
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-mediated post-Ugi transformations for the construction of diverse heterocyclic scaffolds
Upendra K.
Sharma
,
Nandini
Sharma
,
Dipak D.
Vachhani
and
Erik V.
Van der Eycken
*
Laboratory for Organic & Microwave-Assisted Chemistry (LOMAC), Department of Chemistry, University of Leuven (KU Leuven), Celestijnenlaan 200F, B-3001, Leuven, Belgium. E-mail: erik.vandereycken@chem.kuleuven.be
First published on 5th February 2015
Abstract
The Ugi-4CR is by far one of the most successful multicomponent reactions leading to high structural diversity and molecular complexity. However, the reaction mostly affords a linear peptide backbone, enabling post-Ugi transformations as the only solution to rigidify the Ugi-adduct into more drug like species. Not surprisingly, the development of these transformations, leading to new structural frameworks, has expanded rapidly over the last few years. As expected, palladium-catalyzed reactions have received the foremost attention, yet other metals, particularly gold complexes, are fast catching up. This tutorial review outlines the developments achieved in the past decade, highlighting the modifications that are performed in a sequential or domino fashion with emphasis on major concepts, synthetic applications of the derived products as well as mechanistic aspects.
 Upendra K. Sharma | Upendra K. Sharma received his PhD degree (2011) in organic chemistry under the supervision of Dr Arun K. Sinha at CSIR-Institute of Himalayan Bioresource Technology, Palampur, India. Thereafter, he worked as Assistant Professor (Chemistry) at National Institute of Technology (NIT), Jalandhar, India. Later on, he joined the research group of Prof. Erik Van der Eycken, University of Leuven, Belgium as Erasmus Mundus postdoctoral fellow and now continuing in the same lab as F+ postdoctoral fellow. His research interests include the development of new synthetic methods for biologically relevant molecules, multicomponent reactions, and C–H activation. |
 Nandini Sharma | Nandini Sharma received her PhD degree (2012) in organic chemistry under the supervision of Dr Arun K. Sinha at CSIR-Institute of Himalayan Bioresource Technology, Palampur, India. In October 2012, she joined the research group of Prof. Erik V. Van der Eycken at University of Leuven, Belgium as F+ Postdoctoral fellow. Her current research focuses on multicomponent reactions and their application in diversity-oriented synthesis of heterocyclic frameworks, transition metal catalysis and novel strategies for C–H activation. |
 Dipak D. Vachhani | Dipak D. Vachhani received his PhD degree (2013) in organic chemistry from the University of Leuven, Belgium, under the supervision of Professor Erik V. Van der Eycken. Thereafter, he spent one year as post-doctoral fellow (PDM-short) in the same lab working in the area of transition metal-catalyzed C–H activation and multicomponent reactions. Currently, he has been working as a research scientist in an Indian pharmaceutical industry. |
 Erik V. Van der Eycken | Erik Van der Eycken received his PhD degree (1987) in organic chemistry from the University of Ghent, with Professor Maurits Vandewalle. During 1988 to 1992 he worked as a scientific researcher at the R&D laboratories of AGFA-Gevaert, Belgium, and moved back to the University of Ghent in 1992. In 1997 he became a Doctor Assistant at the University of Leuven (KU Leuven). He spent time as a visiting scientist at the University of Graz (C. Oliver Kappe), at The Scripps Research Institute (K. Barry Sharpless), and at Uppsala University (Mats Larhed, Anders Hallberg). He was appointed Professor at the University of Leuven (KU Leuven) in 2007. |
Key learning points• Applications and mechanistic rationale to explain the compatibility of various metals for post-Ugi transformations.• Recent developments, scope and potential of new applications. • Rise of gold-catalysis in post-Ugi transformations. • Access to a diverse range of natural product like structures and molecules of pharmaceutical importance. • Rapid construction of complex heterocycles via cascade reactions along with high regio-, chemo- and diastereoselectivity in some cases. |
Introduction
State-of-the-art in the field of medicinal as well as combinatorial chemistry includes three main features: (1) suitably functionalized drug-like “privileged structures” having specific spatial architecture and rigidity, (2) short synthetic pathways comprising reliable, clean, and high yielding reactions, and (3) an overall “diversification strategy” resulting ideally in “libraries of libraries” of scaffolds resembling natural products or drugs.1 In this context, isocyanide-based multicomponent reactions2 (IMCRs) have been widely explored because of their unmatched capability to generate molecular complexity and diversity to expand the chemical space. From the past twenty years, the Ugi four-component reaction (Ugi-4CR)3 has remained as one of the highly investigated reactions for generating multifunctional adducts, owing to the mildness of the reaction conditions, the wide application scope and the high variability (four diversity points) associated with it (Fig. 1). Moreover, it provides an opportunity for a myriad of post-transformations depending on the functional groups introduced during the MCR, thus leading to the synthesis of several pharmacologically important heterocyclic scaffolds, mostly in two operational steps.4 Inevitably, a number of post-Ugi transformations such as Ugi-deprotection-cyclization (UDC), acid–base catalyzed cyclizations, cycloadditions, macrolactonizations, SNAr, SN2 reactions, aryl couplings, ring closing metathesis, radical cyclizations etc. are reported (Fig. 1) and many of them have been covered elegantly in several reviews.4–7 However, none of the earlier reviews has focused specifically on metal-catalyzed post-Ugi transformations directed towards the synthesis of diverse heterocyclic scaffolds. Moreover, since the last decade, a considerable number of reports dealing with this topic have been published.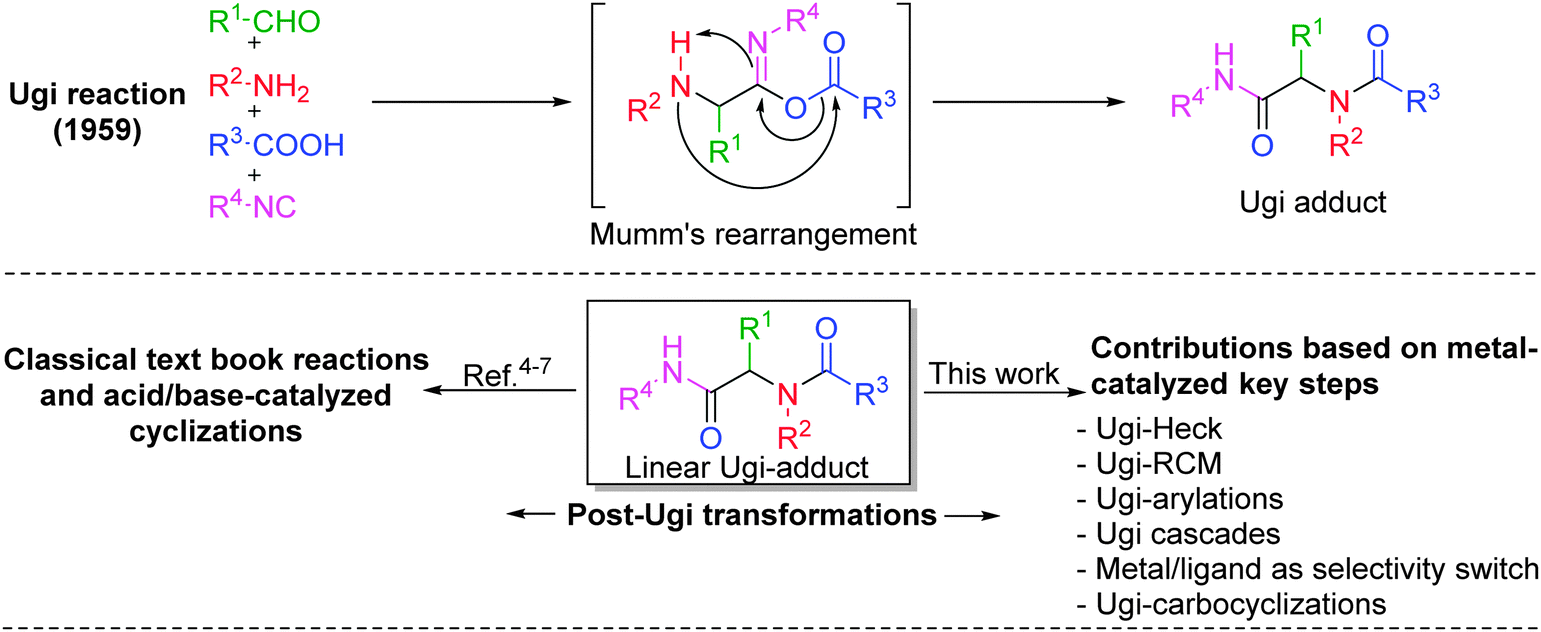 | ||
| Fig. 1 Ugi-4CR and post-transformations for the generation of diverse heterocycles. | ||
In this tutorial review, we wish to present an overview of post-Ugi transformations performed in a sequential or domino fashion. Only metal-catalyzed processes as well as their combinations with elementary organic reactions leading to the de novo formation of the heterocyclic core shall be covered. Owing to the large extent of available literature about metal-catalyzed post-Ugi transformations, we have mainly restricted our review to developments in the last ten year. Methodologies such as Ugi–Click directed towards triazole synthesis7 are kept out due to space limitations. For the sake of clarity, we have divided this review into the following sections: (a) Ugi–Heck, (b) Ugi-RCM, (c) Ugi-arylations, (d) Ugi-cascades, (e) metal–ligand as regio-/chemo-selectivity switch and (f) carbocyclizations (Fig. 2). Among the various metals used for post-Ugi modifications, it is no surprise that Pd-catalysis has a key role, though other metals like Cu, Ag, Au, In and Ru are fast catching up and quite astounding discoveries have been made in the past few years especially with Au-complexes, which will be presented in detail.
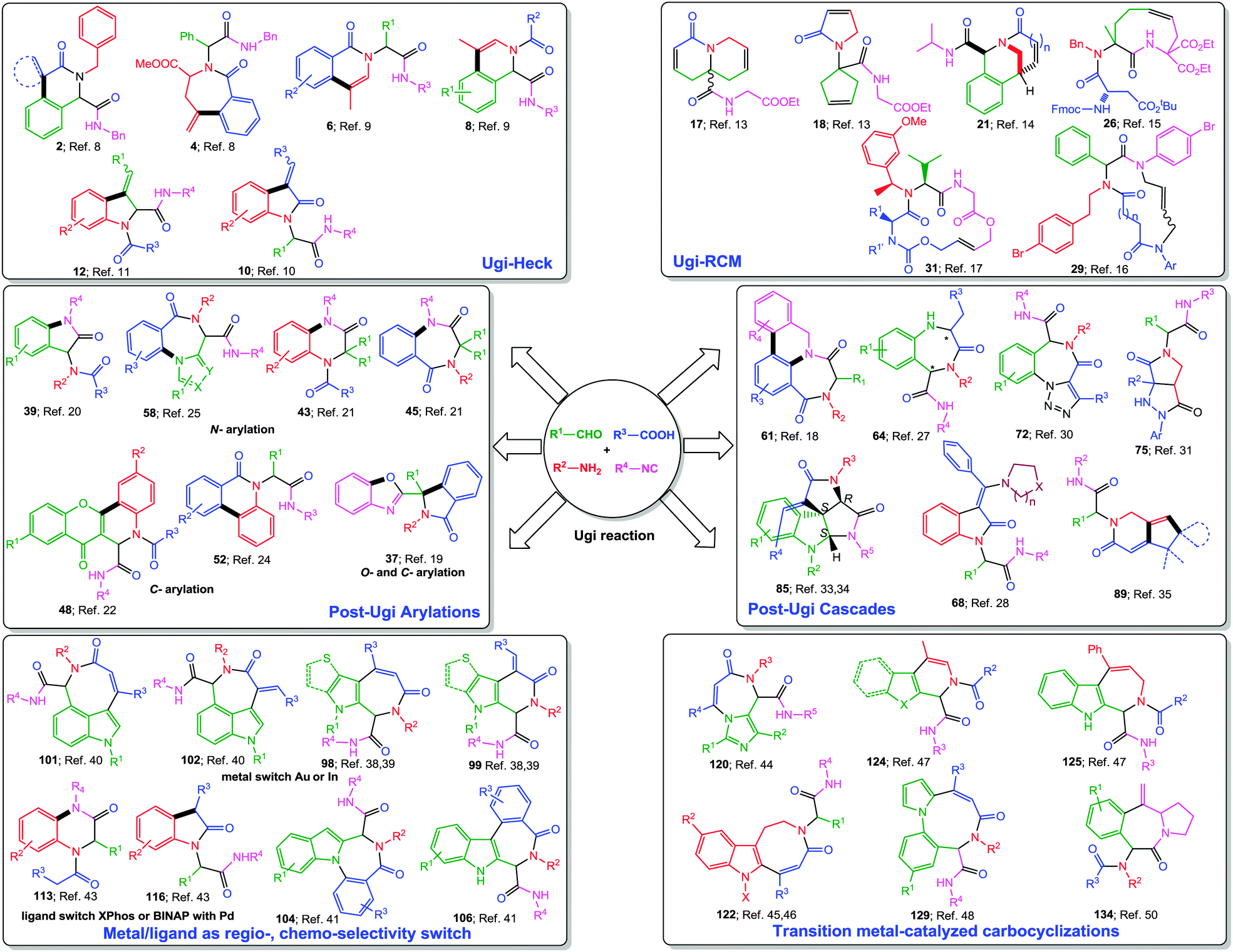 | ||
| Fig. 2 Overview of transition metal-catalyzed post-Ugi transformations. | ||
1. Ugi–Heck sequence
Since the last decade, advancements in generating heterocyclic frameworks via modification of the linear Ugi adduct have undergone an enormous and meaningful upturn. In this section, we have chosen some interesting reports where association of a versatile Pd-catalyzed carbon–carbon bond forming process, the Heck reaction, with the Ugi reaction has been employed for the synthesis of pharmacologically important scaffolds.In a first report of its type, the synthesis of a series of constrained nitrogen containing scaffolds (isoquinolines and benzazepines) has been reported by Gracias and co-workers8via a one-pot sequential Ugi–Heck cyclization approach, whereby the ring size and periphery features of the products were effectively controlled based on the choice of the bi-functional starting materials. Utilizing o-bromobenzaldehyde with unsaturated acids as alkene source, isoquinolines 2 with an olefin-containing spirocyclic system or an α,β-unsaturated system were obtained using Pd(OAc)2 as catalyst, PPh3 as ligand, Et3N as base in MeCN at 125 °C under microwave irradiation for 1 h (Scheme 1, path A). Access to seven-membered lactam 4 was achieved using allyl glycine methyl ester as amine counterpart, with o-halobenzoic acid under similar conditions (Scheme 1, path B). Subsequently, the methodology was extended to solid phase in a two-step sequence using Wang resin coupled Fmoc-protected allyl glycine, affording the required compound in a moderate yield of 33–44%. However, under solid-phase conditions, Heck cyclization requires two-folds of Pd catalyst and PPh3 along with the crucial presence of Bu4NCl in the reaction mixture as compared to solution phase.
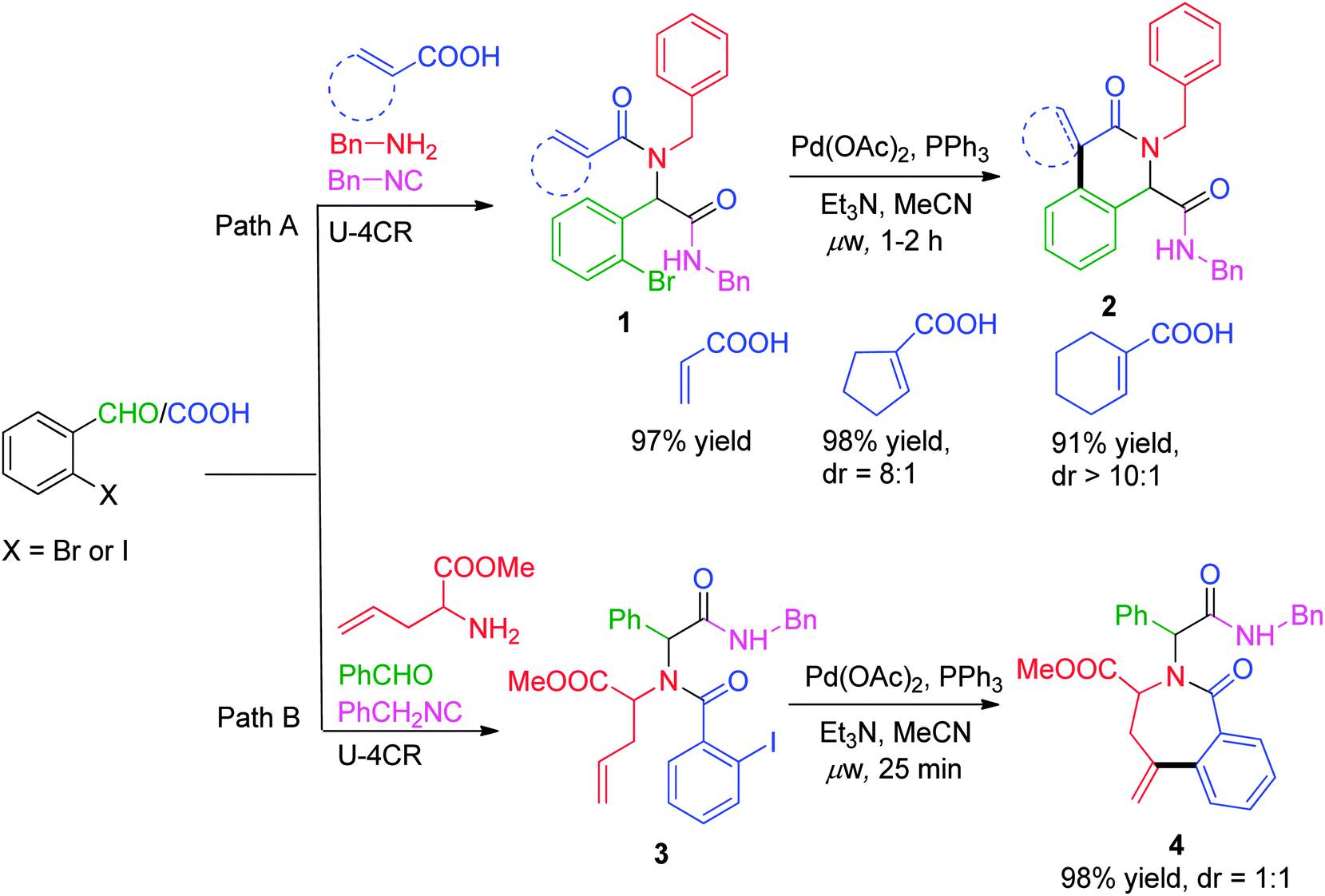 | ||
| Scheme 1 Synthesis of isoquinolines and benzazepines. | ||
In concurrence to the above report, an efficient two-step procedure for the synthesis of isoquinoline scaffolds 6 and 8 in a combinatorial format via an Ugi–Heck reaction sequence has been elaborated by Yang and co-workers.9 The first step in this sequence is a Pd-catalyzed intramolecular Heck reaction followed by a Pd-catalyzed double bond isomerisation of the Ugi adduct generated by reacting N-allylamine either with o-halobenzoic acids or with o-halobenzaldehydes (Scheme 2). The catalytic system consisting of Pd(OAc)2, PCy3 and N-methyldicyclohexylamine as unique base in DMA provided access to a variety of isoquinolines in good to excellent yields. However, in contrast to the method reported by Gracias and co-workers,8 the reaction met with failure under microwave irradiation as double bond isomerization did not proceed completely under the tested conditions.
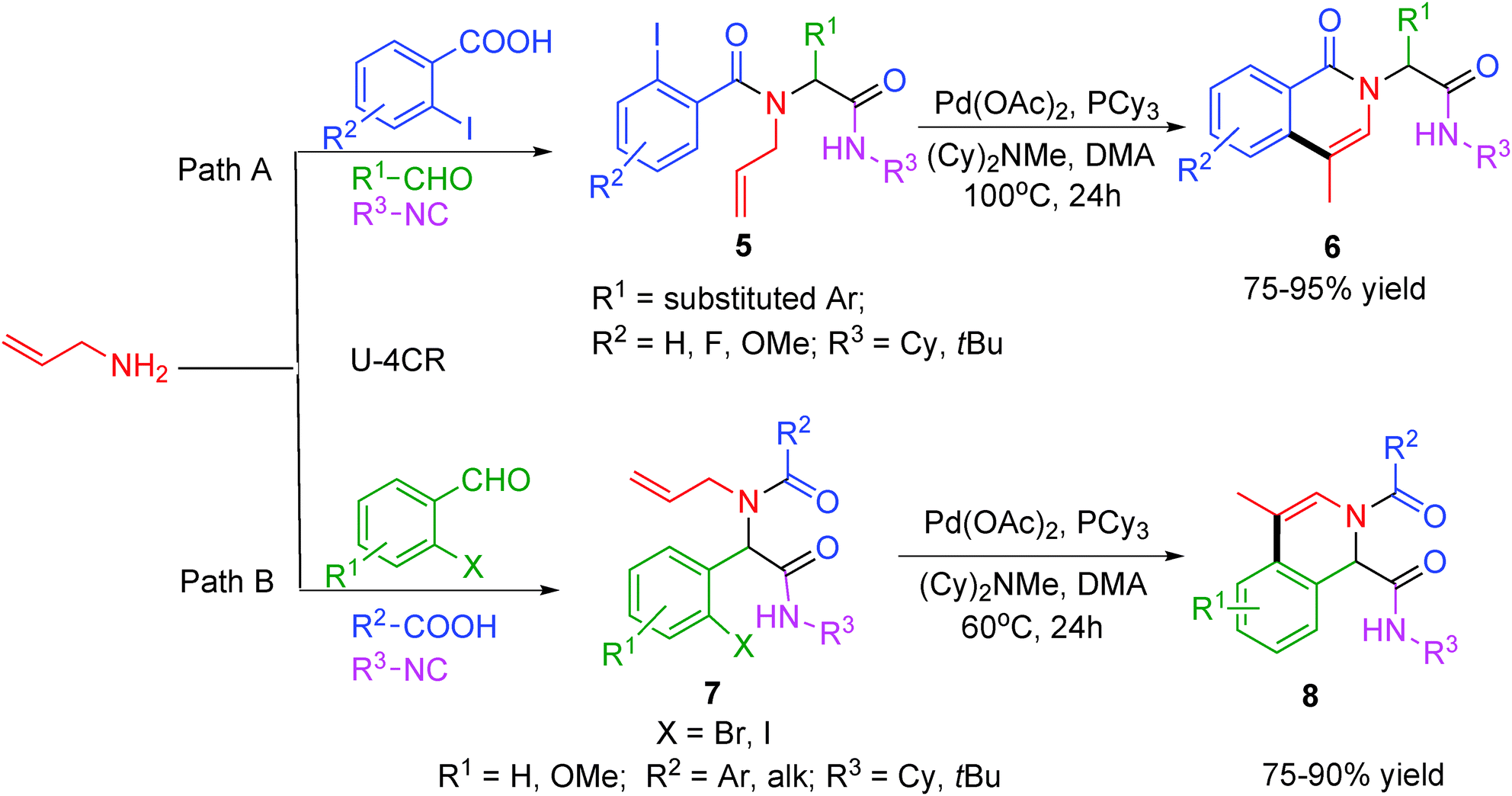 | ||
| Scheme 2 Synthesis of isoquinolines via Ugi–Heck. | ||
Given the pharmacological importance of the indole scaffold, Umkehrer and co-workers10 developed an efficient Ugi–Heck combination for combinatorial library production of indol-2-ones 10 bearing four points of diversity using acrylic acids10 (Scheme 3, path A) as one of the Ugi substrates. In this novel one-pot solution phase synthesis, the starting materials for Ugi reaction were stirred at 50 °C for 24 h in CF3OH (polar protic). Afterwards, the solvent was changed to MeCN and 10 mol% of Pd-catalyst was added which provided the desired products in moderate yields after 16–24 h of stirring at 80 °C. As an update of their work, the authors have reported a similar strategy for the efficient synthesis of dihydroindoles 12 using acrylic aldehydes11 as starting materials (Scheme 3, path B). The method also allows access to 1H-indoles 1311 (Scheme 3, path B) by involving the simultaneous use of formic acid with cinnamaldehyde in the above reaction. Though, the reaction affords low to moderate yields (15–38%), it tolerates well the use of ‘convertible’ isocyanides4 (1-cyclohexen-1-yl and 4-phenyl-1-cyclohexen-1-yl isocyanides) which can be easily cleaved under acidic conditions. In a related work,12 3-arylideneindolin-2-ones 10 were obtained in good yields starting from 2-aminophenols via Ugi-4CR and microwave-accelerated intramolecular Heck reaction (IMHR) using 3–5 mol% Pd(OAc)2/(±) BINAP as catalyst. The 2-hydroxyphenol moiety after the Ugi step was converted into the corresponding triflate using NaH and PhNTf2 before performing the Heck cyclization (Scheme 3, path A). The use of aryl triflates allowed significant shortening of the reaction time than that required for aryl bromides10 along with the generation of more structural diversity on the aromatic ring of the 2-oxindole skeleton.
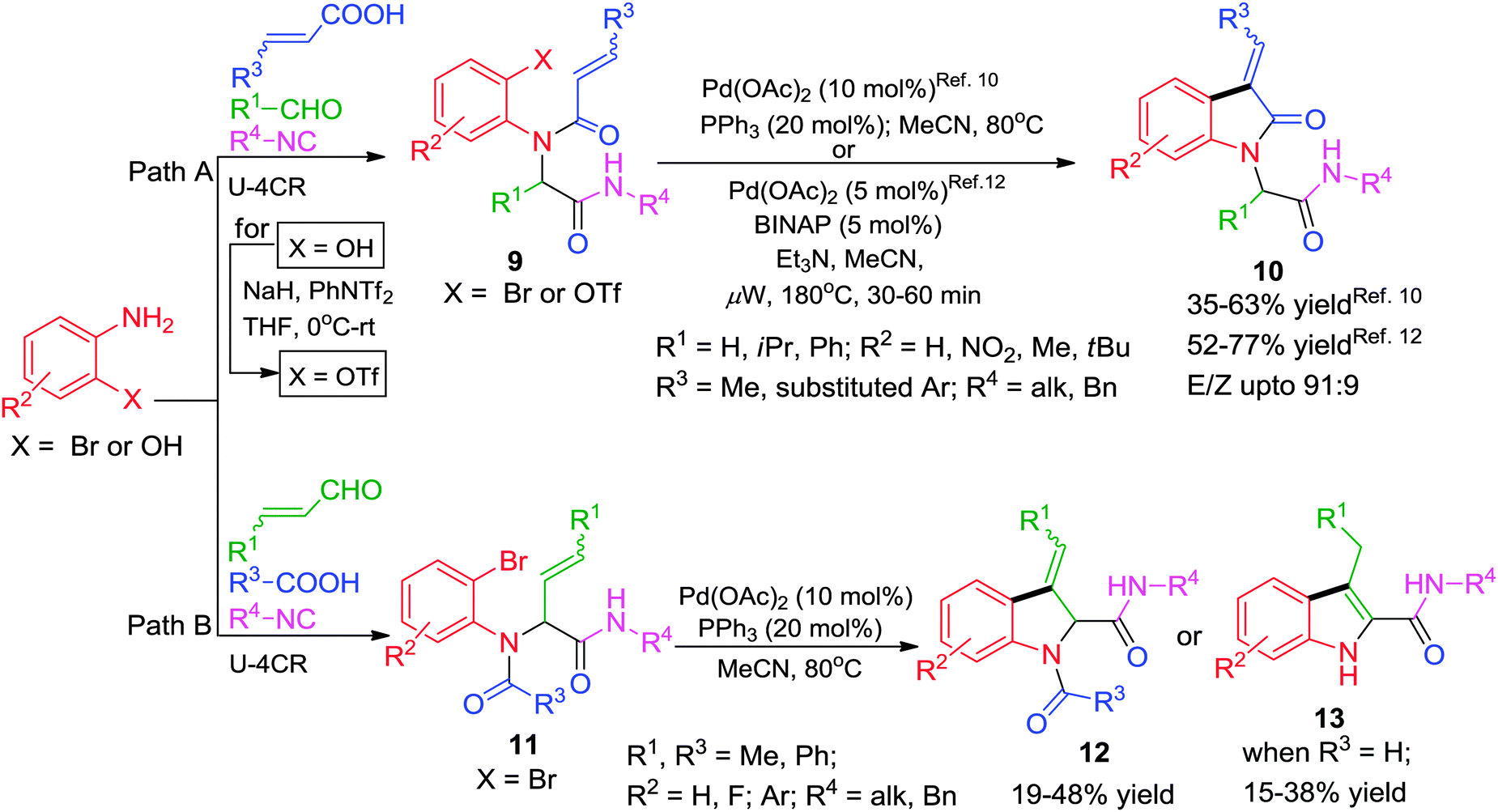 | ||
| Scheme 3 Synthesis of dihydroindoles and indolones via one-pot Ugi–Heck. | ||
2. Ugi-ring closing metathesis (RCM) approach
There is little doubt that the Ugi-RCM has become one of the powerful synthetic tools for the construction of small, medium-sized as well as macrocyclic rings. The attractiveness of this reaction is associated with the utilization of two bifunctional starting materials bearing both a terminal olefinic bond; providing the possibility for a subsequent RCM. Utilizing Grubbs' catalyst carbon, oxygen, nitrogen, and sulfur containing dienes can be cyclized to form functionalized cycloalkenes.13–17 In recent years, this approach has been widely utilized for synthesizing libraries of diversity-oriented templates, which resemble natural products or find multiple applications in peptidomimetics. A recent review by Orru and co-workers7 deals with this topic and the reader is advised to read both reviews to have a better overview.In 2004, Krelaus and Westermann reported the synthesis of peptide-like bicyclic lactams via a sequence of an Ugi 3-component 4-center reaction-RCM approach (Scheme 4).13 Ugi reaction of a two-centered olefinic amino acid (which in turn was derived from ring opening of pyroglutamic acid), allyl amine and isocyanide proceeded well in methanol to provide lactam 14. Subsequent treatment of 14 with Grubbs' catalyst gave bicyclic product 15 in 81% yield. In addition, an alternate route to obtain lactams was also developed by utilizing Ugi reaction to the tetraolefinic amide 16, setting up a direct double RCM which resulted in an equal amount of both products 17 and 18 (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) via two possible modes of cyclization (Scheme 4).
:1) via two possible modes of cyclization (Scheme 4).
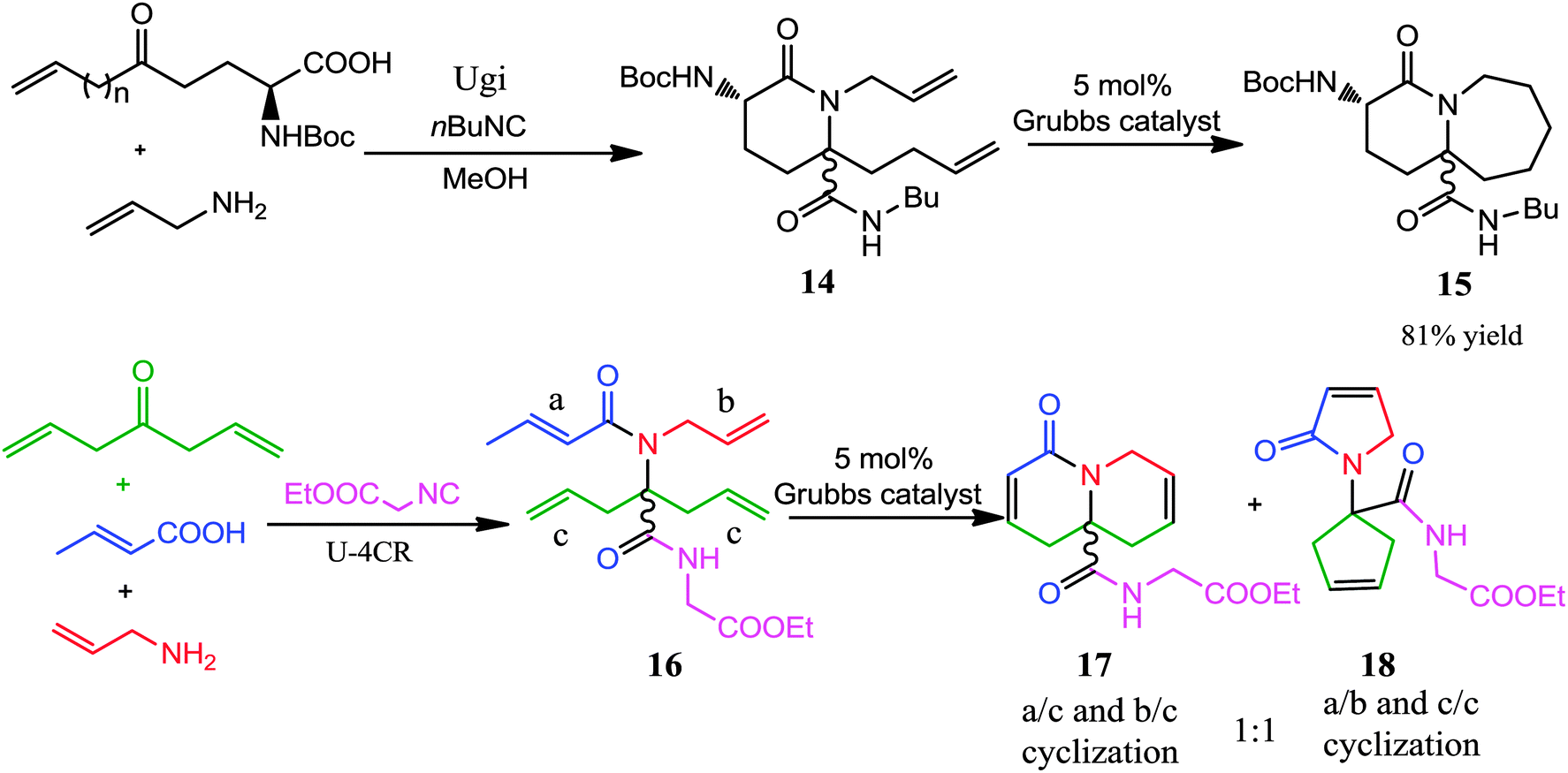 | ||
| Scheme 4 The Ugi-RCM sequence leading to bicyclic lactams. | ||
In another report, Judd and co-workers14 successfully exploited a sequential Ugi-RCM-Heck microwave-assisted approach for the diastereoselective synthesis of a series of bridged bicyclic lactams 21 by utilizing β,γ-unsaturated amide 19 as bifunctional substrates. X-ray diffraction studies revealed that the bicyclic products contain varying degrees of pyramidalization of the bridgehead nitrogen atom, which resulted in their enhanced reactivity towards biological nucleophiles. Further, the nature of the Ugi substrates was altered to access other cyclization manifolds leading to different heterocyclic systems 22–24 in good yields and high diastereoselectivity (Scheme 5).
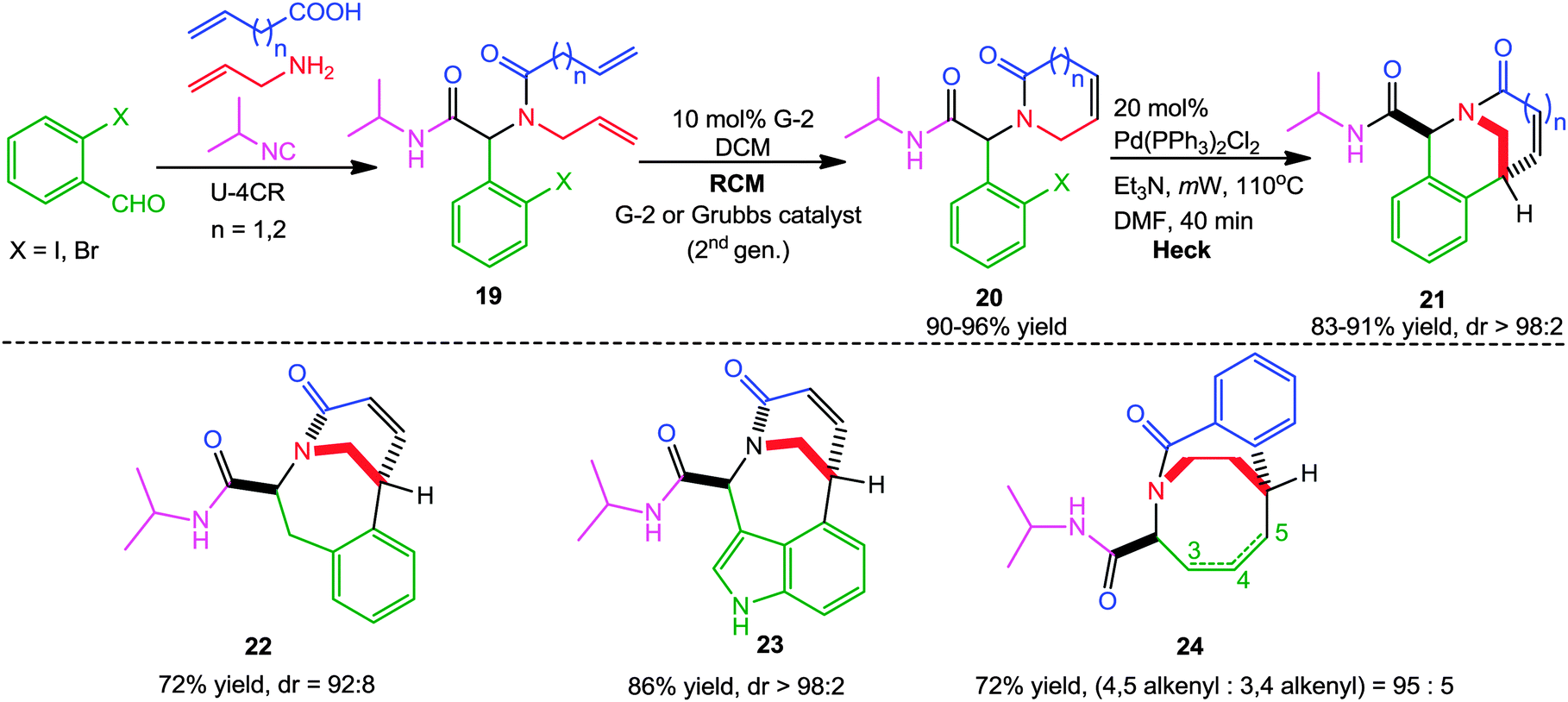 | ||
| Scheme 5 Ugi-RCM-Heck sequence for the generation of bridged bicyclic lactams. | ||
Later on, Banfi and co-workers successful extended this strategy for designing of medium-sized (9–12 membered) cyclic peptidomimics 27.15 Ugi condensation of commercially available protected amino acids with diethyl 2-allyl-2-isocyanomalonate and a preformed imine resulted in diene 25. Subsequent RCM with first generation Grubb's catalyst generated 26, which after decarboxylation and coupling with a protected dipeptide yielded several conformationally biased cyclic pentapeptides, containing the RGD (arginine/glycine/aspartic acid) sequence, built around a tetrahydroazoninone scaffold (Scheme 6). These peptides are of great pharmaceutical interest owing to their recognition by a special category of receptors, the so-called integrins. The synthetic scheme is depicted in Scheme 6, where the N-benzyl group represents a diversity element that can easily be varied.
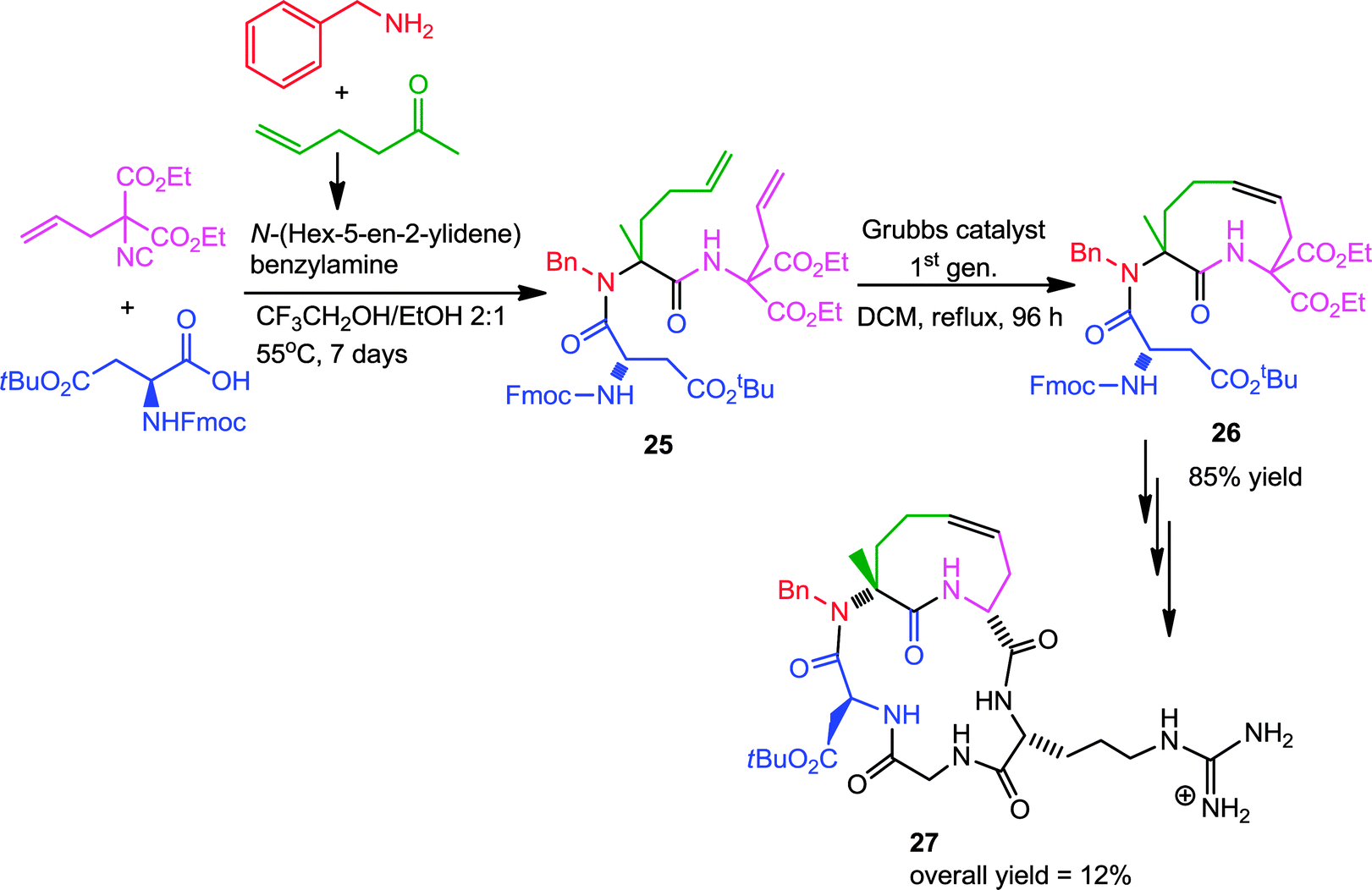 | ||
| Scheme 6 Post-Ugi-RCM based synthesis of cyclic RGD-pentapeptide. | ||
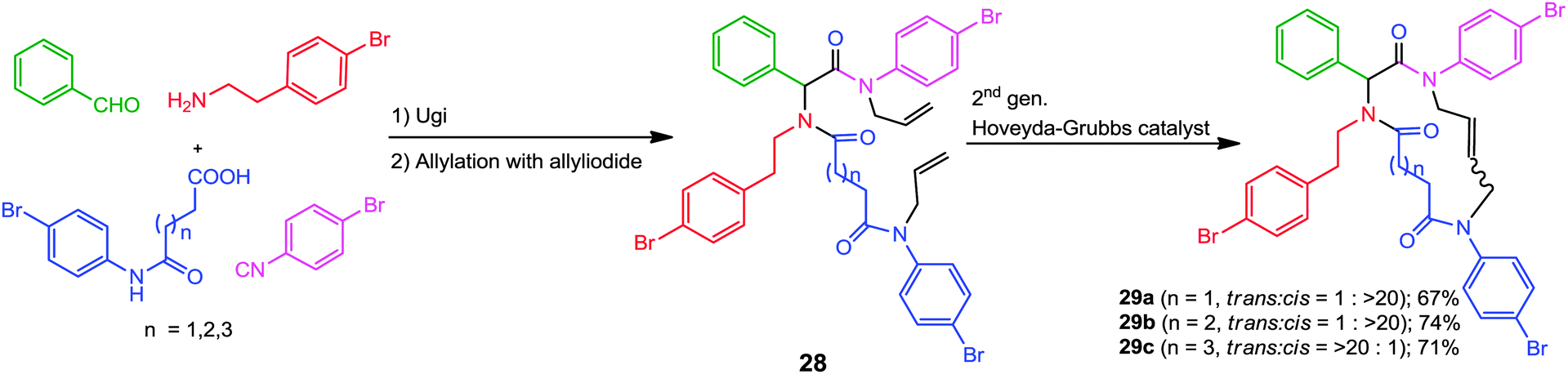
Likewise, for the synthesis of macrocyclic (more than 12 membered) rings, Oikawa and co-workers have developed a straightforward approach based on the ring closing metathesis of allylated Ugi adducts 28.16 These mimics were synthesized in good yields via a three-step procedure involving an Ugi-4CR, the chemoselective introduction of the allyl groups on the Ugi products, followed by RCM. The 13- and 14-membered cycles 29a and 29b respectively were obtained in cis-geometry while the trans-isomer was predominant for the 15-membered cycle 29c (Scheme 7).
 | ||
| Scheme 7 Ugi-RCM for the synthesis of 12–15 membered macrocycles. | ||
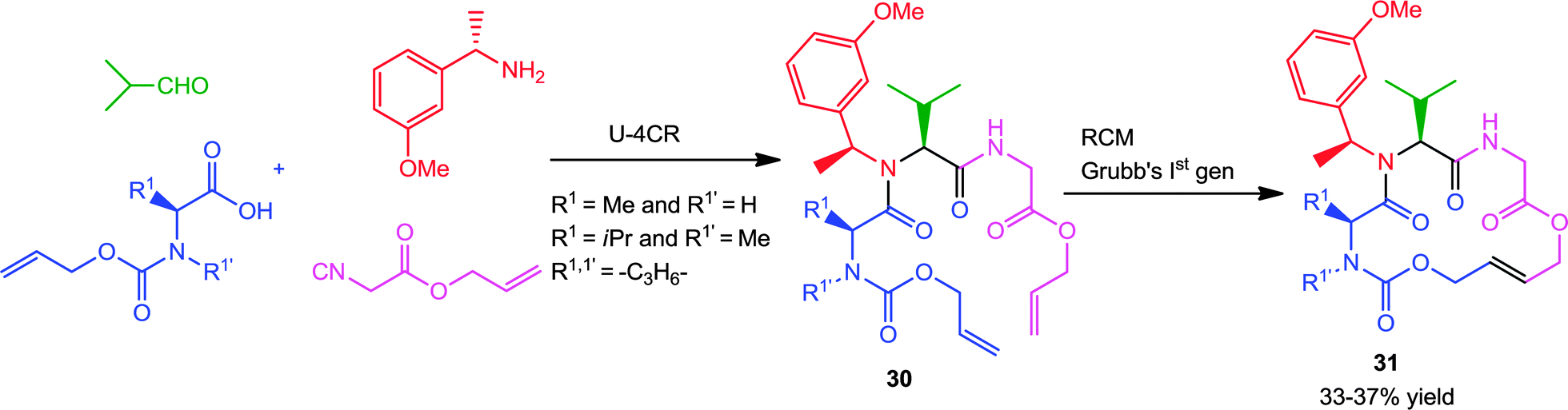
Another interesting procedure by Kazmaier and co-workers17 involves the stereoselective Ugi-RCM approach utilizing allyl isocyanoacetate and different alloc-protected amino acids as bifunctional substrates for synthesizing 16-membered macrocycles 31 (Scheme 8). With (S)-amino acids and (S)-m-methoxy-1-phenylethylamine as chiral components, the Ugi-products 30 were obtained with moderate to high diastereoselectivity. Subsequent RCM with Grubbs' 1st generation catalyst afforded the products in moderate yields, favoring the E-isomer (>95% trans).
 | ||
| Scheme 8 Stereoselective Ugi-RCM towards 16-membered macrocycles. | ||
Based on the above discussions, it is evident that Ugi-RCM is a highly suitable approach to reduce the fluxionality of the peptide backbone via synthesis of cyclic constrained peptidomimetics. However, poor stereoselectivity of the product is still a major issue which needs to be addressed.
3. Ugi-intramolecular arylations
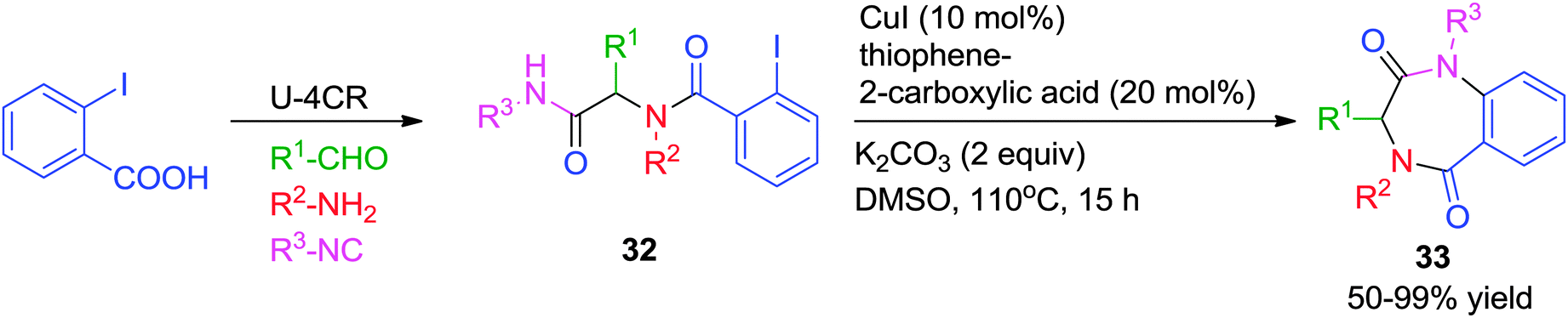
In order to increase the versatility and the complexity-generating power, intramolecular (C-, O- and N-) arylation reactions have been combined with the Ugi-4CR for the construction of pharmaceutically relevant cyclic scaffolds. In comparison to other transition-metals, both Pd- and Cu-catalyzed arylations leading to the formation of 5-, 6-, and to some lesser extent, 7-membered rings have been much investigated.In line with this concept, Zhu and co-workers18 described an efficient methodology for the construction of two different heterocyclic scaffolds from the linear Ugi-adduct 32 by exploiting the different catalytic behavior of Pd- and Cu-catalysts. While Pd triggered a domino intramolecular N-arylation/C–H activation/aryl–aryl bond-forming process (which will be discussed in the post-Ugi cascade section), Cu promoted only the intramolecular N-arylation leading to the formation of 1,4-benzodiazepine-2,5-diones 33. The optimized reaction conditions involved the use of CuI as catalyst and thiophene-2-carboxylic acid as a ligand in DMSO at 110 °C for 15 h (Scheme 9). Notably, it was found that the outcome of the reaction was significantly influenced by the choice of base and solvent.
 | ||
| Scheme 9 Cu(I)-catalyzed synthesis of 1,4-benzodiazepine-2,5-diones. | ||
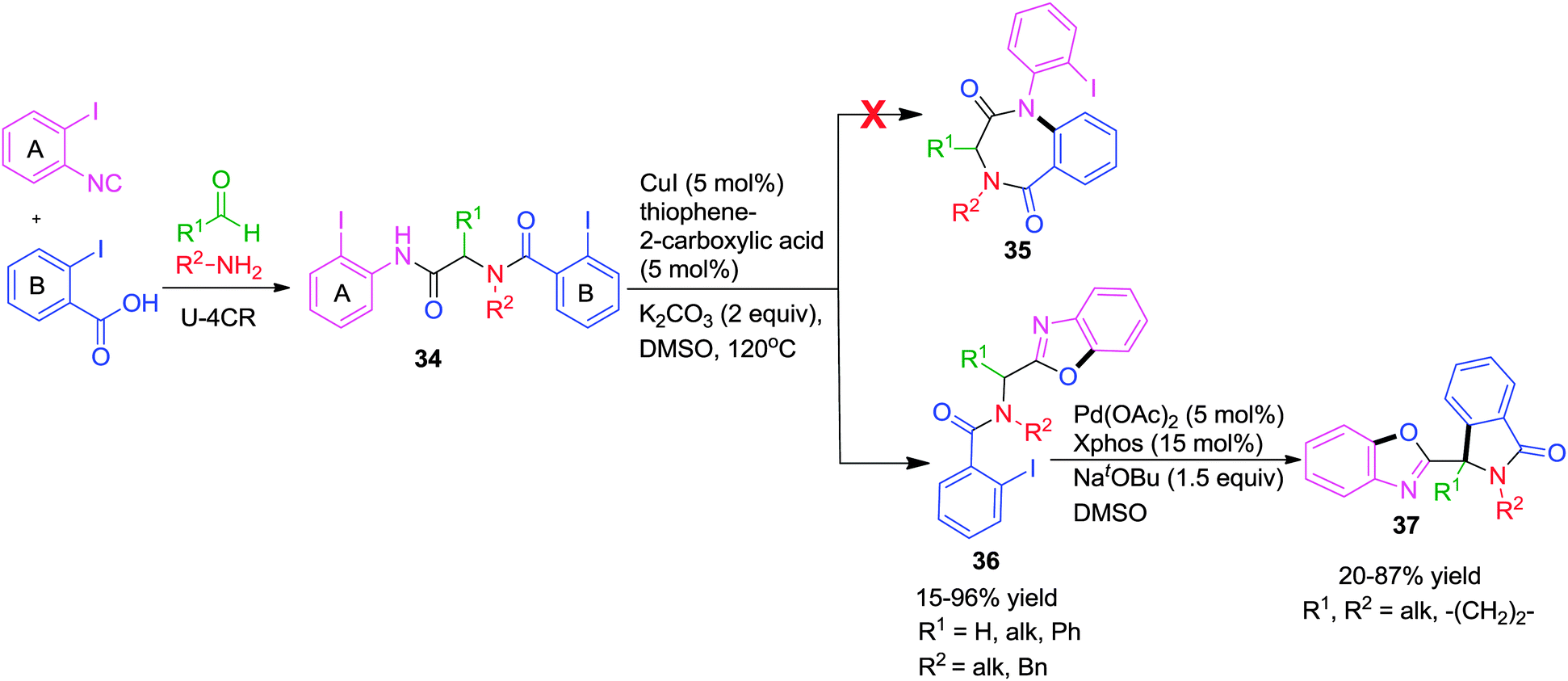
As an extension of this work, the authors described a highly efficient post-Ugi transformation for the synthesis of 3-substituted benzoxazolylisoindolinones 37via two consecutive metal-catalyzed intramolecular reactions.19 Starting from Ugi-adduct 34 with two aryl iodide units, a regio-specific Cu-catalyzed O-arylation followed by a Pd-catalyzed C-arylation afforded the desired products in good to excellent yields. Due to presence of two aryl halides, two reaction pathways were expected with Cu(I) catalysis (Scheme 10). However under the previously optimized conditions,18 benzoxazole 36 was obtained exclusively and no alternative intramolecular N-arylation leading to benzodiazepinedione 35 was observed. The authors deduced that the oxidative addition of ring A aryl iodide to Cu(I) was occurring preferentially over the ring B counterpart although being more electronically poor ring B ought to be prone to oxidative addition. Substituted benzoxazolylisoindolinones 37 were obtained in moderate yields from 36 using a catalytic amount of Pd(OAc)2/X-Phos in DMSO at 120 °C.
 | ||
| Scheme 10 A sequential Cu/Pd-catalyzed intramolecular O- and C-arylation for the synthesis of benzoxazolylisoindolinones. | ||
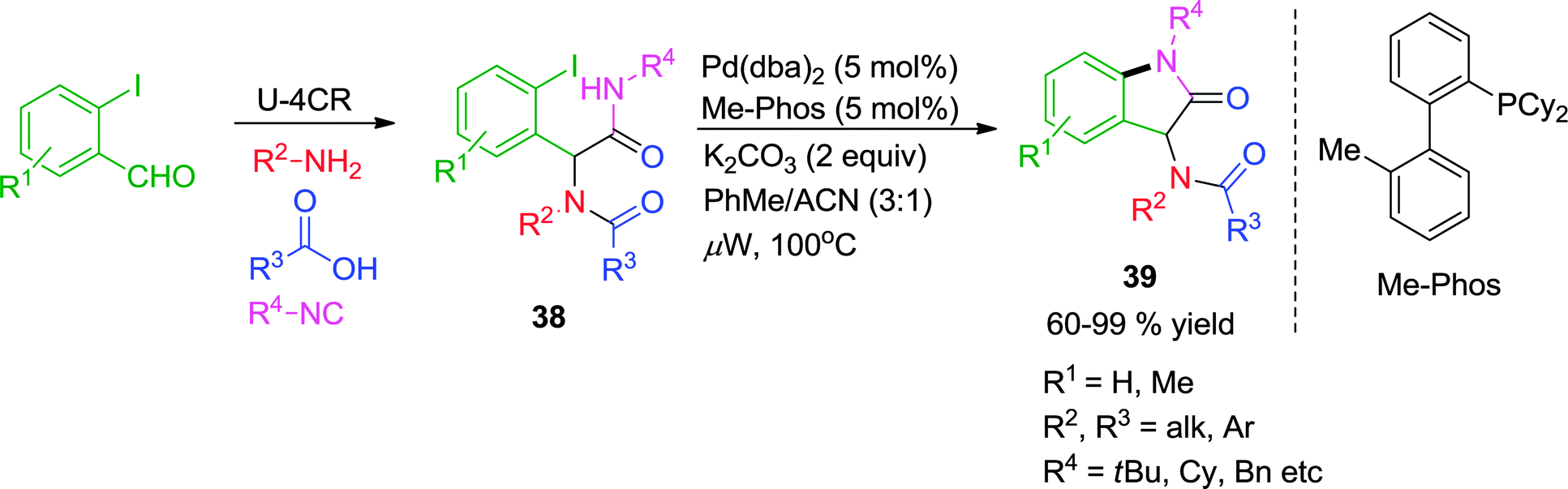
Later on, the authors have reported an efficient methodology to access functionalized oxindoles 39via Pd-catalyzed intramolecular amidation (intramolecular Buchwald–Hartwig reaction) of Ugi substrates 38 under microwave irradiation (Scheme 11).20 Initially, the cyclization was tried with a Cu-catalyst; however the yield could not be increased beyond 42%. Thereafter, the reaction was optimized under using Pd(dba)2 as the catalyst and Me-Phos as the ligand of choice, giving the 2-oxindoles in moderate to excellent yields (60–99%). Sterically hindered amides and iodides were well tolerated and no competitive C–H activation process leading to formation of dihydrophenanthridine was observed in the course of reaction.
 | ||
| Scheme 11 Pd(II)-catalyzed post-Ugi intramolecular amidation for the synthesis of oxindoles. | ||
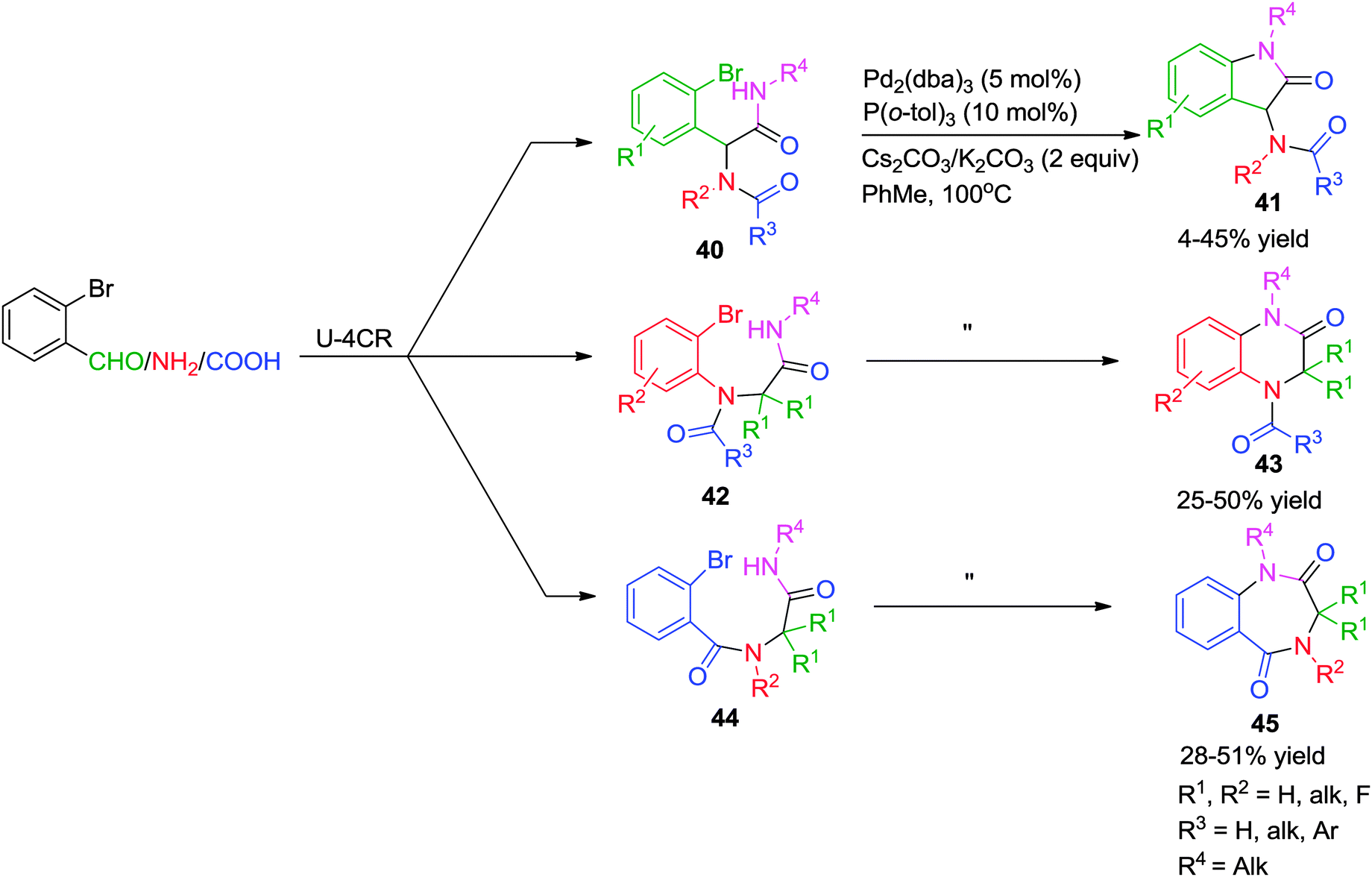
In pursuit of generating a small array of N-heterocyclic compounds, Kalinski and co-workers have synthesized indol-2-ones 41, quinoxalin-2-ones 43 and benzodiazepine-2,5-diones 45via a Pd-assisted post-Ugi N-arylation strategy.21 Varying the initial starting materials (Scheme 12) for the Ugi reaction, three different types of products (5, 6, and 7-membered ring systems) were obtained using the same catalytic system consisting of 5 mol% of a Pd-catalyst, 10 mol% of P(o-tol)3, 2 equiv. of carbonate base (Cs2CO3 for aliphatic- and K2CO3 for benzylic isocyanides) in toluene at 100 °C. Functionalized 2-bromobenzaldehydes and 2-bromoanilines were well tolerated enabling further construction of the scaffold. However, the overall yields are rather moderate (≤50%).
 | ||
| Scheme 12 Pd-catalyzed post-Ugi intramolecular N-arylation for the synthesis of indol-2-ones, quinoxalin-2-ones and benzodiazepine-2,5-diones. | ||
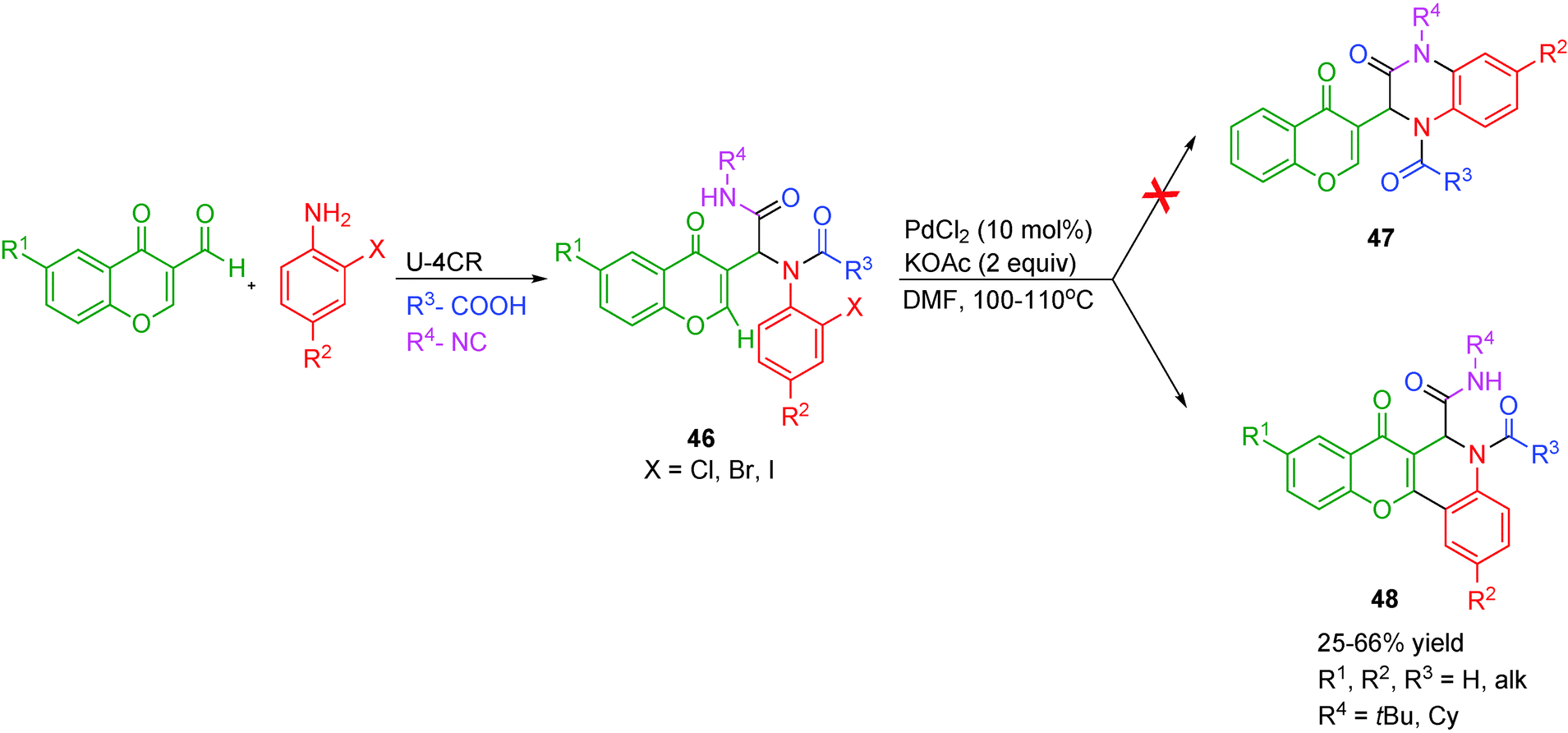
An interesting example of a ligand-free Pd-catalyzed intramolecular C-arylation of chromones has been reported by Bandyopadhyay and co-workers22 for the synthesis of 1,4-benzopyrano[3,2-c]quinolines 48. Two possible modes of cyclization are possible with the Ugi adduct 46: (i) N-arylation between o-halophenyl and the amide to form ketopiperazine 47, or (ii) C-arylation at the C-2 or C-3 position of the chromone with the o-halophenyl group. Under the optimized conditions of 10 mol% of PdCl2 catalyst, KOAc and DMF at 100–110 °C under argon, the latter was favored in this case (Scheme 13). The detrimental effect of ligands, additive (pivalic acid) or Pd(OAc)2 on product yields was assumed to be the result of steric crowding across the Pd-center which in turn hindered attaining the required conformation for cyclization.
 | ||
| Scheme 13 Ligand-free Pd-catalyzed intramolecular C-arylation of chromones for the synthesis of 1,4-benzopyrano[3,2-c]quinolines. | ||
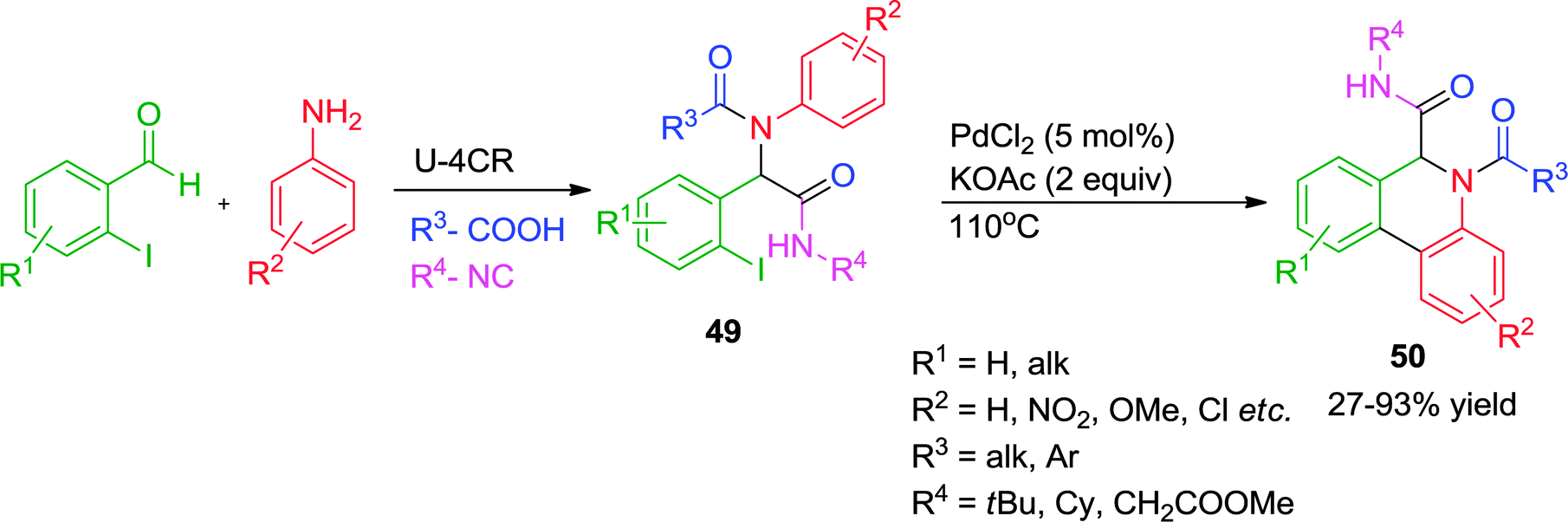
Another Pd-catalyzed ligand-free approach for a post-Ugi C–H arylation process has been reported by Zhu and co-workers23 where the synthesis of dihydrophenanthridines 50 has been achieved in good to excellent yields (Scheme 14). A variety of substituted anilines, regardless of their electron-donating or -withdrawing character, could be used. Interestingly, the alternative reaction pathway leading to the formation of oxindoles20via an intramolecular N-arylation process was not observed under the optimized conditions, indicating that cyclization via Pd-catalyzed C–H functionalization might be a kinetically fast process compared to other elementary reactions. Additionally, when two protons were present in similar steric environment the more acidic C–H bond was found to be prone towards selective functionalization.
 | ||
| Scheme 14 A ligand free Pd-catalyzed post-Ugi C–H arylation process for the synthesis of dihydrophenanthridines. | ||
Concurrently, an independent report from Yang and co-workers24 disclosed an efficient approach to synthesize two types of quinoline scaffolds (52 and 56versus54) via an Ugi-4CR/Pd-catalyzed C–H arylation sequence using o-iodobenzaldehydes, o-iodoanilines and o-iodobenzoic/heteroacids, respectively as pre-functionalized components for the arylation process. In comparison with the method reported by Zhu and co-workers23 the authors have established the importance of structure of the ligand on the outcome of the cyclization. In most of the cases, dppf (1,1′-bis(diphenylphosphino)ferrocene) was found to work well for annulations with Pd(OAc)2, K2CO3, nBu4NBr and DMF at 80 °C. However, when the Ugi adduct lacks an electron-withdrawing group or in case of electronically rich iodobenzoic acids 51, the oxidative capability of the Ugi-product to the metal was decreased. As a result, electron-rich PCy3 ligand was needed to promote annulations (Scheme 15). The methodology offers a wide substrate scope, short reaction time and excellent product yields.
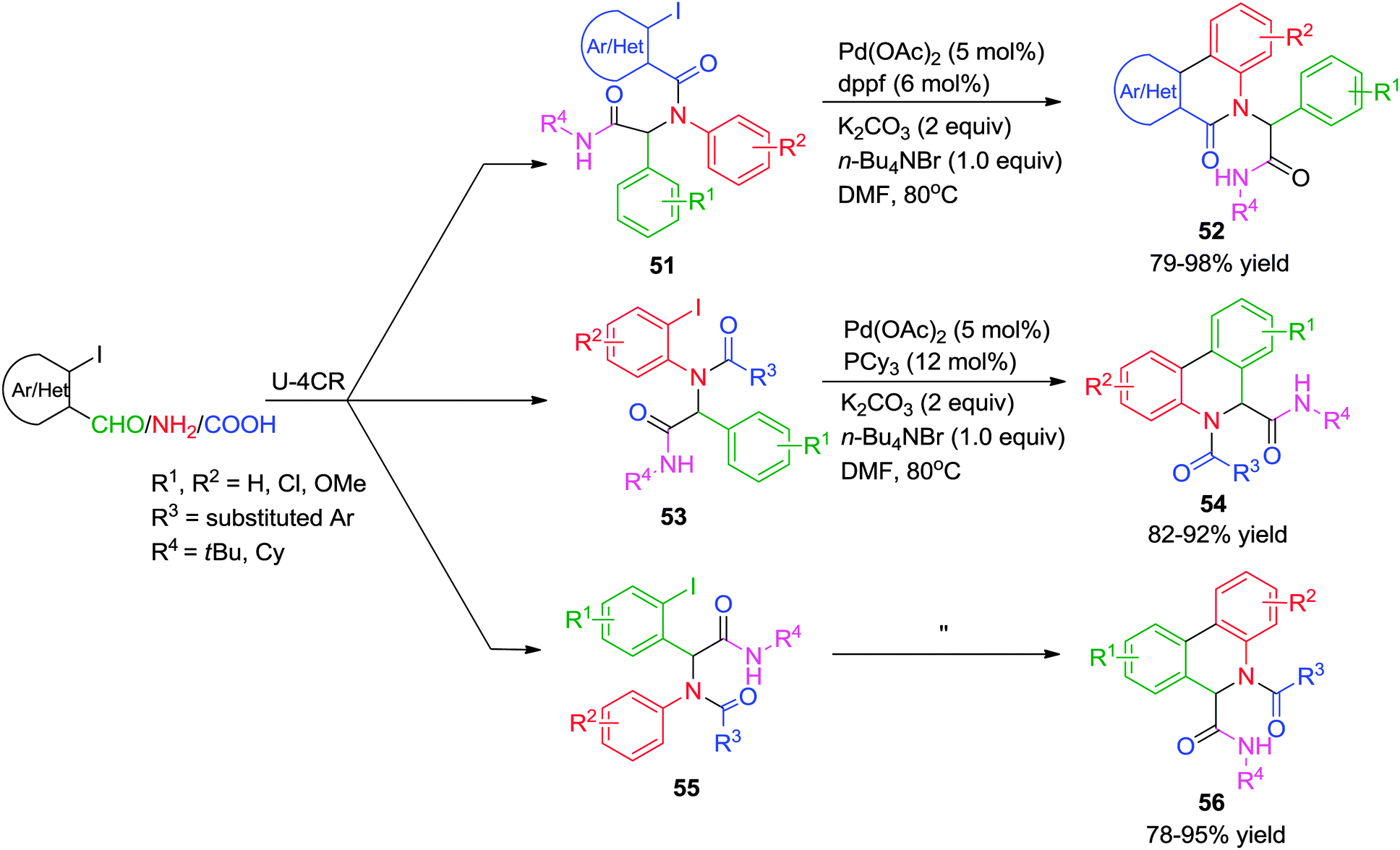 | ||
| Scheme 15 A ligand dependent approach (dppf vs. PCy3) to structurally diverse quinolines via an Ugi-C–H arylation. | ||
Recently, an expedient post-Ugi diversity-oriented intramolecular Ullmann coupling strategy has been reported by Van der Eycken and co-workers for the synthesis of 4H-benzo[f]imidazo[1,4]diazepin-6-ones (58) under microwave irradiation (Scheme 16).25 A wide range of Ugi substrates derived from imidazole-4-carbaldehyde were subjected to intramolecular coupling utilizing CuI as catalyst in DMSO at a ceiling temperature of 100 °C for 30 min. The presence of fused ring systems in the products, analogous to several commercially available imidazobenzodiazepinone drugs, the employment of a cheap Cu-catalyst and the excellent product yields enhance the utility of the methodology.
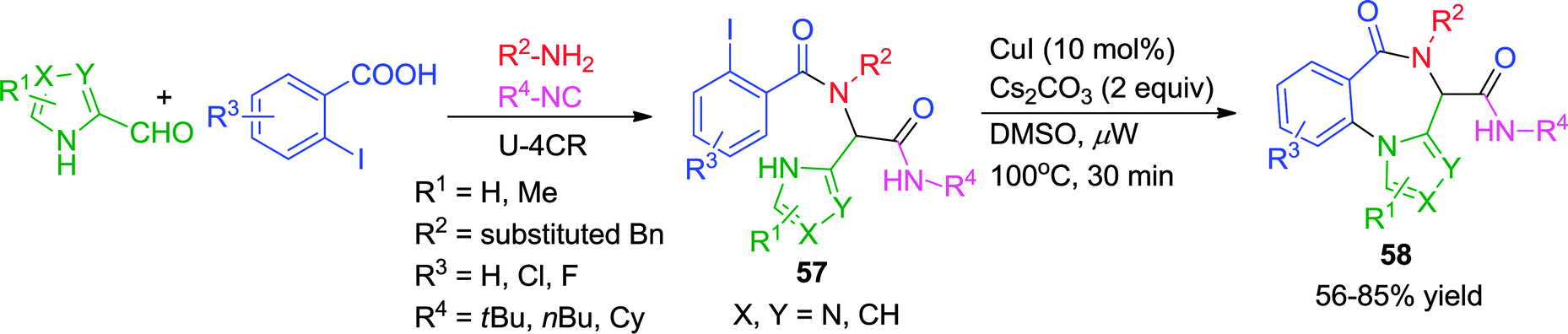 | ||
| Scheme 16 Synthesis of 4H-benzo[f]imidazo[1,4]diazepin-6-ones via Ullmann coupling. | ||
4. Post-Ugi cascades
Cascade transformations are step-economical processes which offer enhancement of molecular complexity and diversity starting from simpler molecules. Herein, the construction of complex new heterocyclic scaffolds can be factually achieved in a predesigned manner by virtue of organometallic elementary reactions where reactive functionalities are generated en route. They are often accompanied by a high degree of chemo-, regio-, and diastereoselectivity. Some noteworthy examples from the literature employing transition metals will be highlighted in this section.As described earlier, Zhu and co-workers uncovered a distinct catalytic behavior of Pd- and Cu-catalysts on the same Ugi adduct.18 While Cu promoted the simple intramolecular N-arylation reaction (Scheme 9), Pd-triggered the domino intramolecular N-arylation/C–H activation/aryl–aryl bond forming process to provide 5,6-dihydro-8H-5,7a-diazacyclohepta[jk]phenanthrene-4,7-diones 61 from Ugi-adduct 59 (Scheme 17). It was ascertained that the presence of two iodide functions is necessary for the sequence and attempts to stop the reaction after intramolecular N-arylation proved futile, although the reaction was shown to involve benzodiazepinedione 60 as intermediate (Scheme 17). The scope of the methodology was extended for the construction of azaphenanthrenes fused to 8-, 9- 10-, 12- and 13-membered lactams. Notably, this is the first report for the generation of medium-rings and macrocycles via intramolecular Buchwald–Hartwig amidation.
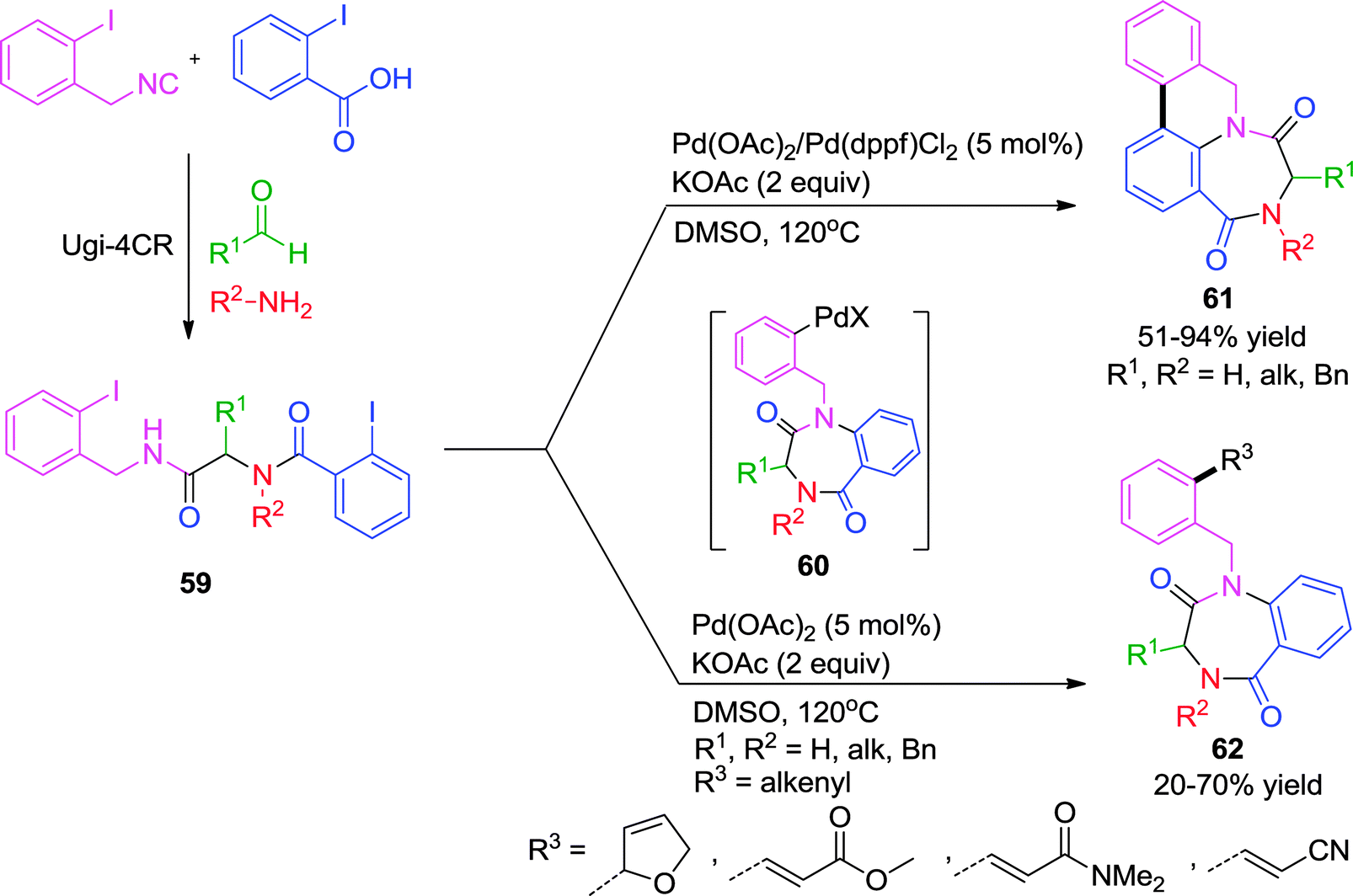 | ||
| Scheme 17 Pd-catalyzed cascade intramolecular and intermolecular C-arylations. | ||
In a subsequent report26 by the same group, this direct C–H arylation was effectively interrupted for an intermolecular Heck reaction in the presence of a suitable trapping agent. Whereas phenylboronic acid, morpholine or 4-benzyloxazolidinone failed to interrupt the process, potassium ferrocyanide, vinyltributylstannane and dihydrofuran were effective as anion-capturing agents. Further, it was observed that ligandless conditions are essential for this tandem process since the presence of triphenylphosphine and other biphenylphosphine ligands favors the intramolecular C–H arylation over intermolecular Heck reaction. This transformation is a rare example where intramolecular N-arylation occurred under ligandless conditions leading to the formation of a seven-membered benzodiazepinedione 62 (Scheme 17). Mechanistically, it was proposed that the two aryl halides were not differentiated in the oxidative addition step leading to a bis-arylpalladium halide intermediate, which might adopt a conformation conductive for cyclization, thus making the intramolecular N-arylation kinetically competitive relative to the other coupling process. An intramolecular N-arylation would generate a second Pd(II) intermediate 60, from which two divergent pathways are possible i.e. intramolecular C-arylation 61 and intermolecular Heck reaction 62 as shown in Scheme 17.
Andreana and co-workers27 have described a two-step route for the synthesis of 1,2,4,5-tetrahydro-1,4-benzodiazepin-3-ones starting from α,β-unsaturated acids and o-nitrobenzaldehyde or o-nitrobenzylamine as bifunctional Ugi-substrates, with various isonitriles. The reaction sequence involves a Fe/NH4Cl-catalyzed reduction of the nitro functionality followed by a 7-exo aza-Michael cyclization. The authors observed that the reduction of o-nitro group was not feasible at room temperature and that the aza-Michael cyclization was sluggish under conventional heating. To address this issue, the authors devised optimal reaction conditions under microwave irradiation at a ceiling temperature of 150 °C for 30 min in a 3:1 ratio of EtOH/H2O to give diversely substituted 1,2,4,5-tetrahydro-1,4-benzodiazepin-3-ones 64 and 66 in moderate to good yields with satisfactory diastereoselectivity (Scheme 18). However, acrylic acid proved to be an unsuccessful substrate for cyclization. Interestingly, a change in the intensity of microwave has a pronounced effect as 6-exo-aza-Michael cyclization was observed instead of desired 7-exo aza-Michael addition.
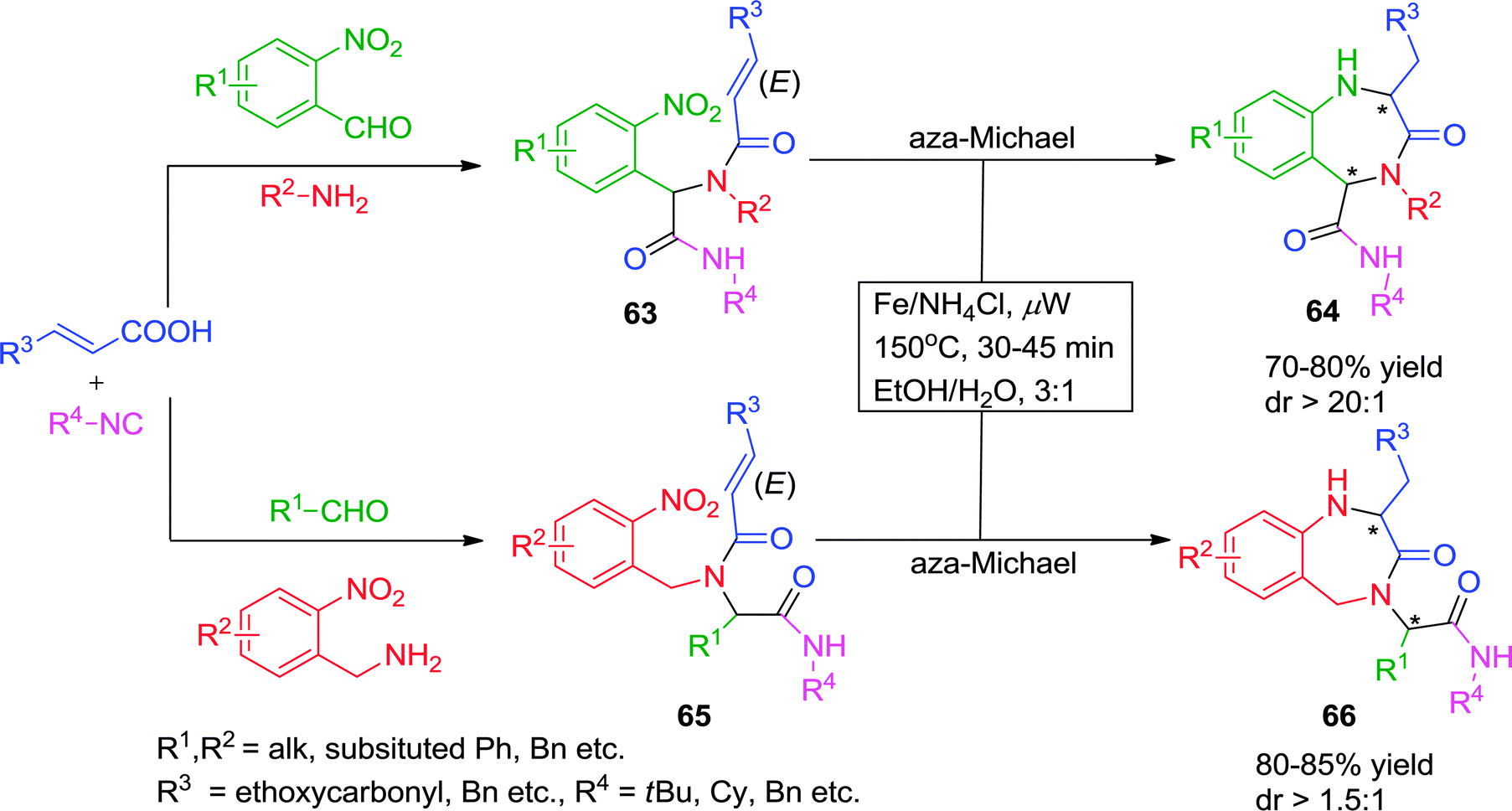 | ||
| Scheme 18 A post-Ugi access to 1,2,4,5-tetrahydro-1,4-benzodiazepin-3-ones. | ||
Subsequently, Balalaie and co-workers reported an efficient approach for the stereoselective synthesis of (Z)-3-(aminoarylmethylene)-oxindoles via a Pd-catalyzed Ugi-carbopalladative cyclization-Buchwald reaction sequence.28 A range of diversely substituted benzaldehydes was reacted with 2-iodoaniline, phenylpropiolic acid and isonitrile in methanol to obtain Ugi adduct 67. 3-(Aminoarylmethylene)-oxindoles 68 were obtained in good yields by reacting 67 with various secondary amines using Pd(OAc)2/tri-(2-furyl)phosphine as the catalytic system in refluxing toluene for 12 h (Scheme 19, path A). Overall, the reaction accomplishes both intramolecular C–C bond formation as well as intermolecular C–N bond formation across a C–C triple bond in a domino fashion. On the basis of known Pd-chemistry, the reaction follows: (a) oxidative addition of Pd(0) to the haloarene; (b) insertion of Pd on the alkyne moiety (carbopalladation); (c) nucleophilic addition of the secondary amine; (d) reductive elimination to afford the final product 68 with regeneration of the Pd(0). The Z-configuration of the products was confirmed using NMR-spectroscopy with a diagnostic signal for the Z-isomer observed at δ 5.56–5.64 (H-4 of the oxindole). Moreover, NOE experiments were also used by the authors to confirm the structure as Z-isomer.
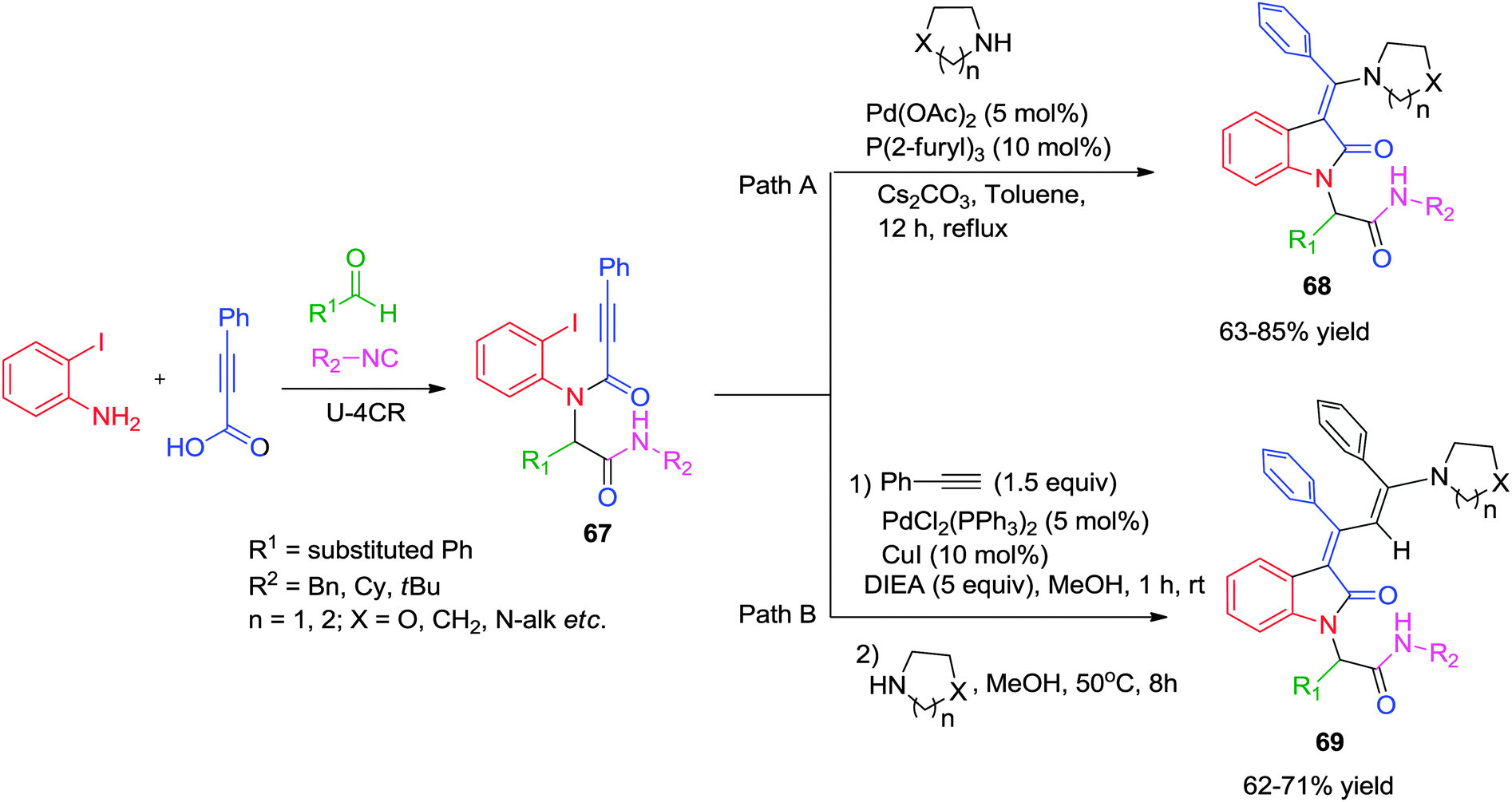 | ||
| Scheme 19 Access to (Z)-3-(aminoarylmethylene)-oxindoles and 3-arylidene-2-oxindoles via a cascade reaction sequence. | ||
Due to the widespread occurrence of the 3-arylidene-2-oxindole subunit in natural products and owing to their significant biological activities, this skeleton has always kept the interest of organic chemist. In one such example, Balalaie and co-workers have reported a six component sequential reaction for the synthesis of 3-arylidene-2-oxindoles via an Ugi–Heck carbocyclization/Sonogashira/nucleophilic addition (Scheme 19, path B).29 This sequential reaction proceeds in three steps, namely (a) an Ugi-4CR leading to N-substituted 2-alkynamides 67; (b) a Heck carbocyclization/Sonogashira cross-coupling of N-(2-iodophenyl) alkynamides with phenylacetylene in a domino process, and finally (c) the nucleophilic addition to the activated triple bond of the intermediate dihydroindolones (E and Z isomers) to form the desired 3-arylidene-2-oxindoles 69 as a Z-isomer.
Inspired by the developments in the field of Cu-catalyzed heterocyclic synthesis, Van der Eycken and co-workers demonstrated a diversity-oriented approach for the synthesis of triazolo[1,5-a][1,4]benzodiazepinones 72 by employing a post-Ugi Cu-catalyzed tandem azide–alkyne cycloaddition/Ullmann C–N coupling strategy (Scheme 20).30 The intramolecular trapping of the N-Cu intermediate 71 formed through Cu-catalyzed [3+2] cycloaddition, led to the formation of the C–N bond in a cascade manner via Ullmann coupling. The methodology is a step economical approach for the synthesis of triazole fused benzodiazepinones 72 with moderate to good yields, four-point diversity and compatibility with a variety of functional groups. However, Ugi adducts derived from alkyl-substituted propiolic acids needed higher catalyst loading for full conversion. Other limitations are the inability of 2-chloro benzaldehyde and bulky alkyl isocyanide derived Ugi adducts to participate in the reaction.
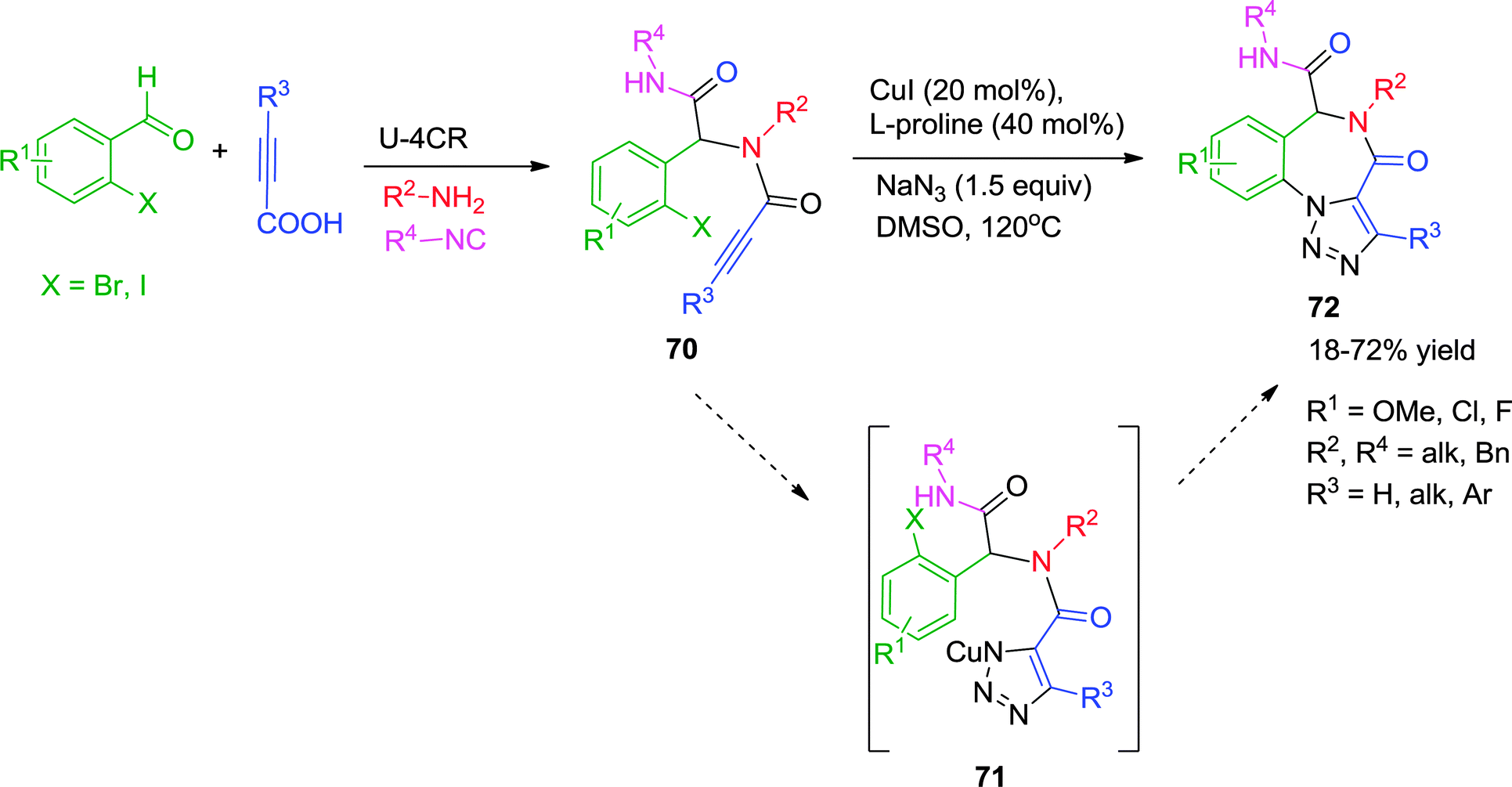 | ||
| Scheme 20 Synthesis of triazolo[1,5-a][1,4]benzodiazepinones via post-Ugi tandem alkyne–azide cycloaddition-Ullmann coupling. | ||
Earlier, Santos et al. have synthesized pyrazolidinones via a post-Ugi Cu-triggered cycloaddition/aerobic oxidation cascade (Scheme 21) employing α-hydrazonocarboxylic acid and allylamine as representative Ugi-substrates.31 The first step is an intramolecular [3+2] cycloaddition between the hydrazone and the alkene functionality in the Ugi adduct 73, which is triggered by the copper acetate or acetic acid. The resulting pyrazoline is oxidized by the Cu(II) salt into intermediate 74 after addition of water. This might undergo ring opening leading to azo or hydrazono derivatives 76, or further oxidation occurred without ring opening giving access to fused pyrazolidinones 75. The process is step-economic with wide functional group tolerance providing the desired products in good yields and with high diversity.
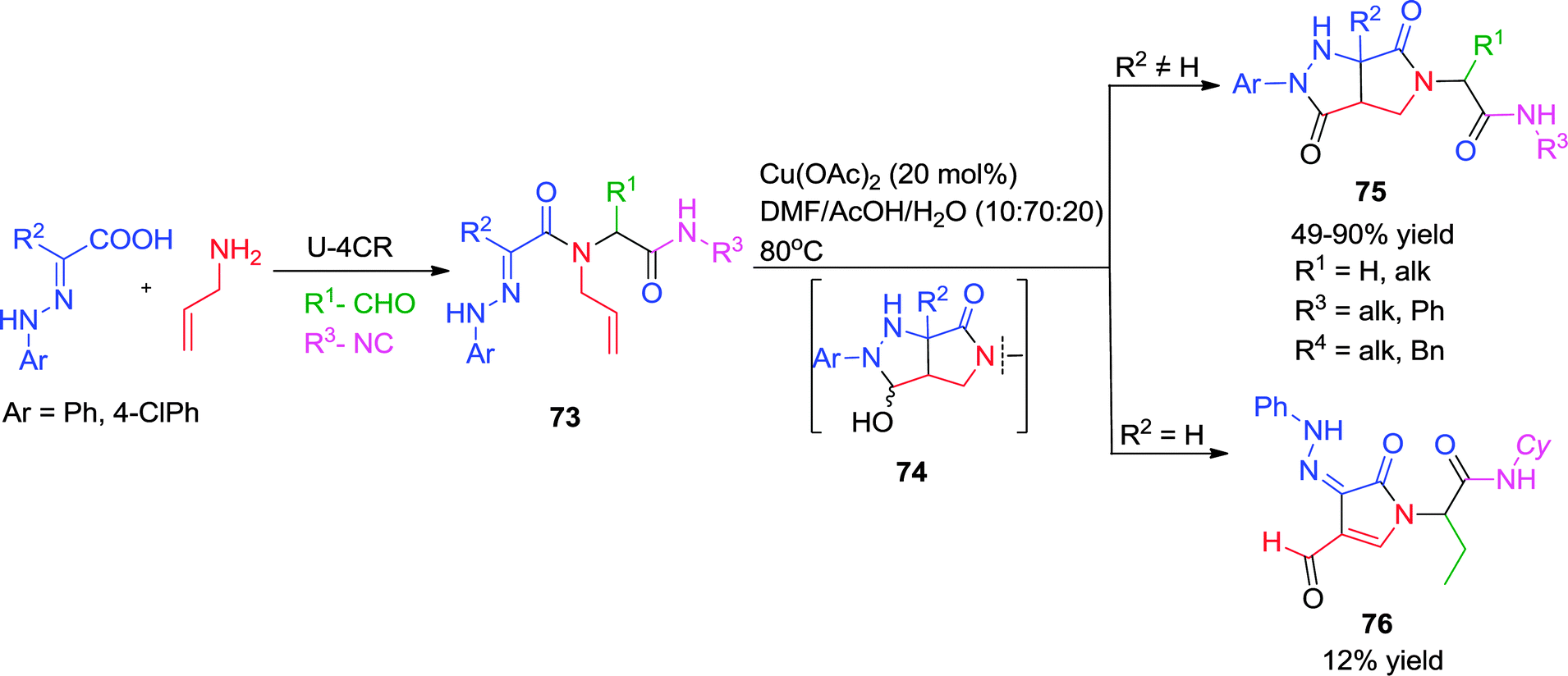 | ||
| Scheme 21 Novel access to fused pyrazolidinones via post-Ugi Cu(II)-catalyzed intramolecular [3+2] cycloaddition-oxidation cascade. | ||
Spiroindolines are privileged motifs frequently encountered within a large family of alkaloids that include communesines and perophoramidines, displaying pronounced cytotoxic and insecticidal activities.32–34 To access such alkaloids in a one pot cascade fashion and securing diversity is highly desirable from the viewpoint of sustainable chemistry. The first one-pot multicomponent attempt towards such complex alkaloids was reported by El Kaïm and co-workers32 whereby spirocyclization was achieved using a stoichiometric amount of Cu(II) salt in refluxing DBU–THF system. The reaction probably involves a peptidyl radical 78 generated from the enolate by a Cu(II)-triggered oxidation. A 5-exo-dig cyclization was followed by further oxidation of the α-aminoalkyl radical 79 to form an iminium cation, which was then trapped by the vicinal amide moiety (Scheme 22). The cyclization failed when an aliphatic substituent was present at the peptidic position (by use of an aliphatic aldehyde in the Ugi coupling). Electron-donating and electron-withdrawing aromatic substituents as well as hindered amides afforded the product in good yields.
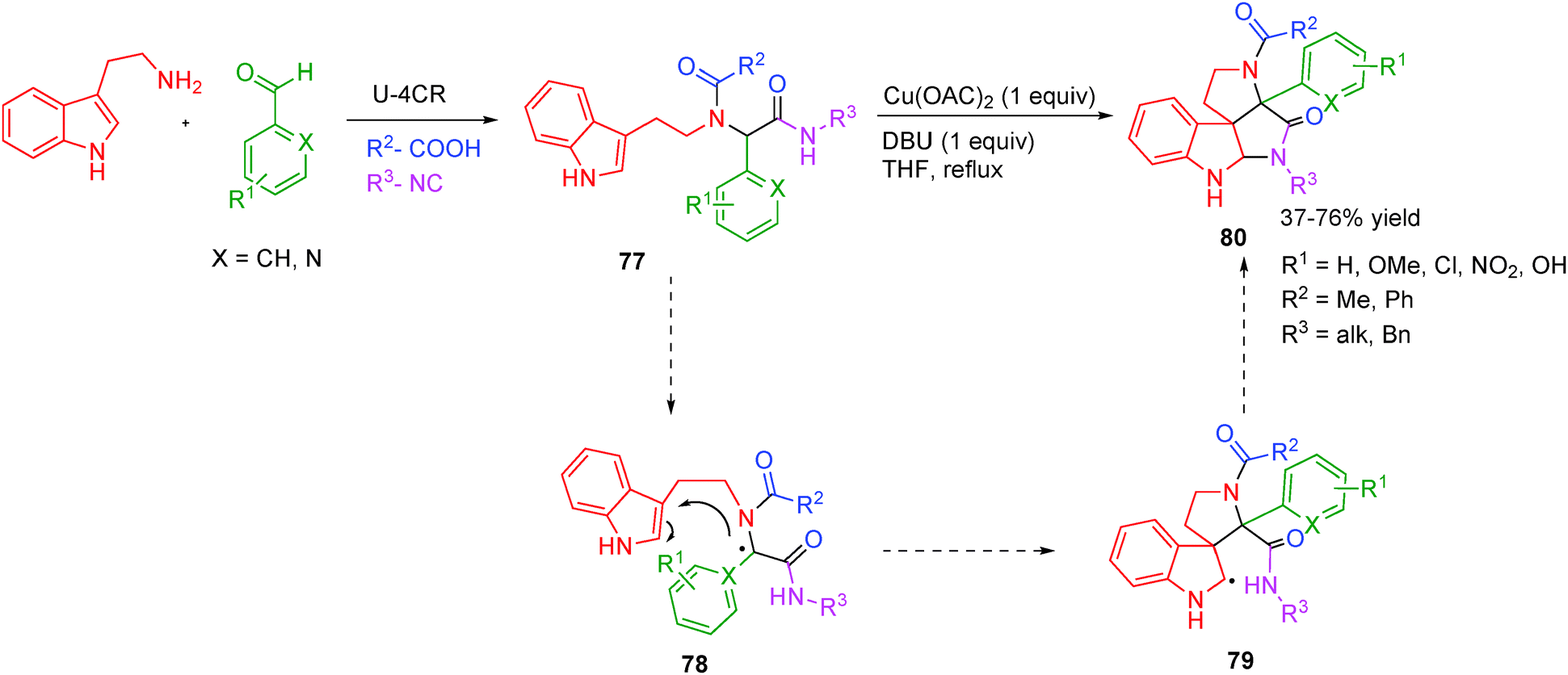 | ||
| Scheme 22 A post Ugi-4CR access to spiroindolines by Cu(II)-catalysis (only radical intermediates are shown). | ||
Subsequently, Van der Eycken and co-workers effectively exploited the catalytic activity of π-acidic gold salts involving triple bond transformations by disclosing a diversity-oriented approach for the synthesis of fused polycyclic spiroindolinones.33,34 The developed post-Ugi, Au(I)-catalyzed diastereoselective domino cyclization process involved a “branched-handed” pre-cyclization architecture resulting from the chiral center present in the Ugi adduct 81. The expected outcome of the reaction was an indoloazepinone through an endo-dig cyclization and rearrangement sequence. Instead an exo-dig cyclization followed by intramolecular trapping of the spiro intermediate occurred, resulting in the diastereoselective formation of a tetracyclic spiroindolinone 85 (Scheme 23).
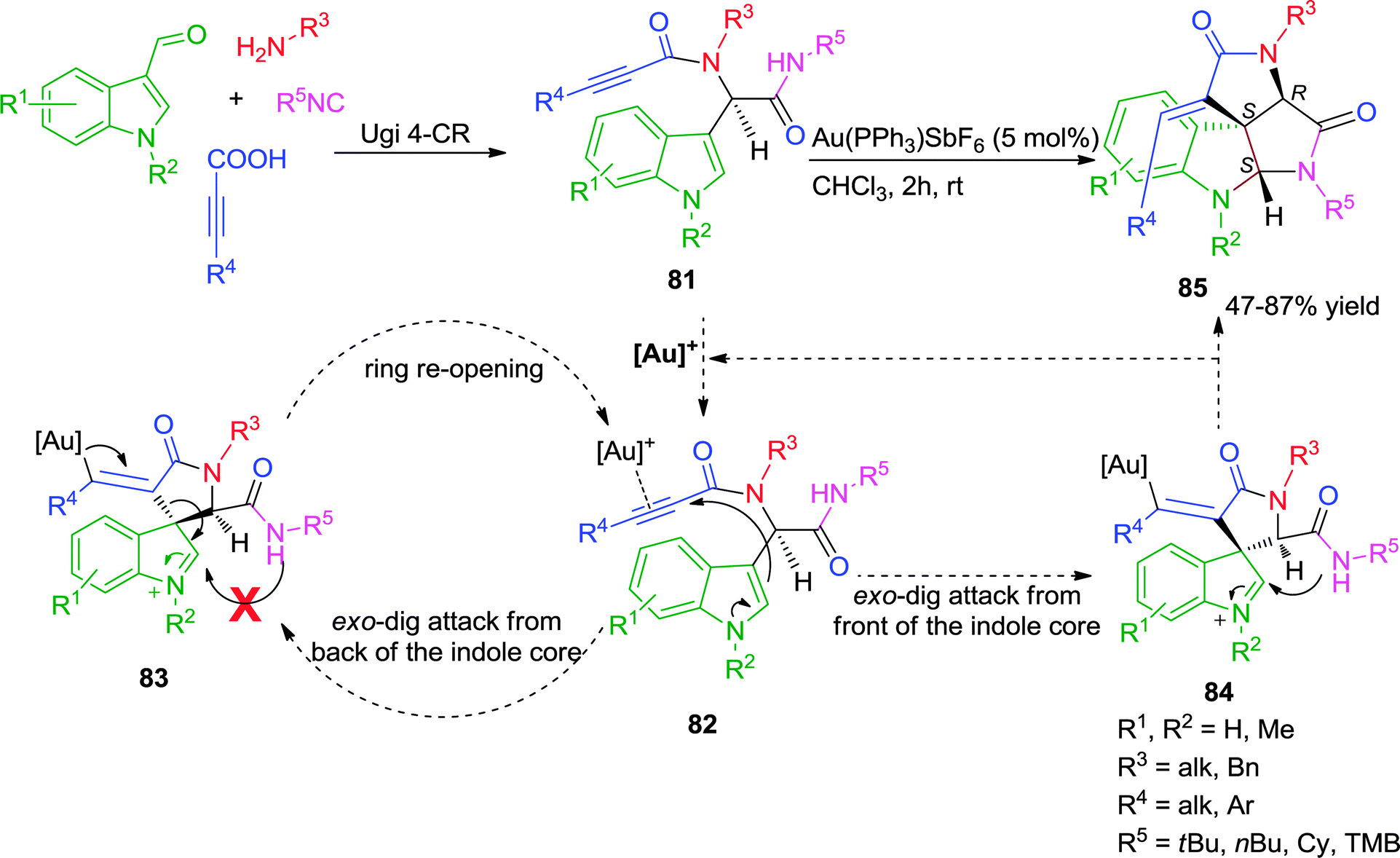 | ||
| Scheme 23 Synthesis of fused polycyclic spiroindolinones employing a Au(I)-catalyzed post-Ugi diastereoselective cascade. | ||
The observed diastereoselectivity of the reaction was explained by the mechanism which involves the activation of the triple bond of the Ugi-adduct 81 by cationic gold, followed by the nucleophilic attack (with two possible pathways) of the C-3 position of the indole. Attack from the back side of the indole core would result in intermediate 83. However trapping of the intermediate iminium ion in 83 by the secondary amide, is sterically impossible resulting in ring re-opening. In contrast, when the attack occurs from the front side of the indole core intermediate 84 is formed, and trapping of the iminium ion is possible giving rise to spiroindolinone 85, possessing two newly formed chiral centers (S,S). Thus, the chiral center already present in the Ugi adduct 81, directs this domino cyclization diastereoselectivity (Scheme 23). This study presents a rare example of attack on the α-position of an alkyne conjugated with an amide and trapping of the iminium intermediate by a sterically hindered tert-butyl amide.
Further, a dual σ–π activation by a Au(I)-catalysts has also been explored in a post-Ugi transformation by Van der Eycken and co-workers.35 Au(IPr)OTf efficiently catalyzed the regioselective cascade cyclization of N-propynylbutynamide 86via Csp3–H functionalization for the synthesis of (spiro)cyclopentapyridinones 89 (Scheme 24). Conversely, when the Ugi-adducts 86 bearing an n-butyl group (R2 = n-butyl) were subjected to the optimized reaction conditions, cyclopentapyridinones 89 were formed in moderate yields alongside the formation of pyrazinones. This anomaly could be explained by the cyclization of less sterically hindered n-butylamide onto the activated terminal alkyne. Mechanistically, the first step is the formation of Au-acetylide which upon π-activation of the butynamide, directs the 6-endo-dig-cyclization, resulting in the formation of the Au–vinylidene intermediate 88. This highly reactive species may undergo facile C–H insertion, which upon protodeauration forms the final product 89 (Scheme 24).
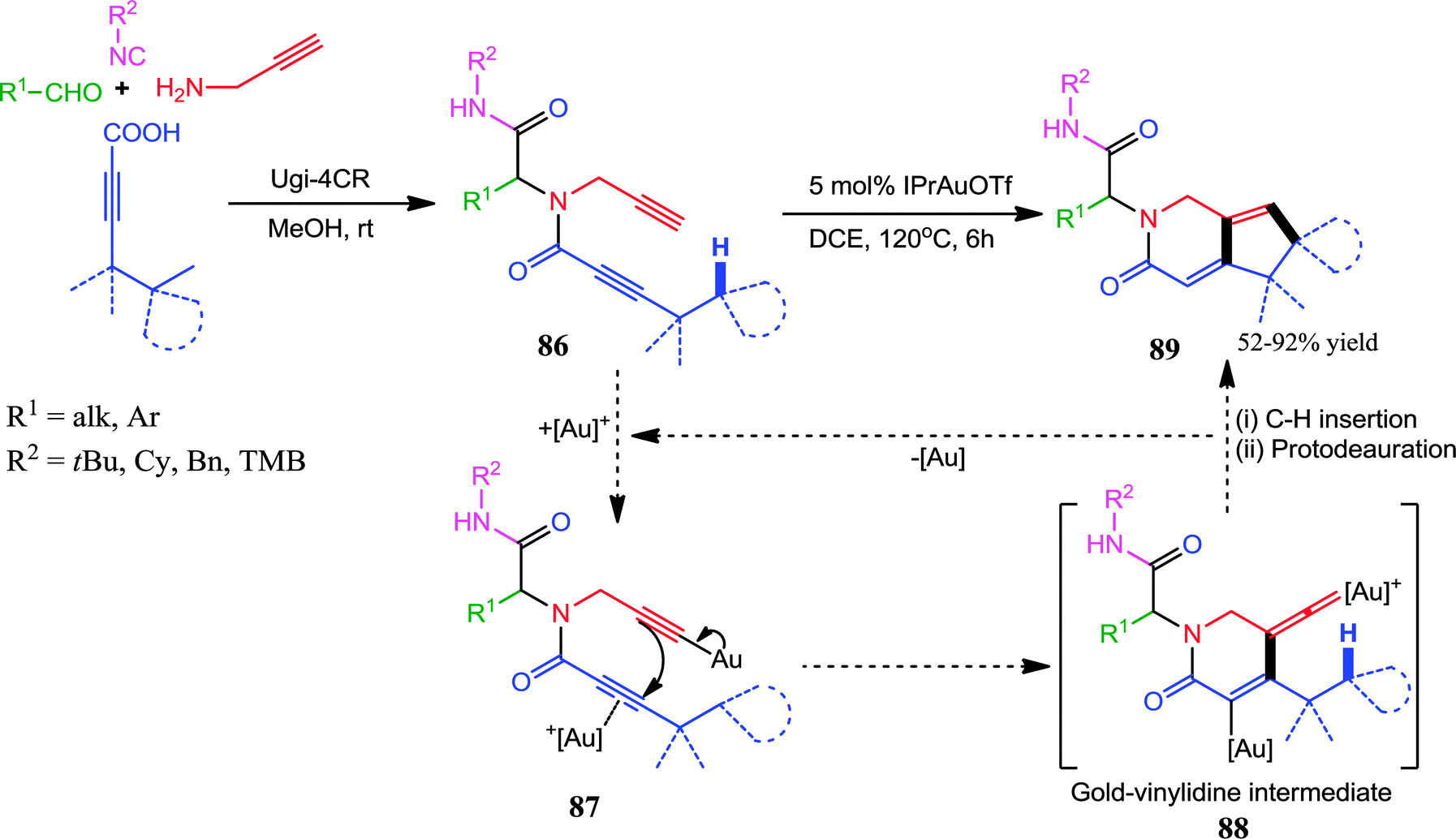 | ||
| Scheme 24 Au(I)-catalyzed regioselective cascade cyclization of N-propynylbutynamides via Csp3–H functionalization for the synthesis of (spiro)cyclopentapyridinones. | ||
Recently, the same group has disclosed a novel access to spiro[indoline-3,2′-pyrrole]-2,5′-diones via Pd-catalyzed post-Ugi cascade cyclization approach involving Buchwald–Hartwig/Michael reaction sequence (Scheme 25).36 The method allows the easy construction of a library of spirooxindoles 90 in moderate to good yields from Ugi adducts 91 generated from readily available precursors. The choice of ligand was critical as only Xantphos was found effective for the aforementioned domino transformation which emphasizes the importance of the flexible coordination environment of the ligand backbone to promote this domino sequence effectively. The protocol worked equally well when alkynoic acids were replaced with α,β-unsaturated acids (such as cinnamic acids or atropic acid) or 2-halobenzoic acids leading to diversely substituted spirooxindoles.
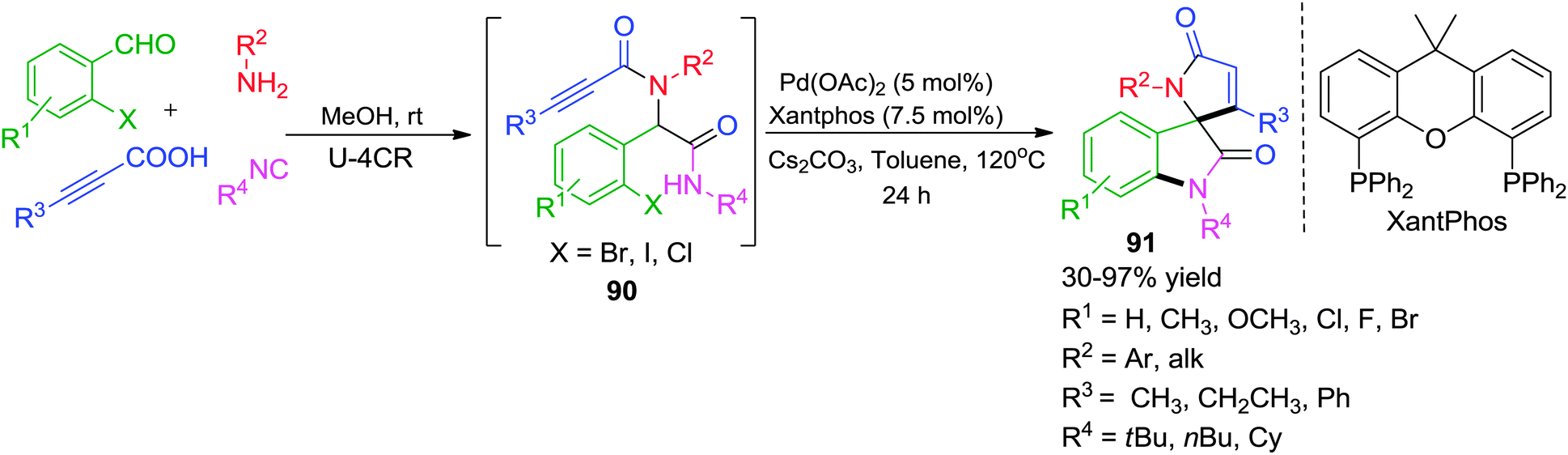 | ||
| Scheme 25 A facile access to functionalized spiro[indoline-3,2′-pyrrole]-2,5′-diones via post-Ugi domino Buchwald–Hartwig/Michael. | ||
In 2012, Chauhan and co-workers37 have described a microwave-assisted ligand-free Pd-catalyzed cascade reaction for the synthesis of isoquinolin-1(2H)-one derivatives. Ugi adduct 92 was reacted with 1.2 equiv. of isocyanide in the presence of 10 mol% of Pd(OAc)2 and Cs2CO3 in DMF at a ceiling temperature of 150 °C under microwave irradiation to give isoquinolinones in moderate to good yields (Scheme 26). To the best of our knowledge, this is the first report describing a cascade reaction involving isocyanide insertion with intramolecular cyclization, followed by a Mazurciewitcz–Ganesan type sequence, leading to the formation of 96. However, the reaction is highly specific as only tert-butyl isocyanide was effective to undergo insertion and cyclization reaction.
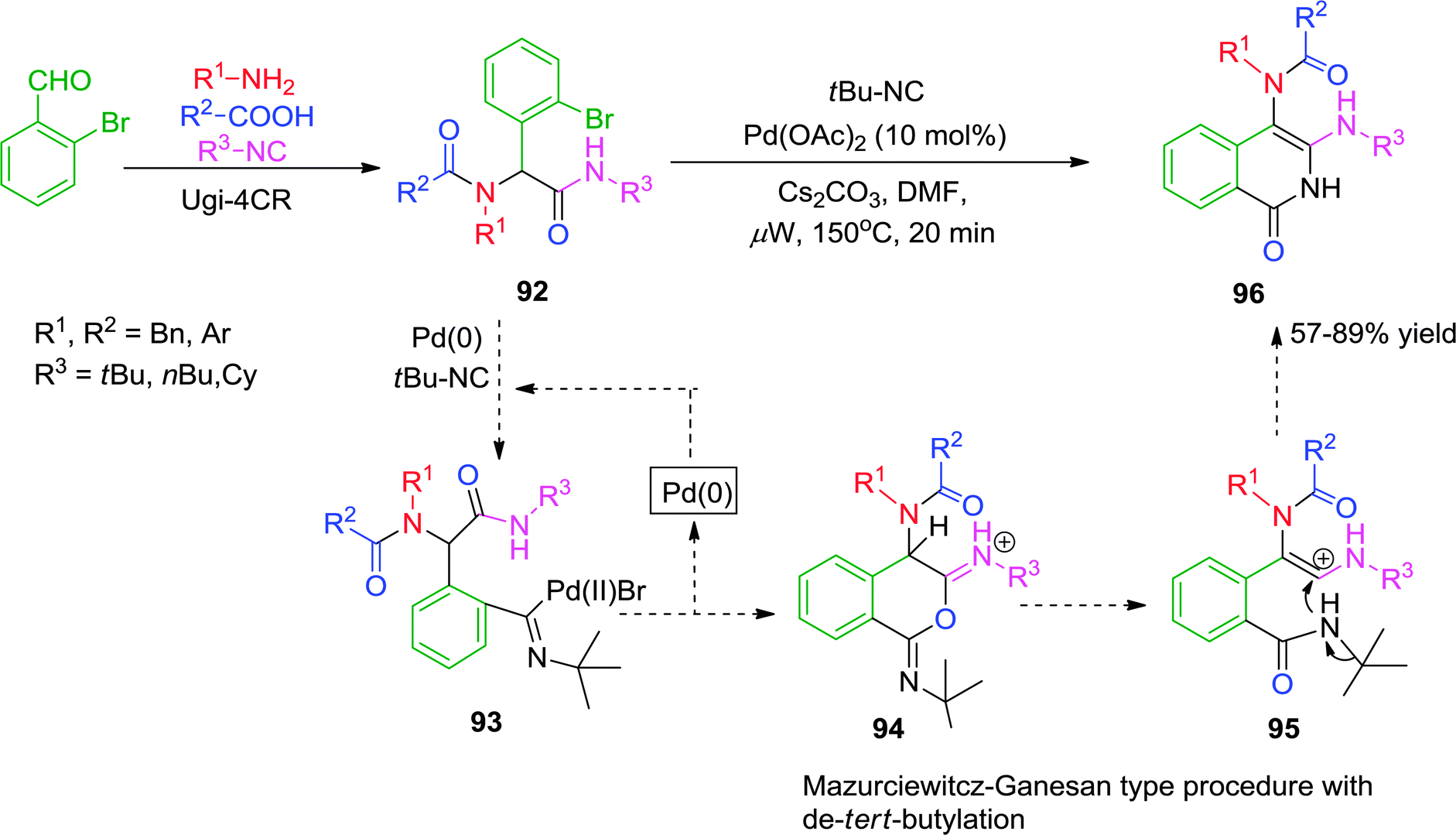 | ||
| Scheme 26 Synthesis of isoquinolin-1(2H)-one derivatives. | ||
5. Metal/ligand as regio-, chemo-selectivity switch
Despite the high efficiency of the hitherto discussed reaction sequences, in most of the processes the functionalized Ugi-adducts were designed for participation in a single chemical process. Consequently, each of the two-step sequences generally led to a single set of heterocyclic systems. The power of an Ugi-4CR/post-transformation strategy would be reinforced if the same Ugi-adduct could be diverged to different heterocyclic scaffolds by exploiting its multifunctionality using different metal catalysts or ligands.Towards this end, Van der Eycken and co-workers38 have reported a diversity-oriented regio-selective intramolecular hydroarylation for the synthesis of 6- and 7-membered pyrrole-fused heterocycles employing Au(I)- and Pt(II)-catalysis, respectively (Scheme 27). It is well known that transition metals can coordinate strongly with the π-electron cloud of alkynes. In this case, coordination of the metal with the alkyne in Ugi adduct 97 is followed by nucleophilic attack of the pyrrole. When Au(PPh3)OTf is used as catalyst, the nucleophilic attack of the pyrrole on the activated alkyne occurs in an exo-dig fashion followed by a 1,2-shift, which upon deprotonation and protodeauration forms pyrrolopyridinones 99. Alternatively, when PtCl2 is used as catalyst, the nucleophilic attack of the pyrrole on the activated alkyne occurs in an endo-dig fashion leading to the formation of pyrroloazepinones 98 (Scheme 27). As the cationic nature of Au activates the alkyne to a larger extent than PtCl2, significantly shorter reaction times were observed for exo-dig cyclization. The yields of the products were strongly influenced by steric factors as was evident from the fact p-methoxyphenyl substituted alkyne failed to undergo endo-dig cyclization. Also, tosyl-protected pyrrole was found unreactive towards cyclization. Subsequently, as an update of their work, the authors extended the above approach employing other heterocycles such as thiophene, benzothiophene and indole.39 However, with these heterocycles only 7-membered azepinones were obtained, as thiophene failed to undergo exo-dig cyclization with Au(I). Furthermore, Pt(II) was found ineffective to activate benzothiophene or indole due to their less nucleophilic character. The unexpected endo-dig cyclization by cationic Au catalyst leading to the formation of 7-membered azepinones was ascribed to its bulky nature.
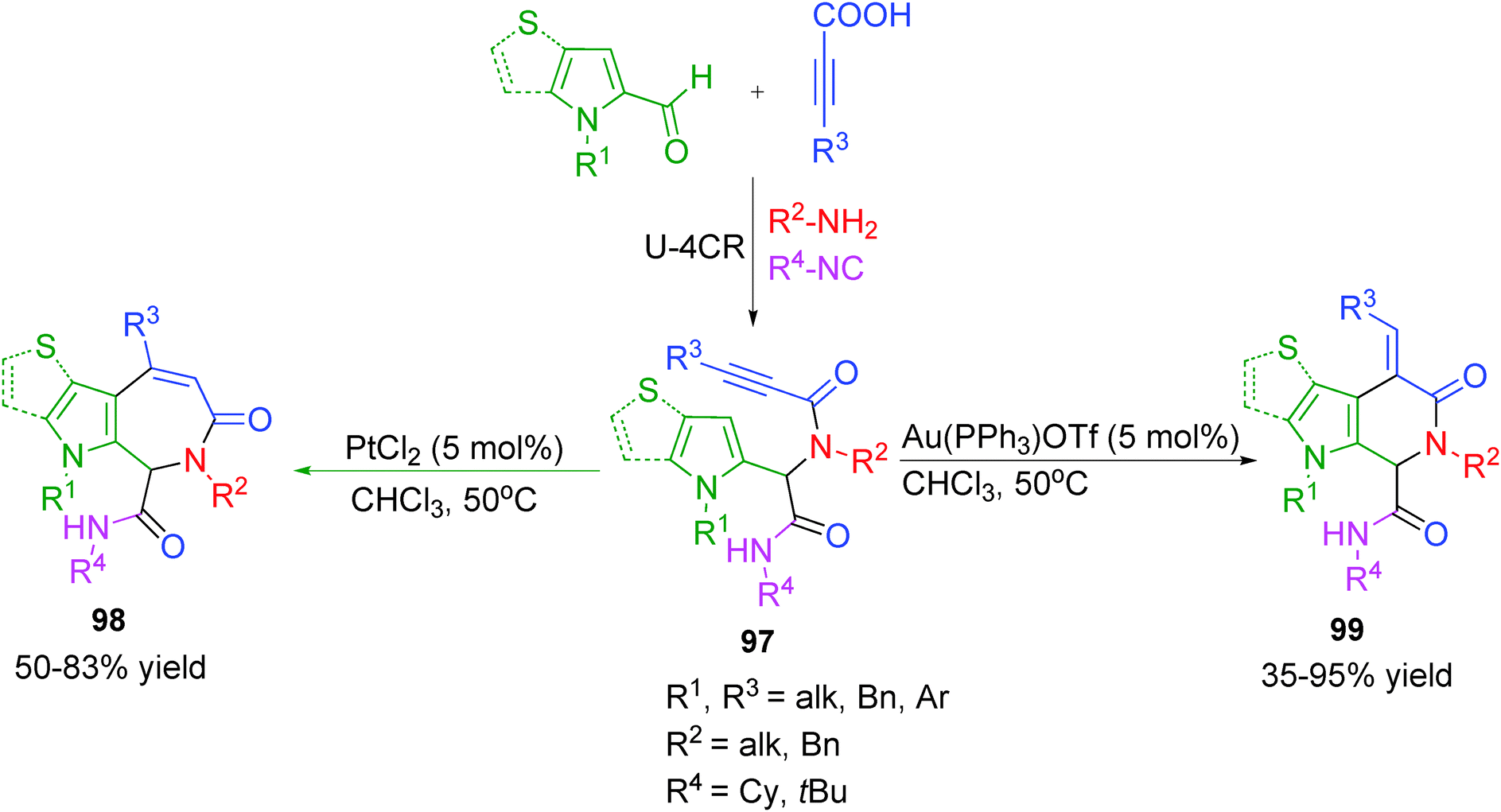 | ||
| Scheme 27 Regioselective synthesis of pyrrolopyridinones and pyrroloazepinones via Au/Pt-switch. | ||
Another notable report by the same group40 involves the regioselective hydroarylation approach for the synthesis of azepinoindolones 102 and azocinoindolones 101. Employing either an In(III)- or Au(I)-catalyst, the ring closure was directed towards an exo-dig or endo-dig cyclization respectively, resulting in the formation of 7- and 8-membered rings (Scheme 28). Various substituents on the amine, the alkyne (including the bulky phenyl group) and the indole were well tolerated. The one limitation of the methodology is the inability of N-tosyl indole to undergo endo-dig cyclization. Interestingly, the use of propargyl amines in Ugi reaction provided exclusively exo-dig cycloisomerized products under both catalytic systems.
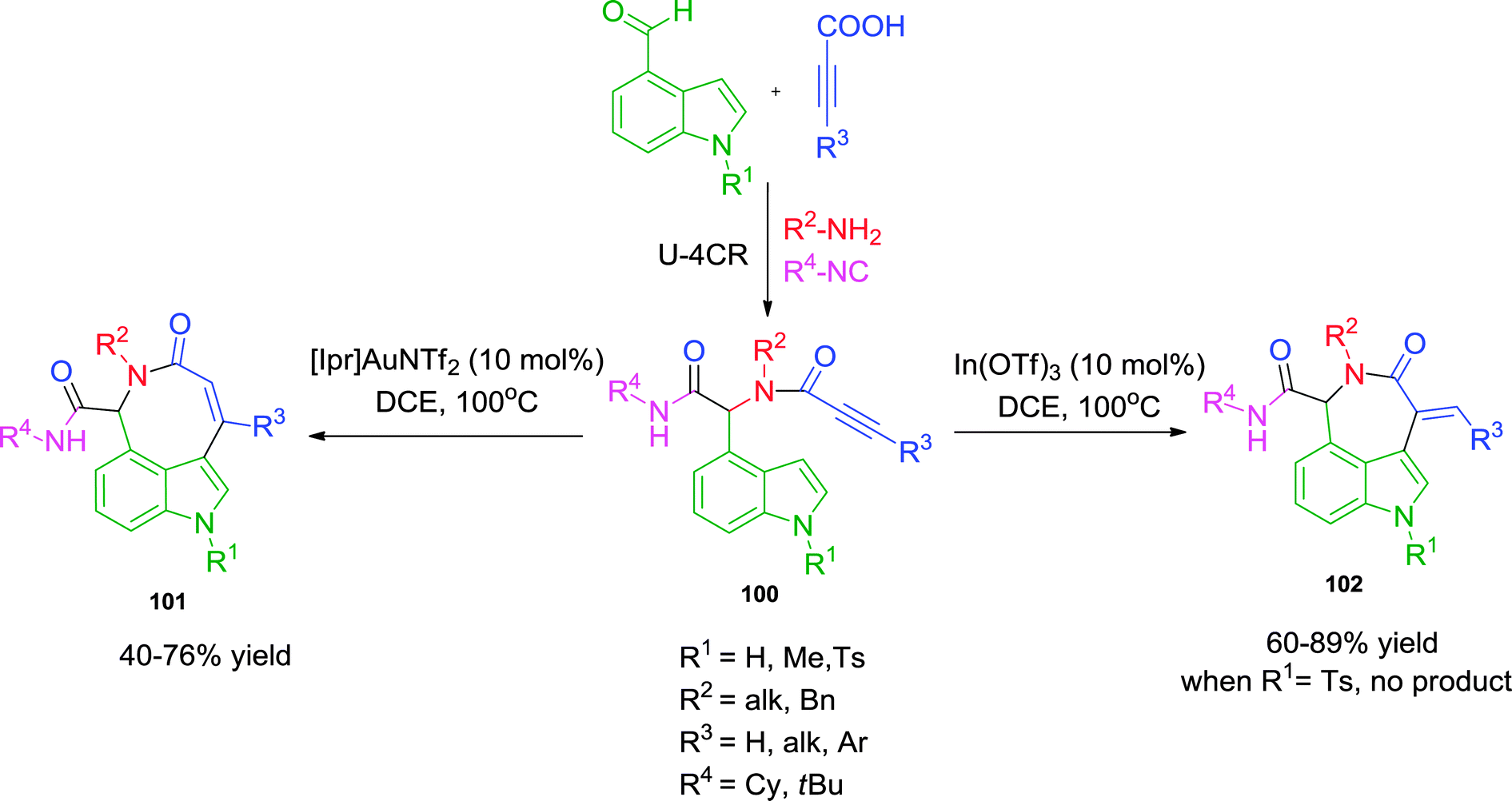 | ||
| Scheme 28 Regioselective synthesis of azepinoindolones and azocinoindolones. | ||
It is evident that an indole nucleus carries three possible reactive sites, with the potential for divergent synthesis. Exploiting the distinctive nucleophilicity and affinity of the various reactive sites in its Ugi adducts with an appropriate choice of the reaction conditions, would allow selective cyclization to form different heterocycles. In this context, Liu and co-workers41 have presented an interesting methodology for the synthesis of three distinct sets of indole based heterocycles under microwave irradiation. When substrate 103 was subjected to Cu(I)-catalysis using 5 mol% CuI in the presence of 10 mol% L-proline and K2CO3 in DMSO at ceiling temperature of 90 °C for 40 min, N1-arylation products 104 were obtained. With the Pd(OAc)2–PPh3 catalytic system in dioxane–MeCN at 110 °C, intramolecular C3-arylated products 106 were preferentially obtained (Scheme 29). Interestingly, treatment of 103 with the inorganic base Cs2CO3 exclusively provided the C–H arylation product 105. Notably, the reaction provides a mild, metal-free approach for the activation of sp3-hybridized C–H bond. A variety of electron-rich and electron-poor indole-2-carbaldehydes, iodobenzoic acids, amines and isonitriles participated in the coupling reactions to give good to excellent yields.
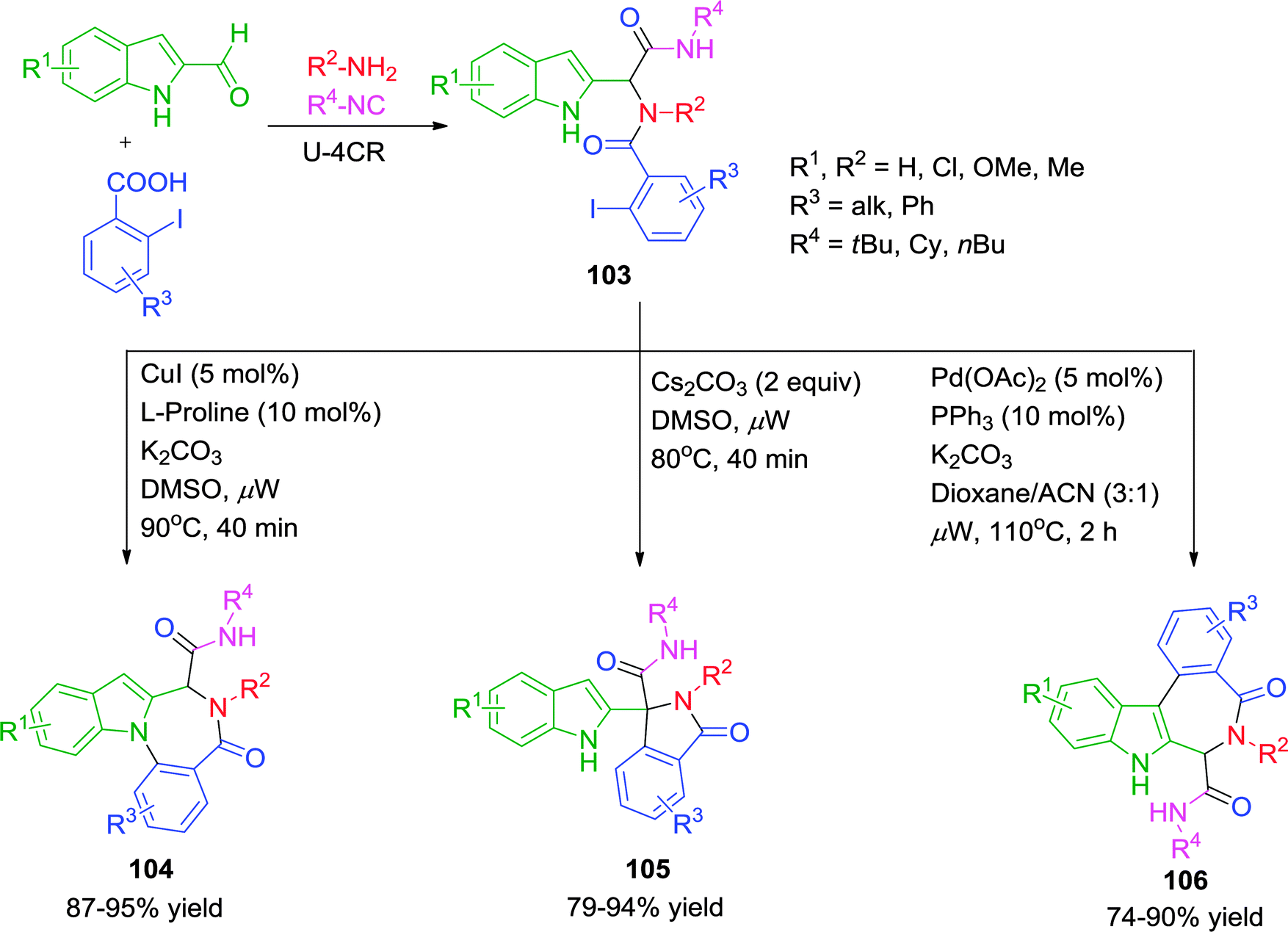 | ||
| Scheme 29 Synthesis of indole-based heterocycles employing metals as regioselectivity switch. | ||
In a further extension of their work,42 when o-iodoanilines were taken as pre-functionalized substrates in the Ugi adduct 107, a different set of heterocyclic compounds was obtained depending on the reaction conditions. 5,6-Dihydroindolo[1,2-a]quinoxalines 108 were synthesized via a Cu-catalyzed N-arylation pathway, whereas 6,7-dihydroindolo[2,3-c]quinolones 109 were formed by Pd-catalyzed C-arylation in good to excellent yields under microwave irradiation (Scheme 30). Besides high regioselectivity, these protocols are advantageous in terms of simple starting materials, broad substrate scope, operational simplicity and short reaction times.
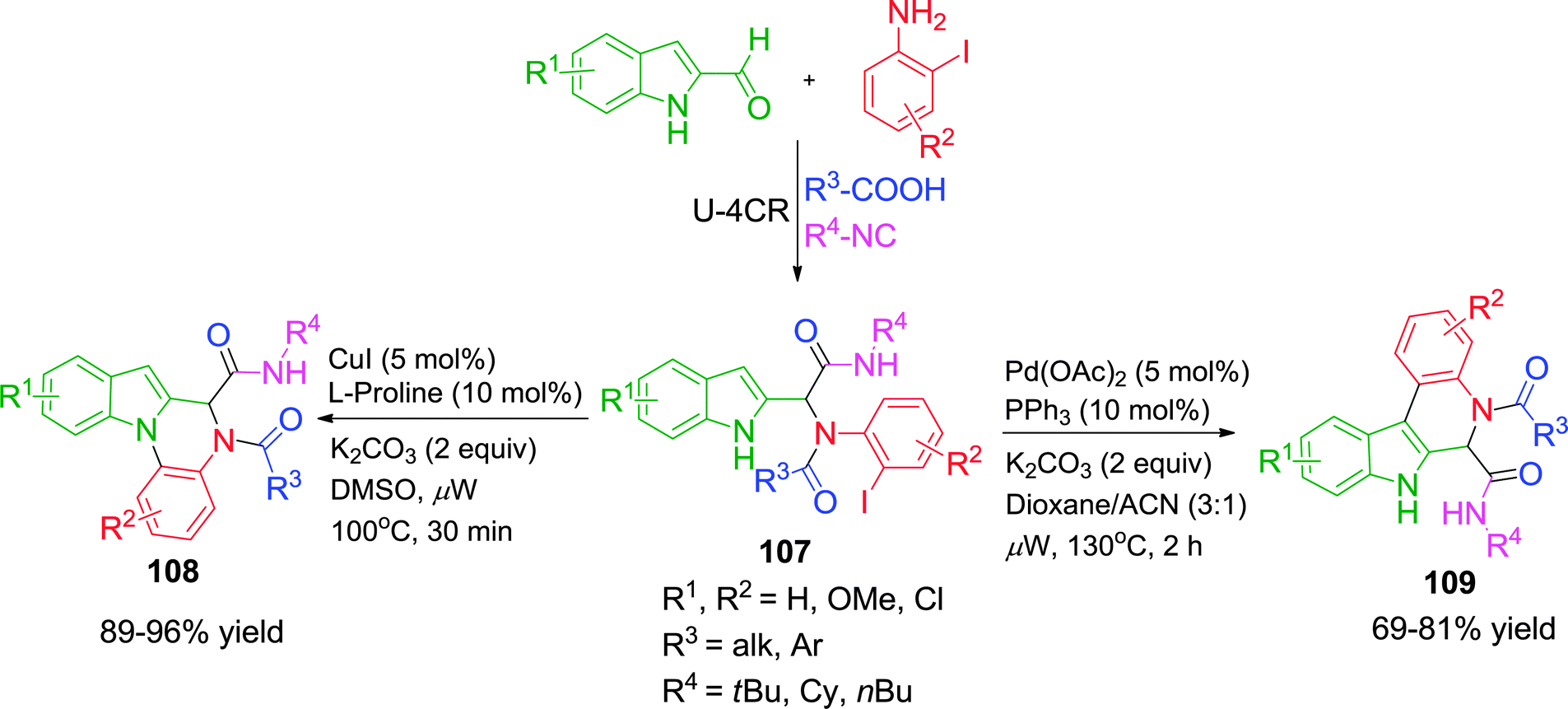 | ||
| Scheme 30 Regioselective synthesis of 5,6-dihydroindolo[1,2-a]quinoxalines and 6,7-dihy-droindolo[2,3-c]quinolones. | ||
Earlier in 2009, Zhu and co-workers have demonstrated the influence of ligands on the efficiency and outcome of the reaction in a Pd-catalyzed transformation with multifunctional substrates.43 Herein, linear amides 110 prepared by Ugi-4CR were converted to 3,4-dihydroquinoxalin-3-ones or 2-(2-oxoindolin-1-yl)acetamides via Pd-catalyzed intramolecular N-arylation and C-arylation processes. While microwave irradiation was important for the reaction efficiency, the choice of ligand determined the reaction pathway. Using monodentate XPhos as supporting ligand, cyclization of 110 afforded the 3,4-dihydroquinoxalin-3-ones 113, while in the presence of bidentate BINAP, under otherwise identical conditions, intramolecular C–H arylation of the tertiary amide occurred to furnish the oxindole 116 (Scheme 31).
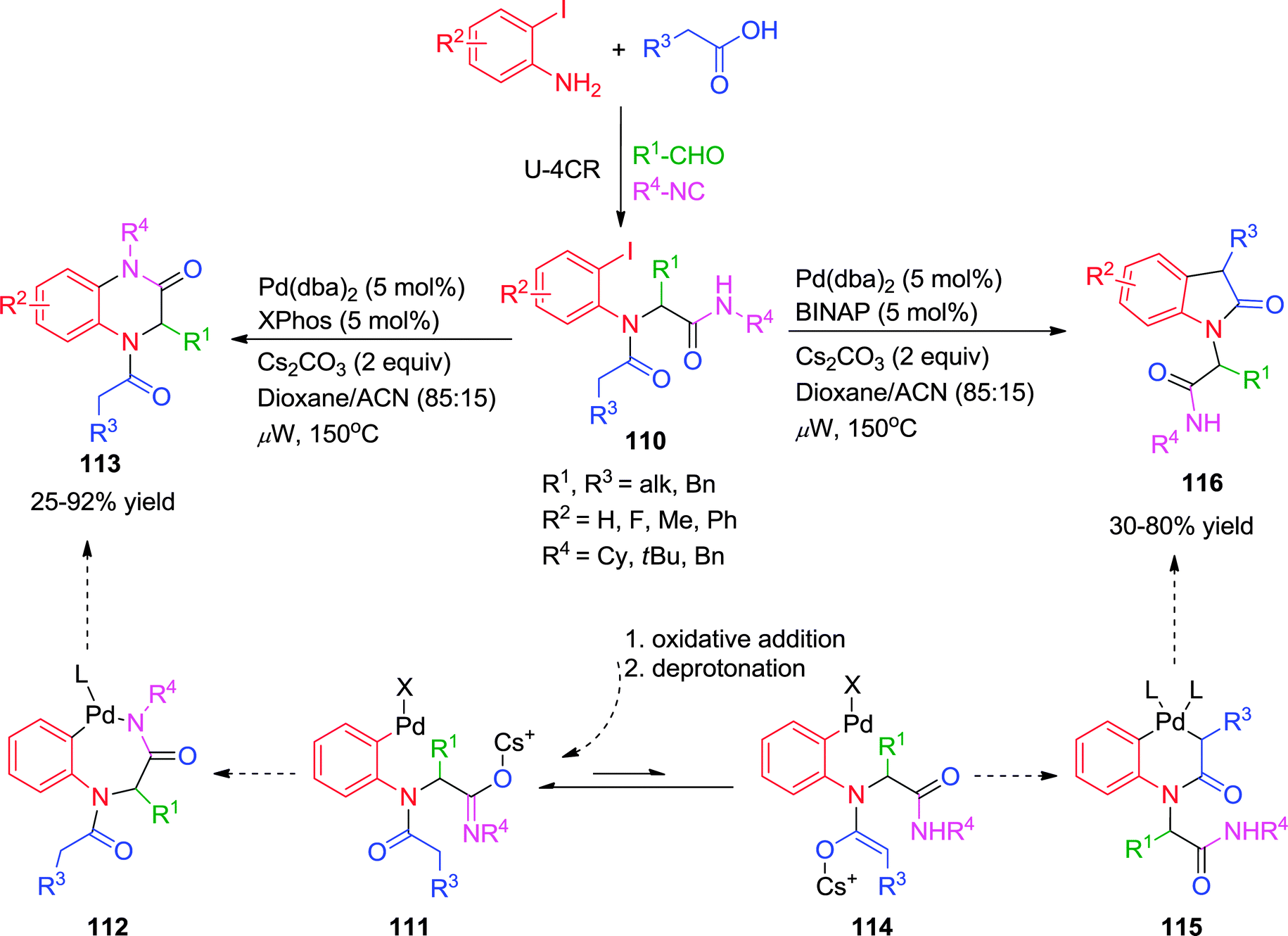 | ||
| Scheme 31 Regioselective synthesis of 5,6-dihydroindolo[1,2-a]quinoxalines and 6,7-dihy-droindolo[2,3-c]quinolones. | ||
Mechanistically, the oxidative addition of the aryl iodide to the Pd(0) species is a common step for both the N-arylation and the C-arylation reaction and thus has no consequence on the subsequent reaction pathways. With regard to the reductive elimination step of the N-arylation, it is known that this step is faster for Pd-complexes with bidentate ligands than for those with monodentate ligands.43 However, the reverse was happening is this case. The authors have hypothesized that the trans-metalation step determined the reaction outcome. So it was assumed that the N–H of the secondary amide would be deprotonated first in the presence of a weak base (Cs2CO3) leading to intermediate 111. When XPhos was used as a ligand, the availability of a vacant coordination site of the metal would facilitate the coordination of the amidate to Pd, thus accelerating the trans-metalation step from 111 to 112. Reductive elimination of 112 would then afford the quinoxalinone 113. On the other hand, with sterically hindered secondary amide (tert-butyl amide) in the presence of BINAP, the coordination of the secondary amide or the attack of the amidate on the tetra-coordinated Pd(II)-species 111 leading to 112 would be slowed down for steric reasons. Consequently, intramolecular proton shift from complex 111 could become competitive leading to the formation of enolate 114. Trans-metalation would subsequently take place to afford palladacycle 115, which upon reductive elimination would produce oxindole 116.
6. Transition metal-catalyzed carbocyclizations
Transition metal-catalyzed carbocyclizations have proven to be very powerful synthetic tools for the construction of carbocycles with various ring size. Coinage metal (Au, Ag, Cu) salts act as π-electrophilic Lewis acids and activate alkynes toward nucleophilic attack, allowing the formation of new C–C and C–heteroatom bonds. Recently, homogenous Au-catalyzed carbocyclization and heteroannulation strategies have become a particularly active research area. Apparently significant reports, especially those employing Au-complexes, for the diverse synthesis of heterocycles, will be discussed under this section. In one report, Van der Eycken and co-workers44 elaborated a diversity-oriented approach for the synthesis of imidazo-[1,4]diazepin-7-ones via a Au(PPh3)BF4-catalyzed heteroannulation approach. π-Coordination of the cationic Au with the alkyne in Ugi-adduct 117 generated intermediate 118 (Scheme 32). This is followed by nucleophilic attack of the imidazole to generate a 7-membered intermediate 119via endo-dig cyclization, which upon deprotonation and protodeauration forms the requisite imidazodiazepinone 120. A wide range of substituents on the alkyne, the isonitrile, the imidazole and the amine gave good to high yields. However, bulky substituents on the alkyne had a considerable effect on the reaction rate and required a two-fold catalyst loading for smooth conversion.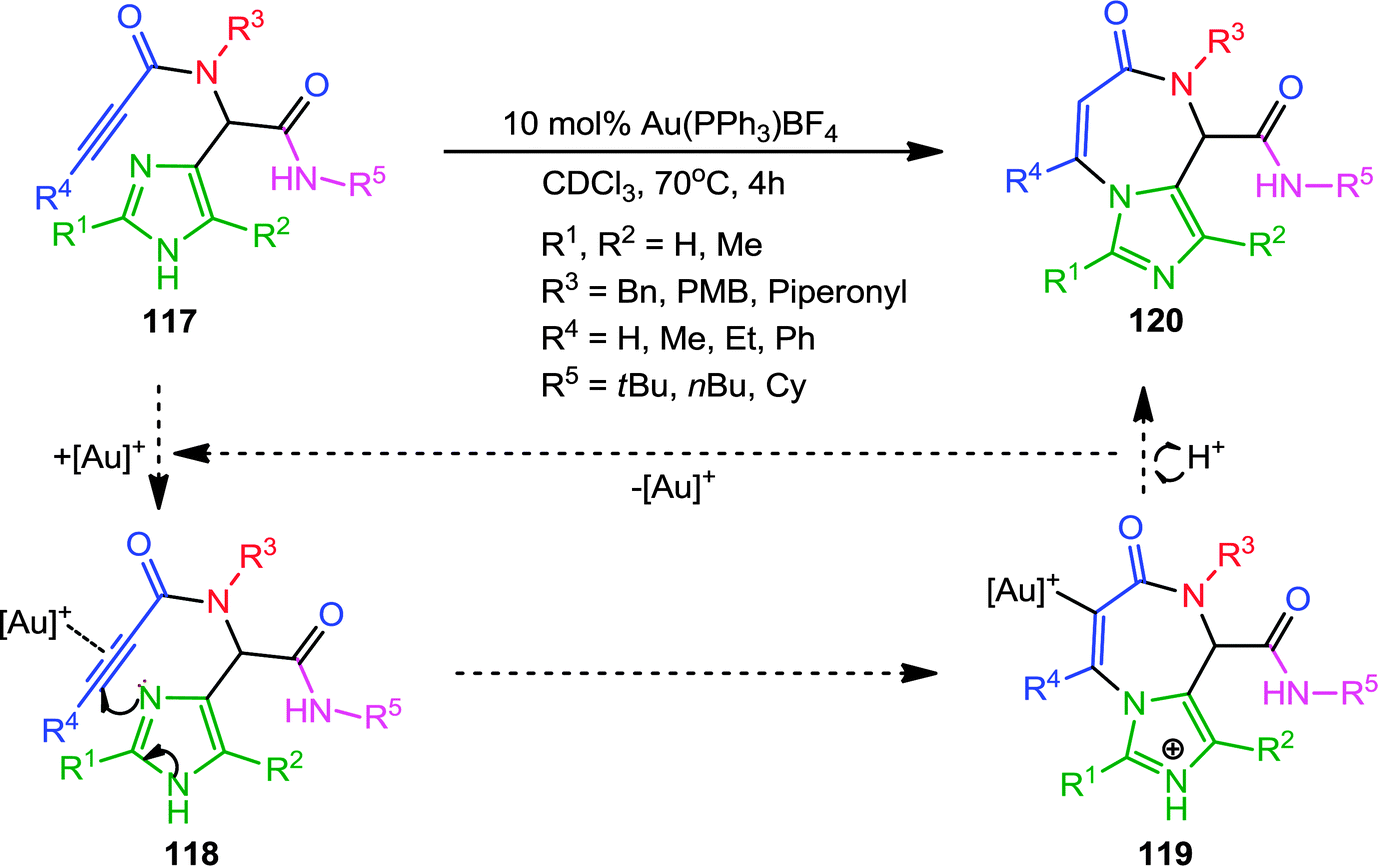 | ||
| Scheme 32 Synthesis of imidazo-[1,4]diazepin-7-ones via Au(I)-catalyzed heteroannulation. | ||
In 2012, the same group has presented a sequential Ugi-gold catalyzed carbocyclization approach for the diversification of the indole core.45 The protocol involved mild reaction conditions, good to excellent yields, 4-point diversity generated by Ugi-4CR and selective endo-dig cyclization using a cationic Au-complex. Mechanistically, the nucleophilic attack of the C-3 position of the indole core on the activated alkyne gives a spiro-intermediate, which upon 1,2-shift, deprotonation and protodeauration forms the final product 122 (Scheme 33). Interestingly, both diastereoisomers obtained from the Ugi reaction with L-tryptophan methylester were cyclized to afford the corresponding pure indoloazocines in good yields. However, due to steric hindrance bulky phenyl and tert-butyl substituents on the alkyne were found incompatible. Later, an improved version of this protocol was reported46 where Au(PPh3)SbF6 is used instead of the previously described Au(PPh3)OTf. This catalytic system is superior in terms of reaction yields, wide functional group compatibility including bulky substitution on alkynes (Scheme 33) with an exception for the tert-butyl group.
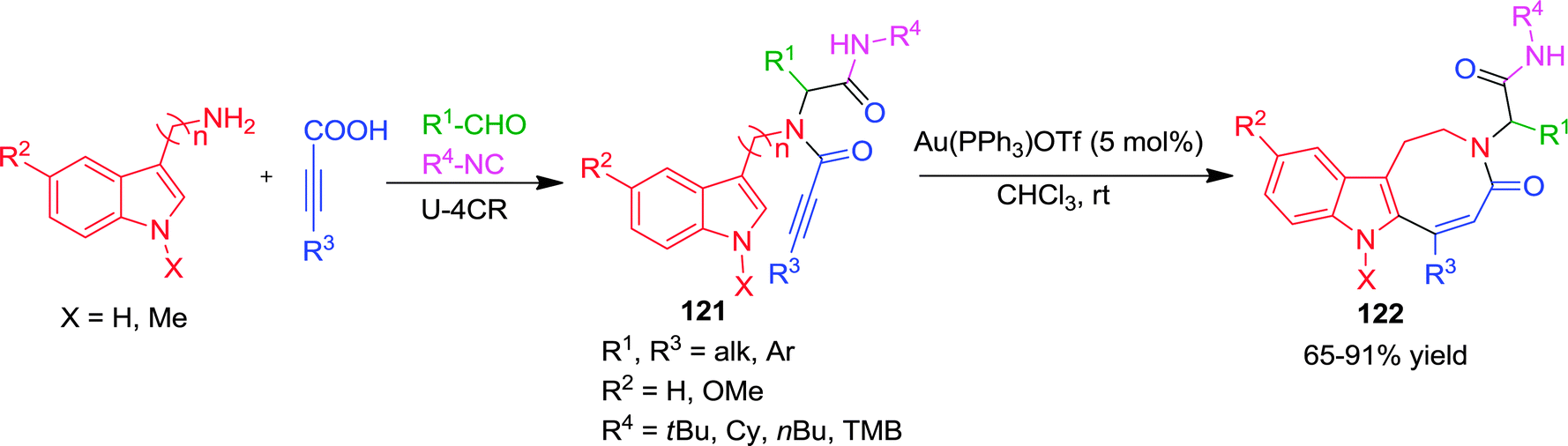 | ||
| Scheme 33 Synthesis of indoloazecinones via Au-catalysis. | ||
In 2013, Van der Eycken and co-workers47 have described a Au(I)-catalyzed intermolecular carbocyclization leading to the formation of pyrrolopyridines, pyridoindoles and azepinoindoles under mild reaction conditions. Ugi-adduct of the type 123 were effectively cyclized using low catalyst loading of 2 mol% AuCl in chloroform at room temperature without compromising the yields. Surprisingly, only terminal alkynes were effective towards the synthesis of pyrrolopyridines 124via a 6-exo-dig cyclization followed by isomerization of the double bond. When indole-2-carbaldehyde was used with internal alkynes, azepinoindoles 125 were formed via 7-endo-dig cyclization. The reaction was strongly dependent on the nucleophilicity of the heterocycle used, though no rationale for exo- and endo-selectivity has been discussed. Also, no product formation was observed with thiophene containing Ugi-adduct due to poisoning of the gold catalyst with sulfur. In another variant of this methodology,39 alkynoic acids were used as alkyne source for the Ugi reaction. Only seven membered ring formation was observed by endo-dig cyclization, resulting in the formation of azepinoindoles and azepinobenzothiophenes 127 using cationic Au(PPh3)OTf as catalyst (Scheme 34). Also, thiophene containing Ugi-adducts worked well under the optimized conditions to give the desired products in high yields.
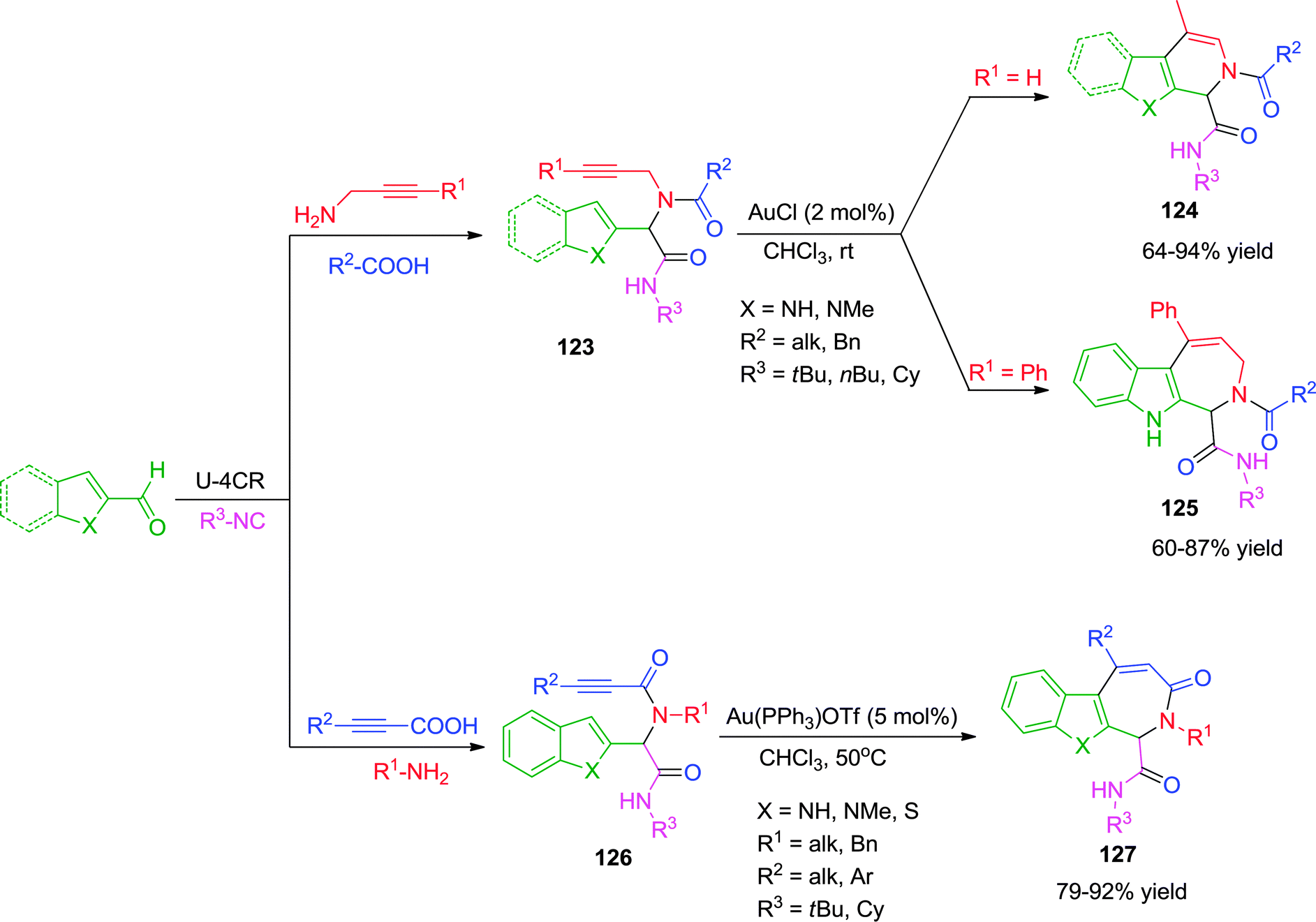 | ||
| Scheme 34 Synthesis of pyrrolopyridines, pyridoindoles, azepinoindoles and azepinobenzothiophenes using Au(I)-catalysis. | ||
Recently, Van der Eycken and co-workers have described an efficient approach for the construction of a fused 9-membered ring system (diazoninone framework) using a sequential Ugi/Au-catalyzed intramolecular carbocyclization (Scheme 35).48 It is well known that medium-sized rings are difficult to synthesize for enthalpic and entropic reasons. Here the authors have exploited the high nucleophilicity of the C-2 position of the pyrrole ring of Ugi-adduct 128. Using 10 mol% of Au(PPh3)OTf as catalyst in chloroform at 50 °C, nine-membered ring analogues of diazoninones were obtained in moderate to good yields. Mechanistically, nucleophilic attack on the Au(I)-activated alkyne occurred in an intramolecular endo-dig mode to generate the 9-membered ring 129. No product formation was observed with indole containing Ugi-adduct due to decomposition under the reaction conditions.
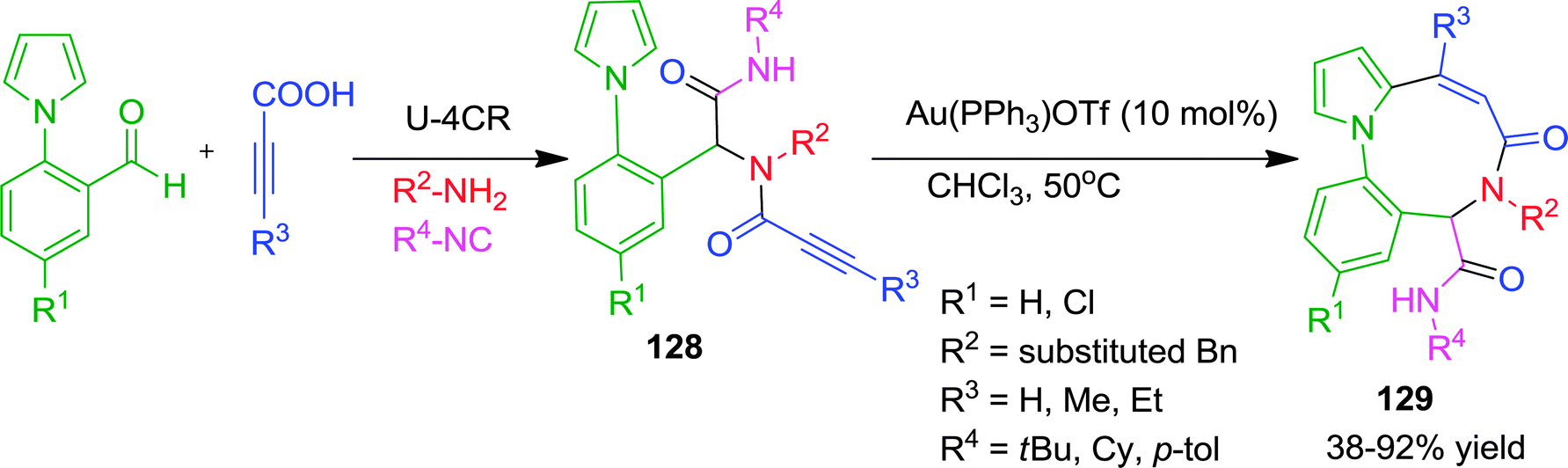 | ||
| Scheme 35 Synthesis of diversely substituted diazoninones via Au-catalyzed carbocyclization. | ||
Earlier in 2008, Riva and co-workers have reported a Pd(0)-catalyzed post-Ugi transformation via SN2′ cyclization to give functionalized N-acyl-2-vinylpyrolidines 131 in overall good yields but with moderate diastereoselectivity.49 A specially designed isocyanide for this type of carbocyclization was synthesized from the corresponding alcohol (Scheme 36).
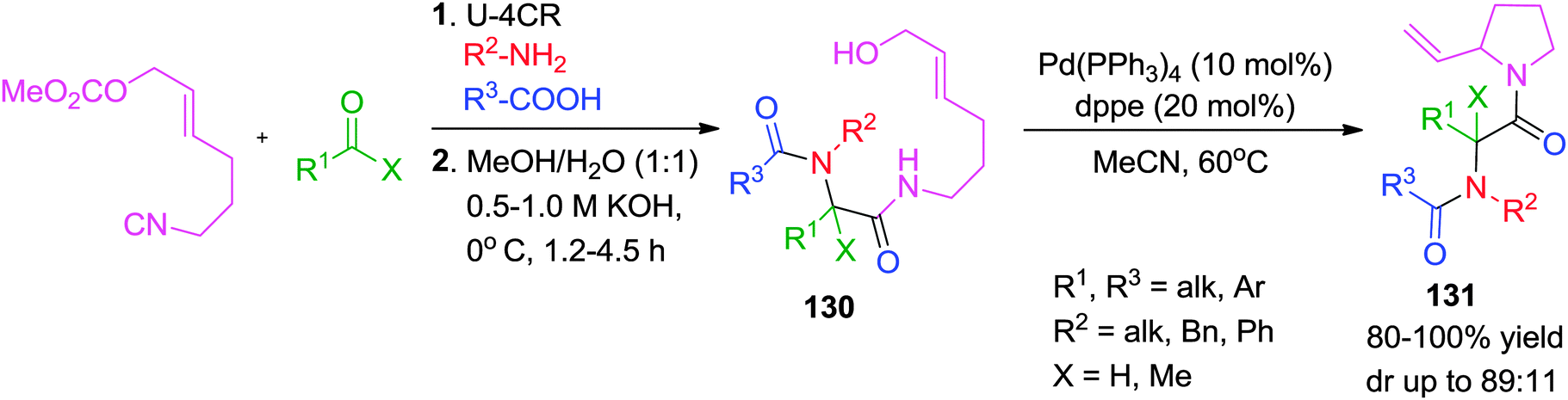 | ||
| Scheme 36 Synthesis of diversely substituted N-acyl-2-vinylpyrolidines. | ||
In a further extension of their work, the same group has reported a Pd-catalyzed synthesis of tricyclic N-heterocyclic compounds 134 from Ugi adduct 132via sequential SN2′ carbocyclization-Heck reaction in a one-pot, two-step manner.50 Using the previously developed Pd(PPh3)4-dppe catalytic system,49 the first cyclization involves a Pd(0)-mediated SN2′ cyclization followed by intramolecular Heck cyclization. The specially designed isocyanide49 has methyl carbonate protected allylic alcohol that acts as a leaving group in the SN2′ cyclization. Use of Cs2CO3 as base was found crucial for the reproducibility and completion of the reaction. Also, the Heck cyclization is highly dependent on the relative configuration and nature of the halogen atom of the starting haloarene. However, the authors were not able to perform the whole reaction as a one pot process because of the presence of excess base-which is necessary to facilitate the Heck reaction but which was found to be incompatible with the SN2′ cyclization (Scheme 37).
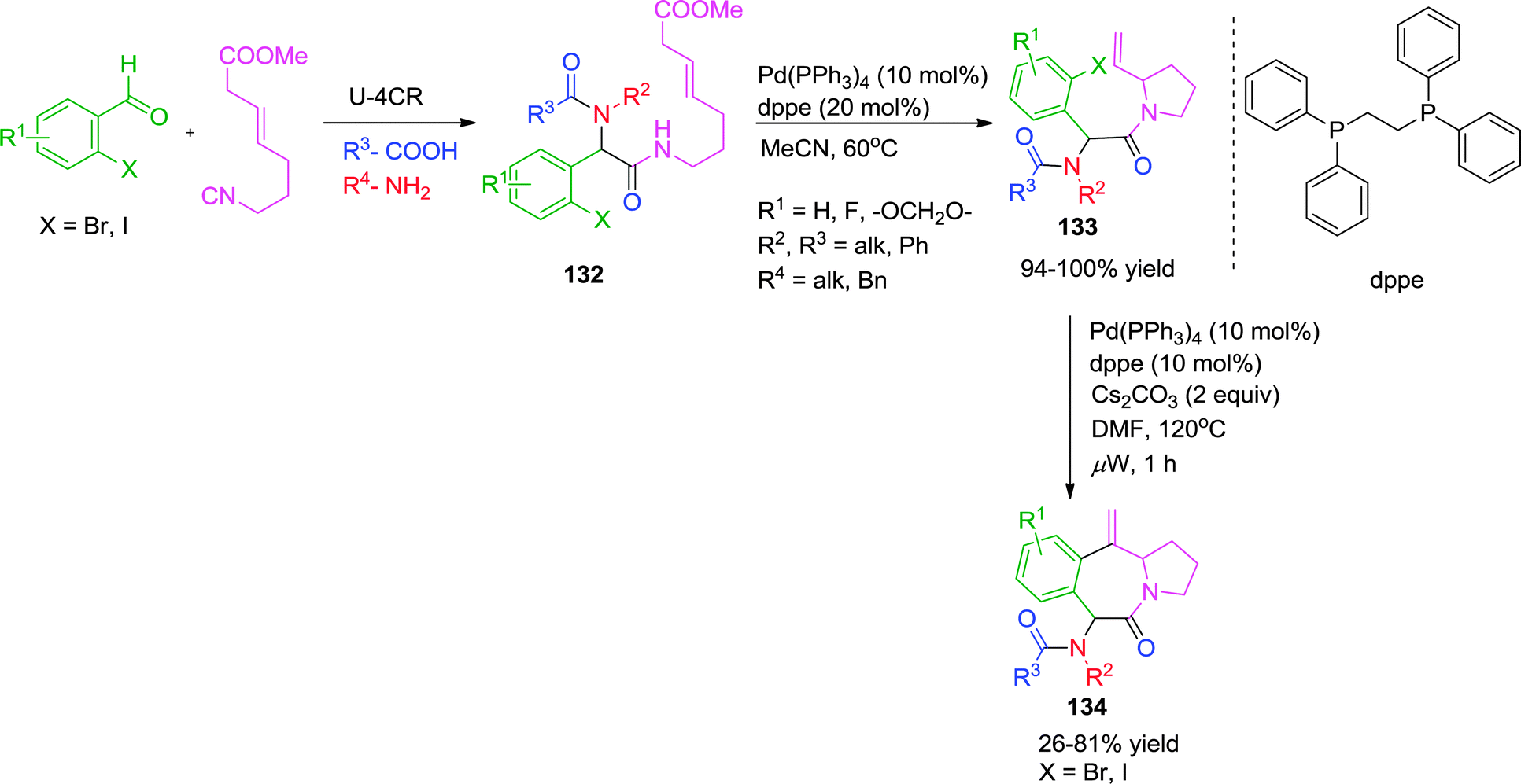 | ||
| Scheme 37 Pd-catalyzed one-pot two-step synthesis of tricyclic N-heterocycles via sequential SN2′ carbocyclization-Heck reaction. | ||
Conclusion
In conclusion, it has been amply demonstrated that transition metal-catalyzed transformations enable to rigidify linear Ugi-adducts into more drug like species via post-Ugi transformations. The high chemo-, regio- and stereo-selectivity obtained in many examples show the potential of this strategy and open a huge exploration field. So far, Pd-catalyzed heteroannulations have occupied the front seat with most efforts made towards post-Ugi–Heck transformations and arylation approaches. The development of novel transformations for generating synthetically challenging structures via tuning the catalytic system, the ligands and the reactivity of the substrates as well as the investigation of the mechanistic aspects, are challenges in this field. We anticipate that this tutorial review, which highlights and discusses the rise of transition-metals as an emerging field, would help researchers to better understand the chemistry behind metal-catalyzed post-Ugi transformations towards the synthesis of diverse heterocyclic scaffolds. Not to forget, such reactions have reached an impressive level of performance and versatility within just a few years, particularly with gold catalysis.References
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed.
- A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed and references cited therein.
- (a) I. Ugi, Angew. Chem., Int. Ed. Engl., 1962, 1, 8–20 CrossRef; (b) I. Ugi and C. Steinbruckner, Chem. Ber., 1961, 94, 734–742 CrossRef CAS.
- J. Zhu and H. Bienaymé, Multicomponent Reactions, Wiley-VCH, Weinheim, 2005 Search PubMed.
- L. Banfi, A. Basso and R. Riva, Top. Heterocycl. Chem., 2010, 23, 1–39 CAS.
- C. Hulme and J. Dietrich, Mol. Diversity, 2009, 13, 195–207 CrossRef CAS PubMed.
- G. Koopmanschap, E. Ruijter and R. V. A. Orru, Beilstein J. Org. Chem., 2014, 10, 544–598 CrossRef PubMed.
- V. Gracias, J. D. Moore and S. W. Djuric, Tetrahedron Lett., 2004, 45, 417–420 CrossRef CAS PubMed.
- Z. Xiang, T. Luo, K. Lu, J. Cui, X. Shi, R. Fathi, J. Chen and Z. Yang, Org. Lett., 2004, 6, 3155–3158 CrossRef CAS PubMed.
- M. Umkehrer, C. Kalinski, J. Kolb and C. Burdack, Tetrahedron Lett., 2006, 47, 2391–2393 CrossRef CAS PubMed.
- C. Kalinski, M. Umkehrer, J. Schmidt, G. Ross, J. Kolb, C. Burdack, W. Hiller and S. D. Hoffmann, Tetrahedron Lett., 2006, 47, 4683–4686 CrossRef CAS PubMed.
- W.-M. Dai, J. Shi and J. Wu, Synlett, 2008, 2716–2720 CrossRef CAS PubMed.
- R. Krelaus and B. Westermann, Tetrahedron Lett., 2004, 45, 5987–5990 CrossRef CAS PubMed.
- T. P. Ribelin, A. S. Judd, I. Akritopoulou-Zanze, R. F. Henry, J. L. Cross, D. N. Whittern and S. W. Djuric, Org. Lett., 2007, 9, 5119–5122 CrossRef CAS PubMed.
- L. Banfi, A. Basso, G. Damonte, F. De Pellegrini, A. Galatini, G. Guanti, I. Monfardini, R. Riva and C. Scapolla, Bioorg. Med. Chem. Lett., 2007, 17, 1341–1345 CrossRef CAS PubMed.
- M. Oikawa, S. Naito and M. Sasaki, Heterocycles, 2007, 73, 377–392 CrossRef CAS PubMed.
- U. Kazmaier, C. Hebach, A. Watzke, S. Maier, H. Mues and V. Huch, Org. Biomol. Chem., 2005, 3, 136–145 CAS.
- G. Cunny, M. Bois-Choussy and J. Zhu, J. Am. Chem. Soc., 2004, 126, 14475–14484 CrossRef PubMed.
- A. Salcedo, L. Neuville and J. Zhu, J. Org. Chem., 2008, 73, 3600–3603 CrossRef CAS PubMed.
- F. Bonnaterre, M. Bois-Choussy and J. Zhu, Org. Lett., 2006, 8, 4351–4354 CrossRef CAS PubMed.
- C. Kalinski, M. Umkehrer, G. Ross, J. Kolb, C. Burdack and W. Hiller, Tetrahedron Lett., 2006, 47, 3423–3426 CrossRef CAS PubMed.
- J. Ghosh, P. Biswas, S. Maiti, T. Sarkar, M. G. B. Drew and C. Bandyopadhyay, Tetrahedron Lett., 2013, 54, 2221–2225 CrossRef CAS PubMed.
- F. Bonnaterre, M. Bois-Choussy and J. Zhu, Beilstein J. Org. Chem., 2008, 4(10), 1–6 Search PubMed.
- Z. Ma, Z. Xiang, T. Luo, K. Luo, Z. Xu, J. Chen and Z. Yang, J. Comb. Chem., 2006, 8, 696–704 CrossRef CAS PubMed.
- Z. Li, L. Legras, A. Kumar, D. D. Vachhani, S. K. Sharma, V. S. Parmar and E. V. Van der Eycken, Tetrahedron Lett., 2014, 55, 2070–2074 CrossRef CAS PubMed.
- A. Salcedo, L. Neuville, C. Rondot, P. Retailleau and J. Zhu, Org. Lett., 2008, 10, 857–860 CrossRef CAS PubMed.
- R. A. De Silva, S. Santra and P. R. Andreana, Org. Lett., 2008, 10, 4541–4544 CrossRef CAS PubMed.
- M. Bararjanian, S. Hosseinzadeh, S. Balalaie, H. R. Bijanzadeh and E. Wolf, Tetrahedron Lett., 2011, 52, 3329–3332 CrossRef CAS PubMed.
- M. Bararjanian, S. Balalaie, F. Rominger, B. Movassagh and H. R. Bijanzadeh, J. Org. Chem., 2010, 75, 2806–2812 CrossRef CAS PubMed.
- D. D. Vachhani, A. Kumar, S. G. Modha, S. K. Sharma, V. S. Parmar and E. V. Van der Eycken, Eur. J. Org. Chem., 2013, 1223–1227 CrossRef CAS.
- A. D. Santos, L. El Kaïm, L. Grimaud and C. Ronsseray, Beilstein J. Org. Chem., 2011, 7, 1310–1314 CrossRef PubMed.
- L. El Kaïm, L. Grimaud, X.-F. Le Goff, M. Menes-Arzate and L. D. Miranda, Chem. Commun., 2011, 47, 8145–8147 RSC.
- S. G. Modha, A. Kumar, D. D. Vachhani, J. Jacobs, S. K. Sharma, V. S. Parmar, L. V. Meervelt and E. V. Van der Eycken, Angew. Chem., Int. Ed., 2012, 51, 9572–9575 CrossRef CAS PubMed.
- A. Kumar, D. D. Vachhani, S. G. Modha, S. K. Sharma, V. S. Parmar and E. V. Van der Eycken, Beilstein J. Org. Chem., 2013, 9, 2097–2102 CrossRef PubMed.
- D. D. Vachhani, M. Galli. J. Jacobs, L. V. Meervelt and E. V. Van der Eycken, Chem. Commun., 2013, 49, 7171–7173 RSC.
- N. Sharma, Z. Li, U. K. Sharma and E. V. Van der Eckyen, Org. Lett., 2014, 16, 3884–3887 CrossRef CAS PubMed.
- V. Tyagi, S. Khan, A. Giri, H. M. Gauniyal, B. Sridhar and P. M. S. Chauhan, Org. Lett., 2012, 14, 3126–3129 CrossRef CAS PubMed.
- S. G. Modha, A. Kumar, D. D. Vachhani, S. K. Sharma, V. S. Parmar and E. V. Van der Eycken, Chem. Commun., 2012, 48, 10916–10918 RSC.
- A. Kumar, D. D. Vachhani, S. G. Modha, S. K. Sharma, V. S. Parmar and E. V. Van der Eycken, Synthesis, 2013, 2571–2582 CAS.
- A. Kumar, Z. Li, S. K. Sharma, V. S. Parmar and E. V. Van der Eycken, Chem. Commun., 2013, 49, 6803–6805 RSC.
- L. Zhang, F. Zhao, M. Zheng, Y. Zhai, J. Wang and H. Liu, Eur. J. Org. Chem., 2013, 5710–5715 CrossRef CAS.
- L. Zhang, F. Zhao, M. Zheng, Y. Zhai and H. Liu, Chem. Commun., 2013, 49, 2894–2896 RSC.
- W. Erb, L. Neuville and J. Zhu, J. Org. Chem., 2009, 74, 3109–3115 CrossRef CAS PubMed.
- A. Kumar, Z. Li, S. K. Sharma, V. S. Parmar and E. V. Van der Eycken, Org. Lett., 2013, 15, 1874–1877 CrossRef CAS PubMed.
- S. G. Modha, D. D. Vachhani, J. Jacobs, L. V. Meervelt and E. V. Van der Eycken, Chem. Commun., 2012, 48, 6550–6552 RSC.
- D. D. Vachhani, A. Kumar, S. G. Modha, S. K. Sharma, V. S. Parmar and E. V. Van der Eycken, Synthesis, 2015 DOI:10.1055/s-0034-1379894.
- A. Kumar, D. D. Vachhani, S. G. Modha, S. K. Sharma, V. S. Parmar and E. V. Van der Eycken, Eur. J. Org. Chem., 2013, 2288–2292 CrossRef CAS.
- Z. Li, A. Kumar, D. D. Vachhani, S. K. Sharma, V. S. Parmar and E. V. Van der Eycken, Eur. J. Org. Chem., 2014, 2084–2091 CrossRef CAS.
- L. Banfi, A. Basso, V. Cerulli, G. Guanti and R. Riva, J. Org. Chem., 2008, 73, 1608–1611 CrossRef CAS PubMed.
- R. Riva, L. Banfi, A. Basso, V. Cerulli, G. Guanti and M. Pani, J. Org. Chem., 2010, 75, 5134–5143 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |