
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An overview of nanoparticles commonly used in fluorescent bioimaging†
Otto S.
Wolfbeis
Institute of Analytical Chemistry, Chemo- and Biosensors, University of Regensburg, 93040 Regensburg, Germany. E-mail: otto.wolfbeis@ur.de
First published on 26th January 2015
Abstract
This article gives an overview of the various kinds of nanoparticles (NPs) that are widely used for purposes of fluorescent imaging, mainly of cells and tissues. Following an introduction and a discussion of merits of fluorescent NPs compared to molecular fluorophores, labels and probes, the article assesses the kinds and specific features of nanomaterials often used in bioimaging. These include fluorescently doped silicas and sol–gels, hydrophilic polymers (hydrogels), hydrophobic organic polymers, semiconducting polymer dots, quantum dots, carbon dots, other carbonaceous nanomaterials, upconversion NPs, noble metal NPs (mainly gold and silver), various other nanomaterials, and dendrimers. Another section covers coatings and methods for surface modification of NPs. Specific examples on the use of nanoparticles in (a) plain fluorescence imaging of cells, (b) targeted imaging, (c) imaging of chemical species, and (d) imaging of temperature are given next. A final section covers aspects of multimodal imaging (such as fluorescence/nmr), imaging combined with drug and gene delivery, or imaging combined with therapy or diagnosis. The electronic supplementary information (ESI) gives specific examples for materials and methods used in imaging, sensing, multimodal imaging and theranostics such as imaging combined with drug delivery or photodynamic therapy. The article contains 273 references in the main part, and 157 references in the ESI.
 Otto S. Wolfbeis | Otto S. Wolfbeis was a Full Professor of Analytical and Interface Chemistry at the University of Regensburg from 1995 to 2012. He has authored numerous papers on optical (fiber) chemical sensors (mainly for oxygen), fluorescent probes, labels (mainly for proteins), and chemical and enzymatic assays, on nanomaterials (such as upconversion nanoparticles) for use in sensing schemes, and in methods of fluorescence (including fluorescence lifetime imaging). He has acted as the (co)organizer of several conferences related to fluorescence spectroscopy (MAF) and to chemical sensors and biosensors (Europtrode). Several of his optical sensors have been commercialized. His current h-index is 81. He served in the board of Angewandte Chemie (Wiley), is the editor in chief of Microchimica Acta (Springer) and one of the three editors of Meth. Appl. Fluorescence (IOPP). Also see: http://www.wolfbeis.de. |
1. Introduction
The term imaging can be understood in many ways. Imaging is a kind of photography in most people's perception. Scientific imaging goes far beyond this. Images can additionally be created by diverse methods such as (near) infrared and Raman spectroscopy, nuclear magnetic resonance (often referred to as magnetic resonance imaging; MRI), radioimaging using respective nuclides, CT imaging, positron emission tomography, electrochemical imaging using rastering electrodes, by mechanical methods such as AFM, and by even more sophisticated scanning methods such as laser ablation ICP-MS MALDI-MS and the like. It has become accepted that virtually any method yielding a 2-dimensional picture (that, ideally, is presented in pseudo-colors) can be referred to as “imaging”. Many of these methods are destructive or require extensive sample preparation, but others are not and therefore well applicable to living systems or intact tissues. The use of nanoparticles as contrast agents for in vivo bioimaging using MRI probably is the largest single field of applications but this topic is not covered in this review. The purpose of this review is to give the reader an overview of the wealth of nanomaterials that do exist for use in fluorescent imaging, and to assist in making decision as to which material may be selected to solve a specific problem.Fluorescence (and phosphorescence‡) based imaging has found particular interest because these spectroscopies are sensitive, selective, rich in contrast, and versatile. The past 20 years also have experienced an enormous increase in resolution that has arrived at the single nanometer scale. One may first differentiate between two kinds of fluorescent imaging. The first involves imaging based on intrinsically fluorescent (bio)chemical species (such as NADH in tissues, crude oil in – and on – seawater, or chlorophyll in all kinds of plants and the open sea). The second covers methods for imaging of samples or cells that have been made fluorescent by adding synthetic fluorescent probes, labels, nanoparticles or nanosensors. The use of such probes is, in fact, indispensible in order to detect species that are not amenable to direct fluorometric imaging (such as of pH) but this also implies the risk of local perturbation by the probe or the material added.
The acquisition of images of biological matter by using fluorescent probes or fluorescent labels and nanomaterials is generally referred to as bioimaging and forms a large field of its own. Letting aside conventional (light) microscopy and MRI, fluorescence imaging probably is the most widespread method in biosciences. Respective pictures are attractive, easily comprehensive, and can be found in a good fraction of research papers and magazines. Reviews cover topics such as fluorescent nanostructures for bioimaging,1 quantum dots for bio-imaging2,3 and single molecule imaging (“one quantum dot at a time”),4 gold nanoclusters with tunable fluorescence as bioimaging probes,5 aggregation-induced emission-based fluorescent nanoparticles,6 nanocomposite particles for bioapplications including imaging,7 on nanoparticles in drug delivery, therapeutics, diagnostics and imaging,8 on quantum dots and polymer hybrid composites as fluorescent switches and turn-on probes for sensing anions,9 on aspects of deep tissue microscopy and optical imaging,10 on controlled synthesis, spectra and bioapplications of lanthanide-doped luminescent nanoprobes,11 on advances in (NP-based) fingerprint imaging,12 on the intersection of CMOS microsystems and upconversion NPs for bioimaging and bioassays,13 to mention only a few. A review by Bünzli14 on lanthanide luminescence for biomedical analyses and imaging contains a section 5.6. on improving sensitivity by using certain nanoparticles. Others are cited later in the respective sections and in the ESI.†
The term fluorescence does not imply a single spectroscopic method but rather includes a variety of techniques in that images can be acquired by measurement of intensity, decay time (lifetime) and polarization, but also by studying effects caused by resonance energy transfer, (dynamic) quenching, or photo-induced electron transfer. Optical imaging was limited to resolutions of a few 100 nm until about 1995, but substantial thrust in terms of resolution resulted from the availability of fluorescent methods of imaging on the nanoscale by methods such as STED, PALM, or STORM, all based on laser technology. Chemo- and bioluminescence can also generate images15 but chemiluminescence requires the addition of reagents and usually is unidirectional in that an increase in the concentration of an analyte can only be monitored. Both methods are time-dependent.
When focusing now on bioimaging based on synthetic fluorescent probes and nanoparticles, one may differentiate between three techniques. (1) In the most simple one, a strong fluorophore or fluorescent nanoparticles are internalized into cells so that they can be imaged. The only purpose of such fluorophores and nanomaterials is to render cells or tissue fluorescent. They do not possess (and are not expected to possess) affinity for a specific site, nor do they respond (like indicator probes) to the presence of chemical species such as certain ions or organic molecules. (2) The second technique is referred to as “targeted bioimaging”. It enables specific domains or species to be detected, very much like immunostaining or fluorescence in situ hybridization. In order to accomplish this, fluorophores or nanoparticles are applied whose surface has been properly functionalized, for example with receptors, ligands, antibodies or oligomers so as to recognize the specific counterpart. Examples include targeting of tumor markers, genes, mitochondria, membranes, or the amyloidic plaques in Alzheimer-associated tissue. (3) The third technique is making use of probes and nanomaterials with sensing capability. This enables (bio)chemical species to be imaged that are to not intrinsically fluorescent. Examples include imaging of the distribution of chemical species such as pH values, glucose, calcium(II) or oxygen in the living and metabolizing cell, if not in tumor cells or in cells exposed to candidate drugs. This group also involves nanosensors for temperature. Representative examples for each of these techniques will be presented in Section 5.
2. Fluorescent nanomaterials and nanoparticles versus molecular fluorophores, labels and probes
The availability of nanomaterials for purposes of imaging has generated a variety of methods for imaging, with features including improved brightness (defined as absorbance times quantum yield), inertness to their microenvironment and a more even distribution (unless targeted imaging of certain domains is desired, of course). Nanoparticles (NPs), in contrast to molecular probes, often are not cytotoxic and do not suffer from nonspecific binding by cellular biomacromolecules or unwanted sequestration. Binding of molecular probes by cellular proteins (or sites) can affect both the optical properties of the probe and even the function of the protein or the binding site. Dyed NPs, or intrinsically fluorescent NPs, in contrast, are virtually inert and do hardly interact with cellular proteins nor are their optical properties affected by the proteins outside. Not surprisingly, all known NPs have photostabilities that are distinctly better than those of molecular probes. Many NPs can be easily internalized into cells and tissues (depending on charge and surface chemistry; positively charged facilitates internalization) and can be even targeted to specific sites. Compared to fluorescent proteins one notes the more simple handling of NPs and more predictable results. Many kinds of NPs are commercially available. The simplicity of loading or labeling with fluorophores or NPs is a particular issue if hundreds of cell lines are being handled simultaneously, for example in high-throughput screening. Second-harmonic generation (which results in low background noise) is more easily accomplished with NPs as demonstrated in the review by Dempsey et al.16 on respective nanocrystals for in vivo imaging, in particular on nanodiamonds, nonlinear crystals, quantum dots and SERS NPs. Finally, it is fair to say that nanosensors have calibration plots that are quite similar if not identical if acquired in vitro and in vivo. Molecular probes, in contrast, are quite different in that respect. It is a matter of fact that one must never use a calibration plot that was established in plain buffer solution to quantify a parameter with data acquired in a cellular system.3. Kinds of nanomaterials often used in bioimaging, and their specific features
A complete coverage of all the nanomaterials used so far in bioimaging would by far exceed the frame of this review but rather fill a book. The following is a discussion of the nanomaterials most often used for purposes of bioimaging. These include NPs made from silica and organically modified silica, hydrophobic and hydrophilic organic polymers, semiconducting organic polymers, quantum dots, carbonaceous nanomaterials including carbon (quantum) dots, carbon nanoclusters and nanotubes, nanodiamonds, upconversion materials, metal particles, metal oxides and others. The discussion on the potential cytotoxity of NPs is going on, and numerous studies have been performed to investigate the potentially harmful or the perturbing effect of NPs on physiological systems.173.1. Fluorescently doped silicas and sol–gels
Silica nanoparticles (SiNPs) were among the first ones to be used in bioimaging. Excessive literature on their uses does exist.18 The group of Wiesner has reviewed19 the state of the art in SiNPs for use in sensing/imaging and has described numerous kinds of SiNPs, among them the one-pot synthesis of PEGylated mesoporous and fluorescent SiNPs possessing a single pore, tunable sizes of around 9 nm, and narrow size distributions.20 In fact, mesoporous silica (in addition to materials related to “controlled porous glass”) are more often used now because they can be heavily loaded with fluorescent dyes, photosensitizers or diagnostic reagents as will also be shown in Section 6. Fluorescent mesoporous silica can be obtained via hydrothermal reaction and functionalized, if desired, with reactive siloxanes.21 Such particles possess low cytotoxicity and excellent cell imaging capability.SiNPs can be easily doped with various kinds of organic, metal–organic and metallic fluorophores, and emission wavelengths range from 300 to 1000 nm, with a trend towards NPs possessing longwave (>600 nm) emissions22 because the fluorescence of NPs at wavelengths of >500 nm is often interfered by the autofluorescence of cells. Color, decay times and size are widely tunable, and dopants can be hydrophobic, hydrophilic, or ionic. SiNPs also may be coated with fluorophores but attention has to be paid to possible aggregation effects that usually are accompanied by self-quenching. Two-photon excitation has been demonstrated but this depends on the kind of fluorescent label. SiNPs and their aggregates of >300 nm in size cause strong light scattering. SiNPs are fairly well biocompatible (i.e., not harmful to cells and tissues). Cell permeability depends on zeta potential. If negatively charged, they hardly pass cell membranes. They are virtually nontoxic, easily excreted (unless particle sizes exceed 50 nm), but also quickly coated by intracellular proteins and attacked by the immunosystem. Their surface can be easily modified with a variety of coatings and using standard silica (and sol–gel) surface chemistry. Particles do not swell but tend to aggregate in the presence of bivalent ions unless inert shells are deposited on the surface. Mesoporous silica structures warrant strongly increased surface areas and enable high loading of cargo for cellular imaging and targeting. The state of the art in designing and characterizing fluorophore-doped SiNPs for bioapplications has been reviewed.23
SiNPs and other NPs are often doped with luminescence lanthanides. Their use in bioimaging has been reviewed by Chen et al.,24 and the fabrication of down-converting and up-converting luminescent probes for optical imaging by Zhang et al.25 Lanthanide-based fluorophores have attractive features such as (a) relatively long decay times which facilitate time-resolved fluorometry and, thus, efficient background suppression; (b) fairly narrow emissions; (c) single-photon and two-photon excitation; and (d) upconversion luminescence and downconversion luminescence. Lanthanide doped SiNPs are obtained by incorporating lanthanide ions, or by grafting the SiNPs with complexes such as Eu(III), Tb(III) or Gd(III).26 Such fluorescent NPs have decay times between 0.35 and 1.87 ms (which facilitates gated spectroscopy), do not photobleach, and display the typical narrow emission bands of lanthanide ions. Other examples for lanthanide doped NPs (also with materials other than silica) will be given in the following sections and in the tables. In addition to silica, NPs made from titania and zirconia and doped with organic and inorganic fluorophores also are known but less often used. They are treated in Section 3.11 (on Other Nanomaterials).
Sol–gels and organically modified sol–gels (ormosils) may also be used for forming NPs. A large variety of materials are known. Their porosity can be governed by the proper choice of materials and by varying experimental conditions such as acid or base catalysis. Sol–gels are prepared by polycondensation of tetraalkoxysilanes, and ormosils by copolymerization of mixtures of tetraalkoxysilanes with alkyl-alkoxysilanes in varying ratios, or from alkyl-alkoxysilanes only. A large variety of materials is known depending on whether mono-, di- or trialkylsilanes of general formula (R1)x(R2O)3−xSi–R1 (where R1 is alkyl or aryl, and R2 is alkyl) are being used. The NPs usually are made fluorescent by non-covalent doping with various kinds of fluorophores. Unless pore sizes are small, covalent immobilization is advised, however. Colors, decay times and particle size are widely tunable. As with SiNPs and others, fluorescence at >500 nm is often interfered by autofluorescence of biomatter. Beads and bead aggregates of >300 nm in size cause strong light scattering. Sol–gel NPs are well biocompatible which can be improved by PEGylation (see Section 4). Cell permeability depends on zeta potential in that NPs are hardly cell permeable if negatively charged. Sol–gel NPs are nontoxic and – if small enough – are easily excreted. Their surface can be modified ex vivo with a variety of coatings, and the surface chemistry of silica and sol–gels is well established. Particles do not swell but tend to aggregate in the presence of bivalent ions if their zeta potential is negative. Photobleaching of the dopant dye can be an issue even though this is often weaker that in the case of dyes that are molecularly distributed in cells.
3.2. Hydrophilic polymers
Hydrophilic materials for use in NP-based imaging include the various kinds of hydrogels, but also natural products such as cellulose. Nanogels are soft and usually water soluble. If NPs are to be formed, they have to be crosslinked or mixed with another material in order to form hybrid materials. Hydrogels are well permeable to ions and hydrophilic organic species such as glucose or amino acids, but not to large biomolecules usually. Typical polymers include polyacrylamide (PAA), polyurethanes, poly(hydroxyethyl methacrylamide) (pHEMA), certain poly(ethylene glycols) or specialty polymers such as Pluronic™ [a commercial poly(ethylene glycol)-co-poly-ethyleneoxide)] widely used in NP-assisted drug delivery. The design of fluorescent organic nanostructures for bioimaging has been reviewed.27 Depending on the kind of polymer and the degree of crosslinking, such NPs undergo substantial (and ionic strength-dependent) swelling in water, but they do not tend to aggregate. The emission wavelengths of respective NPs can be adjusted to almost any wavelength between 300 and 1000 nm via the dopant, usually an organic or metal–organic fluorophore whose color, fluorescence, decay time and size are widely tunable.NPs made from organic monomers can be crosslinked to a various extent, and this determines many of their properties including swellability and solubility. The fluorescence of particles at <500 nm often interfered by autofluorescence of most cells. NPs prepared from hydrogels are biocompatible, cell permeable (depending, as always, on the zeta potential), nontoxic, slowly excreted, fairly quickly coated by intracellular proteins and attacked by the immunosystem. Many of them can be degraded by intracellular enzymes. Their surface cannot be easily chemically modified with additional coatings. Functionalities such as amino groups are better introduced by adding a functional monomer to the main monomer and then to initiate radical polymerization. Techniques are known to prepare organic polymer core–shell NPs. Fluorescent (and other) dopants tend to leach into the aqueous environment of the particle unless firmly retained (electrostatically or covalently). Amino-modified cellulose was applied in a luminescent sensor for high-resolution imaging of pH in vivo.28 pH values were imaged by detecting the green fluorescence of the pH probe fluorescein covalently linked to aminocellulose. A ruthenium phenanthroline complex was incorporated into poly-acrylonitrile beads to give a pH-independent red reference signal. The beads were immobilized in a polyurethane hydrogel on a thin transparent support. Both in vitro and in vivo experiments revealed the versatility of the method during physiological and chronic cutaneous wound healing. The method was later extended to simultaneously image extracellular wound pH and oxygenation in vivo.29 The same pH beads were used, and poly(styrene-co-acrylonitrile) particles dyed with Pd(II)-meso-tetraphenyl-tetrabenzo-porphyrin were added to give a near-infrared signal that depends on local oxygen partial pressure.
A typical recent example of the use of a PAA hydrogel is provided by nanoparticles containing free amino groups that were prepared by copolymerization of acrylamide and methylene-bisacrylamide with 3-aminopropyl-acrylamide and labeling the terminal amino groups with pH probes such as fluorescein, Oregon Green, Alexa 633, and others. The resulting sensor NPs cover a wide range of pH (4.0–8.0) which is needed in certain situations even in cellular imaging.30 In addition to the widely used crosslinked polyacrylamides (PAAs), other acrylamides including polymethacrylamide or poly(N-alkylacrylamides) were employed. For example, core–shell microgels containing indicators were fabricated31 by two-stage free radical precipitation polymerization of N-isopropylacrylamide. The shell of the microgel exhibits a low critical solution temperature and undergoes a transition from a swollen state to a de-swollen state, associated with a hydrodynamic radius of ∼450 nm at 25 °C (in vitro) and of ∼190 nm at 37 °C (in vivo). The microgel readily enters the cytosol which makes it a potential candidate for the delivery of indicator probes into the cytosol.
