
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescent probes for the bioimaging of small anionic species in vitro and in vivo
Trent D.
Ashton
a,
Katrina A.
Jolliffe
b and
Frederick M.
Pfeffer
*a
aCentre for Chemistry and Biotechnology, School of Life and Environmental Sciences, Deakin University, Pigdons Rd, Waurn Ponds, Victoria, Australia. E-mail: fred.pfeffer@deakin.edu.au
bSchool of Chemistry, School of Chemistry (F11), The University of Sydney, Sydney, New South Wales, Australia
First published on 12th February 2015
Abstract
The ability to spatiotemporally identify the formation of specific anionic species, or track changes in their concentration inside living systems, is of critical importance in deciphering their exact biological roles and effects. The development of probes (also called bioimaging agents and intracellular sensors) to achieve this goal has become a rapidly growing branch of supramolecular chemistry. In this critical review the challenges specific to the task are identified and for a select range of small anions of environmental and biological relevance (fluoride, chloride, iodide, cyanide, pyrophosphate, bicarbonate, hydrosulphide, peroxynitrite, hypochlorite and hypobromite) a comprehensive overview of the currently available in vitro and in vivo probes is provided.
 Trent D. Ashton | Trent Ashton received his PhD in organic chemistry from Monash University's Victorian College of Pharmacy campus (now Monash Institute of Pharmaceutical Sciences) in 2008. After a year working for Prof. Michael Pollastri at Boston University he returned to Monash to work for Peter Scammells. He originally joined the Pfeffer lab at Deakin University in 2010 to work on small molecule agents for the treatment of type II diabetes in conjunction with Verva Pharmaceuticals. In addition to supramolecular anion recognition chemistry his current research interests include the development of class selective HDAC inhibitors to target metabolic disorders. |
 Katrina A. Jolliffe | Katrina (Kate) Jolliffe received her BSc (Hons 1) in 1993 and PhD in 1997 from the University of New South Wales. She then held positions at Twente University, The Netherlands; the University of Nottingham, UK and the Australian National University before taking up an ARC QEII research fellowship at The University of Sydney in 2002. In 2007 she became a Senior lecturer at the same institution and was promoted to Associate Professor in 2008 and to full Professor in 2009. She is currently Head of the School of Chemistry at The University of Sydney. Kate's research interests encompass elements of synthetic organic chemistry, supramolecular chemistry and peptide chemistry. |
 Frederick M. Pfeffer | Fred Pfeffer received his BSc (Hons 1) in 1996 and PhD in 2001 from Deakin University. He then worked for three years at Trinity College Dublin, first as a lecturer then as postdoctoral fellow in the group of Prof Thorfinnur Gunnlaugsson. In 2004 he returned to Australia as lecturer and in 2010 was appointed Senior lecturer. Research interests include many aspects of supramolecular chemistry including anion recognition and sensing. A research focus is the synthesis and use of fused [n]polynorbornane scaffolds as preorganising elements for further applications including host |
1. Introduction
1.1 Overview
The study of anion recognition is now a relatively mature science in line with the closely related field of cation recognition.1–5 Over the last 10–15 years sustained effort from the supramolecular chemistry community has refined the fundamental principles relating to how a host interacts with a negatively charged guest.1–17Similarly anion sensing has matured and an array of effective molecular detectors, operating by means of well understood principles, are now available.11,18–32 An excellent recent tutorial review by Gale (see also other articles in this special issue) neatly highlights the strategies that are now widely employed in the detection and/or quantification of anionic species.33 While the field has matured, challenges still exist for the detection of anions in water; the heavily hydrated nature of these species in aqueous environments makes strong binding difficult and also hinders their reactivity.7,12,26,34
As the study of anion recognition and sensing has advanced supramolecular chemists have applied their fundamental knowledge to the detection of anions of biological significance.14,21,35–46 Indeed the rise of anion recognition as a field of study was in no small part due to the fact that the majority of intracellular operations involve anionic species.47 A natural extension of such efforts is the detection or sensing of biologically relevant anions in a biologically relevant setting such as inside living cells or in living organisms.30,35,43,45,48–60 Thus the field of anion imaging has emerged and supramolecular chemists now find themselves planning and executing the synthesis of reporters to selectively detect and indicate the presence of anions inside living cells and organisms. Probes capable of achieving this feat are amongst the most powerful resources available for elucidating the exact biological role of the target anion. While the number of probes capable of efficiently communicating an anion recognition event from such a venue is growing, it is still small when compared to the large number of intracellular sensors/probes for cationic species61 (see also other articles in this special issue). As such the field provides fertile ground for both emerging and established researchers alike.
1.2 Challenges
The ideal anion sensor functioning in vitro or in vivo must satisfy a demanding set of criteria (outlined in brief in Table 1)62–64 and it is clear from this list that an imaging agent must ‘do more’ than a sensor. Key challenges include (i) selecting a suitable fluorophore, (ii) choosing an effective switching mechanism and (iii) catering for the biological environment in which the probe must function (see additional discussion for these three points below). Few, if any, of the currently available probes satisfy all of these criteria and given that sensors that are truly selective for specific anions in water have only emerged in the last 10–15 years it is no surprise that the development of selective anion sensors for bioimaging applications is currently at the forefront of applied supramolecular chemistry.| For recognition/sensing in water | For recognition/sensing in vitro and in vivo |
|---|---|
| Selective for the target in water | Selective for target in cells/small organisms |
| Strong binding/signalling and low detection limit | Sensitive at relevant biological concentrations (e.g. Cl−vs. ONOO−) |
| Water soluble | Water soluble yet amphiphilic for cell permeability. |
| Localise in relevant compartment | |
| Non-toxic | |
| “Switch on” or ratiometric | “Switch on” or ratiometric |
| Large extinction coefficient, quantum yield and Stokes shift. | Large extinction coefficient, quantum yield and Stokes shift. |
| Red or NIR emissive | |
| Photostable and metabolically stable |
While well-known fluorophores (such as rhodamine, fluorescein, BODIPY and cyanine)48,64,74–77 are common in anion imaging studies, several unconventional fluorophores (e.g. Si, Se, Ge and Te rhodamines78,79 and squaraine-rotaxanes54) have successfully been used in recent years. An increasing number of effective probes also employ lanthanide based luminescence for signal transductance.45,56,80–83
A probe can also be classified according to the electronic event by which fluorescence is “switched” or modulated (for example ICT/PET modulation, FRET, heavy ion effect, and excimer formation). Many sensors—known as chemodosimeters—have been designed such that a chemical reaction controls this modulation30,33,35,49,86 and it is logical that these strategies have been adopted by those pursuing the goal of in vitro and in vivo imaging. While the chemodosimeter approach is by far the most popular (and is excellent for the selective detection of a specific species) the chemodosimeter is, in most instances, irreversibly transformed to the signalling moiety and as such true spatiotemporal information cannot be gleaned. Continued effort from the research community is required to achieve the goal of tracking rather than trapping the anionic species of interest. Another very popular approach to modulating fluorescence is the displacement approach.27,46,87 The luminophore is quenched by a species that is non-covalently attached and the luminophore–quencher combination is chosen such that the target anion interacts with the quencher more strongly than it does with the luminophore. Hence the quencher is displaced, the luminophore is released and fluorescence is “switched on”.
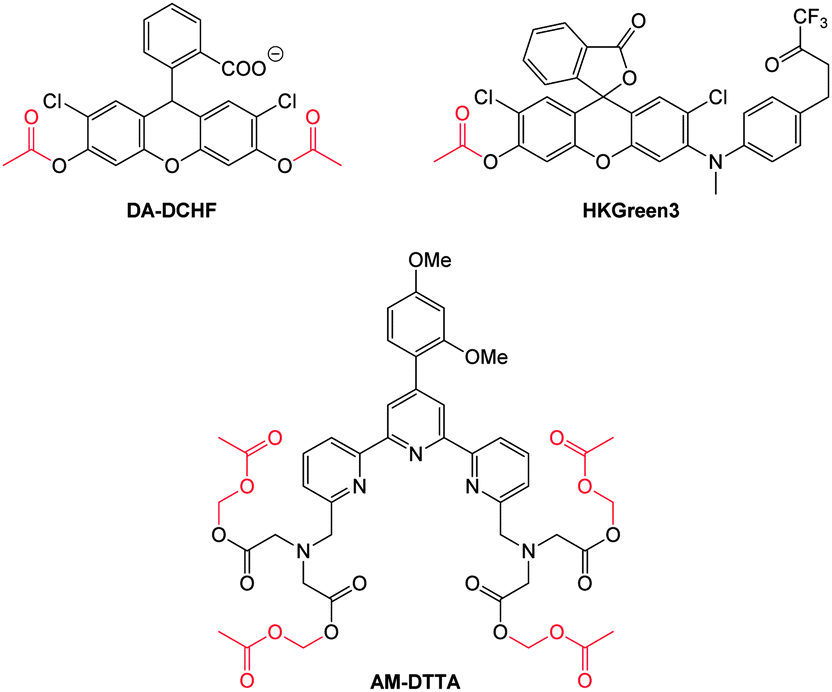 | ||
| Fig. 1 Examples of peroxynitrite probes DA-DCHF, HKGreen3 and AM-DTTA in which intracellular uptake and subsequent trapping was performed using a lipophilic ester (highlighted in red) that was cleaved in vivo by intracellular esterases. | ||
Ideally once the probe is inside the living entity it should localise in the most relevant sub-cellular compartment. Guidelines to predict the likely compartmentalisation of new probes are not unequivocally established63,95 and colocalisation studies with well-established dyes are generally required. Nevertheless some general trends exist: (i) cationic probes gravitate to the mitochondria96,97 as the mitochondrial membrane is negatively polarised and (ii) weakly basic probes accumulate within the more acidic lysosomes.98 These general guidelines have also been adopted in the field of anion bioimaging, for example, the recently described probes for hypochlorite Rh-TPP and Rh-Py (Fig. 2)99 employ a triphenylphosphonium and pyridinium appendage respectively for mitochondrial localisation. The intracellular sensor for hydrosulfide Lyso-NHS used a morpholine substituent for lysosomal localisation.100
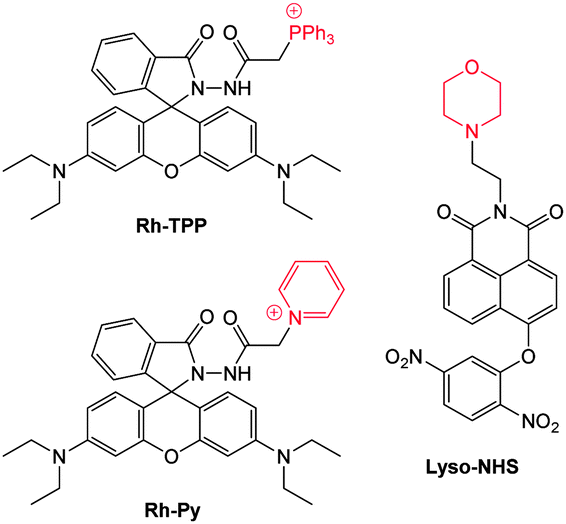 | ||
| Fig. 2 Examples of probes that localise in the mitochondria (Rh-TPP, Rh-Py) and lysosome (Lyso-NHS). | ||
1.3 Structure of this review
In conjunction with a comprehensive listing of recent examples (the majority of examples are from the last 5 years) the broad concept of this review is to provide both a “why” and “how to” target the specific anion of interest. For each of the anions covered herein a justification of the cellular relevance is first provided—even anions of obvious environmental importance (such as cyanide and fluoride) have considerable relevance and interest for intracellular studies (see Section 2). Also covered are a number of anions that are of relevance primarily at a cellular level (see Section 3), for example bicarbonate plays a critical role in living systems as a measure of CO2 uptake/respiration (hypercapnia/hypercapnia = CO2/HCO3− poisoning respiratory acidosis). Similarly, reactive oxygen and nitrogen species (such as hypochlorite ClO− and peroxynitrite ONOO−) have critical in vivo roles and elevated levels of these species are associated with many disease states (see Section 4). Where possible, examples have been grouped by the means (mechanism) by which sensing is achieved and also whether the probes are: intensity modulated (“switch off” or “switch on”) or ratiometric (wavelength modulation). The terms fluorescent probe, anion imaging agent and intracellular sensor are all used interchangeably.2. Anions of environmental and biological relevance
There now exists a number of excellent sensors for anions such as fluoride and cyanide;35,37,39,101 widely recognised as anions of environmental concern. While not commonly appreciated, these anions also have significant relevance in a biological setting and a number of intracellular probes have been developed for their detection. Chloride has a more passive, nonetheless important, role in the environment and, like iodide, plays an important physiological role.102 Anions covered in this section are fluoride, chloride, iodide and cyanide.2.1 Fluoride
Fluoride is a very well-known anion due to its use in drinking water and toothpaste to prevent dental caries and osteoporosis.103 Nevertheless excess fluoride is responsible for a number of deleterious conditions including dental and skeletal fluorosis and is now linked to cancer and neurotoxicity.104–106 Probes capable of selectively indicating fluoride in vitro and in vivo may assist in clarifying the exact biological roles of this anion.Given its “Janus” behaviour the recognition and sensing of fluoride has been a focus of supramolecular chemists.37,39,101,107,108 Two approaches that have been widely used in the design of both sensors and bioimaging agents are (i) deprotonation (Section 2.1.1) and (ii) desilylation (Section 2.1.2). Deprotonation, mediated by the strongly basic fluoride anion, leading to enhanced ICT of a luminophore, was one of the first means by which this anion was detected,18,109–112 and while the approach has been used for imaging, this design is prone to interference from other basic anions (such as acetates). By far the most common approach employs the fluoride mediated desilylation reaction of chemodosimeters that have been designed with a silyl ether.
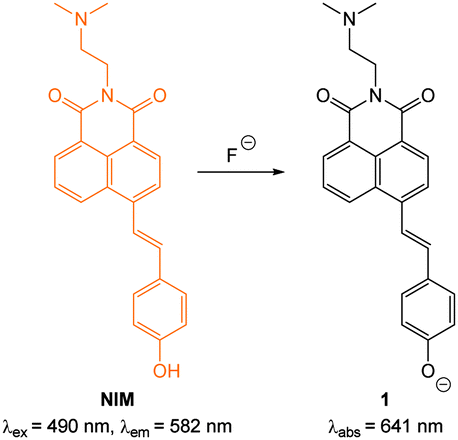 | ||
| Fig. 3 Structure, and deprotonation, of NIM with fluoride. | ||
The ratiometric hydroxynaphthalene probe 2 (Fig. 4) was reported by Liu and Ke in 2014.114 A PEG cyanoacrylate was included to enhance ICT and also balance solubility. The probe was selective to fluoride (no significant fluorescent changes were elicited by AcO−) and the emission intensity ratio I490/I450 nm could be used to quantitate fluoride up to 10 equivalents with a limit of detection (LOD) of 8.5 μM. The probe was cell permeable, non-toxic to prostate cancer (PC3) and epithelial cervical cancer (HeLa) cells and located in the cytoplasm of these cells (confirmed using the red nuclear stain propidium iodide—PI). In PC3 cells a clear change in emission colour was observed when cells pre-treated with 2 were exposed to fluoride.
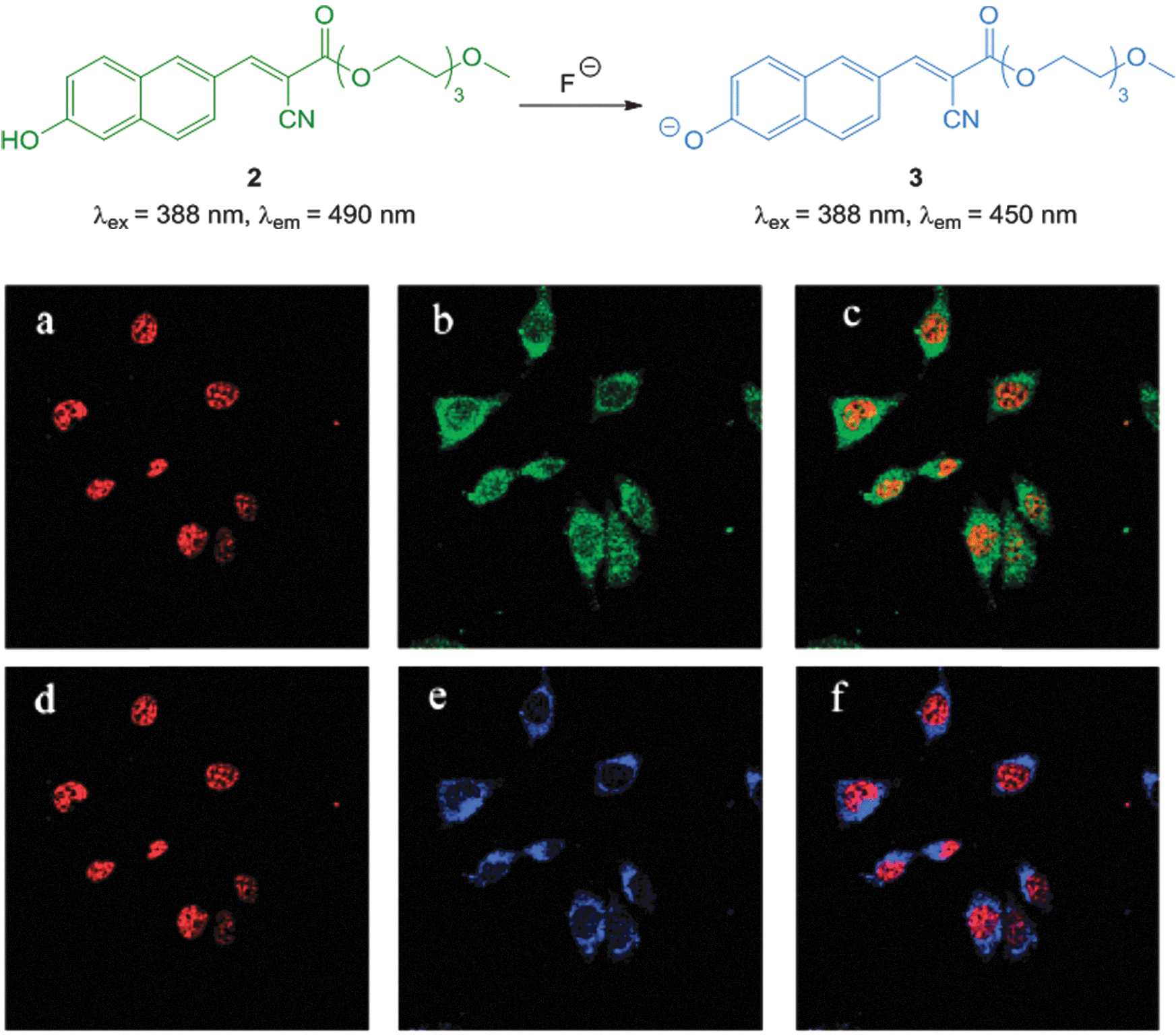 | ||
| Fig. 4 Top: structure of hydroxynaphthalene 2. Bottom: PC3 cells incubated with 2 (10 μM) and nucleus stain propidium iodide (a) red channel image; (b) green channel image of (a); (c) overlay of (a) and (b). PC3 cells incubated with 2, NaF and propidium iodide (d) red channel image; (e) blue channel image of (d); (f) overlay of (d) and (e). Image reproduced with permission.114 | ||
A recent report by Mahapatra (2014) outlined the ratiometric BODIPY azaindole 4 (Fig. 5) which was synthesised in three steps.115 In solution studies (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 CH3CN:H2O) the strong emission at 512 nm (λex = 350 nm) decreased upon addition of fluoride (and to a similar extent acetate) as weak emission at 425 nm increased and the ratio F425/F512 was used to determine F− concentration (<200 equivalents). The N–H of indole has been used previously for the recognition and sensing of fluoride anions116,117 and for probe 4 deprotonation significantly enhanced ICT and a clear change in fluorescence emission was recorded in murine macrophages (RAW264.7) upon addition of fluoride.
:3 CH3CN:H2O) the strong emission at 512 nm (λex = 350 nm) decreased upon addition of fluoride (and to a similar extent acetate) as weak emission at 425 nm increased and the ratio F425/F512 was used to determine F− concentration (<200 equivalents). The N–H of indole has been used previously for the recognition and sensing of fluoride anions116,117 and for probe 4 deprotonation significantly enhanced ICT and a clear change in fluorescence emission was recorded in murine macrophages (RAW264.7) upon addition of fluoride.
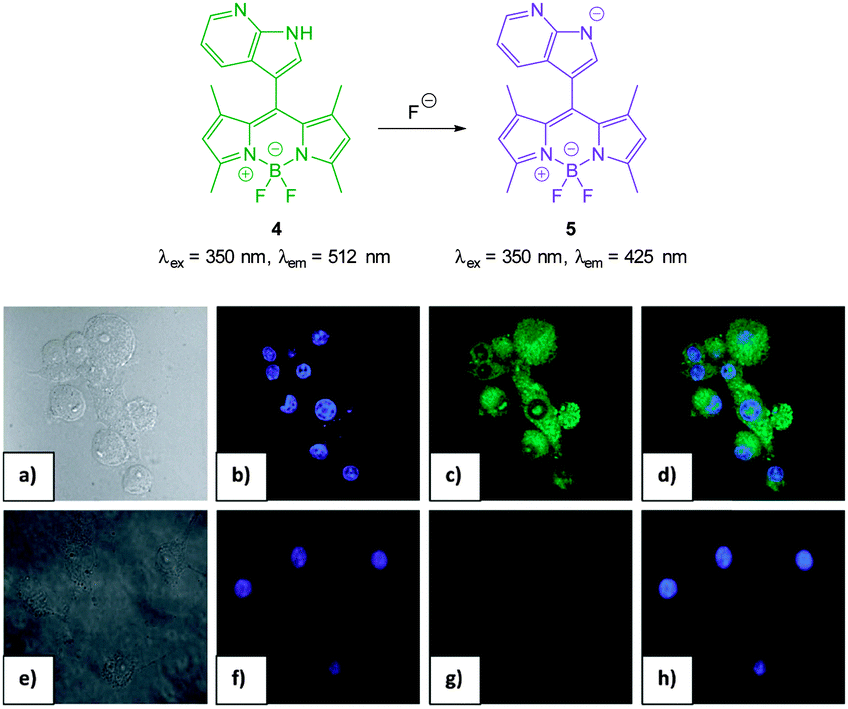 | ||
| Fig. 5 Top: structure, and deprotonation, of probe 4 by F−. Bottom: images of RAW264.7 cells. (a) Bright field image; (b) nuclei stained with 4′,6-diamidino-2-phenylindole (DAPI); (c) cells treated with probe 4; (d) overlay image of (b) and (c); (e) bright field image of probe 4 and F−; (f) probe 4 and F− (g) nuclei with DAPI; (h) overlapping image of (f) and (g). Image reproduced with permission.115 | ||
In 2013 Chellappa reported the rhodamine based probe RDF-1 (Fig. 6) that operates by means of deprotonation leading to spirocycle ring opening.118 A strong “switch on” fluorescence response at 557 nm was observed in the presence of F−. In HeLa cells RDF-1 was non-toxic and within 10 minutes of NaF addition significant fluorescence enhancement was observed.
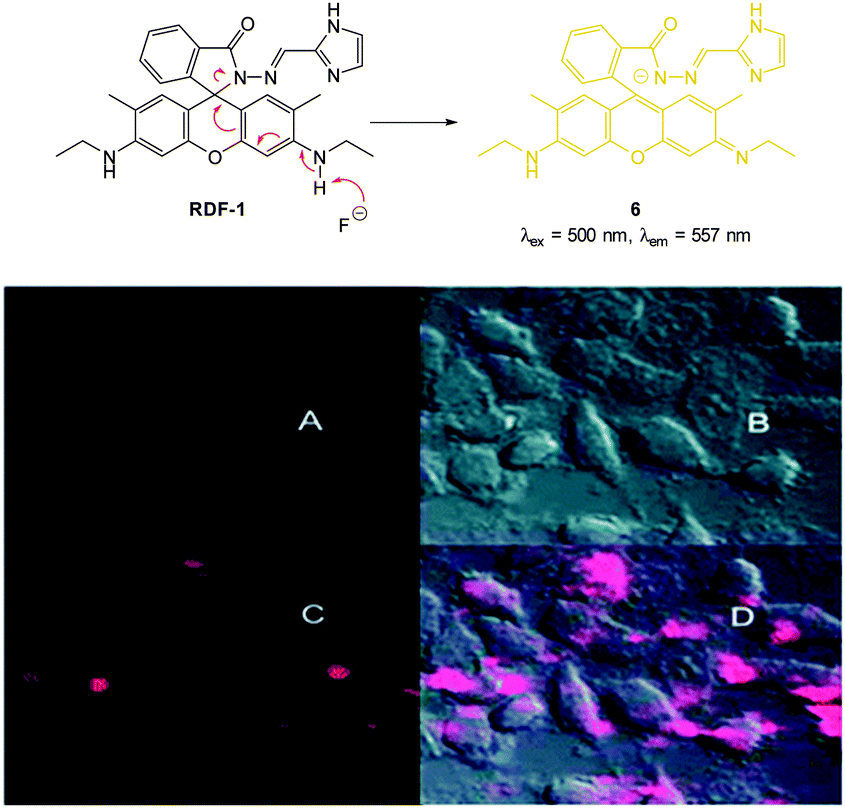 | ||
| Fig. 6 Top: structure and reaction of rhodamine probe RDF-1 with F−. Bottom: (A) fluorescence image of HeLa cells incubated with RDF-1; (B) corresponding bright-field image; (C) fluorescence image of HeLa cells incubated with RDF-1 and NaF; (D) overlaid images of HeLa cells (B and C). Image reproduced with permission.118 | ||
One of the earliest “switch on” probes functioning by means of desilylation, a TBDPSO-coumarin (TBPCA, Fig. 7), was published by Park and Hong in 2009.120 The probe readily entered cells and was retained with no toxicity. Images in human epithelial lung carcinoma A549 cells show clear blue fluorescence upon exposure to NaF.
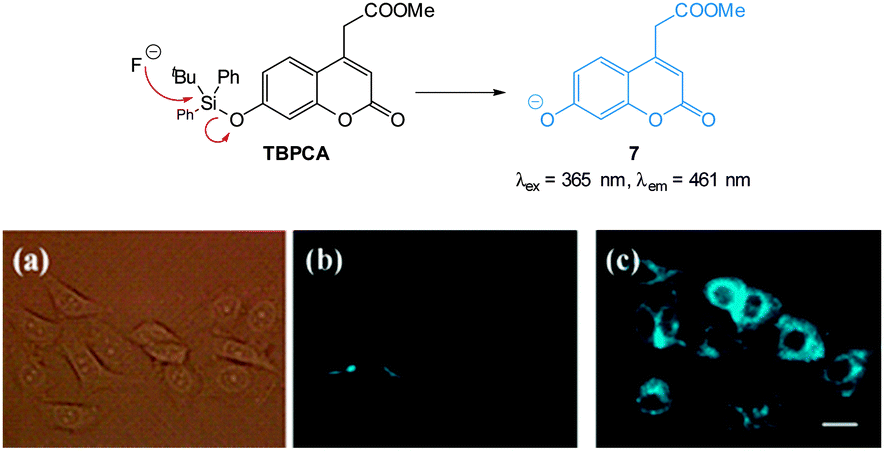 | ||
| Fig. 7 Structure and reaction of coumarin TBPCA with fluoride. (a) Bright-field image of A549 cells with TBPCA (b) fluorescence image with TBPCA without NaF (c) fluorescence image with TBPCA and NaF. Scale = 20 μm. Reproduced with permission.120 | ||
The “switch on”, red emitting probe 8 (Fig. 8) was recently published by Zhu (2014).121 Synthesis of the masked ICT fluorophore involved aldol type reaction of the potential electron donor 4-OTBPS-benzaldehyde with the electron withdrawn 2-dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran (TCF). For probe 8 a linear emission “switch on” at 612 nm was observed upon reaction with fluoride and a LOD = 0.07 mM was determined. The absorption spectra could be used to quantify the amount of fluoride present in solution due to the linear relationship between the increase at 596 nm and the decrease at 438 nm. Imaging was performed in live HeLa cells and 10 μM NaF was readily visualised using fluorescence microscopy.
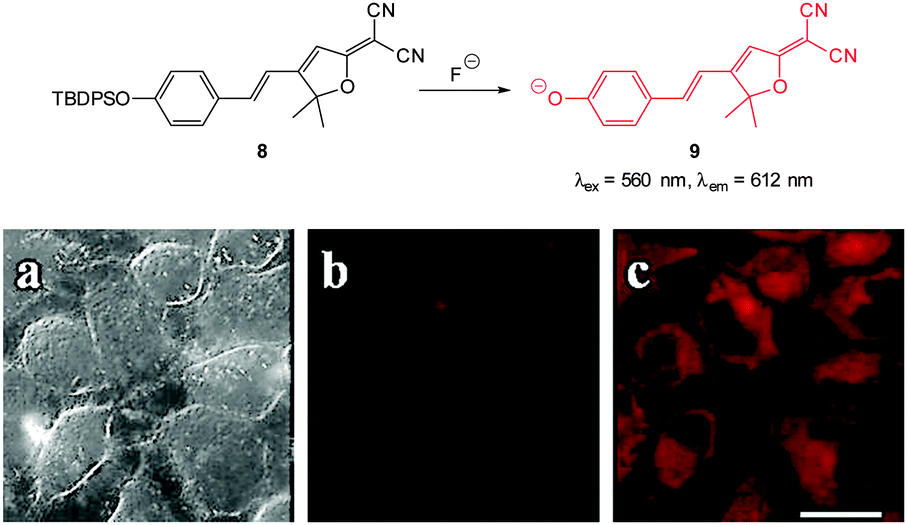 | ||
| Fig. 8 Top: structure and reaction of “switch on” fluoride probe 8. Bottom: fluorescence images of HeLa cells incubated with probe 8 (a) bright-field transmission (b) red channel with no NaF (c) red channel with NaF. Scale = 20 μm. Image reproduced with permission.121 | ||
The highly selective, ratiometric, benzothiazolium hemicyanine 10 (Fig. 9) developed by Ma, Du and Zhang (2011)122 could monitor fluoride concentration using the ratio F500/F558. A limit of detection (0.08 nM) was identified and in live RAW264.7 macrophages a distinct ratiometric fluorescence response was observed upon addition of buffered NaF. The probe was also shown to penetrate rapidly (<5 minutes) and was non-toxic.
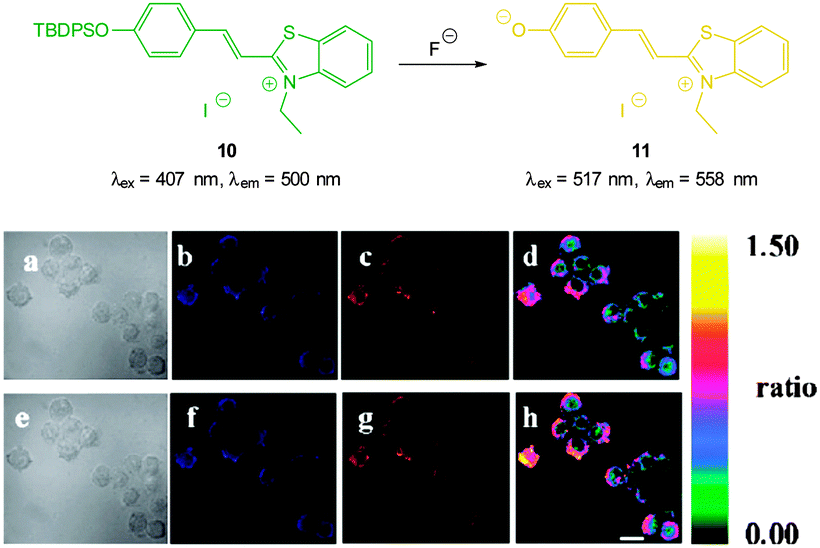 | ||
| Fig. 9 Top: structure and reaction of benzothiazolium hemicyanine 10 with F−. Bottom: images of RAW264.7 macrophages incubated with 10 and no added F− (a) bright-field, (b) blue channel at 490 ± 20 nm, (c) orange channel at 560 ± 20 nm, and (d) ratio image from (c) and (b). Images of RAW264.7 macrophages incubated with 6 after addition of NaF (e) bright-field, (f) blue channel at 490 ± 20 nm, (g) orange channel at 560 ± 20 nm, and (h) ratio image from (g) and (f). Scale = 20 μm. Image reproduced with permission.122 | ||
A variation on the desilylation probe was reported by Zhang in 2013.123 In the presence of fluoride, desilylation of the functionalised naphthalimide chemodosimeter 12 (Fig. 10) was immediately followed by fragmentation to give the conjugate base of 4-hydroxynaphthalimide 13. In solution studies a linear fluorescence “switch on” (λem = 560 nm) was realised (20-fold increase in one hour with only 1.0 equivalent of F−; LOD = 0.35 μg L−1). After incubation of probe 12 with human epithelial carcinoma cells (A549) addition of a solution of NaF elicited a distinct green fluorescent response.
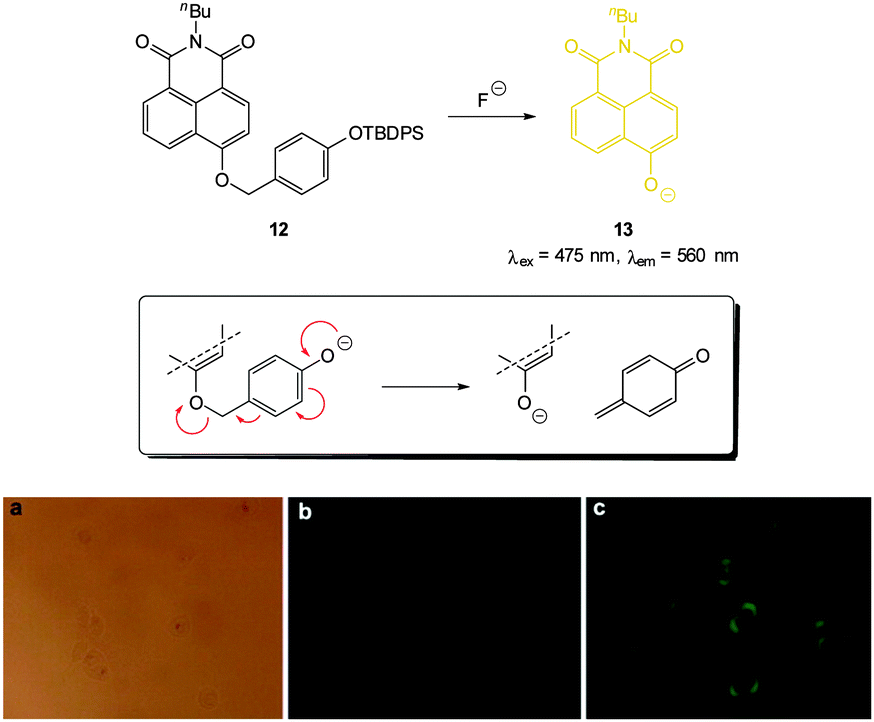 | ||
| Fig. 10 Top: functionalised naphthalimide probe 12 and its reaction with F−; inset shows fragmentation mechanism. Bottom: (a) bright-field image of A549 cells incubated with 12 (20 μM) for 24 h (a) bright field (b) without NaF (c) with NaF (50 mM). Image reproduced with permission.123 | ||
With an eye to enhanced solubility the PEG-thiourea-fluorescein probe PEG-FITC-Si (Fig. 11) was synthesised by Zeng and Wu (2013)88 in two steps from the corresponding fluorescein isocyanate. Again in the presence of fluoride a dramatic increase in fluorescence (λex = 490 nm, λem = 526 nm) was observed (LOD = 19 ppb). Imaging was successfully achieved in HeLa and murine fibroblasts (L929) only 15 minutes following addition of fluoride (100 μM) with perinuclear probe localisation.
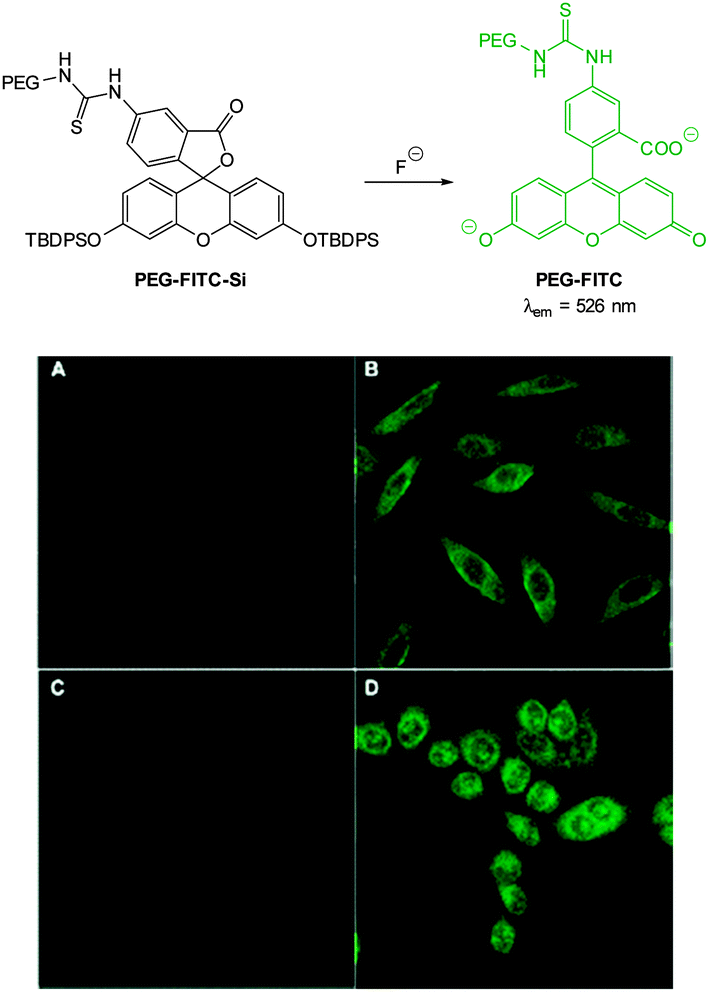 | ||
| Fig. 11 Top: structure of fluoride chemodosimeter PEG-FITC-Si. Bottom: fluorescence imaging of L929 (top) and HeLa (bottom) cells incubated with the sensor before (A and C) and after (B and D) treatment with NaF. Image reproduced with permission.88 | ||
The TBSO-benzothiazole BBTGA (Fig. 12), also deliberately designed for biocompatibility by conjugating glucosamine to improve solubility, was reported by Wang in 2013.89 A linear 30-fold fluorescence enhancement (λem = 508 nm) was noted 5 minutes after the addition of NaF in PBS buffer. The probe was water soluble, non-toxic and when a buffered solution of NaF (0.1 mM) was added to human nasopharyngeal epidermal carcinoma (KB) cells that had been pre-treated with a dilute solution of BBTGA strong fluorescence was observed.
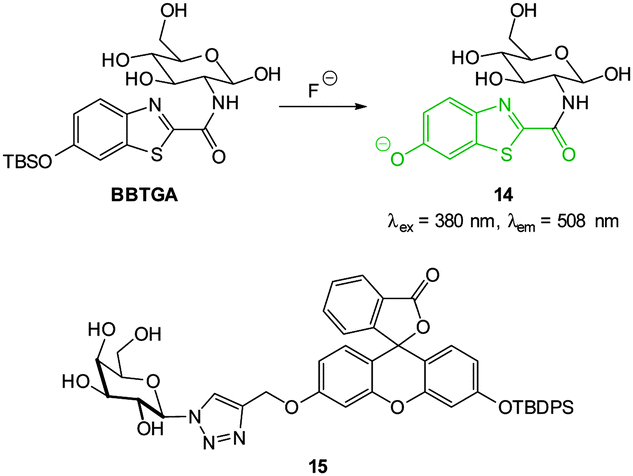 | ||
| Fig. 12 Structure of BBTGA and 15, showing reaction of BBTGA with F−. | ||
Again with solubility and biocompatibility in mind the carbohydrate conjugate probe 15 (Fig. 12) was synthesised by Du (2011) using the well-known copper assisted azide alkyne cycloaddition (CuAAC).124 A very strong (160 fold), linear, fluorescence “switch on” response (λem = 520 nm) was observed with increasing NaF (from 0 to 1.4 mM, probe concentration 50 μM) and a limit of detection of 10.5 μM was identified. A strong fluorescence response was observed upon addition of NaF solution to Hep2G cells that had been incubated with 15 (Fig. 13).
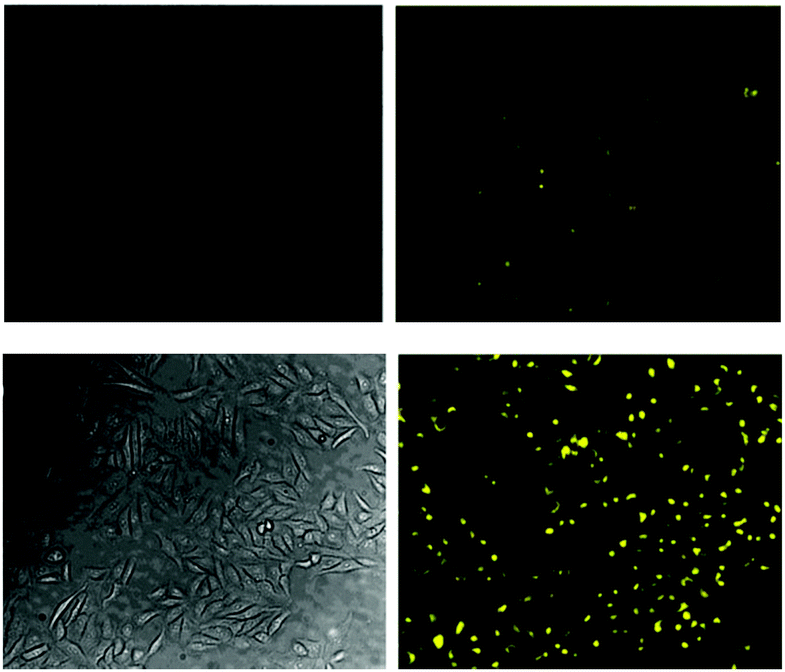 | ||
| Fig. 13 Images of HepG2 cells (left) brightfield and (right) fluorescence with (above) only probe 15, (below) probe 15, and NaF. Image reproduced with permission.89 | ||
The pyrene dimer 16 (Fig. 14) containing a disilane (Si–Si) bond was designed and constructed by Li and Shen (2012).125 Ratiometric measurement in solution (THF:H2O) was possible as the well-known pyrene excimer fluorescence at 470 nm ceased upon reaction of the probe with fluoride and only monomer emission at 378 nm was present. Up to 6 equivalents of F− could be measured using F378/F470. Loading of 16 into HeLa cells was performed using polylactic acid nanoparticles and upon exposure to fluoride a clear change in emission was detected.
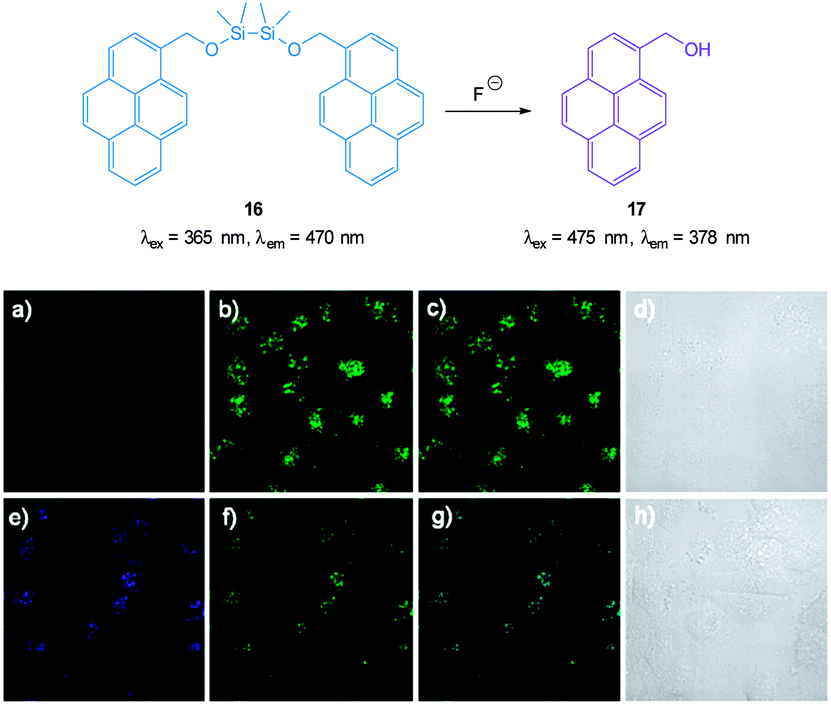 | ||
| Fig. 14 Top: structure and reaction of pyrene dimer 16 with F−. Bottom: images of live HeLa cells incubated with probe 16; (a) emission 410–440 nm (blue channel) (b) emission 440–600 nm (green channel), (c) overlaid a and b, (d) bright-field transmission image. The above cells after addition of 100 μM F− (e) emission 410–440 nm, (f) 440–600 nm, (g) overlaid e and f, (h) bright-field transmission image. Image reproduced with permission.125 | ||
In 2012 Lee, Kim and Ahn published an interesting variant on the desilylation probe.126 Upon desilylation the carefully functionalised aminonaphthalene P1 (Fig. 15) reacts in an additional intramolecular process to give the extended aminocoumarin 20. A fluorescence “switch on” (λem = 595 nm) was observed in both murine metastatic melanoma (B16F10) cells and also in live zebrafish. Imaging was accomplished using two photon microscopy (TPM) and the lower excitation energy associated with this technique is perfect for in vivo research to understand how fluoride is distributed in a whole body context. In the zebrafish, increased concentrations of F− in the tail and abdomen were observed at t = 2 h versus t = 30 min.
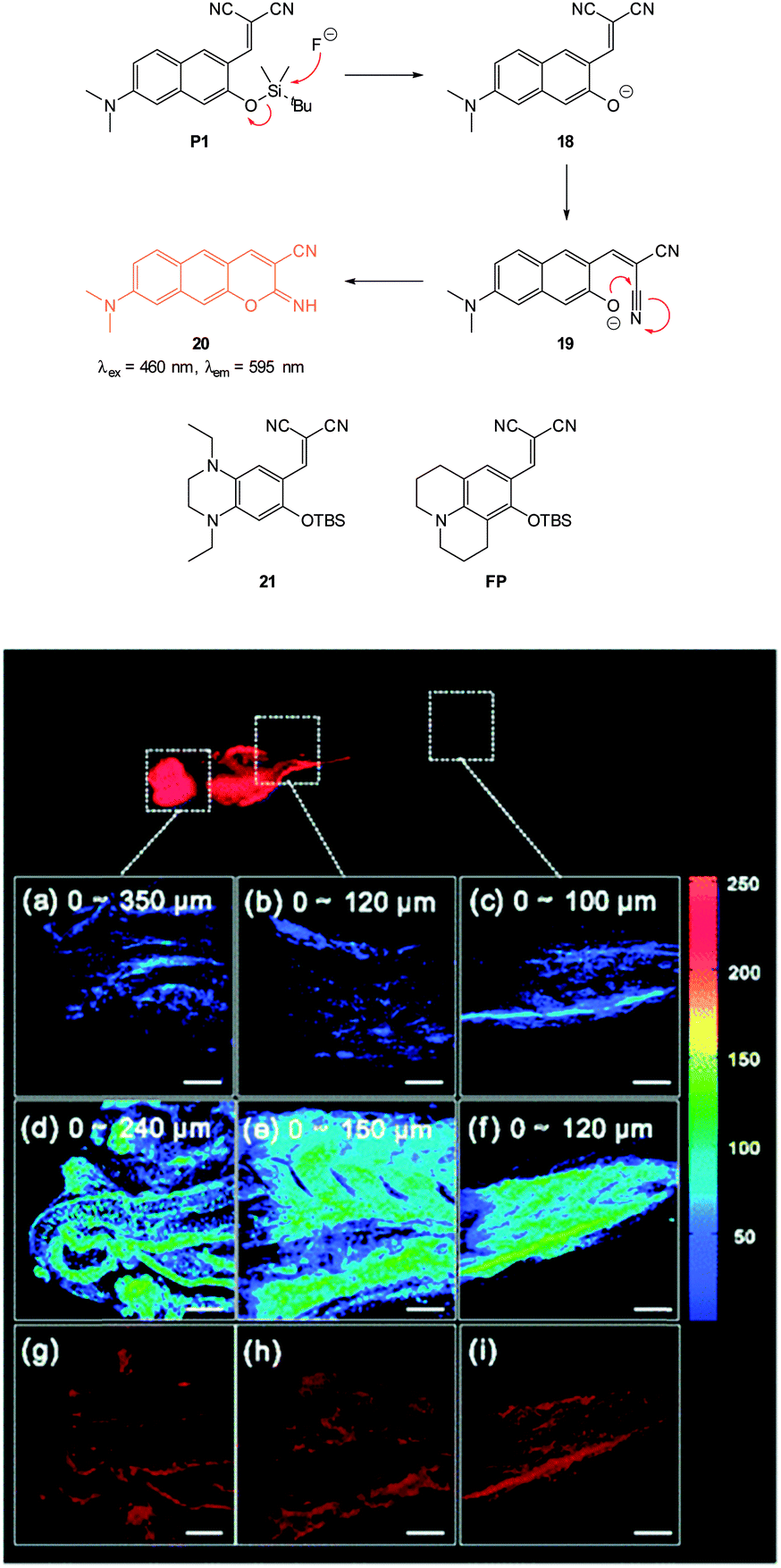 | ||
| Fig. 15 Top: structure and reaction of aminonaphthalene probe P1 with F− to form extended coumarin 20. Bottom: accumulated TPM images for three zebrafish parts: (a–c) probe P1 alone (d–i) probe P1 followed by F− (a–f) intensity data; (g–i) fluorescence images constructed by image stacking for 0–350 μm depth, with a 2 μm imaging depth step. Scale = 50 μm. View area: 300 μm2. Image reproduced with permission.126 | ||
A recent report (2014) by Song also outlined a dicyanoacrylate “switch on” probe (21, Fig. 15) in which a desilylation cascade approach was used to create a red fluorescent iminocoumarin.127 The probe itself was non-fluorescent and for the product a Stokes shift of more than 140 nm was recorded. Using fluorescence microscopy the probe was shown to be readily internalised and was capable of indicating the presence of fluoride in living human keratinocyte (HaCaT) cells (not shown).
The related probe FP (Fig. 15) from the group of Peng (2014),128 also relies on an additional reaction occurring post Si cleaving. The probe was synthesised from the corresponding quinolinecarbaldehyde and the final product of the reaction sequence is a highly fluorescent (ϕF = 0.84, λex = 441 nm, λem = 485 nm) aminobenzopyranimine. The probe was shown to be relatively non-toxic, localised in the mitochondria of both breast cancer (MCF-7) and fibroblast-like (COS-7) cells and fluorescence was dramatically “switched on” when the cells were treated with dilute solutions of NaF (Fig. 16).
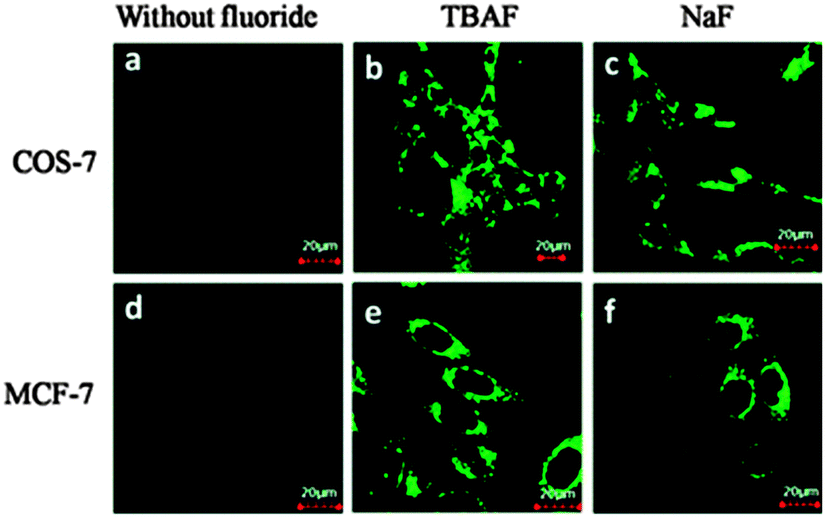 | ||
| Fig. 16 Images of COS-7 and MCF-7 cells incubated with FP before (a and d) and after (b, c, e, and f) treatment with TBAF or NaF. Image reproduced with permission.128 | ||
 | ||
| Fig. 17 Top: structure and reaction of Se BODIPY 22 with F−. Bottom: images of living HepG2 cells Incubated with (a) probe 22 for 1 h; (b) F− for 1 h then probe 22 for 1 h. Images (c) and (d) are bright field images of (a) and (b), respectively. Image reproduced with permission.129 | ||
2.2 Chloride
Chloride is the most abundant anion in living organisms and its transport across cellular membranes is essential for a number of physiological processes including the maintenance of cell volume, acidification of internal compartments and even electrical excitability. Impaired transport of this anion due to a of genetic mutation in a cAMP-regulated Cl− channel defines the condition known as cystic fibrosis (CF).130 Indeed, the pursuit of biologically active chloride transporters to remedy this condition is an important current goal for supramolecular chemists.131–138Other than some recent developments (Section 2.2.3) the strategy employed in the design of chloride imaging agents relies on halide mediated collisional quenching (Section 2.2.1) and as such the majority of Cl− probes are “switch off”. Nevertheless by attaching such probes to “constant” fluorophores several ratiometric probes have been successfully designed and used (Section 2.2.2).
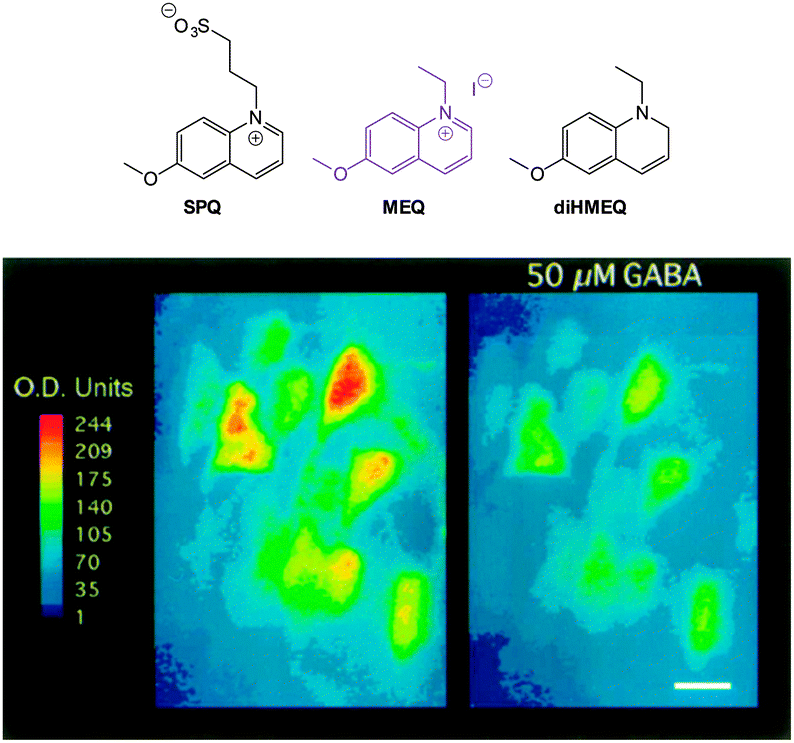 | ||
| Fig. 18 Top: structure of SPQ, MEQ and the cell permeable diHMEQ probes for Cl−. Bottom: images of MEQ-loaded neurons (left) before and (right) 20 min after GABA application. Scale = 15 μm. Image reproduced with permission.146 | ||
The identification of N-methylacridinium-9-carboxamide MACA (λem = 500 nm, Fig. 19) and also the bisacridinium lucigenin (λem = 506 nm, Fig. 19) as longer wavelength variants was a welcome development.149 However, while these compounds have been used successfully in vesicle/liposome based studies150 they were shown to be unstable in cell based studies.149
 | ||
| Fig. 19 Acridine and quinolinium “switch off” probes MACA and lucigenin. | ||
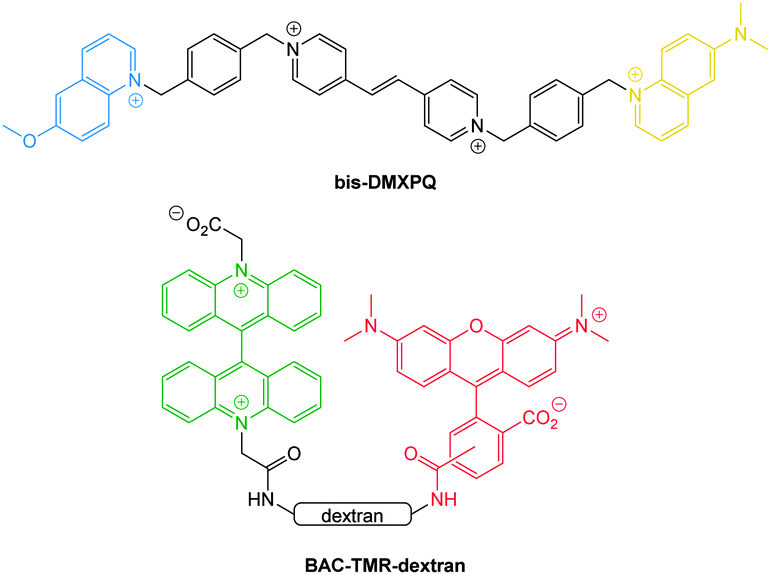 | ||
| Fig. 20 Ratiometric probes bis-DMXPQ and BAC-TMR-dextran used for chloride imaging. | ||
More recently, the ratiometric probe MQAF (Fig. 21) was reported by Tang (2012).153 The structure consisted of the “switch off” methoxyquinolium combined with aminofluorescein and monitoring at two channels [chloride sensitive emission λex = 318 nm, λem = 436 nm and insensitive λex = 494 nm, λem = 519 nm] gave accurate measurements of Cl− concentration. This probe was successfully used in ventricular myocytes to illustrate that induced ischemia results in increased Cl− concentration. In 2014 the same group published the ratiometric methoxyquinolium dansyl combination (MQDS, Fig. 19) and the ratio λem = 440 nm against λem = 560 nm was used to monitor chloride concentration.154 Imaging of liver cancer cells (HepG2) was performed and intracellular chloride concentration was successfully monitored as the extracellular levels in the surrounding media were deliberately increased (Fig. 21).
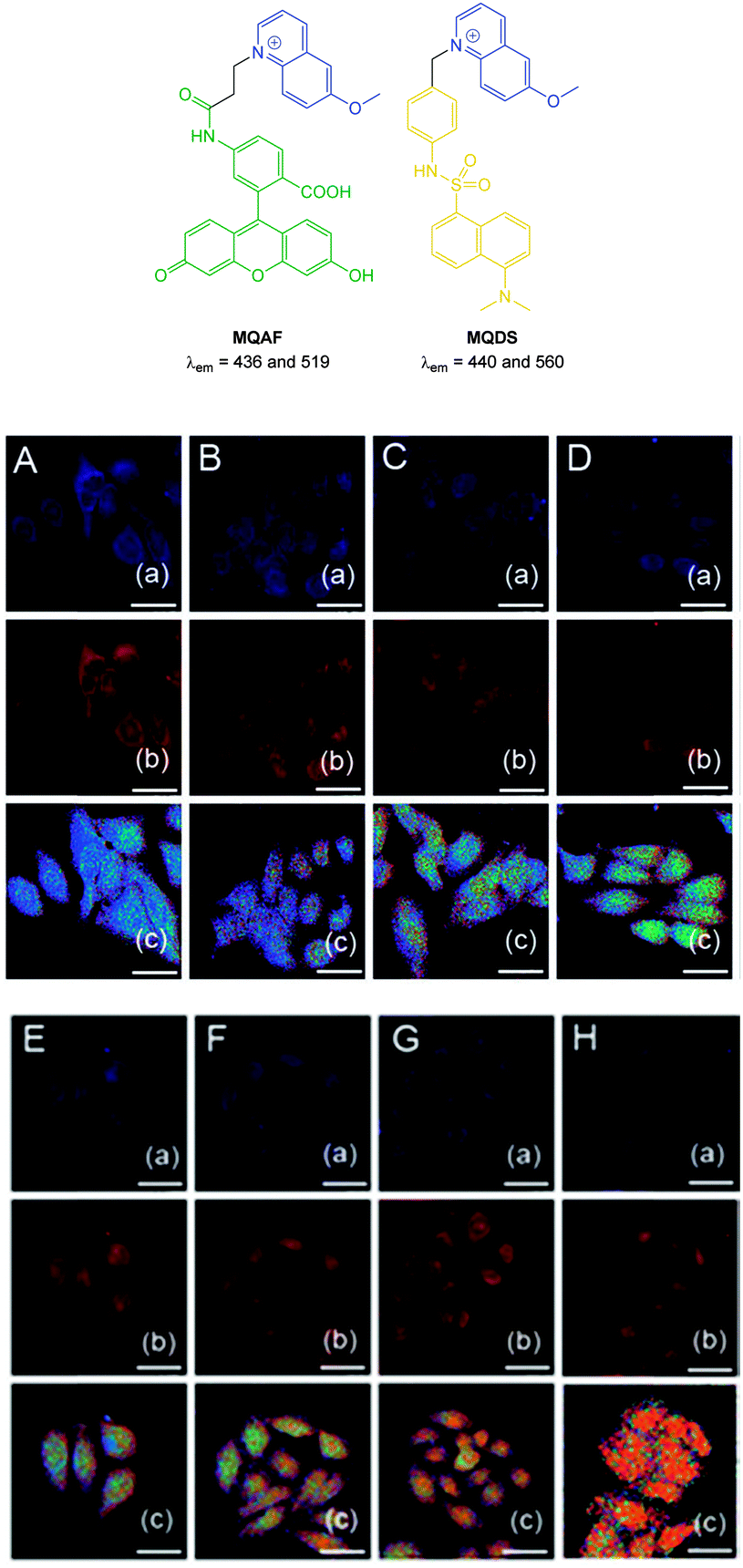 | ||
| Fig. 21 Top: structures of ratiometric quinolium conjugates MQAF and MQDS. Bottom: images of HepG2 cells loaded with MQDS exposed to: A–H, 0–140 mM chloride. Top in each row is blue channel, middle is red channel and the bottom is a ratio image (Fred/Fblue). Scale = 25 μm. Image reproduced with permission.154 | ||
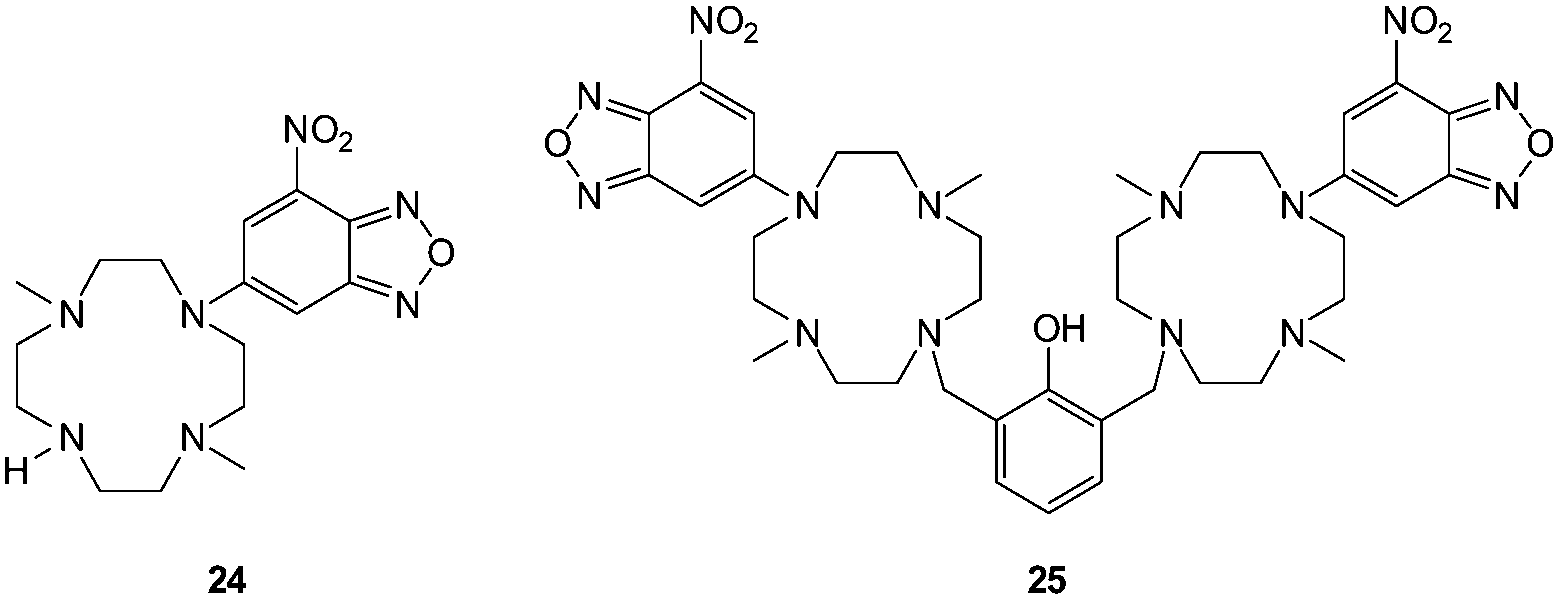 | ||
| Fig. 22 NBD-cyclam ligand 24 and related ditopic 25. | ||
Another new, and very interesting, class of chloride probes are the squaraine-rotaxanes developed by Smith (Fig. 23).157,158 The squaraines have been somewhat overlooked as a biologically compatible fluorophore due to their susceptibility to hydrolysis. In contrast, hydroxysquaraines have been found to be much more stable and for rotaxane 26 (Fig. 23) interaction with chloride shifts the surrounding macrocycle slightly along the squaraine “axle” which in turn leads to fluorescence modulation. While not yet demonstrated in an intracellular setting squaraine-rotaxane probes are red emissive, ratiometric (in acetone λem = 698 nm decreases and λem = 665 nm increases) and, unlike the first generation of probes they are selective for chloride (I− is not bound; Br− binding is 10 fold weaker than that of Cl−) and thus have tremendous potential for further development in an intracellular setting.
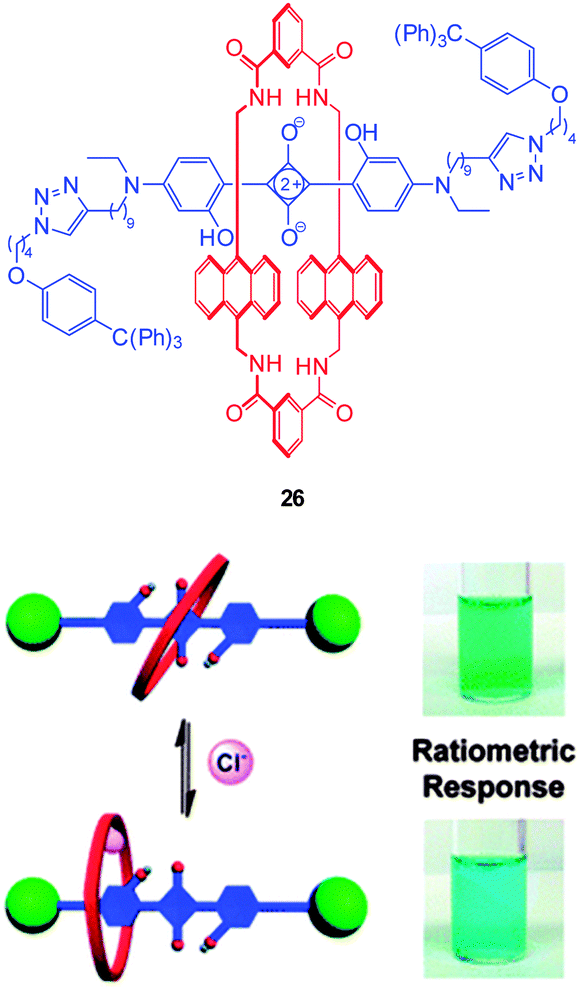 | ||
| Fig. 23 Fluorescent Cl− sensor 26 based on a squaraine-rotaxane shuttle. Image reproduced with permission.166 | ||
2.3 Iodide
In the environment iodide occurs naturally in minerals (alutarite and iodargyrite) and it is interesting that AgI is used by humans as a as a nucleation agent in “cloud seeding” programs due a similarity in crystal structure to that of water ice.159 Iodide is also a common additive to table salt as deficiencies can lead to the condition known as goitre. Indeed the consumption of trace amounts of iodide is essential for human health—the anion is transported to, and accumulated in, the thyroid gland for incorporation into the iodine containing hormones.160,161Selective probes for iodide bioimaging are rare. It is interesting to note that the early probes for chloride such as SPQ and MEQ (see Fig. 18) were actually more sensitive to iodide than chloride,102,140,141 nevertheless the far greater concentration of chloride resulted in minimal interference from iodide.
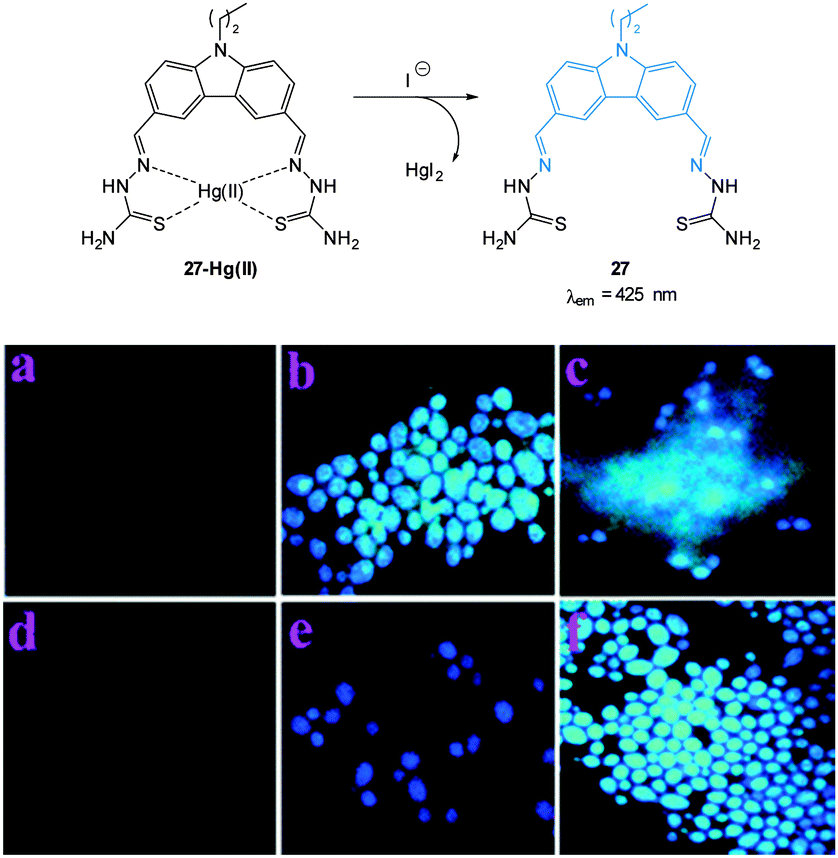 | ||
| Fig. 24 Top: structure and reaction of iodide probe 27-Hg(II); bottom: fluorescence images of Candida albicans (a) cells only, (b) cells with 27, (c) cells with 27 after addition of Hg(II) (5 μM), (d) cells with 27 and Hg(II) (25 μM), (e) cells with the preformed 27-Hg(II) complex and KI, (f) same as (e) after 10 min. Image used with permission.162 | ||
2.4 Cyanide
Cyanide has a long history of use in industry and is also well known as an environmental poison but for cystic fibrosis (CF) sufferers −CN has a particularly sinister role. Infection with Pseudomonas aeruginosa (PA) is common amongst patients with CF and PA is a cyanogenic bacteria (synthesises −CN)163 and in vivo cyanide functions as a potent inhibitor of cellular respiration.164 Indeed PA-mediated cyanogenesis has an acknowledged role in the pathogenesis of CF lung disease.165 Less common, but more problematic is infection with Burkholderia cepacia complex (Bcc) which is typically multidrug resistant and is also cyanogenic.165,166 Hence diagnostics for −CN in vivo would be welcome for rapid identification of these problematic lung infections. Other sources of in vivo cyanide come from cyanogenic glycosides produced by some plants as part of their innate defence system (for example in almond seeds) and these glycosides can be enzymatically hydrolysed to produce free −CN in living tissues.167A number of strategies exist for the detection of cyanide but the most common approaches for imaging −CN rely on its (i) nucleophilicity or (ii) affinity for copper ions.40,168 The nucleophilicity of cyanide has been exploited in the design of chemodosimeters (see Section 2.4.1). Typically, nucleophilic attack of −CN at a chemodosimeter incorporating a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (aldehyde), CNR, CC–CN or related functionality results in a product in which conjugation at some point of the probe is broken and in turn the fluorescence response is modulated. The other successfully used approach involves the displacement of copper (Section 2.4.2). These probes are functionalised with copper chelating groups such that when Cu(II) is introduced the resultant assembly exists in quenched form (heavy/transition metal ion induced quenching). Cyanide has a very high affinity for copper and is capable of selectively displacing the fluorophore. A very stable Cu(CN)2 species is formed and the fluorophore is liberated—“switched on”.
O (aldehyde), CNR, CC–CN or related functionality results in a product in which conjugation at some point of the probe is broken and in turn the fluorescence response is modulated. The other successfully used approach involves the displacement of copper (Section 2.4.2). These probes are functionalised with copper chelating groups such that when Cu(II) is introduced the resultant assembly exists in quenched form (heavy/transition metal ion induced quenching). Cyanide has a very high affinity for copper and is capable of selectively displacing the fluorophore. A very stable Cu(CN)2 species is formed and the fluorophore is liberated—“switched on”.
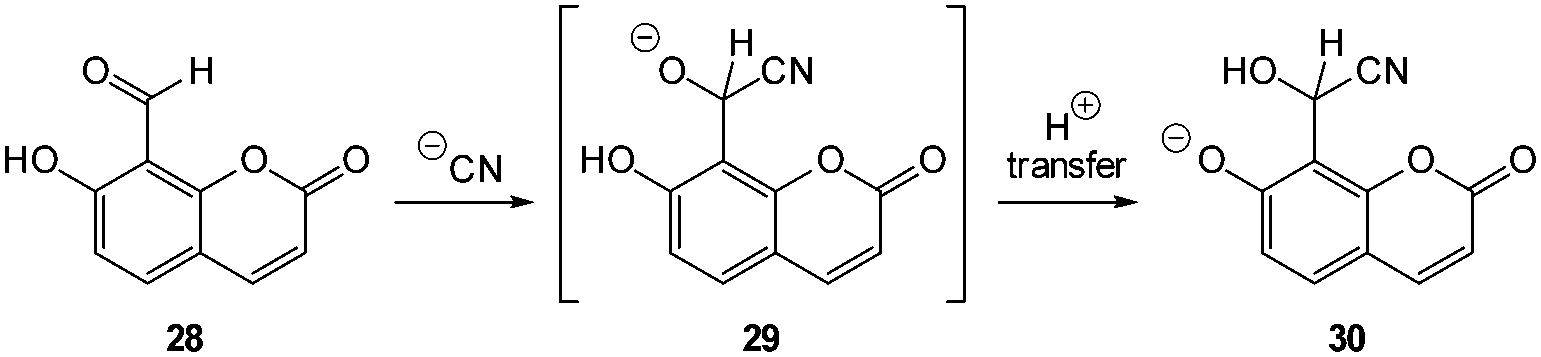 | ||
| Fig. 25 Coumarin based ESIPT sensor 28 for the detection of cyanide. | ||
Yoon, designed the ESIPT hydroxyfluorescein aldehyde probes (both mono and di were synthesised; dialdehyde 31 shown in Fig. 26) for microfluidic sensing of cyanide as well as both in vitro and in vivo probes for cyanide.171,172 Formation of the phenoxide in this instance leads to spirocyclic ring opening and strong fluorescence “switch on” (λem = 520 nm). Using dialdehyde 31 the imaging of cyanide in BALB/c nude mouse model was accomplished (Fig. 26) in vivo.172 This probe was able to detect, in the lungs of the mice, increased levels of cyanide due to infection caused by PA and Bcc. The probe itself did not cause adverse effects when injected as a DMSO solution directly into the lungs of the mice.
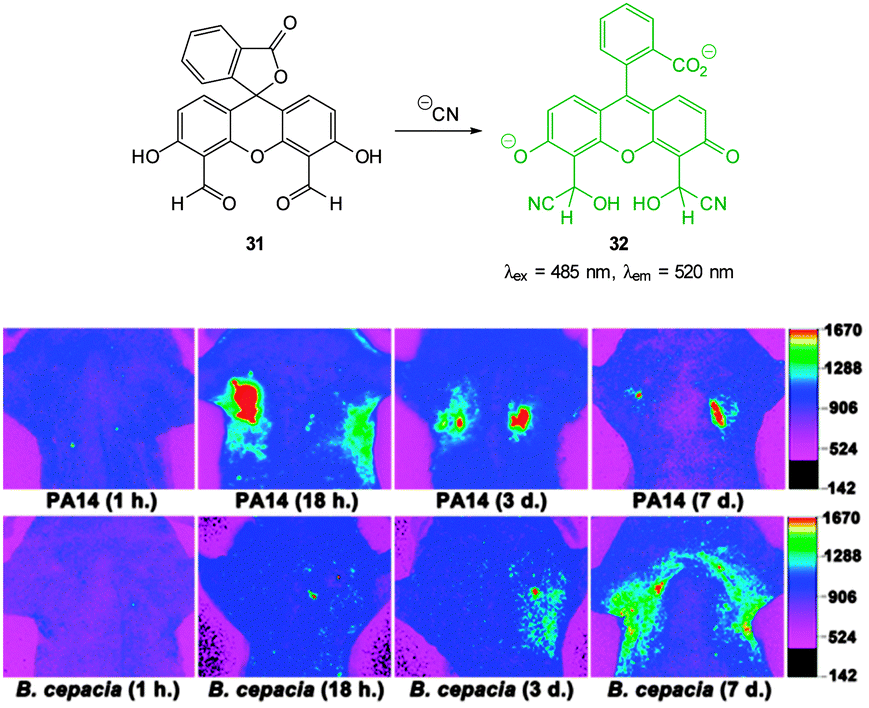 | ||
| Fig. 26 Top: fluorescein dialdehyde chemodosimeter 31 for −CN. Bottom: in vivo images of −CN in the lungs after various incubation times after infection with PA and Bcc. Colour images were reconstructed from inverted fluorescence images. Image reproduced with permission.172 | ||
The salicylaldehyde functionalised fluorene ESIPT probe FSal (Fig. 27) was reported by Malik in 2014.173 The sensor could detect cyanide in solution at very low concentration (<0.1 ppb) by reaction to form the corresponding cyanohydrin leading to a strong fluorescence ‘switch-on’ (λex = 329 nm, λem = 520 nm). The probe was capable of imaging cyanide (as tetrabutylammonium cyanide, TBACN) in human neuroblasts (SH-SY5Y) and the probe was both highly selective and non-toxic.
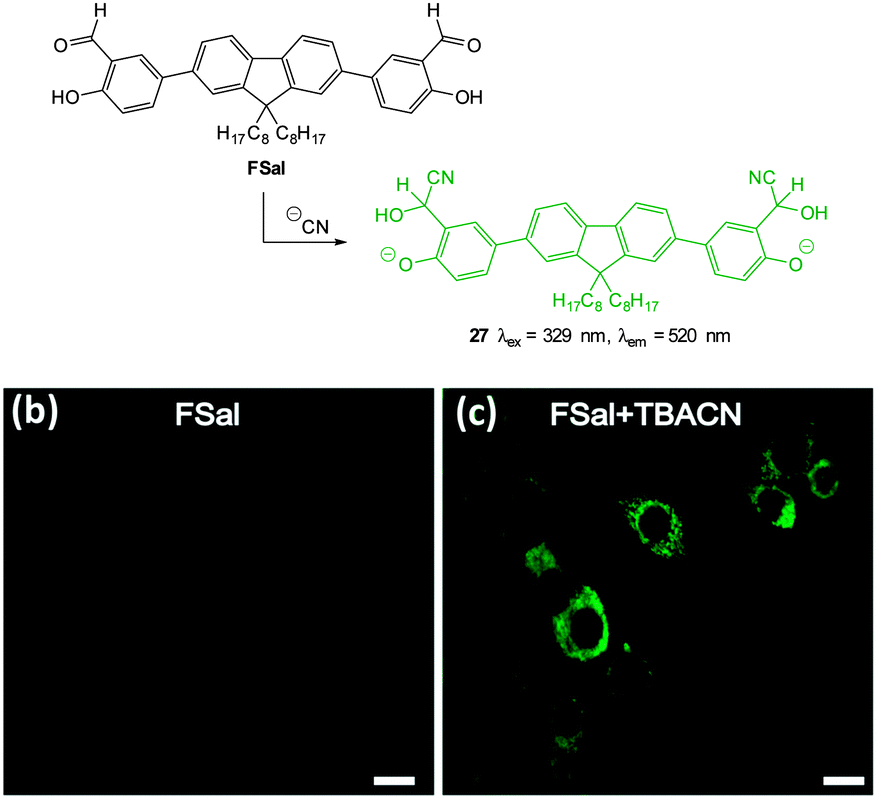 | ||
| Fig. 27 Top: structure and reaction of fluorene based FSal with −CN. Bottom: images of SH-SY5Y neuronal cells which were incubated with (left) FSal alone and (right) FSal then TBACN. Image reproduced with permission.173 | ||
The “switch off” BODIPY dialdehyde 34 (Fig. 28) was reported by Ravikanth in 2013.174 Using NMR spectroscopy the probe was clearly shown to react with two equivalents of −CN and fluorescence (λem = 554 nm) was quenched with the addition of 2.2 equivalents of the anion. In human breast adenocarcinoma cells (MDA-MB-231) the probe was non-toxic and the intense green fluorescence of the probe was quenched upon treatment of the cells with −CN.
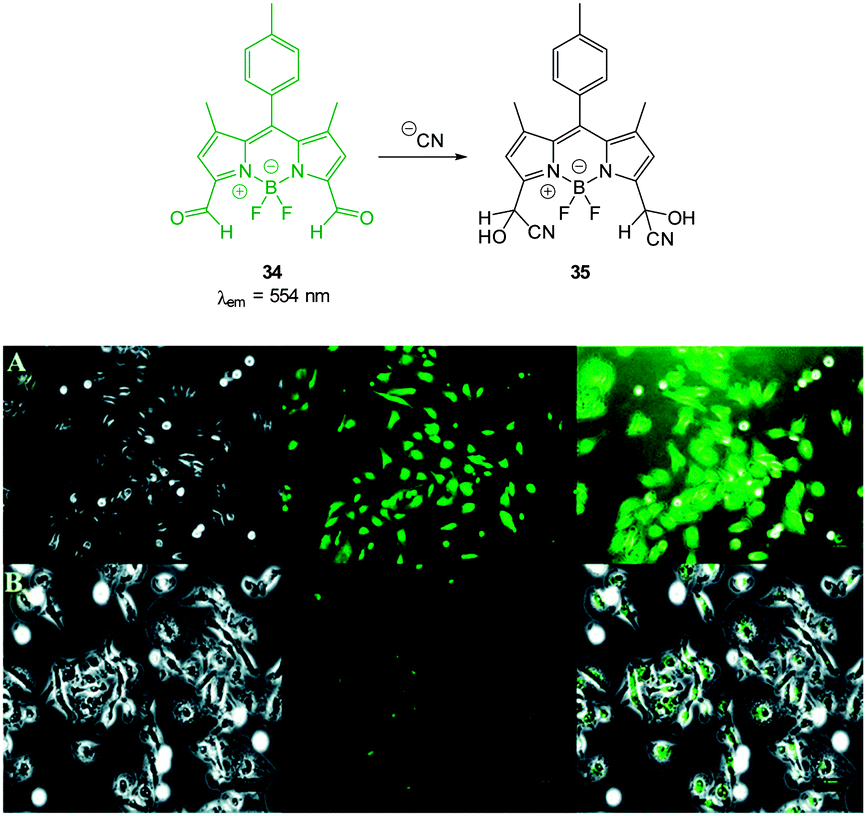 | ||
| Fig. 28 Top: structure and reaction of BODIPY dialdehyde 34 with −CN. Bottom: images of MDA-MB-231 cells (A) treated with 34 only and (B) after incubation with 34 and −CN. Left is bright field, centre is fluorescence and right is overlay image. Scale = 50 μm. Image reproduced with permission.174 | ||
The ratiometric aminocoumarin probe Coum-1 (Fig. 29) reported by Li (2012)175 possesses a reactive dicyanoacrylate appendage (readily installed using the reaction of malononitrile with the corresponding coumarin aldehyde). A distinct response (both colourimetric and fluorescent) was observed following the conjugate addition reaction of cyanide to the alkene. The diminished length of the ICT system leads to a shift in both absorption and emission maxima and using excitation at λ = 447 nm ratiometric measurement of cyanide could be performed [initial coumarin (ϕF = 0.45, λem = 585 nm), product Coum-CN (ϕF = 0.33, λem = 495 nm)]. An impressive 470 fold increase in F495/F585 was realised with the addition of only 1.0 equivalent of −CN. The probe was successfully used for the detection of cyanide in HeLa cells by comparing emission from the red and green channels.
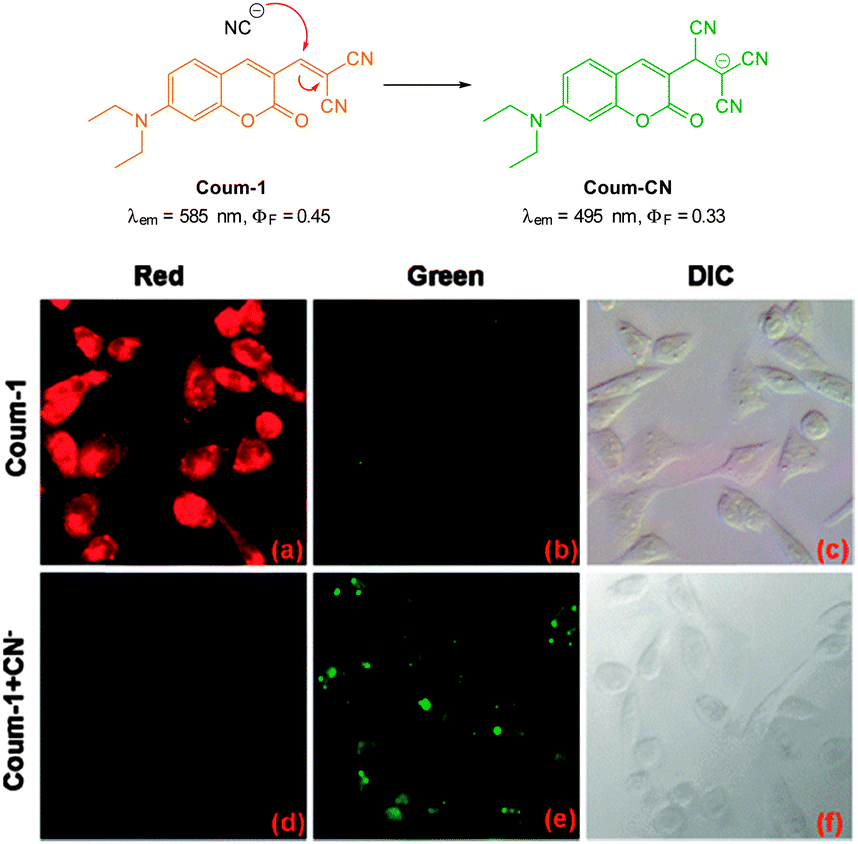 | ||
| Fig. 29 Top: structure and chemodosimetric reaction of Coum-1 with −CN. Bottom: images of HeLa cells incubated with Coum-1 with and without −CN; (a, d) red channel (b, e) green channel and (c, f) brightfield differential interference contrast (DIC). Image reproduced with permission.175 | ||
Also relying on the conjugate addition of −CN the BODIPY chemodosimeter 36 (Fig. 30) was developed by Jang (2012).176 The reaction of cyanide with the dicyanoethylene appendage interrupts the ICT and as a consequence both visible and fluorescent properties were modulated. Interference from fluoride was noted in CH2Cl2 but in water the strong solvation of fluoride rendered it less competitive. A clear “switch on” response (λex = 480 nm, λem = 520 nm) was observed in the cytoplasm of HeLa cells that had been incubated with the probe for 20 min then treated with NaCN.
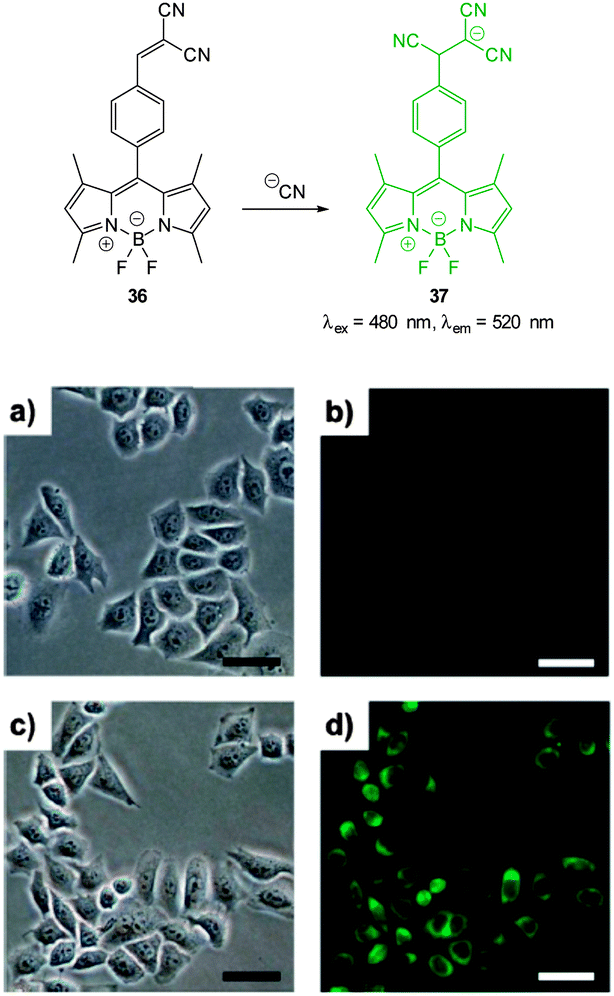 | ||
| Fig. 30 Top: reaction of dicyanoethylene BODIPY 36 with −CN. Bottom: brightfield (a, c) and fluorescence (b, d) images of HeLa cells with only 36 (a, b) then 20 min after treatment with NaCN (c, d). Scale = 50 μm. Image reproduced with permission.176 | ||
A “switch-on” phenothiazine–hemicyanine probe Phc (Fig. 31) was reported by Yang and Li (2014).177 The cyanide anion readily attacked the CN bond of the indolium and a 20 fold enhancement in fluorescence (λem = 488 nm) was observed when solutions of the probe were exposed to only 3.0 equivalents of −CN. The probe was selective amongst other anions tested and was used in both human breast cancer (GES) and HeLa cells to demonstrate a quick (15 min) “switch on” effect in the presence of in vitro cyanide. Furthermore in adult zebra fish exposed to Phc and cyanide (30 μM) a strong fluorescent response was observed, particularly in the gills and abdomen.
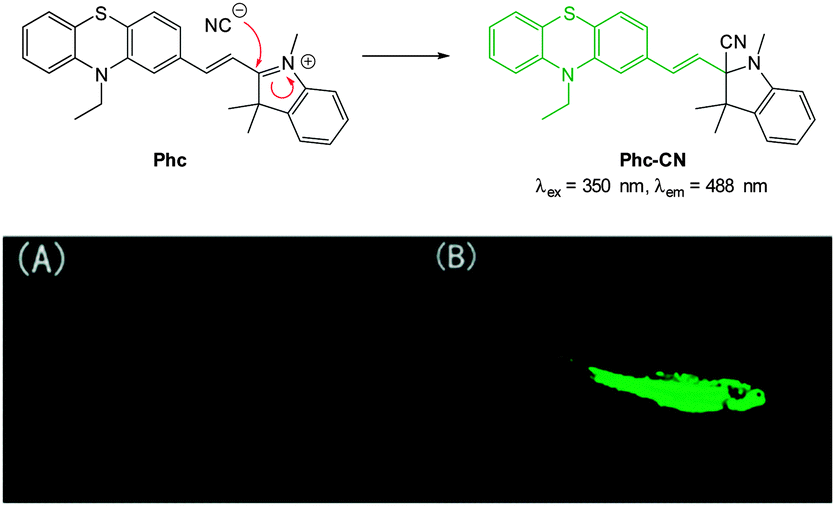 | ||
| Fig. 31 Top: reaction of −CN with indolium Phc. Bottom: images of adult zebrafish under 390 nm light: (A) fish incubated with Phc; (B) fish incubated with Phc and −CN. Image reproduced with permission.177 | ||
The related red-emitting phenazine(di)cyanine-based chemodosimeters PMI and PDMI (Fig. 32) were reported by Hua (2014)178 and again these probes rely on nucleophilic attack of cyanide on a indolium cation. As PDMI has two indolium appendages an excess of −CN was required before the fluorescence “switch on” (λex = 425 nm, λem = 580 nm) occurred. In contrast, for PMI an instantaneous response was observed (λex = 530 nm, λem = 620 nm). Of interest, given that aldehydes are also used as a reactive group for −CN, 1H NMR spectroscopy was used to monitor the intact CHO even as an excess of cyanide was added. The probe located in the cytoplasm In HeLa cells (confirmed using co staining) and a clear “switch on” response to cyanide was noted within 30 minutes of exposure.
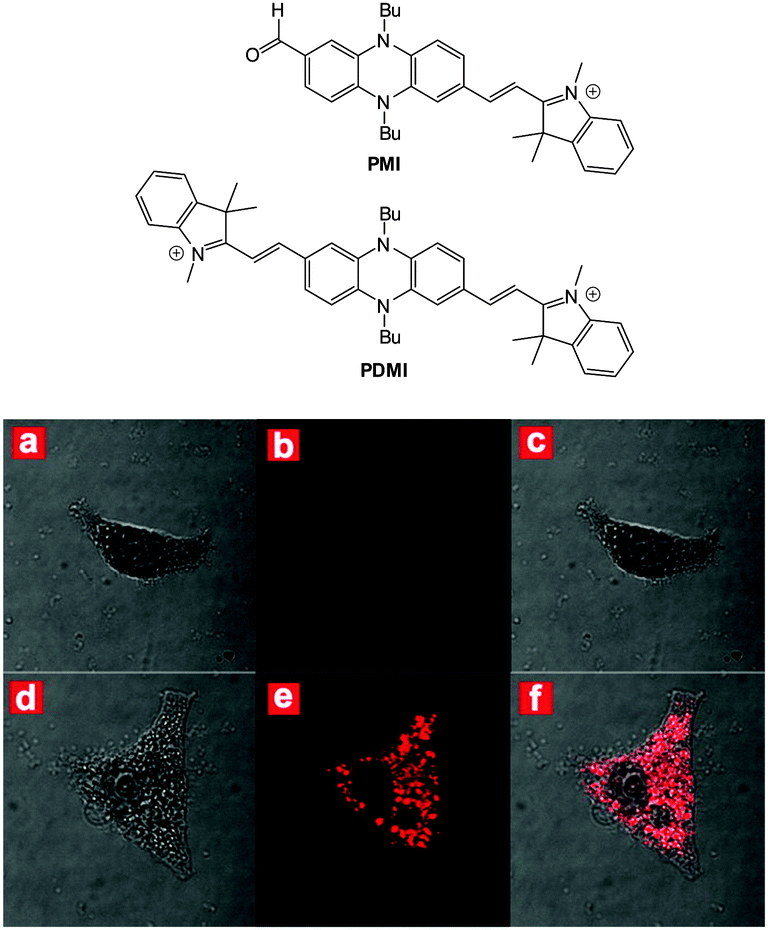 | ||
| Fig. 32 Top: structures of PMI and PDMI. Bottom: images of HeLa cells incubated with PMI: bright-field (a), dark-field (b) and merged (c). Images of HeLa cells incubated with PMI then −CN: bright-field (d), dark-field (e) and merged (f). Image reproduced with permission.178 | ||
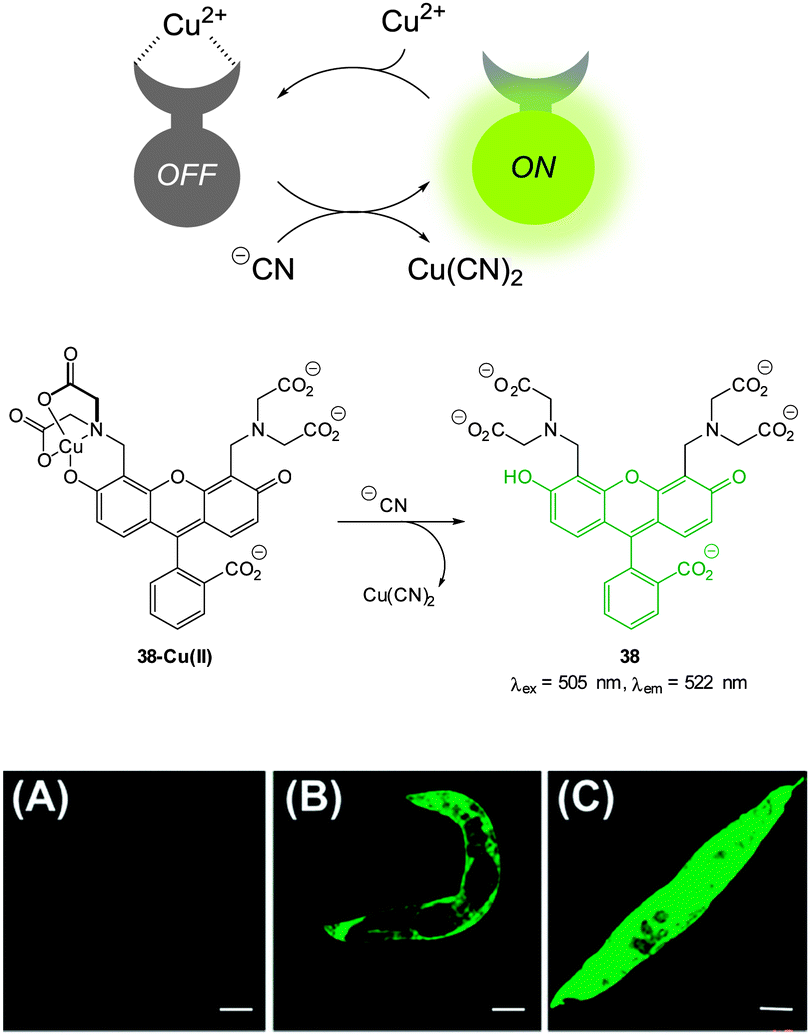 | ||
| Fig. 33 Top: schematic representation of the Cu(II) displacement approach to sensing cyanide. Middle: structure and displacement reaction of 38-Cu(II). Bottom: fluorescence images of (A) young adult nematodes previously incubated with 38 then incubated with Cu(II), (B) nematodes previously incubated with 38 and Cu(II) then incubated with one equiv. of −CN or (C) with ten equiv. of −CN. Scale = 50 mm. Image reproduced with permission.179 | ||
In additional work from the group of Yoon, the NIR emissive cyanine fluorophore (λex = 680 nm, λem = 748 nm) functionalised with picolylamino groups 39 (Fig. 34) was synthesised in a short overall sequence from commercially available cyanine IR-780.181 In the presence of Cu(II) no significant fluorescence was observed (ϕF < 0.01) and initial solution based studies confirmed selective, “switch on” sensing (ϕF = 0.65, λem = 748 nm) of cyanide. Again using the C. elegans nematode as a model organism, aqueous NaCN was readily detected in vivo. Of significant interest when the nematodes were infected with P. aeruginosa (PA14) labeled with green fluorescent protein, the −CN that the bacteria are known to produce was also detected in vivo. Such a result neatly conveys the significance of such small fluorescent probes for medically relevant assays.
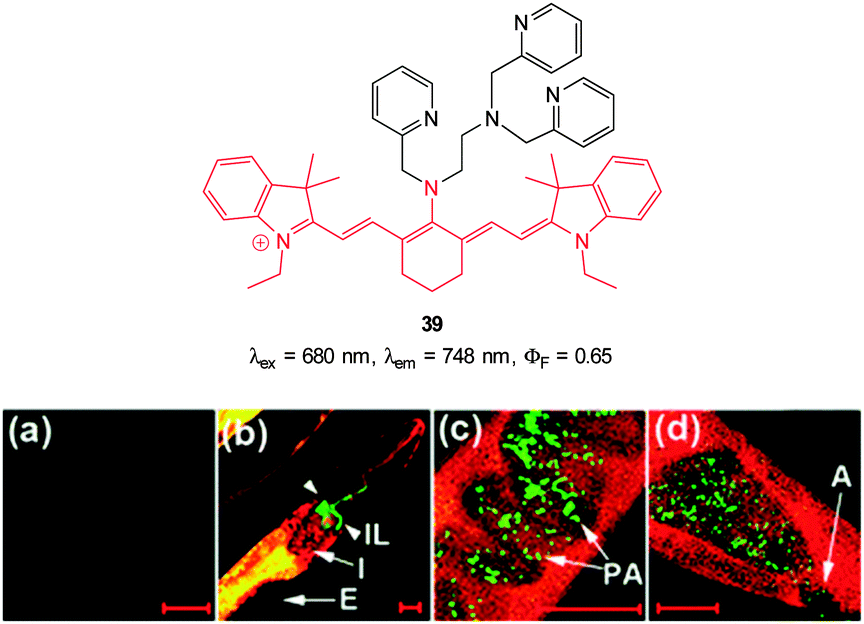 | ||
| Fig. 34 Top: cyanine based ligand 39. Bottom: imaging of cyanide in C. elegans infected with P. aeruginosa (GFP). Prior to incubation with 39-Cu(II)C. elegans nematodes were fed for 2 d on non-infectious E. coli OP50 (a) or GFP-labelled PA14 (b–d). (b) the anterior end, (c) the medial part, (d) the posterior. (I = intestine; IL = intestinal lumen; I = intestine; E = eggs; PA = PA14-GFP; A = anus). Scale = 20 μm. Image reproduced with permission.181 | ||
The Cu(II) displacement approach has also been used by Ghosh and Das to develop a “switch on” probe for cyanide, however, in this instance the emitting species (λex = 380 nm, λem = 583 nm) was a phosphorescent DPA-functionalised iridium complex 40 (Fig. 35).182 Detection of cyanide was achieved inside live HeLa cells within 2 minutes using cells pre-incubated with 40-Cu(II) then exposed to a 0.2 ppm aqueous solution of −CN.
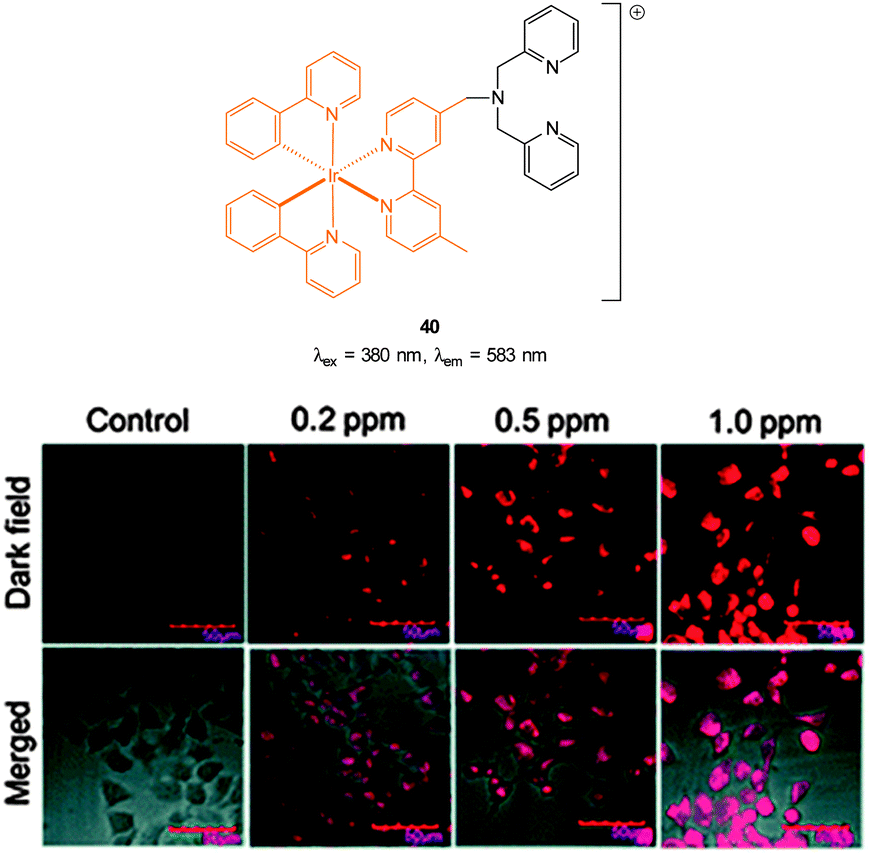 | ||
| Fig. 35 Structure of DPA functionalised Ir complex 40. Bottom: images of live HeLa cells in the presence of 40-Cu(II) acquired after treatment with increasing concentrations of NaCN. Image reproduced with permission.182 | ||
A Cu(II) displacement probe operating by both a colour change and also fluorescent enhancement was reported by Kim.183 Coumarin imine 41 (Fig. 36) was synthesised in four steps from m-anisidine with the last step involving condensation with 2-aminophenol. A crystal structure of the stable non-fluorescent 41-Cu(II) (ϕF = 0.02) confirmed that the metal was complexed by both oxygen atoms and the imine nitrogen atom as shown in Fig. 36. Similar to the previous examples the probe operates by means of Cu(II) displacement, however, unlike the Cu(II) complex, the free coumarin imine 41 is prone to hydrolysis (cyanide actually enhances the rate of hydrolysis) and ultimately it is the coumarin aldehyde 42 (λex = 479 nm, λem = 514 nm, ϕF = 0.65) that functions as the reporting species. Again, excellent selectivity for cyanide amongst a selection of anions was reported (−CN detected at 10−8 M). No adverse effects were noted when human hepatoma cell line HepG2 cells were treated with the complex and a strong intracellular fluorescence response was detected when cells were treated with solutions of KCN.
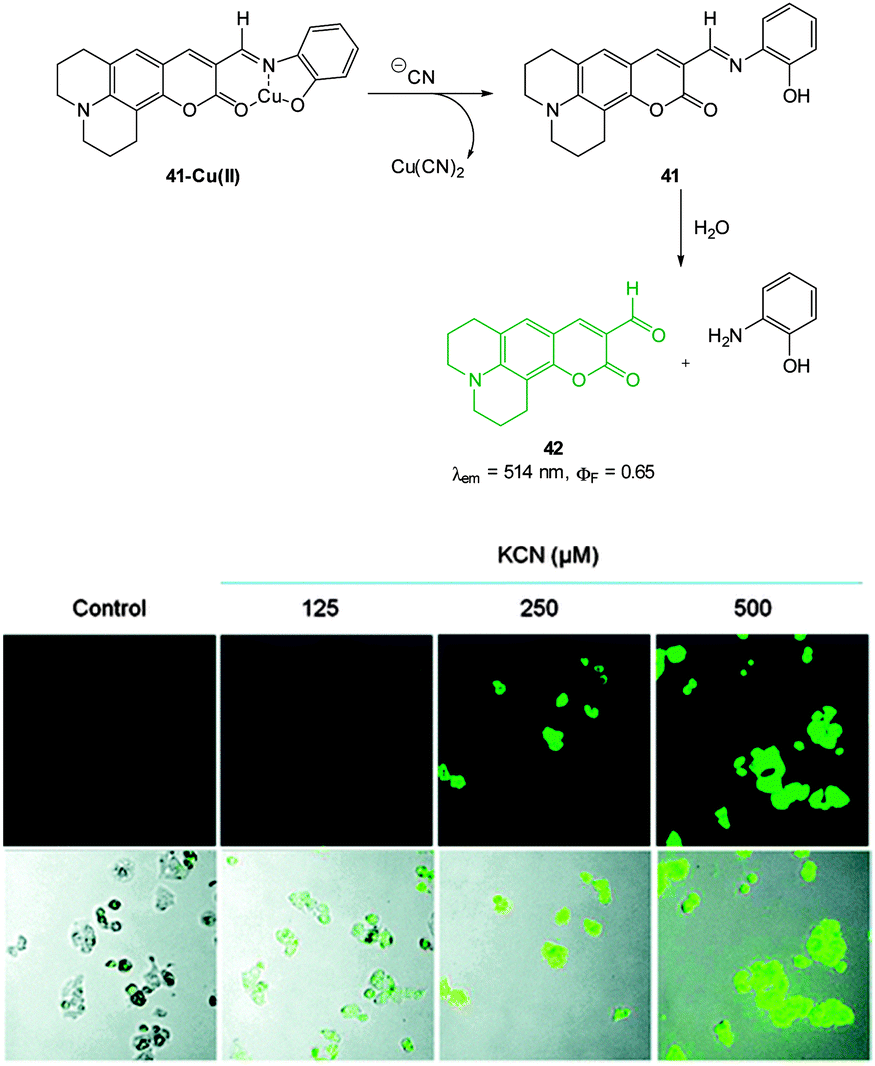 | ||
| Fig. 36 Top: structure, CN mediated displacement, and hydrolysis of 41-Cu(II). Bottom: images of human HepG2 cells pre-treated with 41-Cu(II) (1.0 μM) acquired 1 min after addition of KCN (125, 250, and 500 μM). Image reproduced with permission.183 | ||
An interesting copper displacement probe for cyanide was devised by Zheng (2014).184 The non-fluorescent Cu(II) schiff base complex of benzimidazole hydroxynaphthalene 43 (Fig. 37) was itself formed by displacing Zn(II) from the corresponding, highly fluorescent Zn complex (both the Cu and the Zn complexes were characterised by means of X-ray diffraction). Displacement of copper from the 43-Cu(II) complex was effected by cyanide to give the fluorescent free 43 (λex = 366 nm, λem = 425 nm). Displacement of Cu(II) was also effected by S2− and it should be noted that this anion (known to have an affinity for Cu) is often omitted from the standard suite of anions used to evaluate the selectivity of many Cu based probes. In HeLa cells incubated with the Zn(II) complex addition of Cu(II) quenched the fluorescence, however, the corresponding addition of −CN to restore fluorescence was not performed.
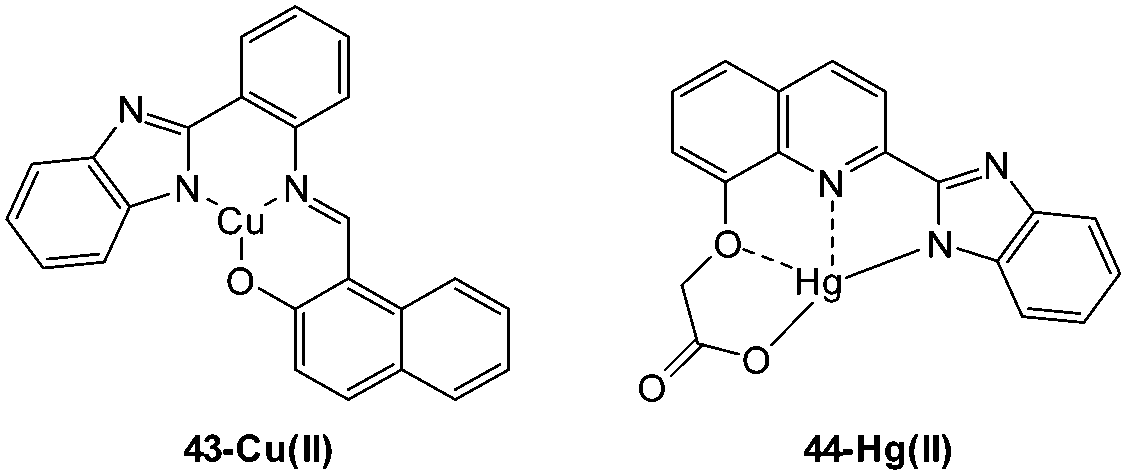 | ||
| Fig. 37 Structure of the naphthalene–benzimidazole complex 43-Cu(II) developed by Zheng184 and the 44-Hg(II) complex from Zhang and Liu.185 | ||
The Hg(II) complex of benzimidazole 44 (Fig. 37) was described by Zhang and Liu (2013)185 for the intracellular sensing of −CN. The complex is non-fluorescent (heavy metal effect) and in the presence of cyanide a strong fluorescence “switch on” (λex = 345 nm, λem = 467 nm) was observed due to displacement of Hg(II). Unfortunately both sulphide and also iodide displaced the cation to give an equivalent response. While these competitors might ultimately limit in vivo applications (toxicity was also not evaluated), successful in vitro sensing of cyanide was demonstrated when HeLa cells that had been incubated with 44-Hg(II) were treated with cyanide.
 | ||
| Fig. 38 Top: structure of −CN probe 45. Bottom: images of 45 in HeLa cells with (left) 30 μM 45 and (right) 30 μM 45 and 30 μM NaCN. Upper images: fluorescence. Lower images: bright-field. Image reproduced with permission.186 | ||
3. Anions of biological relevance
Many crucial intracellular processes involve anionic species47 and the dysregulation of these species is known to accompany a number disease states. Indeed, the dysregulation of intracellular pyrophosphate (PPi) levels is associated with many conditions including cancer (see Section 3.1). Imaging agents for specific anionic targets in vitro and in vivo can be used to confirm the exact biological role of these anions and importantly they can also function as diagnostics for specific medical conditions. Anions covered in this section are: pyrophosphate, bicarbonate and hydrosulfide.3.1 Pyrophosphate
Pyrophosphate (PPi) is produced or used in many cellular metabolic processes, such as ATP hydrolysis and DNA/RNA polymerisation reactions. Intracellular PPi concentrations can provide information on important cellular processes and have recently been suggested as a means of cancer diagnosis.187 The concentration of PPi in other physiological fluids, such as synovial fluid and urine, can also be used to identify diseases such as chondrocalcinosis or calcium pyrophosphate dihydrate (CPPD) crystal deposition disease.188This knowledge has led to the recent development of numerous colourimetric and fluorescent sensors for PPi (see also the review by Yoon in this special issue).14,36,189 However, significant challenges remain in the development of such probes, due to the difficulties associated with binding PPi in water and distinguishing it from related polyphosphates such as ATP. As many of the fluorescent sensors developed for PPi to date are “switch-off” or only exhibit weak fluorescence they are of only limited use in bioimaging applications. Nevertheless, there are several examples where such compounds have been successfully used to image the presence of cellular PPi and there are a handful of recent examples where “switch on” sensors have been developed and effectively used in imaging applications.
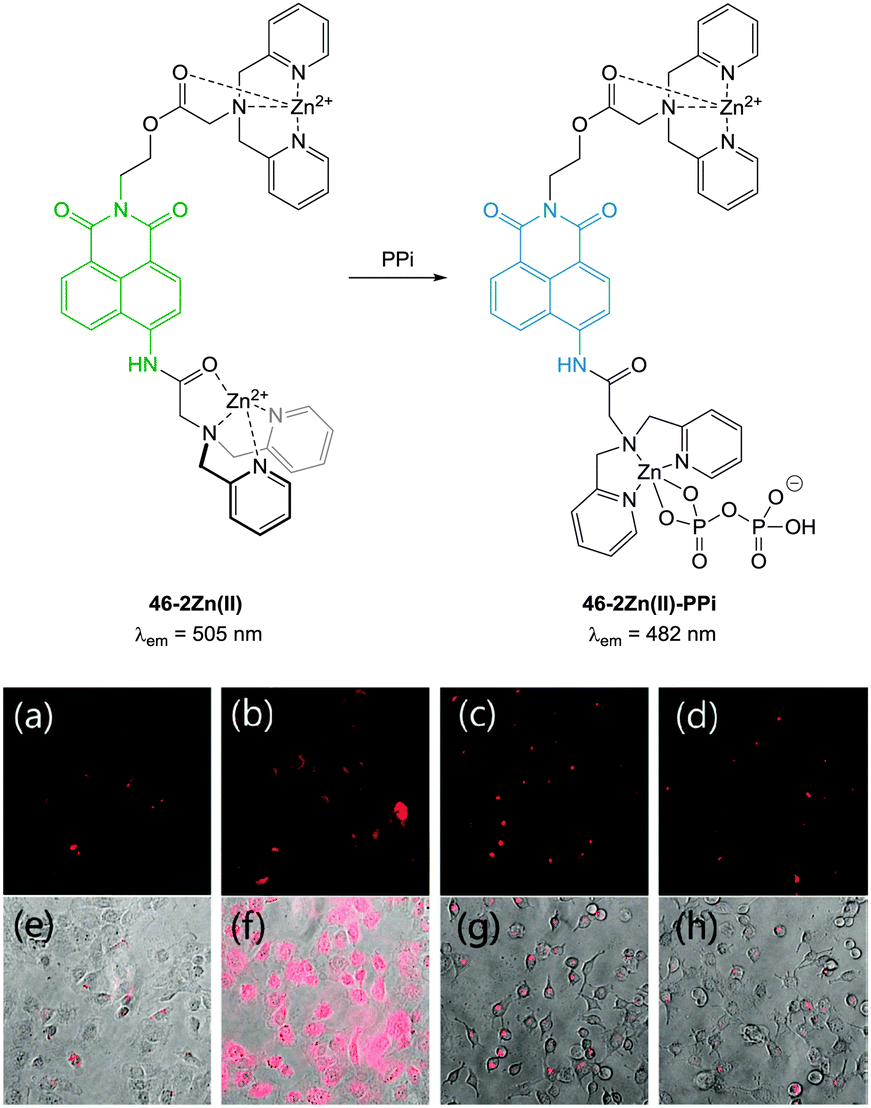 | ||
| Fig. 39 (a) Fluorescence image of C2C12 cells treated with 46-2Zn(II) (1.0 μM). (b) Fluorescence images of C2C12 cells treated with 46 (1.0 μM) and Zn(II) (5.0 μM). (c) Fluorescence images of C2C12 cells treated with 46 (1.0 μM), Zn(II) (5.0 μM) and PPi (0.5 mM). (d) Fluorescence images of C2C12 cells treated with 46 (1.0 μM), Zn(II) (5.0 μM) and PPi (1.0 mM). (e–h) Bright field images of (a–c) respectively. Image reproduced with permission.190 | ||
Hong and co-workers reported the three step synthesis of the NIR emissive benzothiazolium hemicyanine ligand 47 (Fig. 40), which readily forms a bis Zn(II) complex as a PPi binding site.191 In aqueous buffer (pH 7.4) Probe 47-2Zn(II) showed weak emission at 548 nm (λex = 500 nm, ΦF = 0.08) and addition of PPi (1.0 equivalent) “switched on” emission (ΦF = 0.10) with a bathochromic shift to 558 nm. While ATP also gave a measurable response, 47-2Zn(II) was used to image PPi uptake in a C2C12 myoblast cell line with a clear increase in intracellular fluorescence observed 30 minutes following addition of 2.5 equivalents of PPi. Importantly, the cells remained viable as determined using Hoechst nuclear stain and the probe had good cell permeability.
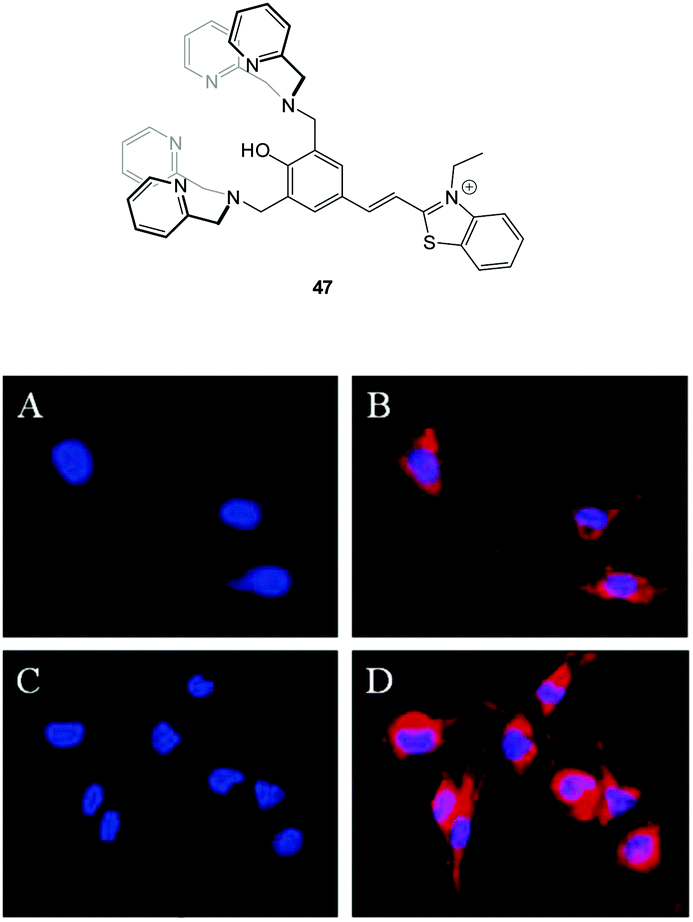 | ||
| Fig. 40 Fluorescence live-cell pseudo-color images of C2C12 myoblast cells. (A and C) Cells stained by Hoechst nuclear dye. (B) Cells incubated with Hoechst nuclear dye and then with 47-2Zn(II). (D) Cells incubated with Hoechst nuclear dye followed by 47-2Zn(II), then Na4P2O7. Emission collected at (A, C) blue channel and (B, D) Cy3 channel upon excitation at (A, C) 350 ± 25 nm and (B, D) 543 ± 11 nm. Image reproduced with permission.191 | ||
:5 mixture of DCCA and Cu(ClO4)2 fluorescence “switch on” was observed, and peaked after 15 equivalents PPi (ΦF = 0.48). The enhancement was attributed to the displacement of one of the ligands from the Cu(II) complex. This probe was evaluated in KB cells and almost no intracellular fluorescence was observed for the [DCCA]2Cu complex alone. After incubation with PPi, a significant increase in cellular fluorescence was observed within 30 minutes with signals localized in the perinuclear area of the cytosol, indicating a subcellular localisation of PPi and good cell membrane permeability of the [DCCA]2Cu complex.
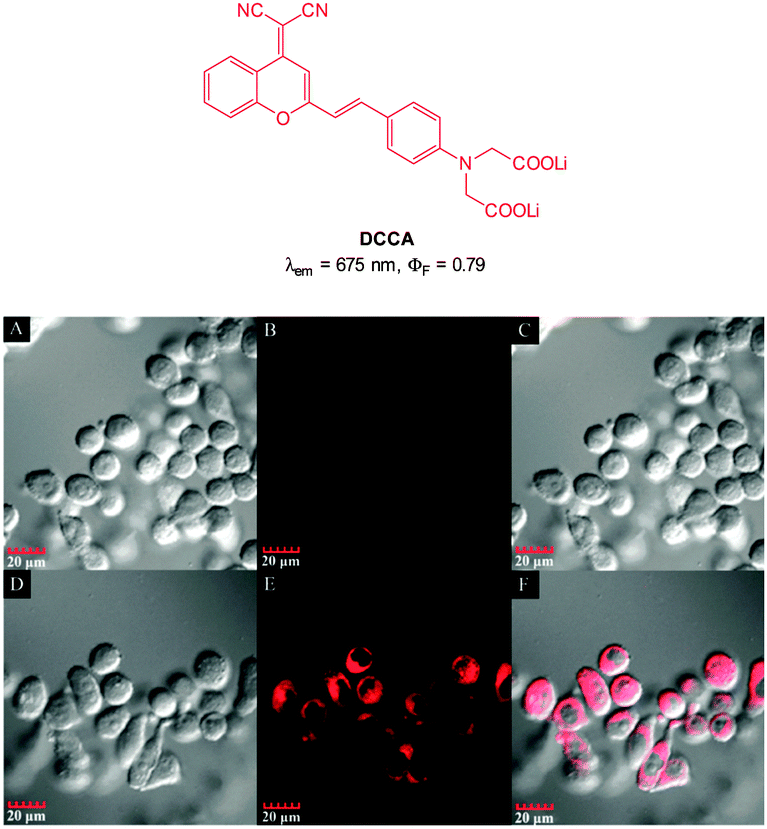 | ||
| Fig. 41 Top: structure of copper ligand DCCA. Bottom: confocal fluorescence images in KB cells: (A–C) cells incubated with DCCA2–Cu(II) alone. (D–F) Cells incubated with DCCA2–Cu(II), then K4P2O7. Emission collected at 630–730 nm, excitation at 405 nm. Bright field (A and D), fluorescence (B and E) and overlap field (C and F). Image reproduced with permission.192 | ||
The imino-thiophenyl calix[4]arene derivative 48 (Fig. 42) has been used to image both Zn(II) and PPi in HeLa cells.193 Ligand 48 was prepared in three steps from p-tert-butylcalix[4]arene and upon addition of Zn(II) in a fluorescence turn-on response (λex = 390 nm, λem ∼ 450 nm) was observed. Subsequent addition of PPi resulted in complete quenching of the emission, attributed to the displacement of Zn(II) from the 48-Zn(II) complex as a result of the higher binding affinity of PPi for Zn(II). In HeLa cells incubated with 10 μM 48, very low fluorescence intensity was observed. After subsequent incubation with ZnSO4/pyrithione for 20 minutes, the cells exhibited highly intense blue fluorescence (4 times higher than with 48 alone). Further treatment with PPi resulted in a decrease in fluorescence intensity (1.5 times higher than 48 alone).
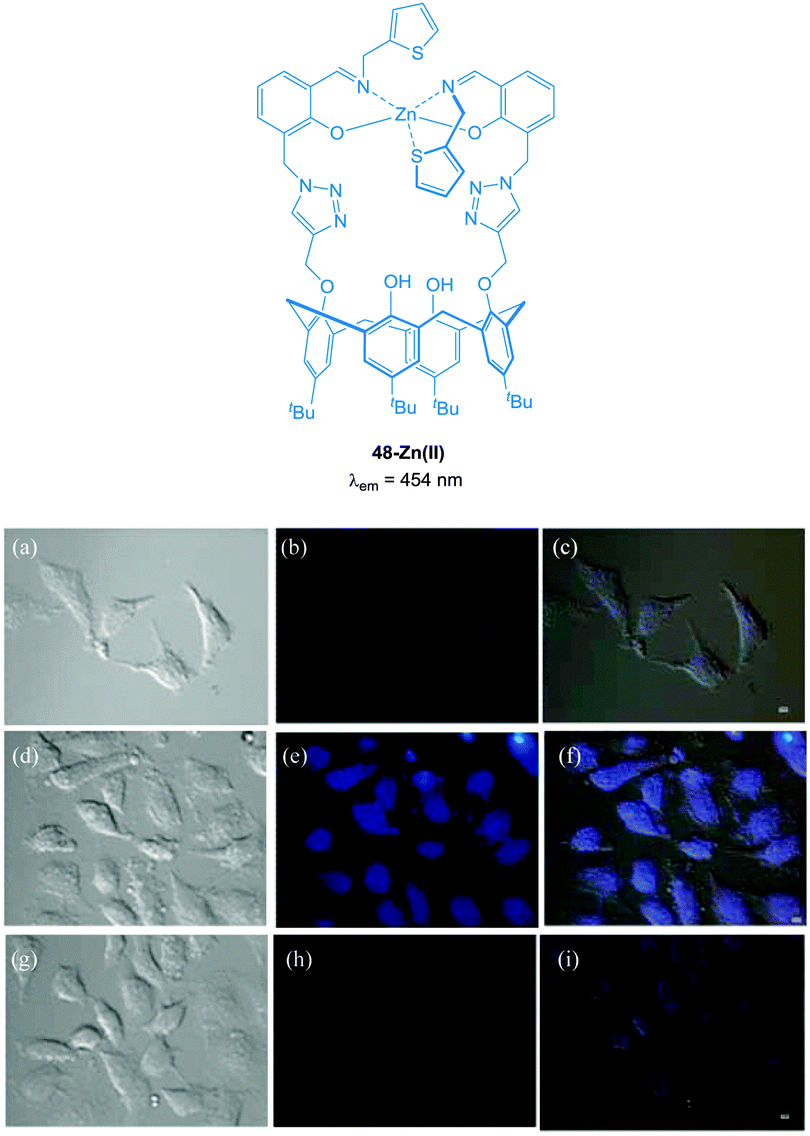 | ||
| Fig. 42 Fluorescence images in HeLa cells (λex ∼ 358 nm and λem ∼ 461 nm); (a) DIC image of cells treated with 48 (10 μM); (b) fluorescence image of (a); (c) merged image of (a) and (b). (d) DIC image of cells treated with 48 then Zn(II)/pyrithione (1:1) solution; (e) fluorescence image of (d); (f) merged image of (d) and (e). (g) DIC image of cells treated with [48 + Zn(II)] followed by PPi; (h) fluorescence image of (g); (i) merged image of (g) and (h). Scale = 10 μm. Image reproduced with permission.193 | ||
The bis Zn(II) complex of the pyridine-naphthalene based SPHN (Fig. 43) has been reported as a PPi selective fluorescent chemosensor.194 Compound SPHN was readily prepared by condensation of the bis-glycine adduct of 2,6-diaminopyridine and 2-hydroxy-1-naphthaldehyde. In 7:3 CH3CN:aqueous HEPES buffer the addition of ZnCl2 to SPHN resulted in a “switch-on” of fluorescence (λex = 400 nm; λem = 450 nm, ΦF = 0.940) which was suggested to be a result of the formation of a 1:2 L:Zn(II) complex. The addition of PPi to SPHN-2Zn(II) led to quenching of this fluorescence as a result of displacement of Zn(II) from SPHN and importantly, a selective response for PPi was observed in the presence of ATP. In HeLa cells preincubated with exogenous Zn(II) the addition of SPHN elicited a fluorescence response, however, when PPi was added at the same time as SPHN, significantly lower levels of fluorescence were observed.
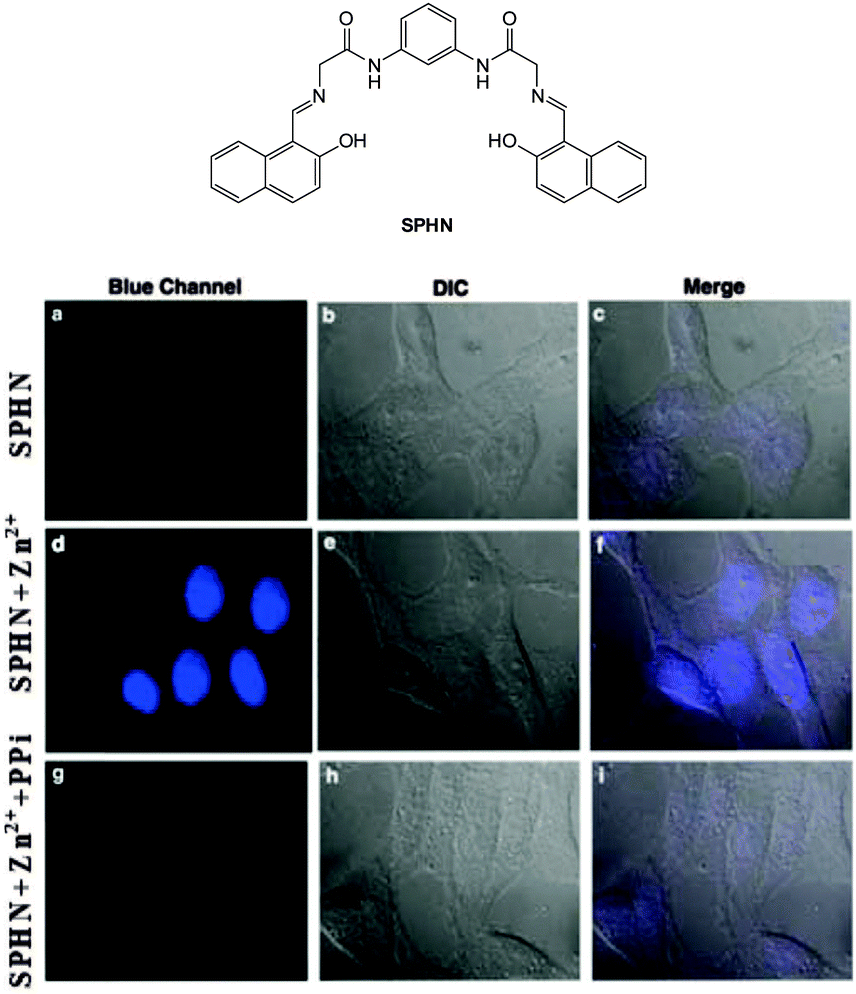 | ||
| Fig. 43 HeLa cells showed intense blue fluorescence in the presence of both SPHN and Zn2+ (d) and did not show any fluorescence in the absence of Zn2+ (a) and in the presence of PPi (g). Corresponding differential interference contrast (DIC) images (e, b and h) and merge images (f, c and i) of the cells are shown. Image reproduced with permission.194 | ||
More recently, Rissanen and co-workers have reported a terpyridine–Zn(II) complex 49-ZnCl2 (Fig. 44) capable of the detection of nanomolar PPi concentrations in water, together with the first example of a small molecule probe to image native PPi concentrations in cells (i.e. without the addition of exogenous PPi).195 The complex was prepared by mixing 4′-(4-N,N′-dimethylaminophenyl)-2,2′:6′,2′′-terpyridine in a 1:1 ratio with ZnCl2 and an X-ray crystallographic structure, confirmed the formation of a 1:1 complex. While 49-ZnCl2 is fluorescent in the solid state, in water the fluorescence is quenched. The addition of PPi to a solution of 49-ZnCl2 in 0.01 M HEPES buffer resulted in an approximately 500 fold increase in fluorescence (λex = 440 nm; λem = 591 nm), attributed to the formation of a 1:3 complex between PPi and 49. Cellular imaging was performed in HeLa cells. Cells were treated with 10 μM 49-ZnCl2 for 30 min and bright orange-yellow emission was observed that allowed mapping of PPi concentration in different parts of the cells with the maximum emission observed in the nuclei as well as the cytoplasmic membranes.
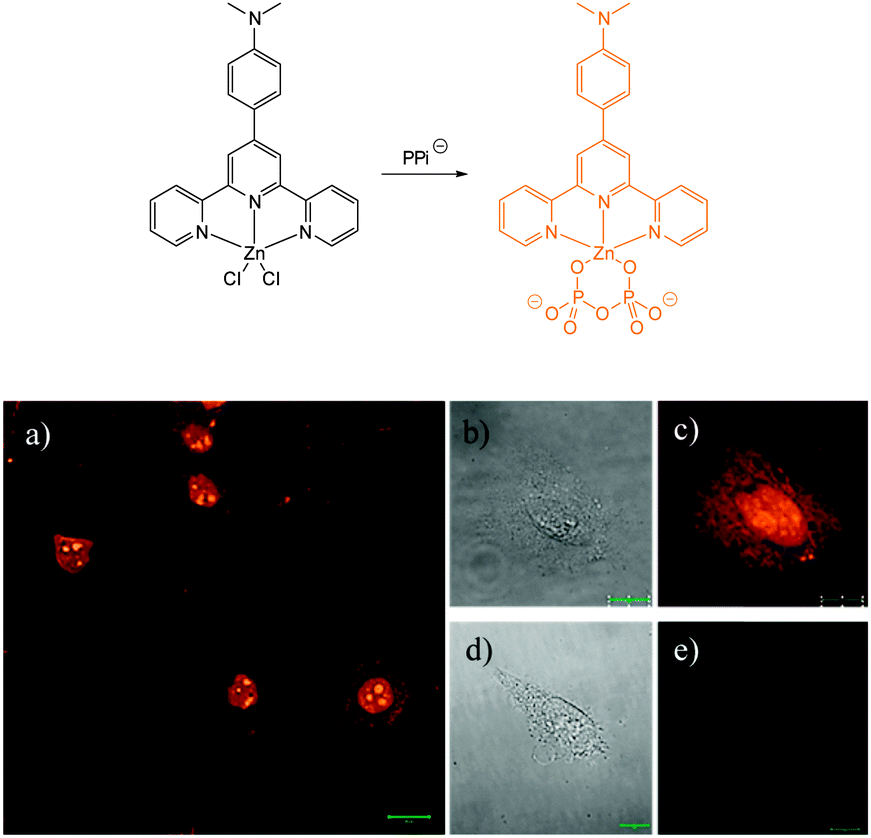 | ||
| Fig. 44 Confocal fluorescence microscopy images of (a) HeLa cells incubated with probe 49-ZnCl2 (50 μM), (c) single HeLa cell, and (e) single HeLa cell without staining (control). Differential interference contrast images of (b) single HeLa cell incubated with probe 49-ZnCl2 (50 μM) and (d) single HeLa cell without staining (control). Image reproduced with permission.195 | ||
3.2 Bicarbonate
The bicarbonate anion is the primary species responsible for maintaining cellular acid–base homeostasis. The enzyme carbonic anhydrase (CA) produces HCO3− inside cells from dissolved CO2 and dysregulation of CA is associated with a number of tumour types.196 The bicarbonate anion also plays a role in physiological processes such as cyclic AMP regulation, osteoporosis and kidney disease.197 Unfortunately only indirect methods have been available for measuring bicarbonate in cells including total H14CO3− concentration or estimates based on pH; each of these are prone to significant error and do not provide spatiotemporal information. As such a more direct means for the bioimaging of this anion using fluorescent probes would be a welcome advance.The dialkynylbenzene probe 50 for the imaging of bicarbonate in vitro was reported by Murphy, Wong and Lee in 2011 (Fig. 45).198 A NIR multiphoton approach was used for excitation and probe emission also tailed into the NIR region. Solution studies identified strong binding of bicarbonate (logK = 7.13) and a four-fold enhancement in fluorescence intensity (λem = 450 nm) was observed as well as a redshift of 30 nm. Binding was tentatively assigned to the δ+ of the amide N leading to enhanced electron transfer (1:2 H:G binding stoichiometry supported this theory). In solution, binding of citrate was also observed (logK = 7.83), nevertheless, in vitro imaging of HCO3− was performed in both HeLa and A549 cells (λex = 900 nm and λem = 400–650 nm) and after 3 h the probes had localised in the cytoplasm and the emission profiled matched the 30 nm red shift observed in solution indicating binding of the target anion.
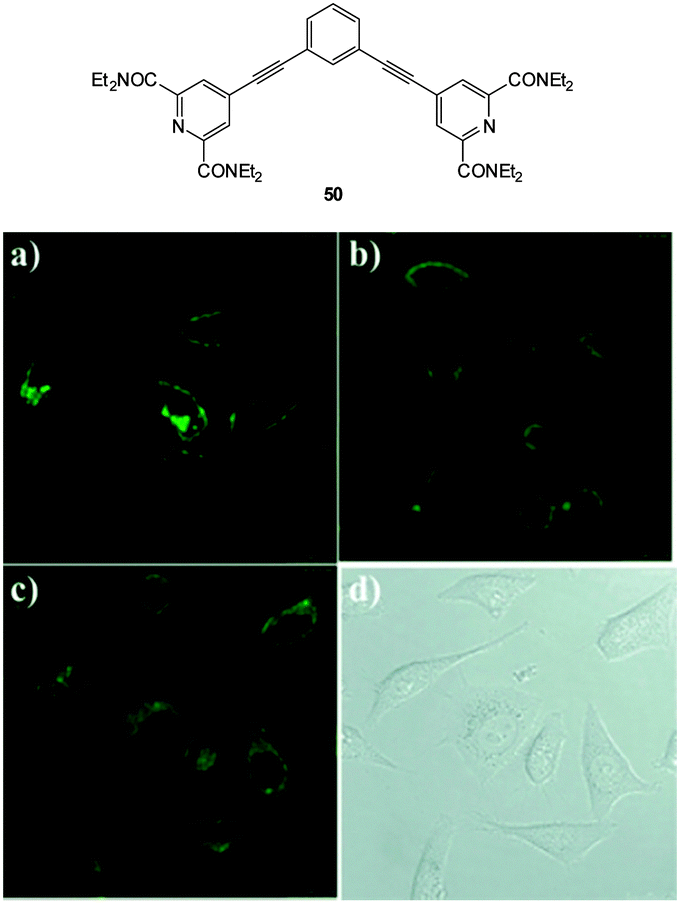 | ||
| Fig. 45 Top: structure of dialkynylbenzene probe 50. Bottom: in vitro cytoplasmic staining microscopy with 50 (λex = 900 nm, λem = 450–750 nm) in A549 cells after (a) 1 h, (b) 6 h, and (c) 12 h. (d) Bright-field image of panel (c). Image reproduced with permission.198 | ||
With the aim of sensing bicarbonate and other oxyanions (such as malate and citrate) “in cellulo” a large body of research effort has been undertaken by the group of Parker using luminescent lanthanide probes.83,199 The cellular uptake, localisation, stability and toxicity of these probes have all been studied in detail.45,81,200–202 These sensors have sharp emission bands (ΔJ = 1, 2, 3 and 4) and as they rely on energy transfer from a chromophore/sensitiser (normally incorporated as part of the ligand) to the metal the emission can be modulated by the target anion disturbing either the metal excited state or the ligand singlet or triplet states (for excellent overviews of the photophysical properties of lanthanides and how they can be manipulated for sensing and imaging see the excellent recent reviews by Meade,82 Parker,83 and Pierre203). Lanthanide complexes are capable of selectively binding anions if both the ligand and metal are judiciously selected. Typically, but not always, the mode of action involves displacement of a bound water (q) by the target anion leading to a modulated emission profile in which the ΔJ = 2 band (e.g. Eu at ca. 620 nm) is considerably altered whereas other bands such as ΔJ = 1 (e.g. Eu at ca. 590 nm) are not. Changes in these two distinct outputs allows the probe to function in a very useful ratiometric fashion.82,83,203 Combinations of individual Tb and Eu complexes (one complex is responsive, the other is not) can also function in a ratiometric manner.204
The detection of bicarbonate by the lanthanide complexes 51-Eu(III) and 52-Eu(III) containing the 1-azaxanthone-4-carboxyl sensitiser was reported by Parker in 2011 and in a follow up study in 2012 (Fig. 46).205,206 The probes were non-toxic and indicated the presence of bicarbonate (HCO3− formed by exposing the cells to a CO2 atmosphere) in the mitochondria of a number of cell lines including A549, MCF-7 and HeLa cells. While solution based binding studies indicated little selectivity of the probes over the common carboxylate interferents citrate and lactate, in cells the concentration of bicarbonate is typically 10 fold greater than lactate and 100 times greater than citrate. Of additional interest the same ligands with terbium were unresponsive and could serve as in internal control for ratiometric imaging. Direct detection in A549 cells and selectivity for bicarbonate was confirmed as when the known carbonic anhydrase inhibitor acetazolamide was introduced to the cells little fluorescence was observed. Binding of the bicarbonate anion modulates either triplet sensitiser to metal energy transfer or the lanthanide excited state directly and ultimately leads to significant enhancement of the ΔJ = 2 band at ∼620 nm (λex = 340 nm).
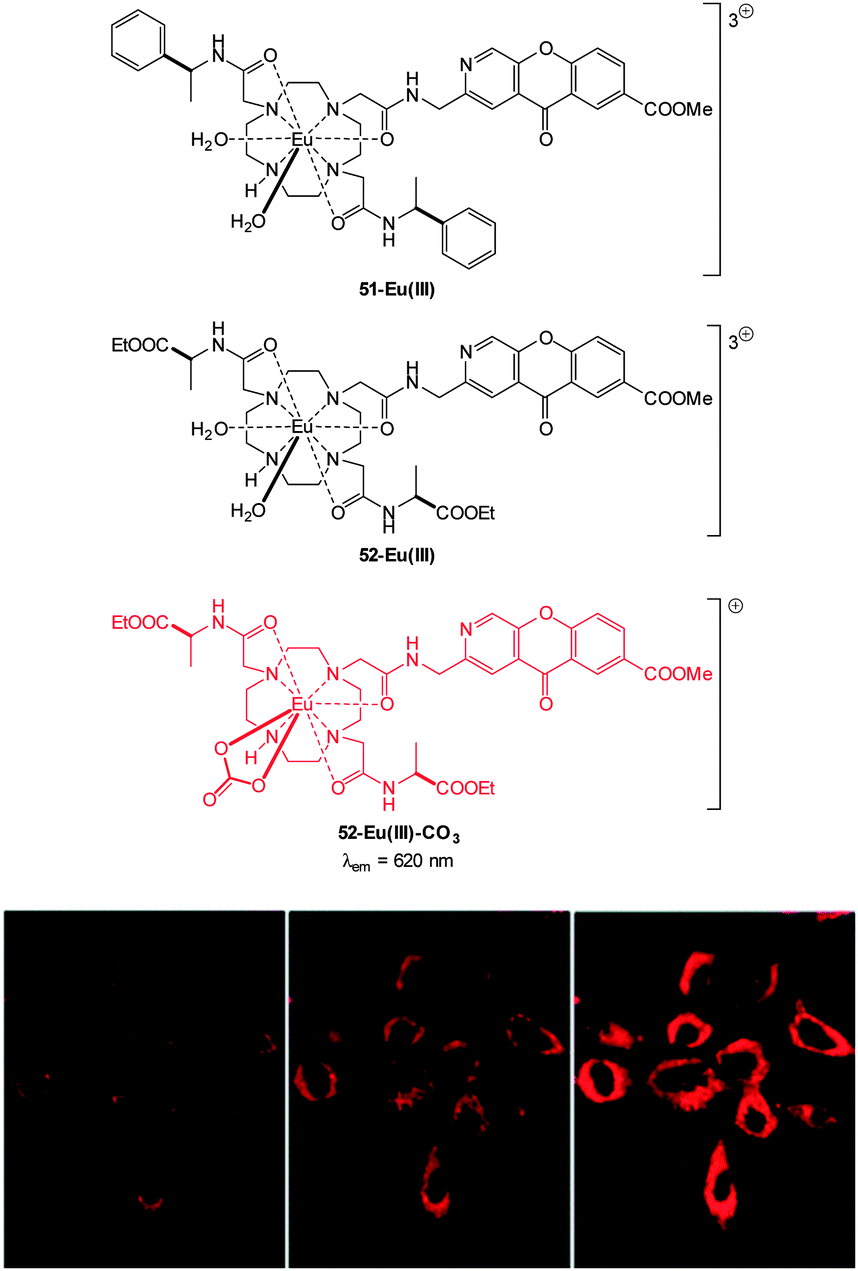 | ||
| Fig. 46 Top: structure of HCO3− sensitive probes 51-Eu(III), 52-Eu(III) and the bound 51-Eu(III)-CO3. Bottom: images of HeLa cells incubated with 52-Eu(III), localised in the mitochondrial region under 3, 4 and 5% CO2 atmosphere. Image reproduced with permission.211 | ||
3.3 Hydrosulphide
Hydrogen sulphide (H2S), along with NO and CO, is considered to be the third gaseous signalling agent, or gasotransmitter.207 Endogenous hydrogen sulphide is produced from homocysteine (Hcy) by the cytosolic enzymes; cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CGL).208 In addition hydrogen sulphide is also produced from cysteine (Cys) by 3-mercaptopyrate sulfurtransferase (3MST) mediated metabolism; 3MST is present in both the cytosol and mitochondria.208 Considering the first and second pKa values for H2S are 7.05 and 15 respectively (at 25 °C and pH 7.4 the ratio of H2S/HS−/S2− can be calculated as 30:70:0.00002 respectively).207 As such, for the purpose of this review we can assume that appreciable quantities of HS− are present. Nevertheless the challenge of targeting HS− is complicated as pH varies between subcellular compartments and as such the H2S/HS− ratio will also vary accordingly.
Hydrogen sulphide has been linked to a number of physiological processes such as inflammation, angiogenesis, respiration, ischaemic reperfusion injury as well as oxidative stress.208,209 As such the development of HS− releasing drugs is an active area of research.210
Due to the rapid catabolism of HS− by sulphide quinone oxidoreductase (SQR), persulfide dioxygenase (SDO), thiosulfate reductase (TR) and sulphite oxidase (SO),208,211,212 any probe must react quickly and emit brightly (high quantum yield (ϕF) and molar absorptivity (ε)). This formidable challenge is made more difficult by the fact that the typical concentration of HS− in blood are in the order of 10−6 M to 10−9 M and 30–300 μM in tissues have been reported.207,212 There is still debate over the physiologically active form of hydrogen sulphide,212 therefore a selective means to detect low concentrations of the anionic form could unlock some of the secrets of its remarkable biology.
The development of fluorescent indicators to sense HS− in living systems is a rapidly developing field.52,213–216 Given the large number of examples this review will highlight bioimaging agents that either (i) present significant advances in sensitivity, (ii) target particular cellular locations, (iii) emit in the NIR range and (iv) have been demonstrated to be applicable to in vivo studies.
For probes that target hydrosulphide, three strategies are commonly employed to modulate fluorescence. The first two are chemodosimeter approaches that rely on HS− as a nucleophile or HS− mediated reduction. The third strategy is sulphide mediated metal displacement. Nucleophilic reactions (typically SNAr or nucleophilic addition) are used to restore ICT or remove a group involved in a PET process, displace a trigger or interrupt a conjugated system. Anionic HS− is a superior nucleophile compared to thiols (pKa > 8.5) at physiological pH. When two proximal electrophiles are present interference from endogenous sulphur species such as cysteine (Cys), homocysteine (Hcy) and glutathione (GSH) is further circumvented. Reduction of an azide is an often utilised approach as the azide can be introduced directly to a fluorophore bearing an arylamine as a component of an ICT system, or as a sulfonylazide. Reduction of nitro groups, hydroxyl amines, and N-oxides are related examples of this strategy, however the reduction of the nitro-group invariably suffers from poor reaction kinetics. The displacement of a metal, typically Cu(II) or Zn(II), from a chelating ligand has also been widely used for bioimaging HS−. There are obvious parallels between this approach and the probe design for −CN selective probes (see Section 2.4).
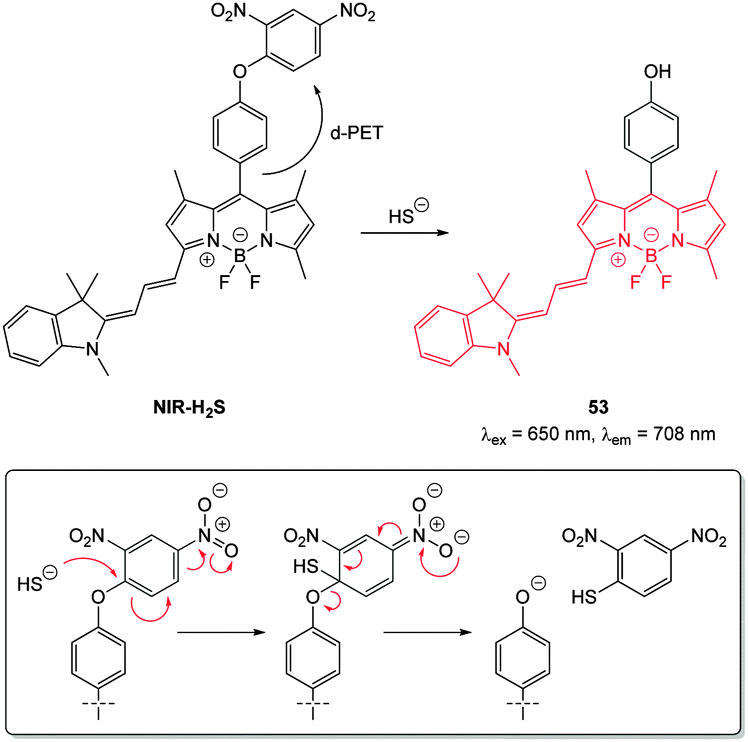 | ||
| Fig. 47 Structure and reaction of cyanine–BODIPY hybrid probe NIR-H2S with HS−; INSET shows fragmentation mechanism. | ||
Zheng and Cui (2014) functionalised nile red with this trigger to give a NIR-emissive (λex = 488 nm, λem = 655 nm) HS− responsive probe NR-HS (Fig. 48).218 Upon reaction with HS− the quantum yield of NR-HS (ΦF = 0.05 in simulated physiological conditions) increased to 0.32 as nile red was regenerated. Maximum fluorescence was obtained after 20 minutes with a limit of detection of 270 nM. Fluorescence microscopy experiments were carried out using MCF-7 cells and NR-HS responded to exogenous HS−.
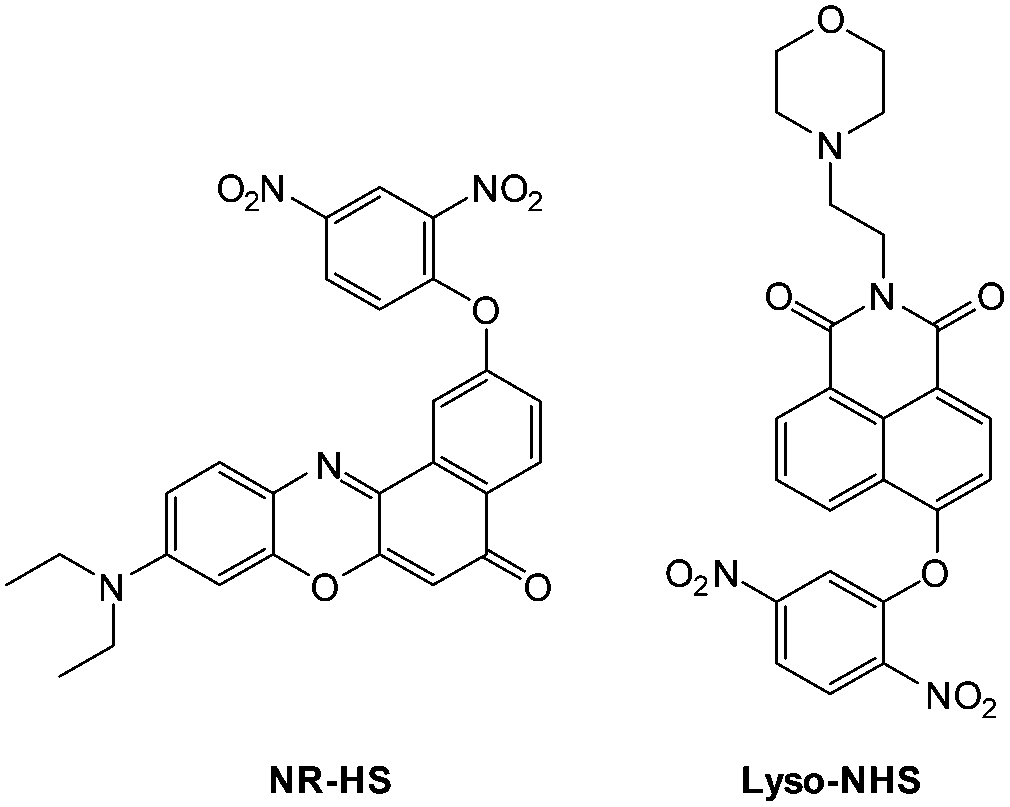 | ||
| Fig. 48 Structures of NR-HS and Lyso-NHS. | ||
The 2,5-dinitrophenylether trigger was incorporated onto a 4-hydroxynaphthalimide fluorophore by Liu et al. to give Lyso-NHS (Fig. 48). The probe also contained a basic morpholine substituent (Lyso-NHS pKa = 3.12) which was responsible for compartmentalisation of the sensor into the lysosome (Fig. 49).100 The reaction of HS− with Lyso-NHS led to a significant increase in fluorescent intensity (λex = 450 nm, λem = 555 nm) with a maximal response after 20 minutes. The probe had a nanomolar (480 nM) detection limit and was used to visualise exogenous HS− in MCF-7 cell lysosomes (confirmed by co-staining with neutral red (NR) a known lysosomal stain) and was not toxic (MTT assay) at the concentrations used.
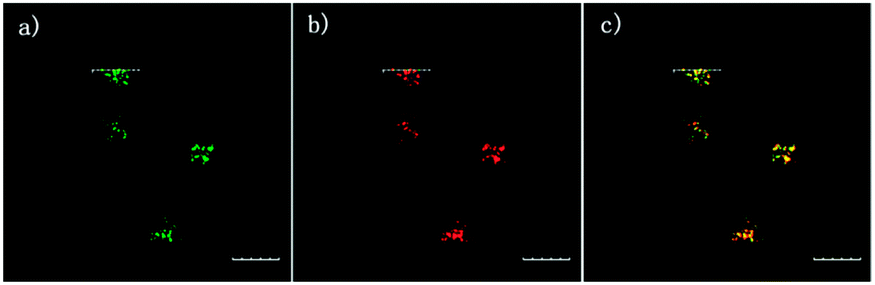 | ||
| Fig. 49 Colocalisation images of Lyso-NHS in MCF-7 cells. (a) Lyso-NHS with HS− (green channel. (b) Neutral red (red channel). (c) Merged images of (a) and (b). Image reproduced with permission.100 | ||
Also using the 2,5-dinitrophenylether trigger, the group of Govindaraju synthesised the HS− probe DNOPCy (Fig. 50).219 Dislodging the 2,5-dinitrophenyl group of DNOPCy (λem = 555 nm) gave Cy-quinone which has a red-shifted emission maximum at λ = 695 nm. This change in fluorescence emission was ideal for ratiometric detection and visualisation of exogenous NaSH in human embryonic kidney cells (HEK293T) was successfully accomplished.
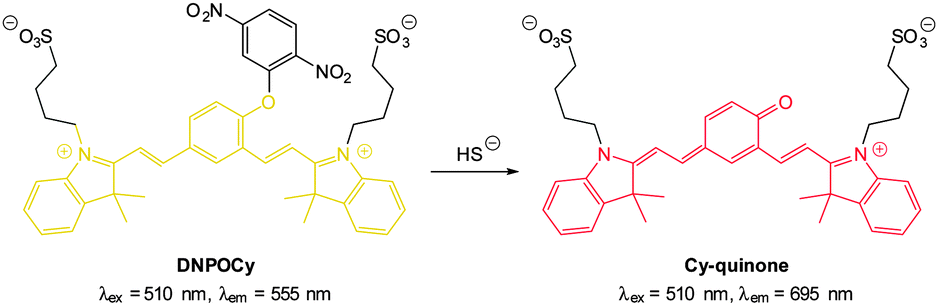 | ||
| Fig. 50 NIR emissive HS− probe DNOPCy. | ||
Through the judicious placement of a proximal aldehyde, Feng (2014) developed the dinitrophenyl ether probe 54 (Fig. 51) that reacted fully within two minutes of exposure to HS− to give maximum fluorescence.220 The reaction generates the modified HMBT ESIPT fluorophore 55 (λex = 450 nm, λem = 555 nm, ΦF = 0.18) and HS− at concentrations as low as 48 nM were detected in simulated physiological conditions. This probe was successfully used for the visualisation of exogenous HS− in HeLa cells.
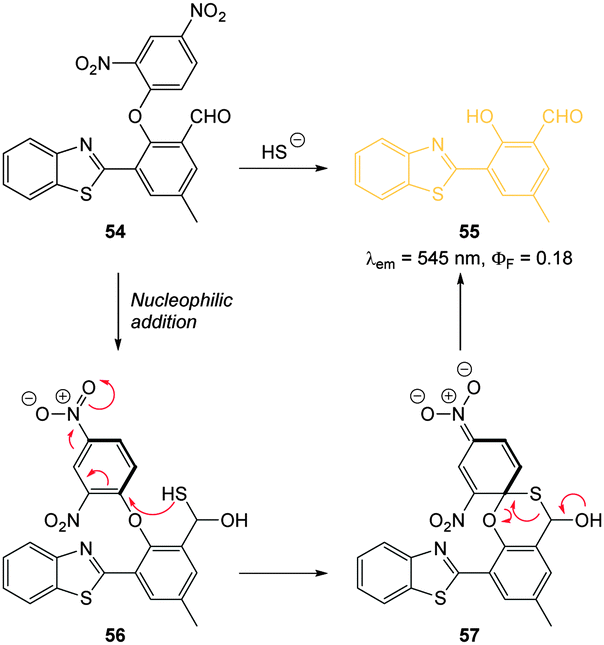 | ||
| Fig. 51 Structure and reaction mechanism of benzothiazole probe 54 with HS−. | ||
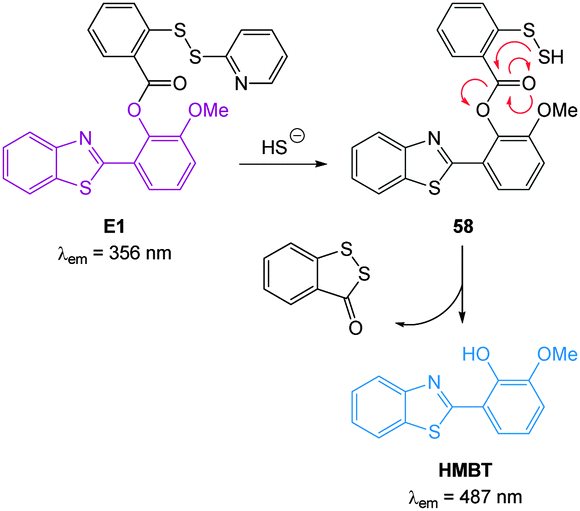
In 2012 Qian and co-workers reported a rapidly reacting HBMT based HS− activated fluorescent probe (Scheme 1).221 Probe E1 can undergo thiol exchange with both HS− and thiols, however only the persulfide generated from HS− can cyclise to release the fluorescent HBMT (λex = 295 nm, λem = 487 nm). Probe E1 was weakly fluorescent due to PET from the pendant dithiol and reacted rapidly (2 min) with HS− with an detection limit of ca. 120 nM. Once again this probe readily detected exogenous HS− in HeLa cell.
 | ||
| Scheme 1 Structure and reaction mechanism of E1 with HS−. | ||
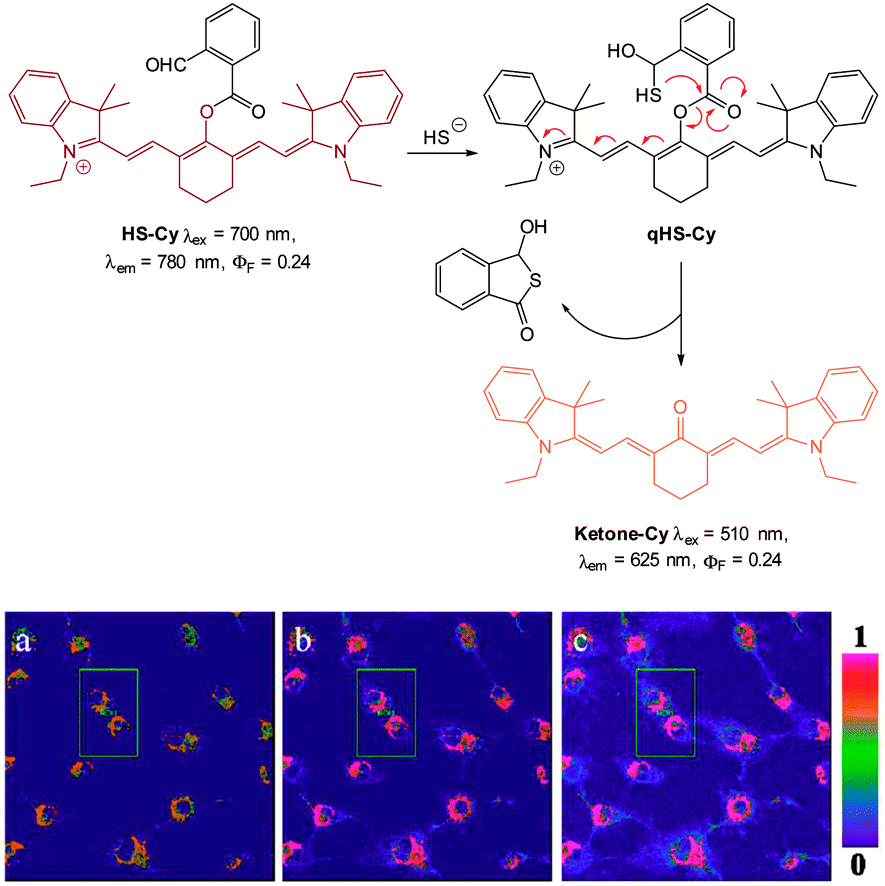
In 2013 the Tang group reported the HS− responsive ratiometric probe HS-Cy (Fig. 52) which incorporates a proximal electrophile.222 Nucleophilic addition of HS− at the aldehyde of HS-Cy leads to a rapid (within 3 min) loss of fluorescence emission (λem = 780 nm, ΦF = 0.24). This decrease in fluorescence was speculated to be caused by a PET process from the free hydroxyl or sulfhydryl groups of the intermediate qHS-Cy. The intramolecular cyclisation between the free sulfhydryl and the ester the releases the ketone cyanine (ketone-Cy) resulting in a 155 nm blue-shifted emission (λem = 625 nm). The cyclisation step is a much slower process, with the emission at 625 nm increasing slowly over 35 min. In the mitochondria of HepG2 cells (pH 8.0) the nucleophilic addition and substitution occurred within 30 s and 5 min respectively. The fluorescence intensity ratio F625/F780 rises from 0.01 to 24.8 following the addition HS−. Probe HS-Cy was used to image exogenous HS− in HepG2 cells and could detect endogenous HS− in A549 cells stimulated with sodium nitroprusside (SNP). A decrease in the F625/F780 ratio was observed when an inhibitor (DL-propargylglycine, PPG) of the HS− producing enzymes (CBS and CES) was added to the cells, confirming HS− as the analyte responsible for the response.
 | ||
| Fig. 52 Top: structure and reaction of HS-Cy with HS−. Bottom: confocal fluorescence ratiometric images of endogenous HS− in living A549 cells. A549 cells loaded with 5 mM HS-Cy for 30 min (a). Cells were pre-stimulated with SNP, then incubated with HS-Cy for 10 min (b) and 20 min (c). Scale = 50 mm. Image reproduced with permission.222 | ||
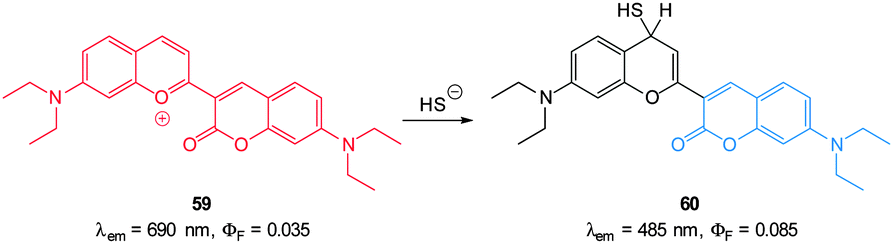
Guo and co-workers reported a fast reacting flavylium derived ratiometric HS− probe 59 (Fig. 53).223 Nucleophilic attack of HS− on this NIR emissive probe (λex = 450 nm, λex = 690 nm) disrupts conjugation and the fluorescent product is essentially a substituted aminocoumarin (λex = 485 nm). In pure PBS buffer, the reaction was complete in 20 s, making probe 59 one of the fastest probes yet reported. Upon treatment with HS− a 694-fold increase in the ratio F485/F690 (0.07–83.90) was noted allowing a detection limit of 140 nM. While this probe did react with mercaptoethanol, selectivity for HS− over cysteine and GSH was apparent. It was suggested that electrostatic repulsion between the benzopyrylium ion and the protonated amines of Cys and GSH prevents addition. Probe 59 was found to be non-toxic (MTT assay) and was subsequently used for the ratiometric imaging of exogenous HS− in HeLa cells.
 | ||
| Fig. 53 Structure and reaction of 59 with HS−. | ||
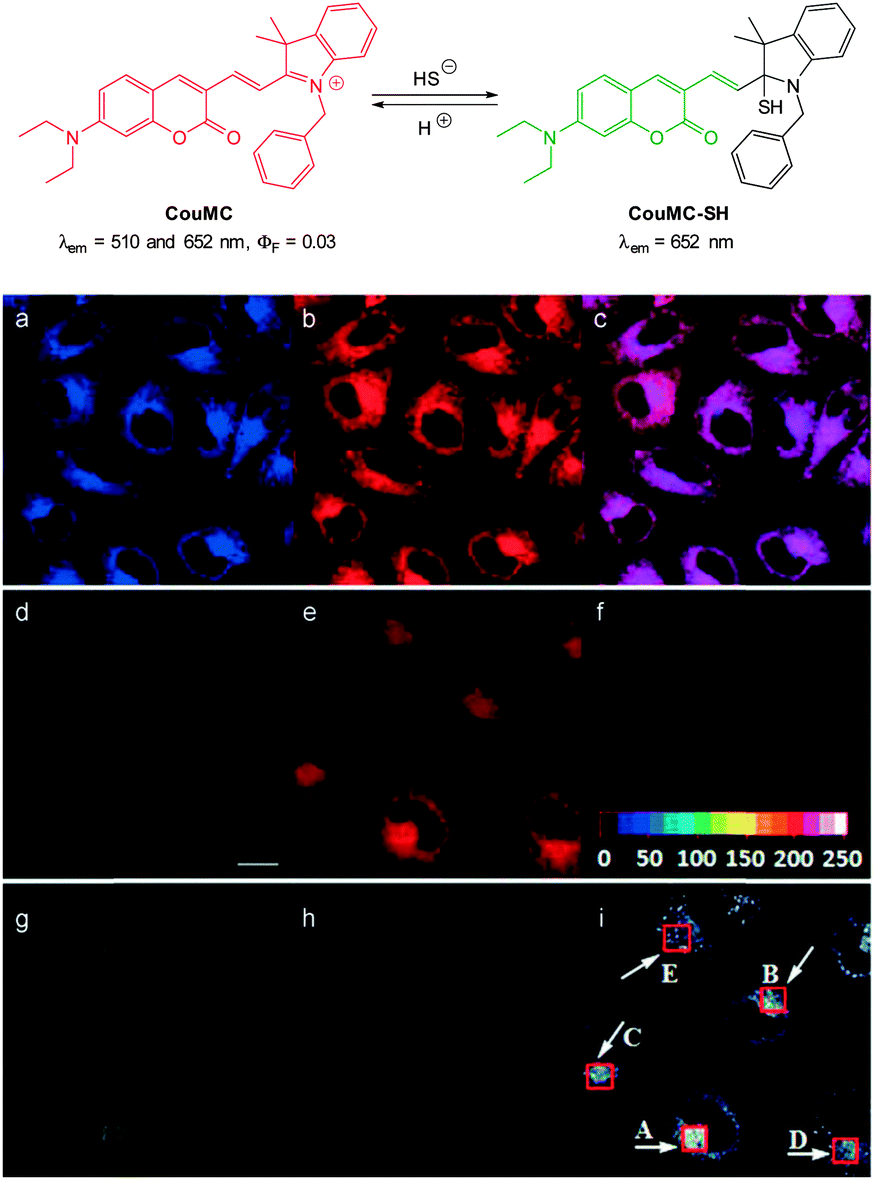
The mitochondria selective HS− probe CouMC (Fig. 54) was reported by the groups of He and Guo in 2013.224 The coumarin-hemicyanine probe CouMC has two fluorescent emissions (λem = 510 and 652 nm) with the red emission, which corresponds to the full conjugated system, being the more intense. Nucleophilic attack of HS− at the indolium CN interrupts the conjugated system of CouMC and the resultant truncated π-system of CouMC-SH exhibits coumarin-like fluorescence (λem = 510 nm). Maximum fluorescence emission was achieved in 30 s in simulated physiological conditions. The red fluorescence could be reinstated when the media containing CouMC-SH was acidified to pH 2.5; CouMC itself, was stable over the pH range of 2.5 to 8.0. In vitro ratiometric imagining studies with CouMC in MCF-7 cells revealed high localisation in the mitochondria (colocalisation with Deep Red 633). The intracellular reaction of the probe with HS− occurred rapidly (<80 s) and as such this probe may find utility in the spatiotemporal tracking of this anion.
 | ||
| Fig. 54 Top: structure and reversible reaction of CouMC with HS−. Bottom: (a–c) fluorescence images of MCF-7 cells co-stained by CouMC and Mito marker Deep Red 633. (a) Pseudocoloured image obtained with band path of 660–750 nm (λex = 488 nm); (b) image from band path of 665–750 nm upon excitation of Deep Red (λex = 633 nm); (c) overlay of (a) and (b). (d–i) Fluorescence imaging of MCF-7 cells (λex = 488 nm). (d–f) Images of cells stained by CouMC; (g–i) images of cells preincubated with CouMC followed by NaSH. (d, g) Green-channel images collected with band path of 500–560 nm; (e, h) red-channel images collected with band path of 640–700 nm; (f, i) ratiometric image scale bar in (d): 20 mm. Image reproduced with permission.224 | ||
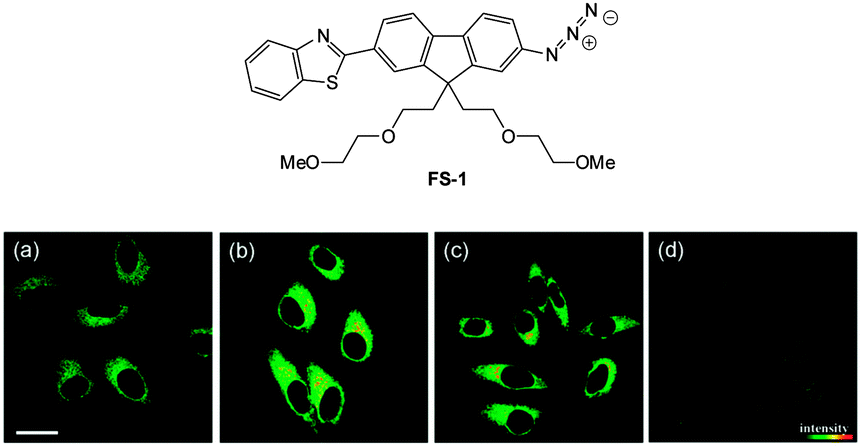 | ||
| Fig. 55 Top: structure of hydrosulphide probe FS-1. Bottom: (a) two-photon microscopy images of HeLa cells labelled with FS-1. (b–d) Cells pre-treated with cysteine (b), GSH (c), or PMA (d) before labelling with FS-1. The TPEF was collected at 400–680 nm upon excitation at 750 nm. Scale = 30 mm. Image reproduced with permission.225 | ||
In 2012 Han and co-workers reported the NIR-emitting ratiometric cyanine-azide probe Cy-N3 (Fig. 56).227 Upon treatment with NaSH the azide probe (λex = 625 nm, λem = 710 nm ΦF = 0.11) is reduced to give Cy-NH2 (λem = 750 nm, ΦF = 0.12) with a concomitant increase in molar absorptivity (ε660 = 130000 M−1 cm−1). The maximum fluorescence response was achieved in 20 min which was superior to related examples at that time. The ratio of emission intensities (F750/F710) increased from 0.6–2.0 with the addition of 10 equivalents of NaSH, and a detection limit of 80 nM was established. The probe was successfully used to detect endogenous HS− in live RAW264.7 macrophages (stimulated using PMA). The fluorescence emission intensity also increased in cells treated with NaSH and Cy-N3 was also used to study the time dependent decomposition of the HS− releasing agent 5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione (ADT-OH) in fetal bovine serum.
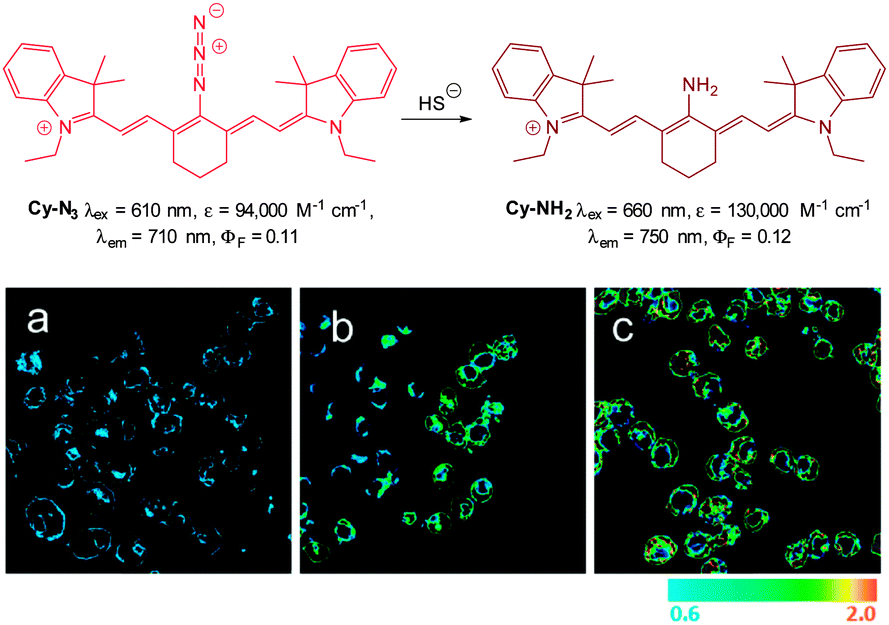 | ||
| Fig. 56 Top: structure and reaction of Cy-N3 with HS−. Bottom: ratiometric fluorescence images (F750/F710) of living RAW264.7 macrophage cells with Cy-N3. Images displayed in pseudo colour represent the ratio of emission intensities. (a) Cells were pretreated with PMA before loading Cy-N3. (b) Cells incubated with Cy-N3. (c) RAW264.7 cells incubated with NaSH and Cy-N3 was added. Image reproduced with permission.227 | ||
In 2013 the groups of Xu and Peng independently reported the red emitting dicyanomethylenebenzopyran probe DCMC-N3 (Fig. 57).228,229 Probe DCMC-N3 is essentially non-fluorescent, however, upon reaction with HS− fluorescence was “switched on” (λem = 670 nm in 1:1 PBS buffer:DMSO or λem = 655 nm in 1:1 phosphate buffer:MeCN). The Xu group showed that DBMC-N3 could be used to indicate the presence of exogenously administered HS− in human umbilical vein endothelial cells (HUVEC). Similarly, preliminary experiments by the Peng group used DBMC-N3 to image exogenous HS− in HeLa cells. The Peng group also utilised the favourable properties of DCMC-N3 (NIR, large Stokes shift and good Φδmax = 50 GM at 820 nm in DMSO) to visualise the presence of HS− in MCF-7 cells using two photon microscopy. Furthermore, a skin-pop injection of probe DCMC-N3 and NaSH (25 equiv.) into ICR mice revealed that the probe could be used in vivo as an enhanced fluorescent response (λex = 530 nm, λem = 655 ± 20 nm) was observed. The development of the fluorescent response over 4 h is shown in Fig. 57.
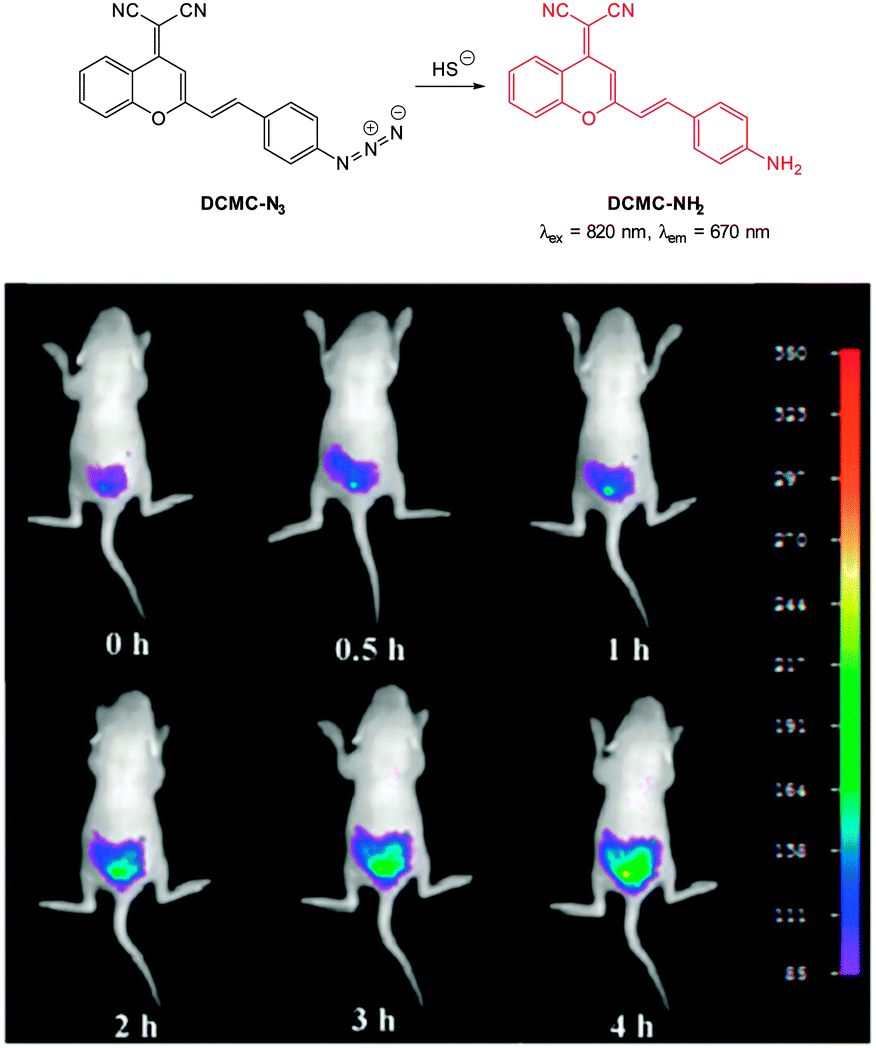 | ||
| Fig. 57 Top: structure and reaction of probe DCMC-N3 with sulphide. Bottom: representative fluorescence images of mice (pseudo-colour) given a skin popping injection of probe DCMC-N3 and then injected with NaSH. Images were taken after incubation of NaSH for different times (0, 0.5, 1, 2, 3 and 4 h). Image reproduced with permission.229 | ||
Through a simple two-step azidation procedure Ma (2012) converted cresyl violet to the ratiometric azide probe 61 (Fig. 58).230 Both 61 (λem = 566 nm, ΦF = 0.54) and cresyl violet (λem = 620 nm, ΦF = 0.44) were strongly fluorescent (λem = 620 nm). The emission ratio (F620/F566) ranged from 0.34–10.5 upon addition of HS− and a detection limit of 100 nM was determined. Probe 61 was used to visualise exogenous HS− added to MCF-7 cells, and the emission could be quenched with addition of ZnCl2 to the cells, confirming that switching is a result of azide reduction. The utility of 61 to visualise HS−in vivo was demonstrated in live zebrafish. As the concentration of HS− was increased (10–500 μM) fluorescence emission ratio (F620/F566) of the probe (10 μM) ranged from 0.73–1.64 with no visible fluorescence decrease on standing for elongated times, indicating that probe 61 was stable in vivo.
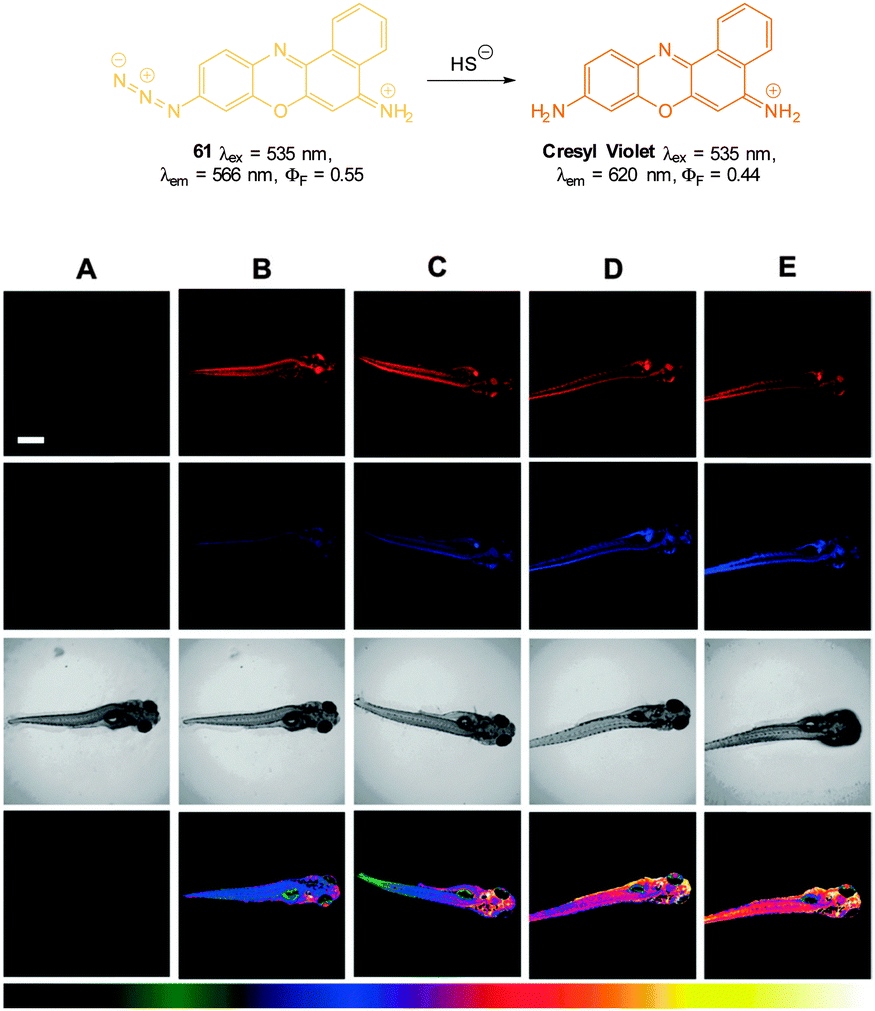 | ||
| Fig. 58 Top: structure and reaction of 61 with HS−. Bottom: fluorescent images of HS− in living 5-day-old zebrafish. (A) Zebrafish only (control). The zebrafish were treated with 61 and then with various concentrations of NaSH: (B) 0 mM, (C) 10 mM, (D) 100 mM, and (E) 500 mM. First row (570–600 nm), second row (640–670 nm) and third row (corresponding DIC images). The images of the fourth row are the ratio channel. The bottom color strip represents pseudocolor correlation with HS− concentration. Scale = 500 mm. Image reproduced with permission.230 | ||
Chang in 2013 reported probe SF7-AM; which incorporated AM-esters to facilitate passive diffusion into cells. Intracellular ester hydrolysis gave the free carboxylates and the probe was subsequently retained within the cells (Fig. 59).231 The inclusion of two azide triggers to the rhodamine-based fluorophore gave enhanced sensitivity and SF7-AM was used as a tool to study vascular endothelial growth factor (VEGF) stimulated HUVEC HS− production as a model for angiogenesis.
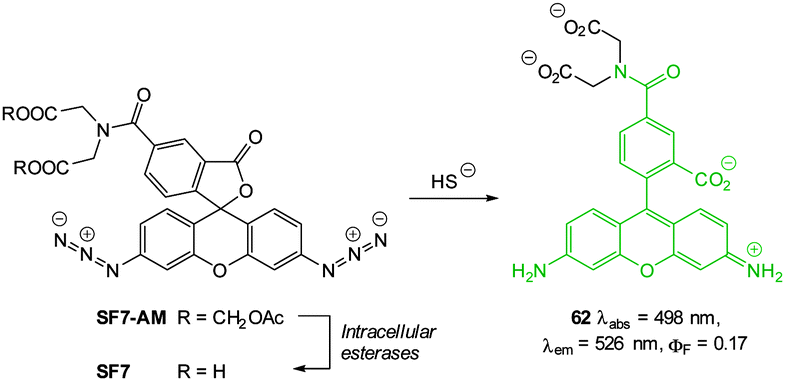 | ||
| Fig. 59 Structure, intracellular esterase and sulphide reactions of SF7-AM. | ||
As an advancement on their Lyso-NHS probe (Fig. 48) Cui and Xu developed the lysosome targeting probe Lyso-AFP (Fig. 60).232 When HS− mediated reduction of the essentially non-fluorescent azide (ΦF = 0.012) occurred an increase in fluorescence response (ΦF = 0.263, λex = 426 nm, λem = 535 nm) was observed over 20 min. As with Lyso-NHS the pendant morpholine of Lyso-AFP lead to lysosomal localisation.
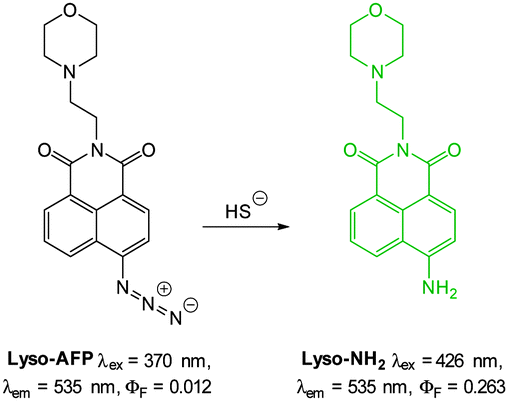 | ||
| Fig. 60 Structure and reaction of Lyso-AFP. | ||
Another lysosome specific probe; rhodamine-based SulpHensor (Fig. 61) was described by Yang (2014).233 No fluorescent response at physiological pH was observed for this probe upon treatment with HS−. The acidic lysosome environment is required to open the spirocycle which results in a weakly fluorescent species (λex = 540 nm, λem = 550 nm, ΦF = 0.05). Subsequent azide reduction by HS− greatly enhances the ICT fluorescent emission at 550 nm (ΦF = 0.05) and a limit of detection of 500 nM for HS− was determined. The HS−/H+ induced fluorescent response of SulpHensor was demonstrated in vitro using HeLa cells. Lysosomal accumulation was confirmed by co-staining with LysoTracker green and comparing the intensity profiles (Fig. 61) from the red (SulpHensor) and green channels (LysoTracker) over a selected region.
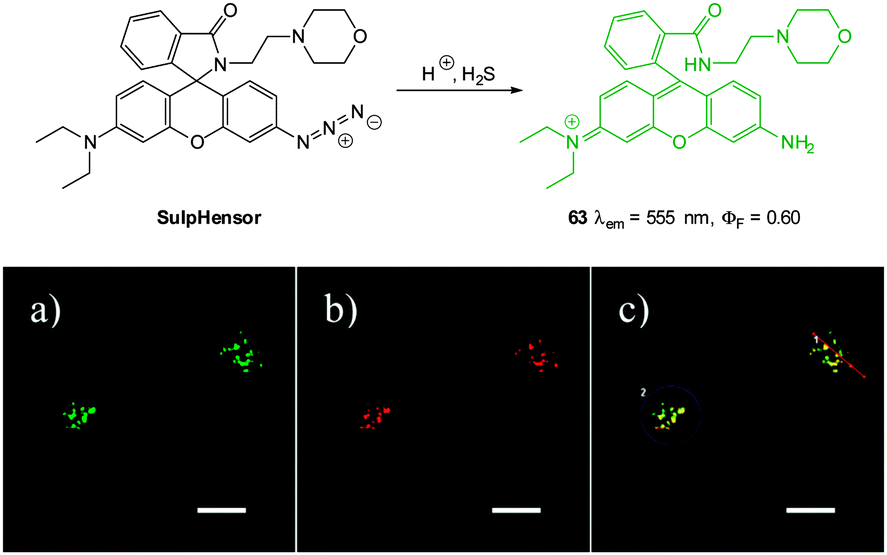 | ||
| Fig. 61 Top: structure and reaction of SulpHensor. Bottom: HeLa cells stained with (a) LysoTracker Green (Ch1, green) and (b) SulpHensor (Ch2, red) with NaSH solution (c) overlay of (a) and (b). Scale = 10 μm. Image reproduced with permission.233 | ||
Bae et al. employed the 4-azidobenzylcarbamate trigger in their two photon fluorescent probe SHS-M2 (Fig. 62). Hydrosulphide mediated reduction initially yields the aminobenzylcarbamate which subsequently fragments to give the observed products. In addition, the attachment of a pendant triphenylphosphonium moiety lead to mitochondrial accumulation in astrocytes.234 Upon reduction with HS− the emission wavelength of SHS-M2 (λem = 464, ΦF = 0.24) was red-shifted by 81 nm (λem = 545 nm, ΦF = 0.12) and despite a decrease in quantum yield the two photon cross section was improved (Φδmax = 17–55 GM). The probe was successfully used to detect mitochondrial HS− in HeLa cells; metabolic precursors (GSH or Cys) of HS− were added prior to the SHS-M2 and an increase in the F545/F464 ratio was recorded. The probe was also used to demonstrate the relationship between the DJ-1 gene and CBS mediated HS− production in astrocytes as a model of Parkinson's disease.
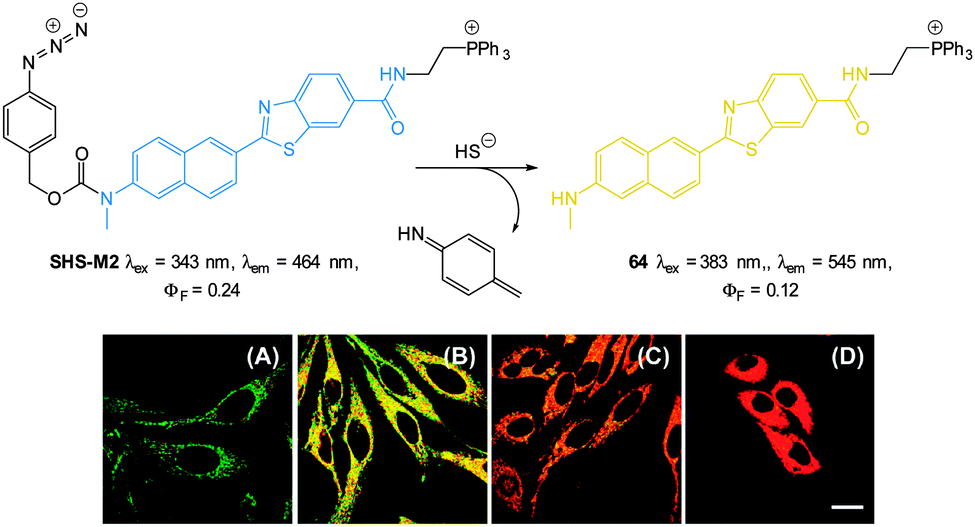 | ||
| Fig. 62 Top: two-photon fluorescent probe SHS-M2 developed by Kim. Bottom: (A–D) Pseudocoloured ratiometric TPM images F545/F464 of (A, D) HeLa cells with (A) SHS-M2 or (D) SHS-M2 and (B, C) HeLa cells pretreated with (B) GSH or (C) cysteine prior to SHS-M2. Image reproduced with permission.234 | ||
The 4-aminonaphthalimide-based HS− probes NAP-1 and AcSH-2 also bearing the 4-azidobenzylcarbamate trigger were independently reported by Zhao and Song in 2014.235,236 Reaction of NAP-1 (AcSH-1) with HS− gives the fluorescent aminonaphthalimide and a corresponding red-shift in emission (from λem = 474 nm to λem = 540 nm).235 A low limit of detection for HS− was determined (50 nM at pH 7.0 and 110 nM at pH 7.4) and the probe was used by Song for monitoring endogenous HS− production in sodium nitroprusside stimulated MCF-7 cells (Fig. 63) as well as quantifying HS− levels in murine hippocampus. Zhao used AcSH-2 to visualise exogenous HS− in MCF-7 cells using TPM (λex = 800 nm, F530/F468 = 0.38–2.90). It was also evident that the N-(2-hydroxylethyl)-imide of AcSH-2 promoted mitochondrial localisation.236
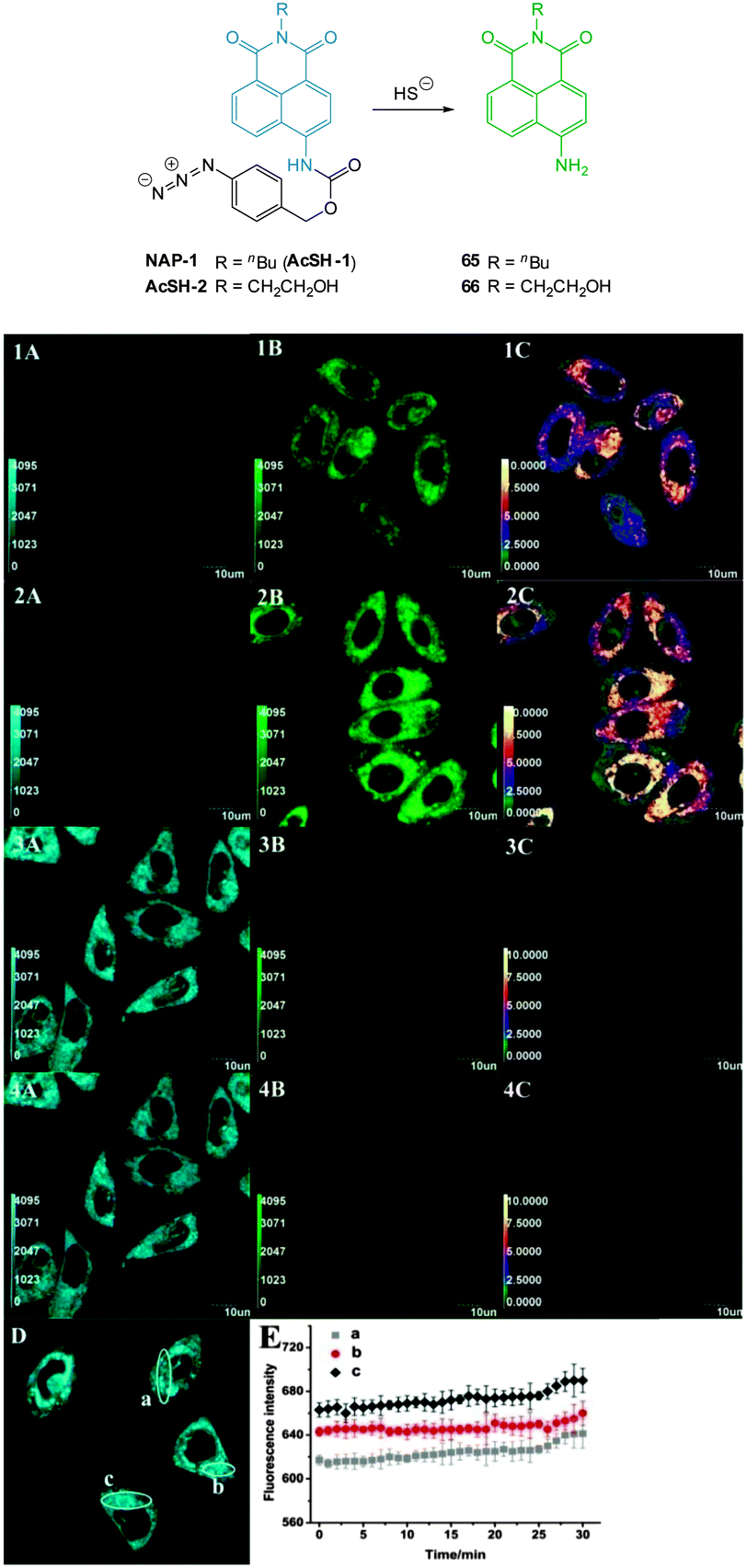 | ||
| Fig. 63 Confocal fluorescence images of endogenous HS− in living MCF-7 cells with NAP-1. Cells were pre-stimulated with SNP, then with NAP-1 (1A, 1B, 1C). Cells were pre-stimulated with SNP, and then with NAP-1 (2A, 2B, 2C). Cells were pre-treated with DL-propargylglycine (PPG), and then with NAP-1 (3A, 3B, 3C). Cells in (3A, 3B, 3C) were thereafter treated with SNP (4A, 4B, 4C). Cells were incubated with NAP-1 alone (D). The average fluorescence intensity from the regions of interest a, b and c in (D) was recorded with 60 s intervals (E) (n = 3). Ratiometric images (F530/F468) generated by the Olympus software (1C, 2C, 3C, 4C). Scale bars = 10 μm. Image reproduced with permission.235 | ||
Chen and co-workers built the m-nitrophenyloxycyanine Cy-NO2 (Fig. 64) as a d-PET based probe for HS−.237 The 3-nitrophenylether attached to the cyanine framework could accept an electron from the excited state of the cyanine fluorophore. Reduction of the nitro group with HS− disrupts the d-PET process resulting in significantly enhanced emission (from ΦF = 0.05 to ΦF = 0.11, λex = 755 nm, λem = 789 nm). The probe localised in the cytosol of RAW264.7 macrophages and was used to detect exogenous HS−. A significant drawback associated with unassisted nitro group reduction is that it is a kinetically slow process, and in this instance fluorescence intensity peaked only after 60 min, even at 37 °C.
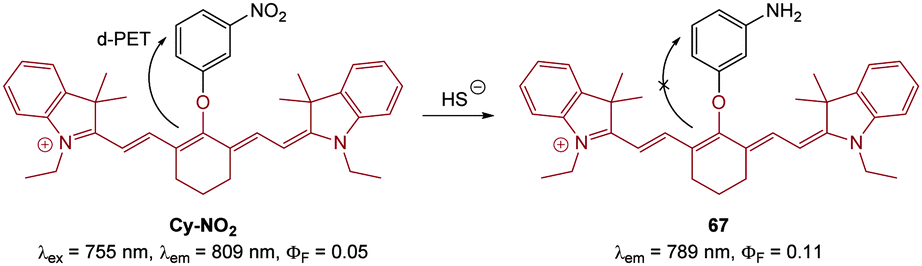 | ||
| Fig. 64 Structure and reaction of hydrosulphide probe Cy-NO2. | ||
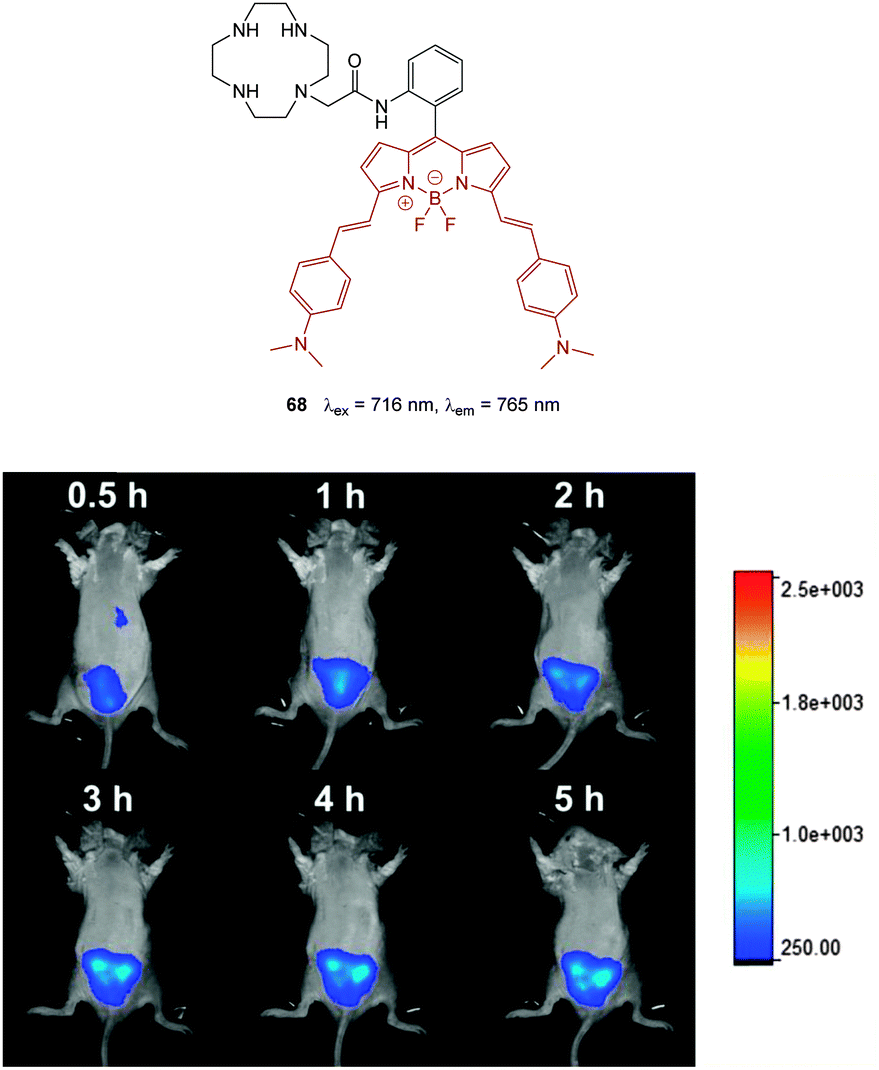 | ||
| Fig. 65 Top: NIR emissive cyclen dosimeter 68 by Deng and Huang. Bottom: representative fluorescence images of mice (pseudocolour) that were injected with 68-Cu(II) (40 mm), followed by Na2S. The images were recorded after the injection of Na2S within 5 h. Image reproduced with permission.238 | ||
In order to keep this section to a reasonable length and highlight the most relevant recent examples it was not possible to include all recent examples of probes functioning by way of (i) nucleophilicity100,218,219,223,224,239–247 (ii) reduction (arylazides,226–228,231–235,248–258 sulfonylazides,259–261 nitro groups,262,263 hydroxyl amines,264 and N-oxides265) as well as (iii) displacement53,266–269 and readers are referred to these citations for additional information.
4. Reactive anions
In addition to the well-known oxidative stress caused by reactive oxygen and nitrogen species, it is becoming increasingly apparent that these moieties are involved in a wide range of physiologically essential processes (such as cellular migration and circadian rhythm).270 Reactive oxygen/nitrogen species can be neutral, anionic, radicals or even radical anions (Table 2) and as many of these species exist only fleetingly they are exceptionally challenging targets for small molecular probes.50,271| Neutral | Anionic | Radical anion | Radical |
|---|---|---|---|
| a A percentage of this species can also be found in neutral form (HOBr pKa = 8.7). | |||
| Hydrogen peroxide HOOH | Peroxynitrite ONOO− | Superoxide O2˙− | Nitric oxide NO˙ |
| Singlet oxygen 1O2 | Hypochlorite ClO− | Hydroxyl HO˙ | |
| Hypobromite BrO−a |
Nitrogen dioxide NO2˙ | ||
| Peroxy radicals ROO˙ | |||
At physiological pH (ca. 7.4) several of these species exist to a considerable extent as an anion (peroxynitrite, hypochlorite and hypobromite) and can therefore be considered biologically relevant anionic targets. Given their diverse roles interest in detecting these species in vitro and in vivo has been strong and several reviews on the topic are available.34,60,271,272
4.1 Peroxynitrite
Peroxynitrite is formed from the rapid reaction of two other RO/N species: superoxide O2˙− and nitric oxide NO˙ (Fig. 66) and its detection in living systems is hampered by a number of factors including (i) its reactivity (it is a potent nucleophile and is a more powerful oxidant than superoxide) with a number of in vivo targets such as proteins, nucleic acids and lipids (especially those containing thiols) and (ii) degradation to other highly reactive species (including both the hydroxyl radical and the nitrogen dioxide radical).57,273 The pKa of ONOOH is 6.8 and as a result ∼80% of the species is found in the anionic form at pH 7.4. Typically the anion adopts a cisoid structure that is responsible for its ‘relative’ stability compared to the parent acid.274 | ||
| Fig. 66 Formation and cisoid structure of peroxynitrite. | ||
Elevated levels of this species are associated with cardiovascular and neurodegenerative disorders, metabolic diseases, inflammation, pain, and cancer.275,276 Hence the development of probes for its detection in vivo is an important task en route to fully understanding its diverse physiological roles.57
Peroxynitrite has long been known as a cellular oxidant and hence sensors to monitor this anion emerged last century.272 Probes for peroxynitrite rely on the same reactivity that the anion exhibits in vivo; ONOO− is both nucleophilic (reacts with CO2in vivo to form carbonate277) and highly oxidative (by one and two electron processes) and is a known nitrating agent.273 All three of these reaction types has been employed in the design of imaging agents for the species however probes that employ the oxidation power of ONOO− are by far the most prominent (Section 4.1.1). Amongst these, probes based on the oxidation of B, Se, and Te form an interesting subset (Section 4.2.2). Common interferents with the detection of ONOO− are typically other ROS in particular hypochlorite (ClO−) and nitroxide radical (˙NO).
Early anion sensors for peroxynitrite inside cells include the “switch on” probes dichlorodihydrofluorescein DCHF and dihydrorhodamine DHR-123 (Fig. 67).278–280 While many imaging studies, and even flow cytometry assays,281 were performed using these, (and similar) probes they are somewhat non-selective and in some instances light sensitive.272
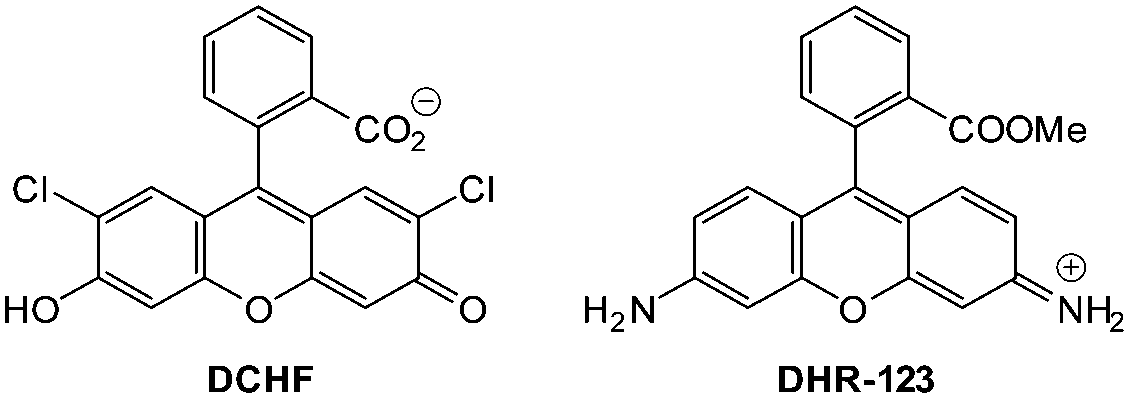 | ||
| Fig. 67 Early chemodosimeter based agents for peroxynitrite; dihydrodichlorofluorescein (DCHF) and dihydrorhodamine (DHR-123). | ||
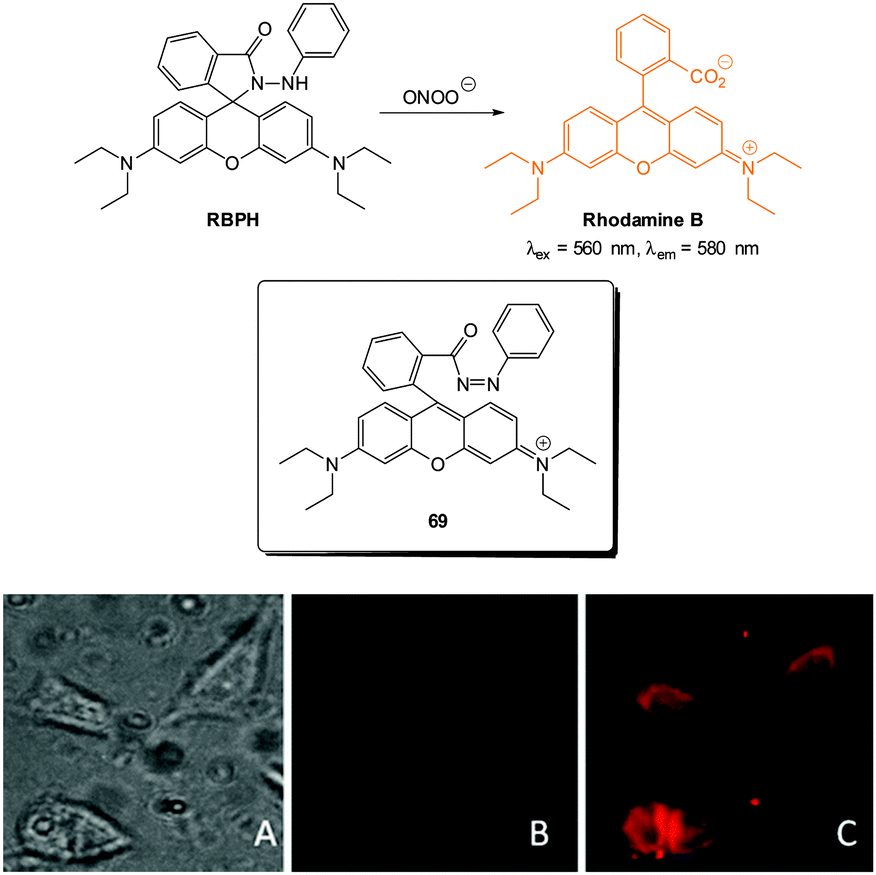 | ||
| Fig. 68 Top: structure and reaction of RBHP with ONOO−; inset shows the diazo intermediate 69 that forms prior to fragmentation. Bottom: (A) bright-field image of MCF-7 cells incubated with RBPH. (B) Corresponding fluorescence image (C) fluorescence image of MCF-7 cells incubated with RBPH then peroxynitrite. Image reproduced with permission.282 | ||
A strategy developed by Yang involves ONOO− nucleophilic attack on a reactive trifluoromethyl ketone group followed by formation of a spirohemiacetal via a dioxirane intermediate.283 Three separate probes have been developed: employing dichlorofluorescein HKGreen1,,284 BODIPY HKGreen2,285 and rhodol HKGreen392 fluorophores (Fig. 69). For these chemodosimeters fluorescence is quenched in the ketone form but following spirohemiacetal formation (and for HK1 and HK3 fragmentation) fluorescence is “switched on”. For HKGreen2 a 21-fold enhancement of fluorescence (λem = 539 nm) was observed upon reaction with just one equivalent of ONOO−. The probes react to a small extent with HO˙ but not the common ClO− interferent. Evaluation in cells was performed for all probes (Results for HKGreen3 in RAW264.7 macrophages shown in Fig. 69). This cell line is known to produce ROS/RNS, including ONOO−, in response to immunological and inflammatory stress and after stimulation of endogenous ONOO− production using bacterial endotoxin lipopolysaccharide (LPS), interferon-γ (IFN-γ) and phorbol 12-myristate 13-acetate (PMA) strong fluorescence was noted. No fluorescence enhancement was noted when ONOO− production was inhibited by (2,2,6,6-tetramethylpiperidin-1-yl)oxy (TEMPO) a superoxide scavenger or aminoguanidine (AG) an inhibitor of NO synthase286 confirming that endogenous production of ONOO− was being detected.
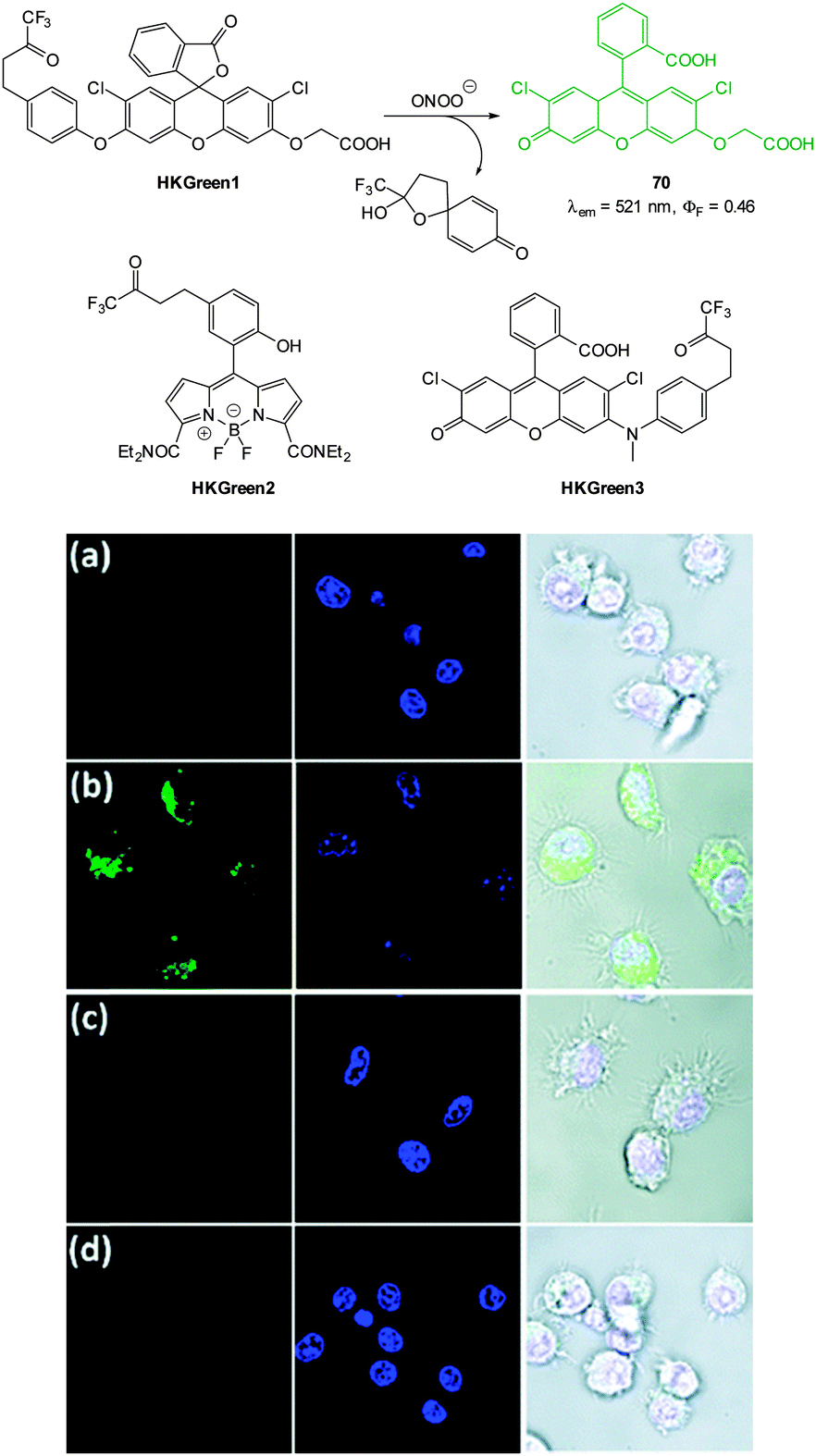 | ||
| Fig. 69 Top: structure of HKGreen1, 2 and 3 and reactivity of BODIPY based chemodosimeters HKGreen1 with peroxynitrite to form 70. Bottom: images of live RAW264.7 cells were treated with stimulants to elicit endogenous ONOO− production then stained with HKGreen-3 (green channel) and Hoechst 33342 (blue channel). (a) Cells without stimulation. (b) Cells stimulated with LPS, IFN-γ, and PMA. (c) Cells pre-treated with TEMPO, and then stimulated with LPS, IFN-γ, and PMA. (d) Cells pre-treated with AG, then stimulated with LPS, IFN-γ, and PMA. Image reproduced with permission.92 | ||
In a more recent article from the Yang group a functionalised rhodol chemodosimeter HKGreen-4 (Fig. 70) was described in which the oxidation of electron rich aromatics was exploited.287 The “switch on” probe (290-fold increase in λem = 535 nm) was capable of discriminating between peroxynitrite and other reactive oxygen species including hydroxyl radical and hypochlorite. HKGreen4 also performed in the presence of CO2, a molecule known to react quickly with ONOO−. The probe was water soluble and was successfully used with either conventional or two photon microscopy to image endogenous ONOO− production in E. coli challenged RAW264.7 cells. In conjunction with a number of enzymatic inhibitors the probe was used to further confirm the generation of endogenous ONOO− production in response to the presence of E. coli. Generation of ONOO− is thought to be an immune effector for bacterial clearance, and the results of this study suggest that the ONOO− formation in E. coli challenged macrophages is enzymatically regulated.
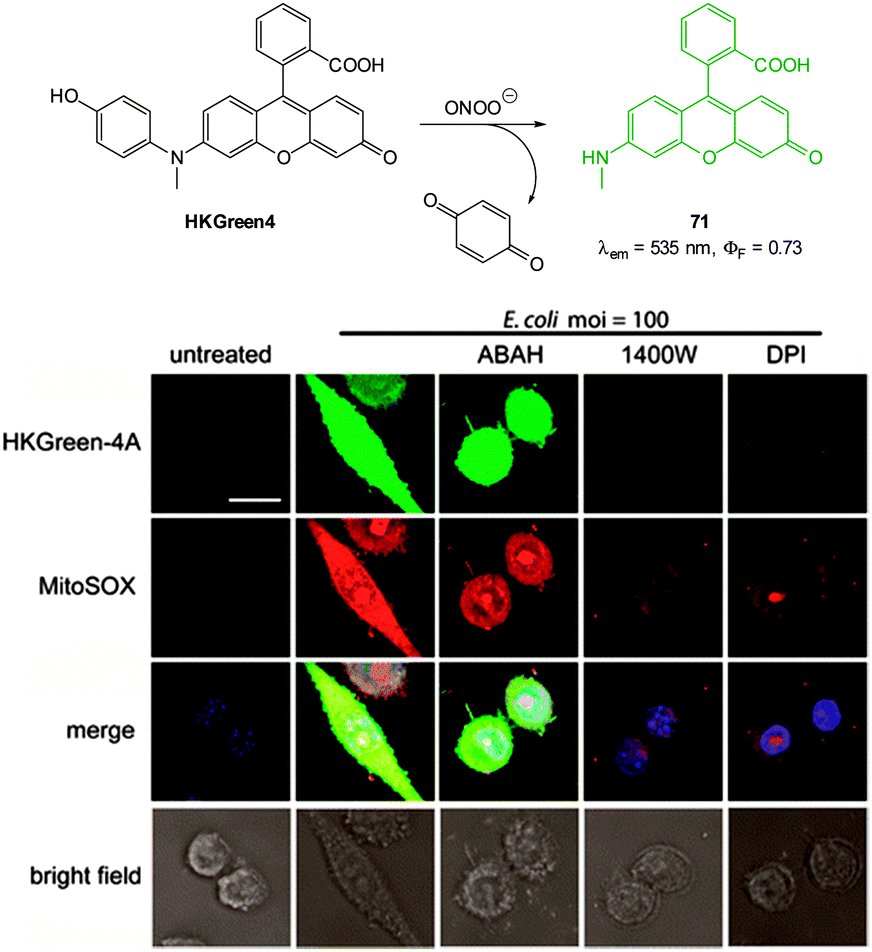 | ||
| Fig. 70 Top: structure and reaction of HKGreen4 with ONOO−. Bottom: images of RAW264.7 macrophages. Cells were co-treated with heat-killed E. coli and various enzyme inhibitors then stained with HKGreen4A, MitoSOX, and Hoechst 33342 before imaging. Aminobenzoic acid hydrazide (ABAH) is an inhibitor of myeloperoxidase that produces ClO−; 1400 W and DPI are inhibitors for iNOS and NOX, respectively. The merged images are overlays of all fluorescence channels. Scale = 10 μm. Image reproduced with permission.287 | ||
The “switch on” coumarin pyridinium probe C-Py-1 was reported by Yu in 2014 (Fig. 71).288 The probe was selective for peroxynitrite amongst other reactive oxygen/nitrogen species with a 25-fold enhancement of emission at λem = 493 nm. The emitting species was identified (1H NMR spectroscopy and ESI-MS) to be the coumarin aldehyde 72 formed from oxidative cleavage of the alkene in the presence of ONOO−. Excellent cell membrane permeation and low cytotoxicity were noted and the probe was successfully applied to the imaging of endogenous ONOO− in RAW264.7 cells. In vitro fluorescence was “switched on” within 30 min of the addition of lipopolysaccharide (LPS) to the cells to stimulate endogenous production of ONOO−.
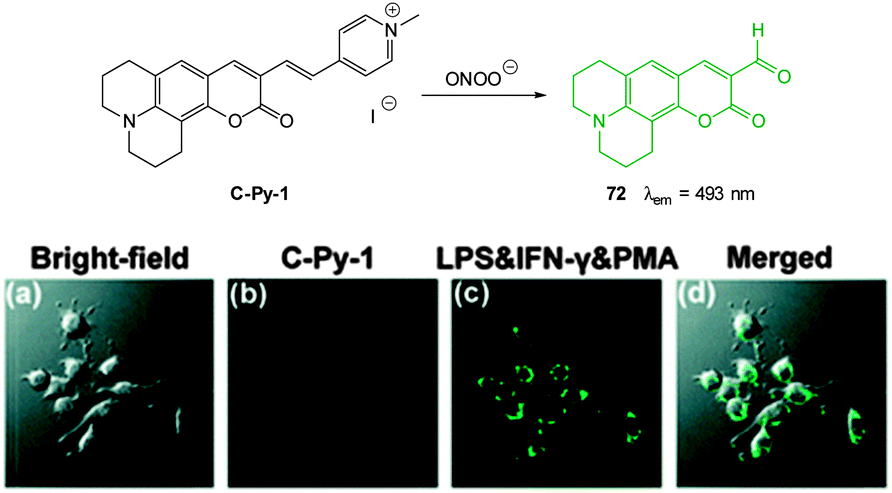 | ||
| Fig. 71 Top: structure and reaction of ONOO− probe C-Py-1. Bottom: images of RAW264.7 cells. (a) Bright-field image; (b) cells stained with C-Py-1; (c) cells were stained with LPS and IFN-γ then PMA and finally with C-Py-1; (d) merged image of (a) and (c). Scale = 10 μm. Image reproduced with permission.288 | ||
The related hemicyanine probe CHCN (Fig. 72) has recently been published by Yoon (2015).289 Oxidative cleavage of the alkene gives coumarin aldehyde 73 and 1,3,3-trimethyloxindole; both products confirmed using ESMS and 1H NMR spectroscopy. This probe indicated ONOO− in a linear ratiometric fashion (F515/F635 increase), had a low LOD (49.7 nM) and displayed excellent selectivity—only a very slight response to a large excess of hypochlorite was noted. The probe was shown to operate successfully in RAW264.7 macrophages when the cells were stimulated to produce ONOO− using a number of agents (LPS, IFN-γ and PMA). No response was observed when inhibitors of endogenous ONOO− production were introduced (TEMPO and aminoguanidine).
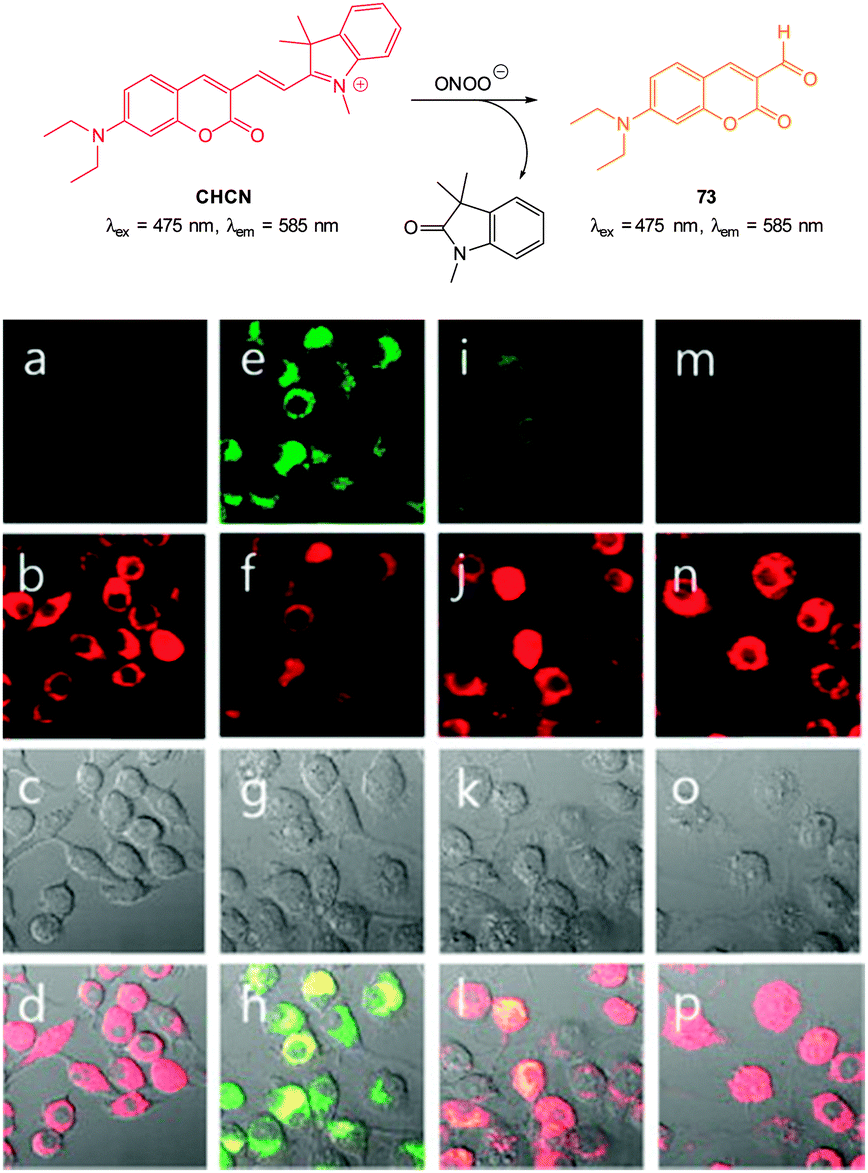 | ||
| Fig. 72 Top: structure and reaction of coumarinindolium probe CHCN. Bottom: images of RAW264.7 cells for endogenous ONOO− during phagocytic immune response. The cells were stained with CHCN. (a) Control; (e) LPS, IFN-γ and PMA; (i) LPS, IFN-γ and PMA then AG; (m) LPS, IFN-γ and PMA then TEMPO. Green channel (a, e, i, m); red channel (b, f, j, n); images (c, g, k, o) are differential interference contrast; images (d, h, l, p) are merged red and green channels. Image reproduced with permission.289 | ||
A multichannel probe PN600 (Fig. 73) that can clearly distinguish ONOO− from ClO− has been developed by Yang (2012).290 While both hypochlorite and peroxynitrite can oxidise the coumarin PN600 to the corresponding aminophenol 74, only ONOO− is powerful enough to oxidise 74 to the red emitting iminoquinone 75. Due to differing excitation yet similar emission wavelengths either of OCl− or the ONOO− could be inferred by monitoring emission at 620 nm and selectively exciting at 355, 465 or 575 to identify which fluorophore had formed and hence which oxidant was present. In human glioma cell line U87 the probe was shown to quickly penetrate the cell membrane and was non-toxic (MTT assay). Using 3-morpholinosydnonimine (SIN-1), a compound known to decompose to NO˙ which in turn forms ONOO−,291 a clear fluorescent response was observed.
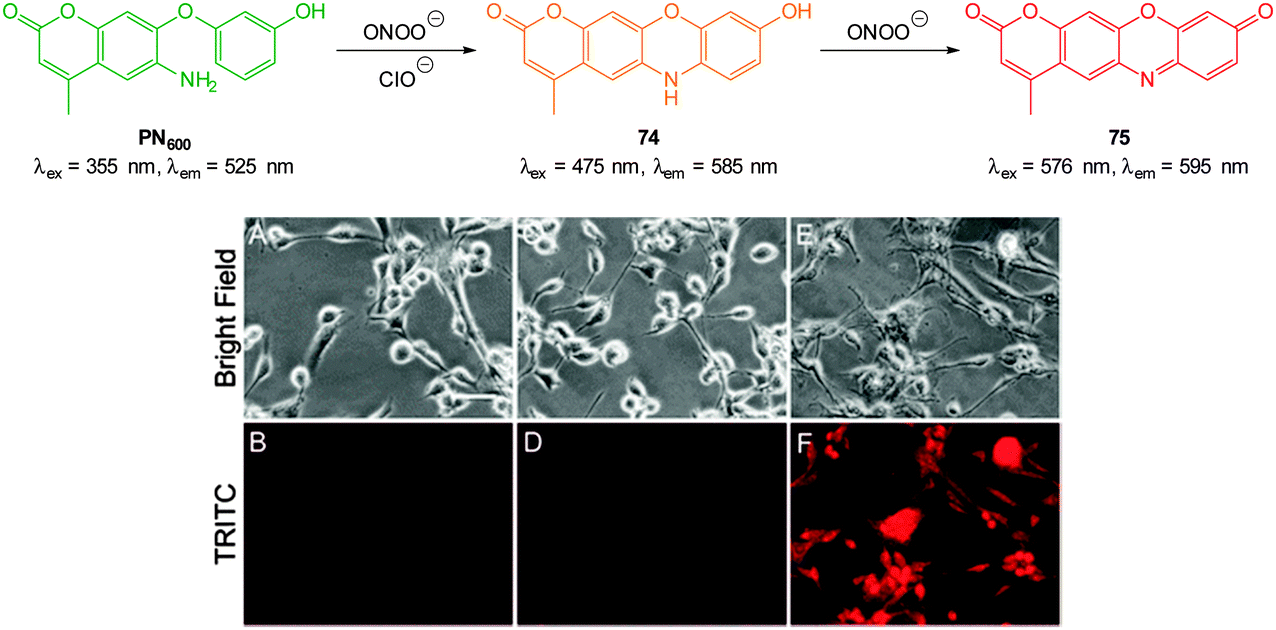 | ||
| Fig. 73 Top: probe PN600 showing reaction with both ONOO− and ClO− to form 74, however only ONOO− can oxidise 74 to the red iminoquinone 75. Bottom: live-cell imaging using PN600. Phase contrast and fluorescence images of human glioma cells treated with (A, B) PN600, (C, D) PN600 + ClO−, and (E, F) PN600 + SIN-1. Scale = 50 μm. Image reproduced with permission.290 | ||
Probes containing boron have recently emerged for the cellular sensing of peroxynitrite.292 The nucleophilicity of ONOO− leads to initial formation of a C–O–B bond in place of the original C–B bond (Scheme 2). This insertion is followed by hydrolysis to afford C–O–H. The process is similar to the established hydroboration/oxidation using alkaline H2O2 however the reaction of boronates with ONOO− is nearly 1 × 107 times faster than the reaction with H2O2 and hence discrimination between these species is possible.293
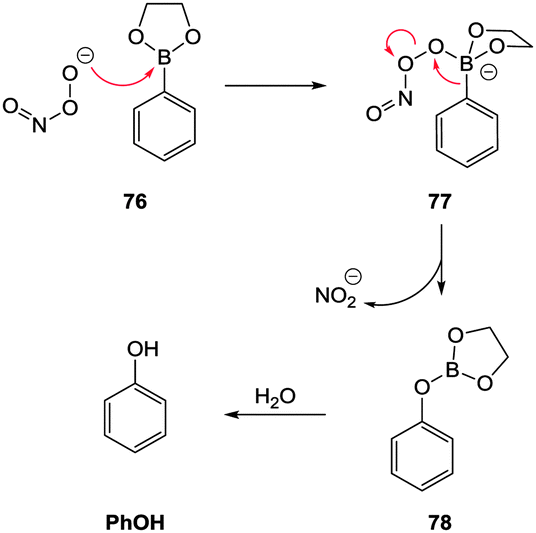 | ||
| Scheme 2 Conversion of arylboronates to phenols using ONOO−. | ||
The naphthalimide-based “switch off” probe 80 was reported by James in 2014 (Fig. 74).294 The fluorescent D-fructose complex (λem = 525 nm) of 79 in which PET from the spacer N is prevented was “switched off” by ONOO− mediated oxidation to the corresponding phenol 81. The product again has a free amine and PET quenches fluorescence. All other ROS except ClO− did not elicit a significant response. In RAW264.7 macrophages ONOO− production was stimulated by a number of immunological factors (again including LPS) and the “switch off” was easily visualised. When TEMPO or aminoguanidine was added to the stressed cells prior to addition of the probe fluorescence was maintained due to a lack of endogenous ONOO−.
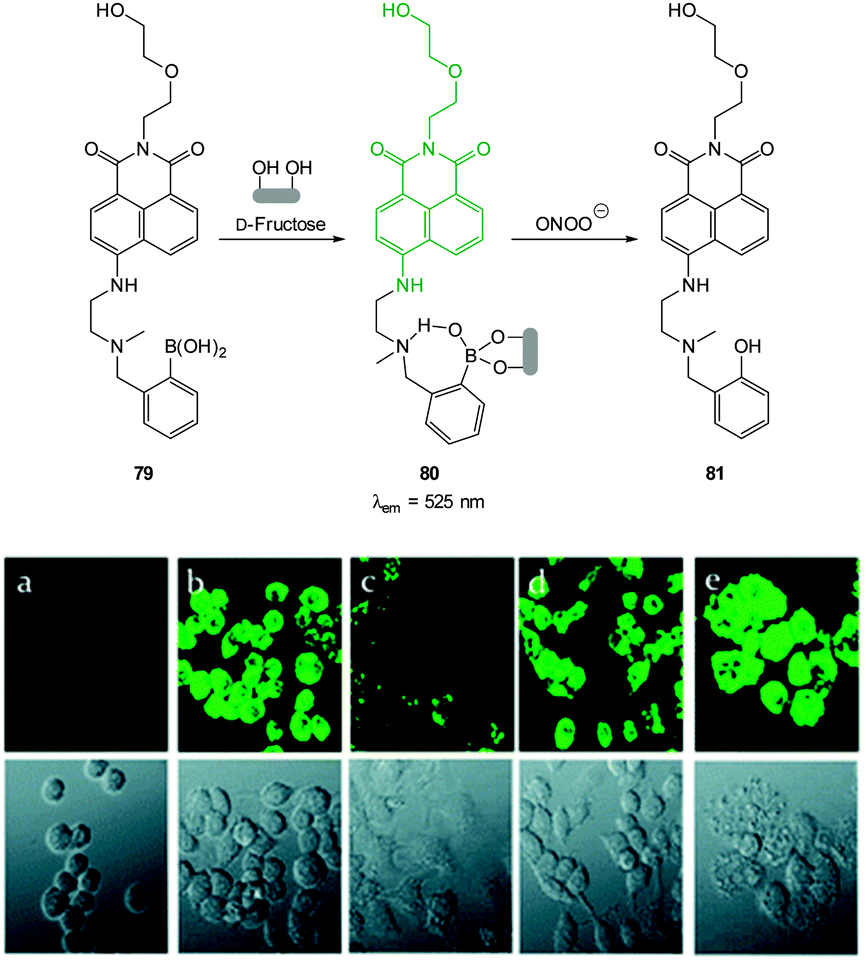 | ||
| Fig. 74 Structure and three stage reaction of naphthalimide boronate 79 with D-fructose and ONOO−. Bottom: fluorescent imaging for endogenous ONOO− in RAW264.7 cells. The probe 80 was formed by mixing 79 and D-fructose in situ. (a) Blank without probe; (b) probe 80 only; (c) probe 80 and LPS, IFN-γ, PMA; (d) c + aminoguanidine; (e) c + TEMPO. Image reproduced with permission.294 | ||
In 2012 Han reported the “switch on” dioxaborolane pyrene probe PyBor (Fig. 75) which was synthesised in three steps from pyrene.295 Reaction with ONOO− was complete in seconds (PyBor is converted to PyOH) and was accompanied by a significant increase in quantum yield from ϕF = 0.08 to 0.60 (PyOH, λex = 347, λem = 410 nm). While slightly sensitive to H2O2, ClO− and BrO− co-staining confirmed PyBor localised in the cytoplasm of RAW264.7 cells and intense intracellular fluorescence was observed when cells that had been incubated with PyBor were treated with LPS, IFN-γ and PMA. When the cells were pre-treated with aminoguanidine, only weak fluorescence was detected.
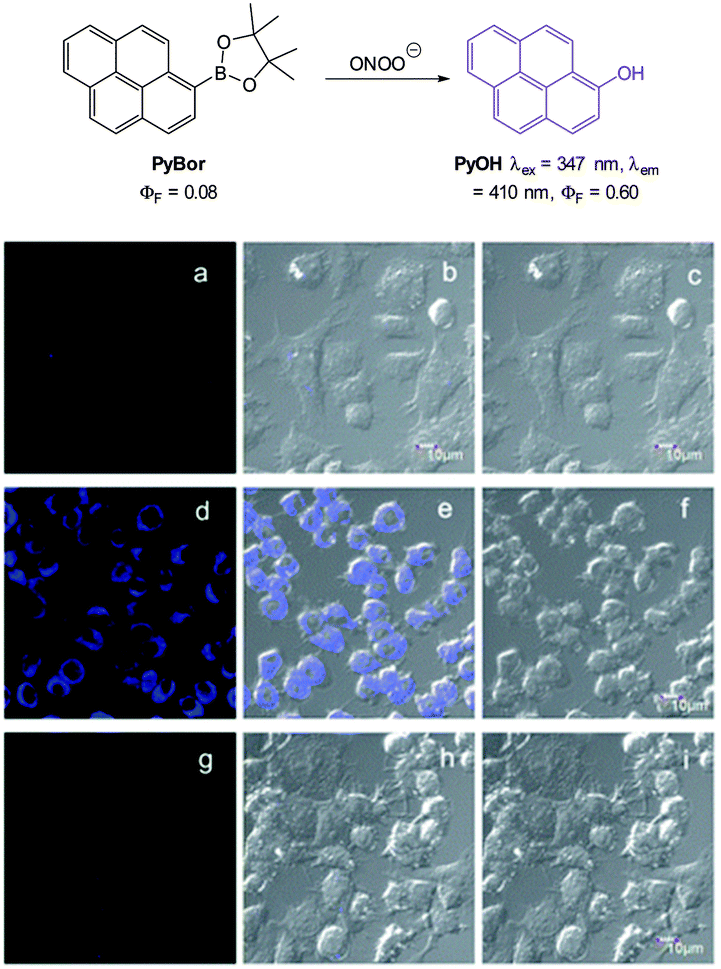 | ||
| Fig. 75 Top: structure and reaction of PyBor with ONOO−. Bottom: images of RAW264.7 macrophages treated with PyBor in the absence or presence of stimulants for 30 min. (a–c) Controls; (d–f) LPS and IFN-γ then PMA; (g–i) AG, LPS, IFN-γ and PMA. (a, d, g) fluorescence Images; (b, e, h) overlay images; (c, f, i) bright-field images. Image reproduced with permission.295 | ||
O form (readily accomplished by ONOO−) electron donation is “switched off” and fluorescence is quenched. The second probe, Cy-PSe297 is a “switch on” probe that uses PET from the dibenzylselenoether to quench BODIPY fluorescence. When oxidised to SeO, electron transfer is inhibited and fluorescence is “switched on”. The third, Cy-NTe298 another “switch on” probe also operates using this principle. The Te component required six steps to construct prior to coupling with commercially available cyanine chloride. Both Cy-PSe and Cy-NTe are easily oxidised by ONOO− and also easily reduced by glutathione (GSH) which makes them mimics of the Se containing glutathione peroxidase enzymes (GPx). In the non-oxidised state the metals quench fluorescence by PET, however PET is not possible in the oxidised forms and strong fluorescence enhancement is observed (23-fold for Cy-PSe and 13-fold for Cy-NTe). The successful “switch on” oxidation, “switch off” reduction cycles for both Cy-PSe and Cy-NTe were demonstrated in RAW264.7 cells by successive treatment with LPS then an ROS scavenger glutathione S-transferase (GST) to “switch off” fluorescence. Mitochondrial localisation of Cy-NTe was reported as was a lack of toxicity (MTT assay) and the switching behaviour of the probe was sensitive enough to be visualised in vivo using BALB/c mice.
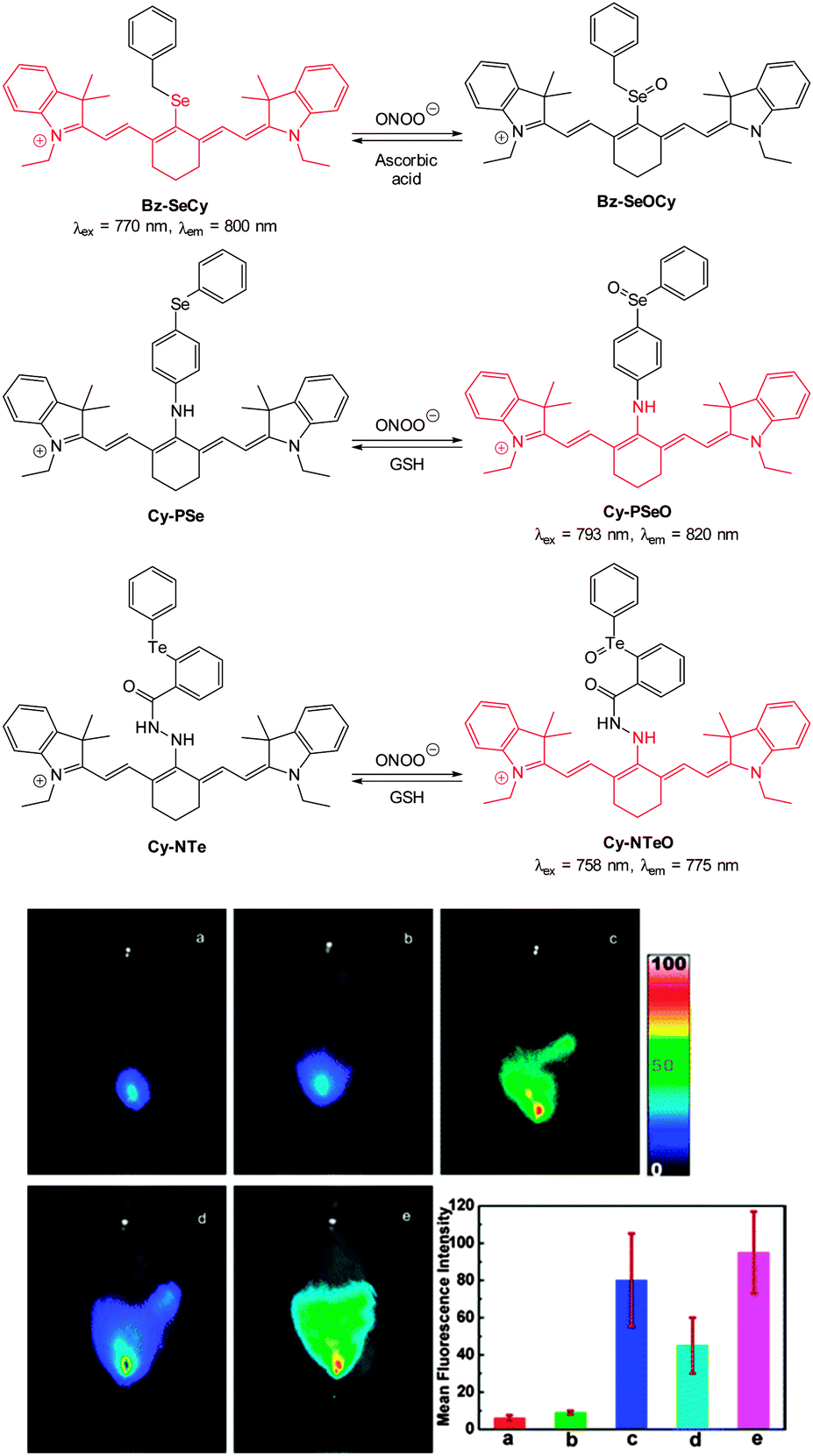 | ||
| Fig. 76 Top: structure of BzSe-Cy, Cy-PSe and Cy-NTe probes and their oxidation products. Bottom: imaging of redox cycles between peroxynitrite and GSH in peritoneal cavity of BALB/c mice. (a) Cy-NTe and L-cysteine were injected in the i.p. cavity. (b) Mice injected i.p. with Cy-NTe. (c) Mice injected with LPS, IFN-γ then PMA followed by Cy-NTe (d) mice treated as described in (c), then injected i.p. with AG, GST and L-cysteine (e) mice treated as described in (d), then injected with SIN-1. Bar graph indicates total number of photons from the entire peritoneal cavity of the mice (a–e). Image reproduced with permission.298 | ||
The selenium containing “switch off” BODIPY probe BOD-Se (Fig. 77) was reported by Han in 2012.299 The authors proposed that, in light of the long time required to elicit a response, oxidation of the Se itself by ONOO− did not modulate fluorescence but that the product containing SeO was hydrolysed (Fig. 78) to liberate a new selenium free florescent BODIPY 82. The BOD-Se probe itself was highly fluorescent (λem = 572 nm, ϕF = 0.96) and a colour change from red to blue was noted upon exposure to ONOO− with an isosbestic point at 567 nm. Fluorescence intensity decreased more than 200-fold along with a shift of the emission maximum (λem = 680 nm, ϕF = 0.05). Again murine macrophage RAW264.7 cells were stressed using LPS/IFN-γ and PMA to induce endogenous production of ONOO− prior to imaging.
