
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDendritic glycopolymers based on dendritic polyamine scaffolds: view on their synthetic approaches, characteristics and potential for biomedical applications
Dietmar
Appelhans
*a,
Barbara
Klajnert-Maculewicz
*b,
Anna
Janaszewska
b,
Joanna
Lazniewska
b and
Brigitte
Voit
*ac
aLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Straße 6, 01069 Dresden, Germany. E-mail: voit@ipfdd.de; applhans@ipfdd.de; aklajn@biol.uni.lodz.pl
bDepartment of General Biophysics, Faculty of Biology and Environmental Protection, University of Lodz, Pomorska 141/143, 90-236 Lodz, Poland
cOrganic Chemistry of Polymers, Technische Universität Dresden, 01062 Dresden, Germany
First published on 18th December 2014
Abstract
In this review we highlight the potential for biomedical applications of dendritic glycopolymers based on polyamine scaffolds. The complex interplay of the molecular characteristics of the dendritic architectures and their specific interactions with various (bio)molecules are elucidated with various examples. A special role of the individual sugar units attached to the dendritic scaffolds and their density is identified, which govern ionic and H-bond interactions, and biological targeting, but to a large extent are also responsible for the significantly reduced toxicity of the dendritic glycopolymers compared to their polyamine scaffolds. Thus, the application of dendritic glycopolymers in drug delivery systems for gene transfection but also as therapeutics in neurodegenerative diseases has great promise.
 Dietmar Appelhans | Dr Dietmar Appelhans completed his PhD study in 1994 under the supervision of Prof. Christian Reichardt at the Philipps-Universität. He then joined the group of Prof. Hans-Jörg Adler at the Technische Universität Dresden, Germany (1994), as a research scientist. Since 1999, he has been engaged in the group of Prof. Brigitte Voit at the Leibniz-Institut für Polymerforschung Dresden e.V. Since 2013 he has been in the department head of “Bioactive and Responsive Polymers”. His current research topics are synthesis and application of dendritic polymers, of polymeric vesicles for biomedical applications and material science, and of hydrogels for drug release and (fluidic) microsystems. He is a (co)author of more than 100 peer review articles in international journals, several patents and 3 book chapters. |
 Barbara Klajnert-Maculewicz | Barbara Klajnert-Maculewicz received her PhD in biophysics from the University of Lodz (Poland) in 2002. She was a post-doctoral fellow at McMaster University, Ontario, Canada, from 2004 to 2005. Then she returned to the University of Lodz where she obtained habilitation (in 2009) and professorship (in 2013). She is an External Scientific Member in Leibniz-Institut für Polymerforschung, Dresden e.V. in Germany. She has published 2 books, 9 book chapters, and over 90 articles. Her research interests are focused on the study of biological properties and biomedical applications of dendrimers. |
 Anna Janaszewska | Anna Janaszewska received her PhD in biophysics from the University of Lodz (Poland) in 2003. Since 2008 she has been a post-doc in the Department of General Biophysics, University of Lodz. Her research focused on the cytotoxicity (in vitro and in vivo) of different groups of dendrimers and their applications in medicine, especially as carriers of anticancer drugs, and antiamyloid agents in neurodegenerative disorders. Now she is involved in studies on dendrimers as carriers of photosensitizers in PDT therapy. |
 Joanna Lazniewska | Joanna Lazniewska obtained her PhD in biophysics from the University of Lodz in 2014. Her research focused on the toxicity of phosphorus-containing dendrimers against neural cell lines. Additionally, she was involved in studies concerning potential application of dendrimers as delivery platforms for anti-cancer siRNA. Her interests are concentrated on the safety of nanomaterials and their usage in biomedicine. |
 Brigitte Voit | Brigitte Voit received her PhD in Macromolecular Chemistry in 1990 from University Bayreuth and, after a postdoc at Eastman Kodak working on hyperbranched polymers, her habilitation degree in 1996 from Technische Universität München. In 1997 Brigitte Voit was appointed Head of the Institute of Macromolecular Chemistry at the Leibniz Institute of Polymer Research (IPF) Dresden, as well as Professor of “Organic Chemistry of Polymers” at Technische Universität Dresden (TU Dresden). Since 2002, she has been heading, in addition, the IPF Dresden as Scientific Director. Her major research interest is in the synthesis of new functional polymer architectures covering topics like dendritic polymers, responsive hydrogels, functional block and graft copolymers, as well as biofunctional and photolabile polymers. |
1. Introduction
The decoration of various macromolecular scaffolds with carbohydrate ligands leads to the fabrication of diverse multivalent glycoconjugates and glycopolymers (Fig. 1).1–12 Their most common use is for triggering and inhibiting a large number of biological phenomena13 induced by an individually arranged recognition carbohydrate motif onto dendritic, polymeric, self-assembled, and other molecular scaffolds,14–16 but also onto nanoparticle and solid surfaces.17–19 This is strongly motivated by the fact that multivalent carbohydrate–protein interactions are mainly involved in a large variety of intercellular recognition processes, including, for example, bacterial and viral adhesion, the evaluation of immune response, targeting drugs, cell growth regulation, cell differentiation, cell–cell interactions, and cancer cell aggregation, but also the metastatic spread of cancer.20–23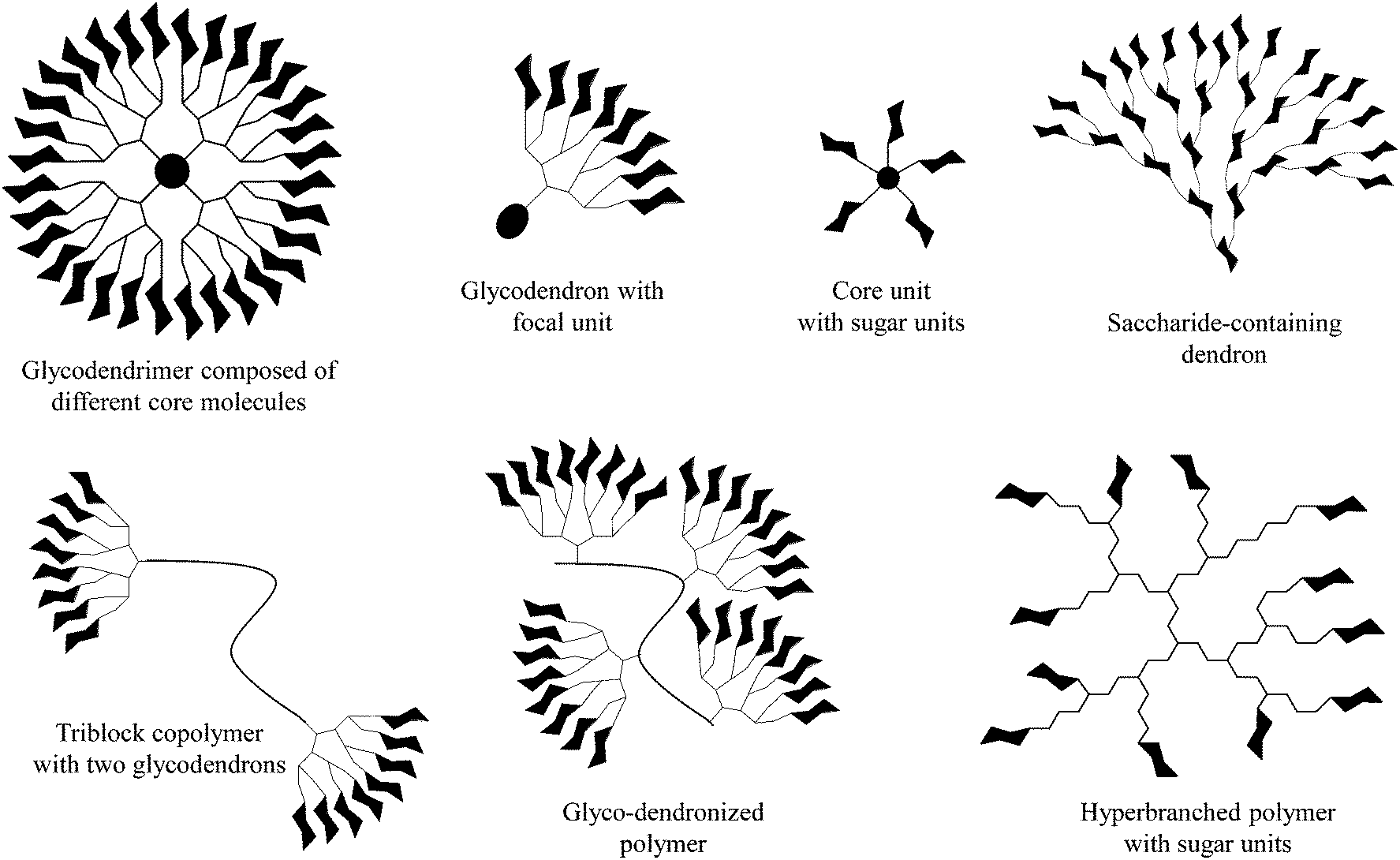 | ||
| Fig. 1 Selected dendritic glycoconjugates usable as (anti-adhesive) dendritic glycoconjugates, drug delivery systems, and polymeric therapeutics. | ||
To explore and exploit the desired carbohydrate–protein interactions in the presence of isolated biomolecules (e.g. protein receptors) and biological entities (cells or tissue), the molecularly and spatially arranged carbohydrate residues on dendritic scaffolds were continuously optimized and verified to better mimic and match the complementary binding voids of protein receptors over the last few years. Thus, the inhibition of plant24–27 and human28,29 lectins, including some other protein receptors on the cell membrane surface of bacteria and viruses,30–36 can be triggered depending on the size and generation of dendritic glycoarchitectures and their surface composition and arrangement of the multivalent carbohydrate shell. For example, this is highlighted by the preferred occupation of the dendritic cell surface DC-SIGN or other mannose-binding proteins by clustered oligomannose dendrons for preventing the cellular uptake of HIV-1 in dendritic cells.37 Additionally, those glycodendrons can be conjugated to carrier proteins for establishing vaccines for producing body's own antibodies that can undergo molecular interactions of the gp-120 binding domain in HIV-1. This strategy may be suited for capturing HIV-1 in dendritic cells (Fig. 2).37 Moreover, recent progress has been achieved in establishing efficient glycodendrimers as antibacterial agents for humans34,38 as alternatives to multi-resistant drug molecules such as, for example, ciprofloxacin and ampicillin.34 In this regard, mannosylated lysine dendrons, additionally equipped with a 6-aminohexanoic acid linker between mannose units and surface groups of lysine dendrons, had been identified to be the better antagonists against Escherichia coli FimH than the unmodified mannosylated lysine dendron (Fig. 3).38
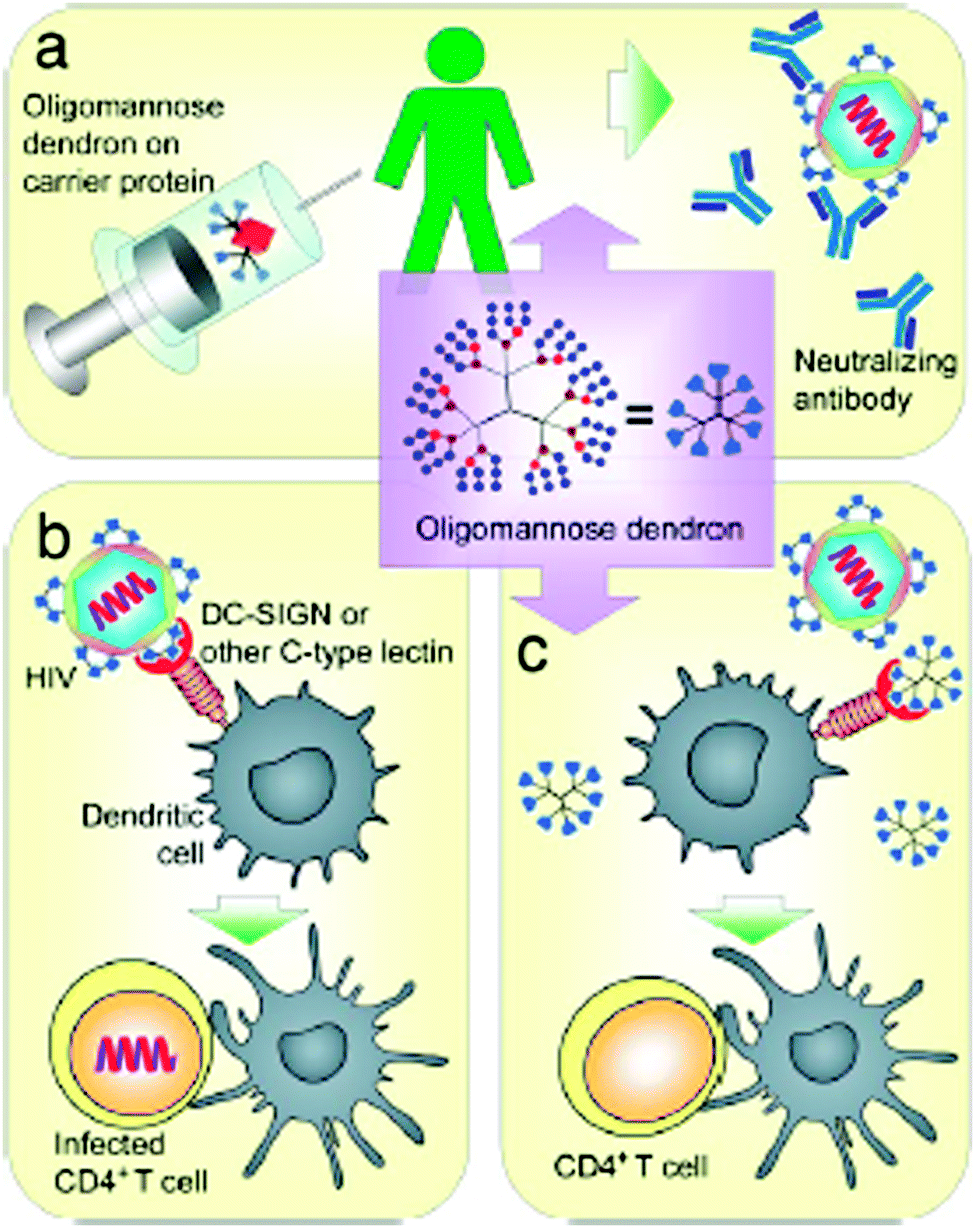 | ||
| Fig. 2 Possible biological molecular interactions of oligomannose dendrons as (anti-adhesive) dendritic glycoconjugates to suppress HIV-1 infection. (a) Possible conjugation to proteins and use as vaccines. (b) HIV-1 infection by HIV-1 binding to dendritic cell-surface DC-SIGN or other mannose-binding proteins to enhance CD4+ T cell infection. (c) Possible inhibition of the binding of HIV-1 to the dendritic cell-surface DC-SIGN or other mannose-binding proteins to prevent dendritic cell-enhanced CD4+ T cell infection. Reproduced with permission from “S.-K. Wang, P.-H. Liang, R. D. Astronomo, T.-L. Hsu, S.-L. Hsieh, D. R. Burton and C.-H. Wong, Targeting the carbohydrates on HIV-1: Interaction of oligomannose dendrons with human monoclonal antibody 2G12 and DC-SIGN Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3690–3695”. Copyright 2008 National Academy of Sciences. USA. | ||
 | ||
| Fig. 3 Anti-adhesive dendritic glycoconjugate – Highly flexible mannose-coated lysine-based glycodendron as an antagonist against Escherichia coli FimH. Reproduced with permission from “A. Papadopoulos, T. C. Shiao and R. Roy, Mol. Pharmaceutics, 2012, 9, 394–403”. Copyright 2012 American Chemical Society. | ||
There is still a high need to identify key factors determining the interactions of multivalent dendritic glycoconjugates for their successful inhibition of protein24–36 and carbohydrate39–41 cell receptors, but also for being able to develop (simplified) models.14,42 Summarizing the most important key factors recently presented in some reviews,1,4,5,18,43,44 one can state that a complex structure–activity relationship between the multivalent dendritic glycoconjugate and the complementary binding voids of the (protein) receptors exists. This means that not only are the mono-, disaccharide or branched oligosaccharide units used responsible for a high affinity against cell receptors but also the composition of the linker (aliphatic, aromatic or heterocycles or combination of them and other molecular subunits) between (oligo-)saccharide units and dendritic scaffold has a significant influence on the final affinity. In some cases linker subunits such as aromatic or triazole units in dendritic glycoconjugates contribute to the enhancement of the binding events towards carbohydrate-binding (cell) protein receptors. For other binding events, (highly) flexible multivalent oligosaccharide units chemically fixed to a more rigid dendritic scaffold had been found to be effective. One can state that in general highly adaptable dendritic glycoconjugates are involved in the molecular recognition of the carbohydrate motifs by the various lectins or cell receptors. On the other hand, the success of highly adaptable dendritic glycoconjugates also depends on (I) their simultaneous binding access to dimeric or higher assembled units of receptor molecules (e.g. lectins) (Fig. 4) and on (II) the ability to cluster randomly distributed membrane receptors on the cell surface for inducing signal transduction (Fig. 5).1,2 Such triggered signal transduction is a helpful tool to initiate the production of antibodies46 or to deliver other antigens in cells.47 Thus, the interaction properties of multivalent dendritic glycoconjugates, based on dendron and dendrimer scaffolds, against isolated lectins and cell membrane receptors can be characterized by either a low or a high multivalency (e.g. avidity) and by a strong binding tendency that can lead to receptor clustering (Fig. 5).2,45
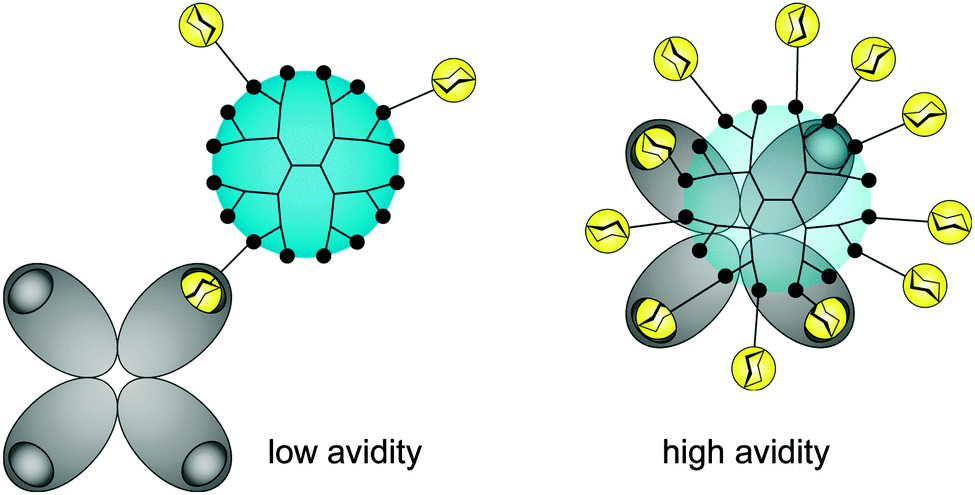 | ||
| Fig. 4 Composition of anti-adhesive dendritic glycoconjugate and their valency (accessible sugar units) dictate the multiple binding sites in oligomeric protein receptors (e.g. lectins). | ||
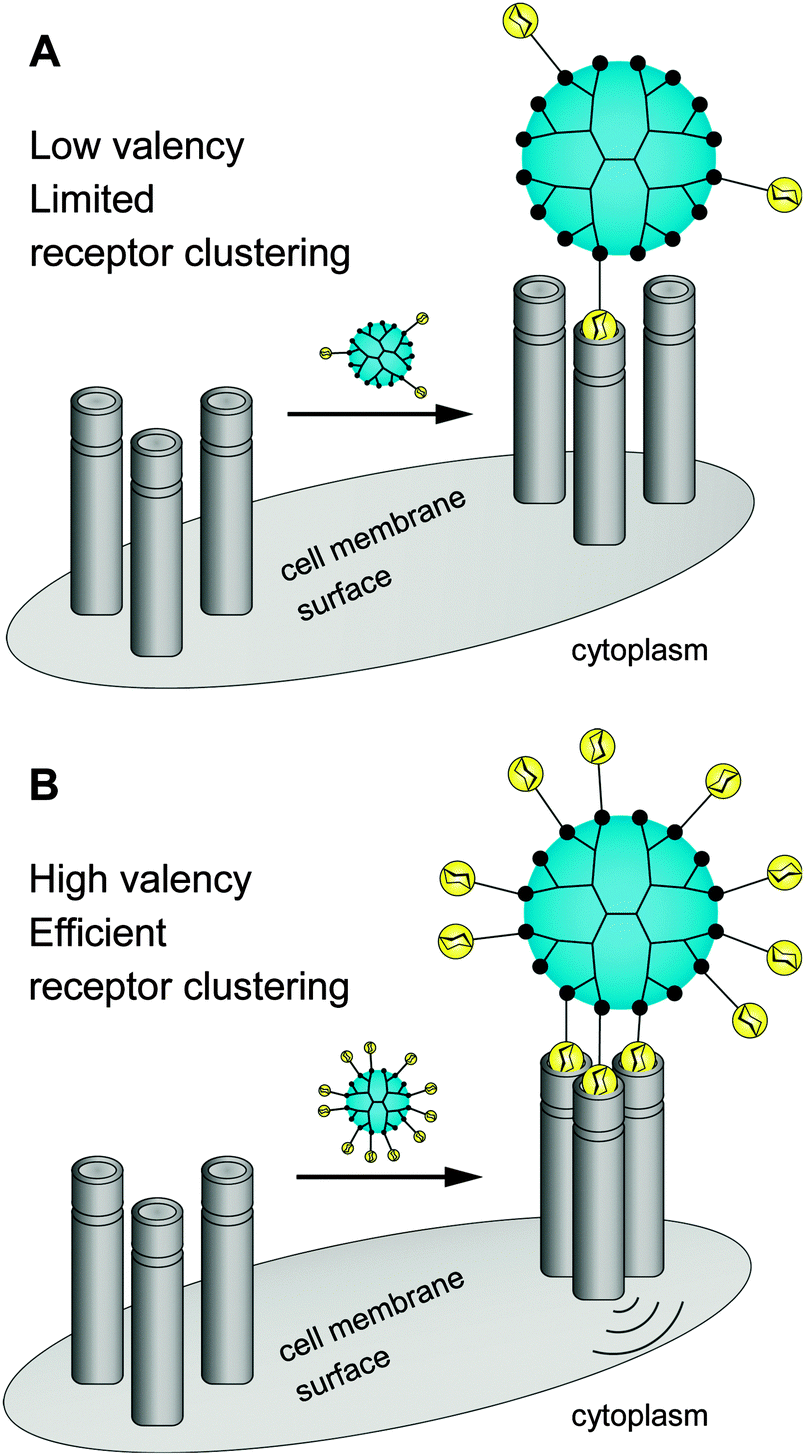 | ||
| Fig. 5 Dendritic glycoconjugates are able to bridging multiple surface receptors, clustering them, and initiating signal transduction. | ||
Overall these outstanding interaction profiles of large dendritic glycoconjugates against various cell membrane receptors allow us to use them as drugs per se in different therapeutic fields as functional antigens,6–8,46 antitumor vaccines,6–8,46 antivirals,37 antibacterial/-microbials,35,38 anticancer drugs for hepatic cancer,48,49 antiangiogenics,50 anti-influenza drugs,51,52 for inhibition of various toxins,35,36 and for triggering the fibroblast growth factor activity,53 but they are also applicable as diagnostic tools in cancer and for the detection of protein receptors and viruses.6–8
For the design and fabrication of the dendritic glycoconjugates various dendritic scaffolds or smaller branched core molecules were used as follows: polyamine dendrons and dendrimers (poly-L-lysine (Lys), polyamides, poly(amidoamine) (PAMAM), poly(propyleneimine) (PPI), polypeptides), hyperbranched poly(ethyleneimine) (PEI), dendritic polyesters from Boltorn, silane scaffolds, cyclodextrin and cyclopeptide cores, the benzene core, the hexaphenylbenzene core, the porphyrin core, the cyclotriphosphazene core, the tetraphenylmethane core, and others.
Under the shadow of drugs per se glycodendrimers can be generally considered as (anti-adhesive) drugs for initiating the inhibition of lectins, viruses and bacteria and other molecular interactions (Fig. 4 and 5).1,33,35 On the other hand dendritic glycoconjugates, carbohydrate-containing dendronized polymers, and hybrid materials of various polysaccharides over the last year have been also established in various application fields. Thus they have been used as drug delivery systems,10,54–95 biosensors,15,16,19,96–101 and imaging agents,1,18,102–106 but also as a base in biohybrid structures obtained through non-covalent and biological conjugation,91,92,107,108 as sugarballs for H-bond-active therapeutics and diagnostics for brain disease,109–118 as supramolecular structures by (defined) self-assembly119–121 or host–guest interactions,122 and in thin film technology for introducing specific interactions with small analyte molecules or proteins.123,124 The PPI, Lys, and PAMAM polyamine dendrimers, but also branched poly(ethyleneimines) (PEI) are the dominating dendritic core scaffolds to realize the desired dendritic glycopolymers for these growing research fields (Fig. 6–8). Carbohydrate decoration of dendritic polymers was developed as an alternative way to PEGylation, which is so far the dominating choice to establish highly biocompatible delivery systems and therapeutics for in vitro and in vivo application.125–133
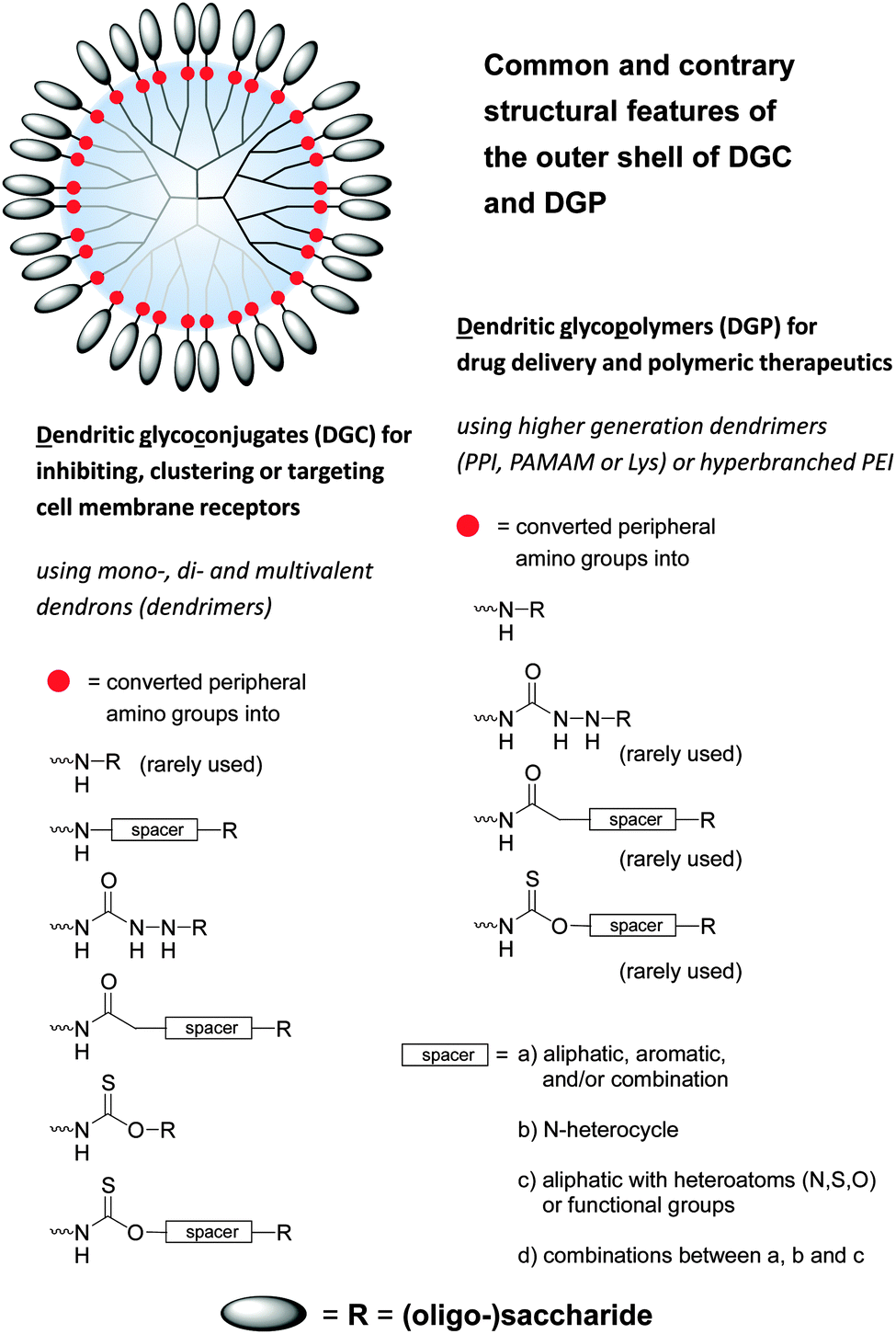 | ||
| Fig. 6 Common and contrary structural features of dendritic polyamine scaffolds decorated with various (oligo-)saccharide units. Dendritic polyamine scaffolds are dendrons and dendrimers (PPI, PAMAM, Lys, polyamide) and also hyperbranched structures (PEI) with peripheral amino groups. Dendritic glycoconjugates are considered as open shell architectures following the declaration in the text (Section 3) and Fig. 7. One main structural feature of dendritic glycopolymers is the direct coupling of (oligo-)saccharide units to the dendritic polyamine scaffold in opposite to (anti-adhesive) dendritic glycoconjugates. | ||
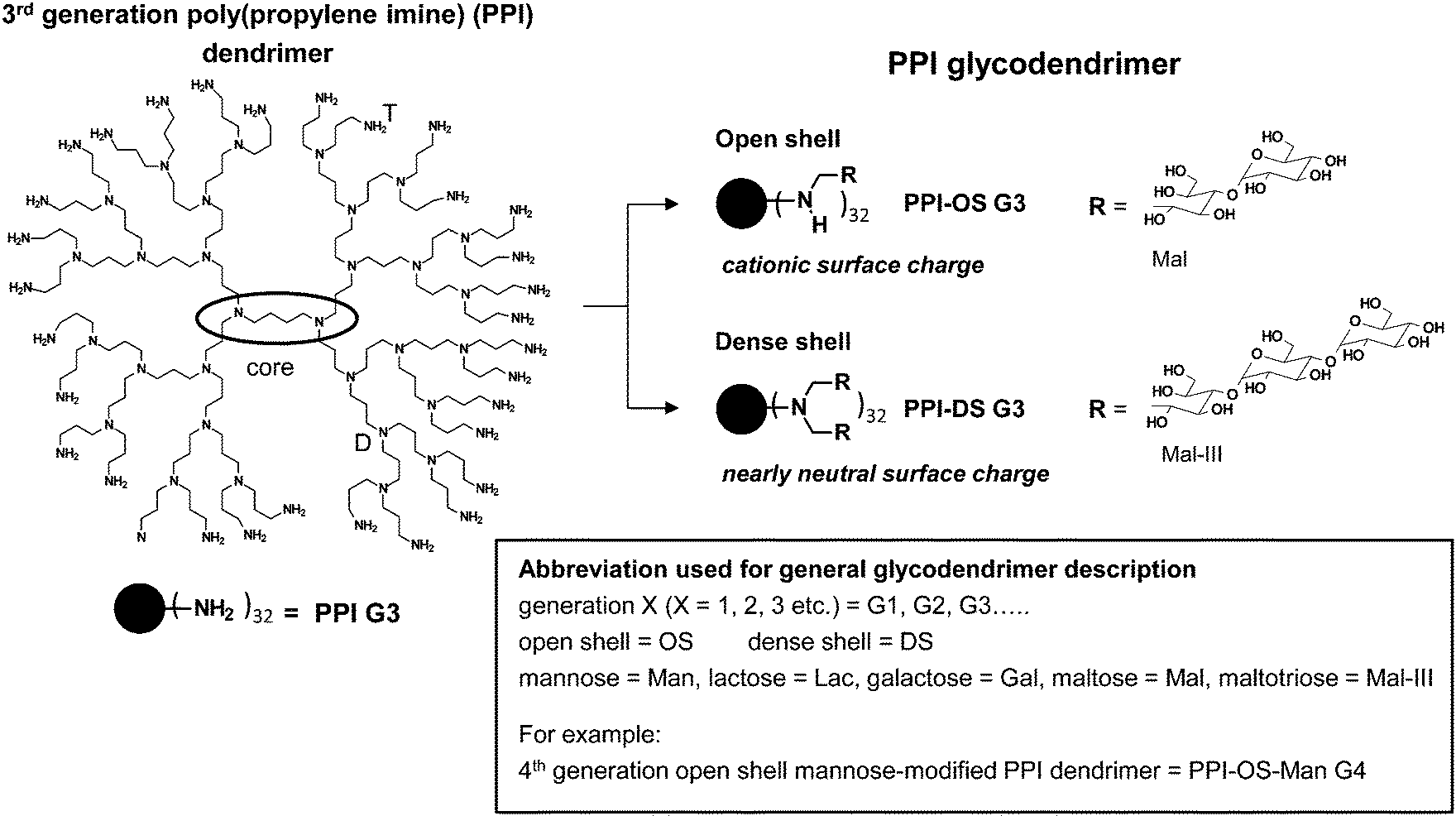 | ||
| Fig. 7 Characteristics of open shell and dense shell PPI glycodendrimers. Open shell is characterized by peripheral amino group wearing maximal one (oligo-)saccharide unit in PPI-OS-Mal G3, while peripheral amino groups in dense shell of PPI-DS-Mal G3 possess two chemically coupled (oligo-)saccharide units.† | ||
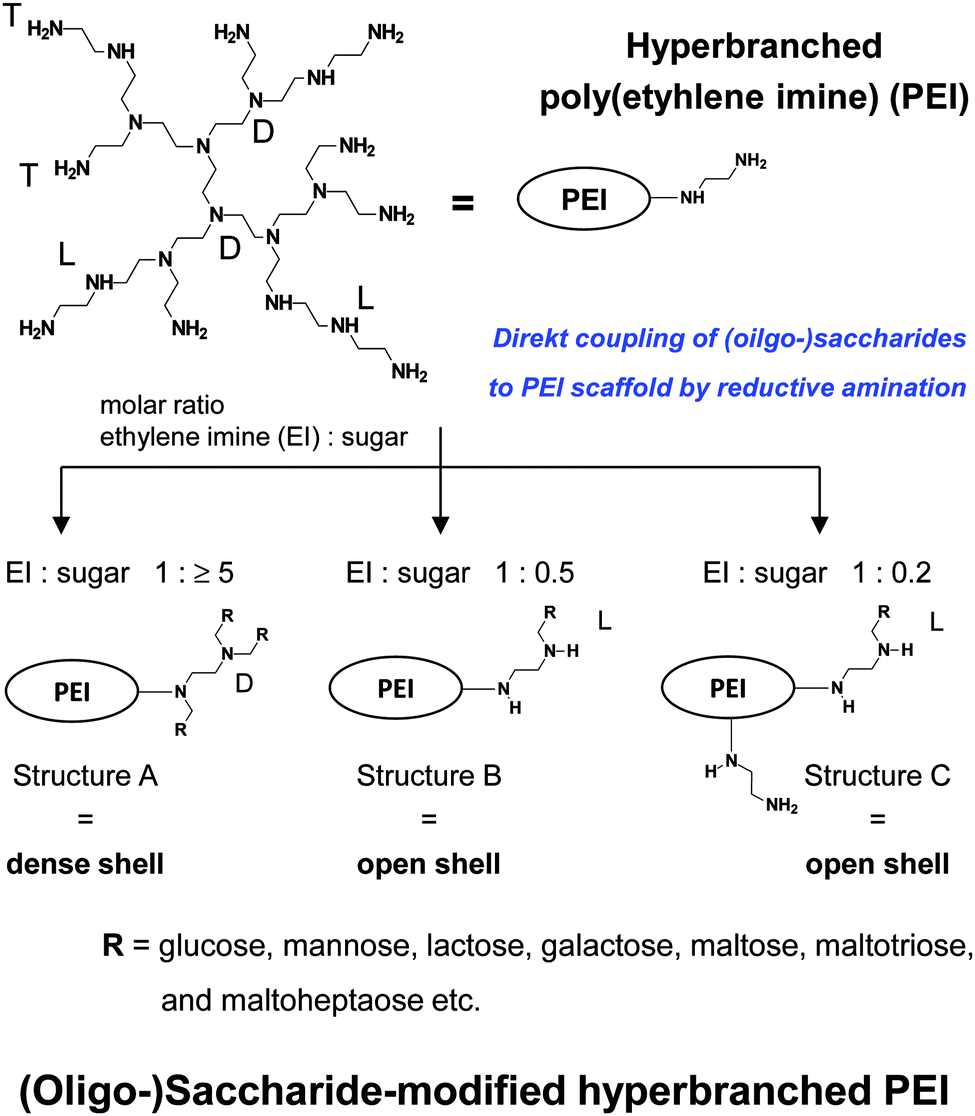 | ||
| Fig. 8 Synthesis and characteristics of (oligo-)saccharide-modified hyperbranched PEI. Decreasing cationic surface charge: PEI > PEI with structure C > PEI with structure B > PEI with structure A. | ||
This review will elaborate and highlight the recent progress of dendritic glycopolymers based on polyamine scaffolds (preferred PPI; less PAMAM (Fig. 9) and PEI) with regard to their use in various biomedical application fields (complexation and biological properties, anti-Alzheimer agents, anti-prion agents, drug delivery). Firstly, the synthetic approaches of these dendritic glycopolymers are very concisely presented and the newest synthetic development is shortly marked. Additionally, the biological and delivery properties of carbohydrate-modified dendritic scaffolds are compared with non-carbohydrate-modified dendritic polymers to better identify their potential use in the field of biomedical applications. Moreover, the driving forces of the molecular interactions of dendritic glycopolymers against drugs, analyte molecules, proteins or amyloidogenic peptides will be emphasized.
 | ||
| Fig. 9 Chemical structure of 2nd generation poly(amidoamine) (PAMAM) dendrimer with 16 terminal amino groups used for the fabrication of mannose- and α-cyclodextrin-modified 2nd generation PAMAM dendrimer.86,88 | ||
2. Synthetic aspects of dendritic glycopolymers
Recent progress in the design and fabrication of glycodendrimers is strongly interwoven with newly established synthetic approaches towards linear and dendritic polymers,134–140 especially of dendrimers, and to generate larger dendrimer generations with the lowest synthetic efforts.137,141–145 Under the term “click reaction”146 various easily performed and highly efficient chemical conjugation strategies for the synthesis of dendritic scaffolds have been successfully used such as yne–azide, thiol–ene, thiol–yne, amine–epoxy, but also the surface functionalization of dendritic polymers137,141–145 benefits from these organic reactions. More recently, straightforward approaches have been developed using at least two click reactions for the synthesis of the dendritic scaffolds142,144,145 as well as for the decoration of dendrimers with various carbohydrates.147–150 A kind of onion peel glycodendrimer, using several times the same click reaction for the fabrication of larger dendrimer generation, has been established by the groups of Hawker, Malkoch and Dondoni (Fig. 10),149 while the group of Roy realized a similar highly multivalent onion peel dendrimer by using only one time an efficient click reaction.151 An efficient orthogonal approach by using thiol–ene and SN2 reactions was recently introduced to accelerate the growth of multifunctional dendrimers and dendritic glycoconjugates.152 Here, the group of Schlaad introduced carbohydrates as side groups in a polymeric backbone for the first time by UV irradiation or by day light.153,154 Moreover, a photo cyclization reaction was also adaptable to introduce carbohydrate-modified dendrons along a polymeric backbone with a high density of carbohydrate functionalization.155 | ||
| Fig. 10 Various onion peel dendrimers with different surface groups.149 Reproduced from ref. 149 with permission from John Wiley and Sons. | ||
Various reviews1,4–8,14–16,20,30–34,40,43,44,47,156–166 can be referred to for elaborating the recent progress in the design and fabrication of dendritic glycoconjugates and the variations achieved with regard to linker chemistry and size, shape, functionality and flexibility or rigidity of the dendritic scaffold, especially for the design and fabrication of anti-adhesive dendritic glycoconjugates (Fig. 2 and 4). One can state that all conventional and modern synthetic tools are applicable for the chemical coupling of carbohydrates to any dendritic scaffold and to transform functional groups in carbohydrate units and their derivatives ready for the functionalization of dendritic scaffolds. As a few highlights are emphasized (I) various solid phase approaches,38,167,168 combined with enzymatic support,169 to fabricate very complex oligosaccharide architectures, (II) the chemical and enzymatic post-modification of dendritic scaffolds to introduce final carbohydrate units105,170–172 or peptide recognition sequences173 and (III) the chemical conjugation of antigens as part of the linker units between the dendritic scaffold and the complex oligosaccharide scaffold.8,37,46,93,94 But also sulfur chemistry can accelerate and strengthen the design, fabrication and application of dendritic glycoconjugates.43
In comparison to the often intensive synthetic efforts for the fabrication of dendritic glyco-scaffolds as anti-adhesive drugs, the synthetic pathways to establish dendritic glycopolymers, for example, as drug delivery systems10,54–95 and therapeutics and for diagnostics in the field of brain disease109–118 are directed preferentially to use fast and easily available conversion steps to achieve carbohydrate modification of dendritic PPI, PAMAM, PEI and Lys scaffolds. The most favorite conjugation tool for the carbohydrate modification of dendritic structures is the reductive amination by using (I) NaBH3CN in sodium borate buffer at about pH 8 and room temperature,54,57,62–65,67,70,76,82,174 (II) borane*pyridine complex in sodium borate buffer at about pH 8 for several days at 50 °C (ref. 10, 11, 55, 56, 60, 61, 77 and 108–124) or (III) simple use of acetate buffer at pH 4 and room temperature or at pH 4 and 60 °C for several hours78,79,83 to convert the intermediate enamine into secondary and tertiary amino groups bearing the desired carbohydrate units (Fig. 7 and 8). For the carbohydrate modification of PEI another important synthetic tool has been established by converting hyperbranched PEI with various carbohydrate phenyl isothiocyanates (Fig. 6).66,68,69,73 For preparing PAMAM glycodendrimers as a drug delivery system85–94 various carbohydrate units with acid, lactone or phenyl isothiocyanate groups were converted with the peripheral amino groups of the dendritic PAMAM scaffold into the desired dendritic glycoconjugates (Fig. 6). More recently, hydrazide-modified PAMAM dendrimers were used for the direct conjugation of reducing saccharides under the preservation of the cyclic scaffold of the saccharides, but with less efficiency in increasing the generation number (Fig. 6).175 The introduction of α-, β- or γ-cyclodextrin units on amino-functionalized dendritic PAMAM scaffolds follows under the principle of SN2 reactions.92 Thus, various carbohydrates (glucose, maltose, maltotriose, maltohexaose, maltoheptaose, galactose, lactose, mannose, mannobiose, cellobiose, tetragalactose, sialic acid, N-acetyl glucosamine, lacto-N-difucohexaose or cyclodextrin) have been successfully introduced. Interestingly, biologically active galactose units were preferentially realized on dendritic scaffolds by the introduction of the disaccharide lactose. After attaching lactose under reductive amination or phenylisothiocyanate conditions, the cyclic form of the galactose rings remains and allows for the specific galactose molecular recognition in the course of ligand-mediated drug targeting.62,65,67–72 In contrast to this, mannose and galactose, when they are directly introduced on the dendritic PPI and Lys scaffold under reductive amination conditions, they do not retain their active cyclic ring conformation as targeting ligands against cell lines.78,79,83,95 However, the mannose units, but also sialic acid, were established on the dendritic PPI scaffold surface in the active form by multi-step reactions.80,83,176
One final consideration is directed to the efficiency of the applied one-pot reactions mentioned above to establish the desired dendritic glycoconjugates.10,54–95,108–118 The conversion of dendritic polyamine scaffold based on amidation, phenyl isothiocyanate derivatives and lactone derivatives is more or less quantitative (Fig. 6) when following the experimental protocols. These results are impressively supported by the design and fabrication of other dendritic glycoconjugates applicable as anti-adhesive drugs.9,25–27,159,177 Reductive amination is an easily applicable synthetic method to couple carbohydrate units on the dendritic polyamine scaffold, while the control over the attached numbers of carbohydrate units on the dendritic polyamine scaffold can be partly challenging when only aiming for monosubstitution of primary amino groups. In this case optimization is needed by adjusting the equivalents of carbohydrate units (≤1 equivalent per peripheral amino group) or the reductive agent for the desired composition of dendritic glycoconjugates (Fig. 7 and 8).10,78,79,83,119,178 Moreover, reductive amination is also highly efficient to generate dense carbohydrate shells around dendritic polyamine scaffolds of hyperbranched PEI and PPI and Lys dendrimers,10,55,108–111,179,180 where, finally, the former peripheral primary amino group is converted into a tertiary amino group bearing two carbohydrate units (Fig. 7 and 8). This type of dendritic glycopolymers is only available under the use of excess carbohydrate units (10–40 equivalents per amino group) and excess reductive agent.10,55,108–111,179 This kind of densification of carbohydrate units on dendritic polyamine scaffolds is only comparable with the recently described work of Malkoch and Dondoni.150 In their case peripheral alkyne groups on a polyester dendrimer surface were used to introduce two carbohydrate units on one alkyne by thiol–yne conversion conditions. With their method less excess carbohydrate units (4 equivalents per yne group and ≤0.3 equivalent photo initiator per yne group) under UV irradiation are needed to fabricate this specific dense shell glycoarchitecture.150
3. Characteristics of dendritic glycopolymers
The knowledge about the interaction characteristics of dendritic glycopolymers in solution is essential for their successful use as a drug delivery system and therapeutics and in diagnostics in the field of neurodegenerative disease. Furthermore, it is also desirable to get insight into in vitro interactions towards biologically active molecules (e.g. proteins or nucleic acids). Therefore, the molecular characteristics of dendritic glycopolymers10,54–95,108–124 will be presented here and will be compared to those of the highly elaborate dendritic glycoconjugates applied as anti-adhesive drugs.19,29–37For both, the well-known anti-adhesive dendritic glycoconjugates1,6–8,20,30–38,46,48–53 and the often less defined dendritic glycopolymers used in other biomedical application fields,10,54–124 the terms open shell and dense shell dendritic glycopolymers will be used as a common feature to describe the molecular characteristics (Fig. 6–8). Both terms, dense and open shell dendritic glycopolymers, were recently introduced by the group of Appelhans and Voit.10,55,56,108–111,114 It is easily described by the degree of (oligo-)saccharide functionalization of dendritic polyamine scaffolds (PPI dendrimers and hyperbranched PEI). In the “dense shell” architecture the primary amino groups of the dendritic polyamine scaffolds are converted into tertiary amino groups bearing two chemically coupled (oligo-)saccharide units (disubstitution) (Fig. 7).10,109 Additionally, in the case of hyperbranched PEI, secondary amino groups are also converted into tertiary amino groups bearing a single saccharide unit (Fig. 8).10 In contrast to this the “open shell” architecture is characterized by the conversion of primary amino groups into secondary amino groups only bearing one chemically attached (oligo-)saccharide unit (monosubstitution) (Fig. 7 and 8).10,109 Most of the dendritic glycopolymers used as drug delivery systems and therapeutics and diagnostics for neurodegenerative disease,10,54–59,62–76,78–107,110,113,114,119–124 belong to the type of open shell dendritic glycopolymers, while some perfectly branched glycodendrimers, mainly applied as sugarballs in the field of neurodegenerative disease109–118 and in drug delivery system,56,60,61,77 possess a dense shell. Most of the dense shell glycoarchitectures used in biomedical applications are based on PPI dendrimer scaffolds.109–118
One specific key issue of the reductive amination is that the reducing unit of mono-, di- and oligosaccharides is directly connected to the primary and secondary amino groups in the dendritic polyamine scaffolds. Therefore, there is usually no spacer between the coupled mono-, di- and oligosaccharide units and the corresponding dendritic polyamine scaffold (Fig. 6). The situation is similar using the conversion of lactones for glycosylation.90 With these synthetic tools a preferred shielding effect against the dendritic polyamine scaffold in terms of surface charge reduction can be achieved10,55,56,109–111 and the complexation properties of the core are better supported.79,80,84,108,181–183 On the other hand, the introduction of targeting ligands is also possible.62,70 Overall, these specific dendritic glycopolymers10,54–95,108–124 are commonly characterized by (oligo-)saccharide units directly linked to the dendritic polyamine scaffold (Fig. 7 and 8) or linked via a short alkyl80,83 and aromatic66,68,69,74 spacer (Fig. 6).
In contrast to this, the well-known anti-adhesive dendritic glycoconjugates1,6–8,20,30–38,46,48–53 can be assigned to an open shell architecture since most of their peripheral functional groups possess only one (oligo-)saccharide unit (Fig. 6). Secondly, the (oligo-)saccharide units are preferentially linked via a spacer to the peripheral functional groups of various dendritic architectures (Fig. 6) to establish highly accessible molecular recognition carbohydrate units on the dendritic scaffolds for undergoing desired protein–carbohydrate interactions. The molecular composition of the spacer can vary widely (Fig. 6). The nature of the spacer depends on the targeted protein receptors (isolated or integrated in the cell membrane) and the type of biological entities (viruses or bacteria) to be investigated. Finally, different dendritic molecular scaffolds ranging from very rigid to highly flexible were successfully applied for this purpose (Fig. 2, 3 and 6). Not only small branched core molecules or dendrons,1,4 but also larger dendrimers are reported for enhanced binding affinities at very low concentration in the nanomolar range of anti-adhesive dendritic glycoconjugates. Overall, the perfect interplay of these different molecular parameters is a pivotal point to achieve highly multivalent dendritic glycoconjugates that can adapt to the chemical and biological space of protein and carbohydrate receptors in various biological environments. Most of the open shell anti-adhesive dendritic glycoconjugates have different structural and molecular features than the above mentioned open shell dendritic glycopolymers used for drug delivery systems.
Further attention is now directed to the molecular characteristics of open and dense shell dendritic glycopolymers, based on PPI dendrimer and PEI,10,55,56,60,61,76–84,108–124 used as a drug delivery system and as sugar balls for therapeutics and diagnostics in neurodegenerative diseases. Playing with the degree of mono-, disaccharide and oligosaccharide functionalization on those dendritic polyamine scaffolds, surface charge or charge density of PPI glycodendrimers is tunable from positive for open shell glycodendrimers to neutral for dense shell glycodendrimers.109,114Open and dense shell PPI glycodendrimers can be in addition decorated with anionic sulfate groups.114,184,185Dense and open shell dendritic glycopolymers based on PEI (Fig. 8) possess pH-dependent cationic surface charge and charge density over a broad pH range [2 to isoelectric point (8–9)] tailored by the given (oligo-)saccharide architectures A, B and C.10,55,57–59,62–75 Architecture A, meaning dense shell architecture, has the lowest cationic charge and architecture C, meaning an open shell architecture with even remaining primary amines, possesses the largest cationic charge within this series. Overall, these dendritic glycoarchitectures preferentially exist as non-aggregated macromolecules in aqueous solution and under physiological conditions.10,55,109,118 The molecular sizes of both types of dendritic glycopolymers are in the lower nanometer range: ≤8 nm for the largest generation of PPI glycodendrimers109,118 and ≤12 nm for glycoarchitectures based on a PEI core with a molecular weight of 25 kDa.10,186 A surprising result is that the diameter of dendritic glycopolymers (PPI as well as PEI based) does not change by varying the pH.186
SAXS and DLS studies verified that open and dense shell PEI glycoarchitectures can be considered as core–shell architectures.186 This implies that the oligosaccharide units of the open and dense shell are mainly located in the outer sphere of these PEI glycoarchitectures. Moreover, the (oligo-)saccharide decoration of PEI induces an enlargement of the dendritic scaffold itself.10,186 Core–shell PEI glycoarchitectures10,55,56,186 can theoretically undergo ionic as well as H-bond-driven interactions, depending on the shell density. These specific molecular characteristics are of importance for the complexation and delivery of drugs,10,55,56,181,182 but can also lead to morphological transformation of anionic vesicles into worm-like networks.187
In line with this, dense shell PPI glycodendrimers can be also ascribed as core–shell architecture as supported by various studies.109,111,114,118 Polyelectrolyte titration experiments prove that anionic polyelectrolytes are not able to penetrate the dense maltose shell of PPI glycodendrimers and thus cannot compensate the cationic charge of the PPI core molecule. Only very small molecules such as water or other nutrients are capable of diffusing through the dense oligosaccharide shell of glycodendrimers. Furthermore, the cationic charge of the dendritic PPI scaffold in dense shell glycodendrimer was only determinable by pH streaming potential measurements,110 but not by zeta potential measurements under physiological conditions.108 In general, dense shell PPI glycodendrimers can be considered as amphiphilic macromolecules with a cationic core and a neutral and H-bond-active shell (Fig. 11).
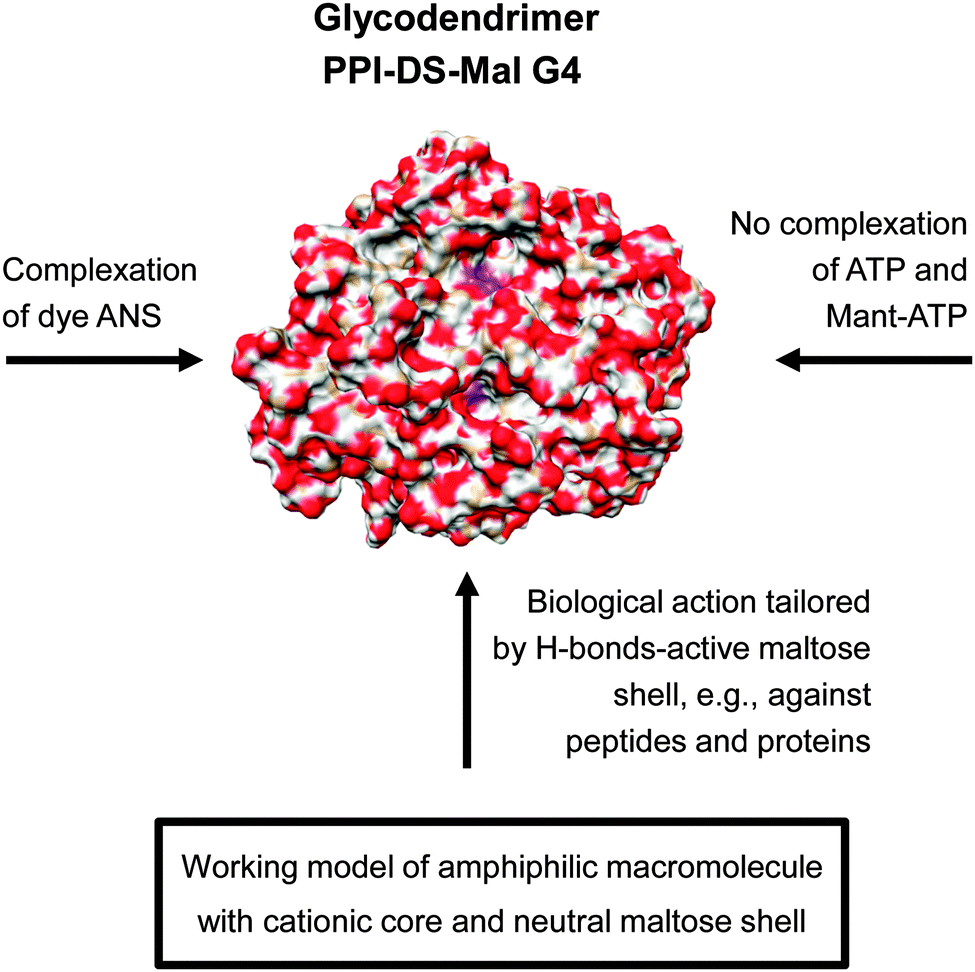 | ||
| Fig. 11 Molecular modelling showing the radial distribution of maltose units had an high impact in understanding the molecular interaction of dense shell glycodendrimer PPI-DS-Mal G4.114 The various molecular interactions of PPI-DS-Mal G4 are outlined considering the low permeable sugar shell for limited ionic interactions with cationic PPI core.109–111 Adapted with permission from “J. M. McCarthy, B. Rasines Moreno, D. Filippini, H. Komber, M. Marek, M. Cernescu, B. Brutschy, D. Appelhans and M. S. Rogers, Biomacromolecules, 2013, 14, 27–37”. Copyright 2013 American Chemical Society. | ||
Thus, preferably H-bond-driven surface interactions of dense oligosaccharide shell can be assumed for the biological interactions of dense shell glycodendrimers.109–118 In line with this, molecular modelling and theoretical calculations confirmed a dense oligosaccharide shell for this class of glycodendrimers where only few oligosaccharide units are back folded and wrapped by the dendritic PPI scaffold (Fig. 11: indicated by a purple color in the PPI-DS-Mal G4 image). For this purpose dense shell glycodendrimers were modeled in a water droplet.114 On the other hand open shell PPI glycodendrimers are characterized by a cationic surface charge as well as an H-bond-active surface.108,110
4. Interactions of dendritic glycopolymers with proteins
Interaction of dendrimers with biological systems determines their potential biomedical application. This also includes their toxicity towards cells. Thus, it is of crucial importance to find the right balance between favorable and detrimental biological effects evoked by dendrimers. Undoubtedly, interactions of dendritic nano-sized molecules with proteins present a biomedical potential. For instance, dendrimers can inhibit fibrillation of proteins involved in neurodegenerative disorders188,189 or bind surface proteins of pathogens limiting the spread of infection.190Since modification of cationic surface groups of dendrimers with neutral or anionic moieties decreases their cytotoxicity,191,192 the ability of maltose-modified dense shell PPI glycodendrimers (G1–G4) to interact with human serum albumin (HSA) was examined and compared to naked PPI.109 Surprisingly enough, the results indicated that there is no significant difference in the strength of interactions between unmodified or sugar-coated dendrimers and HSA. PPI G1 and maltose-modified PPI G1 and G2 did not interact with the studied protein. Naked PPI G2 interacted weakly with HSA, while unmodified and maltose-coated dendrimers PPI G3 and G4 exhibited strong interactions with HSA. These data also point out that dendrimer–HSA interactions are generation-dependent and begin from G3. It is likely that higher generations possess proper size, globular shape and rigidity to effectively bind to the protein molecule. Moreover, it is proposed that besides electrostatic interactions between positively charged dendrimers and negatively charged HSA, carboxylate groups and oxygen atoms of the amide groups of the proteins participate in forming hydrogen bonds.109
Further studies on the impact of PPI dendrimers on protein properties were conducted using unmodified PPI G3, as well as PPI G4, including open and dense shell glycodendrimers decorated with maltose. A model protein, liver alcohol dehydrogenase (LADH) was applied to study the interactions with the dendrimers.193 This protein possesses two tryptophan (Trp) residues, one on the outside (Trp-15), and one buried inside the protein structure (Trp-314). Changes in the protein structure near Trp-15 and to a much higher degree near Trp-314 are reflected in changes in fluorescence spectra. Moreover structural reorganization in the proximity of Trp-314 can be detected based on room temperature tryptophan phosphorescence (RTTP).194 The results showed us that none of the tested dendrimers quenched Trp fluorescence of LADH, indicating that the fragment of protein in the proximity of Trp is not involved in the interactions with dendrimers. RTTP analysis revealed that all PPI caused similar decrease in RTTP along with increasing concentration. Only in the concentration range of 10–25 μM unmodified PPI G4 had stronger impact on the tested parameter than the glycodendrimers. The decrease in RTTP points out that the dendrimers interact with the protein and that the native fold of LADH became more flexible as a result of dendrimer binding. Moreover, a stronger effect of the naked PPI dendrimer at some concentration range indicates an involvement of electrostatic forces in dendrimer–protein association. However, similar influence on RTTP of all dendrimers at other concentrations implies that other types of bonds, such as H-bonding, van der Waals and hydrophobic forces also play a role in PPI dendrimer–LADH interactions. The fact that no changes were detected in the fluorescence, while a decrease in RTTP was observed, is likely to result from a much longer lifetime of phosphorescence than fluorescence. Circular dichroism analysis revealed small changes in the secondary structure of LADH dependent on the type of PPI dendrimer added. Unmodified PPI dendrimer caused the strongest increase in β-sheet content and a small decrease in α-helix structures. On the contrary, dense shell PPI fully coated with maltose had no impact on the secondary structure of LADH. The results indicate that electrostatic interactions between the protein and cationic peripheral amino groups of PPI dendrimers are responsible for alterations in the secondary structure. Furthermore, upon addition of all dendrimers, an increase in the hydrodynamic diameter of the molecules/complexes in solution was observed; in the case of uncoated PPI even formation of larger aggregates was evident and the formed complexes were shown to be stable for 12 hours. Addition of dendrimers had little influence on zeta potential of complexes. In summary, the results demonstrate that all types of tested dendrimers are able to interact with LADH starting from generation 3, although unmodified PPI G3 possesses the greatest affinity to the protein.193
Influence of open shell (OS), dense shell (DS) and naked PPI G3 on the thermal stability of a model protein hen white egg lysozyme (HWEL) was also examined and compared with the impact on the protein of anionic PAMAM G3.5. Moreover, the availability of lysozyme Trp to fluorescence quenchers in the presence of dendrimers was studied. HWEL is characterized by a positive net charge under physiological conditions, although it contains both positively and negatively charged areas. To assess the effects of dendrimers on the thermal stability of HWEL differential scanning calorimetry (DSC) and circular dichroism (CD) methods were employed. The results show some changes in the melting temperature of HWEL after addition of dendrimers. DSC analysis revealed that the biggest effect was observed after addition of uncoated PPI dendrimer, while PPI-DS had no influence on the studied parameter. CD analysis demonstrated the greatest impact of PAMAM G3.5 on the content of α-helix upon heating, while for PPI dendrimers the effect was dependent on the degree of surface modification (PPI > PPI-OS > PPI-DS). The changes in the protein secondary structures (α-helix and β-sheet contents) upon heating were again most pronounced in the presence of PAMAM G3.5 and the lowest in the presence of PPI-DS. These effects are likely to stem from electrostatic interactions between positively charged lysozyme and a negatively charged dendrimer. However, HWEL also possesses anionic areas which enable binding of cationic PPI dendrimers. Furthermore, CD spectra of lysozyme in near-UV, which reflect changes in the environment of aromatic acid residues, were altered to the greatest degree upon addition of PAMAM G3.5. Additionally, all tested dendrimers have shown to decrease the accessibility of lysozyme tryptophan residues to quenchers, indicating interaction between dendritic macromolecules and parts of the protein where Trp residues are located (near the surface). Generally, PPI-DS with the neutral surface can bind to HWEL, but mainly by hydrogen bonds and not electrostatic interactions like the rest of the studied dendrimers. Due to the nature of interactions, weaker in comparison to other dendrimers, the dense shell PPI glycodendrimer does not change the secondary structure of the protein. On the other hand, the strength of interaction between unmodified PPI and lysozyme and PAMAM G3.5 and the protein is very similar due to electrostatic interactions with negatively or positively charged regions of lysozyme, respectively.195
5. Biological properties and biocompatibility of dendritic glycopolymers
The following section is intended to be a critical review of biological properties in vitro and in vivo of some dendritic glycopolymers, including their potential to cross blood–brain-barrier.Toxicity in vitro
Dendritic polymers are excellent candidates for nanomedical applications. Unfortunately, their use might be limited due to high cytotoxicity. Cytotoxicity of dendritic polymers is dependent on the number and nature of functional surface groups, so they can exhibit high toxicity if they possess cationic terminal groups and show slight or no toxic effects when their surface is anionic or neutral.196,197 Appropriate modifications of the dendritic polymers surface can significantly reduce their toxicity. Coating the surface with sugar units is one of the methods available to obtain less toxic compounds.200Modification of amino-terminated PPI dendrimers, as a particular subgroup of dendritic polymers, by coupling two maltose units to one surface amino group via a simple one-pot method, resulted in a very significant reduction of toxicity.109 For comparison reasons two types of dendrimers were studied: cationic PPI dendrimers with open and globular shape, flexible dendritic scaffolds and back-folding properties and nearly neutral dense shell PPI glycodendrimers interacting preferentially by hydrogen bonding. The most important effect was the lack of hemolytic activity, under the experimental conditions at concentrations of 3 and 6 mg mL−1, demonstrated by modified dendrimers PPI-DS-Mal G1 and PPI-DS-Mal G3. Whereas in the case of unmodified amino-terminated PPI dendrimers for the same concentrations the level of hemolysis was 50 and 70% for G1 and 55 and 85% for G3, respectively. This can be explained by the formation of a densely organized maltose shell on the PPI dendrimers surface that separated erythrocytes from the toxic PPI cores. These results are in agreement with previous studies in which modification of PPI G4 dendrimers with mannose and galactose, considered as open shell cationic PPI glycodendrimers (Fig. 7), also diminished hemolytic activity. Additionally, the analyzed mannosylated dendrimer possessed only minor cytotoxic activity against VERO cells, since only a few percent reduction of cell viability was observed for concentrations that exceeded 100 μg mL−1.79
Further studies of amino-terminated and maltotriose-modified PPI dendrimers confirmed that hemotoxicity of dendrimers was concentration-, generation-, and time-dependent.198 For the biological research three types of the G3 PPI dendrimers were used: unmodified and modified with approximately 35% (open shell) and 90% (dense shell) maltotriose units. The study was carried out in the presence of human serum albumin (HSA) or human plasma or in whole blood. Maltotriose-modified PPI dendrimers were characterized by lack of hemolytic activity similarly as other sugar modified dendrimers. In addition they had a minor impact on lymphocyte proliferation and platelet aggregation. The unmodified PPI dendrimer, however, was found to be the most hemolytic because its 60 μM concentration caused even 80% of hemolysis after 24 hours of incubation whereas the same concentration of open and dense shell dendrimers modified with maltotriose caused only 12 and 38% of hemolysis, respectively. The presence of human serum albumin (HSA) or human plasma or whole blood significantly reduced (up to 90%) the extent of hemolysis observed by all analyzed dendrimers. Interestingly, the increase in the degree of surface modification was not proportional to the decrease in hemolysis which was confirmed by changes in the erythrocytes morphology. From echinocytic transformations through cell aggregation to cluster formation, erythrocyte's shape was also dependent on the dendrimer's type and concentration. The untreated cells had their normal physiological biconcave disc shape and neither aggregates nor echinocytes were observed, whereas 24 hour incubation with 30 μM concentration of the unmodified PPI dendrimer and dense shell PPI glycodendrimer led to cluster formation. Additionally, unmodified PPI dendrimers significantly inhibited lymphocyte proliferation even at low 1 μM concentration, in contrast to glycodendrimers that only slightly inhibited cell proliferation. The sugar modification of PPI dendrimers impressively reduced their ability to induce platelets' aggregation, in the case of PPI-DS-Mal-III G3 even to zero, whereas the aggregation caused by the unmodified PPI dendrimer was comparable with that of trypsin, even at the lowest dendrimer concentrations.
These results are in agreement with previous studies in which PPI-OS-Gal G3 and G4 (Fig. 7) in comparison to unmodified PPI dendrimers showed negligible hemotoxicity. Modification with galactose units significantly reduced hemolysis to 10 and 7.1% for both generations, respectively.78
Influence of maltotriose modification on the cytotoxicity of PPI G3 has been studied as a continuation of the research that aims to define the biological properties of sugar-modified PPI dendrimers.199 Cytotoxicity profiles of unmodified amino-terminated PPI G3 and maltotriose-modified dendrimers, PPI-OS-Mal-III G3 and PPI-DS-Mal-III G3 (Fig. 7), were compared with acid-terminated PAMAM G3.5 and amino-terminated PAMAM G4 dendrimers. Modified PPI dendrimers revealed minor cytotoxicity against a normal Chinese hamster ovary CHO cell line and unexpectedly greater cytotoxicity against a moderately doxorubicin and cisplatin resistant human ovarian carcinoma SKOV3 cell line. As predicted, anionic acid terminated PAMAM G3.5 were found to be less toxic than cationic amino-terminated PAMAM G4. The anionic PAMAM G3.5, open shell PPI glycodendrimer and the dense shell PPI glycodendrimer were negligibly toxic towards the CHO cell line in a concentration range of 0.1–300 μM, so the IC50 value could not be calculated, whereas only very low concentration (<8 μM) of cationic dendrimers was needed to achieve the IC50. The IC50 data for the SKOV3 cell line confirmed that this moderately resistant cell line was less susceptible to cationic amino-terminated PPI G3 and PAMAM G4 dendrimers than the nonresistant CHO cell line. At the same time, for SKOV3 cell line open shell and dense shell PPI dendrimers were sufficient to evaluate the IC50 value at concentrations of 100 and 145 μM, respectively, while for non-toxic anionic PAMAM G3.5 it was impossible to calculate the IC50 value.
Other studies also reported high toxicity of unmodified PPI G3 and PAMAM G4 dendrimers for three cancer lines B16F10, CCRF and HepG2 and lack of toxicity for PAMAM G3.5 towards any cell lines in the studied concentration range.200 Similarly to studies for PPI-Mal III G3, functionalization of PPI dendrimers with glycine, phenylalanine, mannose and lactose resulted in a reduction of cytotoxicity in comparison to the unmodified PPI G4 dendrimer. For PPI G4 dendrimers modified with glycine, phenylalanine, and lactose, IC50 values were one hundred times lower and in the case of mannose modification still fifty times lower compared to the unmodified PPI G4 dendrimer.201
Mechanism of cytotoxicity of unmodified PPI dendrimers is believed to be related to the generation of reactive oxygen species (ROS) and damage of the mitochondria. Therefore, an additional study was performed to examine the ability of PPI glycodendrimers to induce ROS generation, changes in mitochondrial membrane potential and generation of apoptotic cell death. The obtained results were in good agreement with previous cytotoxicity findings. Open shell PPI and dense shell PPI dendrimer caused ROS generation, changes in mitochondrial membrane potential and enhanced the amount of apoptotic and necrotic cells in the SKOV3 cell line.199
In summary PPI glycodendrimers do not show toxic effects towards normal cells that are characteristic targets for the unmodified amino-terminated PPI or PAMAM dendrimers. At the same time PPI glycodendrimers exhibit higher cytotoxicity against cancer cells. This observation supports the conclusion that the analyzed glycodendrimers may be suitable for medical applications as anticancer agents.
Another example of the protective effect of maltotriose modification is provided by DNA damage and repair studies.202 Using comet assay different PPI glycodendrimers G3 have been characterized and checked in terms of genotoxicity. As expected, open shell and dense shell PPI glycodendrimers showed weakest cytotoxicity towards peripheral blood mononuclear cells (PBMCs) due to the surface modification of PPI G3. However the most important finding was the lack of influence of the modification degree of the analyzed dendrimers on the DNA damage. Even substitution of approximately 40% amino-terminal groups by maltotriose residues already significantly reduced cytotoxicity of PPI dendrimers and highly limited their genotoxicity. The dense shell PPI glycodendrimer was nontoxic in the whole tested concentration range (0.05–5 mg mL−1), whereas the open shell PPI glycodendrimer in the same concentration range induced slight increase of PBMCs cell viability, particularly for the highest concentration. The distributions of PBMC cells exposed to PPI glycodendrimers according to their DNA damage (% DNA in comet tail) was studied using the alkaline version of the comet assay. Open shell and dense shell PPI glycodendrimers at the concentration of 0.5 mg mL−1 increased the comet fractions of damaged DNA up to 3.98 and 3.35% accordingly, in respect to the 2% DNA damage level observed in untreated control cells. For comparison unmodified amino-terminated PPI dendrimers caused 10.7% DNA damage level. Both unmodified and modified PPI dendrimers revealed an influence on nucleus DNA. However, due to positive surface charge, the influence of amino-terminated and open shell PPI dendrimers was the strongest. The increase of DNA damage in the comet tail can be interpreted as a result of induction of DNA single-strand breaks caused by these dendrimers and/or as the formation of abasic sites, which can be transformed into strand breaks in the alkaline comet assay. The increase of DNA condensation in the comet head observed for unmodified and open shell PPI dendrimers might be due to strong binding of these dendrimers to DNA. This observation correlates well with the results described earlier: both, the increase of the DNA level in the comet tail and DNA condensation in the comet head, are believed to be an effect of DNA strands wrapping around the PPI dendrimer molecule; and DNA condensation preventing its repair might lead to cell death.203 Despite this, the revealed small amount of damaged DNA leakage from the comet head and relatively high cell mortality caused by amino-terminated PPI dendrimers indicated that genotoxicity does not seem to be the main reason of PBMC cell death.
Previously described studies have demonstrated that maltose and maltotriose modification significantly reduced toxicity within the series of PPI dendrimers. The unique property of higher cytotoxicity towards to the moderately doxorubicin and cisplatin resistant human ovarian carcinoma SKOV3 cell line in comparison to the Chinese hamster ovary CHO cell line which does not demonstrate resistance to majority of anticancer agents, made these dendrimers per se potentially interesting for an anticancer therapy.204 Thus, a preliminary evaluation of the clinical value of treating cells of chronic lymphocytic leukemia (CLL) patients with G3 unmodified amino-terminated and maltotriose-modified dense shell PPI dendrimers was carried out. Finding an explanation for the selective toxicity of PPI glycodendrimers towards to cancer cells was essential. Knowing that DNA damage probably is not the main reason leading to PBMCs cells death, an additional study was performed to examine the ability of PPI glycodendrimers to induce changes in mitochondrial membrane potential and apoptotic cell death. Peripheral blood mononuclear cells (PBMCs) collected from untreated CLL patients and healthy donors were used for an in vitro study.204 Chronic lymphocytic leukemia (CLL) was selected since it is the most common leukemia in Europe and North America that usually affects people over 60, but recently is more frequently observed also among younger people. Prognosis of survival time and the course of the disease depend on the type of leukemia. In order to achieve prolonged life of the patients more effective medicines with fewer side effects are sought. Lower cytotoxicity of the PPI glycodendrimer against normal cells and higher against leukemic cancer cells in the concentration range 4–10 mg mL−1 has been demonstrated (Fig. 12). The 24 and 48 hour incubation of leukemic cells with unmodified and modified PPI dendrimers resulted in an increasing number of apoptotic cells along with the higher concentration of the dendrimer and was significantly higher than the percentage of spontaneous apoptotic leukemic cells. Interestingly, the dense shell PPI glycodendrimer (Fig. 12: PPI-DS-Mal-III G3) after 48 hours of incubation induced apoptosis, which is more pronounced than the unmodified PPI dendrimer. The IC50 data confirmed apoptotic action of both analyzed dendrimers which exert significant inhibitory effects on the viability of leukemic cells (Fig. 12). After 48 hour incubation, concentrations of 0.15 and 10 mg mL−1 of the unmodified and dense shell PPI dendrimer, respectively, were sufficient to evaluate the IC50 value, while for normal cells the IC50 value was the same for the unmodified PPI dendrimer and due to low toxicity it was impossible to calculate IC50 for the modified glycodendrimer. The presented results distinctly indicated that the surface modification of PPI G3 dendrimers clearly makes glycodendrimers much more suitable for biomedical applications than unmodified PPI G3 dendrimers.
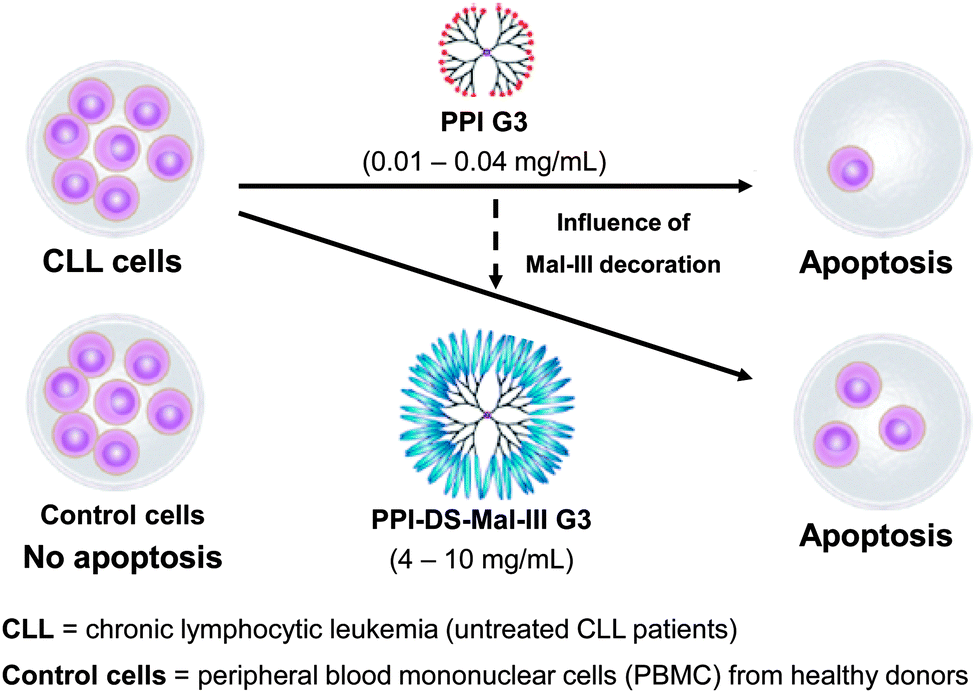 | ||
| Fig. 12 Selective apoptosis of CLL cells and no apoptosis of PBMC control cells by using PPI glycodendrimers in opposite to pure PPI dendrimers which are toxic against normal and cancer cell lines. Reproduced with permission from “I. Franiak-Pietryga, E. Ziolkowska, B. Ziemba, D. Appelhans, B. Voit, M. Szewczyk, J. Gora-Tybor, T. Robak, B. Klajnert and M. Bryszewska, Mol. Pharmaceutics, 2013, 10, 2490–2501”. Copyright 2013 American Chemical Society. | ||
Substitution of terminal amino groups by maltose was another method used for PPI dendrimer modification.178 Dendritic polymers functionalized with the disaccharide maltose, similar to those functionalized with the maltotriose, are non-toxic against normal cells and toxic to several cancer cell lines. Five types of the PPI G3 dendrimers were used: unmodified, maltose-modified open and dense shell and maltotriose-modified open and dense shell. Research was carried out on peripheral blood mononuclear cells (PBMCs) collected from healthy donors and three cancer cell lines: CEM-SS (human T4-lymphoblastoid), U87 (human astroglioma) and MEC1 (B-chronic lymphocytic leukemia). Maltose-modified PPI dendrimers were characterized by lack of toxic activity against normal PBMCs cells, similar to the maltotriose-modified dendrimers, and possessed higher toxicity against all three cancer cell lines: CEM-SS, U87 and MEC1. Among the analyzed glycodendrimers, the maltose-modified open shell PPI dendrimer was the most toxic. Moreover maltotriose- and maltose-modified PPI dendrimers not only reduced cancer cell viability but also induced apoptosis and inhibited their proliferation.
Mechanism of cytotoxicity of unmodified PPI dendrimers is believed to be related to the generation of reactive oxygen species and damage of the mitochondria leading to the cell death due to apoptosis. The obtained results allow us to presume that the mechanism of action and interaction of maltose- and maltotriose-modified dendrimers with the cancer cells might be similar. This observation correlates well with the results described earlier on the mechanism of the toxic effect of PAMAM and PPI dendrimers on human macrophages at the molecular level.205,206
Therefore, identification and understanding of molecular mechanisms of action of glycodendrimers in a tumor cellular environment is so critical especially when their application in antitumor strategies or as diagnostic agents is considered. Toxicity and mechanism of action of two fluorescently labeled open and dense shell PPI-Mal G4 glycodendrimers were tested on several melanoma cell lines (MJS, SK28 and A375).207 Cutaneous melanoma was chosen as one of the most aggressive types of cancer. Prognosis of 5 year survival time depends on the stage of disease and represents 50% for patients with lymph node involvement and 10–20% for patients with distant metastases. The dense shell maltose-modified PPI glycodendrimer was found to internalize in the three different melanoma cell lines more efficiently than in normal cells. Although the viability of cells exposed to increasing concentrations of this glycodendrimer was not lower than 90% up to the concentration of 64 μM. The overall finding was that in all three cancer cell lines glycodendrimers used more than one pathway for their internalization and there was a specific pattern of these pathways for each glycodendrimer in each cell line. For example, only 38% of dense shell glycodendrimer internalized via the non-conventional (non-clathrin, non-cholesterol) pathways in MJS cells, while in SK28 cells 100% of this dendrimer entered as a result of these mechanisms of internalization. The cholesterol-dependent route was found to be the major internalization pathway for open shell glycodendrimer in primary melanoma MJS cells. The most important finding of this study is in fact that PPI glycodendrimers immediately are endocytozed in all cancer lines and are able to cross the cell membrane.
Toxicity in vivo
Most studies of biological properties of PPI glycodendrimers were performed in vitro, and only a few studies have been carried out in vivo. One of them is particularly dedicated to the toxicity of three types of PPI G3 dendrimers: unmodified, open and dense shell maltotriose-modified dendrimers in rats.208 The PPI dendrimers demonstrated dose- and sugar-modification-degree dependent toxicity. As predicted, surface modification results in lowering or completely suppressing the toxic effect of the dendrimer's terminal amino groups, similar to that observed as previously in the case of in vitro studies. A higher dose of unmodified PPI dendrimer caused toxicity, whereas sugar-modified dendrimers revealed minor or lack of toxicity in vivo under any studied concentration. During the animal study, body weight, food and water consumption and urine excretion were analyzed daily. On the 4th, 11th, 25th and 40th day of the experiment, blood from rats was collected to investigate biochemical and hematological parameters such as glucose, creatinine, alanine aminotransferase (AlAT), aspartate aminotransferase (AspAT), amylase, uric acid (UA), white blood cells (WBCs), red blood cells (RBCs), hemoglobin (HGB), hematocrit (HCT), platelets and many others. The condition of the animals was monitored regularly. The open field locomotor activity test was performed on the 4th, 11th, 25th and 40th day of the experiment. In the first experiment, all analyzed dendrimers were orally administered at different doses (1 and 4 mg per kg body weight (b.w.) per day) for 10 days. In the second experiment, glycodendrimers were administered also at a higher dose of 16 mg per kg b.w. per day, whereas unmodified PPI dendrimers, due to their toxicity, were kept at a dose of 4 mg per kg b.w. per day, also for 10 days. Then the treatment was followed by a 30 day recovery period without dendrimer administration.Unmodified PPI dendrimers caused not only changes in the behavior of rats, like a decrease in food and water consumption and lower body weight gain, but also deviation from the standards in hematological and biochemical profiles. However, all disturbances returned to normal levels during the recovery period. Also the side effects observed during treatment with higher doses of the open shell glycodendrimer were not permanent. Probably, this recovery was due to the fact that the dendrimers were excreted via the renal system and did not accumulate in the body for long time. Similar to this in vivo study other in vivo results obtained from open shell G4 (Fig. 7), i.v. administration of mannose- and lactose-modified PPI dendrimers to mice, also indicated that the analyzed nanoparticles accumulated in liver, pancreas, heart, and kidneys but only for a certain length of time and they did not affect these organs by causing irreversible damages or their malfunction.209 Importantly, the unmodified PPI dendrimer at 4 mg per kg b.w. per day dose was four times more toxic to rats than the open shell glycodendrimer at the same dose, whereas the dense shell glycodendrimer was harmless to animals.208 One more time it has been confirmed that surface glyco modification reduces toxicity of the amino-terminated PPI dendrimer, even if only approximately 25% of the amino groups are substituted by maltotriose residues.
As a continuation of the studies on the in vivo effect of unmodified and maltotriose-modified PPI dendrimers, an endogenous level of histamine and spermidine, representatives of biogenic amines and polyamines, upon dendrimer administration has been checked.210 Biogenic amines and polyamines participate in all vital system functions and their levels are important determinants of an organism's condition. Both biogenic amines and polyamines are called local hormones and play a major role in the organism, as they influence all their activities. Histamine is a neurotransmitter but it also plays a crucial role in inflammation processes and in immune responses. Polyamines are involved in cell growth or differentiation. Therefore, even small changes in the level of amines and polyamines are a factor for adverse action of the analyzed compound. It has been checked whether repeated administration of PPI G3, PPI-OS-Mal-III G3, and PPI-DS-Mal-III G3 influences the endogenous level of spermidine, a natural derivative of diaminobutane, and histamine. All analyzed dendrimers were administered at a dose of 4 mg per kg body weight per day for 10 days. Column chromatography on Cellex P, followed by spectrofluorimetric assays of o-phthaldialdehyde-amine condensation products, was employed to analyze tissue spermidine and histamine levels outside the central nervous system, while a radioenzymatic assay was used to measure the histamine level in the brain. A change in histamine concentration, which increased over five times in the small intestine in rats administrated with unmodified PPI dendrimers was most evident, whereas for the modified glycodendrimers all values were similar to the control ones. This enormous increase of the histamine level in small intestine may indicate rapidly developing inflammatory response with infiltration of mast cells and other histamine producing cells caused by the toxic unmodified PPI dendrimer.211 Moreover, this result is in agreement with the observation of high increase of leukocytes found in the unmodified PPI dendrimer-treated rats.208 The level of histamine in the brain decreased only approximately 10% in the case of all analyzed dendrimers. Also changes in spermidine concentration were less distinct than for histamine. Summarizing, a higher dose of the unmodified PPI dendrimer caused changes in biogenic amines content whereas sugar-modified dendrimers revealed minor or lack of influence on the biogenic amine level. Therefore these studies confirmed that the surface glyco-modification significantly reduces toxicity and side effects of in vivo administration of PPI dendrimers.
Additionally, a comparative biodistribution of radiolabelled open shell PPI-OS-Man G4 and PPI-OS-Lac G4, i.v. administrated to female Balb mice, was carried out to evaluate the selective targeting properties of these specific Man- and Gal-containing dendritic glycoconjugates to the liver and lung tissues. Both glycodendrimers were preferentially accumulated in the liver where PPI-OS-Lac, containing the terminal Gal units, showed a slightly higher accumulation rate (∼30% after 6 h) than PPI-OS-Man G4 (∼22% after 6 h). In contrast to this, PPI-OS-Man G4 also accumulated in the kidney with a level of ∼22% after 6 h, while the larger and high-molecular weight PPI-OS-Lac G4 is only nominally captured in the kidney (3.5% after 6 h). Surprisingly, Gal-containing PPI-OS-Lac G4 outlined no real accumulation in the lung (<1%). Accumulation of both glycodendrimers in the liver is explainable by the presence of lectin receptors on the membrane surface of the liver. This biodistribution study also shows us that the requested cyclic conformation of Gal unit in the glycodendrimer PPI-OS-Lac G4 is not a guarantee for a successful selective targeting to the tissue lung. Other unknown (biological) key features of PPI-OS-Lac G4 have to be fulfilled to overcome the biological barrier of lung cells. Finally, the biodistribution study revealed that Man- and Gal-containing dendritic glycoconjugates are usable for selective liver targeting, while naked PPI G4 is preferentially accumulated in the kidney.209
In vivo studies with hyperbranched PEI grafted with oligosaccharides maltose or maltotriose at various degrees (OM-PEIs) are another interesting example of surface modification influencing biocompatibility and changes in pharmacokinetic properties of dendritic macromolecules.56 Overall survival and animal welfare, hepatotoxicity, immune stimulation, erythrocyte aggregation, and the efficacy of DNA delivery in vivo were analyzed. In the experiment, all analyzed polymers were administered at different doses (10, 30 and 100 μg per injection) for 24 and 72 hours to mice. Repeated treatment with higher-degree oligomaltose-grafted PEI (in opposite to non-grafted polymers) caused no weight loss but also reduced lethality and, as it was assessed by serum levels of liver enzymes, eliminated hepatotoxicity. The partially maltotriose-grafted PEI or PEI-based DNA complexes demonstrated dose- and sugar-modification-degree dependent immunostimulatory effects (TNF-α, IFN-γ) and erythrocyte aggregation. In vivo transfection experiments revealed a strong dependence of the OM-PEI architecture on DNA delivery. Summarizing, different patterns of maltose- or maltotriose-grafting on hyperbranched PEI, similar to sugar-modification of PPI dendrimers, also improve both biocompatibility and in vivo efficacy.
Crossing blood–brain-barrier
The biological properties in vivo are closely related to the issue of crossing blood–brain-barrier (BBB). Previous studies have shown the ability of glycodendrimers to cross various biobarriers, namely, PPI glycodendrimers were immediately endocytosed in studied cancer lines and were able to cross cell membranes.207 In addition, PPI glycodendrimers showed selective toxicity against cancer cells.178,198,204 Therefore, the combination of successful crossing of BBB and being potential therapeutic agents would give the chance to develop new therapeutics for brain diseases. A selective BBB is composed principally of specialized capillary endothelial cells fitted with highly restrictive tight junctions. This prevents the passage of therapeutic particles from the blood to the central nervous system (CNS). There are however pathways and mechanism for nanoparticles to cross the BBB that rely on the large surface area of the lipid membranes of the endothelium, transport proteins (carriers), specific receptor- or adsorptive-mediated endocytosis and transcytosis. Nevertheless, most drugs for brain diseases enter the brain via endothelium by adsorptive transcytosis.212Therefore, the next study was devoted to the analysis of the biodistribution of fluorescein-conjugated PPI-OS-Mal-III G3 and PPI-DS-Mal-III G3 in rats and its ability to cross BBB.213 Dendrimers were administrated intraperitoneally once a day, throughout ten days. The dendrimers administrated have shown to be able to enter rat's important organs; moreover their tissue concentration was organ and shell type dependent. The highest amounts of both glycodendrimers were found in liver and kidneys. Accumulation in those tissues after repeated administration was observed despite the fact that three hours after the last injection both dendrimers have not been observed in blood plasma. Agashe et al. also demonstrated that the PPI-OS-Man G4 and PPI-OS-Lac G4 glycodendrimers accumulated in mice's liver and kidneys.209
Quantity of PPI-OS-Mal-III G3 and PPI-DS-Mal-III G3 dendrimers in other tissues did not exceed 4% of a single dose administered to rats, probably due to the rapid excretion by the kidneys. The most important finding of this study was the ability of analyzed glycodendrimers to cross the BBB and to diffuse into the brain. The other prominent result was that cationic open shell PPI glycodendrimer PPI-OS-Mal-III G3 penetrated BBB easier than the PPI dendrimer with neutral dense shell structure (PPI-DS-Mal-III G3). The authors proposed that both glycodendrimers entered the brain via the mechanism of adsorptive transcytosis, which is in good agreement with results obtained by Ku et al.214 PEGylated PAMAM conjugated with fluorescein-doped magnetic silica nanoparticles also penetrated the BBB by the transcytosis of vascular endothelial cells in the absence of destruction by loosening of the endothelial junction or by dissolving the endothelial membrane. Additional TEM study confirmed that the endothelial junctions were still compact and the endothelial membrane was intact.214
In summary PPI glycodendrimers demonstrated the desired low in vitro toxicity109,178,198,199,204,207 and high in vivo biocompatibility.208,210,213 Other dendritic glycopolymers based on PAMAM, Lys or PEI also outlined low in vitro toxicity as found in the case of PPI glycodendrimers. Therefore dendritic glycopolymers based on dendritic polyamine scaffolds can be used as nanomaterials in biomedical applications, since they show a similar strong interaction profile than their cationic dendritic polyamine scaffold but exhibit a much superior biocompatibility. Preferentially their use as a drug delivery system is of high promise, but it will be also interesting to search for other biomedical applications of dendritic glycopolymers in diverse areas such as active compounds in neurodegenerative disorders and inflammatory processes, or for achieving antimicrobial activity.
6. Effects of dendritic glycopolymers in neurodegenerative disease
Prion diseases are fatal neurodegenerative disorders that occur in a variety of mammals. In humans they include Creutzfeldt–Jakob disease (CJD), variant Creutzfeldt–Jakob disease (vCJD), fatal familial insomnia, Gerstmann–Sträussler–Scheinker syndrome, and kuru disease. The diseases occur after conversion of cellular prion protein (PrPC) into a pathogenic, infectious form (PrPSc). PrPSc self-propagates and it aggregates into amyloids. The process leads to rapid neuronal loss and eventually death. Currently no therapy for prion diseases exists. There are however unceasing attempts to find a compound that would be an effective therapeutic agent. Preventing the conversion of PrPC into PrPSc and clearance of PrPSc are two basic therapeutic strategies that are considered. Dendrimers join in a group of compounds that are potentially promising in curing prion disorders. Superfect, a commercially available dendritic structure used as a transfection agent, cleared PrPSc forms in infected neuroblastoma cells.215 This finding motivated further tests of other types of dendrimers. Cationic dendrimers (PAMAM and PPI) were the most potent, whereas neutral hydroxy-terminated PAMAM dendrimers had only minor effects. Therefore electrostatic interactions between charged amino acids and charged surface groups were postulated to be the main forces of these interactions. The same dendrimers, which effectively cleared PrPSc, were known previously from interacting strongly with other proteins.216 Since maltose-coated PPI dendrimers have shown to maintain the ability to interact with proteins, further investigations focused on their influence on the process of fibril formation by a prion peptide PrP 185–208. Fibrillation of this peptide was chosen as a model of the amyloidogenic process. It was demonstrated that PPI-DS-Mal G1, PPI-DS-Mal G2, and PPI-DS-Mal G3 at higher concentrations prevented fibril formation. On the contrary, lower concentrations accelerated the fibril formation process. The proposed mechanism is that dendrimers break the formed fibrils in a different way depending on the concentration. If the process of breakage runs slowly, as in the case of low doses of maltose-modified PPI, new ends can be created, which are then extended and form new fibrils. On the other hand, when the breakage of fibrils is fast, as it is in the case of high concentrations of dendrimers, all fibrils are destroyed to monomers. The last process is obviously desirable.109 However, speeding up the process of fibril formation can also have a protective effect, since short fragments, called protofibrils, were shown to be most toxic.217 Other possible mechanisms of fibril formation inhibition involve binding of peptide monomers or blocking of fibril ends by dendrimers which prevents fibril extension (Fig. 13). The mechanism of breaking fibrils by sugar-modified dendrimers (PPI-DS-Mal G4 and PPI-DS-Mal III G4) was further confirmed by EPR studies.112 Performing experiments, that were analogous to the first attempts by Supattapone et al.,215 was the next logical step in studying neutral PPI glycodendrimers. It turned out that the sugar modification of the surface groups did not abolish the antiprion activity. PPI-DS-Mal G2, PPI-DS-Mal G3, PPI-OS-Mal G4, PPI-DS-Mal G4, and PPI-DS-Mal-III G4 effectively reduced the level of PrPSc in infected ScN2a cells.110 Moreover they cleared the pre-existing aggregates in homogenates from infected mice brains. It has been postulated that dendrimers mediate in the denaturation of PrPSc. Elimination of PrPSc from brain homogenates was earlier observed e.g. for cationic phosphorus dendrimers,218 but the finding that cationic surface groups are not essential for anti-prion activity is important from the toxicological point of view. It has been demonstrated that not only cationic polymers, but also non-toxic glycodendrimers can inhibit the prion infection. However, each type of dendrimers reduces PrPSc in a prion strain dependent manner. Dendrimers with cationic surface groups (PPI G3, PAMAM G5 and PPI-OS-Mal G4) are more potent against a wider range of prion strains than PPI-DS-Mal G4.113 Strain-specific properties are probably governed by PrPSc conformation and the glycosylation pattern that differs between strains. It makes dendrimers a potential diagnostic tool in differentiating between protein strains (Fig. 14). Interestingly, anionic glycodendrimers with sulfate groups on the surface are also able to reduce the level of PrPSc in a prion strain-dependent manner.114 Here, cationic dendrimers may interact with negatively charged groups of PrPSc, while anionic dendrimers can interact with pockets of cationic charges. Earlier similar phenomena were found for the interactions of bovine serum albumin with anionic and cationic PAMAM dendrimers.219 It seems that the density of surface groups is more important than the charge. The ability to interact with PrPSc increases when the surface groups are densely packed. McCarthy et al. explored in a detailed manner the mechanism of anti-prion activity of PPI-DS-Mal G4.115 This dendrimer inhibits conversion from PrPC to PrPSc in dendrimer-pre-treated prion strains infected N2a cells. Several pathways can be involved in this: interfering with short-lived intermediates of the conversion, disturbing PrPSc trafficking, and altering PrPSc structure so it is not capable to initiate PrPC misfolding. Interestingly, PPI-DS-Mal G4 does not interact directly with PrPC within the cells. This is considered to be a positive result, since the PrPC role is not fully understood yet, so no detectable effect on PrPC means that the dendrimer can stop formation of PrPSc with minimum toxicity to the cell.116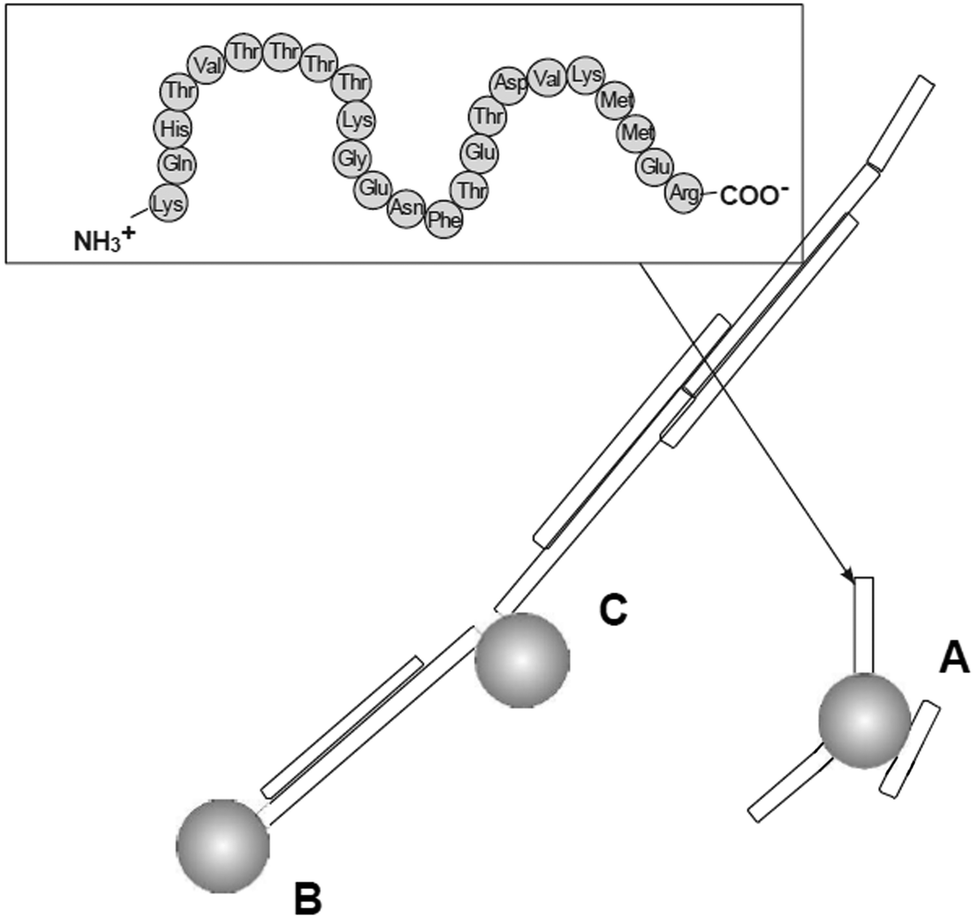 | ||
| Fig. 13 Three main mechanisms of possible anti-amyloid activity of dendrimers: (A) interaction with peptide monomer, (B) blocking fibril ends, (C) breaking fibrils. Chemical structure of the PrP 185–208 peptide is shown in the frame (grey spheres are the dendrimers).109 The disaggregation process is strongly depending on the molar ratio of amyloidogenic peptide and PPI glycodendrimer to fabricate different morphologies of aggregates (Fig. 15). Reproduced from ref. 109 with permission from Wiley-VCH. | ||
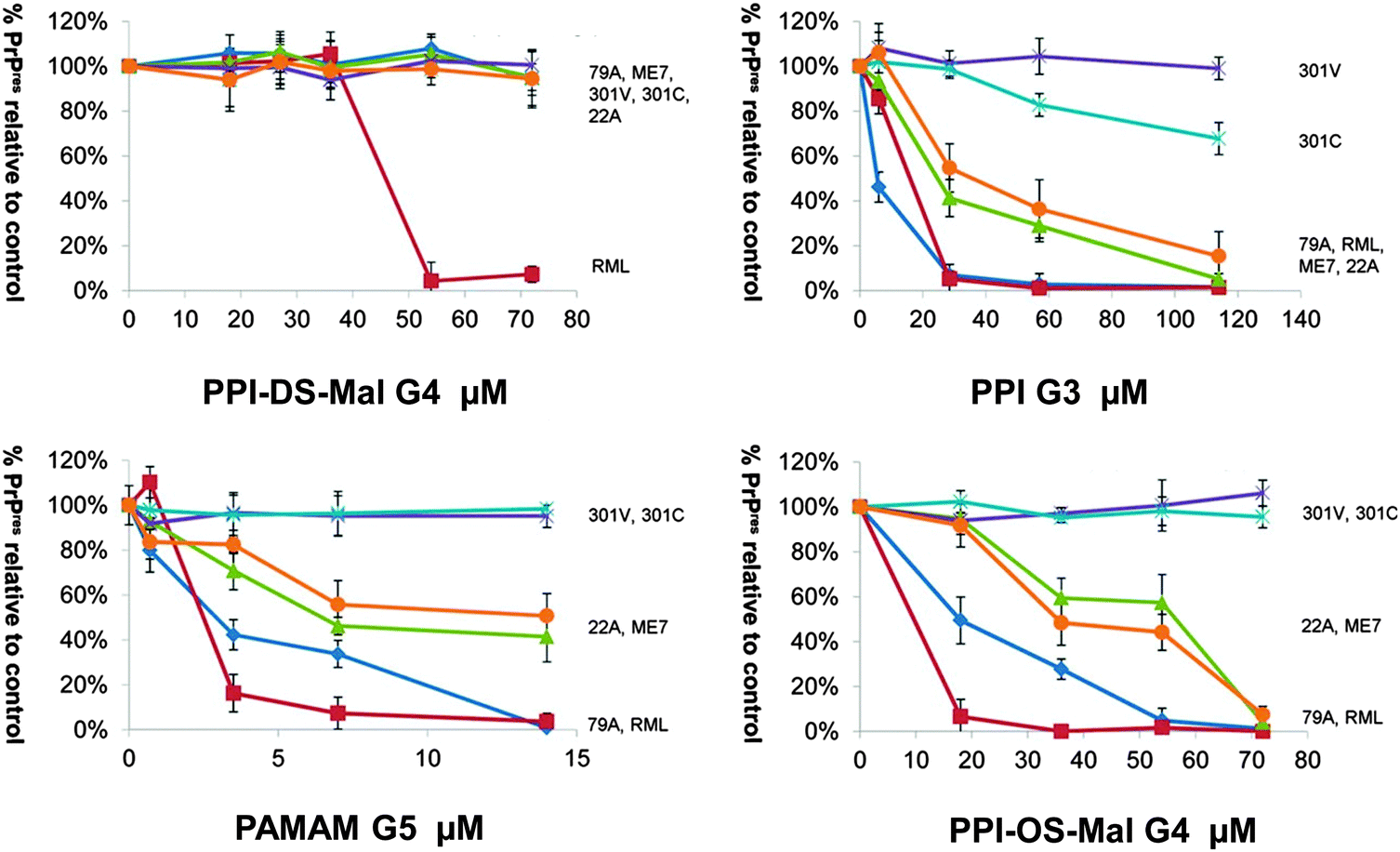 | ||
Fig. 14 Differentiation of prion strains – immunoblot images of dendrimer treated brain homogenates were analyzed by densitometry for % PrPres levels which remained after dendrimer treatment and protease digestion, calculated relative to a non dendrimer treated control. Legend:  79A; 79A;  RML; RML;  ME7; ME7;  301V; 301V;  301C; 301C;  22A. Error bars represent SD; n = 2 biological repeats. * Statistically significant difference between prion strains at the concentrations indicated (p < 0.01). The mean levels of PrPres remaining for each prion strain after treatment with 72 μM PPI-DS-Mal G4, 114 μM PPI G3, 14 μM PAMAM G5 or 54 μM PPI-OS-Mal G4 were compared using a one-way ANOVA. Prion strains 79A, RML, 301V and 301C can be statistically differentiated. 22A and ME7 can be differentiated from the other prion strains but not from one another. Reproduced from ref. 113 with permission from Wiley-VCH. 22A. Error bars represent SD; n = 2 biological repeats. * Statistically significant difference between prion strains at the concentrations indicated (p < 0.01). The mean levels of PrPres remaining for each prion strain after treatment with 72 μM PPI-DS-Mal G4, 114 μM PPI G3, 14 μM PAMAM G5 or 54 μM PPI-OS-Mal G4 were compared using a one-way ANOVA. Prion strains 79A, RML, 301V and 301C can be statistically differentiated. 22A and ME7 can be differentiated from the other prion strains but not from one another. Reproduced from ref. 113 with permission from Wiley-VCH. | ||
The therapeutic potential of dendrimers is not limited to prion diseases. Dendrimers have been shown to possess an in vitro effect on Aβ peptides. These peptides are the main components of fibrillar amyloid plaques found in brains of individuals suffering from Alzheimer's disease. Various dendrimers (PAMAM, PPI, phosphorus) inhibited in vitro fibrilization of a fragment of Aβ (Aβ(1-28)) and reduced toxicity caused by intermediate oligomeric species.188,220,221 PPI-DS-Mal G3 and PPI-DS-Mal G4 dendrimers were proved to interfere with the fibrillization of Aβ(1-40). However, the mode of the action was generation-dependent.111 PPI-DS-Mal G4 blocked fibril formation by generating amorphous aggregates, whereas PPI-DS-Mal G3 generated clumped fibrils (Fig. 15). These two modes of the action had serious consequences on the toxicity of the final product. Amorphous aggregates were found to be toxic, while clumped fibrils are non-toxic.
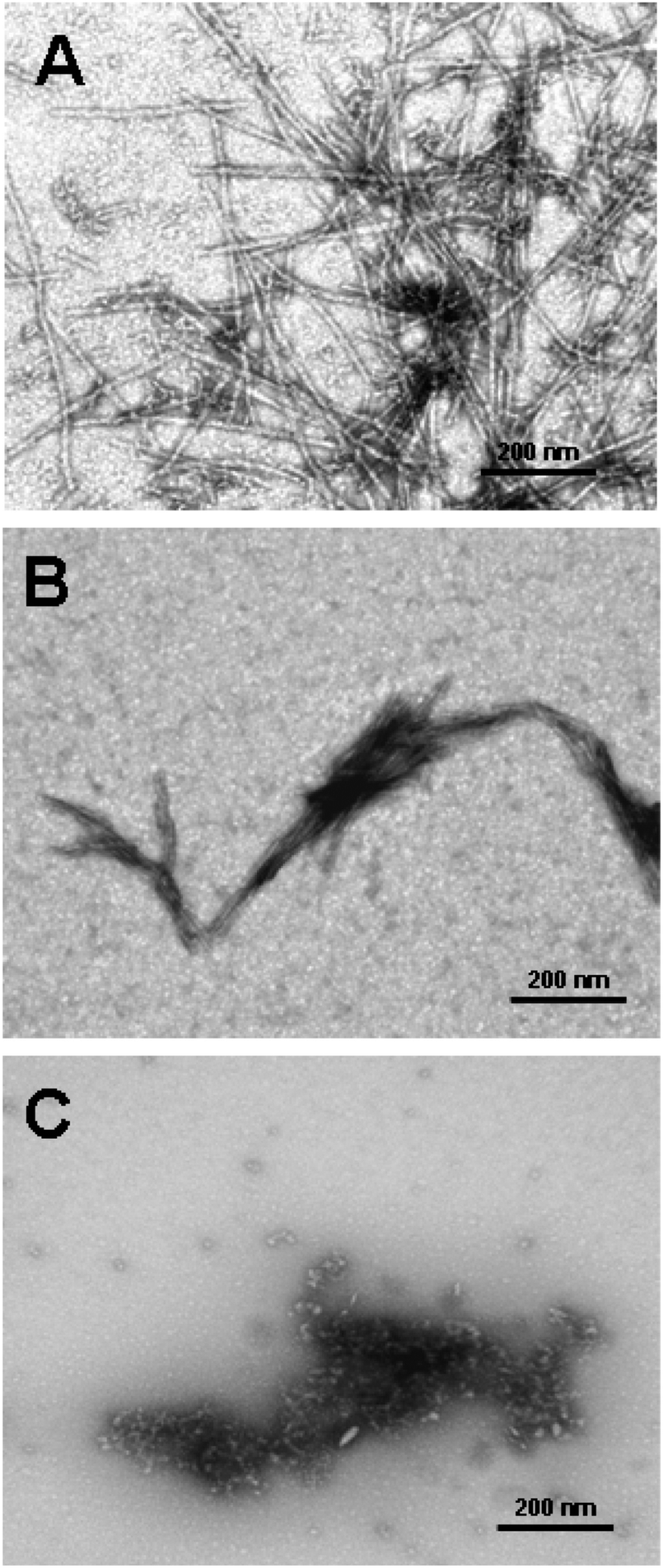 | ||
| Fig. 15 Transmission electron micrographs of: Aβ(1-40) incubated at pH 7.4 (coexistence of fibrils and globular oligomers) (A); Aβ(1-40) icubated at pH 7.4 in the presence of minor PPI-DS-Mal G3 (detection of clumped fibrils) (B); Aβ(1-40) incubated at pH 7.4 in the presence of PPI-DS-Mal G4 (dendrimer–peptide ratio 1, detection of granular non-fibrilar, amorphous aggregates) (C). Aβ(1-40) and Aβ(1-40)–dendrimer complexes were incubated for 12 hours at pH 7.4 and 37 °C before the preparation of the microscopy grids. Reproduced with permission from “O. Klementieva, N. Benseny-Cases, A. Gella, D. Appelhans, B. Voit and J. Cladera, Biomacromolecules, 2011, 12, 3903–3909”. Copyright 2011 American Chemical Society. | ||
The first and till now only in vivo study that reported both the phenomena of crossing the blood-brain-barrier and potential applicability of PPI glycodendrimers as candidates for antiamyloidogenic agents was done by Klementieva et al.118 They used a wide panel of glycodendrimers: PPI-DS-Mal G3, PPI-DS-Mal G4, PPI-OS-Mal G3, and PPI-OS-Mal-III G3. Influence of analyzed glycodendrimers on the cytotoxicity induced by Aβ(1-42) and human brain extracted Aβ peptides was tested in a human neuroblastoma cell line (SH-SY5Y). In the animal study dendrimers were administrated once a day, for five or ten days in the case of short term experiments and for one month in the case of a long-term experiment, in doses of 10 mg per kg b.w per day and 1 mg per kg b.w per day of open and dense shell glycodendrimers, respectively. As expected maltose-modified PPI dendrimers were able to reduce the toxicity of Aβ(1-42). Nevertheless, only dense shell maltose-modified dendrimers of both generations reduced the toxicity of Alzheimer's disease brain extracts in SH-SY5Y neuroblastoma cells. Moreover, at low concentrations those dendrimers possessed capacity to accelerate Aβ(1-42) peptide fibril formation. The analyzed glycodendrimers were able to cross not only cell membranes, but also the BBB. Moreover, both open and dense shell maltose-modified PPI glycodendrimers were able to modify the Aβ profile of APP/PS1 mice (rodent model of Alzheimer's disease). The open shell PPI glycodendrimer was found to be harmful for mice under chronic administration and caused cognitive decline in nontransgenic mice, while dense shell PPI glycodendrimers revealed positive effects which will promote further studies with these dense shell entities in the future.
7. Dendritic glycopolymers as drug delivery systems
Nowadays dendritic polymers are one of the promising and tunable nanomaterials222–226 for therapeutics and diagnostics. Especially, the advantageous (physicochemical) key features of dendritic polymers in drug delivery are well documented in several papers.222,223,226–230 Some specific features of dendrimers that make them highly attractive for in vitro and in vivo delivery of drugs are: tunable surface charges,231 ability to cross biological barriers,232–235 high water-solubility, ability to enhance solubility of poorly water-soluble drugs,232 high loading capacity of drugs,223 stability and biocompatibility and tailored and fine-tuned pharmacokinetics and pharmacodynamics achieved by a suitable design of the molecular architectures.236–239 Further specific dendrimer features for successful biological actions are nano-sized dimensions suitable for the EPR effect (enhanced permeability and retention effect) along leaky blood vessel, e.g., to accumulate in tumors = passive targeting,223,226 active targeting capability by introducing ligand for interacting with over-expressed receptor targets,232,240–243 possibility of different kind of administration forms,244 minimizing drug's degradability,222,244 and others. Overall, the molecular composition inside and outside of the dendritic scaffold and the huge number of functional surface groups of dendritic polymers are mainly responsible for their physicochemical and biological features.Recent progress in dendritic glycopolymers as drug delivery systems will be briefly highlighted here. But firstly the general complexation/interaction properties of dendritic glycopolymers against various analyte and drug molecules are viewed to get further insight into the structure–property relationship of dendritic glycopolymers based on dendritic polyamine scaffolds.
Generally, the interaction and complexation properties of (oligo-)saccharide-modified dendritic polyamines10,54–59,62–75,76–78,84,89,91,92,109,181–183 are mainly tailored by their pH-dependent cationic (surface) charges to undergo ionic interactions with various anionic low- and high-molecular weight drugs (e.g. dye molecules, ATP, si-RNA, DNA). Here, two tendencies are recognizable. Dendritic glycopolymers based on PAMAM dendrimers and PEI scaffolds are preferentially used for the formation of (defined) polyplexes using various RNA and DNA macromolecules,54–59,62–75,89,91,92 while dendritic glycoarchitectures based on PPI dendrimers are mainly used for the complexation of (non-)charged low-molecular weight analytes and drugs.77–84,109,183,245
Complexation and interaction of dendritic glycopolymers with low-molecular weight compounds
A closer view on the complexation properties of PPI glycodendrimers outlines the following issues. First, dense shell PPI glycodendrimers possess very limited ionic interaction properties against small anionic analytes.109,183,245 Thus, a low number of ANS molecules can be complexed by PPI glycodendrimers,109 while ATP and Mant-ATP molecules do not undergo any interactions with these dendritic glycoarchitectures (Fig. 11).246 These anionic nucleic acids are too bulky to drain the dense shell of PPI dendrimers in comparison to smaller anionic ANS molecules. In contrast to this, very small anionic Re clusters (≤1 nm) are complexed by dense shell PPI glycodendrimers.247 Second, cationic open shell PPI glycodendrimers are able to complex anionic nucleic acids, ATP, ADP, AMP, fludarabine and Mant-ATP, at various conditions, and form stable complexes.183,245 Therefore, complexation of the nucleic acids mainly depends on pH and NaCl content.183,245 Complexes with ATP are not degradable by alkaline phosphatase, when using G4 of the PPI glycodendrimer.245 The open shell in PPI glycodendrimers has here the function of a protective shell in the presence of enzymes.Finally, the group of Jain focused on the complexation and interaction of (very) hydrophobic and less water-soluble drugs where ionic interactions of open shell PPI glycodendrimers78,84 play a minor role (Fig. 16).78–84Open shell PPI glycodendrimers are able to (slightly) increase the complexation of those drugs caused by the presence of the saccharide shell and to induce a better sustained release of drugs for several hours or days than their parental counterparts.78–84 Various surface-modified PPI dendrimers (shell (partially) decorated with PEG,248,249 lactoferrin,250 polysorbate 80,251 thiamine,252 luteinizing hormone,253 lipoproteins,254 arginine255 or tuftsin as cell-penetrating peptide256) were also successfully tested for sustained release of poor water-soluble drugs (e.g. doxorubicin, docetaxel, paclitaxel, famotidine, primaquine). Again, the additional surface modification of PPI dendrimers also induces better shielding/densification and (partly) back-folding properties of surface groups to explain sustained release of drugs.248–256 Furthermore, ionic interactions are in minority248–250 and the formation of partially aggregated but still under physiological conditions soluble complexes251–255 is responsible for their positive use in biological applications. Knowing that ionic interactions are the most favorable molecular action of parental PPI dendrimers against drugs,222 while hydrophobic drugs' (indomethacin, famotidine, amphotericin B) complexation by the parental PPI dendrimer are tailored by pH and functional groups of hydrophobes,257 similar interaction features for the open shell PPI glycodendrimers (Fig. 7) can be assumed.78–84 Surprisingly, complexation towards low-molecular weight compounds of other open shell glycodendrimers, e.g. based on PAMAM and the Lys dendrimer core, are rarely encountered. The galactose-modified Lys dendrimer is able to better complex chloroquine phosphate than the un-modified Lys dendrimer.258 Similar complexation behavior for chloroquine phosphate was observed when polyelectrolyte complexes, consisting of a Lys dendrimer and anionic chondronitin sulfate, were used.259Open shell PAMAM glycodendrimers are also capable of ANS complexation.260
 | ||
| Fig. 16 Drugs used in in vitro and in vivo studies, stabilized by open shell PPI glycodendrimer. | ||
Further insights into the complexation and interaction properties of core–shell glycoarchitectures, based on PEI cores, against several vitamins (B1–B3, B6 and B12) and drugs (ATP, pantoprazole and acid-containing estradiol derivative) were recently obtained in pure water.10,182 In this study it was found that molecular interactions between core–shell glycoarchitectures and analytes are mainly tailored by the interaction characteristics of the analytes themselves. These analyte molecules have to match the pronounced ionic interaction characteristics of cationic PEI core–shell structures, otherwise no or low interactions are present between analyte molecules and the dendritic glycopolymer.181,182 Thus, moderate and strong interactions are only given when anionic vitamins182 and drugs10,182 (B3, B12, pantoprazole, acid-containing estradiol derivative and ATP) are mixed with cationic PEI core–shell glycoarchitectures. Moreover, it was found that open and dense shell glycoarchitecture will facilitate the interaction between anionic analytes and cationic PEI scaffold compared to the non-modified parental PEI scaffold, while the glyco shell itself does not contribute to any preferential kind of molecular interactions (e.g. H-bonds) against analyte molecules.182 In addition cationic, amphiphilic and neutral vitamins do not undergo any significant interactions with cationic PEI core–shell glycoarchitectures.
Generally, dendritic glycopolymers stabilize low-molecular-weight drugs better in various solutions and allow for a more sustained release compared to the parental dendritic scaffolds. From these results one cannot distinguish between drugs that are completely encapsulated by dendritic glycoarchitectures as postulated for other dendritic scaffolds222,223 and aggregates consisting of dendrimer-complexed drugs. In the following the term “dendritic glycopolymer-stabilized drugs” will be used to describe the glycopolymer drug delivery systems not distinguishing between drugs located within the dendritic scaffold, those located more in the shell and larger dendrimer/drug aggregates.182,222
Complexation and interaction of dendritic glycopolymers with high-molecular weight compounds
Parental PPI dendrimers have been shown to interact with genetic material forming dendriplexes of different characteristics depending on the generation of the dendrimers and the type of the oligonucleotides. Since one of the potential anti-HIV treatments is gene therapy, interactions between PPI dendrimers and anti-HIV oligonucleotides, as well as properties of formed complexes have been investigated in detail.261 PPI G1, PPI G2 and PPI G3 formed complexes with SREV, ANTI-TAR, and GEM91 anti-HIV oligonucleotides (ODNs) at different molar ratios as assessed by fluorescence polarization. With increasing generation number of PPI dendrimers lower amount of PPI dendrimers is needed to undergo desired formation of stable complexes with ODNs (e.g. 15–20 of PPI G3 for one ODN). These differences could result from different sizes and features of dendrimers (diameters, molecular weight, number of surface charges, shapes). The most effective binding with PPI dendrimers was achieved with SREV, while GEM91 was characterized by the worst binding efficiency. The lengths of oligonucleotides do not play a significant role in interactions with dendrimers. It is, however, likely that nucleotide sequence and the ability to form secondary structures may have an influence on the binding efficiency of ODNs with PPI. Zeta potentials of saturated complexes were slightly negative for PPI G2 and PPI G3 or close to neutral for PPI G1. It is generally assumed that cationic compounds permeate cell membrane more efficiently than neutral or negatively charged compounds.262 Nevertheless, it is possible that the positive surface charge is not the main decisive factor of successful internalization and transfection, since commercially available transfection reagents such as Lipofectin and Lipofectamine are also characterized by negative zeta potentials. Hydrodynamic diameters for all measured PPI complexes amounted to approximately 250 nm, which is an appropriate size for transfection. Studies on dendriplexes morphology using TEM revealed the presence of big aggregates (even up to 800 nm), which may, however, be a result of a sample preparation procedure.261Dendriplexes, composed of the same ODNs and open shell PPI G3 dendrimers modified with maltose (PPI-OS-Mal G3) and maltotriose (PPI-OS-Mal-III G3), have also been examined. In this case one molecule of ODNs bound 4–5 molecules of dendrimers. These dendriplexes were also shown to be stable for more than 20 hours. The hydrodynamic diameter of dendrimers alone was 5.7 and 6 nm for PPI-OS-Mal G3 and PPI-OS-Mal-III G3, respectively. The biggest dendriplexes were formed by both dendrimers with SREV and PPI-Mal G3 with AT and GEM91 (150–200 nm). PPI-Mal-III G3 with AT and GEM91 generated smaller structures of a diameter of 50–100 nm. The size of complexes was independent of the length of ODNs and relayed to a minor degree on the dendrimer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ODNs molar ratio. The zeta potential after addition of dendrimers increased from about 24 mV to about (−18) to (−14) mV for all tested complexes. Detailed analysis of the morphology of dendriplexes revealed that PPI-OS-Mal G3 and PPI-OS-Mal-III G3 form rod-like structures in complex with GEM91. It is suggested that these nanorod structures are formed by a one-directional self-assembling process. On the other hand, PPI-OS-Mal G3/AT dendriplexes existed as monomeric units as well as long (up to 1 μm) fibrils. Similar results were observed for PPI-OS-Mal-III G3/AT. Both dendrimers, complexed with SREV, formed another type of structure, namely, 3D square-like nanostructures. It is assumed that in this case the self-assembling process is oriented in three axes. These data correlated with molecular modelling, corroborating formation of described nanostructures.263
:ODNs molar ratio. The zeta potential after addition of dendrimers increased from about 24 mV to about (−18) to (−14) mV for all tested complexes. Detailed analysis of the morphology of dendriplexes revealed that PPI-OS-Mal G3 and PPI-OS-Mal-III G3 form rod-like structures in complex with GEM91. It is suggested that these nanorod structures are formed by a one-directional self-assembling process. On the other hand, PPI-OS-Mal G3/AT dendriplexes existed as monomeric units as well as long (up to 1 μm) fibrils. Similar results were observed for PPI-OS-Mal-III G3/AT. Both dendrimers, complexed with SREV, formed another type of structure, namely, 3D square-like nanostructures. It is assumed that in this case the self-assembling process is oriented in three axes. These data correlated with molecular modelling, corroborating formation of described nanostructures.263
Drzewinska et al. showed that complexation of all three ODNs with naked PPI, PPI-OS-Mal G3 and PPI-OS-Mal-III G3 protected the oligonucleotides against nucleases contained in cell medium FBS. Furthermore, it was demonstrated that the surface oligosaccharide shell of PPI is necessary for the prevention of ODN hydrolysis by endonuclease S1. This implies that the sugar layer surrounding the dendrimer poses additional “shielding” isolating ODNs from endonuclease S1 degradation. But none of the studied dendrimers protected ODNs against digestion at low pH (∼4.5).264
One of the important aspects of nucleic acids delivery to cells by dendrimers is their interaction with glucosaminoglycans (GAGs). GAGs are components of the extracellular matrix. Due to their negative charge, they can displace oligonucleotide cargo from dendriplexes, thus seriously affecting the transfection process. For this reason, Szewczyk et al.265 studied interactions between complexes formed by PPI G3 (naked, maltose- and maltotriose-modified) and anti-HIV ODNs (AT, SREV and GEM91) and four GAGs (heparin, heparan sulfate, chondroitin sulfate, and hyaluronic acid). Addition of heparin, possessing the highest negative charge of all GAGs, to unmodified PPI–ODNs complexes had no or only a slight effect up to 10 μg mL−1 as assessed by the fluorescence polarization method. Larger concentrations, however, led to a pronounced decrease in fluorescence polarization, down to the value of free ODNs (at the concentration of 20 μg mL−1), indicating a complete dissociation of complexes. In the case of studied glycodendrimers, application of heparin resulted in gradual decomposition of dendriplexes up to the concentration of 2–3 μg mL−1. The different behavior of naked PPI and sugar-modified dendrimers in the presence of heparin may result from several factors. First, uncoated PPI, possessing more positive charges, bind ODNs stronger than the PPI glycodendrimer. Second, maltose and maltotriose units may affect the interaction properties of the cationic scaffold in the glycodendrimer, and last, glycodendrimer–ODNs complexes were formed at lower molar ratios in comparison to dendriplexes with naked PPI. Heparan sulfate also caused a progressive decrease in fluorescence polarization (complexes breakdown) but the value of free ODNs was reached only at about 500 μg mL−1. In contrast to heparin and heparan sulfate, chondroitin sulfate and hyaluronic acid had no or minor influence on the stability of tested dendriplexes. These results were corroborated by electrophoretic mobility studies of ODNs bound to PPI in the absence and presence of GAGs. The obtained results indicate that under physiological conditions only dendriplexes formed by oligosaccharide-modified PPI and ODNs are destroyed by heparin, while naked PPI–ODNs complexes are not affected. In future transfection experiments interaction of dendriplexes with GAGs can lead to effects which negatively influence the transfection process. First, GAGs can lead to disassociation of complexes (in the presence of heparin), which can result in ODNs destruction. Second, GAGs change size and charge of dendriplexes, which can hamper internalization and trafficking of complexes. Third, association with GAGs can modify the intracellular fate of dendriplexes. Thus, finding a perfect delivery platform is a challenging task. Considering that sugar-coated dendrimers possess better protective properties against nucleases and exhibit lower cytotoxicity198,208 than naked PPI, they can be considered as better candidates for drug delivery platforms. On the other hand, while naked PPI–ODNs complexes are unaffected by physiological concentrations of heparin in the absence of cell medium, glycodendrimer–ODNs complexes undergo dissociation in the presence of these GAGs.265
Dendritic glycoconjugates for targeted drug delivery
Recent developments are directed to establish a dendritic nanocarrier with targeting ligands222,223,226,266 to reduce adverse effects of drugs during treatment. For this purpose various different biological ligands (e.g. folic acid, RGD peptide, various (oligo-)saccharides, polysorbate 80, methotrexate, lactoferrin, transferrin, thiamine, antibody and cell-penetrating peptides) were chemically and biologically connected to various dendritic scaffolds, but also to linear polymers267 and nanoparticles.268 Similarly, also glycosylated carrier conjugates269 with targeting moieties address all therapeutic applications as known from non-glycosylated carrier systems (cancer therapy, organ imaging, photodynamic therapy vaccine delivery and delivery of therapeutic agents etc.). It was shown that glycosylated nanocarriers are able to undergo site specific delivery and present an alternative way to administrate drugs into specific cells and organs.269Following the above considerations, open shell PPI glycodendrimers are the preferential molecular entities to solubilize and deliver various low molecular-weight drugs to specific biological entities (cells or organs).78–84 To facilitate the penetration of various biological barriers, e.g., membranes of cancer cells and macrophages, dendritic PPI scaffolds were modified with mannose, galactose and sialic acids as targeting ligands. The main application of the dendrimer-stabilized drugs78,80–84 (Fig. 16) was directed to prevent infection of human immunodeficiency virus (HIV),80,81,83 malaria,78 and tuberculosis79 or to suppress inflammation processes,84 for example, in liver.
T-lymphocytes, dendritic cells, monocytes, and macrophages are the target cells for the general attack of HIV. Monocytes, macrophages and dendritic cells are generally considered as depots to distribute the HIV in human, but also, for example, other viruses. Thus, for the treatment of virus infections any therapeutic agent has to be administrated to these cells in addition to other organs like the liver, lung or brain. Contrary to the anti-adhesive glycodendrimers which block the receptors of macrophages or dendritic cells for the virus uptake/entrance (Fig. 2 and 4), the same or other sugar receptors (mannose, galactose, sialic acid and oligomannose) and further receptors on macrophages and dendritic cells will be used for the targeted uptake of dendritic glycopolymer-stabilized anti-HIV drugs (efavirenz, lamivudine and zidovudine)80,81,83 (Fig. 16). In all cases better cellular uptake of the drug by dendritic glycopolymer-stabilized drugs was achieved in comparison to other control systems (parental PPI dendrimer or other PPI dendrimer derivatives). Furthermore, cell viability of macrophages after the uptake of dendritic glycopolymer-stabilized drugs was also not impaired for at least 48 h. Biological activity of the anti-HIV drug lamivudine, considered in its active triphosphate derivative as a nucleoside analogue reverse transcriptase inhibitor, was successfully proven when using the open shell mannose-decorated PPI dendrimer. In the recent biodistribution study of Gajbhiye et al.83 it was explicitly shown that only the coincidental decoration of mannose and sialic acid on the PPI dendrimer surface guarantees an enhanced accumulation of the anti-HIV drug zidovudine, also acting as a nucleoside analogue reverse transcriptase inhibitor, in the lymph node, while the cationic sialic acid- or mannose-decorated PPI dendrimers outline a reduced uptake of zidovudine of about one third and less in lymph nodes as found in the cases of mannose- and sialic acid decorated PPI dendrimers. The motivation of this study was to show that both mannose and sialic acid units may induce a dual targeting profile recognized by the sugar receptors of lymph nodes. Besides this, galactose-78 and fucose-modified84 PPI dendrimers were successfully used to enhance the targeting of the organ liver for the administration of the drugs primaquine phosphate and sulfasalazine, respectively. This biological behavior is preferentially achieved by the prolonged blood circulation of dendritic glycopolymer-stabilized drugs.
However, the previous use of dense shell PPI glycodendrimers and PEI glycoarchitectures as a drug delivery system for low molecular-weight drugs only focused on the delivery of HIV-derived antigens to immature and mature dendritic cells (DC).60,61,76,77 The molecular uptake of drugs and carrier systems into immature and mature DC is hampered by the biological membrane of DC, while the cellular uptake of small and larger molecules is still facilitated in the case of monocytes.61 To overcome the non-interacting properties of dense shell PPI glycodendrimers against ATP molecules246 (Fig. 11) few PEG-spacered amino groups have been introduced in the outer shell of the dendritic glycoarchitecture to undergo the desired electrolyte complexation with anionic HIV-derived peptides (Nef, Gp160 and P24).77 This forced molecular interaction on the cell membrane surface of immature DC allowed the capturing of anionic peptides in DC when excess of modified dense shell PPI glycodendrimers was used, while the administration of pure anionic peptides resulted in a low uptake of those peptides in DC. A similar concept was used for dense shell PEI glycoarchitectures.60,61 Here, the conjugation of HIV-derived peptides was realized by the conjugation of a spacered Ni(II)–NTA-His6-tag (Fig. 17) where the pH-sensitive conjugation unit can be smoothly destroyed ≤pH 6 (ref. 60) to release those peptides in the endosome or lysosome of DC.61 A similar molecular uptake mechanism in immature DC was found as in the case of PEG-spacered amino group modified dense shell PPI glycodendrimers.77 An excess molar ratio of Ni(II)–NTA modified dense shell PEI glycoarchitectures against His6-tagged anionic peptide Gp160 is needed to capture anionic peptide in DC.61 Moreover, both dendritic glycopolymer architectures, upon being captured in immature DC, do not hamper the maturation process of immature DC and their migration properties, and thus, these glycopolymers may be suitable materials for future DC-based immunotherapy.61,77
 | ||
| Fig. 17 Schematic illustration of Ni(II)–NTA-spacered dendritic glycopolymer (Ni(II)–NTA–DGP) interactions with HIV-derived peptide His6-Gp160. Reproduced with permission from “N. Hauptmann, M. Pion, R. Wehner, M.-A. Muñoz-Fernández, M. Schmitz, B. Voit and D. Appelhans, Biomacromolecules, 2014, 15, 957–967”. Copyright 2014 American Chemical Society. | ||
Finally, PPI dendrimer conjugated dextran nanomaterials were successfully used as potential vectors for the delivery of the anti-cancer agent, doxorubicin hydrochloride, to lung epithelial cancer cells in vitro.82 Moreover, prolonged blood circulation and enhanced accumulation in tumor tissues, triggered by the EPR effect, of nanoparticle-stabilized drug were also determined.82
Summarizing the previously described results, various dendritic glycopolymer-stabilized drugs with and without targeting properties have been established over the last years.77–84 Interestingly, the application of these dendritic glycoconjugates is mainly directed to address the treatment of infectious diseases induced by bacteria and viruses.
A future challenge is a better control over the number of drug molecules in those nanocarriers. This may be solved by the chemical coupling of drugs, for example, to the dendritic scaffold or to the sugar units. Zhang et al.90 combined several advantageous strategies in a ligand-mediated drug delivery system. Besides using a biocompatible dendritic glycopolymer-conjugated drug material, the drug methotrexate was selectively conjugated to sugar units via ester bond formation, while the conjugated drug methotrexate itself also takes over the function of folic acid enhancing the binding avidity to folate-binding proteins on the surface of KB cancer cells. The molecular composition of methotrexate is similar to the folic acid.90 These dendritic glyco-drug conjugates show the potential as anti-cancer nanocarriers for the specific targeting and killing of folate receptor-expressing tumor cells.
Finally, one can state that dendritic glycopolymers, preferentially based on PPI dendrimer cores, are attractive, biocompatible alternative drug delivery systems with (highly) adaptable properties against cells and organs in comparison to other surface-engineered PPI dendrimer nanocarriers.248–256
Dendritic glycoconjugates as targeting non-viral vectors
Some sugar-decorated dendritic polyamine scaffolds usable as non-viral vectors of DNA and RNA are also characterized by selective targeting properties against cells and organs.54–59,62–75,85–89,90,92 Two dendritic polyamine scaffolds, PAMAM dendrimers and hyperbranched PEI, have attracted the highest attention in this specific application field.A closer view on the work of Arima and Uekama reveals an impressive interplay of the molecular composition of sugar-decorated PAMAM dendrimer conjugates with cyclodextrins as non-viral vectors.85–89,92 The initial work of Roessler et al. showed the characterization of the interaction of polyplexes (dendrimer/DNA) in the presence of β-cyclodextrin (β-CD) where β-CD concentration also affects the physico-chemical properties of those polyplexes. Moreover, the improvement of in situ transfection hints to the power of novel dendritic polyamine scaffolds decorated with different cyclodextrins (α-, β- and γ-CD).270 Low numbers of α-, β- and γ-CD attached to the PAMAM dendrimer surface allowed the enhancement of gene transfection in comparison to those of parental PAMAM dendrimers.270 Using 3rd generation PAMAM dendrimer with 2 α-CD moieties biodistribution of polyplexes with DNA 12 h after intravenous administration in mice showed that DNA is efficiently delivered in the spleen, liver and kidney with negligible changes in blood composition.271 Especially, higher gene transfection in the spleen was determined after the administration protocol of polyplexes (12 h). After an optimization process 2nd generation PAMAM dendrimer with 2 α-CD moieties proved to be superior to commercially available TransFast™ and Lipofectin™ for in vitro and in vivo gene transfection.85,271
Arima and Uekama further fabricated α-CD-G2 and α-CD-G3 hybrid materials additionally decorated with mannose, galactose or lactose units as targeting ligands in receptor-mediated uptake processes. To realize mannose receptor-mediated non-viral delivery systems,86,88 α-D-mannopyranosylphenyl isothiocyanate was used to synthesize non-viral vectors with increasing numbers of mannose (up to 8 units) in the outer shell of α-CD-G2 (Man-α-CD-G2). Surprisingly, with increasing Man units, formation of polyplexes with Man-α-CD-G2 decreased, while polyplexes with 8 Man units in Man-α-CD-G2 were enzymatically degraded. Moreover, only a weak binding ability of Man-α-CD-G2 to the mannose-receptor on the cell membrane surface was determined despite having partially enhanced gene transfer activity with increasing Man units in Man-α-CD-G2. This can be explained by the too short or too rigid spacer between Man units and the dendritic PAMAM scaffold where, finally, mannose is not recognized by the mannose-receptor binding proteins under in vitro conditions. Despite the unexpected in vitro behavior of Man-α-CD-G2 with about 3.3 mannose units, in vivo gene transfer activity of non-viral vectors 12 h after intravenous injection to the tail vein of mice was determined. Surprisingly, Man-α-CD-G2 outlined much higher gene transfection compared to the pure 2nd generation PAMAM dendrimer and α-CD-G2 at 12 h after injection.
In line with this, galactose-decorated α-CD-G2 (Gal-α-CD-G2), prepared by the conversion of α-CD-G2 with α-D-galactopyranosylphenyl isothiocyanate, were also tested as non-viral vectors for the treatment of liver diseases.272 As found in the case of Man-α-CD-G2, Gal-α-CD-G2 showed no selective targeting properties, meaning no receptor-mediated cellular uptake against hepatocytic cells, but some gene transfer activity could be detected under in vitro conditions.272 Further efforts have been done in this direction by Arima and colleagues.273,274 Dendritic polyamine scaffolds α-CD-G2 and α-CD-G3 were converted with the lactone of lactose to introduce the desired galactose for undergoing desired asialoglycoprotein receptor-mediated cellular uptake of polyplexes in vitro and in vivo studies. For this purpose, only an average binding of 2.6 lactose units on α-CD-G2 was needed to show hepatocyte-specific gene transfer activity in vivo, consistent with in vitro transfection results, using HepG2 cells.273 The knowledge gained from these studies may help to develop more efficient ligand-modified dendritic non-viral vectors with few α-CD units in the future.
Similarly, efforts have been undertaken to establish targeting sugar-modified hyperbranched PEI (TSPEI) as non-viral vectors in gene therapy for better overcoming biological barriers of lung and liver tissues, but also of dendritic cells, macrophages and other cells (hepatocyte cells, fibroblast membranes or airway epithelial cells).62–75 In the field of TSPEI similar observations were made as found in the case of sugar-decorated PAMAM dendrimers with α-cyclodextrin units: with increasing degree of substitution of PEI functional amino groups by sugar units (e.g. galactose)67 decreasing gene transfer activity was determined. Second, the presence of targeting sugar units on PEI scaffolds led to a decreased, the same or partially better transfection efficiency compared to the parental PEI, depending on the degree of substitution, but in all cases significantly better biological actions of the TSPEI were observed.62,66,74
Considering the results from non-targeting sugar-modified hyperbranched PEI (NTSPEI) as non-viral vectors54–59 similar results with all their positive and negative facets are also available as found for TSPEI. In addition, interesting points of NTSPEI can be extracted from their results. First, the molecular weight of PEI should be preferentially smaller than 25000 g mol−1 in order to minimize in advance any adverse effects of integrated components in the drug delivery system on biological systems.55,56,58,59 Second, passive targeting of various polyplexes to tumor tissues is also given triggered by the EPR effect,4,54,56,58 partly accompanied by higher uptake of polyplexes in tumor tissue. Third, some interesting results on in vitro/in vivo NTSPEI results are provided by the groups of Appelhans and Aigner:55,56 polyplexes based on NSTPEI and siRNA are more stable in increasing concentration of extracellular matrix material heparin than polyplexes composed of the parental PEI. In contrast, other reports stated that the polyplexes are destabilized significantly when the sugar decoration is slightly increased on PEI or PAMAM scaffolds. Biodistribution of polyplexes with siRNA and NTSPEI in nude mice revealed that preferred uptake of these polyplexes is given in the lung and spleen, but less in the muscle, kidney and liver.55 This biodistribution is slightly unusual when comparing other biodistribution of polyplexes or the drug delivery system. In addition, tailoring the DNA polyplex administration56 preferred uptake and molecularly active Luciferase assays are only observable by intraperitoneal injection and no uptake was observed at all in the case of intravenous injection. Moreover, the highest gene transfer activity is recognizable by using the dense shell architecture of PEI (Fig. 18: structure A = PEI-(2-Mal)) which possesses the lowest cationic charge in this series.56 This is in contrast to the general opinion that too low cationic charge and too high sugar decoration in non-viral vectors do not lead to high transfection efficiency. Finally one has to state that each vector system based on PEI or PAMAM scaffolds has its specific features that have to be optimized to be successfully applied in future gene therapy.
 | ||
| Fig. 18 In vivo efficacy of various (OM-)PEI–DNA complexes as indicated by luciferase transgene expression. Luciferase activity was determined 24 h (black bars) and 48 h (grey bars) after intravenous (A) or intraperitoneal (B) complex injection. Most efficient dendritic glycopolymer is maltose-modified PEI with structure A [PEI-(2-Mal)] considered as dense shell in Fig. 8. Reproduced with permission from “D. Gutsch, D. Appelhans, S. Höbel, B. Voit and A. Aigner, Mol. Pharmaceutics, 2013, 10, 4666–4675”. Copyright 2013 American Chemical Society. | ||
8. Conclusions
In this review, we have outlined the beneficial aspects of dendritic glycopolymers designed for application in the fields of drug delivery systems, polymeric therapeutics and diagnostics in brain diseases. In these specific research fields preferential dendritic polyamine scaffolds have been used and decorated with different mono-, di- and oligosaccharides. To understand the biological interactions of these dendritic glycopolymers in vitro and in vivo, examples are given for the interplay of dendritic glycopolymers with low and high molecular weight drugs, peptides, proteins and polynucleotides. These complexation studies reveal the specific characteristics of open and dense shell dendritic glycopolymers. One large advantage of all these oligosaccharide-decorated dendritic polyamine scaffolds is their high in vitro and in vivo biocompatibility compared to the parent dendritic polyamine macromolecules but still paired with the potential for specific (bio)molecular interactions.Overall, the interplay of surface composition, charge density, size of dendritic polyamine scaffold, and shell density of dendritic glycopolymers dictate their final complexation and (biological) interaction characteristics against drug and analyte molecules and larger biological molecules and entities, but also the ability to cross biological barriers, especially the blood–brain-barrier.
Especially, open shell dendritic glycopolymers, based on dendritic PPI and PEI cores, are very suited for the delivery of anionic drugs and poor water-soluble drugs, while open shell dendritic glycopolymers, based on dendritic PAMAM and PEI cores, are mainly selected for the transport of RNA and DNA macromolecules to cells and tissues. In this context targeting sugar-modified dendritic polyamine scaffolds partially facilitate the cellular uptake of drugs in specific cell lines (e.g. cancer or dendritic cells) and tissues (e.g. lung or liver). Thus cationic and H-bonds-active open shell dendritic glycopolymers exhibit a great potential in the delivery of various drugs as well as biomacromolecules and can be designated as promising alternative delivery systems to (PEGylated) dendritic nanocarrier systems.
The biological use of dense shell glycodendrimers is still exclusively directed to sugar-decorated dendritic PPI scaffolds. This specific macromolecular architecture is characterized by a neutral surface charge and an H-bond-active sugar shell. This kind of “sugarball” can be used as an anti-Alzheimer and anti-prion agent, but is also applicable as drug to inhibit prion strain infections of prion protein-containing cells. In a first in vivo study dense shell glycodendrimers have been proven to be involved in retaining the memory ability of mice infected by Alzheimer Aβ peptides.
The intensive studies carried out so far demonstrate clearly that there is a complex interplay between the molecular parameters of dendritic glycopolymers and their specific biological interactions. Therefore, the potential of these specific macromolecular architectures in biomedical applications can only be exploited with a deep understanding of these interactions and, finally, a careful design of the dendritic, multifunctional structure. However, the findings so far demonstrate the specific advantages of dendritic glycopolymers and their general suitability in therapy. The gained knowledge paves the way for the design and fabrication of a more sophisticated dendritic glycoarchitecture and to finally translate them into nanomedicine.
Acknowledgements
The authors thank Mr Johannes Fingernagel and Mr David Simon for technical support (drawing figures and searching for literature) of this review.References
- Y. M. Chabre and R. Roy, Chem. Soc. Rev., 2013, 42, 4657–4708 RSC.
- L. L. Kiessling and J. C. Grim, Chem. Soc. Rev., 2013, 42, 4476–4491 RSC.
- S. R. S. Ting, G. Chen and M. H. Stenzel, Polym. Chem., 2010, 1, 1392–1412 RSC.
- A. Martínez, C. Ortiz Mellet and J. M. García Fernández, Chem. Soc. Rev., 2013, 42, 4746–4773 RSC.
- K. Hatano, K. Matsuoka and D. Terunuma, Chem. Soc. Rev., 2013, 42, 4574–4598 RSC.
- P. Niederhafener, J. Šebestík and J. Jež, J. Pept. Sci., 2008, 14, 2–43 CrossRef PubMed.
- P. Niederhafener, J. Šebestík and J. Jež, J. Pept. Sci., 2008, 14, 44–65 CrossRef PubMed.
- P. Niederhafener, M. Reiniš, J. Šebestík and J. Jež, J. Pept. Sci., 2008, 14, 556–587 CrossRef PubMed.
- N. Jararaman, Chem. Soc. Rev., 2009, 38, 3463–3483 RSC.
- D. Appelhans, H. Komber, M. Abdul Quadir, A. Loos, S. Schwarz, S. Richter, A. Aigner, M. Müller, R. Haag and B. Voit, Biomacromolecules, 2009, 10, 1114–1124 CrossRef CAS PubMed.
- S. Boye, D. Appelhans, V. Boyko, S. Zschoche, H. Komber, P. Friedel, P. Formanek, A. Janke, B. Voit and A. Lederer, Biomacromolecules, 2012, 13, 4222–4235 CAS.
- T. Satoh, Soft Matter, 2009, 5, 1972–1982 RSC.
- P. Albersheim and B. S. Valent, J. Cell Biol., 1978, 78, 627–643 CrossRef CAS.
- N. Jararaman, K. Maiti and K. Naresh, Chem. Soc. Rev., 2013, 42, 4640–4656 RSC.
- K. Gorityala, J. Ma, X. Wang, P. Chen and X.-W. Liu, Chem. Soc. Rev., 2013, 42, 4728–4745 RSC.
- Y. Chen, A. Star and S. Vidal, Chem. Soc. Rev., 2013, 42, 4532–4542 RSC.
- F. Santoyo-Gonzáles and F. Hernández-Mateo, Chem. Soc. Rev., 2013, 42, 3449–3462 Search PubMed.
- M. Marradi, F. Chiodo, I. García and S. Penadés, Chem. Soc. Rev., 2013, 42, 4728–4745 RSC.
- S. Park, J. C. Gildersleeve, O. Blixt and I. Shin, Chem. Soc. Rev., 2013, 42, 4310–4326 RSC.
- M. J. Cloninger, in Molecular Recognition and Polymers: Control of polymer structure and self-assembly, ed. V. Rotello and S. Thayumanavan, John Wiley & Son, 2008, pp. 335–358 Search PubMed.
- P. M. Rudd, M. R. Wormald and R. A. Dwek, Trends Biotechnol., 2004, 22, 524–530 CrossRef CAS PubMed.
- H.-J. Gabius, H. C. Siebert, S. André, J. Jiménez-Barbero and H. Rüdiger, ChemBioChem, 2004, 5, 740–764 CrossRef CAS PubMed.
- E. Gorelik, U. Galili and A. Raz, Cancer Metastasis Rev., 2001, 20, 245–277 CrossRef CAS.
- S. Andre, P. J. C. Ortega, M. A. Perez, R. Roy and H.-J. Gabius, Glycobiology, 1999, 9, 1253–1261 CrossRef CAS PubMed.
- E. K. Woller, E. D. Walter, J. R. Morgan, D. J. Singel and M. J. Cloninger, J. Am. Chem. Soc., 2003, 125, 8820–8826 CrossRef CAS PubMed.
- M. L. Wolfenden and M. J. Cloninger, J. Am. Chem. Soc., 2005, 127, 12168–12169 CrossRef CAS PubMed.
- S. L. Mangold and M. J. Cloninger, Org. Biomol. Chem., 2006, 4, 2458–2465 CAS.
- S. Andre, R. J. Pieters, I. Vrasidas, H. Kaltner, I. Kuwabara, F.-T. Liu, R. M. J. Liskamp and H.-J. Gabius, ChemBioChem, 2001, 2, 822–830 CrossRef CAS.
- V. Percec, P. Leowanawat, H. J. Sun, O. Kulikov, C. D. Nusbaum, T. M. Tran, A. Bertin, D. A. Wilson, M. Peterca, S. Zhang, N. P. Kamat, K. Vargo, D. Moock, E. D. Johnston, D. A. Hammer, D. J. Pochan, Y. Chen, Y. M. Chabre, T. C. Shiao, M. Bergeron-Brlek, S. Andre, R. Roy, H.-J. Gabius and P. A. Heiney, J. Am. Chem. Soc., 2013, 135, 9055–9077 CrossRef CAS PubMed.
- R. Roy, Polym. News, 1996, 21, 226–232 CAS.
- C.-L. Schengrund, Biochem. Pharmacol., 2003, 65, 699–707 CrossRef CAS.
- A. R. Borges and C.-L. Schengrund, Curr. Drug Targets: Infect. Disord., 2005, 5, 247–254 CrossRef CAS.
- N. Sharon, Biochim. Biophys. Acta, 2006, 1760, 527–537 CrossRef CAS PubMed.
- M. Touaibia and R. Roy, Mini-Rev. Med. Chem., 2007, 7, 1270–1283 CrossRef CAS.
- A. Imberty, Y. A. Chabre and R. Roy, Chem. – Eur. J., 2008, 14, 7490–7499 CrossRef CAS PubMed.
- T. R. Branson and W. B. Turnbull, Chem. Soc. Rev., 2013, 42, 4613–4622 RSC.
- S.-K. Wang, P.-H. Liang, R. D. Astronomo, T.-L. Hsu, S.-L. Hsieh, D. R. Burton and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3690–3695 CrossRef CAS PubMed.
- A. Papadopoulos, T. C. Shiao and R. Roy, Mol. Pharmaceutics, 2012, 9, 394–403 CrossRef CAS PubMed.
- R. Roy, M.-G. Baek and K. Rittenhouse-Olson, J. Am. Chem. Soc., 2001, 123, 1809–1816 CrossRef CAS PubMed.
- R. Roy and M.-G. Baek, Rev. Mol. Biotechnol., 2002, 90, 291–309 CrossRef CAS.
- P. Vepŕek, M. Hajdúch, P. Dubák, R. Kuklík, J. Poláková and K. Bezouška, J. Med. Chem., 2006, 49, 6400–6407 CrossRef PubMed.
- N. Seah, P. V. Santaroce and A. Basu, Org. Lett., 2009, 11, 559–562 CrossRef CAS PubMed.
- M. Gringas, Y. M. Chabre, M. Roy and R. Roy, Chem. Soc. Rev., 2013, 42, 4823–4841 RSC.
- V. Martos, P. Castreño, J. Valero and J. de Mendoza, Curr. Opin. Chem. Biol., 2008, 12, 698–706 CrossRef CAS PubMed.
- M. Monsigny, R. Mayer and A.-C. Roche, Carbohydr. Lett., 2000, 4, 35–52 CAS.
- T. C. Shiao and R. Roy, New J. Chem., 2012, 36, 324–339 RSC.
- Y. M. Chabre and R. Roy, in Dendrimer-Based Drug Delivery Systems, ed. Y. Y. Cheng, Wiley-VCH, Weinheim, 2012, pp. 407–438 Search PubMed.
- S. H. Medina, V. Tekumella, M. V. Chevliakov, D. S. Shewach, W. D. Ensminger and M. E. H. El Sayed, Biomaterials, 2011, 32, 4118–4129 CrossRef CAS PubMed.
- S. H. Medina, G. Tiruchinapally, M. V. Chevliakov, Y. Y. Durmaz, R. N. Stender, W. D. Ensminger, D. S. Shewach and M. E. H. El Sayed, Adv. Healthcare Mater., 2013, 2, 1337–1350 CrossRef CAS PubMed.
- S. Shaunak, S. Thomas, E. Gianasi, A. Godwin, E. Jones, I. Teo, K. Mireskandari, P. Luther, R. Duncan, S. Patterson, P. Khaw and S. Brocchini, Nat. Biotechnol., 2004, 22, 977–984 CrossRef CAS PubMed.
- J.-I. Sakamoto, T. Koyama, D. Miyamoto, S. Yingsakmongkon, K. I. P. Hidari, W. Jampangern, T. Suzuki, Y. Suzuki, Y. Esumi, T. Nakamura, K. Hatano, D. Terunuma and K. Matsuoka, Bioorg. Med. Chem., 2009, 17, 5451–5464 CrossRef CAS PubMed.
- H. Oka, T. Onaga, T. Koyama, C.-T. Guo, Y. Suzuki, Y. Esumi, K. Hatano, D. Terunuma and K. Matsuoka, Bioorg. Med. Chem., 2009, 17, 5465–5475 CrossRef CAS PubMed.
- J. L. de Paz, C. Noti, F. Böhm, S. Werner and P. H. Seeberger, Chem. Biol., 2007, 14, 879–887 CrossRef CAS PubMed.
- I.-K. Park, S.-E. Cook, Y.-K. Kim, H.-W. Kim, M.-H. Cho, H.-J. Jeong, E.-M. Kim, J.-W. Nah, H.-S. Bom and C.-S. Cho, Arch. Pharmacal Res., 2005, 28, 1302–1310 CrossRef CAS.
- S. Höbel, A. Loos, D. Appelhans, S. Schwarz, J. Seidel, B. Voit and A. Aigner, J. Controlled Release, 2011, 149, 146–158 CrossRef PubMed.
- D. Gutsch, D. Appelhans, S. Höbel, B. Voit and A. Aigner, Mol. Pharmaceutics, 2013, 10, 4666–4675 CrossRef CAS PubMed.
- C. J. Needham, A. K. Williams, S. A. Chew, F. K. Kasper and A. G. Mikos, Biomacromolecules, 2012, 13, 1429–1437 CrossRef CAS PubMed.
- F.-W. Hu, W.-W. Chen, M.-D. Zhao, H. Yuan and Y.-Z. Du, Gene Ther., 2013, 20, 597–606 CrossRef CAS PubMed.
- L. Jia, Z. Li, D. Zhang, Q. Zhang, J. Shen, H. Guo, X. Tian, G. Liu, D. Zheng and L. Qi, Polym. Chem., 2013, 4, 156–165 RSC.
- N. Hauptmann, M. Pion, M.-A. Muñoz-Fernandez, H. Komber, C. Werner, B. Voit and D. Appelhans, Macromol. Biosci., 2013, 13, 531–538 CrossRef CAS PubMed.
- N. Hauptmann, M. Pion, R. Wehner, M.-A. Muñoz-Fernández, M. Schmitz, B. Voit and D. Appelhans, Biomacromolecules, 2014, 15, 957–967 CrossRef CAS PubMed.
- M. A. Zanta, O. Boussif, A. Adib and J. P. Behr, Bioconjugate Chem., 1997, 8, 839–844 CrossRef CAS PubMed.
- P. Erbacher, T. Bettinger, P. Belguise-Valladier, S. Zou, J. L. Coll, J. P. Behr and J. S. Remy, J. Gene Med., 1999, 1, 210–222 CrossRef CAS.
- T. Bettinger, J.-S. Remy and P. Erbacher, Bioconjugate Chem., 1999, 10, 558–561 CrossRef CAS PubMed.
- F. Leclercq, C. Dubertret, B. Pitard, D. Scherman and J. Herscovici, Bioorg. Med. Chem. Lett., 2000, 10, 1233–1235 CrossRef CAS.
- S. S. Diebold, M. Kursa, E. Wagner, M. Cotten and M. Zenke, J. Biol. Chem., 1999, 274, 19087–19094 CrossRef CAS PubMed.
- K. Kunath, A. von Harpe, D. Fischer and T. Kissel, J. Controlled Release, 2003, 88, 159–172 CrossRef CAS.
- I. Fajac, G. Thevenot, L. Bedouet, C. Danel, M. Riquet, M. Merten, C. Figarella, J. Dall'Ava-Santucci, M. Monsigny and P. Briand, J. Gene Med., 2003, 5, 38–48 CrossRef CAS PubMed.
- S. Grosse, Y. Aron, I. Honore, G. Thevenot, C. Danel, A. C. Roche, M. Monsigny and I. Fajac, J. Gene Med., 2004, 6, 345–356 CrossRef CAS PubMed.
- S. E. Cook, I. K. Park, E. M. Kim, H. J. Jeong, T. G. Park, Y. J. Choi, T. Akaike and C. S. Cho, J. Controlled Release, 2005, 105, 151–163 CrossRef CAS PubMed.
- S. Grosse, G. Thevenot, M. Monsigny and I. Fajac, J. Gene Med., 2006, 8, 845–851 CrossRef CAS PubMed.
- J. Chen, X. Gao, K. Hu, Z. Pang, J. Cai, J. Li, H. Wu and X. Jiang, Biochem. Biophys. Res. Commun., 2008, 375, 378–383 CrossRef CAS PubMed.
- I. Y. Park, I. Y. Kim, M. K. Yoo, Y. J. Choi, M. H. Cho and C. S. Cho, Int. J. Pharm., 2008, 359, 280–287 CrossRef CAS PubMed.
- S. Grosse, G. Thevenot, Y. Aron, E. Duverger, M. Abdelkarim, A. C. Roche, M. Monsigny and I. Fajac, J. Controlled Release, 2008, 132, 105–112 CrossRef CAS PubMed.
- W. Cheng, C. Yang, J. L. Hedrick, D. F. Williams, Y. Y. Yang and P. G. Ashton-Rickardt, Biomaterials, 2013, 34, 3697–3705 CrossRef CAS PubMed.
- Y.-K. Kim, I.-K. Park, H.-L. Jiang, R. Arote, H.-J. Jeong, E.-M. Kim, M.-H. Cho, H.-S. Bom and C.-S. Cho, Key Eng. Mater., 2007, 342–343, 457–460 CAS.
- E. Vacas Córdoba, M. Pion, B. Rasines, D. Filippini, H. Komber, M. Ionov, M. Bryszewska, D. Appelhans and M.-A. Muñoz-Fernández, Nanomedicine: Nanotechnology, Biology and Medicine, 2013, 9, 972–984 CrossRef PubMed.
- D. Bhadra, A. K. Yadav, S. Bhadra and N. K. Jain, Int. J. Pharm., 2005, 295, 221–233 CrossRef CAS PubMed.
- P. V. Kumar, A. Asthana, T. Dutta and N. K. Jain, J. Drug Targeting, 2006, 14, 546–556 CrossRef CAS PubMed.
- T. Dutta and N. K. Jain, Biochim. Biophys. Acta, 2007, 1770, 681–686 CrossRef CAS PubMed.
- T. Dutta, H. B. Agashe, M. Garg, P. Balasubramanium, M. Kabra and N. K. Jain, J. Drug Targeting, 2007, 15, 89–98 CrossRef CAS PubMed.
- A. Agarwal, U. Gupta, A. Asthana and N. K. Jain, Biomaterials, 2009, 30, 3588–3596 CrossRef CAS PubMed.
- V. Gajbhiye, N. Ganesh, J. Barve and N. K. Jain, Eur. J. Pharm. Sci., 2013, 48, 668–679 CrossRef CAS PubMed.
- R. Gupta, N. K. Mehra and N. K. Jain, Eur. J. Pharm. Biopharm., 2014, 86, 449–458 CrossRef CAS PubMed.
- F. Kihara, H. Arima, T. Tsutsumi, F. Hirayama and K. Uekama, Bioconjugate Chem., 2002, 13, 1211–1219 CrossRef CAS PubMed.
- K. Wada, H. Arima, T. Tsutsumi, Y. Chihara, K. Hattori, F. Hirayama and K. Uekama, J. Controlled Release, 2005, 104, 397–413 CrossRef CAS PubMed.
- K. C. Wood, S. R. Little, R. Langer and P. T. Hammond, Angew. Chem., Int. Ed., 2005, 44, 6704–6708 CrossRef CAS PubMed.
- H. Arima, Y. Chihara, M. Arizono, S. Yamashita, K. Wada, F. Hirayama and K. Uekama, J. Controlled Release, 2006, 116, 64–74 CrossRef CAS PubMed.
- H. Arima and K. Motoyama, Drug Delivery Syst., 2010, 25, 598–606 CrossRef CAS.
- Y. Zhang, T. P. Thomas, K.-Y. Lee, M. Li, H. Zong, A. M. Desai, A. Kotlyar, B. Huang, M. M. Banaszak Holl and J. R. Baker Jr., Bioorg. Med. Chem., 2011, 19, 2557–2564 CrossRef CAS PubMed.
- J. Liu, J. Zhou and Y. Luo, Bioconjugate Chem., 2012, 23, 174–183 CrossRef CAS PubMed.
- H. Arima, K. Motoyama and T. Higashi, Adv. Drug Delivery Rev., 2013, 65, 1204–1214 CrossRef CAS PubMed.
- V. Grobarova, V. Benson, D. Rozbesky, P. Novak and J. Cerny, Immunol. Lett., 2013, 156, 110–117 CrossRef CAS PubMed.
- J. J. Garcia-Vallejo, M. Ambrosini, A. Overbeek, W. E. van Riel, K. Bloem, W. W. J. Unger, F. Chiodo, J. G. Bolscher, K. Nazmi, H. Kalay and Y. van Kooyk, Mol. Immunol., 2013, 53, 387–397 CrossRef CAS PubMed.
- P. Agrawal, U. Gupta and N. K. Jain, Biomaterials, 2007, 28, 3349–3359 CrossRef CAS PubMed.
- H. M. Branderhorst, R. Ruijtenbeek, R. M. J. Liskamp and R. J. Pieters, ChemBioChem, 2008, 9, 1836–1844 CrossRef CAS PubMed.
- N. Bogdan, F. Vetrone, R. Roy and J. A. Capobianco, J. Mater. Chem., 2010, 20, 7543–7550 RSC.
- B. M. Cummins, J. Lim, E. E. Simanek, M. V. Pishko and G. L. Cote, Biomed. Opt. Express, 2011, 2, 1243–1257 CrossRef CAS PubMed.
- N. Bogdan, R. Roy and M. Morin, RSC Adv., 2012, 985–991 RSC.
- N. M. Bandaru and N. H. Voelcker, J. Mater. Chem., 2012, 22, 8748–8758 RSC.
- M. Ogiso, J. Kobayashi, T. Imai, K. Matsuoka, M. Itoh, T. Imamura, T. Okada, H. Miura, T. Nishiyama, K. Hatanaka and N. Minoura, Biosens. Bioelectron., 2013, 41, 465–470 CrossRef CAS PubMed.
- P. Krist, L. Vannucci, M. Kuzma, P. Man, K. Sadalapure, A. Patel, K. Bezouska, M. Pospisil, L. Petrus, T. K. Lindhorst and V. Kren, ChemBioChem, 2004, 5, 445–452 CrossRef CAS PubMed.
- J. P. Andre, C. F. G. C. Geraldes, J. A. Martins, A. E. Merbach, M. I. M. Prata, A. C. Santos, J. J. P. de Lima and E. Toth, Chem. – Eur. J., 2004, 10, 5804–5816 CrossRef CAS PubMed.
- A. L. Branco de Barros, S. Fernandes de Andrade, J. Dias de Souza Filho, V. N. Cardoso and R. J. Alves, J. Radioanal. Nucl. Chem., 2013, 298, 605–609 CrossRef CAS PubMed.
- P. Bojarova, R. R. Rosencrantz, L. Elling and V. Kren, Chem. Soc. Rev., 2013, 42, 4774–4797 RSC.
- K.-R. Wang, H.-W. An, R.-X. Rong, Z.-R. Cao and X.-L. Li, Macromol. Rapid Commun., 2014, 35, 727–734 CrossRef CAS PubMed.
- M.-G. Baek and R. Roy, Bioorg. Med. Chem., 2001, 9, 3005–3011 CrossRef CAS.
- F. Ennen, S. Boye, L. Lederer, M. Cernescu, H. Komber, B. Brutschy, B. Voit and D. Appelhans, Polym. Chem., 2014, 5, 1323–1339 RSC.
- B. Klajnert, D. Appelhans, H. Komber, N. Morgner, S. Schwarz, S. Richter, B. Brutschy, M. Ionov, A. K. Tonkikh, M. Bryszewska and B. Voit, Chem. – Eur. J., 2008, 14, 7030–7041 CrossRef CAS PubMed.
- M. Fischer, D. Appelhans, B. Klajnert, M. Bryszewska, B. Voit and M. Rogers, Biomacromolecules, 2010, 11, 1314–1325 CrossRef CAS PubMed.
- O. Klementieva, N. Benseny-Cases, A. Gella, D. Appelhans, B. Voit and J. Cladera, Biomacromolecules, 2011, 12, 3903–3909 CrossRef CAS PubMed.
- M. F. Ottaviani, M. Cangiotti, L. Fiorani, S. Lucchi, T. Wasiak, D. Appelhans and B. Klajnert, Curr. Med. Chem., 2012, 19, 5907–5921 CrossRef CAS.
- J. McCarthy, B. Rasines Moreno, D. Appelhans and M. Rogers, Adv. Healthcare Mater., 2012, 1, 768–772 CrossRef CAS PubMed.
- J. M. McCarthy, B. Rasines Moreno, D. Filippini, H. Komber, M. Marek, M. Cernescu, B. Brutschy, D. Appelhans and M. S. Rogers, Biomacromolecules, 2013, 14, 27–37 CrossRef CAS PubMed.
- J. M. McCarthy, M. Franke, U. K. Resenberger, S. Waldron, J. C. Simpson, J. Tatzelt, D. Appelhans and M. S. Rogers, PLoS One, 2013, 8, e55282 CAS.
- J. M. McCarthy, D. Appelhans and M. S. Rogers, Prion, 2013, 7, 198–202 CrossRef CAS PubMed.
- D. Appelhans, N. Benseny, O. Klementiveva, M. Bryszewska, B. Klajnert and J. Cladera, in Dendrimer in Biomedical Applications, ed. B. Klajnert, L. Peng and V. Cena, RSC, 2013, pp. 1–13 Search PubMed.
- O. Klementieva, E. Aso, D. Filippini, N. Benseny-Cases, M. Carmona, S. Juves, D. Appelhans, J. Cladera and I. Ferrer, Biomacromolecules, 2013, 14, 3570–3580 CrossRef CAS PubMed.
- M. Mkandawire, A. Pohl, T. Gubarevich, V. Lapina, D. Appelhans, G. Rödel, W. Pompe, J. Schreiber and J. Opitz, J. Biophotonics, 2009, 2, 596–606 CrossRef CAS PubMed.
- J. Majoinen, J. Haataja, D. Appelhans, A. Lederer, A. Olsewska, J. Seitsonen, V. Aseyev, E. Kontturi, H. Rosilo, M. Österberg, N. Houbenov and O. Ikkala, J. Am. Chem. Soc., 2014, 136, 866–869 CrossRef CAS PubMed.
- M. Paolino, F. Ennen, H. Komber, M. Cernescu, A. Cappelli, B. Brutschy, B. Voit and D. Appelhans, Polym. Chem., 2012, 3, 3239–3242 RSC.
- M. Paolino, F. Ennen, S. Lamponi, M. Cernescu, B. Voit, A. Cappelli, D. Appelhans and H. Komber, Macromolecules, 2013, 46, 3215–3227 CrossRef CAS.
- A. Richter, A. Janke, S. Zschoche, R. Zimmermann, F. Simon, K.-J. Eichhorn, B. Voit and D. Appelhans, New J. Chem., 2010, 34, 2105–2108 RSC.
- M. Warenda, A. Richter, D. Schmidt, A. Janke, M. Müller, F. Simon, R. Zimmermann, K.-J. Eichhorn, B. Voit and D. Appelhans, Macromol. Rapid Commun., 2012, 33, 1466–1473 CrossRef CAS PubMed.
- L. M. Kaminskas and C. H. J. Porter, Adv. Drug Delivery Rev., 2011, 63, 890–900 CrossRef CAS PubMed.
- Y. Cheng, L. Zhao, Y. Li and T. Xu, Chem. Soc. Rev., 2011, 40, 2673–2703 RSC.
- D. Sethi, M. L. Thakur and E. Wickstrom, Curr. Mol. Imaging, 2012, 1, 3–11 CrossRef CAS.
- A. Dominguez, B. Suarez-Merino and F. Goni-de-Cerio, J. Nanosci. Nanotechnol., 2014, 14, 766–779 CrossRef CAS PubMed.
- R. Duncan, Curr. Opin. Biotechnol., 2011, 22, 492–501 CrossRef CAS PubMed.
- R. Duncan and M. J. Vicent, Adv. Drug Delivery Rev., 2013, 65, 60–70 CrossRef CAS PubMed.
- A. Jain, A. Jain, A. Gulbake, S. Shilpi, P. Hurkat and S. K. Jain, Crit. Rev. Ther. Drug Carrier Syst., 2013, 30, 293–329 CrossRef CAS.
- L. M. Lopez-Marin, E. Tamariz, L. S. Acosta-Torres and V. M. Castano, Curr. Drug Metab., 2013, 14, 531–539 CrossRef CAS.
- Y. Ikeda and Y. Nagasaki, J. Appl. Polym. Sci., 2014, 131, 40293 CrossRef.
- R. H. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- K. Kempe, A. Krieg, C. Remzi Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176–191 RSC.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- A. Carlmark, C. Hawker, A. Hult and M. Malkoch, Chem. Soc. Rev., 2009, 38, 352–362 RSC.
- M. J. Robb and C. J. Hawker, in Synthesis of Polymers, ed. D. A. Schlüter, C. J. Hawker and J. Sakamoto, Wiley-VCH, 2012, pp. 923–971 Search PubMed.
- P. Wu, A. K. Feldman, A. T. Nugent, C. J. Hawker, B. Voit, A. Scheel, J. Pyun, J. M. J. Fréchet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928–3932 CrossRef CAS PubMed.
- A. Carlmark, E. Malström and M. Malkoch, Chem. Soc. Rev., 2013, 42, 5858–5879 RSC.
- P. Antoni, Y. Hed, A. Nordberg, D. Nyström, H. von Holst, A. Hult and M. Malkoch, Angew. Chem., 2009, 121, 2160–2164 CrossRef.
- P. Antoni, M. J. Robb, L. Campos, M. Montanez, A. Hult, E. Malström, M. Malkoch and C. J. Craig, Macromolecules, 2010, 43, 6625–6631 CrossRef CAS.
- X. Ma, Z. Zhou, E. Jin, Q. Sun, B. Zhang, J. Tang and Y. Shen, Macromolecules, 2012, 46, 37–42 CrossRef.
- R. J. Amir, L. Albertazzi, J. Willis, A. Khan, T. Kang and C. J. Hawker, Angew. Chem., Int. Ed., 2011, 50, 3425–3429 CrossRef CAS PubMed.
- M. V. Walter and M. Malkoch, in Synthesis of Polymers, ed. D. A. Schlüter, C. J. Hawker and J. Sakamoto, Wiley-VCH, Weinheim, 2012, pp. 1027–1057 Search PubMed.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed.
- P. Wu, M. Malkoch, J. N. Hunt, R. Vestberg, E. Kaltgrad, M. G. Finn, V. V. Fokin, K. B. Sharpless and C. J. Craig, Chem. Commun., 2005, 5775–5777 RSC.
- A. Dondoni, Chem. – Asian J., 2007, 2, 700–708 CrossRef CAS PubMed.
- M. L. Conte, M. J. Robb, Y. Hed, A. Marra, M. Malkoch, C. J. Hawker and A. Dondoni, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4468–4475 CrossRef CAS PubMed.
- M. Ghirardello, K. Öberg, S. Staderini, O. Renaudet, N. Berthet, P. Dumy, Y. Hed, A. Marra, M. Malkoch and A. Dondoni, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2422–2433 CrossRef CAS.
- R. Sharma, K. Naresh, Y. M. Chabre, R. Rej, N. Saadeh and R. Roy, Polym. Chem., 2014, 5, 4321–4331 RSC.
- N. Kottari, Y. M. Chabre, T. Chieh Shiao, R. Rej and R. Roy, Chem. Commun., 2014, 1983–1985 RSC.
- N. ten Brummelhuis, C. Diehl and H. Schlaad, Macromolecules, 2008, 41, 9946–9947 CrossRef CAS.
- J. Sun and H. Schlaad, Macromolecules, 2010, 43, 4445–4448 CrossRef CAS.
- P. Laurino, R. Kikkeri, N. Azzouz and P. H. Seeberger, Nano Lett., 2011, 11, 73–78 CrossRef CAS PubMed.
- N. Jayaraman, S. A. Nepogodiev and J. F. Stoddart, Chem. – Eur. J., 1997, 3, 1193–1199 CrossRef CAS.
- N. Röckendorf and T. K. Lindhorst, Top. Curr. Chem., 2001, 217, 201–238 CrossRef.
- W. B. Turnbull and J. F. Stoddart, Rev. Mol. Biotechnol., 2002, 90, 231–255 CrossRef CAS.
- R. Roy, Trends Glycosci. Glycotechnol., 2003, 15, 291–310 CrossRef CAS.
- S. A. Nepogodiev and J. F. Stoddart, Adv. Macromol. Carbohydr. Res., 2003, 2, 191–240 CAS.
- H. Sashiwa and S.-I. Aiba, Prog. Polym. Sci., 2004, 29, 887–908 CrossRef CAS PubMed.
- F. Santoyo-Gonzáles and F. Hernández-Mateo, Top. Heterocycl. Chem., 2007, 7, 133–177 Search PubMed.
- Y. M. Chabre and R. Roy, Curr. Top. Med. Chem., 2008, 8, 1237–1285 CrossRef CAS.
- D. Zanini and R. Roy, in Carbohydrate Mimics – Concept and Methods, ed. Y. Chapleur, Wiley-VCH, Weinheim, 1998, pp. 385–415 Search PubMed.
- T. K. Lindhorst, in Bioorganic Chemistry – Highlights and New aspects, ed. U. Diederichsen, T. K. Lindhorst, B. Westermann and L. A. Wessjohann, Wiley-VCH, 1999, pp. 133–150 Search PubMed.
- S. J. Danishefsky and J. Y. Roberge, in Glycopeptides and related Compounds – Synthesis, Analysis and Applications, ed. D. G. Large and C. D. Warren, Marcel Dekkers, Inc., 1997, pp. 245–294 Search PubMed.
- M. Llinares and R. Roy, Chem. Commun., 1997, 2119–2120 RSC.
- E. A. B. Kantchev, C.-C. Chang, S.-F. Cheng, A.-C. Roche and D.-K. Chang, Org. Biomol. Chem., 2008, 6, 1377–1385 CAS.
- T. Matsushita, S. Handa, K. Naruchi, F. Garcia-Martin, H. Hinou and S.-I. Nishimura, Polym. J., 2013, 45, 854–862 CrossRef CAS.
- D. Zanini and R. Roy, Bioconjugate Chem., 1997, 8, 187–192 CrossRef CAS PubMed.
- M. M. Palcic, H. Li, D. Zanini, R. S. Bhella and R. Roy, Carbohydr. Res., 1997, 305, 433–442 CrossRef CAS.
- A. K. Choudhury, M. Kitaoka and K. Hayashi, Eur. J. Org. Chem., 2003, 2462–2470 CrossRef CAS.
- T. Matsushita, I. Nagashima, M. Fumoto, T. Ohta, K. Yamada, H. Shimizu, H. Hinou, K. Naruchi, T. Ito, H. Kondo and S.-I. Nishimura, J. Am. Chem. Soc., 2010, 132, 16651–16656 CrossRef CAS PubMed.
- I. Y. Park, I. Y. Kim, M. K. Yoo, Y. J. Choi, M. H. Cho and C. S. Cho, Int. J. Pharm., 2008, 359, 280–287 CrossRef CAS PubMed.
- X. Liu, J. Liu and Y. Luo, Polym. Chem., 2012, 3, 310–313 RSC.
- T. Dutta, M. Garg, V. Dubey, D. Mishra, K. Singh, D. Pandita, A. K. Singh, A. K. Ravi, T. Velpandian and N. K. Jain, Nanotoxicology, 2008, 2, 62–70 CrossRef CAS.
- K. Aoi, K. Itoh and M. Okada, Macromolecules, 1995, 28, 5391–5393 CrossRef CAS.
- B. Ziemba, I. Franiak-Pietryga, M. Pion, D. Appelhans, M.-Á. Muñoz-Fernández, B. Voit, M. Bryszewska and B. Klajnert-Maculewicz, Int. J. Pharm., 2014, 461, 391–402 CrossRef CAS PubMed.
- H. Baigude, K. Katsuraya, K. Okuyama, S. Tokunaga and T. Uryu, Macromolecules, 2003, 36, 7100–7106 CrossRef CAS.
- H. Baigude, K. Katsuraya, K. Okuyama, Y. Yachi, S. Sato and T. Uryu, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3622–3633 CrossRef CAS.
- N. Polikarpov, D. Appelhans, P. Welzel, A. Kaufmann, P. Dhanapal, C. Bellmann and B. Voit, New J. Chem., 2012, 36, 438–451 RSC.
- S. Tripp, D. Appelhans, C. Striegler and B. Voit, Chem. – Eur. J., 2014, 20, 8314–8319 CrossRef CAS PubMed.
- A. Szulc, D. Appelhans, B. Voit, M. Bryszewska and B. Klajnert, J. Fluoresc., 2013, 23, 349–356 CrossRef CAS PubMed.
- R. D. Kensinger, B. C. Yowler, A. J. Benesi and C.-L. Schengrund, Bioconjugate Chem., 2004, 15, 349–358 CrossRef CAS PubMed.
- R. D. Kensinger, B. J. Catalone, F. C. Krebs, B. Wigdahl and C.-L. Schengrund, Antimicrob. Agents Chemother., 2004, 48, 1614–1623 CrossRef CAS.
- A. F. Thünemann, R. Bienert, D. Appelhans and B. Voit, Macromol. Chem. Phys., 2012, 213, 2362–2369 CrossRef.
- A. Köth, D. Appelhans, B. Tiersch and J. Koetz, Soft Matter, 2011, 7, 10581–10584 RSC.
- T. Wasiak, M. Ionov, K. Nieznanski, H. Nieznanska, O. Klementieva, M. Granell, J. Cladera, J.-P. Majoral, A.-M. Caminade and B. Klajnert, Mol. Pharmaceutics, 2012, 9, 458–469 CrossRef CAS PubMed.
- K. Milowska, T. Gabryelak, M. Bryszewska, A.-M. Caminade and J.-P. Majoral, Int. J. Biol. Macromol., 2012, 50, 1138–1143 CrossRef CAS PubMed.
- H. Oka, T. Onaga, T. Koyama, C.-T. Guo, Y. Suzuki, Y. Esumi, K. Hatano, D. Terunuma and K. Matsuoka, Bioorg. Med. Chem. Lett., 2008, 18, 4405–4408 CrossRef CAS PubMed.
- V. Gajbhiye, P. Vijayaraj Kumar, R. Kumar Tekade and N. Jain, Curr. Pharm. Des., 2007, 13, 415–429 CrossRef CAS.
- F. Wang, X. Cai, Y. Su, J. Hu, Q. Wu, H. Zhang, J. Xiao and Y. Cheng, Acta Biomater., 2012, 8, 4304–4313 CrossRef CAS PubMed.
- M. Ciolkowski, I. Halets, D. Shcharbin, D. Appelhans, B. Voit, B. Klajnert and M. Bryszewska, New J. Chem., 2012, 36, 1992–1999 RSC.
- J. B. Ross, C. J. Schmidt and L. Brand, Biochemistry, 1981, 20, 4369–4377 CrossRef CAS.
- M. Ciolkowski, B. Palecz, D. Appelhans, B. Voit, B. Klajnert and M. Bryszewska, Colloids Surf., B, 2012, 95, 103–108 CrossRef CAS PubMed.
- K. Jain, P. Kesharwani, U. Gupta and N. K. Jain, Int. J. Pharm., 2010, 394, 122–142 CrossRef CAS PubMed.
- S. Svenson and D. A. Tomalia, Nanoparticulates as drug carriers, Imperial College Press, London, 2006, p. 298 Search PubMed.
- B. Ziemba, I. Halets, D. Shcharbin, D. Appelhans, B. Voit, I. Pieszynski, M. Bryszewska and B. Klajnert, J. Biomed. Mater. Res., Part A, 2012, 100, 2870–2880 CrossRef PubMed.
- A. Janaszewska, K. Maczynska, G. Matuszko, D. Appelhans, B. Voit, B. Klajnert and M. Bryszewska, New J. Chem., 2012, 36, 428–437 RSC.
- N. Malik, R. Wiwattanapatapee, R. Klopsch, K. Lorenz, H. Frey, J. W. Weener, E. W. Meijer, W. Paulus and R. Duncan, J. Controlled Release, 2000, 65, 133–148 CrossRef CAS.
- H. B. Agashe, T. D. Dutta, M. Garg and N. K. Jain, J. Pharm. Pharmacol., 2006, 58, 1491–1498 CrossRef CAS PubMed.
- B. Ziemba, G. Matuszko, D. Appelhans, B. Voit, M. Bryszewska and B. Klajnert, Biopolymers, 2012, 97, 642–648 CrossRef CAS PubMed.
- B. H. Zinselmeyer, S. P. Mackay, A. G. Schatzlein and I. F. Uchegbu, Pharm. Res., 2002, 19, 960–967 CrossRef CAS.
- I. Franiak-Pietryga, E. Ziolkowska, B. Ziemba, D. Appelhans, B. Voit, M. Szewczyk, J. Gora-Tybor, T. Robak, B. Klajnert and M. Bryszewska, Mol. Pharmaceutics, 2013, 10, 2490–2501 CrossRef CAS PubMed.
- J. H. Kuo, M. S. Jan and H. W. Chiu, J. Pharm. Pharmacol., 2005, 57, 489–495 CrossRef CAS PubMed.
- J. H. Kuo, M. S. Jan and Y. L. Lin, J. Controlled Release, 2007, 120, 51–59 CrossRef CAS PubMed.
- A. Filimon, L. E. Sima, D. Appelhans, B. Voit and G. Negroiu, Curr. Med. Chem., 2012, 19, 4955–4968 CrossRef CAS.
- B. Ziemba, A. Janaszewska, K. Ciepluch, M. Krotewicz, W. A. Fogel, D. Appelhans, B. Voit, M. Bryszewska and B. Klajnert, J. Biomed. Mater. Res., Part A, 2011, 99, 261–268 CrossRef PubMed.
- H. B. Agashe, A. K. Babbar, S. Jain, R. K. Sharma, A. K. Mishra, A. Asthana, M. Garg, T. Dutta and N. K. Jain, Nanomedicine, 2007, 3, 120–127 CrossRef CAS PubMed.
- K. Ciepluch, B. Ziemba, A. Janaszewska, D. Appelhans, B. Klajnert, M. Bryszewska and W. A. Fogel, J. Physiol. Biochem., 2012, 68, 447–454 CrossRef CAS PubMed.
- S. H. He, World J. Gastroenterol., 2004, 10, 309–318 CAS.
- N. J. Abbott, L. Rönnbäck and E. Hansson, Nat. Rev. Neurosci., 2006, 7, 41–53 CrossRef CAS PubMed.
- A. Janaszewska, B. Ziemba, K. Ciepluch, D. Appelhans, B. Voit, B. Klajnert and M. Bryszewska, New J. Chem., 2012, 36, 350–353 RSC.
- S. Ku, F. Yan, Y. Wang, Y. Sun, N. Yang and L. Ye, Biochem. Biophys. Res. Commun., 2010, 394, 871–876 CrossRef CAS PubMed.
- S. Supattapone, H. O. Nguyen, F. E. Cohen, S. B. Prusiner and M. R. Scott, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14529–14534 CrossRef CAS.
- D. Shcharbin, B. Klajnert and M. Bryszewska, J. Biomater. Sci., Polym. Ed., 2005, 16, 1081–1093 CrossRef CAS PubMed.
- M. Stefani and C. M. Dobson, J. Mol. Med., 2003, 81, 678–699 CrossRef CAS PubMed.
- J. Solassol, C. Crozet, V. Perrier, J. Leclaire, F. Beranger, A. M. Caminade, B. Meunier, D. Dormont, J. P. Majoral and S. Lehmann, J. Gen. Virol., 2004, 85, 1791–1799 CrossRef CAS.
- B. Klajnert, L. Stanislawska, M. Bryszewska and B. Palecz, Biochim. Biophys. Acta, 2003, 1648, 115–125 CrossRef CAS.
- B. Klajnert, M. Cortijo-Arellano, J. Cladera and M. Bryszewska, Biochem. Biophys. Res. Commun., 2006, 345, 21–28 CrossRef CAS PubMed.
- B. Klajnert, J. Cladera and M. Bryszewska, Biomacromolecules, 2006, 7, 2186–2191 CrossRef CAS PubMed.
- R. K. Tekade, P. V. Kumar and N. K. Jain, Chem. Rev., 2009, 109, 49–87 CrossRef CAS PubMed.
- S. Mignani and J.-P. Majoral, New J. Chem., 2012, 37, 3337–3357 RSC.
- S. Mignani, S. E. Kazzouli, M. Bousmina and J.-P. Majoral, Prog. Polym. Sci., 2013, 38, 993–1008 CrossRef CAS PubMed.
- S. Mignani, S. E. Kazzouli, M. Bousmina and J.-P. Majoral, Chem. Rev., 2014, 114, 1327–1342 CrossRef CAS PubMed.
- P. Kesharwani, K. Jain and N. K. Jain, Prog. Polym. Sci., 2014, 39, 268–307 CrossRef CAS PubMed.
- J. R. Baker, Am. Soc. Hematol., 2009, 1, 708–719 CrossRef PubMed.
- S. H. Medina and E. H. El-Sayed, Chem. Rev., 2009, 109, 3141–3157 CrossRef CAS PubMed.
- S. Svenson, Eur. J. Pharm. Biopharm., 2009, 71, 445–462 CrossRef CAS PubMed.
- Y. Li, J. Wang, M. G. Wientjes and J. L. S. Au, Adv. Drug Delivery Rev., 2012, 64, 29–39 CrossRef CAS PubMed.
- U. Boas and M. H. Heegaard, Chem. Soc. Rev., 2004, 33, 43–63 RSC.
- A. Dolenc, J. Kristi, S. Baumgartner and O. Planinsek, Int. J. Pharm., 2009, 376, 204–212 CrossRef CAS PubMed.
- P. S. Narayan, S. Pooja, A. Khushboo, T. Diwakar, S. Ankit and A. K. Singhai, Int. J. Pharm. Life Sci., 2010, 382–388 Search PubMed.
- M. Ina, Drug Delivery Syst., 2011, 1, 70–74 Search PubMed.
- M. Calderon, M. A. Quadir, M. Strumia and R. Haag, Biochimie, 2010, 92, 1242–1251 CrossRef CAS PubMed.
- C. C. Lee, J. A. Mackay, J. M. J. Fréchet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS PubMed.
- D. Q. McNermy, P. R. Leroueil and J. R. Baker, Nanomedicine: Nanotechnology, Biology and Medicine, 2010, 2, 249–259 CrossRef PubMed.
- T. Garg, O. Singh, S. Arora and R. S. R. Murthy, Int. J. Pharm. Sci. Rev. Res., 2011, 7, 211–220 CAS.
- W. Wijagkanalan, S. Kawakami and M. Hashida, Pharm. Res., 2011, 28, 1500–1519 CrossRef CAS PubMed.
- N. K. Jain, V. Misra and N. K. Mehra, Expert Opin. Drug Delivery, 2013, 10, 353–367 CrossRef CAS PubMed.
- C. C. Anajwala, G. K. Jani and S. M. V. Swamy, Int. J. Pharm. Sci. Nanotechnol., 2010, 3, 1043–1056 CAS.
- R. Misra, S. Acharya and S. K. Sahoo, Drug Discovery Today, 2010, 15, 842–850 CrossRef CAS PubMed.
- R. Sinha, G. J. Kim, S. Nie and D. M. Shin, Mol. Cancer Ther., 2006, 5, 1909–1917 CrossRef CAS PubMed.
- S. Mignani, S. E. Kazzouli, M. Bousmina and J.-P. Majoral, Adv. Drug Delivery Rev., 2013, 65, 1316–1330 CrossRef CAS PubMed.
- A. Szulc, D. Appelhans, B. Voit, M. Bryszewska and B. Klajnert, New J. Chem., 2012, 36, 1610–1615 RSC.
- B. Klajnert and D. Appelhans, unpublished results.
- M. Kuhlbeil, H. Stephan, H.-J. Pietzsch, G. Geipel, D. Appelhans, B. Voit, J. Hoffmann, B. Brutschy, Y. V. Mironov, K. A. Brylev and V. E. Fedorov, Chem. – Asian J., 2010, 5, 2507–2514 CrossRef PubMed.
- V. Gajbhiye, P. V. Kumar, A. Sharma and N. K. Jain, Curr. Nanosci., 2008, 4, 267–277 CrossRef CAS.
- V. Gajbhiye, P. Vijayaraj Kumar, R. K. Tekade and N. K. Jain, Eur. J. Med. Chem., 2009, 44, 1155–1166 CrossRef CAS PubMed.
- B. D. Kurmi, V. Gajbhiye, J. Kayat and N. K. Jain, J. Pharm. Sci., 2011, 100, 2311–2320 CrossRef CAS PubMed.
- V. Gajbhiye and N. K. Jain, Biomaterials, 2011, 32, 6213–6225 CAS.
- S. K. Patel, V. Gajbhiye and N. K. Jain, J. Drug Targeting, 2012, 20, 841–849 CrossRef CAS PubMed.
- V. Shah, O. Taratula, O. B. Garbuzenko, O. R. Taratula, L. Rodriguez-Rodriguez and T. Minko, Clin. Cancer Res., 2013, 19, 6193–6204 CrossRef CAS PubMed.
- A. Jain, K. Jain, N. K. Mehra and N. K. Jain, J. Nanopart. Res., 2013, 15, 2003 CrossRef.
- K. Jain and N. K. Jain, J. Nanosci. Nanotechnol., 2014, 14, 5075–5087 CrossRef CAS PubMed.
- T. Dutta, M. Garg and N. K. Jain, Eur. J. Pharm. Sci., 2008, 34, 181–189 CrossRef CAS PubMed.
- U. Gupta, H. B. Agashe and N. K. Jain, Eur. J. Pharm. Sci., 2007, 10, 358–367 CAS.
- P. Agrawal, U. Gupta and N. K. Jain, Biomaterials, 2007, 28, 3349–3359 CrossRef CAS PubMed.
- D. Bhadra, S. Bhadra and N. K. Jain, Pharm. Res., 2005, 23, 623–633 CrossRef PubMed.
- K. Tsutsumiuchi, K. Aoi and M. Okada, Polym. J., 1999, 31, 935–941 CrossRef CAS.
- E. Pedziwiatr-Werbicka, M. Ferenc, M. Zaborski, B. Gabara, B. Klajnert and M. Bryszewska, Colloids Surf., B, 2011, 83, 360–366 CrossRef CAS PubMed.
- L. Albertazzi, M. Fernandez-Villamarin, R. Riguera and E. Fernandez-Megia, Bioconjugate Chem., 2012, 23, 1059–1068 CrossRef CAS PubMed.
- J. Maly, E. Pedziwiatr-Werbicka, M. Maly, A. Semeradtova, D. Appelhans, A. Danani, M. Zaborski, B. Klajnert and M. Bryszewska, Curr. Med. Chem., 2012, 19, 4708–4719 CrossRef CAS.
- J. Drzewinska, D. Appelhans, B. Voit, M. Bryszewska and B. Klajnert, Biochem. Biophys. Res. Commun., 2012, 427, 197–201 CrossRef CAS PubMed.
- M. Szewczyk, J. Drzewinska, V. Dzmitruk, D. Shcharbin, B. Klajnert, D. Appelhans and M. Bryszewska, J. Phys. Chem. B, 2012, 116, 14525–14532 CrossRef CAS PubMed.
- M. Nahar, T. Dutta, S. Murugesan, A. Asthana, D. Mishra, V. Rajkumar, M. Tare, S. Saraf and N. K. Jain, Crit. Rev. Ther. Drug Carrier Syst., 2006, 23, 259–318 CrossRef CAS.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- K. E. Sapsford, W. Russ Algar, L. Berti, K. Boeneman Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- K. Jain, P. Kesharwani, U. Gupta and N. K. Jain, Biomaterials, 2012, 33, 4166–4186 CrossRef CAS PubMed.
- H. Arima, F. Kihara, F. Hirayama and K. Uekama, Bioconjugate Chem., 2001, 12, 476–484 CrossRef CAS PubMed.
- F. Kihara, H. Arima, T. Tsutsumi, F. Hirayama and K. Uekama, Bioconjugate Chem., 2003, 14, 342–350 CrossRef CAS PubMed.
- K. Wada, H. Arima, T. Tsutsumi, F. Hirayama and K. Uekama, Biol. Pharm. Bull., 2005, 28, 500–505 CAS.
- H. Arima, S. Yamashita, Y. Mori, Y. Hayashi, K. Motoyama, K. Hattori, T. Takeuchi, H. Jono, Y. Ando, F. Hirayama and K. Uekama, J. Controlled Release, 2010, 146, 106–117 CrossRef CAS PubMed.
- K. Motoyama, Y. Mori, S. Yamashita, Y. Hayashi, H. Jono, Y. Ando, F. Hirayama, K. Uekama and H. Arima, J. Inclusion Phenom. Macrocyclic Chem., 2011, 70, 333–338 CrossRef CAS.
Footnote |
| † Authors of this RSC review strongly recommend the nomenclature of “poly(propylene imine) dendrimers” suggested by D. A. Tomalia and M. Rookmaker published, in Polymer Data Handbook, ed. J. E. Mark, Oxford University Press, New York, 2nd edn, 2009, pp. 979–982. The nomenclature for Tomalia-type PAMAM dendrimers and other dendrimer architectures can be adopted also for PPI dendrimers to provide a commonly usable nomenclature and description of all dendrimer structures for predicting the right number of surface groups of a perfect dendrimer. In the literature, most nomenclature of PPI dendrimers is addressed with a higher generation number (+1) in comparison to PAMAM dendrimers. Here, in this review authors use the nomenclature of PPI dendrimers suggested by D. A. Tomalia and M. Rookmaker. This means we present results obtained by 1st to 4th generation PPI dendrimers described in the previous literature as 2nd to 5th generation PPI dendrimers, while in our previous studies, but also in many studies by others the old version of nomenclature for PPI dendrimers (2nd to 5th generation PPI dendrimers) has been used. We motivate all to use the right nomenclature for PPI dendrimers in their future studies. |
| This journal is © The Royal Society of Chemistry 2015 |