The Pluronic™ hydrogel (see above) was used to fabricate nanosensors for fluorescent imaging of physiological pH values.32 Features include (a) very small diameters (12 nm); (b) biocompatibility due to the use of a hydrogel kind of material, and (c) the lack of toxicity. The nanosensors were incorporated into an agar film to enable continuous monitoring of the pH value of bacterial cultures, and thus of their growth. Dually responding nanosensor particles were reported that were prepared from an organic–inorganic composite (Pluronic™ reinforced with silica) and used for simultaneous imaging of oxygen and pH in the cellular cytosol.33Fig. 1 shows a schematic of the preparation of the dual nanosensor for oxygen and pH, the architecture of the NPs, and the chemical structures of the probes used. Other multiple (bio)sensors, i.e. sensors capable of two or more analytes simultaneously have been reviewed,34 but only a moderate fraction of them makes use of nanomaterials.
 | ||
| Fig. 1 Structure of a nanosensor for dual sensing of oxygen and pH. Its core consists of Pluronic F-127, a nonionic, surfactant triblock copolymer composed of a central hydrophobic chain of poly(propylene oxide) flanked by two hydrophilic chains of poly(ethylene glycol) (PEG) and reinforced with silica. The NPs are capped with PEG. The oxygen probe (PtTBTBP) and the reference fluorophore TFPP are located in the core, and the pH probe (FITC) is conjugated to the terminal ends of the PEG capping. Reproduced from ref. 33 with permission (2014) of the Am. Chem. Soc. | ||
Nanogels (like NPs) are of interest in being extremely soft materials that take up water in fraction between 10 and 90%. The gels are well permeable to hydrophilic species and can be made fluorescent by simple labeling with inert labels and made responsive by attaching a fluorescent probe. Nanogel particles are well suited to image pH values inside cells.35 In a method termed CLARITY, nanoporous hydrogel-hybridized forms of intact mouse brain were prepared and crosslinked to a three-dimensional network of hydrophilic polymers.36 They are optically transparent and permeable macromolecules. Tissue imaging is said to reveal local circuit wiring, cellular relationships, subcellular structures, protein complexes, nucleic acids and neurotransmitters. CLARITY also enables intact-tissue in situ hybridization, immunohistochemistry in non-sectioned tissue, and antibody labelling. Fluorophore-labeled polymeric nanogels for sensing temperature (T) have attracted much interest because they pave the way to sense T inside cells. The topic has been extensively reviewed.37
3.3. Hydrophobic organic polymers
Polystyrene nanoparticles (PS-NPs) are highly hydrophobic and can be doped with apolar fluorophores with emission peak wavelengths that range from the near UV to beyond 1000 nm. Their color, decay times and size also are widely tunable. Doping with lipophilic materials is preferred because ionic probes are poorly soluble in hydrophobic NPs from which the probes tend to leach out. As in all kinds of NPs for use in imaging, any fluorescence occurring at below 500 nm is interfered by the autofluorescence of biomatter. An (organic) dopant fluorophore can photobleach if exposed to strong laser light. PS-NPs are fairly biocompatible (i.e., not harmful to cells and tissue), fairly well cell permeable, nontoxic, and their excretion is slow. If placed inside cells, they are only slowly coated by intracellular proteins and hardly attacked by the immunosystem. The modification of their surface is limited to certain functional groups. Functionalities (such as amino groups) are best introduced by the addition of co-reagents containing such groups to the monomer before starting emulsion polymerization. Post-modification and additional coating are rather difficult. PS-NPs do not measurably swell in water and do not readily aggregate, but this depends on their charge and zeta potential. One of the first nanomaterials for sensing purposes consisted of (pH-insensitive) fluorescent PS beads coated with polyaniline whose absorbance is pH dependent over a large range of pH values. Depending on the actual pH value, the coating screens off the emission of the beads.38 PS-NPs are well permeable to gases but impermeable to charged species including proteins. Particles with an average diameter of 85 nm were loaded with an oxygen-quenchable luminescent ruthenium complex and then used to image oxygen inside cells following 2-photon excitation.39Polyacrylonitrile (PAN) NPs can be doped with fluorophores with emission wavelengths ranging from 300 to 1000 nm, preferably with hydrophilic dyes. Color, decay times and size widely tunable. Beads and aggregates of >300 nm in size cause light scattering. PAN is fairly biocompatible (not harmful to cells and tissue) and fairly well cell permeable, nontoxic, easily excreted but slowly coated by intracellular proteins. It is hardly attacked by the immunosystem. Its surface is rather inert and cannot be readily modified once the particles have been formed, usually by precipitation by adding water to a solution of PAN in dimethylformamide. PAN particles do not swell but there is a tendency to aggregation. Fluorophore-doped PAN-NPs were applied, for example, to referenced imaging of pH and temperature with sub-μm spatial resolution.40
Biocompatible fluorescent organic NPs with tunable photoluminescence were obtained via one-pot oxidation of polydopamine and subsequently utilized for cell imaging,41 and water dispersible red fluorescent organic NPs for use in cell imaging were reported by Luo.42 Quantum-dots conjugated to dopamine function as redox coupled assemblies and can be applied to in vitro and intracellular pH sensing.43 Other polymers include poly(vinyl butyral)44 that was labeled with a perylene dyes that is easily taken up without coating and does not display in vitro cytotoxicity on human cancer cells. Hu et al.45 have introduced a class of organic nanocomposites with functionalities for both fluorescence imaging and magnetic therapy. Magnetic NPs (5 nm in diameter) were incorporated into the amphiphilic block copolymer poly(styrene-b-allyl alcohol) that was labeled with pyrene. The fluorescence of the resulting NPs (200 nm i.d.) was exploited when imaging cancer cells, while magnetically controlled mechanical damage of cell membranes represents a way for cancer cell treatment referred to as magnetolytic therapy. Magnetic field induced heating may pave, in future, the way to hyperthermal cancer therapy. This is schematically shown in Fig. 2. For numerous other examples, see Section 6.
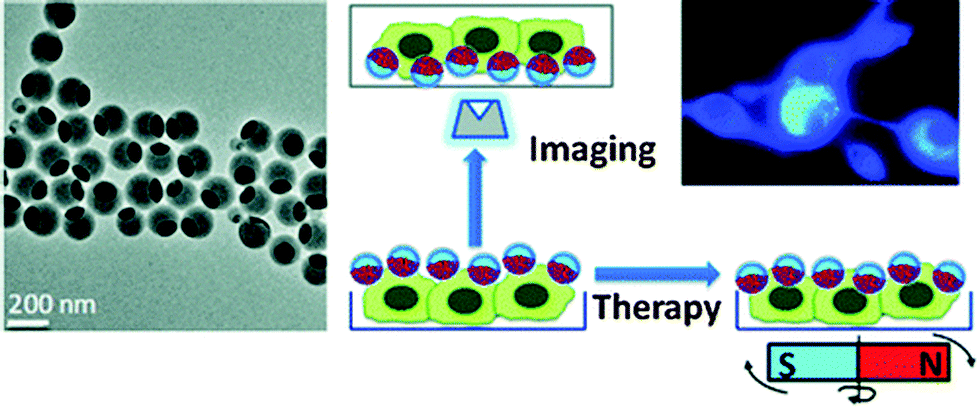 | ||
| Fig. 2 Left: TEM of fluorescent organic nanobeads containing magnetic NPs. Right: bimodal use of the nanobeads for purposes of imaging cancer cells (top) and magnetically induced lysis of cell membranes. From ref. 45 with permission (2014) of the Am. Chem. Soc. | ||
3.4. Semiconducting (organic) polymer dots (P-dots)
These come in addition to more conventional (dyed) NPs such as those made from polystyrene, polyacrylamide etc. The polymer usually is prepared from aromatic precursors possessing polymerizable double or triple bonds. Particles (as needed for imaging) are prepared by either emulsion polymerization or nanoprecipitation.46 Doping with fluorophores is not needed. The backbone of conjugated polymers behaves like an array of light-harvesting units that exhibit a larger optical cross section compared to small organic molecule dyes. Photobleaching was not reported so far. Fine-tuning of the conjugated polymer structure and the polymeric encapsulation matrix leads to fluorescent probes with specific spectral properties and targeting capability. P-dots display strong fluorescence that often extends far into the NIR, are highly inert and do not swell in water. Little is known about biocompatibility, internalization, and excretion from tissues. Their uses in imaging and therapy have been reviewed.47Fluorescent nanodots consisting of semiconducting polymer blends can be attached to peptides (such as chlorotoxin)48 and then can be used for targeted imaging (of malignant brain tumors, for example) in clinical diagnosis. By coupling the pH-indicator fluorescein to P-dots, a material is obtained that displays two fluorescence peaks, one being pH sensitive, the other not so that it can acts as an internal reference. Fully reversible pH sensing was demonstrated49 for the pH 5.0 to 8.0 range. Intracellular pH values were determined by imaging of HeLa cells following the uptake of the P-dots by endocytosis. Tetraphenylethene-based fluorescent organic NPs undergo aggregation-induced emission inside cells and this was monitored via cell imaging.50 The fluorescence of conjugated polymers, particularly if anionic, can be quenched by ions such as Cu(II).51 Semiconducting P-dots (20 to 50 nm) can also serve as photoacoustic probes for real-time imaging of reactive oxygen species in living mice tissue where they accumulate quite readily.52
3.5. Carbon dots
Carbon dots (C-dots53), first reported in 2006, are said to be clusters of carbon atoms (for definitions see ref. 54) with diameters of typically 2 to 8 nm, but also contain substantial fractions of oxygen and hydrogen if not nitrogen. They do not measurably swell in aqueous solution but aggregation was occasionally observed. C-dots can be made strongly fluorescent and need not be doped or labeled. Their emission color can be tuned to some extent by varying the experimental conditions of synthesis. Both the excitation and emission spectra are very wide and usually extend from the UV to the red (650 nm), a fact that virtually excludes their use in multiplexing. A fine review on the synthesis and photophysical properties (and uses in bioimaging) of C-dots is available.55 It includes the very true statement that “C-dots have a much more comprehensive definition compared to graphene quantum dots.” The QYs of C-dots range from 5 to 30%.Fig. 3 shows the emission spectra of carbon dots at different excitation wavelengths from 330 to 475 nm. Their strongest fluorescence is blue, but longwave excitation (at >460 nm) induces green to yellow emission. A review on the synthesis, properties and applications of C-dots contains an interesting section on the origins of their excitation wavelength dependent emission, and particularly the controversial upconverted luminescence.56 The emissions also are likely to be pH-dependent. Single-particle fluorescence intensity fluctuation (“blinking”) has been reported recently.57 Decay times are in the nanosecond time regime and do not vary much. The fluorescence of C-dots can be of the upconversion and the down-conversion type.
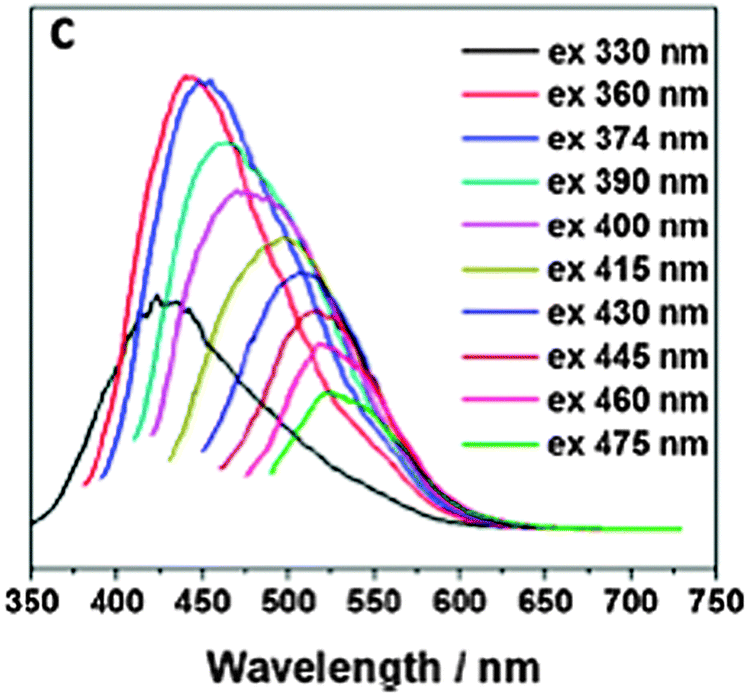 | ||
| Fig. 3 Wavelength dependence of aqueous solutions of carbon dots in water at excitation wavelengths between 330 and 475 nm. Reprinted from ref. 58 with permission (2014) of the Am. Chem. Soc. | ||
C-dots, and carbon nanoparticles in general, can be single-photon excited and multi-photon excited. C-dot-based inorganic–organic nanosystems were applied, for example, to two-photon imaging of pH variation in living cells and tissues.59 C-dots are fairly well biocompatible (i.e., not harmful to cells and tissue within a few days), fairly well cell permeable, not known to be toxic, easily excreted, weakly interacting with proteins, and hardly attacked by the immunosystem. Functional surface engineering for purposes of bioconjugation and imaging is more difficult than in the case of Q-dots but possible.60 They neither swell nor photobleach. Their fluorescence is pH dependent and quenched by iodide61 (and probably by other notorious quenchers too). A recent review covers the subject.62
In terms of synthesis, both top-down and bottom-up approaches are known. The resulting C-dots, in fact, always contain substantial fractions of oxygen (up to 50%) and also nitrogen if a nitrogen-containing substance such as an amino acid is added during synthesis. And yet, they are often termed – mainly by Chinese authors – graphene quantum dots even though graphene by definition consists of C and H only and is nonfluorescent. Examples where the application of “graphenes” is claimed but materials other than sp2-graphenes have been used include, for example, intracellular fluorescence imaging with a “graphene”-based fluorescent probe,63 and the use of highly biocompatible “graphene” nanosheets for cellular imaging.64 A particularly confusing example is represented65 by an article entitled The in vivo and in vitro toxicity of graphene quantum dots that has nothing to do with graphene (which is free of oxygen by definition and non-fluorescent). The authors have prepared the highly fluorescent (!) graphene material by oxidation (!) and also claim it to possess a particularly high oxygen content (!).
C-dots have been prepared from numerous organic materials and natural products containing carbon in various form, one example being58 the preparation of 3 nm blue fluorescent C-dots from cow milk by heating it to 180 °C for 2 h. The particles can be used to image U87 cells. If C-dots are doped with nitrogen,66 they are even more strongly fluorescent. Both hydrophilic and hydrophobic C-dots are known. Hydrophilic materials are preferred in imaging.67 Hydrophobic materials are less often used but also available by microwave synthesis.68 Raw C-dots are mainly prepared by microwave induced thermal carbonization of molecular precursors such as glucose (and other carbohydrates), citrate, (poly)glycols, often in the presence of a nitrogen source (such as tryptophan or EDTA). Surface passivated (und, therefore, bright) C-dots can be directly synthesized by microwave induced pyrolysis of glycerol in the presence of 4,7,10-trioxa-1,13-tridecanediamine.69 C-dots can also be isolated from soot, or prepared from glucose with P2O5 at room temperature.70 C-dots may be rendered to more strongly fluorescent by alkali or acid-assisted ultrasonic treatment.71
Photoluminescent C-dots have also been produced by laser ablation of graphite followed by oxidation with nitric acid and functionalization with diamine-terminated poly(ethylene glycol).72 They show multicolor fluorescence.73 C-dots can be produced inexpensively and on a large scale. Fluorescence is conferred or improved by chemical treatment (or passivation) of the surface, for example by oxidation, doping with inorganics, or capping. Water dispersible C-dots with tunable photoluminescence can also be synthesized74via hydrothermal oxidation of nanodiamonds and were subsequently utilized for cell imaging. Carbon nanoparticles (10 nm i.d.) for use in fluorescent bioimaging can be obtained now75 on the milligram to gram scale by carbohydrate carbonization (even though in our experience this method is difficult to reproduce not the least because of an inadequate experimental part). Table 1 gives examples of CNPs (prepared on the gram scale) along with colors of emission (from blue to red). Red-fluorescent CNPs are preferred because autofluorescence of biomatter is weaker in this spectral range (Fig. 4).
| FCNP | Molecular mass or particle size | Composition (C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H:N:O; in %) :H:N:O; in %) |
Emission peak and (excitation) wavelength | Molar absorbance at excitation wavelength | Quantum yield (%) |
|---|---|---|---|---|---|
| FCNPblue | 400–2200 Da | 65:6:8:21 |
440 nm (370 nm) | ∼2 × 103 | 6–30 |
| FCNPgreen | 2500–14000 Da, 2–4 nm |
75:10:5:10 |
500 nm (400 nm) | ∼5 × 104 | 14 |
| FCNPyellow | 1–4 nm | 50:15:2:33 |
560 nm (425 nm) | ∼4 × 103 | 12 |
| FCNPred | ∼4–10 nm | 70:5:1:24 |
600 nm (385 nm) | ∼7 × 105 | 7 |
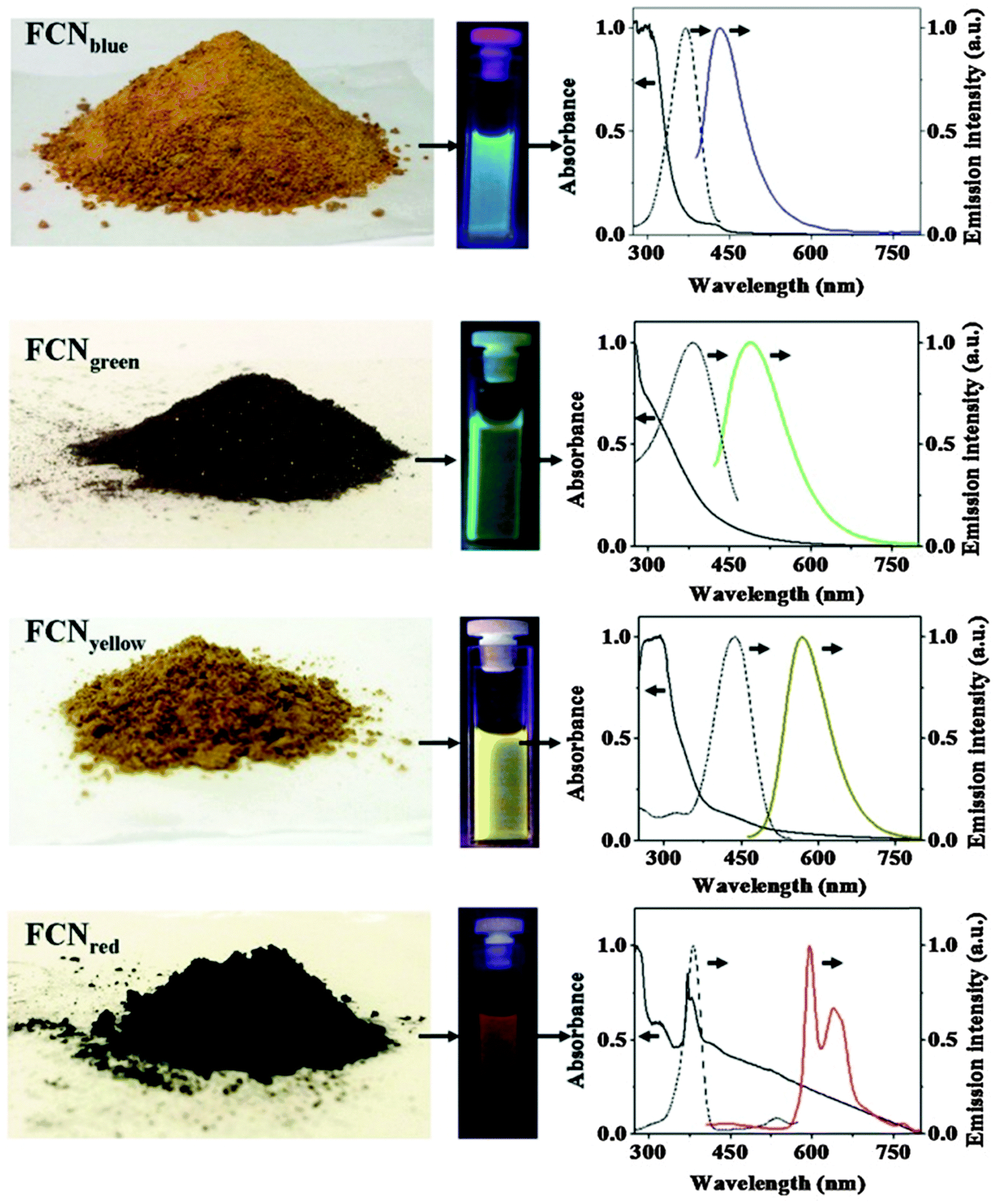 | ||
| Fig. 4 Images of gram scale solid samples of fluorescent carbon nanoparticles (FCNPs), of their solutions under appropriate excitations, and their absorption (—), excitation (⋯) and emission (color lines) spectra. Emission spectra have been measured by exciting at 370 nm for FCNPblue, by exciting at 400 nm for FCNPgreen, by exciting at 425 nm for FCNPyellow and by exciting at 385 nm for FCNPred. From ref. 75 with permission (2014) by Nature Publ. Group. | ||
Both C-dots with invariable and with continuously tunable emission are known.76 They enable ratiometric sensing of pH values via the ratio of the intensities of the excitation-independent and pH-independent blue emission and the excitation-dependent and pH dependent full-color emissions. Ratiometric (blue and green) fluorescent nanosensors have been described77 that are based on water soluble carbon nanodots with multiple sensing capacities. This, however, is an euphemism for poor selectivity in that the dots respond to temperature, pH, and to Fe(III) ions, all of which mutually interfere.
3.6. Other carbonaceous materials
Other nanosized carbon allotropes include fullerenes (C70 mainly; these being much smaller than C-dots), and the larger species including carbon nanotubes (CNTs; single walled and multiwalled), nanodiamonds, graphene (which is nonfluorescent), and the oxidized species graphene oxide, reduced graphene oxide, graphite oxide, graphene quantum dots (often synonymously used for C-dots; see Section 3.5.) and the like. Other authors refer to their materials as graphene (or graphene dots) even if the material was prepared by reduction of graphite oxide (by Hummers method) and still contains large fractions of oxygen. It may be better termed “reduced graphite oxide”. There is much confusion.The fluorescence of graphite oxide78 (like that of C-dots79) is sensitive to pH. Like C-dots, such carbon nanomaterials need not be doped with fluorophores and are extremely photostable. The colors of emission of all known variants depend on the wavelength of excitation. Excitation in the UV (350–380 nm) often results in good brightness and blue fluorescence, but excitation wavelengths can be as long as 650 nm and fluorescence then occurs in the near IR as is shown below. The decay times of fluorescence are in the order of nanoseconds. Nano-sized fluorescent graphite oxides (nano-GOs) with different size distribution were prepared via a one-pot hydrothermal route using ultra-small graphite powder as a starting material and subsequently separated using dialysis tubes with different molecular weight cutoff.80 Such nano-GOs were found to be readily internalized by A549 cells and then located in the cytoplasm. They display size-dependent photoluminescence (green, yellow, red) and excellent biocompatibility.
Graphene81 and (color-tunable) fullerenes82 have not often been used for purposes of imaging because of their lack of (or rather weak) fluorescence. Fullerene C70 (unlike C60 which is nonfluorescent) displays normal and delayed fluorescence which is strongly quenched by oxygen83 and highly dependent on temperature.84 Gong et al.85 have isolated red, green and blue fluorescent hollow carbon NPs from chromatographic fractions and demonstrated them to be excellent (multi-color) probes for cellular imaging.
Stabilized graphene oxides were applied to cellular imaging,63 some after having been made highly biocompatible86 or stabilized and biofunctionalized.87 Blue and green fluorescent carbon NPs derived with vitamin B1 are also described to be very bright and applicable to cell imaging.88 Luo et al. have reviewed applications of carbon-based quantum dots for fluorescence imaging of cells and tissues.89 Fluorescent graphene quantum dots (GQDs) have been synthesized90 that display both upconverted and excitation-independent downconverted photoluminescence. Doping of GQDs with almost any kind of heteroatom including nitrogen,91 boron,92 sulfur,93 fluorine94 and chlorine95 enhances their brightness. By coupling them to photocatalytically active rutile (TiO2/GQD) and anatase (TiO2/GQD) systems, the complete visible spectrum of sunlight can be harnessed. Strong two-photon-induced fluorescence was reported96 for photostable, biocompatible nitrogen-doped graphene quantum dots (N-GQDs; possibly C-dots?) for cellular and deep-tissue imaging. Their two-photon absorption cross-section reaches 48000 Göppert-Mayer (GM) units, which far surpasses that of many organic dyes. It is comparable to that of the high performance semiconductor Q-dots and represents the highest value ever reported for carbon-based nanomaterials. The penetration depth in phantom tissue revealed an imaging depth as deep as 1.8 mm. Zhu et al.97 have presented a study on surface chemistry based routes to modulate the photoluminescence of GQDs, how to govern the fluorescence mechanism to induce up-conversion fluorescence, and on bioimaging applications.
Single-walled and multi-walled CNTs are fluorescent in the NIR but have low quantum yield.98 The one-dimensional electronic structure of nanotubes results in sharp interband transitions in the absorption spectra of SWNTs, and in photoluminescence in the NIR region (800–1600 nm). These wavelengths include the tissue-transparent region of the electromagnetic spectrum. Their other properties are comparable to those of C-dots. Like C-dots, they do not photobleach. While traceable in living cells, their cytotoxicity is still not refuted,99,100 but carbon nanotubes encapsulated by a DNA oligonucleotide remain functional in live cells for up to three months.101 CNTs have been applied to Raman imaging, near-infrared (NIR) fluorescence imaging and photoacoustic imaging but still are less often used than some other fluorescent nanomaterials. The group of Strano102 has reviewed advances in molecular recognition based on single-walled CNTs and respective nanoengineered platforms. These were used to fluorescently sense species such as ATP, NO, H2O2, and glucose in cells.
Three-dimensional tracking of single-walled CNTs with an orbital tracking microscope was demonstrated.103 The technique was applied to determine the viscosity regimes within live HeLa cells, and this was used to spatially map corral volumes (0.27–1.32 μm3), to determine active transport velocity (455 nm s−1), and to calculate local viscosities (54–179 cP) within the cell. The NIR emission of CNTs (with their second window at 950–1400 nm) is attractive for in vivo fluorescence imaging due to its deep penetration depth in tissues and low tissue autofluorescence. Genetically engineered multifunctional M13 phages were shown to assemble single-walled CNTs and ligands for targeted fluorescence imaging of tumors.104
Nanodiamonds (NDs) are being produced, as powders, by detonation synthesis on a commercial scale. They have low or no cytotoxicity.105,106 NDs doped with nitrogen are particularly bright. Synthetic NDs form fluorescent centers by thermal annealing and then have rather longwave peaks of excitation (∼560 nm) and emission (∼700 nm). The intrinsic red fluorescence is strong enough to detect a single 35 nm ND in a cell.107 Others types of NDs possess green fluorescence (peaking at 531 nm) and represent a promising alternative to semiconductor quantum dots (see above) because they are photostable, hardly toxic, easily excreted, and can be fairly easily bioconjugated. Hydrophobic derivatives of NDs possessing blue emission have been obtained by modifying the surface with long-chain alkyl groups.108 NDs do not swell, and their size can be hardly tuned by chemical means.
3.7. Metal chalcogenide quantum dots (classical Q-dots)
These are typically made from combinations of zinc(II), cadmium(II), selenide and sulfide. Numerous additional components and dopants are known, and sophisticated methods have been developed to modify surfaces and to create additional shells. Q-dots have experienced an unprecedented success in imaging because of their unique properties. They need not be doped with fluorophores because their fluorescence results from a photonic quantum effect. The color of emission and their size are well tunable and interdependent. Decay times (in the low ns time domain), in contrast, do not vary much. Q-dots display Gaussian emission spectra (with FWHMs of typically 30 nm) and therefore have multiplexing capacity (like upconverting NPs but unlike C-dots). All Q-dots require photoexcitation at <500 nm where biomatter often strongly absorbs (this causing an inner filter effect), and fluorescence intensity strongly varies over time (“blinks”; however, non-blinking Q-dots have been described recently). The quantum yield (QY) of Cd/Zn based Q-dots is rather high (0.3–1.0) which is distinctly better than the QYs of upconversion NPs (see Section 3.8), for example. Dots and aggregates of >300 nm in size cause strong light scattering. Single photon, 2-photon, and recently,109 3-photon excitation and imaging have been demonstrated, and cross sections can be as large as 60000 GM units.
Q-dots are cell toxic unless coated with inert shells, but passivation and reduced toxicity of CdS dots also were accomplished by coating them with DNA.110 They are fairly well cell permeable but clearance from tissue is difficult, partially because of their interaction with thiol groups of cysteines in proteins.111 Their surface chemistry is well established, and several kinds of surface-modified Q-dots are commercially available. They do not swell or photobleach but fluorescence depends on temperature. They are uniquely suited for high resolution and multiplexed imaging of cells. Good reviews cover aspects such as on applications to fluorescence spectroscopy and imaging,112 rendering them biocompatible,113 on the cytotoxicity of cadmium-based Q-dots,114 or on nucleic acid-passivated Q-dots acting as biomolecular templates of varying form and function.115 Recent work includes the application of Q-dot nanosensors to fluorescence lifetime imaging microscopy of intracellular pH,116 or of Q-dots loaded with fluorescent liposomes in order to perform fluorescence resonance energy transfer studies and NIR in vivo imaging of mouse tissues.117 The fluorescence decay time of Q-dots becomes pH-dependent on coating them with NIR fluorescent dyes.118 The fluorescence of certain Q-dots is quenched by Zn(II) and Cd(II) ions,119 and this paves the way for imaging such ions intracellularly. Q-dots may additionally be doped with other metal ions to give, for example, brightly fluorescent Mn-doped ZnS, Mn-doped ZnSe, or Cu-doped InZnS particles (10–80 nm in diameter) which represent a new class of fluorescent nanoparticles with low toxicity.120
Aside from Q-dots composed of Zn(II), Cd(II), sulfide and selenide there are numerous other kinds of such particles. It is difficult to keep track with the variety of materials that have been presented in recent years. NPs with quantum effects can also consist of group III–V elements. Some are brightly fluorescent, one example being InP Q-dots functionalized with a Ln(III) chelate and coated with a cell-penetrating peptide for use as bimodal imaging agents (MRI and confocal microscopy).121 The reader is referred to some of the many reviews that exist on the use of Q-dots in bioimaging. Also see Tables S1–S5 in the ESI.†
3.8. Upconversion nanoparticles (UCNPs)
Most UCNPs (also referred to as upconversion nanocrystals) consist of hexagonal NaYF4 nanocrystals doped with trivalent lanthanide ions such as Er(III), Yb(III) or Tm(III). The dopant is the emitter and additional doping with fluorophores is not needed. UCNPs display several emission colors (with at least two strong bands in the visible) whose peak wavelengths depend on the kind of lanthanide dopant. However, single color emitting UCNPs of the NaYbF4 type and emitting in the green,122 red123 and NIR,124 or consisting of lanthanide-doped KMnF3 nanocrystals have also been reported.125 The size of UCNPs is widely tunable (typical sizes ranging from 10 to 100 nm) and affects quantum yields. The control of size and of the emission and excitation spectra is still a challenge.126 The fact that most UCNPs display multi-photon (bicolor) emissions also paves the way to increased resolution in microscopy.127 Their dual (or multiple) emissions often enable referenced (2-wavelength) sensing and imaging but unfortunately the ratio of the two emissions (often green and red) varies with the coating and undergoes a change if hydrophobic particles of the oleate type are converted into hydrophilic particles (as preferably used in bioimaging). The use of upconversion NPs in bioimaging, therapy, drug delivery and bioassays has been reviewed.128 Very recently it was shown129 that coating UCNPs with a layer of silver causes metal-enhanced (plasmon) upconversion and a 30-fold increase in brightness compared to NPs without the silver core. The NPs used for imaging of HeLa cells have a 3-layer core–shell–shell architecture of the type Ag@SiO2@Lu2O3:Gd,Yb,Er. The silver induced plasmon enhancement mechanism in NaYF4:Yb,Er NPs (Maxwell versus Förster) was studied in some detail.130Unlike in the case of carbon dots, the color of the emission of UCNPs is independent of the excitation wavelength which is rather longwave (750–1000 nm). UCNPs with oleate capping (as obtained by the most popular method of synthesis) possess moderate brightness only, but those modified with hydrophilic coatings are much less bright, with QYs that hardly exceed 0.5% in water solution. QYs of 1–3% have also been reported but only for bulk materials or for dried and aggregated particles. The seemingly poor QYs of UCNPs are still acceptable because their anti-Stokes emission allows fluorescence images to be acquired against a black background.
Remarkably, and unlike in the case of Q-dots, the QYs of UCNPs also depend on the power density (Watt cm−2) of the (cw) laser and on particle size, with smaller particles of the same kind displaying smaller QYs.131 It is reminded that the QY of UCNPs is poorly defined.132 If data are given, it shall also be stated whether these refer to a single emission band or the total emission (in either Stokes and anti-Stokes mode). In addition, the size of the particles investigated must be given along with power density, and how the inner filter effect of water solutions (under 980 nm excitation) has been taken into account. UCNPs, if coated with a shell of undoped NaYF4 (and even silica), are much brighter in water than uncoated UCNPs. The group of Resch-Genger133 has determined, in a solid study, the QY of oleic acid-coated UCNPs of type NaYF4:Yb,Er to be 0.35% under well defined experimental conditions. Gargas et al.134 report on seemingly highly attractive nanocrystals (5–8 nm i.d.) for single-molecule imaging. The brightness under single-particle imaging conditions is said to be much higher than that of other compositions. However, the power density applied in the experiments is as high as >106 W cm−2 which is hardly tolerated by any living organism. If such a power density is applied to watery samples, strong local heating will occur. In fact, if excited with conventional power density, the luminescence of these UCNPs is so weak that no spectra can be acquired.
UCNPs may also be applied to optically encode and to multiplexed imaging of cells and microspheres.135 The method may be extended to lifetime-based encoding by exploiting their tunable luminescence lifetimes which are in the microsecond time regime in case of NaYF4:Yb,Tm.136 By exciting a single color band, one can generate more than ten excited state populations with lifetimes ranging from 25.6 μs to 662 μs and decode their well-separated lifetime identities which are independent of both color and intensity.
Host crystals other than NaYF4 have been studied recently. Light management in UCNPs was demonstrated for ultrasmall NaGdF4 nanoparticles core–shell architectures to tune their emission color.137 In another example,138 the brightness of core–shell nanocrystals (NaLuF4:Gd,Yb,Er coated with a shell of NaLuF4:Yb) was found to be remarkably higher than that of inert-shell coated nanocrystals. These particles can be used to image HeLa cells. Even more complex UCNPs of the type NaLuF4:Gd,Yb,Er were synthesized recently and applied to bioimaging.139
UCNPs are perfectly suited for bioimaging because fluorescence is not at all interfered by the autofluorescence of cells.140 Moreover, they are fairly biocompatible and – if small enough – cell permeable.141 They are not known to be toxic and easily excreted. They usually are weakly interacting with proteins but can be well conjugated to them.142,143 UCNPs are hardly attacked by the immunosystem, and their surface can be easily modified. They do not swell but tend to aggregate in the presence of bivalent ions. Photobleaching cannot occur. Their luminescence is highly dependent on temperature144 and (rather unselectively) quenched by several heavy metal ions.145 It is reminded that photoexcitation of UCNPs with lasers of wavelengths above 800 nm can lead to local heating which can represent a substantial source of error in quantitative fluorometry.
If excited with NIR lasers, UCNPs heat up. This can be desired or not. It is not desired in the case of bioimaging, and an excitation wavelength of 915 nm was recommended to reduce laser induced heating of UCNPs of the type NaYbF4:Tm,Er,Ho for deeper in vivo imaging.146 Heating may, however, also be desired because local heating can be exploited in cancer therapy. In a typical application, oleate-capped UCNPs were coated first with a shell of plain silica (also in order to make them more stable in water solution) and then with a layer of silica doped with a blue carbocyanine dye. The optical emission of the upconverting NP was absorbed by the dye to cause a local heating by up to 21 °C and this causes cells to disrupt. Fig. 5 shows a schematic of the preparation of such UCNPs and respective TEMs. Even shorter excitation wavelengths can be used as shown147 with core–shell UCNPs doped with Nd(III) ions as sensitizers. The upconversion effect already occurs at excitation wavelengths of around 800–820 nm, and this can strongly reduce sample heating and overtone IR absorption by water. Others have shifted the excitation wavelength for upconversion to 1490 nm by using LiYF4:Er nanocrystals.148
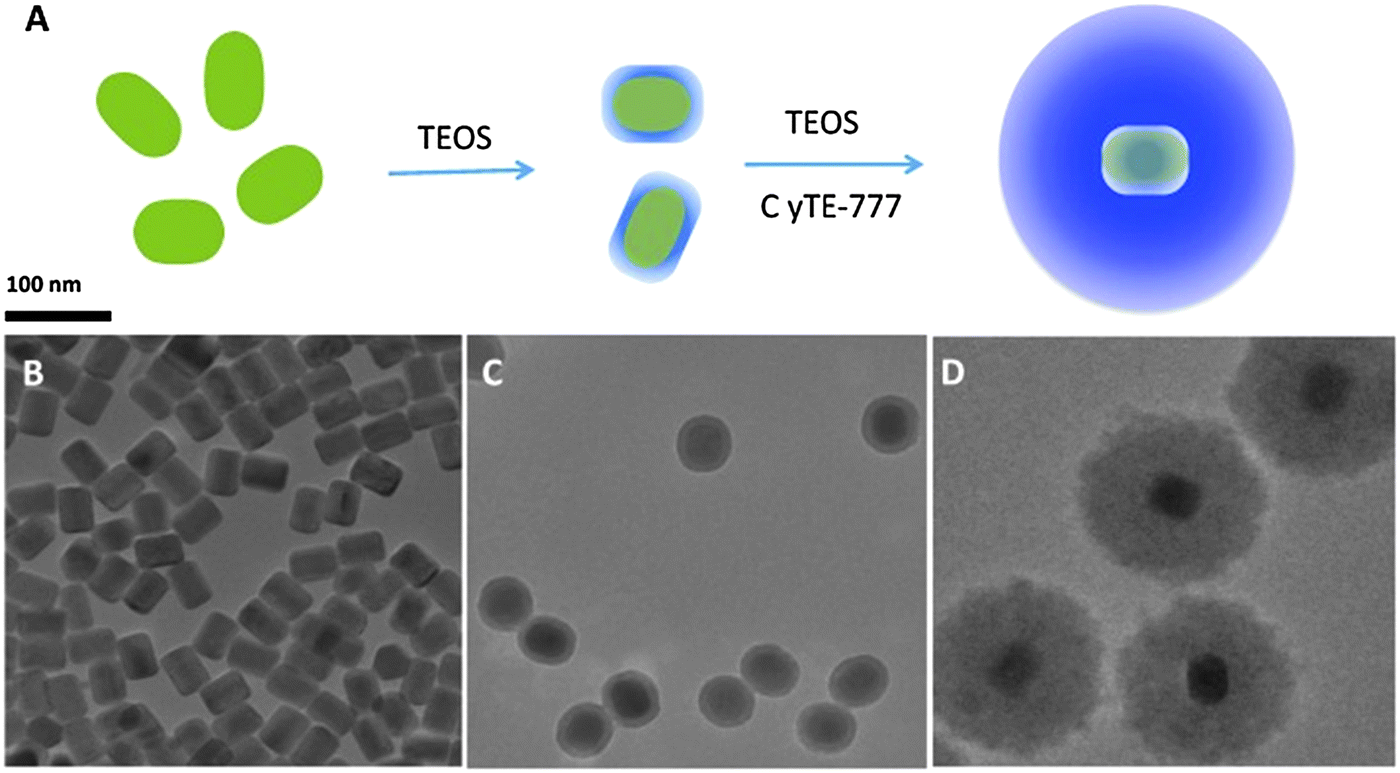 | ||
| Fig. 5 (A) Preparation of UCNPs for use in imaging and hyperthermal cancer treatment. (A) Coating of the green UCNPs first with silica and then with silica doped with the blue and NIR emitting dye Cite-777. (B–D) TEM images of the UNNPs, UCNP@SiO2 core–shell particles, and UCNP@SiO2 particles coated with the dye–silica nanocomposite. From ref. 146 with permission. | ||
Most syntheses yield water-insoluble NPs which first have to be surface-modified so as to enable phase transfer to aqueous solutions. Reviews are available on the design, nanochemistry and applications of UCNPs in theranostics,149 on the surface chemistry of UCNPs, and how to make them hydrophilic.150 Any surface modification has, however, an effect on their luminescence and colloidal properties.151 The upconversion fluorescence of NPs of the type Ag@SiO2@Y2O3:Er can be fine-tuned by the size of the silver core.152 The emission color of lanthanide doped NaYF4 UCNPs also changes during the transformation of crystalline phases from α, a transition state of α mixed with β, and finally to the β phase (from red to yellow and finally to green). Coated with polyethyleneneimine, such particles were used to image cells.153
Examples for applications include the kinetic determination of the activity of the enzyme phospholipase.154 A mixture of a PEGylated phospholipid and a rhodamine-labeled phospholipid was deposited on an UCNP composed of hexagonal NaYF4 doped with 20% Yb(III) and 2% Er(III). The 540 nm emission of the UCNP is used to photoexcite rhodamine B close to the NP. If, however, the phosphodiester is hydrolyzed by the enzyme phospholipase inside a cell, the rhodamine is released from the surface of the UCNP, and this leads to the suppression of the fluorescence of the rhodamine. The red emission peaking at 655 nm is not affected by this process and can serve as an internal standard. Earlier work in the use of UCNPs in chemical sensing and biosensing has been reviewed.155 A recent review covers their application to bioassays and bioimaging.156
3.9. Noble metal nanoparticles
Specific features of such NPs include excellent photostability, water-solubility, size-dependent colors, the lack of swelling, sharp contrast, the ease of characterization by means such as TEM or SEM, and an established surface chemistry (often thiol-based) which is useful if targeted imaging or biosensing/imaging is desired. The uptake of engineered gold nanoparticles by mammalian cells has been reviewed by Dykman and Khlebtsov.157 Gold and silver NPs can also be coupled to plasmonic detection quite readily or used to generate fluorescence patterns through differential release of fluorescent polymers.158Single gold or silver NPs display rather weak fluorescence but were used to image HeLa cells.159 While one-photon luminescence is weak, two-photon luminescence of gold NPs is strong under excitation at 514 and 633 nm.160 Two-photon luminescence imaging of cancer cells down to 75 μm depth and using molecularly targeted gold nanorods161 and of silver NPs162 has been reported. On the other hand, gold NPs can quench fluorescence by phase induced radiative rate suppression.163 It was relatively late when it was discovered that metal clusters made from metallic gold, silver, copper, for example, display strong intrinsic fluorescence.164 Noble metals are preferred for their inertness. The surface of these clusters (gold and silver in particular) can be protected with alkanethiolate monolayers. If properly modified, they enable plain imaging and targeted imaging. While luminescence is often attributed to particle size effects that cause size-dependent fluorescence,165 structural parameters such as surface ligands, valence states of metal atoms and crystallinity of NPs also affect spectra and decay times. Gold NPs and clusters can be composed of a few to millions of atoms.166 Such “quantum” clusters may also be protected or made targetable by coating them with respective proteins.167 For example, gold NPs were functionalized with luminescent ruthenium(II) polypyridyl to endow DNA binding capability and applicability to cellular imaging.168 These structures bind to DNA and undergo rapid cellular uptake, being localized within the cell cytoplasm and the nucleus within 4 h. Various kinds of fluorescent silver nanoclusters (with green, red and yellow luminescences) have been reported by Díez et al.169 In a smart sensing approach towards probing phosphate ions,170 gold nanoclusters (NCs) were capped with 11-mercaptoundecanoic acid and loaded with Eu(III) ions. The red fluorescence of the gold NCs is quenched by the Eu(III) ions, but fluorescence is restored upon addition of phosphate.
3.10. Dendrimers, lipid drops and micelles
Dendrimers (dendrites) are a kind of NPs but much smaller than those treated so far. They can be both hydrophilic and hydrophobic and are easily internalized by cells. Their fluorescence usually originates from a label that has to be attached to the hyperbranched dendrimer skeleton such that no self-quenching does occur. The brightness of dendrimeric NPs can be controlled by size, and color by the terminal fluorophore. Their size exceeds 2 nm in very few cases. Dendrimer chemistry is well established. They have been applied to plain imaging, to targeted imaging, and to sensing/imaging. Features of fluorescent dendrimers include very large molar absorbances that can be in the order of 7000000 M−1 cm−1. The quantum yields of conventional fluorescence range from 0.3 to 0.5 provided that the fluorophores are arranged such that they do not undergo self-quenching.
Nanoscopic fluorescent dendrimers were designed171 that carry up to 96 two-photon chromophores and show very large two-photon absorption cross-sections (up to 56000 GM units). Such organic nanodots are said to represent “brilliant” alternatives to semiconductor quantum dots. A schematic of such a dendrimer particle is shown in Fig. 6. In a typical example of its use in imaging, a dendrimer-based fluorescent pH probe was used to visualize pH values in living HeLa cells.172 In another example,173 an amino-terminated generation-5 dendrimer labeled with a fluorescent marker was integrated into a nanogel and used to track it inside cells by fluorescence microscopy.
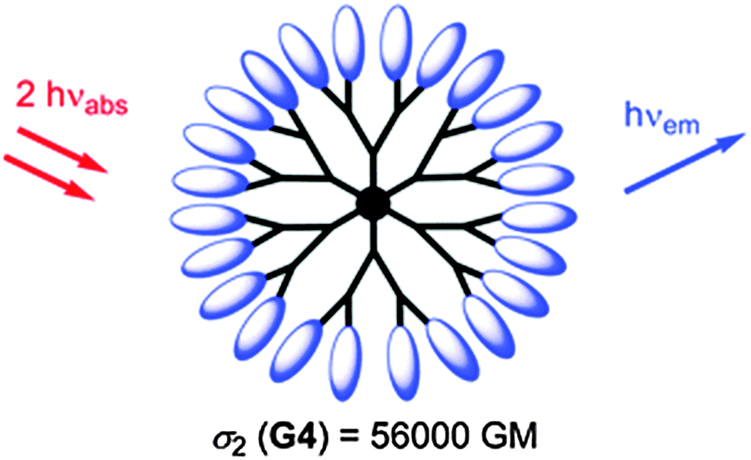 | ||
| Fig. 6 Structure of a hyperbranched dendrimer with terminal fluorophores for use in single-photon and two-photon fluoroimaging. From ref. 171. © Royal Soc. Chem. | ||
Vinogradov's group174 has used smart dendrimers labeled with quenchable probes for oxygen. These are most viable (and commercially available) nanoprobes for imaging of oxygen in blood vessels and (cerebral) tissues. A dendrimer termed Oxyphor G2 [which is a two-layer glutamate dendrimer containing the Pd(II) complex of tetra-(4-carboxyphenyl) benzoporphyrin] is now widely used for phosphorescence based assay and imaging of oxygen. Its chemical structure is shown in Fig. 7. Other (and much larger) Oxyphors are known, some being more lipophilic, others more hydrophilic and even being charged.
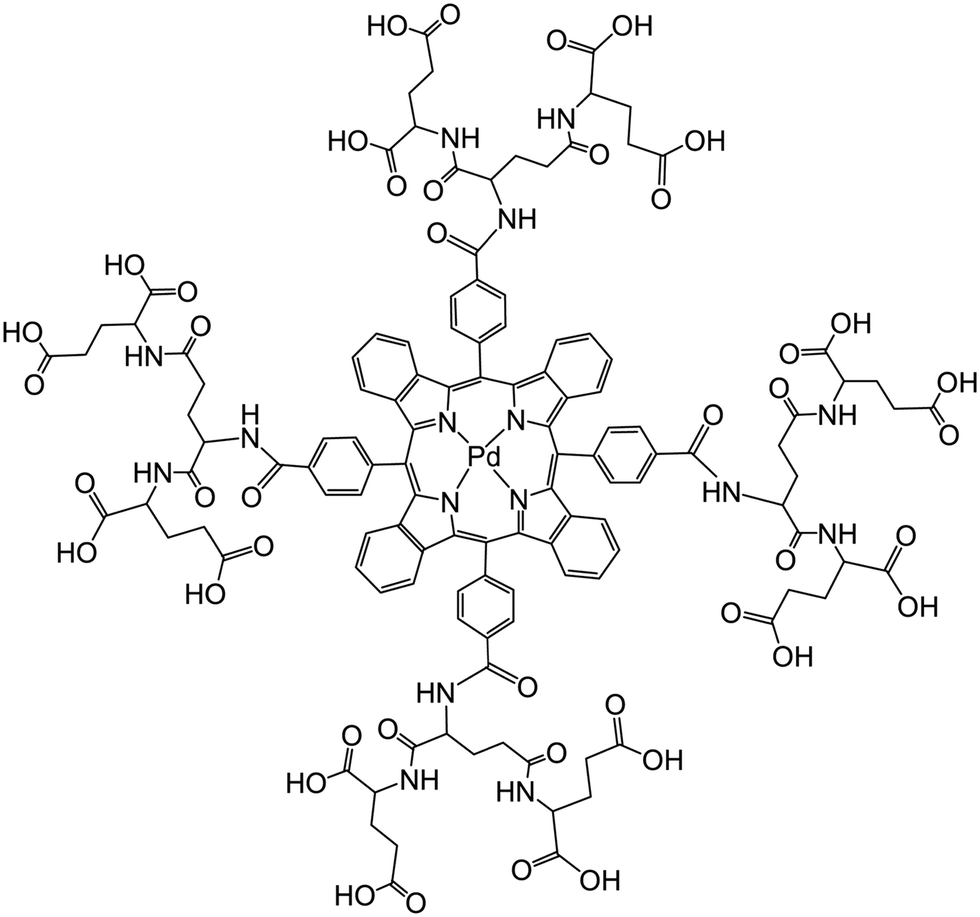 | ||
| Fig. 7 Chemical structure of the dendrimeric nanoparticles used (under the tradename Oxyphore G) for imaging of oxygen. It consists of a Pd(II)benzoporphyrin core and a glutamate-based periphery whose carboxy groups are dissociated which imparts water solubility. From ref. 174 with permission (2014) by Springer Verlag (Berlin). | ||
In addition to dye-labeled dendrimers, there are reports175 on autofluorescent hyperbranched poly(amido amine) NPs and their application to cell imaging. They display blue emission, are said to be nontoxic, and can be recognized by sialo-glycoprotein receptors on the surface of cancer cells. Similarly, a hyperbranched conjugated polyelectrolyte was reported for use in bioimaging.176 It has a core–shell structure, an emission maximum at 565 nm, a quantum yield of 12% and a Stokes shift of 143 nm in water. Its poly(ethylene glycol) shell minimizes nonspecific interaction. Conjugated to the anti-HER2 affibody, it was utilized for targeted cellular imaging of HER2-overexpressed cancer cells. A Q-dot-based ratiometric pH nanosensor with a dendrimer coating was constructed for measurement of physiological pH ranges. It is based on CdSe/CdZnS nanocrystals where the pH probe SNARF was conjugated to the poly(amido amine) dendrimer coating.177 Dendrimer probes for enhanced photostability and localization in fluorescence imaging have been reviewed.178 The group of Tian179 described multifunctional nanomicelles for recognition and precisely targeted NIR-light induced cancer therapy. The nanomicelle encapsulates a pH-activatable fluorescent probe and a robust NIR photosensitizer and is functionalized with an aptamer for targeting viable cancer cells. The fluorescent probe can fluorescently detect the lysosomes for real-time imaging. NIR irradiation causes the generation of reactive oxygen species to trigger lysosomal cell death. The Lovell group reported180 on the development of porphysomes (i.e., nanovesicles formed from self-assembled porphyrin bilayers) with very large extinction coefficients, structure-dependent fluorescence self-quenching, and photothermal properties. Their NIR fluorescence, regenerated on dissociation, results in low-background fluorescence imaging. Porphysomes are biodegradable and induce minimal acute toxicity in mice. Similar to liposomes, the aqueous core of porphysomes can be loaded with drugs or agents. Injected porphysomes accumulate in tumors of xenograft-bearing mice, and laser irradiation induces photothermal tumor ablation.
3.11. Other nanomaterials
Numerous other materials have been described for use as fluorescent NPs, most with the potential of acting as probes for (bio)imaging. These include metal oxides, sulfides, tellurides, fluorides, complex ionic species (including more than one cation and anion). They cannot be discussed here in depth but a selection is given. They usually are covered with additional layers (such as reactive groups and/or PEG) or coated with shells of (inert) solid materials, often silica. The list of conceivable candidates for preparing NPs of various size and optical properties is virtually endless. One typical example is given by Nd(III)-doped LaF3 nanoparticles that can be applied to imaging in the second optical window of biomatter.181 Fluoridated hydroxyapatites (HAps) were doped with Eu(III) or Tb(III) that were shown to be viable NPs for cell imaging.182 They were made water-dispersible by hydrophobic/hydrophilic transformation with the surfactant Pluronic™ F127. Similar NPs were obtained by PEGylation of fluoridated HAp nanorods doped with Ln(III) and used for cell imaging.183 A comparative study was performed on the structure of Tb(III) doped fluorescent HAp nanocrystals184 and showed them to possess good cytocompatibility and cell imaging capability.NPs consisting of a YVO4 matrix and doped with trivalent lanthanides and phosphorus enable multicolor tuning at a single wavelength of excitation.185 Emission wavelengths and intensity ratios can be controlled using the host–activator system and the concentrations of dopants. By coupling luminescent Tb(III) complexes to magnetite (Fe3O4), NPs are obtained186 that display paramagnetism, low cytotoxicity, and high cell uptake. If coated with folic acid, they can be used for in vitro fluorescence and magnetic resonance targeted imaging of cells that overexpress the folate receptor, for example the HeLa cells.
Titanium dioxide (“titania”) is easily prepared and doped, for example with europium(III), and respective TiO2 hollow nanoshells are viable two-photon nanoprobes. If coated with poly(ethylene imine), they adhere to HeLa cervical cancer cells.187 Mesoporous titania was deposited on silver–silica core–shell NPs to result in a nanoarchitecture of type Ag@SiO2@mTiO2. The metal core acts as an enhancer of fluorescence. The particles were loaded with the fluorescent flavin mononucleotide and the fluorescent cancer drug doxorubicin and used for simultaneous bimodal (fluorescence and SERS) imaging of drug delivery.188 Amino-functionalized zirconia NPs (5 nm i.d.) doped with Ln(III) ions were prepared and used for time-resolved fluorescence resonance energy transfer detection of avidin with a detection limit of 3.0 nM. The ZrO2 NPs can specifically recognize cancer cells overexpressed with a urokinase plasminogen activator receptor.189
Fluorescent Ag2S nanoclusters possess tunable photoluminescence that extends from the red to the NIR. They can be made biocompatible and applied to bioimaging by introducing glutathione as the capping reagent.190 Two-photon excitable CaF2 NPs doped with Tm(III) and Yb(III) represent multifunctional nanoprobes for deep-tissue fluorescence bioimaging.191 On the basis of the strong Tm(III) ion emission (at around 800 nm), tissue penetration depths as large as 2 mm have been demonstrated, which is more than 4 times that of the visible emissions in comparable lanthanide-doped CaF2 NPs. These NPs with an outstanding penetration depth, together with the fluorescence thermal sensitivity are well suited as multifunctional nanoprobes for high-contrast and deep-penetrating in vivo imaging. Other sulfides also display quantum dot-like emission, for example 5 nm NPs prepared from lead(II) sulfide which were protected with oleic acid and oleyl amine and made hydrophilic by hydroxylation at the terminal end of the alkyl chains.192
Nanoscale metal–organic frameworks (often handicapped by hydrolytic decomposition) were shown to enable bioimaging193 by incorporation of luminescent or high Z element building blocks. They can serve as viable contrast agents. The pores and channels of such frameworks may also be loaded with active agents and drugs. Colloidal and stable silicon (not silica!) NPs with their red fluorescence and being grafted with hyperbranched polyglycerol are water soluble and display good colloidal stability.194 The transformation of the hydrophobic surface of plain silicon NPs into a hydrophilic surface makes them water-soluble and suitable for specific targeting overexpressed cervical cancer cells and glioblastoma cells. Such particles are said to be “bright” but it is difficult to assess brightness (defined as molar absorbance multiplied with quantum yield) if neither of the two is known. Inorganic semiconductor fluorescent NPs consisting of ultrathin silica carbide (SiC) also display pH-dependent luminescence and can be used195 to probe intracellular pH values in the range from 5.6 to 7.4.
Lipid NPs – like hydrogels NPs – are soft but highly apolar. They resemble hydrophobic dendrimers and can be easily prepared but often lack stability if placed in complex systems. Gravier et al.196 have described lipid NPs loaded with fluorophores (NIR dyes including Indocyanine Green) for use in fluorescence imaging (so called “lipidots”). Multichannel in vivo imaging of lymph nodes in mice was demonstrated for doses as low as 2 pmol of NPs which have very high “molar” absorbance. Their cytotoxicity is very low. An interesting example of a micelle-based NP probe was reported by Zhou et al.197 who have prepared such NPs from micelle-forming block copolymers with tertiary amine and poly(ethylene oxide) segments. If the local pH value is below the pKa of the ammonium groups, the micelles dissociate into unimers which is accompanied by a strong increase in fluorescence due to the suppression of homo-FRET. These NPs are highly sensitive to pH and were applied to target specific endocytic organelles in living cells and to image pH values. The method of pH-induced micellization was extended to a panel of multicolored nanoparticles with a wide emission range (500–820 nm) and different pH transitions.198 Wang et al.199 have shown that ultra-small fluorescent nanosensors for oxygen can be obtained by a rather simple method. The materials have a hydrophobic core capable of firmly hosting hydrophobic luminescent oxygen probes and a shell composed of a long-chain poly(ethylene glycol) which renders them cell-membrane impermeable but yet highly sensitive to oxygen. These NPs are highly stable in aqueous solutions, in cell culture media, and in extracellular fluids such as blood, interstitial and brain fluid. Four kinds of nanosensors were presented, whose excitation spectra cover a wide spectral range (395–630 nm), thus matching many common laser lines, and with emission maxima ranging from 565 to 800 nm, thereby minimizing interference from background luminescence of biomatter.
4. Coatings and surface modifications
Coatings and surface modifications of NPs can serve two purposes. The first is to render NPs fluorescent, the second is to improve the properties of the NPs, for example by making them brighter, better biocompatible, or cell-permeable. NPs can be made fluorescent by attaching fluorophores to their surface by depositing additional coatings which may carry a fluorophore, or by immobilizing a fluorophore on their surface. The layer-by-layer technique (LbL) has turned out to be quite viable200 but the stability may be an issue if the NPs are placed in complex samples such as serum. The LbL technique consists in the deposition of single layers of (polymer) molecules of alternating polarity or charge and works best with alternatively charged ionic materials.Methods for the functionalization of inorganic NPs for bioimaging applications are rather diverse because conjugation of NPs to specific biomolecules enables targeted imaging, can reduce overall toxicity, and improve brightness. In general, such NPs are modified in two steps, the first being surface activation, the second being conjugation.201 The latter knows numerous methods depending on the kind of species to be immobilized. These range from polymers such as poly(ethylene glycol) to proteins, fluorophores, oligomers or other particles. Both physical adsorption and covalent binding have their merits and are widely used. Quality criteria of the final product include colloidal stability in water, biocompatibility, cell permeability, and a brightness that enables even minute quantities of NPs to be discerned from an often strongly fluorescent background.202 Methods for the functionalization of the surface of metal complex-derived NPs for use in molecular imaging have been reviewed.203 One of the most versatile methods consists in the biomimetic modification of the surface of NPs by phospholipids as described in a milestone paper by the Selvin group.204 The approach involves coating the surface of NPs with a monolayer of phospholipids containing different functional terminal groups that can govern polarity and can render the particles water soluble, for example, or conjugatable to biomolecules. These particles have a range of applications in fields such as imaging and quantitative bioanalysis. Specifically, dispersible and functionalized NPs were described for selective imaging of live cancer cells. Also see a related recent article where zwitterionic phospholipid coatings are employed.205
Many fluorescent NPs are simply coated with a shell of silica. This introduces a negative charge on the surface and – more importantly – protects the core from external quenchers. In addition, the silica coating can also be made fluorescent with a second dye and be further (bio)functionalized. Fig. 8 shows a schematic of a typical process. Methods for coating NPs with silica are well established and well reproducible. Biomedical applications of organically modified bioconjugated silica NPs have been reviewed.206
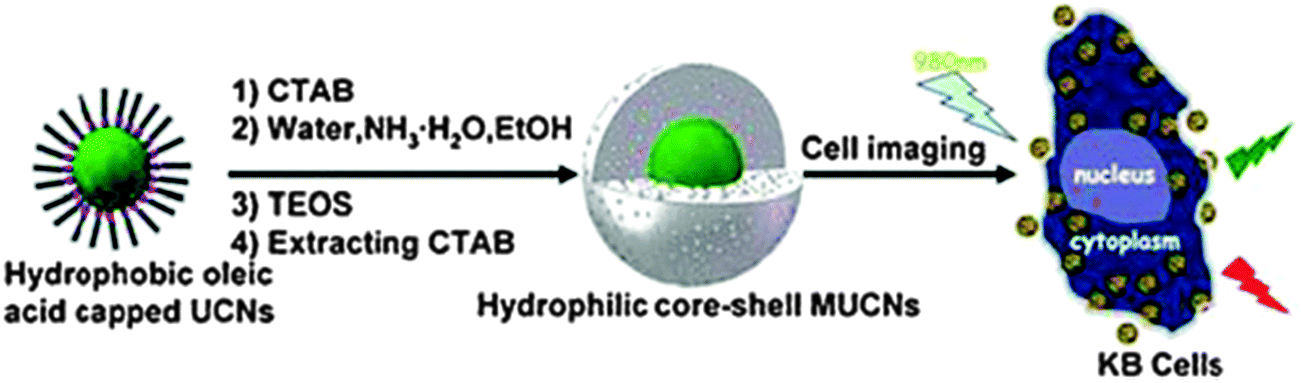 | ||
| Fig. 8 Schematic of the common method for coating oleate-capped upconversion nanoparticles (UCNPs) with a shell of silica, and their subsequent internalization into cells. From ref. 207 with permission. | ||
When deployed in vivo, NPs are typically protected from the immune system by coating them with poly(ethylene glycol) (PEG). A wide variety of strategies are known208 to coat and characterize NPs with PEG. The core materials differ strongly in terms of size, shape, density, loading level, molecular weight, charge and method for purification. However, other hydrogels have been used that work as well. Jin et al.209 have coated upconversion nanocrystals of the type NaYF4:Yb,Er with poly(vinyl pyrrolidone, and then a ligand exchange reaction was performed with polyethylenimine (PEI) and poly(acrylic acid). The coated UCNPs can be dispersed in aqueous medium. The positively charged PEI-coated particles are readily taken up by cells (in comparison to their neutral or negatively charged counterparts). The long-term in vivo biodistribution of polyacrylic acid-coated upconversion nanophosphors was imaged and their toxicity is low.210 Others have coated UCNPs with the hydrogel poly[(N-isopropylacrylamide)-co-(methacrylic acid)].211 Gelatine, another kind of hydrogel, was used to coat CdTe/CdSe Q-dots to make them hydrophilic, more biocompatible and, thus, suitable for bioimaging.212 The cell permeability of particles is strongly improved if their surface is coated with poly-L-lysine.213
UCNPs are hydrophobic if prepared by the most common method. In order to make them water-soluble, Liras et al.214 have capped the surface of UCNPs with a polymer by replacing the original oleate ligand by multidentate thiolate grafting. The side chains of the coating extend into the solution and render the UCNPs water-dispersible. The resulting nanohybrids exhibit an emission brighter by a factor of 10 in organic solvent and by a factor of 2 even in water, and their fluorescence is highly thermoresponsive. A review is available on the functionalization of inorganic NPs including metal (Au, Ag), metal oxide (Fe3O4), and semiconductor nanocrystals (e.g. quantum dots and magnetic quantum dots) for bioimaging applications.215 Muhr et al.150 have summarized methods for converting hydrophobic UCNPs (as obtained by standard synthetic methods for making particles with low dispersity) to water soluble hydrophilic UCNPs which is almost mandatory if such particles are to be used in bioimaging. The state of the art in the use of upconversion nanophosphors for small-animal imaging,216 and of methods for modification and characterization of UCNPs for use in bioanalytical methods217 have been reviewed.
The delivery of luminescent europium-coated NPs into platelets can be controlled via the local pH values and can occur within a few min.218 Gold NPs were coated with a luminescent Eu(III) complex (EuL) and the pHLIP peptide to give particles of the type pHLIP/EuL@AuNPs. The 13 nm diameter gold NPs act as a scaffold for the attachment of both the luminescent probe and the peptide to target delivery. The NPs enter the platelets under low pH conditions only, typically at pH 6.5, but not at pH 7.4, and this is mediated by pHLIP translocation across the membrane. Luminescence microscopy images clearly show the red luminescence of the europium probe.
If NPs are to be used for purposes of targeting (see Section 5.2), it is mandatory to modify the surface of the NP such that it can recognize its target. It is known, for example, that the coating of a surface with triphenylphosphonium groups (using reagents such as MitoP™) will cause the particle to accumulate in the proximity of mitochondria.219 This was exploited to visualize oxygen tension at various sites on the microscale.220 Depending on the kind of surface (silica, polylysine, or triphenylphosphonium groups), the polystyrene nanosensors are located in the extracellular or intracellular space, or near mitochondria. A good example for the architecture of a NP that can target cancer cells is provided by the work of Cho et al.221 who have made rhodamine-dyed NPs (∼35 nm i.d.) highly biocompatible by coating the surface with phosphocholine and lectin as shown in Fig. 9. Such particles have a high affinity for sialic acid as overexpressed on the surface of tumor cells.
 | ||
| Fig. 9 Architecture of a nanoparticle capable of targeting cancer cells. The surface is rendered biocompatible and negatively charged by coating it with phosphocholine groups. Immobilization of lectin via biotin–streptavidin interaction results in particles that have extremely high affinity for sialic acid that is overexpressed on the surface of cancer cells. The images show, from left to right, a TEM and fluorescence images of non-cancerous and cancerous cells. From ref. 221 with permission (2014) of the Am. Chem. Soc. | ||
5. Specific examples on the use of nanoparticles
5.1. Plain fluorescence imaging
In this case, the particles are simply added to the cell culture, tissue, or vascular system to be imaged, usually by, but not limited to microscopy. The only purpose of the particles is to render cells fluorescent, and therefore they are expected to display a large brightness, to be detectable in even deep regions of tissue or in blood, not to be toxic, and not to respond to their microenvironment (such as its local pH). NPs that work in the optical window of biomatter, i.e. in the 600 to 900 nm range, are strongly preferred, while probes with UV excitation suffer from inner filter effects on both the excitation and emission, and from strong UV, blue and green background fluorescence of most biomatter. Water-dispersible and fluorescent organic NPs that are made more biocompatible by PEGylation and exploiting the phenomenon of aggregation induced emission enhancement have found particular interest in the past few years with respect to cell imaging applications.222 Quite a number of articles have been published in recent years on the use of NPs for plain imaging, and a selection is presented in Table S1 in the ESI.†5.2. Targeted bioimaging
If specific chemical groups or domains are to be detected, a molecular probe (indicator), a labeling reagent (so to visualize functional groups such as thiols), or a domain-recognizing probe (that can target mitochondria or the glycolipids in Alzheimer-associated tissue, for example) is being added. If the species to be targeted is an intracellular protein, Q-dots have been almost exclusively used in the past,223 but UCNPs and C-dots are on the rise. Such methods are compromised, however, by nonspecific binding, difficulty of intracellular delivery, or endosomal trapping of the NPs. Targeted bioimaging includes classical (not particle-based) methods of intracellular immunostaining and in situ hybridization. Biju224 has reviewed methods for chemical modifications and bioconjugate reactions of nanomaterials such as silica NPs, gold NPs, gold quantum clusters, semiconductor Q-dots, carbon nanotubes, fullerene and graphene for use in (targeted) sensing and imaging.Internalization is facilitated by modifying the surface with poly(ethylene glycol) (PEG) and/or coating with a cell penetrating peptide (which also avoids endosomal sequestration) and, finally, with a receptor. The art of delivering bioconjugated quantum dots to their targets and their uses for nonspecific extracellular and intracellular imaging have been reviewed.225 Biocompatibility and internalization are critical issues, and particles usually are coated with PEG to impart or improve biocompatibility during functional live-cell imaging. It was shown more recently that sulfobetaine zwitterionic organic species on quantum dots serve the same purpose.226 Specifically, bidentate zwitterionic dihydrolipoic acid–sulfobetaine ligands represent a favorable alternative to PEG-coated NPs since they combine small size, low nonspecific adsorption, and stability over time and a wide range of pH values and salinity. In addition to using antibody-modified NPs, nanomaterials functionalized with DNA aptamers have been applied to targeted imaging of specific sites inside cells.227
Tumor cells are best recognized by targeting their folate receptor. It was targeted with an aggregation-enhanced fluorescent silica nanoprobe and used for both one-photon and two-photon excited bioimaging.228 The two-photon technique can overcome the limitations caused by fluorescence quenching due to high chromophore loading and provided 3D cellular-level resolution imaging of up to 350 μm deep in a HeLa tumor. Hyaluronated fullerene with its strong NIR fluorescence also allows for high-resolution fluorescence imaging of tumor sites in vivo.229 Dually targeting upconversion nanocrystals were obtained230 by attaching 3-aminophenylboronic acid and hyaluronated fullerene to them. The two ligands warrant specific targeting of cancer cells. In vivo single-cell pharmacokinetic imaging of PARP-1 inhibitors and model drug behavior was demonstrated231 under varying conditions. Cancer cells were visualized through expression of H2B-apple (580 nm; red) and TAMs were visualized by blue and fluorescent NPs internalized into endosomes. The method is said to allow for gaining a better insight into drug action in vivo.
Work on the use of DNA aptamer modified nanomaterials for analysis of intracellular components and metabolites (also including aspects of imaging) has been reviewed.232 A most useful method for targeted imaging mRNA has been presented by the Mirkin group.233 Their term “multiplexed nanoflare” stands for a NP agent capable of simultaneously detecting two distinct mRNA targets inside a living cell. These probes are spherical and consist of polynucleotides conjugated to gold NPs as shown in Fig. 10. The oligomers are densely packed and highly oriented, many of which are hybridized to a reporter with a distinct fluorophore label and each complementary to its corresponding mRNA target. If multiplexed nanoflares are exposed to their targets, they provide a sequence specific signal in both extra- and intracellular environments. One of the targets can be used as an internal control, improving detection by accounting for cell-to-cell variations in NP uptake and background. Compared to single-component nanoflares, these structures allow for a precise determination of relative levels of mRNA in individual cells, but also have applications in cell sorting. In yet another new scheme for targeting, bioorthogonal (non-natural) chemistry was used to amplify NP binding and to enhance the sensitivity of cell detection. Antibodies against biomarkers were modified and used as scaffolds to couple NPs to live cells.234 The group of Han235 has reviewed the use of upconversion NPs as tools for multiscale targeted bioimaging. Other examples are given in Table S2 in the ESI.†
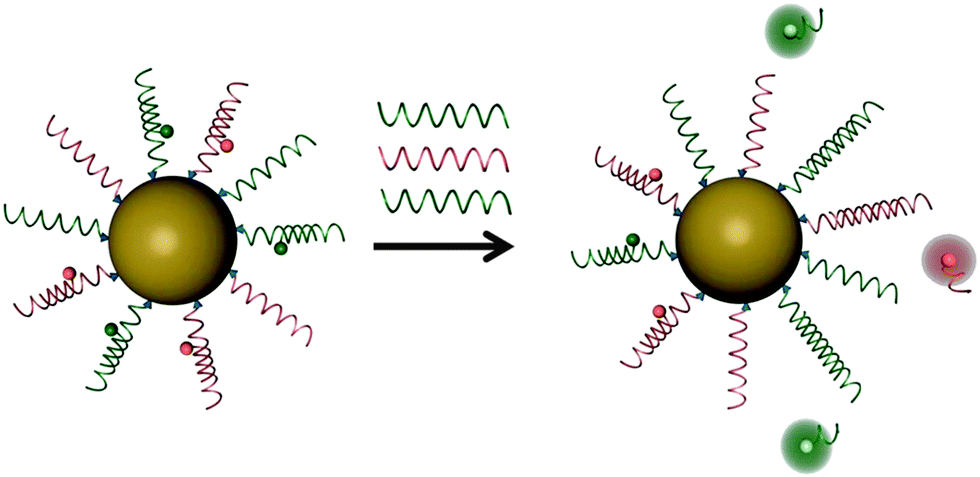 | ||
| Fig. 10 Schematic representation of target detection of mRNA by multiplexed nanoflares. The multiplexed nanoflares bind different target nucleic acids (shown in red and green), displacing the corresponding flare. Once the flare is released, the fluorophore is no longer quenched by the gold surface, and an increase in fluorescence can be measured. Using two different fluorophores, the ratio of each target can be determined in cells. From ref. 233 with permission (2014) by the Am. Chem. Soc. | ||
5.3. Imaging of chemical species
There is substantial interest in visualizing chemical parameters such as pH values or chemical species such as glucose, calcium(II) or oxygen in the living and metabolizing cell, if not in tumor cells or in cells exposed to candidate drugs. Nanosensors have the unique feature of not being affected by intracellular species that often bind to molecular probes and thereby may change their binding constants or act as quenchers. The use of fluorescent NPs for intracellular sensing has been reviewed by the Hall group.236 | ||
| Fig. 11 Pseudocolor images of pH values on an inflammated wound. Bluish colors indicate fairly intact skin (with pH values between 5 and 6), whilst green and red colors represent areas of sustained inflammation and partial granulation. From ref. 237. | ||
 | ||
| Fig. 12 RGB image (A) and ratiometric image (B) of the distribution of oxygen in NRK cells using nanosensors for oxygen. The brightness of the red luminescence (resulting from the probe for oxygen) and the green luminescence (of the reference dye) are stored in 2 different memories and can be used to calculate the ratio for each single pixel. It can be plotted in pseudo-colours (right) to reflect the local oxygen partial pressure. From ref. 241. © Royal Soc. Chem. | ||
5.4. Imaging of temperature
Conventional methods for measurement of temperature (T) cannot be applied to cells and other nanostructures. In fact, the use of molecular or NP-based optical probes is the only way to intracellularly sense T. A review on this topic has appeared.37 One may differentiate between two kinds of nanomaterials that can be used to sense T over time. The first makes use of one or two fluorophores of which at least one displays a highly T-dependent luminescence. If a single probe is used, the determination of lifetime is the (referenced) method of choice. If two fluorophores are used, one acts as the probe while the other serves as a reference or as a partner in a FRET system. An Eu(III) chelate probe in a silica matrix was claimed to possess luminescence that strongly depends on T in the physiological range.242 The second type is based on the use of fluorescent NPs displaying intrinsic fluorescence, examples being Q-dots,243 C-dots,244 or upconverting nanocrystals which have the additional advantage of displaying two emission bands of different sensitivity to T.245 The group of Uchiyama246 was the first to report on nanomaterial-based intracellular sensing of temperature by using a fluorescent polymeric thermometer and fluorescence lifetime imaging microscopy. The spatial and temperature resolutions were at the diffraction limited level (200 nm) and 0.18–0.58 °C. The intracellular temperature distribution indicated that the nucleus and centrosome of a COS7 cell showed a significantly higher temperature than the cytoplasm and that the temperature gap between the nucleus and the cytoplasm differed depending on the cell cycle.It appears that NPs prepared from a poly(methyl methacrylate)-co-1,2-bis(trimethoxysilyl)decane composite and containing a red-luminescent europium(III) complex display the best sensitivity to T at present.247 The NPs also contain a green-emitting and virtually T-independent reference dye. The ratio of the green and a red fluorescence under single-wavelength excitation is highly dependent on T in the 25–45 °C range, with a sensitivity of −4.0% per °C. Given their small size (20–30 nm) and biocompatibility (due to the presence of an outer layer of silica), such NPs are likely to be useful nanoprobes for imaging of T inside cells. Silica NPs doped with the T-probe Ru(bpy) were also used248 to image intracellular T but have a smaller sensitivity (expressed as the signal change ΔS per °C). An interesting method, albeit not applied to imaging, is exploiting the T-sensitive fluorescence of NaYF4:Er,Yb3 upconversion NPs, where the intensity ratio of the two green bands of the Er(III) dopant changes with temperature.249 It was applied to measure T inside HeLa cells. Representative other nanomaterials and their features in terms of optical sensing of T are complied in Table S6 in the ESI.†
6. Multimodal imaging, and imaging combined with drug and gene delivery, or with (photodynamic) therapy
One very exciting trend at present involves a combination of fluorescent imaging with other methods of imaging, mainly magnetic resonance imaging (MRI), plasmonic imaging, and positron emission tomography (PET) imaging. This is referred to as bimodal or multimodal imaging. Another one involves the combination of fluorescent imaging with drug or gene delivery but these rely on the release of a diagnostically active species. A third (and closely related one) involves fluorescent imaging combined with photodynamic therapy (PDT) or photothermal therapy (PTD) where agents may be, or may not be, released from the NPs. These methods shall be discussed in the following sections, again mainly from a nanomaterials point of view.6.1. Multimodal imaging
Fluorescent NPs, magnetic NPs, and plasmonic NPs are the three most common nanoparticles when looking at the particle functionality. Their use in multimodal imaging has been briefly reviewed.250 Depending on the kind of multimodal imaging, a second species is required to be present in the NP in addition to a fluorescent probe that will give a signal in MRI or PET. This section gives examples in addition to those already described earlier in this review (search for bimodal or multimodal). A typical example251 is provided by the use of a nanocomposite consisting of magnetic iron oxide NPs and single-walled CNTs. These heterostructured complexes were utilized as bioimaging agents after encapsulation with oligonucleotides with the sequence d(GT)15 and enrichment using a 0.5 Tesla magnetic array. The resulting nanotube complexes show distinct NIR fluorescence, Raman scattering, and visible/NIR absorbance. Macrophage cells that engulf the DNA-wrapped complexes were imaged using MRI and NIR mapping.Dendrimer based bimodal imaging with longwave emitting fluorophores (these are preferred for the reasons indicated before) was shown for a fluorinated dendron conjugated to a cyanine dye for bimodal MRI and NIR fluorescence imaging.252 Bimodal upconversion fluorescence and X-ray imaging was also accomplished253 by using hexagonal phase NaLuF4:Gd/Yb/Er nanorods, and blood vessels of lung were visualized with the aim to improve the diagnosis of lung and pulmonary vascular diseases. It is advised to read the critical comment by van Veggel et al.254 on the use of lanthanide-doped NPs for fluorescence and MRI. Dual mode nanoprobes (without obvious cytotoxicity) for targeted bimodal fluorescent imaging and MRI of MCF-7 breast cancer cells were obtained255 by co-encapsulation of an NIR-emissive conjugated polymer and lipid-coated iron oxides (IOs) in a hydrophilic poly(lactic-co-glycolic-acid)–poly(ethylene glycol) composite and coating the 180 nm particles with folic acid.
An important recent finding is the effect of the size and the phase on the multimodality of co-encapsulated magnetic photon-upconverting polymeric NPs for use in bimodal (fluorescent/magnetic resonance) imaging.256 In another sophisticated approach, the large potential of multimodal functionality was impressively demonstrated257 by showing that NPs of the layer architecture UCNP@mSiO2–Ln(dbm)4 (where mSiO2 stands for mesoporous silica, Ln for any trivalent ion out of Eu, Sm, Er, Nd, Yb; and dbm stands for the organic ligand complexing the Ln ion). Both downconversion and upconversion luminescence are found, and this results in multicolor emission (covering the spectral region from 450 nm to 1700 nm) under visible-light excitation and 980 nm excitation, respectively. The mesoporous materials were applied to in vitro imaging based on Eu(III) luminescence (under 405 nm excitation), and to small animal imaging based on Tm(III) luminescence (under 980 nm excitation). In addition, the Gd(III) dopant causes T1 signal enhancement and thus makes them potential contrast agents for MRI. This is schematically shown in Fig. 13. Other multifunctional nanocomposites include those for dual luminescence imaging, especially those showing both upconversion and downconversion luminescence.258
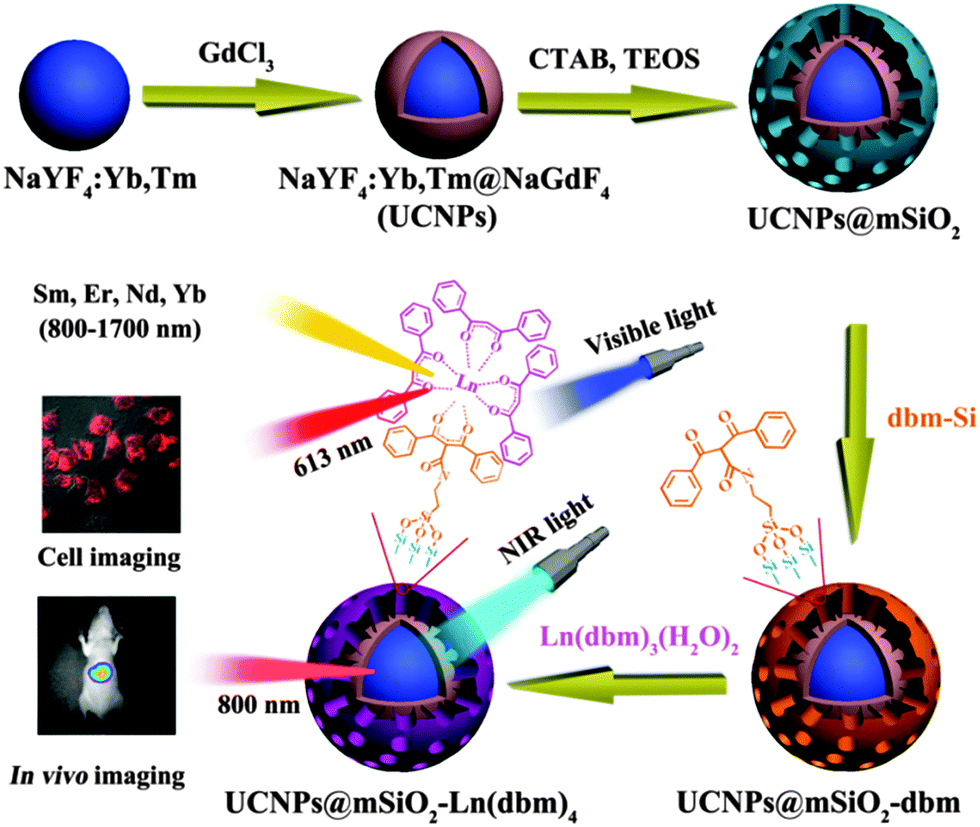 | ||
| Fig. 13 Schematic of the preparation and structure of multifunctional mesoporous NPs containing both upconversion and magnetic nanophosphors (with an architecture of the type NaYF4:Yb,Tm@NaGdF4) and covered with a conventional luminescent lanthanide complex (Ln-dbm) for use in upconversion and downconversion luminescence imaging and as T1-weighed MRI contrast agents. From ref. 257 © RSC. | ||
Bimodal surface enhanced Raman spectroscopy along with 3D fluorescence imaging based on the use of NPs is rather new. In the first approach of that kind,259 a rhodamine dye was conjugated to gold NPs which then were applied to live endothelial cells and revealed inhomogeneous distribution in the cytoplasm. Table S7 in the ESI† summarizes other representative examples of NPs for use in multimodal imaging.
6.2. Imaging combined with drug delivery, gene delivery or chemotherapy
Theranostics with the cancer drug cis-platin has attracted much interest. Nanoparticles consisting of an upconversion fluorescent core and a porous silica shell were applied, for example, to deliver cis-platin to tumors. Such NPs may even be used in synergistic chemo- and radiotherapy by radiosensitization of the cis-platin in the shell, and in magnetic/luminescent bimodal imaging.260 The design of multifunctional upconversion NPs coated with a polymer nanocomposites for use in the delivery of cis-platin and in biomedical imaging was also described.261 In a series of articles, the groups of Qu and Ren have impressively shown how fluorescence nanoparticles prepared from various materials can be applied to simultaneous imaging and delivery of genes and drugs. In one example, polycation-functionalized and water-soluble gold nanoclusters were constructed that act as a platform for simultaneous gene delivery and cell imaging.262 In another, lanthanide-modified hollow mesoporous nanoparticles were also shown to serve this purpose.263 Dextranes, in crosslinked form, can also be used to prepare NPs for use in in vitro drug release by loading dextran–chitosan NPs with the drug curcumin.264 C-dots were shown to be useful nanocarriers for gene delivery and bioimaging. They were prepared by pyrolysis of glycerol in the presence of polyethylenimine which imparts better brightness.265 Advances in lanthanide ion-based upconversion nanomaterials for drug delivery have been reviewed recently.266 Further examples are given in Table S8 in the ESI.†In impressive work on multimodally functional NPs, chemo-, radio- and photodynamic therapy and simultaneous MRI/UCL imaging have been demonstrated for a single kind of NP.267 This represents an exciting leap forward. By integrating upconversion NPs and mesoporous silica into a single platform, diagnostic/therapeutic functions were combined to provide a more advanced way for the efficient theranostics of cancer. Specifically, multifunctional Gd(III)-UCNPs with a mesoporous silica shell were constructed for the co-delivery of a radio-/photo-sensitizer hematoporphyrin (HP) and the radiosensitizer and chemodrug docetaxel. Upon NIR excitation and X-ray irradiation, a tumor was eliminated by the synergistic chemo-/radio-/photodynamic tri-modal therapy under the assistance of simultaneous magnetic and luminescent bimodal imaging. In other exciting work,268 upconversion NPs of the type BaGdF5; 10 nm in size were shown (a) to act as carriers for the drug doxorubicin; (b) to act as multimodal probes for simultaneous imaging (optical/magnetic/X-ray/CT) and drug release which can be triggered by low pH. This exciting approach was applied to kill HeLa cells.
6.3. Imaging combined with photodynamic or photothermal therapy
The heating effect exerted by cw-laser light as used for photoexcitation of upconversion NPs is the result of the absorbance of light by such NPs and by their aqueous microenvironment because water has a weak overtone absorption at above 850 nm. This was exploited in a bimodal scheme for UCNP-based imaging and photothermal therapy (PDT).269 Similarly, in vivo optical imaging and PDT of tumors were accomplished with upconversion NPs with a NaYF4:Yb,Er core and a NaGdF4 shell combined with the photosensitizer chlorin e6. Tumors were observed both in the luminescence images and via MRI. In vivo PDT was simultaneously triggered by irradiation with 980 nm light.270 Related studies were performed where PDT is based on the multicolor emission capability of upconversion NPs at a single excitation wavelength that can simultaneously activate two photosensitizers. Mice were photodynamically treated by direct injection of the NPs into melanoma tumors or by intravenous injection of NPs conjugated to a tumor-targeting agent.271Dextran based NPs dyed with Indocyanine Green can be used for NIR imaging and PDT in vitro.272 The dye is retained by electrostatic interactions, and the NPs are well biocompatible and readily internalized because the dextran is block copolymerized with PEG. Even triple-functional core–shell structured NPs were reported more recently.273 The NPs were covalently grafted with a photosensitizer for luminescent, magnetic resonance imaging and PDT in vivo. Representative examples of NPs that have been used for these purposes are summarized in Table S9 in the ESI.†
7. Conclusions and outlook
The state of the art in fluorescent imaging is impressive. This is due to the progress made in materials science and in spectroscopy and microscopy. The number of nanomaterials, mainly nanoparticles, for use in fluorescent imaging is entangling, however, and a newcomer may find it difficult to make the appropriate selection. Depending on the kind of application, different kinds of criteria may apply when selecting particles. Nanoparticles always are preferred in intracellular studies, and often preferred (over microparticles) in extracellular studies. Table 2 gives an overview of figures of merit for the classes of NPs that are more widely used. The following criteria apply when selecting nanoparticles for use in fluorescence imaging:| Kind of NP | Smallest size (nm) | Particles best characterized bye | Exc/em wavelengths (nm) | Brightness | Photo-stability | Surface chemistry | Toxicity | Imaging & drug delivery | Imaging and PDT |
|---|---|---|---|---|---|---|---|---|---|
| a Semiconducting (organic) polymer dots. b Carbon dots. c Quantum dots based on the use of heavy metal ions. d Upconversion nanoparticles. e TEM: transmission electron microscopy; DLS: dynamic light scattering; AFM: atomic force microscopy; XPS: X-ray photoelectron spectroscopy (for elemental analysis); HRTEM: high-resolution TEM; SEM: scanning electron microscopy; ICP-AES: inductively-coupled plasma atomic emission spectroscopy. f Depending on the dye used. g Usually two or more emission bands. h No reliable data found. | |||||||||
| Silica NPs (dye doped) | 15 | TEM, DLS | From UV to NIRf | High | Fair | Facile | Low | Less suited | Less suited |
| Porous silicas (dye doped) | 20 | TEM, DLS | From UV to NIR | High | Fair | Facile | Low | Well suited | Well suited |
| Organic polymer dots (dye doped) | 15 | DLS | From UV to NIR | High | Fair | Fair | Low | Hardly demonstrated but likely | Hardly demonstrated but likely |
| P-dotsa | 25 | DLS, AFM | (320–450)/(400–600) | Moderate | High | Difficult | —h | Not yet demonstrated | Not yet demonstrated |
| C-dotsb | 2–3 | AFM, XPS, HRTEM | (350–500)/(450–700) | Good | High | Complex | Said to be low | Possibly suited | Possibly suited |
| Q-dotsc | 8 | SEM, TEM, DLS, | (330–450)/(420–700) | High | High | Moderately facile | Low if properly coated | Not recommended | Not recommended |
| UCNPsd | 8 | TEM, DLS, XRD, ICP-AES | (780–980)/(350–NIR)g | Poor | High | Fairly facile | Low | Well suited | Well suited |
| Gold NP clusters | 20 | SEM, TEM, UV/Vis | (500–570)/(560–680) | Moderate | High | Facile | Low | Well suited | Well suited |
(a) Plain imaging requires bright particles that are non-toxic and – if this is needed – well cell permeable. Candidate materials include upconversion nanocrystals, C-dots, Q-dots (if properly coated), dyed particles consisting of (mesoporous) silica or polystyrene, and semiconductor P-dots.
(b) Targeted (cellular) imaging requires bright particles that are not (cell-)toxic and, if needed, cell permeable. As their surface has to be coated with a ligand binding to the target (the receptor), their surface has to be modified which is easy in the case of silica particles and silica-coated upconversion materials, also with C-dots and particles made from PAA, but less so with P-dots and with most hydrophobic organic particles.
(c) NPs for use in chemical sensing are supposed, first of all, to be selective and, ideally, hardly affected by temperature. Biocompatibility is needed in the case of imaging biosystems, of course. Sensors working in the optical (spectral) window of biomatter (i.e. between 600 and 900 nm) are preferred. Such sensors should also be calibratable in vivo.
(d) NPs for use in imaging of temperature (T) are, first of all, expected to be completely inert except for the T-dependence of their luminescence. Depending on the application, biocompatibility and cell permeability may be required. They are supposed to respond to T with a large signal change which results in a good resolution. In medical and bioapplications, the resolution is supposed to be better than ±0.2 °C in the 20–55 °C range. Other nanosensors are supposed to work over a wide range of Ts, and this will not require such a good resolution.
(e) NPs for use in multimodal imaging require nanomaterials of high sophistication. Such NPs are expected to display bright fluorescence and, in parallel, to be applicable to a second kind of imaging, MRI for example. Hence, additional functional materials have to be incorporated into the core or the shell of the NPs however without compromising other properties including size and cell viability.
(f) NPs for use in optical imaging along with (cancer) therapy also require nanomaterials of high sophistication. Particles are preferably covered with porous shells that can be loaded with a drug, a gene or a photodynamic or a photothermal agent. Drugs and genes can be released once the NPs have arrived at the site of action. Controlled release can be induced by light or by changes in the local pH value, for example.
Given the number of conceivable applications of nanoparticles in the very large medical field (cancer diagnosis and therapy mainly), in bioanalysis, biology, the military, security (from documents to banknotes) and in various kinds of technical nanosystems, it is difficult to make general recommendation but it is hoped that readers will find this review to be of help when making decisions as to what material to be chosen.
References
- M. Chen and M. Yin, Prog. Polym. Sci., 2014, 39, 365–395 CrossRef CAS PubMed.
- J. Li and J. J. Zhu, Analyst, 2013, 138, 2506–2515 RSC.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, A. M. Wu, S. S. Gabhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- F. Pinaud, S. Clarke, A. Sittner and M. Dahan, Nat. Methods, 2010, 7, 275–285 CrossRef CAS PubMed.
- (a) S. Palmal and N. R. Jana, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 102–110 CrossRef CAS PubMed; (b) C. Zhou, S. Yang, J. Liu, M. Yu and J. Sheng, Exp. Biol. Med., 2013, 238, 1199–1209 CrossRef CAS PubMed.
- X. Zhang, X. Zhang, L. Tao, Z. Chi, J. Xu and Y. Wei, J. Mater. Chem. B, 2014, 2, 4398–4414 RSC.
- Nanocomposite Particles for Bio-applications, ed. T. Trindade and A. L. D. da Silva, Pan Stanford Pte. Publ., Singapore, 2011, ISBN 978-981-4267-78-6 Search PubMed.
- S. Parveen, R. Misra and S. K. Sahoo, Nanomed.: Nanotechnol., Biol. Med., 2012, 8, 147–166 CrossRef CAS PubMed.
- G. Yang, P. Shen, K. Tan and Y. Xia, Microchim. Acta, 2014, 181, 607–613 CrossRef CAS.
- V. Ntziachristos, Nat. Methods, 2010, 7, 603–614 CrossRef CAS PubMed.
- Y. Liu, D. Tu, H. Zhu and X. Chen, Chem. Soc. Rev., 2013, 42, 6924–6958 RSC.
- P. Hazarika and D. A. Russell, Angew. Chem., Int. Ed., 2012, 51, 3524–3531 CrossRef CAS PubMed.
- L. Wei, S. Doughan, Y. Han, M. V. DaCosta, U. J. Krull and D. Ho, Sensors, 2014, 14, 16829–16855 CrossRef CAS PubMed.
- J. C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- T. Ozawa, H. Yoshimura and S. B. Kim, Anal. Chem., 2013, 85, 590–609 CrossRef CAS PubMed.
- W. P. Dempsey, S. E. Fraser and P. Pantazis, BioEssays, 2014, 34, 351–360 CrossRef PubMed.
- Y. Zhang, Y. Bai, J. Jia, N. Gao, Y. Li, R. Zhang, G. Jiang and B. Yan, Chem. Soc. Rev., 2014, 43, 3762–3809 RSC.
- K. Wang, X. He, X. Yang and H. Shi, Acc. Chem. Res., 2013, 46, 1367–1376 CrossRef CAS PubMed.
- A. Burns, P. Sengupta, T. Zedayko, B. Baird and U. Wiesner, Small, 2006, 2, 723–726 CrossRef CAS PubMed.
- K. Ma, H. Sai and U. Wiesner, J. Am. Chem. Soc., 2012, 4, 13180–13183 CrossRef PubMed.
- H. Zhou, X. Lv, L. Zhang, A. Gong, A. Wu, Z. Liang, G. Peng and H. Lin, J. Mater. Chem. C, 2014, 2, 9625–9630 RSC.
- (a) H. S. Mader, X. Li, S. M. Saleh, M. Link, P. Kele and O. S. Wolfbeis, Ann. N. Y. Acad. Sci., 2008, 1130, 218–223 CrossRef CAS PubMed; (b) S. M. Saleh, R. Ali and O. S. Wolfbeis, Microchim. Acta, 2011, 174, 429–434 CrossRef CAS.
- S. W. Bae, W. Tan and J. I. Hong, Chem. Commun., 2012, 48, 2270–2282 RSC.
- X. Chen, Y. Liu and D. Tu, Lanthanide Doped Luminescent Nanomaterials, Nanomedicine and Nanotoxicology, Springer Verlag, Berlin, 2014, ch. 7, pp. 145–164 DOI:10.1007/978-3-642-40364-4_7.
- Y. Zhang, W. Wei, G. K. Das and T. T. Y. Tan, J. Photochem. Photobiol., C, 2014, 20, 71–96 CrossRef CAS PubMed.
- S. L. C. Pinho, H. Faneca, C. F. G. C. Geraldes, M. H. Delville, L. D. Carlos and J. Rocha, Biomaterials, 2012, 33, 925–935 CAS.
- M. Chen and M. Yin, Prog. Polym. Sci., 2014, 39, 395 CrossRef PubMed.
- S. Schreml, R. J. Meier, O. S. Wolfbeis, M. Landthaler, R. M. Szeimies and P. Babilas, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2432–2437 CrossRef CAS PubMed.
- S. Schreml, R. J. Meier, M. Kirschbaum, O. S. Wolfbeis, M. Landthaler and P. Babilas, Theranostics, 2014, 4, 721–735 CrossRef CAS PubMed.
- H. H. Sun, K. Almdal and T. L. Andresen, Chem. Commun., 2011, 47, 5268–5270 RSC.
- X. Zhou, F. Su, Y. Tian and D. R. Meldrum, PLoS One, 2014, 9, e88185, DOI:10.1371/journal.pone.0088185.
- X. Wang, R. J. Meier and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2013, 52, 406–409 CrossRef CAS PubMed.
- X. Wang, J. A. Stolwijk, T. Lang, M. Sperber, R. J. Meier, J. Wegener and O. S. Wolfbeis, J. Am. Chem. Soc., 2012, 134, 17011–17014 CrossRef CAS PubMed.
- M. I. J. Stich, L. H. Fischer and O. S. Wolfbeis, Chem. Soc. Rev., 2010, 39, 3102–3114 RSC.
- H. Peng, J. A. Stolwijk, L. Sun, J. Wegener and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2010, 49, 4246–4249 CrossRef CAS PubMed.
- K. Chung, J. Wallace, S. Kim, S. Kalyanasundaram, A. S. Andalman, T. J. Davidson, J. J. Mirzabekov, K. A. Zalocusky, J. Mattis, A. K. Denisin, S. Pak, H. Bernstein, C. Ramakrishnan, L. Grosenick, V. Gradinaru and K. Deisseroth, Nature, 2013, 497, 332–337 CrossRef CAS PubMed.
- X. Wang, O. S. Wolfbeis and R. J. Meier, Chem. Soc. Rev., 2013, 42, 7834–7869 RSC.
- E. Pringsheim, D. Zimin and O. S. Wolfbeis, Adv. Mater., 2001, 13, 819–822 CrossRef CAS.
- X. Wang, D. E. Achatz, C. Hupf, M. Sperber, J. Wegener, S. Bange, J. M. Lupton and O. S. Wolfbeis, Sens. Actuators, B, 2013, 188, 257–262 CrossRef CAS PubMed.
- X. Wang, R. J. Meier and O. S. Wolfbeis, Adv. Funct. Mater., 2012, 22, 4202–4207 CrossRef CAS.
- X. Zhang, S. Wang, L. Xu, L. Feng, Y. Ji, L. Tao, S. Li and Y. Wei, Nanoscale, 2012, 4, 5581–5584 RSC.
- M. Luo, Bull. Korean Chem. Soc., 2014, 35, 1732–1736 CrossRef CAS.
- I. L. Medintz, M. H. Stewart, S. A. Trammell, K. Susumu, J. B. Delehanty, B. C. Mei, J. S. Melinger, J. B. Blanco-Canosa, P. E. Dawson and H. Mattoussi, Nat. Mater., 2010, 9, 676–684 CrossRef CAS PubMed.
- D. Posavec, A. Dorsch, U. Bogner, G. Bernhardt and S. Nagl, Microchim. Acta, 2011, 173, 391–399 CrossRef CAS.
- S. H. Hu and X. H. Gao, J. Am. Chem. Soc., 2010, 132, 7234–7237 CrossRef CAS PubMed.
- K. Li and B. Liu, J. Mater. Chem., 2012, 22, 1257–1264 RSC.
- R. Tang and X. Feng, Can. Chem. Trans., 2013, 1, 78–84 CrossRef CAS.
- C. Wu, S. J. Hansen, Q. Hou, J. Yu, M. Zeigler, Y. Jin, D. R. Burnham, J. D. McNeill, J. M. Olson and D. T. Chiu, Angew. Chem., Int. Ed., 2011, 50, 3430–3434 CrossRef CAS PubMed.
- Y. H. Chan, C. Wu, F. Ye, Y. Jin, P. B. Smith and D. T. Chiu, Anal. Chem., 2011, 833, 1448–1455 CrossRef PubMed.
- X. Zhang, M. Liu, B. Yang, X. Zhang and Y. Wei, Colloids Surf., B, 2013, 112, 81–86 CrossRef CAS PubMed.
- L. Sun, D. Hao, W. Shen, Z. Qian and C. Zhu, Microchim. Acta, 2012, 177, 357–364 CrossRef CAS.
- K. Pu, A. J. Shuhendler, J. V. Jokerst, J. Mei, S. S. Gambhir, Z. Bao and J. Rao, Nat. Nanotechnol., 2014, 9, 233–239 CrossRef CAS PubMed.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- M. Bacon, S. J. Bradley and T. Nann, Part. Part. Syst. Charact., 2014, 31, 415–428 CrossRef CAS.
- Y. Song, S. Zhu and B. Yang, RSC Adv., 2014, 4, 27184–27200 RSC.
- H. Li, Z. Kang, Y. Liu and S. T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC.
- S. K. Das, Y. Liu, S. Yeom, D. Y. Kim and C. I. Richards, Nano Lett., 2014, 14, 620–625 CrossRef CAS PubMed.
- L. Wang and H. S. Zhou, Anal. Chem., 2014, 86, 8902–8905 CrossRef CAS PubMed ; and ref. 4, 17 and 21–25 cited therein.
- B. Kong, A. W. Zhu, C. Q. Ding, X. M. Zhao, B. Li and Y. Tian, Adv. Mater., 2012, 24, 5844–5848 CrossRef CAS PubMed.
- C. Ding, A. Zhu and Y. Tian, Acc. Chem. Res., 2014, 47, 20–30 CrossRef CAS PubMed.
- F. Du, F. Zeng, Y. Ming and S. Wu, Microchim. Acta, 2013, 180, 453–460 CrossRef CAS.
- P. G. Luo, S. Sahu, S. T. Yang, S. K. Sonkar, J. Wang, H. Wang, G. E. LeCroy, L. Cao and Y. Sun, J. Mater. Chem. B, 2013, 1, 2116–2127 RSC.
- C. Peng, W. B. Hu, Y. T. Zhou, C. H. Fan and Q. Huang, Small, 2010, 6, 1686–1692 CrossRef CAS PubMed.
- K. P. Liu, J. J. Zhang, F. F. Cheng, T. T. Zheng, C. M. Wang and J. J. Zhu, J. Mater. Chem., 2011, 21, 12034–12040 RSC.
- Y. Chong, Y. Ma, H. Shen, X. Tu, X. Zhou, J. Xu, J. Dai, S. Fan and Z. Zhan, Biomaterials, 2014, 35, 5041–5048 CrossRef CAS PubMed.
- C. F. Hu, Y. L. Liu, Y. H. Yang, J. H. Cui, Z. R. Huang, Y. L. Wang and L. F. Yang, J. Mater. Chem. B, 2013, 1, 39 RSC.
- J. C. G. Esteves da Silva and H. M. R. Goncalves, Trends Anal. Chem., 2011, 30, 1327–1336 CrossRef CAS PubMed.
- S. Mitra, S. Chandra, T. Kundu, R. Banerjee, P. Pramanik and A. Goswami, RSC Adv., 2012, 2, 12129–12131 RSC.
- C. Liu, P. Zhang, F. Tian, W. Li, F. Li and W. Liu, J. Mater. Chem., 2011, 21, 13163–13167 RSC.
- P. Zuo, D. Xiao, M. Gao, J. Peng, R. Pan, Y. Xia and H. He, Microchim. Acta, 2014, 181, 1309–1316 CrossRef CAS.
- H. Li, X. He, Y. Liu, H. Huang, S. Lian, S. Lee and Z. Kang, Carbon, 2011, 49, 605–609 CrossRef CAS PubMed.
- Y. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- H. P. Liu, T. Ye and C. D. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473–6475 CrossRef CAS PubMed.
- X. Zhang, S. Wang, C. Zhu, M. Liu, Y. Ji, L. Feng, L. Tao and Y. Wei, J. Colloid Interface Sci., 2013, 397, 39–44 CrossRef CAS PubMed.
- S. K. Bhunia, A. Saha, A. R. Maity, S. C. Ray and N. R. Jana, Sci. Rep., 2013, 3, 1473, DOI:10.1038/srep01473.
- H. Nie, M. Li, Q. Li, S. Liang, Y. Tan, L. Sheng, W. Shi and S. X. Zhang, Chem. Mater., 2014, 26, 3104–3112 CrossRef CAS.
- S. Qu, H. Chen, X. Zheng, J. Cao and X. Liu, Nanoscale, 2013, 5, 5514–5518 RSC.
- S. Kochmann, T. Hirsch and O. S. Wolfbeis, J. Fluoresc., 2012, 22, 849–855 CrossRef CAS PubMed.
- H. Goncalves, A. Duarte and J. C. G. Esteves da Silva, Biosens. Bioelectron., 2010, 26, 1302 CrossRef CAS PubMed.
- X. Zhang, S. Wang, M. Liu, B. Yang, L. Feng, Y. Ji, L. Tao and Y. Wei, Phys. Chem. Chem. Phys., 2013, 15, 19013–19018 RSC.
- G. Gollavelli and Y. C. Ling, Biomaterials, 2012, 33, 2532–2545 CrossRef CAS PubMed.
- J. Jeong, J. Jung, M. Choi, J. W. Kim, S. J. Chung, S. Lim, H. Lee and B. H. Chung, Adv. Mater., 2012, 24, 1999–2003 CrossRef CAS PubMed.
- C. Baleizão, S. Nagl, M. Schaeferling, M. N. Berberan-Santos and O. S. Wolfbeis, Anal. Chem., 2008, 80, 6449–6457 CrossRef PubMed.
- C. Baleizao, S. Nagl, S. M. Borisov, M. Schaeferling, O. S. Wolfbeis and M. N. Berberan-Santos, Chem. – Eur. J., 2007, 13, 3643–3651 CrossRef CAS PubMed.
- X. Gong, Q. Hu, M. C. Paau, Y. Zhang, S. Shuang, C. Dong and M. M. F. Choi, Nanoscale, 2014, 6, 8162–8170 RSC.
- K. P. Liu, J. J. Zhang, F. F. Cheng, T. T. Zheng, C. M. Wang and J. J. Zhu, J. Mater. Chem., 2011, 21, 12034–12040 RSC.
- B. J. Hong, O. C. Compton, Z. An, I. Eryazici and S. T. Nguyen, ACS Nano, 2012, 6, 63–73 CrossRef CAS PubMed.
- S. K. Bhunia, N. Pradhan and N. R. Jana, ACS Appl. Mater. Interfaces, 2014, 6, 7672 CAS.
- P. G. Luo, F. Yang, S. T. Yang, S. K. Sonkar, L. Yang, J. J. Broglie, Y. Liu and Y. Sun, RSC Adv., 2014, 4, 10791–10807 RSC.
- S. Zhuo, M. Shao and S. T. Lee, ACS Nano, 2012, 6, 1059–1064 CrossRef CAS PubMed.
- (a) Y. Li, Y. Zhao, H. H. Cheng, Y. Hu, G. Q. Shi, L. M. Dai and L. T. Qu, J. Am. Chem. Soc., 2012, 134, 15 CrossRef CAS PubMed; (b) C. F. Hu, Y. L. Liu, Y. H. Yang, J. H. Cui and Z. R. Huang, J. Mater. Chem. B, 2013, 1, 39 RSC; (c) Q. Liu, B. D. Guo, Z. Y. Rao, B. H. Zhang and J. R. Gong, Nano Lett., 2013, 13, 2436 CrossRef CAS PubMed; (d) J. Ju and W. Chen, Biosens. Bioelectron., 2014, 58, 219 CrossRef CAS PubMed.
- (a) L. Zhang, Z. Y. Zhang, R. P. Liang, Y. H. Li and J. D. Qiu, Anal. Chem., 2014, 86, 4423 CrossRef CAS PubMed; (b) Z. T. Fan, Y. C. Li, X. H. Li, L. Z. Fan, S. X. Zhou, D. C. Fang and S. H. Yang, Carbon, 2014, 70, 149 CrossRef CAS PubMed.
- D. Qu, M. Zheng, P. Du, Y. Zhou, L. G. Zhang, D. Li, H. Q. Tan, Z. Zhao, Z. G. Xie and Z. C. Sun, Nanoscale, 2013, 5, 12272 RSC.
- Q. Feng, Q. Q. Cao, M. Li, F. C. Liu, N. J. Tang and Y. W. Du, Appl. Phys. Lett., 2013, 102, 013111 CrossRef PubMed.
- X. M. Li, S. P. Lau, L. B. Tang, R. B. Ji and P. Z. Yang, J. Mater. Chem. C, 2013, 1, 7308 RSC.
- Q. Liu, B. Guo, Z. Rao, B. Zhang and J. R. Gong, Nano Lett., 2013, 13, 2436–2441 CrossRef CAS PubMed.
- S. Zhu, J. Zhang, S. Tang, C. Qiao, L. Wang, H. Wang, X. Liu, B. Li, Y. Li, W. Yu, X. Wang, H. Sun and B. Yang, Adv. Funct. Mater., 2012, 22, 4732–4740 CrossRef CAS.
- Z. Liu, K. Yang and S. T. Lee, J. Mater. Chem., 2011, 21, 586–598 RSC.
- P. Cherukuri, C. J. Gannon, T. K. Leeuw, H. K. Schmidt, R. E. Smalley, S. A. Curley and R. B. Weisman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18882–18886 CrossRef CAS PubMed.
- S. Dang, Q. Liu, X. Zhang, K. He, C. Wang and X. Fang, J. Nanosci. Nanotechnol., 2012, 12, 4478–4484 CrossRef CAS PubMed.
- D. A. Heller, S. Baik, T. E. Eurell and M. S. Strano, Adv. Mater., 2005, 17, 2793–2799 CrossRef CAS.
- B. Mu, J. Zhang, T. P. McNicholas, N. F. Reuel, S. Kruss and M. S. Strano, Acc. Chem. Res., 2014, 47, 979–988 CrossRef CAS PubMed.
- N. F. Reuel, A. Dupont, O. Thouvenin, D. C. Lamb and M. S. Strano, ACS Nano, 2012, 6, 5420–5428 CrossRef CAS PubMed.
- H. Yi, D. Ghosh, M. H. Ham, J. Qi, P. W. Barone, M. S. Strano and A. M. Belcher, Nano Lett., 2012, 12, 1176–1183 CrossRef CAS PubMed.
- H. Huang, E. Pierstorff, E. Osawa and D. Ho, Nano Lett., 2007, 7, 3305–3314 CrossRef CAS PubMed.
- R. Lam, M. Chen, E. Pierstorff, H. Huang, E. J. Osawa and D. Ho, ACS Nano, 2008, 2, 2095–2102 CrossRef CAS PubMed.
- C. C. Fu, H. Y. Lee, K. Chen, T. S. Lim, H. Y. Wu, P. K. Lin, P. K. Wei, P. H. Tsao, H. C. Chang and W. Fann, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 727–732 CrossRef CAS PubMed.
- V. N. Mochalin and Y. Gogotsi, J. Am. Chem. Soc., 2009, 131, 4594–4595 CrossRef CAS PubMed.
- J. H. Yu, S.-H. Kwon, Z. Petrášek, O. K. Park, S. W. Jun, K. Shin, M. Choi, Y. I. Park, K. Park, H. B. Na, N. Lee, D. W. Lee, J. H. Kim, P. Schwille and T. Hyeo, Nat. Mater., 2013, 12, 359–366 CrossRef CAS PubMed.
- N. Ma, J. Yang, K. M. Stewart and S. O. Kelley, Langmuir, 2007, 23, 12783–12787 CrossRef CAS PubMed.
- M. Cao, C. Cao, M. Liu, P. Wang and C. Zhu, Microchim. Acta, 2009, 165, 341–346 CrossRef CAS.
- E. Petryayeva, W. R. Algar and I. L. Medintz, Appl. Spectrosc., 2013, 67, 215–251 CrossRef CAS PubMed.
- S. J. Rosenthal, J. C. Chang, O. Kovtun, J. R. McBride and I. D. Tomlinson, Chem. Biol., 2011, 18, 10–24 CrossRef CAS PubMed.
- N. Chen, Y. He, Y. Su, X. Li, Q. Huang, H. Wang, X. Zhang, R. Tai and C. Fan, Biomaterials, 2012, 33, 1238–1244 CrossRef CAS PubMed.
- N. Ma, G. Tikhomirov and S. O. Kelley, Acc. Chem. Res., 2010, 43, 173–180 CrossRef CAS PubMed.
- A. Orte, J. M. Alvarez-Pez and M. J. Ruedas-Rama, ACS Nano, 2013, 7, 6387–6395 CrossRef CAS PubMed.
- C. Ye, Y. Wang, C. Li, J. Yu and Y. Hu, Microchim. Acta, 2013, 180, 117–125 CrossRef CAS.
- R. Tang, H. Lee and S. Achilefu, J. Am. Chem. Soc., 2012, 134, 4545–4548 CrossRef CAS PubMed.
- X. Luo, W. Wu, F. Deng, D. Chen, S. Luo and C. Au, Microchim. Acta, 2014, 181, 1361–1367 CrossRef CAS.
- A. R. Maity, S. Palmal, S. K. Basiruddin, N. S. Karan, S. Sarkar, N. Pradhan and N. R. Jana, Nanoscale, 2013, 5, 5506–5513 RSC.
- G. J. Stasiuk, S. Tamang, D. Imbert, C. Poillot, M. Giardiello, C. Tisseyre, E. L. Barbier, P. H. Fries, M. de Waard, P. Reiss and M. Mazzanti, ACS Nano, 2011, 5, 8193–8201 CrossRef CAS PubMed.
- X. M. Li, R. Wang, F. Zhang and D. Y. Zhao, Nano Lett., 2014, 14, 3634–3639 CrossRef CAS PubMed.
- G. Tian, Z. Gu, L. Zhou, W. Yin, X. Liu, L. Yan, S. Jin, W. Ren, G. Xing, S. Li and Y. Zhao, Adv. Mater., 2012, 24, 1226–1231 CrossRef CAS PubMed.
- J. Wang, F. Wang, C. Wang, Z. Liu and X. G. Liu, Angew. Chem., Int. Ed., 2011, 50, 10369–10372 CrossRef CAS PubMed.
- J. Wang, F. Wang, C. Wang, Z. Liu and X. G. Liu, Angew. Chem., Int. Ed., 2011, 50, 10369–10372 CrossRef CAS PubMed.
- L.-D. Sun, Y.-F. Wang and C.-H. Yan, Acc. Chem. Res., 2014, 47, 1001–1009 CrossRef CAS PubMed.
- H. Liu, C. T. Xu and S. Andersson-Engels, Opt. Express, 2014, 22, 17782–17790 CrossRef PubMed.
- Y. Yang, Microchim. Acta, 2014, 181, 263–294 CrossRef CAS.
- D. Yin, C. Wang, J. Ouyang, X. Y. Zhang, Z. Jiao, Y. Feng, K. Song and M. Wu, ACS Appl. Mater. Interfaces, 2014, 6, 18480–18488 CAS.
- D. Lu, S. K. Cho, S. Ahn, L. Brun, C. J. Summers and W. Park, ACS Nano, 2014, 8, 7780–7792 CrossRef CAS PubMed.
- M. Pollnau, D. R. Gamelin, S. R. Luthi, H. U. Guedel and M. P. Hehlen, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3337 CrossRef CAS.
- J.-C. Boyer and F. C. J. M. van Veggel, Nanoscale, 2010, 2, 1417–1419 RSC.
- C. Würth, J. Pauli, C. Lochmann, M. Spieles and U. Resch-Genger, Anal. Chem., 2012, 84, 1345–1352 CrossRef PubMed.
- D. J. Gargas, E. M. Chan, A. D. Ostrowski, S. Aloni, M. V. P. Altoe, E. S. Barnard, B. Sanii, J. J. Urban, D. J. Milliron, B. E. Cohen and P. J. Schuck, Nat. Nanotechnol., 2014, 9, 300–305 CrossRef CAS PubMed.
- H.-H. Gorris and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2013, 52, 3584–3600 CrossRef CAS PubMed.
- Y. Lu, J. Zhao, R. Zhang, Y. Liu, D. Liu, E. M. Goldys, X. Yang, P. Xi, A. Sunna, J. Lu, Y. Shi, R. C. Leif, Y. Huo, J. Shen, J. A. Piper, J. P. Robinson and D. Jin, Nat. Photonics, 2014, 8, 32–36 CrossRef CAS.
- M. Quintanilla, F. Ren, D. Ma and F. Vetrone, ACS Photonics, 2014, 1, 662–669 CrossRef CAS.
- Y. Ou, D. Yin, X. Cao, C. Wang, K. Song, B. Liu, L. Zhang, Y. Han and M. Wu, Dalton Trans., 2014, 43, 14001–14008 RSC.
- D. Yin and X. Cao, Dalton Trans., 2014, 43, 14001–14008 RSC.
- H. S. Mader, P. Kele, S. M. Saleh and O. S. Wolfbeis, Curr. Opin. Chem. Biol., 2010, 14, 582–596 CrossRef CAS PubMed.
- E. Hemmer, F. Vetrone and K. Soga, Mater. Res. Soc., 2014, 39, 960–964 CrossRef CAS.
- S. Wilhelm, T. Hirsch, W. M. Patterson, E. Scheucher, T. Mayr and O. S. Wolfbeis, Theranostics, 2012, 3, 239–248 CrossRef PubMed.
- R. B. Liebherr, T. Soukka, O. S. Wolfbeis and H. H. Gorris, Nanotechnology, 2012, 23, 485103 CrossRef PubMed.
- A. Sedlmeier, D. E. Achatz, L. H. Fischer, H. H. Gorris and O. S. Wolfbeis, Nanoscale, 2012, 4, 7090–7096 RSC.
- S. M. Saleh, R. Ali and O. S. Wolfbeis, Chem. – Eur. J., 2011, 17, 14611–14617 CrossRef CAS PubMed.
- Q. Zhan, J. Qian, H. Liang, G. Somesfalean, D. Wang, S. He, Z. Zhang and S. Andersson-Engels, ACS Nano, 2011, 5, 3744–3757 CrossRef CAS PubMed.
- (a) Y. Wang, G. Liu, L. Sun, J. Xiao, J. Zhou and C. Yan, ACS Nano, 2013, 7, 7200–7206 CrossRef CAS PubMed; (b) H. Wen, H. Zhu, X. Chen, T. F. Hung, B. Wang, G. Zhu, S. F. Yu and F. Wang, Angew. Chem., Int. Ed., 2013, 52, 13419–13423 CrossRef CAS PubMed.
- G. Y. Chen, T. Y. Ohulchanskyy, A. Kachynski, H. Agren and P. N. Prasad, ACS Nano, 2011, 5, 4981–4986 CrossRef CAS PubMed.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- V. Muhr, Th. Hirsch, S. Wilhelm and O. S. Wolfbeis, Acc. Chem. Res., 2014, 47, 3481–3493 CrossRef CAS PubMed.
- S. Wilhelm, M. Kaiser, C. Würth, J. Heiland, C. Carrillo-Carrion, V. Muhr, O. S. Wolfbeis, W. J. Parak, U. Resch-Genger and T. Hirsch, Nanoscale, 2015, 7, 1403–1410 RSC.
- F. Zhang, G. B. Braun, Y. Shi, Y. Zhang, X. Sun, N. O. Reich, D. Zhao and G. Stucky, J. Am. Chem. Soc., 2010, 132, 2850–2851 CrossRef CAS PubMed.
- Y. Hu, Y. Sun, Y. Li, S. K. Sun, J. Huo and X. J. Zhao, RSC Adv., 2014, 4, 43653–43660 RSC.
- Y. Cen, Y. Wu, X. Kong, S. Wu, R. Yu and X. Chu, Anal. Chem., 2014, 86, 7119–7127 CrossRef CAS PubMed.
- D. E. Achatz, R. Ali and O. S. Wolfbeis, Top. Curr. Chem., 2011, 300, 29–50 CrossRef CAS.
- M. V. DaCosta, S. Doughan, Y. Han and U. J. Krull, Anal. Chim. Acta, 2014, 832, 1–33 CrossRef CAS PubMed.
- L. A. Dykman and N. G. Khlebtsov, Chem. Rev., 2014, 114, 1258–1288 CrossRef CAS PubMed.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- H. He, C. Xie and J. Ren, Anal. Chem., 2008, 80, 5951–5957 CrossRef CAS PubMed.
- A. Tcherniak, S. Dominguez-Medina, W. Chang, P. Swanglap, L. S. Slaughter, C. F. Landes and S. Link, J. Phys. Chem. C, 2011, 115, 15938–15949 CAS.
- N. J. Durr, T. Larson, D. K. Smith, B. A. Korgel, K. Sokolov and A. Ben-Yakar, Nano Lett., 2007, 7, 941–945 CrossRef CAS PubMed.
- J. Jiang, H. Gu, H. Shao, E. Devlin, G. C. Papaefthymiou and J. Y. Ying, Adv. Mater., 2008, 20, 4403–4407 CrossRef CAS.
- E. Dulkeith, M. Ringler, T. A. Klar, J. Feldmann, A. Muñoz Javier and W. J. Parak, Nano Lett., 2005, 5, 585–589 CrossRef CAS PubMed.
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- L. Shang, S. Dong and G. U. Nienhaus, Nano Today, 2011, 6, 401–418 CrossRef CAS PubMed.
- J. Zheng, C. Zhou, M. Yu and J. Liu, Nanoscale, 2012, 4, 4073–4083 RSC.
- P. L. Xavier, K. Chaudhari, A. Baksi and T. Pradeep, Nano Rev., 2012, 3, 14767, DOI:10.3402/nano.v3i0.14767.
- R. B. P. Elmes, K. N. Orange, S. M. Cloonan, D. C. Williams and T. Gunnlaugsson, J. Am. Chem. Soc., 2011, 133, 15862–15865 CrossRef CAS PubMed.
- (a) L. Díez and R. H. A. Ras, Nanoscale, 2011, 3, 1963 RSC; (b) I. Díez, M. I. Kanyuk, A. P. Demchenko, A. Walther, H. Jiang, O. Ikkala and R. H. A. Ras, Nanoscale, 2012, 4, 4434–4437 RSC.
- S. N. Ding, C. M. Li, B. H. Gao, O. Kargbo, N. Wan, X. Chen and C. Zhou, Microchim. Acta, 2014, 181, 1957–1963 CrossRef CAS.
- O. Mongin, T. R. Krishna, M. H. V. Werts, A.-M. Caminade, J.-P. Majoral and M. Blanchard-Desce, Chem. Commun., 2006, 915–917 RSC.
- L. Albertazzi, B. Storti, L. Marchetti and F. Beltram, J. Am. Chem. Soc., 2010, 132, 18158–18167 CrossRef CAS PubMed.
- M. Gonçalves, D. Maciel, D. Capêlo, S. Xiao, W. Sun, X. Shi, J. Rodrigues, H. Tomas and Y. Li, Biomacromolecules, 2014, 15, 492–499 CrossRef PubMed.
- A. Devor, S. Sakadžić, M. A. Yaseen, E. Roussakis, P. Tian, H. Slovin, I. Vanzetta, I. Teng, P. A. Saisan, L. E. Sinks, A. M. Dale, S. A. Vinogradov and D. A. Boas, Neuromethods, 2014, 85, 225–253 Search PubMed.
- W. Yang, C. Pan, X. Liu and J. Wang, Biomacromolecules, 2011, 12, 1523–1531 CrossRef CAS PubMed.
- K.-Y. Pu, J. Shi, L. Cai, K. Li and B. Liu, Biomacromolecules, 2011, 12, 2966–2974 CrossRef CAS PubMed.
- R. C. Somers, R. M. Lanning, P. T. Snee, A. B. Greytak, R. K. Jain, M. G. Bawendi and D. G. Nocera, Chem. Sci., 2012, 3, 2980–2985 RSC.
- Y. Kim, S. H. Kim, M. Tanyeri, J. A. Katzenellenbogen and C. M. Schroeder, Biophys. J., 2013, 104, 1566–1575 CrossRef CAS PubMed.
- J. Tian, L. Ding, H. Ju, Y. Yang, X. Li, Z. Shen, Z. Zhu, J. Yu and C. J. Yang, Angew. Chem., Int. Ed., 2014, 53, 9544–9548 CrossRef CAS PubMed.
- J. F. Lovell, C. S. Jin, E. Huynh, H. Jin, C. Kim, J. L. Rubinstein, W. C. W. Chan, W. Cao, L. V. Wang and G. Zheng, Nat. Mater., 2011, 10, 324–332 CrossRef CAS PubMed.
- U. Rocha, K. U. Kumar, C. Jacinto, I. Villa, F. Sanz-Rodríguez, M. de la Cruz, A. Juarranz, E. Carrasco, F. C. J. M. van Veggel, E. Bovero, J. García Solé and D. Jaque, Small, 2014, 10, 1141–1154 CrossRef CAS PubMed.
- J. Hui, X. Zhang, Z. Zhang, S. Wang, L. Tao, Y. Wei and X. Wang, Nanoscale, 2012, 4, 6967–6970 RSC.
- X. Zhang, J. Hui, B. Yang, Y. Yang, D. Fan, M. Liu, L. Tao and Y. Wei, Polym. Chem., 2013, 4, 4120–4125 RSC.
- X. Li, H. Zeng, L. Teng and H. Chen, Mater. Lett., 2014, 125, 78–81 CrossRef CAS PubMed.
- F. Wang, X. Xue and X. Liu, Angew. Chem., Int. Ed., 2008, 47, 906–909 CrossRef CAS PubMed.
- B. Wang, J. Hai, Q. Wang, T. Li and Z. Yang, Angew. Chem., Int. Ed., 2011, 50, 3063–3066 CrossRef CAS PubMed.
- S. Sandoval, J. Yang, J. G. Alfaro, A. Liberman, M. Makale, C. E. Chiang, I. K. Schuller, A. C. Kummel and W. C. Trogler, Chem. Mater., 2012, 24, 4222–4230 CrossRef CAS PubMed.
- Y. Wang, L. Chen and P. Liu, Chem. – Eur. J., 2012, 18, 5935–5943 CrossRef CAS PubMed.
- Y. Liu, S. Zhou, D. Tu, Z. Chen, M. Huang, H. Zhu, E. Ma and X. Chen, J. Am. Chem. Soc., 2012, 134, 15083–15090 CrossRef CAS PubMed.
- C. Wang, Y. Wang, L. Xu, D. Zhang, M. Liu, X. Li, H. Sun, Q. Lin and B. Yang, Small, 2012, 8, 3137–3142 CrossRef CAS PubMed.
- N. Dong, M. Pedroni, F. Piccinelli, G. Conti, A. Sbarbati, J. Ramírez-Hernández, L. Martínez Maestro, M. C. Iglesias-de la Cruz, F. Sanz-Rodriguez, A. Juarranz, F. Chen, F. Vetrone, J. A. Capobianco, J. G. Solé, M. Bettinelli, D. Jaque and A. Speghini, ACS Nano, 2011, 5, 8665–8671 CrossRef CAS PubMed.
- D. Zherebetskyy, M. Scheele, Y. Zhang, N. Bronstein, C. Thompson, D. Britt, M. Salmeron, P. Alivisatos and L.-W. Wang, Science, 2014, 344, 1380–1382 CrossRef CAS PubMed.
- J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS PubMed.
- P. Das and N. R. Jana, ACS Appl. Mater. Interfaces, 2014, 6, 4301–4305 CAS.
- J. H. Guo, S. J. Xiong, X. L. Wu, J. C. Shen and P. K. Chu, Biomaterials, 2013, 34, 9183–9189 CrossRef CAS PubMed.
- J. Gravier, F. P. Navarro, T. Delmas, F. Mittler, A.-C. Couffin, F. Vinet and I. Texier, J. Biomed. Opt., 2011, 16, 096013 CrossRef PubMed.
- K. J. Zhou, Y. G. Wang, X. N. Huang, K. Luby-Phelps, B. D. Sumer and J. M. Gao, Angew. Chem., Int. Ed., 2011, 50, 6109–6114 CrossRef CAS PubMed.
- K. J. Zhou, H. M. Liu, S. R. Zhang, X. N. Huang, Y. G. Wang, G. Huang, B. D. Sumer and J. M. Gao, J. Am. Chem. Soc., 2012, 134, 7803–7811 CrossRef CAS PubMed.
- X. Wang, J. A. Stolwijk, M. Sperber, R. J. Meier, J. Wegener and O. S. Wolfbeis, Methods Appl. Fluoresc., 2013, 1, 035002 CrossRef.
- R. R. Costa and J. F. Mano, Chem. Soc. Rev., 2014, 43, 3453–3479 RSC.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, New York, 2013 Search PubMed.
- N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2011, 44, 925–935 CrossRef CAS PubMed.
- C. R. Maldonado, L. Salassa, N. Gomez-Blanco and J. C. Mareque-Rivas, Coord. Chem. Rev., 2013, 257, 2668–2688 CrossRef CAS PubMed.
- L. Li, R. Zhang, L. Yin, K. Zheng, W. Qin, P. R. Selvin and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 6121–6125 CrossRef CAS PubMed.
- C. Yao, P. Wang, L. Zhou, R. Wang, X. Li, D. Zhao and F. Zhang, Anal. Chem., 2014, 86, 9749–9757 CrossRef CAS PubMed.
- K. Paulkumar, R. Arunachalam and G. Annadurai, Int. J. Nanotechnol., 2011, 8, 653–663 CrossRef CAS.
- L. Sun, T. Liu, Y. Qiu, J. Liu, F. Li, L. Shi and O. S. Wolfbeis, Microchim. Acta, 2014, 181, 775–782 CrossRef CAS.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- J. Jin, Y. J. Gu, C. W. Y. Man, J. Cheng, Z. Xu, Y. Zhang, H. Wang, V. H. Lee, S. H. Cheng and W. Wong, ACS Nano, 2011, 5, 7838–7847 CrossRef CAS PubMed.
- L. Xiong, T. Yang, Y. Yang, C. Xu and F. Li, Biomaterials, 2010, 27, 7078–7085 CrossRef PubMed.
- Y. Dai, P. Ma, Z. Cheng, X. Kang, X. Zhang, Z. Hou, C. Li, D. Yang, X. Zhai and J. Lin, ACS Nano, 2012, 6, 3327–3338 CrossRef CAS PubMed.
- R. G. Aswathy, B. Sivakumar, D. Brahatheeshwaran, T. Ukai, Y. Tomofumi, M. Yasuhiko, T. Maekawa and S. D. Kumar, Mater. Express, 2011, 1, 291–298 CrossRef CAS PubMed.
- X. H. Wang, H. S. Peng, Z. Chang, L. L. Hou, F. T. You, F. Teng, H. W. Song and B. Dong, Microchim. Acta, 2012, 178, 147–152 CrossRef CAS.
- M. Liras, M. González-Béjar, E. Peinado, L. Francés-Soriano, J. Pérez-Prieto, I. Quijada-Garrido and O. García, Chem. Mater., 2014, 26, 4014–4022 CrossRef CAS.
- N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2011, 44, 925–935 CrossRef CAS PubMed.
- J. Zhou, Z. Liu and F. Li, Chem. Soc. Rev., 2012, 41, 1323–1349 RSC.
- A. Sedlmeier and H. H. Gorris, Chem. Soc. Rev., 2015 10.1039/C4CS00186A.
- A. Davies, D. J. Lewis, S. P. Watson, S. G. Thomas and Z. Pikramenou, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1862–1867 CrossRef CAS PubMed.
- (a) T. Murase, T. Yoshihara and S. Tobita, Chem. Lett., 2012, 41, 262–263 CrossRef CAS; (b) E. T. Chouchani, C. Methner, S. M. Nadtochiy, A. Logan, V. R. Pell, S. Ding, A. M. James, H. M. Cochemé, J. Reinhold, K. S. Lilley, L. Partridge, I. M. Fearnley, A. J. Robinson, R. C. Hartley, R. A. J. Smith, T. Krieg, P. S. Brookes and M. P. Murphy, Nat. Med., 2013, 19, 753–759 CrossRef CAS PubMed.
- X. Wang, H. Peng, L. Yang, F. You, F. Teng, L. Hou and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2014, 53, 12471–12475 CAS.
- J. Cho, K. Kushiro, Y. Teramura and M. Takai, Biomacromolecules, 2014, 15, 2012–2018 CrossRef CAS PubMed.
- X. Zhang, X. Zhang, B. Yang, Y. Zhang, M. Liu, Y. Chen and Y. Wei, Colloids Surf., B, 2014, 113, 435–441 CrossRef CAS PubMed.
- Y. Choi, K. Kim, S. Hong, H. Kim, Y. J. Kwon and R. Song, Bioconjugate Chem., 2011, 22, 1576–1586 CrossRef CAS PubMed.
- V. Biju, Chem. Soc. Rev., 2014, 43, 744–764 RSC.
- V. Biju, T. Itoh and M. Ishikawa, Chem. Soc. Rev., 2010, 39, 3031–3056 RSC.
- E. Muro, T. Pons, N. Lequeux, A. Fragola, N. Sanson, Z. Lenkei and B. Dubertret, J. Am. Chem. Soc., 2010, 132, 4556–4557 CrossRef CAS PubMed.
- H. Xing, N. Y. Wong, Y. Xiang and Y. Lu, Curr. Opin. Chem. Biol., 2012, 16, 429–435 CrossRef CAS PubMed.
- X. Wang, A. R. Morales, T. Urakami, L. Zhang, M. V. Bondar, M. Komatsu and K. D. Belfield, Bioconjugate Chem., 2011, 22, 1438–1450 CrossRef CAS PubMed.
- D. S. Kwag, K. Park, K. T. Oh and E. S. Lee, Chem. Commun., 2013, 49, 282–284 RSC.
- X. Wang, C. X. Yang, J. T. Chen and X. P. Yan, Anal. Chem., 2014, 86, 3263–3267 CrossRef CAS PubMed.
- G. M. Thurber, K. S. Yang, T. Reiner, R. H. Kohler, P. Sorger, T. Mitchison and R. Weissleder, Nat. Commun., 2013, 4, 1504 CrossRef PubMed.
- H. Xing, N. Y. Wong, Y. Xiang and Y. Lu, Curr. Opin. Chem. Biol., 2012, 16, 429–435 CrossRef CAS PubMed.
- A. E. Prigodich, P. S. Randeria, W. E. Briley, N. J. Kim, W. L. Daniel, D. A. Giljohann and C. A. Mirkin, Anal. Chem., 2012, 84, 2062–2066 CrossRef CAS PubMed.
- J. B. Haun, N. K. Deveraj, S. A. Hilderbrand, H. Lee and R. Weissleder, Nat. Nanotechnol., 2010, 5, 660–665 CrossRef CAS PubMed.
- X. Wu, G. Chen, J. Shen, Z. Li, Y. Zhang and G. Han, Bioconjugate Chem., 2015 DOI:10.1021/bc5003967.
- M. J. Ruedas-Rama, J. D. Walters, A. Orte and E. A. H. Hall, Anal. Chim. Acta, 2012, 751, 1–23 CrossRef CAS PubMed.
- S. Schreml, R. J. Meier, J. Cattani, D. Flittner, S. Gehmert, O. S. Wolfbeis, M. Landthaler and P. Babilas, Exp. Dermatol., 2012, 21, 942–970 CrossRef PubMed.
- R. Arppe, T. Näreoja, S. Nylund, L. Mattsson, S. Koho, J. M. Rosenholm, T. Soukka and M. Schaeferling, Nanoscale, 2014, 6, 6837–6843 RSC.
- K. Zhou, H. Liu, S. Zhang, X. Huang, Y. Wang, G. Huang, B. D. Sumer and J. M. Gao, J. Am. Chem. Soc., 2012, 134, 7803–7811 CrossRef CAS PubMed.
- X. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC.
- X. D. Wang, H. H. Gorris, J. A. Stolwijk, R. J. Meier, D. B. M. Groegel, J. Wegener and O. S. Wolfbeis, Chem. Sci., 2011, 2, 901–906 RSC.
- H. Peng, M. I. J. Stich, J. Yu, L. Sun, L. H. Fischer and O. S. Wolfbeis, Adv. Mater., 2010, 22, 716–719 CrossRef CAS PubMed.
- L. M. Maestro, E. M. Rodriguez, F. S. Rodriguez, M. C. Iglesias-de la Cruz, A. Juarranz, R. Naccache, F. Vetrone, D. Jaque, J. A. Capobianco and J. G. Solé, Nano Lett., 2010, 10, 5109–5115 CrossRef CAS PubMed.
- P. Yu, X. Wen, Y. R. Toh and J. Tang, J. Phys. Chem. C, 2012, 116, 25552–25557 CAS.
- A. Sedlmeier, D. E. Achatz, L. H. Fischer, H. H. Gorris and O. S. Wolfbeis, Nanoscale, 2012, 4, 7090–7096 RSC.
- K. Okabe, N. Inada, C. Gota, Y. Harada, T. Funatsu and S. Uchiyama, Nat. Commun., 2012, 3, 705 CrossRef PubMed.
- H. Peng, S. Huang and O. S. Wolfbeis, J. Nanopart. Res., 2010, 12, 2729–2733 CrossRef CAS.
- L. Yang, H. S. Peng, H. Ding, F. T. You, L. L. Hou and F. Teng, Microchim. Acta, 2014, 181, 743–749 CrossRef CAS.
- F. Vetrone, R. Naccache, A. Zamarrón, A. J. de la Fuente, F. Sanz-Rodríguez, L. M. Maestro, E. M. Rodriguez, D. Jaque, J. G. Solé and J. A. Capobianco, ACS Nano, 2010, 4, 3254–3258 CrossRef CAS PubMed.
- N. C. Bigall, W. J. Parak and D. Dorfs, Nano Today, 2012, 7, 532–563 CrossRef PubMed.
- J. H. Choi, F. T. Nguyen, P. W. Barone, D. A. Heller, A. E. Moll, D. Patel, S. A. Boppart and M. S. Strano, Nano Lett., 2007, 7, 861–867 CrossRef CAS PubMed.
- Z. Wang, X. Yue, Y. Wang, C. Qian, P. Huang, M. Litzak, G. Niu, F. Wang, P. Rong, D. O. Kiesewetter, Y. Ma and X. Chen, Adv. Healthcare Mater., 2014, 3, 1326–1333 CrossRef CAS PubMed.
- S. Zeng, H. Wang, W. Lu, Z. Yi, L. Rao, H. Liu and J. Hao, Biomaterials, 2014, 35, 2934–2941 CrossRef CAS PubMed.
- F. C. J. M. van Veggel, C. Dong, N. J. J. Johnson and J. Pichaandi, Nanoscale, 2012, 4, 7309–7321 RSC.
- K. Li, D. Ding, D. Huo, K. Y. Pu, N. N. P. Thao, Y. Hu, Z. Li and B. Liu, Adv. Funct. Mater., 2012, 22, 3107–3115 CrossRef CAS.
- M. Challenor, P. Gong, D. Lorenser, M. J. House, R. C. Woodward, T. S. Pierre, M. Fitzgerald, S. A. Dunlop, D. D. Sampson and K. S. Iyer, Dalton Trans., 2014, 43, 16780–16787 RSC.
- L. Sun, X. Ge, J. Liu, Y. Qiu, Z. Wei, B. Tian and L. Shi, Nanoscale, 2014, 6, 13242–13252 RSC.
- Y. Ding, X. Zhang, H. Zhu and J.-J. Zhu, J. Mater. Chem. C, 2014, 2, 946 RSC.
- A. Jaworska, T. Wojcik, K. Malek, U. Kwolek, M. Kepczynski, A. A. Ansary, S. Chlopicki and M. Baranska, Microchim. Acta, 2014, 182, 119–127 CrossRef PubMed.
- W. Fan, B. Shen, W. Bu, F. Chen and J. Shi, J. Am. Chem. Soc., 2013, 135, 6494–6503 CrossRef CAS PubMed.
- P. Ma, H. Xiao, X. Li and J. Lin, Adv. Mater., 2013, 25, 4898–4905 CrossRef CAS PubMed.
- Y. Tao, Z. Li, E. Ju, J. Ren and X. Qu, Nanoscale, 2013, 5, 6154 RSC.
- Z. Li, Y. Tao, S. Huang, N. Gao, J. Ren and X. Qu, Chem. Commun., 2013, 49, 7129 RSC.
- A. Anitha, V. G. Deepagan, V. V. Divya Rani, D. Menon, S. V. Nair and R. Jayakumar, Carbohydr. Polym., 2011, 1158–1164 CrossRef CAS PubMed.
- C. Liu, P. Zhang, X. Zhai, F. Tian, W. Li, J. Yang, Y. Liu, H. Wang, W. Wang and W. Liu, Biomaterials, 2012, 33, 3604–3613 CrossRef CAS PubMed.
- D. Yang, Z. Hou, Z. Cheng, C. Li and J. Lin, Chem. Soc. Rev., 2015 10.1039/C4CS00155A.
- W. Fan, B. Shen, W. Bu, F. Chen, Q. He, K. Zhao, S. Zhang, L. Zhou, W. Peng, Q. Xiao, D. Ni, J. Liu and J. Shi, Biomaterials, 2014, 32, 8992–9002 CrossRef PubMed.
- D. Yang, Y. Dai, J. Liu, Y. Zhou, Y. Chen, C. Li, P. Ma and J. Lin, Biomaterials, 2014, 35, 2011–2023 CrossRef CAS PubMed.
- G. Shan, R. Weissleder and S. A. Hilderbrand, Theranostics, 2013, 3, 267–274 CrossRef CAS PubMed.
- Y. I. Park, H. M. Kim, J. H. Kim and T. Hyeon, Adv. Mater., 2012, 24, 5755–5761 CrossRef CAS PubMed.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, Nat. Med., 2012, 18, 1580–1585 CrossRef CAS PubMed.
- P. Liu, C. Yue, B. Shi, G. Gao, M. Li, B. Wang, Y. Ma and L. Cai, Chem. Commun., 2013, 49, 6143–6145 RSC.
- X. F. Qiao, J. C. Zhou, J. W. Xiao, Y. F. Wang, L. D. Sun and C. H. Yan, Nanoscale, 2012, 4, 4611–4623 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cs00392f |
| ‡ The common term fluorescence is used here for the sake of simplicity even if the term phosphorescence may apply. |
| This journal is © The Royal Society of Chemistry 2015 |