
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cucurbiturils: from synthesis to high-affinity binding and catalysis
Khaleel I.
Assaf
and
Werner M.
Nau
*
Jacobs University Bremen, Campus Ring 1, 28759 Bremen, Germany. E-mail: w.nau@jacobs-university.de
First published on 15th October 2014
Abstract
In the wide area of supramolecular chemistry, cucurbit[n]urils (CBn) present themselves as a young family of molecular containers, able to form stable complexes with various guests, including drug molecules, amino acids and peptides, saccharides, dyes, hydrocarbons, perfluorinated hydrocarbons, and even high molecular weight guests such as proteins (e.g., human insulin). Since the discovery of the first CBn, CB6, the field has seen tremendous growth with respect to the synthesis of new homologues and derivatives, the discovery of record binding affinities of guest molecules in their hydrophobic cavity, and associated applications ranging from sensing to drug delivery. In this review, we discuss in detail the fundamental properties of CBn homologues and their cyclic derivatives with a focus on their synthesis and their applications in catalysis.
 Khaleel I. Assaf | Khaleel I. Assaf was born in Amman, Jordan, in 1985. He graduated with a MSc degree from the Hashemite University, Jordan, in 2009, under the mentorship of Musa El-Barghouthi. He started his PhD studies in 2010 under the supervision of Werner Nau at Jacobs University Bremen, Germany. His research interests lie in the areas of computational and supramolecular chemistry. |
 Werner M. Nau | Werner M. Nau was born in Fulda, Germany, in 1968. He started his chemistry studies in 1987 at the University of Würzburg, Germany, obtained his MSc in 1991 at St. Francis Xavier University, Canada, in spectroscopy, and his PhD at the University of Würzburg in 1994 with Waldemar Adam in organic chemistry. Following his postdoc at the University of Ottawa, Canada, in 1994/95 with J. C. “Tito” Scaiano in photochemistry, he completed his habilitation in physical chemistry with J. Wirz at the University of Basel, Switzerland, where he became Assistant Professor in 2000. Since 2002, he has been a Professor of Chemistry at Jacobs University Bremen, Germany, and, since 2012, Dean. He has held visiting professorships at St. Francis Xavier University, the University of Wisconsin–Madison, USA, the University of Cambridge, UK, and the Institute of Chemical Research of Catalonia (ICIQ), Spain. His research combines principles of supramolecular chemistry and organic photochemistry with spectroscopy and is directed towards applications in bioanalytical chemistry, specializing on fluorescent dyes and probes. |
1. Introduction
The chemistry of cucurbit[n]urils (CBn) is a rapidly developing field (Fig. 1), which has been regularly reviewed from different perspectives.1–7 Most recently, the synthesis of functionalized derivatives has progressed, and among the manifold applications, cucurbituril-catalyzed reactions are receiving increasing attention, which defines two focus areas of this review. The factors which govern their molecular recognition and high-affinity binding properties are continuously unfolding, such that we also update information pertinent in this fundamental area.8–11 Biochemical and medicinal-chemical aspects12–32 are excluded from this review, as are applications in chemosensing,33–48 which in part have been recently reviewed elsewhere.49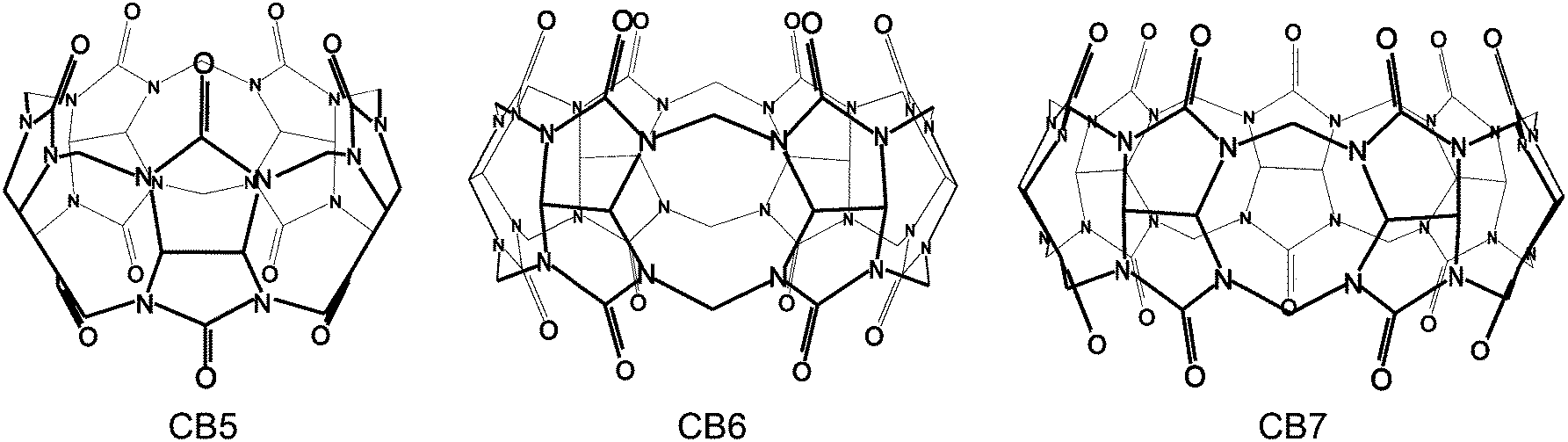 | ||
| Fig. 1 Molecular structures of the three smallest CBn homologues. | ||
2. Synthesis
Synthesis of cucurbituril homologues
In 1905, the parent cucurbituril (CB6) was synthesized by Behrend and coworkers as a sparingly soluble “condensation product”.50 Until today, cucurbiturils are produced by variations of the old synthesis, which involves acid-catalyzed condensation of glycoluril (1) and formaldehyde (Fig. 2). The molecular structure of CB6 was uncovered by Mock and coworkers in 1981; Mock also coined the name “cucurbituril”, due to the resemblance of its structure to a pumpkin, which in turn belongs to the botanical family cucurbitaceae.51 The group of Kim as well as that of Day varied the reaction conditions (e.g., 80–100 °C, HCl or 9 M H2SO4, 10–100 h), which proved to be essential to successfully isolate other homologues, including CB5, CB6, CB7, CB8, and CB10·CB5; CB6 remained the major product.52–54 The precise reaction mechanism of CBn was investigated in great detail by the group of Isaacs.55–58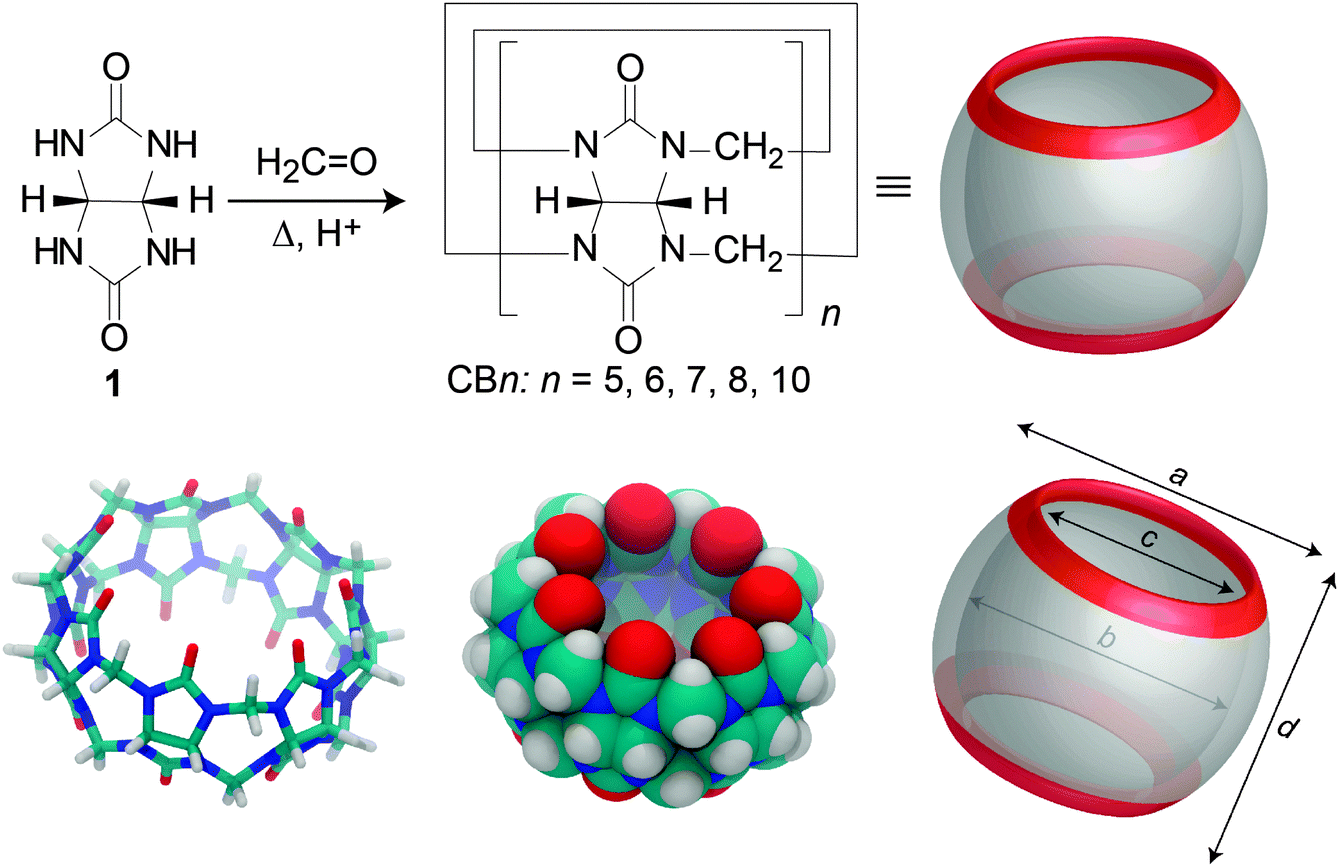 | ||
| Fig. 2 (Top) Synthesis of CBn homologues by condensation of glycoluril (1) and formaldehyde under acidic conditions. (Bottom) Different representations of the CB7 structure. | ||
Recently, the structure of the yet largest CBn member (CB14) has been reported with 14 normal glycoluril units linked by 28 methylene bridges (Fig. 3).59 The twisted CB14 provided important information that larger CBn are actually being formed in the course of CBn synthesis.
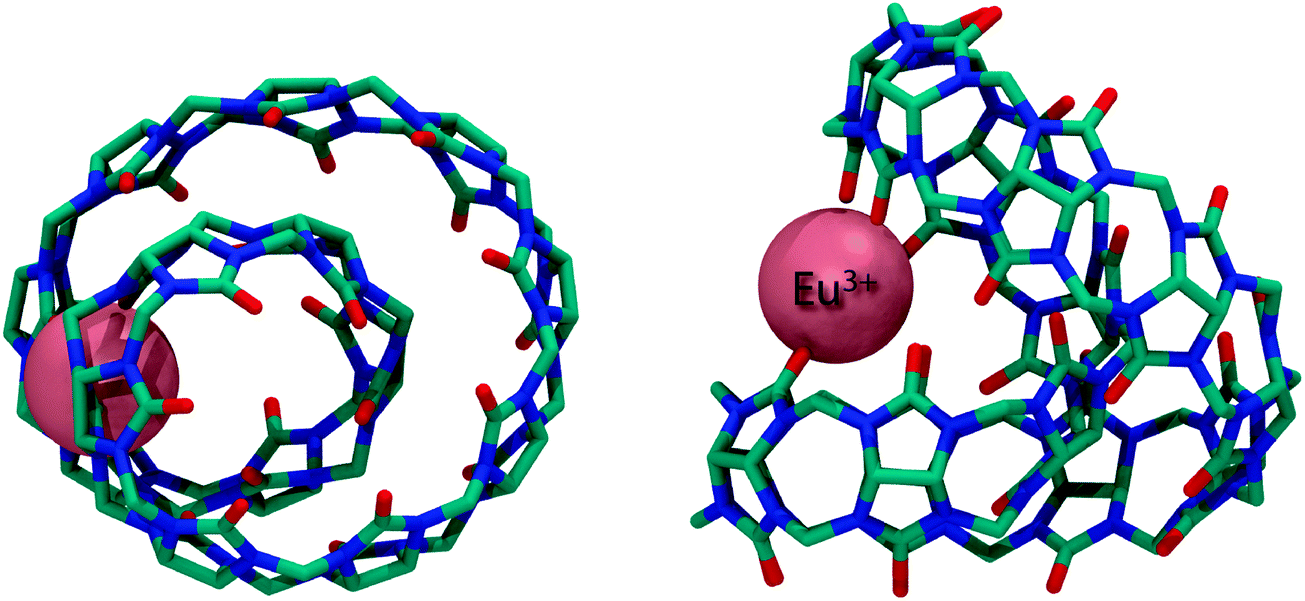 | ||
| Fig. 3 Top (left) and side (right) view of the X-ray crystal structure of the Eu3+ CB14 complex; hydrogen atoms removed for clarity. | ||
It should be noted that CBn preparation and purification often introduces various impurities, always water and acid, frequently acetone and methanol, and, depending on the isolated homologue, ammonium and alkali metal salts.53 A number of techniques have been used to assess the purity (specifically the CBn content) of CBn samples, such as NMR, ITC, and TGA. Kaifer and coworkers have described a practical method to assay the purity of CB7 and CB8 based on UV-Vis titrations with cobaltocenium, which forms highly stable complexes with CB7 and CB8.60
Synthesis of cucurbituril derivatives
Functionalized CBn, inverted-CBn, nor-sec-CBn, and various congeners have also been discovered (Table 1 and Fig. 4), with essentially a new member reported in every year since 2000. Due to their enhanced cavity size and solubility, the new members display a range of novel applications, including their use for supramolecular vesicles, fluorescence sensing, drug delivery, catalysis, etc.22,25,31,44,48,61–65| Year | Compound name | Research group |
|---|---|---|
| 1905 | CB6 (not structurally identified) | Behrend50 |
| 1981 | CB6 | Mock51 |
| 1992 | Me10CB5 | Stoddart66 |
| 2000 | CB5,7,8 | Kim52 |
| 2001 | CycH5CB5, CycH6CB6 | Kim67 |
| 2002 | Ph2CB6, CB10·CB5 | Nakamura, Day54,68 |
| 2003 | (HO)2nCBn, cucurbituril analogues, and partially substituted CBn | Isaacs, Kim, Day69–72 |
| 2004 | Me4CB6, Me6CB6, hemicucurbit[n]uril (n = 6 and 12) | Day, Tao, Keinan, Miyahara73–75 |
| 2005 | CB10, iCBn (n = 6 and 7), hemicucurbit[6]uril | Isaacs, Kim, Buschmann76–79 |
| 2006 | ns-CB10 | Isaacs80 |
| 2007 | (±)-Bis-ns-CB6, Me12CB6 | Isaacs, Tao81,82 |
| 2008 | Partially substituted CBn: MenCBn (n = 5 and 6), and CycHnCB6 | Tao83–86 |
| 2009 | Partially substituted CycPnCB6 | Tao87 |
| 2010 | Bambus[6]uril (Bu6) | Sindelar88 |
| 2011 | (No new member reported) | |
| 2012 | Monofunctionalized CB6, monofunctionalized CB7 | Scherman, Isaacs89–91 |
| 2013 | CB14, norbornahemicucurbiturils, dimeric cucurbit[6]uril, and cyclohexylhemicucurbit[6]uril | Tao, Sindelar, Isaacs, Aav59,92–94 |
| 2014 | Monosubstituted CB6 at methylene bridge (mPhCB6) | Sindelar95 |
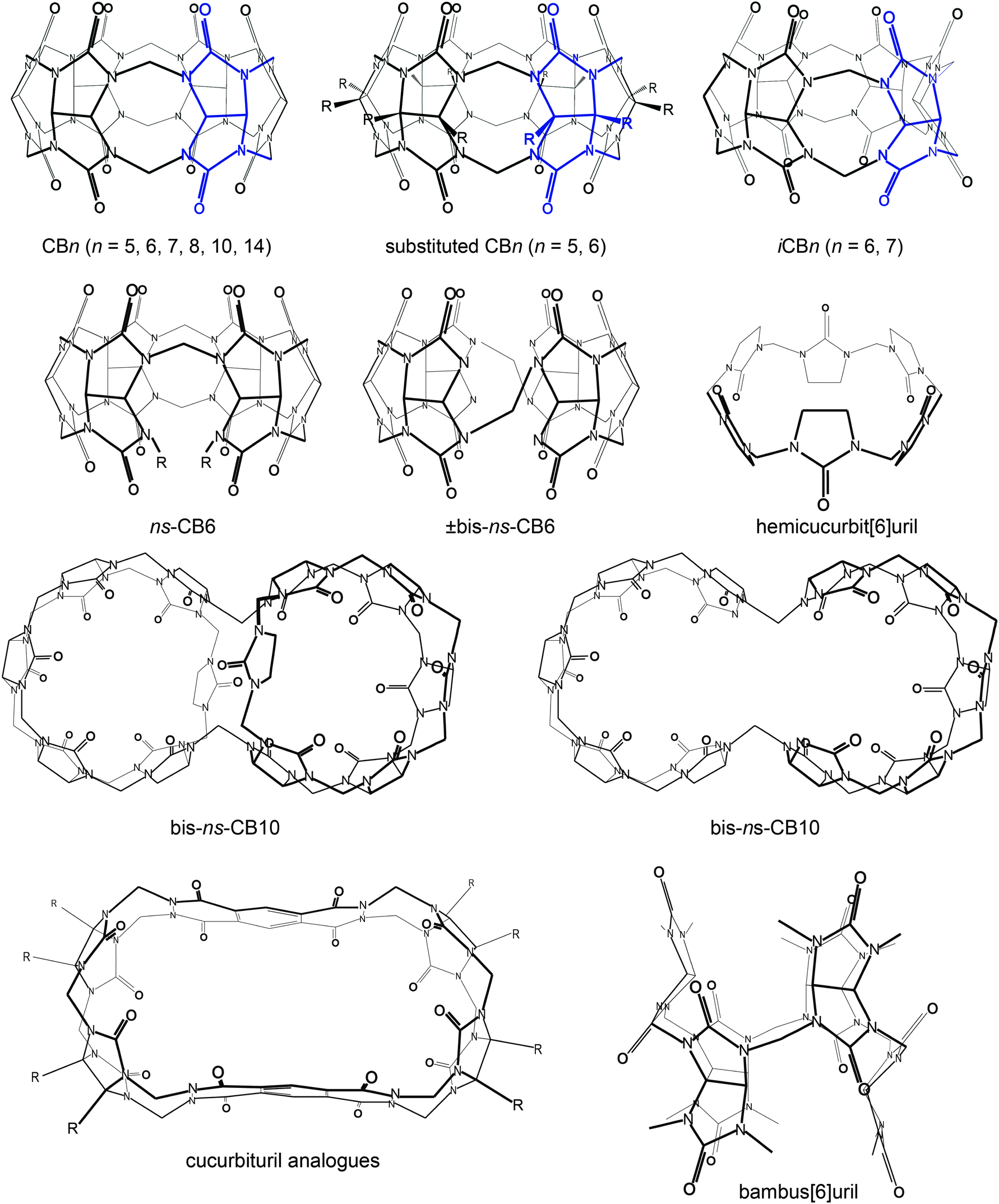 | ||
| Fig. 4 Molecular structures of different cucurbit[n]urils and related cyclic macrocycles; blue parts indicate repetition units varying with n. | ||
The first modified CBn was reported by Stoddart in 1992, who synthesized the fully equatorially methylated CB5 (Me10CB5).66 Later, the group of Kim isolated the fully substituted cyclohexanecucurbit[n]urils (CycH5CB5 and CycH6CB6), that are soluble in organic solvents and, surprisingly, also 170 times better soluble in water than parent CB6.67,96 Many partially substituted CBn and mixtures thereof have been obtained (Scheme 1 and Fig. 5).53,68–73,82,84,85,87,96–101 The reactions of substituted glycolurils so far reported favor invariably the formation of the smaller CBn homologues, CB5 and CB6. In fact, no fully substituted CB7 or CB8 have been isolated. Since alkylation of CBn does not enable functionalization, Kim introduced in 2003 a direct method to produce fully hydroxylated CBn by oxidation of unsubstituted CBn under harsh conditions (hot K2S2O8). The resulting prehydroxyl–CBn could be easily modified to provide tailored CBn derivatives with different functional groups, frequently accompanied by an enhanced solubility in common organic solvents.72
