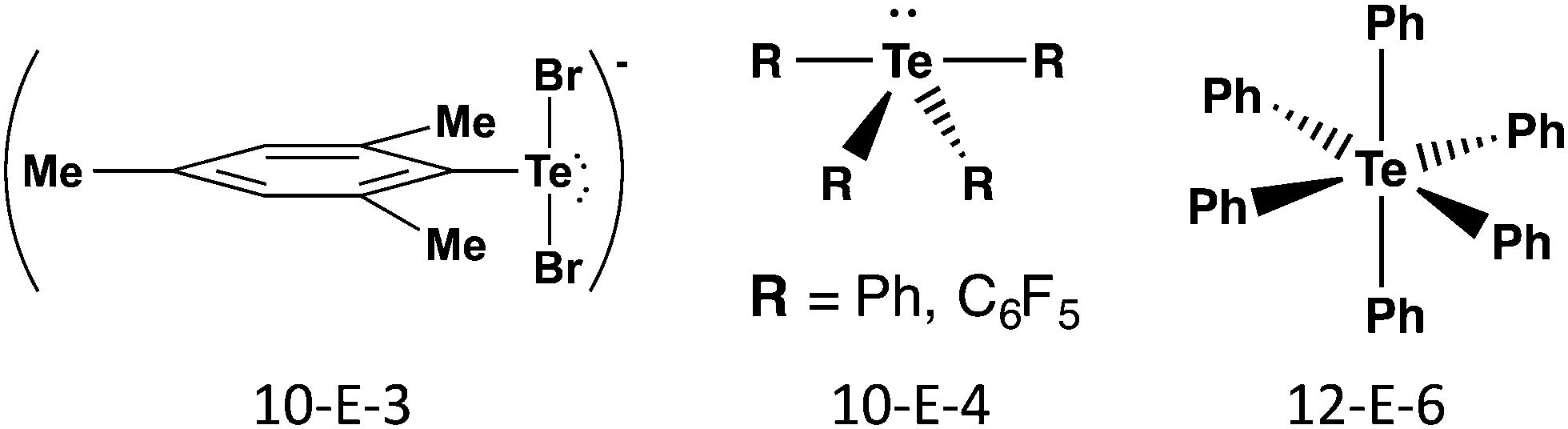 | ||
| Scheme 1 Classification and illustrative examples of hypervalent tellurium species containing 3c–4e bonds. | ||
The so-called Rundle–Pimentel model, which does not require the expansion of the electron octet, is currently accepted to describe this interaction (Scheme 2) (ref. 2d, ESI†). Simple examples of 3c–4e bonding are provided by the organotellurium ions [R2Te–Te(R)–TeR2]+ (R = Mes)12 and [PhTe–Te(Ph)–TePh]− (ref. 13) for which there are no sulfur or selenium analogues (ref. 2a, ESI†). In both ions, the Te–Te bond distances of ca. 3.0 Å are substantially longer than a single bond (ca. 2.70 Å). The monoanion (PhTe)3− is a tellurium analogue of the triiodide anion I3−, which is often used as a textbook example of 3c–4e bonding; the long Te–Te bonds are the result of the delocalisation of the bonding electron pair over all three Te atoms (Scheme 2).
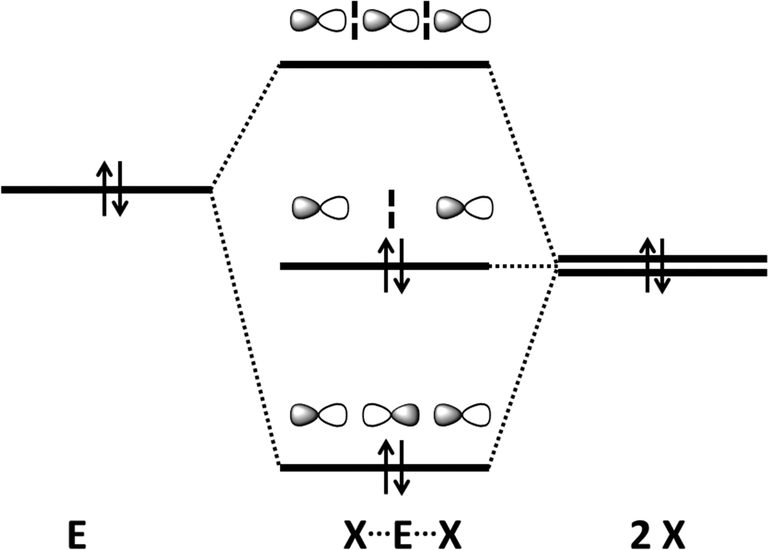 | ||
| Scheme 2 A simplified MO description of 3c–4e bonding.2 | ||
Hypervalent interactions have been compared using selenium- and tellurium-containing peri-substituted acenaphthenes [Acenap(EPh)(E′Ph)].14a For selenium, charge-transfer “spoke” adducts (X-X-ER2, 10-X-2, 1) are formed. By contrast, in the case of tellurium, only “seesaw” insertion adducts (X-ER2-X, 10-E-4, 2) have been observed (Scheme 3). The combination of NMR spectroscopic and DFT techniques has been used to study the nature of interactions between formally non-bonded, but close-lying, tellurium atoms. The exceptionally large through-space JTeTe coupling constants observed for peri-substituted compounds were attributed to 3c–4e bonding due to hyperconjugative n2 → σ* interactions.14b The JTeTe coupling constants also exhibit a clear conformational dependence.14c
 | ||
| Scheme 3 10-X-2 (1) and 10-E-4 (2) adduct formation in peri-substituted compounds. | ||
The proclivity for tellurium to engage in hypervalent interactions is also illustrated by the tendency of telluronium ions R3Te+ to act as σ-acceptor ligands. The most compelling examples of this behaviour involve installation of a telluronium group on a naphthalene skeleton with an electron-donor substituent in the peri position. This type of interaction may involve either main group or transition-metal donors.15,16 For example, telluronium boranes exhibit a strong fluoride ion affinity that can be attributed to n2(F) → σ*(Te–C) interaction in the complex 3 (Scheme 4). Compared to the sulfur analogue, the increased polarizability and electropositivity of tellurium enhance the ionic component of the fluoride-group 16 interaction; concomitantly, the larger size of tellurium allows for a stronger covalent component of this contact. These effects combine to produce elevated Lewis acidity for the R3Te+ centres that may be advantageous for applications in anion recognition or catalysis.15 In the transition-metal complex 4 (Scheme 4) the Pd–Te distance is <10% longer than the sum of the covalent radii implying a strong interaction. DFT calculations reveal a highly polar Pd–Te bond which, according to NBO analysis, involves a palladium lone pair with d character that donates to a vacant σ*(Te–C) orbital.16
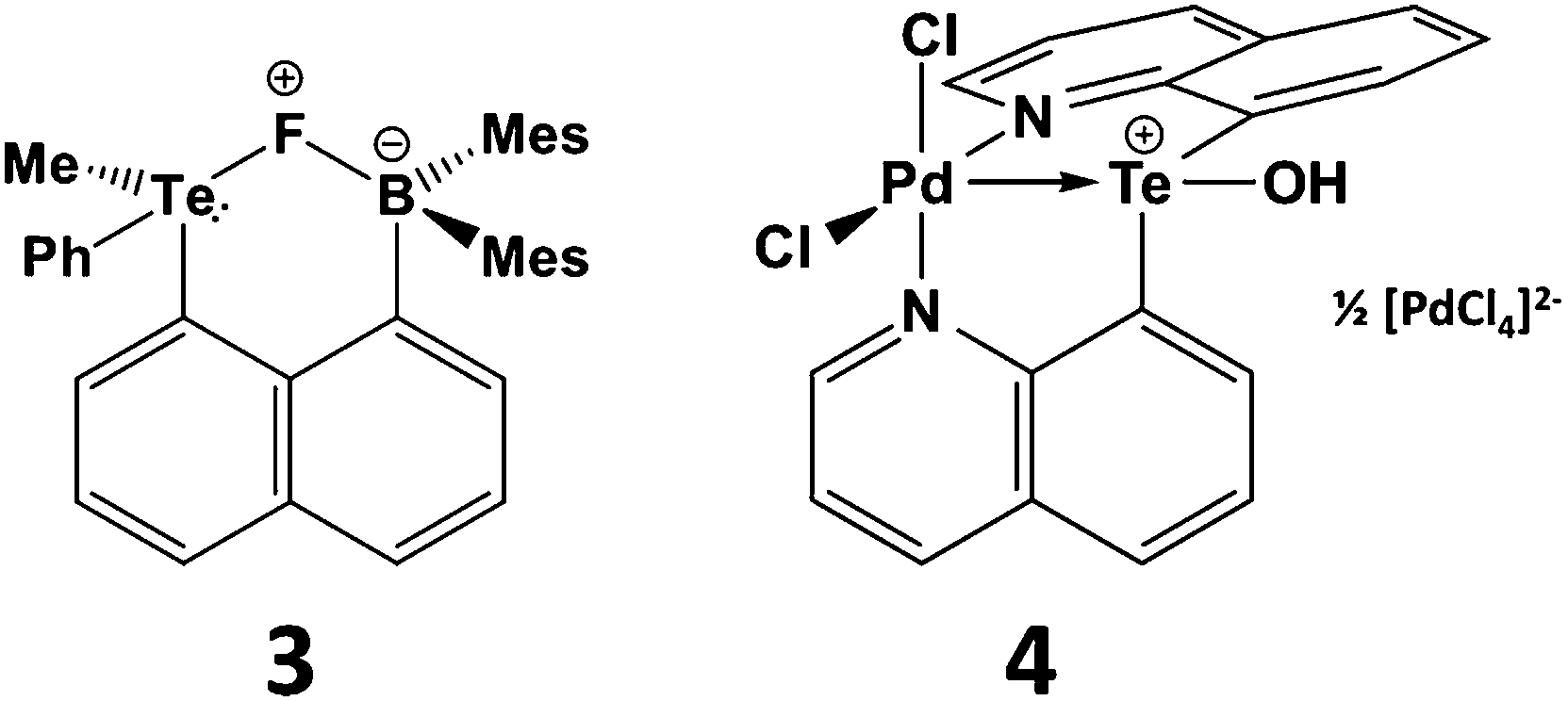 | ||
| Scheme 4 n2 → σ*(Te–C) interactions in peri-substituted telluronium salts. | ||
An intriguing example of hypervalency is provided by the complex [{Ir(TeCl4)(TeCl3)}2(Te10)] (5) (Fig. 4), in which the Te10 molecule is stabilised in a metal halide lattice (cf. examples in Section 2).17 The neutral Te10 species in 5 consists of two nearly linear 3c–4e bonding arrangements bridging two four-membered rings. The bond lengths in the Te4 ring are in the narrow range 2.779–2.820 Å, while those in the connecting 3c–4e units are 2.937 and 3.072 Å. The formal charge distribution in neutral Te10 is represented by the formula (Te+0.50)4(Te0)4(Te−)2.
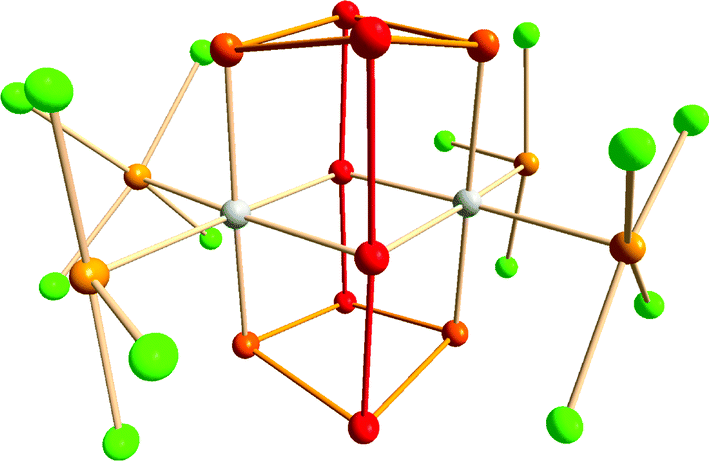 | ||
| Fig. 4 Molecular structure of [{Ir(TeCl4)(TeCl3)}2(Te10)] (5).16 The tellurium atoms and the connections involved in the 3c–4e bonding are displayed as red, other tellurium atoms in the Te10 cage as orange, exocyclic tellurium atoms in the TeCln (n = 3, 4) units as light orange, and chlorine atoms as green. | ||
Hypervalency is also involved in the bonding of some polyatomic chalcogen cations and anions (ref. 2b and c, ESI†). Whereas sulfur, selenium, and tellurium all form homopolyatomic cations with general formulae of E42+ and E82+, certain tellurium cations have no analogues among the lighter congeners, e.g. the cations (Te7)n2n+ and (Te8)n2n+ which have extended structures (ref. 2b, ESI†). In another departure from sulfur and selenium chemistry, tellurium also forms cations with charges that deviate from 2+, e.g. Te64+ (6) which adopts an elongated trigonal prismatic structure (Fig. 5a).18 DFT calculations show that the Te–Te bond lengths in 6 can be explained by the interaction of two Te32+ fragments through 6c–2e π*–π* bonds (Fig. 5b) (ref. 2a, ESI†). The elongation of the prism is caused by the lowering of energy of the occupied a2” orbital, which is antibonding with respect to the three bonds parallel to the C3 axis.
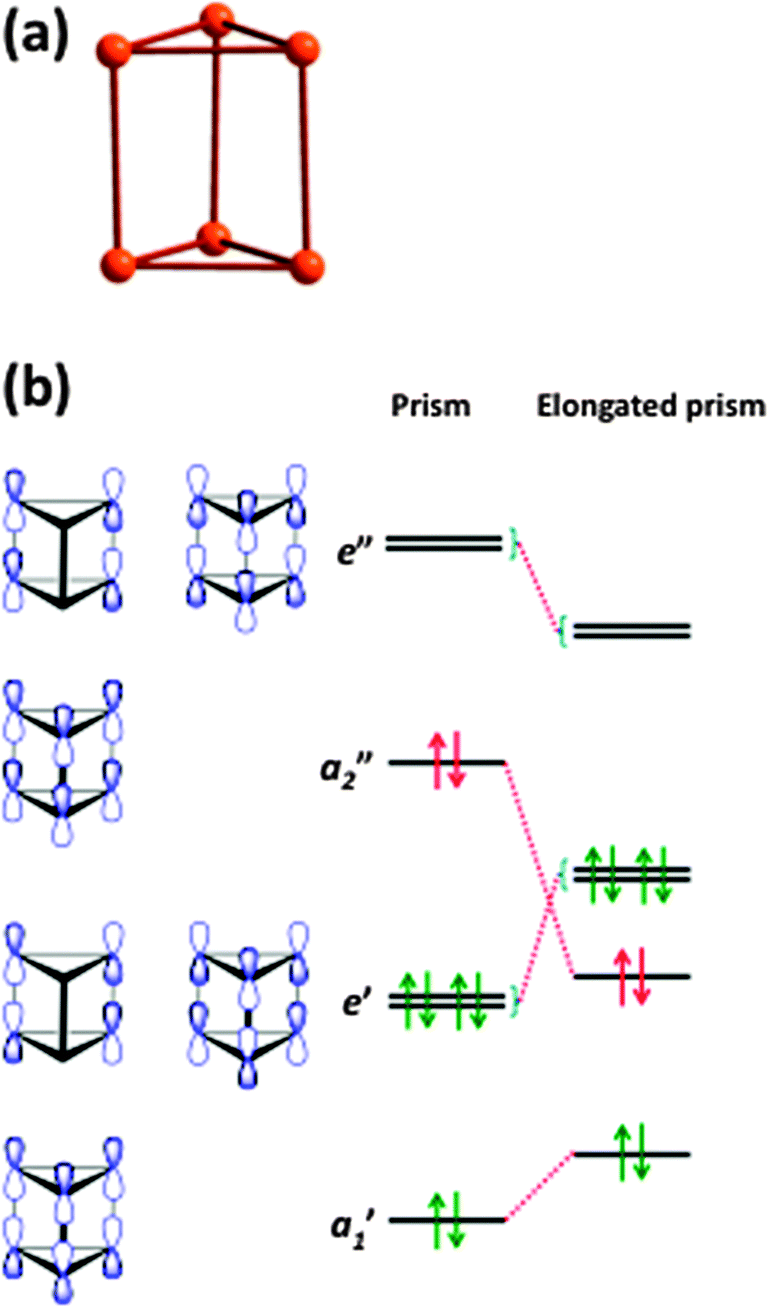 | ||
| Fig. 5 (a) Structure of the Te64+ cation (6) and (b) molecular orbitals of 6 (see references cited in ref. 2a, ESI†). | ||
Tellurium also exhibits marked differences from sulfur and selenium in the types of homoatomic anions that it engenders (ref. 2c, ESI†). In addition to unbranched chains En2−, tellurium forms unique spirocyclic dianions, e.g. Te72− (7) in [Re6Te8](Te7) (Fig. 6a)19a and Te82− (8) in [K(15-crown-5)2]2(Te8) (Fig. 6b).19b These dianions are best considered as complexes of the Te2+ cation Te,Te-chelated by two acyclic Tex2− dianions (x = 3 or 4). The rich structural chemistry of polytellurides is sometimes obscured by the deceptively simple stoichiometry of the salts (ref. 2a and c, ESI†). These anions can contain classical bent TeTe22−, linear TeTe24−, T-shaped TeTe34−, or square-planar TeTe46− units involving 3c–4e bonds and SBIs, which result in the formation of network structures as illustrated for (Te6)n− (9) in Fig. 6c.
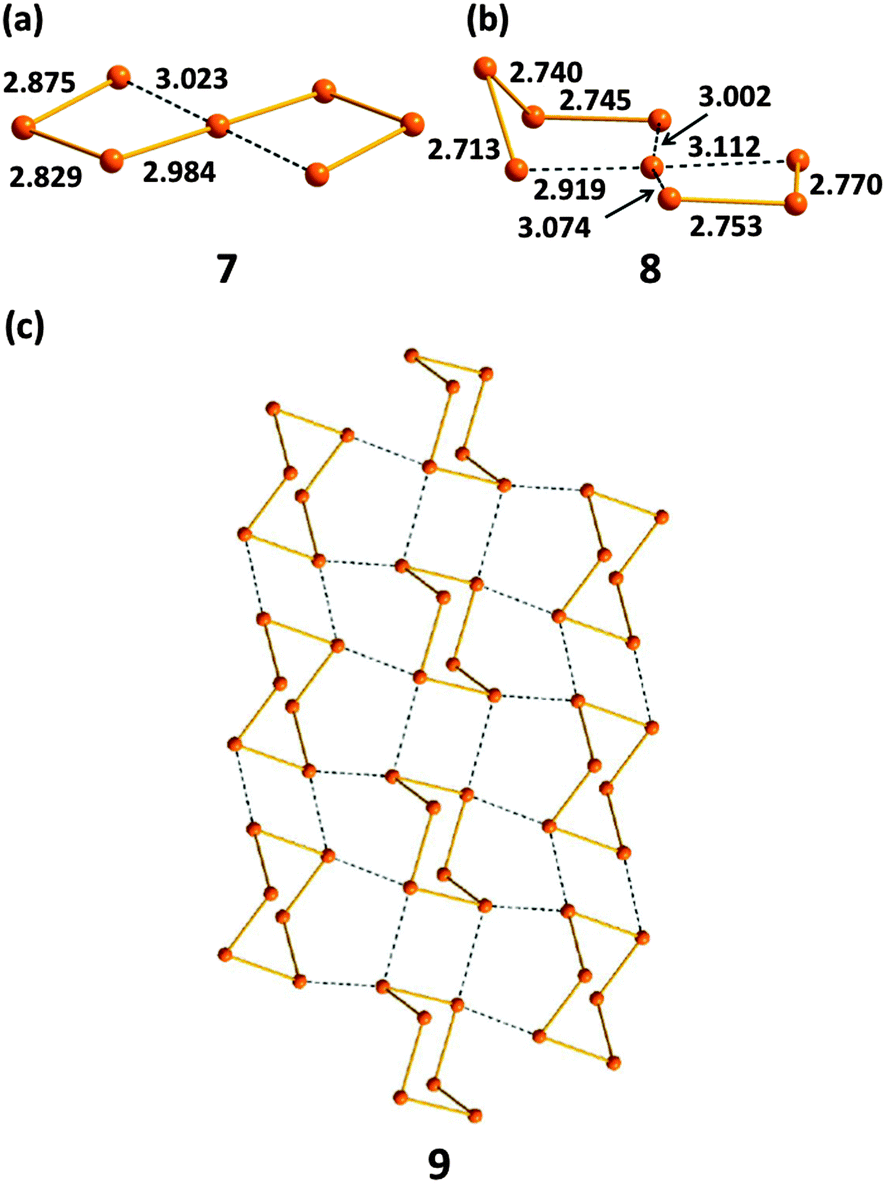 | ||
| Fig. 6 The structures of (a) spirocyclic Te72− (7), (b) spirocyclic Te82− (8), (c) polymeric (Te6)n− (9) (see references cited in ref. 2a, ESI†). Bond distances are in Å. | ||
4 Secondary bonding: intra- and inter-molecular interactions
The concept of a secondary bond describes interactions which result in interatomic contacts that are longer than covalent single bonds, but shorter than the sum of van der Waals radii (vdW radius for Te = 2.20 Å).4 All orbital interactions, as well as electrostatic and dispersion contributions (i.e. weak interactions caused by temporary dipoles) need to be taken into account for the complete description of secondary bonds. In different contexts, these interactions have also been called soft–soft, closed-shell, nonbonding, semi-bonding, non-covalent, weakly bonding, or σ-hole interactions (ref. 2e and f, ESI†). In the case of tellurium compounds, a σ-hole describes a region of positive electrostatic potential, which is located on the opposite side of one of the covalent bonds of tellurium and is directed towards a negatively charged atom resulting in a non-covalent intra- or inter-molecular interaction. The different nature of SBIs can be exemplified by noting that they are predominantly covalent in telluradiazoles,20 electrostatic in isotellurazole N-oxides,21a and mainly due to dispersion interactions in bis(alkynyl)tellurides.21bSecondary bonding interactions (SBIs) bear a qualitative relationship with hypervalent interactions, but the former are much weaker than the latter. The significance of SBIs increases when descending group 16 and, consequently, the influence of secondary bonds can often be invoked to explain the structures and properties of tellurium compounds that differ from those of sulfur and selenium analogues. The secondary bond is a consequence of a n2(D) → σ*(E–X) interaction in which the lone pair of a donor atom D interacts with the antibonding σ* orbital of the heavy atom (E) and a more electronegative atom (X). This leads to a 3c–4e arrangement, which is of variable strength, but may approach that of a hypervalent single bond (Fig. 7) (ref. 2f, ESI†). Since the polarisability of atoms increases down the periodic table, the energy difference between the σ(E–X) and σ*(E–X) orbitals diminishes. This furnishes stronger SBIs for tellurium compared to those of selenium and sulfur. The 3c–4e nature of the secondary bond requires that E⋯D is co-linear with the E–X bond, as shown in Fig. 7. Consequently, the orientations of the bonds around tellurium correspond to those predicted by the VSEPR model.
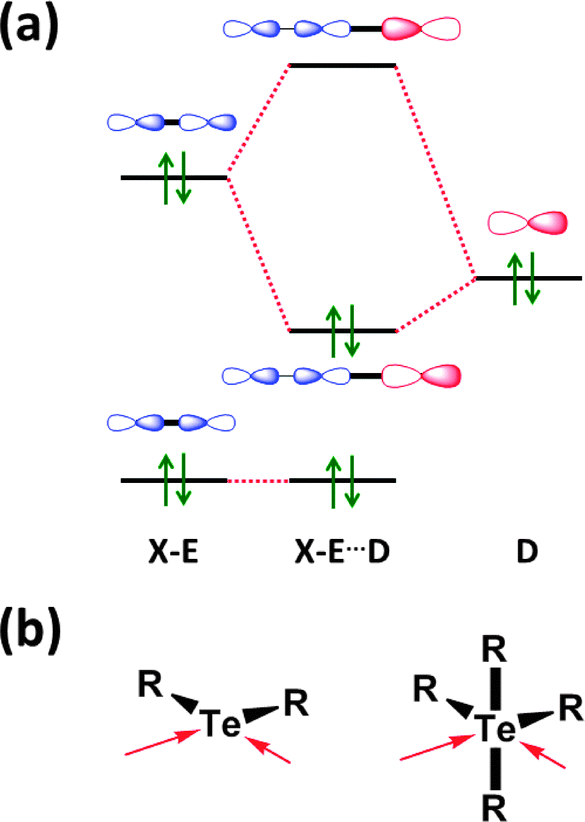 | ||
| Fig. 7 (a) Simplified depiction of orbital interactions in secondary bonding and (b) the preferred directions of the secondary bonds around tellurium(II) and tellurium(IV) (ref. 2f, ESI†). | ||
The expansion of the bonding environment of tellurium as a result of SBIs is nicely illustrated by the solid-state structure of tellurium dioxide. In contrast to the monomeric structure of gaseous sulfur dioxide SO2 (10) and the polymeric chains formed by solid selenium dioxide (11), tellurium dioxide (12) is composed of a three-dimensional network with tellurium in a distorted octahedral environment (Scheme 5) in which two Te⋯O close contacts are SBIs. The ability to participate in π-bonding with other p-block elements is dramatically diminished for tellurium (Section 5).
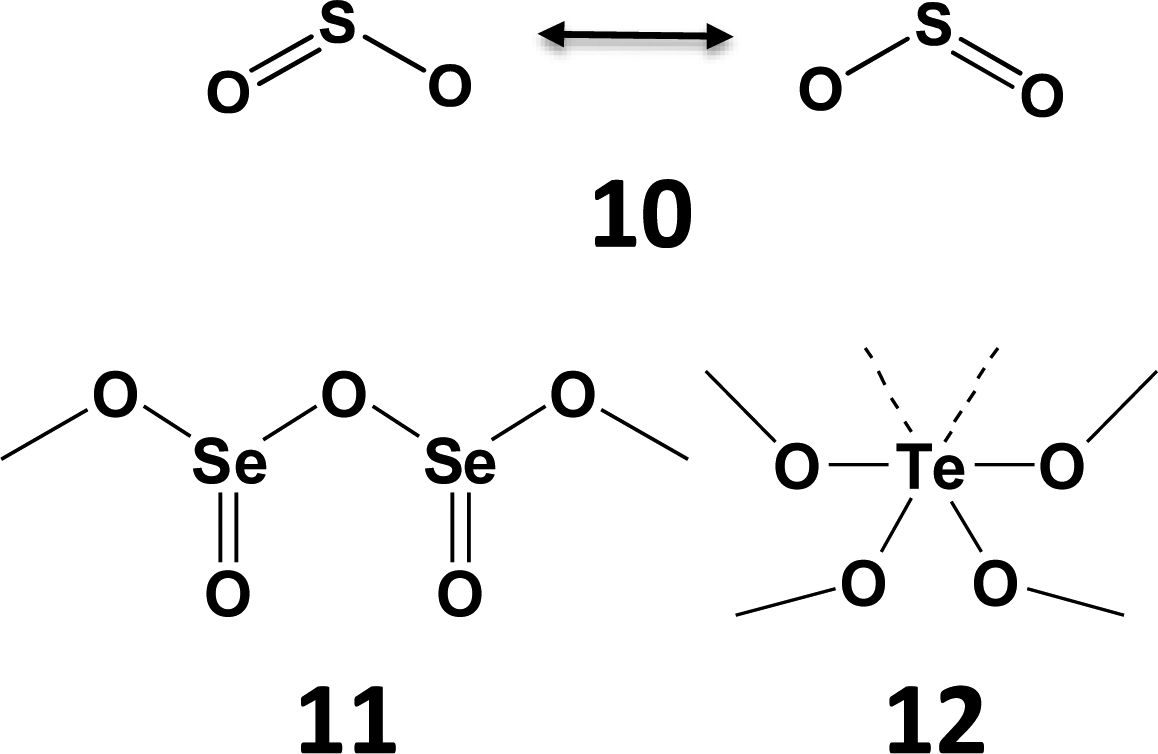 | ||
| Scheme 5 The structures of SO2 (10), SeO2 (11), and TeO2 (12). | ||
4.1 Intramolecular interactions
SBIs can be either intra- or inter-molecular. Intramolecular heteroatom interactions with tellurium can stabilise reactive functional groups in organic compounds and are also relevant in biological systems. As a simple example, aryl tellurium halides, e.g. PhTeX (X = Cl, Br), are unstable with respect to disproportionation. However, this class of organotellurium(II) compound is stabilised thermodynamically by an n2(N) → σ*(Te–Cl) interaction in (2-Me2NCH2C6H4)TeCl (13) (Scheme 6). The N⋯Te contact of 2.362 Å in 13 is longer than a single bond, but substantially shorter than the van der Waals distance; concomitantly the Te–Cl bond is elongated (2.536 Å) owing to partial occupation of the σ*(Te–Cl) orbital.22a The available metrical data for these heteroatom-stabilised derivatives indicate that the Te–X bond becomes longer as the strength of the n2(N) → σ*(Te–X) interaction increases. Intramolecular N⋯Te coordination is also a feature of the stabilisation of the first monomeric telluroxide (2-MeNCH2C6H4)2Te![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (Section 5).22b
O (Section 5).22b
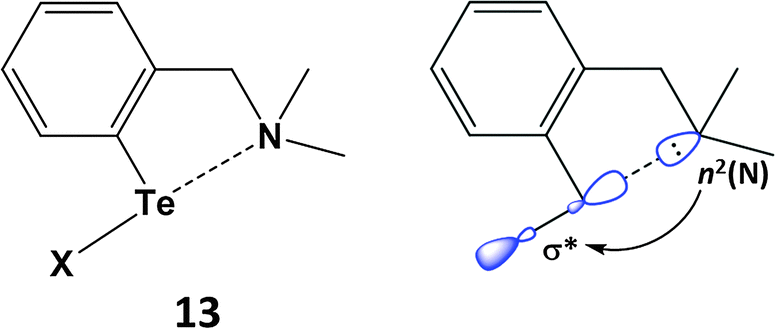 | ||
| Scheme 6 Stabilisation of ArTeX via an n2(N) → σ*(Te–X) (X = Cl, Br) interaction. | ||
The significance of SBIs for biological activity was demonstrated by exploring the catalytic function of organic ditellurides in the reduction of H2O2 using PhSH as a co-substrate.22c It was found that that while intramolecular Te⋯N SBIs enhance the reduction rate, an increase in the strength of this interaction has an adverse effect on the rate of reaction. The formation of a tellurenyl sulfide containing a 3c–4e PhS⋯Te⋯N moiety was observed during the reduction and it was suggested that a strong Te⋯N interaction stabilises the tellurenyl sulfide and thus precludes a nucleophilic attack of PhSH at sulfur, which is an important step in the catalytic cycle.22c
4.2 Intermolecular interactions
Intermolecular SBIs may produce materials with novel conducting or magnetic properties, as exemplified by benzo-2,1,3-chalcogenadiazoles, which can be used as building blocks in the construction of supramolecular materials (ref. 2f, ESI†). Their strong, directional SBIs involve two donor–acceptor n(N)2 → σ* interactions. In the case of tellurium, these SBIs are similar in magnitude to hydrogen-bonding. In contrast to the monomeric structures adopted by thia- and selena-diazoles in the solid state, benzo-1,2,5-telluradiazole (14) is associated into infinite ribbon chains with Te⋯N SBIs of 2.682(7)–2.720(7) Å (Fig. 8a) (ref. 2f, ESI†).20 The steric hindrance in the dibromo derivative (15) restricts the supramolecular association and discrete dimers with Te⋯N SBIs of 2.697(8) Å are formed (ref. 2f, ESI†).20 The supramolecular network is supplemented by very weak N⋯H and Te⋯Br interactions between dimeric molecules in the same plane (Fig. 8b).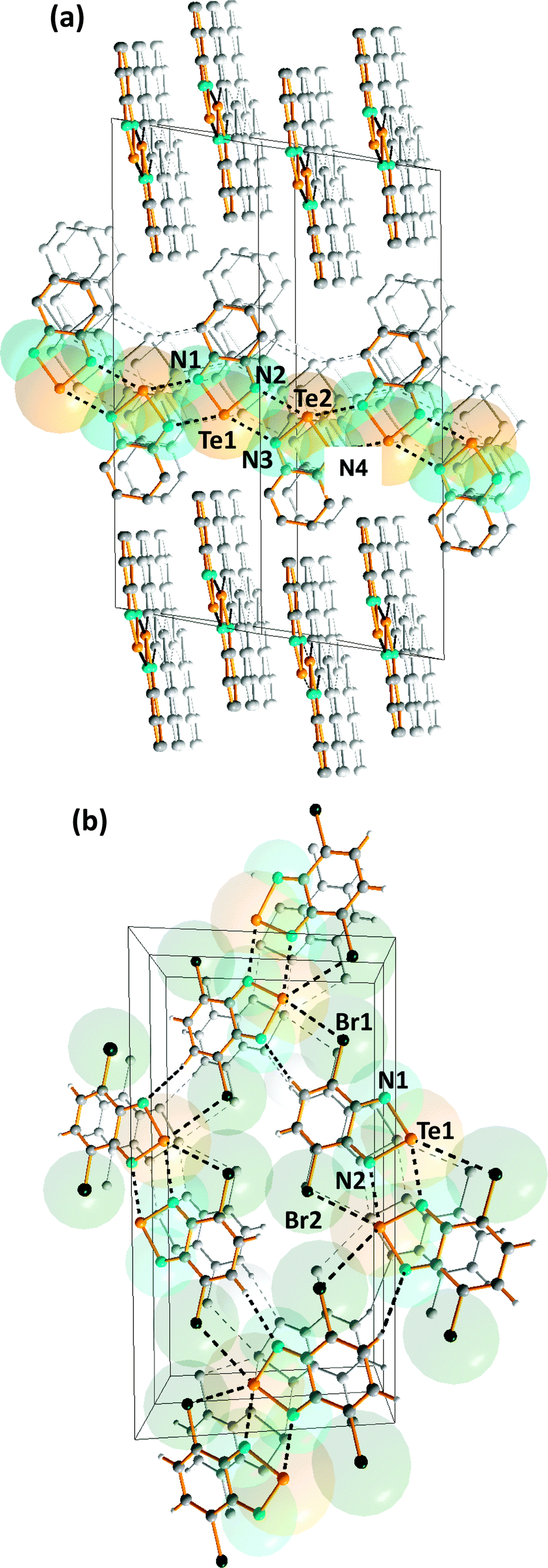 | ||
| Fig. 8 Supramolecular lattices of (a) benzo-1,2,5-telluradiazole (14) and (b) 3,5-dibromobenzo-1,2,5-telluradiazole (15) (ref. 2f, ESI†).20a | ||
In addition to optical communications, non-linear optical (NLO) materials are of interest in a number of industrial applications, e.g. gas-sensing and the detection of hazardous materials. 1,2,5-Telluradiazoles may act as building blocks in materials that exhibit NLO activity. A moderate steric repulsion must be introduced within the supramolecular ribbon-polymers to distort the structure and create a non-centrosymmetric crystal with second-order NLO properties. The second harmonic generation efficiency of telluradiazoles is modest owing to the antiparallel orientation of the molecular dipoles in the crystal, but 5-benzoylbenzo-2,1,3-telluradiazole was found to form acentric crystals thus facilitating the potential design of more efficient NLO materials (ref. 2f, ESI†).20
SBIs are also involved in the products of one-electron oxidation of dialkyl ditellurides. The initial oxidation process generates the radical cation (RTe–TeR)˙+, which dimerises to rectangular (RTe)42+ [R = Et, nPr, iPr, Ph]. The long Te⋯Te contact of 3.284 Å in (EtTe)42+ (16) results from a weak π*–π* interaction (Fig. 9), which removes electron density from the antibonding π* orbital of (EtTe–TeEt)˙+ generating short Te–Te bonds of 2.653 Å in the monomeric units.23a A monomeric radical cation with bulky substituents (R = 2,6-MesC6H3) has recently been structurally characterised (d(Te–Te) = 2.662 Å).23b
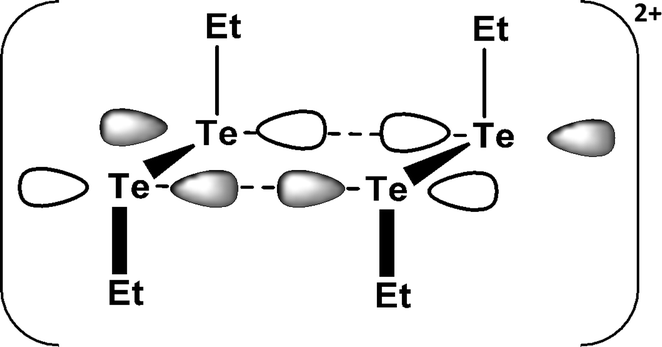 | ||
| Fig. 9 The π*–π* interaction in (EtTe)42+ (16).23a | ||
5 Multiple bonding
The increasingly poor np-2p π-overlap for heavier group 16 elements: S (n = 3) > Se (n = 4) > Te (n = 5) is evinced in significantly weaker π-bonds between tellurium and second row elements compared to those involving the lighter chalcogens. In this section the structural consequences of this trend will be illustrated with selected examples taken from chalcogen–carbon, chalcogen–nitrogen and chalcogen–oxygen chemistry. This behaviour also extends to the third row as manifested by the structures and reactivities of chalcogen–phosphorus compounds.The reluctance of tellurium to engage in π-bonding is evident from the paucity of compounds containing the TeC< functionality, moreover those that have been isolated as monomers exhibit both thermal instability and photosensitivity. Examples of telluroketones TeCR2 are scarce (ref. 2g, ESI†); they display a strong tendency to dimerise, despite the protective influence of two bulky substituents on the carbon atom. The first stable telluroketone, 1,1,3,3-tetramethylindantellone, was isolated in the 1990s and the structure of the weakly coordinated η1,σ-tungsten pentacarbonyl complex (17), a dark purple compound, was subsequently reported (Scheme 7).24 The carbon–tellurium bond length [d(C–Te) = 1.987(5) (4) Å] in 17 is close to the calculated value of 1.968 Å for the model telluroketone Me2CTe. By contrast, all other metal complexes of telluroketones or telluroaldehydes adopt a η2,π-bonding mode.
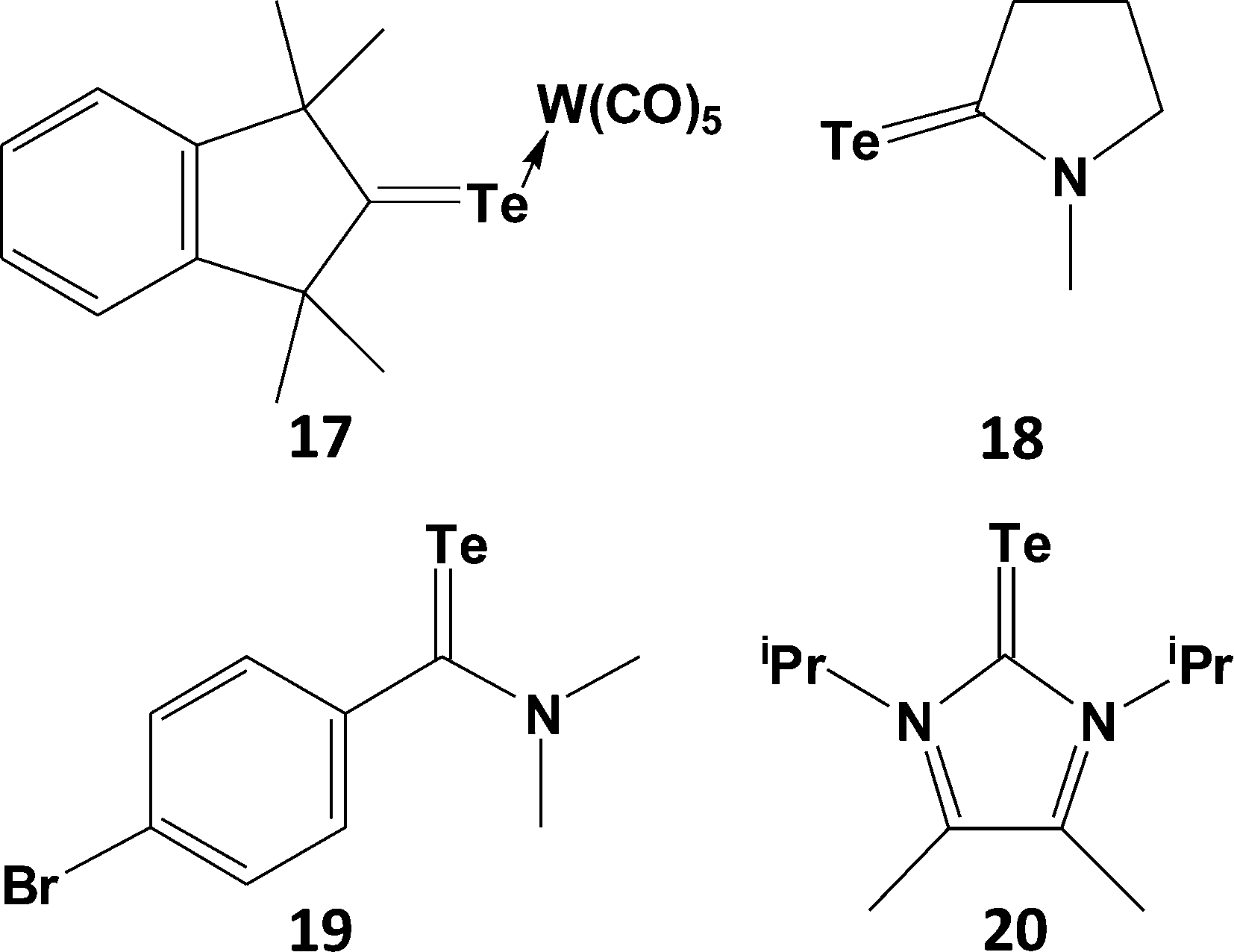 | ||
| Scheme 7 Telluroketones and telluroamides. | ||
Although telluroamides TeC(NR2)R′ exhibit higher thermal stabilities than telluroketones due to the resonance effect of the nitrogen substituent (vide infra), they are markedly less stable than their selenium analogues (ref. 2h, ESI†). In 1997 the first crystal structures of telluroamides, e.g.18, revealed C–Te bond distances in the range 2.04–2.05 Å, mid-way between single and double-bond values.25a More recently, the aromatic telluroamide 19 was shown to have a similar C–Te bond length (2.056(4) Å).25b These structural parameters indicate that resonance structure B is a major contributor to the bonding in telluroamides (Scheme 8). The elongation of the C–Te bond afforded by the introduction of amino substituents on carbon is further illustrated by the value of 2.087(4) Å found for the tellurourea 20.25c
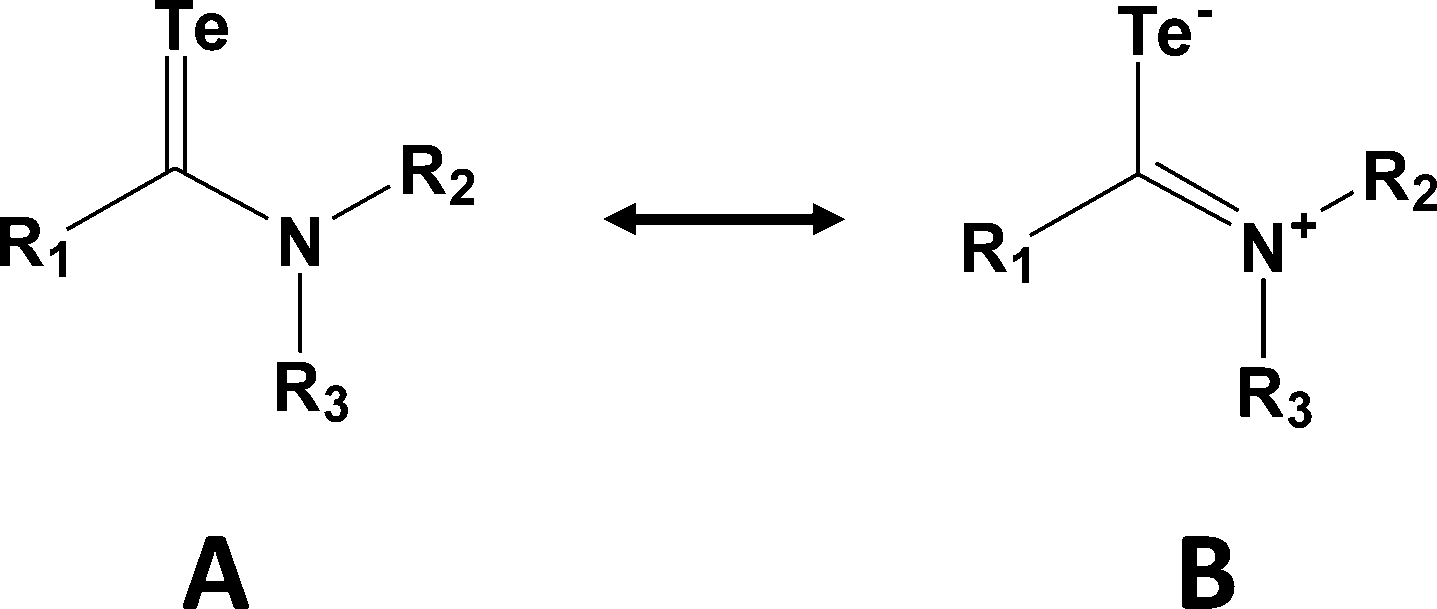 | ||
| Scheme 8 Resonance structures for telluroamides. | ||
Telluroaldehydes TeCHR are limited to one bulky group on the carbon atom, consequently they show an even stronger tendency than telluroketones to dimerise to give four-membered rings, 1,3-ditelluretanes. A compelling example of this behaviour is the reductive elimination of a telluroaldehyde fragment in the reaction of a vanadium alkylidene–telluride complex (21) with an excess of tellurium (Scheme 9). The extruded monomer TeCHtBu spontaneously dimerises to give a four-membered 1,3-C2Te2 ring (23), as well as the five-membered 1,2,4-tritelluroalane (24) presumably formed by Te insertion.26
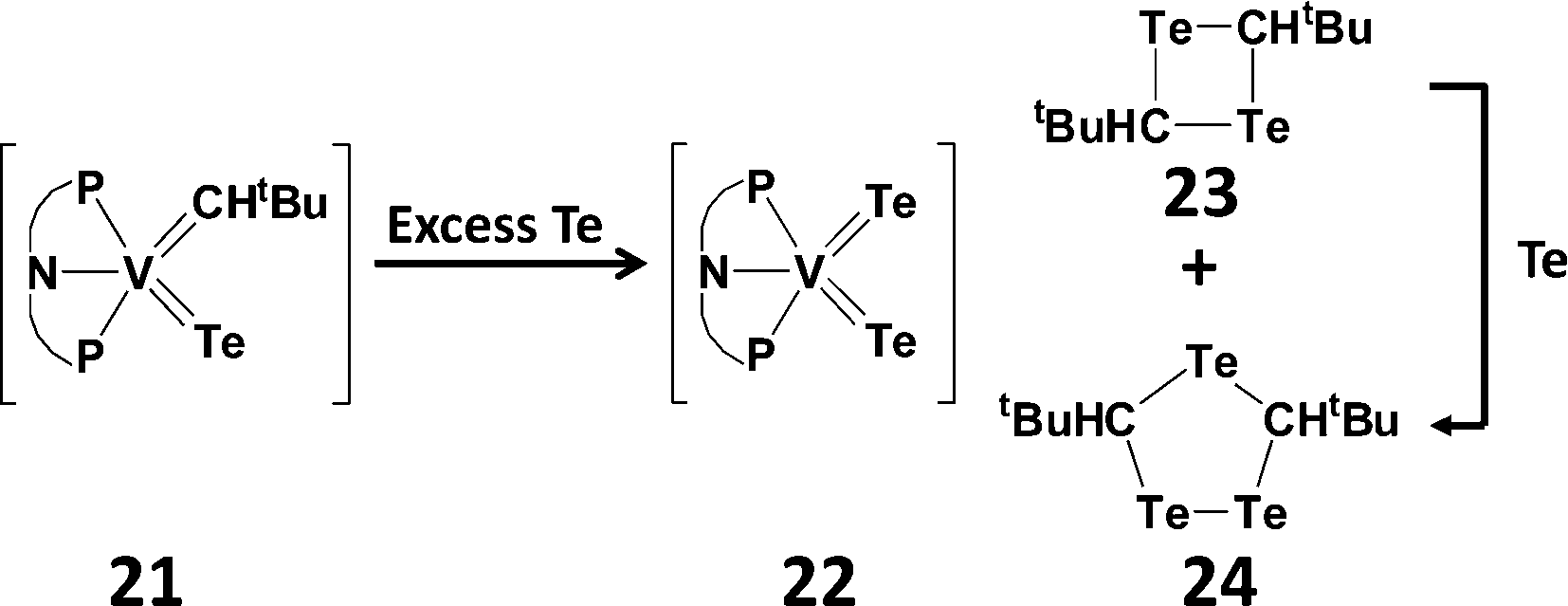 | ||
| Scheme 9 Dimerisation of a transient telluroaldehyde [PNP = N(2-PiPr2-4-methylphenyl)2].26 | ||
The structures and reactivities of the series of chalcogen(IV) diimides E(NR)2 (E = S, Se, Te) provide a persuasive demonstration of the relative weakness of tellurium–nitrogen π-bonding (ref. 2i, ESI†). All sulfur(IV) and selenium(IV) diimides are monomeric, although the latter are thermally unstable in solution with respect to decomposition to give cyclic selenium imides.27 By contrast, tellurium(IV) diimides adopt dimeric structures with different conformations in the solid state (Scheme 10).28
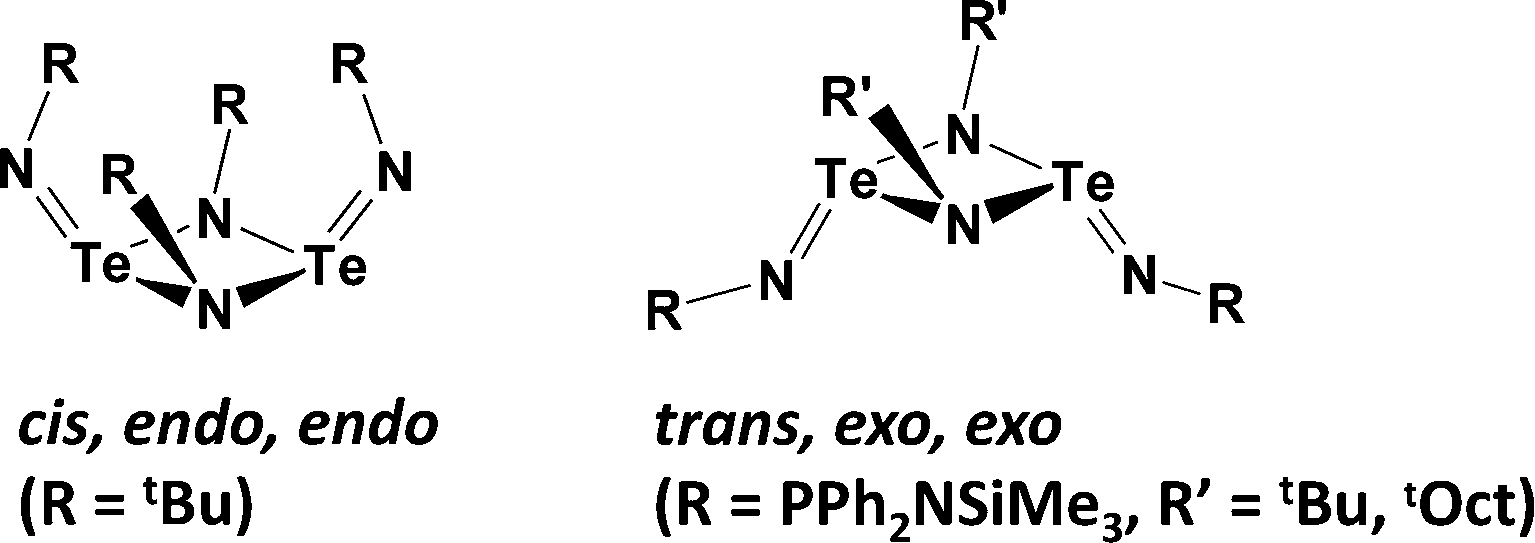 | ||
| Scheme 10 The two major conformations of tellurium(IV) diimide dimers. | ||
These experimental observations can be rationalised through consideration of the calculated dimerisation energies for the [2+2] cycloaddition of two E(NR)2 monomers (E = S, Se, Te; R = H, Me, tBu, SiMe3). As illustrated in Fig. 10, this process is strongly endothermic for sulfur(IV) diimides, approximately thermoneutral for selenium(IV) diimides, and markedly exothermic for tellurium(IV) diimides (ref. 2i, ESI†).
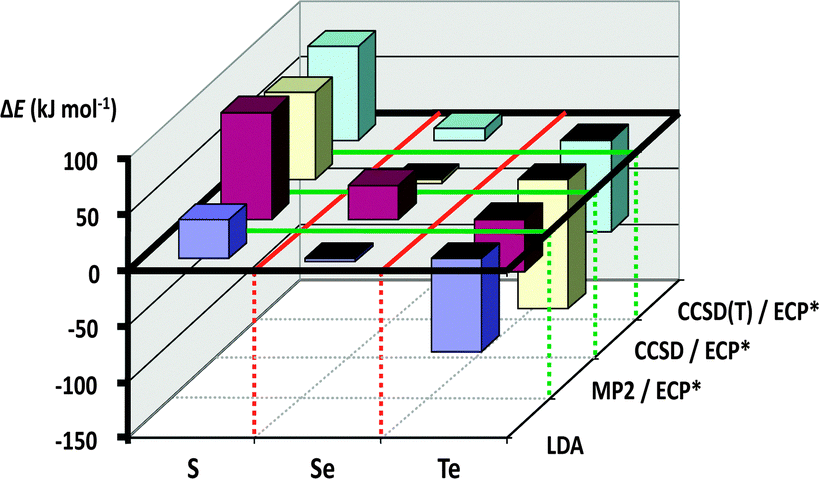 | ||
| Fig. 10 Cyclodimerisation energies of E(NMe2)2 (E = S, Se, Te)29 (reproduced with permission by Taylor & Francis). | ||
The increasing tendency for the heavier chalcogen imides to undergo dimerisation is further illustrated in the structures of hybrid imido–oxo compounds RNEO. The sulfur derivatives (E = S) are monomeric (25), whereas the selenium analogue OSe(μ-NtBu)2SeO is a dimer (26) in the solid state.27 The corresponding imidotelluroxane has only been obtained as a bis-adduct of the tetramer [OTe(μ-NtBu)2Te(μ-O)]2 with the strong Lewis acid B(C6F5)3 (27) (Scheme 11); this adduct formation presumably blocks the production of the energetically favoured polymer (tBuNTeO)∞.30
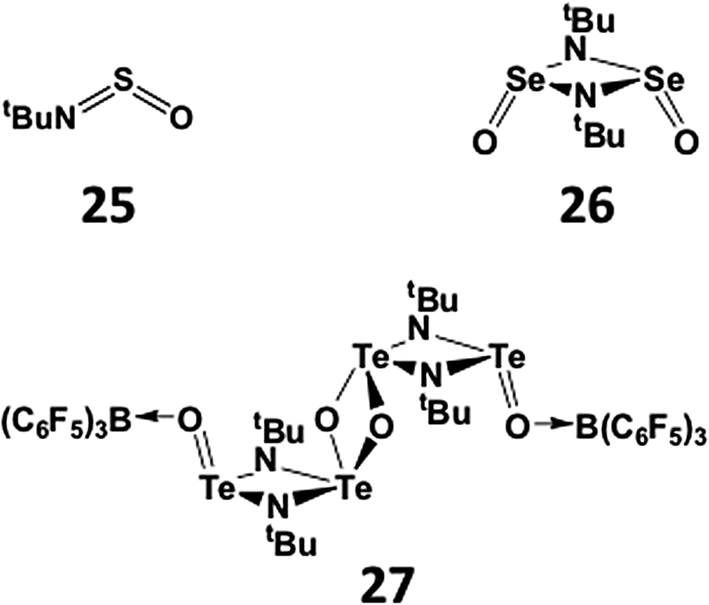 | ||
| Scheme 11 Hybrid imido–oxo compounds of sulfur, selenium, and tellurium. | ||
Binary chalcogen nitrides provide a noteworthy illustration of the individual behaviour of tellurium. The tetrachalcogen tetranitrides E4N4 (E = S, Se) both adopt an intriguing cage structure with two weak transannular E⋯E interactions.4 In distinct contrast, the tellurium analogue assumes the empirical composition Te3N4 for which a μ3-nitrido structural motif was suggested in a review31a and, subsequently, substantiated by the structural determination of the Lewis acid adduct Te6N8(TeCl4)4 (28) (Fig. 11).31b
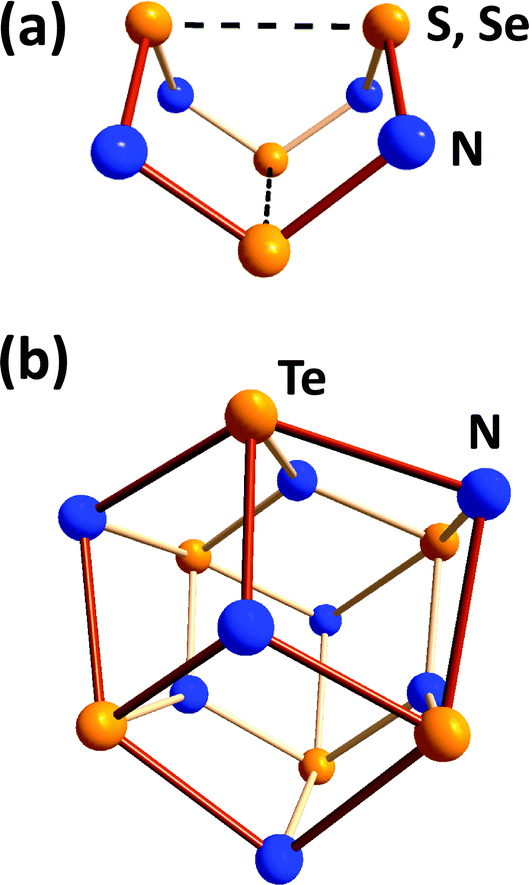 | ||
| Fig. 11 Molecular structures of (a) E4N4 (E = S, Se) and (b) the core Te6N8 unit in Te6N8(TeCl4)4 (28). | ||
The three-dimensional polymeric structure of tellurium dioxide (12) with only single Te–O bonds (Scheme 5) was discussed in Section 3. In a similar vein the structural chemistry of organic compounds incorporating TeO functionalities, e.g. diorganotellurium oxides R2TeO, diorganotellurones R2TeO2, organotellurinic acids RTe(O)OH, and organotelluronic acids RTe(O)(OH)3, provides numerous instances of the facile dimerisation of TeO bonds (ref. 2j, ESI†). For example, in the solid state diphenyltellurium oxide Ph2TeO (29) is comprised of unsymmetrical Te2O2 rings in which two monomer units [|d(TeO)| = 1.89(1) Å] are weakly associated via Te⋯O SBIs [|d(TeO)| = 2.55(2) Å]. In a further departure from the structures of the lighter chalcogen analogues Ph2EO (E = S or Se), these dimeric units are united by even longer Te⋯O SBIs [|d(TeO)| = 3.77(2) Å] leading to a one-dimensional polymer (Fig. 12a). In the diaryltellurium oxide (2-Me2NCH2C6H4)2TeO (30) which has a formal tellurium–oxygen double bond [|d(TeO)| = 1.829(1) Å],22b dimerisation is precluded by two intramolecular Te⋯N SBIs (Fig. 12b).
 | ||
| Fig. 12 Molecular structures of (a) Ph2TeO (29) and (b) (2-Me2NCH2C6H4)2TeO (30) (ref. 2j, ESI†). | ||
The well-established octahedral structure of telluric (orthotelluronic) acid Te(OH)6 is in marked contrast to the tetrahedral arrangement of ligands in selenic and sulfuric acids E(O)2(OH)2 (E = S, Se).4 This fundamental structural disparity has a profound influence on the properties of these oxo acids. Orthotelluronic acid is a weak acid (pKa(1) = 7.68, pKa(2) = 11.29), whereas selenic acid is fully dissociated in aqueous solution with respect to loss of the first proton and pKa(2) = 1.92 for the second dissociation. This behaviour is paralleled in some measure by organochalcogonic acids REO3H (E = S, Se, Te). Sulfonic and selenonic acids [PhE(O)2OH (E = S, Se)] are both strong acids, although the latter are thermally unstable. The first telluronic acid was recently shown to be a dimer [2,6-Mes2C6H3Te(μ-O)(OH)3]2 (31) with a central Te2O2 ring (Fig. 13);32 the average Te–O bond length is 1.938(3) Å, cf. 1.912(6) Å in Te(OH)6.
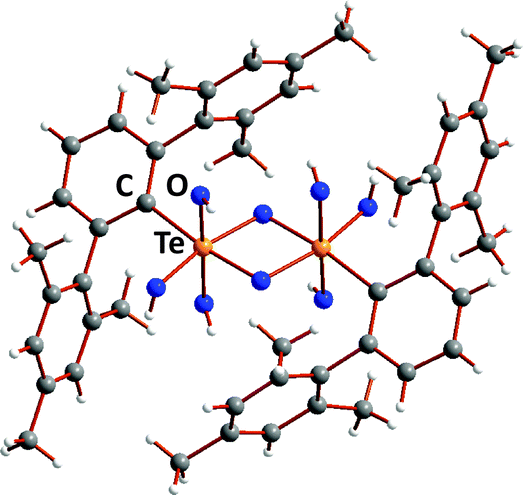 | ||
| Fig. 13 Molecular structure of telluronic acid [2,6-Mes2C6H3Te(μ-O)(OH)3]2 (31).32 | ||
DFT calculations of the energies of the various structural arrangements of chalcogonic acids PhEO3H, i.e. tetracoordinated (meta), pentacoordinated (meso) and hexacoordinated (ortho), provide a convincing rationalisation of the experimental observations.32 As illustrated in Fig. 14, the metachalcogonic acid with two EO bonds is significantly more stable than the more highly coordinated alternatives for E = S or Se, whereas the parachalcogonic acid with only single E–O bonds is decidedly preferred for E = Te; the latter has approximately the same energy as the dimeric paratelluronic acid observed in the solid state.
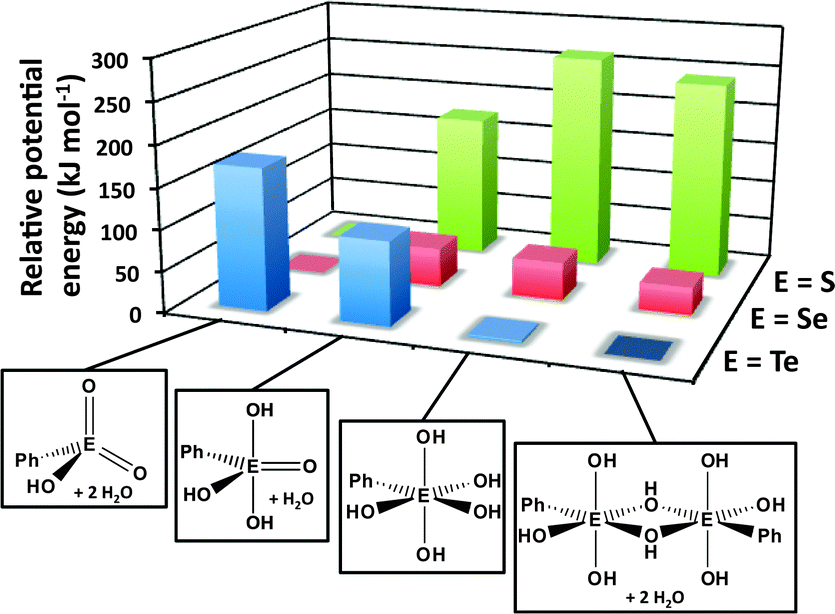 | ||
| Fig. 14 Relative potential energies of meta-, meso-, ortho-, and para-phenyl chalcogonic acids (DFT/B3PW91 calculations for E = S, Se, Te).32 The most stable structure for each triad is assigned a potential energy of zero. | ||
The structures and reactivities of phosphorus–tellurium compounds also exhibit notable differences from those of their sulfur or selenium analogues For example, the lability of the phosphorus–tellurium bond in trialkylphosphine tellurides (tellurophosphoranes) R3PTe has been used effectively in their application as tellurium-transfer reagents, e.g. for the generation of semi-conducting metal tellurides.31a However, stable coinage metal complexes with iPr3PTe in which this phosphine telluride exhibits the ability to act as a bridging ligand reflecting the softness of tellurium have been structurally characterised. For example, the cation in [Ag(iPr3PTe)2]SbF6 is a coordination polymer containing a spirocyclic arrangement of Ag2Te2 rings, whereas the selenium analogue [Ag(iPr3PSe)2]+ is comprised of linear Se–Ag–Se units that exhibit weak Ag⋯Se contacts.33
The tendency of R3PTe to undergo reversible Te transfer in the presence of free R3P is well-known. The mechanism of this process has been investigated recently for the monotelluride TeiPr2CH2PiPr2 (32), which contains P(V) and P(III) sites in the same molecule.34a Variable temperature and variable concentration 31P NMR spectroscopy have shown that 32 undergoes rapid intermolecular tellurium exchange with an activation energy of 21.9 ± 3.2 kJ mol−1 (Fig. 15), cf. a value of ca. 20.4 kJ mol−1 obtained from DFT calculations.34a In a recent book chapter on dynamic NMR spectroscopy this process has been described as “an excellent example of intermolecular exchange” since it provides a clear demonstration that the rates depend linearly on concentration.34b
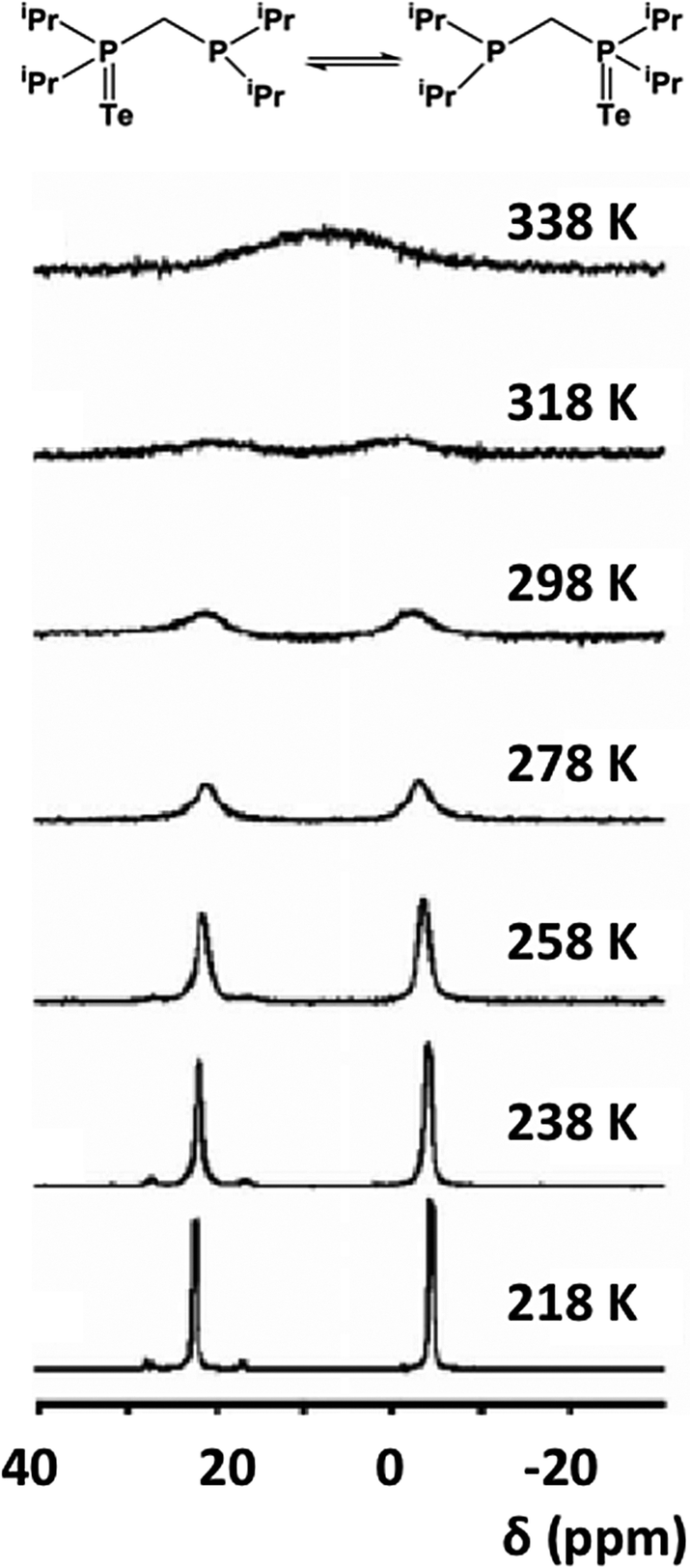 | ||
| Fig. 15 Variable-temperature 31P NMR spectra of 32 in d8-toluene.34 (Copyright © 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
Organophosphorus–chalcogen compounds of the type [ArP = E(μ-E)]2 [33a, Ar = 4-MeOC6H4, E = S, Lawesson's reagent (LR); 33b, Ar = Ph, E = Se, Woollins' reagent (WR)] (Scheme 12) are commercially available and used in organic synthesis, e.g. for the conversion of carbonyl (CO) to CE (E = S, Se) functionalities (ref. 2k, ESI†). The pyridine-stabilised monomer PhP(=Se)2·pyr has very recently been structurally characterised.35 However, extensive attempts to prepare tellurium analogues by the reaction of ArPCl2 with various tellurium sources, e.g. sodium ditelluride Na2Te2 in THF, gave only the six-membered rings (ArPTe)3 (34a, Ar = Ph3C; 34b, Ar = 2,4,6-tBu3C6H2) (Scheme 13) with P–Te single bonds and/or phosphorus-rich rings of the type (ArP)nTe (n = 2, 3, 4).36 No evidence for stable heterocycles that incorporate both terminal (exo) and bridging (endo) Te atoms, i.e. an RP = Te(μ-Te) unit, was forthcoming.
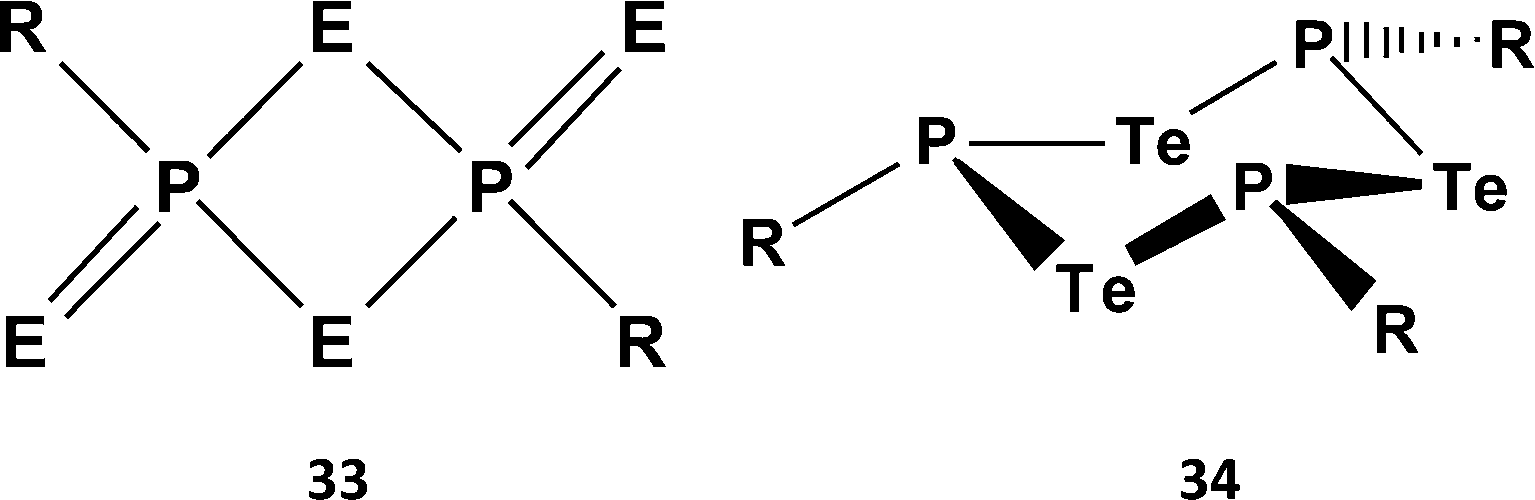 | ||
| Scheme 12 Lawesson's and Woollins' reagents (33) and (RPTe)3 rings (34). | ||
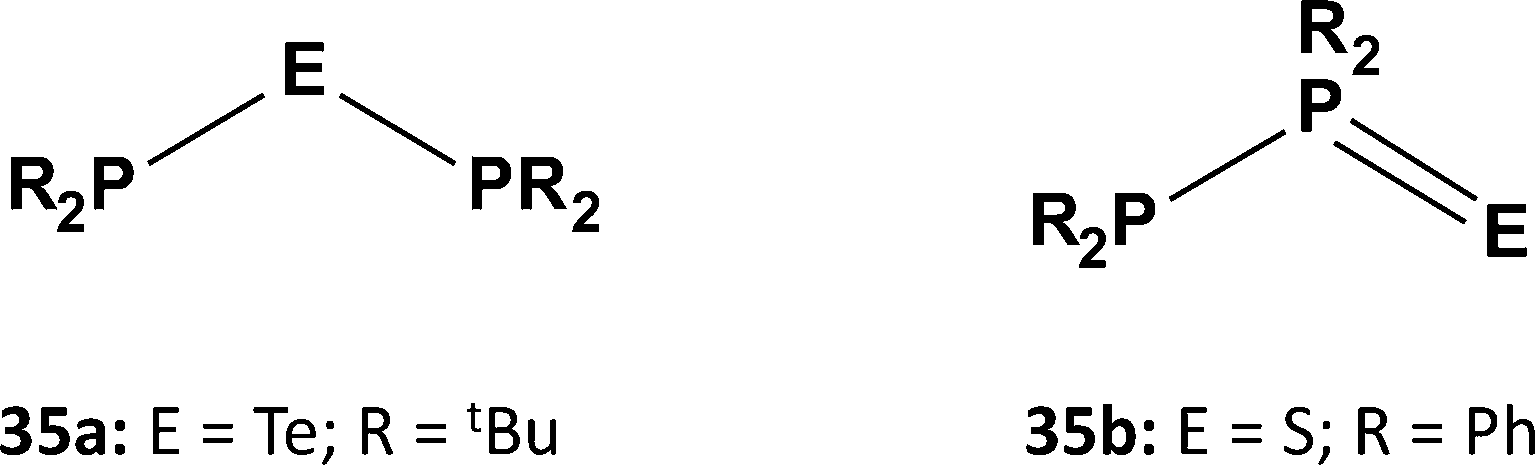 | ||
| Scheme 13 Two isomers of (RP2)2E (E = Te, S). | ||
A similar contrast in behaviour between tellurium and sulfur systems is exhibited by acyclic phosphorus–chalcogen compounds of the composition E(PR2)2. In 1980 the reaction of tBu2PCl with Na2Te was shown to provide a high-yield synthesis of the symmetrical monotelluride tBu2P–Te–PtBu2 (35a, E = Te; Scheme 13), which exhibits a singlet in the 31P NMR spectrum [1J(PTe) = 451 Hz].37a By contrast, it was recently found that treatment of R2PCl (R = Ph, Cy) with Li2S in MeCN produces the unsymmetrical monosulfides R2P–PR2S (35b; Scheme 13), which give rise to two well-separated doublets in the 31P NMR spectra [1J(PP) = 247 Hz (R = Ph) and 301 Hz (R = Cy)]; however, the monosulfides 35b rearrange to the symmetrical ligands (35a, E = S) upon coordination to ruthenium.37b
DFT calculations for the series Me3PE (E = O, S, Se, Te) provide some insights into the nature of the formal PE bond, as well as a rationale for the thermodynamic lability of the tellurium derivative.38 The phosphorus–chalcogen bond in these chalcogenides is comprised of a σ and two π components that can be represented by resonance structures C and D (Fig. 16). The two π contributions result from hyperconjugative back-donation from the chalcogen p orbitals to σ* orbitals on the R3P fragment. The σ-bond component is largest for oxygen and decreases dramatically down the series O > S > Se > Te, whereas the π-bond orders are only attenuated slightly. The combination of these bonding effects results in calculated PE bond energies of approximately −544, −337, −266 and −184 kJ mol−1, thus accounting for the thermal and photochemical instability of compounds with terminal PTe bonds.
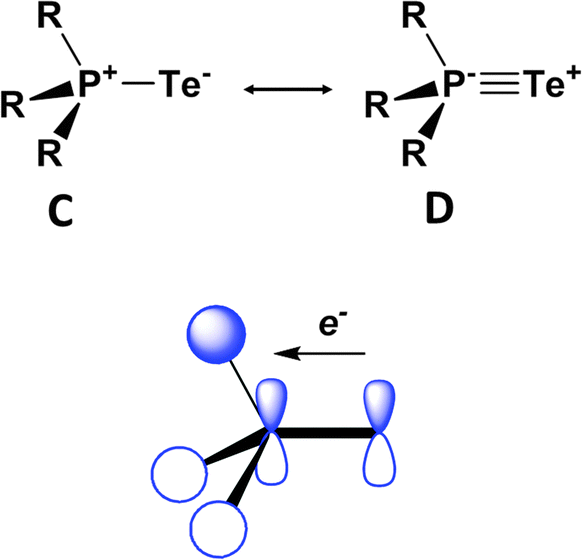 | ||
| Fig. 16 Resonance structures and one of the π-back bonds for Me3PE.38 | ||
6 Lewis acid behaviour of tellurium halides and tellurium cations
An intriguing and unique feature of the chemistry of trialkylphosphine tellurides (Section 5) is the oxidative coupling engendered by ferrocenium salts which produces dications [R3P–Te–Te–Te–PR3]2+ (R = iPr, tBu) (36).39 These dications may be viewed as either (a) phosphine-stabilised Te32+ dications (36a) or (b) complexes of the Te2+ dication with two TePR3 ligands (36b) (Scheme 14). On the basis of the observed bond lengths |d(P–Te)| = 2.492(3)–2.505(4) Å and |d(Te–Te)| = 2.713(1)–2.715(2) Å in [tBu3P–Te–Te–Te–PtBu3][SbF6]2, the authors concluded that the former description is more appropriate; however, computational support for this viewpoint is lacking. Several complexes of the Te2+ dication with monodentate electron-donor ligands, e.g. N-heterocyclic carbenes (NHCs), as well as bidentate ligands SPPh2(CH2)3PPh2S,40 dppe and 2,2′-bipyridyl have recently been characterised (vide infra) (ref. 2j, ESI†). | ||
| Scheme 14 Bonding schemes for the dication [(R3PTe)2Te]2+ (36). | ||
Chalcogen dihalides EX2 (E = S, Se, Te; X = Cl, Br, I) exhibit an increasing tendency along the series to disproportionate to a mixture of the element, E2X2 and EX4. Sulfur dichloride SCl2 may be stored for months at low temperatures, whereas solutions of SeCl2 are only stable for ca. 1 day in the coordinating solvent THF.41 Although TeCl2 is only available as an in situ reagent generated by reduction of TeCl4 with Me3SiSiMe3, the adduct tmtu·TeCl2 (tmtu = tetramethylthiourea) is well-known and has been used in metathetical reactions (ref. 2l, ESI†).
Since tellurium dihalides are potentially versatile reagents in both inorganic and organic transformations, as well as a potential source of the Te2+ dication, there has been renewed interest in coordination complexes of TeX2 (ref. 2l, ESI†). For example, TeI2 is stabilised by chelation in the adduct Dipp2BIAN·TeI2 (Dipp = 2,6-diisopropylphenyl; BIAN = bis(aryl)iminoacenaphthene).42 The reagent bipy·TeCl2 (bipy = 2,2′-bipyridine) has been used to synthesise diaryl tellurides (ref. 2l, ESI†) and phosphorescent tellurophenes (37) via a transmetallation process (Scheme 15).43 The phosphorescent property of tellurophene 37 is attributed in part to the near-degeneracy of the triplet state (T3) and the singlet excited state (S1), which are separated by ca. 1 eV in the S and Se analogues.43
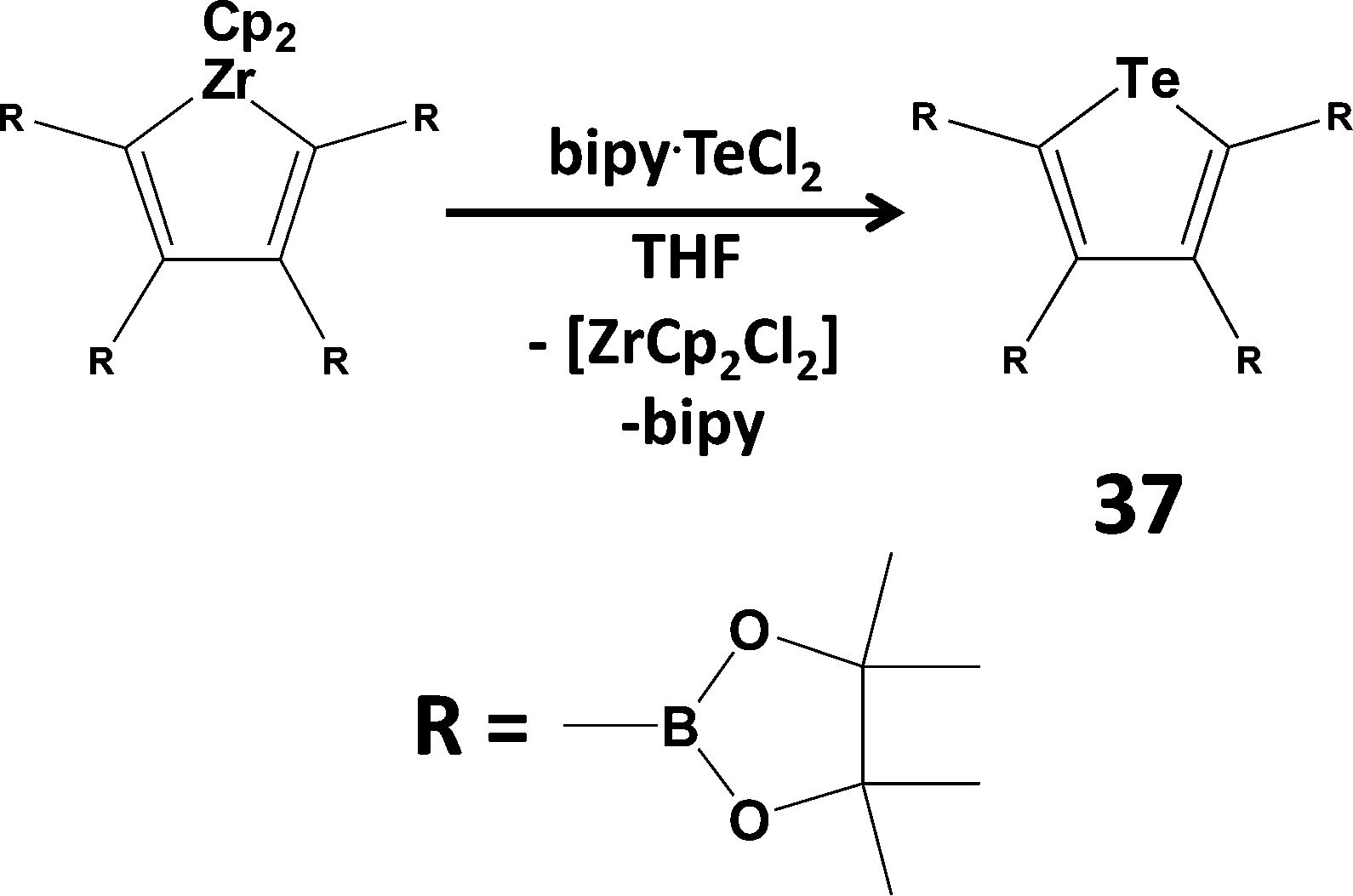 | ||
| Scheme 15 Preparation of the phosphorescent tellurophene 37. | ||
Although it has not proved possible to generate the Te2+ dication from tellurium dihalide complexes, two successful synthetic strategies have been reported recently: (a) the generation of the transient tellurium(II) triflate Te(OTf)2 (OTf = trifluoromethanesulfonate) using a ligand-exchange methodology (Scheme 16a)44a to give the dppe complex 38a or (b) reaction of chlorocyclopropenium salts with Te(SiMe3)2 to give carbene-stabilised Te2+ salts 38b (Scheme 16b).44b DFT calculations confirm the presence of two lone pairs on the two-coordinate tellurium centre in both 38a and 38b with most of the positive charge on the phosphorus atoms or the cyclopropenium rings, respectively.44
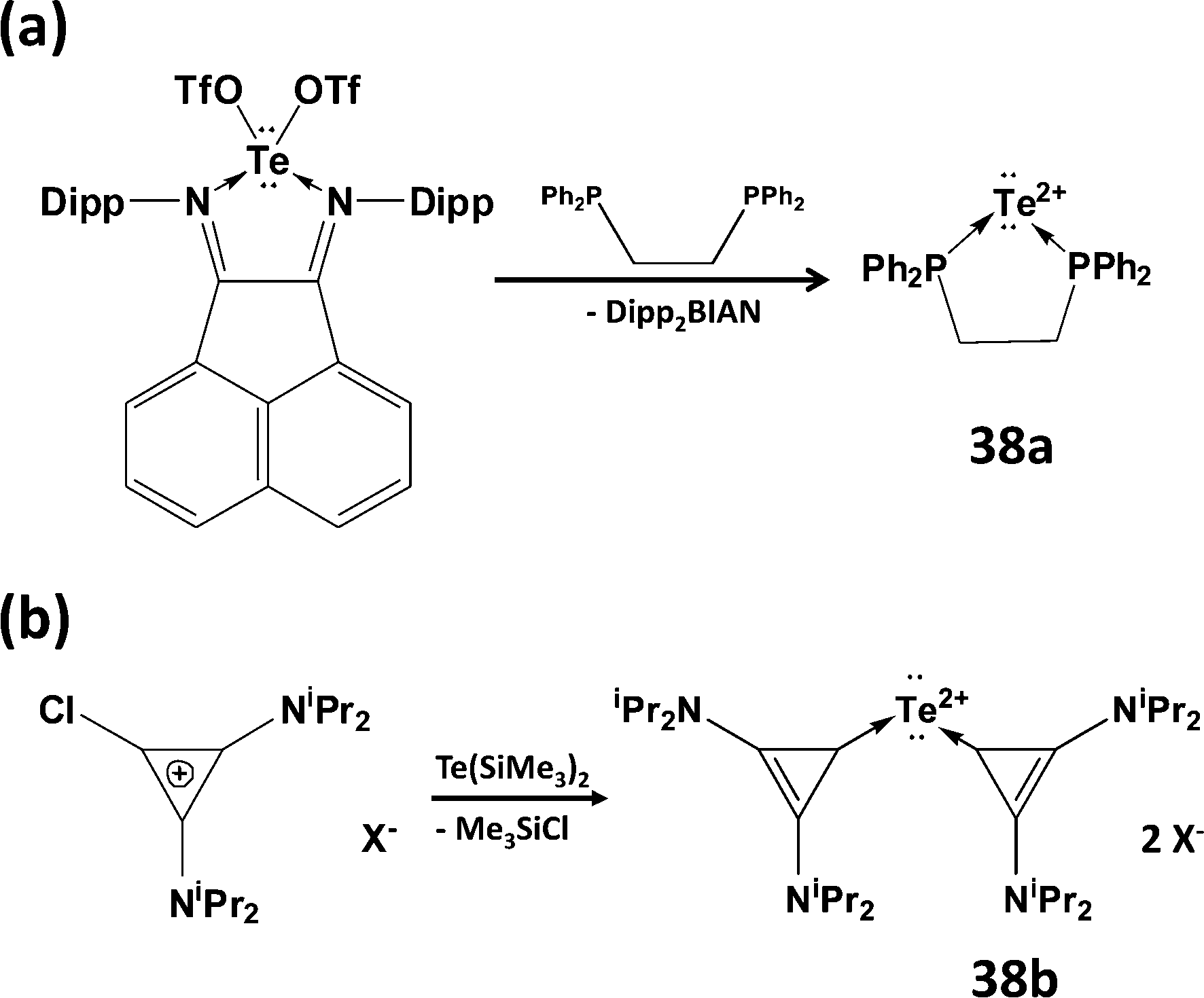 | ||
| Scheme 16 Synthetic approaches to complexes of the dication Te2+. | ||
Organotellurium(II) cations RTe+ are also long sought-after reagents. However, like tellurium(II) dihalides, the obvious precursors RTeX (X = Cl, Br, I) are unstable with respect disproportionation (Section 4.1). An exception to this instability is the compound of stoichiometry “PhTeI”, which is formed as a violet-black solid by reaction of PhTe–TePh with I2; in the solid state it exists as the tetramer Ph4Te4I4 (39) (Scheme 17a); the individual PhTeI units are linked through weak Te⋯Te bonds.45 By contrast, the selenium analogue is comprised of a charge-transfer complex between the diselenide PhSeSePh and I2 that forms the centrosymmetric structure 40via very weak Se⋯I contacts (Scheme 17b). The structural difference between 39 and 40 is analogous to that between the “seesaw” insertion adducts (2) and the charge-transfer “spoke” adducts (1) demonstrated by peri-substituted acenaphthylenes (Scheme 3).
 | ||
| Scheme 17 Structures of (a) Ph4Te4I4 (39) and (b) (Ph2Se2·I2)2 (40). | ||
The tetrameric structure of 39 is easily disrupted upon addition of PPh3 to give the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct Ph3PTe(Ph)I [d(P–Te) = 2.568(2) Å].45 However, this monomeric complex does not serve as a viable source of PhTe+. The monohalides RTeX can also be stabilised (a) by the use of highly bulky groups (kinetic stabilisation) or (b) via intramolecular coordination of a heteroatom substituent attached to an aryl ring (thermodynamic stabilisation) (Scheme 6).46 For example, stoichiometric reactions of SO2Cl2, Br2 or I2 with the bulky aryl ditelluride BbtTe–TeBbt (Bbt = 2,6-[bis(trimethylsilyl)methyl]-4-[tris(trimethylsilyl)methyl]phenyl) produces aryltellurium(II) halides BbtTeX (X = Cl, Br, I) which can be isolated as red-purple, blue or green solids, respectively.46 Interestingly, halogenation of ditellurides with less bulky aryl substituents generates mixed-valent ditellurium dihalides ArX2TeIV–TeIIAr (Ar = 2,6-dimesitylphenyl) (Section 1).5 The latter can be used as a source of aryltellurenyl cations stabilised by two-electron donors, e.g. PPh3 (41), NHC (42) (Scheme 18),46,47 or via [1+4] cycloaddition with 2,3-dimethyl-1,3-butadiene.46
:1 adduct Ph3PTe(Ph)I [d(P–Te) = 2.568(2) Å].45 However, this monomeric complex does not serve as a viable source of PhTe+. The monohalides RTeX can also be stabilised (a) by the use of highly bulky groups (kinetic stabilisation) or (b) via intramolecular coordination of a heteroatom substituent attached to an aryl ring (thermodynamic stabilisation) (Scheme 6).46 For example, stoichiometric reactions of SO2Cl2, Br2 or I2 with the bulky aryl ditelluride BbtTe–TeBbt (Bbt = 2,6-[bis(trimethylsilyl)methyl]-4-[tris(trimethylsilyl)methyl]phenyl) produces aryltellurium(II) halides BbtTeX (X = Cl, Br, I) which can be isolated as red-purple, blue or green solids, respectively.46 Interestingly, halogenation of ditellurides with less bulky aryl substituents generates mixed-valent ditellurium dihalides ArX2TeIV–TeIIAr (Ar = 2,6-dimesitylphenyl) (Section 1).5 The latter can be used as a source of aryltellurenyl cations stabilised by two-electron donors, e.g. PPh3 (41), NHC (42) (Scheme 18),46,47 or via [1+4] cycloaddition with 2,3-dimethyl-1,3-butadiene.46
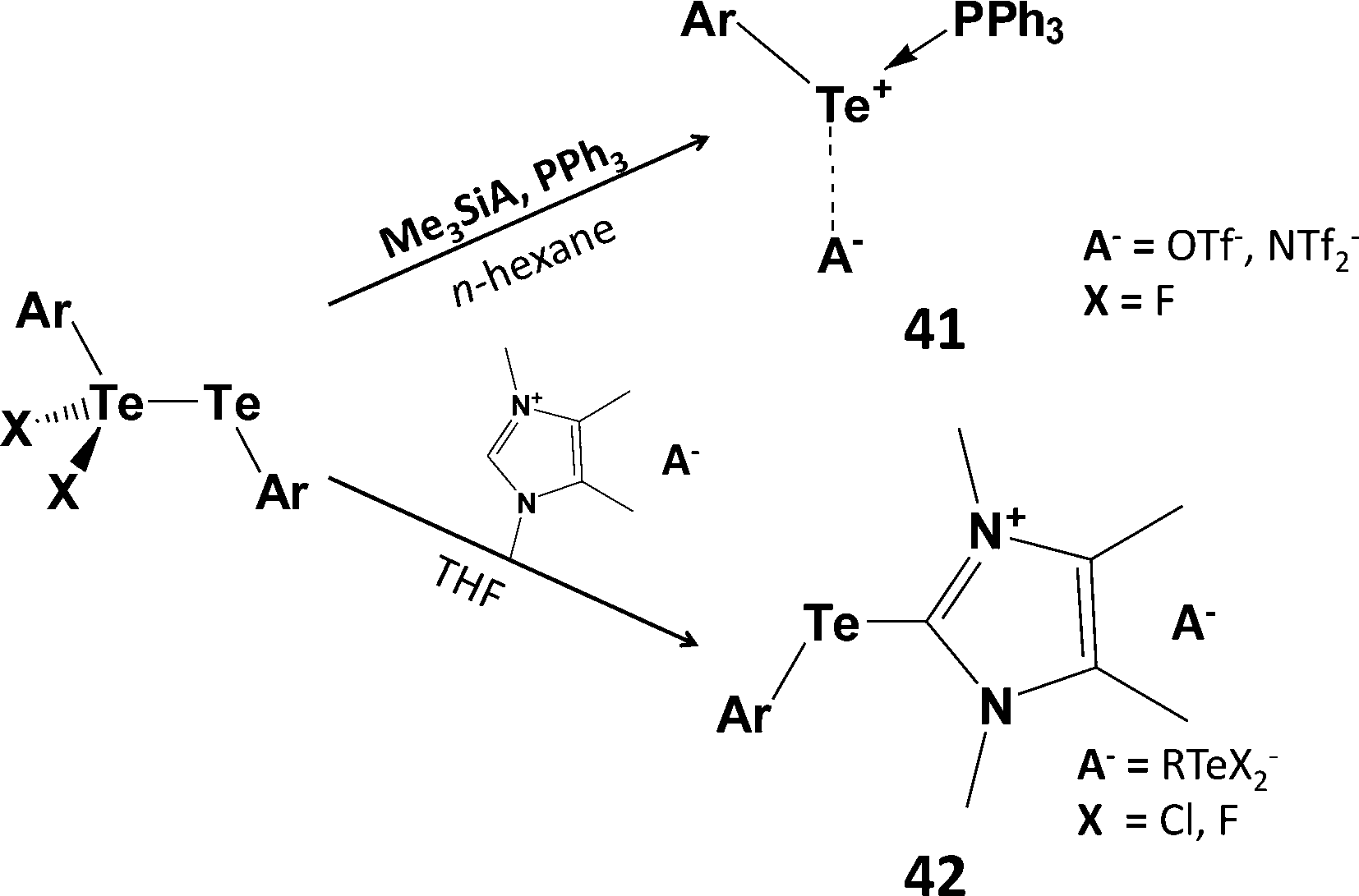 | ||
| Scheme 18 Synthesis of phosphine and carbene adducts of ArTe+; 41 (Ar = Bbt = 2,6-[bis(trimethylsilyl)methyl]-4-[tris(trimethylsilyl)methyl]phenyl; 42, Ar = 2,6-Mes2C6H3). | ||
7 Summary and conclusions
An understanding of the fundamental chemistry of tellurium is essential for the development of novel applications of tellurium reagents in both inorganic and organic chemistry. In addition, such knowledge may lead to the discovery of new functional materials with unusual properties. With reference to selected examples of the singular features of tellurium compounds from the contemporary literature, this tutorial review has attempted to provide a background to that understanding through consideration of the concepts of hypervalency, three-centre bonding, inter- and intra-molecular secondary bonding interactions, σ- and π-bond energies, and the Lewis acid behaviour of tellurium halides and cations. From a fundamental perspective the difficulties of working with thermally labile tellurium compounds are often compensated by the discovery of unexpected chemistry. In the future, exciting technological applications of the unique properties of tellurium compounds in a number of areas can be predicted confidently, inter alia as metal telluride nanowires for use as thermoelectric or photovoltaic materials and infrared detectors.48 The discovery of suitable solvents for the synthesis of these materials, e.g. thiol–amine mixtures10b or ionic liquids49 will facilitate such studies. Interesting developments in the production of low-bandgap polymers through the incorporation of tellurophene rings can also be anticipated.50Acknowledgements
Continuing financial support from NSERC (Canada) and Academy of Finland is gratefully acknowledged. The authors are thankful to Prof. I. Vargas-Baca (McMaster University) and Prof. J. S. Ritch (University of Winnipeg) for insightful comments on the penultimate draft of this article.References
- J. Ibers, Nat. Chem., 2009, 1, 508 CrossRef CAS PubMed.
- See ESI†.
- M. Miyasato, M. Minoura and K. Akiba, Angew. Chem., Int. Ed., 2001, 40, 2674–2676 CrossRef CAS.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson Education Ltd., 4th edn, 2012 Search PubMed.
- J. Beckmann, M. Hesse, H. Ploeschner and K. Seppelt, Angew. Chem., Int. Ed., 2007, 46, 8277–8280 CrossRef CAS PubMed.
- D. H. Webber and R. L. Brutchey, Chem. Commun., 2009, 5701–5703 RSC.
- A. Nordheider, T. Chivers, R. Thirumoorthi, K. S. Athukorala Arachchige, A. M. Z. Slawin, J. D. Woollins and I. Vargas-Baca, Dalton Trans., 2013, 42, 3291–3294 RSC , and references cited therein.
- J. Schaefer, A. Steffani, D. A. Plattner and I. Krossing, Angew. Chem., Int. Ed., 2012, 51, 6009–6012 CrossRef CAS PubMed , and references cited therein.
- (a) W. S. Sheldrick and M. Wachhold, Angew. Chem., Int. Ed. Engl., 1995, 34, 450–451 CrossRef CAS; (b) H.-J. Deiseroth, M. Wagener and E. Neumann, Eur. J. Inorg. Chem., 2004, 4755–4758 CrossRef CAS; (c) A. Günther, A. Isaeva, A. I. Baranov and M. Ruck, Chem. – Eur. J., 2011, 6382–6388 CrossRef PubMed.
- (a) J. Lu, Y. Xie, F. Xu and L. Zhu, J. Mater. Chem., 2002, 12, 2755–2761 RSC; (b) D. H. Webber, J. J. Buckley, P. D. Antunez and R. L. Brutchey, Chem. Sci., 2014, 5, 2498–2502 RSC.
- F. Sladky, B. Bildstein, C. Rieker, A. Gieren, H. Betz and T. Hübner, J. Chem. Soc., Chem. Commun., 1985, 1800–1801 RSC.
- J. Jeske, W.-W. du Mont and P. G. Jones, Angew. Chem., Int. Ed. Engl., 1997, 36, 2219–2221 CrossRef CAS.
- A. C. Hillier, S.-Y. Liu, A. Sella and M. R. J. Elsegood, Angew. Chem., Int. Ed., 1999, 38, 2745–2747 CrossRef CAS.
- (a) F. R. Knight, K. S. A. Arachchige, R. A. M. Randall, M. Bühl, A. M. Z. Slawin and J. D. Woollins, Dalton Trans., 2012, 41, 3154–3165 RSC, and references cited therein (b) M. Bühl, F. R. Knight, A. Křístková, I. M. Ondík, O. L. Malkina, R. A. M. Randall, A. M. Z. Slawin and J. D. Woollins, Angew. Chem., Int. Ed., 2013, 52, 2495–2498 CrossRef PubMed; (c) F. R. Knight, L. M. Diamond, K. S. Athukorala Arachchige, P. Sanz Camacho, R. A. M. Randall, S. E. Ashbrook, M. Büchl, A. M. Z. Slawin and J. D. Woollins, Chem. – Eur. J. DOI:10.1002/chem.201405599.
- H. Zhao and F. P. Gabbaï, Nat. Chem., 2010, 2, 984–990 CrossRef CAS PubMed.
- T.-P. Lin and F. P. Gabbaï, Angew. Chem., Int. Ed., 2013, 52, 3864–3868 CrossRef CAS PubMed.
- A. Günther, M. Heise, F. R. Wagner and M. Ruck, Angew. Chem., Int. Ed., 2011, 50, 9987–9990 CrossRef PubMed.
- R. C. Burns, R. J. Gillespie, W.-C. Luk and D. R. Slim, Inorg. Chem., 1979, 18, 3086–3094 CrossRef CAS.
- (a) F. Klaiber, W. Petter and F. Hulliger, J. Solid State Chem., 1983, 46, 112–120 CrossRef CAS; (b) B. Schreiner, K. Dehnicke, K. Maczek and D. Fenske, Z. Anorg. Allg. Chem., 1993, 619, 1414–1418 CrossRef CAS.
- (a) A. F. Cozzolino, I. Vargas-Baca, S. Mansour and A. H. Mahmooudkhani, J. Am. Chem. Soc., 2005, 127, 3184–3190 CrossRef CAS PubMed; (b) A. F. Cozzolino, P. J. W. Elder, L. M. Lee and I. Vargas-Baca, Can. J. Chem., 2013, 91, 338–347 CrossRef CAS , and references cited therein.
- (a) J. Kübel, P. J. W. Elder, H. A. Jenkins and I. Vargas-Baca, Dalton Trans., 2010, 39, 11126–11128 RSC; (b) C. Bleiholder, D. B. Werz, H. Köppel and R. Gleiter, J. Am. Chem. Soc., 2006, 128, 2666–2674 CrossRef CAS PubMed.
- (a) P. Rakesh, H. B. Singh and R. J. Butcher, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o214 CAS , and references cited therein; (b) T. M. Klapötke, B. Krumm and M. Scherr, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 1347–1354 CrossRef; (c) G. Mugesh, A. Panda, S. Kumar, S. D. Apte, H. B. Singh and R. J. Butcher, Organometallics, 2002, 21, 884–892 CrossRef CAS.
- (a) B. Müller, H. Poleschner and K. Seppelt, Dalton Trans., 2008, 4424–4427 RSC; (b) O. Mallow, M. A. Khanfar, M. Malischewski, P. Finke, M. Hesse, E. Lork, T. Augenstein, F. Breher, J. R. Harmer, N. V. Vasilieve, A. Zibarev, A. S. Bogomyakove, K. Sepplet and J. Beckmann, Chem. Sci., 2015, 6, 497–504 RSC.
- M. Minoura, T. Kawashima, N. Tokitoh and R. Okazaki, Chem. Commun., 1996, 123–124 RSC , and references cited therein.
- (a) G. M. Li, R. A. Zingaro, M. Segi, J. H. Reibenspies and T. Nakajima, Organometallics, 1997, 16, 756–762 CrossRef CAS; (b) Y. Mutoh, T. Murai and S. Yamago, J. Organomet. Chem., 2007, 692, 129–135 CrossRef CAS PubMed; (c) N. Kuhn and G. Henkel, Chem. Ber., 1993, 126, 2047–2049 CrossRef CAS.
- U. J. Kilgore, J. A. Karty, M. Pink, X. Gao and D. J. Mindiola, Angew. Chem., Int. Ed., 2009, 48, 2394–2397 CrossRef CAS PubMed.
- T. Maaninen, R. Laitinen and T. Chivers, Chem. Commun., 2002, 1812–1813 RSC , and references cited therein.
- T. Chivers, X. Gao and M. Parvez, J. Am. Chem. Soc., 1995, 117, 2359–2360 CrossRef CAS.
- R. Laitinen, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 777–782 CrossRef CAS.
- G. Schatte, T. Chivers, H. M. Tuononen, R. Suontamo, R. Laitinen and J. Valkonen, Inorg. Chem., 2005, 44, 443–451 CrossRef CAS PubMed.
- (a) T. Chivers, J. Chem. Soc., Dalton Trans., 1996, 1185–1194 RSC; (b) W. Mosa, C. Lau, M. Möhlen, B. Neumüller and K. Dehnicke, Angew. Chem., Int. Ed., 1998, 37, 2840–2842 CrossRef.
- J. Beckmann, J. Bolsinger, P. Finke and M. Hesse, Angew. Chem., Int. Ed., 2010, 49, 8030–8032 CrossRef CAS PubMed.
- C. Daniliuc, C. Druckenbrodt, C. G. Hrib, F. Ruthe, A. Blaschette, P. G. Jones and W. W. du Mont, Chem. Commun., 2007, 2060–2062 RSC.
- (a) P. J. W. Elder, T. Chivers and R. Thirumoorthi, Eur. J. Inorg. Chem., 2013, 2867–2876 CrossRef CAS , and references cited therein; (b) A. D. Bain, in Modern NMR Techniques for Synthetic Chemistry, ed. J. Fisher, CRC Press, 2014, ch. 2, pp. 15–61 Search PubMed.
- L. Ascherl, A. Nordheider, K. S. Athukorala Arachchige, D. B. Cordes, K. Karaghiosoff, M. Bühl, A. M. Z. Slawin and J. D. Woollins, Chem. Commun., 2014, 50, 6214–6216 RSC.
- A. Nordheider, T. Chivers, O. Schön, K. Karaghiosoff, K. S. Athukorala Arachchige, A. M. Z. Slawin and J. D. Woollins, Chem. – Eur. J., 2014, 20, 704–712 CrossRef CAS PubMed , and references cited therein.
- (a) W.-W. du Mont, Angew. Chem., Int. Ed. Engl., 1980, 19, 554–555 CrossRef; (b) P. E. Sues, A. J. Lough and R. H. Morris, Chem. Commun., 2014, 50, 4707–4710 RSC.
- N. Sandblom, T. Ziegler and T. Chivers, Can. J. Chem., 1996, 74, 2363–2371 CrossRef CAS , and references cited therein.
- N. Kuhn, H. Schumann and R. Boese, J. Chem. Soc., Chem. Commun., 1987, 1257–1258 RSC.
- W. Levason, G. Reid, M. Victor and W. Zhang, Polyhedron, 2009, 28, 4010–4016 CrossRef CAS PubMed.
- A. Maaninen, T. Chivers, M. Parvez, J. Pietkäinen and R. S. Laitinen, Inorg. Chem., 1999, 38, 4093–4097 CrossRef CAS.
- G. Reeske and A. H. Cowley, Chem. Commun., 2006, 4856–4858 RSC.
- G. He, W. T. Delgado, D. J. Schatz, C. Merten, A. Mohammadpour, L. Mayr, M. J. Ferguson, R. McDonald, A. Brown, K. Shankar and E. Rivard, Angew. Chem., Int. Ed., 2014, 53, 4587–4591 CrossRef CAS PubMed.
- (a) J. W. Dube, M. M. Hänninen, J. L. Dutton, H. M. Tuononen and P. J. Ragogna, Inorg. Chem., 2012, 51, 8897–8903 CrossRef CAS PubMed; (b) A. Kozma, J. Petuškova, C. W. Lehmann and M. Alcazaro, Chem. Commun., 2013, 49, 4145–4147 RSC.
- P. D. Boyle, W. I. Cross, S. M. Godfrey, C. A. McAuliffe, R. G. Pritchard, S. Sarwar and J. M. Sheffield, Angew. Chem., Int. Ed., 2000, 39, 1796–1798 CrossRef CAS.
- K. Sugamata, T. Sasamori and N. Tokitoh, Eur. J. Inorg. Chem., 2012, 775–778 CrossRef CAS , and references cited therein.
- J. Beckmann, P. Finke, S. Heitz and M. Hesse, Eur. J. Inorg. Chem., 2008, 1921–1925 CrossRef CAS.
- H. Yang, S. W. Finefrock, J. D. A. Caballero and Y. Wu, J. Am. Chem. Soc., 2014, 136, 10242–10245 CrossRef CAS PubMed.
- K. Ding, H. Lu, Y. Zhang, M. L. Snedaker, D. Liu, J. A. Maciá-Agulló and G. D. Stucky, J. Am. Chem. Soc., 2014, 136, 15465–15468 CrossRef CAS PubMed.
- Y. S. Park, Q. Wu, C.-Y. Nam and R. H. Grubbs, Angew. Chem., Int. Ed., 2014, 53, 10691–10695 CrossRef CAS PubMed , and references cited therein.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cs00434e |
| This journal is © The Royal Society of Chemistry 2015 |