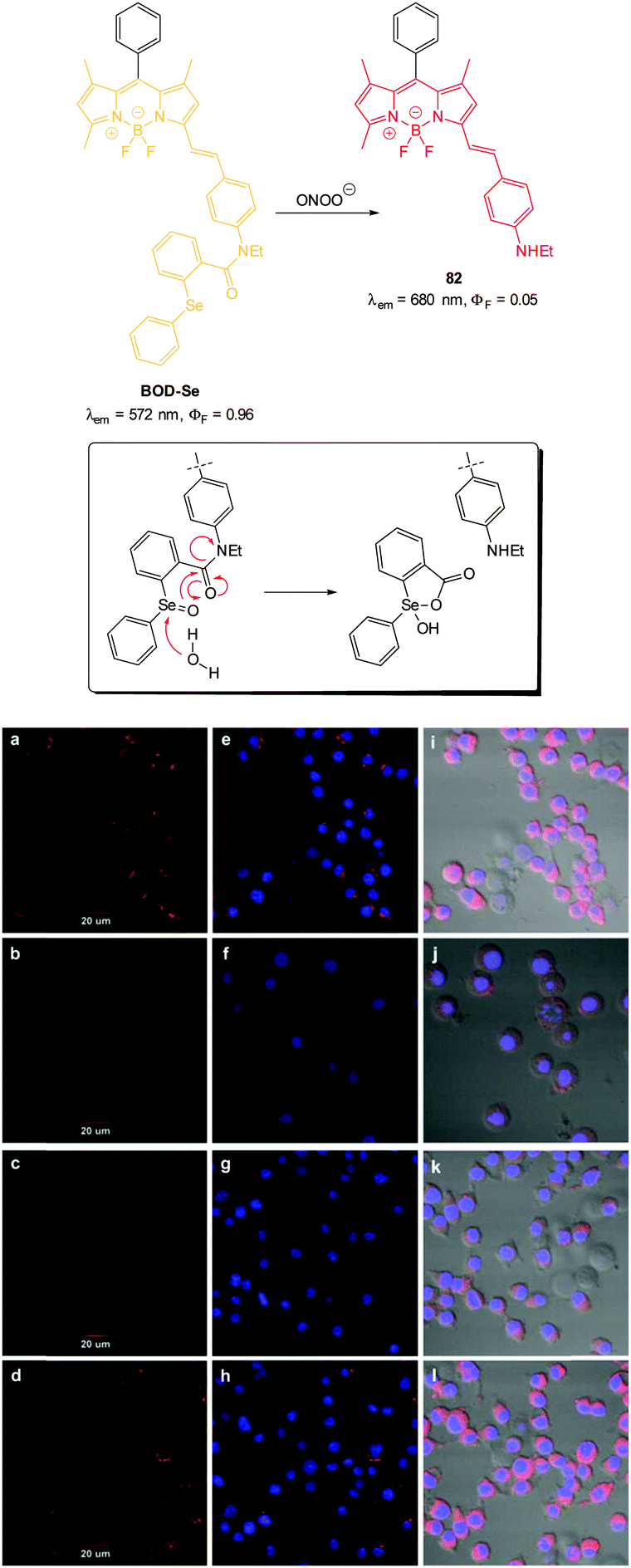 | ||
| Fig. 77 Top: structure and reaction of BOD-Se with ONOO−; inset: proposed tandem hydrolysis/cyclisation of oxidised BOD-Se leading to fragmentation. Bottom: images RAW264.7 cells incubated with BOD-Se then treated with (a, e, i) controls. (b, f, j) SIN-1 (c, g, k) LPS, IFN-γ then PMA. (d, h, l) AG, LPS and IFN-γ then PMA (e–h) overlay showing BOD-Se fluorescence and Hoechst dye; (i–l) bright-field overlay of BOD-Se and Hoechst dye. Image reproduced with permission.299 | ||
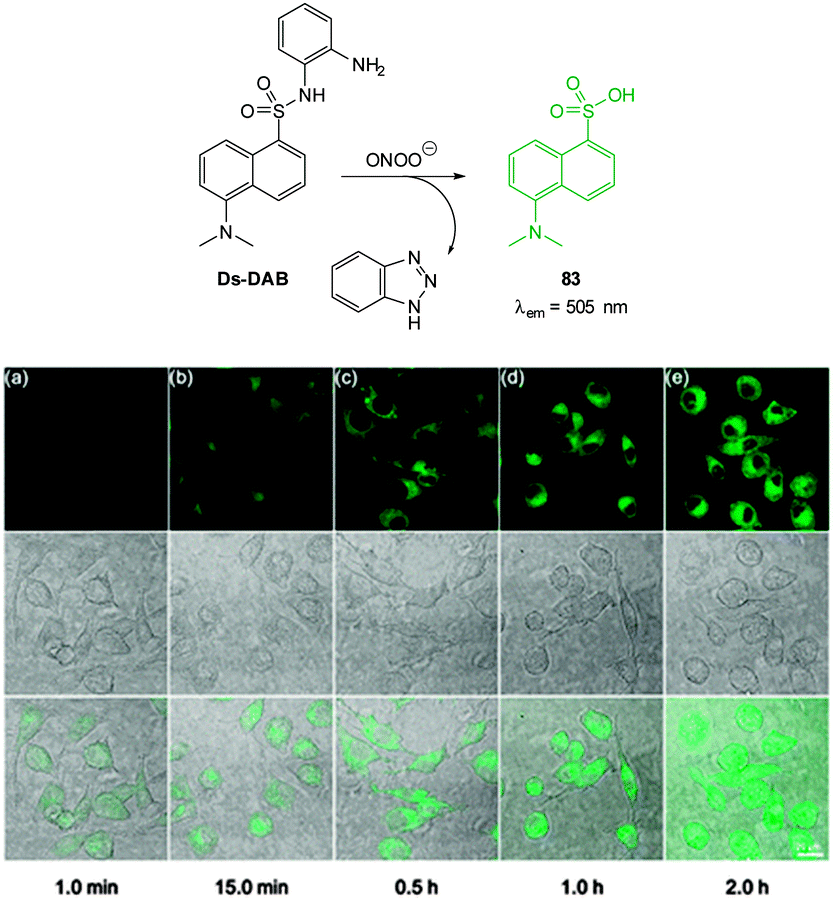 | ||
| Fig. 78 Top: structure and reaction of Ds-DAB with peroxynitrite. Bottom: in vitro detection of ONOO− in RAW264.7 macrophages. Cells were treated with LPS then PMA and subsequently incubated with Ds-DAB for the time stated prior to imaging. Top = fluorescence, middle = bright-field, bottom = overlay; scale = 20 μm. Image reproduced with permission.300 | ||
A probe for nitrative stress (NiSPY-3, Fig. 79) was rationally designed by Nagano (2006)301 and is an interesting example in which nitration leads to fluorescence “switch on” (λex = 505 nm, λem = 520 nm) based on an a-PET process. The probe was selective for peroxynitrite and was successfully used to visualise exogenous ONOO− in live HeLa cells.
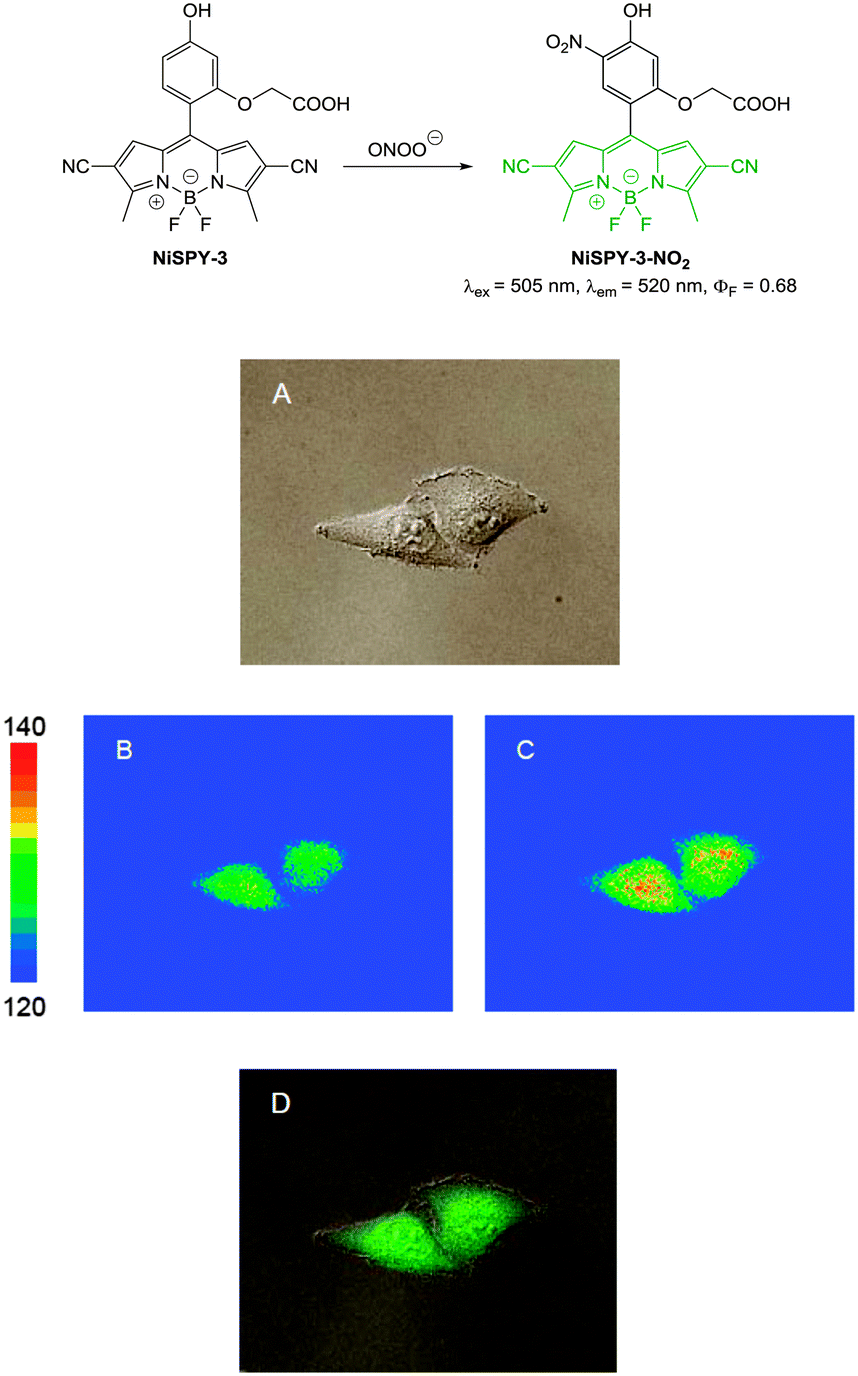 | ||
| Fig. 79 Top: structure of NiSPY-3 and its reaction with ONOO−. Bottom: differential interference contrast (A), pseudo-colour fluorescence (B, C) and merged images (D) of HeLa cells loaded with NiSPY-3. Pseudo-colour images were obtained before (B) and after (C) addition of peroxynitrite. Image reproduced with permission.301 | ||
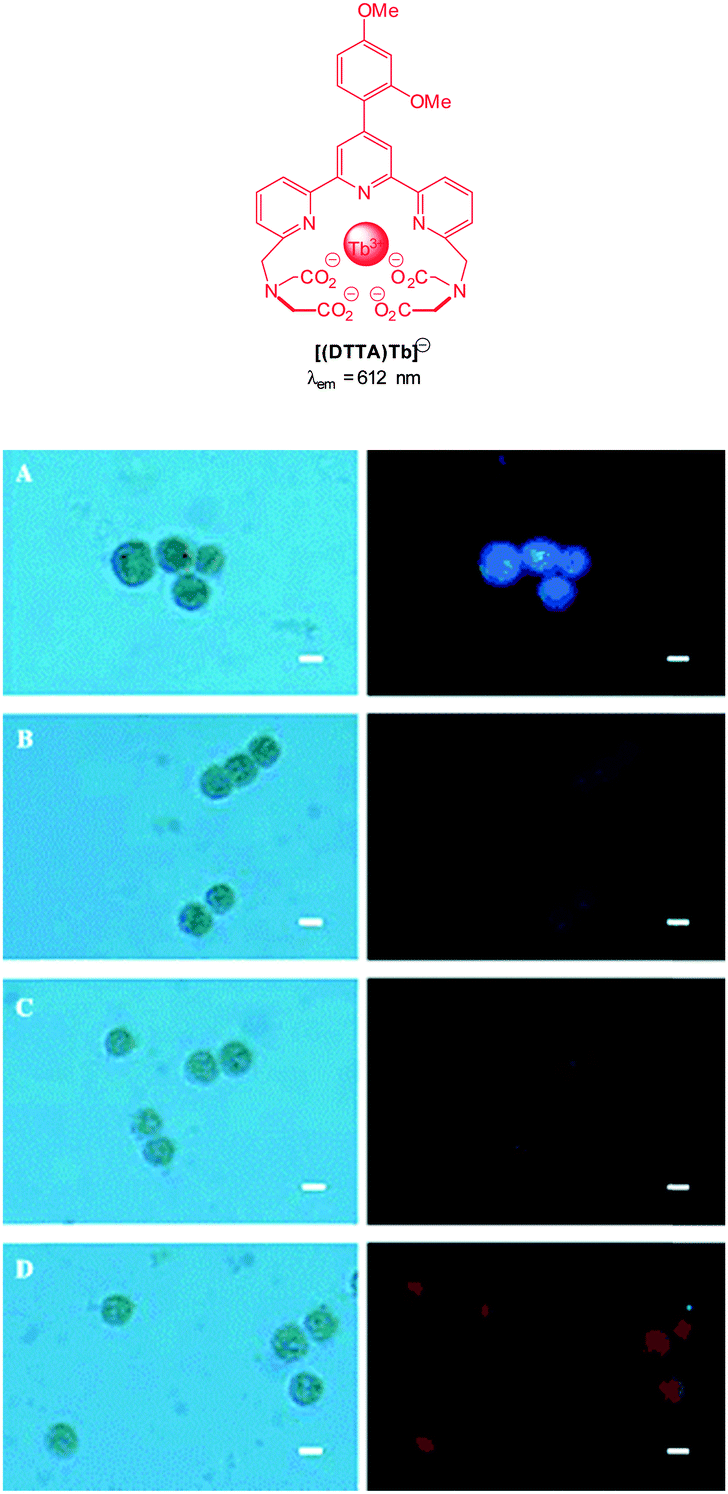 | ||
| Fig. 80 Top: structure of the DTTA-Tb(III) complex. Bottom: bright-field (left) and normal luminescence (right) images of the [Eu(III)/Tb(III)(DTTA)]-loaded HeLa cells in the presence of ONOO− generated from SIN-1 (A: negative control; B: 100 μM; C: 200 μM; D: 500 μM of SIN-1). Scale = 10 μm. Image reproduced with permission.93 | ||
An interesting example of a probe for the sensing of ONOO− by means of luciferin bioluminescence was reported in 2013 by Bonini using the peroxy-caged luciferin PCL-1 (Fig. 81).302 Unfortunately, cell images were not shown in this instance. The boronic acid based chemodosimeter PCL-1 (originally designed and used by Chang for the in vivo bioimaging of H2O2 in mice)303 reacts far more rapidly with ONOO− than it does with H2O2 to form luciferin which in turn forms oxyluciferin with concomitant emission of light (λem = 560 nm).
 | ||
| Fig. 81 Structure and reaction of luciferin based probe PCL-1 with ONOO−. | ||
4.2 Hypochlorite
Hypochlorite plays an important physiological role defending against invading pathogens and endogenous hypochlorite is produced from the reaction of H2O2 with Cl− and is catalysed by the heme enzyme myeloperoxidase (MPO). Over stimulation of the MPO immune response, and increased hypochlorite levels can lead to host tissue damage and inflammation that is associated with a number of serious disorders such as cancer, neurodegeneration, arthritis and cardiovascular disease.| H2O2 + Cl⊖ → H2O + ClO⊖ | (1) |
The majority of chemodosimeter for hypochlorite rely on oxidation; indeed ClO− is well known, even at a household level, as a strong oxidising agent. The most common triggers include the oxidation of p-hydroxy and p-aminophenyl ethers to give quinones or iminoquinones (Section 4.2.1) although the p-aminophenyl ethers typically elicit a larger response to ClO− and are more selective over ONOO− (versus p-hydroxy). Oximes provide a pathway for non-radiative decay and are also readily react with hypochlorite (Section 4.2.2). As for probes that target ONOO−, the oxidation of S, Se, and Te has been employed in spirocyclic ring opening triggers for fluorescein and rhodamine dyes. This class of probe (notably the Se and Te based examples) are conveniently reversible as they can be reduced back to the parent structure by thiols (e.g. Cys, GSH) or hydrosulfide.
An extension on the Nagano fluorescein system was reported in 2007 by Libby,306 whereby the p-aminophenyl ether moiety was attached to a water soluble sulfonaphthofluorescein (SNAPF, Fig. 82). Oxidation with hypochlorite resulted in the expulsion of the aminoquinone moiety to reveal a fluorescein derivative with an extended π-system (λem = 676 nm). SNAPF was then used to detect hypochlorite produced by human neutrophils stimulated with PMA (Fig. 83). When a MPO inhibitor was co-administered in stimulated cells no fluorescence was observed, indicating that MPO produced hypochlorite was being detected. SNAPF was also used to detect hypochlorite in stimulated mouse macrophages transgenic for human MPO (h-MPOTg) and stimulated human MPO-containing macrophages. Using SNAPF the successful bioimaging of hypochlorite generated in vivo was also performed in h-MOPTg mice with thioglycollate induced peritonitis. The SNAPF probe was administered 24 hours after thioglycollate stimulation, and after a further hour a 1.4-fold increase in fluorescence was observed.
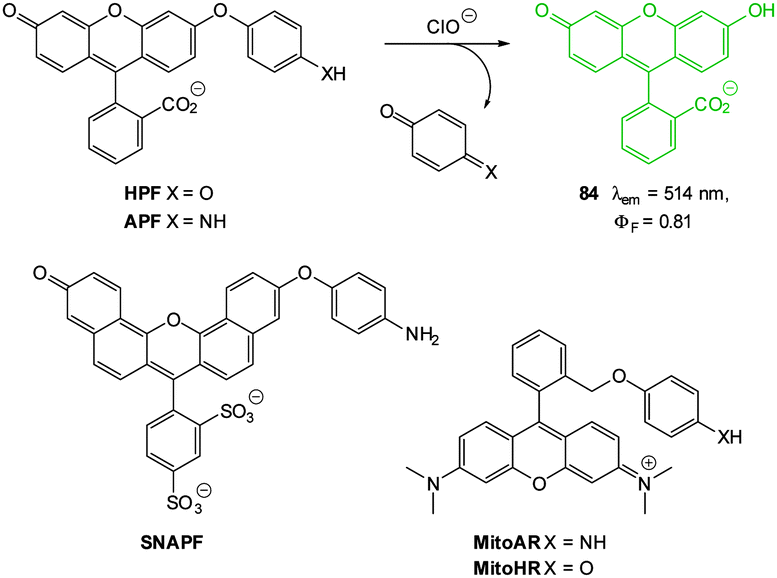 | ||
| Fig. 82 Structures of “switch on” probes for ClO− that function by oxidative cleavage. | ||
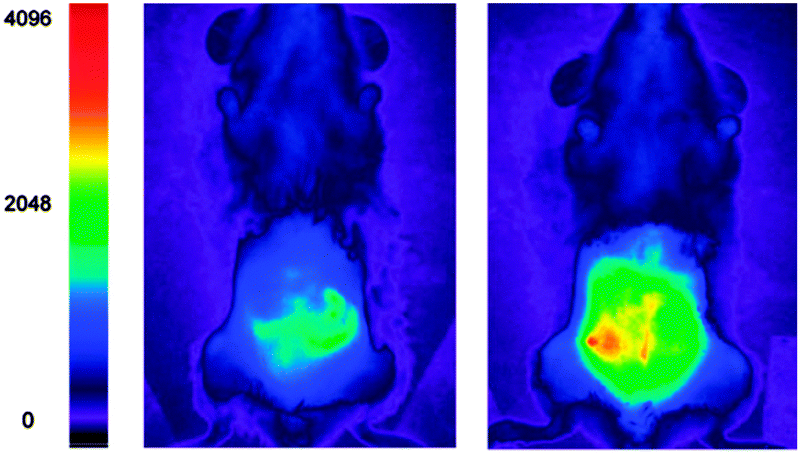 | ||
| Fig. 83 In vivo demonstration of SNAPF: (left) h-MPOtg mouse with thioglycollate induced peritonitis pre-SNAPF injection; (right) the same animal post-injection of SNAPF. No visible fluorescence was observed when SNAPF was injected into mice that had been injected with saline solution as opposed to thioglycollate, nor was there any increase in fluorescence of a wavelength-matched control dye, SNAPF, when injected into mice with peritonitis compared to saline-injected animals (n = 4 per group). Image reproduced with permission.306 | ||
In conjunction with a rhodamine fluorophore the p-aminophenyl ether trigger (MitoAR, Fig. 82) was also exploited by Nagano (2007).307 It was envisaged that the inherent positive charge of the ring open rhodamine would lead to accumulation of the probe within the mitochondria of living cells. In this instance the phenyl ether was located in the 2-position of the phenyl substituent in an effort to facilitate PET to the xanthene. Hypochlorite mediated oxidation of the p-aminophenyl ether correlated with a large increase in fluorescence response (λem = 574 nm) but only moderate selectivity over HO˙ was noted. Nevertheless, MitoAR was used to monitor the MPO-catalysed production of mitochondrial ROS in HL-60 cells using H2O2 stimulation (Fig. 84).
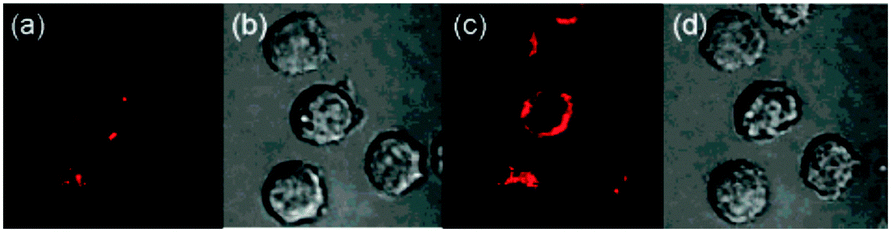 | ||
| Fig. 84 Detection of endogenous ClO− generation in mitochondria using MitoAr: (a, b) fluorescence and bright-field images of HL-60 cells loaded with MitoAR (c, d) 10 min after addition of H2O2. Image reproduced with permission.307 | ||
Yuan et al. successfully employed the 4-amino-3-nitrophenyl moiety as a hypochlorite responsive PET trigger to modulate the luminescence of terpyridine polyacid lanthanide complexes, ANMTTA-Tb(III) and ANMTTA-Eu(III) (Fig. 85).308 Oxidation of the 4-amino-3-nitrophenyl substituent gave the benzofurazan-1-oxide (BFO), which reacted with a further equivalent of ClO− to restore the luminescent terbium and europium complexes HTTA-Tb(III) and HTTA-Eu(III). Probe ANMTTA-Eu(III) was used for the time-gated spatiotemporal luminescence visualisation of exogenous hypochlorite in HeLa and in LPS/IFN-γ/PMA-stimulated RAW264.7 macrophages. Addition of the MPO inhibitor, ABAH, to cells resulted in no fluorescence emission.
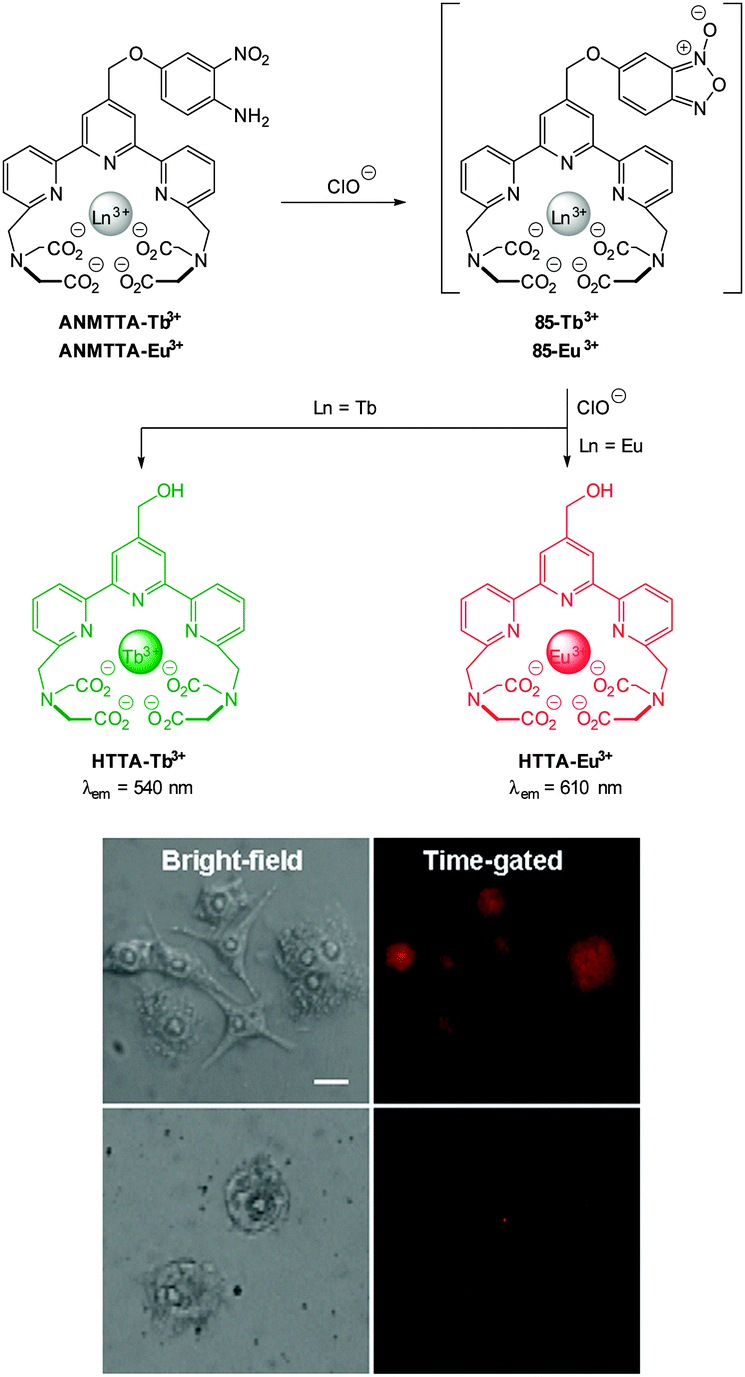 | ||
| Fig. 85 Top: structures of ANMTTA-Tb(III), ANMTTA-Eu(III) and their reaction with ClO−. Bottom: time-gated luminescence images of the ANMTTA-Eu(III) loaded RAW264.7 macrophage cells after stimulation with LPS/IFN-γ/PMA (top) or stimulated with LPS/IFN-γ/PMA/4-aminobenzoic acid hydrazide (ABAH). Scale bar: 10 μm. Image reproduced with permission.308 | ||
An elegantly designed system HKOCl-1 (Fig. 86), related to the aforementioned probes, was reported by Yang in 2008.309 This example used a BODIPY fluorophore which was suitably quenched (ΦF < 0.01) by a p-methoxyphenol substituent by means of PET. Oxidative demethylation of HKOCl-1 to the quinone (HKOCl) was effected by hypochlorite; the greater oxidation potential of the quinone makes PET unfavourable and fluorescence is “switched on” (λem = 541 nm). This process was selective for hypochlorite, with ONOO− eliciting a response only at a 10-fold higher concentration. The probe was used for the visualisation of live MPO producing RAW264.7 murine macrophages stimulated using lipopolysaccharide (LPS), interferon-γ (IFN-γ) and PMA. Stimulated cells showed an increase in the fluorescent output compared to the control. Additionally, when cells were stimulated in the presence of 2,2,6,6-tetramethylpiperidinooxy (TEMPO), much weaker fluorescent was observed. TEMPO is a known superoxide (O2˙−) scavenger which is an intermediate in hypochlorite synthesis from the MPO system.
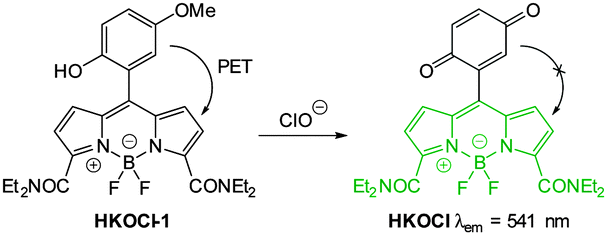 | ||
| Fig. 86 Structure and reaction of HKOCl-1 with ClO−. | ||
In 2012 Yao reported the dihydrofluorescein based probe FCN2 (Fig. 87) which, unlike preceding examples, is triggered by oxidation dealkylation rather than spirocycle ring opening.310 Ether cleavage from FCN2 restores the ICT of the dihydrofluorescein system (86) and results in a 1643-fold increase in fluorescent intensity (λem = 485 nm, ΦF = 0.71). FCN2 was completely soluble in aqueous solution and, although some autooxidation was noted (over 24 h), reacted rapidly enough (30 min) to be applicable for in vitro (NIH3T3 cells) and in vivo applications (larval and adult zebrafish). The fluorescent response generated from the treatment of 3 month old zebrafish with exogenous hypochlorite and FCN2, indicated the accumulation of hypochlorite in gall bladder, intestine, eye, liver and eggs.
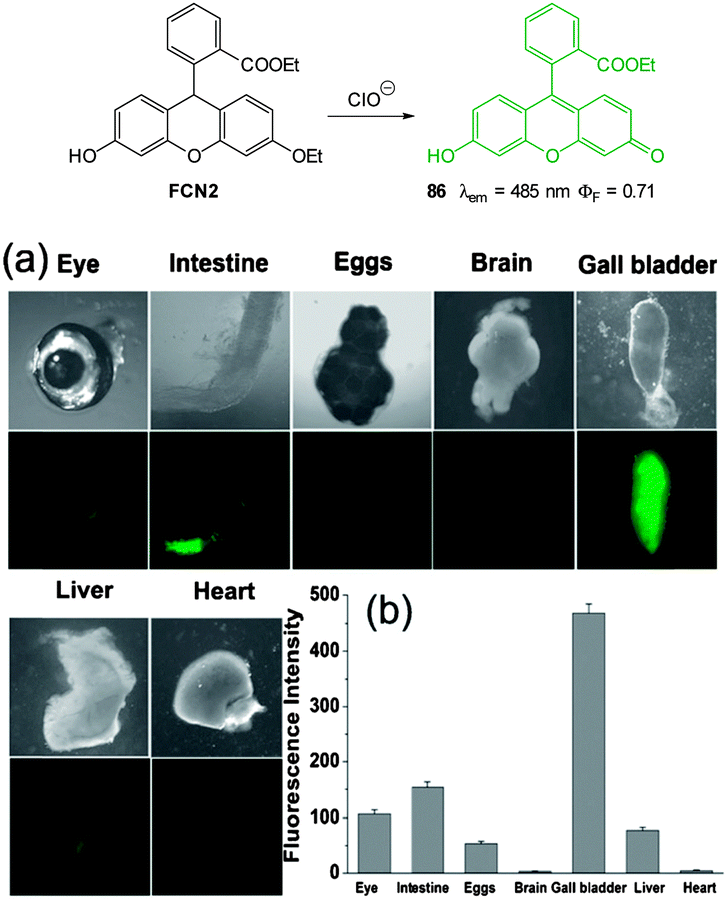 | ||
| Fig. 87 Adult zebrafish were (a) exposed to ClO− in E3 embryo media then incubated with FCN2. (b) The average emission intensities of isolated organs. Image reproduced with permission.310 | ||
Peng and co-workers reported the oxidation-activated “enhanced PET” BODIPY-based probe BClO (Fig. 88) which was synthesised in an exquisite two step procedure from 2,4-dimethylpyrrole.311 The PET quenching was mediated by a pendant pyrrole unit which was envisaged to provide “enhanced PET” compared to single electron donors (such as heavy metal containing probes). The resultant probe, BClO was essentially non-fluorescent at λem = 505 nm (ΦF = 0.006), however, oxidation with hypochlorite afforded the pyrrol-3-one BOClO in which PET was restricted and a 56-fold “switch on” fluorescence response was observed (ΦF = 0.347). In solution studies BClO was highly sensitive to hypochlorite with a limit of detection of 0.56 nM. Facilitated by the remarkable sensitivity of BClO the authors determined the basal hypochlorite levels in MCF-7 and HeLa cancer cell lines. When incubated with BClO (1 μM) for 20 min at 37 °C, both MCF-7 and HeLa displayed an increased fluorescence response (in relation to healthy COS-7 and RAW264.7 cells) corresponding to intracellular ClO− concentrations of 9.45 nM and 8.23 nM respectively. In both of the cancer cell lines visualised, pre-treatment with the antioxidant GSH or 4-aminobenzoic acid hydrazide (ABAH, an MPO inhibitor) resulted in a significant drop in fluorescence response. The authors suggest that it may be possible to use BClO in a diagnostic capacity; differentiating healthy and cancer cells based on endogenous hypochlorite concentration. The utility of BClO was further demonstrated by the detection of hypochlorite produced in MCF-7 cells stimulated with elesclomol (an ROS generating anticancer agent).
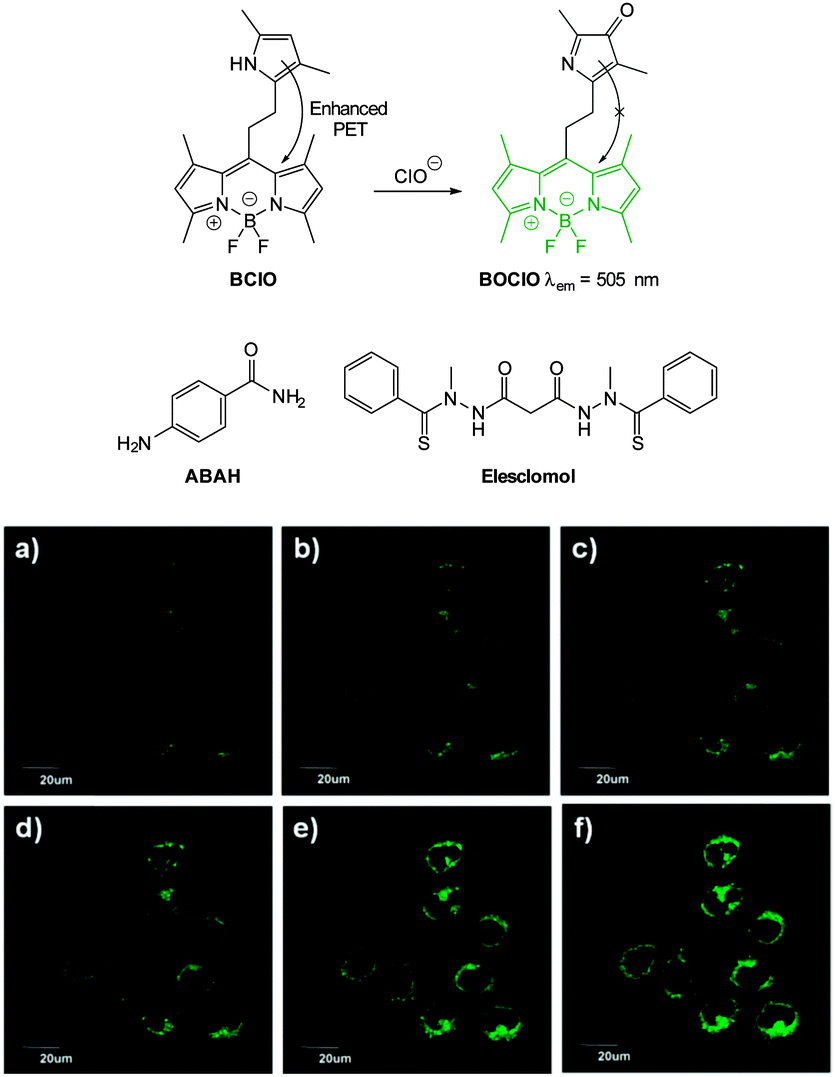 | ||
| Fig. 88 Top: structure and reaction of BClO with hypochlorite. Middle: structures of ABAH and elesclomol. Bottom: live MCF-7 cells were cultured with BClO then confocal fluorescence images were recorded after the addition of elesclomol at different time points: (a) 0, (b) 30, (c) 60, (d) 90, (e) 120, and (f) 180 min. Image reproduced with permission.311 | ||
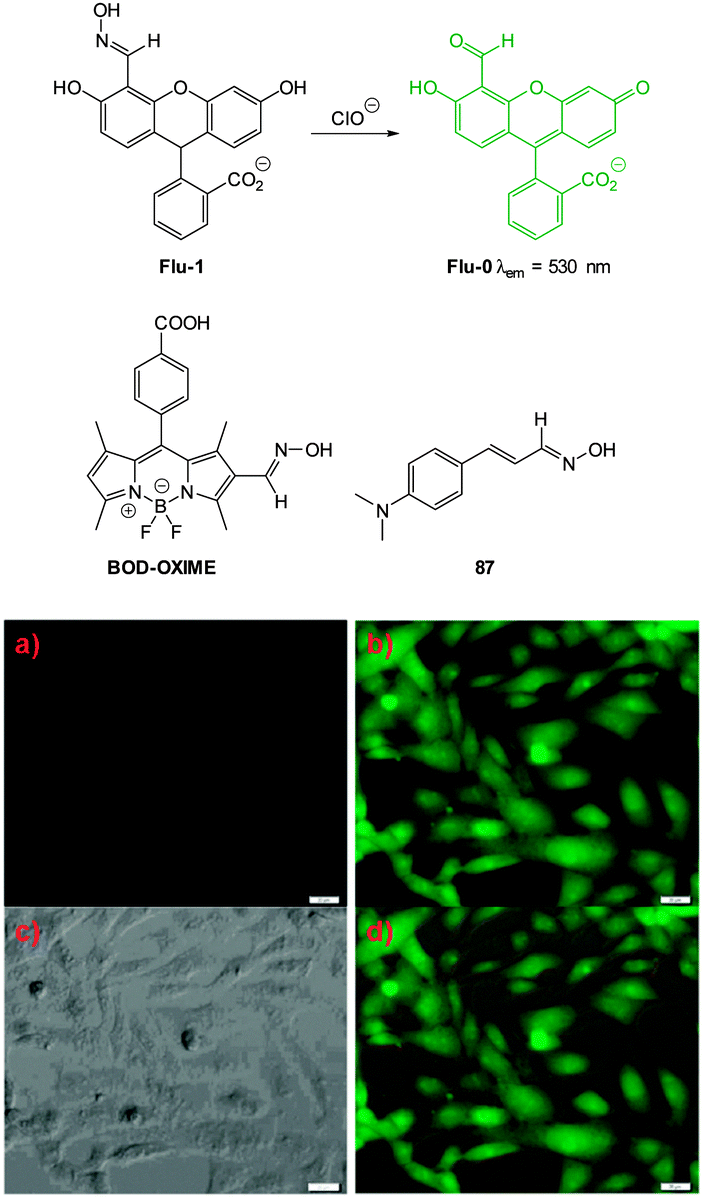 | ||
| Fig. 89 Top: structure, and reaction of, Flu-1 with ClO−. Middle: structures of BODOXIME and 87. Bottom: (a) image of HeLa cells incubated with Flu-1 (20 μM) for 30 min. (b and c) Fluorescence and bright-field images of cells pre-treated with Flu-1 then incubated with ClO− for 30 min. (d) Overlay image of (b) and (c). Image reproduced with permission.313 | ||
Wu, Zeng and Wu (2013) employed the same oxime/aldehyde strategy using a BODIPY fluorophore.313 The water soluble BOD-OXIME (Fig. 89) was poorly fluorescent (ΦF = 0.04) but reacted in a dose dependant “switch on” manner (LOD = 17.7 nm) with hypochlorite to give BOD-CHO which exhibited extraordinary fluorescence in aqueous solutions (ΦF = 0.96, λem = 525 nm). The visualisation of both exogenous (ClO−) and endogenous hypochlorite (PMA stimulation) in RAW264.7 macrophages was successfully demonstrated. Selectivity for ClO−in vitro was excellent as no emission was observed when ABAH or taurine (a ClO− scavenger) were added to the PMA stimulated cells.
A recent report from the Kumar group in 2014 identified 4-dimethylaminocinnimaldehyde oxime 87 (ΦF = 0.008, Fig. 89) as a selective “switch on” probe for hypochlorite.314 Oxidation with hypochlorite gave the nitrile oxide and this group participates in an extended ICT system leading to strong emission at 556 nm (ΦF = 0.51). The formation of the nitrile oxide was confirmed by spectroscopic means (1H and 13C NMR) and by in situ trapping using 2-butene to give the 4,5-dihydroisoxazole. The ability of probe 87 to visualise endogenous and exogenous hypochlorite was demonstrated using the brain resident murine macrophages (BV2 microglial) and C6 glial cell lines (Fig. 90). Addition of exogenous NaOCl and also LPS stimulated endogenous hypochlorite production led to increased fluorescence.
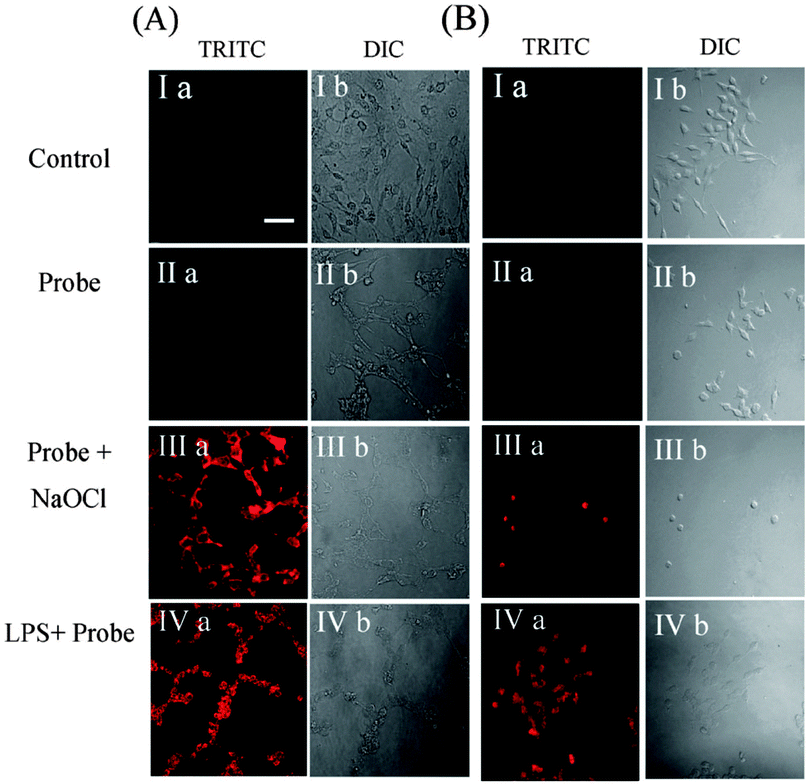 | ||
| Fig. 90 Fluorescence and DIC images of C6 glial (A) and BV2 microglial (B) cell lines. (Ia) Control of C6 glial and BV2 microglial cell lines respectively. (Ib) DIC images of Ia. (IIa) Images of C6 glial and BV2 microglial cells with 87. (IIb) DIC images of IIa. (IIIa) Red fluorescence images of C6 glial and BV2 microglial cells with 87 and NaOCl. (IIIb) DIC images of IIIa. (IVa) Red fluorescence images of C6 glial and BV2 microglial cells with 87 and LPS. (IVb) DIC images of IVa. Scale = 50 nm. Image reproduced with permission.314 | ||
The first ratiometric fluorometric probe for hypochlorite was developed by Yuan using an analogous approach to those above.315 The probe consists of a diaminomaleonitrile derived imine 88 of aminocoumarin aldehyde 89 (Fig. 91) The weakly fluorescent imine (λem = 585 nm, ΦF = 0.02) reacted with hypochlorite to give the parent aldehyde and a bathochromic shift to an emission maximum centred at λem = 505 nm (ΦF = 0.59) was observed. At physiological pH a marked increase in emission ratio (F505/F585) from 0.12 to 28.2 was observed after the probe was treated with hypochlorite. Probe 88 was successfully used to visualise exogenous hypochlorite in MCF-7 cells.
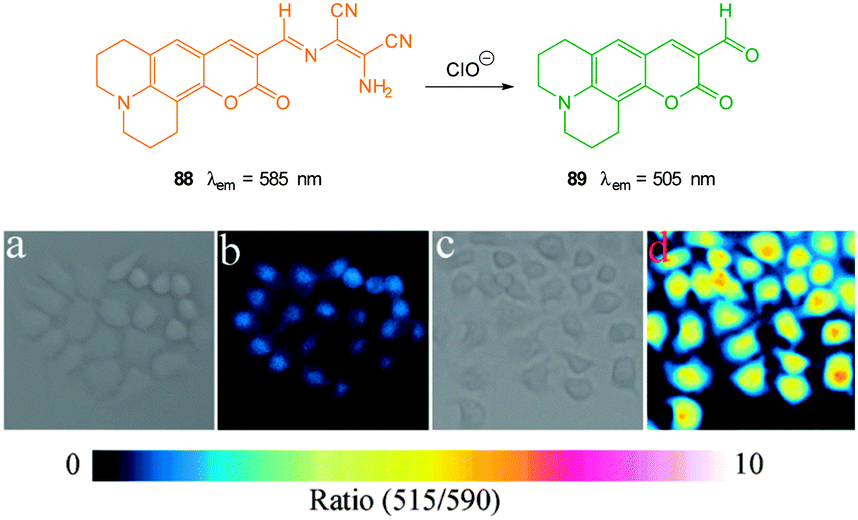 | ||
| Fig. 91 Top: structure and reaction of aminocoumarinimine 88 with ClO− (a) bright-field image of live MCF-7 cells incubated with only probe 88 (b) fluorescence ratio image (F505/F585) of (a); (c) bright-field image of live MCF-7 cells incubated with probe 88 then with ClO−; (d) fluorescence ratio image (F505/F585) of (c). Image reproduced with permission.315 | ||
In an extension of this work, Nagano (2011) reported the silylrhodamine analogue MMSiR (Fig. 92).317 The isosteric replacement of oxygen with silicon resulted in a significant red-shift in the emission wavelength to the near-infrared (NIR) region (λem = 670 nm, ϕF = 0.31). Similar to HySOx, the MMSiR probe was also used in the imaging of phagocytosis. In addition the hydrophilic probe wsMMSiR was synthesised and applied to the in vivo imagining of hypochlorite in a mouse peritonitis model (Fig. 93). In this study C57BL/6 mice were treated with an intraperitoneal injection of zymosan to stimulate neutrophil invasion of the peritoneal cavity. Injection of wsMMSiR and PMA resulted in enhanced fluorescence emission in the abdomen.
 | ||
| Fig. 92 Structure and spirocycle ring opening reactions of HySOx and MMSiR with hypochlorite. | ||
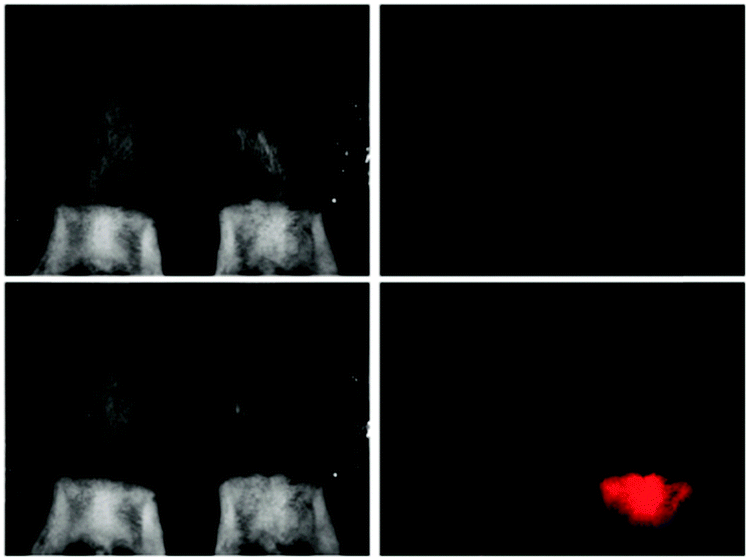 | ||
| Fig. 93 Comparison of white light (left panels) and fluorescence (right panels) images of unstimulated mouse (left) and the peritonitis model mouse stimulated with zymosan and PMA (right). wsMMSiR and PMA were successively administered by intraperitoneal injection. Images were obtained just before (top) and 60 min after (bottom) PMA injection. Image reproduced with permission.317 | ||
Using a more traditional rhodamine fluorophore, Yoon (2007) developed both a thio- and selenoester trigger for selective ClO− sensing.318 Upon reaction with hypochlorite, the non-fluorescent R19-S, R19-Se and R101-S undergo ring opening and fluorescence is “switched on” at λem = 550 nm, 545 nm and 585 nm respectively (Fig. 94). Probe R19-S was used to visualise hypochlorite production in phagocytes and microbial hypochlorite generation in intestinal epithelia of Drosophila melanogaster.
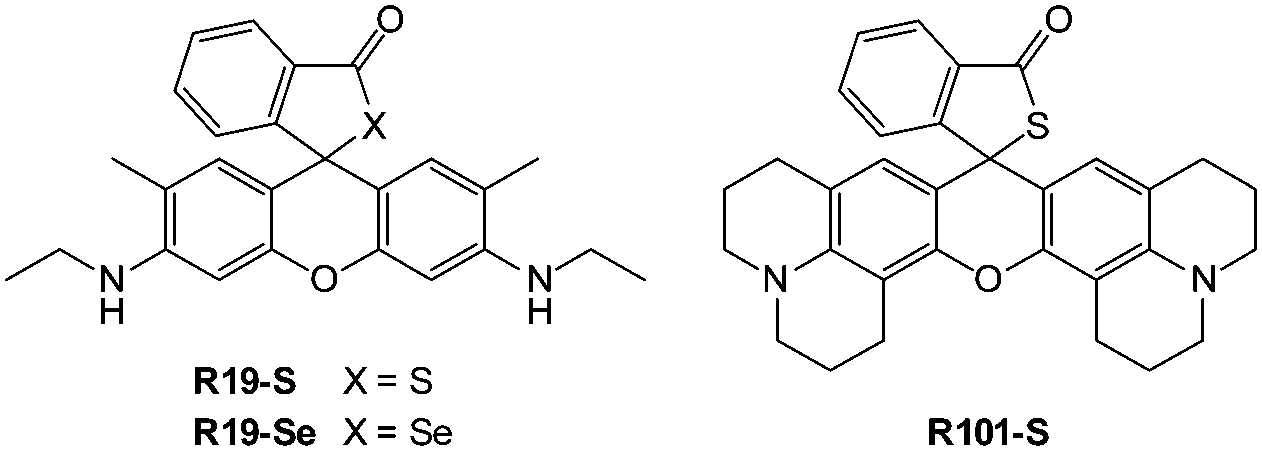 | ||
| Fig. 94 Structure of the rhodamine probes developed by Yoon containing S and Se triggers. | ||
In the last few years a number of diacylhydrazine–rhodamine probes have been reported. These probes rely on selective oxidation by hypochlorite to give a diacyl diimide, then subsequent hydrolysis reveals the fluorescent rhodamine. An early example of this approach was reported by Ma and co-workers although high pH was required (pH 12).319 Modifications to the benzoyl substituent can be used to tailor probes which are more suited for imaging purposes. For example, by using a pendant alkoxyquinoline moiety (RHQ, Fig. 95) Goswami and co-workers were able to image endogenous hypochlorite in human peripheral blood mononuclear cells (PBMCs).320 A similar fluorescein based system (90 RO and 91 RS, Fig. 95) from the Li group was used to monitor hypochlorite in Rhodobacter ferrooxidans prokaryotes as a potential model for hypochlorite induced stress.321
In the last few years a number of diacylhydrazine–rhodamine probes have been reported. These probes rely on selective oxidation by hypochlorite to give a diacyl diimide, then subsequent hydrolysis reveals the fluorescent rhodamine. An early example of this approach was reported by Ma and co-workers although high pH was required (pH 12).319 Modifications to the benzoyl substituent can be used to tailor probes which are more suited for imaging purposes. For example, by using a pendant alkoxyquinoline moiety (RHQ, Fig. 95) Goswami and co-workers were able to image endogenous hypochlorite in human peripheral blood mononuclear cells (PBMCs).320 A similar fluorescein based system (90 RO and 91 RS, Fig. 95) from the Li group was used to monitor hypochlorite in Rhodobacter ferrooxidans prokaryotes as a potential model for hypochlorite induced stress.321
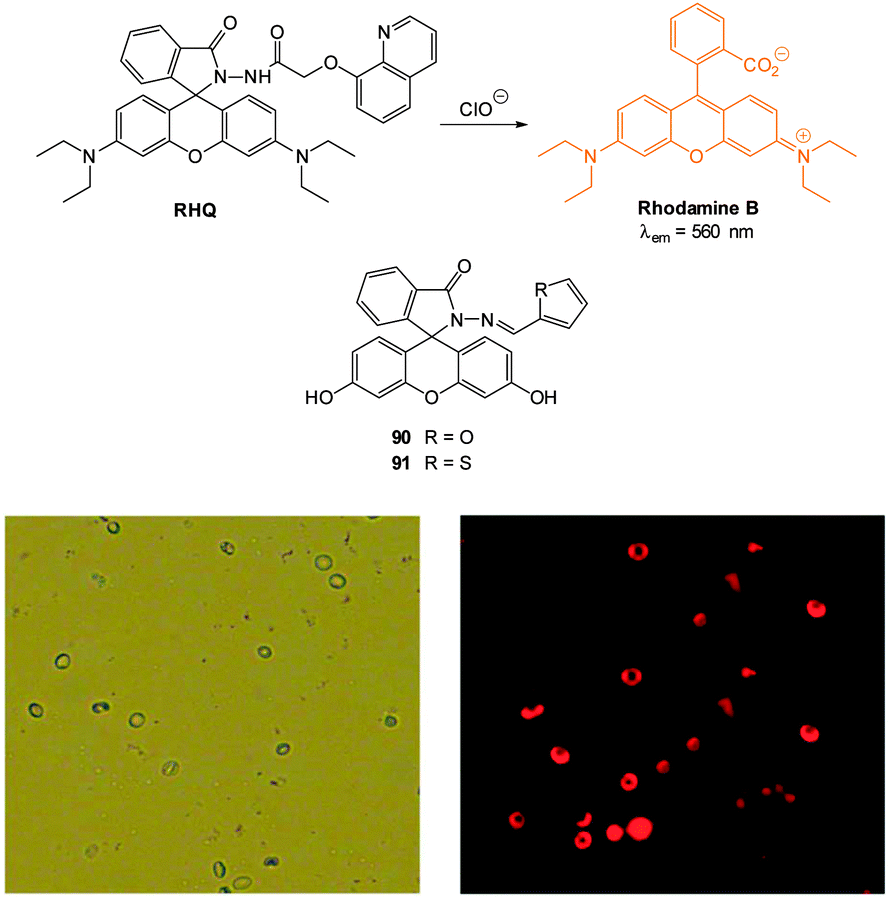 | ||
| Fig. 95 Top: structure of RHQ probe and related probes 90 and 91. Bottom: (left) bright field image of PBMCs (40×), (right) fluorescence image of PBMCs (40×) treated with RHQ. Image reproduced with permission.320 | ||
In 2014 Hou et al. employed this trigger in the mitochondria targeting probes Rh-TPP and Rh-Py (Fig. 96).99 The incorporation of a phosphonium (Rh-TPP) or pyridinium ion (Rh-Py) onto the probe facilitated the imaging of exogenous hypochlorite in the mitochondria of HeLa cells.
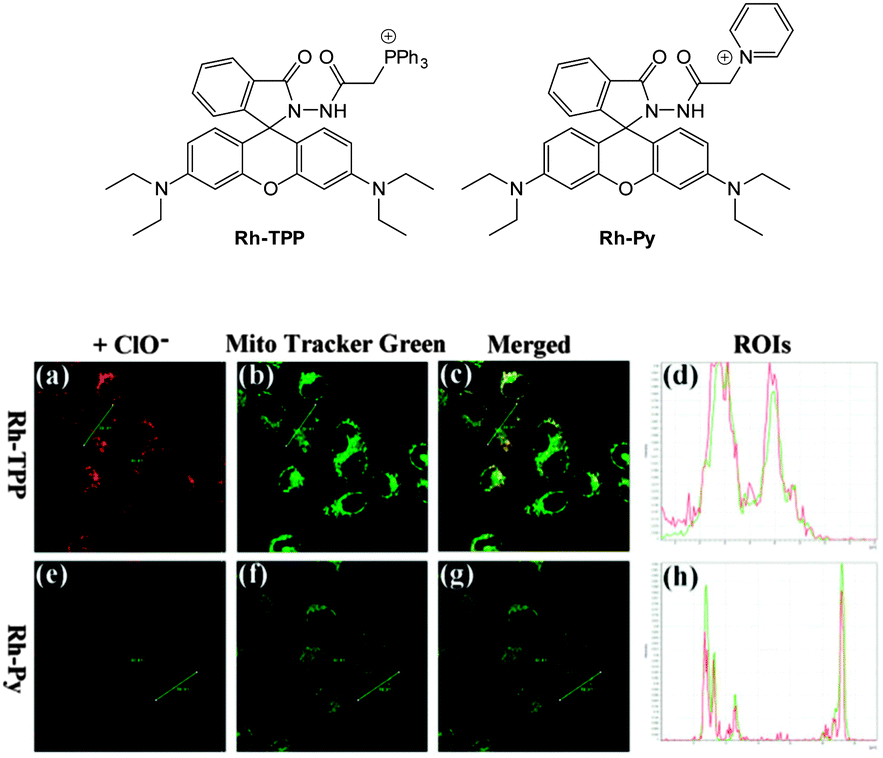 | ||
| Fig. 96 Top: structure of mitochondria targeting probes Rh-TPP and Rh-Py Bottom: HeLa cells were stained with Rh-TPP or Rh-Py, incubated with ClO−, then Mito Tracker Green was added. (a) and (e): Fluorescence images of Rh-TPP and Rh-Py with ClO−; (b) and (f): the fluorescence images of Mito Tracker Green; (c) and (g): merged images; (d) and (h): intensity profile of ROIs across HeLa cells. Red lines represent the intensity of synthetic probes and green lines represent the intensity of Mito Tracker Green. Scale = 25 μm. Image reproduced with permission.99 | ||
The ability of Rh-TPP and Rh-Py to visualise hypochlorite in vivo was also demonstrated (Fig. 97). Nude mice were injected with either probe followed by an injection with a ClO− solution. In each case a persistent fluorescent response was elicited from hypochlorite.
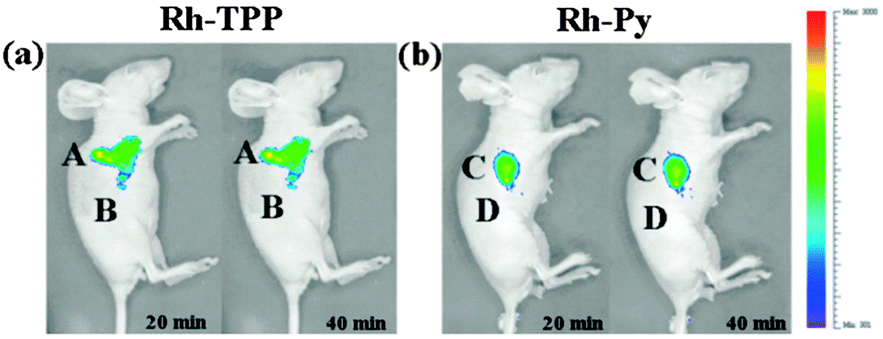 | ||
| Fig. 97 Representative fluorescence images (pseudocolor) of nude mice given a skin-pop injection of (a) Rh-TPP and (b) Rh-Py (region A and C, respectively) and a subsequent skin-pop injection of ClO−. Representative fluorescence images (pseudocolor) of nude mice given a skin-pop injection of (a) Rh-TPP and (b) Rh-Py region B and D, respectively). Images were taken after incubation for 20 and 40 min, respectively. Image reproduced with permission.99 | ||
A hydroxamic acid variant of this approach was outlined by Shin and Tae in 2013.322 In this case it was proposed that the hydroxamic acid was oxidised to the corresponding ring opened acyl nitroso group and it was anticipated that this product was rapidly hydrolysed to rhodamine 19 (λem = 547 nm). Probe 92 (Fig. 98) could detect hypochlorite concentration at ca. 25 nM. Of interest, when the exocyclic amines had an additional ethyl substituent, or were replaced by hydroxyl groups (fluorescein) the probes did not respond to hypochlorite, even when a 20-fold excess was administered.
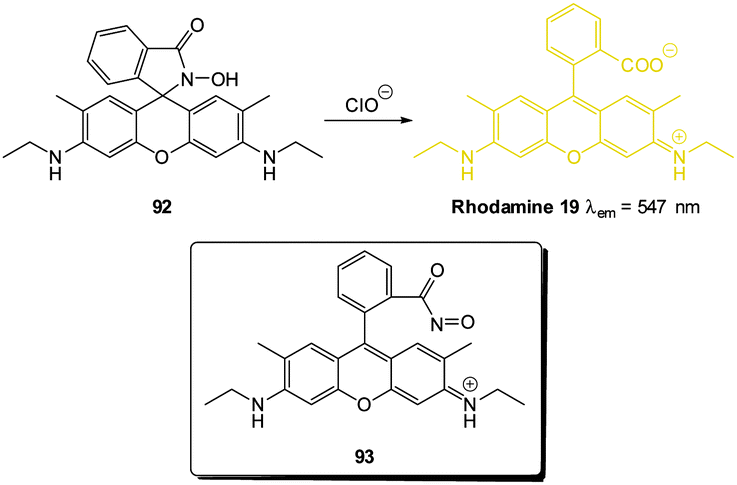 | ||
| Fig. 98 Structure and reaction of 92 with hypochlorite. Inset shows proposed acylnitroso intermediate 93. | ||
Probe 92 could be used to detect exogenous hypochlorite in A549 lung cancer cells with a clear dose responsive fluorescence “switch on” with increasing ClO− concentration (Fig. 99). Furthermore 92 was successfully used for the in vivo detection of exogenous hypochlorite added to live zebrafish.
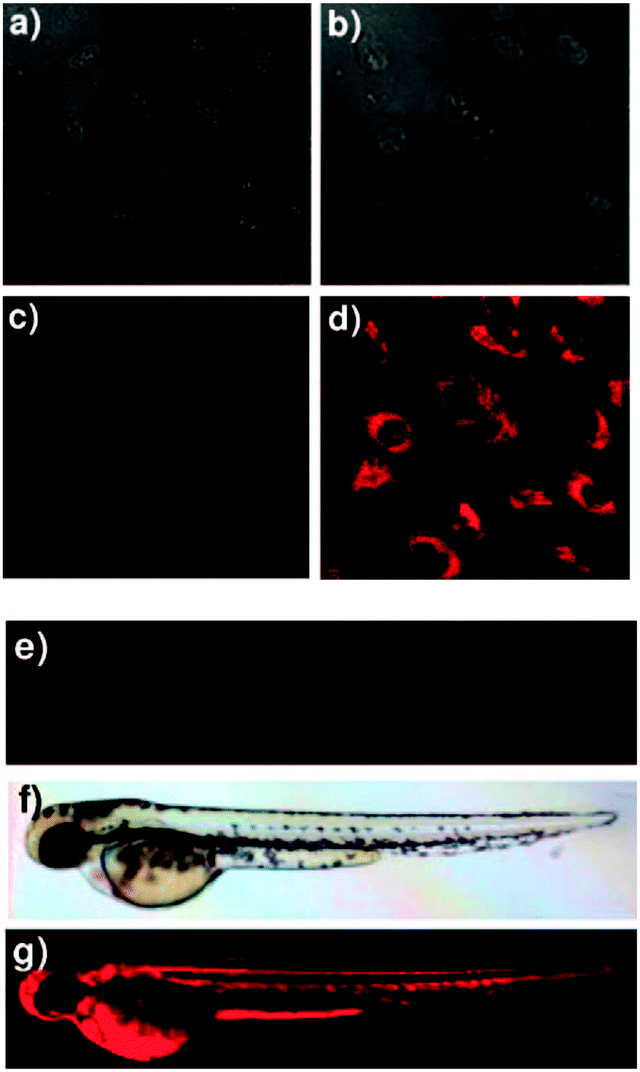 | ||
| Fig. 99 Bright field image of A549 cells treated with (a) 92 in the absence of ClO−. (b) With both ClO− and 92. Fluorescence image of A549 cells treated with (c) 92 in the absence of ClO−. (d) Both ClO− and 83. Fluorescence images of zebrafish treated with (e) 92 in the absence of ClO−. (f) Bright field image of zebrafish treated with 92 and ClO− (g) fluorescence images of zebrafish treated both 92 and ClO−. Image reproduced with permission.322 | ||
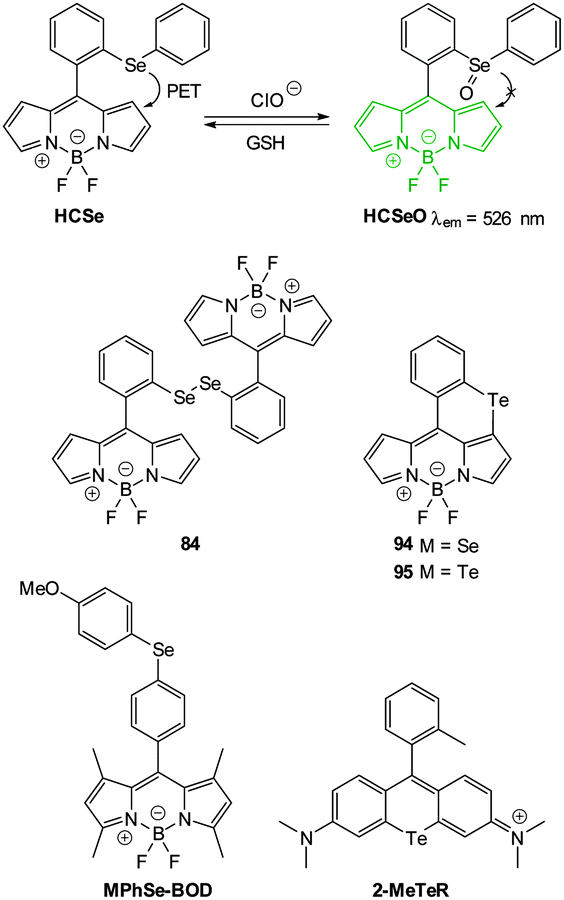 | ||
| Fig. 100 Structures of Se and Te containing probes for ClO−. | ||
Structurally similar annulated BODIPY chalcogenides (discovered somewhat serendipitously during the synthesis of dichalcogenides such as 93) were reported by Churchill in 2013.324 These unexpected annulated heterocycles were formed by base induced SEAr reaction of the dipyrrole dichalcogenide (93) and their structure confirmed by means of an X-ray structure of the selenide derivative (94). It is of note that the diselenide 93 has been utilised as a reversible superoxide probe.325 Despite not being applied to a living system, the annulated tellurium BODIPY (95) was shown to be at least 62-fold selective for ClO− over other ROS (O2˙−, H2O2, tBuOOH, ClO−, HO˙, and tBuO˙) under physiological conditions (0.1 M PBS/EtOH (99:1 v/v), pH 7.5). Following oxidation with hypochlorite, PET from the tellurium was blocked and fluorescence was “switched on” (λem = 597 nm, ΦF increased from 0.06 to 0.23). Similar to HCSe, telluride oxidation could be reversed by treatment with GSH.
MPhSe-BOD is weakly fluorescent (ΦF = 0.13, λem = 510 nm) due to a Se modulated PET process.326 Hypochlorite oxidation interrupts the PET process and restores strong fluorescence (MPhSeO-BOD, ΦF = 0.96). The reverse process (reduction) is selectively performed by HS−. The probe was used to visualise endogenously produced hypochlorite in PMA stimulated RAW264.7 macrophages. The role of hypochlorite was confirmed by comparison to taurine and xanthine–xanthine oxidase controls. Imaging of the redox cycling between hypochlorite and HS− was also demonstrated in RAW264.7 cells.
The tellurium containing rhodamine system (2-MeTeR, ΦF < 0.001) presented by Nagano in 2012 has the advantage that its oxidised form (2-MeTeOR) is red emissive (λem = 686 nm, ΦF = 0.18).327 However, this probe suffers from interference from other ROS such as HO˙ and ONOO−. Nevertheless the probe was used to visualise endogenously produced ROS in human promyelocytic leukemia (HL-60) cells following stimulation with H2O2. As the added H2O2 was consumed by MPO and/or the reaction was reversed by intracellular reductants, the increased fluorescent intensity was short lived. Fluorescence was restored following a second addition of H2O2 (Fig. 101). Fluorescence intensity was also reduced when the cells were treated with aminobenzoic acid hydrazide (ABAH).
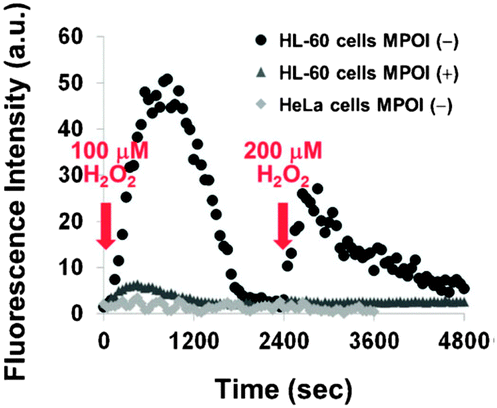 | ||
| Fig. 101 Time courses of the change in fluorescence intensity observed with 2-MeTeR for H2O2-stimulated or unstimulated HL-60 cells or H2O2-stimulated HeLa cells. H2O2 (100 μM and 200 μM final concentration) was added at 100 s and 2400 s, respectively. The myeloperoxidase inhibitor (MPOI) is aminobenzoic acid hydrazide (ABAH; 2 mM). The fluorescence intensity was measured at 690 nm with excitation at 660 nm. Image reproduced with permission.327 | ||
In 2013 the Han group reported the 4-aminonaphthalimide-based hypochlorite sensor NI-Se, shown in Fig. 102.328 While related to the aforementioned examples, the selenide was non-fluorescent (ΦF = 0.04) and oxidation gave the fluorescent selenoxide NI-SeO (λem = 523 nm, ΦF = 0.45). Unlike the previously described selenide–selenoxide systems fluorescence was not modulated by a PET mechanism. Instead, the 2-(phenylselenyl)benzyl moiety induced an excited state configurational twist which was not present in the oxidised NI-SeO. The reduction of the selenoxide could be reversed by the addition of HS− to the system; this process was repeated up to six times with a decrease in fluorescence intensity of 50%. Cellular imagining of NI-Se was performed using mouse macrophage RAW264.7 cells stimulated with LPS and PMA and enhanced fluorescence was observed. Weak fluorescence was also observed when cells were pre-treated with salicylhydroxamic acid (SHA), a known inhibitor of MPO. A similar result was obtained with the use of a ROS scavenger glutathione S-transferase (GST, EC: 2.5.1.18). Co-staining with Hoechst 33342 revealed that probe was located mainly in the cytosol.
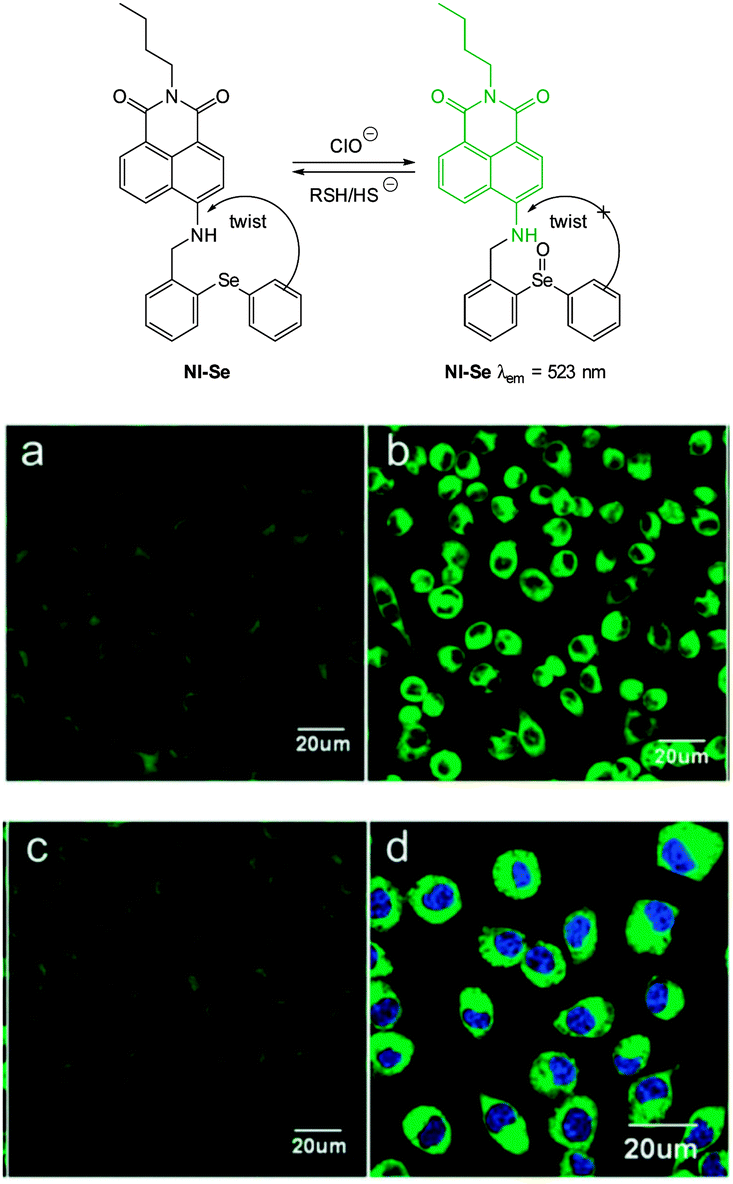 | ||
| Fig. 102 Top: structure and reaction of NI-Se with hypochlorite. Bottom: fluorescence images of ClO− in living RAW264.7 cells. (a–c) Cells were pretreated with LPS. (a) Incubated with NI-Se. (b) Incubated with NI-Se and then with PMA. (c) Incubated with SHA and co-incubated with PMA, then incubated with NI-Se. (d) RAW264.7 cells loaded with Hoechst 33342 and NI-Se. Image reproduced with permission.328 | ||
NI-Se was also used in living mice for the in vivo imaging of hypochlorite produced in a LPS model of acute inflammation (Fig. 103). Similar to the in vitro study, when LPS was injected, followed by NI-Se, an enhanced signal was collected compared to the control (NI-Se only). Injection of HS− resulted in a decrease in fluorescence emission.
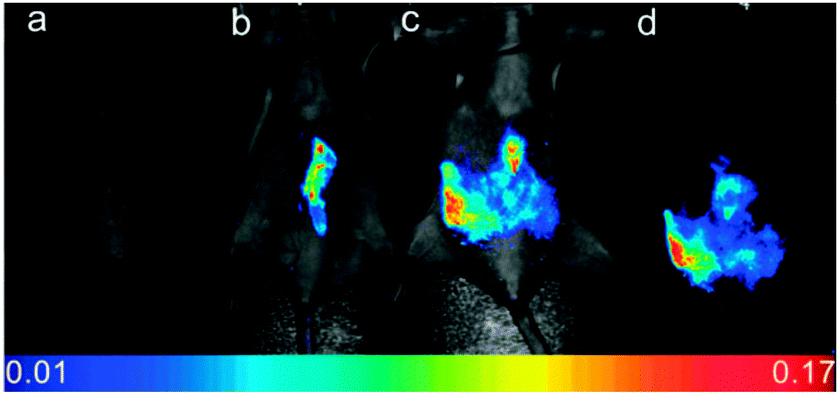 | ||
| Fig. 103 Fluorescent images of ClO− production and HS− reduction in the peritoneal cavity of the mice with NI-Se. (a) Control, neither LPS nor NI-Se was injected; (b) saline was injected in the intraperitoneal (i.p.) cavity of mice, followed by i.p. injection of NI-Se; (c) LPS was injected into the peritoneal cavity of the mice, followed by i.p. injection of NI-Se. (d) An additional HS− was injected in parallel to (c). Image reproduced with permission.328 | ||
A new strategy for the detection of hypochlorite involving a selenide containing probe was presented by Li et al. in 2013.329 The partially reduced coumarin derivatives CM1 and CM2, (Fig. 104) each bearing a 3-phenylselenyl moiety were synthesised and both were non-fluorescent (ΦF < 0.001). In this instance the selenoxide produced from hypochlorite oxidation underwent a cope-type elimination to give the corresponding fluorescent reporters 96 (λem = 480 nm, ΦF = 0.036) and 97 (λem = 468 nm, ΦF = 0.047). The utility of probe CM1 was demonstrated in vitro, by the quantitative determination of exogenous hypochlorite in NIH3T3 cells. CM1 was also used to indicate the endogenous formation of hypochlorite in H2O2 stimulated HL-60 human progranulocytic leukemia cell lines and RAW264.7 macrophages stimulated with lipopolysaccharide (LPS). In both of these instances there was a significant enhancement of the fluorescent response in relation to the controls.
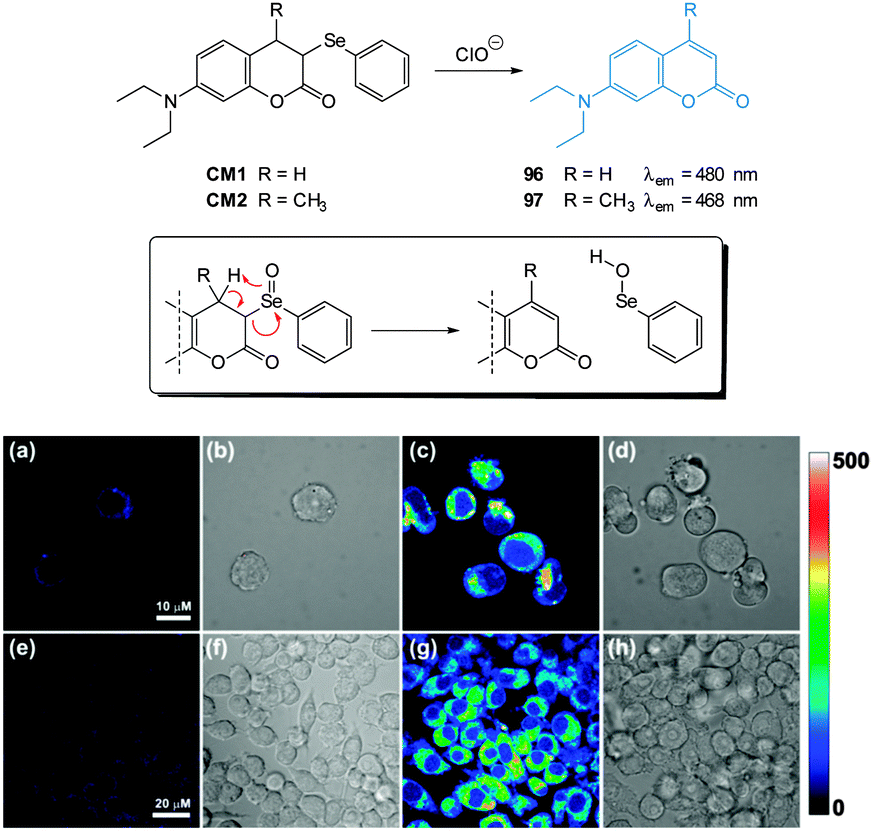 | ||
| Fig. 104 Top: structure of CM1 and CM2 and the mechanism of their reaction with ClO−. Bottom: (a) fluorescence images of HL-60 cells with CM1 (b) bright-field images of cells in panel (a). (c) After addition of H2O2 (d) bright-field images of cells in panel (c). (e) Fluorescence images of RAW264.7 cells with CM1 (f) bright-field images of cells in panel (e). (g) Cells loaded with CM1 then LPS (h) bright-field images of cells in panel (g). Image reproduced with permission.329 | ||
A PET-based Ru(bpy)32+ based probe for the monitoring of ClO−/HS− redox cycle was developed by the Sun group in 2014 (Fig. 105).330 A pendent phenothiazine (PTZ) was attached to one of the 2,2′-bipyridine ligands, which suppressed fluorescence emission from the complex. Upon oxidation of the PTZ sulfur with ClO− the fluorescence emission was restored (λem = 605 nm, λex = 450 nm, ΦF = 0.39). This process could be reversed with HS− in a recyclable fashion (12 cycles). The authors were able to visualise this ClO−/HS− redox cycle in live mice; weak fluorescence was observed after administration of probe 98 and this response was modulated during alternating additions of ClO− and HS−.
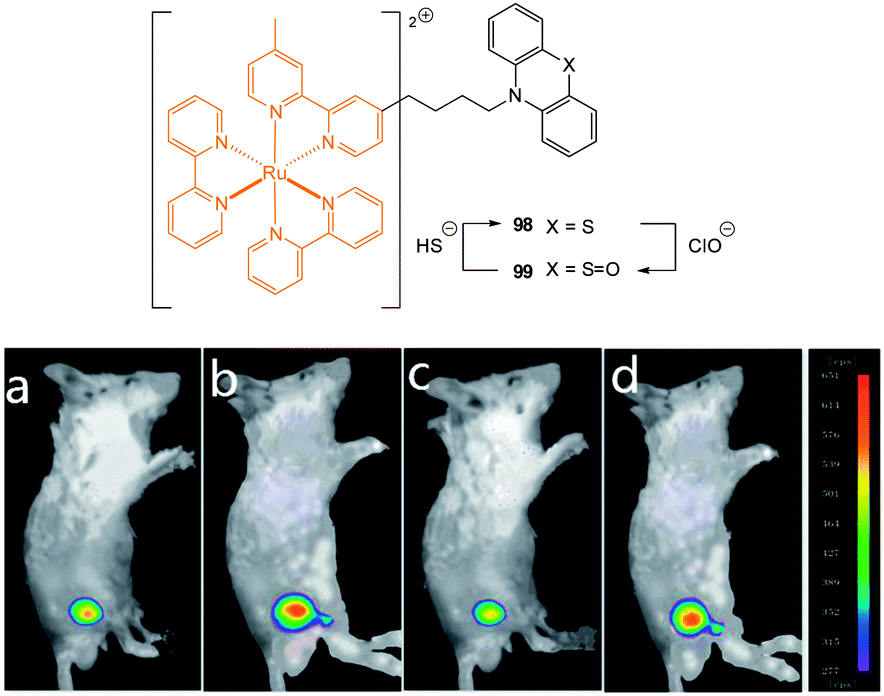 | ||
| Fig. 105 The luminescence imaging of the redox cycle between ClO− and HS− in live mice: (a) 98 was loaded into the leg cortex of the mice; (b) ClO− was loaded in the same position; (c) HS− was loaded in the same position; and (d) another ClO− was loaded in the same position. Image reproduced with permission.330 | ||
The groups of Wang and Peng reported a cyanine–phenothiazine hybrid probe, PTZ-Cy2 which was sensitive to both hypochlorite and HO˙.331 The sulphur atom of the non-fluorescent probe was initially oxidised by OCl− and/or HO˙ to give the fluorescent sulfoxide OPTZ-Cy2 which gave a pink emission (λem = 595 nm). Continued addition of OCl− to OPTZ-Cy2 led to a blue shifted emission (λem = 470 nm) resulting from the degradation of the conjugated cyanine alkene to the aldehyde OPTA. This blue shifted emission was not seen during HO˙ addition, rather, excessive HO˙ lead to a decrease in the total emission indicating decomposition. PTZ-Cy2 was used to visualise the ROS in PMA stimulated HeLa cells (Fig. 106). Co-staining with MitoTracker Deep Red FM indicates that fluorescence resulting from PTZ-Cy2 and OPTZ-Cy2 was localised in the mitochondria. Non-mitochondrial fluorescence was ascribed to the non-charged OPTA diffusing away from the mitochondria.
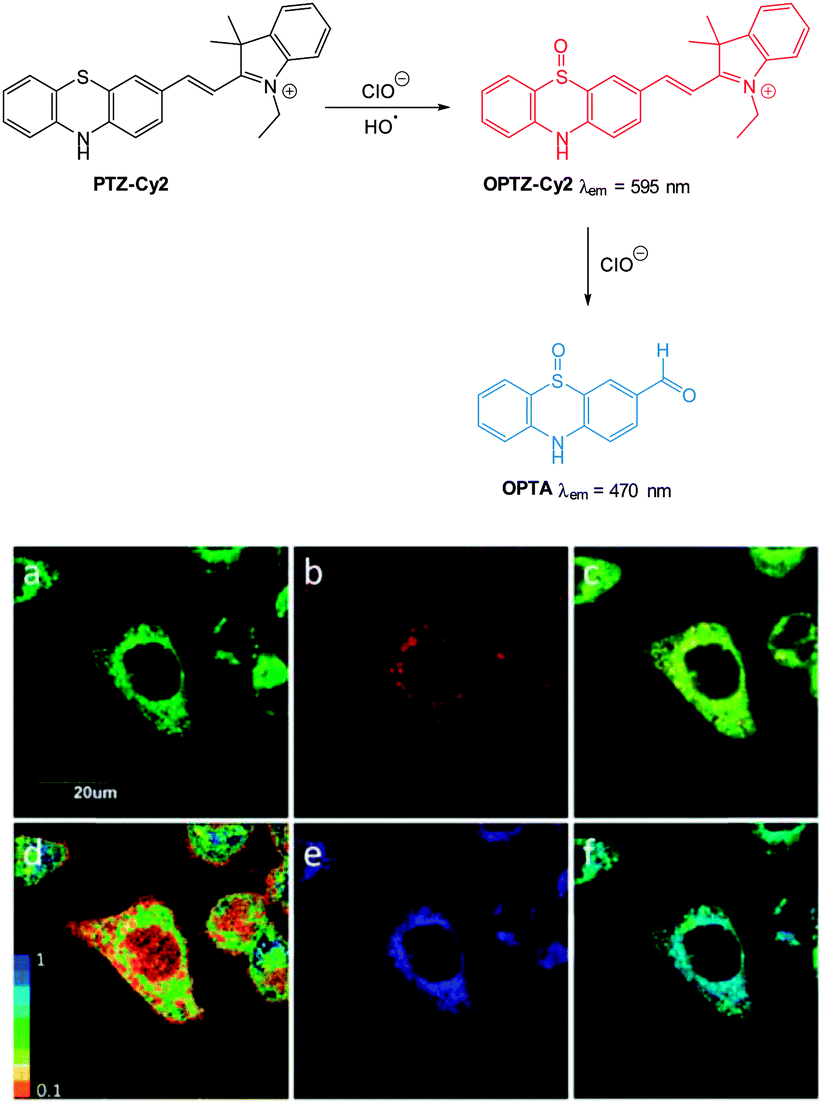 | ||
| Fig. 106 Top: structure and reaction of PTZ-Cy2 with ClO−. Bottom: HeLa cells pre-treated with PMA then PTZ-Cy2 plus MitoTracker Deep Red FM. (a) Emission of PTZ-Cy2 at 470 nm; (b) emission of PTZ-Cy2 at 590 nm; (c) merged image of (a) and (b) with bright-field image; (d) pseudocolour ratiometric images (F470/F590); (e) fluorescence image of MitoTracker Deep Red FM (λem = 690 nm); (f) overlay of (a) and (e). Image reproduced with permission.331 | ||
 | ||
| Fig. 107 Representative fluorescent images (pseudocolour) of in vivo ClO− production from the peritoneal cavity of the mice with 100 during an LPS-mediated inflammatory response: (a) negative control, neither LPS nor 100 was injected; (b) saline was injected in the intraperitoneal (i.p.) cavity of mice, followed by i.p. injection of 100; (c) LPS was injected into the peritoneal cavity of the mice, followed by i.p. injection of 100. Image reproduced with permission.332 | ||
Ma (2010) described the PET system 9-AEF consisting of an anthracene linked by an alkene to ferrocene (Fig. 108).333 The electron rich ferrocene instigates PET to the anthracene and fluorescence is quenched (ΦF < 0.001). Hypochlorite could oxidise the alkene, and in the product (not characterised) PET is unfavoured and anthracene-like emission was observed (λem = 441 nm, ΦF = 0.12). It should be noted that no single product was identified, nevertheless, the response to hypochlorite was dose dependant and the probe was successfully used to image hypochlorite in HeLa cells. No fluorescence was observed in cells containing 9-AEF unless they were pre- or post-treated with hypochlorite.
 | ||
| Fig. 108 Structure of hypochlorite sensitive probe 9-AEF as described by Ma.333 | ||
4.3 Hypobromite
In an analogous manner to hypochlorite, hypobromous acid/hypobromite can be produced endogenously by macrophages and eosinophils from the reaction between H2O2 and bromide which is catalysed by eosinophil peroxidase (EPO) in response to invading pathogens. Overproduction of HOBr can lead to diseases such as cancer, arthritis, cardiovascular disease and asthma. There is a demonstrated correlation between clinical severity in asthma patients and serum EPO levels. While the fraction of HOBr/BrO− is likely to be high (the pKa for HOBr is 8.7–8.8 at 25 °C) a small percentage of the anionic species is likely to exist at physiological pH and in certain compartments such as mitochondria a considerable percentage of BrO− is likely. The design of fluorescent probes for in vitro of detection of this species has recently received attention. | ||
| Fig. 109 Top: structure of mCy-TemOH and Cy-TemOH and their reaction with BrO−. Bottom: ratiometric fluorescence images (F632/F550) of living RAW264.7 cells with mCy-TemOH (a)–(d). pseudo color images represent the ratio of emission intensities. (a) Control. (b) mCy-TemOH-loaded cells incubated with H2O2, EPO and KBr. (c) mCy-TemOH loaded, EPO, and H2O2 treated cells incubated with glutathione S-transferase, then ascorbic acid. (d) Cells exposed to a second dose of EPO, KBr and H2O2. (e) and (f) Confocal microscopy imaging for subcellular locations of mCy-TemOH in living RAW264.7 cells. (e) Cells incubated with mCy-TemOH (red) and co-staining with Hoechst dye (blue). (f) Overlay of (e) and its bright-field image. Image reproduced with permission.334 | ||
The full cyanine probe, Cy-TemOH, exhibited NIR absorption (λabs = 702 nm) and emission (λem = 755 nm). Cy-TemOH also responded to the HOBr/ascorbic acid redox cycle but suffered from severe bleaching after three cycles. Nevertheless, Cy-TemOH was successfully used to visualise the redox cycle in RAW264.7 cells. Both Cy-TemOH and mCy-TemOH localised in the cytoplasm and cells remained viable (MTT assay).
The same group then developed a NIR-reversible BODIPY-based probe which could monitor the redox cycle between HOBr and HS−.335 The probe, diMPhSe-BOD (Fig. 110) absorbs strongly (ε672 = 22770 M−1 cm−1) but was weakly fluorescent (λem = 711 nm, ΦF = 0.00083) due to the electron transfer (PET) from the diarylselenides. Oxidation with HOBr leads to a blue-shifted emission (λem = 635 nm) and 118-fold increase in the F635/F711 ratio. The reverse reaction is accomplished by HS− (selectively over other RSS such as Cys, Hcys and GSH). It is also notable that the emission ratio generated from the reaction with HOBr was ca. 6-fold greater than with ClO−. The HOBr/HS− redox cycle could be visualised in RAW264.7 cells and using diMPhSe-BOD HOBr and H2S could be detected at concentrations as low as 50 and 100 nM respectively.
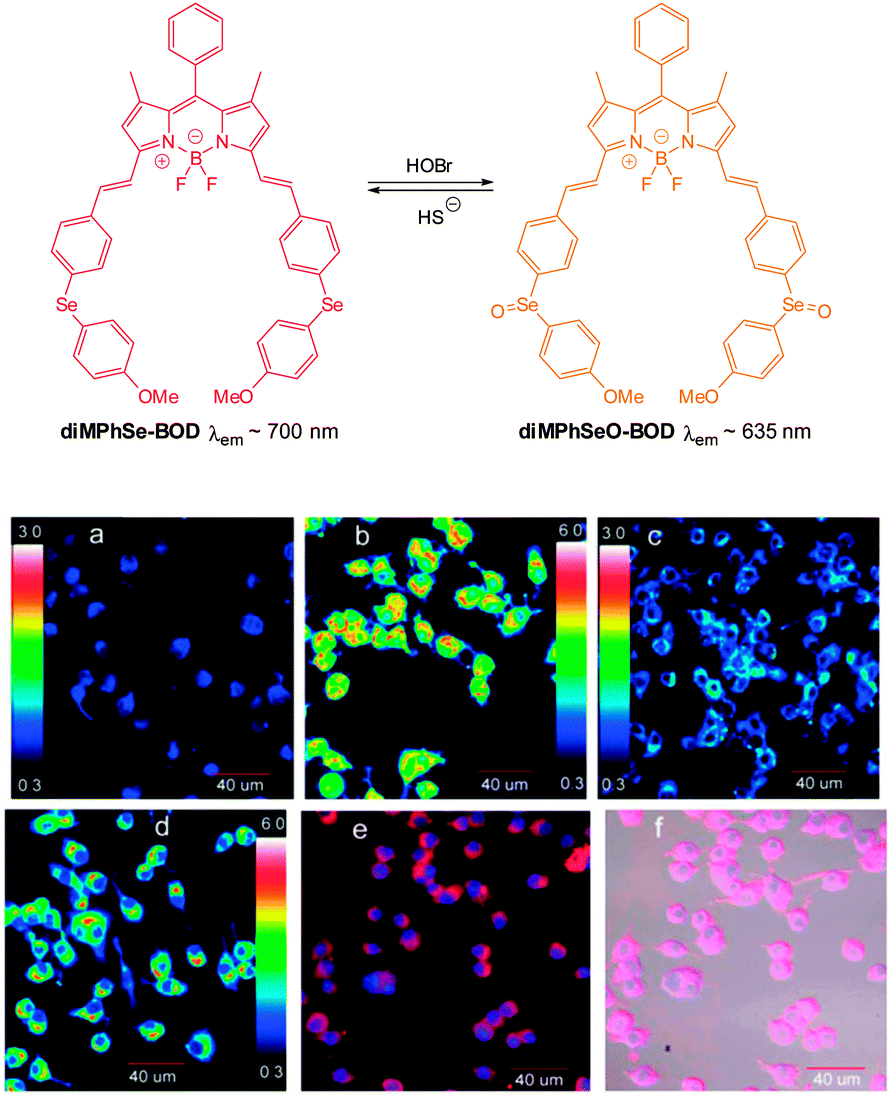 | ||
| Fig. 110 Confocal fluorescence images of the redox cycles between HOBr and H2S in RAW264.7 cells. Macrophage cells. (a) Control; (b) diMPhSe-BOD-loaded cells with H2O2, EPO, and KBr; (c) cells in (b) after addition of HS−; (d) (c) was treated with a second dose of H2O2, EPO and KBr; (e) overlay of images showing fluorescence from diMPhSe-BOD and Hoechst dye; (f) overlay of bright-field, diMPhSe-BOD, and Hoechst dye images. Image reproduced with permission.335 | ||
5. Conclusion and future work
One of the key purposes of this review was to highlight that the strategies employed for the design of small fluorescent anion sensors can be applied to in vitro and in vivo bioimaging.As such further developments in fluorophore design and strategies for signal modulation336 are likely to have immediate imaging applications. The emerging use of multiphoton excitation is already having an impact. Developments in the use of fluorescence lifetime337 will also broaden the range of approaches that can be used. Probes that are compatible with a super-resolution approach would also be beneficial for the ultimate aim of pinpointing the origin and/or fate of the species of interest.
Despite some impressive examples, many of the probes described herein do not meet the highly demanding criteria set out for an “ideal probe” in Section 1.2. Complete selectivity for the analyte of interest is not always realised and probes that are not dependent on a chemical transformation are currently the most prone to interference. Indeed it is a truly formidable challenge to design a selective, reversible, recognition process that operates by means of non-covalent forces in an exceptionally competitive media! Further refinement of fundamental recognition principles and the ability to readily synthesise (or assemble) sophisticated structures is required to overcome this hurdle. While the selectivity of chemodosimeters is often excellent the reactions are typically irreversible and hence they are incapable of mapping in a truly spatiotemporal manner.
Furthermore, despite the obvious potential that these probes offer, many are destined to be research tools only; the widespread use of fluorescent probes in clinical settings is hampered by expensive operational set-up and sample by sample analysis. Nevertheless, several of the probes included herein have been successfully used in flow systems147,171,281 and with the current rapid progress in the field of micro-fluidics and micro-optics the goal of cost effective, “smartphone style”, point of care diagnostics using selective fluorescent probes is sure to be realised.338–342
It is also interesting to note that for many small anions (Cl−, I−, HCO3−, and BrO−) the complete list of probes is very short and hence any progress would be welcome. Nevertheless, the field of anion sensing is rapidly advancing, and for the researchers involved in this field these challenges should be seen as nothing more than an excellent opportunity to further the fundamental understanding of our natural world.
Glossary of cell lines
| RAW264.7 | Murine/mouse macrophages |
| PC3 | Prostate cancer |
| HUVEC | Human umbilical vein endothelial cell |
| NIH3T3 | Murine/mouse fibroblast |
| HeLa | Epithelial cervical cancer |
| MCF-7 | Breast cancer |
| HEK-293 | Human embryonic kidney cells |
| L929 | Murine/mouse fibroblast |
| A549 | Adenocarcinomic human alveolar basal epithelial cells |
| COS-7 | Fibroblast-like |
| B16-F10 | Murine/mouse skin melanoma |
| U266 | Human myeloma |
| HepG2 | Hepatocellular carcinoma |
| PMN | Human polymorphonuclear neutrophils |
| BV2 microglial | Brain resident murine macrophages |
| MDA-MB-231 | Human breast adenocarcinoma |
| SH-SY5Y | Human neuronal neuroblasts |
| HL-60 | Human promyelocytic leukemia |
| GES | Human breast cancer cells |
Acknowledgements
The authors TDA, KJ and FP would like to thank the Australian Research Council (ARC DP140100227) and TDA and FP also thank the Strategic Research Centre for Chemistry and Biotechnology (Deakin University) for financial support.References
- Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, John Wiley and Sons, Chichester, 2012 Search PubMed.
- Recognition of Anions, ed. R. Vilar, Springer-Verlag, Berlin, 2008 Search PubMed.
- Anion Receptor Chemistry, ed. J. L. Sessler, P. A. Gale and W.-S. Cho, The Royal Society of Chemistry, Cambridge, 2006 Search PubMed.
- Encyclopedia of Supramolecular Chemistry, ed. J. L. Atwood and J. W. Steed, Marcel Dekker, New York, 2004 Search PubMed.
- Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. García-España, Wiley-VCH, New York, 1997 Search PubMed.
- M. M. G. Antonisse and D. N. Reinhoudt, Chem. Commun., 1998, 443–448 RSC.
- S. Kubik, C. Reyheller and S. Stuwe, J. Inclusion Phenom. Macrocyclic Chem., 2005, 52, 137–187 CrossRef CAS PubMed.
- E. A. Katayev, Y. A. Ustynyuk and J. L. Sessler, Coord. Chem. Rev., 2006, 250, 3004–3037 CrossRef CAS PubMed.
- F. P. Schmidtchen, Coord. Chem. Rev., 2006, 250, 2918–2928 CrossRef CAS PubMed.
- P. Blondeau, M. Segura, R. Perez-Fernandez and J. de Mendoza, Chem. Soc. Rev., 2007, 36, 198–210 RSC.
- J. W. Steed, Chem. Soc. Rev., 2009, 38, 506–519 RSC.
- S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC.
- F. P. Schmidtchen, Chem. Soc. Rev., 2010, 39, 3916–3935 RSC.
- A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS PubMed.
- M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC.
- P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205–241 RSC.
- M. J. Langton and P. D. Beer, Acc. Chem. Res., 2014, 47, 1935–1949 CrossRef CAS PubMed.
- H. D. P. Ali, P. E. Kruger and T. Gunnlaugsson, New J. Chem., 2008, 32, 1153–1161 RSC.
- T. Gunnlaugsson, A. P. Davis and M. Glynn, Chem. Commun., 2001, 2556–2557 RSC.
- H. Miyaji and J. L. Sessler, Angew. Chem., Int. Ed., 2001, 40, 154–157 CrossRef CAS.
- T. Gunnlaugsson, A. P. Davis, J. E. O'Brien and M. Glynn, Org. Lett., 2002, 4, 2449–2452 CrossRef CAS PubMed.
- R. Martinez-Manez and F. Sancenon, Chem. Rev., 2003, 103, 4419–4476 CrossRef CAS PubMed.
- C. Suksai and T. Tuntulani, Chem. Soc. Rev., 2003, 32, 192–202 RSC.
- F. P. Schmidtchen, in Anion Sensing, ed. I. Stibor, 2005, pp. 1–29 Search PubMed.
- C. Suksai and T. Tuntulani, in Anion Sensing, ed. I. Stibor, 2005, pp. 163–198 Search PubMed.
- T. Gunnlaugsson, M. Glynn, G. M. Tocci, P. E. Kruger and F. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094–3117 CrossRef CAS PubMed.
- B. T. Nguyen and E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118–3127 CrossRef CAS PubMed.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- M. E. Moragues, R. Martinez-Manez and F. Sancenon, Chem. Soc. Rev., 2011, 40, 2593–2643 RSC.
- J. Du, M. Hu, J. Fan and X. Peng, Chem. Soc. Rev., 2012, 41, 4511–4535 RSC.
- L. E. Santos-Figueroa, M. E. Moragues, E. Climent, A. Agostini, R. Martinez-Manez and F. Sancenon, Chem. Soc. Rev., 2013, 42, 3489–3613 RSC.
- H. T. Ngo, X. J. Liu and K. A. Jolliffe, Chem. Soc. Rev., 2012, 41, 4928–4965 RSC.
- P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015 10.1039/C4CS00179F.
- X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590–659 CrossRef CAS PubMed.
- K. Kaur, R. Saini, A. Kumar, V. Luxami, N. Kaur, P. Singh and S. Kumar, Coord. Chem. Rev., 2012, 256, 1992–2028 CrossRef CAS PubMed.
- S. K. Kim, D. H. Lee, J.-I. Hong and J. Yoon, Acc. Chem. Res., 2009, 42, 23–31 CrossRef CAS PubMed.
- M. Cametti and K. Rissanen, Chem. Commun., 2009, 2809–2829 RSC.
- T. W. Hudnall, C.-W. Chiu and F. P. Gabbai, Acc. Chem. Res., 2009, 42, 388–397 CrossRef CAS PubMed.
- C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed.
- F. Wang, L. Wang, X. Chen and J. Yoon, Chem. Soc. Rev., 2014, 43, 4312–4324 RSC.
- Q. Xu, C. Jin, X. Zhu and G. Xing, Chin. J. Org. Chem., 2014, 34, 647–661 CrossRef CAS.
- D. Zhang, J. R. Cochrane, A. Martinez and G. Gao, RSC Adv., 2014, 4, 29735–29749 RSC.
- M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015 10.1039/C4CS00280F.
- Y. Zhou and J. Yoon, Chem. Soc. Rev., 2012, 41, 52–67 RSC.
- E. J. New, D. Parker, D. G. Smith and J. W. Walton, Curr. Opin. Chem. Biol., 2010, 14, 238–246 CrossRef CAS PubMed.
- X. J. Liu, H. T. Ngo, Z. J. Ge, S. J. Butler and K. A. Jolliffe, Chem. Sci., 2013, 4, 1680–1686 RSC.
- J. L. Sessler, P. A. Gale and W.-S. Cho, in Anion Receptor Chemistry, ed. J. L. Sessler, P. A. Gale and W. -S. Cho, The Royal Society of Chemistry, 2006, pp. 1–26 Search PubMed.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2012, 113, 192–270 CrossRef PubMed.
- L. M. Hyman and K. J. Franz, Coord. Chem. Rev., 2012, 256, 2333–2356 CrossRef CAS PubMed.
- Y. Urano, Curr. Opin. Chem. Biol., 2012, 16, 602–608 CrossRef CAS PubMed.
- N. Kumar, V. Bhalla and M. Kumar, Coord. Chem. Rev., 2013, 257, 2335–2347 CrossRef CAS PubMed.
- J. Wang, L. Long, D. Xie and Y. Zhan, J. Lumin., 2013, 139, 40–46 CrossRef CAS PubMed.
- L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- E. Pershagen and K. E. Borbas, Coord. Chem. Rev., 2014, 273–274, 30–46 CrossRef CAS PubMed.
- S. F. Peteu, R. Boukherroub and S. Szunerits, Biosens. Bioelectron., 2014, 58, 359–373 CrossRef CAS PubMed.
- K. Wang, H. Peng and B. Wang, J. Cell. Biochem., 2014, 115, 1007–1022 CrossRef CAS PubMed.
- X. Wu and W. Zhu, Chem. Soc. Rev., 2015 10.1039/C4CS00152D.
- S. Wang, N. Li, W. Pan and B. Tang, TrAC, Trends Anal. Chem., 2012, 39, 3–37 CrossRef CAS PubMed.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- R. Y. Tsien, Annu. Rev. Neurosci., 1989, 12, 227–253 CrossRef CAS PubMed.
- R. Y. Tsien, in Methods in Cell Biology, ed. D. Lansing Taylor and Y.-L. Wang, Academic Press, 1989, pp. 127–156 Search PubMed.
- S. A. Hilderbrand, Live Cell Imaging, 2009, pp. 17–45 Search PubMed.
- S. A. Hilderbrand and R. Weissleder, Curr. Opin. Chem. Biol., 2010, 14, 71–79 CrossRef CAS PubMed.
- R. Weissleder and V. Ntziachristos, Nat. Med., 2003, 9, 123–128 CrossRef CAS PubMed.
- J. O. Escobedo, O. Rusin, S. Lim and R. M. Strongin, Curr. Opin. Chem. Biol., 2010, 14, 64–70 CrossRef CAS PubMed.
- W. R. Zipfel, R. M. Williams and W. W. Webb, Nat. Biotechnol., 2003, 21, 1369–1377 CrossRef CAS PubMed.
- F. Helmchen and W. Denk, Nat. Methods, 2005, 2, 932–940 CrossRef CAS PubMed.
- P. Klán and J. Wirz, Photochemistry of Organic Compounds, John Wiley & Sons, Ltd, 2009, pp. 25–72 Search PubMed.
- K. Baathulaa, Y. Xu and X. Qian, J. Photochem. Photobiol., A, 2010, 216, 24–34 CrossRef CAS PubMed.
- P. Abbyad, W. Childs, X. Shi and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20189–20194 CrossRef CAS PubMed.
- P. Horváth, P. Šebej, T. Šolomek and P. Klán, J. Org. Chem., 2015 DOI:10.1021/jo502213t.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC.
- X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2011, 112, 1910–1956 CrossRef PubMed.
- H. Zheng, X.-Q. Zhan, Q.-N. Bian and X.-J. Zhang, Chem. Commun., 2013, 49, 429–447 RSC.
- Y. Q. Sun, J. Liu, X. Lv, Y. Liu, Y. Zhao and W. Guo, Angew. Chem., Int. Ed., 2012, 51, 7634–7636 CrossRef CAS PubMed.
- S. T. Manjare, Y. Kim and D. G. Churchill, Acc. Chem. Res., 2014, 47, 2985–2998 CrossRef CAS PubMed.
- X. Wang, H. Chang, J. Xie, B. Zhao, B. Liu, S. Xu, W. Pei, N. Ren, L. Huang and W. Huang, Coord. Chem. Rev., 2014, 273–274, 201–212 CrossRef CAS PubMed.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 CrossRef CAS PubMed.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2013, 114, 4496–4539 CrossRef PubMed.
- S. J. Butler and D. Parker, Chem. Soc. Rev., 2013, 42, 1652–1666 RSC.
- A. Demchenko, J. Fluoresc., 2010, 20, 1099–1128 CrossRef PubMed.
- J. Fan, M. Hu, P. Zhan and X. Peng, Chem. Soc. Rev., 2013, 42, 29–43 RSC.
- D.-G. Cho and J. L. Sessler, Chem. Soc. Rev., 2009, 38, 1647–1662 RSC.
- X. Lou, D. Ou, Q. Li and Z. Li, Chem. Commun., 2012, 48, 8462–8477 RSC.
- F. Zheng, F. Zeng, C. Yu, X. Hou and S. Wu, Chem. – Eur. J., 2013, 19, 936–942 CrossRef CAS PubMed.
- B. Ke, W. Chen, N. Ni, Y. Cheng, C. Dai, H. Dinh and B. Wang, Chem. Commun., 2013, 49, 2494–2496 RSC.
- R. Y. Tsien, Nature, 1981, 290, 527–528 CrossRef CAS.
- J. P. Crow, Nitric oxide, 1997, 1, 145–157 CrossRef CAS PubMed.
- T. Peng and D. Yang, Org. Lett., 2010, 12, 4932–4935 CrossRef CAS PubMed.
- C. Song, Z. Ye, G. Wang, J. Yuan and Y. Guan, Chem. – Eur. J., 2010, 16, 6464–6472 CrossRef CAS PubMed.
- F. Di Virgilio, T. H. Steinberg, J. A. Swanson and S. C. Silverstein, J. Immunol., 1988, 140, 915–920 CAS.
- R. Horobin, J. Stockert and F. Rashid-Doubell, Histochem. Cell Biol., 2013, 139, 623–637 CrossRef CAS PubMed.
- F. Rashid and R. W. Horobin, Histochemistry, 1990, 94, 303–308 CAS.
- S. Trapp and R. Horobin, Eur. Biophys. J., 2005, 34, 959–966 CrossRef CAS PubMed.
- S. Trapp, G. Rosania, R. Horobin and J. Kornhuber, Eur. Biophys. J., 2008, 37, 1317–1328 CrossRef CAS PubMed.
- J. T. Hou, M. Y. Wu, K. Li, J. Yang, K. K. Yu, Y. M. Xie and X. Q. Yu, Chem. Commun., 2014, 50, 8640–8643 RSC.
- T. Liu, Z. Xu, D. R. Spring and J. Cui, Org. Lett., 2013, 15, 2310–2313 CrossRef CAS PubMed.
- Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511–5571 CrossRef CAS PubMed.
- C. D. Geddes, K. Apperson, J. Karolin and D. J. S. Birch, Anal. Biochem., 2001, 293, 60–66 CrossRef CAS PubMed.
- H. S. Horowitz, J. Public Health Dent., 2003, 63, 3–8 CrossRef PubMed.
- S. Ayoob and A. K. Gupta, Crit. Rev. Environ. Sci. Technol., 2006, 36, 433–487 CrossRef CAS.
- E. Bassin, D. Wypij, R. Davis and M. Mittleman, Cancer Causes Control, 2006, 17, 421–428 CrossRef PubMed.
- A. L. Choi, G. Sun, Y. Zhang and P. Grandjean, Environ. Health Perspect., 2012, 120, 1362–1368 CrossRef CAS PubMed.
- E. Galbraith and T. D. James, Chem. Soc. Rev., 2010, 39, 3831–3842 RSC.
- Z. Guo, I. Shin and J. Yoon, Chem. Commun., 2012, 48, 5956–5967 RSC.
- T. Gunnlaugsson, P. E. Kruger, T. C. Lee, R. Parkesh, F. M. Pfeffer and G. M. Hussey, Tetrahedron Lett., 2003, 44, 6575–6578 CrossRef CAS.
- T. Gunnlaugsson, P. E. Kruger, P. Jensen, F. M. Pfeffer and G. M. Hussey, Tetrahedron Lett., 2003, 44, 8909–8913 CrossRef CAS PubMed.
- L. S. Evans, P. A. Gale, M. E. Light and R. Quesada, Chem. Commun., 2006, 965–967 RSC.
- S. Camiolo, P. A. Gale, M. B. Hursthouse and M. E. Light, Org. Biomol. Chem., 2003, 1, 741–744 CAS.
- X. Zheng, W. Zhu, D. Liu, H. Ai, Y. Huang and Z. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 7996–8000 CAS.
- R. Liu, Y. Gao, Q. Zhang, X. Yang, X. Lu, Z. Ke, W. Zhou and J. Qu, New J. Chem., 2014, 38, 2941–2945 RSC.
- A. K. Mahapatra, R. Maji, K. Maiti, S. S. Adhikari, C. Das Mukhopadhyay and D. Mandal, Analyst, 2014, 139, 309–317 RSC.
- F. M. Pfeffer, K. F. Lim and K. J. Sedgwick, Org. Biomol. Chem., 2007, 5, 1795–1799 CAS.
- J. L. Sessler, D.-G. Cho and V. Lynch, J. Am. Chem. Soc., 2006, 128, 16518–16519 CrossRef CAS PubMed.
- G. Sivaraman and D. Chellappa, J. Mater. Chem. B, 2013, 1, 5768–5772 RSC.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., 2006, pp. 16–366 Search PubMed.
- S. Y. Kim, J. Park, M. Koh, S. B. Park and J.-I. Hong, Chem. Commun., 2009, 4735–4737 RSC.
- B. Zhu, H. Kan, J. Liu, H. Liu, Q. Wei and B. Du, Biosens. Bioelectron., 2014, 52, 298–303 CrossRef CAS PubMed.
- B. Zhu, F. Yuan, R. Li, Y. Li, Q. Wei, Z. Ma, B. Du and X. Zhang, Chem. Commun., 2011, 47, 7098–7100 RSC.
- Z. Luo, B. Yang, C. Zhong, F. Tang, M. Yuan, Y. Xue, G. Yao, J. Zhang and Y. Zhang, Dyes Pigm., 2013, 97, 52–57 CrossRef CAS PubMed.
- G. Wei, J. Yin, X. Ma, S. Yu, D. Wei and Y. Du, Anal. Chim. Acta, 2011, 703, 219–225 CrossRef CAS PubMed.
- L. Gai, H. Chen, B. Zou, H. Lu, G. Lai, Z. Li and Z. Shen, Chem. Commun., 2012, 48, 10721–10723 RSC.
- D. Kim, S. Singha, T. Wang, E. Seo, J. H. Lee, S.-J. Lee, K. H. Kim and K. H. Ahn, Chem. Commun., 2012, 48, 10243–10245 RSC.
- P. Hou, S. Chen, H. Wang, J. Wang, K. Voitchovsky and X. Song, Chem. Commun., 2014, 50, 320–322 RSC.
- S. Zhang, J. Fan, S. Zhang, J. Wang, X. Wang, J. Du and X. Peng, Chem. Commun., 2014, 50, 14021–14024 RSC.
- W. Gong, R. Su, L. Li, K. Xu and B. Tang, Chin. Sci. Bull., 2011, 56, 3260–3265 CrossRef CAS.
- T.-Y. Chen and T.-C. Hwang, Physiol. Rev., 2008, 88, 351–387 CrossRef CAS PubMed.
- P. A. Gale, Acc. Chem. Res., 2011, 44, 216–226 CrossRef CAS PubMed.
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426–4430 CrossRef CAS PubMed.
- C. J. E. Haynes and P. A. Gale, Chem. Commun., 2011, 47, 8203–8209 RSC.
- P. R. Brotherhood and A. P. Davis, Chem. Soc. Rev., 2010, 39, 3633–3647 RSC.
- A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC.
- J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843–3862 RSC.
- G. W. Gokel and N. Barkey, New J. Chem., 2009, 33, 947–963 RSC.
- B. A. McNally, W. M. Leevy and B. D. Smith, Supramol. Chem., 2007, 19, 29–37 CrossRef CAS PubMed.
- A. S. Verkman, Am. J. Physiol., 1990, 259, C375–C388 CAS.
- S. Jayaraman, L. Teitler, B. Skalski and A. S. Verkman, Am. J. Physiol., 1999, 277, C1008–C1018 CAS.
- C. D. Geddes, K. Apperson, J. Karolin and D. J. S. Birch, Dyes Pigm., 2001, 48, 227–231 CrossRef CAS.
- Molecular Probes Handbook, A Guide to Fluorescent Probes and Labeling Technologies, ed. I. Johnson, Life Technologies Corporation, 2010 Search PubMed.
- A. S. Verkman, in Physiology and Pathology of Chloride Transporters and Channels in the Nervous System, ed. F. J. Alvarez-Leefmans and E. Delpire, Academic Press, San Diego, 2010, pp. 109–123 Search PubMed.
- D. Arosio and G. M. Ratto, Front. Cell. Neurosci., 2014, 8, 258, DOI:10.3389/fncel.2014.00258.
- J. Biwersi and A. S. Verkman, Biochemistry, 1991, 30, 7879–7883 CrossRef CAS.
- J. R. Inglefield and R. D. Schwartz-Bloom, J. Neurosci. Methods, 1997, 75, 127–135 CrossRef CAS.
- B. Pilas and G. Durack, Cytometry, 1997, 28, 316–322 CrossRef CAS.
- Y. Kovalchuk and O. Garaschuk, Cold Spring Harb. Protoc., 2012 DOI:10.1101/pdb.prot070037.
- J. Biwersi, B. Tulk and A. S. Verkman, Anal. Biochem., 1994, 219, 139–143 CrossRef CAS PubMed.
- B. A. McNally, A. V. Koulov, B. D. Smith, J.-B. Joos and A. P. Davis, Chem. Commun., 2005, 1087–1089 RSC.
- S. B. J. V. A. S. Jayaraman, Am. J. Physiol., 1999, 276, C747–C757 CAS.
- N. D. Sonawane, J. R. Thiagarajah and A. S. Verkman, J. Biol. Chem., 2002, 277, 5506–5513 CrossRef CAS PubMed.
- P. Li, T. Xie, N. Fan, K. Li and B. Tang, Chem. Commun., 2012, 48, 2077–2079 RSC.
- P. Li, S. Zhang, N. Fan, H. Xiao, W. Zhang, W. Zhang, H. Wang and B. Tang, Chem. – Eur. J., 2014, 20, 11760–11767 CrossRef CAS PubMed.
- S. Amatori, G. Ambrosi, M. Fanelli, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli, R. Pontellini, P. Rossi and M. A. Varrese, Chem. – Eur. J., 2012, 18, 4274–4284 CrossRef CAS PubMed.
- S. Amatori, G. Ambrosi, E. Borgogelli, M. Fanelli, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli, P. Rossi and A. Tassoni, Inorg. Chem., 2014, 53, 4560–4569 CrossRef CAS.
- J. J. Gassensmith, S. Matthys, J.-J. Lee, A. Wojcik, P. V. Kamat and B. D. Smith, Chem. – Eur. J., 2010, 16, 2916–2921 CrossRef CAS PubMed.
- C. G. Collins, E. M. Peck, P. J. Kramer and B. D. Smith, Chem. Sci., 2013, 4, 2557–2563 RSC.
- P. J. DeMott, Atmos. Res., 1995, 38, 63–99 CrossRef CAS.
- C. D. Geddes, Meas. Sci. Technol., 2001, 12, R53 CrossRef CAS.
- G. Dai, O. Levy and N. Carrasco, Nature, 1996, 379, 458–460 CrossRef CAS PubMed.
- A. K. Mahapatra, J. Roy, P. Sahoo, S. K. Mukhopadhyay and A. Chattopadhyay, Org. Biomol. Chem., 2012, 10, 2231–2236 CAS.
- P. Castric, Curr. Microbiol., 1994, 29, 19–21 CrossRef CAS.
- J. L. Way, Annu. Rev. Pharmacol. Toxicol., 1984, 24, 451–481 CrossRef CAS PubMed.
- J. R. Govan and V. Deretic, Microbiol. Rev., 1996, 60, 539–574 CAS.
- B. Ryall, X. Lee, J. Zlosnik, S. Hoshino and H. Williams, BMC Microbiol., 2008, 8, 108 CrossRef PubMed.
- Cyanide in Water and Soil: Chemistry, Risk, and Management, ed. D. A. Dzombak, R. S. Ghosh and G. M. Wong-Chong, CRC Press, Boca Raton, Florida, 2005 Search PubMed.
- Z. Xu, X. Chen, H. N. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 127–137 RSC.
- K.-S. Lee, H.-J. Kim, G.-H. Kim, I. Shin and J.-I. Hong, Org. Lett., 2007, 10, 49–51 CrossRef PubMed.
- J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
- S. K. Kwon, S. Kou, H. N. Kim, X. Chen, H. Hwang, S.-W. Nam, S. H. Kim, K. M. K. Swamy, S. Park and J. Yoon, Tetrahedron Lett., 2008, 49, 4102–4105 CrossRef CAS PubMed.
- S. W. Nam, X. Chen, J. Lim, S. H. Kim, S. T. Kim, Y. H. Cho, J. Yoon and S. Park, PLoS One, 2011, 6, e21387 CAS.
- M. K. Bera, C. Chakraborty, P. K. Singh, C. Sahu, K. Sen, S. Maji, A. K. Das and S. Malik, J. Mater. Chem. B, 2014, 2, 4733 RSC.
- S. Madhu, S. K. Basu, S. Jadhav and M. Ravikanth, Analyst, 2013, 138, 299–306 RSC.
- X. Cheng, R. Tang, H. Jia, J. Feng, J. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2012, 4, 4387–4392 CAS.
- C.-H. Lee, H.-J. Yoon, J.-S. Shim and W.-D. Jang, Chem. – Eur. J., 2012, 18, 4513–4516 CrossRef CAS PubMed.
- M. Sun, S. Wang, Q. Yang, X. Fei, Y. Li and Y. Li, RSC Adv., 2014, 4, 8295–8299 RSC.
- L. Yang, X. Li, Y. Qu, W. Qu, X. Zhang, Y. Hang, H. Ågren and J. Hua, Sens. Actuators, B, 2014, 203, 833–847 CrossRef CAS PubMed.
- S. Y. Chung, S. W. Nam, J. Lim, S. Park and J. Yoon, Chem. Commun., 2009, 2866–2868 RSC.
- C. D. Sifri, J. Begun and F. M. Ausubel, Trends Microbiol., 2005, 13, 119–127 CrossRef CAS PubMed.
- X. Chen, S. W. Nam, G. H. Kim, N. Song, Y. Jeong, I. Shin, S. K. Kim, J. Kim, S. Park and J. Yoon, Chem. Commun., 2010, 46, 8953–8955 RSC.
- U. Reddy G, P. Das, S. Saha, M. Baidya, S. K. Ghosh and A. Das, Chem. Commun., 2013, 49, 255–257 RSC.
- H. S. Jung, J. H. Han, Z. H. Kim, C. Kang and J. S. Kim, Org. Lett., 2011, 13, 5056–5059 CrossRef CAS PubMed.
- W. Cao, X.-J. Zheng, D.-C. Fang and L.-P. Jin, Dalton Trans., 2014, 43, 7298–7303 RSC.
- Y.-Y. Guo, X.-L. Tang, F.-P. Hou, J. Wu, W. Dou, W.-W. Qin, J.-X. Ru, G.-L. Zhang, W.-S. Liu and X.-J. Yao, Sens. Actuators, B, 2013, 181, 202–208 CrossRef CAS PubMed.
- M. Lee, J. H. Moon, K. M. K. Swamy, Y. Jeong, G. Kim, J. Choi, J. Y. Lee and J. Yoon, Sens. Actuators, B, 2014, 199, 369–376 CrossRef CAS PubMed.
- S. Xu, M. He, H. Yu, X. Cai, X. Tan, B. Lu and B. Shu, Anal. Biochem., 2001, 299, 188–193 CrossRef CAS PubMed.
- A. E. Timms, Y. Zhang, R. G. G. Russell and M. A. Brown, Rheumatology, 2002, 41, 725–729 CrossRef CAS PubMed.
- S. Lee, K. K. Y. Yuen, K. A. Jolliffe and J. Yoon, Chem. Soc. Rev., 2015 10.1039/c1034cs00353e.
- J. F. Zhang, S. Kim, J. H. Han, S.-J. Lee, T. Pradhan, Q. Y. Cao, S. J. Lee, C. Kang and J. S. Kim, Org. Lett., 2011, 13, 5294–5297 CrossRef CAS PubMed.
- D.-N. Lee, A. Jo, S. B. Park and J.-I. Hong, Tetrahedron Lett., 2012, 53, 5528–5530 CrossRef CAS PubMed.
- W. Zhu, X. Huang, Z. Guo, X. Wu, H. Yu and H. Tian, Chem. Commun., 2012, 48, 1784–1786 RSC.
- R. K. Pathak, K. Tabbasum, A. Rai, D. Panda and C. P. Rao, Anal. Chem., 2012, 84, 5117–5123 CrossRef CAS PubMed.
- S. Goswami, A. K. Das, B. Pakhira, S. Basu Roy, A. K. Maity, P. Saha and S. Sarkar, Dalton Trans., 2014, 43, 12689–12697 RSC.
- S. Bhowmik, B. N. Ghosh, V. Marjomaki and K. Rissanen, J. Am. Chem. Soc., 2014, 136, 5543–5546 CrossRef CAS PubMed.
- M. Leppilampi, P. Koistinen, E.-R. Savolainen, J. Hannuksela, A.-K. Parkkila, O. Niemelä, S. Pastoreková, J. Pastorek, A. Waheed, W. S. Sly, S. Parkkila and H. Rajaniemi, Clin. Cancer Res., 2002, 8, 2240–2245 CAS.
- W. S. Sly and P. Y. Hu, Annu. Rev. Biochem., 1995, 64, 375–401 CrossRef CAS PubMed.
- W. Wu, H.-K. Kong, H. Li, Y.-M. Ho, Y. Gao, J. Hao, M. B. Murphy, M. H.-W. Lam, K.-L. Wong and C.-S. Lee, Eur. J. Org. Chem., 2011, 5054–5060 CrossRef CAS PubMed.
- D. Parker, Aust. J. Chem., 2011, 64, 239–243 CrossRef CAS.
- B. S. Murray, E. J. New, R. Pal and D. Parker, Org. Biomol. Chem., 2008, 6, 2085–2094 CAS.
- E. J. New and D. Parker, Org. Biomol. Chem., 2009, 7, 851–855 CAS.
- S. J. Butler, L. Lamarque, R. Pal and D. Parker, Chem. Sci., 2014, 5, 1750–1756 RSC.
- A. Thibon and V. Pierre, Anal. Bioanal. Chem., 2009, 394, 107–120 CrossRef CAS PubMed.
- M. S. Tremblay, M. Halim and D. Sames, J. Am. Chem. Soc., 2007, 129, 7570–7577 CrossRef CAS PubMed.
- D. G. Smith, G. L. Law, B. S. Murray, R. Pal, D. Parker and K. L. Wong, Chem. Commun., 2011, 47, 7347–7349 RSC.
- D. G. Smith, R. Pal and D. Parker, Chem. – Eur. J., 2012, 18, 11604–11613 CrossRef CAS PubMed.
- P. Nagy, Z. Palinkas, A. Nagy, B. Budai, I. Toth and A. Vasas, Biochim. Biophys. Acta, 2014, 1840, 876–891 CrossRef CAS PubMed.
- K. R. Olson, J. A. Donald, R. A. Dombkowski and S. F. Perry, Respir. Physiol. Neurobiol., 2012, 184, 117–129 CrossRef CAS PubMed.
- A. Stein and S. M. Bailey, Redox Biol., 2013, 1, 32–39 CrossRef CAS PubMed.
- J. L. Wallace, Trends Pharmacol. Sci., 2007, 28, 501–505 CrossRef CAS PubMed.
- P. Kamoun, Amino Acids, 2004, 26, 243–254 CrossRef CAS PubMed.
- J. I. Toohey, Anal. Biochem., 2011, 413, 1–7 CrossRef CAS PubMed.
- R. E. Hansen and J. R. Winther, Anal. Biochem., 2009, 394, 147–158 CrossRef CAS PubMed.
- M. Vendrell, D. Zhai, J. C. Er and Y. T. Chang, Chem. Rev., 2012, 112, 4391–4420 CrossRef CAS PubMed.
- V. S. Lin and C. J. Chang, Curr. Opin. Chem. Biol., 2012, 16, 595–601 CrossRef CAS PubMed.
- B. Peng and M. Xian, Asian J. Org. Chem., 2014, 3, 914–924 CrossRef CAS PubMed.
- X. Cao, W. Lin, K. Zheng and L. He, Chem. Commun., 2012, 48, 10529–10531 RSC.
- C. Tang, Q. Zheng, S. Zong, Z. Wang and Y. Cui, Sens. Actuators, B, 2014, 202, 99–104 CrossRef CAS PubMed.
- D. Maity, A. Raj, P. K. Samanta, D. Karthigeyan, T. K. Kundu, S. K. Pati and T. Govindaraju, RSC Adv., 2014, 4, 11147 RSC.
- Z. Huang, S. Ding, D. Yu, F. Huang and G. Feng, Chem. Commun., 2014, 50, 9185–9187 RSC.
- Z. Xu, L. Xu, J. Zhou, Y. Xu, W. Zhu and X. Qian, Chem. Commun., 2012, 48, 10871–10873 RSC.
- X. Wang, J. Sun, W. Zhang, X. Ma, J. Lv and B. Tang, Chem. Sci., 2013, 4, 2551 RSC.
- J. Liu, Y. Q. Sun, J. Zhang, T. Yang, J. Cao, L. Zhang and W. Guo, Chem. – Eur. J., 2013, 19, 4717–4722 CrossRef CAS PubMed.
- Y. Chen, C. Zhu, Z. Yang, J. Chen, Y. He, Y. Jiao, W. He, L. Qiu, J. Cen and Z. Guo, Angew. Chem., Int. Ed., 2013, 52, 1688–1691 CrossRef CAS PubMed.
- S. K. Das, C. S. Lim, S. Y. Yang, J. H. Han and B. R. Cho, Chem. Commun., 2012, 48, 8395–8397 RSC.
- A. R. Lippert, E. J. New and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 10078–10080 CrossRef CAS PubMed.
- F. Yu, P. Li, P. Song, B. Wang, J. Zhao and K. Han, Chem. Commun., 2012, 48, 2852–2854 RSC.
- Y. Zheng, M. Zhao, Q. Qiao, H. Liu, H. Lang and Z. Xu, Dyes Pigm., 2013, 98, 367–371 CrossRef CAS PubMed.
- W. Sun, J. Fan, C. Hu, J. Cao, H. Zhang, X. Xiong, J. Wang, S. Cui, S. Sun and X. Peng, Chem. Commun., 2013, 49, 3890–3892 RSC.
- Q. Wan, Y. Song, Z. Li, X. Gao and H. Ma, Chem. Commun., 2013, 49, 502–504 RSC.
- V. S. Lin, A. R. Lippert and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7131–7135 CrossRef CAS PubMed.
- Q. Qiao, M. Zhao, H. Lang, D. Mao, J. Cui and Z. Xu, RSC Adv., 2014, 4, 25790 RSC.
- S. Yang, Y. Qi, C. Liu, Y. Wang, Y. Zhao, L. Wang, J. Li, W. Tan and R. Yang, Anal. Chem., 2014, 86, 7508–7515 CrossRef CAS PubMed.
- S. K. Bae, C. H. Heo, D. J. Choi, D. Sen, E. H. Joe, B. R. Cho and H. M. Kim, J. Am. Chem. Soc., 2013, 135, 9915–9923 CrossRef CAS PubMed.
- L. Zhang, S. Li, M. Hong, Y. Xu, S. Wang, Y. Liu, Y. Qian and J. Zhao, Org. Biomol. Chem., 2014, 12, 5115–5125 CAS.
- X. L. Liu, X. J. Du, C. G. Dai and Q. H. Song, J. Org. Chem., 2014, 79, 9481–9489 CrossRef CAS PubMed.
- R. Wang, F. Yu, L. Chen, H. Chen, L. Wang and W. Zhang, Chem. Commun., 2012, 48, 11757–11759 RSC.
- H. Wu, S. Krishnakumar, J. Yu, D. Liang, H. Qi, Z. W. Lee, L. W. Deng and D. Huang, Chem. – Asian J., 2014, 9, 3604–3611 CrossRef CAS PubMed.
- W. Sun, W. Li, J. Li, J. Zhang, L. Du and M. Li, Tetrahedron, 2012, 68, 5363–5367 CrossRef CAS PubMed.
- W. Chen, S. Chen, B. Zhou, H. Wang, X. Song and H. Zhang, Dyes Pigm., 2015, 113, 596–601 CrossRef CAS PubMed.
- H. Chen, W. Lin, H. Cui and W. Jiang, Chem. – Eur. J., 2015, 21, 733–745 CrossRef CAS PubMed.
- B. Peng, W. Chen, C. Liu, E. W. Rosser, A. Pacheco, Y. Zhao, H. C. Aguilar and M. Xian, Chem. – Eur. J., 2014, 20, 1010–1016 CrossRef CAS PubMed.
- L. Yuan and Q. P. Zuo, Chem. – Asian J., 2014, 9, 1544–1549 CrossRef CAS PubMed.
- T. Liu, X. Zhang, Q. Qiao, C. Zou, L. Feng, J. Cui and Z. Xu, Dyes Pigm., 2013, 99, 537–542 CrossRef CAS PubMed.
- X. Pei, H. Tian, W. Zhang, A. M. Brouwer and J. Qian, Analyst, 2014, 139, 5290–5296 RSC.
- F. Zheng, M. Wen, F. Zeng and S. Wu, Polymer, 2013, 54, 5691–5697 CrossRef CAS PubMed.
- S. El Sayed, C. d. l. Torre, L. E. Santos-Figueroa, E. Pérez-Payá, R. Martínez-Máñez, F. Sancenón, A. M. Costero, M. Parra and S. Gil, RSC Adv., 2013, 3, 25690 RSC.
- B. Chen, C. Lv and X. Tang, Anal. Bioanal. Chem., 2012, 404, 1919–1923 CrossRef CAS PubMed.
- C. S. Lim, S. K. Das, S. Y. Yang, E. S. Kim, H. J. Chun and B. R. Cho, Anal. Chem., 2013, 85, 9288–9295 CrossRef CAS PubMed.
- G. J. Mao, T. T. Wei, X. X. Wang, S. Y. Huan, D. Q. Lu, J. Zhang, X. B. Zhang, W. Tan, G. L. Shen and R. Q. Yu, Anal. Chem., 2013, 85, 7875–7881 CrossRef CAS PubMed.
- B. Chen, W. Li, C. Lv, M. Zhao, H. Jin, H. Jin, J. Du, L. Zhang and X. Tang, Analyst, 2013, 138, 946–951 RSC.
- C. Yu, X. Li, F. Zeng, F. Zheng and S. Wu, Chem. Commun., 2013, 49, 403–405 RSC.
- K. Zheng, W. Lin and L. Tan, Org. Biomol. Chem., 2012, 10, 9683–9688 CAS.
- G. Zhou, H. Wang, Y. Ma and X. Chen, Tetrahedron, 2013, 69, 867–870 CrossRef CAS PubMed.
- T. Chen, Y. Zheng, Z. Xu, M. Zhao, Y. Xu and J. Cui, Tetrahedron Lett., 2013, 54, 2980–2982 CrossRef CAS PubMed.
- J. Zhang and W. Guo, Chem. Commun., 2014, 50, 4214–4217 RSC.
- J. Zhou, Y. Luo, Q. Li, J. Shen, R. Wang, Y. Xu and X. Qian, New J. Chem., 2014, 38, 2770 RSC.
- K. Liu and S. Zhang, Tetrahedron Lett., 2014, 55, 5566–5569 CrossRef CAS PubMed.
- L. E. Santos-Figueroa, C. de laTorre, S. El Sayed, F. Sancenón, R. Martínez-Máñez, A. M. Costero, S. Gil and M. Parra, Eur. J. Org. Chem., 2014, 1848–1854 CrossRef CAS PubMed.
- K. Wang, H. Peng, N. Ni, C. Dai and B. Wang, J. Fluoresc., 2014, 24, 1–5 CrossRef CAS PubMed.
- K. Sun, X. Liu, Y. Wang and Z. Wu, RSC Adv., 2013, 3, 14543–14548 RSC.
- S. I. Reja, N. Kumar, R. Sachdeva, V. Bhalla and M. Kumar, RSC Adv., 2013, 3, 17770–17774 RSC.
- D. Zhu, L. Xue, G. Li, Y. Che and H. Jiang, Org. Chem. Front., 2014, 1, 501 RSC.
- W. Xuan, R. Pan, Y. Cao, K. Liu and W. Wang, Chem. Commun., 2012, 48, 10669–10671 RSC.
- Z. Wu, Y. Feng, B. Geng, J. Liua and X. Tang, RSC Adv., 2014, 4, 30398–30401 RSC.
- S. Chen, P. Hou, J. W. Foley and X. Song, RSC Adv., 2013, 3, 5591 RSC.
- X. Li, Y. Gong, K. Wu, S. H. Liang, J. Cao, B. Yang, Y. Hu and Y. Han, RSC Adv., 2014, 4, 36106 RSC.
- Z. Dong, X. Le, P. Zhou, C. Dong and J. Ma, New J. Chem., 2014, 38, 1802–1808 RSC.
- A. Zhu, Z. Luo, C. Ding, B. Li, S. Zhou, R. Wang and Y. Tian, Analyst, 2014, 139, 1945–1952 RSC.
- B. C. Dickinson and C. J. Chang, Nat. Chem. Biol., 2011, 7, 504–511 CrossRef CAS PubMed.
- X. Chen, X. Tian, I. Shin and J. Yoon, Chem. Soc. Rev., 2011, 40, 4783–4804 RSC.
- B. Kalyanaraman, V. Darley-Usmar, K. J. A. Davies, P. A. Dennery, H. J. Forman, M. B. Grisham, G. E. Mann, K. Moore, L. J. Roberts Ii and H. Ischiropoulos, Free Radical Biol. Med., 2012, 52, 1–6 CrossRef CAS PubMed.
- R. Radi, J. Biol. Chem., 2013, 288, 26464–26472 CrossRef CAS PubMed.
- J.-H. M. Tsai, J. G. Harrison, J. C. Martin, T. P. Hamilton, M. van der Woerd, M. J. Jablonsky and J. S. Beckman, J. Am. Chem. Soc., 1994, 116, 4115–4116 CrossRef CAS.
- C. Szabo, H. Ischiropoulos and R. Radi, Nat. Rev. Drug Discovery, 2007, 6, 662–680 CrossRef CAS PubMed.
- P. B. J. S. L. L. Pacher, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS PubMed.
- M. G. Bonini, R. Radi, G. Ferrer-Sueta, A. M. D. C. Ferreira and O. Augusto, J. Biol. Chem., 1999, 274, 10802–10806 CrossRef CAS PubMed.
- S. L. Hempel, G. R. Buettner, Y. Q. O'Malley, D. A. Wessels and D. M. Flaherty, Free Radical Biol. Med., 1999, 27, 146–159 CrossRef CAS.
- H. Possel, H. Noack, W. Augustin, G. Keilhoff and G. Wolf, FEBS Lett., 1997, 416, 175–178 CrossRef CAS.
- N. W. Kooy, J. A. Royall, H. Ischiropoulos and J. S. Beckman, Free Radical Biol. Med., 1994, 16, 149–156 CrossRef CAS.
- A. Negre-Salvayre, N. Augé, C. Duval, F. Robbesyn, J.-C. Thiers, D. Nazzal, H. Benoist and R. Salvayre, Methods Enzymology, Academic Press, 2002, pp. 62–71 Search PubMed.
- G. Ambikapathi, S. Kempahanumakkagari, B. Ramappa Lamani, D. Kuramkote Shivanna, H. Bodagur Maregowda, A. Gupta and P. Malingappa, J. Fluoresc., 2013, 23, 705–712 CrossRef CAS PubMed.
- D. Yang, Y.-C. Tang, J. Chen, X.-C. Wang, M. D. Bartberger, K. N. Houk and L. Olson, J. Am. Chem. Soc., 1999, 121, 11976–11983 CrossRef CAS.
- D. Yang, H.-L. Wang, Z.-N. Sun, N.-W. Chung and J.-G. Shen, J. Am. Chem. Soc., 2006, 128, 6004–6005 CrossRef CAS PubMed.
- Z.-N. Sun, H.-L. Wang, F.-Q. Liu, Y. Chen, P. Kwong, H. Tam and D. Yang, Org. Lett., 2009, 11, 1887–1890 CrossRef CAS PubMed.
- R. B. R. Muijsers, E. van den Worm, G. Folkerts, C. J. Beukelman, A. S. Koster, D. S. Postma and F. P. Nijkamp, Br. J. Pharmacol., 2000, 130, 932–936 CrossRef CAS PubMed.
- T. Peng, N. K. Wong, X. Chen, Y. K. Chan, D. H. Ho, Z. Sun, J. J. Hu, J. Shen, H. El-Nezami and D. Yang, J. Am. Chem. Soc., 2014, 136, 11728–11734 CrossRef CAS PubMed.
- J. T. Hou, J. Yang, K. Li, Y. X. Liao, K. K. Yu, Y. M. Xie and X. Q. Yu, Chem. Commun., 2014, 50, 9947–9950 RSC.
- X. Zhou, Y. Kwon, G. Kim, J.-H. Ryu and J. Yoon, Biosens. Bioelectron., 2015, 64, 285–291 CrossRef CAS PubMed.
- Q. Zhang, Z. Zhu, Y. Zheng, J. Cheng, N. Zhang, Y. T. Long, J. Zheng, X. Qian and Y. Yang, J. Am. Chem. Soc., 2012, 134, 18479–18482 CrossRef CAS PubMed.
- F. Martin-Romero, Y. Gutiérrez-Martin, F. Henao and C. Gutiérrez-Merino, J. Fluoresc., 2004, 14, 17–23 CrossRef CAS.
- J. Zielonka, A. Sikora, M. Hardy, J. Joseph, B. P. Dranka and B. Kalyanaraman, Chem. Res. Toxicol., 2012, 25, 1793–1799 CrossRef CAS PubMed.
- A. Sikora, J. Zielonka, M. Lopez, J. Joseph and B. Kalyanaraman, Free Radical Biol. Med., 2009, 47, 1401–1407 CrossRef CAS PubMed.
- X. Sun, Q. Xu, G. Kim, S. E. Flower, J. P. Lowe, J. Yoon, J. S. Fossey, X. Qian, S. D. Bull and T. D. James, Chem. Sci., 2014, 5, 3368 RSC.
- F. Yu, P. Song, P. Li, B. Wang and K. Han, Analyst, 2012, 137, 3740–3749 RSC.
- K. Xu, H. Chen, J. Tian, B. Ding, Y. Xie, M. Qiang and B. Tang, Chem. Commun., 2011, 47, 9468–9470 RSC.
- F. Yu, P. Li, G. Li, G. Zhao, T. Chu and K. Han, J. Am. Chem. Soc., 2011, 133, 11030–11033 CrossRef CAS PubMed.
- F. Yu, P. Li, B. Wang and K. Han, J. Am. Chem. Soc., 2013, 135, 7674–7680 CrossRef CAS PubMed.
- B. Wang, F. Yu, P. Li, X. Sun and K. Han, Dyes Pigm., 2013, 96, 383–390 CrossRef CAS PubMed.
- K.-K. Lin, S.-C. Wu, K.-M. Hsu, C.-H. Hung, W.-F. Liaw and Y.-M. Wang, Org. Lett., 2013, 15, 4242–4245 CrossRef CAS PubMed.
- T. Ueno, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2006, 128, 10640–10641 CrossRef CAS PubMed.
- N. A. Sieracki, B. N. Gantner, M. Mao, J. H. Horner, R. D. Ye, A. B. Malik, M. E. Newcomb and M. G. Bonini, Free Radical Biol. Med., 2013, 61, 40–50 CrossRef CAS PubMed.
- G. C. Van de Bittner, E. A. Dubikovskaya, C. R. Bertozzi and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21316–21321 CrossRef CAS PubMed.
- J. C. Morris, J. Phys. Chem., 1966, 70, 3798–3805 CrossRef CAS.
- K. Setsukinai, Y. Urano, K. Kakinuma, H. J. Majima and T. Nagano, J. Biol. Chem., 2003, 278, 3170–3175 CrossRef CAS PubMed.
- J. Shepherd, S. A. Hilderbrand, P. Waterman, J. W. Heinecke, R. Weissleder and P. Libby, Chem. Biol., 2007, 14, 1221–1231 CrossRef CAS PubMed.
- Y. Koide, Y. Urano, S. Kenmoku, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 10324–10325 CrossRef CAS PubMed.
- Y. Xiao, R. Zhang, Z. Ye, Z. Dai, H. An and J. Yuan, Anal. Chem., 2012, 84, 10785–10792 CrossRef CAS PubMed.
- Z.-N. Sun, F.-Q. Liu, Y. Chen, P. K. H. Tam and D. Yang, Org. Lett., 2008, 10, 2171–2174 CrossRef CAS PubMed.
- Y. Zhou, J. Y. Li, K. H. Chu, K. Liu, C. Yao and J. Y. Li, Chem. Commun., 2012, 48, 4677–4679 RSC.
- H. Zhu, J. Fan, J. Wang, H. Mu and X. Peng, J. Am. Chem. Soc., 2014, 136, 12820–12823 CrossRef CAS PubMed.
- X. Cheng, H. Jia, T. Long, J. Feng, J. Qin and Z. Li, Chem. Commun., 2011, 47, 11978–11980 RSC.
- G. Wu, F. Zeng and S. Wu, Anal. Methods, 2013, 5, 5589–5596 RSC.
- S. I. Reja, V. Bhalla, A. Sharma, G. Kaur and M. Kumar, Chem. Commun., 2014, 50, 11911–11914 RSC.
- L. Yuan, W. Lin, J. Song and Y. Yang, Chem. Commun., 2011, 47, 12691–12693 RSC.
- S. Kenmoku, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 7313–7318 CrossRef CAS PubMed.
- Y. Koide, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, J. Am. Chem. Soc., 2011, 133, 5680–5682 CrossRef CAS PubMed.
- X. Chen, K. A. Lee, E. M. Ha, K. M. Lee, Y. Y. Seo, H. K. Choi, H. N. Kim, M. J. Kim, C. S. Cho, S. Y. Lee, W. J. Lee and J. Yoon, Chem. Commun., 2011, 47, 4373–4375 RSC.
- X. Chen, X. Wang, S. Wang, W. Shi, K. Wang and H. Ma, Chem. – Eur. J., 2008, 14, 4719–4724 CrossRef CAS PubMed.
- S. Goswami, S. Das, K. Aich, P. K. Nandi, K. Ghoshal, C. K. Quah, M. Bhattacharyya, H.-K. Fun and H. A. Abdel-Aziz, RSC Adv., 2014, 4, 24881–24886 RSC.
- X. Jin, L. Hao, Y. Hu, M. She, Y. Shi, M. Obst, J. Li and Z. Shi, Sens. Actuators, B, 2013, 186, 56–60 CrossRef CAS PubMed.
- Y.-K. Yang, H. J. Cho, J. Lee, I. Shin and J. Tae, Org. Lett., 2009, 11, 859–861 CrossRef CAS PubMed.
- S.-R. Liu and S.-P. Wu, Org. Lett., 2013, 15, 878–881 CrossRef CAS PubMed.
- S. T. Manjare, J. Kim, Y. Lee and D. G. Churchill, Org. Lett., 2014, 16, 520–523 CrossRef CAS PubMed.
- S. T. Manjare, S. Kim, W. D. Heo and D. G. Churchill, Org. Lett., 2014, 16, 410–412 CrossRef CAS PubMed.
- B. Wang, P. Li, F. Yu, P. Song, X. Sun, S. Yang, Z. Lou and K. Han, Chem. Commun., 2013, 49, 1014–1016 RSC.
- Y. Koide, M. Kawaguchi, Y. Urano, K. Hanaoka, T. Komatsu, M. Abo, T. Terai and T. Nagano, Chem. Commun., 2012, 48, 3091–3093 RSC.
- Z. Lou, P. Li, Q. Pan and K. Han, Chem. Commun., 2013, 49, 2445–2447 RSC.
- G. Li, D. Zhu, Q. Liu, L. Xue and H. Jiang, Org. Lett., 2013, 15, 2002–2005 CrossRef CAS PubMed.
- F. Liu, Y. Gao, J. Wang and S. Sun, Analyst, 2014, 139, 3324–3329 RSC.
- F. Liu, T. Wu, J. Cao, H. Zhang, M. Hu, S. Sun, F. Song, J. Fan, J. Wang and X. Peng, Analyst, 2013, 138, 775–778 RSC.
- L. Yuan, W. Lin, Y. Yang and H. Chen, J. Am. Chem. Soc., 2012, 134, 1200–1211 CrossRef CAS PubMed.
- S. Chen, J. Lu, C. Sun and H. Ma, Analyst, 2010, 135, 577–582 RSC.
- F. Yu, P. Song, P. Li, B. Wang and K. Han, Chem. Commun., 2012, 48, 7735–7737 RSC.
- B. Wang, P. Li, F. Yu, J. Chen, Z. Qu and K. Han, Chem. Commun., 2013, 49, 5790–5792 RSC.
- E. M. Stennett, M. A. Ciuba and M. Levitus, Chem. Soc. Rev., 2014, 43, 1057–1075 RSC.
- M. K. Kuimova, Phys. Chem. Chem. Phys., 2012, 14, 12671–12686 RSC.
- A. F. Coskun and A. Ozcan, Curr. Opin. Biotechnol., 2014, 25, 8–16 CrossRef CAS PubMed.
- X. Fan and I. M. White, Nat. Photonics, 2011, 5, 591–597 CrossRef CAS PubMed.
- D. A. Basiji, W. E. Ortyn, L. Liang, V. Venkatachalam and P. Morrissey, Clin. Lab. Med., 2007, 27, 653–670 CrossRef PubMed.
- A. Ozcan, Lab Chip, 2014, 14, 3187–3194 RSC.
- H. Zhu, S. Mavandadi, A. F. Coskun, O. Yaglidere and A. Ozcan, Anal. Chem., 2011, 83, 6641–6647 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |