
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal catalysis in confined spaces
Stefan H. A. M.
Leenders
,
Rafael
Gramage-Doria
,
Bas
de Bruin
and
Joost N. H.
Reek
*
Homogeneous, Supramolecular and Bio-inspired Catalysis Group, Van 't Hoff Institute for Molecular Science (HIMS), University of Amsterdam (UvA), Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: J.N.H.Reek@uva.nl
First published on 23rd October 2014
Abstract
Transition metal catalysis plays an important role in both industry and in academia where selectivity, activity and stability are crucial parameters to control. Next to changing the structure of the ligand, introducing a confined space as a second coordination sphere around a metal catalyst has recently been shown to be a viable method to induce new selectivity and activity in transition metal catalysis. In this review we focus on supramolecular strategies to encapsulate transition metal complexes with the aim of controlling the selectivity via the second coordination sphere. As we will discuss, catalyst confinement can result in selective processes that are impossible or difficult to achieve by traditional methods. We will describe the template-ligand approach as well as the host–guest approach to arrive at such supramolecular systems and discuss how the performance of the catalyst is enhanced by confining it in a molecular container.
 Stefan H. A. M. Leenders | Stefan Leenders (1987) studied chemistry at the Radboud University Nijmegen and obtained his MSc degree (cum laude) in 2011. He is currently doing his PhD studies at the University of Amsterdam under the supervision of Prof. Joost Reek. His current research interests are the assembly of supramolecular systems and their applications in catalysis. |
 Rafael Gramage-Doria | Rafael Gramage-Doria (1985) received his PhD in 2012 from the University of Strasbourg under the guidance of Dr Dominique Matt and Prof. Dominique Armspach. His work in the area of metallocyclodextrin chemistry was recognized with the thesis prize from the University of Strasbourg. Then, he was awarded a NWO-Rubicon grant to pursue postdoctoral studies with Prof. Joost Reek at the University of Amsterdam to study supramolecular architectures in transition-metal catalysis and in 2014, he joined the group of Prof. Takashi Ooi at ITbM-Nagoya University focusing on ion-pair catalysis. In 2015, he will become CNRS Associate Researcher at the University of Rennes. |
 Bas de Bruin | Bas de Bruin (1971) did his PhD research at the RUN in Nijmegen (Prof. A. W. Gal, 1999). He did his postdoc at the MPI für Bio-Anorganische Chemie in Mülheim a/d Ruhr (Prof. K. Wieghardt), after which he returned to the RUN as an assistant professor. In 2005 he moved to the University of Amsterdam (UvA, The Netherlands), where he was promoted to an associate professor in 2008 and to a full professor in 2013. He is involved in various research activities, including radical organometallic chemistry, EPR spectroscopy, olefin oxygenation, polymer synthesis, mechanistic studies and computational catalysis, with a focus on the development of new (bio-inspired) catalytic transformations. |
 Joost N. H. Reek | Joost Reek (1967) did his PhD in Nijmegen with a thesis on supramolecular chemistry (Prof. Nolte). After a postdoctoral fellowship in Sydney, Australia with Prof. Crossley, he joined the group of Prof. van Leeuwen at the University of Amsterdam (UvA, The Netherlands) as an assistant professor (1998) were he started his research activities focusing on transition metal catalysis. He was promoted to an associate professor in 2003 and in 2006 he became a full professor. In 2009 he started a spin-off company (InCatT) to explore the commercial potential of new supramolecular strategies for combinatorial approaches in (asymmetric) catalysis. In 2013 he became the director of the van ‘t Hoff institute for molecular science. His current research activities include transition metal catalysis, supramolecular catalysis and catalysis for green energy applications. |
1. Introduction
Catalysis has grown to play a prominent role in science as it enables the preparation of chemicals and materials in an atom economical and efficient manner.1 As a consequence, far less waste is produced in catalytic processes compared to reactions that use stoichiometric reagents. In addition, new selective catalytic processes facilitate short-cuts in total synthesis, which again is favorable in terms of efficiencies. These benefits translate into overall more economical processes and as such, catalysis is well implemented in the bulk and fine chemical industry. Homogeneous transition metal catalysis has been well developed over the past decades and is nowadays applied in both the bulk and fine chemical industry. One of their advantages over heterogeneous catalysts is that the properties of transition metal complexes can be tuned by changing the ligands that are coordinated to the metal. This is especially important for reactions for which it is difficult to achieve (high) selectivities. However, despite substantial progress in the field, there are still many reactions for which the selectivity and activity cannot be controlled to a useful extent. Consequently, new tools that allow the control of the selectivity of a reaction are more than welcome. In that perspective, it is interesting to look at enzymes, nature's catalysts, which typically show a superb selectivity and reactivity. Their mode of function is complex, multiple and far from completely understood.2 However, by now we have sufficient knowledge on the working mechanisms of enzymes to formulate some general principles that may be translated into synthetic systems. One of the most obvious differences between enzymes and transition metal catalysts is their size. Enzymes are generally 50–100 times larger than metal complexes, and the large protein surrounding the active site often provides a well-defined confined space (second coordination sphere) around the active center. This can be mimicked with bio-inspired supramolecular chemistry. Indeed, the field of supramolecular chemistry has now evolved to such an extent that it becomes an accessible tool for the formation of synthetic ‘cages catalysts’. Encapsulating a metal complex in a supramolecular container within the confined space can impose steric restrictions on the catalyst-bound substrate to mimic the second coordination sphere effects of a protein matrix around the active site of a metallo-enzyme. This can result in reaction pathways that are different from those of the free catalyst, leading to remarkably enhanced selectivities of the caged catalysts compared to their non-encapsulated analogues.So far, research has been mostly focused on the development of new cage-like structures that have an interior that can be utilized for purely cage-catalyzed chemical transformations (i.e. a cage-shaped ‘organic catalyst’ without a catalytically active transition metal included in the cage cavity). A given substrate that is brought into the confined space of this ‘cavity’ experiences a series of ‘confinement effects’, creating a different environment around the substrate than in the bulk solvent. Often the encapsulated substrate molecule can only adapt specific conformations as it has to adjust to the size and shape of the cavity. This also limits its motion, and restricts the number of possible reaction pathways. Usually it also results in a reduced activation entropy of the reaction. The proximity and orientation of the reactive groups can be restricted, which affects the selectivity of a reaction (e.g. formation of low-entropy products, such as ring-compounds). In some cases, the substrate or the reaction intermediate is forced to adopt a high-energy (and low entropy) conformation leading to increased reactivity,3 which can effectively lower the free-energy reaction barrier, thus accelerating the reaction. Furthermore, the transition state of the effective reaction pathway can be stabilized by attractive interactions between the catalyst and the surrounding cavity, decreasing the overall energy barrier of the reaction. A number of elegant capsular catalysts that display unusual capsule-driven selectivity and/or enhanced activity in catalyzed reactions have been reported.4–6 For example, the Diels–Alder reactions within the octahedral coordination capsules, developed by Fujita and co-workers,6f,7 led to products that are not formed in the bulk, demonstrating the capsule-directed selectivity effect.8,9 Raymond and co-workers showed that the rate of the acid-catalyzed hydrolysis reaction of orthoesters is dramatically increased by the microenvironment of the cage, and occurs even in a basic reaction medium,10 demonstrating the potential of the strategy to enhance rates.
In the current review we focus on transition metal catalyzed transformations that take place in molecular containers. Similar to purely cage-catalyzed chemical transformations mediated by a cage-shaped ‘organic catalyst’, substrate pre-organization is expected to be of crucial relevance for such encapsulated transition metal catalysts. In addition, the confined space can have an effect on certain specific reaction steps that occur at the metal site. In many reactions the transition metal shuttles through various oxidation states, thereby changing their coordination environment and geometry. For example, in a typical palladium catalyzed cross coupling, the metal cycles via square planar Pd(II) and tetrahedral Pd(0). Constraints imposed by the second coordination sphere could lead to (de-)stabilization of either one of these states. In addition, substrate coordination to the metal, as well as the rotational freedom of the substrate could also be controlled by encapsulation, leading to new tools to control selectivity of a transition metal catalyzed reaction.
In Section 2 of this review we first show some examples involving a ligand and its metal complex covalently bound to the molecular container. These examples illustrate the benefits of having a capsule surrounding transition metal catalysts. Section 3 discusses the ligand-template approach, in which a ligand not only coordinates to the active metal center, but is also used as the template to form a cage-shaped second coordination sphere around the metal complex. The advantages of encapsulating metalloporphyrins in catalysis are described in Section 4, and examples of the host–guest approach in which a catalyst is confined in a preformed cavity-shaped container are discussed in Section 5. Finally, in Section 6, some supramolecular ways of binding catalysts in a protein environment to induce new enantioselectivity are addressed.
2. Metal encapsulation by covalent anchoring
Locating an active metal center in, or close to a molecular cavity can provide benefits in terms of activity and selectivity. First approaches in creating such systems dealt with covalent anchoring of metal complexes to synthetic receptors. The synthesis of such species is generally time-consuming and mostly limited to the molecular cavities that are readily available, such as resorcin[n]arenes, calix[n]arenes or cyclodextrins. There are some interesting examples that clearly show how the environment of a catalytic system can discriminate between different pathways occurring within the catalytic cycle. The group of Rebek, Jr. reported an example of a palladium complex attached to a cavitand, which is able to distinguish between different substrates in allylic alkylation (Fig. 1).11 In the presence of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two substrates that differ in size, the catalyst–cavitand system preferably forms one of the two corresponding palladium allyl species, which was proven by mass spectroscopy. This indicates that the capsular catalyst is already selective during the oxidative addition of the substrate, something that is not observed for the non-capsular catalyst studied in control reactions. Upon nucleophilic attack, the conventional palladium catalyst gives rise to a statistical mixture of products whereas the capsular catalyst produces the smallest product with a 9:1 selectivity.
:1 mixture of two substrates that differ in size, the catalyst–cavitand system preferably forms one of the two corresponding palladium allyl species, which was proven by mass spectroscopy. This indicates that the capsular catalyst is already selective during the oxidative addition of the substrate, something that is not observed for the non-capsular catalyst studied in control reactions. Upon nucleophilic attack, the conventional palladium catalyst gives rise to a statistical mixture of products whereas the capsular catalyst produces the smallest product with a 9:1 selectivity.
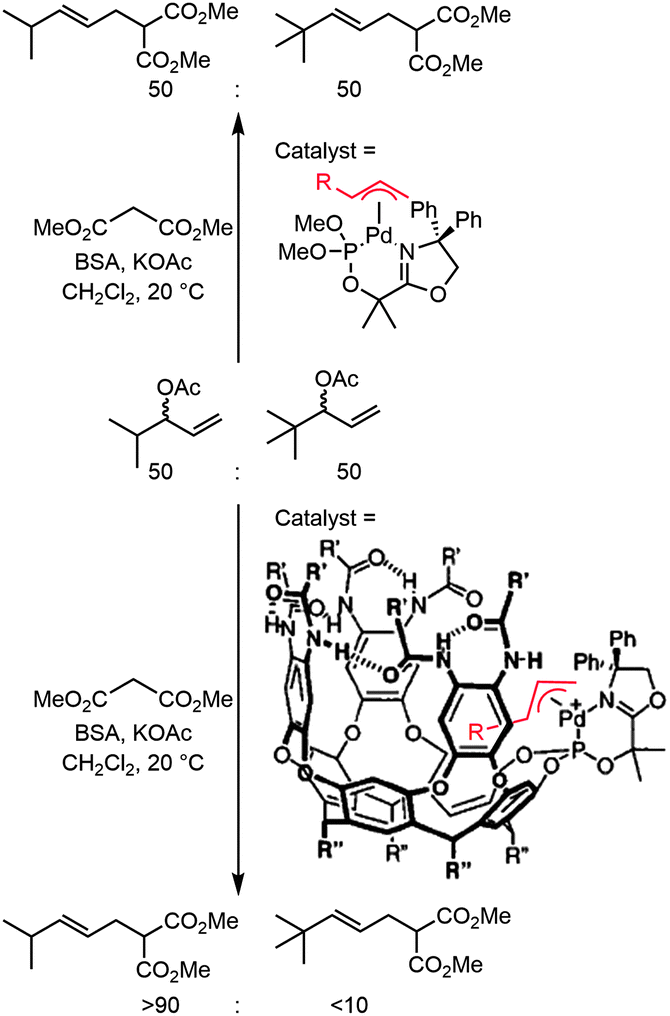 | ||
| Fig. 1 Nucleophilic attack on the Pd-allylic species yielded new substrate selectivity when a cavitand was used to create a second coordination sphere around the catalyst (BSA: N,O-bis(trimethylsilyl)acetamide). | ||
In a similar way, Sollogoub and co-workers nicely showed a gold–carbene catalyst in which the selectivity was controlled by the α- and β-cyclodextrin cavities attached to the catalyst (Fig. 2).12 Whereas the smaller α-cyclodextrin gold–carbene catalyst gives a 1:0.65 (1:2) ratio of both five-membered ring products, the bigger β-cyclodextrin analogue yielded the six-membered cyclic product as the major one (1:0:3.3, 1:2:3). Clearly, the selectivity of the reaction is controlled by the second coordination sphere that is surrounding the catalyst.
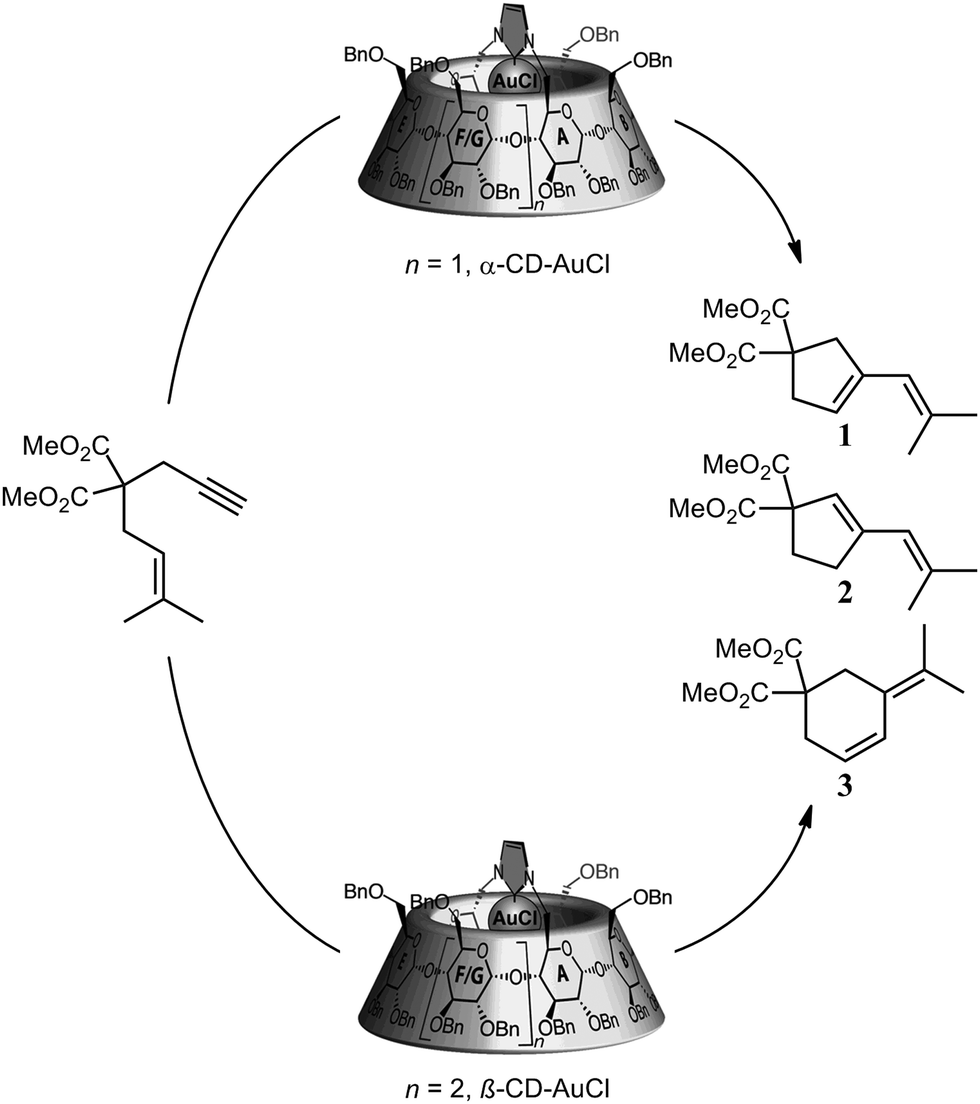 | ||
| Fig. 2 Product-selectivity controlled by the size of the cyclodextrin around the gold carbene catalyst. | ||
More recently, Matt and co-workers described the use of monophosphine–rhodium complexes embedded in α- and β-cyclodextrin cavities in the asymmetric hydroformylation of styrene (Fig. 3).13 The cavity-functionalized ligands are bulky, thus enforcing formation of encapsulated complexes that are only coordinated to a single phosphine ligand. The shape and bulk of the ligand prevents coordination of a second phosphine ligand to the rhodium center. Interestingly, both a high regio-selectivity (98%) and high enantiomeric excess (ee up to 95%) could be achieved with this capsular catalyst. The enantioselectivity is controlled by the chiral cyclodextrin environment around the rhodium complex. While a detailed insight is currently lacking, these examples clearly demonstrate that the covalent attachment of a cavity-shaped host to a metal complex can be effectively used to tune the selectivity of a catalytic reaction.
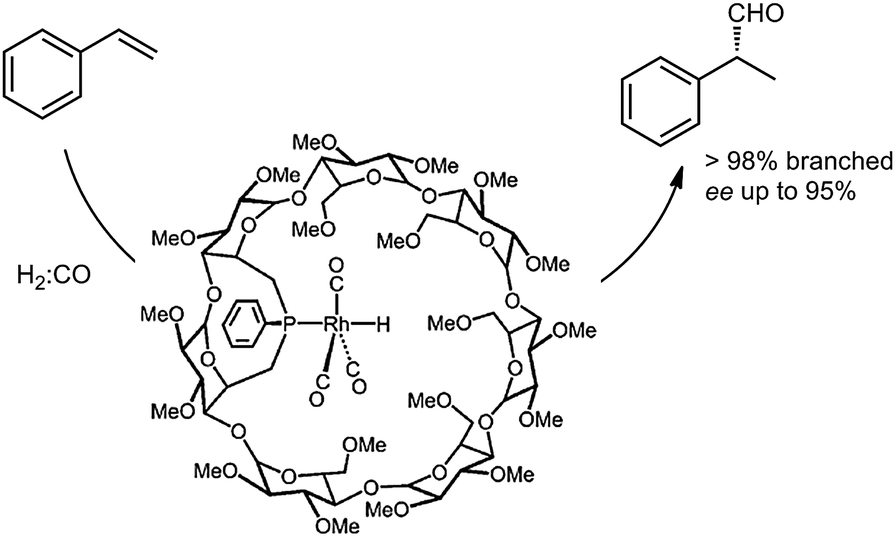 | ||
| Fig. 3 Monophosphine–rhodium complex confined in a cyclodextrin, applied in the asymmetric hydroformylation of styrene. | ||
As an alternative approach to the above described covalent attachment of cavities and catalysts, self-assembly can commendably be used to construct catalysts embedded in a host, thus providing a well-defined surrounding or a second coordination sphere. Examples of such a self-assembly approach for second sphere formation are described in the following sections.
3. Ligand-template approach for transition metal complex encapsulation
Reek and co-workers have introduced the ligand-template approach as a new strategy that leads to catalyst encapsulation. In this approach a ligand-template has a dual function: (1) it coordinates to the transition metal that is the active site, and (2) it functions as a template for the assembly of the cage around the active site.6i,14,15 The most successful example of a ligand-template is meta-tris-pyridylphosphine (4) that coordinates via its phosphorus atom to the catalytic active metal complex (e.g. [RhH(CO)3]) and via its pyridine groups to three zinc(II)-porphyrin building blocks (Fig. 4). As such, a narrow cavity is obtained via self-assembly, surrounding the catalytically active metal complex and enforcing mono-phosphorus coordination. With this approach in hand, different building blocks were studied to investigate how the size and shape of the generated cavity influence the reactivity and selectivity of the catalyst. If ligand 4 is used in combination with smaller building blocks such as zinc(II)salen, zinc(II)salphen and bis-(thiosemicarbazonato)zinc(II) complexes, the conformational flexibility is too large to enforce exclusive formation of well-defined encapsulated species.14c,e,f Also, if instead of the meta-tris-pyridylphosphine ligand (4) the para-analogue is used as the template ligand, bis-phosphorus coordinated complexes are formed as a result of the open structure of the assembly.14d The second coordination sphere formed via the coordination of zinc-porphyrins (e.g.5) leads to the proper cavity, allowing fine tuning via the phenyl groups of the porphyrin. The application of zinc-phthalocyanines results in spacious cages, imposing little restriction on the active site in the cavity. This ligand-template strategy has been mainly exploited in the rhodium-catalyzed hydroformylation reaction of non-substituted alkenes.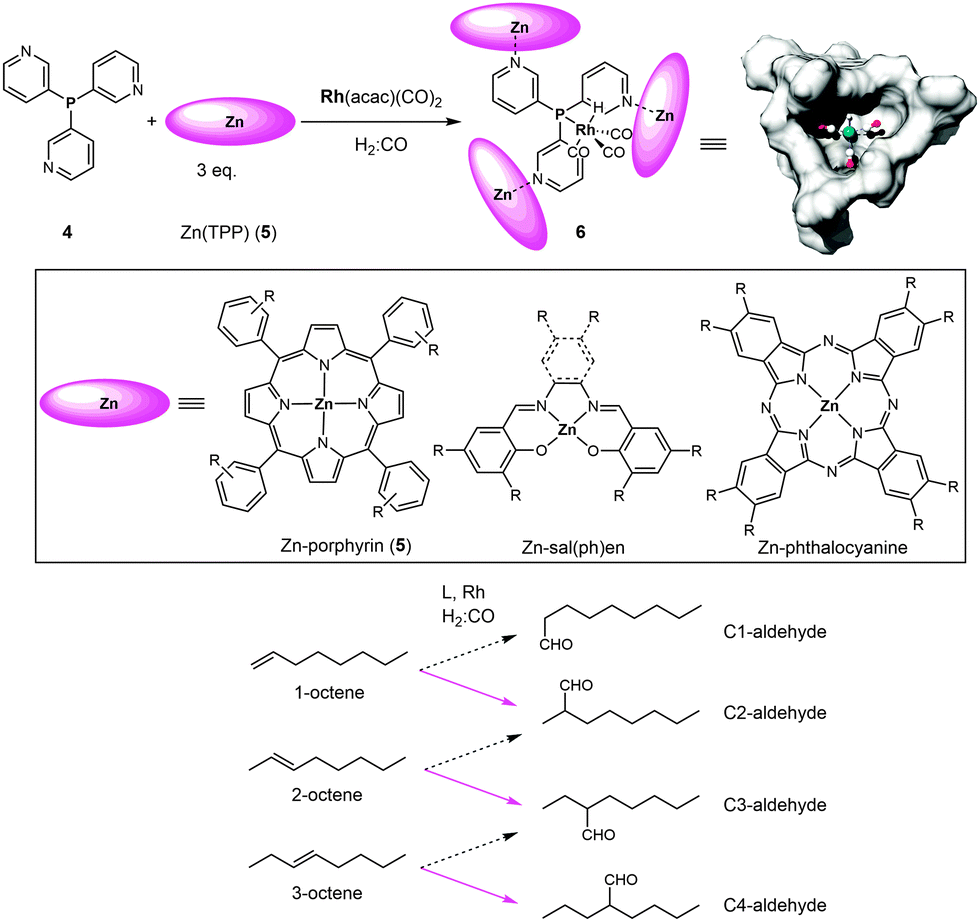 | ||
| Fig. 4 Ligand-template approach for the encapsulation of hydroformylation catalysts. The solid pink arrows indicate the major products obtained with the encapsulated rhodium catalyst 6. | ||
When 6 was employed as a catalyst in the hydroformylation of alkenes, it gave rise to unusual selectivities. For 1-octene the branched aldehyde was the main product formed, which is difficult to obtain with traditional ligands. For internal alkenes the main product formed was the one with the formyl group at the inner carbon atom of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (e.g. in the case of 1-octene, 2-octene and 3-octene the major aldehyde formed is the one where the CHO group is located at the C2, C3 and C4 positions, respectively; see Fig. 4, bottom). Molecular modeling using density functional theory (DFT) together with detailed experimental studies revealed that the selectivity is determined during the hydride migration step (depicted in Fig. 5). Some of the pathways are effectively blocked as the transition state for the inserted alkene requires substantial reorganization of the capsule, which has a high energy penalty.16 Thus the formation of the C3-alkyl species is favored (Fig. 5c), leading to a higher selectivity towards the C3-aldehyde.
C double bond (e.g. in the case of 1-octene, 2-octene and 3-octene the major aldehyde formed is the one where the CHO group is located at the C2, C3 and C4 positions, respectively; see Fig. 4, bottom). Molecular modeling using density functional theory (DFT) together with detailed experimental studies revealed that the selectivity is determined during the hydride migration step (depicted in Fig. 5). Some of the pathways are effectively blocked as the transition state for the inserted alkene requires substantial reorganization of the capsule, which has a high energy penalty.16 Thus the formation of the C3-alkyl species is favored (Fig. 5c), leading to a higher selectivity towards the C3-aldehyde.
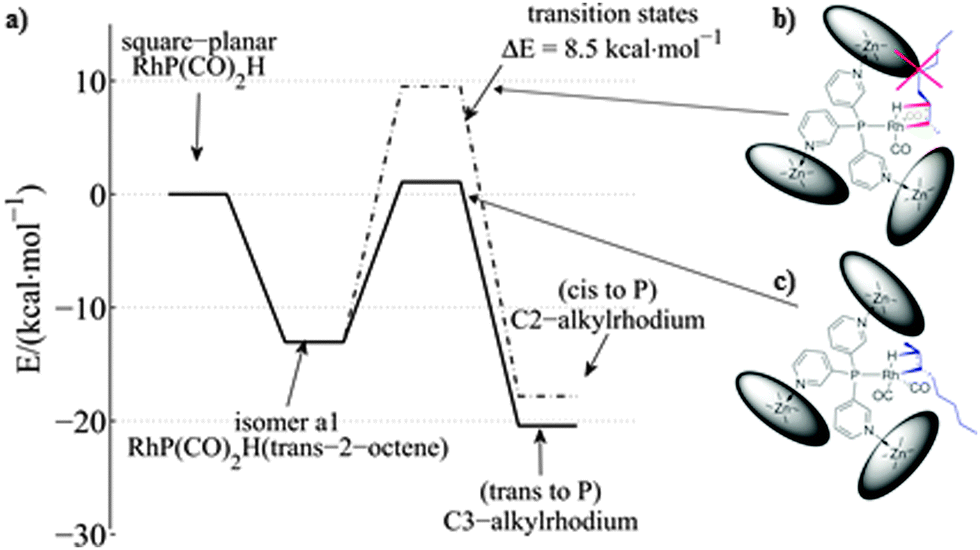 | ||
| Fig. 5 Energy profiles for the hydride migration step (a) that leads towards the more stable C3-alkylrhodium species (c) vs. the C2-alkylrhodium species (b). Reprinted with permission from ref. 16. Copyright © 2013, Nature Publishing Group. | ||
This example shows that substrate rotation at the active site is controlled by the second coordination sphere and evidences a substrate preorganization feature, which is quite similar to substrate preorganization imposed by the hydrophobic cavity of an enzyme. Interestingly, the X-ray structure of 4·53 shows C–H⋯π interactions between adjacent porphyrin buildings blocks in the assembly, which are disrupted to accommodate the transition state that leads to the minor product. As a consequence, small changes to the porphyrin building block can lead to large changes in selectivity. Importantly, by using zinc-phthalocyanines as building blocks instead of porphyrins, the cavity generated around the rhodium active site is much larger, and this leads to a reversal of the selectivity from C3 to C2 aldehydes (Fig. 6). This represents the first example in which the catalyst is the same, and where the selectivity is completely controlled by a synthetic second coordination sphere around it. In addition, the selectivity of the hydroformylation of internal alkenes is extremely difficult to control by traditional ligand design strategies. As such, this supramolecular tool adds new opportunities in transition metal catalysis. It is noteworthy to mention that the selectivity of the encapsulated hydroformylation catalyst 6 can be maintained at high temperatures (75–80 °C) by changing the syngas ratio from 1:1 (H2:CO) to 1:2 (high partial CO pressure), which is important when considering industrial applications.17
 | ||
| Fig. 6 Encapsulated rhodium catalyst with porphyrins (a) and phthalocyanines (b) as templates and the confined space where hydroformylation occurs favoring the C3-aldehyde (c) and favoring the C2-aldehyde (d). Reprinted with permission from ref. 16. Copyright © 2013, Nature Publishing Group. | ||
This strategy can be further applied to other metal-catalyzed transformations. For example, 4·53 was used in the palladium catalyzed Heck reaction, which appeared faster compared to classical triphenylphosphine systems, although this was mainly due to shorter incubation times.14a
To extend the ligand-template approach to asymmetric hydroformylation of internal alkenes, Reek and co-workers reported the use of bulky chiral pyridine-based phosphoramidite ligands in combination with zinc(II)-templates for the encapsulation of transition metal catalysts.18 These monodentate ligands showed an exceptional supramolecular control of the ligand coordination in a rhodium hydrido complex for hydroformylation. Upon addition of a zinc template, in situ high-pressure NMR and IR studies revealed a change in coordination mode of the ligand from an equatorial to an axial position, trans to the hydride (Fig. 7). Application of these supramolecular ligands in asymmetric hydroformylation of challenging internal unfunctionalized alkenes proved that this unusual coordination induced by the supramolecular capsule is reflected in higher activity and enantioselectivity. The non-supramolecular cis-complex gives a poor conversion of 2-octene of only 12% and a moderate enantiomeric excess (ee%) of only 25% of the C3-aldehyde. The supramolecular system, which enforces formation of the trans-complex, has a clearly enhanced performance: a conversion of 56% and ee% of 45%. The stereoselectivities obtained with this system left room for improvement, and hence it was anticipated that higher selectivities should be attainable when using a more rigid self-assembled system.
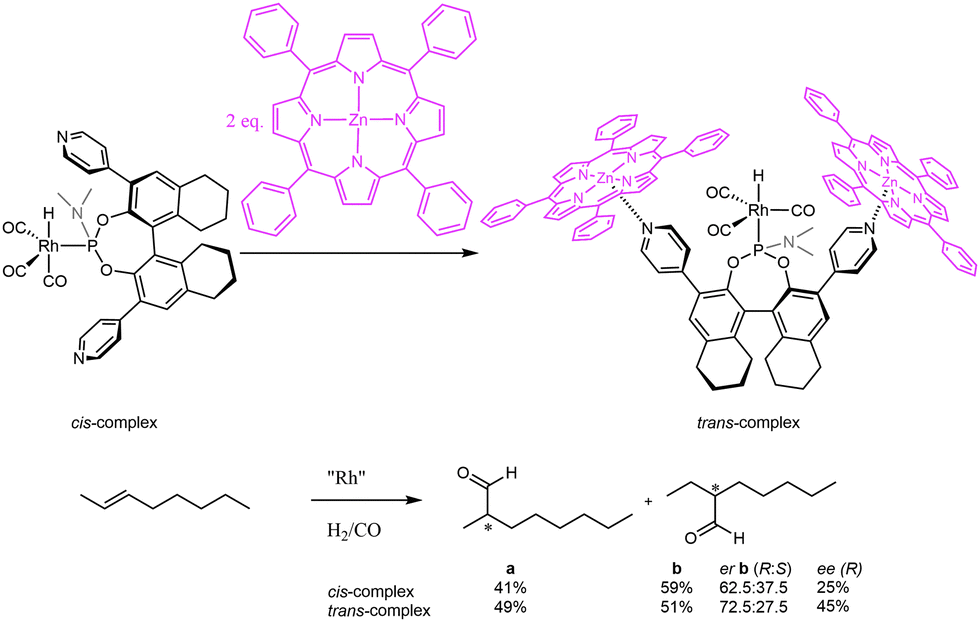 | ||
| Fig. 7 Phosphoramidite ligands used in combination with porphyrins to induce enantioselectivity in the hydroformylation of internal alkenes. | ||
This led the authors to design a new chiral, box-shaped catalyst (7, Fig. 8), which is based on a similar chiral pyridylphosphoramidite template ligand building block.19 By employing bis-zinc-salphen platforms, rigid molecular boxes are formed with a specific chiral second coordination sphere around the bis-chelated rhodium catalyst. The fact that the active species is spatially confined in a chiral cavity leads to high regioselectivity towards the formation of the internal aldehydes and results in high enantioselectivities (e.g. an enantiomeric ratio up to 93:7 for the C3 aldehyde was obtained from cis-2-octene).
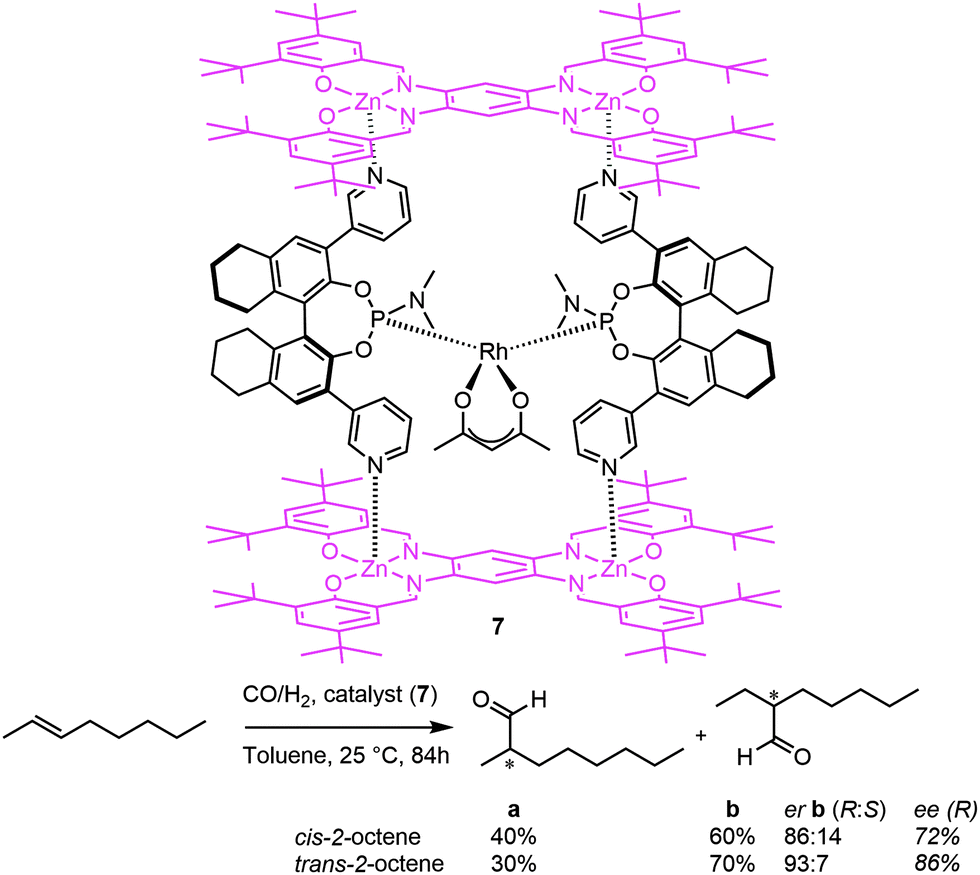 | ||
| Fig. 8 A self-assembled chiral rhodium catalyst employed in the asymmetric hydroformylation of internal alkenes. | ||
The ligand template approach has been further extended to functionalized hybrid bidentate ligands, BIAN ligands and xanthene based phosphorus ligands.20 It resulted in interesting new ways of controlling the selectivity in gold-catalyzed coupling reactions, palladium catalyzed co-polymerization and in asymmetric hydroformylation, but in these cases the effects were probably more due to changes in the steric properties of the ligand rather than to encapsulation effects. Overall, these examples show that the ligand-template approach is a very powerful strategy that leads to new ways of controlling reactions that are difficult to control otherwise. In the current examples the strategy uses the orthogonal binding properties of the soft phosphine donor and the hard pyridine donor, but many other interactions could be used for this. The next section will discuss different orthogonal approaches for capsule formation around porphyrin based catalysts.
4. Confined metalloporphyrins in catalysis
Inspired by enzymes containing metalloporphyrins, such as cytochrome P450, synthetic metalloporphyrins behaving as catalysts for transition metal catalysis have attracted a lot of attention. Metallo-porphyrins are active catalysts for a variety of reactions, and substitutions on the porphyrin backbone can be used for electronic fine-tuning. The incorporation of different metals in the central core makes them suitable for different reactions, for example epoxidation of alkenes. Manganese(III) porphyrin (or salen) catalysts have been thoroughly investigated as epoxidation catalysts, and the active species is believed to be a mononuclear manganese(V)-oxo species.21 However, the catalytic productivity is hampered due to the formation of μ-oxo-bridged dimeric porphyrin species, leading to a loss of activity. Therefore formation of supramolecular assemblies to prevent dimer formation by site-isolation of a single metallo-porphyrin is an interesting strategy to increase the stability and the turnover number (TON) of the catalyst.The group of Nolte reported on the confinement of an active and selective manganese catalyst in a cavity by capping a porphyrin scaffold with a glycoluril clip (Fig. 9).22 The resulting cavity with a diameter of 9 Å is able to bind nitrogen-donors in an axial fashion; which induces catalysis at the opposite site, in the binding cavity. This does require the use of a bulky nitrogen donor. When using a small pyridine (py) donor, pyridine binding to manganese actually occurs inside the cavity, so that catalysis occurs at the outside where inactive dimeric species can still be formed during the reaction. However, the bulkier tert-butylpyridine (tbpy) binds from the outside, and hence the vacant site for catalysis is fully isolated and protected towards formation of undesired μ-oxo-bridged manganese(IV)-porphyrin dimeric structures. This feature dramatically increases the catalyst activity and stability when applied in the oxidation of α-pinene, cis-stilbene and trans-stilbene. Next to this, a remarkable cavity-induced selectivity towards cis-epoxide was observed for cis-stilbene. This approach was further intensively exploited in the epoxidation of polybutadiene where the catalyst moves along the polymer.23 Further catalyst development has been explored by introducing urea functionalities at the outside of the cage. In this system the epoxidation takes place in the cavity, regardless of which pyridine is added. Consequently, higher activities and selectivities in the cis-epoxidation of polybutadiene were achieved.24
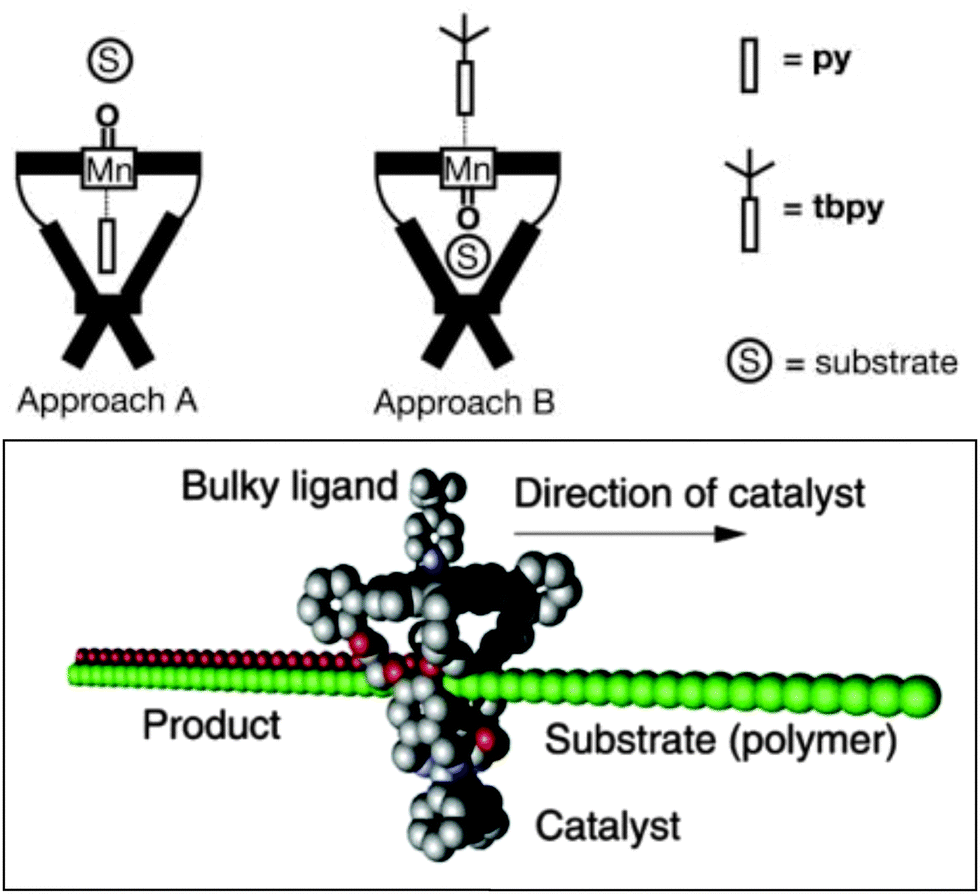 | ||
| Fig. 9 A site-isolated active manganese within a cavity (top) and its application in the epoxidation of polybutadiene (bottom). Reprinted with permission from ref. 23. Copyright © 2003, Nature Publishing Group. | ||
Preventing the formation of unreactive dimeric manganese(IV)-porphyrin species during the catalysis can also be achieved by encapsulating pyridine-functionalized metalloporphyrins in self-assembled molecular squares, as was reported by Nguyen, Hupp and co-workers (Fig. 10).25 The metallo-supramolecular square 8, constructed from four zinc-porphyrins at the sides and four rhenium complexes at the corners, is able to bind the manganese(III)-porphyrin 8 with a high association constant (ca. 106 M−1). Encapsulated catalyst 8·10 was used in the epoxidation of styrene showing a tenfold increase in stability (turnover number) extending its lifetime from ten minutes to more than three hours. Such numbers can be even surpassed if the metallo-supramolecular square is used to bind the tetra-pyridine-manganese(III)-porphyrin 9, which displays an even higher binding constant (ca. 107 M−1). It was noticed that upon dilution of the manganese catalyst (which should further inhibit bimolecular degradation pathways), TON values up to 7 × 103 and 21 × 103 for 8·10 and 8·9, respectively, were reached.
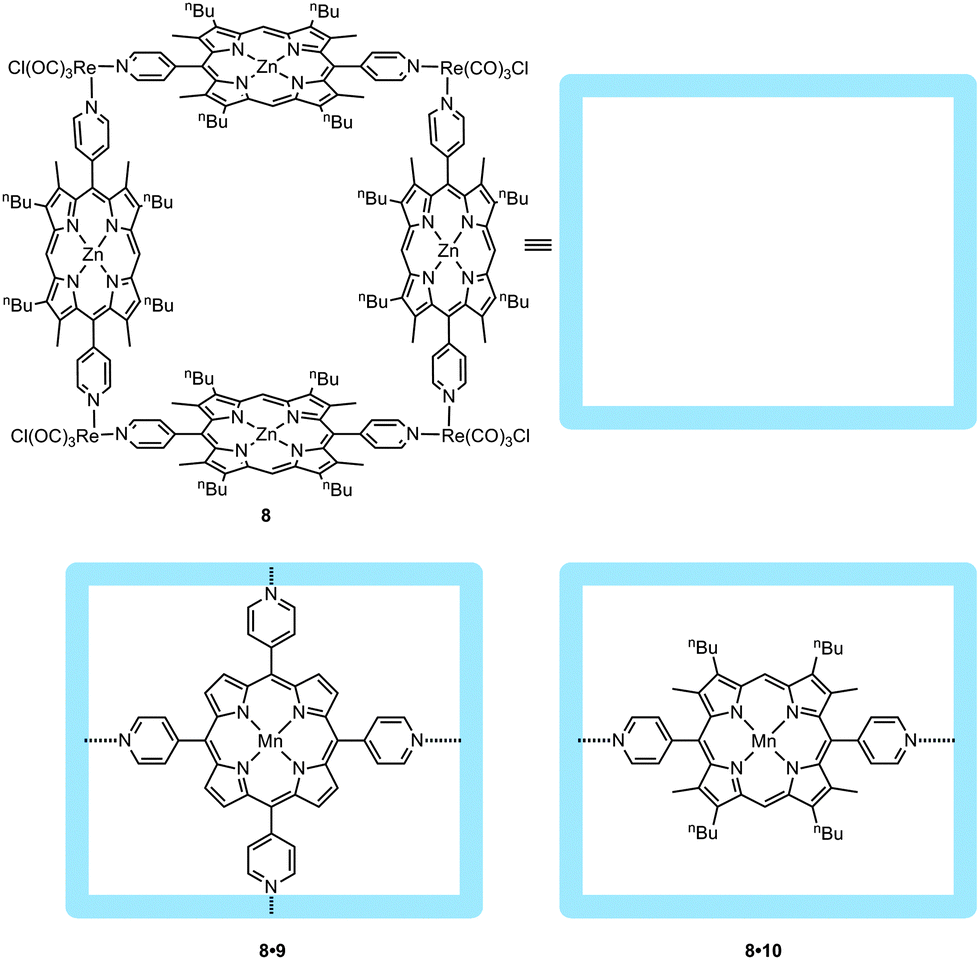 | ||
| Fig. 10 Metallo-supramolecular square 8 as designed by Hupp et al. (top). Through pyridine–zinc interactions, 9 and 10 are embedded in the square (bottom). | ||
The environment around the catalyst inside the cavity could be further confined by pairwise embedding chiral pyridylester 11 in the metallo-supramolecular square 8 as depicted in Fig. 11. The binding of these guests restricts the cavity size and influences the substrate selectivity in the catalytic epoxidation of olefins. In this manner, 8·10·112 reacts with the smaller substrate, cis-stilbene (12) seven times faster compared to the larger substrate 13 and four times faster than 14. Unfortunately, no enantioselectivity was observed due to free rotation of the zinc panels, indicating that the additional guests in the cavity do not influence the transition states of the catalyst. It, however, does impose a stabilizing factor on the catalyst. This demonstrates that different reactivities for different substrate sizes can be imposed on the catalyst due to the restriction of space.
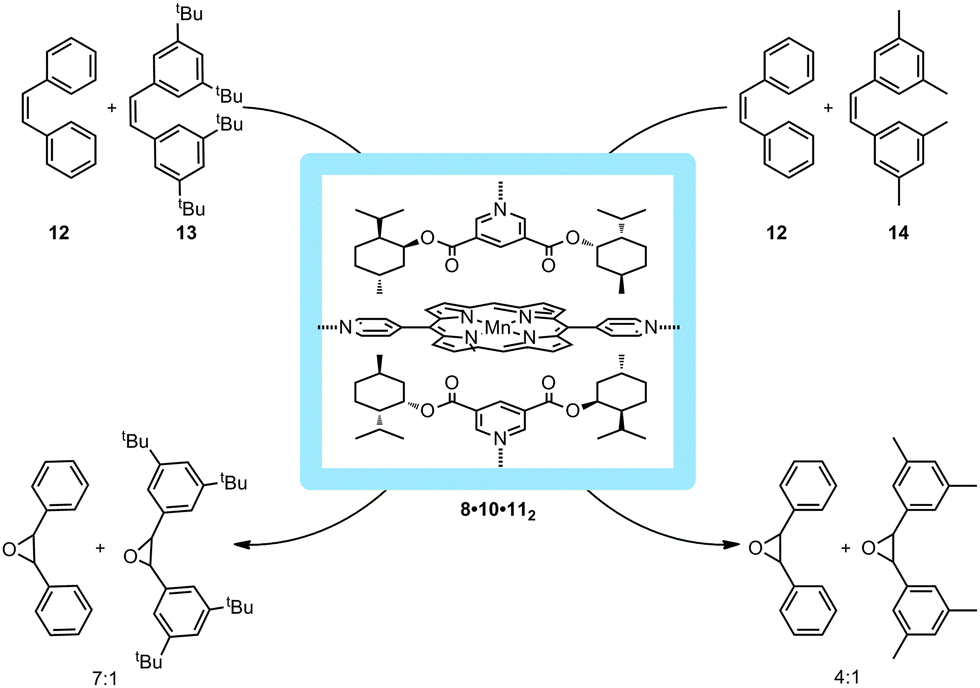 | ||
| Fig. 11 Increasing the bulk around the catalyst with molecular square 8 proved to induce substrate selectivity based on steric restrictions. | ||
The same groups demonstrated the control of substrate- and enantio-selectivity via the utilization of a rigid, metallo-supramolecular box composed of twelve zinc-porphyrins, held together by four tin-porphyrins. In this assembly two catalytically active manganese-porphyrins were embedded.26 The self-assembled molecular box could be formed by stepwise addition of the components or by mixing all building blocks in one pot. The axial ligands of the tin porphyrin building blocks should be sufficiently bulky to allow selective formation of the assembly with the catalyst in the middle. Also, these axial ligands give the ability to fine-tune the second coordination sphere around the manganese catalyst in the cavity. Catalyst encapsulation proved to invoke substrate selectivity and cis-stilbene (12) was shown to be converted to its corresponding epoxide more than five times faster than the sterically larger tetra(tert-butyl)stilbene 13 (Fig. 12). In this case the porphyrin planes cannot freely rotate anymore, (in contrast to metallo-supramolecular square 8) and the tin-porphyrins can bear chiral ligands. These features enabled enantioselective transformations with this system demonstrated by the oxidation of thioether 15. This yielded the corresponding sulfoxide with an enantiomeric excess of 12%. Despite the poor enantioselectivity, which is probably due to the small size of the axial chiral ligand on the tin-porphyrin, these results prove that chirality transfer via a second coordination sphere is feasible. Furthermore, this enantiomeric excess was only observed when the catalyst was embedded in the self-assembled supramolecular box and could be reversed by changing the chirality of the ligand attached to the tin-porphyrin that is located in the box. Although no detailed mechanistic studies for these systems have been reported, in the commonly accepted olefin epoxidation mechanism the selectivity is believed to be determined by the approach of the alkene to the active manganese(-salen) catalyst.27 It is therefore likely that such an approach is controlled to some extent by the cage effect imposed by these supramolecular systems, which explains the selectivity observed.
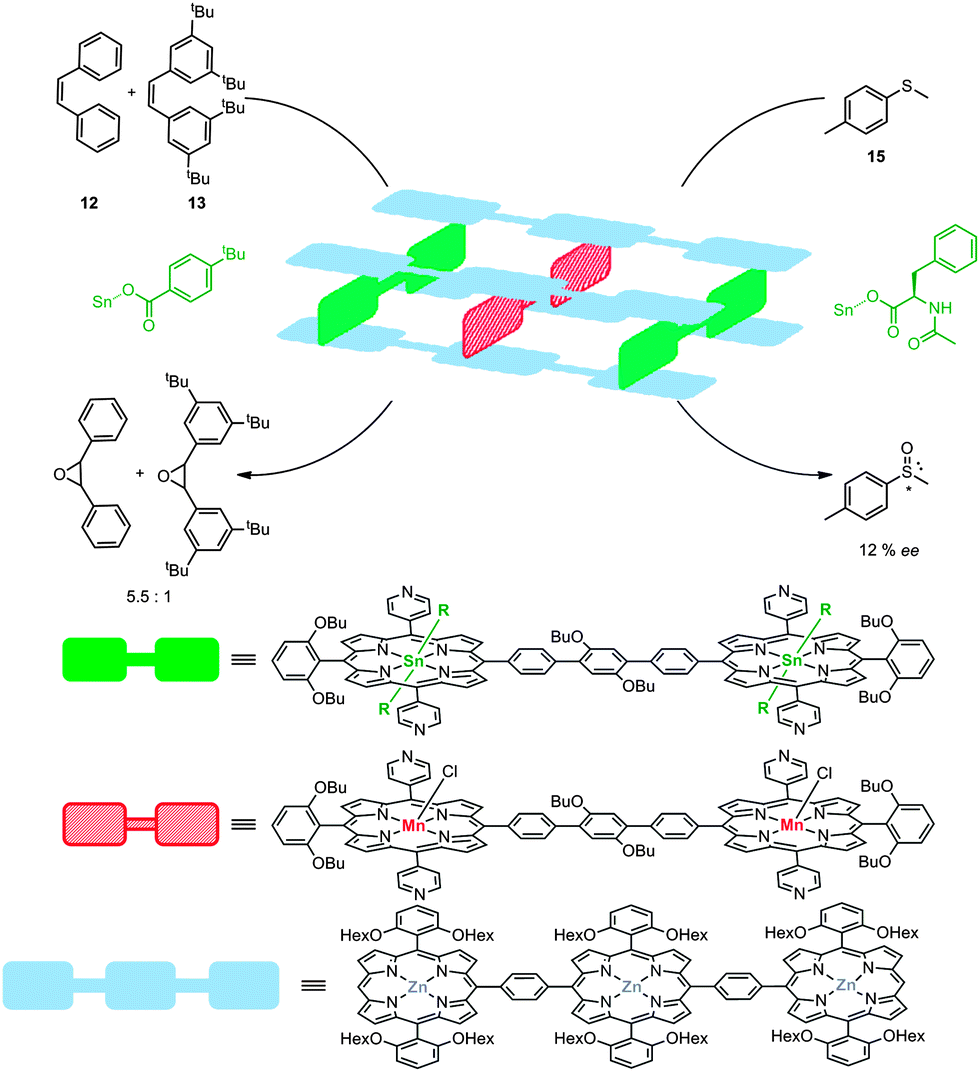 | ||
| Fig. 12 Multi-component assembly based on various porphyrin blocks. Chiral ligands attached to the tin porphyrin lead to chiral induction in the oxidation of sulfide 15. | ||
Bimetallic deactivation pathways are also very common in radical-type transition metal catalysis, and therefore site isolation of such catalysts may lead to enhanced life times. For example, cyclopropanation reactions mediated by cobalt(II) catalysts proceed via carbene–radical species, stabilized on a cobalt(II)-porphyrin scaffold, which reacts with alkenes to form the product. However, depending on the nature of the substrate that is used, the radical can become delocalized. As a result of having discrete spin density at a more remote carbon atom, partial loss of the ‘steric’ control of the catalysts over the substrate radical can lead to undesired radical–radical coupling leading to C–C bond formation, thus leading to catalyst deactivation (Fig. 13).28,29
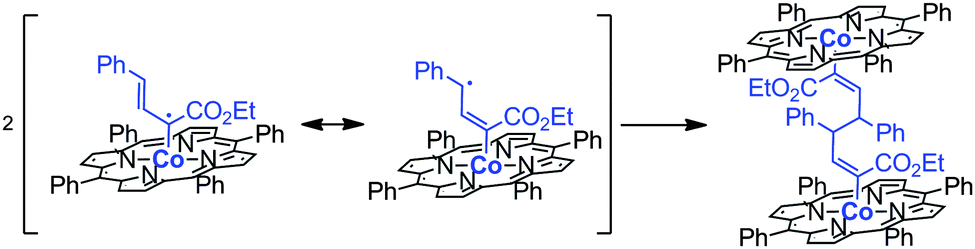 | ||
| Fig. 13 Dimerization pathway that leads to an inactive intermediate during the metalloradical-mediated catalysis. | ||
To prevent such dimerization to occur, supramolecular encapsulation of a cobalt-porphyrin catalyst was studied by de Bruin and co-workers. Inspired by Nitschke's cubes, formed by a self-assembly process of six zinc-porphyrins held together by eight iron complexes at the corners,30 de Bruin and co-workers managed to prepare a larger analogue by using bigger zinc-porphyrin scaffolds. This resulted in a molecular flask (16) that is able to encapsulate a single catalytically active tetra-pyridyl-cobalt(II)-porphyrin 17 (16·17, Fig. 14).31 Interestingly, metallo-radical-trapping experiments performed with an encapsulated Co(II)-porphyrin catalyst and EDA (18, EDA = ethyl diazoacetate; one of the reagents in cyclopropanation catalysis) in the presence of 1,4-cyclohexadiene (19, a hydrogen-atom-transfer reagent) indicate that EDA indeed reacts within the self-assembled cube leading to 16·20. Thus, molecular flask 16·17 represents an improved catalyst that prevents unreactive dimerization pathways by site-isolation and as such displays longer life times compared to the non-encapsulated version.
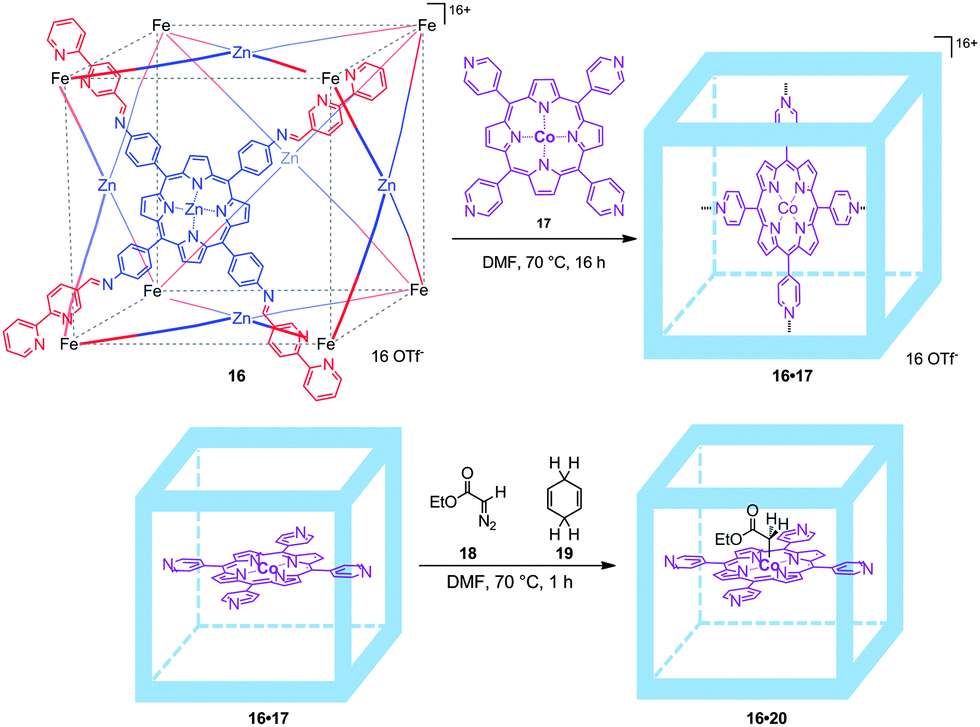 | ||
| Fig. 14 Cobalt encapsulated catalyst 16·17 (top) and radical-trapping experiments. The cage prevents binuclear radical-type deactivation processes (bottom). | ||
Indeed, in the cobalt-catalyzed cyclopropanation of styrene with diazo compounds (Fig. 15) the encapsulated cobalt(II)-porphyrin catalyst 16·17 is active even after four hours and reaches comparable activity to the best cobalt(II)-porphyrin used for such transformations to date (Zhang's catalyst),32 whereas the non-encapsulated catalysts showed only short life times (conversions stopped after one hour). In addition, the trans–cis selectivity for such transformation is different when performed inside the cage: 63:37 (16·17) vs. 75:25 (17). The encapsulated catalyst was also studied in the intramolecular reaction of 21 giving rise to a mixture of isomers 22-E and 22-Z. For this reaction 16·17 produces the highest yields of all available non-encapsulated Co(II)-porphyrin catalysts, and encapsulation also resulted in altered regioselectivity. For example, when using conventional cobalt(II)-tetraphenylporphyrin as a catalyst the E:Z ratio is close to 40:60 whereas upon catalyst encapsulation there is an improved preference for the Z isomer (E:Z ratio of 16:84).
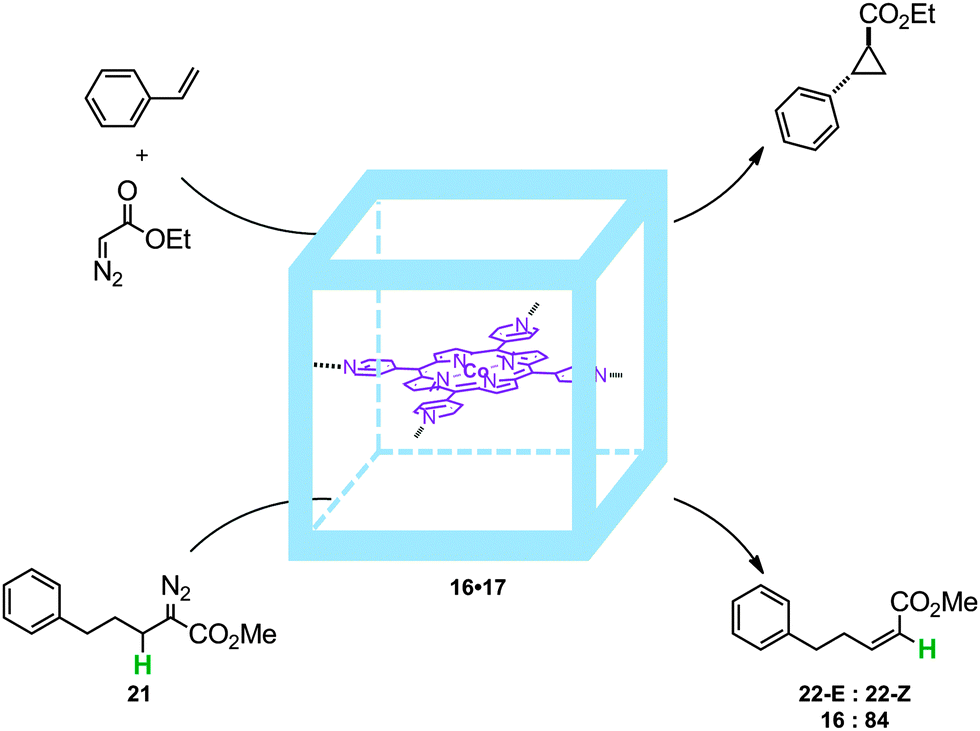 | ||
| Fig. 15 Molecular container 16 can be used to stabilize a cobalt-porphyrin catalyst, giving high TONs in cyclopropanation reactions. The encapsulation also leads to a change in selectivity regarding the E:Z ratio of 22. | ||
By changing the anion of the molecular container from triflate (OTf) to triflimide (NTf2), the system could be dissolved in water/acetone (5:1) mixtures. In this reaction medium TON values of more than 300 for the cyclopropanation of styrene with EDA were obtained with the confined catalyst, which outperformed non-encapsulated cobalt(II)-porphyrins.33 The supramolecular encapsulated catalyst was compatible with different alkenes, giving high yields for styrene analogues with electron-donating or electron-withdrawing substituents. Limited reactivity was observed with methacrylates, bulky alkenes and bulky diazo substrates. The restricted space inside the molecular container was further exploited by studying size-selective transformations via competitive experiments. In these experiments, styrene and a bulky alkene were competing for the reaction with a diazo-reagent (Fig. 16). Interestingly, the encapsulated cobalt(II)-porphyrin catalyst 16·17 preferentially cyclopropanates the smaller styrene substrate, whereas non-encapsulated catalysts give an equal distribution of the small and large products. This shows that the second coordination sphere gives rise to size selectivity, which is difficult to achieve by modifications to the first coordination sphere around a catalyst. Although this confinement around the catalytic center shows substrate selectivity, the current system cannot control the cis/trans selectivity. The two zinc porphyrin building blocks still available for coordination of axial ligands may provide a supramolecular handle to further confine the catalyst such that it becomes even more selective.
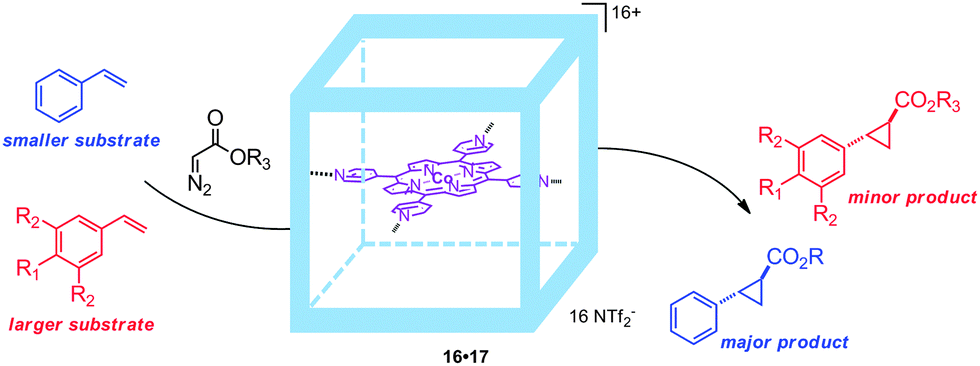 | ||
| Fig. 16 Embedding the catalyst 17 inside a second sphere gives rise to substrate selectivity, due to the steric requirements of the substrate. | ||
5. Catalytically active host–guest complexes
In the examples shown in the previous section the catalyst was incorporated in capsules and containers by self-assembly, leading to well-defined systems in which the orientation of the catalyst is fixed by specific metal–ligand coordination bonds between the catalyst and the cage. An alternative approach is the preparation of a preformed self-assembled host in which the catalyst can be bound as a guest, using weak interactions such as π–π stacking, hydrogen bonding, ionic and dipolar interactions, the ‘hydrophobic effect’, and entropic binding based on the replacement of multiple solvent molecules by a single guest in the cavity (i.e. the catalyst). It is of course of key importance that the catalyst remains bound inside the molecular container during catalysis. Furthermore, the system should be capable of co-encapsulating the catalyst and the substrate(s). Self-assembled supramolecular flasks formed in aqueous media are a particular promising class of systems enabling encapsulation of different chemical entities due to a combination of hydrophobic and ionic effects. In recent years, many water-soluble supramolecular flasks have been reported, but only few of them have been employed in metal complex encapsulation aiming for catalysis. The groups of Raymond and Bergman explored the use of an M4L6 anionic tetrahedral capsule (23),6g,h which is formed by six bis-catecholamide struts and four octahedral gallium(III) centers. The highly negatively charged (−12) and homochiral (ΔΔΔΔ and ΛΛΛΛ) capsule accommodates positively charged organometallic guests (Fig. 17), and as such, the capsule can be used to bind cationic metal complexes. For example, it has been used as a mediator for stoichiometric C–H activation using an encapsulated cationic iridium complex.34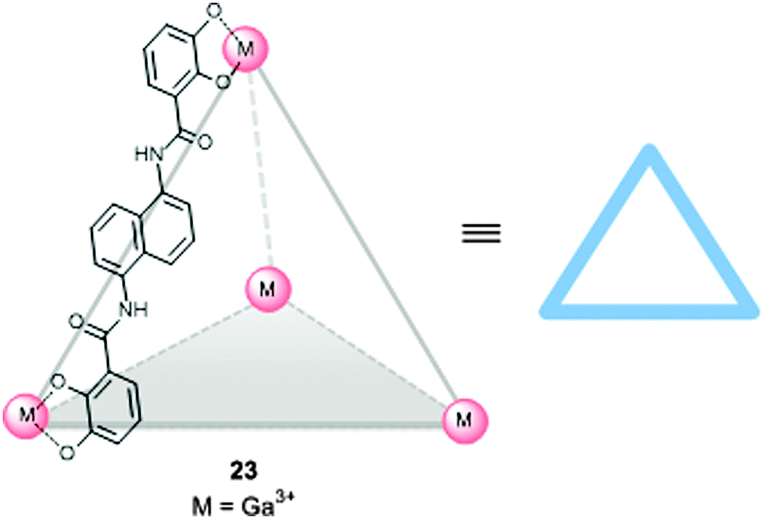 | ||
| Fig. 17 Water soluble self-assembled tetrahedral cage 23 that can bind cationic metal complexes in the cavity. | ||
The application of this capsule in controlling the properties of transition-metal catalysts was proven with different types of metal complexes. A series of bisphosphine rhodium-diene cations were encapsulated and the hydrogenation of the cyclooctadiene ligand yielded the active catalyst in the form of a hydrated bisphosphine complex (Rh(PMe3)2(D2O)2, 24).35 Whereas the hydrated complex itself was not encapsulated because it has a too high solubility in water, in situ hydrogenation of the cyclooctadiene ligand yielded the kinetically trapped active catalyst 23·24. This active species is fully ejected from the cavity after twelve hours and the system should therefore be used within this timeframe, for example for fast isomerization reactions of allylic substrates (Fig. 18). While the free catalyst showed conversion of different allylic alcohols and ethers to their corresponding aldehydes or enol ethers, the encapsulated catalyst 23·24 showed substrate selectivity, controlled by the aperture of the container. Based on the size of the substrate, only prop-2-en-1-ol (25) and its methyl ether (26) were isomerized by the encapsulated catalyst. This contrasts with the non-encapsulated catalyst that is able to isomerize larger and sterically more hindered substrates (like 27). It was furthermore shown that the capsule also protects the catalyst. For instance, while crotyl alcohol usually inhibits the free catalyst, the encapsulated rhodium-catalyst (23·24) is still able to convert allyl alcohols to the aldehydes in the presence of this inhibitor.
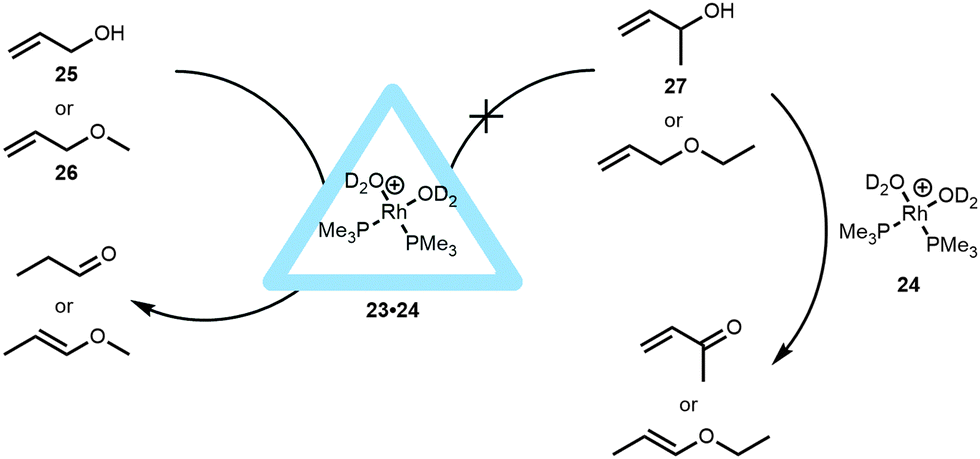 | ||
| Fig. 18 Rhodium encapsulated catalyst 23·24 and its catalytic behavior in allylic isomerization compared to non-encapsulated rhodium catalyst. | ||
[RuCp(PMe3)(MeCN)2]+ (28) was also sequestrated within the M4L6 tetrahedral cage (23·28, Fig. 19).36 Within the assembly, a water-solvated ruthenium species was expected to form in D2O, however no exchange of acetonitrile with water occurred and the ruthenium complex was bound quantitatively inside the cavity of 23. Such ruthenium complexes are known to isomerize allylic alcohols towards the corresponding aldehydes or ketones. In fact, the supramolecular ruthenium catalyst 23·28 provides TON > 1000 and a very long lifetime in the isomerization of 3-buten-2-ol (27), values that are much higher than those obtained for the non-encapsulated ruthenium catalyst, even if the latter is applied in organic media. Kinetic studies were performed, revealing that the encapsulated ruthenium catalyst does not display product inhibition. In fact, the system seems to accelerate as the reaction reaches completion. This also results in an increase in the pseudo-first-order rate constant near the end of the reaction. Based on kinetic analysis and competition experiments with an additional allyl ether, it was suggested that substrate-inhibition occurs due to the binding of a second olefin to the catalyst–substrate complex during the catalysis. It is speculated that, as the substrate concentration lowers, less of this olefin inhibition occurs and thus the rate constant slightly increases. Although no intermediates were observed, this example clearly shows that catalyst encapsulation alters the kinetics for the formation of some intermediates during the catalytic cycle. Similar to the encapsulated rhodium catalyst 23·24, the supramolecular ruthenium catalyst 23·28 also showed substrate selectivity (Fig. 19). For instance, the larger 1-phenylprop-2-en-1-ol does not react with the encapsulated 23·28, likely because of its big size. It is noteworthy that 3-buten-2-ol (27) can now be isomerized to the ketone which was not possible with the rhodium analogue (23·24, Fig. 18). This indicates that the aperture of the cage itself plays no role in this substrate and that it is more likely that the catalyst has a different orientation inside the capsule. Importantly, these experiments show that the cage still allows small substrates to come in contact with the catalyst.35,36
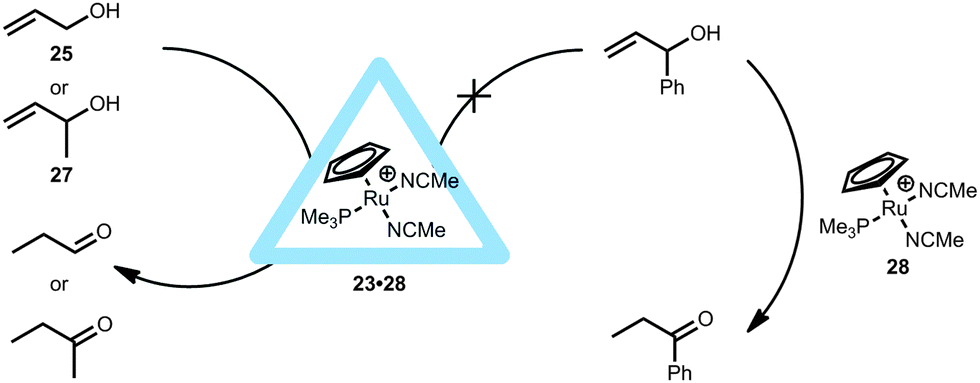 | ||
| Fig. 19 Allylic isomerization within 23·28 compared to the non-encapsulated ruthenium catalyst showing substrate selectivity. | ||
The supramolecular capsule 23 was also used in intramolecular cyclization reactions when monophosphine gold complexes were encapsulated leading to 23·29.37 The supramolecular cage 23 drives the equilibrium of the gold complexes to the cationic form and thus (Me3P)Au+ is encapsulated, regardless of the anion (Cl−, Br− or NTf2−) present in solution. The encapsulated gold(I) complex 23·29 was applied in the hydroalkoxylation of allenol 30 (Fig. 20). The various free (non-encapsulated) gold(I) complexes showed different yields (11–87%) depending on the gold–anion bond strengths (Me3PAuBr gave the poorest yield); whereas the encapsulated gold catalyst 23·29 gave a reasonable yield (48%) for the exo-hydroalkoxylated product, regardless of the counterion used. The M4L6 tetrahedron itself does not catalyze the reaction. Also, performing the reaction while the pocket is blocked with a strong binding guest (PEt4+), resulted in a yield similar to the control reaction indicating that an encapsulated gold(I) species is the active catalyst. Further comparison of the encapsulated gold complex 23·29 with Me3PAuBr showed that the reaction rate is accelerated by a factor of eight and the TON is increased to 67. Comparable to the case with the encapsulated 23·24 and 23·28 catalysts, this is not an example in which the fundamental transformations at the metal center are controlled by the confined space, as the same products are seen without the capsule. These systems, however, show that the supramolecular cage controls the coordination sphere around the gold(I) complex, and can act as a phase transfer reagent to enhance the reaction rate.
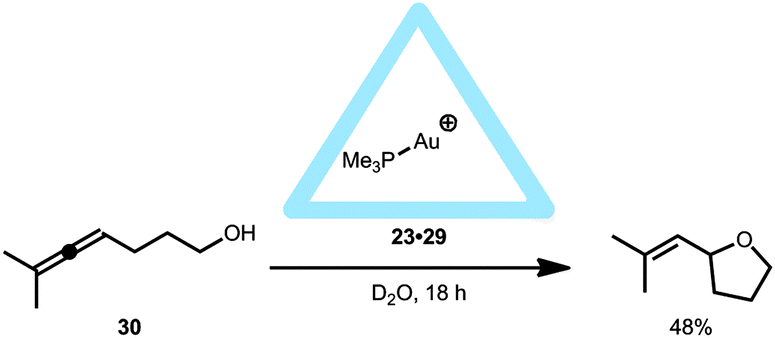 | ||
| Fig. 20 Catalytic behavior of encapsulated 23·29 in the intramolecular hydroalkoxylation reaction of 30. | ||
Interestingly, the gold-encapsulated catalyst 23·29 did provide a different product distribution compared to the free complex when applied in the cyclo-isomerization of enyne 31. With 23·29 a remarkable change in product distribution was observed (Fig. 21).38 It is believed that, if the reaction takes place outside the cage, the well solvated gold carbene species 3239,40 undergoes a nucleophilic attack of water to form the hydroalkoxylated species 34. However, when the reaction takes place inside the cage, less water is available due to the hydrophobic cavity, and the activated species has time to undergo cyclo-isomerization to form product 33. Although the transition state leading to both products is probably the same, the hydrophobic environment within the capsule makes the nucleophilic attack of water energetically less favored compared to the non-encapsulated system. Hence, the pathway towards the intramolecular rearrangement is more accessible within the encapsulated gold(I) catalyst 23·29. Furthermore, the selectivity of the reaction remained the same, independent of the gold precursor used (Me3PAuCl or Me3PAuBr), indicating that the encapsulated cationic Me3PAu+ complex is the active species.
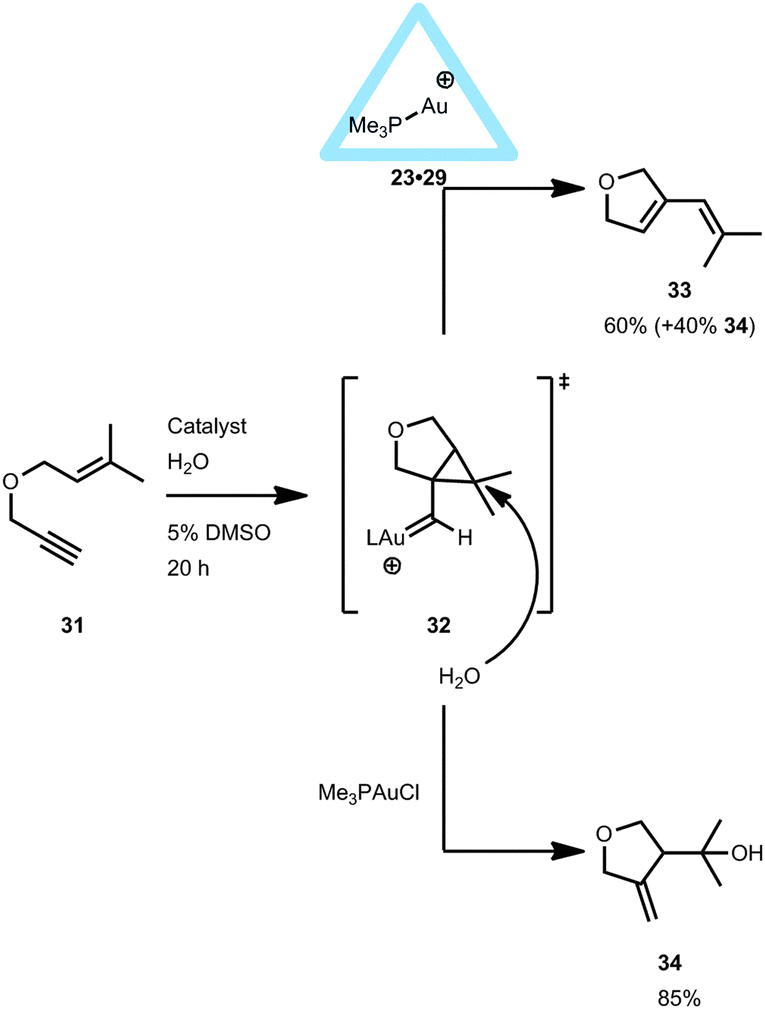 | ||
| Fig. 21 Catalytic behavior of encapsulated 23·29 in the intramolecular cyclo-isomerization reaction of 31 compared to non-encapsulated gold catalysts. | ||
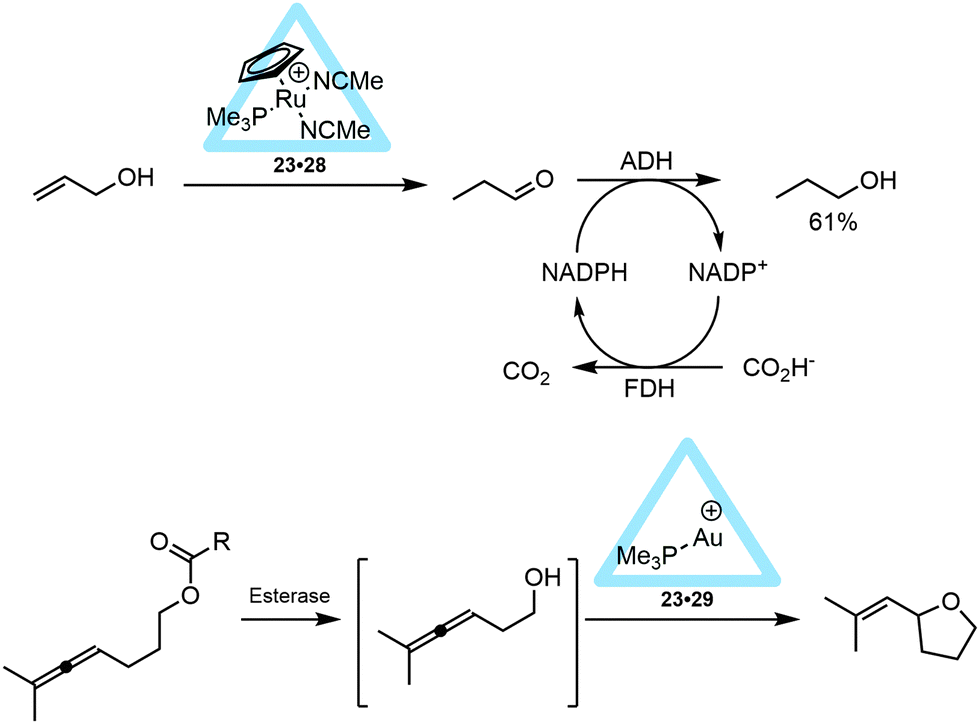
Metal complex encapsulation can lead to protection of the catalyst from degradation. This was nicely demonstrated by combining the previously discussed encapsulated 23·28 (and 23·29) catalyst with enzymes such as esterases, lipases and alcohol dehydrogenases (ADH and FDH) to perform cascade reactions. In a one-pot reaction the ruthenium-encapsulated catalyst 23·28 was used in combination with ADH and FDH, enabling the conversion of an allylic alcohol to the aliphatic alcohol (Fig. 22, top).41 In this reaction the metal catalyzed reaction precedes the enzyme catalyzed transformation. Alternatively, when the gold encapsulated catalyst 23·29 was used in a tandem reaction with an esterase, the enzymatic reaction takes place before the metal catalyzed reaction.41 The protection of the cationic gold complex in the supramolecular container is crucial as the free gold complex inhibits the esterase. In the overall reaction an ester was hydrolyzed by an esterase or lipase to give allenol, which was subsequently cyclized by the encapsulated 23·29 catalyst (Fig. 22, bottom). These examples further illustrate the potential of metal encapsulation as it allows the combination of different catalysts for cascade transformations that cannot be combined otherwise.
 | ||
| Fig. 22 Tandem reactions using a combination of metal encapsulated and enzyme catalysts in water. | ||
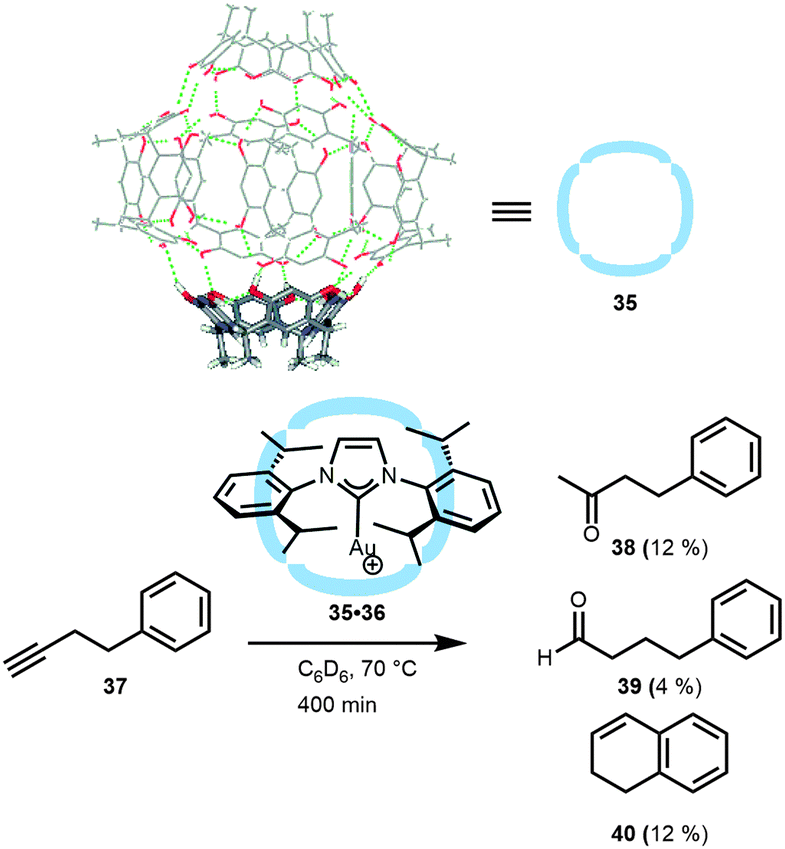
Reek, Scarso and co-workers explored the use of water–hydrogen-bonded hexameric capsules (35)42 based on readily available resorcin[4]arenes. These resorcin[4]arene building blocks form self-assembled hexameric capsules in water-saturated organic solvents and have been demonstrated to encapsulate a variety of neutral and cationic guests.43 Reek and Scarso demonstrated that these capsules can also be used to encapsulate gold complexes (Fig. 23).44 Upon encapsulation of the cationic gold(I) carbene complex, the triflate anion was separated and not bound in the cavity. The confined gold(I) catalyst 35·36 was explored in the hydration of butyne 37, which normally gives Markovnikov addition of water (38), or forms 1,2-dihydronaphthalene 40 under anhydrous conditions. Thus, the non-encapsulated (i-Pr-NHC)Au(OTf) results in the quantitative formation of the Markovnikov product 38 within 30 min. Although the encapsulation of the gold catalyst slowed down the reaction (5% conversion after 30 min, 28% after 400 min), a new interesting distribution of products was observed. In contrast to the free gold catalyst, the encapsulated analogue yielded a small amount of linear aldehyde 39 (4%), next to 12% of 38 and, interestingly, the formation of 1,2-dihydronapthalene 40 (12%) was observed. Thus far the origin of the change in selectivity remained somewhat unclear. Probably, the molecular container may impose a reaction barrier for water to enter the cavity, thus slowing down the Markovnikov addition, or the capsule could force an unusual geometry of the substrate–metal complex inside the container, thus favoring the intramolecular reaction. In the latter case, the second coordination sphere disfavors certain reaction pathways, which may suggest that it should be possible to force the formation of other products, like the 5-membered ring (5-exo-dig product),45 by changing the shape of the cavity in which the metal catalyzed reaction takes place.
 | ||
| Fig. 23 Capsule 35 and implications in catalysis of encapsulated 35·36. | ||
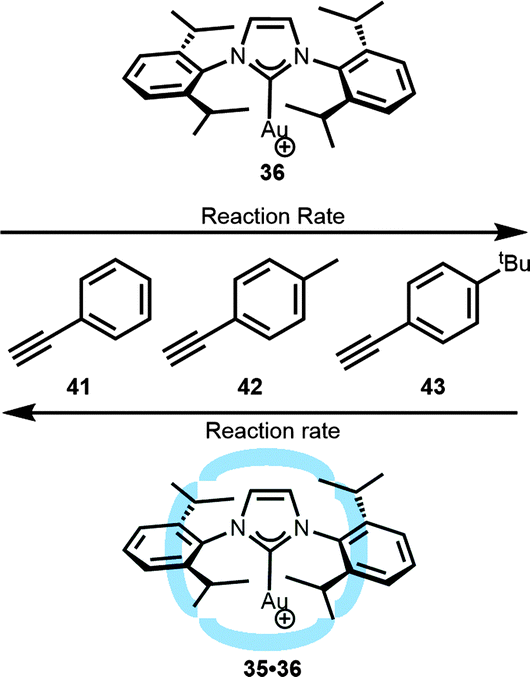
Also other substrates were used for the hydration reaction, and a decrease in reaction rate was noted when the catalyst was enclosed in the hexameric cage.46 The difference in the rate of various substrates that differ in the size was translated into substrate-selectivity controlled by the cage. An interesting rate increase was observed when aliphatic cyclic functionalized alkyne (ethynylcyclohexane) was compared to linear alkynes (1-octyne and 1-dodecyne). A plausible explanation is that, due to its smaller and more rigid shape, the cyclohexane moiety fits better in the void of the container than the linear alkynes. The better fit results in a shift of the equilibrium to the substrate bound species, giving rise to a higher rate. The effect of the host on the guest is more clear when aromatic alkynes are used (41–43, Fig. 24). In these cases, the non-encapsulated gold catalyst shows higher reactivity for the larger substituted (and more electron rich) alkynes following the order 41 < 42 < 43. The host–guest complex shows the reverse substrate selectivity. The second coordination sphere gives rise to a relative higher rate for the smaller and non-substituted aromatic substrate overruling the natural selectivity that was based on the electronic properties of the substrate.
 | ||
| Fig. 24 Substrate-selectivity observed in the hydration of alkynes with encapsulated 35·36 catalyst. | ||
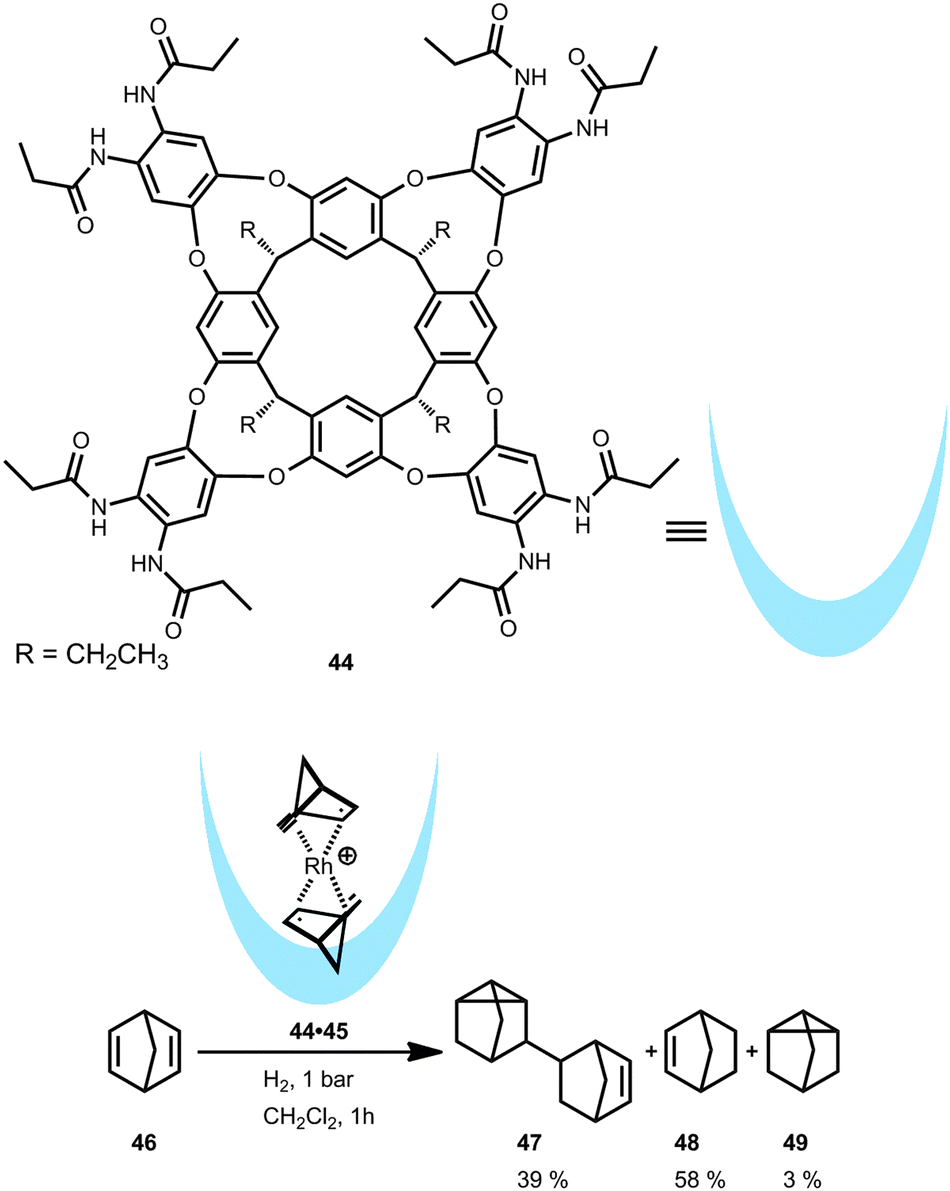
In contrast to the hexameric cage, a self-folding cavitand forms when an amide-functionalized resorcin[4]arene (44) is used. As demonstrated by Ballester and co-workers, this cavitand is able to bind a [Rh(nbd)2]+ (nbd = norbornadiene) complex which was studied in the catalytic hydrogenation of 46 (Fig. 25).47 Due to the size and shape of the cavitand 44 and the rhodium complex, only one part of the encapsulated catalyst 44·45 is exposed to the outside forming a dichloromethane-solvated species in solution. This partial exposure stabilizes the rhodium complex when pressurized with hydrogen and prevents the formation of rhodium(0)-black which is typically observed with non-encapsulated rhodium(I) complexes. Cavitand 44 stabilizes intermediates that are not present when the metal complex is free in solution. In the hydrogenation of 46, the non-encapsulated [Rh(nbd)2]+ provides dimeric product 47 in more than 80% yield. In contrast, the encapsulated rhodium catalyst 44·45 results in a different product distribution, namely 47, 48 and 49 in a 39:58:3 ratio (Fig. 25, bottom). A plausible explanation for the formation of the dimeric product 47 is leaching of the catalyst from the cavity. However, the major product 48 is likely formed because the transition state of the dimerization is hampered by the molecular container.
 | ||
| Fig. 25 Encapsulated 44·45 catalyst and its performance in hydrogenation of norbornadiene. | ||
Clearly, all these examples show that the formation of host–guest complexes is a viable method to control the second coordination sphere around metal complex and can be used in catalysis. However, finding the proper fit for a guest inside the host still remains a challenge. Furthermore it requires that a substrate can be co-encapsulated with the active site in the cavity.
6. Synthetic catalysts encapsulated in a protein cavity
Instead of using nature as a source of inspiration, proteins can also be used to encapsulate a transition metal catalyst. Intuitively, such cages can be used for enantioselective transformations as the protein cage is chiral. As such, the main focus of this approach has been the implementation of achiral catalysts in chiral protein hosts, with the particular advantage that the environment can be optimized by protein engineering. Various reviews have already extensively discussed the different approaches for embedding catalysts in protein cavities (e.g. the covalent, dative or supramolecular approach).48 Therefore we will only provide a short overview of the supramolecular approaches as these examples clearly show how a synthetic transition metal catalyst can be enhanced with a second sphere by simply mixing the components. In this respect, Ward and co-workers have exploited the strong binding of biotin to streptavidin by functionalizing metal catalysts with a biotin functionality.49 Through the ligand-template approach the streptavidin could be used as a template to form the second coordination sphere around an achiral catalyst (Fig. 26). In this way ruthenium, rhodium and iridium based piano stool complexes were embedded in the natural capsules and used as artificial transfer hydrogenases for the asymmetric hydrogenation of ketones.50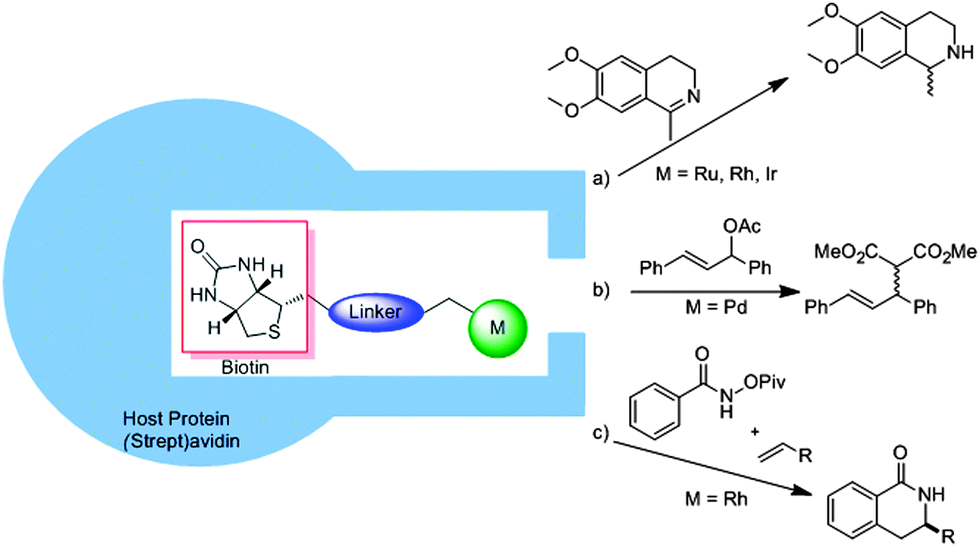 | ||
| Fig. 26 The affinity of biotin to streptavidin can be used to embed an achiral catalyst in a second coordination sphere, thereby inducing chirality in different reactions. | ||
In the hydrogenation of a prochiral imine, high enantioselectivities were induced due to the chiral molecular container (Fig. 26a).51 An embedded iridium catalyst yielded the best selectivity, converting the product with 96% ee (R) or 78% ee (S), depending on which mutant of the streptavidin was used. Embedding a palladium catalyst with this supramolecular binding approach in a protein environment gave a catalyst suitable for asymmetric allylic alkylation (Fig. 26b).52 The screening of different biotin–palladium complexes with different (strept)avidin mutants gave an optimum alkylation of 1,3-diphenylallylacetate with a conversion of 95% and an enantiomeric excess of 90%. This system could further be employed in the rhodium catalyzed C–H activation of a protected benzohydroxamic acid as shown in Fig. 26c.53 For this reaction a carboxylate moiety near the catalyst proved to be important and again, selective mutations in the protein yielded high regio and enantiomeric ratios (er, 91:9). Different substrates could be converted with this system showing that small substrate variations can still be accommodated in the cavity.
Another way to encapsulate transition metal catalysts in a protein matrix was demonstrated by the group of Watanabe. In this example a chromium–salphen catalyst was embedded in an apo-myoglobin protein.54 The binding is based on hydrophobic interactions and induced chirality on the metal catalyst, as was shown by the oxidation of thioanisole (50) with an ee of 4% (R) for the non-modified myoglobin and 13% (S) for a mutated myoglobin. Further optimization led to an ee of 33% (S) and 24% (R) when the structure of the Schiff base catalyst was changed and manganese was used (Fig. 27a).55 Serum albumin can also be used to introduce new metal complexes inside a protein cage as was shown by the group of Gross. By employing manganese corroles, as depicted in Fig. 27a (52), inside human serum albumin (HSA) an ee of 74% (S) was obtained in the asymmetric sulfoxidation of thioanisole.56 This albumin type of protein molecular container also binds sulfonated copper phthalocyanine, as reported by the group of Jiao.57 Bovine serum albumin (BSA) was shown to induce enantioselectivity on a copper catalyst. Utilization of the encapsulated catalyst in a Diels–Alder reaction with a pyridine functionalized dienophile and cyclopentadiene resulted in enantioselectivities up to 98% (Fig. 27b, endo/exo ratio of the product was not influenced).
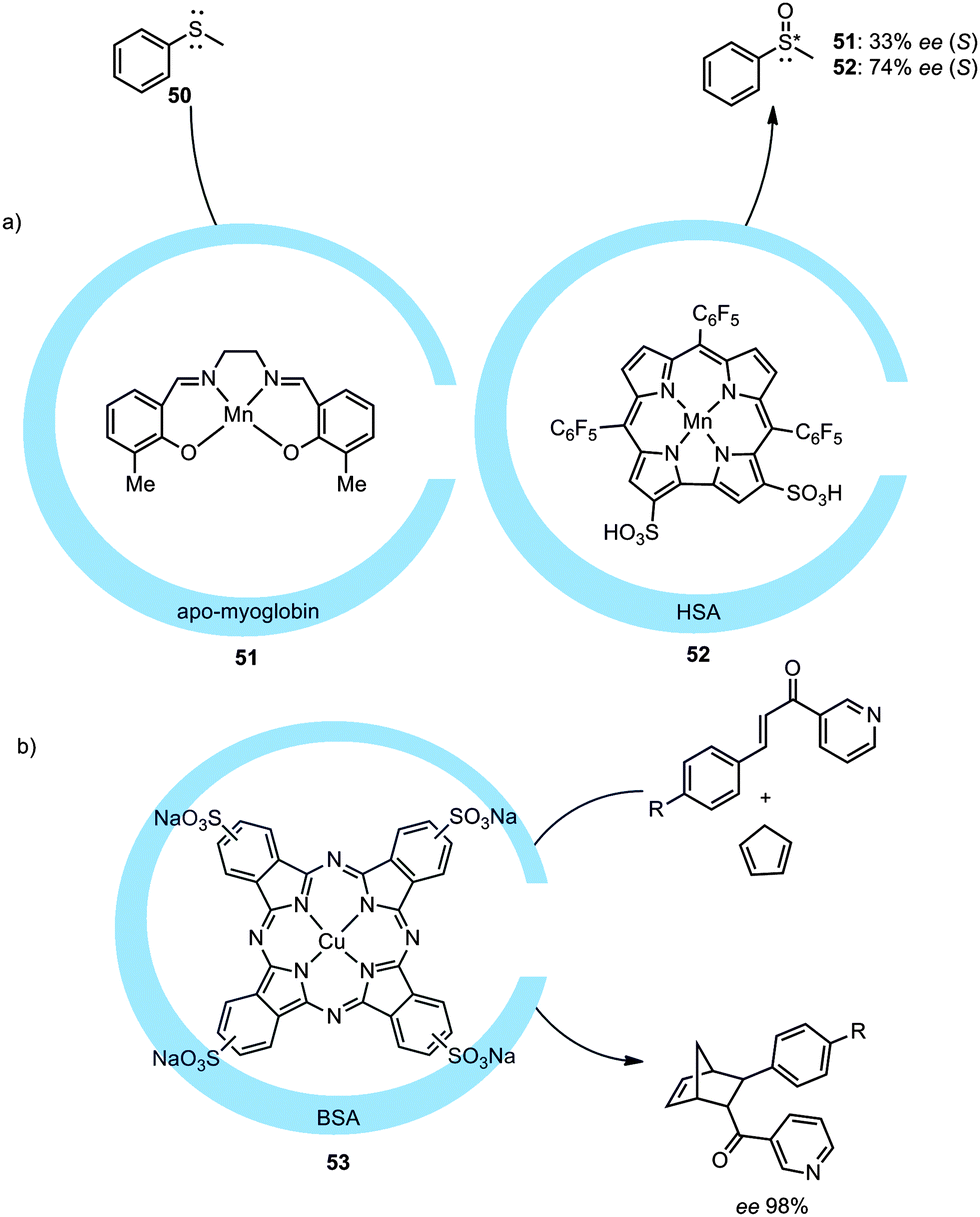 | ||
| Fig. 27 Encapsulation of different metal complexes in the protein environment and their use in catalysis (BSA = bovine serum albumin, HSA = human serum albumin). | ||
These examples show that nonchiral catalysts can be bound by various strategies in a protein molecular container and it has been proven to be a good way to induce enantioselectivity in catalysis. This new approach to find the right combination of the host protein mutant and the guest metal complex adds a valuable tool to our pallet in the search for enatioselective catalysts.
7. Conclusions and perspectives
Activity, selectivity and stability are crucial parameters of a transition metal catalyst that are traditionally controlled by the ligands directly bound to a catalytically active metal, i.e. the first coordination sphere. Metal catalysts can, however, be encapsulated in various types of molecular containers, providing tools to control these parameters via the second coordination sphere. This review summarizes different ways to encapsulate catalysts in well-defined, homogeneous confined spaces. Encapsulation through covalent linkage, the template-ligand approach or hosting the catalyst as a guest in a molecular container or a protein-cavity has been demonstrated. As discussed in the various examples given in this review, such strategies can change certain steps in the catalytic cycle, giving rise to new kinetic profiles and altered selectivities. One of the main features observed in the examples discussed here is a higher stability of the encapsulated catalyst as the molecular container protects the active site from decomposition pathways, like self-deactivation through bridged species in epoxidation and cyclopropanation reactions. Furthermore, the capsular environment can induce substrate selectivity based on the size of the substrate and the aperture of the container, thus yielding high selectivity for the conversion of one substrate in a mixture of substrates. Although we are just at the beginning of exploring the possibilities in this area, it is clear that a molecular container can affect new regio- and enantio-selectivities in catalysis, which can sometimes be difficult to achieve by traditional means. Concluding from the examples provided in this review, the confinement of the catalyst has a clear and positive effect on the catalytic activities and/or selectivities. However, although these approaches are promising, the precise mode of operation is frequently poorly understood, while this understanding is essential for the future development of new capsules by rational design. It is relevant to note that microporous materials like metal–organic frameworks (MOFs)58 or porous organic polymers (POPs)59 have also been demonstrated to be excellent hosts for the confinement of catalysts. One of the challenges in this field is to predict the selectivity displayed by a predesigned encapsulated catalyst, be it in solution or in the solid state in the form of a MOF. This prediction requires detailed knowledge of the effects prompted by a confined space imposed by a capsule around a catalyst on its elementary reaction steps, such as oxidative addition, migratory insertion and reductive elimination. Obviously, understanding how these elementary steps are influenced by a surrounding cage is important in order to predict how cage effects can be exploited to increase the activity and selectivity of a catalyst, and may allow us to affect the rate determining steps at will. With this in mind, the next generation of confined catalysts should become a well-accepted new tool to arrive at transition metal catalysts with superior properties.Acknowledgements
Financial support was provided by the University of Amsterdam and the Netherlands Organization for Scientific Research-Chemical Sciences (NWO-CW: a Rubicon grant to R.G.-D., a TOP grant to J.N.H.R. and a VICI grant to B. de B.).Notes and references
- (a) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed; (b) R. Noyori, Nat. Chem., 2009, 1, 5–6 CrossRef CAS PubMed; (c) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed; (d) B. Cornils and W. A. Herrmann, Applied Homogeneous Catalysis with Organometallic Compounds, Wiley-VCH, Weinheim, Germany, 2nd edn, 2002 Search PubMed; (e) R. A. Sheldon, I. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (f) G. Rothenberg, Catalysis, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- D. Ringe and G. A. Petsko, Science, 2008, 320, 1428–1429 CrossRef CAS PubMed.
- (a) D. L. Pompliano, A. Peyman and J. R. Knowles, Biochemistry, 1990, 29, 3186 CrossRef CAS; (b) S. Lüdtke, P. Neumann, K. M. Erixon, F. Leeper, R. Kluger, R. Ficner and K. Tittmann, Nat. Chem., 2013, 5, 762–767 CrossRef PubMed; (c) M. Fujihashi, T. Ishida, S. Kuroda, L. P. Kotra, E. F. Pai and K. Miki, J. Am. Chem. Soc., 2013, 135, 17432–17443 CrossRef CAS PubMed; (d) N. Wu, Y. R. Mo, J. L. Gao and E. F. Pai, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2017–2022 CrossRef CAS PubMed; (e) W. J. Albery and J. R. Knowles, Angew. Chem., Int. Ed. Engl., 1977, 16, 285–293 CrossRef CAS PubMed.
- (a) M. M. Conn and J. Rebek Jr., Chem. Rev., 1997, 97, 1647–1668 CrossRef CAS PubMed; (b) J. Rebek Jr., Chem. Soc. Rev., 1996, 25, 255–264 RSC; (c) F. Hof, S. L. Craig, C. Nuckolls and J. Rebek Jr., Angew. Chem., Int. Ed., 2002, 41, 1488–1508 CrossRef CAS; (d) A. Lutzen, Angew. Chem., Int. Ed., 2005, 44, 1000–1002 CrossRef PubMed; (e) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC; (f) D. Ajami and J. Rebek Jr., Acc. Chem. Res., 2013, 46, 990–999 CrossRef CAS PubMed; (g) J. Rebek Jr., Angew. Chem., Int. Ed., 2005, 44, 2068–2078 CrossRef PubMed; (h) M. Yoshizawa and M. Fujita, Bull. Chem. Soc. Jpn., 2010, 83, 609–618 CrossRef CAS; (i) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed; (j) Z. Laughrey and B. C. Gibb, Chem. Soc. Rev., 2011, 40, 363–386 RSC.
- T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC.
- (a) Z. Dong, Q. Luo and J. Liu, Chem. Soc. Rev., 2012, 41, 7890–7908 RSC; (b) H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS PubMed; (c) M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed; (d) B. Breiner, J. K. Clegg and J. R. Nitschke, Chem. Sci., 2011, 2, 51–56 RSC; (e) Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349–358 CrossRef CAS PubMed; (f) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed; (g) M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650–1659 CrossRef CAS PubMed; (h) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 351–360 CrossRef PubMed; (i) A. W. Kleij and J. N. H. Reek, Chem. – Eur. J., 2006, 12, 4218–4227 CrossRef CAS PubMed; (j) D. M. Vriezema, M. C. Aragones, J. A. A. W. Elemans, J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS PubMed; (k) B. C. Pemberton, R. Raghunathan, S. Volla and J. Sivaguru, Chem. – Eur. J., 2012, 18, 12178–12190 CrossRef CAS PubMed; (l) R. Gramage-Doria and J. N. H. Reek, ChemCatChem, 2013, 5, 677–679 CrossRef CAS.
- (a) S.-Y. Yu, T. Kusukawa, K. Biradha and M. Fujita, J. Am. Chem. Soc., 2000, 122, 2665–2666 CrossRef CAS; (b) M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469–471 CrossRef CAS.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed.
- (a) T. Murase, S. Horiuchi and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2866–2867 CrossRef CAS PubMed; (b) S. Horiuchi, T. Murase and M. Fujita, Chem. – Asian J., 2011, 6, 1839–1847 CrossRef CAS PubMed.
- (a) M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CrossRef CAS PubMed; (b) M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 11423–11429 CrossRef CAS PubMed.
- C. Gibson and J. Rebek Jr., Org. Lett., 2002, 4, 1887–1890 CrossRef CAS PubMed.
- M. Guitet, P. Zhang, F. Marcelo, C. Tugny, J. Jiménez-Barbero, O. Buriez, C. Amatore, V. Mouriès-Mansuy, J.-P. Goddard, L. Fensterbank, Y. Zhang, S. Roland, M. Ménand and M. Sollogoub, Angew. Chem., Int. Ed., 2013, 52, 7213–7218 CrossRef CAS PubMed.
- M. Jouffroy, R. Gramage-Doria, D. Armspach, D. Sémeril, W. Oberhauser, D. Matt and L. Toupet, Angew. Chem., Int. Ed., 2014, 53, 3937–3940 CrossRef CAS PubMed.
- (a) V. F. Slagt, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. Van Leeuwen, Angew. Chem., Int. Ed., 2001, 40, 4271–4274 CrossRef CAS; (b) V. F. Slagt, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2004, 126, 1526–1536 CrossRef CAS PubMed; (c) A. W. Kleij, M. Lutz, A. L. Spek, P. W. N. M. van Leeuwen and J. N. H. Reek, Chem. Commun., 2005, 3661–3663 RSC; (d) A. W. Kleij, M. Kuil, D. M. Tooke, A. L. Spek and J. N. H. Reek, Inorg. Chem., 2005, 44, 7696–7698 CrossRef CAS PubMed; (e) I. Jacobs, A. C. T. van Duin, A. W. Kleij, M. Kuil, D. M. Tooke, A. L. Spek and J. N. H. Reek, Catal. Sci. Technol., 2013, 3, 1955–1963 RSC; (f) V. Bocokic, M. Lutz, A. L. Spek and J. N. H. Reek, Dalton Trans., 2012, 41, 3740–3750 RSC.
- M. Kuil, T. Soltner, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2006, 128, 11344–11345 CrossRef CAS PubMed.
- V. Bocokić, A. Kalkan, M. Lutz, A. L. Spek, D. T. Gryko and J. N. H. Reek, Nat. Commun., 2013, 4, 2670 Search PubMed.
- T. Besset, D. W. Norman and J. N. H. Reek, Adv. Synth. Catal., 2013, 355, 348–352 CAS.
- (a) R. Bellini, S. H. Chikkali, G. Berthon-Gelloz and J. N. H. Reek, Angew. Chem., Int. Ed., 2011, 50, 7342–7345 CrossRef CAS PubMed; (b) R. Bellini and J. N. H. Reek, Chem. – Eur. J., 2012, 18, 7091–7099 CrossRef CAS PubMed.
- T. Gadzikwa, R. Bellini, H. L. Dekker and J. N. H. Reek, J. Am. Chem. Soc., 2012, 134, 2860–2863 CrossRef CAS PubMed.
- (a) R. Bellini and J. N. H. Reek, Eur. J. Inorg. Chem., 2012, 4684–4693 CrossRef CAS; (b) R. Gramage-Doria, R. Bellini, J. Rintjema and J. N. H. Reek, ChemCatChem, 2013, 5, 1084–1087 CrossRef CAS; (c) J. Flapper and J. N. H. Reek, Angew. Chem., Int. Ed., 2007, 45, 8590–8592 CrossRef PubMed; (d) T. S. Koblenz, H. L. Dekker, C. G. de Koster, P. W. N. M. van Leeuwen and J. N. H. Reek, Chem. Commun., 2006, 1700–1702 RSC; (e) T. S. Koblenz, H. L. Dekker, C. G. de Koster, P. W. N. M. van Leeuwen and J. N. H. Reek, Chem. – Asian J., 2011, 6, 2444–2462 CrossRef CAS PubMed; (f) T. S. Koblenz, H. L. Dekker, C. G. de Koster, P. W. N. M. van Leeuwen and J. N. H. Reek, Chem. – Asian J., 2011, 6, 2431–2443 CrossRef CAS PubMed.
- T. Linker, Angew. Chem., Int. Ed. Engl., 1997, 36, 2060–2062 CrossRef CAS.
- J. A. A. W. Elemans, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Chem. Commun., 2000, 2443–2444 RSC.
- P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Nature, 2003, 424, 915–918 CrossRef CAS PubMed.
- C. Monnereau, P. H. Ramos, A. B. C. Deutman, J. A. A. W. Elemans, R. J. M. Nolte and A. E. Rowan, J. Am. Chem. Soc., 2010, 132, 1529–1531 CrossRef CAS PubMed.
- M. L. Merlau, M. del Pilar Mejia, S. T. Nguyen and J. T. Hupp, Angew. Chem., Int. Ed., 2001, 40, 4239–4242 CrossRef CAS.
- S. J. Lee, S.-H. Cho, K. L. Mulfort, D. M. Tiede, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2008, 130, 16828–16829 CrossRef CAS PubMed.
- L. Kürti, M. M. Blewett and E. J. Corey, Org. Lett., 2009, 11, 4592–4595 CrossRef PubMed.
- H. Lu, W. I. Dzik, X. Xu, L. Wojtas, B. de Bruin and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 8518–8521 CrossRef CAS PubMed.
- W. I. Dzik, X. Xu, X. P. Zhang, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2010, 132, 10891–10902 CrossRef CAS PubMed.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS PubMed.
- M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2013, 19, 10170–10178 CrossRef CAS PubMed.
- X. Cui, X. Xu, L. Wojtas, M. M. Kim and X. P. Zhang, J. Am. Chem. Soc., 2012, 134, 19981–19984 CrossRef CAS PubMed.
- M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2014, 20, 4880–4884 CrossRef CAS PubMed.
- D. H. Leung, D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., 2004, 116, 981–984 ( Angew. Chem., Int. Ed. , 2004 , 43 , 963–966 ) CrossRef.
- D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 2746–2747 CrossRef CAS PubMed.
- C. J. Brown, G. M. Miller, M. W. Johnson, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2011, 133, 11964–11966 CrossRef CAS PubMed.
- Z. J. Wang, C. J. Brown, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 7358–7360 CrossRef CAS PubMed.
- W. M. Hart-Cooper, K. N. Clary, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2012, 134, 17873–17876 CrossRef CAS PubMed.
- V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268–4315 CrossRef CAS PubMed.
- E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed.
- Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100–105 CrossRef CAS PubMed.
- (a) L. MacGillivray and J. Atwood, Nature, 1997, 389, 469–472 CrossRef CAS PubMed; (b) E. S. Barrett, T. J. Dale and J. Rebek Jr., J. Am. Chem. Soc., 2008, 130, 2344–2350 CrossRef CAS PubMed.
- (a) A. Shivanyuk and J. Rebek Jr., Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7662–7665 CrossRef CAS PubMed; (b) L. Avram and Y. Cohen, Org. Lett., 2002, 4, 4365–4368 CrossRef CAS PubMed.
- A. Cavarzan, A. Scarso, P. Sgarbossa, G. Strukul and J. N. H. Reek, J. Am. Chem. Soc., 2011, 133, 2848–2851 CrossRef CAS PubMed.
- C. Nevado and A. M. Echavarren, Chem. – Eur. J., 2005, 11, 3155–3164 CrossRef CAS PubMed.
- A. Cavarzan, J. N. H. Reek, F. Trentin, A. Scarso and G. Strukul, Catal. Sci. Technol., 2013, 3, 2898–2901 CAS.
- M. A. Sarmentero, H. Fernández-Pérez, E. Zuidema, C. Bo, A. Vidal-Ferran and P. Ballester, Angew. Chem., Int. Ed., 2010, 49, 7489–7492 CrossRef CAS PubMed.
- (a) T. Ueno, H. Tabe and Y. Tanaka, Chem. – Asian J., 2013, 8, 1646–1660 CrossRef CAS PubMed; (b) P. J. Deuss, R. den Heeten, W. Laan and P. C. J. Kamer, Chem. – Eur. J., 2011, 17, 4680–4698 CrossRef CAS PubMed; (c) M. R. Ringenberg and T. R. Ward, Chem. Commun., 2011, 47, 8470–8476 RSC; (d) J. Steinreiber and T. R. Ward, Coord. Chem. Rev., 2008, 252, 751–766 CrossRef CAS PubMed.
- T. R. Ward, Acc. Chem. Res., 2011, 44, 47–57 CrossRef CAS PubMed.
- (a) C. Letondor, N. Humbert and T. R. Ward, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4683–4687 CrossRef CAS PubMed; (b) A. Pordea, M. Creus, C. Letondor, A. Ivanova and T. R. Ward, Inorg. Chim. Acta, 2010, 363, 601–604 CrossRef CAS PubMed.
- M. Dürrenberger, T. Heinisch, Y. M. Wilson, T. Rossel, E. Nogueira, L. Knörr, A. Mutschler, K. Kersten, M. J. Zimbron, J. Pierron, T. Schirmer and T. R. Ward, Angew. Chem., Int. Ed., 2011, 50, 3026–3029 CrossRef PubMed.
- J. Pierron, C. Malan, M. Creus, J. Gradinaru, I. Hafner, A. Ivanova, A. Sardo and T. R. Ward, Angew. Chem., Int. Ed., 2008, 47, 701–705 CrossRef CAS PubMed.
- T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500–503 CrossRef CAS PubMed.
- M. Ohashi, T. Koshiyama, T. Ueno, M. Yanase, H. Fujii and Y. Watanabe, Angew. Chem., Int. Ed., 2003, 42, 1005–1008 CrossRef CAS PubMed.
- T. Ueno, T. Koshiyama, S. Abe, N. Yokoi, M. Ohashi, H. Nakajima and Y. Watanabe, J. Organomet. Chem., 2007, 692, 142–147 CrossRef CAS PubMed.
- A. Mahammed and Z. Gross, J. Am. Chem. Soc., 2005, 127, 2883–2887 CrossRef CAS PubMed.
- M. T. Reetz and N. Jiao, Angew. Chem., Int. Ed., 2006, 45, 2416–2419 CrossRef CAS PubMed.
- (a) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; (b) A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC; (c) P. Valvekens, F. Vermoortele and D. De Vos, Catal. Sci. Technol., 2013, 3, 1435 RSC; (d) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 6011–6061 RSC.
- (a) P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS; (b) Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
| This journal is © The Royal Society of Chemistry 2015 |