
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dendritic nanocarriers based on hyperbranched polymers
Indah Nurita
Kurniasih
,
Juliane
Keilitz
and
Rainer
Haag
*
Institute of Chemistry and Biochemistry, Freie Universität Berlin, Takustr. 3, 14195 Berlin, Germany. E-mail: haag@chemie.fu-berlin.de; Fax: +4930838452611
First published on 18th May 2015
Abstract
Hyperbranched polymers are obtained through one-step polymerization reactions and exhibit properties that are very similar to those of perfect dendrimer analogues. Therefore, hyperbranched polymers are a suitable alternative for perfect dendrimers as building blocks for dendritic nanocarrier systems. With regard to using soluble hyperbranched polymers as carrier systems, their flexible chains are a major benefit as they can adopt and compartment guest molecules. Upon encapsulation, the properties of the host decides the fate of the guest, e.g., solubility, but the host can also shield a guest from the environment and protect it, e.g., from degradation and deactivation. With regard to the advantages of using hyperbranched polymers as nanocarrier systems and their scalable synthesis, we will discuss different types of hyperbranched polymers and their application as nanocarrier systems for drugs, dyes, and other guest molecules.
 Indah Nurita Kurniasih | Dr Indah Nurita Kurniasih obtained her BSc degree in Chemistry at the Diponegoro University, Indonesia. In 2007, she completed her MSc in Polymer Science at the Freie Universität Berlin where she also received her PhD degree under the supervision of Rainer Haag in 2013. Her current research interests include self-assembly, supramolecular architectures and drug delivery systems, with particular focus on hyperbranched polymers. |
 Juliane Keilitz | Dr Juliane Keilitz carried out her graduate research in the group of Prof. Dr Rainer Haag at the Freie Universität Berlin working on polymer-supported homo- and heterogeneous catalysts and received her PhD in 2010. In the same year, she joined Prof. Dr Mark Lautens' group at the University of Toronto (Canada) as a DFG postdoctoral fellow working on enantioselective rhodium-catalyzed domino transformations. In 2012 she returned to Germany and worked as a postdoctoral fellow in Prof. Haag's group before she became an Assistant Editor at Wiley-VCH in 2014. |
 Rainer Haag | Prof. Dr Rainer Haag obtained his PhD with A. de Meijere at the University of Göttingen in 1995. After postdoctoral work with S. V. Ley, University of Cambridge (UK), and G. M. Whitesides, Harvard University, Cambridge (USA), he completed his habilitation at the University of Freiburg in 2002. He then became associate professor at the University of Dortmund and in 2004 was appointed full Professor of organic and macromolecular chemistry at the Freie Universität Berlin. His main research interests are the mimicry of biological systems by functional dendritic polymers, with particular focus on applications in nanomedicine. |
Introduction
Nanocarriers are nanoparticulate materials that are able to bind and/or encapsulate nanosized objects to protect, transport, and ideally release them at a given time or location. Their size ranges from 1–100 nm and they have been constructed from a wide range of materials, including organic (e.g., polymers) as well as inorganic materials (e.g., metal, metal oxide, and nonmetal oxide). Further functionalisation can be used to control and adjust the nanocarrier properties.There is a vast variety of guest molecules that have been transported with nanocarrier systems, including drugs, dyes, imaging agents, catalysts, metal nanoparticles, and enzymes; however, most research done in the field is focussed on medical applications, i.e., drug treatment as well as diagnostic purposes.1,2 Other fields are the stabilization of catalysts, e.g., metal nanoparticles and enzymes.
The major and most important advantages of nanocarriers that apply to all fields of application, is their size in the nanometre range as well as their large surface-to-volume ratio, which favour the adsorption and/or incorporation of payloads. The nanoparticles' size is also helpful with regard to the separation of any nanocarrier system (with and without cargo) from small molecules. A number of easy separation techniques are available, e.g., (membrane) filtration, precipitation, and gel permeation chromatography. All of these make use of the difference in size of small molecules versus nanoparticulate objects.3
With regard to using soluble macromolecules as carrier systems, their flexible chains are a major benefit as they can adopt and compartiment guest molecules. Upon encapsulation, the properties of the host decide about the fate of the guest. The host can shield a guest from the environment and protect it, e.g., from degradation and deactivation. For instance, with the help of an appropriate nanocarrier, hydrophobic drugs can be solubilised and transported in water. The carrier might even be able to transport the drug to a certain site in the body, where it is finally released and can act its cure. Upon encapsulation, it is not only the drug that is protected from its environment, but also the system to which the drug is applied. Usually, the encapsulation of drugs and, ideally, their targeted release, leads to a reduced systemic toxicity of the drugs, which is a consequence of the shielding, but also caused by the fact that smaller amounts of drug have to be applied to achieve the same effect.
Additionally, the high density of functional groups allows to tune the size, solubility, and biocompatibility as well as to attach targeting groups or imaging probes.4,5 Also, more than one guest, e.g., two different drugs or a drug and an imaging agent, can be encapsulated, which might allow for the real-time monitoring of the distribution of a drug in the body.
Furthermore, the particle size is an important factor for the drug transport, because renal excretion through the kidneys as well as the capture by the immune system has to be prevented in order to increase blood circulation times and achieve a therapeutic effect. The threshold of renal clearance is about 45 kDa (4–6 nm in hydrodynamic diameter)6,7 and the immune system especially acts on highly charged and large particles (more than several hundreds of nanometres). Therefore, the ideal size of a nanocarrier for drug delivery purposes is between 10 and 200 nm and it should not bear a charge, i.e., a positive charge.8,9 However, one has to keep in mind, that the carrier system at a certain point should also be excreted from the body. In this context, it is desirable to develop big nanocarrier systems that can degrade after their duty is done. It is self-evident that the resulting fragments, like the carrier itself, have to be biocompatible and nontoxic.
Further advantages of macromolecular nanocarriers are their ability to enhance the uptake of drugs or imaging agents by passive and active targeting.10,11 Passive targeting is possible through the so-called enhanced permeation and retention (EPR) effect:12 tumour and inflamed tissue often exhibits a more porous and disruptive endothelial cell layer due to their rapidly forming vasculature. This allows the entry of large molecules, which accumulate due to the less developed lymphatic drainage system. In contrast, small-molecular compounds easily diffuse into diseased and healthy tissue, in which they can cause severe side effects. Active targeting can be achieved by the attachment of targeting moieties to the surface of the nanocarrier.
The properties of the outer functional groups also dictate the pharmacokinetic behaviour and distribution profile in the body. Additionally, the introduction of (bio)degradability is a field of intense research.
Another example is catalysts, which can be soluble in their non-solvents when they are encapsulated in a carrier. This is not only attractive for being able to use cheaper and more environmentally benign solvents, but might also have an influence on their catalytic activity. Here, the high surface-to-volume ratio as well as the high density of functional groups can have a beneficial influence on the stabilization and activity of catalysts. However, it is usually not desired to transport and release a catalyst, but rather to bind and protect it. Ideally, the application of nanocarriers in this field enables the recovery and recycling of an often expensive catalyst. In some cases an additional beneficial effect was observed due to the high local concentration of catalytic sites at the surface of a dendritic polymer. This so-called dendritic effect can result in higher reaction rates as well as better selectivities as compared to reactions with the corresponding monomeric catalysts.
An important advantage of using macromolecules as carriers for catalysts is the facilitated separation of the polymer-stabilized/bound catalysts from the reaction medium. Applicable methods are, e.g., precipitation, membrane filtration, liquid–liquid extraction, centrifugation, and size exclusion chromatography. The goal is the recycling of expensive catalysts as well as the easy purification of the reaction products from catalyst residues, which can be toxic metal residues. The leaching of catalyst from these polymeric systems is a problem in these applications and needs further improvement.
The two major approaches for the formation of nanocarriers from soluble macromolecules are (1) the self-assembly of amphiphilic block copolymers to form micelles and (2) the synthesis of a multifunctional core molecule that can be equipped with a functional shell (Fig. 1). With the first approach, core–shell structures are obtained, which exhibit a hydrophobic interior surrounded by a hydrophilic shell. This structure can easily encapsulate hydrophobic guest in its interior driven by hydrophobic interactions. A related type of macromolecular self-assembly is vesicles, in which the amphiphile forms a bilayered structure with an aqueous core and a hydrophilic outer shell, which is in contact with the surrounding water. These structures are able to transport hydrophilic guests in the aqueous core as well as hydrophobic guest in the bilayer.
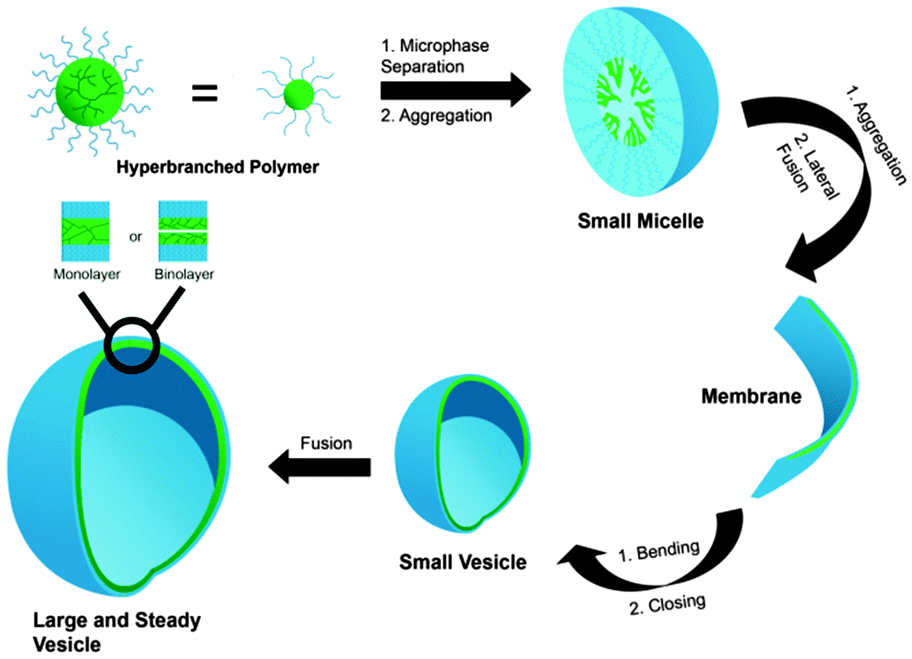 | ||
| Fig. 1 Schematic representation of amphiphilic hyperbranched polymers forming micelles, membrane, and vesicle. Modified from ref. 15. With permission of WILEY. | ||
There are certain drawbacks associated with the use of amphiphilic block copolymers for the construction of self-assembled nanocarriers. First of all, they are very sensitive to their surrounding, i.e., ionic strength, pH, and temperature of the medium or the presence of enzymes.13,14 The second, and most prominent disadvantage is the fact that once they are diluted to a concentration below their critical micelle concentration, micelles usually fall apart if they are not especially stabilized, e.g., by cross-linking of the core and/or shell.
Dendritic polymers are divided into two major classes: perfect dendrimers and hyperbranched polymers. Dendrimers are perfectly branched architectures with a degree of branching of 100% and consequently exhibit a polydispersity index of 1, meaning that they are monodisperse.16–18 Compared with nanocarriers based on amphiphilic block copolymers that exhibit low membrane fluidity, dendritic polymers show a superior stability and toughness, lower permeability and good fluidity owing to less entanglement among the polymer chains. A drawback is their tedious multistep synthesis, which makes them rather expensive and limits their availability. In contrast, randomly branched polymers, so-called hyperbranched polymers, are obtained through fast and cheap one-step polymerization reactions and exhibit properties very similar to those of their perfect dendrimer analogues. Therefore, hyperbranched polymers are a good alternative for perfect dendrimers and their use as nanocarriers will be outlined in more detail in the following.
Many types of hyperbranched polymers are available, e.g., polyesters, polyethers, polyamines and polyamides.19 In addition to carriers solely consisting of the hyperbranched polymer, other architectures are available, e.g., core–shell and core–multishell polymers, linear–hyperbranched hybrid copolymers, and hypergrafted polymers as well as core–shell structures with a hyperbranched core surrounded by a hyperbranched shell (Fig. 2).
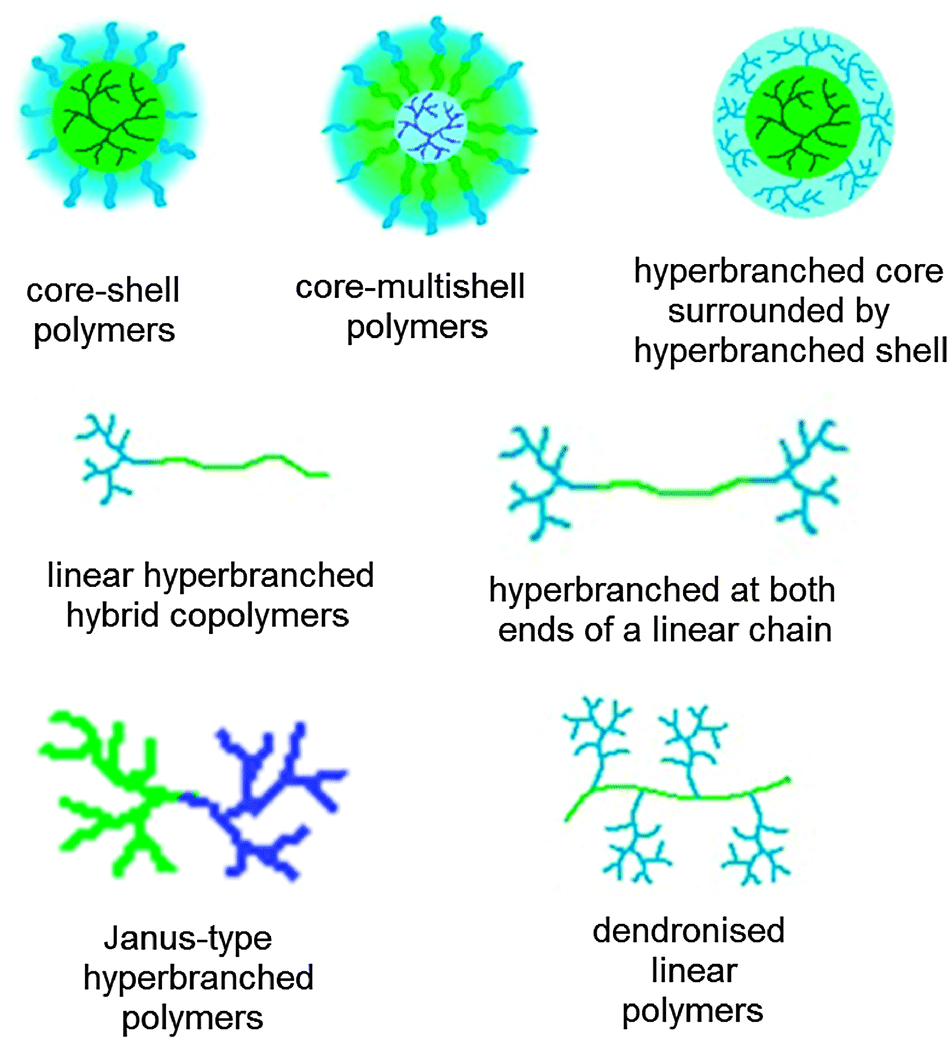 | ||
| Fig. 2 Different architectures of nanocarriers and nanocarrier building blocks containing hyperbranched polymers. The colours indicate a polarity gradient (blue hydrophilic, green hydrophobic). | ||
There are two major ways to transport guest molecules: by covalent binding to the carrier or by encapsulation. In both cases, the guest molecule has to be released at the desired site of action. For covalently bound guest molecules, which are mostly used in drug delivery,20 this involves the cleavage of the bond between the drug precursor and the carrier by an external trigger, e.g., pH, redox potential, enzymatic cleavage, irradiation, and temperature changes.21 Since the active drug is only generated upon release, its precursor cannot do any harm beforehand; however, this approach requires the development of prodrugs as well as carriers that possess appropriate functional groups for coupling, which complicates their synthesis significantly. The alternative is the noncovalent encapsulation of guest molecules by physical interactions. Theoretically, one type of carrier can be used to transport a range of guest molecules, which makes it a more modular approach. This review will concentrate on the second approach and highlight recent advances and developments achieved in the area of noncovalent drug encapsulation.
The transport properties of dendritic systems are influenced by three factors: (1) the core of the carrier, determining its shape; (2) the internal structure, determining the dimension of the internal domain and the interactions between guest molecules and the host; and (3) the outer shell, determining the solubility properties as well as the accessibility of the carriers' interior, thereby influencing the loading as well as release properties.22 The incorporation of guest molecules into macromolecular nanocarriers is based on noncovalent interactions, for example, hydrophobic interactions, hydrogen bonding, π–π interactions between the guest molecule and the nonpolar core, and electrostatic interactions.
The interior of the nanocarrier determines the amount and size as well as the polarity of the guest molecules that can be incorporated. However, the interior does not necessarily have to be hydrophobic in order to encapsulate hydrophobic guests. In case of water-soluble carriers the encapsulation mechanism can also be based on hydrotropic interactions between guest and host.23 Even more important is the influence of the shell properties (size, ionization, density) on the encapsulation. Nowadays, the adjustment of the shell to improve the loading capacity as well as release behaviour has become an important area of research. Also, the shell controls the protein corona that will form, when exposed to biological fluids.
Nanocarriers based on dendritic polymers exhibit an excellent (in vivo) stability; however, for many applications the release of encapsulated guests is desirable. This is especially interesting for drug delivery and can be achieved by different mechanisms, e.g., by diffusion through the polymer matrix, swelling followed by diffusion, or in response to the environment. Such a response can be based on a specific stimulus of chemical, biochemical, or physical origin that leads to a change of the structural composition or conformation of the nanocarrier. Possible triggers are pH, temperature, salt concentration, redox potential, light, ultrasound, or the presence of enzymes.9 The changes can be decomposition, ionization, isomerisation, or depolymerisation.
In the following chapter, recent results obtained in the synthesis, modification, and application of water soluble nanocarrier systems based on hyperbranched polymers will be presented. Special attention is given to the different ways of functionalisation and modification that have been investigated in order to achieve the desired nanocarrier properties. The following chapter is divided according to different types of hyperbranched polymers and the special features of each polymer will be introduced. We will concentrate on nanocarriers constructed from soluble macromolecules, thereby excluding hybrid nanocarriers with solid cores (e.g., with a metal, metal oxide, mesoporous, and magnetic core). Also, we focus on water-soluble systems. However, the reader will realize that it is easily possible to solubilise a wide range of nanoparticles simply by attaching an appropriate functional shell.
Nanocarriers based on different hyperbranched polymer types
Hyperbranched polyethers
Because they are not perfectly branched, hPGs possess two types of hydroxy functionalities: linear monohydroxy and terminal dihydroxy units with the linear ones deriving from the imperfect branching. Statistically, the linear hydroxy groups are closer to the core, whereas the terminal ones are located at the periphery of the molecule.30 Therefore, the hydrophobicity and/or hydrophilicity of hPGs can be tailored by post-synthetic chemical modification of the specific linear or terminal hydroxy groups. Such a fine-tuning can be achieved by applying standard hydroxy group chemistry, thereby transforming them, e.g., to azides, alkynes, and amines, which offers promising options for the preparation of amphiphilic core–shell structures.30,31
Our group reported a series of nanocarriers based on hyperbranched polyglycerol, exhibiting a core–(multi)shell architecture (Fig. 3). In solution (e.g., water and organic solvents), these amphiphilic core–shell nanocarriers either exist as unimolecular micelles or they form multimolecular aggregates. These system are able to increase the solubilisation of drugs and dyes such as nimodipine, pyrene, and Nile red in water.32–38 However, recent findings indicate that the core–multishell (PG-C18-PEG350) nanocarrier forms a multidomain structure that facilitates aggregation (see below).
 | ||
| Fig. 3 (a) Different modification of hyperbranched polyglycerol as nanocarrier systems for drugs/dye delivery applications. (b) Structure of core–multishell (PG-C18-PEG350) based on SANS data and cryo TEM. Modified from ref. 35, 37, 39–42 and 44 with permission from The American Chemical Society, Elsevier B. V. and The Royal Society of Chemistry. | ||
Because many drugs are hydrophobic compounds that cannot be easily administered to the patient, nanocarriers, which are able to encapsulate and transport hydrophobic molecules, are of great interest. When using unimolecular hPG nanocarriers, the introduction of hydrophobic units into the polymer structure is often the only means to enable the transport of hydrophobic compounds. Two major approaches are available, which either introduce the hydrophobicity directly in the core or on the periphery of the hPG core. Kurniasih et al. reported on a series of core–shell architectures based on hPG, in which they made use of the different types of hydroxy groups (linear and branched) of hPG.37 The linear groups in the core were selectively functionalized with aromatic and perfluorinated groups, whereas the peripheral groups were regioselectively PEGylated to improve the solubility of the nanocarrier system. The resulting nanocarrier systems encapsulated Nile red and pyrene by different mechanisms and were also able to release the guest molecules. Pyrene was found to be solubilised in the core of the unimolecular carrier and its release was achieved in the presence of an enzyme, which selectively cleaved the biphenyl esters in the core of the polymer. In contrast, Nile red was solubilised in polymer aggregates, which broke down to single molecules upon dilution, leading to the release of the guest; a behaviour that is similar to that of polymeric micelles. The different encapsulation mechanisms and controlled release properties make these nanocarrier systems promising candidates for the simultaneous delivery of two hydrophobic drugs, which is a current need in combination therapy, e.g., for cancer treatment.37
The second approach of introducing hydrophobicity was inspired by the lipid bilayer of liposomes. These artificial vesicles have a polar core (water) surrounded by a lipid bilayer. In analogy, Haag and co-workers developed core–multishell architectures with polarity gradient from the core to the shell (Fig. 3b).35,43 These so-called core–multishell nanoparticles exhibit a polar interior (hPG), a hydrophobic inner shell, and a hydrophilic outer shell. They are able to solubilise both hydrophobic and hydrophilic guest molecules such as Nile red and Congo red. A careful investigation of the loading behaviour in dependence on the guest and the concentration revealed that the dye loading triggers an aggregation of the core–multishell nanocarriers giving aggregates with radii of 100–120 nm. Such results underline the necessity of systematically studying the influence of various facts that might influence the size of nanocarrier/guest assemblies. Ballauff et al. recently analyzed the structure of these core–multishell nanocarriers using small-angle neutron scattering (SANS) with contrast variation.44 In the nonpolar solvent toluene, the nanocarriers form rather compact structures. A fractal structure with a correspondingly rough surface is found here as well, which is explained by the backfolding of the hydrophilic mPEG chains in the nonpolar environment. In water the nanocarriers associate to form well-defined nanoparticle clusters with an ellipsoidal shape.
This finding is explained by a local demixing of the hydrophobic parts of the polymer multidomain nanoparticles. Therefore, these particles carry hydrophobic patches on their surface which lead to aggregation.
Burt and co-workers also synthesized core–shell structures, whose hPG core was functionalized with C8/10 chains, whereas the periphery was derivatized with methoxy poly(ethylene glycol).45,46 In one study, these polymers were further functionalized with carboxylate groups to bind and deliver the anticancer drug cisplatin.45 A higher amount of carboxylate groups was shown to result in a higher binding efficiency for cisplatin. At mildly acidic pH, the release of cisplatin from the nanocarriers with a high amount of carboxylate groups was significantly slower than from those with a lower density of carboxylates. This offers a convenient handle to tune the release kinetics. The carboxylate functionalized polymers showed good biocompatibility and the drug-loaded analogues were shown to successfully inhibit the proliferation of model bladder cancer cells. In another approach, the same type of polymers were functionalized with amine groups at their periphery.46 The resulting nanocarriers were loaded with docetaxel and also tested in a mouse model of bladder cancer. Upon treatment, no apparent signs of local or systemic toxicity were observed and the nanocarrier-drug formulation showed a significantly higher effectivity in reducing the tumour growth.
Another way of introducing functionality into a hyperbranched carrier was demonstrated by Nowag et al., who presented the functionalisation of hPG derivatives with ethylethylene diamine moieties (Fig. 6).47,48 These groups were introduced to achieve Cu-binding by the formation of square planar bidentate complexes. These interactions are reversible and enabled the pH-dependent release of Cu. These efficient copper nanocarriers were also able to cross an artificial blood brain barrier. Copper levels in the brain supposedly have an influence on Alzheimer's disease and the developed nanocarriers were tested as a treatment option. The carriers itself were shown to have a low toxicity, which makes them good candidates for balancing Cu levels in neurodegenerative diseases (Fig. 4).
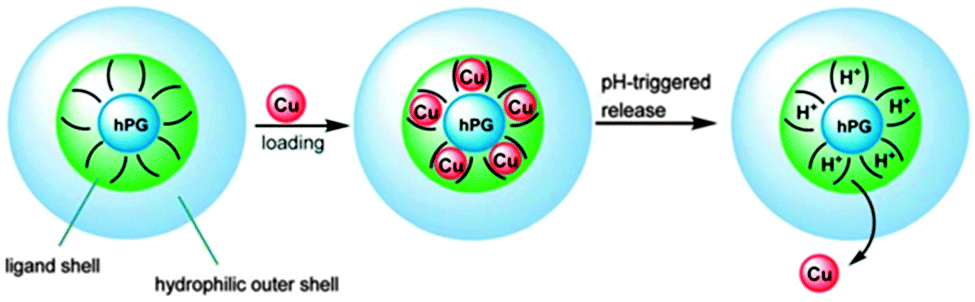 | ||
| Fig. 4 Nanocarriers based on hyperbranched polyglycerols for the transport and release of copper ions. Modified from ref. 47 with permission from The Royal Society of Chemistry. | ||
Wyszogrodzka et al. introduced a convergent approach to biocompatible polyglycerol “click” dendrons for the synthesis of modular core–shell architectures. By applying the Williamson ether synthesis followed by an ozonolysis/reduction procedure, glycerol based dendrons were prepared up to the fourth generation. These structures could be used to observe the structure-transport relation.49 Similarly, a rather robust modular approach for the construction of nanocarrier systems was developed by Zimmerman and co-workers, who generated water-soluble alkyne-functionalized hPGs (Fig. 5).50 They were used for the preparation of potential biological imaging agents. Investigated examples include monofunctionalised fluorescein or biotin hPGs, gold nanoparticles, and unimolecular acid-labile micelles that can be used for the incorporation of nonpolar guest molecules. In this work, the alkyne functional groups served as the universal handle for the modular synthesis, because they could be reacted with any molecule bearing an azide group in a simple click reaction.
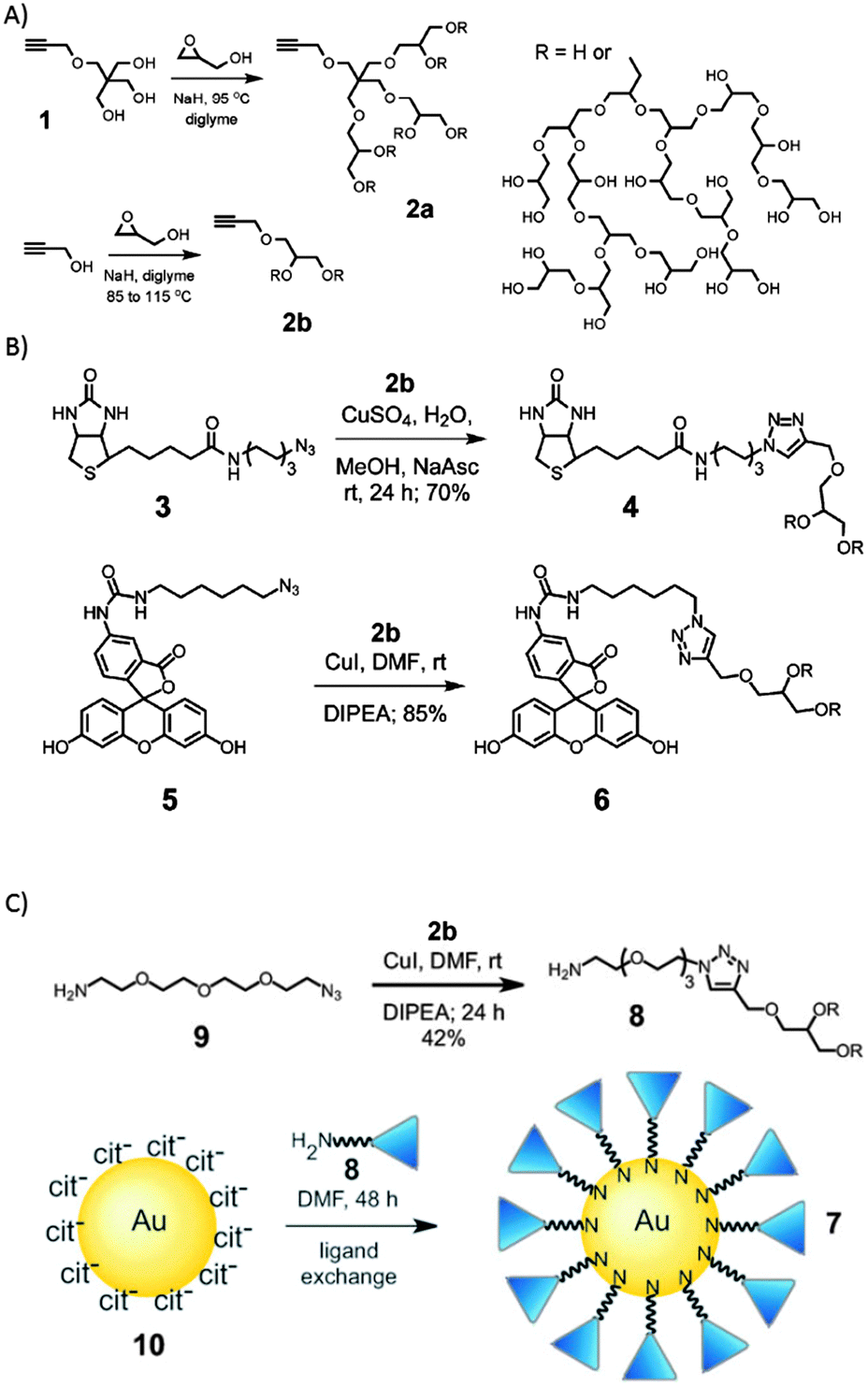 | ||
| Fig. 5 Clickable nanocarrier system based on hPGs. (A) Hyperbranched polymerization to form hPG, (B) click chemistry of hPG and (C) synthesis of hPG coated gold nanoparticles. Modified from ref. 50 with permission from The Royal Society of Chemistry. | ||
To introduce bigger cavities into the hPG-based unimolecular carriers while at the same time generating a more dense shell, Burakowska et al. used hPG as the core and the outer shell, both of which were separated by a long aliphatic hydrophobic inner shell.39 These globular nanocarriers possess the ability to encapsulate polar guest molecules such as Rose bengal and Congo red, but also unpolar compounds such as nimodipine, Nile red, and pyrene. It was shown that nonpolar guest molecules are transported by the unimolecular mechanism, whereas polar guest molecules induced the formation of aggregates with diameters of about 70 nm. The transport capacity was much higher with the aggregates (10 guest molecules per nanocarrier) than compared to the unimolecular system (1.5 guest molecules per nanocarrier).
The transport and delivery of DNA and RNA for therapeutic purposes is a particularly challenging area of research. Here different criteria are necessary to achieve a good transport and at the same time guarantee a high biocompatibility. On the one hand, a sufficiently high density of cationic groups is required to bind the RNA or DNA guest molecules, but on the other hand it is exactly this high density of positive charge that induces a high toxicity of the carrier after the release of the guest. Therefore, it is desirable to be able to precisely control the amount of charge on the nanocarrier. Fischer et al. reported the synthesis of responsive core–shell architectures with a star-like oligoamine shell,51 which were used for siRNA complexation. The variation of shell length and nitrogen content was investigated. In contrast to polyethylenimine (PEI) and other known polyamines used for gene delivery, these novel compounds carry charges only on their surface or shell, whereas the core consists of a highly biocompatible noncharged aliphatic polyether. This overall reduced amount of charge significantly reduced the toxicity of the carriers. The in vitro gene silencing properties were found to be similar to those of positive control samples with a comparable low toxicity. Because of their high biocompatibility, these nanocarrier systems are potential candidates for in vivo applications.
Another important aspect about the use of nanocarriers is the controlled release of the guest molecules under certain external stimuli or at a certain location. The introduction of functional groups that can be cleaved upon the influence of a specific trigger, is a promising strategy to achieve such a controlled release. Haag and co-workers synthesized photolabile cross-linked hPG nanocarriers (Fig. 6).40,41 These nanocarriers were used to selectively and efficiently bind DNA and ionic compounds. In case of the ionic guest molecules it was found that the stability of the resulting host–guest complex depends on the counterion of the guest molecule, but can also be tuned by modification of the different polymer building blocks, in particular the outer shell. The photo-triggered degradation of the nanocarriers was used to release the encapsulated guest molecules.40 In case of the DNA-binding nanocarriers, the binding efficiency depended on the length and density of the conjugated amines. Irradiation of the complexes led to photolytic cleavage and release of the guests.41 These examples show that the use of photolabile nanocarriers could be of particular relevance for biomedical applications, in which the selective binding of guest molecules as well as their controlled triggered release at a certain site is of crucial importance.
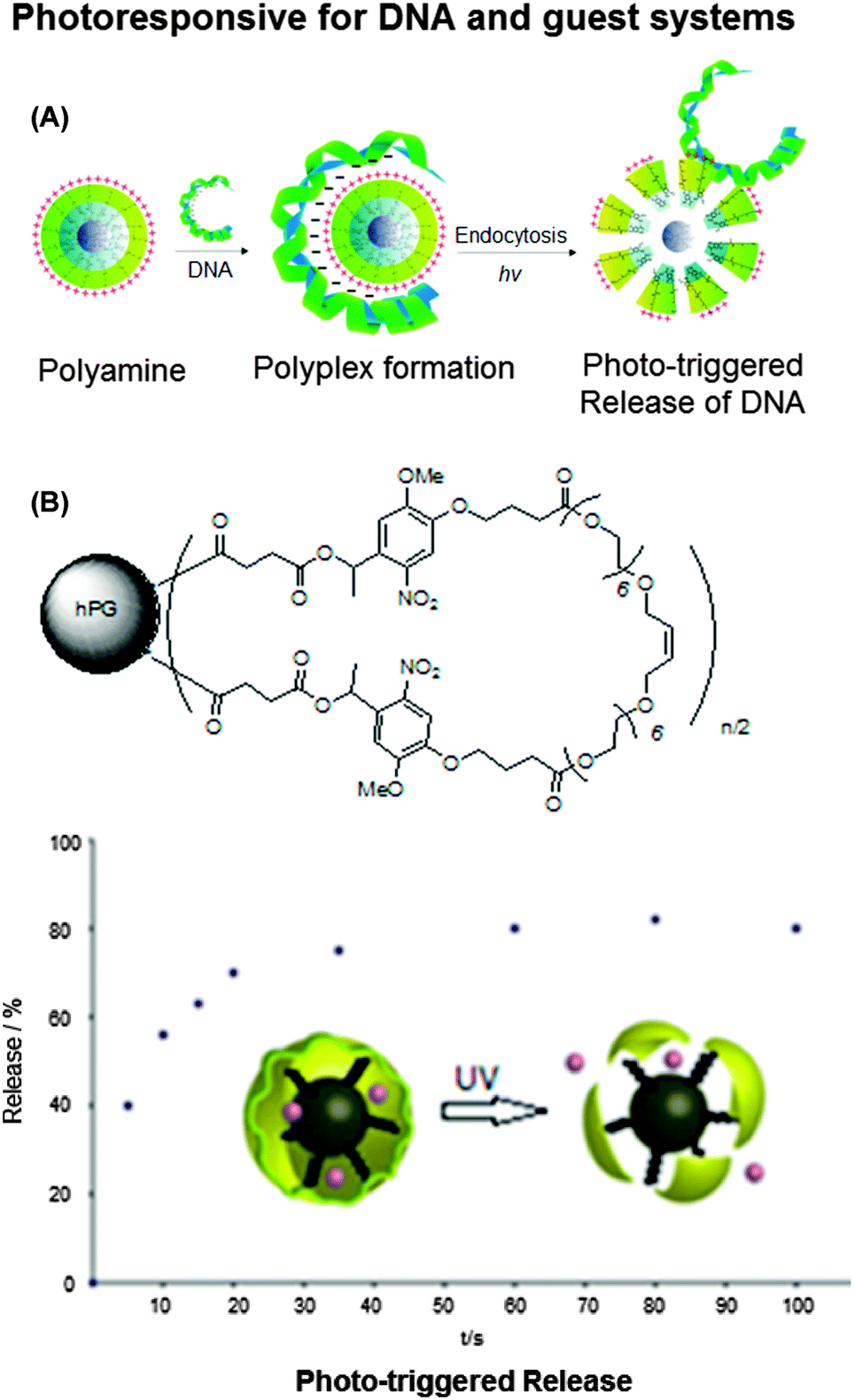 | ||
| Fig. 6 Photoresponsive hyperbranched polyglycerols. (A) Photodegradable hPG amine for reversible DNA complexation and (B) structure of cyclized photoresponsive nanocarrier encapsulation of guest molecules (rose bengal) and release of rose bengal sodium salt from polymer by UV irradiation at λ = 350 nm (upper), and the scheme of DNA–polymer complex formation and release (lower). Modified from ref. 40 and 41 with permission from WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
B.-S. Kim, Y. Shibasaki, and co-workers developed a light-responsive micellar system using spiropyran (SP), a well-known photochromic molecule, as the covalently bound core molecule of hPGs (SP-hPG).52 In aqueous medium, these systems self-assembled to polymeric micelles. However, upon exposure to UV irradiation, the initially hydrophobic SP isomerised to form the hydrophilic zwitterionic merocyanine, leading to the disassembly of the micellar structures. This structural change of the micelles was reversible upon exposure to visible light irradiation. The prospective of the SP-hPG micelle for using it as a smart drug delivery system was investigated using pyrene as a model hydrophobic guest. The study demonstrated the successful loading of the nanocarrier with pyrene and its release upon external UV irradiation. The in vitro cytotoxicity tests with the SP-hPG polymers showed excellent biocompatibility and no toxicity of the SP-hPG micelles.
The same group also reported a one-pot synthesis of a hyperbranched copolymer based on polyglycerol with poly(ethoxyethyl glycidyl ether) (PEEGE) side chains, denoted PEEGE-b-(hPG-co-PEEGE).53 The micelle formation as well as the ability to encapsulate a guest molecule (pyrene) were investigated. Upon a decrease of the pH, the hydrophobic PEEGE moieties were deprotected to yield hydrophilic polyglycerol, leading to the disassembly of the micelles and the release of the guest molecules (Fig. 7). Cytotoxicity tests showed a high biocompatibility, which makes these nanocarriers promising potential drug delivery agents.
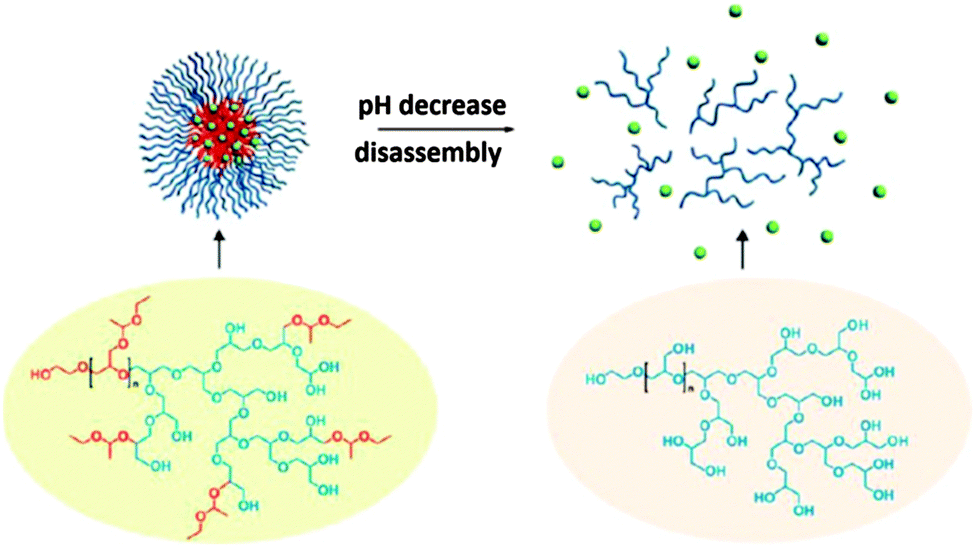 | ||
| Fig. 7 The disassembly of the pyrene-loaded polymeric micelle under acidic conditions. Reprinted with permission from ref. 53; Copyright 2013 American Chemical Society. | ||
I. Kim et al. reported multibranched pentablock copolymers with an inner PEG block, with hyperbranched polyglycerol attached on both ends, followed by poly(L-lactide) [poly(L-lactide)-b-hPG-b-poly(ethylene glycol)-b-hPG-b-poly(L-lactide)].54 The multibranched pentablock copolymers exhibited a good biocompatibility, rendering them suitable biomaterials for various applications in biological medicine areas. The self-assembly of the pentablock copolymers into spherical micelles was well controllable and micelles with average diameters in the range of 60–140 nm were obtained and demonstrated to be easily loaded with drug. The anticancer drug doxorubicin was encapsulated into the copolymer system, and its controlled release was achieved under various pH conditions.
For drug delivery applications, the targeted transport is also a desirable feature of nanocarriers. A targeting ability can be introduced by functionalisation with a targeting moiety. H. Kong and co-workers synthesized bioactive hPGs for the association with stem cells to fit them with the capability of targeting their site of action such as inflamed endothelium.55 The overall process would be analogous to the mobilization of leukocytes to the injured endothelium.
Since the size of a nanocarrier in drug delivery determines where it ends up in the body, how long it stays in the blood stream, and how fast it is cleared from the body, it is highly attractive to develop synthetic procedures that give access to particles from the same type of material in a wide range of different sizes. Sisson et al. reported high-molecular-weight hPG analogues, constructed by the cross-linking of existing hPG macromonomers in miniemulsion.56,57 A nanoreactor template was used to control the size of the resulting nanogels, whereas the cross-linking was achieved by a “click” type Huisgen alkyne/azide cycloaddition reaction. Nanogels in the size range of 20–90 nm were obtained. These systems were used to either covalently link or physically encapsulate dye or drug molecules. The physical encapsulation was performed in dispersion before the cross-linking polymerization.
Steinhilber et al. improved this process by introducing inverse nanoprecipitation approach, leading to hydrogel nanocarriers in the size range of 100–1000 nm (Fig. 8).58 Biodegradability was achieved by introducing benzacetal groups as pH-cleavable linkers. It was shown that at mildly acidic conditions (pH 5), e.g., as in inflamed and tumour tissue, the acetal groups are readily cleaved, resulting in the rapid degradation of the hydrogel network. Under neutral conditions, however, they remained stable. These scaffolds were used to encapsulate the therapeutically relevant enzyme asparaginase with an efficacy of 100%. After release, the structure and function of the enzyme were maintained and it showed full activity. The goal of a follow-up project was to avoid the use of metal ions in the synthesis of these hydrogels, because they could promote the formation of free radicals and damage sensitive biomolecules and living cells. This was achieved by using droplet-based microfluidics in combination with the bioorthogonal strain-promoted azide–alkyne cycloaddition.59 Again, benzacetal linkers were used to introduce degradability under mildly acidic conditions. To achieve a homogeneous and efficient encapsulation of cells, these had to be incorporated upon hydrogel formation. The pH-triggered release of the living cells was shown and it was found that the encapsulation as well as the release had no influence on the cell viability.
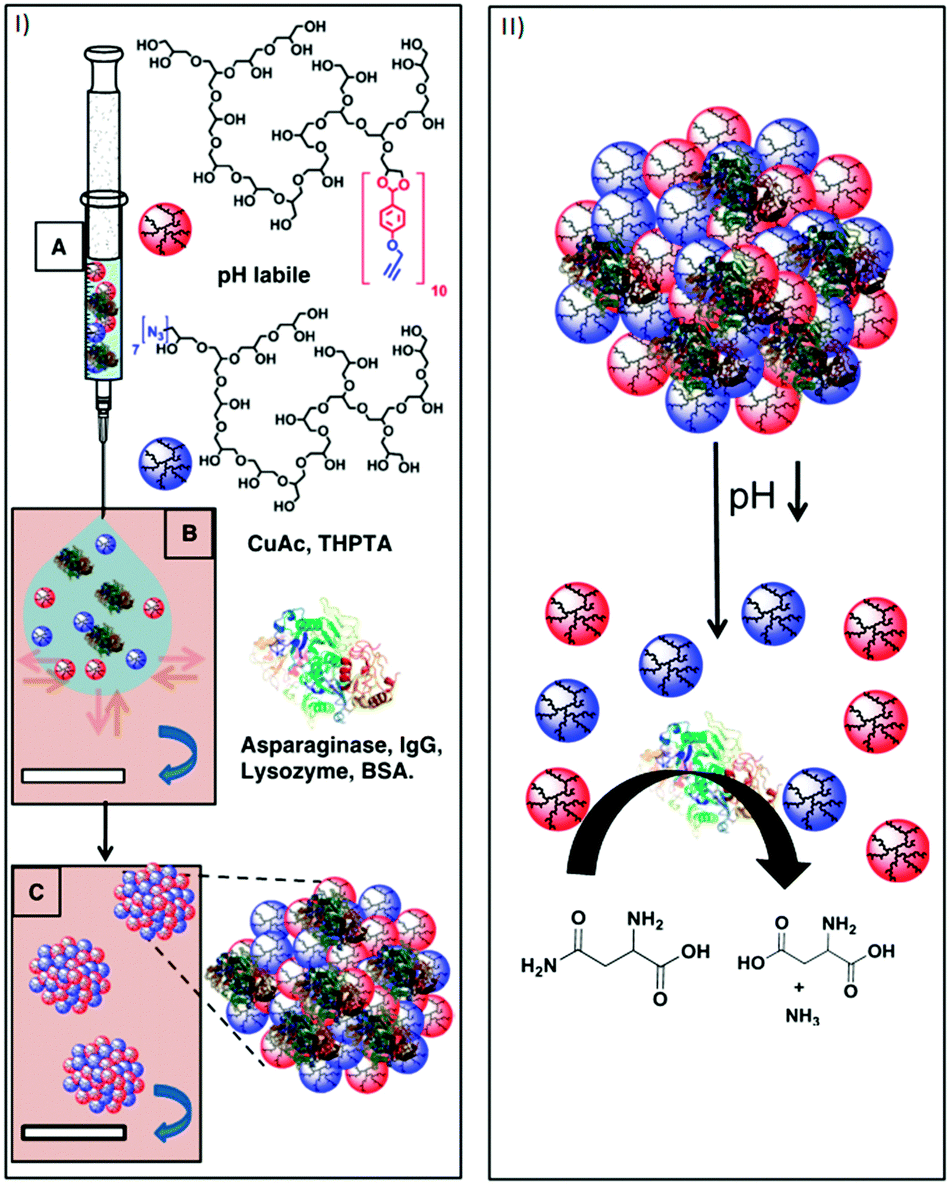 | ||
| Fig. 8 (I) Schematic representation of the nanoprecipitation process. (A) Injection of an aqueous solution of alkyne-functionalized polyglycerol macromonomers (red spheres), azide functionalized polyglycerol macromonomers (blue spheres), and the proteins asparaginase, IgG, lysozyme, and BSA (3D structure of α-helixes and β-turns). (B) Particle templation by diffusion of aqueous phase (blue arrow) into acetone phase (violet). (C) Particle gelation by azide alkyne [2+3] cycloaddition. (II) Representation of the degradation-triggered release of a therapeutically relevant enzyme and its catalytic conversion of asparagine to aspartic acid under ammonia generation. Reprinted with permission from ref. 55; Copyright 2013 Elsevier B. V. | ||
Recently, hyperbranched nanogels for enzyme encapsulation have been reported by C. Wu et al.60 The oxidative cross-linking of hyperbranched polyglycerol (hPG) functionalized with phenol groups hydrogels are successfully catalyzed by horseradish peroxidase. The hydrogel properties can be adjusted by tuning the crosslinking. They are cytocompatible and show potential for encapsulating living cells for regenerative therapy. The gel formation can be triggered by glucose and well controlled under various environment conditions.
When nanocarriers are used for catalytic applications, the requirements are a little bit different, because the catalyst should be encapsulated and stay in the nanocarrier. At the same time, substrates that should be converted by the catalyst have to be able to reach the catalytic site, whereas the products have to be released by the carrier. Keilitz et al. employed hPG-based nanocarrier systems for the synthesis and stabilization of metal nanoparticles, e.g., gold, palladium, and platinum.61–63 Again, core–(multi)shell nanocarriers with a hydrophilic hPG core, a hydrophilic outer shell, and in some cases a hydrophobic inner shell were used. The platinum and palladium nanoparticles supported by these polymeric nanocarriers were shown to be active catalysts for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond hydrogenations.63 The size and stability of the metal nanoparticles depended on the metal to polymer ratio used during their synthesis. A lower metal loading gave smaller particles with higher activity. Additionally, it was shown that the surface of the platinum particles is not blocked by the polymeric stabilizer and it was still possible to functionalize the nanoparticles with chirality-inducing cinchonidine for asymmetric hydrogenation reactions.61
C double bond hydrogenations.63 The size and stability of the metal nanoparticles depended on the metal to polymer ratio used during their synthesis. A lower metal loading gave smaller particles with higher activity. Additionally, it was shown that the surface of the platinum particles is not blocked by the polymeric stabilizer and it was still possible to functionalize the nanoparticles with chirality-inducing cinchonidine for asymmetric hydrogenation reactions.61
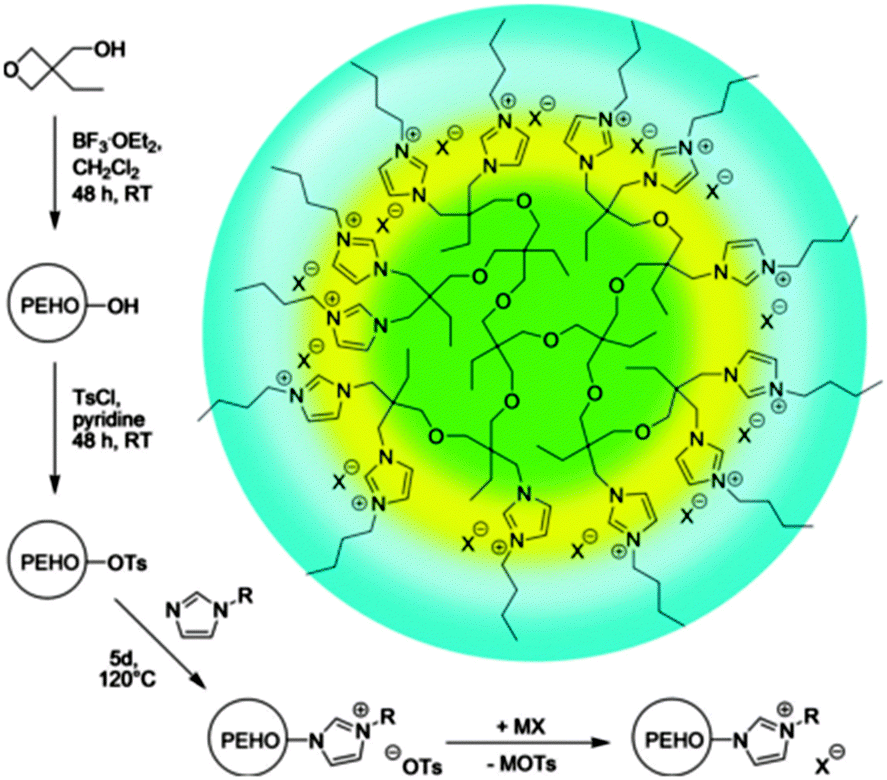 | ||
| Fig. 9 Routes to nanostructured hyperILs with onion-like topology and facile polarity design. Ts = p-toluenesulfonyl (tosyl). Reprinted from ref. 64, Copyright 2013, with permission from WILEY-VCH. | ||
Hyperbranched polyesters
Drug carrier systems that are not degradable may lead to an accumulation in different organs, causing toxic effects or inflammation. Due to this reason, the (bio)degradability of polymeric as well as unimolecular micelles is a desirable property and a crucial parameter for drug delivery applications. Hyperbranched polyesters are an example of biodegradable drug carrier systems. Additional advantages are their low toxicity as well as their nonionic and nonimmunogenic properties.A. M. Nyström et al. reported nanocarrier systems based on hydrophobic Boltorn hyperbranched polyester cores (H30 and H40) with hydrophilic poly(ethylene glycol) shells (PEG5k and PEG10k).66,67 Boltorn is a hyperbranched aliphatic polyester based on 2,2-bis(methylol) propionic acid (bis-MPA) as the monomer unit. Toxicological tests did not indicate toxicity of the resulting nanostructures. Doxorubicin (DOX) was successfully encapsulated in these hyperbranched carriers and encapsulation efficiencies of more than 30% were achieved. The carriers with lower generation Boltorn and shorter PEG chains exhibited a higher capacity for doxorubicin encapsulation. The sustained release of doxorubicin over a prolonged time was achieved and the drug exhibited a higher cytotoxicity compared to free doxorubicin. Additionally, it was shown that the DOX-loaded nanocarriers were internalized into cancer cells.
Similar amphiphilic hyperbranched copolymers with the ability to target bone were synthesized by X. Zhu and co-workers. The hydrophobic hyperbranched polyester core was equipped with a hydrophilic mPEG shell and alendronate (ALE) targeting moieties.68 These amphiphilic polymers were tested as carriers for bone-targeted drug delivery (Fig. 10). In aqueous solution, H40-star-PEG/ALE was found to form micelles, which was confirmed by transmission electron microscopy (TEM) and dynamic light scattering (DLS) measurements. Testing of the H40-star-PEG/ALE micelles with regard to cytotoxicity and hemolysis showed a high biocompatibility of the nanocarriers. Doxorubicin was encapsulated into the H40-star-PEG/ALE micelles as a model anticancer drug. The efficient uptake of the resulting DOX-loaded micelles into cells was shown and a high anticancer activity against HN-6 human head and neck carcinoma cells was found. Since alendronate strongly binds to hydroxyapatite (HA), which was confirmed in a HA binding assay, these nanocarriers can be used to target bone structures for the treatment of skeletal metastases.
 | ||
| Fig. 10 Synthesis of H40-star-PEG/ALE micelles and their application for bone-targeted drug delivery. Reprinted with permission from ref. 68; Copyright 2013 American Chemical Society. | ||
Further biodegradability was introduced by D. Yan et al. by adding labile linkages between core and shell. They synthesized amphiphilic multiarm copolymers with a Boltorn core and hydrophilic polyphosphate arms, which were connected through disulfide linkages that can be cleaved in the presence of reducing agents such as glutathione (GSH).69 The Boltorn H40-star-poly(L-lactide)-disulfide-poly(2-ethoxy-2-oxo-1,3,2-dioxaphos-pholane) (H40-star-PLA-SS-PEP) were able to self-assemble in aqueous solution, thereby forming micelles with an average diameter of 70 nm. An in vitro evaluation showed that upon exposure to a reductive environment, the hydrophilic shell of the micelles could be detached, resulting in a fast release of the encapsulated drug (Fig. 11). An investigation of the intracellular drug delivery against a HELA human cervical carcinoma cell line showed, that the release was faster in HELA cells pretreated with glutathione monoester than in non-pretreated cells. This resulted in a higher cellular proliferation inhibition in glutathione-pretreated cells compared to non-pretreated cells as it was shown with a cytotoxicity assay. The introduction of biodegradability together with responsiveness against a reductive environment is a promising approach to increase the efficiency of hydrophobic drugs.
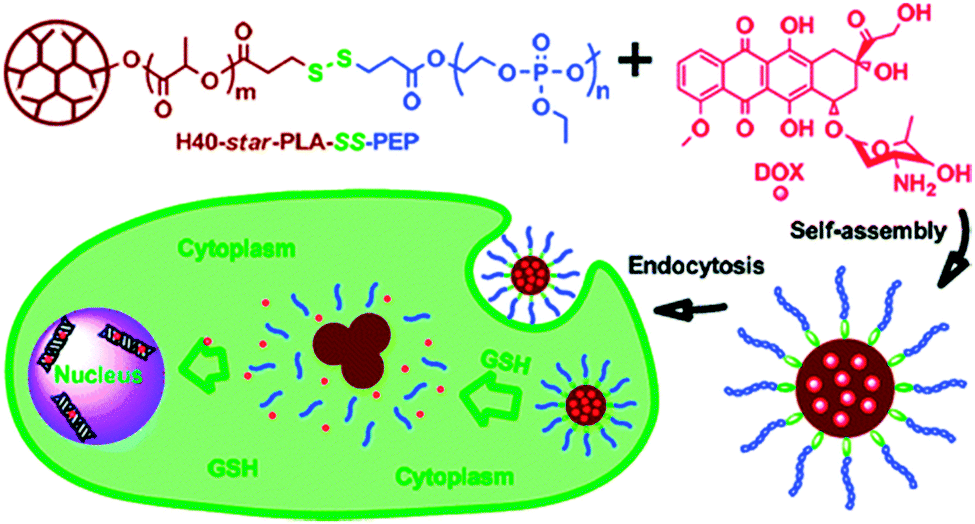 | ||
| Fig. 11 The self-assembly of H40-star-PLA-SS-PEP with doxorubicin. Reprinted with permission from ref. 69; Copyright 2011 American Chemical Society. | ||
Hydrogels with attractive drug release characteristics were prepared by A. Khademhosseini et al., using hyperbranched polyester macromers.70 These nanocarrier systems were used for the encapsulation of the hydrophobic drug dexamethasone within the hydrophobic cavities of the hydrogel and a sustained release of the entrapped drug was reported. The properties of the hydrogel could be tuned by changing the cross-linking density. With regard to an application in cellular therapies (e.g. tissue engineering) the cell adhesion, spreading, and proliferation on the hydrogel surface was investigated and found to be better on the highly cross-linked, and therefore mechanically stiff, hyperbranched polyester hydrogels compared their softer analogues. These results might help in the development of materials that can be tailored to specific functions in tissue engineering applications.
Hyperbranched polyethylene
Hyperbranched polyethylene with globular shape in aqueous solution and molecular dimensions in the nanometre ranges have received broad interest for applications including controlled drug delivery and release, phase transfer, and as unimolecular nanocarriers.Nanocarriers based on hyperbranched polyethylene core and polyethylene glycol shell have been synthesized by Z. Guan and co-worker. Nile red has been used as guest molecules to studied the transport capacity of the nanocarrier systems. In the light-scattering, fluorescence and UV-VIS spectroscopic studies with Nile red in aqueous solution, unimolecular micellar properties of the nanocarrier systems were observed. These molecular nanocarriers can be promising candidates for drug delivery and controlled drug release.71,72
The first tandem coordination, ring-opening, hyperbranching polymerization of ethylene for the efficient synthesis of a unimolecular core–shell nanocarrier was presented by C. S. Popeney et al.73 Their nanocarriers consisted of a dendritic polyethylene (hPE) core with a grafted hPG shell. Due to the high hydrophilicity of hPG, the nanocarrier was water-soluble. It was shown that the core–shell copolymer transports the hydrophobic guests pyrene and Nile red by a unimolecular transport mechanism and achieves an efficient transport of the hydrophobic dye Nile red into living cells under biologically relevant high dilution conditions, which was not possible with the amphiphile-derived micelle (Fig. 12). This work clearly demonstrated the advantages of unimolecular transport systems for the delivery of hydrophobic guests under highly dilute conditions, because this cannot be achieved with self-assembled micellar nanocarriers. Thus, these unimolecular core–shell nanocarriers are promising candidates for the formulation and delivery of poorly water-soluble active agents across cellular membranes.
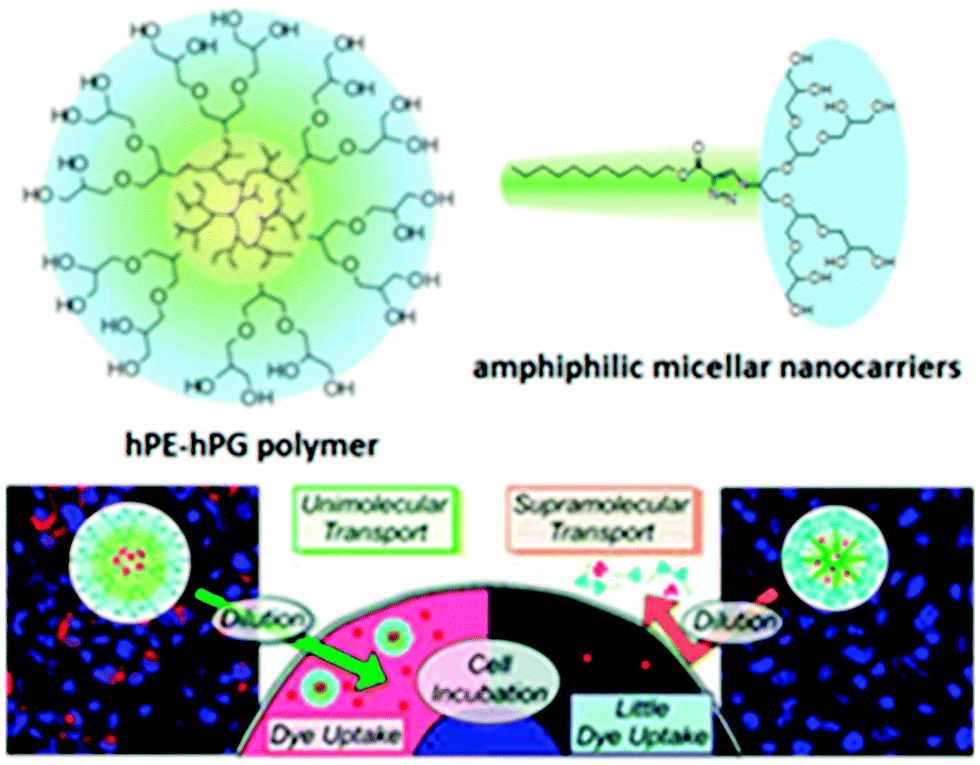 | ||
| Fig. 12 Image of tumour cells showing the fluorescence of Nile red (red) and DAPI nuclear stain (blue) for cells incubated with hPE–hPG unimolecular nanocarriers compared to cells treated with the amphiphile-derived micellar nanocarriers at the same concentration. Reprinted with permission from ref. 73; Copyright 2012 American Chemical Society. | ||
Zhang et al. also showed the encapsulation of coumarin 153 in unimolecular micelles of amphiphilic core–shell polymers obtained by coupling chain walking copolymerization and ATRP (atom-transfer radical polymerisation).74 Oligo(ethyleneglycol)methacrylate (OEGMA) was used as the comonomer to provide both water solubility and thermosensitivity to the unimolecular dendritic polyethylene amphiphiles. The temperature-sensitive phase transition behaviour of these core–shell amphiphiles was investigated by Rayleigh scattering. With increasing hydrophilicity of the polymers, the lower critical solution temperature (LCST) of the polymer solution increases.
Hyperbranched polystyrene
Hyperbranched polystyrene-based nanocarriers were grafted with poly(N-isopropylacrylamide) (PNIPAm) chains by S.-Q. Chen (Fig. 13).75 In THF, these systems exist as star-like polymers. Upon replacement of THF with water, the nanocarriers remained stable as unimolecular carriers; however, the water-insoluble polystyrene collapses, thereby enabling the encapsulation of hydrophobic chemicals. Due to the PNIPAm arms, which is a thermoresponsive polymer, these systems exhibited thermally induced shrinkage with an LCST of around 29–32 °C. As a hydrophobic model compound, pyrene was successfully encapsulated.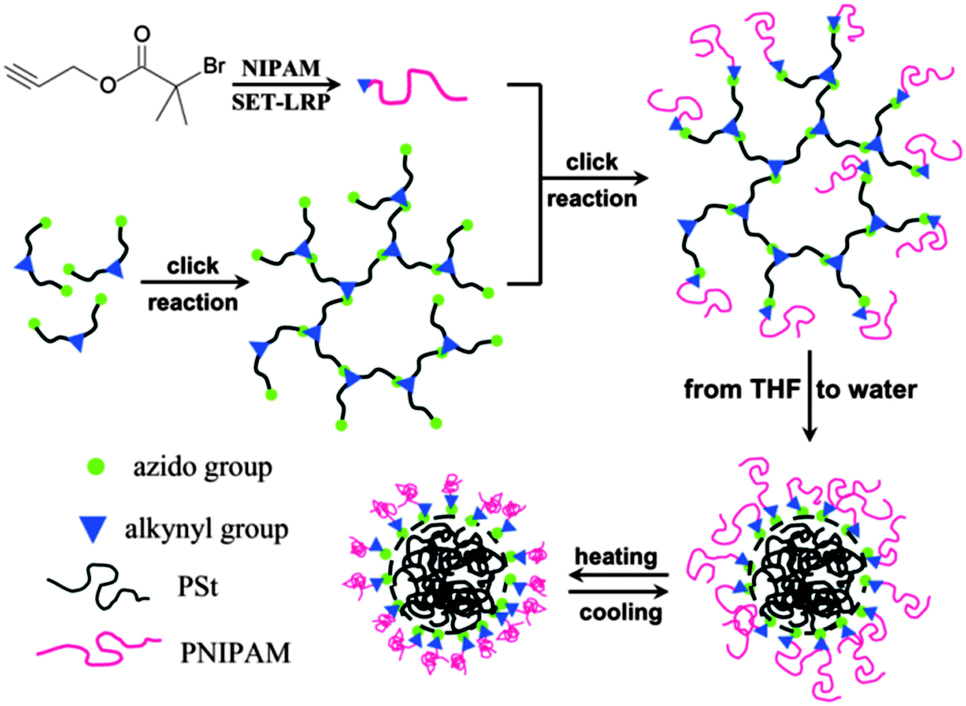 | ||
| Fig. 13 Synthesis strategy for star-like amphiphilic copolymers with lsc–hPS core and PNIPAm grafting chains. Reprinted with permission from ref. 75; Copyright 2013 Wiley Periodicals, Inc. | ||
H. Nagashima et al. reported the synthesis of hyperbranched polystyrenes (hPS) with tunable hydrophobicity/hydrophilicity.76 This was achieved by introducing Cl groups on the surface of the hyperbranched polymer, which were then treated with amines to give hPS–NR3+Cl−. The hydrophilicity/hydrophobicity of the nanocarrier can be tuned by choosing a suitable R group. These systems were employed to synthesize and stabilize platinum nanoparticles, which were used in a biphasic hydrogenation of stilbene and successfully recycled (Fig. 14).
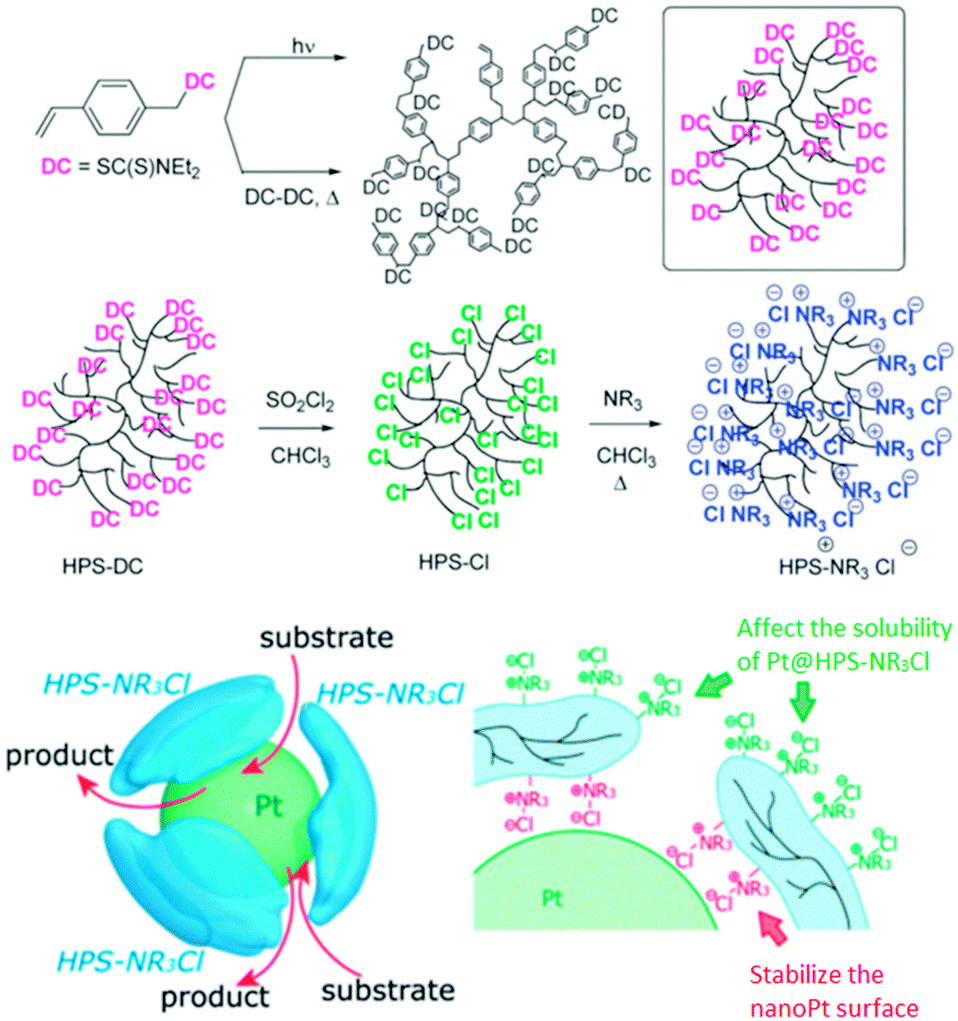 | ||
| Fig. 14 Hyperbranched polystyrene and the stabilization of a platinum nanoparticle by hPS–N(C12H25)3+Cl−. Reproduced from ref. 76 with permission from The Royal Society of Chemistry. | ||
Hyperbranched poly(urea-urethanes)
Biocompatible amphiphilic hyperbranched poly(urea-urethane)s were prepared by T. Loontjens et al. in a three-step one-pot procedure.77 Thereby, the monomer generation, the polymerization, and the functionalisation with mPEG occurs in one reaction vessel. Additionally, the product can be isolated by a simple precipitation step, rendering the overall process very attractive for large-scale syntheses. In water, these polymers formed spherical micelles; the primary micelles had diameters of 12–28 nm, whereas at higher concentration aggregates with a size of about 160 nm were formed. These micelles showed an lower critical solution temperature (LCST) behaviour close to the human body temperature and exhibited low cytotoxicity, which makes them interesting candidates for drug delivery applications.Hyperbranched polyethylenimine
Hyperbranched polyethylene imine (hPEI) can be synthesized by ring-opening polymerization of aziridine. The presence of the high amount of amino groups contributes to the unique properties of these nanocarrier systems such as the ability to simultaneously deliver ionic and apolar guests as well as to release guest molecules upon an external trigger, e.g., pH and solvent. However, the high cellular toxicity of hPEI limits its use for biomedical applications.B. Voit and co-workers studied the interactions of hyperbranched hPEI decorated with various oligosaccharide shells with water-soluble B vitamins (thiamine, riboflavin, cyanocobalamin, niacin, and pyridoxine), an estradiol derivative, and pantoprazole (Fig. 15).78 It was found that the strength of the interaction depends on the properties of the analyte molecules. The strongest driving force for the formation of host–guest complexes are ionic interactions. Additionally, it was shown that the core–shell glyco architecture was a necessary prerequisite for obtaining stable analyte–polymer complexes, because the complexes with pure PEI were not stable under the shear forces applied during ultrafiltration of the aqueous solution without an adjusted pH. This allows the conclusion that robust noncovalent interactions between the guest molecules and the dendritic polyamine scaffold are required to be able to further process these host–guest complexes.
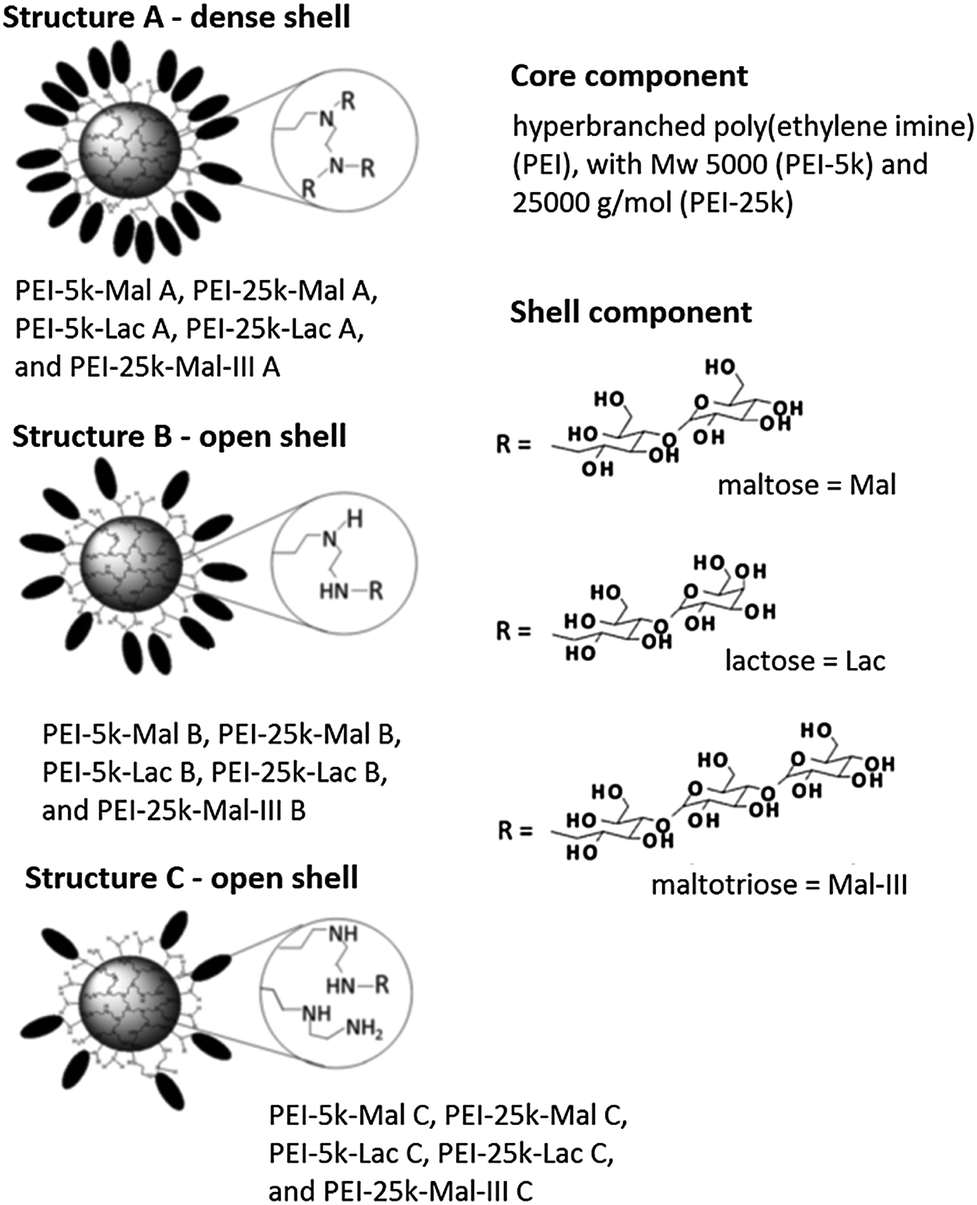 | ||
| Fig. 15 PEI-OS architectures and analytes used in the study by B. Voit and co-workers. Top: the investigated PEI-OS architectures are shown with the three types of oligosaccharides, Mal, Lac, Mal-III, that act as shell components and with two PEI cores, PEI-5k and PEI-25k. The various substructures A, B, and C and the differences in their oligosaccharide shell densities are also illustrated. Reprinted from ref. 78; Copyright 2014, with permission from WILEY-VCH. | ||
Our group synthesized dendritic core–multishell (CMS) architectures based on hPEI as a core, which were later investigated with hPG cores (see above). These nanocarriers enabled the transport of hydrophobic (Nile red, pyrene, nimodipine) and hydrophilic guest molecules (Congo red, Rose Bengal) in both polar and nonpolar solvents (Fig. 16).43 Besides the hPEI core, these nanocarrier systems contained linear amphiphilic building blocks forming a hydrophobic inner shell followed by a hydrophilic monomethyl poly(ethylene glycol) (mPEG) outer shell. This external polar layer provides good solubility in water as well as in organic solvents and a high degree of biocompatibility. In addition, this nanocarrier system needs to self-assemble to bigger aggregates in order to act as a carrier for guest molecules. A unimolecular transport was not observed. Because of their unique properties, these CMS nanoparticles already found various biomedical applications, e.g., for the in vivo targeting of a F9 teratocarcinoma tumour and the modulation of the copper level in eukaryotic cells79,80 as well as for enhancing the skin penetration ability of rhodamine B and Nile red.81 It was also shown that the CMS nanotransporters can benefit from the enhanced permeation and retention effect (EPR effect), leading to an accumulation of the nanocarrier–drug complex in diseased tissue resulting in the more selective delivery of their payload.80
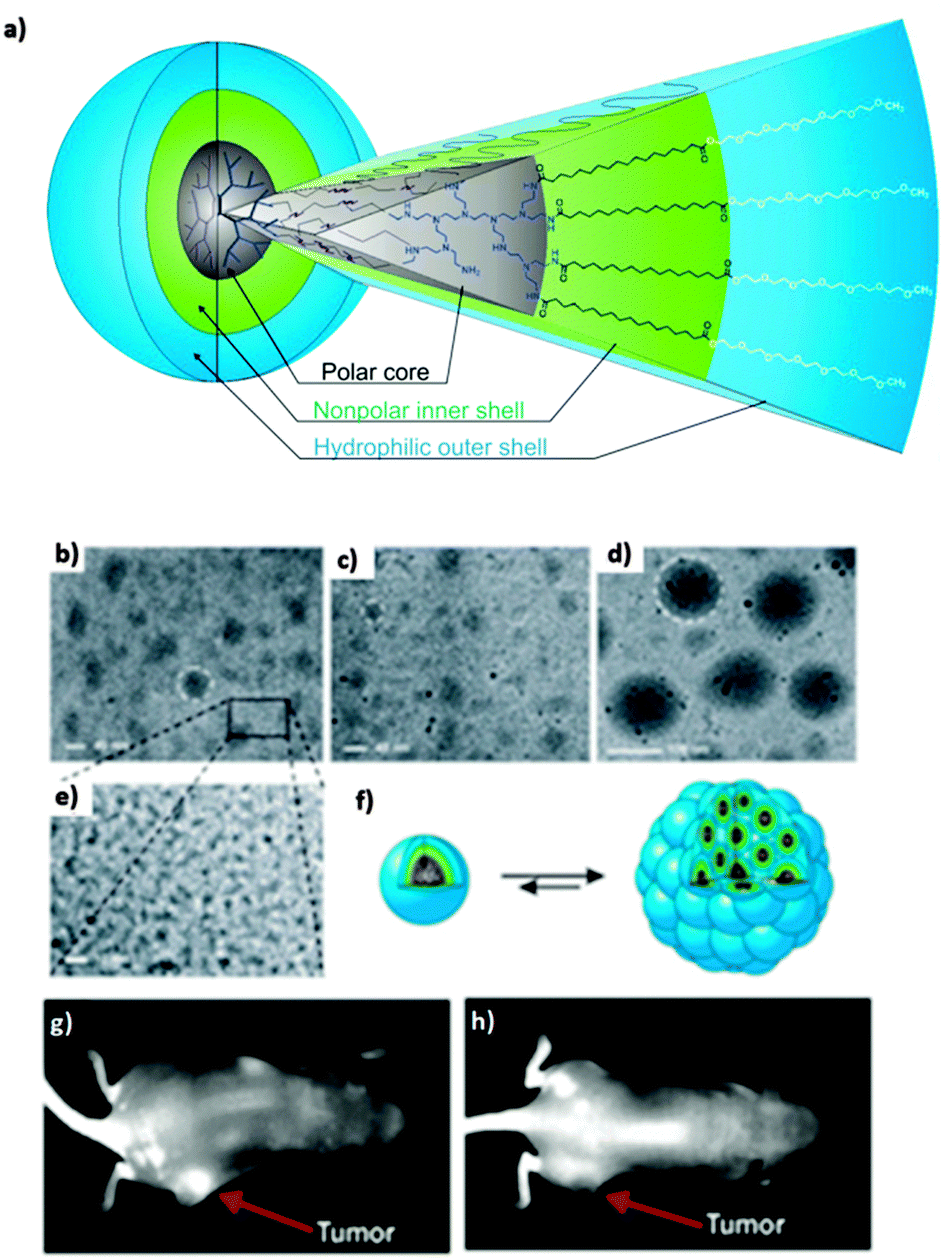 | ||
| Fig. 16 (a) Dendritic core–multishell (CMS) architectures based on hPEI. CryoTEM measurement (b)–(e) of CMS based hPEI with 1 mg mL−1 polymer concentration in pure water. The size of the unimers was determined to be about 5 nm. (b) Aggregate diameters for pure multishell nanocarriers were determined to be in a range of 30–50 nm. (c) After encapsulation of nimodipine the size changed to diameters of 20–30 nm. (d) Encapsulation of β-carotene increased the size of aggregates to 110–130 nm. (e) Enlargement from (d). (f) Model of a unimer (left) and supramolecular aggregate (right). (g) Strong contrast was observed after 6 hours of administration of indotricarbocyanine tetrasulfate (ITCC) dye loaded CMS nanoparticles to F9 tetracarcinoma (tumor tissue) bearing mice. (h) Contrast achieved with free dye after the same time period is not very prominent Reprinted from ref. 43 and 80; Copyright 2007, with permission from WILEY-VCH and Elsevier B. V. | ||
H. Pu et al. also reported a unimolecular micelle that is able to deliver polar (Rose bengal, methyl orange) as well as apolar (pyrene) guest molecules. The macromolecular nanocapsules were based on hyperbranched polyethylenimine (hPEI) with a hybrid shell of poly(ethylene glycol) monomethyl ether (mPEG) and polystyrene (PS).82 Depending on the ratio of mPEG and PS, the nanocapsules are soluble in a wide range of solvents as well as in water. Their structure depends on the chosen solvent. In water, they exist as onion-like particles with three layers (hPEI-PS-PEG), whereas in chloroform a hybrid shell particle (hPEI-PEG/PS) is present. In all cases, these particles exist as unimolecular micelles. Their ability to act as nanotransporters was demonstrated in biphasic mixtures of water and chloroform, in which the nanoparticles can transfer anionic water-soluble guest molecules from the aqueous to the chloroform phase. In aqueous solution, the nanocarriers can be used to capture ionic as well as apolar compounds. It was also shown, that guest molecules can be released upon an external stimulus, which could either be a change in pH or a switch of the solvent.
A novel supramolecular complex based on hPEI was reported by Y. Chen et al.83 The simple mixing of hPEI with a solution of amphiphilic calix[4]arene (AC4), containing four aliphatic chains and four hydroxy groups in chloroform yielded a supramolecular complex, which formed through the noncovalent interaction of amino groups of hPEI with the phenol groups of the calix[4]arene (Fig. 17). The cationic water-soluble dye methyl blue and the anionic water-soluble dye methyl orange were used as model guest molecules to test the performance of hPEI-AC4 as a supramolecular nanocarrier. It was found that the water-soluble methyl orange is incorporated in the core of hPEI-AC4, whereas the cationic water-soluble methyl blue is accommodated within the AC4 moieties on the surface. The encapsulation capacity for methyl orange was pH dependent and reached its highest value at weakly acidic conditions. When the pH value was outside the range of about 4.5 to 9.5, reversible release of the guest was observed. The overall amount of encapsulated methyl orange and methyl blue could be successfully controlled by the ratio of the hydroxy groups of AC4 to the amino groups of hPEI.
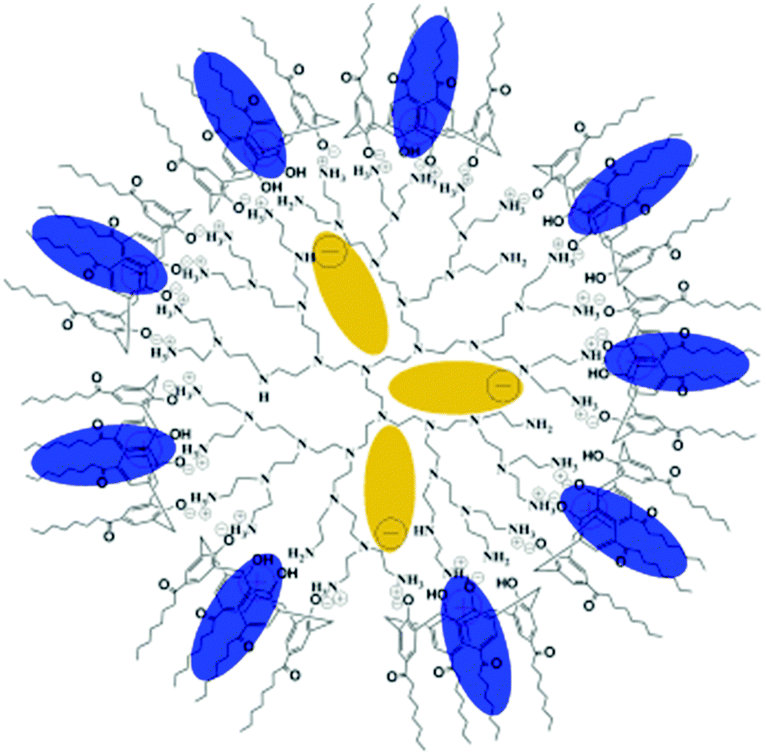 | ||
| Fig. 17 Structure of supramolecular hPEI-AC4 nanocarriers with encapsulated guests in isolated sites. Blue ellipses indicate methyl blue and dark yellow ellipses indicate methyl orange. Reprinted with permission from ref. 83; Copyright 2009 WILEY-VCH. | ||
The attachment of biodegradable and biocompatible polypeptides onto a hyperbranched PEI resulted in water-soluble amphiphilic star-block-copolypeptides as reported by D. Liu et al.84 As the inner hydrophobic shell, polyphenylalanine or polyleucine was used, whereas the outer polyglutamate shell was negatively charged. The polymers formed well-defined supramolecular aggregates with very low critical aggregation constants (cac). In contrast to conventional micelles derived from surfactants or linear amphiphilic block copolymer, these systems are very stable upon dilution. It was shown that various compounds ranging from hydrophobic to hydrophilic guest molecules, can be encapsulated simultaneously.
S.-C. Jiang, Y. Chen, and co-workers reported amphiphilic hyperbranched copolymers based on a hPEI core equipped with generation 1 and 2 dendrons with two and four aliphatic tails, respectively.85 These systems were used for the encapsulation of hydrophilic guests, including methyl orange, Congo red, and Direct Blue 15, into the hydrophilic PEI core and showed higher encapsulation capacities than analogues with a linear shell. This study showed the potential of enhancing the encapsulation capacity by introducing branching units into the outer shell building blocks, thereby generating a much denser outer shell with at the same time greater cavities. Additionally, the presence of bigger cavities enables the encapsulation of bitter guest molecules like Congo red and Direct Blue 15.
A multicompartment release system, which combines the advantages of dendritic architectures and hydrogels was described by N. Polykarpov et al.86 The developed hydrogel contains dendritic glycopolymers as nanocontainers, which can be employed as a delivery system for drug molecules. The glycopolymer is based on a hPEI core with a maltose shell (PEI-Mal) and as a guest molecule adenosine triphosphate disodium salt (ATP) was used. It was shown that the uptake and release of ATP from the hyperbranched host depends on the dendritic glycostructure and the pH and can be fine-tuned. It was possible to achieve a selective release only of ATP or the simultaneous release of ATP and the glycopolymer scaffold. This multicompartment release system has the potential to enhance the desired delivery features in complex biological environments with changing pH values.
Hyperbranched poly(amido amine)s (hPAAs)
Hyperbranched poly(amido amine)s (hPAAs) containing different amounts of β-cyclodextrin (hPAA-CDs) were synthesized by Chen et al. (Fig. 18).87 These nanocarriers were used as nonviral gene delivery agents. It was shown that the cellular uptake of the polymer–DNA complexes was very fast, resulting in transfection efficiencies similar to those observed with pure hPAAs. The inner cavities of the nanocarriers were used to additionally encapsulate drugs. Therefore, these systems have potential application, e.g., for the combination of gene therapy and chemotherapy.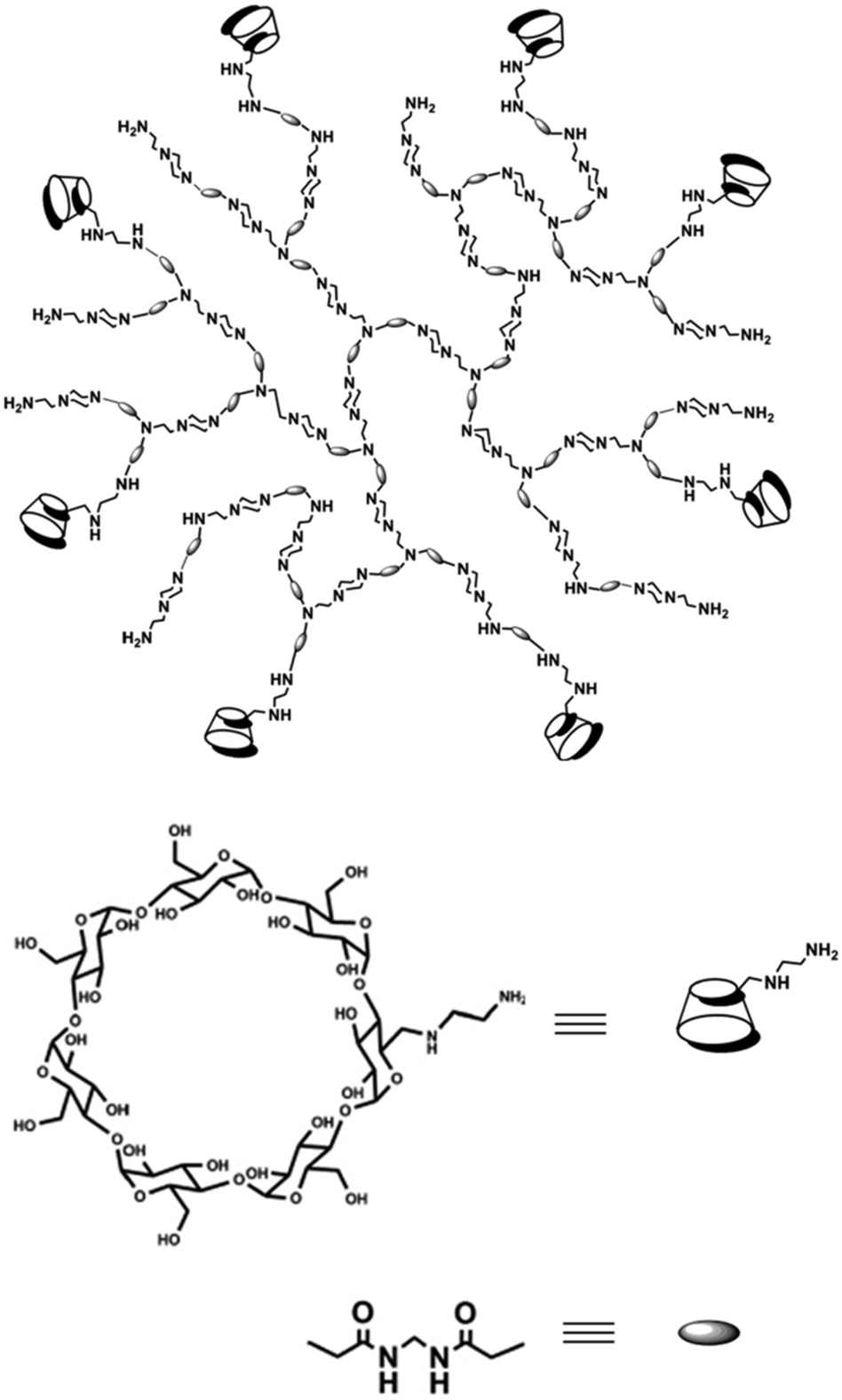 | ||
| Fig. 18 Structure of hPAA-CD polymers. Reproduced from ref. 87; Copyright 2011, with permission from the American Chemical Society. | ||
Y. Liu et al. presented redox-responsive gene delivery systems based on hyperbranched poly(amido amine)s (hPAAs).88 These nanocarrier systems contained a tertiary amino core and different terminal functional groups (amine, PEG, and hydroxy). Although all polymers possessed the same redox-responsive core, the differences of the structure of the terminal functional groups resulted in a clear distinction in terms of biophysical properties and transfection efficiency in vitro. hPAAs terminated with 1-(2-aminoethyl)piperazine were found to be the most efficient for binding DNA. The core was found to degrade rapidly upon cleavage of the disulfide bonds, resulting in improved transfection efficiency. Overall, the study has shown that the degradation of the gene delivery agent plays an important role in achieving high transfection efficiencies and a low cytotoxicity.
Heterogeneous catalysts based on water soluble metal nanoparticles have been developed and exhibited high efficiency and environmental friendly properties.89 These metal nanoparticle catalysts can be stable under shear forces and dilution. Poly(amido amine) (PAMAM) dendrimers have been shown to be perfect nanoreactors and stabilizers for metal nanoparticles.90,91 In contrast to those perfectly branched but costly monodisperse dendrimers, hyperbranched polymers analogous to PAMAM dendrimers called HYPAM are easily accessible.92,93
J.-D. Marty et al. presented the synthesis and characterization of platinum nanoparticles stabilized by hyperbranched poly(amido amine) (HYPAM) (Fig. 19).94 These metal nanoparticles are suitable for use as reusable homogeneous hydrogenation catalyst in water under mild conditions. S. Saliba et al. used core–shell and core–multishell nanocarrier systems based on hyperbranched poly(amido amine) polymers (HYPAM) for the formation and stabilization of ZnO nanoparticles.95 These nanocarrier systems contained a hyperbranched poly(amido amine) core with a core–shell or a core–multishell architecture based on PEG derivatives with poly(ethylene glycol) as the outer shell. Due to this outer shell, the polymer–nanoparticle complexes could be easily redispersed in various solvents with the ZnO nanoparticles showing stable photoluminescent properties.
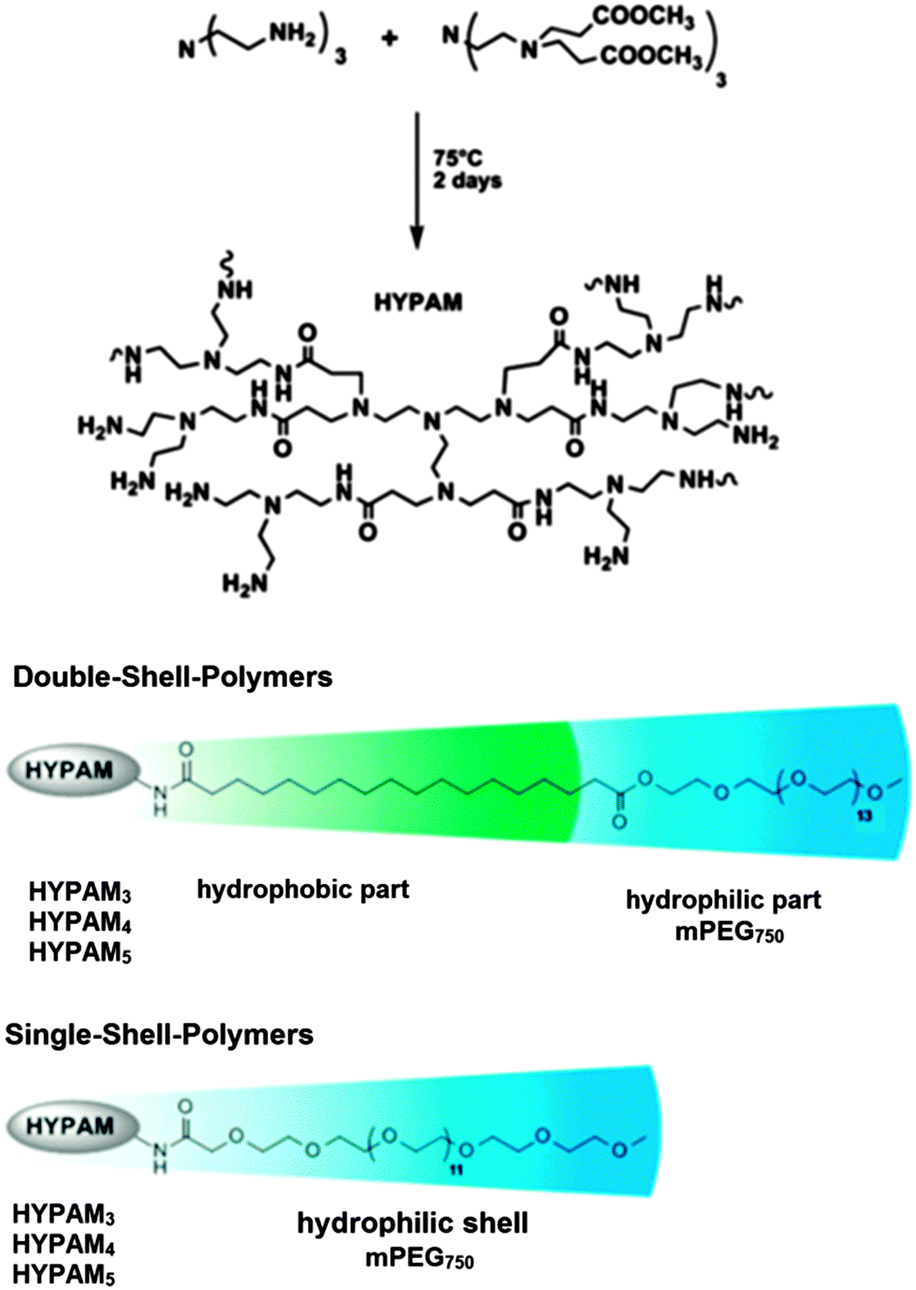 | ||
| Fig. 19 Structure of hyperbranched poly(amido amine) (HYPAM) polymers. Adapted from ref. 94 with permission from the American Chemical Society and The Royal Society of Chemistry. | ||
Branched thermotropic liquid crystals were successfully obtained from ionic interactions between hyperbranched poly(amido amine) (HYPAM) and sodium dodecylsulfate by H. H. Nguyen et al.96 Polarized optical microscopy, differential scanning calorimetry, and small-angle X-ray scattering showed the complex formation of columnar rectangular and lamellar thermotropic mesophases. The liquid crystalline phase was then used for the in situ formation of gold nanoparticles. The templating effect of the liquid crystal mesophase resulted in the formation of isotropic nanoparticles, the size of which was dictated by the local organization of the mesophase and by the molar mass of the hyperbranched complex. The presence of NPs induced strong changes in the local organization of the LC polymer, but nevertheless the obtained hybrid materials remained liquid crystalline.
Hyperbranched polyphosphates
Polyphosphates are an important class of eminent biomaterials. Due to their high biocompatibility, biodegradability, and flexibility, they are widely used in biomedicine. A series of phosphorus-containing dendrimers have been synthesized by A.-M. Caminade and co-workers. They have shown that phosphorus-containing dendrimers are useful for biological purposes, in particular as transfection agents,97 anti-prion (including in vivo)98 and anti-HIV agents,99 against Alzheimer diseases,100 as imaging agents (including in vivo),101 for the activation and multiplication of human monocytes and innate immune natural killer (NK) cells,102 and as vehicle for ocular drug delivery of carteolol.103Since phosphate-based dendrimers are synthesized by multistep reactions, they are rather expensive nanocarriers, and their hyperbranched analogues present a suitable alternative. D. Yan and co-workers constructed full-polyphosphate nanocarriers through the self-assembly of amphiphilic hyperbranched multiarm copolymers.104 The hydrophilic core and the hydrophobic arms of the polymers (called HPHEEP-star-PPEPs) consist of hyperbranched and linear polyphosphates, respectively. In aqueous medium, these copolymers self-assemble to give nanocarriers with controlled sizes from 48 to 74 nm. Their size can be controlled by adjusting the length of the hydrophobic arm. It was shown that the nanocarriers are highly biocompatible and are easily taken up by cells. The treatment of a breast cancer cell line with the chlorambucil-loaded nanocarriers in vitro achieved a higher proliferation inhibition than free chlorambucil.
To further improve the nanocarrier systems, the same group introduced disulfide groups into the backbone of similar amphiphilic copolyphosphates (HPHSEP-star-PEPx).105 The resulting nanosized micellar drug delivery systems exhibited smart redox-responsive behavior (Fig. 20) as well as good biocompatibility and biodegradability. In water, self-assembly forms spherical micelles with tunable sizes from 70 to 100 nm. Under reductive environment, the micelles are destroyed, allowing a triggered drug release. This was demonstrated by using doxorubicin-loaded HPHSEP-star-PEPx nanocarriers against a HELA human cervical carcinoma cell line. It was found that the doxorubicin-loaded nanocarriers showed a higher cellular proliferation inhibition in a reductive environment (glutathione monoester).
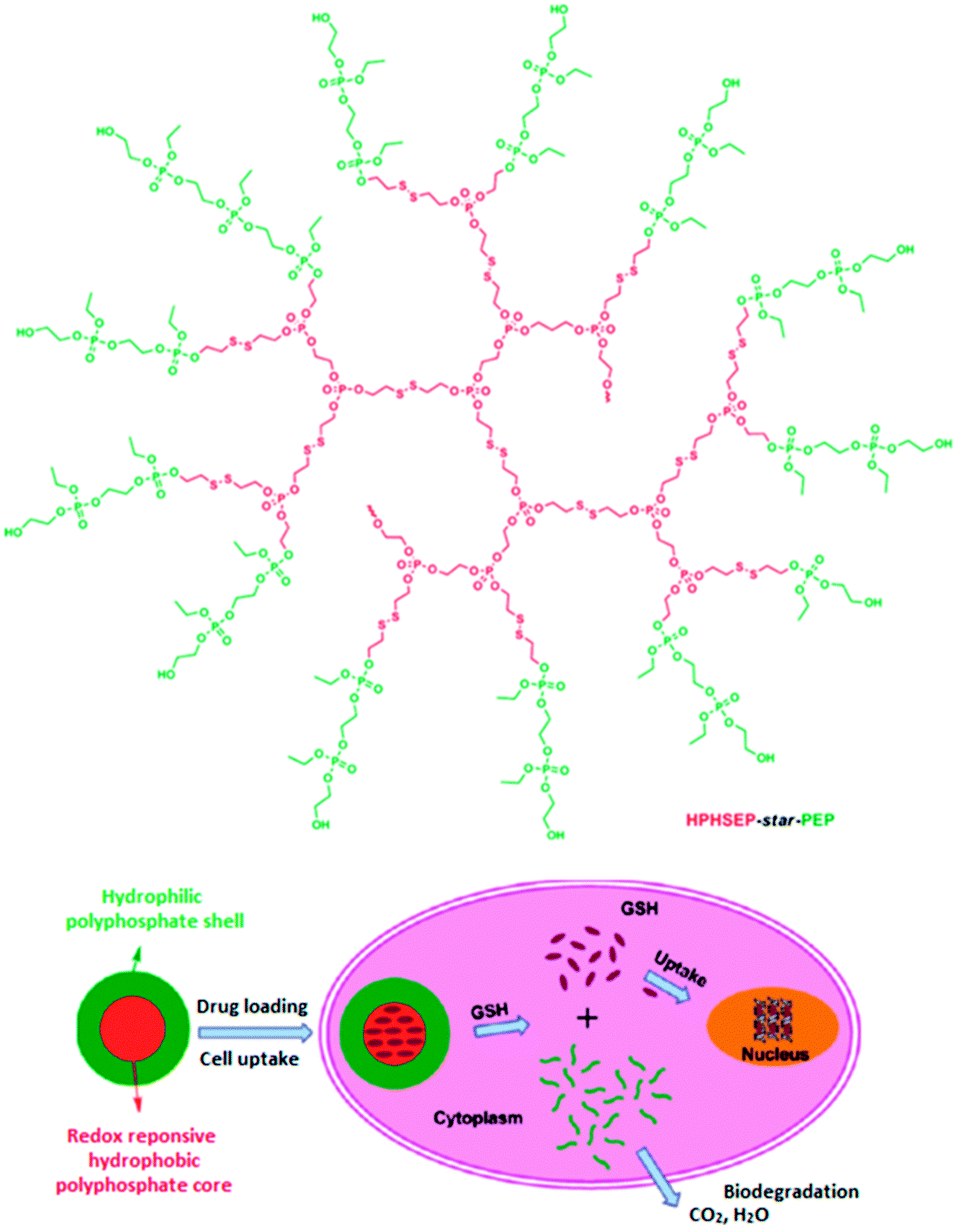 | ||
| Fig. 20 Structure of the amphiphilic hyperbranched copolyphosphates (upper part). Representation of the redox-responsive behavior of HPHSEP-star-PEPx micelles for intracellular drug release triggered by glutathione. Adapted with permission from ref. 105; Copyright 2011 American Chemical Society. | ||
J. Ji and co-workers successfully prepared biocompatible photo-responsive biocompatible micelles consisting of an amphiphilic hyperbranched polyphosphate.106 The light-responsiveness was achieved by attaching 2-diazo-1,2-naphthoquinone-5-sulfonyl chloride (DNQ) groups. In aqueous solution, this polymer forms micelles with average sizes around 180 nm. As a model drug, coumarin 102 was encapsulated and shown to be released in a controlled manner under UV irradiation at 365 nm.
The synthesis of biocompatible and biodegradable nanogels with a branched structure through a miniemulsion process involving an ionic liquid was developed by J. Wang et al. (Fig. 21).107 These nanogels were shown to easily encapsulate doxorubicin and the release could be triggered by an enzyme. The efficient uptake of these nanocarriers was shown for a human breast cancer cell line. Additionally, surface functionalisation was demonstrated by attaching a fluorescein dye and it was found that the nanogels are able to prevent protein adsorption.
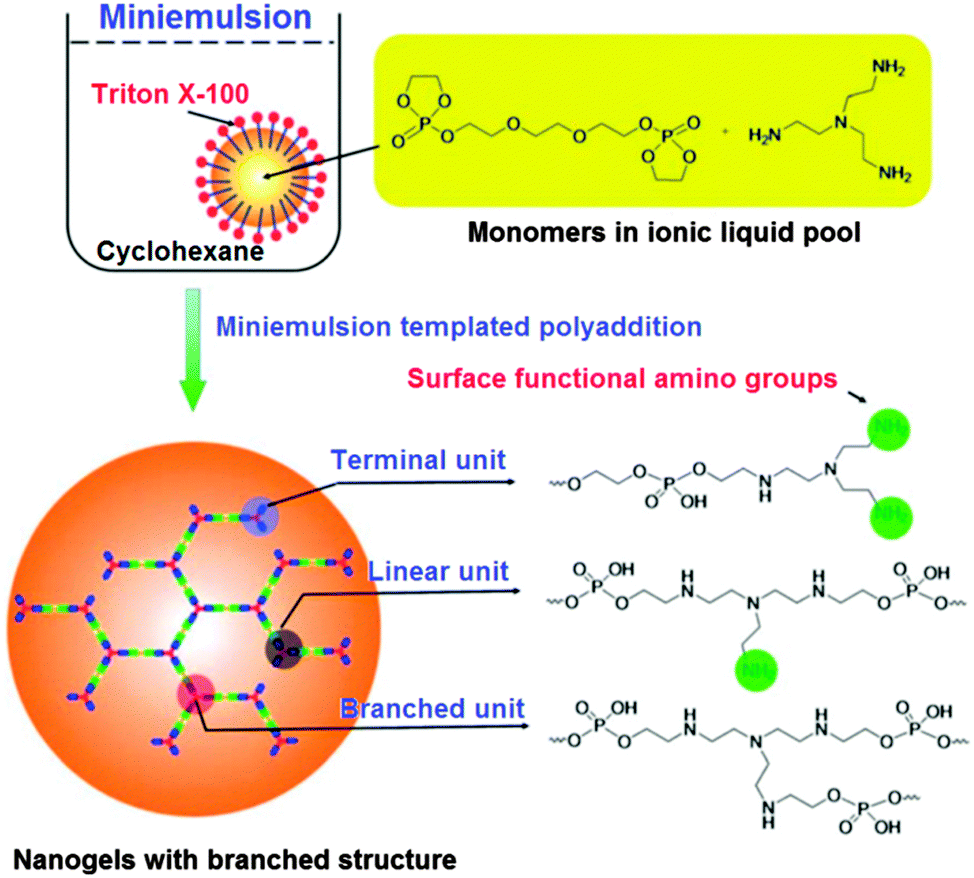 | ||
| Fig. 21 The synthesis of nanogels based on polyphosphates. Reproduced with permission from ref. 107; Copyright 2012 The Royal Society of Chemistry. | ||
Other hyperbranched polymers
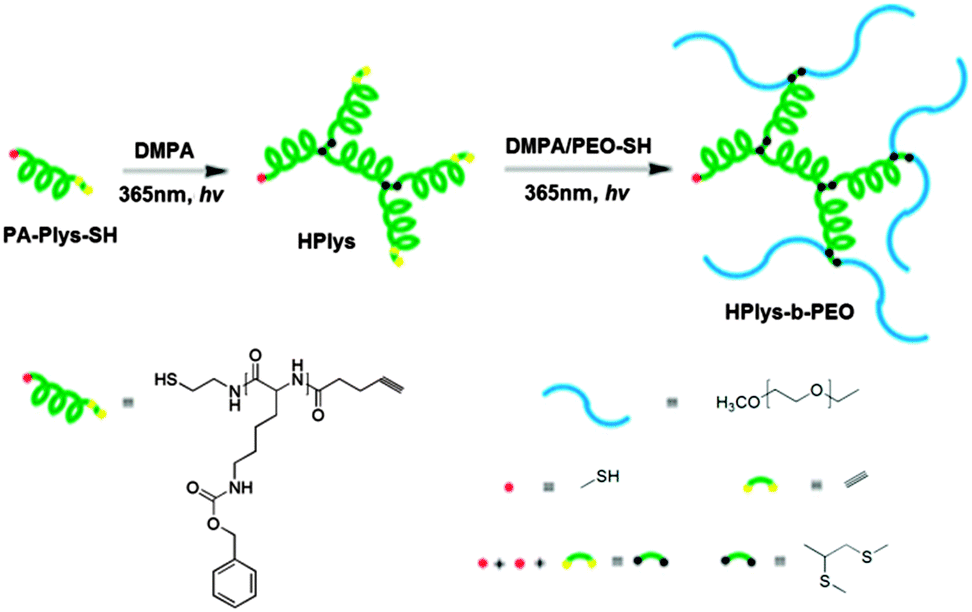 | ||
| Fig. 22 Hyperbranched poly(ε-benzyloxycarbonyl-L-lysine) (hPlys) and the related hPlys-b-PEO block copolymer. Reproduced with permission from ref. 108; Copyright 2013 American Chemical Society. | ||
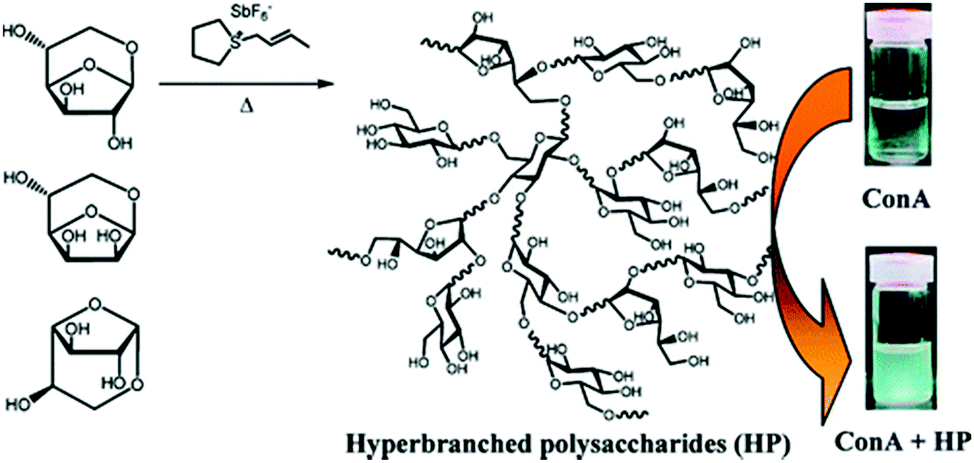 | ||
| Fig. 23 Hyperbranched polysaccharides complexes with Concanavalin A.109 Copyright 2011, with permission from the American Chemical Society. | ||
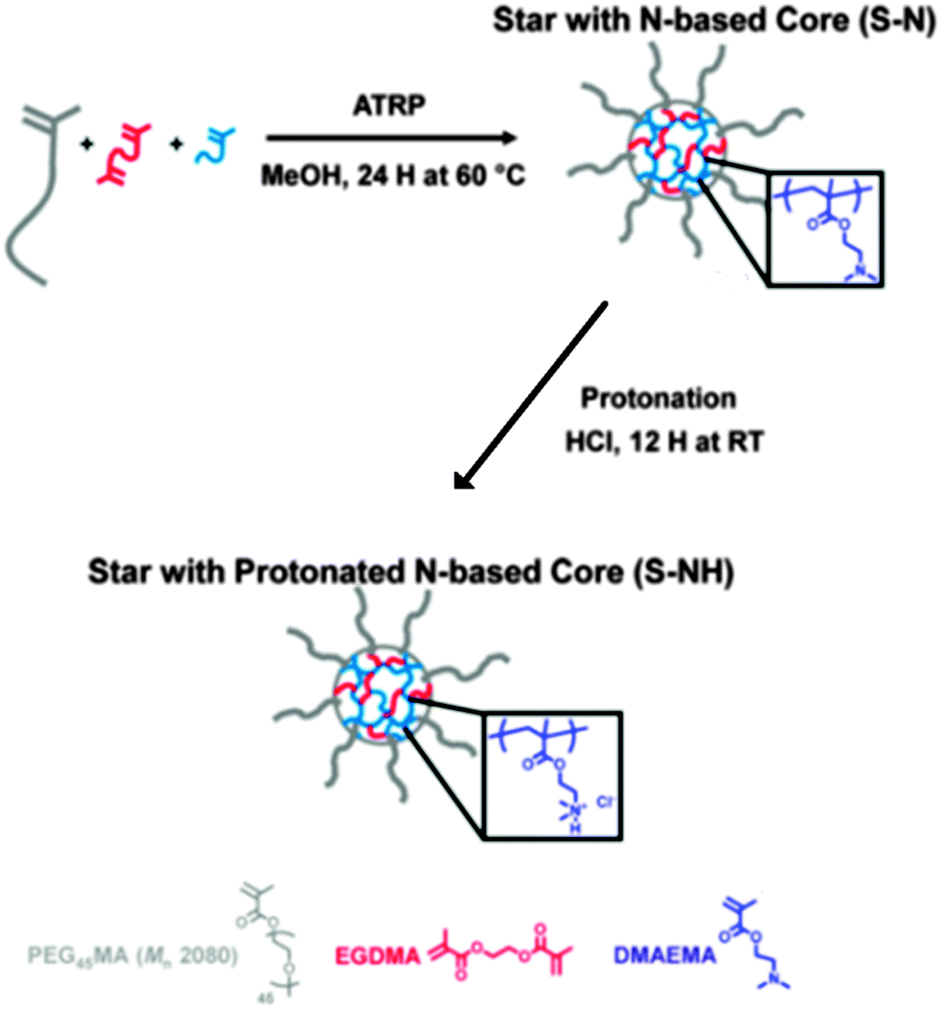 | ||
| Fig. 24 Synthesis of star polymers with a cationic core (S–NH) for siRNA or pDNA delivery by ATRP. Reproduced with permission from ref. 110; Copyright 2013 American Chemical Society. | ||
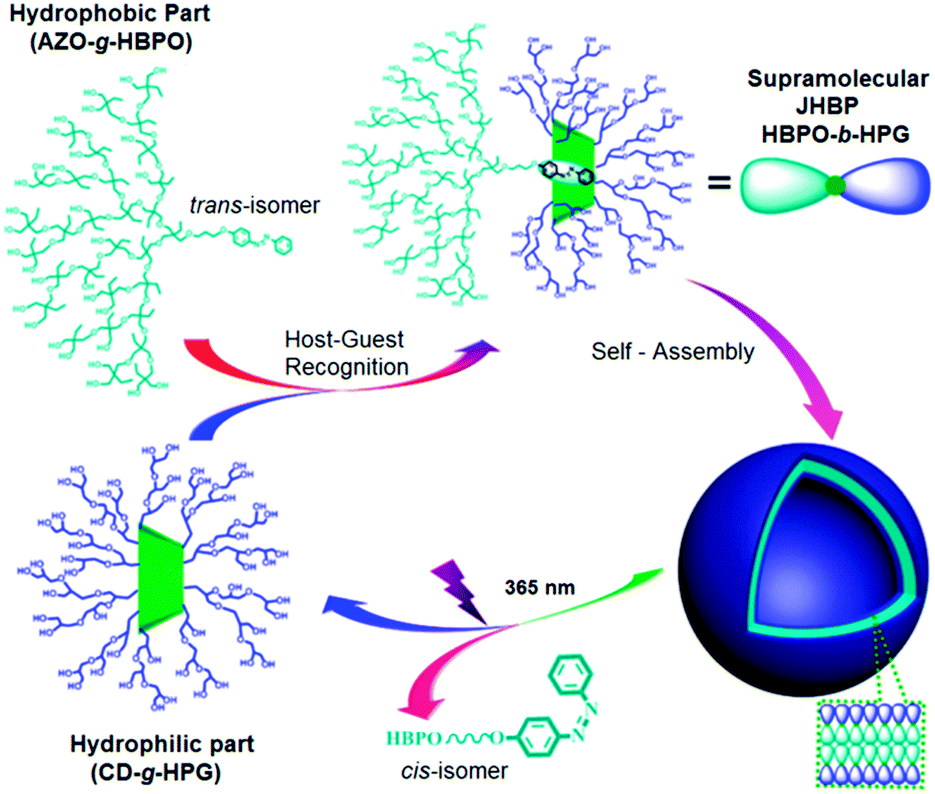 | ||
| Fig. 25 Self-assembly and disassembly processes of the supramolecular Janus hyperbranched polymer of HBPO-b-HPG. Reprinted with permission from ref. 112; Copyright 2013 American Chemical Society. | ||
T. Satoh et al. introduced a hydrophilic nanocapsule based on a hPEI core modified with β-cyclodextrin (CD) and a poly(ethylene oxide) (PEO) shell.113 These nanocarrier systems were shown to efficiently encapsulate the hydrophobic guest molecule pyrene and exhibited unique release properties: the release can be chemically triggered on-demand and depends on the amount of the chemical that is used as the stimulus. Additionally, the carriers showed a high pH sensitivity. The chemoselective release is attributed to a synergic effect of the structural features of the nanocapsule, which a single building block (cyclodextrin or the covalent micelle) does not possess alone. It was reasoned that the synergic effect results from the entrapped pyrene, which is partially exposed from the cyclodextrin moiety and therefore sensitive to the environment.
Conclusions
The choice of the optimal hyperbranched nanocarrier certainly depends on the desired application; however, some requirements apply in general. These are the controllable synthesis (with regard to the reproducibility, size, dispersity, functional groups), an easy functionalisation to adjust the properties, a high loading capacity for the desired guest molecule, good solubility in the required medium, high stability, reasonable price. Also the actual architecture of the nanocarrier or aggregates in water plays a major role. The different types of nanocarrier aggregates as well as the properties of nanocarriers are yet poorly understood and need further investigation.• For the most common application, i.e., drug delivery, the most stringent requirements apply. These are, for example, high biocompatibility of the nanocarrier as well as of all possible degradation products, the degradability and/or elimination of the nanocarrier after the delivery, high stability on the shelf as well as under physiological conditions, targeting of the diseased tissue.
• Further relevant parameters are increased blood circulation times; controlled release mechanism, high uptake efficiency, high loading capacity, low aggregation tendency or controllable aggregation; triggered release; nontoxic, nonimmunogenic, cheap production. The self-assembly behaviour of hyperbranched polymers in the presence of the guest molecules, such as drugs, dye, proteins, DNA/RNA as well as buffer systems/serum also need more investigations to develop more efficient nanocarrier systems under physiological conditions.
• Nanocarrier systems also can be used as carriers for catalysts, the major advantage being facilitated separation of the polymer-stabilized/bound catalyst from the reaction medium. By using nanocarrier systems, expensive catalysts (metals, enzymes) can be recycled in a straight forward manner. The purification of reaction products from catalyst residues can be accommodated.
The future of hyperbranched nanocarriers is bright as they will move more into applications with the ease of synthesis and scale up being the major driving forces. The different topologies, flexibilities and composition of nanocarrier system are expected to extend the categories and functionalities of the here presented functional hyperbranced polymers.
Acknowledgements
The authors thank SFB 1112 and the Helmholtz portfolio Topic on Multimodal Imaging for financial support. The authors also thank Dr Wiebke Fischer for the modification of Fig. 1.Notes and references
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- C. Y. Ang, S. Y. Tan and Y. Zhao, Org. Biomol. Chem., 2014, 12, 4776–4806 CAS.
- C. C. Tzschucke, C. Markert, W. Bannwarth, S. Roller, A. Hebel and R. Haag, Angew. Chem., Int. Ed., 2002, 41, 3964–4000 CrossRef CAS.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- C. M. Paleos, D. Tsiourvas, Z. Sideratou and L.-A. Tziveleka, Expert Opin. Drug Delivery, 2010, 7, 1387–1398 CrossRef CAS PubMed.
- L. W. Seymour, R. Duncan, J. Strohalm and J. Kopeček, J. Biomed. Mater. Res., 1987, 1341–1358 CrossRef CAS PubMed.
- H. Soo Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef PubMed.
- S. K. Hobbs, W. L. Monsky, F. Yuan, W. G. Roberts, L. Griffith, V. P. Torchilin and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4607–4612 CrossRef CAS.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- S. Svenson, Curr. Opin. Solid State Mater. Sci., 2012, 16, 287–294 CrossRef CAS.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 5757–5766 CrossRef CAS PubMed.
- U. Agrawal, M. Gupta, R. S. Jadon, R. Sharma and S. P. Vyas, Drug Delivery Transl. Res., 2013, 3, 479–497 CrossRef CAS PubMed.
- Y. Wang, B. Li, H. Jin, Y. Zhou, Z. Lu and D. Yan, Chem. – Asian J., 2014, 9, 2281 CrossRef CAS PubMed.
- A. D. Schlüter and J. P. Rabe, Angew. Chem., Int. Ed., 2000, 39, 864–883 CrossRef.
- G. R. Newkome, G. N. Moorefield and F. Vögtle, Dendrimers and Dendrons, Concepts, Syntheses, Perspectives, Wiley-VCH, Weinheim, Germany, 2001 Search PubMed.
- D.-L. Jiang and T. Aida, Prog. Polym. Sci., 2005, 30, 403–422 CrossRef CAS.
- D. Yan, C. Gao and H. Frey, Hyperbranched polymers: Syntheses, properties and Applications, J. Wiley Publishers, New York, London, 2011 Search PubMed.
- J. Kopeček, Adv. Drug Delivery Rev., 2013, 65, 49–59 CrossRef PubMed.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS PubMed.
- A. Kowalczuk, R. Trzcinska, B. Trzebicka, A. H. E. Müller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2014, 39, 43–86 CrossRef CAS.
- T. Ooya, J. Lee and K. Park, Bioconjugate Chem., 2004, 15, 1221–1229 CrossRef CAS PubMed.
- R. Tokar, P. Kubisa, S. Penczek and A. Dworak, Macromolecules, 1994, 27, 320–322 CrossRef CAS.
- A. Sunder, R. Hanselmann, H. Frey and R. Mülhaupt, Macromolecules, 1999, 32, 4240–4246 CrossRef CAS.
- A. Sunder, R. Mülhaupt, R. Haag and H. Frey, Adv. Mater., 2000, 12, 235–239 CrossRef CAS.
- R. K. Kainthan, J. Janzen, E. Levin, D. V. Devine and D. E. Brooks, Biomacromolecules, 2006, 7, 703–709 CrossRef CAS PubMed.
- R. K. Kainthan and D. E. Brooks, Biomaterials, 2007, 28, 4779–4787 CrossRef CAS PubMed.
- R. K. Kainthan, C. Mugabe, H. M. Burt and D. E. Brooks, Biomacromolecules, 2008, 9, 886–895 CrossRef CAS PubMed.
- R. Haag, J.-F. Stumbé, A. Sunder, H. Frey and A. Hebel, Macromolecules, 2000, 33, 8158–8166 CrossRef CAS.
- S. Roller, H. Zhou and R. Haag, Mol. Diversity, 2005, 9, 305–316 CrossRef CAS PubMed.
- H. Türk, A. Shukla, P. C. A. Rodrigues, H. Rehage and R. Haag, Chem. – Eur. J., 2007, 13, 4187–4196 CrossRef PubMed.
- S. Xu, Y. Luo, R. Graeser, A. Warnecke, F. Kratz, P. Hauff, K. Licha and R. Haag, Bioorg. Med. Chem. Lett., 2009, 19, 1030–1034 CrossRef CAS PubMed.
- I. N. Kurniasih, H. Liang, J. P. Rabe and R. Haag, Macromol. Rapid Commun., 2010, 31, 1516–1520 CrossRef CAS PubMed.
- E. Fleige, B. Ziem, M. Grabolle, R. Haag and U. Resch-Genger, Macromolecules, 2012, 45, 9452–9459 CrossRef CAS.
- I. N. Kurniasih, H. Liang, V. D. Möschwitzer, M. A. Quadir, M. Radowski, J. P. Rabe and R. Haag, New J. Chem., 2012, 36, 371–379 RSC.
- I. N. Kurniasih, H. Liang, S. Kumar, A. Mohr, S. K. Sharma, J. P. Rabe and R. Haag, J. Mater. Chem. B, 2013, 1, 3569–3577 RSC.
- E. Fleige, K. Achazi, K. Schaletzki, T. Triemer and R. Haag, J. Controlled Release, 2014, 185, 99–108 CrossRef CAS PubMed.
- E. Burakowska and R. Haag, Macromolecules, 2009, 42, 5545–5550 CrossRef CAS.
- E. Burakowska, S. C. Zimmerman and R. Haag, Small, 2009, 5, 2199–2204 CrossRef CAS PubMed.
- W. Fischer, M. A. Quadir, A. Barnard, D. K. Smith and R. Haag, Macromol. Biosci., 2011, 11, 1736–1746 CrossRef CAS PubMed.
- S. F. Haag, E. Fleige, M. Chen, A. Fahr, C. Teutloff, R. Bittl, J. Lademann, M. Schäfer-Korting, R. Haag and M. C. Meinke, Int. J. Pharm., 2011, 416, 223–228 CAS.
- M. R. Radowski, A. Shukla, H. von Berlepsch, C. Boettcher, G. Pickaert, H. Rehage and R. Haag, Angew. Chem., Int. Ed., 2007, 46, 1265–1269 CrossRef CAS PubMed.
- C. Rabe, E. Fleige, V. Karsten, N. Szekely, P. Lindner, W. Burchard, R. Haag and M. Ballauff, Polymer, 2014, 55, 6735–6742 CrossRef CAS.
- L. Ye, K. Letchford, M. Heller, R. Liggins, D. Guan, J. N. Kizhakkedathu, D. E. Brooks, J. K. Jackson and H. M. Burt, Biomacromolecules, 2011, 12, 145–155 CrossRef CAS PubMed.
- C. Mugabe, Y. Matsui, A. I. So, M. E. Gleave, M. Heller, M. Zeisser-Labouebe, L. Heller, I. Chafeeva, D. E. Brooks and H. M. Burt, Biomacromolecules, 2011, 12, 949–960 CrossRef CAS PubMed.
- S. Nowag, C. Frangville, G. Multhaup, J. D. Marty, C. Mingotaud and R. Haag, J. Mater. Chem. B, 2014, 2, 3915–3918 RSC.
- S. Fehse, S. Nowag, M. Quadir, K. S. Kim, R. Haag and G. Multhaup, Biomacromolecules, 2014, 15, 1910–1919 CrossRef CAS PubMed.
- M. Wyszogrodzka and R. Haag, Chem. – Eur. J., 2009, 14, 9202–9204 CrossRef PubMed.
- A. Zill, A. L. Rutz, R. E. Kohman, A. M. Alkilany, C. J. Murphy, H. Kong and S. C. Zimmerman, Chem. Commun., 2011, 47, 1279–1281 RSC.
- W. Fischer, M. Calderon, A. Schulz, I. Andreou, M. Weber and R. Haag, Bioconjugate Chem., 2010, 21, 1744–1752 CrossRef CAS PubMed.
- S. Son, E. Shin and B.-S. Kim, Biomacromolecules, 2014, 15, 628–634 CrossRef CAS PubMed.
- Y. Oikawa, S. Lee, D. H. Kim, D. H. Kang, B.-S. Kim, K. Saito, S. Sasaki, Y. Oishi and Y. Shibasaki, Biomacromolecules, 2013, 14, 2171–2178 CrossRef CAS PubMed.
- J. Wang, M. H. Kim, D. E. Kang, H. Suh and I. Kim, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2553–2564 CrossRef CAS.
- J. H. Jeong, J. J. Schmidt, R. E. Kohman, A. T. Zill, R. J. DeVolder, C. E. Smith, M.-H. Lai, A. Shkumatov, T. W. Jensen, L. G. Schook, S. C. Zimmerman and H. Kong, J. Am. Chem. Soc., 2013, 135, 8770–8773 CrossRef CAS PubMed.
- A. L. Sisson, I. Papp, K. Landfester and R. Haag, Macromolecules, 2009, 42, 556–559 CrossRef CAS.
- A. L. Sisson and R. Haag, Soft Matter, 2010, 6, 4968–4975 RSC.
- D. Steinhilber, M. Witting, X. Zhang, M. Staegemann, F. Paulus, W. Friess, S. Küchler and R. Haag, J. Controlled Release, 2013, 169, 289–295 CrossRef CAS PubMed.
- D. Steinhilber, T. Rossow, S. Wedepohl, F. Paulus, S. Seiffert and R. Haag, Angew. Chem., Int. Ed., 2013, 52, 13538–13543 CrossRef CAS PubMed.
- C. Wu, C. Strehmel, K. Achazi, L. Chiappisi, J. Dernedde, M. C. Lensen, M. Gradzielski, M. B. Ansorge-Schumacher and R. Haag, Biomacromolecules, 2014, 15, 3881–3890 CrossRef CAS PubMed.
- J. Keilitz, S. Nowag, J.-D. Marty and R. Haag, Adv. Synth. Catal., 2010, 352, 1503–1511 CrossRef CAS.
- J. Keilitz, M. R. Radowski, J.-D. Marty, R. Haag, F. Gauffre and C. Mingotaud, Chem. Mater., 2008, 20, 2423–2425 CrossRef CAS.
- M. Schwarze, J. Keilitz, S. Nowag, R. Y. Parapat, R. Haag and R. Schomäcker, Langmuir, 2011, 27, 6511–6518 CrossRef CAS PubMed.
- F. Schüler, B. Kerscher, F. Beckert, R. Thomann and R. Mülhaupt, Angew. Chem., Int. Ed., 2013, 52, 455–458 CrossRef PubMed.
- X. Wang, X. Sun, G. Jiang, R. Wang, R. Hu, X. Xi, Y. Zhou, S. Wang and T. Wang, J. Appl. Polym. Sci., 2013, 128, 3289–3294 CrossRef CAS.
- X. Zeng, Y. Zhang, Z. Wu, P. Lundberg, M. Malkoch and A. M. Nyström, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 280–288 CrossRef CAS.
- X. Zeng, Y. Zhang and A. M. Nyström, Biomacromolecules, 2012, 13, 3814–3822 CrossRef CAS PubMed.
- H. Chen, G. Li, H. Chi, D. Wang, C. Tu, L. Pan, L. Zhu, F. Qiu, F. Guo and X. Zhu, Bioconjugate Chem., 2012, 23, 1915–1924 CrossRef CAS PubMed.
- J. Liu, Y. Pang, W. Huang, X. Huang, L. Meng, X. Zhu, Y. Zhou and D. Yan, Biomacromolecules, 2011, 12, 1567–1577 CrossRef CAS PubMed.
- H. Zhang, A. Patel, A. K. Gaharwar, S. M. Mihaila, G. Iviglia, S. Mukundan, H. Bae, H. Yang and A. Khademhosseini, Biomacromolecules, 2013, 14, 1299–1310 CrossRef CAS PubMed.
- G. Chen and Z. Guan, J. Am. Chem. Soc., 2004, 126, 2662–2663 CrossRef CAS PubMed.
- G. Chen, D. Huynh, P. L. Felgner and Z. Guan, J. Am. Chem. Soc., 2006, 128, 4298–4302 CrossRef CAS PubMed.
- C. S. Popeney, M. C. Lukowiak, C. Bottcher, B. Schade, P. Welker, D. Mangoldt, G. Gunkel, Z. Guan and R. Haag, ACS Macro Lett., 2012, 1, 564–567 CrossRef CAS.
- L. Zhang, J. Su, W. Zhang, M. Ding, X. Chen and Q. Wu, Langmuir, 2010, 26, 5801–5807 CrossRef CAS PubMed.
- C. He, B.-K. Jin, W.-D. He, X.-S. Ge, J. Tao, J. Yang and S.-Q. Chen, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2142–2149 CrossRef CAS.
- K. Kojima, K. Chikama, M. Ishikawa, A. Tanaka, T. Nishikata, H. Tsutsumi, K. Igawa and H. Nagashima, Chem. Commun., 2012, 48, 10666–10668 RSC.
- F. Xiang, M. Stuart, J. Vorenkamp, S. Roest, H. Timmer-Bosscha, M. C. Stuart, R. Fokkink and T. Loontjens, Macromolecules, 2013, 46, 4418–4425 CrossRef CAS.
- S. Tripp, D. Appelhans, C. Striegler and B. Voit, Chem. – Eur. J., 2014, 20, 8314–8319 CrossRef CAS PubMed.
- C. Treiber, M. Abdul Quadir, P. Voigt, M. Radowski, S. Xu, L.-M. Munter, T. A. Bayer, M. Schaefer, R. Haag and G. Multhaup, Biochemistry, 2009, 48, 4273–4284 CrossRef CAS PubMed.
- M. A. Quadir, M. R. Radowski, F. Kratz, K. Licha, P. Hauff and R. Haag, J. Controlled Release, 2008, 132, 289–294 CrossRef CAS PubMed.
- S. Küchler, M. R. Radowski, T. Blaschke, M. Dathe, J. Plendl, R. Haag, M. Schäfer-Korting and K. D. Kramer, Eur. J. Pharm. Biopharm., 2009, 71, 243–250 CrossRef PubMed.
- Y. Liang, D. Wan, X. Cai, M. Jin and H. Pu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 681–691 CrossRef CAS.
- X.-L. Lou, F. Cheng, P.-F. Cao, Q. Tang, H.-J. Liu and Y. Chen, Chem. – Eur. J., 2009, 15, 11566–11572 CrossRef CAS PubMed.
- W. Zhuang, L. Liao, H. Chen, J. Wang, Y. Pan, L. Zhang and D. Liu, Macromol. Rapid Commun., 2009, 30, 920–924 CrossRef CAS PubMed.
- Y. Liu, Y. Fan, X.-Y. Liu, S.-Z. Jiang, Y. Yuan, Y. Chen, F. Cheng and S.-C. Jiang, Soft Matter, 2012, 8, 8361–8369 RSC.
- N. Polikarpov, D. Appelhans, P. Welzel, A. Kaufmann, P. Dhanapal, C. Bellmann and B. Voit, New J. Chem., 2012, 36, 438–451 RSC.
- Y. Chen, L. Zhou, Y. Pang, W. Huang, F. Qiu, X. Jiang, X. Zhu, D. Yan and Q. Chen, Bioconjugate Chem., 2011, 22, 1162–1170 CrossRef PubMed.
- Y. Ping, D. Wu, J. N. Kumar, W. Cheng, C. L. Lay and Y. Liu, Biomacromolecules, 2013, 14, 2083–2094 CrossRef CAS PubMed.
- Catalysis and Electrocatalysis at Nanoparticle Surfaces, ed. A. Wieckowski, E. R. Savinova and C. G. Vayenas, Marcel Dekker, New York, 2003 Search PubMed.
- H. Ye, J. A. Crooks and R. M. Crooks, Langmuir, 2007, 23, 11901–11906 CrossRef CAS PubMed.
- O. M. Wilson, M. R. Knecht, J. C. Garcia-Martinez and R. M. Crooks, J. Am. Chem. Soc., 2006, 128, 4510–4511 CrossRef CAS PubMed.
- N. Pérignon, A.-F. Mingotaud, J.-D. Marty, I. Rico-Lattes and C. Mingotaud, Chem. Mater., 2004, 16, 4856–4858 CrossRef.
- N. Pérignon, J.-D. Marty, A.-F. Mingotaud, M. Dumont and I. M. Rico-Lattes, Macromolecules, 2007, 40, 3034–3041 CrossRef.
- J.-D. Marty, E. Martinez-Aripe, A.-F. O. Mingotaud and C. Mingotaud, J. Colloid Interface Sci., 2008, 326, 51–54 CrossRef CAS PubMed.
- S. Saliba, C. Valverde Serrano, J. Keilitz, M. L. Kahn, C. Mingotaud, R. Haag and J.-D. Marty, Chem. Mater., 2010, 22, 6301–6309 CrossRef CAS.
- H. H. Nguyen, C. Valverde Serrano, P. Lavedan, D. Goudounèche, A.-F. Mingotaud, N. Lauth-de Viguerie and J.-D. Marty, Nanoscale, 2014, 6, 3599–3610 RSC.
- C. Padié, M. Maszewska, K. Majchrzak, B. Nawrot, A. M. Caminade and J.-P. Majoral, New J. Chem., 2009, 33, 318–326 RSC.
- B. Klajnert, M. Cangiotti, S. Calici, M. Ionov, J.-P. Majoral, A. M. Caminade, J. Cladera, M. Bryszewska and M. F. Ottaviani, New J. Chem., 2009, 33, 1087–1093 RSC.
- M. Blanzat, C. O. Turrin, A. M. Aubertin, C. Vidal, A. M. Caminade, J.-P. Majoral, I. Rico-Lattes and A. Lattes, ChemBioChem, 2005, 6, 2207–2213 CrossRef CAS PubMed.
- B. Klajnert, M. Cangiotti, S. Calici, J.-P. Majoral, A. M. Caminade, J. Cladera, M. Bryszewska and M. F. Ottaviani, Macromol. Biosci., 2007, 7, 1065–1074 CrossRef CAS PubMed.
- O. Mongin, A. Pla-Quintana, F. Terenziani, D. Drouin, C. Le Droumaguet, A. M. Caminade, J.-P. Majoral and M. Blanchard-Desce, New J. Chem., 2007, 31, 1354–1367 RSC.
- P. Marchand, L. Griffe, M. Poupot, C. O. Turrin, G. Bacquet, J. J. Fournié, J.-P. Majoral, R. Poupot and A. M. Caminade, Bioorg. Med. Chem. Lett., 2009, 19, 3963–3966 CrossRef CAS PubMed.
- G. Spataro, F. Malecaze, C.-O. Turrin, V. Soler, C. Duhayon, P.-P. Elena, J.-P. Majoral and A.-M. Caminade, Eur. J. Med. Chem., 2010, 45, 326–334 CrossRef CAS PubMed.
- J. Liu, W. Huang, Y. Pang, X. Zhu, Y. Zhou and D. Yan, Biomaterials, 2010, 31, 5643–5651 CrossRef CAS PubMed.
- J. Liu, Y. Pang, W. Huang, Z. Zhu, X. Zhu, Y. Zhou and D. Yan, Biomacromolecules, 2011, 12, 2407–2415 CrossRef CAS PubMed.
- C. Chen, G. Liu, X. Liu, S. Pang, C. Zhu, L. Lv and J. Ji, Polym. Chem., 2011, 2, 1389–1397 RSC.
- Y.-Y. Yuan, J.-Z. Du, W.-J. Song, F. Wang, X.-Z. Yang, M.-H. Xiong and J. Wang, J. Mater. Chem., 2012, 22, 9322–9329 RSC.
- X. Chang and C.-M. Dong, Biomacromolecules, 2013, 14, 3329–3337 CrossRef CAS PubMed.
- N. T. Hoai, A. Sasaki, M. Sasaki, H. Kaga, T. Kakuchi and T. Satoh, Biomacromolecules, 2011, 12, 1891–1899 CrossRef CAS PubMed.
- H. Y. Cho, S. E. Averick, E. Paredes, K. Wegner, A. Averick, S. Jurga, S. R. Das and K. Matyjaszewski, Biomacromolecules, 2013, 14, 1262–1267 CrossRef CAS PubMed.
- W. Tao, Y. LIu, B. Jiang, S. Yu, W. Huang, Y. Zhou and D. Yan, J. Am. Chem. Soc., 2012, 134, 762–764 CrossRef CAS PubMed.
- Y. Liu, C. Yu, H. Jin, B. Jiang, X. Zhu, Y. Zhou, Z. Lu and D. Yan, J. Am. Chem. Soc., 2013, 135, 4765–4770 CrossRef CAS PubMed.
- D. Wan, S. Ohta, T. Kakuchi and T. Satoh, Soft Matter, 2011, 7, 6422–6425 RSC.
| This journal is © The Royal Society of Chemistry 2015 |