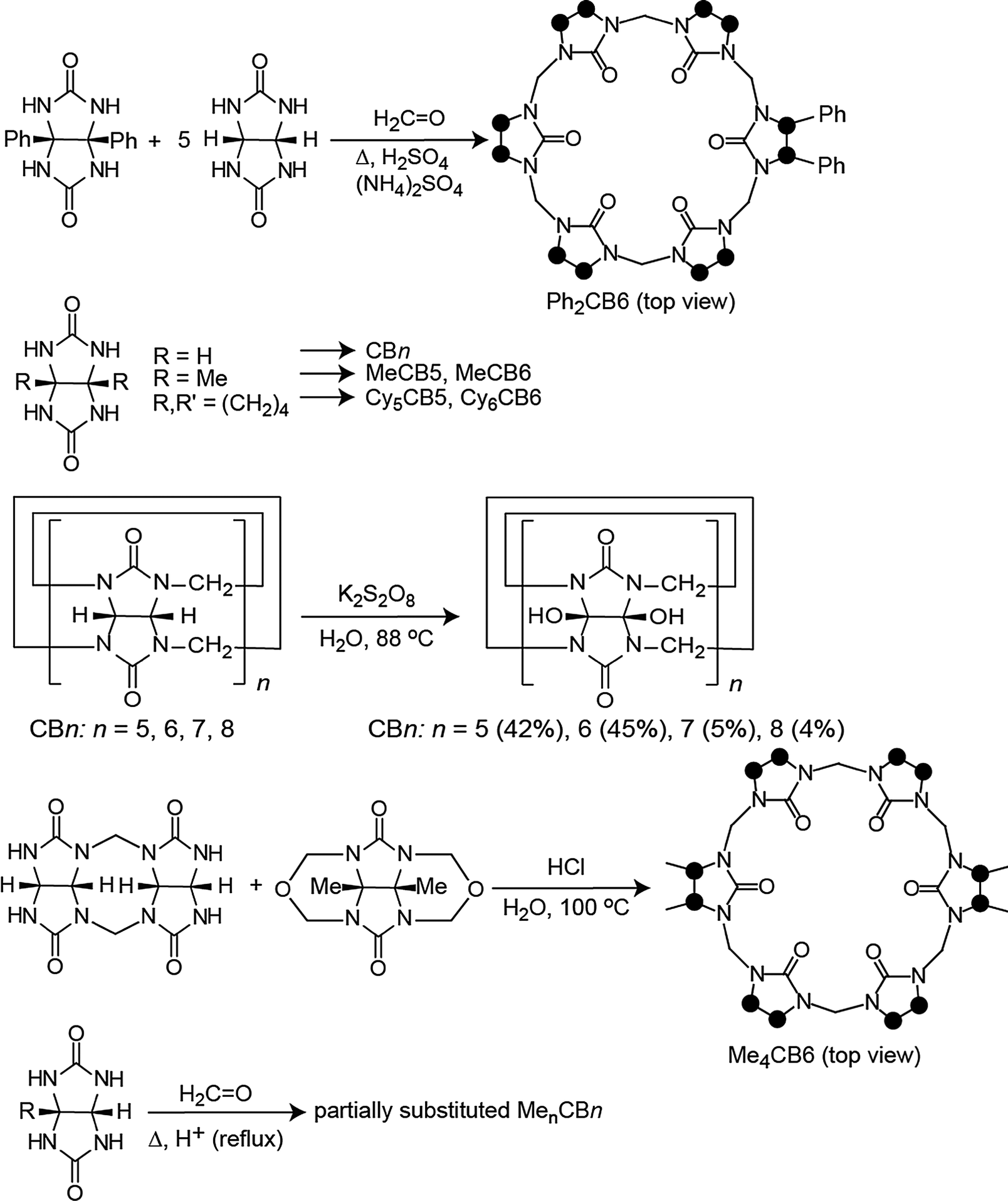 | ||
| Scheme 1 Representative synthetic procedures for different cucurbituril derivatives; see Table 1 for references; dots mark connection points to the second identical half of the symmetric structures. | ||
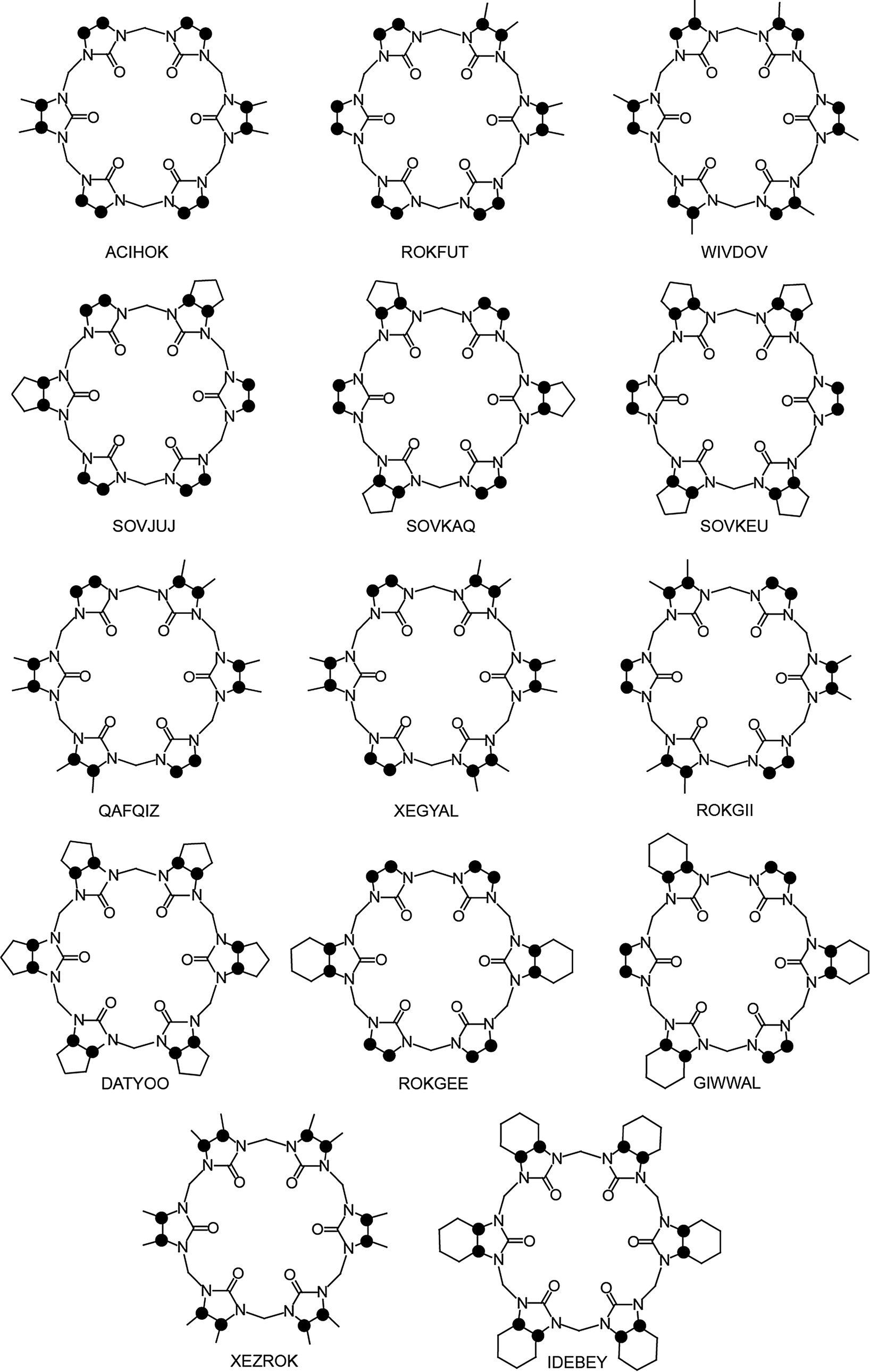 | ||
| Fig. 5 Top views of the reported XRD molecular structures of substituted CB6 derivatives (codes taken from the CCDC data base); see Table 1 for references; dots mark connection points to the second identical half of the symmetric structures. | ||
Isaacs et al. reported the isolation of inverted cucurbit[n]urils (iCBn, Fig. 6), which contain a single inverted glycoluril unit directed towards the inside of the CB cavity.78 iCB6 and iCB7 were isolated either by gel permeation chromatography or by fractional crystallization. Heating of iCB6 or iCB7 in concentrated HCl resulted in the conversion into mixtures of (regular) CBn.102 An indication for the reverse process, interconversion of CB7 to iCB7, has recently been obtained in the gas phase.103 Until now, the inverted derivatives have not found any specific application.
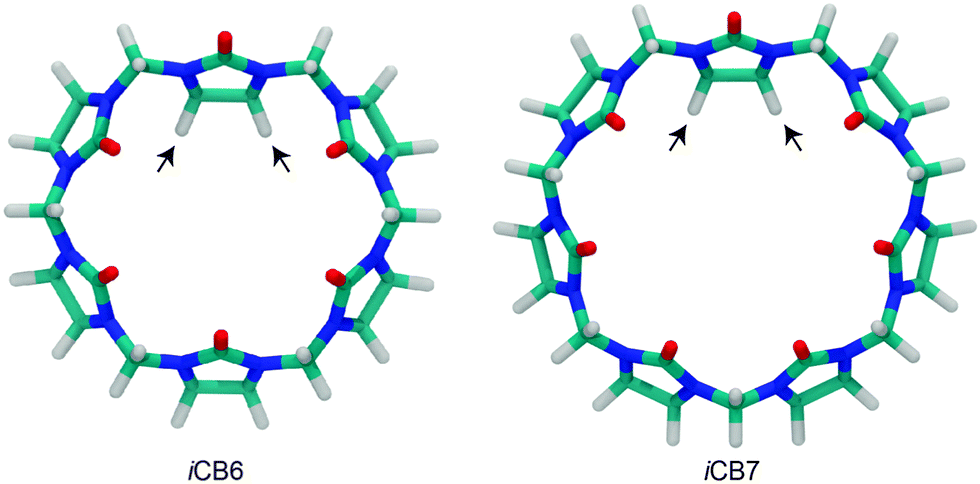 | ||
| Fig. 6 Inverted cucurbiturils, iCBn (n = 6 and 7); arrows indicate inverted glycoluril units; see Fig. 4 for the molecular structure. | ||
Recently, different monofunctionalized CB7 derivatives have been synthesized by Isaacs91 through reaction of the acyclic glycoluril hexamer with a glycoluril bis(cyclic ether) (Scheme 2b). Further, the monofunctionalized CB7 with a chloroalkyl tether can be transformed into the azide functional group, such that other substrates can be conveniently conjugated via click chemistry (Scheme 2b). The high water solubility of the monofunctionalized CB7 (>250 mM) and their host–guest recognition properties will render them solubilization agents for poorly soluble drug molecules and promising candidates for labeling and chemosensing applications as well as surface modification.
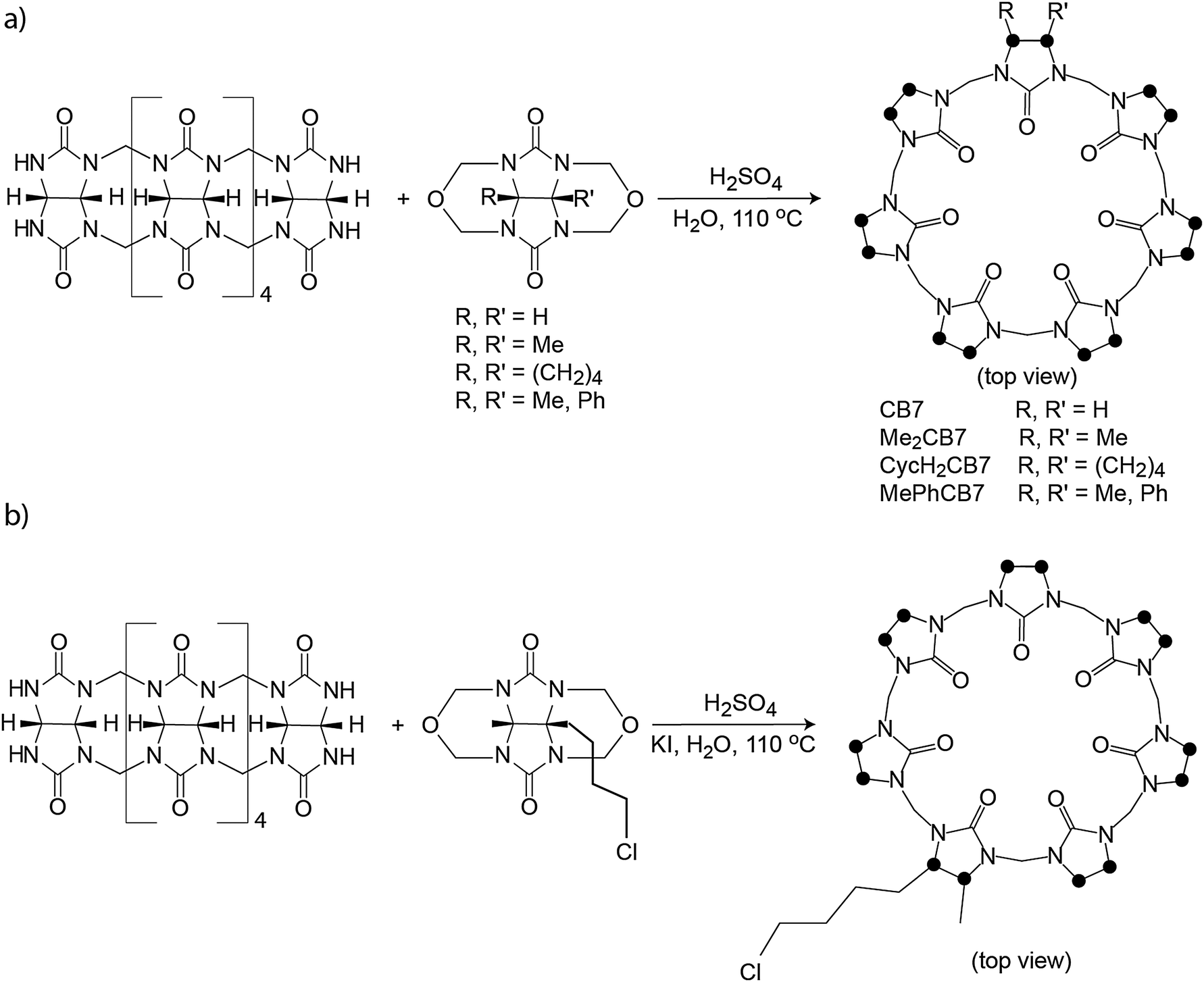 | ||
| Scheme 2 (a) Synthesis of Me2CB7, CycH2CB7, and MePhCB7. (b) Synthesis of a monofunctionalized CB7 derivative; dots mark connection points to the second identical half of the symmetric structures. | ||
A new related macrocycle, prepared by the acid-catalyzed condensation of 2,4-dimethylglycoluril and formaldehyde, was named bambus[6]uril (Fig. 7), Bu6, which combines the structural features of both, cucurbiturils and hemicucurbiturils. Bu6 acts, however, as an anion receptor, e.g., it was shown to bind halide anions through C–H⋯X− interactions.88,104,105
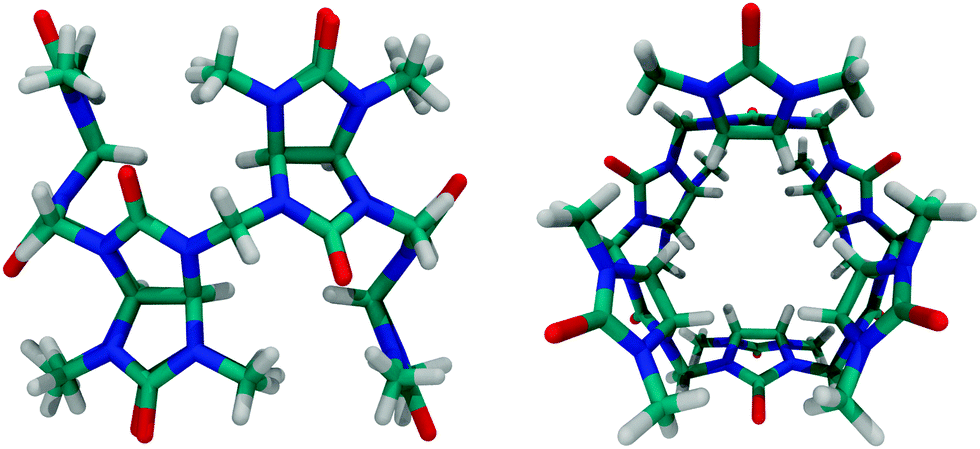 | ||
| Fig. 7 Side and top views of bambus[6]uril; see Fig. 4 for the molecular structure. | ||
By introducing new aldehydes to the reaction mixture of glycoluril and formaldehyde, Sindelar and coworkers have also synthesized the first example for a monosubstituted cucurbituril on the methylene-bridged position (Scheme 3). The most promising result was obtained with 3-phenylpropionaldehyde: (2-phenylethyl)-CB6 was isolated from the crude mixture in 0.2% yield.95
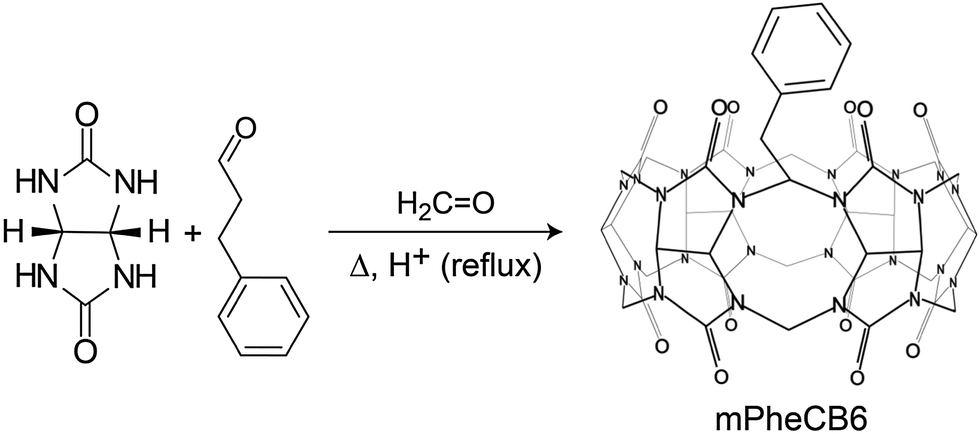 | ||
| Scheme 3 Synthesis of the methylene bridge-monosubstituted cucurbituril mPheCB6. | ||
Other prominent derivatives include bis-nor-sec-CB1080 and nor-sec-CB6,81 which were synthesized by the group of Isaacs. Bis-nor-sec-CB10 has a similar structure as CB10, but lacks two methylene bridges which results in two symmetrically interconnected cavities (Fig. 8). Nor-sec-CB6 (Fig. 4) is structurally comparable to CB6 but lacks a single methylene bridge. The removal of another methylene bridge results in (±)-bis-nor-sec-CB6 (Fig. 4), the first reported chiral member of the CBn family.
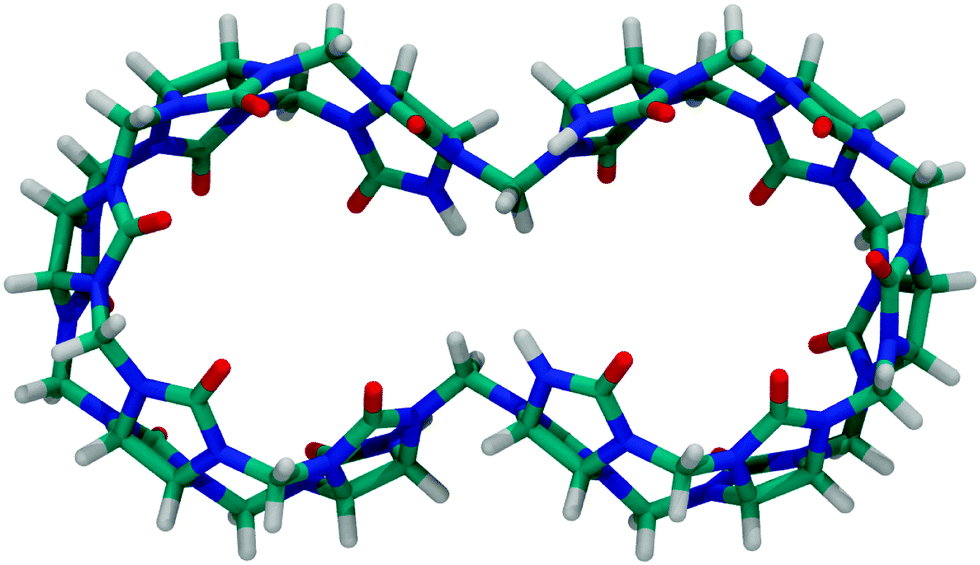 | ||
| Fig. 8 Top view of bis-nor-sec-CB10; see Fig. 4 for the molecular structure. | ||
3. Physical properties and peculiarities of the cucurbituril cavity
The defining structural features of the CBn family are their highly symmetric structure, with negatively charged carbonyl rims and a hydrophobic cavity. The electrostatic potential map of CBn visualizes the high electron density at the carbonyl oxygens (Fig. 9) and clearly illustrates their cation-receptor functionality. Their inner cavity, however, has neither functional groups nor electron pairs pointing towards the inside. It therefore cannot engage in hydrogen-bonding interactions, which – along with the non-dipolar nature of the macrocycle itself – provides an intuitive rationale for the high hydrophobicity of the cavity. | ||
| Fig. 9 Electrostatic potential map (top and side view) of CB7, revealing the negatively charged carbonyl portals (in red). | ||
Polarity and polarizability
Understanding the unique binding of cucurbiturils requires an understanding of the physical microenvironment of the inner cavity. When a guest is encapsulated within the inner cavity of CB, its absorption, fluorescence, and NMR spectra usually change. Such complexation-induced spectral shifts suggest changes in the surrounding electronic and/or magnetic environment. Several solvatochromic dyes have been utilized to probe the cavity of CBn.36,106 These probes are well known to display characteristic changes in their absorption, fluorescence, or phosphorescence spectra as the polarity of the (solvent) environment varies. For example, for CB7, the absorption spectra of rhodamine 6G signal the immersion in a less polar environment when the dye is encapsulated. A polarity similar to that of alcohols (n-octanol, ε = 10.3) has been reported for CB7,107 similar to the polarity determined for the cavities of other water-soluble macrocycles such as cyclodextrins or calixarenes. However, while CBn hosts have a nonpolar inner cavity and are themselves not dipolar, they display a very high quadrupole moment,8 which allows higher-order electrostatic interactions. These have been implicated to account for the equatorial orientation of dipolar guests, such as ketones, in their cavity to achieve optimal quadrupole interactions.8,108 In contrast to the low polarity of the inner cavity of CBn, the portal regions of CBn have expectedly a more polar microenvironment.109Beyond the relatively low polarity, we have shown that the inner cavity of CBn macrocycles has an extremely low polarizability/refractive index, which is closer to the gas-phase than to that of any other known solvent.110,111 The polarizability was initially detected using DBO (2,3-diazabicyclo[2.2.2]oct-2-ene), a neutral azoalkane, as a spectrophotometric probe.110 Its spherical shape and small size (111 Å3) allow it to be fully immersed inside the size-complementary cavity of CB7 (242 Å3) as well as that of other, similarly sized, water-soluble macrocycles, in particular β-cyclodextrin and p-sulfonatocalix[4]arene. A linear correlation between the inverse oscillator strength of the near-UV absorption band of DBO and the polarizability of different solvents was established, which could be subsequently used to interpolate or extrapolate the polarizability of several macrocyclic cavities, including CB7, cyclodextrins, and calixarenes (Table 2). The results showed that the CB7 interior has an extremely low polarizability (0.12), far below that of the two other macrocycles, which showed polarizabilities between those of water and benzene. The low polarizability of the CBn cavity can be rationalized along several lines: (i) there are no bonds or lone electron pairs pointing to the inside, (ii) the hosts are non-aromatic, (iii) any residual electron density at the nitrogens is mesomerically delocalized to the carbonyl oxygens, and, potentially, (iv) the packing is sufficiently loose to avoid close guest-to-wall contacts, e.g., the packing coefficient (PC) of DBO in CB7 is only 46% and therefore relatively loose, leaving some “vacuum”. For further discussion on PC arguments, see below and ref. 8.
| Environment | Refractive index (n)a | Polarizability (P)b |
|---|---|---|
| a Refractive index, converted using the formula P = (n2 − 1)/(n2 + 2). b From the empirical relationship (1/f = 3020–8320P) by using the oscillator strength (f) of DBO chromophore as a solvatochromic probe. c For equatorial chromophore orientation; values for axial chromophore alignment are n = 1.19 and P = 0.12. d Heptakis-(2,6-di-O-methyl)-β-cyclodextrin. e p-Sulfonatocalix[4]arene, for equatorial chromophore orientation; values for axial chromophore alignment are n = 1.43 and P = 0.26. f Value determined using biacetyl as solvatochromic probe, cf.ref. 110. | ||
| Gas phase | 1.000 | 0.000 |
| CB7c | 1.17 | 0.11 |
| Perfluorohexane | 1.252 | 0.159 |
| β-Cyclodextrin | 1.33 | 0.20 |
| H2O | 1.333 | 0.206 |
| SDS micelles | 1.34 | 0.21 |
| n-Hexane | 1.375 | 0.229 |
| Triton XR-100 micelles | 1.38 | 0.23 |
| POPC liposomes | 1.38 | 0.23 |
DMe-β-CD (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex)d :1 complex)d |
1.39 | 0.24 |
| Calixarenee | 1.41 | 0.25 |
| Benzene | 1.501 | 0.295 |
| Diiodomethane | 1.742 | 0.404 |
| Hemicarcerandf | 1.86 | 0.45 |
By exploiting DBO as a fluorescent probe, the low polarizability inside CB7 could be independently confirmed through very long radiative lifetimes, which are theoretically expected to increase as the refractive index of the environment, that is, its polarizability, decreases. Further independent evidence for the unique electronic environment offered by the interior of CB7 has recently been reported by Czar and Jockusch.112 The authors used acridine orange (AOH+) as a probe to compare the gas-phase fluorescent properties of the free dye and its CB7 complex with that in aqueous solution. The excitation maxima of the dye in vacuum and in CB7 were the same, regardless of whether the CB7·AOH+ complex is itself in the gas phase or in aqueous solution. This result supports the finding that the cucurbituril interior is indeed “gas-phase-like”.
Most recently, we have involved NMR to further test the much debated hypothesis of a unique “phase of matter” inside cucurbiturils.45 In detail, we took advantage of the fact that 19F chemical shifts do not strongly depend on magnetic shielding effects (such as 1H NMR shifts), but rather on the polarizability of the immediate surrounding of the F atoms.113,114 In an unconventional approach, SF6 and CF4 were used as “solvato-resonant” probes for the polarizability (P) by establishing a linear empirical correlation between the 19F chemical shift and the polarizability of different solvents as well as the gas phase (Fig. 10). The polarizability of the CB7 cavity, interpolated from the 19F chemical shift of the CB7·SF6 complex, was found to be higher (P = 0.13) than that of the gas phase (P ≡ 0.00) but lower than that of water (P = 0.21), a result which is well in line with previous results obtained by optical spectroscopy.36,106,110,112 CF4 did not bind to CB7, but could be used as a solvato-resonant probe for CB6; in this case, the interpolated polarizability (P = 0.10) was found to be even somewhat lower than the value obtained for SF6 inside CB7.115
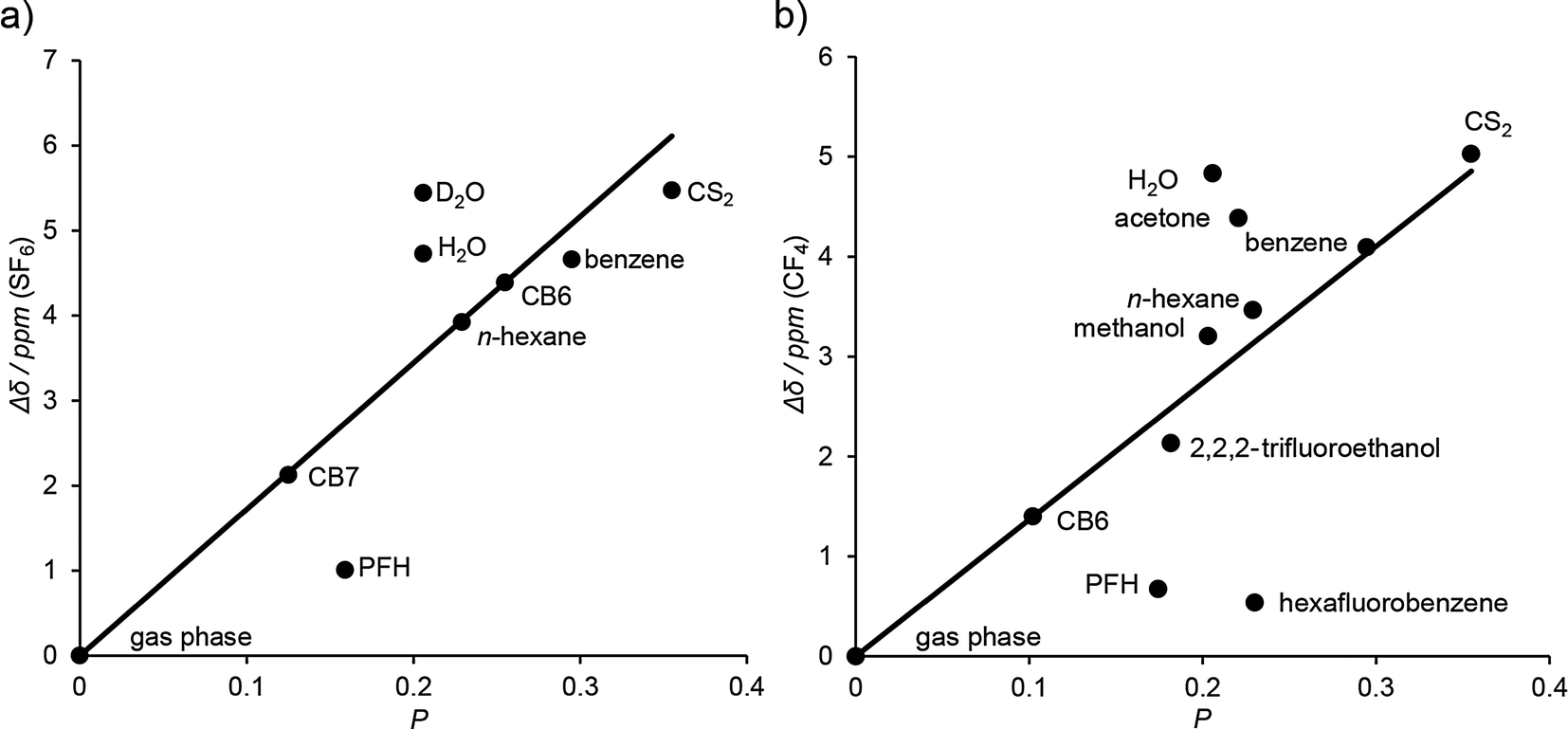 | ||
| Fig. 10 Linear correlation of the 19F chemical shift in the gas phase, different solvents, and in the complexes with CB6 and CB7 versus the polarizability (P) of the environment (a) for SF6 and (b) for CF4 as solvato-resonant polarizability probes. The chemical shifts (down-field) are relative to the gas phase. The polarizabilities of the CB6 and CB7 cavities were interpolated from the correlations, see text and ref. 45. | ||
Structural features
Table 3 lists pertinent structural parameters of common CBn homologues and stereoisomers. In contrast to many other macrocyclic hosts, CBn have a very rigid structure (borne out, for example, by small distortions in various crystal structures)116,117 which renders the definition of the cavity parameters particularly informative. In detail, all CBn, (n = 5–8 and 10), have the same height (d = 9.1 Å) but show large variations in cavity width (Table 3). It can be seen that the cavity width of CB10 is slightly too small to allow equatorial encapsulation of CB5. Indeed, the X-ray structure of the CB10·CB5 complex showed that CB5 sits diagonally to the axis of CB10.54| Outer diameter | Inner cavity | Height | ||
|---|---|---|---|---|
| a | b | c | d | |
| a Values were calculated for the geometry of CB9 optimized by the B3LYP/6-311G** method. b Average determined from the crystal structure with the CCDC code COQWOV, ref. 55. c Range defined by the shortest and longest wall-to-wall distances; determined from the crystal structures of iCB6 and iCB7 with the CCDC codes NEBDII and NEBDEE, respectively, from ref. 78. | ||||
| CB5 | 13.1 | 4.4 | 2.4 | 9.1 |
| CB6 | 14.4 | 5.8 | 3.9 | 9.1 |
| CB7 | 16.0 | 7.3 | 5.4 | 9.1 |
| CB8 | 17.5 | 8.8 | 6.9 | 9.1 |
| CB9a | 19.0 | 10.3 | 8.6 | 9.1 |
| CB10b | 20.0 | 11.7 | 10.0 | 9.1 |
| iCB6c | 10.7–14.4 | 3.8–5.8 | 4.3–3.9 | 9.1 |
| iCB7c | 11.2–16.0 | 5.4–7.3 | 6.7–5.4 | 9.1 |
The key-and-lock principle for macrocyclic hosts requires a complementarity between the guest size and the volume of the binding site to allow the formation of an inclusion complex. It has been shown that the “inner” cavity of CBn (Fig. 11) is best suited to assess this complementarity and to evaluate binding strengths on grounds of PC arguments (see next Section).8 As can be seen from Table 4, the sizes of the inner CBn cavities span a very large range from 68–691 Å3, those of the inverted iCBn diastereoisomers are expectedly slightly smaller.
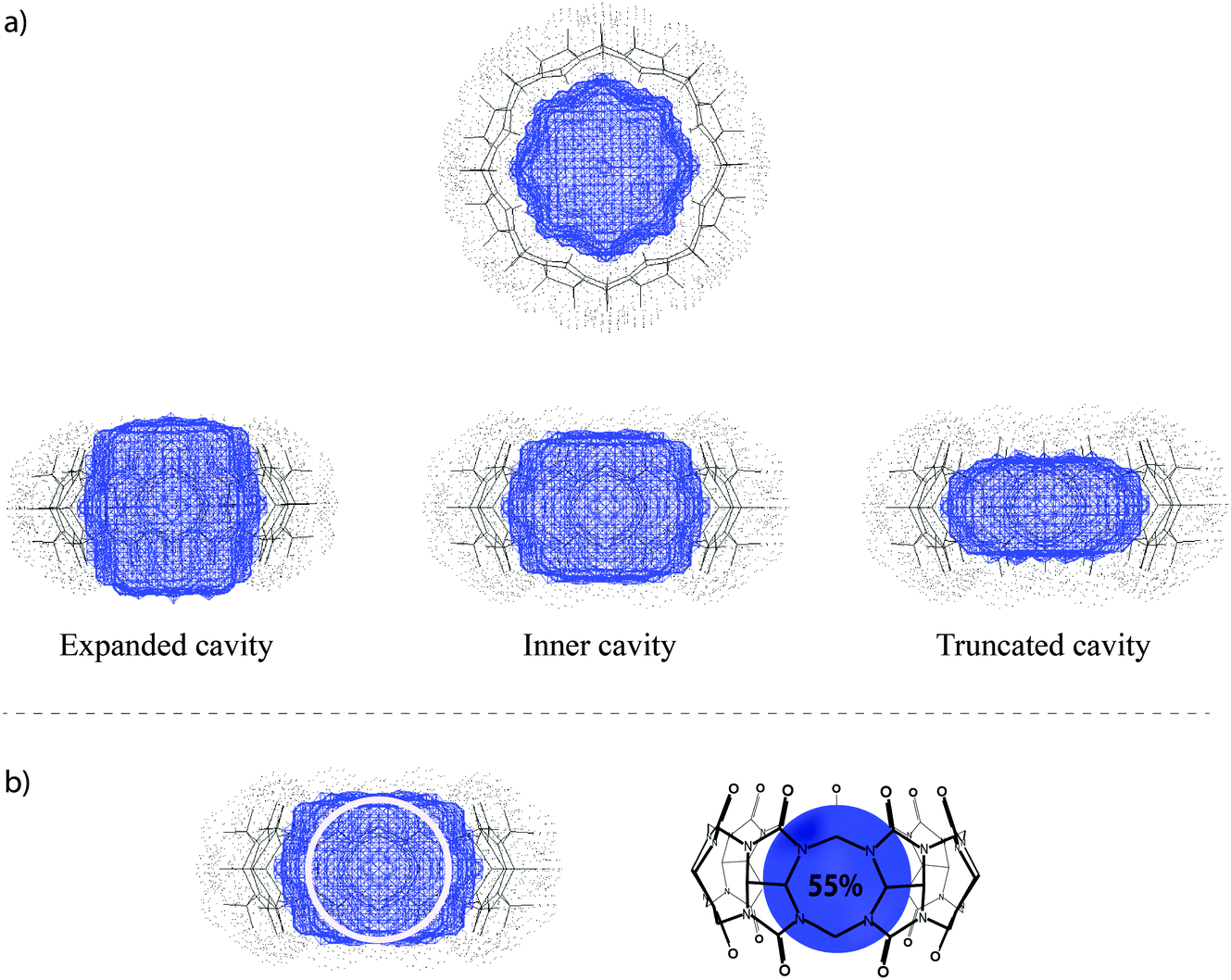 | ||
| Fig. 11 (a) Top and side views of different cavity definitions, taken from ref. 8. Copyright © 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (b) Schematic illustration of an ideal cavity filling of 55% by encapsulation of a spherical guest. | ||
Packing coefficient
Rebek and Mecozzi introduced the packing coefficient (PC), that is, the ratio of the guest size and the host cavity volume, as an estimator of the steric goodness of fit of host–guest inclusion complexes.118 A value of ca. 55% was found to give the best binding affinities for the resulting host–guest complexes, while larger or smaller values were associated with lower affinities. Although the packing rule was originally derived for capsules as hosts, it has proven useful for several other macrocyclic receptors, particularly cucurbiturils.8 It has not been written down yet that the number of 55% is actually a natural one as far as water-soluble host–guest systems are concerned: the packing coefficient of water in water, for example, is also 55%, meaning that each water molecule (V = 17.0 Å3) has on average 45% void space at its disposition. The enthalpic minimum (estimated from the Lennard-Jones potential with ε0 = 1.12σ), lies at 71% for a spherical cavity, which is higher because of the neglect of entropy, the consideration of which lowers the ideal PC value.We have previously demonstrated that PC arguments can be successfully applied to predict the stability of CBn complexes.8 The analysis of packing coefficients for representative sets of known guests with clearly defined hydrophobic binding motifs revealed average values of 47% for CB5, 58% for CB6, 52% for CB7, and 53% for CB8, which are well in line with the ideal packing (55% solution). In general, guests with associated packing coefficients of higher than 75% do not form stable inclusion complexes as a consequence of the associated entropic penalty unless additional interactions (such as ion–dipole ones) are at work. On the other hand, guests with packing coefficients lower than 30% also do not form stable complexes. Therefore, while the absolute PC values are only an empirical estimator, stable inclusion complexes result within the PC range of 45–65%.
Release of high-energy water molecules
The first guest molecules experimentally proven to be encapsulated in the hydrophobic cavity of a cucurbituril (CB6) were water molecules.52 Depending on their cavity volume, CBn can accommodate between 2 (for CB5) to 22 water molecules (for CB10). Estimates for the number of water molecules contained in the CB cavity are given in Table 5.8 These water molecules are of high energy as a consequence of their greatly limited hydrogen bonding and due to weak dispersion interactions with the walls of the weakly polarizable CB cavity (see above).| CBn | Number of water molecules in inner cavity | ΔEpota (kJ mol−1) |
|
|---|---|---|---|
| MD simulation | PC analysis | ||
| a Difference in potential energy for the removal of all cavity water molecules and transfer of those to a spherical cavity in the aqueous bulk, see ref. 120. b Values from ref. 120 are given in square brackets. | |||
| CB5 | 2 [2.0]b | 2 | −41.6 ± 28.8 |
| CB6 | 4 [3.3]b | 4 | −51.1 ± 29.0 |
| CB7 | 7 [7.9]b | 8 | −102.4 ± 31.3 |
| CB8 | 10 [13.1]b | 12 | −66.2 ± 10.7 |
| CB9 | 14 | 16 | — |
| CB10 | 20 | 22 | — |
In aqueous solution, the release of high-energy water from the cavity upon the complexation by a guest contributes a major part to the overall hydrophobic effect (Fig. 12).8,119–121 Biedermann et al. used MD simulations to calculate the relative potential energy gain for the removal of all water molecules from CBn cavities (Table 5).120 The Results showed that the deficiency of hydrogen bonding of the encapsulated water molecules and their absolute number are the determinants for the overall gained energy. With increasing cavity size, the energy of each water molecule decreases due to improved hydrogen-bonding possibilities. The maximum calculated potential energy was obtained for CB7. Indeed, the 242 Å3 large CB7 cavity does not allow the internal water molecules to arrange in a structurally stable H-bond network.
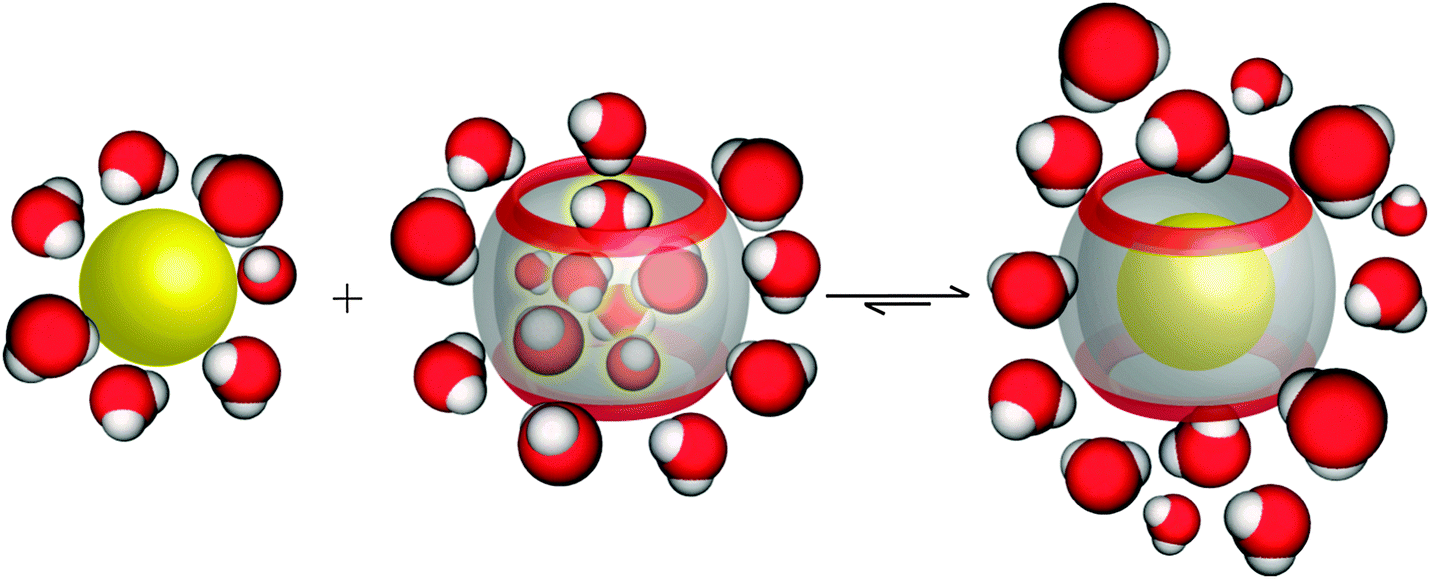 | ||
| Fig. 12 Schematic illustration of the release of high-energy water molecules from the CB7 cavity upon binding of a hydrophobic guest. | ||
However, the energy of each individual internal water molecule is highest for small CBn, such as CB5 and CB6, but the total energy for releasing all water from the inner cavity of CB7 is higher due to the larger number of water molecules (more than twice) that are released upon the complexation process. On the other hand, the large cavity of CB8 allows the optimization of the H-bond network to a degree that is structurally similar to bulk water, with the result that the energy contribution for water release is less from CB8 than from CB7, despite its larger cavity. Kim and coworkers have reported another essential role for water molecules on host–guest recognition.122 After the transfer of the CBn–alkyldiammonium complexes from aqueous solution to the gas phase, it was found that the complex stabilities in the gas phase are different from those in aqueous solutions. The results were attributed to the fact that water molecules affect the ion–dipole interactions between the host and the guest. Indeed, in the absence of solvent, ion–dipole interactions are the main driving force for complexation. In contrast, in solution, the hydrophobic effect, and in particular the release of high-energy water, become dominant.
4. Host–guest Chemistry
The host–guest chemistry of CBn has already been reviewed extensively.1,4,8,9,24,98,123–125 The hydrophobic effect, with emphasis on the release of the high-energy water, as well as ion–dipole and dipole–dipole interactions have been addressed as the main driving forces for the binding of different guests by CBn. In 1983, Mock and coworkers were the first to study the complexation of alkylammonium and alkyldiammonium ions with CB6 in aqueous formic acid and to determine their binding affinities.126 Two years later, Freeman reported the first X-ray diffraction structure of a host–guest complex of a CBn, that of the p-xylylenediammonium ion encapsulated by CB6 (Fig. 13).127 | ||
| Fig. 13 X-ray structure of the p-xylylenediammonium ion encapsulated by CB6, the first X-ray diffraction structure of a CBn complex.127 | ||
The first structure-selectivity relationships between various alkyl- and aryl-substituted ammonium ions and CB6 were also established by Mock.128,129 The extensive studies revealed that CB6 shows a chain length-dependent selectivity (Fig. 14). Diammonium ions with a pentano or hexano bridge bind significantly stronger to CB6 than those with shorter or longer lengths. For 1,6-diammoniumhexane, the alkyl chain contracts to adopt a slightly folded conformation in order to optimize cavity filling and to additionally maximize ion–dipole interactions.130 It was also found that CB6, with a cavity volume of 142 Å3, exhibits size and shape complementarity.44,128,129,131–134 For example, CB6 can form an inclusion complex with p-tolylmethanamine, but not with its ortho or meta isomers. Similarly, we have shown that even though n-pentane, isopentane, and neopentane are equally large (all 96 Å3, PC = 68%), only the first two guests showed sizable binding with CB6, while neopentane does not form an inclusion complex with CB6, likely for kinetic reasons (slow ingression through the tighter portals).44 A consistent result had been obtained by Mock for the neopentylammonium ion.126,128 Moreover, CB6 differentiates between saturated and unsaturated hydrocarbons, with the latter displaying a weaker binding for the same carbon framework.44
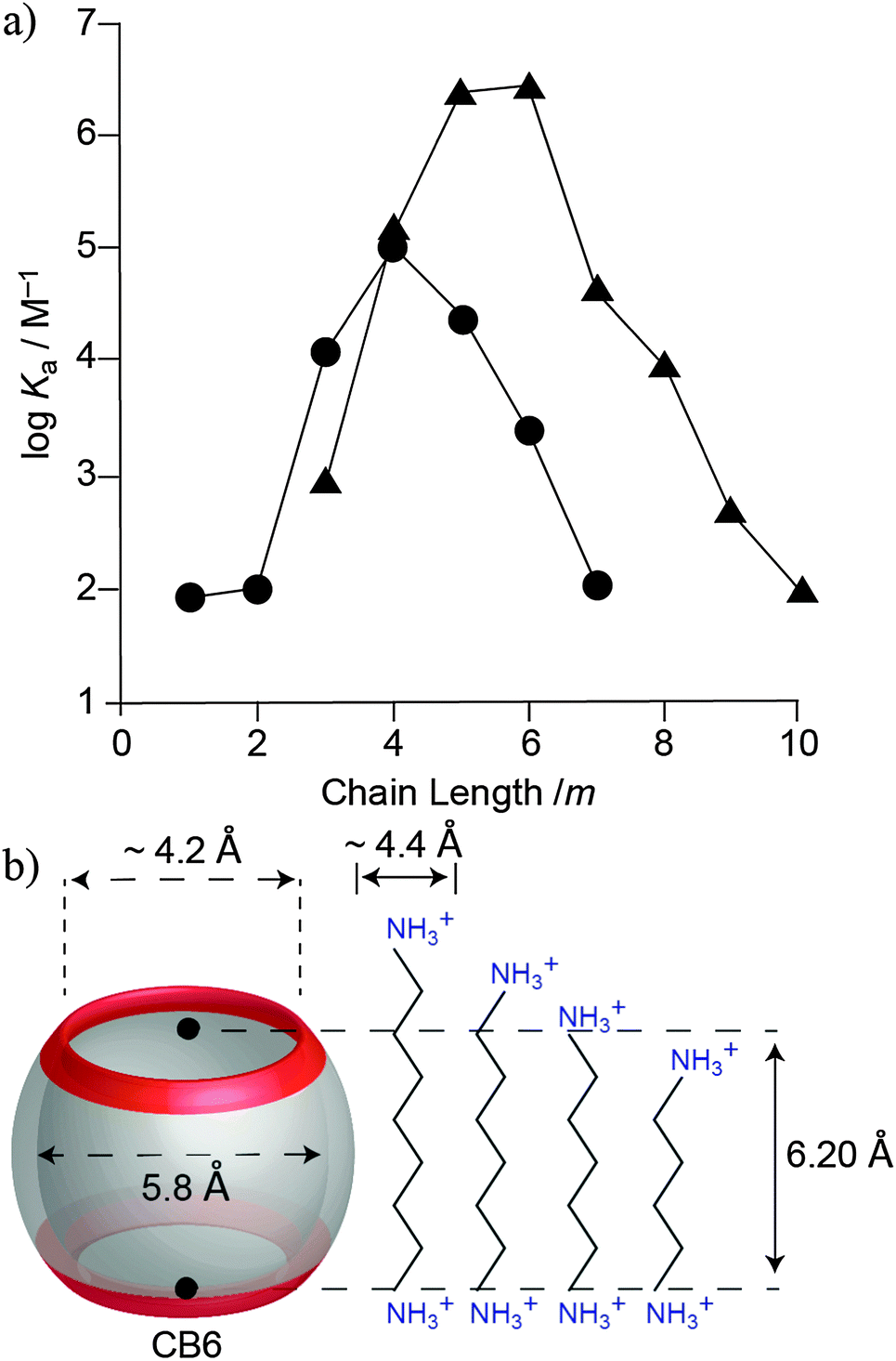 | ||
| Fig. 14 (a) Relationship between the binding constant (logKa) versus chain length m for H(CH2)mNH3+ (●) and +H3N(CH2)mNH3+ (▲). (b) Dimensional comparison between CB6 and various α,ω-alkane diammonium ions. | ||
As is the case for all macrocycles that possess a hydrophobic cavity, the guest hydrophobicity plays also an important role in the stability of CB complexes. For example, an alkylamine with a thioether linkage was found to bind more strongly than its oxygen counterpart, but less than the alkylamine itself. This result has been attributed to the hydrophobicity of an alkyl group being highest, that of an alkoxy group being lowest, and that of an alkylthio group being in between.128 It should be noted that the driving force due to desolvation of convex guests is mainly related to their surface area. It produces a “classical” hydrophobic effect with a favorable entropic component, which is different from the “nonclassical” effect due to the removal of high-energy water from concave cavities, which has a favorable enthalpic signature.121
Recently, we have reported a size-selective binding of alkyl versus perfluoroalkyl chains by CBn.45 CB7 shows preferential binding of perfluoroalkyl residues over normal alkyl groups, which characterizes this macrocycle as being at least as fluorophilic as lipophilic in nature. The 1H and 19F NMR of the ditopic guest 1-(perfluorobutyl)pentane clearly expose this binding preference (Fig. 15). The preferential binding of the fluorinated residue can be reconciled in terms of the sufficiently large cavity size of CB7 (242 Å3), which snugly fits the perfluoroalkyl tail and results in an ideal packing of 55%. In contrast, the smaller CB6 homologue (cavity size 142 Å3) showed a preferential complexation of the alkyl chain of the same guest, because the volume of the perfluoroalkyl group is slightly too large to be accommodated by the smaller host cavity.
 | ||
| Fig. 15 Representative structures illustrating the residue-selective binding of 1-(perfluorobutyl)pentane with (a) CB6 and (b) CB7. | ||
High-affinity binding
CBn bind many size/shape complementary guests with very high affinity (103–1010 M−1). CB7, for example, forms stable 1:1 inclusion complexes with adamantanes135,136 and diamantanes,137,138 with bicyclooctanes,110,135,136 and with ferrocene98,139–143 as well as with cobaltocene143–145 derivatives (Table 6). The reported affinities of such residues can approach146,147 or even exceed137 those of the strongest non-covalent interactions found in nature, in the biotin–avidin pair. The ferrocene–CB7 complex was first reported by Kaifer143 and Kim98 in 2003. Fig. 16 shows two crystallographically independent ferrocene/CB7 complexes that differ in their co-conformation, i.e., the relative alignment of the guest inside the host.148 DFT calculations in the gas phase (B3LYP/6-31G*) predict that the complex with perpendicular orientation should be favored.149 We have attributed this to quadrupole interactions resulting from the high (negative) quadrapole moment of both, CB7 and ferrocene.8 Further studies on ferrocene derivatives led to an extremely high affinity of 1015 M−1 by introducing an ammonium unit (Table 6).150
| Guest | K/M−1 | Guest | K/M−1 |
|---|---|---|---|
| a Unpublished results, by fluorescent indicator displacement. b From ref. 135. c From ref. 136. d From ref. 143. e From ref. 150. f From ref. 137. g Measured in pure D2O. | |||
| A1 | 7 × 109 | C1 | >106 |
| A2 | 2 × 1010 | C2 | 3 × 109 |
| A3 | 2 × 1014 | C3 | 2 × 1012 |
| A4 | 1 × 1014 | C4 | 4 × 1012 |
| A5 | 8 × 1014 | C5 | 3 × 1015 |
| A6 | 2 × 1012 | D1 | 4 × 109 |
| A7 | 5 × 1015 | D2 | 3 × 1011 |
| B1 | 6 × 109 | D3 | 2 × 1015 |
| B2 | 2 × 1014 | D3 | 7 × 1017g |
| B3 | 1 × 1015 | ||
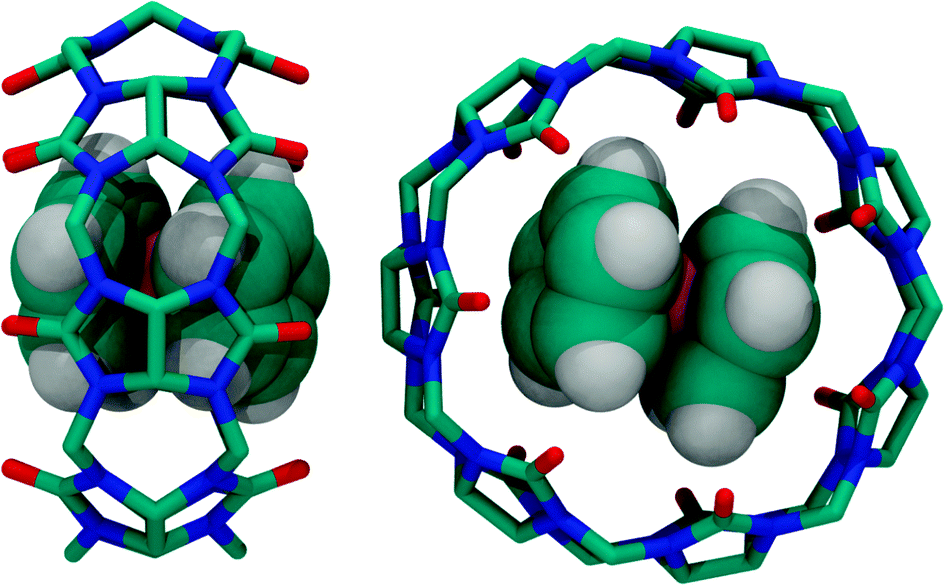 | ||
| Fig. 16 Two different X-ray crystal structures of the complex between ferrocene and CB7 (side view for co-axial co-conformation and top view for orthogonal co-conformation), see ref. 139. | ||
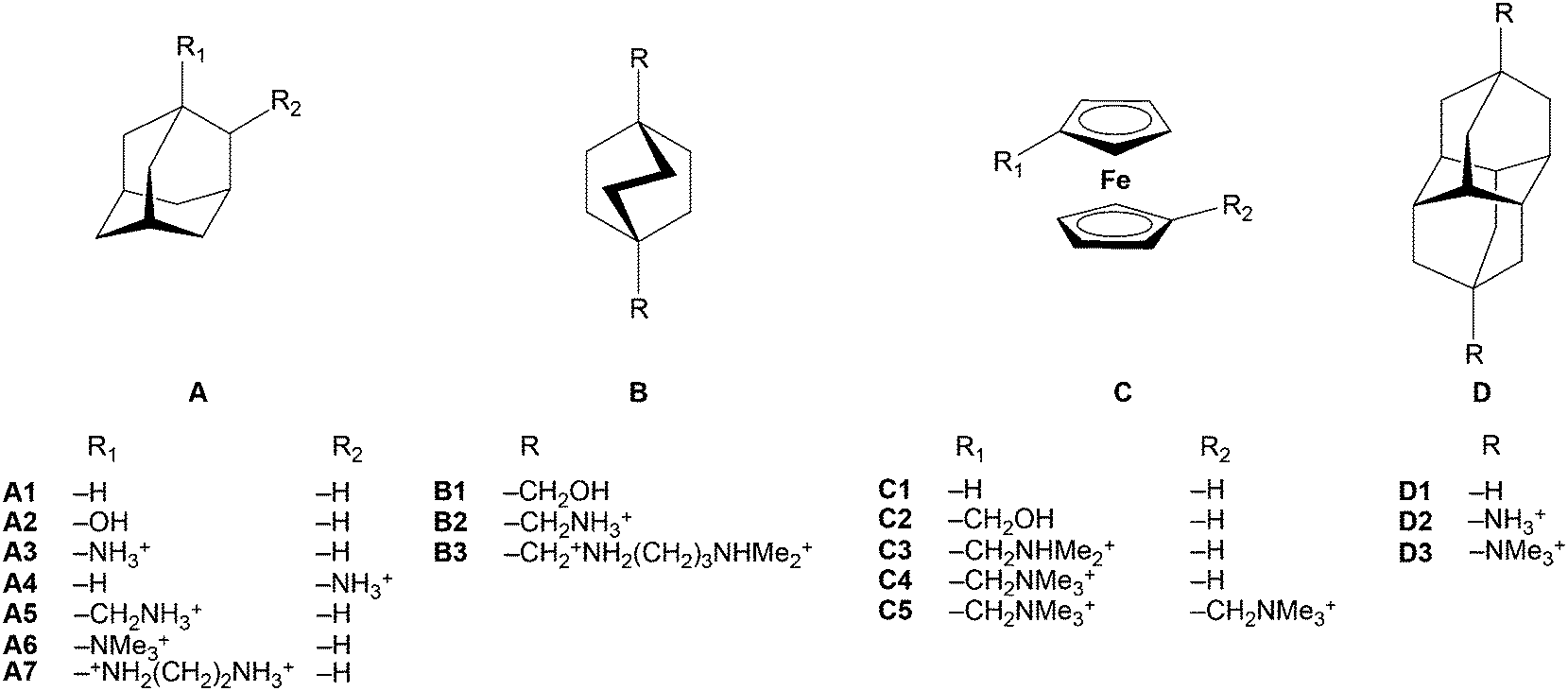 | ||
| Fig. 17 Molecular structures of high-affinity binders to CB7. | ||
Adamantylammonium was also found to form a very stable complex with CB7 (Ka = 4.2 × 1012 M−1).136 Most recently, Isaacs and coworkers have reported the tightest binding that was ever measured for a monovalent molecular recognition event in water, between CB7 and diamantane diammonium ion, with Ka = 7.2 × 1017 M−1 in D2O and 1.9 × 1015 M−1 in 50 mM NaO2CCD3 by using 1H NMR competition experiments.137 The crystal structure of the CB7 complex, Fig. 18, shows the complete inclusion of the diamantane residue in the CB7 cavity. The near perfect size as well as shape complementarity between the rigid CB7 cavity and the diamantane core led to an attomolar dissociation constant.
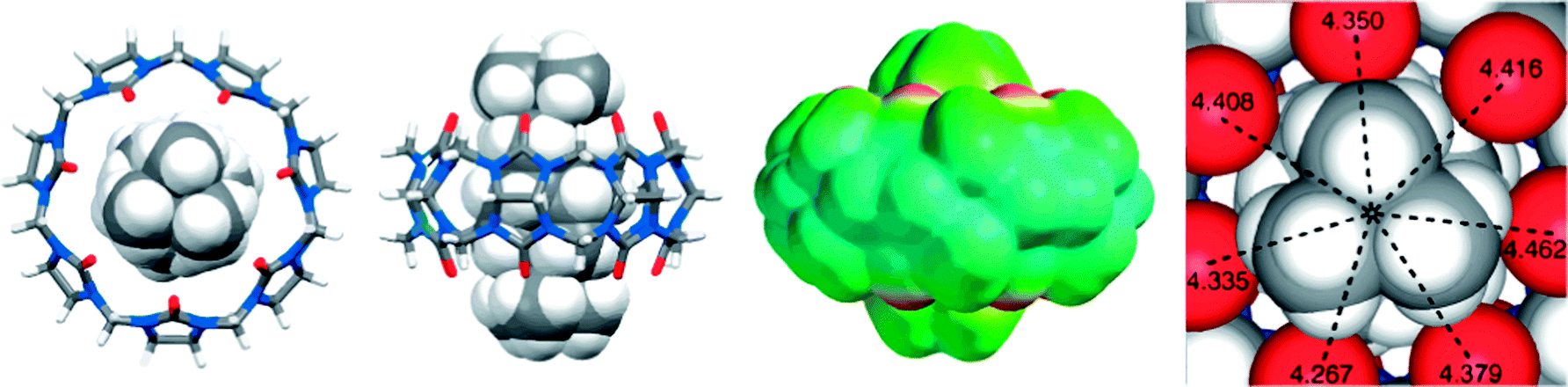 | ||
| Fig. 18 Representations of the X-ray crystal structure of the CB7·diamantane diammonium complex. Taken from ref. 137. Copyright © 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
The combined binding constants for CB7 in Table 6 can be approximately understood – considering only the composite hydrophobic effect and ion–dipole interactions, but ignoring differential dispersion interactions for the known reasons8 – as follows: (1) the removal of all high-energy water molecules alone can account for a binding constant as high as 108 M−1. (2) The desolvation of the guest contributes another factor of 102–104, depending on its hydrophobic surface area. (3) Up to two suitably, ideally centro-symmetrically, placed trimethylammonium groups add another factor of 103 each. This results in the observed range from 1010–1018. Incidentally, the fact that a quaternary trimethylammonium group presents (when measured under identical conditions) a better binding motif than a ternary or primary ammonium group (Table 6) demonstrates nicely that hydrogen bonding interactions present no dominant driving force in the binding of guests to CBn hosts. In fact, hydrogen bonding of guests with a host is in general unlikely to outperform hydrogen bonding with water molecules in aqueous solution.
Interestingly, diamantane in CB7 presents a more densely packed complex (PC = 79%) than adamantane in CB7 (61%). The latter has an essentially perfect packing. Indeed, the binding constant of the diamantane derivative D2 is less than that of the adamantane derivative A3. The tight packing of D3 is structurally reflected in a significantly expanded cavity (increase in the diameter by ∼0.3 Å and increase in the volume from 242 to 258 Å3).137 The fact that D3 shows nevertheless the highest binding constant suggest that the PC value is over-compensated by the positive effect caused by the methylation of both amino groups; the latter causes a central positioning of the positive charge and, presumably, increased Coulombic interactions due to a drop in the apparent dielectric permittivity between the interacting (partial) charges.
In contrast to other supramolecular recognition systems, the thermodynamic data for the complexation of CBn with a variety of guest molecules overcome the enthalpy–entropy compensation trend.135,139,150 The cancellation of enthalpic gain by commensurate entropic changes of opposite sign in the course of binding is common for tight non-covalent complexes, like protein–ligand binding; other macrocycles, e.g., cyclodextrins, also obey the enthalpy–entropy compensation principle.150–155 This establishes a stand-alone feature of CBn macrocycles, which can be attributed, among others, to the dominant role of the release of high-energy water as the enthalpic driving force for complexation.
Most recently, Vàzquez et al. have reported a new method for identifying the quality of high affinity binders to CB7 and CB8 by indicator displacement, in which dyes with a macrocycle-specific optical fingerprint upon encapsulation by CB7 and CB8 were used. This method allows a rapid screening of strongly and selectively binding guest molecules at micromolar concentrations.156
5. Catalysis inside CBn cavities
The potential of CBn to control and catalyze chemical reactions through their distinctive inner cavity4,64,103,125,157–166 has been explored to a considerably lesser extent than that of cyclodextrins as alternative macrocycles.167–173 Nevertheless, several successful examples of CBn-catalyzed or CBn-promoted unimolecular and bimolecular reactions are nowadays available, and have been previously reviewed by Sivaguru.64 Herein, we summarize chemical reactions catalyzed by CBn, and highlight recent achievements (Table 7). Physical phenomena induced by complexation, such as the modulation of photoinduced electron transfer,174 photostabilization,36 complexation-induced pKa shifts,16,18,175–177 solubility enhancements of guests,14,178–182 and adverse effects on chemical reactions, that is inhibition,183–187 are not in the focus in this review, because they have been covered elsewhere.| Year | Reaction | Host (catalyst) | Guest (reactant) | Ref. | Reaction conditions |
|---|---|---|---|---|---|
| 1983 | 1,3-Dipolar cycloaddition | CB6 | 2 and 3 | 157 | Aq. formic acid, 40 °C |
| 2001 | Photodimerization | CB8 | 10 | 188 | hν (300 nm) |
| 2005, 2006 | Photodimerization | CB8 | 20 | 189 and 190 | hν (Hg lamp) |
| 2005, 2007, 2014 | Photodimerization | CB8 | 13–15 | 191–193 | hν (Hg lamp) |
| 2006 | Photodimerization | CB7 | 33 | 194 | hν (365 nm) |
| 2008 | Photodimerization | CB8 | 29, 31 | 195 and 196 | hν (Hg lamp) |
| 2008, 2010, 2011 | Photodimerization | CB8 | 23 | 197–200 | hν (>300 nm) |
| 2008, 2014 | Photodimerization | CB8 | 25 | 201 and 202 | hν (>300 nm) |
| 2009 | Hydrolysis | CB6, CB7 | 7–9 | 166 | |
| 2009, 2011 | Oxidation | CB8 | Alcohols | 162 and 203 | IBX |
| 2010 | Oxidation | CB6 | Alkanes | 204 | Oxovanadium(IV) |
| 2010 | Desilylation | CB7 | 44 | 205 | Ag(I) salts |
| 2010 | Hydrolysis | CB7 | ArCOCl | 184 | |
| 2011 | Photolysis | CB7 | 47 and 50 | 206 | Transition metal ions |
| 2013 | Oxidation | HemiCB6 | 5 | 207 | |
| 2013 | Oxidation | HemiCB6 | Phenols | 208 | IBX |
| 2013 | Retro-Diels–Alder | CB6, CB7, CB8 | Azoalkanes, e.g., 47, 50 | 103 | Gas phase |
| 2014 | Photofragmentation | CB8 | 35 and 39 | 209 | hν (Xe lamp) |
CB6-catalyzed 1,3-dipolar cycloadditions
The dipolar [3+2] cycloaddition between azides (2) and acetylenes (3) was the first reaction reported to be happening inside the CB cavity, particularly inside CB6.157 In fact, it paved the way to a type of catalytic reactions which have later become famous as in situ “click chemistry”.210,211 As can be seen from Scheme 4, the formation of the [1,2,3]-triazole heterocycle 4 was accelerated by a factor of 6 × 104 with regiospecificity. Unambiguously, the strong ion–dipole interactions between the ammonium ions and the CB6 carbonyl rims stabilize the ternary complex. Mock explained the observed rate enhancement in terms of: (1) overcoming of entropic constraints and (2) a strain activation of the bound substrate.157 Carlqvist and Maseras have used quantum-calculations (DFT, B3LYP/6-31G(d)) to provide more details on 1,3-dipolar cycloaddition inside CB6. The calculations, which lack consideration of dispersion interactions, showed no evidence for a transition state stabilization, which was interpreted in favour of the entropy constraints argument.212 Be this as it may, the CB6-catalyzed click reaction has been subsequently extensively used for synthesis of rotaxanes, pseudorotaxanes, and polyrotaxanes.160,213–218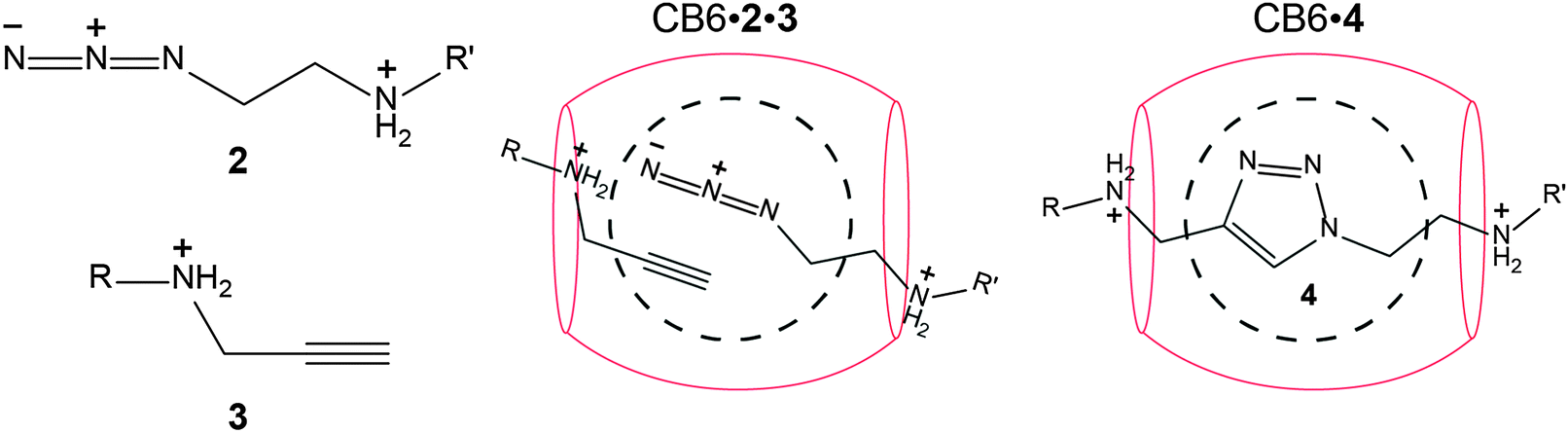 | ||
| Scheme 4 Click reaction of azide (2) and alkyne (3) to form a [1,2,3]-triazole (4) catalyzed by CB6. | ||
Oxidation reactions catalyzed by CBn
The oxidation of alcohols into their corresponding aldehydes by o-iodoxylbenzoic acid (IBX) was investigated in the presence of CB8. The conversion of aryl and allyl alcohols was increased by 30–50% in the presence of CB8.162,203 The pH effect on the conversion was also addressed; maximum conversion was obtained at neutral pH.203 A similar catalytic effect was obtained when the same reaction was conducted in the presence of acetic acid.219 The catalytic role of CB8 is not entirely understood in this case.Reddy et al. studied a series of known reactions mediated by molecular halogens (iodine and bromine) and their solid-state CB6 complexes.220 The solid state complexes were prepared by diffusing the halogen vapors through solid CB6. For example, the iodine-catalyzed Prins cyclization reaction was investigated by using free iodine versus the CB6·I2 solid-state complex. The results revealed that, in the presence of the pre-complexed iodine, the cyclization proceeded in higher yields. It should be noted that the structures of the CB6·I2 and CB6·Br2 inclusion complexes are known,221 while their use as reagents presents a novel application.220
Tao and coworker reported the aerobic oxidation of furan and thiophene (Scheme 5). In aqueous solution, the oxidation was found to take place in the presence of HemiCB6, while no reaction happened in the absence of the host. The protonation of HemiCB6 appears to play a major role, because the oxidation was accelerated at low pH.207 The IBX oxidation of hydroxylbenzyl alcohols was investigated in the absence and presence of HemiCB6. The IBX oxidation of phenols is known to produces a mixture of aldehydes and o-quinones, while the presence of HemiCB6 directed the IBX oxidation towards the phenolic hydroxyl groups to produce aldehydes.208
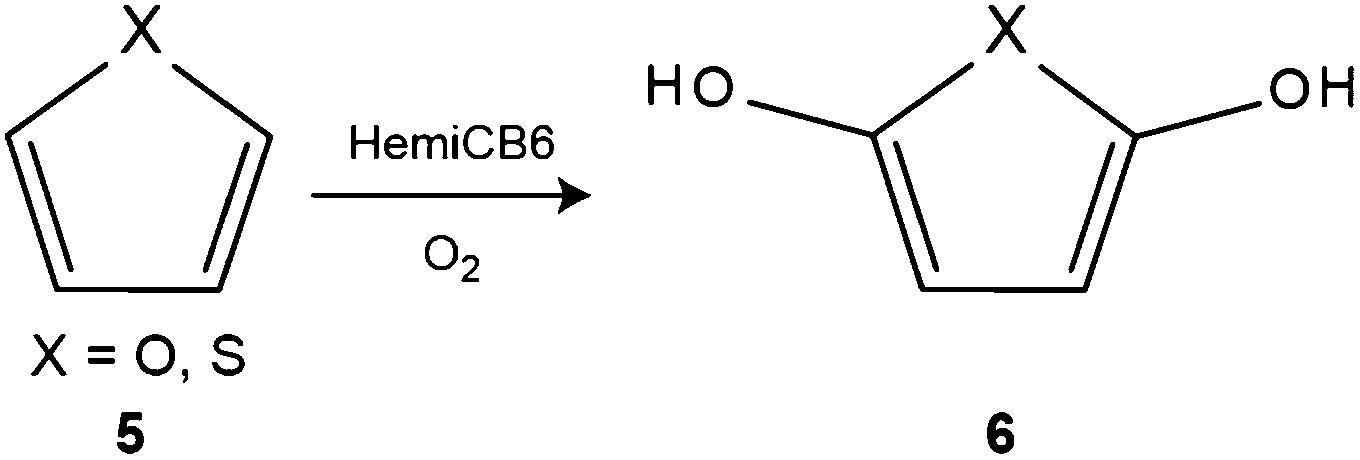 | ||
| Scheme 5 Aerobic oxidation of heterocyclic substrates (5) mediated by hemicucurbit[6]uril. | ||
Hydrolysis reactions catalyzed by CBn
García-Río and coworkers studied the hydrolysis of different substituted benzoyl chlorides in the presence of CB7 and DM-β-CD (methylated β-cyclodextrin).184 The reaction was catalyzed by CB7 and inhibited by DM-β-CD when the benzoyl chloride was carrying electron-donating substituents. For the substrates with electron-withdrawing groups, the reaction was catalyzed by DM-β-CD and inhibited by CB7. For example, the hydrolysis of 4-nitrobenzoyl chloride was inhibited in the presence of CB7 by a factor of 100, while for 4-methoxybenzoyl chloride the hydrolysis was accelerated 5-fold. The results were explained in terms of a differential stabilization of the acylium ion intermediate (in the transition state) by the CB7 carbonyl rim.184Klöck et al. reported the hydrolysis of amides (7), carbamates (8), and oximes (9) in acidic aqueous solutions mediated by CBs (Fig. 19).166 In the presence of CB6 and CB7, the reaction showed a catalytic effect. The hydrolysis of carbamates with a cadaverine moiety, which is known to bind very strongly to CB6 and CB7, was accelerated by a factor of ∼5 (kcat/kuncat) for CB6 and CB7 at pD 0.9 and by a factor of 11.6 for CB7 at pD 1.4. In the case of the mono-N-(tert-butoxy)carbonyl derivative (8), the deprotection rate constant was found to be ∼30 times faster with CB6 than the uncatalysed reaction. Furthermore, the oxime (9) hydrolysis in the presence of CB7 was studied. At high pD values (e.g. pD 5.8), the catalyzed reaction was very fast, with a peak acceleration factor of 285. The catalytic effect was attributed to a complexation-induced pKa shift,15,17,159,160,181,182 which assists the required protonation of the substrate.16,18,176,177,222,223 Unfortunately, since the reaction product binds more strongly than the reactant (e.g., benzaldehyde binds 6 times better than benzaldoxime), product inhibition was observed.
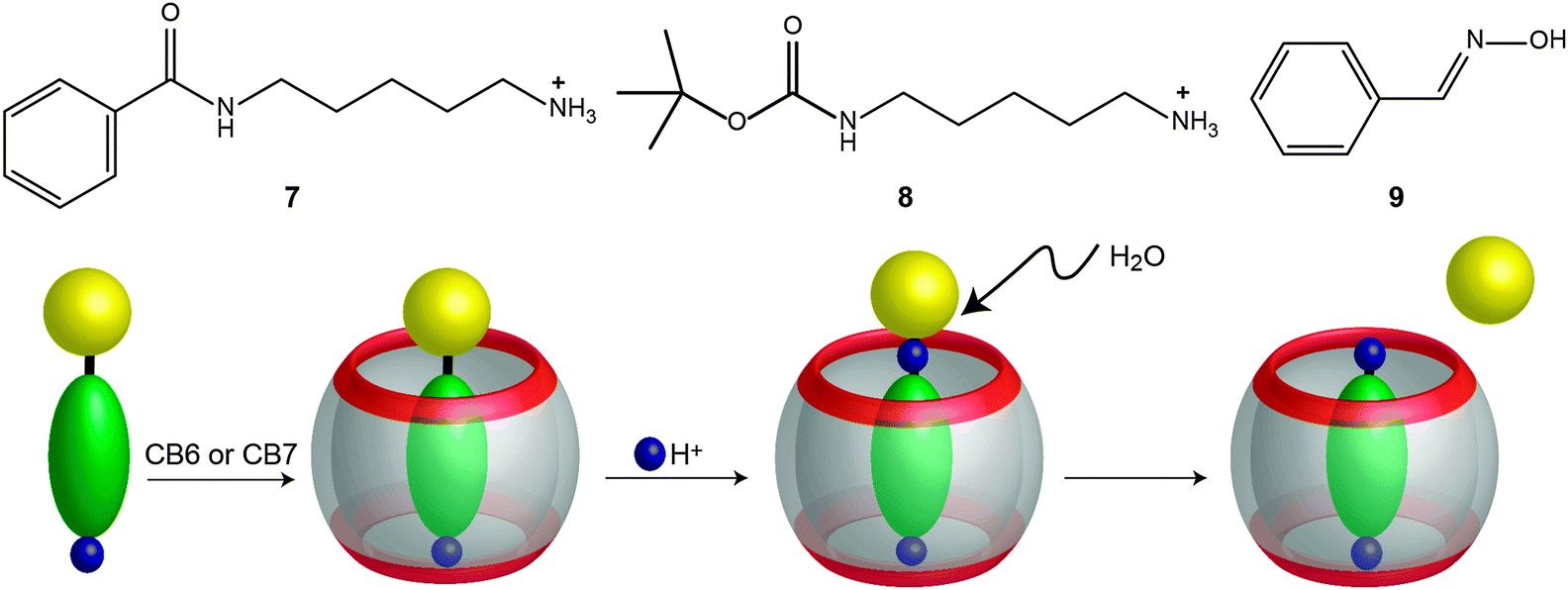 | ||
| Fig. 19 Acidic hydrolysis of an amide (N-benzoyl-cadaverine, 7), a carbamate (mono-N-(tert-butoxy)carbonyl cadaverine, 8), and an oxime (benzaldoxime, 9) mediated by CBn.166 | ||
Photochemical reactions catalyzed or templated by CBn
Complexation by CBn greatly influences the photophysical properties of encapsulated guests34,36,42,175,224 and their excited state processes4,53,183–187 which can be used, in combination with their ability to control the spatial arrangement of two components in their ternary complexes, to design more efficient and more selective photochemical reactions.4,42,64,188,197,198,201 Photochemical applications are facilitated by the fact that CBn are optically transparent in the near UV and the visible region, with the exception of some chromophore-modified derivatives.225–227 In fact, other macrocyclic hosts (e.g., cyclodextrins,168–171 calixarenes,228,229 and the octaacid host230,231) have also been widely used as molecular containers for intermolecular photochemical reactions. Of interest in this context are particularly the larger CBn homologues, CB7 and CB8, which are sufficiently large to accommodate two guests and, thus, to form ternary complexes, but which have only been reported in 2001.52 An overview of the corresponding photocyclization reactions templated by CBn is shown in Scheme 6.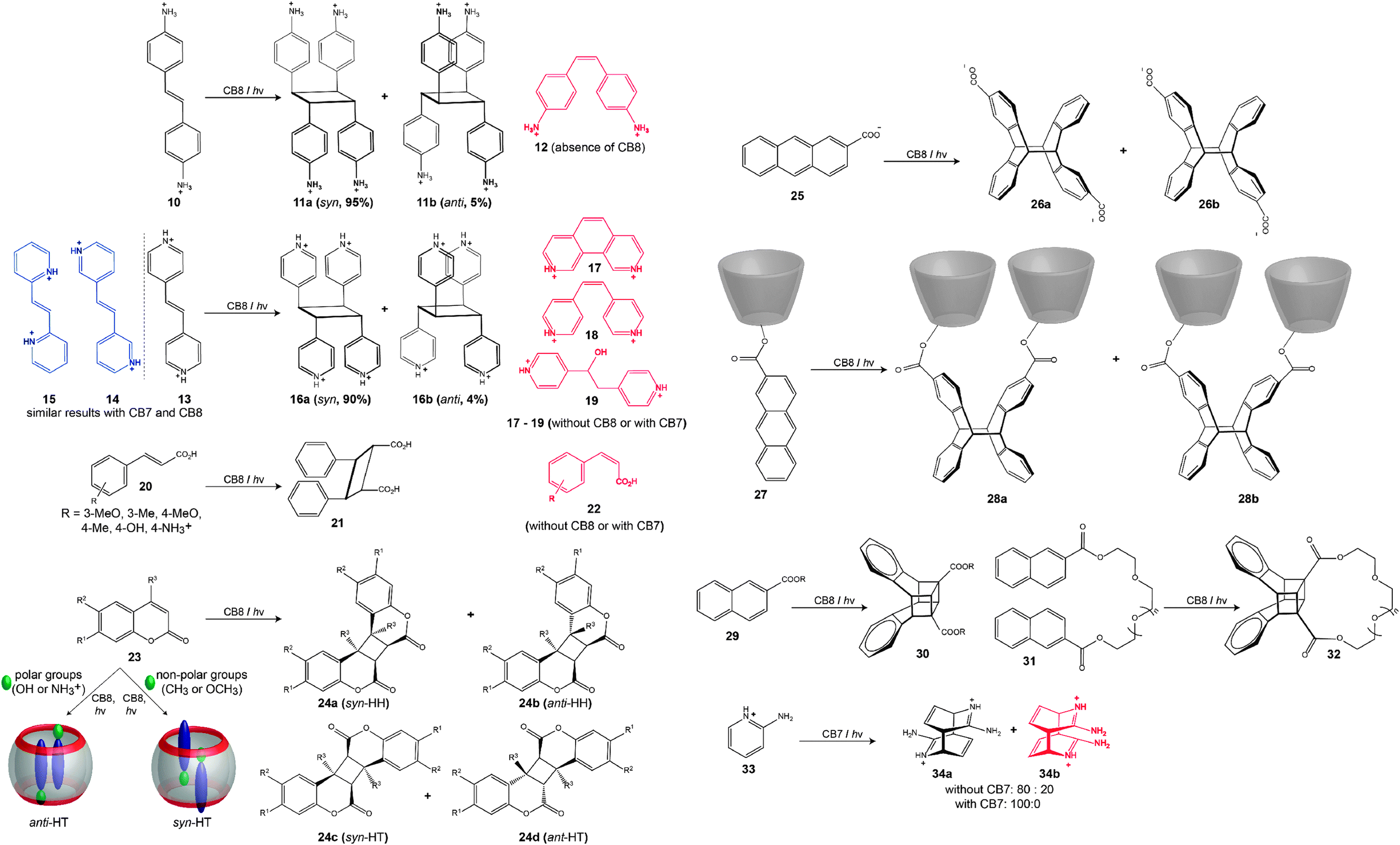 | ||
| Scheme 6 Photocyclization reactions templates by CBn macrocycles, see also Table 7. | ||
The first case of CB-catalyzed photoreactions with high stereoselectivity was reported by Kim and coworkers.188 Two equivalents of (E)-diaminostilbene (10) were shown to form a stable 1:2 complex with CB8 in aqueous solution, and upon UV-irradiation (at 300 nm, 30 min) the ternary complex underwent a [2+2] cycloaddition to yield the syn-adduct (11a) as major product and only a trace amount of the anti-adduct (11b). Interestingly, in the presence of CB8, no formation of the (Z)-isomer (12) was observed, while in the absence of CB8 the (Z)-isomer (12) was the main product.232 Other advantages offered by CBn were their ability to release the product from the cavity by increasing the pH (∼9) and to be recycled after precipitation with excess methanol.188
Ramamurthy and coworkers carried out another impressive set of experiments.189–192 By irradiation of different olefins (13–15) encapsulated in CB8 in aqueous solution, the syn dimer was obtained in 90% yield (16a), while irradiation of the 1:2 host–guest complex in the solid state showed no dimerization even after two days.191,192 When these reactions were carried out without CB8, phenanthroline (17), the cis-isomer (18), and the hydration product (19, major product) were formed instead. Although photodimerization did not happen in the presence of CB7, the formation of 18 was promoted by this smaller host in higher yield (>67%) than in the absence of CB7.191 Symmetrical and unsymmetrical bispyridyl ethylenes, as well as stilbazole derivatives were employed in related photocycloaddition reactions; through a combination of best fit and a minimization of electrostatic repulsion within the CB8 cavity, new reactions could be designed.192 Ramamurthy further extended this chemistry to include the photodimerization of neutral guests, such as trans-cinnamic acid (20).189,190 Upon irradiation of 20 encapsulated by CB8 (in aqueous solution as well as in the solid state), the reaction afforded the syn head-to-head dimer (21), along with the corresponding cis isomer (22).189 The latter was formed as dominant product in the presence of the smaller CB7. Zhang and coworkers have employed the same reaction type to construct covalently attached hyperbranched polymers, which present photosensitive supramolecular polymers with high molecular weight.193
Sivaguru reported the [2+2] cycloaddition of coumarin derivatives (23), both cationic and neutral, in the presence of CB8.197–199,233 It was shown that upon UV excitation (>300 nm) of the 1:2 complex of CB8 and two coumarins, the head-to-tail (HT) adduct was formed as the major product. The type of HT adduct (syn or anti) depended on the nature of substituents; coumarins with polar functional groups led to the anti photoadduct and those with nonpolar groups to the syn counterpart. Later, Sivaguru and coworkers showed that the formation of the 1:2 complex (ternary complex) is the rate determining step for the cycloaddition, in which the formation of the 1:1 complex is kinetically fast and that of the 1:2 complex is actually slow.200 In the case of methylated coumarin (6-methylcoumarin), photodimerization led exclusively to the syn dimer (>99%) with a syn-HH/syn-HT ratio of about 70:30.233 The research groups of Inoue and Kim reported the effect of CB8 on the [4+4] cross-photodimerization of 2-anthracene carboxylate (25) and another 2-anthracene carboxylate linked to α-CD (27).201 In the case of the self-photodimerization of 25, no significant effect on the syn/anti ratio or the head-to-head/head-to-tail ratio was observed in the presence and absence of CB8, e.g., the head-to-tail product remained the major isomer (26a and 26b). In contrast, the cross-photodimerization of 27 inverted the HT/HH ratio form 98:2 to 1:99 by changing the templating host from γ-CD to CB8; accordingly, the products of the reaction with CB8 were 38a and 38b. The [4+4] photodimerization of anthracene in the presence of CB8 has recently been applied to polymer ligation and covalent network formation.202 Another example, for the photodimerization of alkyl-2-naphthoate (29), was reported by Wu and coworkers, which afforded the cubane-type products 30.195,196 The photodimerization of methyl-2-naphthoate occurred efficiently in the presence of CB8 (60% conversion after 2 h), whereas in the free form methyl-2-naphthoate does not react. Similar results were obtained for 2-naphthalene-labeled poly(ethyl glycol) (31); the 1:1 complex was irradiated (>280 nm) for 12 min, sufficient to obtain more than 96% of the dimer (32); no product was formed in the absence of CB8.196
Macartney and coworkers demonstrated that photocycloadditions are not limited to the larger CB8 host, but that they can also be conducted with the intermediary sized CB7.194 In the presence of 50 mol% of CB7, 2-aminopyridine (33, as hydrochloride salt) underwent a [4+4] photodimerization and afforded exclusively the anti–trans isomers (34a) in 90% yield after irradiation for 21 h. In the absence of CB7, a mixture of anti–trans (34a) and syn–trans (34b) was obtained in a 4:1 ratio. Importantly, CB7 was also found to stabilize the photoproduct and, thus, to protect it from thermal re-aromatization. Packing arguments suggest a stronger binding of the photoproduct in this case,8 that is, product inhibition is expected, but actually desirable in this special case.
The pre-assembled 1:2 host–guest complexes of CB8 and benzimidazole (35) underwent photohydrolysis to form a new major photoproduct, 2-aminoformanilide (38) (Scheme 7). When the reaction was carried out in the absence of CB8, the photoreaction of 35 led to dehydrodimerization products (36) and (37). The phototransformation of free thiabendazole (39) also leads to the formation of 40, 41, and 42, while in the presence of CB8 the coupling product 43 was formed as an additional product.209
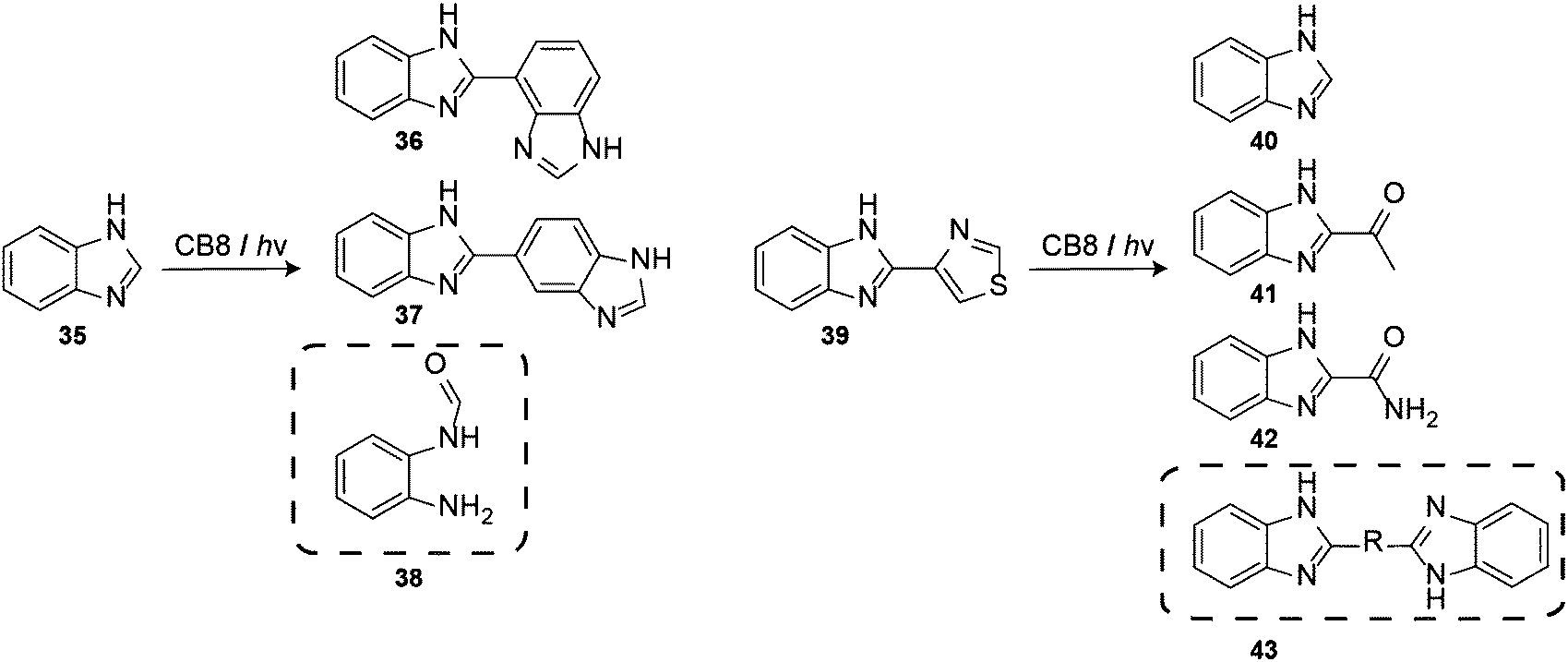 | ||
| Scheme 7 CB8 modulates the phototransformation of (a) benzimidazole (35) and (b) thiabendazole (39); marked products were observed only in the presence of CB8. | ||
Catalysis assisted by metal ions
Due to their electronegative carbonyl rims, CBn macrocycles are well known to bind metal cations at the portal regions.234–242 This property has recently been employed to promote or catalyze organometallic reactions.204–206 Demets and coworkers reported the oxidation of short unbranched alkanes, e.g. n-pentane, in the presence of CB6/oxovanadium(IV) (Fig. 20).204 Upon treatment of the complex with hydrogen peroxide or iodosylbenzene, a mixture of 2-pentanol, 2-pentanone, and 3-pentanone was obtained. The use of Z-cyclooctene, cyclohexane, and styrene as substrates showed no products formation, which was attributed to the fact that such guests do not form complexes with CB6, consistent with a detailed study of hydrocarbon binding to CB6.44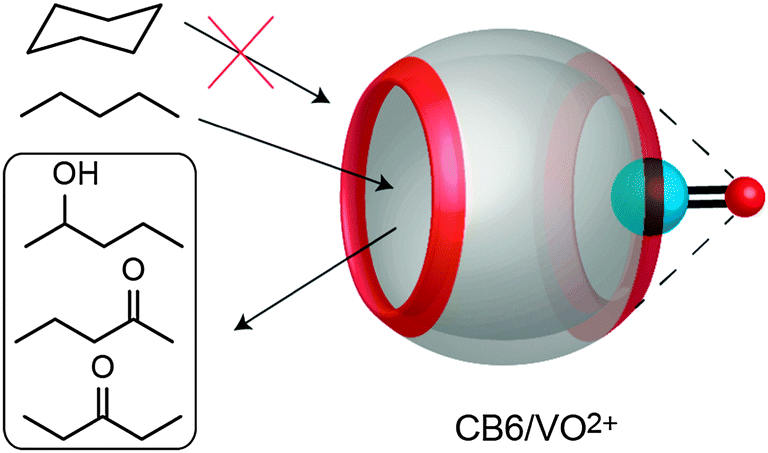 | ||
| Fig. 20 The CB6/oxovanadium(IV) complex catalyzes n-pentane oxidation, but not that of larger alkanes. | ||
Masson and Lu have recently reported a novel catalytic cycle for the desilylation of a trimethylsilylalkynyl derivative (44) assisted by CBn in the presence of Ag(I) salts (Fig. 21).205 The formation of a ternary complex, e.g., Ag+·CB7·trimethylsilylalkynyl, was postulated, which would first lead to trimethylsilanol (46) complexed inside CB7 and an alkynylsilver organometallic complex. It was assumed that the latter is subsequently hydrolyzed to the desilylated alkyne (45) and Ag+.
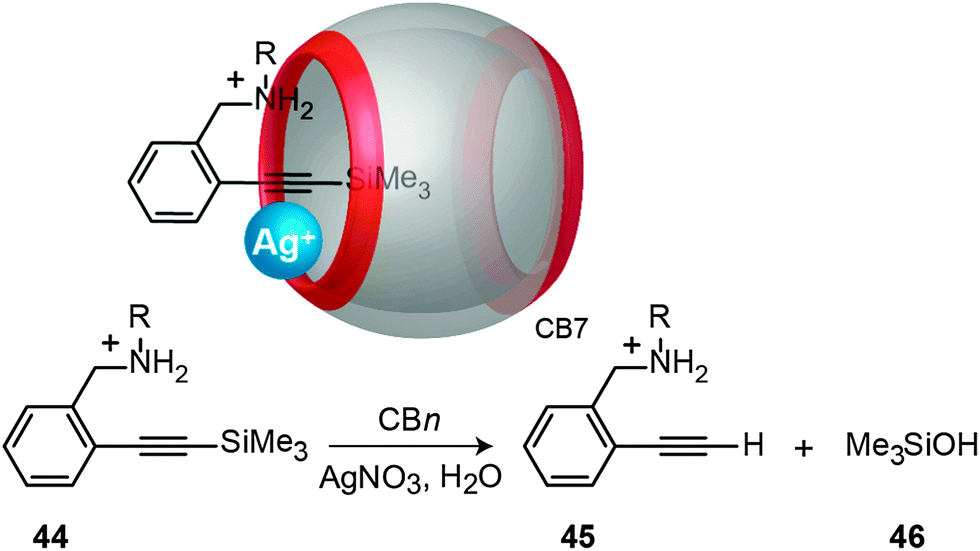 | ||
| Fig. 21 CBn-catalyzed desilylation of trimethylsilylalkynyl derivatives (44) in the presence of Ag(I) salt; shown on top is the postulated ternary complex. | ||
Our group documented a phase-selective photolysis of bicyclic azoalkanes (2,3-diazabicyclo[2.2.1]hept-2-ene (47) and 2,3-diazabicyclo[2.2.2]octa-2-ene (50)) promoted by transition-metal ions coordinated to the CB7 rim (Fig. 22).206 The reaction was selected to afford a photoproduct with a reduced affinity to the host and with a higher solubility in an organic phase than in water. In detail, irradiation of 47 and 50 in their near-UV n,π* absorption band in water is known to cause nitrogen extrusion under formation of bicyclo[2.1.0]pentane (housane) (48) as exclusive photoproduct of 47 and of a 70:30 mixture of 1,5-hexadiene (51) and bicyclo[2.2.0]hexane (52) as photoproducts of 55; the conversion can be conveniently monitored by gas chromatography. In the presence of CB7 and of specific transition metal ions (Fe3+, Co2+, Ni2+, Cu2+, Ag+, also Tl+), the photolysis of DBO afforded a 5-fold to 10-fold excess of the diene product. The chemoselectivity was postulated to occur from the triplet-excited state, populated by heavy-atom-induced intersystem crossing. With the smaller azoalkane 47, surprisingly, cyclopentene (49) was formed as a new photoproduct (41%), but only in the presence of both, CB7 and Ag+. Because the formation of a ternary complex between CB7, Ag+, and 50 could be crystallographically established, it is likely that a new mechanism, photoinduced electron-transfer, leads to cyclopentene. Such redox-active ternary complexes can be considered as metalloenzyme models.
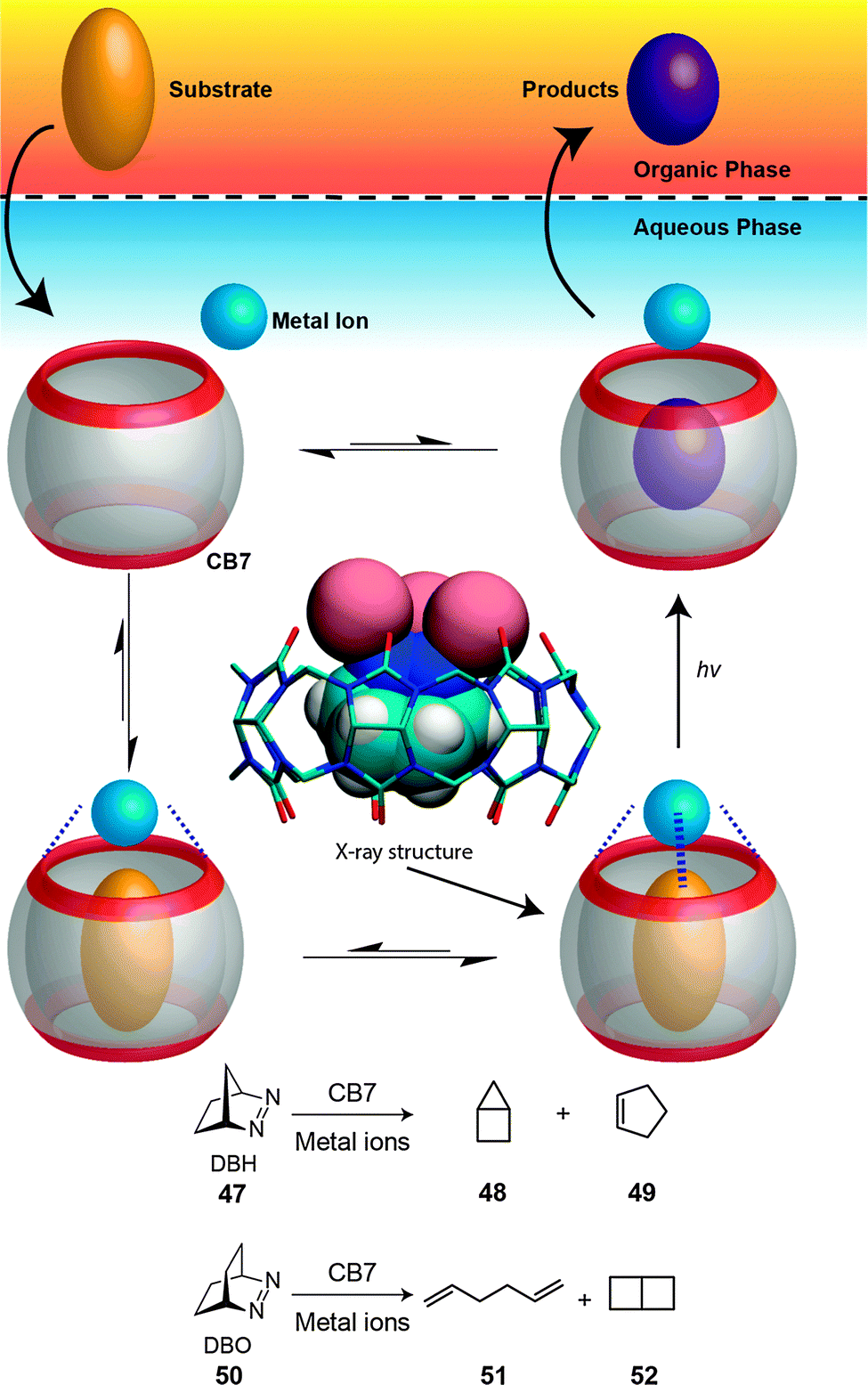 | ||
| Fig. 22 Chemoselective photoreactions of bicyclic azoalkanes (47 and 50) mediated by CB7 in the presence of transition metal ions.206 | ||
Catalysis and reactivity inside the inner phase of CBn in the gas phase
Lee et al. have investigated chemical reactions, retro-Diels–Alder reactions of bicyclic azoalkanes, inside CBn molecular containers in the gas phase by using a mass spectrometric study combined with quantum-chemical calculations.103 Ion mobility experiments were first performed to prove that the formed complexes between CBn (CB6, CB7, and CB8) and the protonated azoalkanes (DBH, DBO, and DBN) are indeed inclusion complexes and not of the exclusion type. It was clearly shown that free CB7 exhibits the same drift time as the host–guest complex (Fig. 23a), which depends directly on the collisional cross section areas of the “empty” and the “filled” CB7. The fact that the cross sections are the same cannot be reconciled with an exclusion complex, because it would display a significantly larger cross section.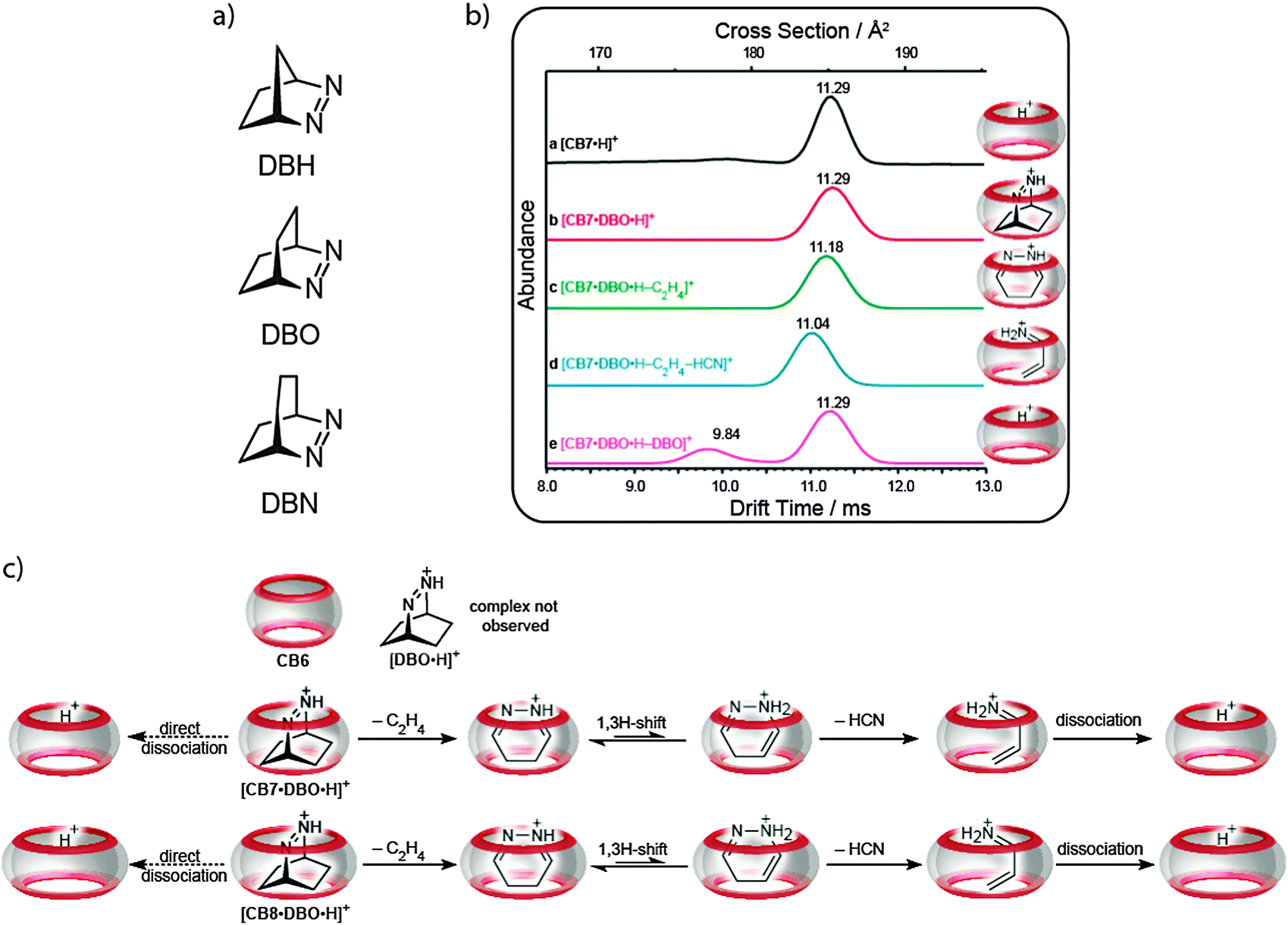 | ||
| Fig. 23 (a) Chemical structures of the bicyclic azoalkanes. (b) Ion mobilograms of different CB7 complexes. (c) Reaction and dissociation pathways of the inclusion complex of DBO inside CBn in the gas phase. | ||
In a systematic approach, we have examined the fragmentation reaction of each viable complex of differently sized CBn and guests (for example CB7, Fig. 23b). After the mass selection, the protonated complexes, [CBn·guest·H]+, were subject to collision-induced dissociation (CID) experiments. This thermal activation of the CBn–azoalkane complexes induced in some cases dominant chemical reactions of the encapsulated guests and the formation of [CBn·fragment·H+] product complexes as opposed to irreversible dissociation of the host–guest complexes; the latter was expected and is observed for many supramolecular assemblies, including cyclodextrin complexes. For example, protonated DBH inside CB7 afforded elimination of ethylene with subsequent dissociation of the intermediary pyrazole complex upon CID. In contrast, when DBH was more tightly packed (inside CB6) or more loosely packed (with CB8), the simple dissociation pathway into guest and host dominated over the inner-phase chemical reaction. Most interestingly, thermal activation of DBO complexed inside CB7 or CB8 (note that no complex was formed for DBO with CB6) led, predominantly, first to elimination of ethylene, followed by elimination of hydrogen cyanide, and, ultimately, dissociation of the protonated propenimine complex (Fig. 23c).
The chemical reactivity of the gas phase complexes was interpreted in terms of three contributing factors: (1) the intrinsic activation energies for chemical reaction of the guest, (2) the constrictive binding displayed by the particular host, and (3) the void space inside the host–guest complex. Modelled Lennard-Jones 12–6 potentials of a spherical guest expanding inside the inner cavity of a rigid host, which included attractive (dispersion) and repulsive van der Waals forces, showed that the chemical reaction is “switched on” when the packing coefficients of the complexed guest falls within the range of 30–50%. In these cases, as shown by DFT calculations, the transition state experiences a dispersive stabilization, because the substrate must expand towards the cavity walls in order to eliminate the nitrogen molecule. These gas-phase reactions do therefore provide a genuine and puristic case of an inner-phase “catalysis” of a chemical reaction.
6. Conclusions and outlook
Looking at only a single decade of active research on cucurbituril homologues, their potential in the areas of synthesis, high-affinity binding, and catalysis is still waiting to be fully explored. With respect to synthesis, several exciting developments have taken place, which include the emergence of monofunctionalized derivatives,90–92,96 also through convergent synthesis routes (Table 1).87,95 The monofunctionalized derivatives offer much potential for the labeling of surfaces and for selective conjugation to biomolecules. Also new homologues are still being reported, such as the recently reported largest member, CB14.59 With respect to high-affinity binding, new host–guest combinations are still pushing the limits, and the highest value, attomolar binding, has just recently been reached.137 Cucurbiturils are, among macrocycles, the hosts with highest (commonly 6 orders of magnitude higher) affinity, paving the way for hitherto unprecedented applications in biology, medicine, and material science. While recent studies point to the importance of the extrusion of high-energy water as an important denominator,119 the quantitative understanding of the underlying factors responsible for the high-affinity binding of cucurbituril homologues remains challenging.243 In contrast, dispersion interactions appear to be of very minor importance for guest binding by cucurbiturils in aqueous solution, which renders these hosts as interesting experimental and theoretical models to dissect the contributions of intermolecular interactions to host–guest binding, for example in the binding of perfluorinated compounds. Another mechanistic trend in recent years involves the use of gas-phase studies to obtain fundamental information on the host–guest binding of cucurbiturils.103,112,122 With respect to catalysis, to address the third thematic area of this review, several case studies are nowadays available (Table 7). Nevertheless, compelling application examples which fully exploit the high affinities of cucurbiturils, their peculiariar binding properties, or the availability of derivatives (including monofunctionalized ones to explore heterogeneous conditions) are still waiting to be uncovered.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG Grant NA-686/5), the Deutscher Akademischer Austausch Dienst (DAAD, graduate fellowships for KIA), and the COST Action CM1005 “Supramolecular Chemistry in Water” for their support.References
- L. Isaacs, Acc. Chem. Res., 2014, 47, 2052 CrossRef CAS PubMed.
- W. M. Nau and O. A. Scherman, Isr. J. Chem., 2011, 51, 492, and consecutive reviews in this special issue Search PubMed.
- N. Vallavoju and J. Sivaguru, Chem. Soc. Rev., 2014, 43, 4084 RSC.
- E. Masson, X. X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Y. Lu, RSC Adv., 2012, 2, 1213 RSC.
- X.-L. Ni, X. Xiao, H. Cong, L.-L. Liang, K. Cheng, X.-J. Cheng, N.-N. Ji, Q.-J. Zhu, S.-F. Xue and Z. Tao, Chem. Soc. Rev., 2013, 42, 9480 RSC.
- X. L. Ni, X. Xiao, H. Cong, Q. J. Zhu, S. F. Xue and Z. Tao, Acc. Chem. Res., 2014, 47, 1386 CrossRef CAS PubMed.
- J. Lü, J.-X. Lin, M.-N. Cao and R. Cao, Coord. Chem. Rev., 2013, 257, 1334 CrossRef.
- W. M. Nau, M. Florea and K. I. Assaf, Isr. J. Chem., 2011, 51, 559 CrossRef CAS.
- Y. H. Ko, I. Hwang, D.-W. Lee and K. Kim, Isr. J. Chem., 2011, 51, 506 CrossRef CAS.
- E. Y. Chernikova, Y. V. Fedorov and O. A. Fedorova, Russ. Chem. Bull., 2012, 61, 1363 CrossRef CAS.
- A. E. Kaifer, Acc. Chem. Res., 2014, 47, 2160 CrossRef CAS PubMed.
- N. J. Wheate, D. P. Buck, A. I. Day and J. G. Collins, Dalton Trans., 2006, 451 RSC.
- H. D. Nguyen, D. T. Dang, J. L. J. van Dongen and L. Brunsveld, Angew. Chem., Int. Ed., 2010, 49, 895 CrossRef CAS PubMed.
- N. Dong, S.-F. Xue, Q.-J. Zhu, Z. Tao, Y. Zhao and L.-X. Yang, Supramol. Chem., 2008, 20, 659 CrossRef CAS.
- C. Kim, S. S. Agasti, Z. J. Zhu, L. Isaacs and V. M. Rotello, Nat. Chem., 2010, 2, 962 CrossRef CAS PubMed.
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764 CrossRef CAS PubMed.
- A. L. Koner, I. Ghosh, N. Saleh and W. M. Nau, Can. J. Chem., 2011, 89, 139 CrossRef CAS.
- N. Saleh, A. L. Koner and W. M. Nau, Angew. Chem., Int. Ed., 2008, 47, 5398 CrossRef CAS PubMed.
- G. Ghale, V. Ramalingam, A. R. Urbach and W. M. Nau, J. Am. Chem. Soc., 2011, 133, 7528 CrossRef CAS PubMed.
- M. Florea and W. M. Nau, Org. Biomol. Chem., 2010, 8, 1033 CAS.
- D. H. Macartney, Future Med. Chem., 2013, 5, 2075 CrossRef CAS PubMed.
- S. Walker, R. Oun, F. J. McInnes and N. J. Wheate, Isr. J. Chem., 2011, 51, 616 CrossRef CAS.
- N. Saleh, M. A. Meetani, L. Al-Kaabi, I. Ghosh and W. M. Nau, Supramol. Chem., 2011, 23, 654 CrossRef CAS.
- D. H. Macartney, Isr. J. Chem., 2011, 51, 600 CrossRef CAS.
- Y. Huang, Z. Tao, S.-F. Xue and Q.-J. Zhu, Chem. J. Chin. Univ., 2011, 32, 2022 CAS.
- N. J. Wheate, V. Vora, N. G. Anthony and F. J. McInnes, J. Inclusion Phenom. Macrocyclic Chem., 2010, 68, 359 CrossRef CAS.
- L. Goldoni, M. Grugni, S. De Munari, M. Cassin and R. Bernardini, Chem. Lett., 2010, 39, 676 CrossRef CAS.
- K. M. Park, K. Suh, H. Jung, D.-W. Lee, Y. Ahn, J. Kim, K. Baek and K. Kim, Chem. Commun., 2009, 71 RSC.
- Y. J. Jeon, S.-Y. Kim, Y. H. Ko, S. Sakamoto, K. Yamaguchi and K. Kim, Org. Biomol. Chem., 2005, 3, 2122 CAS.
- S. Sun, F. Li, F. Liu, J. Wang and X. Peng, Sci. Rep., 2014, 4, 3570 Search PubMed.
- G. Hettiarachchi, D. Nguyen, J. Wu, D. Lucas, D. Ma, L. Isaacs and V. Briken, PLoS One, 2010, 5, e10514 Search PubMed.
- V. D. Uzunova, C. Cullinane, K. Brix, W. M. Nau and A. I. Day, Org. Biomol. Chem., 2010, 8, 2037 CAS.
- F. Biedermann, E. Elmalem, I. Ghosh, W. M. Nau and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 7739 CrossRef CAS PubMed.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941 CrossRef CAS PubMed.
- M. Shaikh, S. D. Choudhury, J. Mohanty, A. C. Bhasikuttan and H. Pal, Phys. Chem. Chem. Phys., 2010, 12, 7050 RSC.
- W. M. Nau and J. Mohanty, Int. J. Photoenergy, 2005, 7, 133 CrossRef CAS.
- J. Mohanty, H. Pal, A. K. Ray, S. Kumar and W. M. Nau, ChemPhysChem, 2007, 8, 54 CrossRef CAS PubMed.
- P. Montes-Navajas and H. Garcia, J. Photochem. Photobiol., A, 2009, 204, 97 CrossRef CAS.
- N. Barooah, J. Mohanty, H. Pal and A. C. Bhasikuttan, Org. Biomol. Chem., 2012, 10, 5055 CAS.
- H. Y. Zhang, L. L. Liu, C. Gao, R. Y. Sun and Q. C. Wang, Dyes Pigm., 2012, 94, 266 CrossRef CAS.
- E. Y. Chernikova, D. V. Berdnikova, Y. V. Fedorov and O. A. Fedorova, Macroheterocycles, 2013, 6, 234 CrossRef CAS.
- G. Parvari, O. Reany and E. Keinan, Isr. J. Chem., 2011, 51, 646 CrossRef CAS.
- C. P. Carvalho, R. Ferreira, J. P. Da Silva and U. Pischel, Supramol. Chem., 2013, 25, 92 CrossRef CAS.
- M. Florea and W. M. Nau, Angew. Chem., Int. Ed., 2011, 50, 9338 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Supramol. Chem., 2014, 26, 657 CrossRef CAS.
- R. N. Dsouza, A. Hennig and W. M. Nau, Chem. – Eur. J., 2012, 18, 3444 CrossRef CAS PubMed.
- W. M. Nau, G. Ghale, A. Hennig, H. Bakirci and D. M. Bailey, J. Am. Chem. Soc., 2009, 131, 11558 CrossRef CAS PubMed.
- F. Biedermann and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 5694 CrossRef CAS PubMed.
- G. Ghale and W. M. Nau, Acc. Chem. Res., 2014, 47, 2150 CrossRef CAS PubMed.
- R. Behrend, E. Meyer and F. Rusche, Justus Liebigs Ann. Chem., 1905, 339, 1 CrossRef.
- W. A. Freeman, W. L. Mock and N.-Y. Shih, J. Am. Chem. Soc., 1981, 103, 7367 CrossRef CAS.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540 CrossRef CAS.
- A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, J. Org. Chem., 2001, 66, 8094 CrossRef CAS PubMed.
- A. I. Day, R. J. Blanch, A. P. Arnold, S. Lorenzo, G. R. Lewis and I. Dance, Angew. Chem., Int. Ed., 2002, 41, 275 CrossRef CAS.
- L. Isaacs, Chem. Commun., 2009, 619 RSC.
- W.-H. Huang, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2008, 130, 8446 CrossRef CAS PubMed.
- A. Chakraborty, A. X. Xu, D. Witt, J. Lagona, J. C. Fettinger and L. Isaacs, J. Am. Chem. Soc., 2002, 124, 8297 CrossRef CAS PubMed.
- L. Isaacs, Isr. J. Chem., 2011, 51, 578 CrossRef CAS.
- X.-J. Cheng, L.-L. Liang, K. Chen, N.-N. Ji, X. Xiao, J.-X. Zhang, Y.-Q. Zhang, S.-F. Xue, Q.-J. Zhu, X.-L. Ni and Z. Tao, Angew. Chem., Int. Ed., 2013, 52, 7252 CrossRef CAS PubMed.
- S. Yi and A. E. Kaifer, J. Org. Chem., 2011, 76, 10275 CrossRef CAS PubMed.
- H.-K. Lee, K. M. Park, Y. J. Jeon, D. Kim, D. H. Oh, H. S. Kim, C. K. Park and K. Kim, J. Am. Chem. Soc., 2005, 127, 5006 CrossRef CAS PubMed.
- M. del Pozo, E. Blanco, E. Fatás, P. Hernández and C. Quintana, Analyst, 2012, 137, 4302 RSC.
- F. Benyettou, I. Milosevic, Y. Lalatonne, F. Warmont, R. Assah, J.-C. Olsen, M. Jouaid, L. Motte, C. Platas-Iglesias and A. Trabolsi, J. Mater. Chem. B, 2013, 1, 5076 RSC.
- B. C. Pemberton, R. Raghunathan, S. Volla and J. Sivaguru, Chem. – Eur. J., 2012, 18, 12178 CrossRef CAS PubMed.
- S. Ghosh and L. Isaacs, J. Am. Chem. Soc., 2010, 132, 4445 CrossRef CAS PubMed.
- A. Flinn, G. C. Hough, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1992, 31, 1475 CrossRef.
- J. Z. Zhao, H.-J. Kim, J. Oh, S.-Y. Kim, J. W. Lee, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 4233 CrossRef CAS.
- H. Isobe, S. Sato and E. Nakamura, Org. Lett., 2002, 4, 1287 CrossRef CAS PubMed.
- C. A. Burnett, J. Lagona, A. X. Wu, J. A. Shaw, D. Coady, J. C. Fettinger, A. I. Day and L. Isaacs, Tetrahedron, 2003, 59, 1961 CrossRef CAS.
- J. Lagona, J. C. Fettinger and L. Isaacs, Org. Lett., 2003, 5, 3745 CrossRef CAS PubMed.
- A. I. Day, A. P. Arnold and R. J. Blanch, Molecules, 2003, 8, 74 CrossRef CAS.
- S. Y. Jon, N. Selvapalam, D. H. Oh, J.-K. Kang, S.-Y. Kim, Y. J. Jeon, J. W. Lee and K. Kim, J. Am. Chem. Soc., 2003, 125, 10186 CrossRef CAS PubMed.
- Y. J. Zhao, S. F. Xue, Q. J. Zhu, Z. Tao, J. X. Zhang, Z. B. Wei, L. S. Long, M. L. Hu, H. P. Xiao and A. I. Day, Chin. Sci. Bull., 2004, 49, 1111 CrossRef CAS.
- S. Sasmal, M. K. Sinha and E. Keinan, Org. Lett., 2004, 6, 1225 CrossRef CAS PubMed.
- Y. Miyahara, K. Goto, M. Oka and T. Inazu, Angew. Chem., Int. Ed., 2004, 43, 5019 CrossRef CAS PubMed.
- S. M. Liu, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 16798 CrossRef CAS PubMed.
- H.-J. Buschmann, A. Zielesny and E. Schollmeyer, J. Inclusion Phenom. Macrocyclic Chem., 2006, 54, 181 CrossRef CAS.
- L. Isaacs, S.-K. Park, S. M. Liu, Y. H. Ko, N. Selvapalam, Y. Kim, H. Kim, P. Y. Zavalij, G.-H. Kim, H.-S. Lee and K. Kim, J. Am. Chem. Soc., 2005, 127, 18000 CrossRef CAS PubMed.
- H.-J. Buschmann, E. Cleve and E. Schollmeyer, Inorg. Chem. Commun., 2005, 8, 125 CrossRef CAS.
- W.-H. Huang, S. M. Liu, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2006, 128, 14744 CrossRef CAS PubMed.
- W.-H. Huang, P. Y. Zavalij and L. Isaacs, Angew. Chem., Int. Ed., 2007, 46, 7425 CrossRef CAS PubMed.
- L.-B. Lu, Y.-Q. Zhang, Q.-J. Zhu, S.-F. Xue and Z. Tao, Molecules, 2007, 12, 716 CrossRef CAS PubMed.
- J.-X. Lin, Y.-Q. Zhang, J.-X. Zhang, S.-F. Xue, Q.-J. Zhu and Z. Tao, J. Mol. Struct., 2008, 875, 442 CrossRef CAS.
- L.-B. Lu, D.-H. Yu, Y.-Q. Zhang, Q.-J. Zhu, S.-F. Xue and Z. Tao, J. Mol. Struct., 2008, 885, 70 CrossRef CAS.
- Z.-C. Tian, X.-L. Ni, X. Xiao, F. Wu, Y.-Q. Zhang, Q.-J. Zhu, S.-F. Xue and Z. Tao, J. Mol. Struct., 2008, 888, 48 CrossRef CAS.
- L. M. Zheng, J. N. Zhu, Y. Q. Zhang, Q. J. Zhu, S. F. Xue, Z. Tao, J. X. Zhang, X. Zhou, Z. B. Wei, L. S. Long and A. I. Day, Supramol. Chem., 2008, 20, 709 CrossRef CAS.
- L.-H. Wu, X.-L. Ni, F. Wu, Y.-Q. Zhang, Q.-J. Zhu, S.-F. Xue and Z. Tao, J. Mol. Struct., 2009, 920, 183 CrossRef CAS.
- J. Svec, M. Necas and V. Sindelar, Angew. Chem., Int. Ed., 2010, 49, 2378 CrossRef CAS PubMed.
- N. Zhao, G. O. Lloyd and O. A. Scherman, Chem. Commun., 2012, 48, 3070 RSC.
- L. P. Cao and L. Isaacs, Org. Lett., 2012, 14, 3072 CrossRef CAS PubMed.
- B. Vinciguerra, L. P. Cao, J. R. Cannon, P. Y. Zavalij, C. Fenselau and L. Isaacs, J. Am. Chem. Soc., 2012, 134, 13133 CrossRef CAS PubMed.
- J. B. Wittenberg, P. Y. Zavalij and L. Isaacs, Angew. Chem., Int. Ed., 2013, 52, 3690 CrossRef CAS PubMed.
- T. Fiala and V. Sindelar, Synlett, 2013, 2443 CAS.
- R. Aav, E. Shmatova, I. Reile, M. Borissova, F. Topić and K. Rissanen, Org. Lett., 2013, 15, 3786 CrossRef CAS PubMed.
- L. Gilberg, M. S. A. Khan, M. Enderesova and V. Sindelar, Org. Lett., 2014, 16, 2446 CrossRef CAS PubMed.
- V. Lewin, J. Rivollier, S. Coudert, D.-A. Buisson, D. Baumann, B. Rousseau, F.-X. Legrand, H. Kouřilová, P. Berthault, J.-P. Dognon, M.-P. Heck and G. Huber, Eur. J. Org. Chem., 2013, 3857 CrossRef CAS.
- A. X. Wu, A. Chakraborty, D. Witt, J. Lagona, F. Damkaci, M. A. Ofori, J. K. Chiles, J. C. Fettinger and L. Isaacs, J. Org. Chem., 2002, 67, 5817 CrossRef CAS PubMed.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621 CrossRef CAS PubMed.
- Y.-J. Zhao, S.-F. Xue, Y.-Q. Zhang, Q.-J. Zhu, Z. Tao, J.-X. Zhang, Z.-B. Wei and L.-S. Long, Acta Chim. Sin., 2005, 63, 913 CAS.
- W.-J. Chen, J.-P. Zeng, Y.-Q. Zhang, Q.-J. Zhu, S. F. Xue, Z. Tao and G. Wei, Chin. J. Inorg. Chem., 2010, 26, 2018 CAS.
- A. I. Day, X. Xiao, Y.-Q. Zhang, Q.-J. Zhu, S.-F. Xue and Z. Tao, J. Inclusion Phenom. Macrocyclic Chem., 2011, 71, 281 CrossRef CAS.
- S. M. Liu, K. Kim and L. Isaacs, J. Org. Chem., 2007, 72, 6840 CrossRef CAS PubMed.
- T. C. Lee, E. Kalenius, A. I. Lazar, K. I. Assaf, N. Kuhnert, C. H. Grun, J. Janis, O. A. Scherman and W. M. Nau, Nat. Chem., 2013, 5, 376 CrossRef CAS PubMed.
- V. Havel, J. Svec, M. Wimmerova, M. Dusek, M. Pojarova and V. Sindelar, Org. Lett., 2011, 13, 4000 CrossRef CAS PubMed.
- A. Révész, D. Schröder, J. Svec, M. Wimmerová and V. Sindelar, J. Phys. Chem. A, 2011, 115, 11378 CrossRef PubMed.
- M. A. Rankin and B. D. Wagner, Supramol. Chem., 2004, 16, 513 CrossRef CAS.
- J. Mohanty and W. M. Nau, Angew. Chem., Int. Ed., 2005, 44, 3750 CrossRef CAS PubMed.
- I. W. Wyman and D. H. Macartney, Org. Biomol. Chem., 2008, 6, 1796 CAS.
- S. Senler, W. Li, M. H. Tootoonchi, S. Yi and A. E. Kaifer, Supramol. Chem., 2014, 26, 677 CrossRef CAS.
- C. Márquez and W. M. Nau, Angew. Chem., Int. Ed., 2001, 40, 4387 CrossRef.
- W. M. Nau, A. Hennig and A. L. Koner, Fluorescence of Supermolecules, Polymers, and Nanosystems, 2008, vol. 4, p. 185 Search PubMed.
- M. F. Czar and R. A. Jockusch, ChemPhysChem, 2013, 14, 1138 CrossRef CAS PubMed.
- R. J. Abraham and D. F. Wileman, J. Chem. Soc., Perkin Trans. 2, 1973, 1521 RSC.
- D. F. Evans, J. Chem. Soc., 1960, 877 RSC.
- SF6 as a solvato-resonant polarizability probe afforded a slightly higher polarizability for the CB6 cavity.
- D. G. Samsonenko, A. V. Virovets, J. Lipkowski, O. A. Geras'ko and V. P. Fedin, J. Struct. Chem., 2002, 43, 664 CrossRef CAS.
- T. V. Mitkina, N. F. Zakharchuk, D. Y. Naumov, O. A. Gerasko, D. Fenske and V. P. Fedin, Inorg. Chem., 2008, 47, 6748 CrossRef CAS PubMed.
- S. Mecozzi and J. Rebek, Jr., Chem. – Eur. J., 1998, 4, 1016 CrossRef CAS.
- F. Biedermann, M. Vendruscolo, O. A. Scherman, A. De Simone and W. M. Nau, J. Am. Chem. Soc., 2013, 135, 14879 CrossRef CAS PubMed.
- F. Biedermann, V. D. Uzunova, O. A. Scherman, W. M. Nau and A. De Simone, J. Am. Chem. Soc., 2012, 134, 15318 CrossRef CAS PubMed.
- F. Biedermann, W. M. Nau and H.-J. Schneider, Angew. Chem., Int. Ed., 2014 DOI:10.1002/anie.201310958.
- S. J. C. Lee, J. W. Lee, H. H. Lee, J. Seo, D. H. Noh, Y. H. Ko, K. Kim and H. I. Kim, J. Phys. Chem. B, 2013, 117, 8855 CrossRef CAS PubMed.
- P. Cintas, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 17, 205 CrossRef CAS.
- W. L. Mock, Supramolecular Chemistry II - Host Design and Molecular Recognition, 1995, vol. 175, p. 1 Search PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844 CrossRef CAS PubMed.
- W. L. Mock and N.-Y. Shih, J. Org. Chem., 1983, 48, 3618 CrossRef CAS.
- W. A. Freeman, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 382 CrossRef.
- W. L. Mock and N.-Y. Shih, J. Org. Chem., 1986, 51, 4440 CrossRef CAS.
- W. L. Mock and N.-Y. Shih, J. Am. Chem. Soc., 1988, 110, 4706 CrossRef CAS.
- T. C. Krasia, S. Khodabakhsh, D. Tuncel and J. H. G. Steinke, Cucurbituril: A Versatile “Bead” for Polyrotaxane Synthesis, Springer-Verlag, Berlin Heidelberg, 2004 Search PubMed.
- W. L. Mock and N.-Y. Shih, J. Am. Chem. Soc., 1989, 111, 2697 CrossRef CAS.
- W. L. Mock and J. Pierpont, J. Chem. Soc., Chem. Commun., 1990, 1509 RSC.
- C. Márquez, R. R. Hudgins and W. M. Nau, J. Am. Chem. Soc., 2004, 126, 5806 CrossRef PubMed.
- A. V. Virovets, V. A. Blatov and A. P. Shevchenko, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 350 CrossRef CAS PubMed.
- S. Moghaddam, C. Yang, M. Rekharsky, Y. H. Ko, K. Kim, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2011, 133, 3570 CrossRef CAS PubMed.
- S. M. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 15959 CrossRef CAS PubMed.
- L. P. Cao, M. Šekutor, P. Y. Zavalij, K. Mlinaric-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988 CrossRef CAS PubMed.
- M. Šekutor, K. Molčanov, L. P. Cao, L. Isaacs, R. Glaser and K. Mlinarić-Majerski, Eur. J. Org. Chem., 2014, 2533 CrossRef.
- W. S. Jeon, K. Moon, S. H. Park, H. Chun, Y. H. Ko, J. Y. Lee, E. S. Lee, S. Samal, N. Selvapalam, M. V. Rekharsky, V. Sindelar, D. Sobransingh, Y. Inoue, A. E. Kaifer and K. Kim, J. Am. Chem. Soc., 2005, 127, 12984 CrossRef CAS PubMed.
- Y. Chen, A. Klimczak, E. Galoppini and J. V. Lockard, RSC Adv., 2013, 3, 1354 RSC.
- I. Hwang, K. Baek, M. Jung, Y. Kim, K. M. Park, D.-W. Lee, N. Selvapalam and K. Kim, J. Am. Chem. Soc., 2007, 129, 4170 CrossRef CAS PubMed.
- A. E. Kaifer, W. Li and S. Yi, Isr. J. Chem., 2011, 51, 496 CrossRef CAS.
- W. Ong and A. E. Kaifer, Organometallics, 2003, 22, 4181 CrossRef CAS.
- V. Logvinenko, T. Mitkina, V. Drebushchak and V. Fedin, J. Therm. Anal. Calorim., 2011, 105, 103 CrossRef CAS.
- D. Sobransingh and A. E. Kaifer, Langmuir, 2006, 22, 10540 CrossRef CAS PubMed.
- N. M. Green, Methods Enzymol., 1990, 184, 51 CAS.
- Y. Pazy, T. Kulik, E. A. Bayer, M. Wilchek and O. Livnah, J. Biol. Chem., 2002, 277, 30892 CrossRef CAS PubMed.
- X. Y. Zhang and W. M. Nau, Angew. Chem., Int. Ed., 2000, 39, 544 CrossRef CAS.
- R. V. Pinjari and S. P. Gejji, J. Phys. Chem. A, 2008, 112, 12679 CrossRef CAS PubMed.
- M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. M. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. M. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737 CrossRef CAS PubMed.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875 CrossRef CAS PubMed.
- K. N. Houk, A. G. Leach, S. P. Kim and X. Y. Zhang, Angew. Chem., Int. Ed., 2003, 42, 4872 CrossRef CAS PubMed.
- Y. Inoue, Y. Liu, L.-H. Tong, B.-J. Shen and D.-S. Jin, J. Am. Chem. Soc., 1993, 115, 10637 CrossRef CAS.
- Y. Inoue, T. Hakushi, Y. Liu, L.-H. Tong, B.-J. Shen and D.-S. Jin, J. Am. Chem. Soc., 1993, 115, 475 CrossRef CAS.
- C.-E. Chang and M. K. Gilson, J. Am. Chem. Soc., 2004, 126, 13156 CrossRef CAS PubMed.
- J. Vàzquez, P. Remón, R. N. Dsouza, A. I. Lazar, J. F. Arteaga, W. M. Nau and U. Pischel, Chem. – Eur. J., 2014, 20, 9897 CrossRef PubMed.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and T. L. Manimaran, J. Org. Chem., 1983, 48, 3619 CrossRef CAS.
- T. C. Krasia and J. H. G. Steinke, Chem. Commun., 2002, 22 RSC.
- S. P. Gromov, A. I. Vedernikov, L. G. Kuz'mina, D. V. Kondratuk, S. K. Sazonov, Y. A. Strelenko, M. V. Alfimov and J. A. K. Howard, Eur. J. Org. Chem., 2010, 2587 CrossRef CAS.
- D. Tuncel, O. Ünal and M. Artar, Isr. J. Chem., 2011, 51, 525 CrossRef CAS.
- Q.-S. Wang, H. Cong, J.-X. Zhang, Z. Tao and S.-F. Xue, Chin. J. Org. Chem., 2011, 31, 1049 CAS.
- Y.-H. Wang, H. Cong, F.-F. Zhao, S.-F. Xue, Z. Tao, Q.-J. Zhu and G. Wei, Catal. Commun., 2011, 12, 1127 CrossRef CAS.
- H. Cong, Q.-S. Wang, J.-X. Zhang, Z. Tao and S.-F. Xue, Chin. J. Org. Chem., 2012, 32, 1093 CrossRef CAS.
- M. Wieland, J.-L. Mieusset and U. H. Brinker, Tetrahedron Lett., 2012, 53, 4351 CrossRef CAS.
- H. Cong, Z.-J. Li, Y.-H. Wang, Z. Tao, T. Yamato, S.-F. Xue and G. Wei, J. Mol. Catal. A: Chem., 2013, 374, 32 CrossRef.
- C. Klöck, R. N. Dsouza and W. M. Nau, Org. Lett., 2009, 11, 2595 CrossRef PubMed.
- R. Breslow, Acc. Chem. Res., 1995, 28, 146 CrossRef CAS.
- A. Nakamura and Y. Inoue, J. Am. Chem. Soc., 2003, 125, 966 CrossRef CAS PubMed.
- C. Yang, G. Fukuhara, A. Nakamura, Y. Origane, K. Fujita, D.-Q. Yuan, T. Mori, T. Wada and Y. Inoue, J. Photochem. Photobiol., A, 2005, 173, 375 CrossRef CAS.
- A. Nakamura and Y. Inoue, J. Am. Chem. Soc., 2005, 127, 5338 CrossRef CAS PubMed.
- Q. A. Wang, C. Yang, G. Fukuhara, T. Mori, Y. Liu and Y. Inoue, Beilstein J. Org. Chem., 2011, 7, 1 CrossRef PubMed.
- D. D. Sternbach and D. M. Rossana, J. Am. Chem. Soc., 1982, 104, 5853 CrossRef CAS.
- R. Breslow and S. D. Dong, Chem. Rev., 1998, 98, 1997 CrossRef CAS PubMed.
- S.-C. Cui, T. Tachikawa, M. Fujitsuka and T. Majima, J. Phys. Chem. C, 2011, 115, 1824 CAS.
- A. L. Koner and W. M. Nau, Supramol. Chem., 2007, 19, 55 CrossRef CAS.
- N. Saleh, Y. A. Al-Soud, L. Al-Kaabi, I. Ghosh and W. M. Nau, Tetrahedron Lett., 2011, 52, 5249 CrossRef CAS.
- M. Shaikh, J. Mohanty, P. K. Singh, W. M. Nau and H. Pal, Photochem. Photobiol. Sci., 2008, 7, 408 CAS.
- Y. Huang, Q.-H. Hu, G.-X. Song, Z. Tao, S.-F. Xue, Q.-J. Zhu, Q.-D. Zhouc and G. Wei, RSC Adv., 2014, 4, 3348 RSC.
- D. Ma, G. Hettiarachchi, D. Nguyen, B. Zhang, J. B. Wittenberg, P. Y. Zavalij, V. Briken and L. Isaacs, Nat. Chem., 2012, 4, 503 CrossRef CAS PubMed.
- N. Dong, X.-L. Wang, J.-P. Pan and Z. Tao, Acta Chim. Sin., 2011, 69, 1431 CAS.
- D. Tuncel, M. Artar and S. B. Hanay, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4894 CrossRef CAS.
- Y. Huang, S.-F. Xue, Z. Tao, Q.-J. Zhu, H. Zhang, J.-X. Lin and D.-H. Yu, J. Inclusion Phenom. Macrocyclic Chem., 2008, 61, 171 CrossRef CAS.
- Z. Miskolczy, M. Megyesi, G. Tárkányi, R. Mizsei and L. Biczók, Org. Biomol. Chem., 2011, 9, 1061 CAS.
- N. Basilio, L. García-Río, J. A. Moreira and M. Pessêgo, J. Org. Chem., 2010, 75, 848 CrossRef CAS PubMed.
- R. B. Wang, L. N. Yuan and D. H. Macartney, Chem. Commun., 2006, 2908 RSC.
- L. S. Berbeci, W. Wang and A. E. Kaifer, Org. Lett., 2008, 10, 3721 CrossRef PubMed.
- H. Cong, C.-R. Li, S.-F. Xue, Z. Tao, Q.-J. Zhu and G. Wei, Org. Biomol. Chem., 2011, 9, 1041 CAS.
- S. Y. Jon, Y. H. Ko, S. H. Park, H.-J. Kim and K. Kim, Chem. Commun., 2001, 1938 RSC.
- M. Pattabiraman, A. Natarajan, L. S. Kaanumalle and V. Ramamurthy, Org. Lett., 2005, 7, 529 CrossRef CAS PubMed.
- M. Pattabiraman, L. S. Kaanumalle, A. Natarajan and V. Ramamurthy, Langmuir, 2006, 22, 7605 CrossRef CAS PubMed.
- M. Pattabiraman, A. Natarajan, R. Kaliappan, J. T. Mague and V. Ramamurthy, Chem. Commun., 2005, 4542 RSC.
- M. V. S. N. Maddipatla, L. S. Kaanumalle, A. Natarajan, M. Pattabiraman and V. Ramamurthy, Langmuir, 2007, 23, 7545 CrossRef CAS PubMed.
- H. Yang, Z. Ma, Z. Q. Wang and X. Zhang, Polym. Chem., 2014, 5, 1471 RSC.
- R. B. Wang, L. N. Yuan and D. H. Macartney, J. Org. Chem., 2006, 71, 1237 CrossRef CAS PubMed.
- L. Lei, L. Luo, X.-L. Wu, G.-H. Liao, L.-Z. Wu and C.-H. Tung, Tetrahedron Lett., 2008, 49, 1502 CrossRef CAS.
- X.-L. Wu, L. Luo, L. Lei, G.-H. Liao, L.-Z. Wu and C.-H. Tung, J. Org. Chem., 2008, 73, 491 CrossRef CAS PubMed.
- N. Barooah, B. C. Pemberton and J. Sivaguru, Org. Lett., 2008, 10, 3339 CrossRef CAS PubMed.
- N. Barooah, B. C. Pemberton, A. C. Johnson and J. Sivaguru, Photochem. Photobiol. Sci., 2008, 7, 1473 CAS.
- B. C. Pemberton, N. Barooah, D. K. Srivatsava and J. Sivaguru, Chem. Commun., 2010, 46, 225 RSC.
- B. C. Pemberton, R. K. Singh, A. C. Johnson, S. Jockusch, J. P. Da Silva, A. Ugrinov, N. J. Turro, D. K. Srivastava and J. Sivaguru, Chem. Commun., 2011, 47, 6323 RSC.
- C. Yang, T. Mori, Y. Origane, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, J. Am. Chem. Soc., 2008, 130, 8574 CrossRef CAS PubMed.
- F. Biedermann, I. Ross and O. A. Scherman, Polym. Chem., 2014, 5, 5375 RSC.
- H. Cong, F.-F. Zhao, J. X. Zhang, X. Zeng, Z. Tao, S.-F. Xue and Q.-J. Zhu, Catal. Commun., 2009, 11, 167 CrossRef CAS.
- S. M. de Lima, J. A. Gómez, V. P. Barros, G. D. Vertuan, M. D. Assis, C. F. D. Graeff and G. J.-F. Demets, Polyhedron, 2010, 29, 3008 CrossRef CAS.
- X. Y. Lu and E. Masson, Org. Lett., 2010, 12, 2310 CrossRef CAS PubMed.
- A. L. Koner, C. Márquez, M. H. Dickman and W. M. Nau, Angew. Chem., Int. Ed., 2011, 50, 545 CrossRef CAS PubMed.
- H. Cong, T. Yamato and Z. Tao, J. Mol. Catal. A: Chem., 2013, 379, 287 CrossRef CAS.
- H. Cong, T. Yamato and Z. Tao, New J. Chem., 2013, 37, 3778 RSC.
- J. Smitka, A. Lemos, M. Porel, S. Jockusch, T. R. Belderrain, E. Tesarova and J. P. Da Silva, Photochem. Photobiol. Sci., 2014, 13, 310 CAS.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522 RSC.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC.
- P. Carlqvist and F. Maseras, Chem. Commun., 2007, 748 RSC.
- D. Tuncel and M. Katterle, Chem. – Eur. J., 2008, 14, 4110 CrossRef CAS PubMed.
- D. Tuncel, Ö. Özsar, H. B. Tiftik and B. Salih, Chem. Commun., 2007, 1369 RSC.
- D. Tuncel, N. Cindir and U. Koldemir, J. Inclusion Phenom. Macrocyclic Chem., 2006, 55, 373 CrossRef CAS.
- D. Tuncel and J. H. G. Steinke, Chem. Commun., 2002, 496 RSC.
- D. Tuncel and J. H. G. Steinke, Chem. Commun., 1999, 1509 RSC.
- G. Celtek, M. Artar, O. A. Scherman and D. Tuncel, Chem. – Eur. J., 2009, 15, 10360 CrossRef CAS PubMed.
- C.-K. Lin and T.-J. Lu, Tetrahedron, 2010, 66, 9688 CrossRef CAS.
- K. R. K. K. Reddy, T. S. Cavallini, G. J. F. Demets and L. F. Silva, New J. Chem., 2014, 38, 2262 RSC.
- H. S. El-Sheshtawy, B. S. Bassil, K. I. Assaf, U. Kortz and W. M. Nau, J. Am. Chem. Soc., 2012, 134, 19935 CrossRef CAS PubMed.
- A. Praetorius, D. M. Bailey, T. Schwarzlose and W. M. Nau, Org. Lett., 2008, 10, 4089 CrossRef CAS PubMed.
- J. Mohanty, A. C. Bhasikuttan, W. M. Nau and H. Pal, J. Phys. Chem. B, 2006, 110, 5132 CrossRef CAS PubMed.
- H.-J. Buschmann and E. Schollmeyer, J. Inclusion Phenom. Mol. Recognit. Chem., 1997, 29, 167 CrossRef CAS.
- T. Minami, N. A. Esipenko, B. Zhang, L. Isaacs and P. Anzenbacher, Jr., Chem. Commun., 2014, 50, 61 RSC.
- T. Minami, N. A. Esipenko, B. Zhang, L. Isaacs, R. Nishiyabu, Y. Kubo and P. Anzenbacher, Jr., J. Am. Chem. Soc., 2012, 134, 20021 CrossRef CAS PubMed.
- D. Lucas, T. Minami, G. Iannuzzi, L. P. Cao, J. B. Wittenberg, P. Anzenbacher, Jr. and L. Isaacs, J. Am. Chem. Soc., 2011, 133, 17966 CrossRef CAS PubMed.
- R. Kaliappan, L. S. Kaanumalle and V. Ramamurthy, Chem. Commun., 2005, 4056 RSC.
- R. Kaliappan, Y. H. Ling, A. E. Kaifer and V. Ramamurthy, Langmuir, 2009, 25, 8982 CrossRef CAS PubMed.
- A. Parthasarathy, L. S. Kaanumalle and V. Ramamurthy, Org. Lett., 2007, 9, 5059 CrossRef CAS PubMed.
- A. Natarajan, L. S. Kaanumalle, S. Jockusch, C. L. D. Gibb, B. C. Gibb, N. J. Turro and V. Ramamurthy, J. Am. Chem. Soc., 2007, 129, 4132 CrossRef CAS PubMed.
- H. Meier, Angew. Chem., Int. Ed. Engl., 1992, 31, 1399 CrossRef.
- B. C. Pemberton, E. Kumarasamy, S. Jockusch, D. K. Srivastava and J. Sivaguru, Can. J. Chem., 2011, 89, 310 CrossRef CAS.
- Y. Zhao, L.-L. Liang, K. Chen, N.-N. Ji, X.-J. Cheng, X. Xiao, Y.-Q. Zhang, S.-F. Xue, Q.-J. Zhu, N. Dong and Z. Tao, Dalton Trans., 2014, 43, 929 RSC.
- W. H. Lei, G. Y. Jiang, Q. X. Zhou, Y. J. Hou, B. W. Zhang, X. X. Cheng and X. S. Wang, ChemPhysChem, 2013, 14, 1003 CrossRef CAS PubMed.
- J.-X. Hu, Y.-F. Hu, X. Xiao, Y.-Q. Zhang, Z. Tao, S.-F. Xue, J.-X. Liu and Q.-J. Zhu, Eur. J. Inorg. Chem., 2013, 3632 CrossRef CAS.
- Z.-W. Gao, X. Feng, L. Mu, X.-L. Ni, L.-L. Liang, S.-F. Xue, Z. Tao, X. Zeng, B. E. Chapman, P. W. Kuchel, L. F. Lindoy and G. Wei, Dalton Trans., 2013, 42, 2608 RSC.
- Y.-F. Hu, K. Chen, J.-X. Liu, R.-L. Lin, W.-Q. Sun, S.-F. Xue, Q.-J. Zhu and Z. Tao, Polyhedron, 2012, 31, 632 CrossRef CAS.
- K. Chen, X. Feng, L.-L. Liang, Y.-Q. Zhang, Q.-J. Zhu, S.-F. Xue and Z. Tao, Cryst. Growth Des., 2011, 11, 5712 CAS.
- X. X. Zhang, K. E. Krakowiak, G. P. Xue, J. S. Bradshaw and R. M. Izatt, Ind. Eng. Chem. Res., 2000, 39, 3516 CrossRef CAS.
- D. Whang, J. Heo, J. H. Park and K. Kim, Angew. Chem., Int. Ed., 1998, 37, 78 CrossRef CAS.
- R. Hoffmann, W. Knoche, C. Fenn and H.-J. Buschmann, J. Chem. Soc., Faraday Trans., 1994, 90, 1507 RSC.
- C. N. Nguyen, T. K. Young and M. K. Gilson, J. Chem. Phys., 2012, 137, 149961 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |