
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrative self-sorting: a versatile strategy for the construction of complex supramolecular architecture
Zhenfeng
He
a,
Wei
Jiang
*a and
Christoph A.
Schalley
*b
aDepartment of Chemistry, South University of Science and Technology of China, Xueyuan Blvd. 1088, Nanshan District, Shenzhen, 518055, P. R. China. E-mail: jiangw@sustc.edu.cn; Fax: +86-755-86245672; Tel: +86-755-88018316
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustraße 3, 14195 Berlin, Germany. E-mail: christoph@schalley-lab.de; Fax: +49 30 838 4 52639; Tel: +49 30 838 52639
First published on 6th November 2014
Abstract
Large protein-sized synthetic supramolecular architecture is rare and certainly has not yet achieved the structural and functional complexity of biomolecules. As multiple, identical copies of a few building blocks are repetitively used, a highly symmetrical architecture results with limitations in function. In marked contrast, functional structures in nature are often assembled with high geometric precision from many different building blocks. They cooperate in a complex way realizing energy conversion, mechanical motion or transport phenomena. Beyond self-assembly, the structurally and functionally complex biomolecular machines rely on self-sorting to correctly position all subunits through orthogonal recognition sites. Mimicking such self-sorting processes is a promising strategy for supramolecular synthesis – resulting in higher structural complexity and promising access to a more sophisticated function. The term “integrative self-sorting” was coined to describe the strategy to form well-defined assemblies with well-controlled subunit positions. The key process is the incorporation of two or more orthogonal binding motifs into at least some of the subunits. Modularity and programmability based on orthogonal yet similar binding motifs generate diversity and complexity. Integrative self-sorting is thus inherently related to systems chemistry. Depending on the individual binding motifs, (multi-)stimuli responsiveness can be achieved. When different recognition events en route to the final assembly occur on significantly different time scales, kinetic pathway selection is observed. In this account, we review the modularity, programmability, and emergent properties of integrative self-sorting, emphasizing its utility and perspective for complex supramolecular architectures.
 Zhenfeng He (left), Wei Jiang (middle) and Christoph Schalley (right) | Zhenfeng He received his BSc degree in chemistry from Anshan Normal University in 2004. After working in Anshan Huifeng Chemicals Co., Ltd, he joined Jilin University and obtained his PhD degree under the supervision of Prof. Lixin Wu in 2012. In April 2013, He joined the Department of Chemistry, South University of Science and Technology of China (SUSTC) as a Research Fellow. His research interests include organic–inorganic hybrid materials, design of complex supramolecular architectures, supramolecular polymers, and functional hybrid supramolecular complexes. |
Wei Jiang received his BSc degree in 2004 from Xi'an Jiaotong University and MSc degree in 2007 from Nankai University with Prof. Yu Liu. He was awarded the Schering prize for his PhD work which he finished in 2010 at Freie Universität Berlin with Prof. Christoph A. Schalley. After postdoctoral research with Prof. Julius Rebek Jr at the Scripps Research Institute in 2011 and 2012, he started his independent research career at the Department of Chemistry, South University of Science and Technology of China (SUSTC). His research is focused on the design and synthesis of novel macrocyclic hosts and their applications in catalysis, sensing, and smart materials. |
Christoph Schalley studied chemistry at the University of Freiburg/Germany and graduated in 1993 from the Technical University Berlin. In his PhD work with Helmut Schwarz at TU Berlin, he received his thorough education as a mass spectrometrist and gas-phase chemist. After postdoctoral work on supramolecular chemistry with Julius Rebek, Jr at The Scripps Research Institute, La Jolla/USA, he combined both topics when he started his own research group as a habilitand at the University of Bonn. In 2005, he was appointed professor of organic chemistry at Free University Berlin. Meanwhile Schalley group's research topics are diverse and comprise supramolecular chemistry in the gas phase as well as in solution and at interfaces. |
1 Introduction
For more than a century, chemists have thrived on developing new synthetic methodologies to assemble molecules by breaking and making strong, kinetically inert covalent bonds. This state-of-the-art organic synthesis1 enables chemists to build almost any organic molecule even with controlled stereochemistry. This is achieved by repeating the cycle of synthetic transformation and purification of the desired product multiple times. The synthesis of complicated and well-defined high molecular weight organic molecules remains nevertheless challenging and time-consuming.Besides covalent synthesis, supramolecular synthesis2–4 and dynamic combinatorial chemistry5–10 have become important tools for the generation of complex molecular architecture.11–13 The reversibility of non-covalent and dynamic covalent bonds enables error correction and with it makes molecules programmable. For more than two decades, supramolecular chemists have generated supramolecular structures with the help of concepts such as self-assembly,14–17 templating18 and self-sorting.19–22 Spectacular architecture has been constructed which even functions as molecular machines or switches.23,24 The majority of supramolecular assemblies are however highly symmetrical due to the use of multiple copies of the same building blocks, thus limiting the diversity and versatility of these structures significantly. As a consequence, the question of how far one could push chemical self-assembly was identified in 2005 as one of the top 25 scientific challenges for the next 25 years.25
In this perspective, the focus is on the construction of complex supramolecular architecture through the strategy of integrative self-sorting.26 First, we discuss a systems view on supramolecular synthesis, followed by an introduction to the characteristics and classifications of self-sorting with a focus on and examples for the role of integrative self-sorting in the construction of large and complex supramolecular architecture. The system properties of integrative self-sorting including kinetic pathway selection are discussed next. Finally, applications of integrative self-sorting to the synthesis of hetero[n]rotaxanes and cooperation with other strategies of supramolecular synthesis will also be addressed. We are convinced that the strategy of integrative self-sorting, together with many other strategies of supramolecular synthesis, will ultimately lead to the assembly of supramolecular architecture with complex functions.
2 A systems view on supramolecular synthesis
Complex chemical systems are traditionally studied via a reductionist approach by analysing their individual components or subsystems separately under otherwise well-controlled conditions. Recently, chemists have shifted this paradigm more and more towards studies of chemical systems as a whole, realizing the collective behaviour to be more than the sum of the properties of the individual parts. The term “systems chemistry”27–29 has recently been coined30 for this approach to the emergent properties of chemical networks. The advent of systems chemistry has also reshaped our ideas on supramolecular synthesis. The final emerging structures are due to a delicate balance of many different non-covalent interactions. Changes in the environment or external stimuli will shift this balance, resulting in a structural and functional response and an adaption of the systems under study to the new environment (Fig. 1). We are convinced that the concepts of supramolecular chemistry have matured into a state in which their application to functional chemical systems with emergent properties is the next great scientific challenge.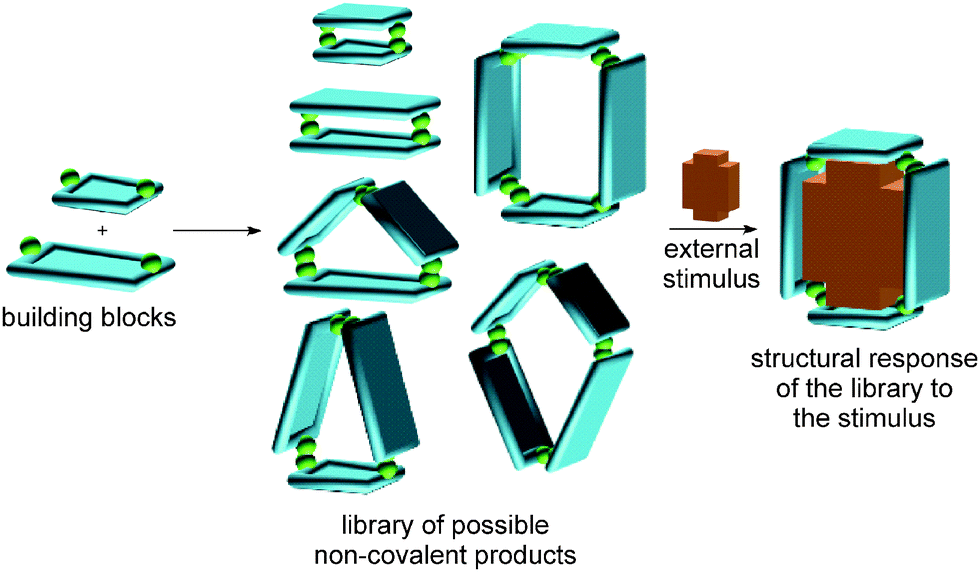 | ||
| Fig. 1 Schematic representation of a structural response of a library of non-covalent complexes generated by the addition of an external stimulus. Similar behaviour is observed in many dynamic combinatorial libraries which share some decisive properties with supramolecules, e.g. the reversibility of bond formation between the building blocks and the potential to correct errors and mismatches. | ||
3 Self-sorting
The term self-sorting31–33 describes the ability of mixtures of different molecules to recognize their mutual counterparts selectively so that specific pairs are formed rather than a library of all possible non-covalent complexes of the compounds present in the mixture. Self-sorting is related to systems chemistry as it is the result of a network of competing recognition events defined by the binding constants between all the possible pairs. Self-sorting systems thus have higher information content and are more organized than an unspecific mixture, because only one or very few complexes are selected out of a larger number of potentially possible assemblies based on the delicate balance of all the competing interactions between the molecules in the mixture. This distinguishes self-sorting from other self-assembly strategies, in which usually identical building blocks are repetitively used. Self-sorting systems are often based on the same type of binding motifs. In principle, this allows cross-interactions between mismatched building blocks to occur. The challenge is then to make even these similar binding motifs orthogonal to each other. While orthogonal binding motifs are of prime importance, factors not directly related to the binding motifs themselves, such as stoichiometry, spacer size and shape, or spacer–spacer interactions, can help to control the final self-sorting outcome.For the details and the exact chronology of the development of self-sorting, the readers are referred to previously published reviews.31–33 Briefly, different cases of self-sorting can be distinguished. First of all, self-sorting can be thermodynamically or kinetically controlled.34 Self-sorting can be also classified into narcissistic (Fig. 2a, left) – all compounds only recognize identical copies of them and form homomeric complexes35 – and social self-sorting (Fig. 2a, right) – pairs of compounds mutually recognize each other and form heteromers.36 Social self-sorting can occur under different circumstances:36 when a host H1 prefers guest G1 over G2 (ΔGH1G1 < ΔGH1G2) and host H2 prefers guest G2 over G1 (ΔGH2G1 > ΔGH2G2), type I social self-sorting is observed in a wide range of stoichiometries of these four molecules, because ΔGH1G1 + ΔGH2G2 < ΔGH1G2 + ΔGH2G1. Often, it is an advantage, when all components in a mixture are quantitatively consumed in the self-sorting process. If such a completive self-sorting37 occurs, the binding energy liberated upon the formation of complex H1G1 in addition to H2G2 supports self-sorting as compared to a situation, in which H1G1 assembles and H2 and G2 remain as free components. The second, less frequently realized type II social self-sorting is encountered, when both hosts H1 and H2 prefer G1 over G2 (ΔGH1G1 < ΔGH1G2 and ΔGH2G1 < ΔGH2G2). In this case, the difference between ΔGH1G1 and ΔGH2G1 must be sufficiently large. In addition, self-sorting only occurs reliably within a quite narrow stoichiometry range and mixtures form, when these stoichiometry requirements are not met.
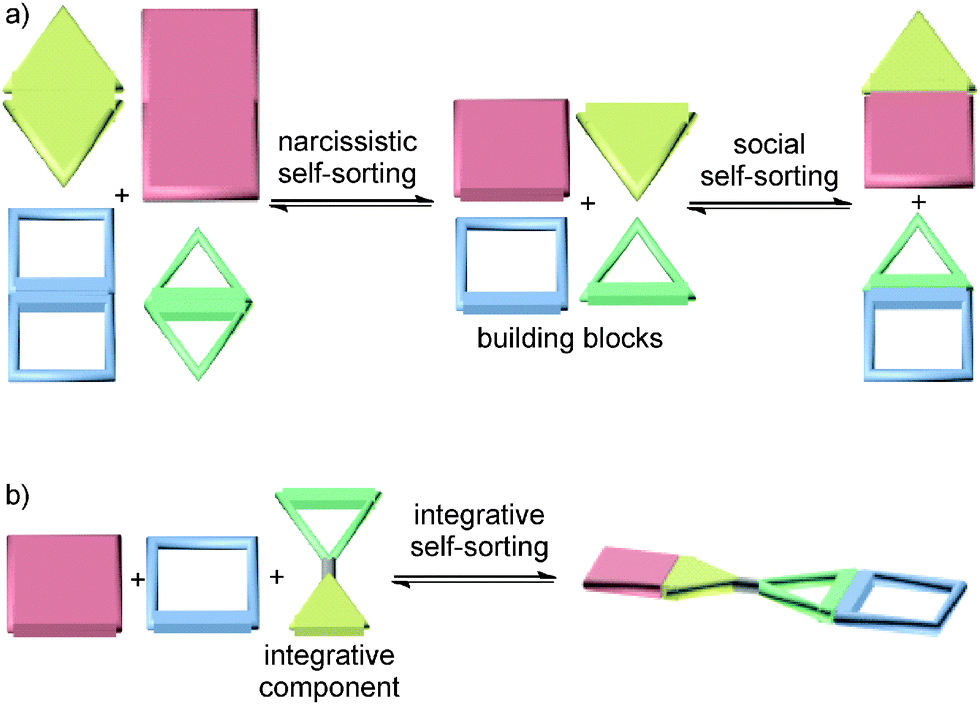 | ||
| Fig. 2 (a) Schematic representations of narcissistic and social self-sorting. (b) Integrative self-sorting with one integrative component bearing two orthogonal binding sites. | ||
4 Integrative self-sorting
If one aims at making use of self-sorting to construct larger, programmed assemblies from a number of different building blocks that are all correctly positioned in the final complex, it is useful to synthesize one or more components integrating binding sites from different motifs. Accordingly, we distinguish non-integrative,38 (Fig. 3a) from integrative (Fig. 3b) self-sorting.26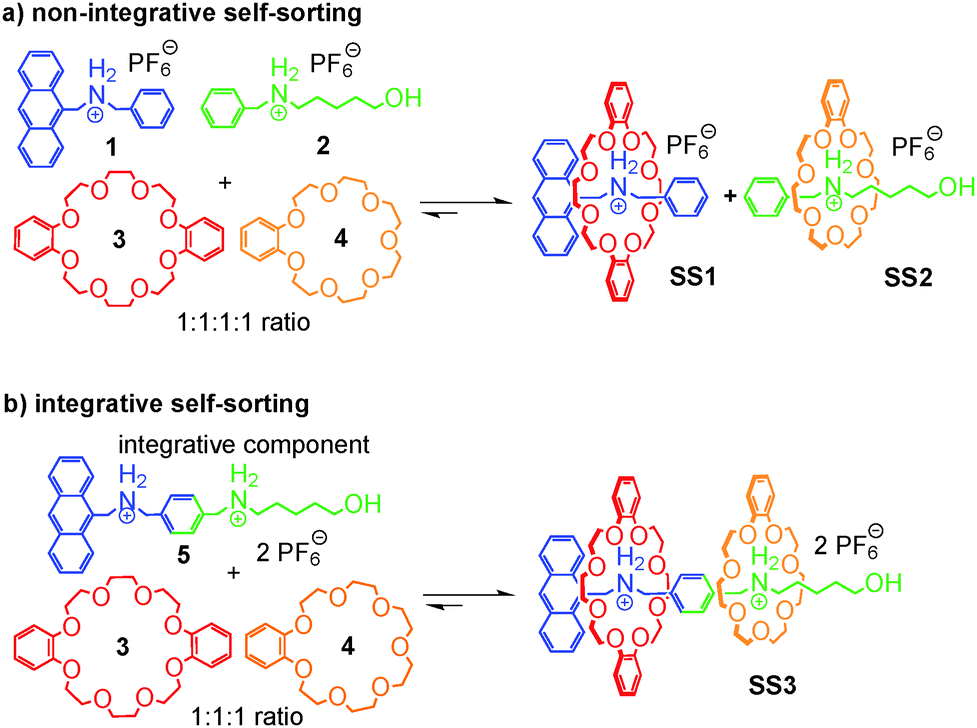 | ||
| Fig. 3 (a) A four-component non-integrative self-sorting system of 1 to 4 sorts into two out of four possible pseudorotaxanes according to the axles' steric demand and crown ether cavity sizes. (b) Integrative self-sorting based on the same motifs generates a sequence-specific hetero[3]pseudorotaxane from 3 to 5. | ||
Fig. 3a illustrates this idea using simple crown/ammonium pseudorotaxanes as an example.26 An equimolar mixture of compounds 1, 2, 3, and 4 self-sorts with high fidelity into the two pseudorotaxanes SS1 and SS2, even though the two binding motifs are very similar. Instead, other assemblies, for example 2@3, are possible, but not realized. The reason for the high fidelity of the self-sorting process is the difference in the steric demand of the phenyl and hydroxypentyl groups. While the latter matches the cavity size of the smaller crown ether, the phenyl group is too bulky to thread through the cavity of 4, preventing pseudorotaxane formation from 1 and 4. In addition, the building block 4 binds more strongly to 2 than 3. Self-sorting is thus driven thermodynamically towards SS1 and SS2.
From this four-component social self-sorting system, an integrative self-sorting complex can be obtained by joining at least one binding site from each motif covalently into one integrative component,39 which acts as a molecular “hub” bringing the other units together into one well-defined assembly. As a first test, the two axles were merged into heteroditopic axle 5. As shown in Fig. 3b, the construction of the final assembly is straightforward by mixing the integrative component 5 with the other two crown ether building blocks 3 and 4 with the correct stoichiometry. The same self-sorting principle as in the four-component system assists the assembly of the final complex. NMR and tandem MS experiments confirm the formation of a sequence-specific hetero[3]pseudorotaxane SS3 with precisely controlled positions of each component.
The strategy of integrative self-sorting can be made modular and programmable by rational design of additional integrative components. Since each of the self-sorting complexes contains two different building blocks, there are four covalent connections possible: 1 and 2, 3 and 4, 2 and 3, or 1 and 4. The heterodivalent integrative components 5, 6, and 7 (Fig. 3 and 4) were made as well as their homodivalent analogues 8–11, which opened further opportunities to program assemblies, when used together with integrative components. As long as integrative components exist in the assembly systems, the two self-sorting motifs SS1 and SS2 will be incorporated into the final assembly. With this rather small library of building blocks, different combinations of subunits lead to a quite impressive series of different architectures minimizing the synthetic effort by modularity and significantly enriching the structural diversity and complexity. Fig. 5 shows the supramolecular architectures SS4–SS11,39 which have been assembled using these building blocks and could unambiguously be characterized by a combination of NMR and tandem MS experiments.40
 | ||
| Fig. 4 The chemical structures of (a) integrative, heterodivalent components 6 and 7 and (b) their homodivalent analogues 8 to 11. | ||
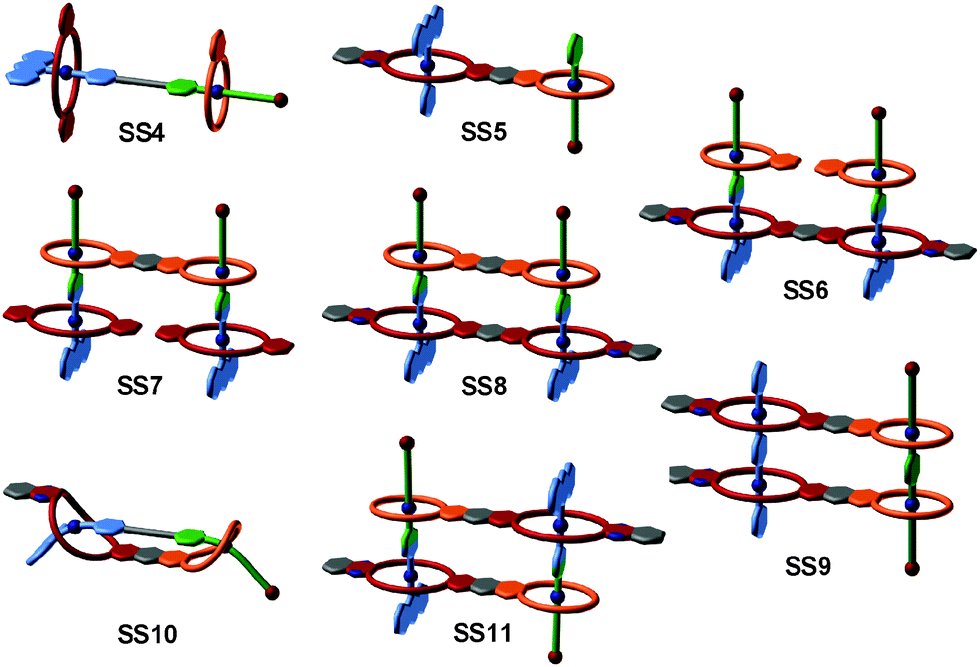 | ||
| Fig. 5 Integrative self-sorting complexes SS4 to SS11. For simplicity, cartoon representations are shown with binding sites colour-coded in analogy to Fig. 4. | ||
Many opportunities exist to extend structural variety even further with the same building blocks: connecting axles with crown ethers lead to the formation of self-sorting hetero-daisy chain pseudorotaxanes.41 Also, one could synthesize integrative components with more than two orthogonal binding sites. Finally, extending the selection of binding motifs will boost the accessible and programmable range of supramolecular structures. This concept will ultimately enable one to access supramolecular architecture with protein- or DNA-level complexity.
Besides the binding sites, the connecting spacers in the integrative components should be well-designed with respect to length, rigidity and potential attractive or repulsive spacer–spacer interactions. Assemblies SS8 and SS10 provide a simple example: axle 5 with its short phenylene spacer assembles with host 7 to form [2 + 2] assembly SS8, while the longer axles 6 (n = 1–3) prefer intramolecular binding to form [1 + 1] assembly SS10. The latter example points to a limitation of integrative self-sorting: as reducing particle number and producing order usually decrease entropy, it will often be difficult to master these entropy effects when very large complexes are to be made. Some ways to escape this problem might be (i) a precise fine-tuning of spacer rigidity, (ii) the use of binding motifs, for which binding leads to entropy increases by, for example, solvent molecule liberation, or (iii) the use of binding motifs with particularly high binding enthalpies. Also, multivalent and cooperative binding42 can certainly be advantageous. The cooperativity of divalent binding in pseudorotaxanes analogous to SS10 of the crown-ammonium type has recently been investigated in detail.43 The binding strength benefits significantly from chelate cooperativity, when the spacer is ideally suited so that spacer–spacer interactions aid complex formation.
This integrative self-sorting strategy is quite versatile and applicable to many other self-sorting systems, among them water-soluble hetero[3]pseudorotaxane SS14 based on cucurbit[n]urils CB[7] and CB[8] (ref. 44) (Fig. 6a), one of the rare cases of type II social self-sorting. The underlying principle is the self-sorting of an equimolar mixture of 12, 13, CB[7], and CB[8] into SS12 and SS13. Integrative component 14 was prepared by connecting 12 and 13 covalently. When mixing 14 with CB[7] and CB[8] in an equimolar ratio, self-sorting automatically leads efficiently to hetero[3]pseudorotaxane SS14 with positional control for each component. This cucurbituril-based concept has been extended to the longer guest chain 15 (Fig. 6b), which not only forms self-sorted complexes, but undergoes multiple, complex re-folding and re-sorting steps upon the addition of external stimuli.45 This process is reminiscent of natural signaling cascades in which a chemical system is responsive to external chemical stimuli leading to an alteration in behaviour including the generation of secondary messengers. Five consecutive steps were implemented using the different host–guest properties of three differently sized cucurbit[n]urils (n = 6 to 8). Very recently, integrative self-sorting based on the host–guest properties of cucurbiturils has also been applied to the construction of supramolecular polymers.46,47
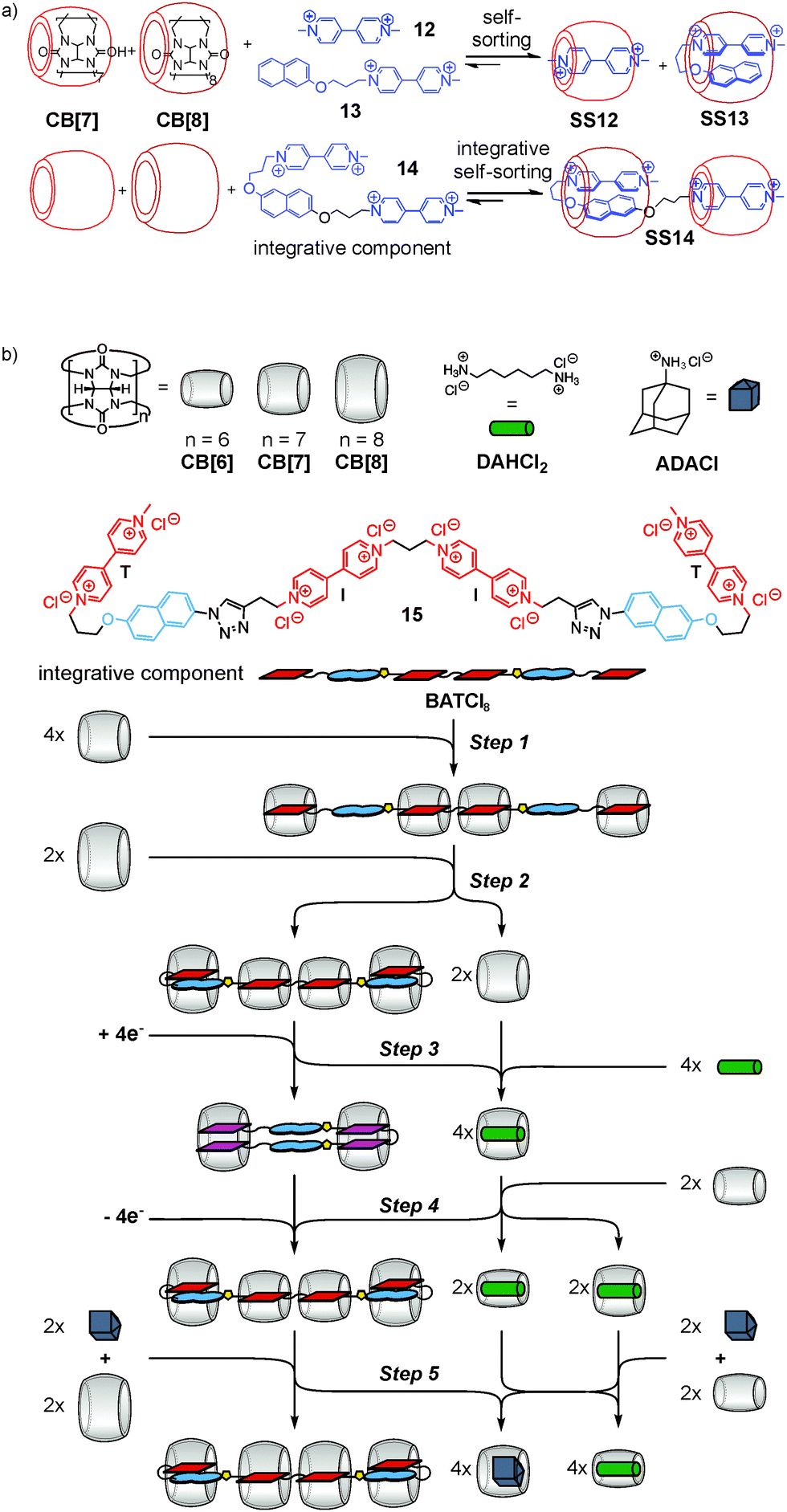 | ||
| Fig. 6 (a) Integrative self-sorting aids the construction of water-soluble hetero[3]pseudorotaxane SS14. (b) A signaling cascade based on self-sorted, stimuli-responsive cucurbituril complexes. Five consecutive refolding and resorting steps have been realized including redox reactions between the viologen dications and the cation radical dimer (steps 3, 4). | ||
Integrative self-sorting is not limited to small discrete supramolecular complexes, but has also been applied to dendrimers48 and side-chain-modified polymers.49 Huang and coworkers50–53 reported a self-sorting supramolecular polymer (Fig. 7) based on the social self-sorting of 3, 16, 17, and 18 that utilizes the binding preferences of crown ether 3 for the secondary ammonium axle 17 and of crown ether 16 for the paraquat derivative 18 to order the monomer sequence along the polymer chain. The two self-sorting complexes SS15 and SS16 constitute the two modules for the design of polymer SS17 with alternating building blocks. Two integrative components 19 and 20 were synthesized by adding flexible spacers between 3 and 18 and between 16 and 17, respectively. Flexible spacers cause low effective molarities, so that intermolecular binding is predominant at high concentrations and SS17 forms.
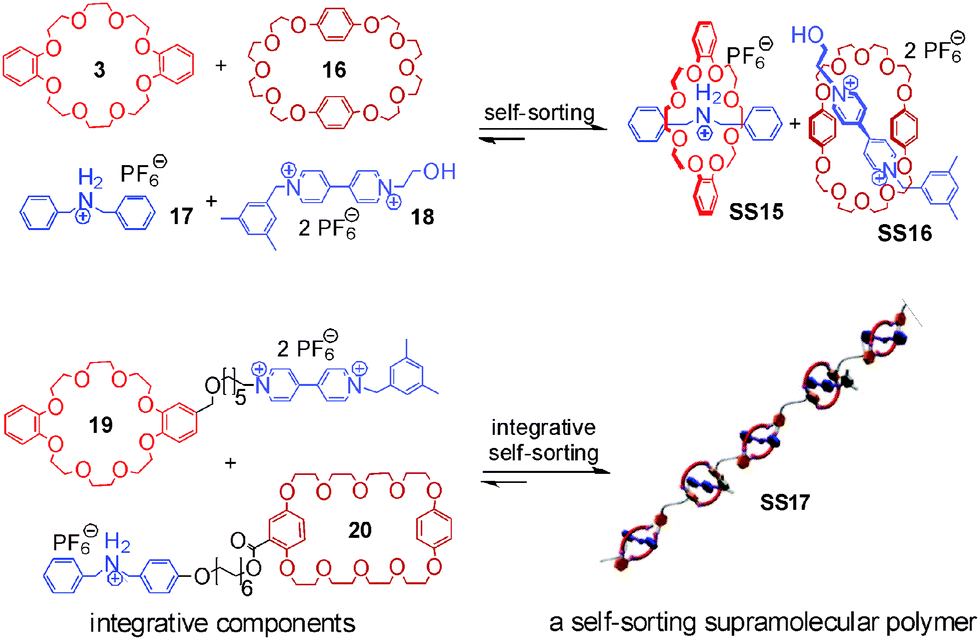 | ||
| Fig. 7 Self-sorting supramolecular polymer SS17.51 Adapted from ref. 51 with the kind permission by American Chemical Society. | ||
In a series of elegant papers, Schmittel and coworkers implemented integrative self-sorting in metallo-supramolecular chemistry. In the equimolar mixture of Cu+, Zn2+, and 21–24, only SS18 and SS19 form out of 20 possible metal complexes (Fig. 8). The high fidelity is attributed to the maximum site occupancy, and steric, electronic, and geometric factors. With this self-sorting system as the basis, the integrative component 27 and two homoditopic ligands 25 and 26 were synthesized. The 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1:1 mixture of Cu+, Zn2+, 27, 25, and 26 in acetonitrile leads to the formation of the unique supramolecular isosceles trapezoid SS20 through integrative self-sorting. This system was later extended by the same group to the construction of several metal-coordinated scalene triangles,54–56 a scalene quadrilateral,57 and a pentagonal macrocycle.58 Remarkably, these supramolecular structures cannot easily be constructed via conventional self-assembly strategies – convincingly demonstrating integrative self-sorting to be a versatile strategy for the construction of diverse supramolecular architecture with high-level complexity.
:2:2:1:1 mixture of Cu+, Zn2+, 27, 25, and 26 in acetonitrile leads to the formation of the unique supramolecular isosceles trapezoid SS20 through integrative self-sorting. This system was later extended by the same group to the construction of several metal-coordinated scalene triangles,54–56 a scalene quadrilateral,57 and a pentagonal macrocycle.58 Remarkably, these supramolecular structures cannot easily be constructed via conventional self-assembly strategies – convincingly demonstrating integrative self-sorting to be a versatile strategy for the construction of diverse supramolecular architecture with high-level complexity.
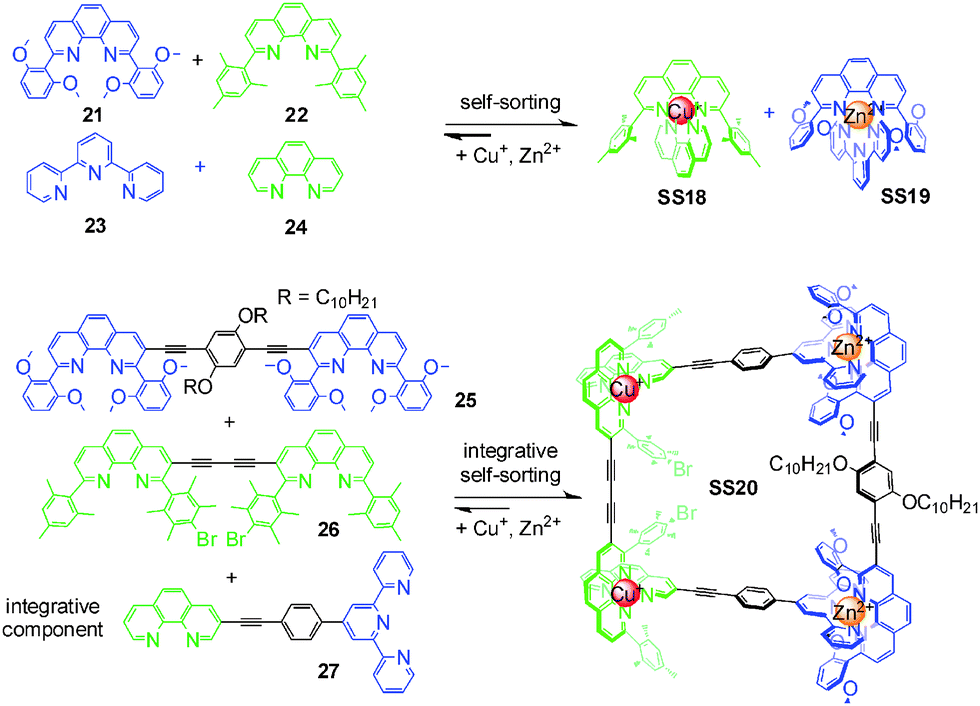 | ||
| Fig. 8 Isosceles trapezoid SS20.37 | ||
5 Guiding assembly pathways during integrative self-sorting
So far, we have considered the final outcome of the self-sorting processes after equilibration. Assemblies may, however, also be kinetically trapped even though one usually assumes non-covalent bonds to form and dissociate quickly. One example is Fujita's selective cross-catenation of metallo-supramolecular PdII-containing macrocycles with sterically different substituents.59 Quite different catenation rates dictate a self-assembly pathway, which initially leads to a hetero[2]catenane, which then slowly converts into a mixture of the two homo[2]catenanes. Another example is the guest exchange in a cylindrical capsule, which requires about three months at room temperature to complete.60 When self-assembly occurs from many copies of the same building blocks, one usually encounters a system, which explores large parts of the accessible potential energy surface and forms a very diverse library of different complexes initially. Over time, this mixture converges by error correction into the most favourable assembly – often a single complex. In marked contrast, self-sorting systems with their orthogonal binding motifs may encounter significantly different kinetic barriers for different assembly pathways. Also, mismatched complexes that are disfavored by thermodynamics may well play a role in the assembly process. Kinetic path selection is therefore likely to occur and is probably rather the rule than the exception. The importance of this aspect becomes immediately clear when considering protein folding as a natural analogue.61 Here, path selection is not only of major importance, but is actively supported in the living cells by chaperone proteins. To design the assembly pathways of supramolecular architectures is certainly a challenging task requiring a comprehensive understanding of all relevant kinetic and thermodynamic parameters.Fig. 9 shows again the simple self-sorting system composed of the two axles 1 and 2 and the two crown ethers 3 and 4. The four possible threading events occur on very different time scales.62 Threading 1 into 4 is so slow on the human time scale that threading is not observed at all. Axle 2 threads into 4 much faster on a many-minute time scale, followed by 1 binding inside the cavity of the larger crown ether 3 on a second time scale. Finally, the narrow axle 2 slips into the larger crown ether 3 on a time scale of about a hundred milliseconds. Even though 28 is not formed in the final equilibrium, it is clearly a prominent intermediate according to mixed-flow microreactor ESI mass spectrometric experiments. The mismatch is then corrected over time until the final mixture of SS1 and SS2 is formed under thermodynamic control.
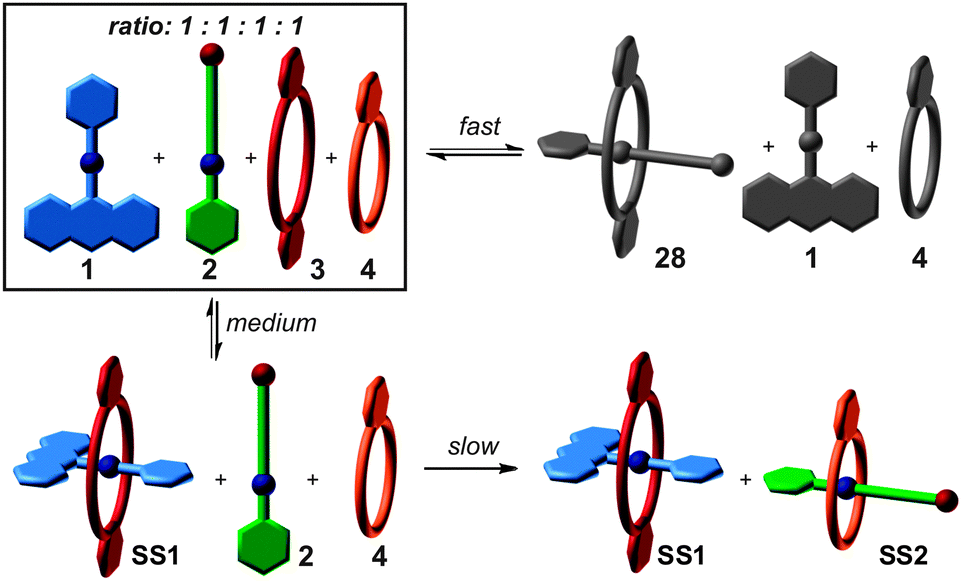 | ||
| Fig. 9 The self-sorting pathway of SS1 and SS2 from the equimolar mixture of 1 – 4. Three binding events occurring on very different time scales (fast = milliseconds, medium = seconds, slow = minutes). Mismatched complex 28 initially forms quickly and prominently as an intermediate, but does not play any role in the final product mixture. | ||
Based on the understanding of the kinetic behaviour of this simple self-sorting system, the formation pathways of integrative self-sorting complexes may even be rationally designed (Fig. 10). The kinetic properties of pseudorotaxanes can be tuned by changing the two end groups of the axles without reducing the fidelity of thermodynamic self-sorting. The design of the integrative component 5 meets the requirements of a productive sorting pathway for the formation of SS3 (Fig. 10a): the anthracene stopper blocks one side of the axle against threading of any crown ether. According to the rates observed for the simple system, the fastest process is thus slipping of the larger crown ether 3 on the green station. As the next step is significantly slower, intermediate 29 accumulates increasing the efficiency of the next step, i.e. transfer of crown 3 from the green to the blue binding site. Again, this medium fast process is followed by an even slower step, and intermediate 30 also accumulates thus rendering the last step efficient, too. The mismatched assemblies 31 and 32 form only with minor amounts and are corrected, when the system approaches its thermodynamic minimum. Consequently, we find a streamlined formation pathway for this hetero[3]pseudorotaxane.
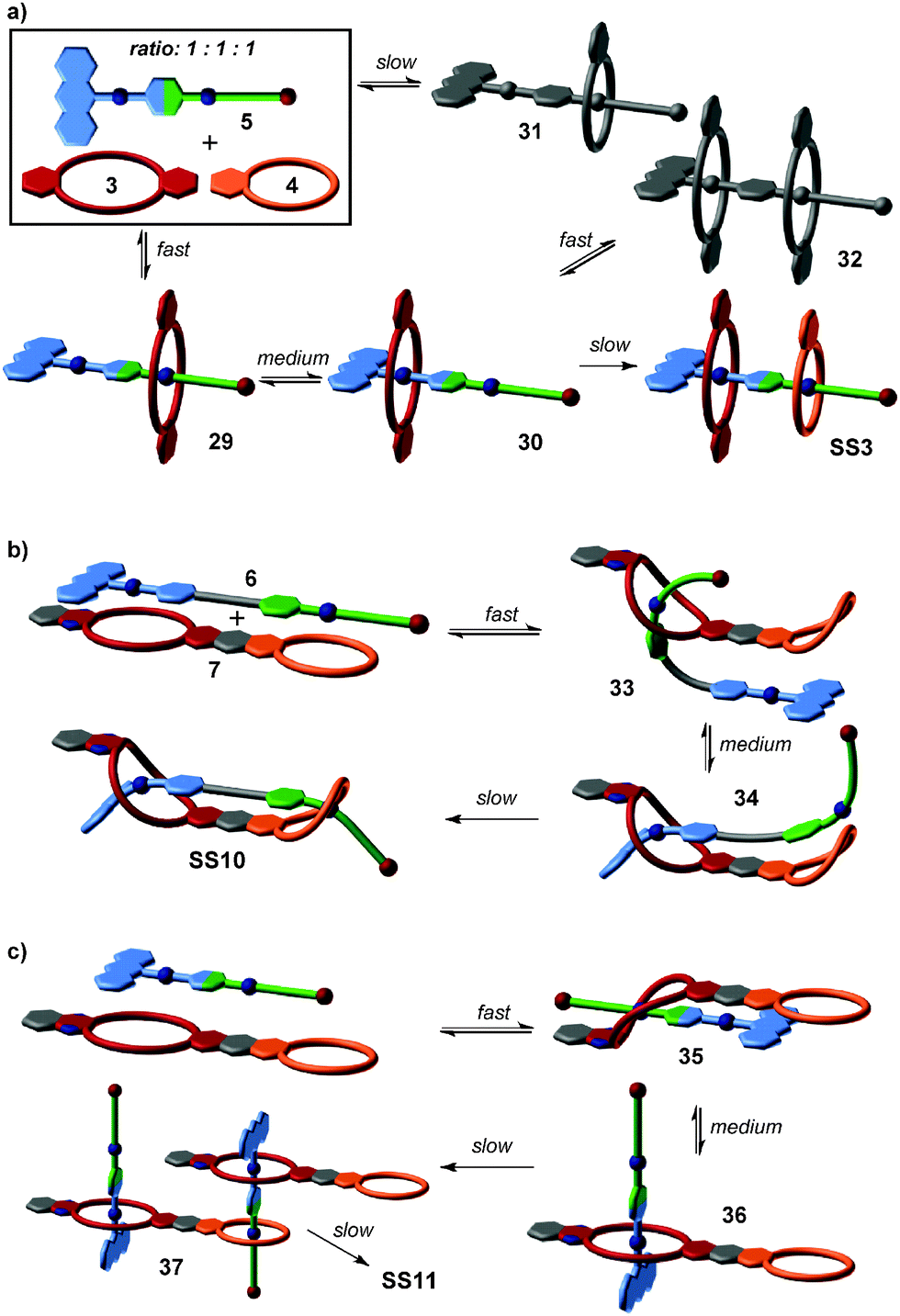 | ||
| Fig. 10 (a) Kinetic path selection en route to SS3. Structures in gray are dead ends requiring error-correction. (b) Stream-lined formation pathway of SS10 as deduced from the research on simpler systems and observed by 1H NMR. (c) Kinetic formation pathway of SS11 as detected by ESI mass spectrometry. | ||
Analogously, the formation of SS10 from 6 and 7 follows the same threading sequence (Fig. 10b). This assembly pathway is the optimal pathway to form the final complex SS10. It is noteworthy that the last two threading steps occur in an intramolecular fashion. The same approach can be transferred to more complicated assemblies SS8 and SS11. ESI mass spectrometry has unraveled SS11 to form by first generating a 1:1 complex of axle 5 and crown heterodimer 7 (Fig. 10c). Again, the larger crown ether wins the initial competition for the terminal green station on the axle in a fast process, then transfers to the blue station. The 1:1 complex accumulates and then dimerizes in slow steps completing assembly formation by threading of the green stations into the smaller crown ethers. This is very different for SS8 for which several isomerization processes have been shown to be involved in assembly formation. In this case, there is no streamlined formation pathway.
In the pseudorotaxanes discussed above, a mismatched assembly (large crown threading onto the narrow axle) indeed guides rather than hampers the formation of the correct assemblies. Such effects will certainly become more and more important, when the assembly size and complexity are increased in order to generate large supramolecular architecture. Multiple non-covalent bonds may cooperate and then significantly limit the reversibility during assembly formation. A good design will therefore require the simultaneous consideration of the appropriate binding motifs and of suitable assembly pathways.
6 Secondary effects on integrative self-sorting
So far, we have reviewed self-sorting systems in which the complexes under study contained orthogonal binding motifs that led to the formation of only one final structure. If one for example considers SS11 (Fig. 5), the simultaneous presence of two different crown ethers in 7 and of two different secondary ammonium binding sites in 5 ensured that the two axles are arranged in an antiparallel way. At the same time, the two crown dimers are also antiparallel to each other. If one exchanges heteroditopic axle 5 against its symmetric homoditopic counterpart 11, it is not a priori clear anymore, whether the two crown ether heterodimers are still arranged in an antiparallel manner. A parallel arrangement would be possible as well and mixtures of both isomers may form (Fig. 11). Lowering the information content in one of the integrative components thus may cause ambiguity.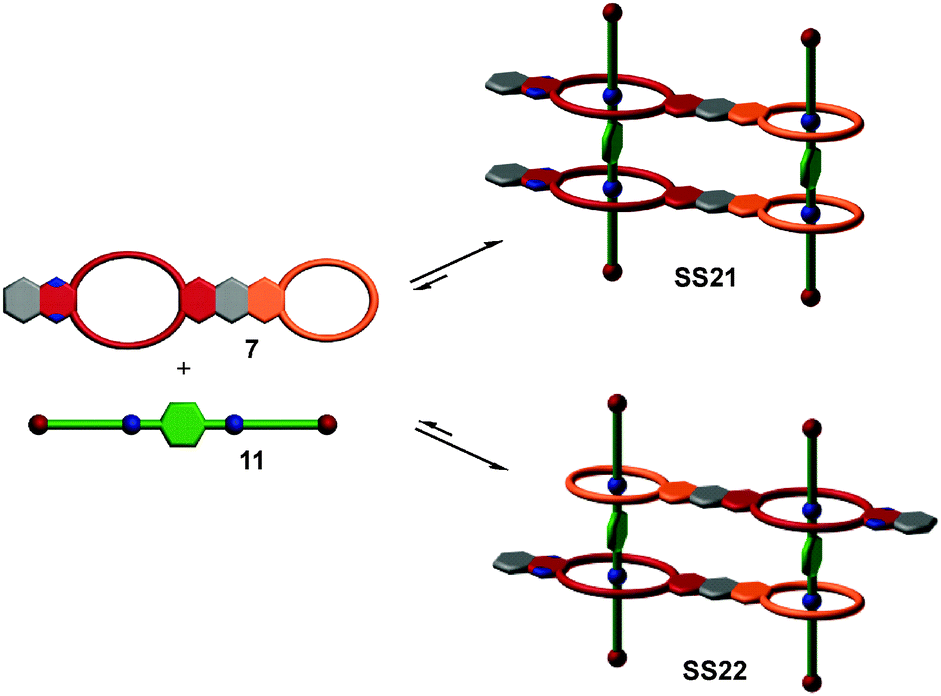 | ||
| Fig. 11 When the symmetrical axle 11 is combined with crown heterodimer 7, two isomeric assemblies may form. Centrosymmetric SS22 is clearly thermodynamically preferred over its mirror-symmetric analogue SS21. | ||
Despite this ambiguity, only one isomer forms:63 the centrosymmetric assembly SS22 with its antiparallel crown dimers is thermodynamically more stable than mirror-symmetric SS21 by 9.6 kJ mol−1 – presumably because of a better arrangement of the host dipoles. We use the term “secondary interactions” for these subtle effects, as they are not directly associated with the programming of the binding sites, but nevertheless can cause a well-defined structural outcome even for integrative self-sorting systems that lack sufficient information for perfect self-sorting. In most cases, such effects will be overruled by the self-sorting originating from suitably programmed, orthogonal binding motifs as the energy differences are smaller for the secondary than for the primary effects. Nevertheless, they should be taken into account when designing self-sorting structures as they may favor, but also disfavor the formation of the desired assembly.
7 Application of integrative self-sorting to the synthesis of hetero[n]rotaxanes
Rotaxanes are an important class of mechanically interlocked molecules64 and have advanced to be workhorses in molecular nanotechnology and the design of molecular machines.23,24 Their synthesis depends on template effects and the self-assembly of appropriate pseudorotaxane precursors. Hetero[n]rotaxanes with two or more different macrocycles are nevertheless still quite rare, although the incorporation of different macrocycles in the same structure promises to broaden the structural variety and with it the implementable function. We therefore discuss in this section, how integrative self-sorting can be used to efficiently synthesize hetero[n]rotaxanes.Self-sorted pseudorotaxanes can be converted to rotaxanes by attaching the appropriate stopper groups that prevent dethreading of the axle – the so-called capping method. Thus, hetero[n]rotaxanes with two or more different macrocycles trapped on the same axle can be conveniently constructed through integrative self-sorting of hetero[n]pseudorotaxanes as the precursors. Fig. 12a shows a simple example: by treating the sequence-specific [3]pseudorotaxane SS3 in CH2Cl2 with benzoic acid anhydride in the presence of Bu3P as the catalyst, cascade-stoppered hetero[3]rotaxane 33 can be synthesized in good yield (70%).26 In the structure of 33, the phenyl groups at the end and the middle of the axle trap the smaller crown ether 4. The larger crown ether 3 can still pass over the central phenyl group, but certainly not over the small crown ether. As a consequence, the larger crown ether is also trapped on the axle. This “stopper cascade” survived heating of the rotaxanes in DMSO-d6 at 80 °C for 2 days and thus provides evidence for the stability of the resulting hetero[3]rotaxanes. On the basis of our research, Chiu and coworkers65 took integrative self-sorting to a higher level under solvent-free conditions. Their system consists of 3, 21-crown-7, an axle analogous to 5, but terminated with alkynes at both ends, one tetrazine stopper, and one diphenyltetrazine stopper. Most interestingly, self-sorting even occurs in the solid state reaction during the threading of the two macrocycles and the stoppering Diels–Alder reaction between alkyne and tetrazine. A hetero[3]rotaxane similar to 38 was obtained.
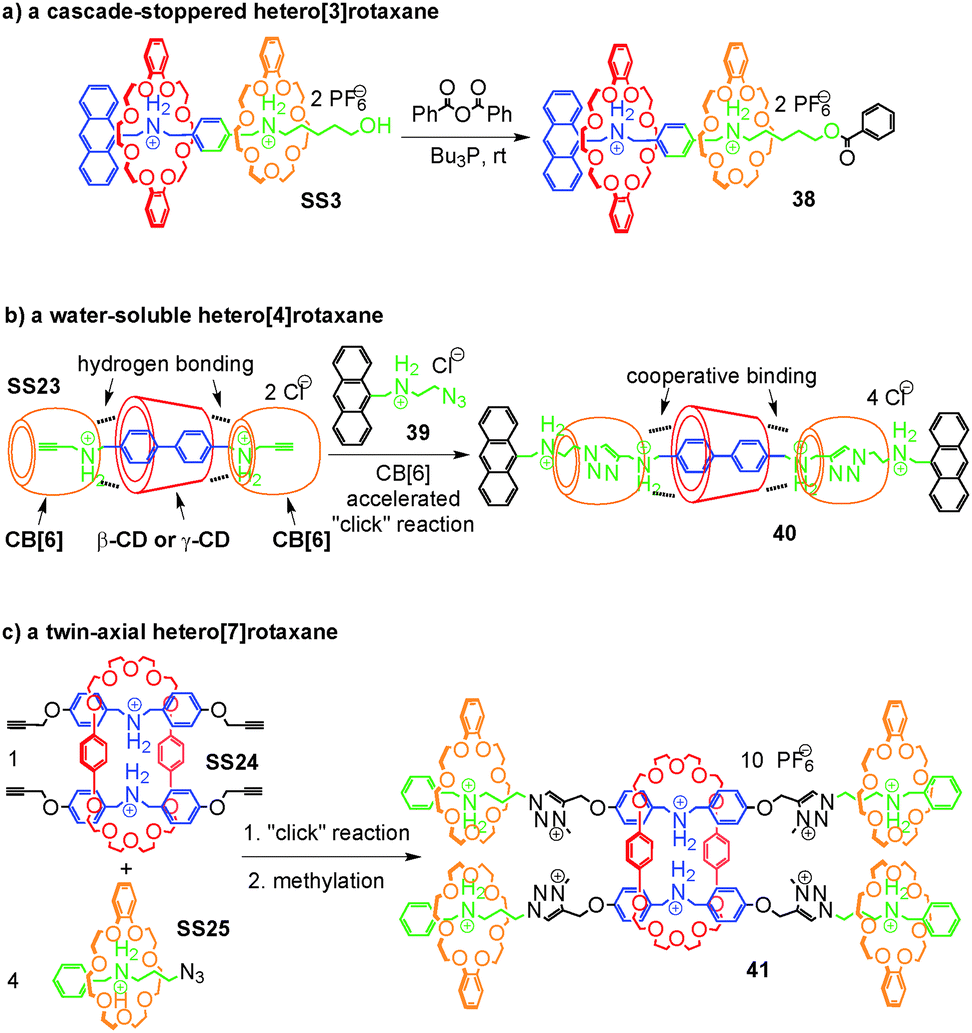 | ||
| Fig. 12 Synthesis of hetero[n]rotaxanes based on integrative self-sorting: (a) a cascade-stoppered hetero[3]rotaxane 38, (b) a water-soluble hetero[4]rotaxane 40 and (c) a twin-axial hetero[7]rotaxane 41. | ||
When the number of macrocycles in hetero[n]pseudorotaxanes increases, the efficiency of integrative self-sorting may decrease since incorporation of many components into one complex is entropically unfavorable. Thus, the yield of the corresponding hetero[n]rotaxane synthesis may be compromised unless positive cooperativity among the building blocks favours heteropseudorotaxane assembly. Recently, Stoddart and co-workers66 reported a clever strategy coined “cooperative capture” to tackle this problem (Fig. 12b). The integrative self-sorting systems they studied involve cucurbit[6]uril (CB[6]) and cyclodextrins (β-CD and γ-CD) as the macrocycles. The formation of self-sorting complex SS23 is based on the following features:67 (a) the cyclodextrin prefers the biphenyl unit of the axle due to its hydrophobic interior cavity. (b) CB[6] instead prefers the propargyl group attached to both sides of the biphenyl spacer of the axle and binds to the secondary ammonium ions with its seam of carbonyl groups. (c) In addition to these preferences, hydrogen bonding occurs between the hydroxyl groups of CD and the carbonyl groups of CB[6] which further stabilize SS23 cooperatively. The formation of this self-sorted hetero[4]pseudorotaxane is then followed by the CB[6]-accelerated alkyne–azide 1,3-dipolar cycloaddition as the stoppering reaction.68 The cooperative binding between CD and CB[6] in SS23 was shown to not just stabilize the complex, but also to further accelerate the stoppering reaction. Remarkably, heating a stoichiometric mixture of all components in water to 80 °C affords the water-soluble hetero[4]rotaxane 40 in quantitative yield within one (β-CD) or five (γ-CD) minutes. The yield did not decrease, even when this concept was extended to the synthesis of a polyhetero[n]rotaxane with 64 macrocycles. The CD–CB[6] pair is not the only example for the strategy of cooperative capture. The same group found later that pillar[5]arene69 and pillar[6]arene70 can replace CDs efficiently in templating rotaxane formation in acetonitrile together with CB[6].
Liu and co-workers71 showed an alternative strategy to tackle the problem of entropic penalty during the synthesis of hetero[n]rotaxanes with many macrocycles (Fig. 12c). In the examples in Fig. 12a and b, the hetero[n]rotaxanes were synthesized via the corresponding integratively self-sorted hetero[n]pseudorotaxanes, suggesting that the integrative components need to be synthesized beforehand. In Liu's strategy, the synthesis of integrative components was merged in one step with the stoppering of pseudorotaxanes, significantly reducing the synthetic effort and avoiding the entropic penalty in integrative self-sorting complexes. They found SS24 and SS25 to self-sort in non-polar solvents on the basis of similar reasons to SS1 and SS2 discussed above. The two pseudorotaxanes carry azide and alkyne groups at the ends of their axles, respectively. Applying the “click” reaction and methylation, the mixture of the two self-sorting complexes SS24 and SS25 can be directly transformed to the twin-axial hetero[7]rotaxane 41 in 42% isolated yield. In this rotaxane, the axle is the integrative component which brings all other components together. The synthesis of the integrative component synchronizes with the construction of the hetero[7]rotaxane. During the synthesis, the intermediate assembly is still dynamic, but is quickly converted into the hetero[7]rotaxanes. This modular approach is very efficient for the synthesis of quite complex hetero[n]rotaxanes. Of course, this strategy is not limited to the synthesis of hetero[n]rotaxanes and can also be applied to the construction of other, non-intertwined integrative self-sorting complexes directly from social self-sorting mixtures.
8 Combining integrative self-sorting with subcomponent self-assembly
To push forward the limits of supramolecular synthesis, it may be interesting to combine the strategy of integrative self-sorting with others. We have already discussed above some examples for template syntheses of (pseudo)rotaxanes and for metallo-supramolecular self-assembly in which the combination of these strategies with integrative self-sorting is beneficial for the generation of well-defined supramolecular architecture. Also, it was shown how homomultivalent components could be used together with integrative components providing rich opportunities for programming new structures. In this section, we extend these examples to a combination of integrative self-sorting with subcomponent self-assembly. Subcomponent self-assembly is a strategy using small and simple building blocks which form the components required for the final assembled structure in a dynamic library. Selection and sorting thus occurs hierarchically on a covalent and a non-covalent level.Recently, Nitschke and coworkers72 reported the formation of a cubic cage structure by elegantly combining their subcomponent self-assembly strategy with integrative self-sorting (Fig. 13). Two self-sorting metal-coordination motifs are needed: to direct subcomponent self-assembly, the tris(pyridylimine) iron(II) coordination is applied, which is orthogonal to the tetrakis(pyridine)platinum(II) motif. In order to achieve integrative self-sorting, the heteroditopic ligand 42 was synthesized as the integrative component to join the two self-sorting complexes into a single self-sorted assembly. The threefold symmetry of tris(pyridylimine)iron(II) and the fourfold symmetry of tetrakis(pyridine)platinum(II) then determine the formation of the cubic cage SS26 when mixing 42, 2-formylpyridine, Fe(CF3SO3)2, AgBF4, and Pt(NCPh)2Cl2 in a 24:24:8:12:6 ratio. Overall, 62 building blocks are brought together through the formation of 96 bonds. Although a very similar cage has been constructed through subcomponent self-assembly alone,73 much less synthetic effort was required in the present case as simpler compounds could be used. Such combinations of integrative self-sorting with other strategies of supramolecular synthesis are – as demonstrated with this example – a suitable approach to increase complexity in a single step from very simple subunits to huge supramolecular architecture. This will help paving the road to higher complexity.
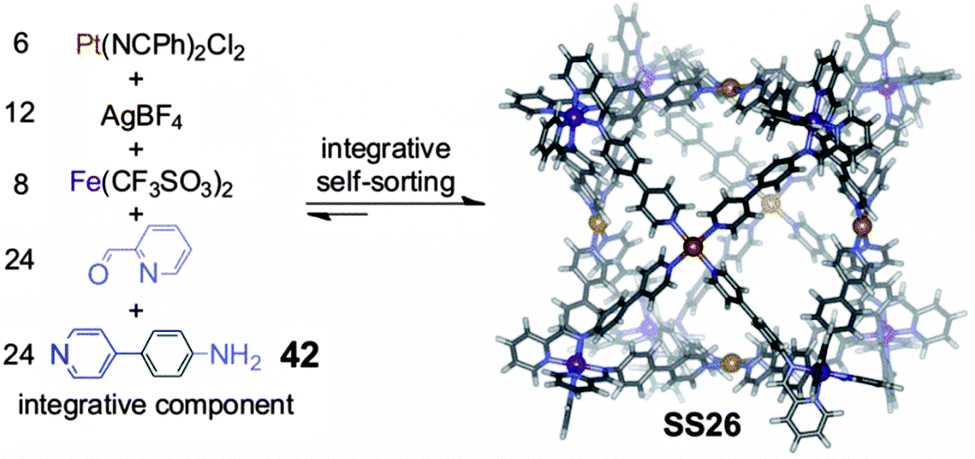 | ||
| Fig. 13 Supramolecular synthesis of a metal-coordinated cubic cage SS26 from very simple building blocks via integrative self-sorting and subcomponent self-assembly. Adapted from ref. 72 with the kind permission by Wiley-VCH. | ||
9 Conclusions and perspectives
Integrative self-sorting is a modular approach to program complex and diverse supramolecular architecture by rational design. Information written into the integrative components allows controlling building block sequences and positions in the final assemblies. Sometimes even stereochemical aspects can be controlled. The basis is usually a quite simple social self-sorting system. Integrative components carrying at least two different binding sites are the key to bring all subunits together in a well-defined structure. Starting from a given simple self-sorting system, there exist many different ways to synthesize integrative components so that extensive LEGO toolboxes can be designed and readily made, providing ample opportunity to generate a large diversity of different structures. Thus, both complexity and diversity can be achieved simultaneously.Integrative self-sorting inherently relates to systems chemistry as demonstrated with, for example, the cucurbituril-based signaling cascade discussed above. Stimuli-responsiveness is already a function of such a system, but more sophisticated function will certainly be implemented in the future by attaching suitable functional groups to the building blocks, which – well positioned in space around the final assembly – cooperate with each other.
The more complex the structures become, the more important solutions to difficulties regarding for example entropy losses become that are due to the fact that large numbers of building blocks are joined within the final assembly. One strategy to solve this problem is certainly the use of cooperative binding between the subunits as exemplified for some hetero[n]pseudorotaxanes given above. Another problem will arise, when we approach protein sizes and complexity: kinetics may play an important role in assembly formation. Kinetic pathway selection will certainly be an issue to be taken into account as seriously as the thermodynamic issues that are related to the orthogonality of binding motifs.
In combination with other concepts in supramolecular synthesis, e.g. subcomponent self-assembly, integrative self-sorting represents a versatile and powerful strategy for the generation of supramolecular architecture. Huge steps from the very simple to the highly complex can be taken, when the component design is appropriate. This has been illustrated using Nitschke's self-assembled cube.
The application of self-sorting to the synthesis of supramolecular architecture will certainly develop further in the future. We are confident that concepts such as integrative self-sorting together with other strategies will provide access to much larger, much more complex and ultimately functional synthetic chemical architecture. Research along these lines may also contribute its share to the understanding of biological phenomena and the construction of smart supramolecular machines and materials. The question raised in 2005 of how far self-assembly might be pushed forward in future finds one answer here: new concepts such as integrative self-sorting help the supramolecular chemist to extend self-assembly beyond its current boundaries. It remains interesting to follow where this process may lead to in the future.
Acknowledgements
We are grateful to all collaborators for their fine contributions and especially Igor Linder for his help with the figures. The Deutsche Forschungsgemeinschaft (SFB 765) and South University of Science and Technology of China (SUSTC) are acknowledged for financial support.Notes and references
- K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44–122 CrossRef CAS.
- G. M. Whitesides, E. E. Simanek, J. P. Mathias, C. T. Seto, D. Chin, M. Mammen and D. M. Gordon, Acc. Chem. Res., 1995, 28, 37–44 CrossRef CAS.
- M. C. T. Fyfe and J. F. Stoddart, Acc. Chem. Res., 1997, 30, 393–401 CrossRef CAS.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS.
- J.-M. Lehn, Chem. – Eur. J., 1999, 5, 2455–2463 CrossRef CAS.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- F. B. L. Cougnon and J. K. M. Sanders, Acc. Chem. Res., 2012, 45, 2211–2221 CrossRef CAS PubMed.
- J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- G. M. Whitesides, E. E. Simanek, J. P. Mathias, C. T. Seto, D. Chin, M. Mammen and D. M. Gordon, Acc. Chem. Res., 1995, 28, 37–44 CrossRef CAS.
- M. C. T. Fyfe and J. F. Stoddart, Acc. Chem. Res., 1997, 30, 393–401 CrossRef CAS.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CAS.
- J. S. Lindsey, New J. Chem., 1991, 15, 153–180 CAS.
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed., 1996, 35, 1155–1196 CrossRef CAS.
- C. A. Schalley, A. Lützen and M. Albrecht, Chem. – Eur. J., 2004, 10, 1072–1080 CrossRef CAS PubMed.
- Templated Organic Synthesis, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim/Germany, 2000 Search PubMed.
- K. Osowska and O. Š. Miljanić, Synlett, 2011, 1643–1648 CAS.
- K. Osowska and O. Š. Miljanić, Angew. Chem., Int. Ed., 2011, 50, 8345–8349 CrossRef CAS PubMed.
- K. Osowska and O. Š. Miljanić, J. Am. Chem. Soc., 2011, 133, 724–727 CrossRef CAS PubMed.
- R. C. Lirag, K. Osowska and O. Š. Miljanić, Org. Biomol. Chem., 2012, 10, 4847–4850 CAS.
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed.
- R. F. Service, Science, 2005, 309, 95 CrossRef CAS PubMed.
- W. Jiang, H. D. F. Winkler and C. A. Schalley, J. Am. Chem. Soc., 2008, 130, 13852–13853 CrossRef CAS PubMed.
- R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 RSC.
- J. R. Nitschke, Nature, 2009, 462, 736–738 CrossRef CAS PubMed.
- G. von Kiedrowski, S. Otto and P. Herdewijn, J. Syst. Chem., 2010, 1, 1–6 CrossRef.
- M. Kindermann, I. Stahl, M. Reimold, W. M. Pankau and G. von Kiedrowski, Angew. Chem., Int. Ed., 2005, 44, 6750–6755 CrossRef CAS PubMed.
- S. Ghosh and L. Isaacs, in Complex Self-Sorting Systems, Dynamic Combinatorial Chemistry: In Drug Discovery, Bioorganic Chemistry, and Materials Science, ed. B. L. Miller, Wiley, Hoboken, 2009, pp. 118–154 Search PubMed.
- M. M. Safont-Sempere, G. Fernandez and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- M. L. Saha and M. Schmittel, Org. Biomol. Chem., 2012, 10, 4651–4684 Search PubMed.
- P. Mukhopadhyay, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2006, 128, 14093–14102 CrossRef CAS PubMed.
- A. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS PubMed.
- P. Mukhopadhyay, A. Wu and L. Isaacs, J. Org. Chem., 2004, 69, 6157–6164 CrossRef CAS PubMed.
- K. Mahata and M. Schmittel, J. Am. Chem. Soc., 2009, 131, 16544–16554 CrossRef CAS PubMed.
- C. Talotta, C. Gaeta, Z. Qi, C. A. Schalley and P. Neri, Angew. Chem., Int. Ed., 2013, 52, 7427–7441 CrossRef PubMed.
- W. Jiang and C. A. Schalley, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10425–10429 CrossRef CAS PubMed.
- W. Jiang and C. A. Schalley, J. Mass Spectrom., 2010, 45, 788–798 CrossRef CAS PubMed.
- B. Zheng, F. Klautzsch, M. Xue, F. Huang and C. A. Schalley, Org. Chem. Front., 2014, 1, 532–540 RSC.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- W. Jiang, K. Nowosinski, N. L. Löw, E. V. Dzyuba, F. Klautzsch, A. Schäfer, J. Huuskonen, K. Rissanen and C. A. Schalley, J. Am. Chem. Soc., 2012, 134, 1860–1868 CrossRef CAS PubMed.
- W. Jiang, Q. Wang, I. Linder, F. Klautzsch and C. A. Schalley, Chem. – Eur. J., 2011, 17, 2344–2348 CrossRef CAS PubMed.
- L. Cera and C. A. Schalley, Chem. Sci., 2014, 5, 2560–2567 RSC.
- Z. Huang, L. Yang, Y. Liu, Z. Wang, O. A. Scherman and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 5351–5355 CrossRef CAS PubMed.
- Q. Zhang and H. Tian, Angew. Chem., Int. Ed., 2014, 53, 10582–10584 CrossRef CAS PubMed.
- S.-Y. Kim, Y. H. Ko, J. W. Lee, S. Sakamoto, K. Yamaguchi and K. Kim, Chem. – Asian J., 2007, 2, 747–754 CrossRef CAS PubMed.
- C. Burd and M. Weck, Macromolecules, 2005, 38, 7225–7230 CrossRef CAS.
- F. Wang, C. Han, C. He, Q. Zhou, J. Zhang, C. Wang, N. Li and F. Huang, J. Am. Chem. Soc., 2008, 130, 11254–11255 CrossRef CAS PubMed.
- F. Wang, B. Zheng, K. Zhu, Q. Zhou, C. Zhai, S. Li, N. Li and F. Huang, Chem. Commun., 2009, 4375–4377 RSC.
- S. Dong, B. Zheng, M. Zhang, X. Yan, X. Ding, Y. Yu and F. Huang, Macromolecules, 2012, 45, 9070–9075 CrossRef CAS.
- S. Dong, X. Yan, B. Zheng, J. Chen, X. Ding, Y. Yu, D. Xu, M. Zhang and F. Huang, Chem. – Eur. J., 2012, 18, 4195–4199 CrossRef CAS PubMed.
- K. Mahata, M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2010, 132, 15933–15935 CrossRef CAS PubMed.
- M. Schmittel and K. Mahata, Chem. Commun., 2010, 46, 4163–4165 RSC.
- M. L. Saha, J. W. Bats and M. Schmittel, Org. Biomol. Chem., 2013, 11, 5592–5595 CAS.
- M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2013, 135, 17743–17746 CrossRef CAS PubMed.
- M. L. Saha, N. Mittal, J. W. Bats and M. Schmittel, Chem. Commun., 2014, 50, 12189–12192 RSC.
- A. Hori, K.-i. Yamashita and M. Fujita, Angew. Chem., Int. Ed., 2004, 43, 5016–5019 CrossRef CAS PubMed.
- W. Jiang, D. Ajami and J. Rebek, Jr., J. Am. Chem. Soc., 2012, 134, 8070–8073 CrossRef CAS PubMed.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS PubMed.
- W. Jiang, A. Schafer, P. C. Mohr and C. A. Schalley, J. Am. Chem. Soc., 2010, 132, 2309–2320 CrossRef CAS PubMed.
- W. Jiang, D. Sattler, K. Rissanen and C. A. Schalley, Org. Lett., 2011, 13, 4502–4505 CrossRef CAS PubMed.
- J.-P. Sauvage and C. Dietrich-Buchecker, Molecular Catenanes, Rotaxanes and Knots, Wiley-VCH, Weinheim, 1999 Search PubMed.
- P.-N. Chen, C.-C. Lai and S.-H. Chiu, Org. Lett., 2011, 13, 4660–4663 CrossRef CAS PubMed.
- C. Ke, R. A. Smaldone, T. Kikuchi, H. Li, A. P. Davis and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 381–387 CrossRef CAS PubMed.
- C. Yang, Y. H. Ko, N. Selvapalam, Y. Origane, T. Mori, T. Wada, K. Kim and Y. Inoue, Org. Lett., 2007, 9, 4789–4792 CrossRef CAS PubMed.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and T. L. Manimaran, J. Org. Chem., 1983, 48, 3619–3620 CrossRef CAS.
- C. Ke, N. L. Strutt, H. Li, X. Hou, K. J. Hartlieb, P. R. McGonigal, Z. Ma, J. Iehl, C. L. Stern, C. Cheng, Z. Zhu, N. A. Vermeulen, T. J. Meade, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 17019–17030 CrossRef CAS PubMed.
- X. Hou, C. Ke, C. Cheng, N. Song, A. K. Blackburn, A. Sarjeant, Y. Y. Botros, Y.-W. Yang and J. F. Stoddart, Chem. Commun., 2014, 50, 6196–6199 RSC.
- Z.-J. Zhang, H.-Y. Zhang, H. Wang and Y. Liu, Angew. Chem., Int. Ed., 2011, 50, 10834–10838 CrossRef CAS PubMed.
- M. M. J. Smulders, A. Jiménez and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681–6685 CrossRef CAS PubMed.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |