 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGas-phase chemistry of molecular containers
Zhenhui
Qi
,
Thomas
Heinrich
,
Suresh
Moorthy
and
Christoph A.
Schalley
*
Institut für Chemie und Biochemie, Freie Universität Berlin, Takustraße 3, 14195 Berlin, Germany. E-mail: christoph@schalley-lab.de; Fax: +49 30 838 55367; Tel: +49 30 838 52639
First published on 24th June 2014
Abstract
The remarkable technical advances in mass spectrometry during the last decades, including soft ionisation techniques, the coupling of electrospray ionisation to flow reactors, and the broad scope of tandem mass spectrometric experiments applicable to mass-selected ions allow investigating the chemistry of molecular capsules in solution as well as in the absence of any environment. With these methods, mass spectrometry is capable of answering many questions starting from providing analytical characterisation data (elemental composition, stoichiometry, etc.) to structural aspects (connectivities, positions of building blocks in supramolecular complexes) and to the examination of solution and gas-phase reactivity including reactions inside molecular containers. The present article reviews this work with a focus rather on the chemical questions that can be answered than on the technical specialities of (tandem) mass spectrometry.
 Zhenhui Qi | Zhenhui Qi received his Master degree under the supervision of Prof. Junqiu Liu and Prof. Jiacong Shen from Jilin University, China in 2009. Then, he obtained his PhD degree at Freie Universität Berlin, Germany, in the group of Prof. Dr Christoph A. Schalley. His research interests focus on mass spectrometry, responsive materials, and functional surfaces. |
 Thomas Heinrich | Thomas Heinrich studied chemistry at Freie Universität Berlin. He obtained his Bachelor degree under the guidance of Rainer Haag and his Master degree in the group of Christoph A. Schalley. Since March 2013 he has been carrying out his doctoral research work in the same group. His research focuses mainly on Supramolecular Chemistry at interfaces where he develops switchable surfaces with the aim of establishing molecular machines. |
 Suresh Moorthy | Suresh Moorthy received both, the Bachelor and Master of Science in Chemistry from Bharathidasan University, Trichy, India. He obtained his PhD from Central Salt and Marine Chemicals Institute, Bhavnagar, India in the area of Supramolecular Chemistry under the direction of Dr Amitava Das. He spent one year Humboldt postdoc Fellowship at Freie Universität Berlin, Germany, with Chris Schalley in 2013. He is currently doing his postdoc at the University of Queensland, Brisbane, Australia. His research interest focuses on photoinduced energy transfer in self-assembled multichromophoric supramolecular systems. |
 Christoph A. Schalley | Christoph Schalley studied chemistry at the University of Freiburg/Germany and graduated in 1993 from TU Berlin. In his PhD work with Helmut Schwarz, he received his education as a mass spectrometrist and gas-phase chemist. After postdoctoral work on supramolecular chemistry with Julius Rebek at The Scripps Research Institute, La Jolla, USA, he combined both topics when starting his own research group at the University of Bonn. In 2005, he was appointed professor of organic chemistry at FU Berlin. The Schalley group's research topics are diverse and comprise supramolecular chemistry in the gas phase as well as in solution and at interfaces. |
1 Introduction
Molecular containers1 create void cavities inside their walls, in which molecules can be encapsulated that are then isolated from the bulk outside the container. Such encapsulation is reminiscent of natural binding pockets that occur in enzymes or ribosomes – in particular when the molecular container is based on non-covalent bonding and can reversibly open and close. Fabricating synthetic molecular containers is a way to affect the guest molecule in a highly controlled and specific manner and to change its reactivity significantly by providing a particular environment to it.2 Since Breslow's pioneering work on cyclodextrins as enzyme mimics,3 the design of synthetic molecular containers has progressed in an amazing way making use of calixarenes and resorcinarenes,4 cucurbiturils,5 carcerands,6 and other scaffolds.7 Among supramolecular containers, those based on hydrogen bonding1,8 and metal coordination9 are the most prominent and meanwhile provide quite huge interior spaces. For example, Fujita's metallo-supramolecular M24L48 containers10 are made from 72 components (48 identical ligands on the edges and 24 metal ions at the vertices of a rhombicuboctahedron) and its molecular weight exceeds 20 kDa. Similarly, the hydrogen-bonded resorcinarene and pyrogallarene hexamers reported by Atwood11 and Mattay12 comprise an inner volume of ca. 1200 Å3. Another interesting aspect of supramolecular containers is their reversible formation. Encapsulation in these capsules therefore creates a mechanical barrier for guest encapsulation and release.13 As non-covalent bonds are sensitive to effects of the environment and potentially also to chemical stimuli added to the capsules, options exist to fine-tune the gating processes and thus to control encapsulation and to alter guest reactivity inside catalytically active containers.14With containers at hand that grow in size as well as their complexity, their characterisation becomes increasingly challenging. For a convincing characterisation, a number of different complementary methods need to be applied. While X-ray crystallography and NMR spectroscopy are currently and will likely continue to be the two most commonly employed analytical methods, mass spectrometry has seen a quite vivid development recently and is now a very valuable technique for the characterisation of supramolecules15 providing the three “S” advantages outlined by McLafferty: specificity, sensitivity and speed.16 The intact ionisation of complete viruses – such as the tobacco mosaic virus with its more than 2100 non-covalently bound protein subunits surrounding a templating RNA strand17 – and intact virus capsids – such as the hepatitis B virus capsids18 – clearly shows how far the limits of the characterisation of molecular capsules by mass spectrometry can be pushed forward.
Beyond analytical characterisation, mass spectrometry also serves for the analysis of solution processes. In particular, electrospray ionisation can be directly coupled to liquid chromatography as well as mixed-flow devices19 or even microfluidics20 (Fig. 1). Studies of both, biological and chemical processes,21 such as protein folding,22 enzyme catalysis,23 the identification of reaction intermediates24 and of error correction processes in self-assembly19 illustrate the potential of mass spectrometry for monitoring processes occurring in solution.
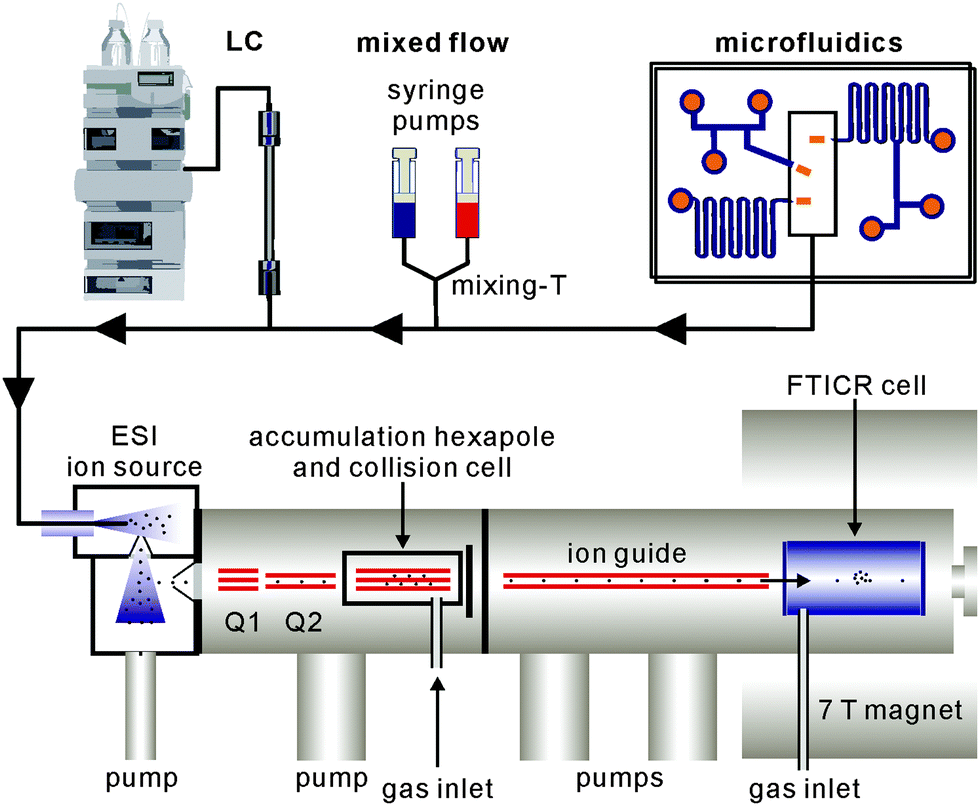 | ||
| Fig. 1 Schematic illustration of various devices that can be interfaced with ESI-MS: liquid chromatography (left), mixed-flow reactors (centre) and microfluidic devices (right). We show here the coupling to an electrospray ionisation Fourier-transform ion-cyclotron-resonance (ESI-FTICR) mass spectrometer. Coupling to other types of ESI instruments is similarly possible. | ||
On the other hand, mass spectrometry is a method for the investigation of completely desolvated ions in the high vacuum. The absence of any environment provides access to the ions' intrinsic properties which differ significantly as compared to their properties in condensed phase. This is particularly true for non-covalent complexes. In addition, reactivity studies on isolated ions in the gas phase occur under conditions which do not allow the dynamic exchange processes between the supramolecules. While one, for example, observes fast guest and building block exchanges in solution, reactivity studies in the gas phase rather reveal intramolecular rearrangements and therefore provide a very different view on reactivity.
Modern mass spectrometers provide a number of different tandem MS experiments that can be used to investigate the structure and reactivity of a desired ion in the gas phase. The prerequisite is a mass-selection step prior to the gas-phase experiment (Fig. 2, top). Different types of mass spectrometers perform this mass-selection differently. The figure shows a quadrupole as an example. Undesired ions (blue and green) formed in the ion source travel on unstable trajectories and do not reach the end of the quadrupole, while the parent ion of interest (red) can further be manipulated after this selection step. Once the desired ions are isolated, different fragmentation experiments (Fig. 2, second row) can be performed, among them collision-induced dissociation (CID), infrared multiphoton dissociation (IRMPD), electron-capture dissociation (ECD) or blackbody infrared dissociation (BIRD). Carefully interpreted fragmentation data contain information on many aspects of the ions under study. For supramolecular complexes, the stoichiometry and arrangement of the non-covalently bound subunits in the complex relative to each other can be acquired. Thermodynamic aspects, e.g. the stability of the complex ions or a ranking of binding energies in the gas phase, can also be elucidated. Furthermore, mechanistic information on the fragmentation pathways can be obtained. Isotopic labelling is often very helpful for the interpretation of the fragmentation data. However, not only fragmentation reactions are possible. In ion trap or Fourier-transform ion-cyclotron-resonance (FTICR) mass spectrometers, also bimolecular reactions with sufficiently volatile neutral reactants can be performed. H/D-exchange reactions are a prominent example (Fig. 2, third row).25 Recently, H/D-exchange experiments even revealed how dynamic supramolecular complexes can be and how mobile their subunits are.26 Finally, ion mobility spectrometry mass spectrometry (IMS-MS; Fig. 2, bottom) is a quickly growing technology, since commercial instruments have become available. Briefly, ions of interest are pulled by a low electric field or a so-called traveling wave through a gas-filled drift tube. The larger the collision cross section of the ion, the later it arrives at the detector. Consequently, the arrival time distributions contain size information. Although these experiments have not seen extensive use in the characterisation of molecular containers, they are capable of separating isomeric ions of the same m/z, if they differ sufficiently in collision cross section.
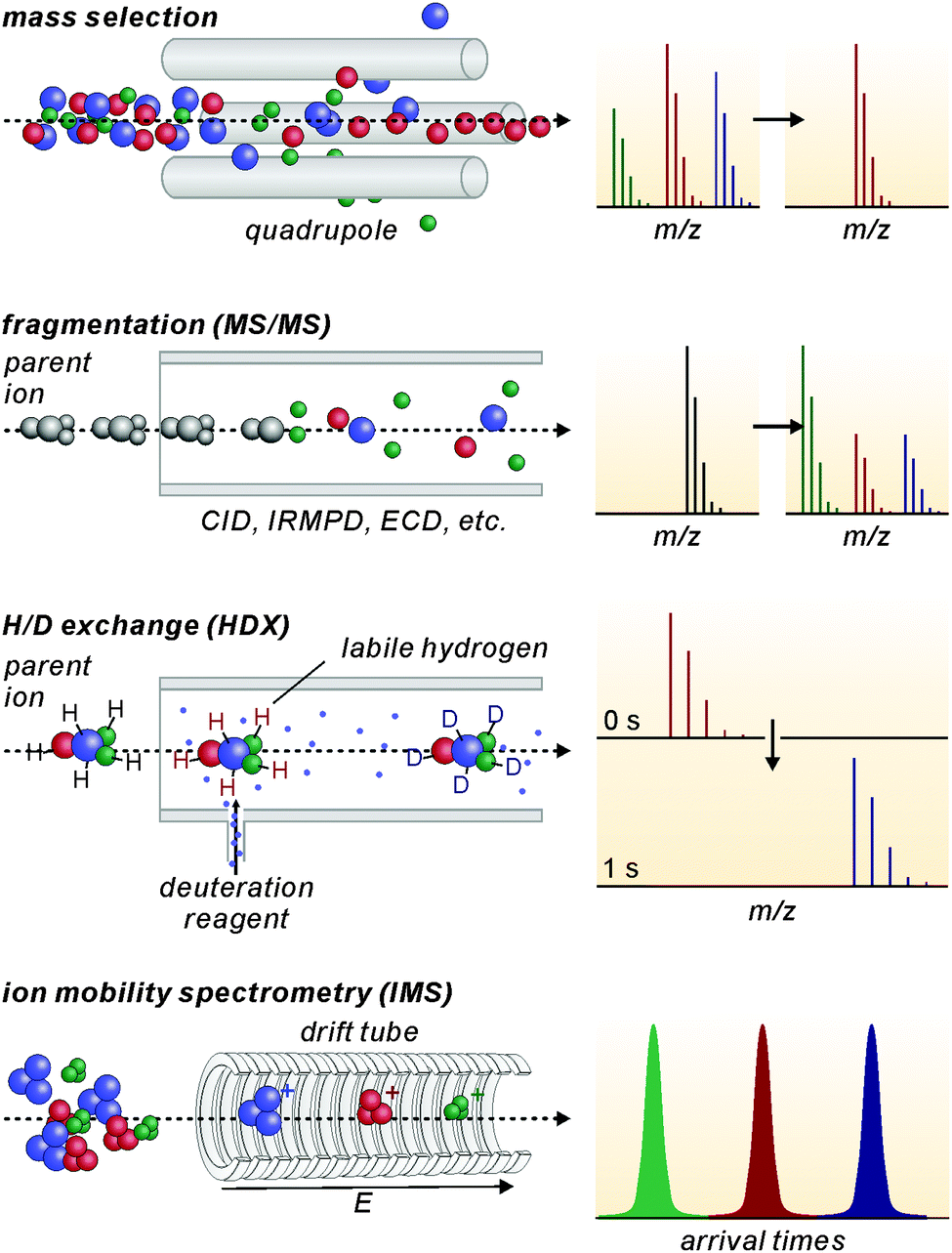 | ||
| Fig. 2 Schematic illustration of some common tandem MS (MS/MS) experiments. After the mass-selection step (top), fragmentation reactions (second row) or bimolecular reactions (third row) can be performed with the isolated parent ions. A separation by the ions' collision cross sections is possible with ion mobility spectrometry mass spectrometry (bottom), in which the ions are pulled through a gas-filled drift tube with low electric fields. | ||
After this more general introduction into the gas-phase chemistry of supramolecular complexes and the methodology used for its examination, this review will focus on the characterisation and investigation of molecular containers, their structure and reactivity by mass spectrometry. The examples are taken mostly, but not exclusively, from the areas of hydrogen-bonded and metallo-supramolecular containers.
2 Analytical and structural characterisation of supramolecular containers
In this section, we focus on metallo-supramolecular and hydrogen-bonded containers. They certainly form the two most prominent groups as both types of interaction, metal coordination and hydrogen bonding, are directional and thus provide geometric control in self-assembly reactions.Metallo-supramolecular cages
An efficient ionisation of containers and their host–guest complexes is the prerequisite for any mass spectrometric experiments to be done with them. Electrospray ionisation (ESI) is certainly the standard ionisation method. For weakly bound metallo-supramolecular complexes, coldspray ionisation (CSI) is very useful.27 This variant of ESI, in which the sample solution and the ion source are cooled, generates ions with lower internal energies that are less prone to fragmentation. Intact cages can thus be produced, which are otherwise already fragmenting to a large degree under common ESI conditions. Another ESI variant is sonic spray ionisation (SCI-MS) which was also used successfully to ionise fragile metallo-supramolecular cages.28 Many metallo-supramolecular assemblies contain transition metal cations and are easily ionised by stripping off counterions. When the ligand itself is the counterion and cannot easily be stripped off – like in Beer's resorcinarene-based polymetallic nanocages29 – an oxidation of one or more metal ions in the sample prior to ionisation can offer an alternative.Often, signals are observed that can be attributed to fragments of the cages. However, there is a second potential reason for their appearance. ESI concentrations are usually in the low μM regime. When metallo-supramolecular complexes start to dissociate at such low concentrations, incomplete assemblies may be observed just because they already exist in the sample solution at this low concentration, not because they are due to fragmentation. Increasing concentration to, e.g. 100 to 300 μM can be advantageous, even if the ion source then requires more extensive cleaning.
Fujita's lab has extensively utilised coldspray ionisation to examine metallo-supramolecular containers. Fig. 3 (top) shows a series of MnL2n cages10,30,31 which were prepared from bent ligands bearing two pyridyl groups and square-planar Pd(II) ions. Characteristic NMR signal shifts indicate complexation of ligand and metal ion, but do not as easily deliver size information unless, for example, extensive DOSY NMR experiments are performed. CSI-MS provides this information easily (Fig. 3, bottom). In all spectra, a charge-state distribution is observed related to different numbers of stripped-off counterions. Well-resolved isotopic patterns are in line with the calculated ones. Not unexpectedly when considering the low ion source temperatures, some of the ions also carry some solvent molecules. Endohedral functionalization of, for example, the M12L24 cage with 24 identical groups such as photo-responsive azobenzene32 or perfluoroalkyl chains33 increases the molecular mass significantly. CSI-MS still delivers conclusive mass information.
 | ||
| Fig. 3 CSI mass spectra of Fujita's MnL2nsphere 1 (n = 6), sphere 2 (n = 12), and sphere 3 (n = 24) metallo-supramolecular capsules. Adapted and reproduced from ref. 10, 30 and 31 with kind permission by the American Association for the Advancement of Science, the Royal Society of Chemistry and Wiley-VCH. | ||
Their ability to selectively encapsulate guests in their void interior space is an intriguing property of molecular containers.8a,34 Nitschke's self-assembled porphyrin-faced M8L6 cubic cage revealed interesting selectivity for the encapsulation of large aromatic guests (Fig. 4).35 Container Ni-1 with its cavity volume of ca. 1340 Å3 can encapsulate a stack of three coronenes or one fullerene molecule. Mass spectrometry provides clear evidence for target complex formation in agreement with NMR spectroscopy. Cage Ni-1 has a binding preference for C70 and a coronene triple over C60. The addition of coronene (3.5 equiv.) or C70 (2 equiv.) to a DMF solution of C60-saturated Ni-1 leads to the complete replacement of C60 as determined by ESI-MS as well as 1H NMR spectroscopy. After the addition of fullerene soot, neither empty cages nor C60-filled containers, but rather cages loaded with C70, C76, C78. C82 and C84 were observed by ESI-MS.
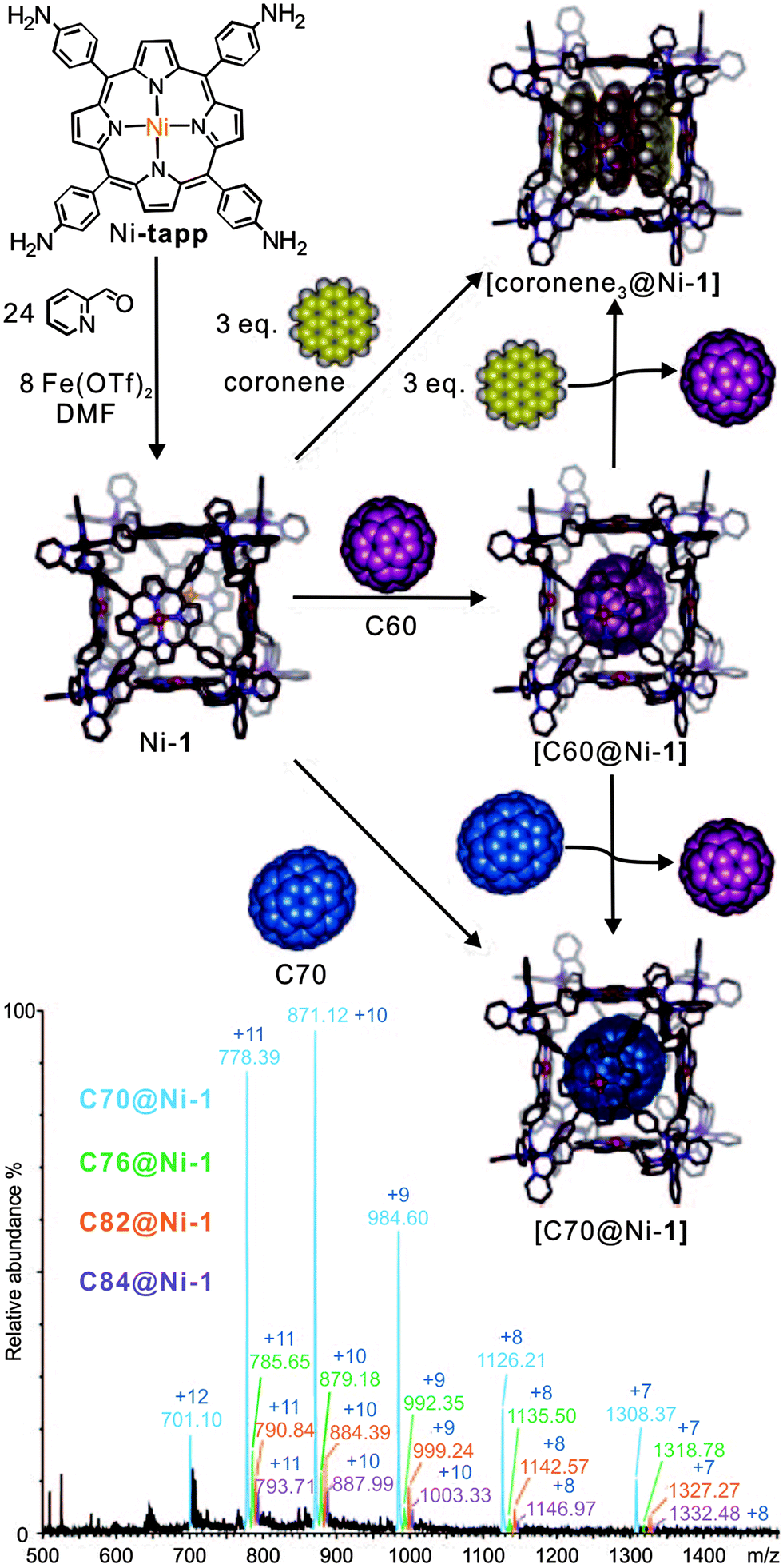 | ||
| Fig. 4 The synthesis of cubic cages Ni-1 through subcomponent self-assembly. The resulting cage hierarchically forms host–guest inclusion with coronene, C60, and C70. C70 and coronene guests bind more strongly than C60. The bottom ESI-MS shows the result of mixing fullerene soot with Ni-1. Adapted and reproduced from ref. 35 with kind permission by Wiley-VCH. | ||
When structural assignments are to be supported by mass spectrometric experiments, specific complexation needs to be distinguished from so-called unspecific binding which frequently occurs in ESI mass spectrometry. Often, changes in sample concentration already provide insight into this matter, but tandem mass spectrometry can also contribute significantly as exemplified by the metallo-supramolecular [7]catenane M4A6B6 in Fig. 5.36 Each edge of the tetrahedron bears one naphthalene diamide (NDI) ligand which can thread through bis-1,5-(dinaphtho)-[38]crown-10 B. Consequently, one expects the M4A6B6 tetrahedron to be the largest complex in the series. Nevertheless, complexes up to M4A6B118+ are observed in the ESI mass spectra and one structure-related question is certainly, how many crown ethers are threaded and thus bound by mechanical bonds. Infrared multiphoton-dissociation (IRMPD) experiments provide an answer: while M4A6B68+ fragments through two competing pathways, i.e. the loss of a crown-ether and the charge-separation-driven dissociation of the tetrahedral scaffold into two M2A3B34+, M4A6B78+ exclusively loses a crown ether first and then continues to fragment just like the M4A6B68+ ion. The loss of the first crown ether from the M4A6B78+ ion is thus energetically more favourable than the crown ether loss from M4A6B68+. The conclusion is that M4A6B68+ bears six threaded crowns which require the tetrahedron to open prior to dissociation, while the seventh crown ether is not threaded and thus easily lost without opening the complex.
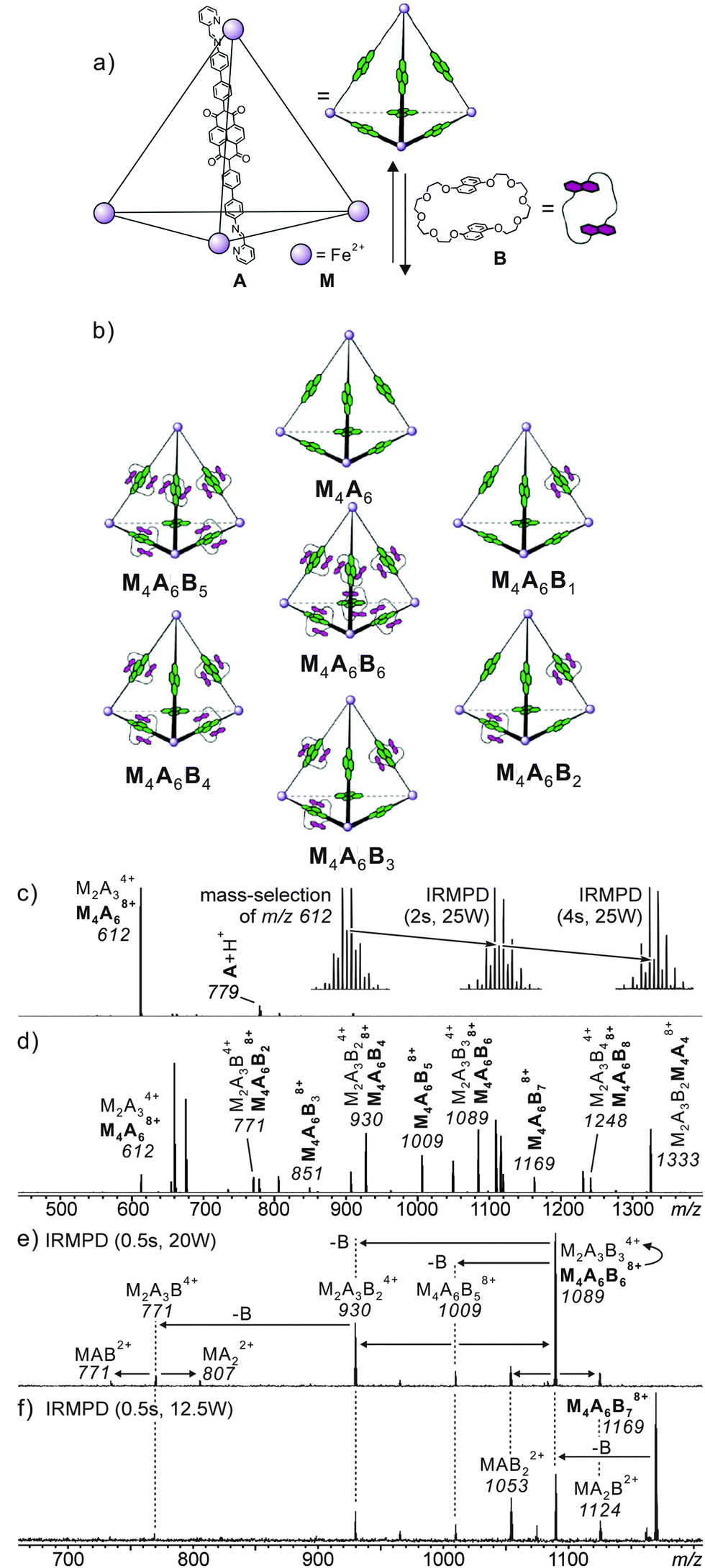 | ||
Fig. 5 (a) Synthesis of M4A6; (b) representation of a dynamic combinatorial library (DCL) formed upon addition of excess crown ether B. (c) ESI-FTICR mass spectrum of M4A6. Insets show isotope patterns of m/z 612 after mass-selection at different time intervals of irradiation in an IR multiphoton dissociation experiment. The gradual growth of the [M2A3]4+ ion (peak spacing of 1/4 amu) at the expense of the [M4A6]8+ parent ion, indicating the imposed [M2A3]4+ ions are the fragmentation product during the ionisation process. The similar fragmentation also occurred for the [M4A6B6]8+ parent ion. (d) ESI-FTICR mass spectrum of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 cage–crown ether mixture. (e,f) Spectra obtained from IRMPD experiments performed with mass-selected [M4A6C6]8+ and [M4A6C7]8+ ions, respectively. Adapted and reproduced from ref. 36 with kind permission by Wiley-VCH. :10 cage–crown ether mixture. (e,f) Spectra obtained from IRMPD experiments performed with mass-selected [M4A6C6]8+ and [M4A6C7]8+ ions, respectively. Adapted and reproduced from ref. 36 with kind permission by Wiley-VCH. | ||
Hydrogen-bonded capsules
Because of its usually somewhat harsher ionisation conditions, matrix-assisted laser desorption/ionisation (MALDI) has very rarely been used with success to ionise hydrogen-bonded containers. One notable example is certainly the barbiturate/melamine stacks made by Reinhoudt et al.37 When hydrogen-bonded containers are to be investigated by the softer ESI-MS, one nevertheless frequently encounters the difficulty that non-competitive, rather unpolar solvents are not the best spray solvents for electrospray ionisation. Vice versa, polar organic solvents that are good spray solvents often lead to the dissociation of the desired complexes. In addition, charge labelling is required, if the capsule is neutral. The encapsulation of charged guests is a convenient way to approach both problems.38 On the one hand, the necessary charge is present inside the cavity of the capsule and stripping off the non-encapsulated counterion is quite easily possible. On the other hand, the non-covalent interactions between the ion inside the container and the container walls are usually stronger than those of an equivalent neutral guest and thus stabilise the container. This usually allows adding at least a few percent of a polar solvent to the sample thus facilitating the electrospray process.Rebek's softballs (Fig. 6) illustrate this.1a Monomers S1–S4 consist of two glycoluril binding sites connected by spacers of different lengths. The corresponding dimers thus bear differently sized cavities. 1H NMR studies in aprotic, unpolar solvents like chloroform or xylene provided evidence for dimer formation as well as the encapsulation of suitable guests. In order to detect the dimeric capsules by ESI-MS, quaternary ammonium ions with weakly coordinating counterions (BF4−, PF6−) were used for ion labelling.39 With the same strategy, other hydrogen-bonded capsules – the tetrameric “footballs”,40 the so-called flexiballs,41 in which the binding sites are connected to a central scaffold in a less rigid fashion by single bonds, cavitand-based capsules large enough to encapsulate host–guest complexes thus forming Matroshka-doll-like molecule-in-molecule-in-molecule complexes42 and tetraurea calixarene capsules43 – were successfully ionised.
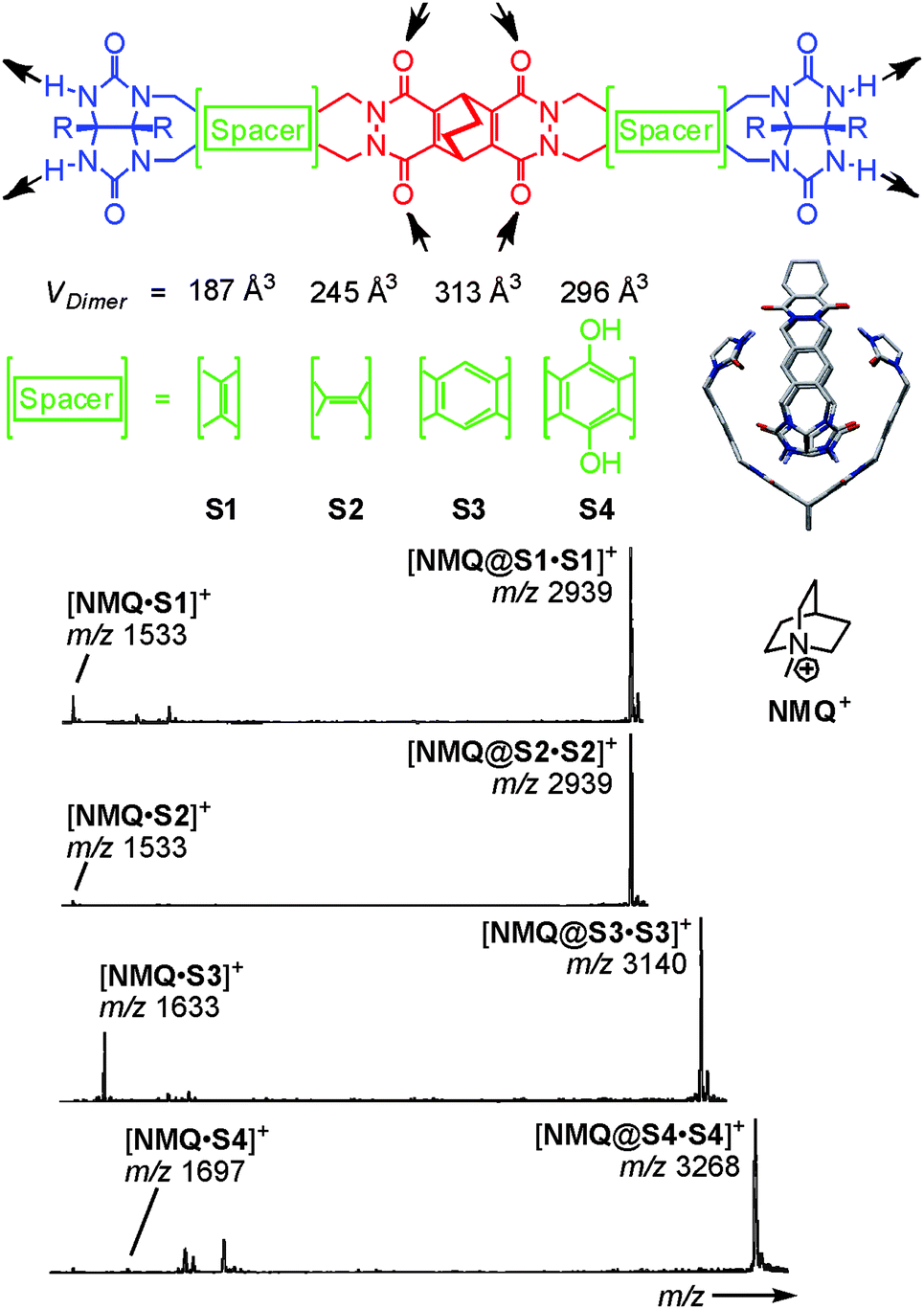 | ||
| Fig. 6 Top: chemical structures of the self-complementary building blocks present in molecular softballs. Arrows: H bond donors and acceptors. Right: force-field-optimized structure of the H-bonded dimeric capsule S3·S3; solubilizing R groups (n-heptylphenyl) and carbon-bound H atoms are omitted for clarity. Bottom: ESI mass spectra of 50 mM chloroform solutions of softballs S1·S1–S4·S4 with 1 eq. of guest cation NMQ+. Adapted and reproduced from ref. 39 with kind permission by Wiley-VCH. | ||
Evidence for intact capsular structures with guests residing inside the capsules' cavities came from a series of experiments that demonstrated (i) guest binding to be size-selective, (ii) capsule formation not to occur for deformed capsule monomers or monomers with protected binding sites and (iii) covalent bond cleavage to be energetically competitive in the gas phase with the loss of the guest cation. The latter fragmentation experiment indicates the barrier of guest loss to be high – well consistent with a steric barrier created by the capsule walls that need to be opened prior to guest release.
Resorcinarene Re and pyrogallarene Py shown in Fig. 7 also form dimeric capsules,44 when a sufficiently small guest cation such as tetramethyl ammonium is encapsulated. With larger guests such as tetrahexyl ammonium or cobaltocenium, hexameric capsules are instead observed in solution and solid state. They bear one resorcinarene or pyrogallarene on each of the six faces of a cube.11,12,45 The resorcinarene hexamers carries eight water molecules on the corners of the cube completing the hydrogen bonding pattern. The cavity volume reaches 1200 Å3. Similar in size, the pyrogallarene hexamers does not require any water molecules to complete H-bonding.
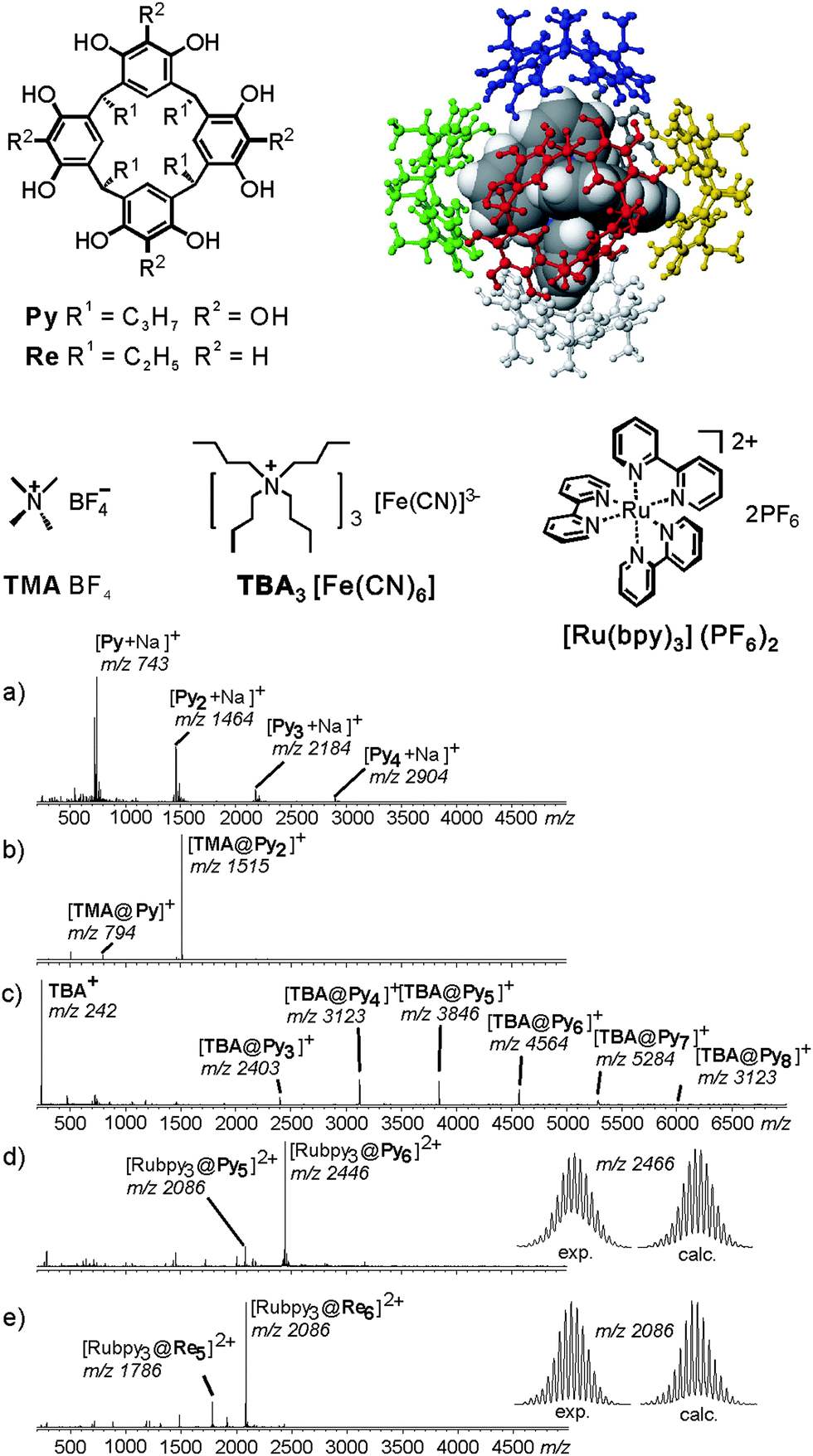 | ||
| Fig. 7 Encapsulation of ionic guests by a pyrogallarene capsule is used in order to investigate the complex by ESI-FTICR. ESI-FTICR mass spectra of (a) Py, (b) after addition of TMABF4, (c) TBA [Fe(CN)6]3− and (d) Ru(bpy)3(PF6−)2. (e) Same ESI-FTICR mass spectrum recorded with Re instead of Py. Insets: experimental and calculated isotope patterns of the hexamer ions [Ru(bpy)3@Py6]2+ and [Ru(bpy)3@Re6]2+. Adapted and reproduced from ref. 46 with kind permission by Wiley-VCH. | ||
Mass spectrometric studies of pyrogallarenes and resorcinarenes clearly reveal the choice of the templating guest cation to be decisive for hexamers formation.46 Electrospray ionisation of Py alone shows a series of unspecific oligomers to form (Fig. 7a). When small cations such as tetramethyl ammonium are added, dimers are the exclusive product (Fig. 7b). Larger cations with unfavourable symmetry like tetrabutyl ammonium lead to the formation of a series of larger oligomers. They include hexameric complexes, but their formation is not specific (Fig. 7c). When a pseudo-octahedral guest such as Ru(bpy)32+ is used which fits the cavity size and the symmetry of the hexameric capsule, the hexamer is almost the only product formed (Fig. 7d). Apparently, the formation of a hexameric capsule requires an appropriate template that fits into the cavity of this capsule with respect to size and symmetry. Control experiments with tetramethylated resorcinarene did not yield any hexamers. Evidence for the capsular structure and the guest dication located inside the capsule came from IRMPD experiments performed with mass-selected hexamers. They show guest release only to occur after the loss of three monomers. This is clearly in contrast to the structure of an empty capsule with the guest cation bound to the outer surface. For such an isomer, one would expect facile loss of the complete hexamers.
3 Monitoring solution reactivity by electrospray ionisation mass spectrometry
After discussing the intact ionisation and providing a few selected examples on the question how the structure of supramolecular containers can be approached by mass spectrometric experiments, we will now address mass spectrometric experiments which monitor reactions of containers in solution. We choose two processes as examples: building block exchanges in metallo-supramolecular containers and self-sorting phenomena in hydrogen-bonded dimeric tetraurea calixarene capsules.Building block exchange processes
Ligand exchange reactions are a characteristic dynamic feature of most metallo-supramolecular assemblies.47 The first case to be discussed here is that of self-assembled metallo-supramolecular squares decorated with G0 to G3 Fréchet dendrons (Fig. 8).48 The hydrophobic dendrons surround a nanometre-sized cavity at the core of the assemblies and eight amide-groups inside this pocket provide hydrogen-bonding sites for guest recognition. The square can form a dynamic library of up to 54 different isomeric squares which differ by the arrangement of the dendrons above and below the square plane. In addition, they can point inwards or outwards due to the torsional angle along the aryl–aryl bond. The corresponding NMR spectra are thus very complicated. However, as all possible isomers for each square have the same elemental composition and thus the same mass, ESI mass spectrometry provides a straightforward characterization.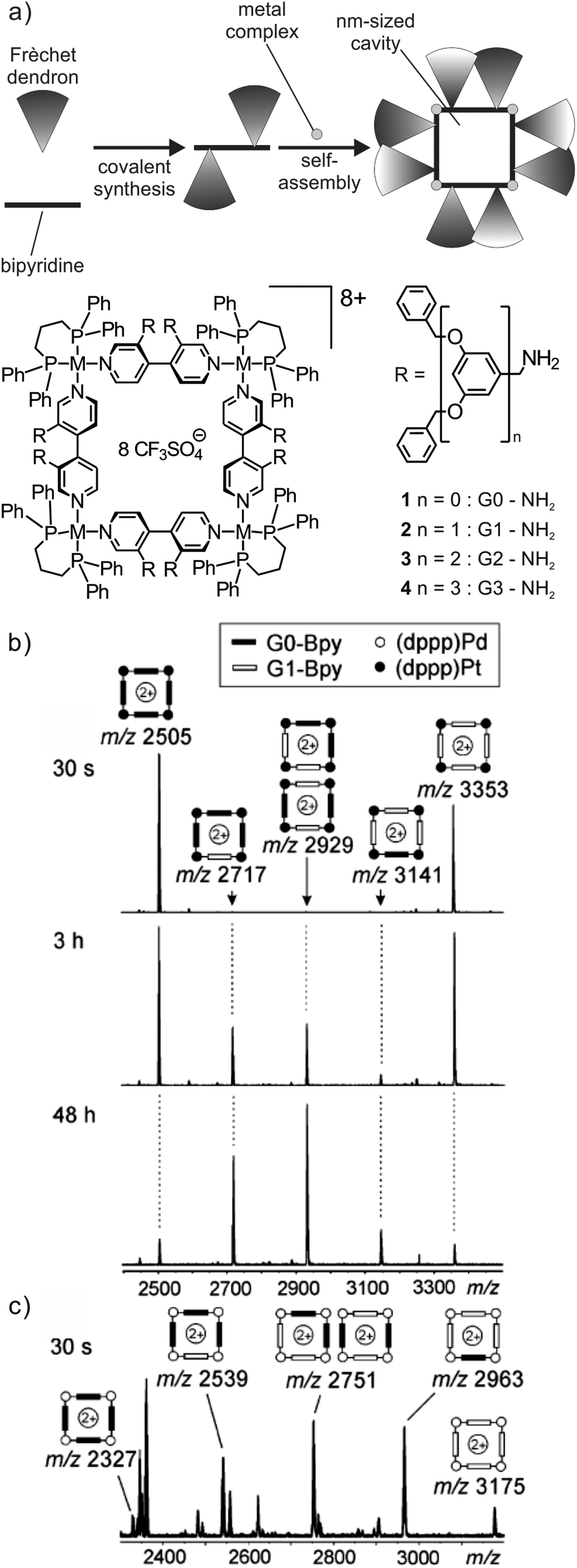 | ||
| Fig. 8 (a) Self-assembled metallo-supramolecular dendrimers: 4,4′-bipyridines bearing Fréchet dendrons assemble with the appropriate metal corners to yield dendron-decorated squares with a nanometre-sized hydrophobic cavity. ESI-FTICR mass spectra of 1:1 mixtures of (b) G0 and G1 Pt(II) squares 30 s, 3 h and 2 days after mixing and (c) G0 and G1 Pd(II) squares 30 s after mixing both squares. Adapted and reproduced from ref. 48 with kind permission from Wiley-VCH. | ||
When two squares with different bipyridine ligands are mixed (e.g. mixing independently formed G0 and G1 (dppp)Pt(II) squares, Fig. 8b), mass spectrometry can also be used to record the dynamic component exchange process. After the initial mixing and ionisation (ca. 30 s), two prominent peaks corresponding to the pre-generated G0 and G1 (dppp)Pt(II) squares are observed. Since the exchange process at the Pt centre is rather slow, equilibrium is reached only after ca. 2 days and no further intensity changes are observed. Owing to the absence of any cooperative interactions between the dendron moieties, all possible forms (G04:G03G11:G02G12:G01G13:G14) are observed in the mass spectra in an almost statistical 1:4:6:4:1. Small deviations from this ratio may be associated with different ESI response factors that are due to differences in desolvation energies. In contrast to the Pt(II) complexes, the corresponding (dppp)Pd(II) squares exhibit a rather fast exchange. A complete equilibration is already obtained after less than 30 s (Fig. 8c).
With a similar protocol, Stang et al. monitored dynamic ligand exchanges between coordination-driven self-assembled supramolecular polygons by ESI-MS even quantitatively using isotope labelling rather than different substituents to distinguish the two initial assemblies.49
Fujita's group employed mass spectrometry to monitor the ligand exchange between large M12L24 spherical complexes.50 This spherical cage is again based on Pd(II)–pyridine interactions. However, this assembly-mode leads to a significantly different kinetic behaviour: as shown in Fig. 9, two spheres, M12LA24 and M12LB24, bear nearly identical ligands which only differ with respect to the endohedral alkyl chains (n-C3H7 and n-C6H13 for LA and LB, respectively). Immediately after mixing, the two independently generated spheres in a 1:1 ratio, two similarly intense signals are observed corresponding to the two homo-24mers. Apparently, ligand exchange is rather slow. Time-dependent mass spectrometric experiments reveal a quite remarkable kinetic stability of the spheres against ligand exchange. Over longer reaction times, the first ligand exchange is observed with rather low abundance and has a half-life of ca. 20 days. In a second experiment, in which a free ligand was added to the previously prepared sphere of the second one, a much faster exchange process operates with a half-life for the first ligand exchange of ca. 23 min. The ligand exchange on tetracoordinate mononuclear metal complex [Pd(py)4](OTf) even occurs with a half-life of 36 s. Consequently, cooperative behaviour due to the fully closed coordination complex slows down the ligand exchange process. These experiments nicely show that ESI mass spectrometry can provide more profound insight into exchange processes in solution and at least produces semi-quantitative kinetic data.
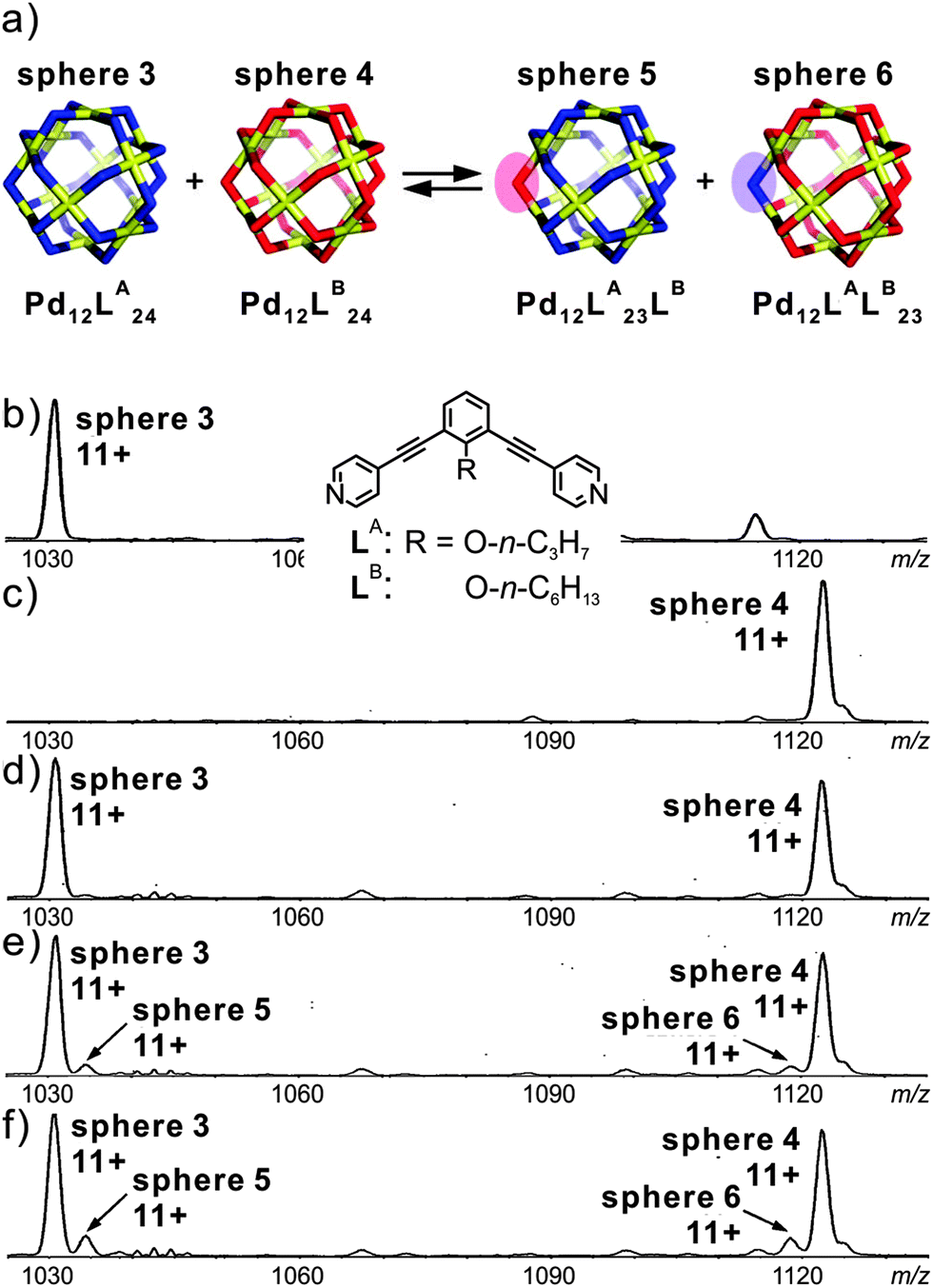 | ||
| Fig. 9 (a) Schematic representation of the ligand exchange between sphere 3 and sphere 4. (b) and (c) CSI-MS spectra (CH3CN, triflate salt) showing the expanded regions of the 11+ signal for sphere 3 and sphere 4. Time dependent spectra recorded after keeping the sample (d) overnight, (e) for 35 h, and (f) for 70 h at room temperature. Reproduced from ref. 50 with kind permission by the American Chemical Society. | ||
Self-sorting of tetraurea calixarene capsules
Recently, Böhmer's and our groups have reported a self-sorting system based on dimeric hydrogen-bonded tetraurea calixarene capsules made from U1–U11.51 As shown in Fig. 10, the dimerisation of eleven tetra-urea calix[4]arenes is controlled by steric factors. Many dimers are unable to form when both monomers bear wrongly positioned loops that would need to catenate upon dimerization. As catenation is not possible without breaking at least one covalent bond, these dimers do not form. Analogously, other monomers carry bulky stopper groups unable to penetrate the loops. Again, a mismatch of looped with stoppered monomers rules out the formation of a number of dimers. Following these simple rules, 35 possible dimers can be formed (Fig. 10b). When stoichiometry is controlled in addition to these steric effects, the number of dimers can further be reduced to six, because U11 can only bind to U1 and thus fully consumes this monomer. Among the remaining monomers, U10 can only bind to U2etc. Finally, only the six dimers highlighted in yellow in Fig. 10 are expected to emerge.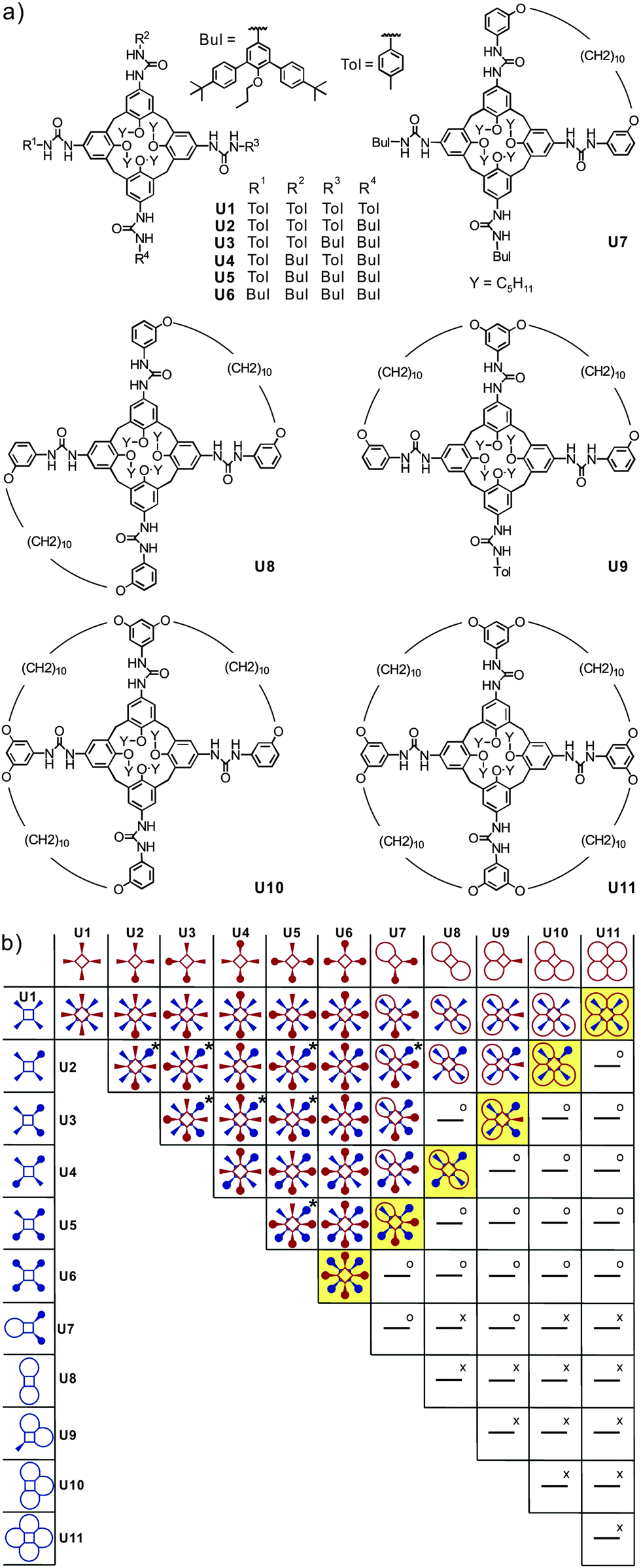 | ||
| Fig. 10 (a) Eleven tetraurea calix[4]arenes U1–U11 and (b) table containing the expected outcome of the self-sorting processes occurring in a stoichiometric mixture of all monomers U1–U11. Dimeric combinations marked “x” are impossible because of overlapping loops, while “o” indicates the impossibility of bulky residues to penetrate these loops. Two or three regioisomers are possible for dimers marked “x”. Adapted and reproduced from ref. 51 with kind permission by Wiley-VCH. | ||
For simpler mixtures of only a selection of two or three of these tetraurea calixarene monomers, NMR spectroscopy is still able to provide clear evidence for such self-sorting mixtures. However, when all monomers are mixed, an analysis by NMR becomes difficult. Using ion labelling with tetraethyl ammonium, mass spectrometry can then be used for the investigation of this mixture. Indeed, self-sorting in solution clearly occurs (Fig. 11). One result is that it takes several days to reach the final equilibrium. However, some minor signals with intensities lower than ca. 5% appear that can be assigned to mismatched dimers, which are not expected to form in a perfect self-sorting system. These signals may be due to a minor amount of unspecific dimerization. When a slight excess of the looped monomers is added, the mismatched dimers vanish and only the signals for the expected capsules are observed. This example illustrates how mass spectrometry helps to analyse self-sorting in rather complex mixtures. It thus adds valuable insight complementary to that obtained with other methods.
 | ||
| Fig. 11 ESI mass spectra of equimolar mixtures of (a) U1·U11, (b) U2·U10, (c) U3·U9, (d) U4·U8, and (e) U5·U7. (f) U6·U6. (g) ESI mass spectrum of an equilibrated mixture of all eleven monomers. (h,i) Spectra of the same mixture with 10% and 20% excess of loop-containing monomers U7–U11, respectively. For ion labelling, each capsule contains one tetraethyl ammonium ion as the guest. Reproduced from ref. 51 with kind permission by Wiley-VCH. | ||
4 Gas-phase experiments aiming at the investigation of intrinsic ion reactivity
In comparison to solution, the high vacuum inside the mass spectrometer provides a very different environment. The absence of solvation strengthens most non-covalent interactions as there is no competition with the solvent molecules. Experiments performed after mass-selecting an ion of interest therefore provide valuable insight into the intrinsic properties of the containers under study. Also, the dynamic subunit exchange processes discussed above are efficiently suppressed in the gas phase and intramolecular reactivity is observed rather than intermolecular exchange processes.52 This part commences with fundamental binding studies on resorcinarene containers followed by a mechanistic analysis of cage contraction processes that occur in the gas phase. Finally, bimolecular reactions, i.e. the gas-phase H/D-exchange will provide mechanistic as well as structural features on resorcinarene host–guest complexes and hydrogen-bonding in resorcinarene and pyrogallarene capsules.Fundamental binding studies: the importance of C–H⋯anion interactions
A number of different methods for the evaluation of gas-phase binding data exist, many of which, however, have not yet been applied to capsules and containers. Schrader and co-workers have examined a series of electrostatically bound calixarene-based capsules with respect to their relative stabilities53 and used collision experiments during which the collision energy was gradually increased. Determining the point at which 50% of the capsules are destroyed (CE50 value) provides a measure for its stability. The ranking of the stability of different capsules gained in the gas-phase correlates quite nicely with that determined from NMR titrations revealing the solvation effects to be of minor importance compared to the non-covalent interactions between the two electrostatically bound monomers.A similar strategy was utilized to unravel the importance of C–H⋯anion interactions54 in resorcinarene cavitands. While resorcinarenes themselves are cation binders, the corresponding cavitands (Fig. 12) turned out to bind anions rather than cations.55 Even weakly bound anions such as PF6− form complexes upon electrospray ionisation with a calculated binding energy of ca. 25 kJ mol−1. Even the complexation of sulphate dianions, which do not exist unsolvated in the gas phase due to an instantaneous electron loss, is observed in complexes such as [SO4@cav1]2− and [SO4@cav12]2−. Obviously, solvation of the sulphate dianion by the cavitand is sufficiently strong to stabilize it efficiently. Likely, multiple C–H⋯anion interactions between the methylene groups at the rim and the anion lead to quite significant binding energies that – according to calculations – need to be in the range of ∼130 to 160 kJ mol−1 for an efficient stabilization of the sulphate dianion.
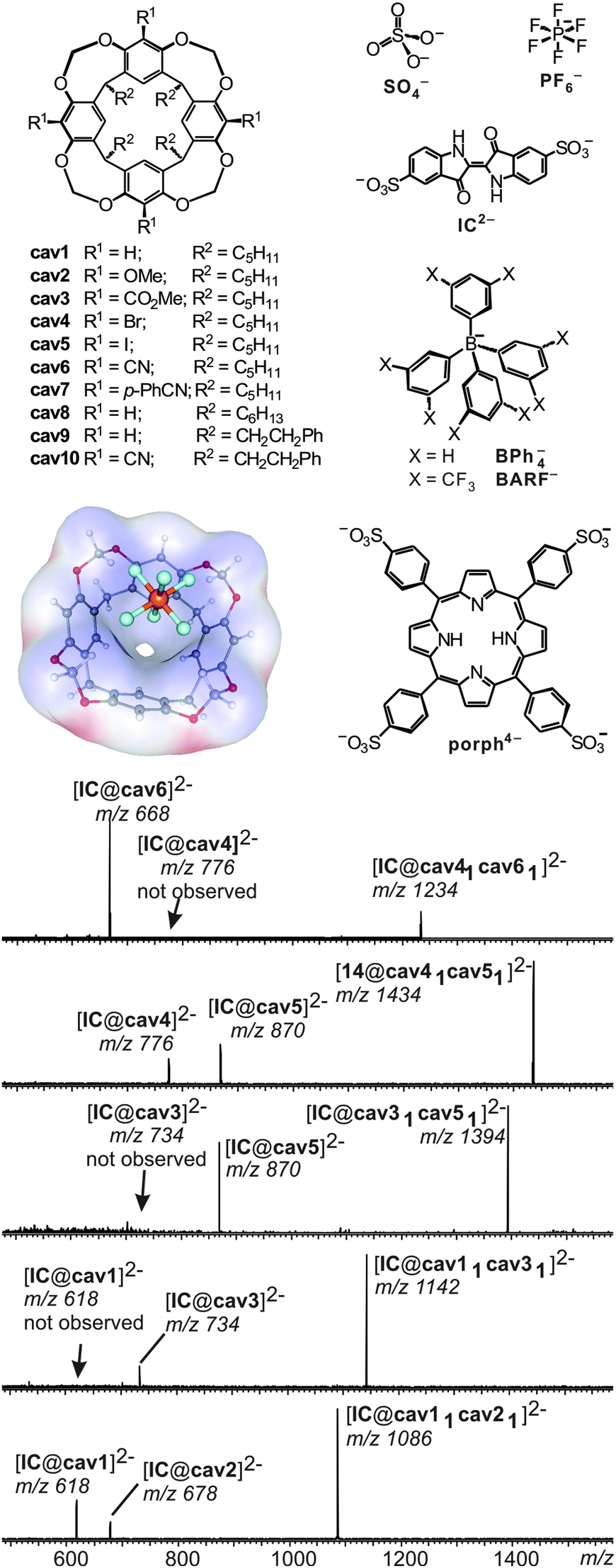 | ||
| Fig. 12 Top, left: anion-binding cavitands cav1–cav10 and B3LYP/6-31++G(d,p)-optimized structure of complex [PF6@cav1]− with the electrostatic potential mapped on the host's van der Waals surface. Top, right: guest anions. Bottom: CID experiments with mass-selected, doubly charged heterodimeric IC2− complexes. Adapted and reproduced from ref. 55 with kind permission by Wiley-VCH. | ||
In order to investigate substituent effects at the cavitand's aromatic rings, indigo carmine IC2− has been used as the guest. This dianion forms homo- and heterodimers when mixed with two differently substituted cavitands. Mass-selection of the heterodimer followed by collision-induced fragmentation allows comparing the two different cavitands incorporated in the complex directly. The less strongly bound cavitand dissociates to a greater extent and thus the relative peak intensities of the two possible 1:1 cavitand/IC2− product ions provide a direct measure of the relative gas-phase binding energy ranking. In agreement with the expected substituent effects, anion binding strengths increase in the series OMe ≤ H ≪ CO2Me ≪ Br ≈ I ≪ CN, i.e. from electron-donating to electron withdrawing substituents. Mass spectrometry consequently provides a means to conduct fundamental binding studies even on complexes that do not exist in solution due to the competition with solvent molecules or ion pairing effects.
Mechanistic analysis of intramolecular processes: a double cage contraction in the gas phase
As outlined above, one unique feature of gas-phase studies with mass-selected supramolecular complex ions in the gas phase is the fact that the dynamic equilibria which govern solution reactivity are efficiently suppressed. Consequently, the intramolecular reactivity of the ions of interest can be examined and offers completely new insight that is difficult to obtain from solution studies – if possible at all.In an earlier study,56 we were able to demonstrate simple metallo-supramolecular squares to fragment into triangles by a backside attack mechanism as shown in Fig. 13. The analysis was based on IRMPD experiments performed with the triply charged square. For fragmentation, the square needs to be opened first giving rise to a linear tetramer. This tetramer could in principle fragment with similar energy demand at several metal–nitrogen bonds along the chain. Nevertheless, the fragmentation is highly selective for the formation of doubly charged M3L32+ triangles and the corresponding singly charged 1:1 ML+ complex. Other fragmentation channels, e.g. the formation of two M2L2 fragments were not observed. The energetic preference for only one of the possible fragmentation reactions can be rationalized by a “backside-attack” mechanism: within the linear tetramer, the free pyridine coordination site first forms a new M–N bond followed by subsequent loss of the ML+ fragment. The binding energy gained by bond formation is thus available in the complex and supports dissociation. Such addition–elimination mechanisms are well-known for ligand exchanges at d8 metal ion complexes. Other pathways within the linear tetramer cannot follow such a backside-attack mechanism due to steric reasons and thus fragmentations at other M–N bonds do not benefit from a preceding bond formation and are thus energetically more demanding. Two conclusions can be drawn from these findings: (i) the strain within the triangle structure must be lower than the binding energy of a M–N bond and (ii) the M3L32+ product is initially a cyclic triangular structure before it continues to fragment. IRMPD experiments with the bowl-shaped M6L4 metallo-supramolecular cage shown in Fig. 13b reveal similar fragmentation mechanisms.57 Two “backside-attack” steps lead to the contraction into smaller cages and finally an M3L2+ cage is formed. Most interestingly, it is what is not observed in the mass spectra which finally leads to this mechanistic analysis.
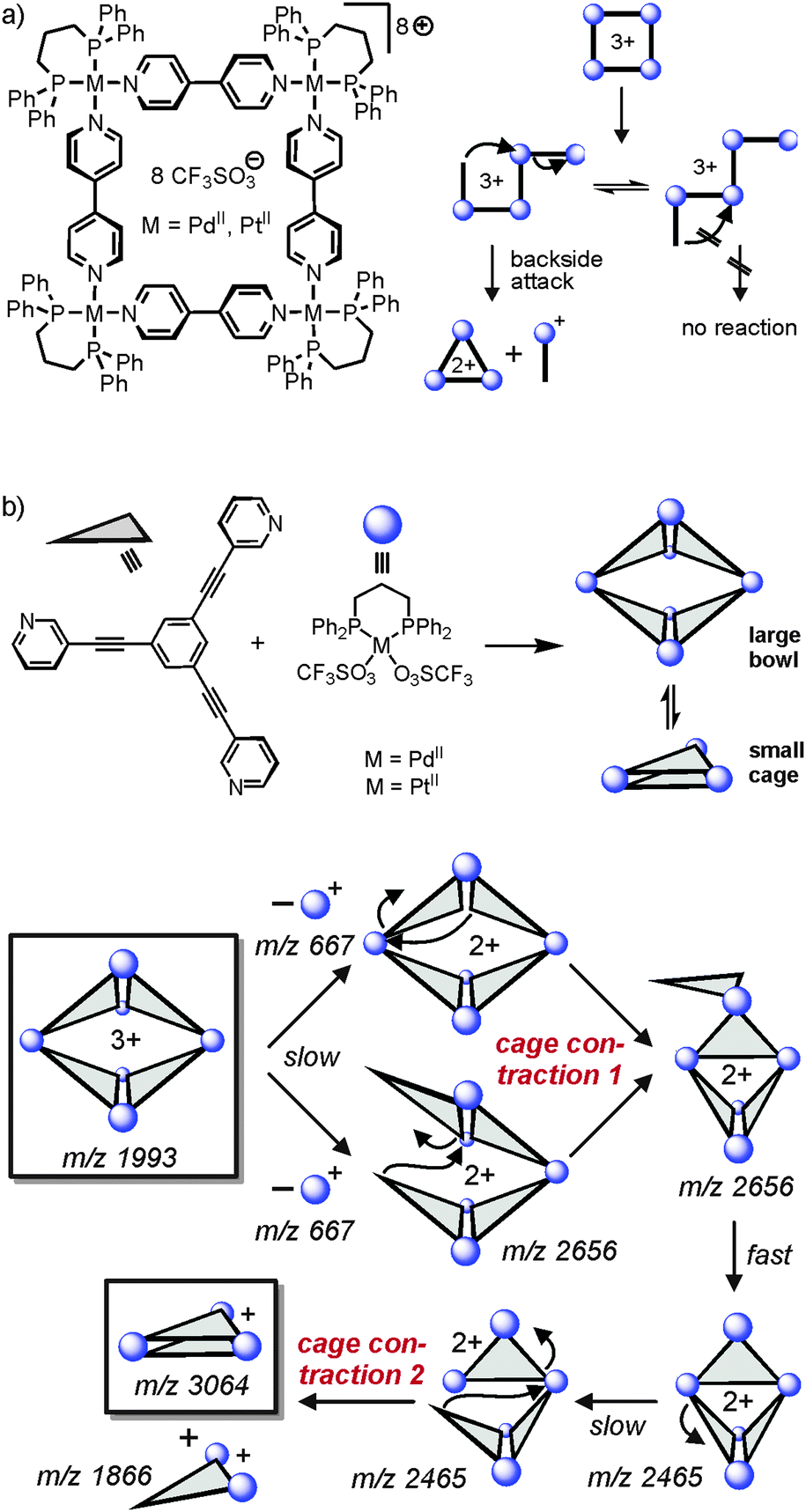 | ||
| Fig. 13 (a) Fragmentation pathway of metallo-supramolecular squares in the gas phase. A “backside-attack” mechanism rationalizes the strong preference for only one dissociation reaction, although several energetically equivalent fragmentation sites exist within the linear M4L43+ complex after ring opening. (b) The same mechanism operates in a double cage contraction process. Adapted and reproduced from ref. 57 with kind permission by the Royal Society of Chemistry. | ||
H/D exchange reactions: a supramolecular analogue of proton transport in water
H/D-exchange (HDX) experiments in the gas phase can provide detailed information on supramolecular structure and reactivity in particular for hydrogen-bonded complex ions.25 Mechanistically, a simple HDX reaction occurs in a sequence of five steps: (i) encounter complex formation between the charged substrate and the neutral deuteration agent, (ii) a proton transfer from the substrate to the deuteration agent, (iii) isotope scrambling, (iv) deuteron back-transfer to the substrate and, finally, (v) dissociation of the product complex and loss of the non-deuterated agent. This simple mechanism depends on the proton affinity difference between the substrate and the agent. The larger this difference, the slower the exchange. If a second functional group is present in the substrate, which may actively mediate the process, H/D-exchanges are often much faster, as so-called relay mechanisms58 become possible.Vainiotalo et al. investigated the H/D-exchange on tetrasulfonylated resorcinarenes resoS1 and resoS2 (Fig. 14) which form 1:1 complexes with the primary, secondary and tertiary ammonium ions mR, dR and tR (R = Me, Et).59 N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S hydrogen bonding is certainly important for binding besides cation-π interactions. In these complexes, no exchange is observed for the resorcinarene OH protons when the complexes were reacted with ND3 – likely because they are involved in hydrogen bonding to the sulfonyl groups of the adjacent aromatic rings. Instead, all of the guests' NH protons do exchange. The exchange rates were determined and amount to k1 = 2.36 × 10−11 cm3 molecule−1 s−1, k2 = 1.71 × 10−11 cm3 molecule−1 s−1 and k3 = 7.66 × 10−12 cm3 molecule−1 s−1 for the three exchange steps of [mEt·resoS1]+ as an example.
S hydrogen bonding is certainly important for binding besides cation-π interactions. In these complexes, no exchange is observed for the resorcinarene OH protons when the complexes were reacted with ND3 – likely because they are involved in hydrogen bonding to the sulfonyl groups of the adjacent aromatic rings. Instead, all of the guests' NH protons do exchange. The exchange rates were determined and amount to k1 = 2.36 × 10−11 cm3 molecule−1 s−1, k2 = 1.71 × 10−11 cm3 molecule−1 s−1 and k3 = 7.66 × 10−12 cm3 molecule−1 s−1 for the three exchange steps of [mEt·resoS1]+ as an example.
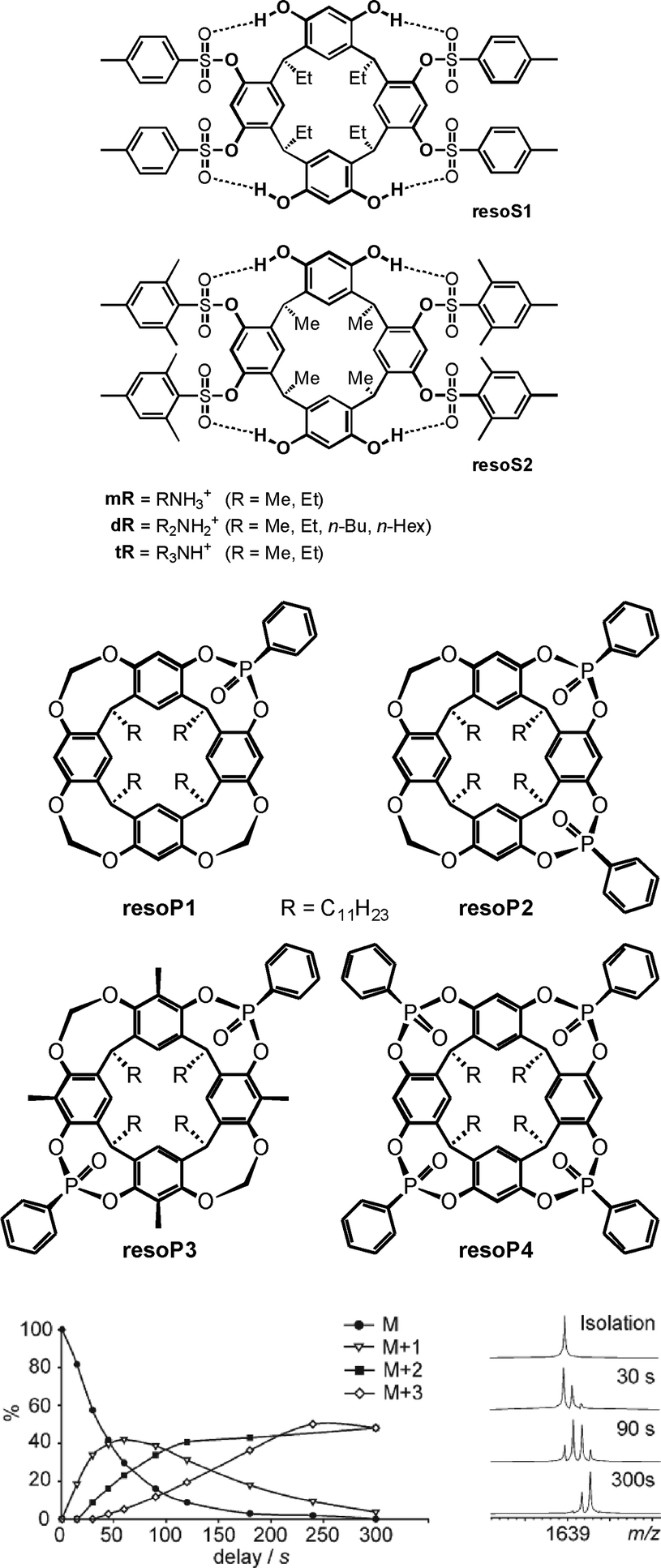 | ||
| Fig. 14 Tetrasulfonylated (top) and phosphonylated (centre) resorcinarenes whose ammonium ion complexes have been investigated by gas-phase H/D-exchange experiments. Bottom: relative abundances of the parent ion and the H/D-exchange products for the [EtNH3·resoP4]+ complex (left) and the corresponding mass spectra after different reaction times (right). ND3 was used as the exchange reagent. Bottom figure adapted and reproduced from ref. 60a with kind permission by the Royal Society of Chemistry. | ||
Similarly, the phosphonylated cavitands resoP1–resoP4 (Fig. 14)60 also bind ammonium ions.61 These cavitands are much more rigid as compared to resoS1 and resoS2 and bear PO groups converging towards the resorcinarene cavity. HDX experiments help to unravel the binding modes for primary, secondary and tertiary ammonium ions. Using ND3 as the deuteration agent, the exchange reaction is fast for [mEt@resoP4]+ with a primary ammonium ion as the guest, while it does not occur at all for the corresponding secondary ammonium ion complex [dEt@resoP4]+. The latter finding is again in contrast to the observation that [dEt@resoP1]+ and [dEt@resoP3]+ undergo a fast exchange of both ammonium NH protons.
Clearly, these data are in agreement with the interpretation that the primary ammonium ion binds only to two PO groups in resoP4, while the third N–H proton is available for exchange. A rearrangement within the complex then leads to the formation of one hydrogen and one deuteron bond and thus a second NH proton is available for the exchange. Similarly, the third proton is exchanged in a third step. A secondary ammonium ion instead forms two hydrogen bonds to two PO groups in resoP4 and is thus protected against any exchange, because no exchangeable NH proton is left. The same ion can however form only one hydrogen bond to resoP1 (only one PO group available) and resoP3 (unsuitable arrangement of the two PO groups). These studies nicely show how H/D-exchange experiments unravel details of the binding mode and at the same time provide insight into dynamic processes occurring in container/guest complexes.
When the dimeric resorcinarene capsule [Cs·reso2]+ shown in Fig. 15 is subjected to H/D-exchange experiments, one would likely expect based on the complexes discussed above that no significant exchange is observed as all OH groups are involved in hydrogen bonding and form a seam of hydrogen bonds which meanders around the whole capsule. In marked contrast to expectation, a rather rapid exchange of all OH protons is instead observed, when the dimer is subjected to gas-phase HDX experiments with methanol-OD.62 This surprising behaviour can be explained easily by invoking a concerted exchange mechanism as sketched in Fig. 15. Insertion of a MeOD molecule at any position of the hydrogen bonding opens the path for simple electron pair migrations around the whole seam. None of the OH protons needs to migrate. Energetically unfavourable charge separation intermediates, which would be the products of a proton shift from the capsule to the exchange reagent, can efficiently be avoided by such a mechanism, as the methanol-OD molecule serves simultaneously as a deuteron donor and proton acceptor. As all OH groups are held in position by the capsule scaffold, a concerted mechanism is feasible reminiscent of the well-known Grotthus-type proton migration in water.63 In control experiments, resorcinarene dimers that are either tetramethylated or encapsulate guests that are too large to fit completely inside do not show such a fast exchange, because the hydrogen bonding seam is disrupted and no concerted mechanism possible.
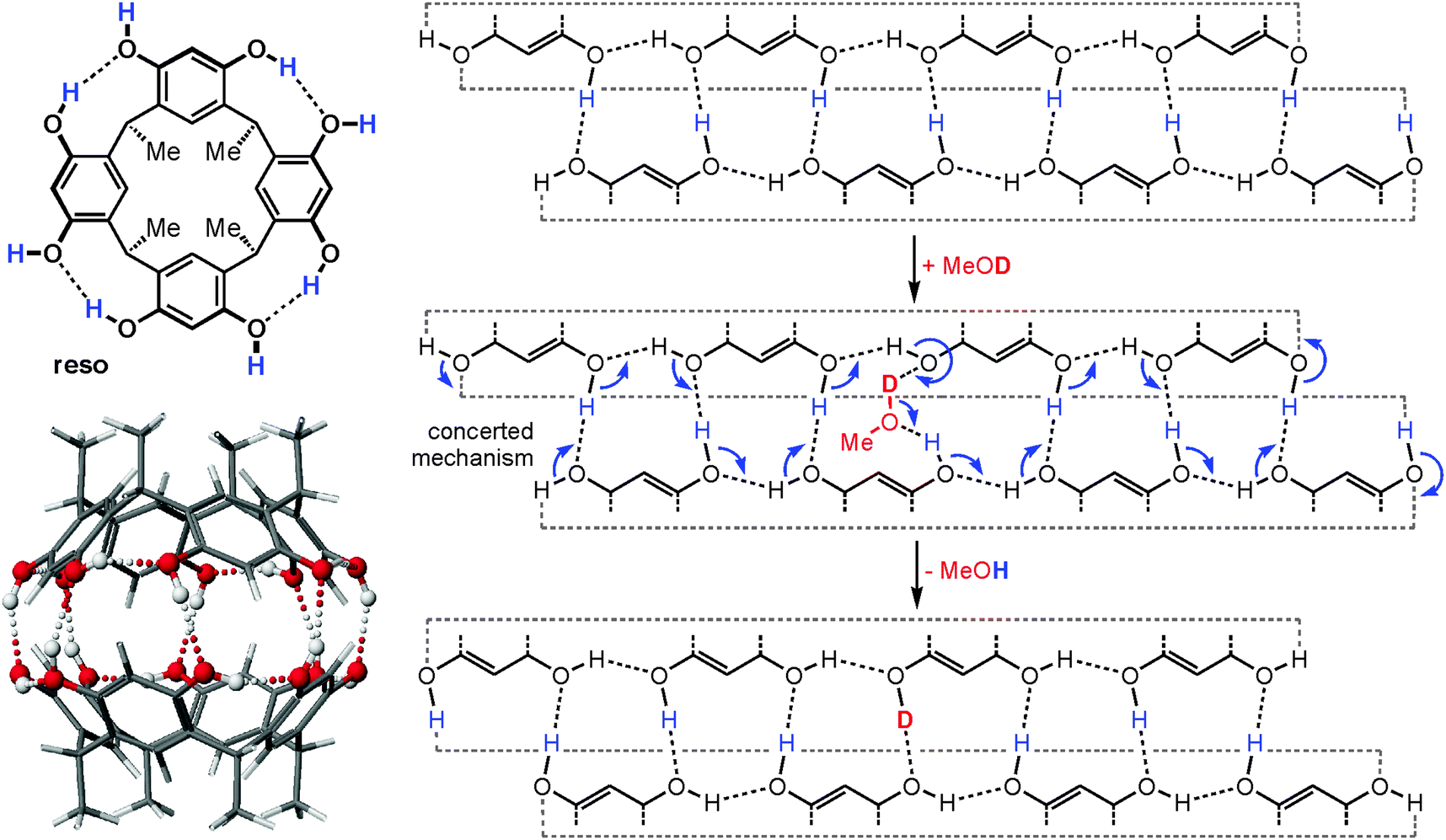 | ||
| Fig. 15 Left: chemical structure of the resorcinarene reso and force-field-optimized structure of the corresponding hydrogen bonded dimer. Right: mercator projection of the hydrogen bonding seam of the dimeric capsule and a concerted mechanism for the H/D-exchange reaction. Reproduced from ref. 62b with kind permission by the Royal Society of Chemistry. | ||
5 Ion mobility spectrometry mass spectrometry
Since ion mobility instruments became commercially available, ion mobility spectrometry mass spectrometry (IMS-MS)64 enjoys increasing popularity and has also been used to address structural questions in supramolecular chemistry.65 Mass-selected ions are subjected into a drift cell, which is filled with a collision gas, often nitrogen, at pressures of a few millibars and are pulled at low velocities through the gas cloud. As the collisions are soft, fragmentation can usually be prevented. The larger the collision cross section of the ion under study, the more collisions it undergoes and the later it arrives at the detector. The arrival time can thus be used as a measure for the collision cross section and thus for the size of the ion. Collision cross sections can also be calculated theoretically quite precisely so that it is often possible to compare the theoretical values obtained for a series of different structures with the experimental values and decide, what the structure of the ion under study is – at least as long as the ions are not too floppy and are structurally well-defined.A number of metallo-supramolecular containers has been investigated by ion mobility experiments. Stang and Bowers et al. applied it successfully to characterize rectangles, triangles, and the cage-like prism shown in Fig. 16.66 In the arrival time distribution of the prism hexacation, three features are observed which can be assigned to the intact M6L36+ prism as well as M4L24+ and M2L2+ fragments which all have the same mass-to-charge ratio. Their assignment is supported by experiments in which the parent ions were collided with different collision voltages prior to the IMS experiments. The harsher the collision conditions, the more prominent the fragments are as expected. Furthermore, the calculated collision cross sections are in good agreement with the arrival times of the three complexes. Similarly, IMS experiments have been applied to giant metallo-supramolecular cube-like structures.67
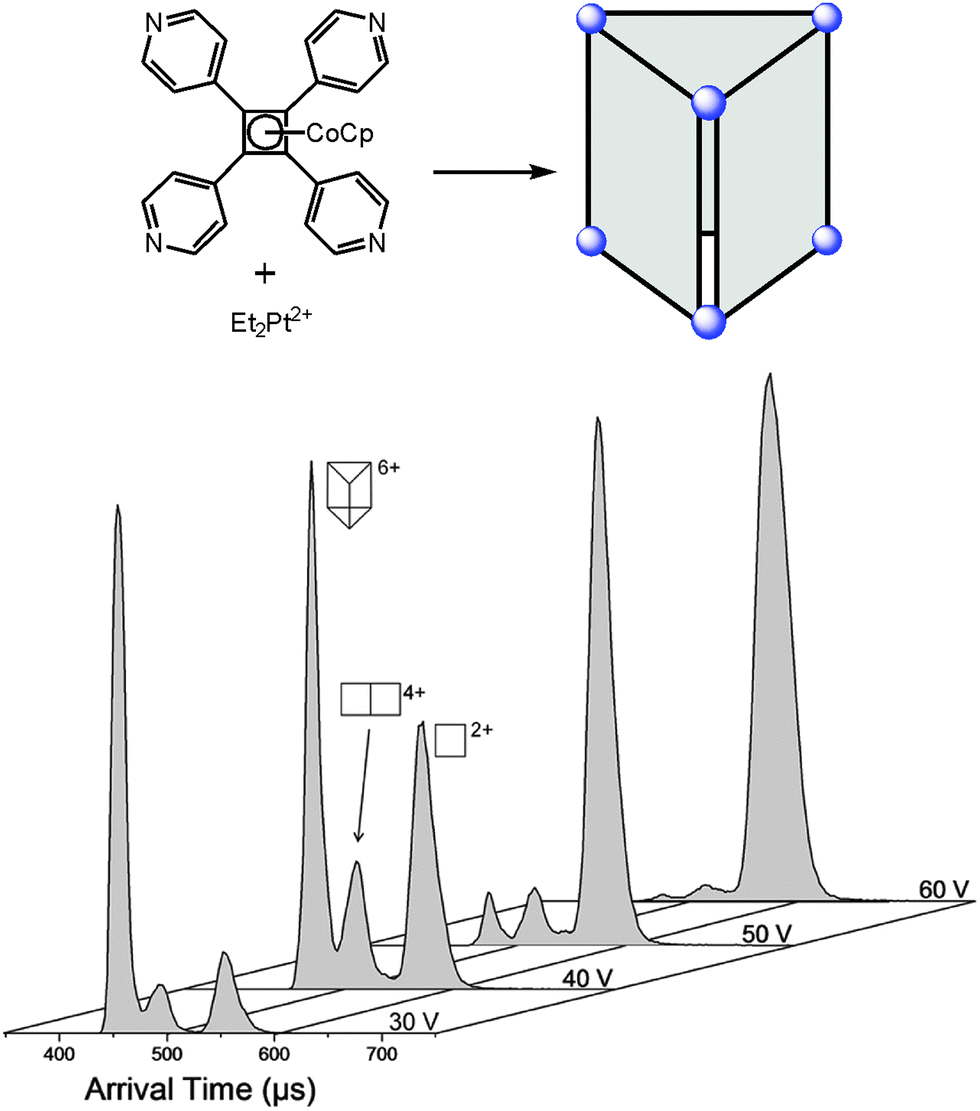 | ||
| Fig. 16 The M6L36+ prism (top) was subjected to IMS experiments. Clearly, several different complexes are observed which have the same m/z. Besides the intact prism hexacation, a quadruply charged M4L24+ and a doubly charged M2L2+ complex exist which can be separated by size in the drift tube. Experimental collision cross sections agree with calculated ones. The assignment is also secured by increasing collision voltages that provoke more and more fragmentation so that the intensity of the intact prism decreases in favour of more prominent fragment signals. It should be noted that the arrival time also depends on the charge state so that more highly charged ions travel faster through the drift cell. Adapted and reproduced from ref. 66 with kind permission by the American Chemical Society. | ||
Barran and Lusby et al. have applied IMS experiments to the trigonal prismatic cages HL1Pt and L2Pt depicted in Fig. 17.68 Both cages can be synthesized as two different isomers depending on the orientation of the bidentate, cyclometallated ligand at the metal centre, i.e. cis-HL1Pt versus trans-HL1Pt and cis-L2Pt versus trans-L2Pt, and the question arises whether it is possible to distinguish the isomers by IMS.
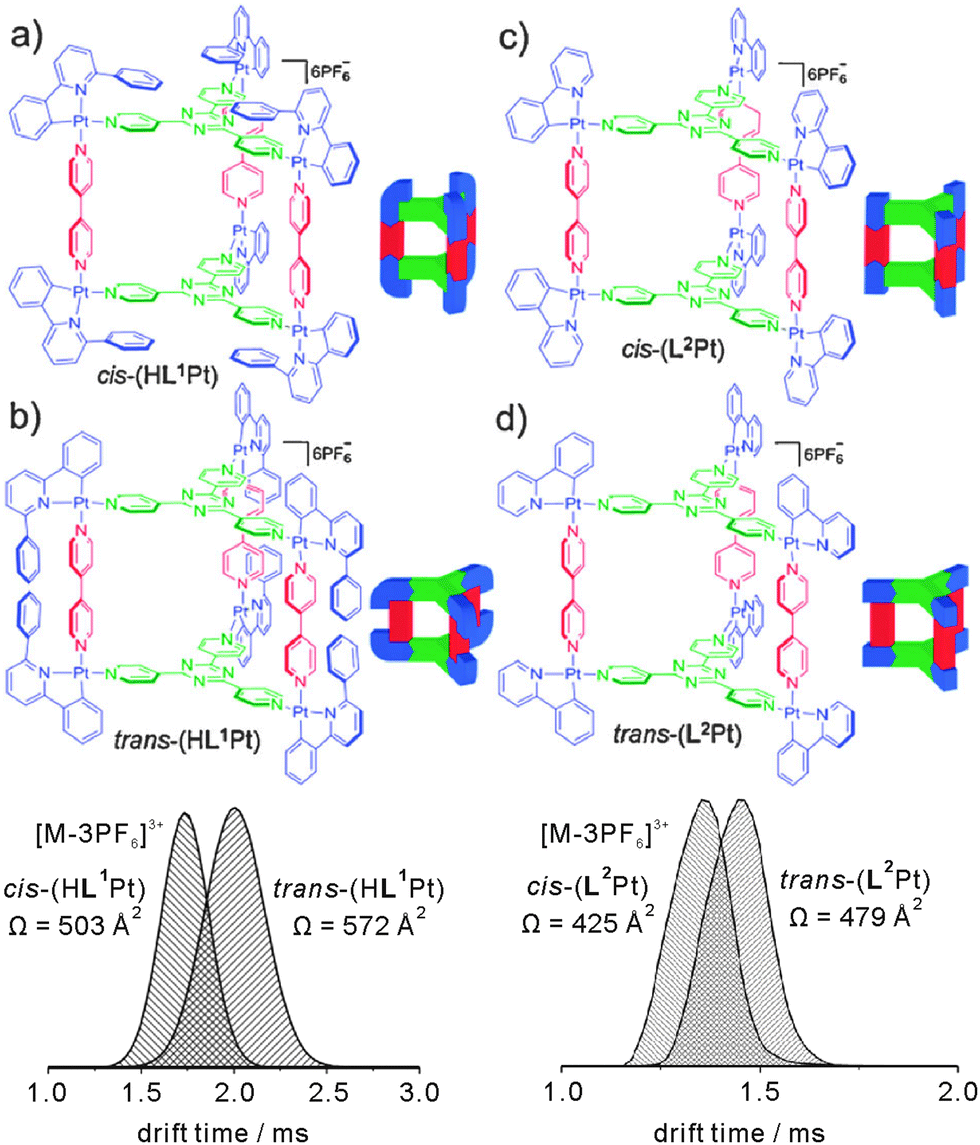 | ||
| Fig. 17 Top: chemical structures of the trigonal prismatic metallo supramolecular cages HL1Pt and L2Pt in their cis- and trans-forms. Bottom: IMS experiments provide sufficient resolution to distinguish both isomers by their collision cross sections. Reproduced from ref. 68 with kind permission by the Royal Society of Chemistry. | ||
For both cages, intact parent ions are successfully generated by electrospray ionisation through stripping off counterions. Fig. 17 shows the arrival time distributions for the [M-3PF6]3+ ions. Clearly, the IMS-MS experiments are capable of distinguishing the two isomers. The triply charged cis-HL1Pt complex was found to be smaller than the trans-HL1Pt cage with collision cross sections of 503 Å2 and 572 Å2, respectively. Even for the (L2Pt) system, in which the two isomers only differ with respect of the relative positions of the pyridine versus phenyl ring of the bidentate ligand, the resolution of the IMS experiment is sufficiently high to separate the two isomers that have collision cross sections of 425 Å2 and 479 Å2.
6 Probing reactions of guest molecules inside containers
The gas-phase studies on containers summarised above all addressed the container itself, its structure, its reactivity or its stability. Although guest molecules are present in many of the above examples, the studies above did not address the inner-phase reactivity – i.e. the reactivity of the guest inside the container and how it is affected by guest encapsulation. In a very recent study, Nau and Kalenius et al. used tandem mass spectrometry in concert with theory to investigate host–guest complexes of cucurbit[n]uril (CBn, n = 6–8) containers with bicyclic azoalkanes 6, 7 and 8 (Fig. 18).69 Usually, electrospray ionisation of such cucurbituril host–guest complexes is readily achieved.70 In the present case, protonation provides the necessary charge. The three cucurbiturils under study have different cavity volumes resulting in different guest selectivities. Nevertheless, they bind a range of differently sized bicyclic azoalkanes thus realizing different packing coefficients.71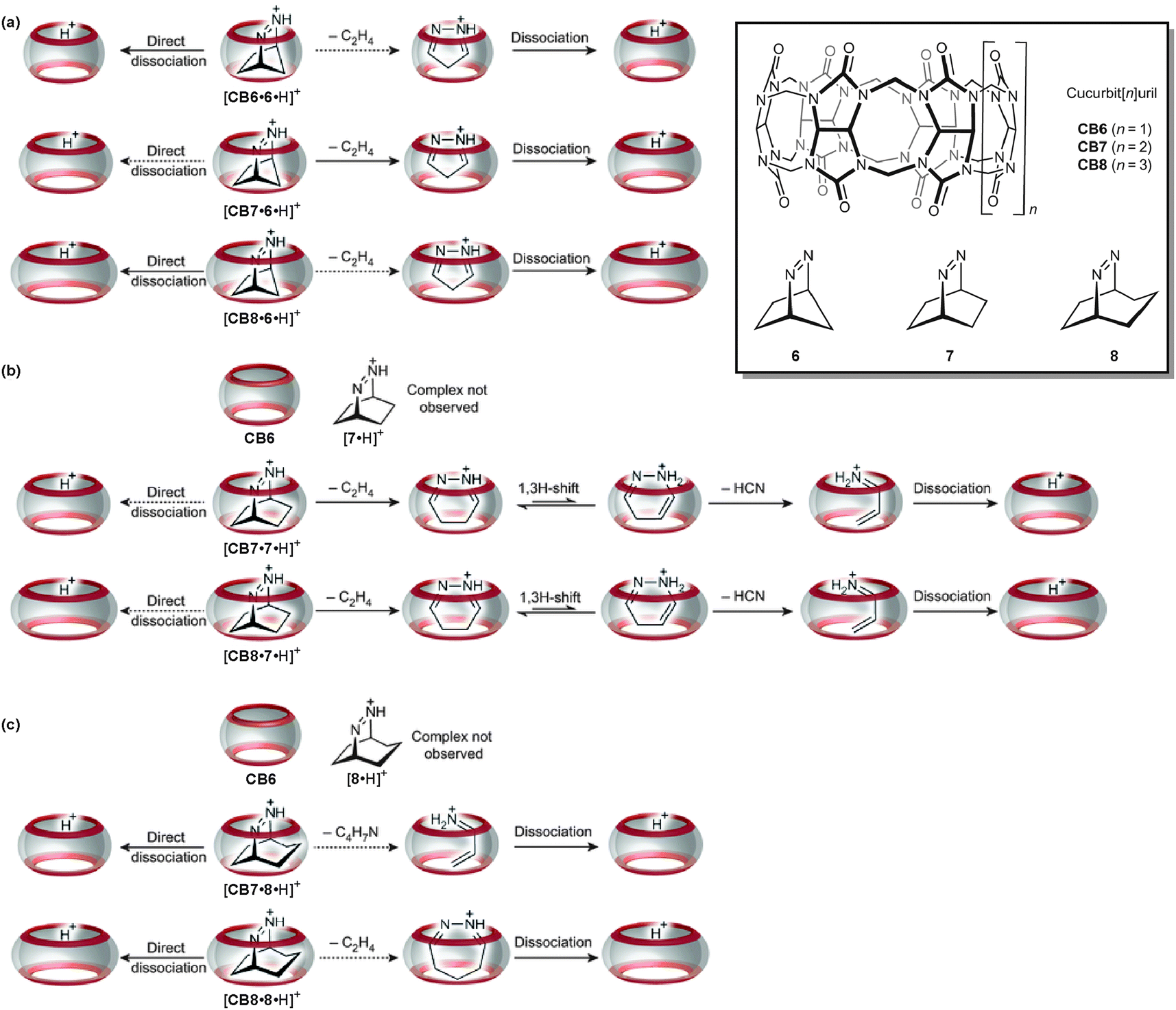 | ||
| Fig. 18 The gas-phase dissociation reactions of different cucurbituril–azoalkane complexes is characterised by a competition between complex dissociation and an inner-phase retro-Diels–Alder reaction. Depending on the size complementarity of the guest's van der Waals volume and the container cavity, reactivity significantly differs. Adapted and reproduced from ref. 69 with kind permission by the Nature Publishing Group. | ||
In collision-induced dissociation experiments with mass-selected complex ions, the fragmentation patterns differ with respect to the size complementarity of host cavity and guest. Two reactions compete with each other: the dissociation of the guest from the complex and the cycloelimination of ethene followed by consecutive rearrangements and/or dissociation reactions of the product complexes formed in the retro-Diels–Alder reaction. The cycloelimination inside the cavity becomes dominant, when the packing coefficients are within a range of 30–50%. Smaller guest cations likely dissociate with a lower activation barrier as they can pass the inwards-bent seams of the cucurbiturils' carbonyl groups more easily. For larger guests, the cycloelimination of ethene, which is expected to have a positive activation volume, is probably hindered. A combination of constrictive binding and the void space inside the container thus emerges as important factors for the inner-phase reactivity.
7 Conclusions
In the present review, we have summarized the current status of how mass spectrometry contributes to the investigation of molecular containers, in particular to our understanding of metallo-supramolecular and hydrogen-bonded containers. The information delivered by mass spectrometry goes far beyond simply providing mass information and widens out knowledge of containers with respect to structural issues, the encapsulation of suitable guests, the stability of container guest complexes as well as their reactivity in solution and in the gas phase. Mass spectrometry is well suited to analyse rather complex mixtures so that self-assembly and self-sorting processes can be monitored. When it comes to the gas-phase chemistry of containers, many different aspects have been studied from the analysis of mechanistic aspects in cage contraction reactions through H/D-exchange processes in hydrogen-bonded capsules to the evaluation of inner-phase reactivity of the encapsulated guest inside the container.The application of gas-phase chemistry to supramolecules is developing quickly. Mass spectrometry will thus also play a more and more important role in characterization of molecular containers. Certainly, many of the studies discussed above are far from economically useful applications and belong to the realm of fundamental science. Nevertheless, they underline how important the development and popularisation of novel methods such as ion mobility mass spectrometry are for the advancement of supramolecular chemistry.
Acknowledgements
We thank all the past and current members of the Schalley group and our cooperation partners for their excellent and unique contributions to our studies on the gas-phase chemistry of molecular capsules.Notes and references
- (a) J. Rebek, Jr., Chem. Soc. Rev., 1996, 25, 255–264 RSC; (b) J. Rebek, Jr., Acc. Chem. Res., 2009, 42, 1660–1668 CrossRef PubMed.
- (a) M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed; (b) M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CrossRef CAS PubMed; (c) T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC; (d) A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC; (e) Z. Dong, Q. Luo and J. Liu, Chem. Soc. Rev., 2012, 41, 7890–7908 RSC; (f) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC; (g) B. Bibal, C. Mongin and D. M. Bassani, Chem. Soc. Rev., 2014, 43, 4179–4198 RSC.
- R. Breslow and P. Campbell, Bioorg. Chem., 1971, 1, 140–156 CrossRef CAS.
- (a) P. Timmerman, W. Verboom and D. N. Reinhoudt, Tetrahedron, 1996, 52, 2663–2704 CrossRef CAS; (b) D. J. Cram and J. M. Cram, Container Molecules and Their Guests, Royal Society of Chemistry, Hertfordshire, 1997 Search PubMed.
- (a) K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267–279 RSC; (b) J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- R. Warmuth and J. Yoon, Acc. Chem. Res., 2001, 34, 95–105 CrossRef CAS PubMed.
- (a) K. Hermann, M. Nakhla, J. Gallucci, E. Dalkilic, A. Dastan and J. D. Badjić, Angew. Chem., Int. Ed., 2013, 52, 11313–11316 CrossRef CAS PubMed; (b) S. Kubik, Chem. Soc. Rev., 2009, 38, 585–605 RSC; (c) L. Cronin and A. Müller, Chem. Soc. Rev., 2012, 41, 7333–7334 RSC; (d) O. Bistri, B. Colasson and O. Reinaud, Chem. Sci., 2012, 3, 811–818 RSC; (e) B. M. Rambo, H.-Y. Gong, M. Oh and J. L. Sessler, Acc. Chem. Res., 2012, 45, 1390–1401 CrossRef CAS PubMed.
- (a) L. Adriaenssens and P. Ballester, Chem. Soc. Rev., 2013, 42, 3261–3277 RSC; (b) S. M. Biros and J. J. Rebek, Chem. Soc. Rev., 2007, 36, 93–104 RSC.
- (a) P. D. Frischmann and M. J. MacLachlan, Chem. Soc. Rev., 2013, 42, 871–890 RSC; (b) S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS PubMed.
- Q.-F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144–1147 CrossRef CAS PubMed.
- L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469–472 CrossRef CAS PubMed.
- T. Gerkensmeier, W. Iwanek, C. Agena, R. Fröhlich, S. Kotila, C. Näther and J. Mattay, Eur. J. Org. Chem., 1999, 2257–2262 CrossRef CAS.
- (a) J. Santamaría, T. Martín, G. Hilmersson, S. L. Craig and J. Rebek, Jr., Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8344–8347 CrossRef; (b) M. Yamanaka, A. Shivanyuk and J. Rebek, Jr., J. Am. Chem. Soc., 2004, 126, 2939–2943 CrossRef CAS PubMed.
- S. Rieth, K. Hermann, B.-Y. Wang and J. D. Badjic, Chem. Soc. Rev., 2011, 40, 1609–1622 RSC.
- (a) C. A. Schalley, Int. J. Mass Spectrom., 2000, 194, 11–39 CrossRef CAS; (b) C. A. Schalley, Mass Spectrom. Rev., 2001, 20, 253–309 CrossRef CAS PubMed; (c) M. Vincenti and A. Irico, Int. J. Mass Spectrom., 2002, 214, 23–36 CrossRef CAS; (d) B. Baytekin, H. T. Baytekin and C. A. Schalley, Org. Biomol. Chem., 2006, 4, 2825–2841 RSC; (e) Z. Qi and C. A. Schalley, Supramol. Chem., 2010, 22, 672–682 CrossRef CAS; (f) F. Yang and D. V. Dearden, Isr. J. Chem., 2011, 51, 551–558 CrossRef CAS; (g) L. Cera and C. A. Schalley, Chem. Soc. Rev., 2014, 43, 1800–1812 RSC.
- F. W. McLafferty, Science, 1981, 214, 280–287 CAS.
- (a) G. Siuzdak, B. Bothner, M. Yeager, C. Brugidou, C. M. Fauquet, K. Hoey and C.-M. Change, Chem. Biol., 1996, 3, 45–48 CrossRef CAS; (b) S. D. Fuerstenau, W. H. Benner, J. J. Thomas, C. Brugidou, B. Bothner and G. Siuzdak, Angew. Chem., Int. Ed., 2001, 40, 541–544 CrossRef CAS.
- M. W. H. Pinkse, C. S. Maier, J.-I. Kim, B.-H. Oh and A. J. R. Heck, J. Mass Spectrom., 2003, 38, 315–320 CrossRef CAS PubMed.
- W. Jiang, A. Schäfer, P. C. Mohr and C. A. Schalley, J. Am. Chem. Soc., 2010, 132, 2309–2320 CrossRef CAS PubMed.
- D. Gao, H. Liu, Y. Jiang and J. M. Lin, Lab Chip, 2013, 13, 3309–3322 RSC.
- D. Fabris, Mass Spectrom. Rev., 2005, 24, 30–54 CrossRef CAS PubMed.
- L. Konermann, J. Pan and Y.-H. Liu, Chem. Soc. Rev., 2011, 40, 1224–1234 RSC.
- D. Schröder, Acc. Chem. Res., 2012, 45, 1521–1532 CrossRef PubMed.
- See, for example: (a) S. Fürmeier and J. O. Metzger, J. Am. Chem. Soc., 2004, 126, 14485–14492 CrossRef PubMed; (b) C. Marquez and J. O. Metzger, Chem. Commun., 2006, 1539–1541 RSC; (c) L. S. Santos and J. O. Metzger, Angew. Chem., Int. Ed., 2006, 45, 977–981 CrossRef PubMed; (d) J. Roithova, Chem. Soc. Rev., 2012, 41, 547–559 RSC; (e) J. Wassenaar, E. Jansen, Z.-J. van, F. M. Bickelhaupt, M. A. Siegler, A. L. Spek and J. N. H. Reek, Nat. Chem., 2010, 2, 417–421 CrossRef CAS PubMed.
- H. D. F. Winkler, E. V. Dzyuba and C. A. Schalley, New J. Chem., 2011, 35, 529–541 RSC.
- (a) D. P. Weimann, H. D. F. Winkler, J. A. Falenski, B. Koksch and C. A. Schalley, Nat. Chem., 2009, 1, 573–577 CrossRef CAS PubMed; (b) H. D. F. Winkler, D. P. Weimann, A. Springer and C. A. Schalley, Angew. Chem., Int. Ed., 2009, 48, 7246–7250 CrossRef CAS PubMed; (c) Z. Qi, C. Schlaich and C. A. Schalley, Chem. – Eur. J., 2013, 19, 14867–14875 CrossRef CAS PubMed; (d) H. D. F. Winkler, E. V. Dzyuba, A. Springer, L. Losensky and C. A. Schalley, Chem. Sci., 2012, 3, 1111–1120 RSC.
- (a) S. Sakamoto, M. Fujita, K. Kim and K. Yamaguchi, Tetrahedron, 2000, 56, 955–964 CrossRef CAS; (b) K. Yamaguchi, J. Mass Spectrom., 2003, 38, 473–490 CrossRef CAS PubMed.
- J. S. Gardner, R. G. Harrison, J. D. Lamb and D. V. Dearden, New J. Chem., 2006, 30, 1276–1281 RSC.
- O. D. Fox, J. Cookson, E. J. S. Wilkinson, M. G. B. Drew, E. J. MacLean, S. J. Teat and P. D. Beer, J. Am. Chem. Soc., 2006, 128, 6990–7002 CrossRef CAS PubMed.
- K. Suzuki, M. Tominaga, M. Kawano and M. Fujita, Chem. Commun., 2009, 1638–1640 RSC.
- M. Tominaga, K. Suzuki, M. Kawano, T. Kusukawa, T. Ozeki, S. Sakamoto, K. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2004, 43, 5621–5625 CrossRef CAS PubMed.
- M. Tominaga, K. Suzuki, T. Murase and M. Fujita, J. Am. Chem. Soc., 2005, 127, 11950–11951 CrossRef CAS PubMed.
- S. Sato, J. Iida, K. Suzuki, M. Kawano, T. Ozeki and M. Fujita, Science, 2006, 313, 1273–1276 CrossRef CAS PubMed.
- C. Schmuck, Angew. Chem., Int. Ed., 2007, 46, 5830–5833 CrossRef CAS PubMed.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS PubMed.
- S. P. Black, A. R. Stefankiewicz, M. M. J. Smulders, D. Sattler, C. A. Schalley, J. R. Nitschke and J. K. M. Sanders, Angew. Chem., Int. Ed., 2013, 52, 5749–5752 CrossRef CAS PubMed.
- P. Timmerman, K. A. Jolliffe, M. C. Calama, J.-L. Weidmann, L. J. Prins, F. Cardullo, B. H. M. Snellink-Ruël, R. H. Fokkens, N. M. M. Nibbering, S. Shinkai and D. N. Reinhoudt, Chem. – Eur. J., 2000, 6, 4104–4115 CrossRef CAS.
- D. P. Weimann and C. A. Schalley, Supramol. Chem., 2008, 20, 117–128 CrossRef CAS.
- C. A. Schalley, J. M. Rivera, T. Martín, J. Santamaría, G. Siuzdak and J. Rebek, Jr., Eur. J. Org. Chem., 1999, 1325–1331 CrossRef CAS.
- C. A. Schalley, T. Martin, U. Obst and J. Rebek, J. Am. Chem. Soc., 1999, 121, 2133–2138 CrossRef CAS.
- B. M. O'Leary, T. Szabo, N. Svenstrup, C. A. Schalley, A. Lützen, M. Schäfer and J. Rebek, Jr., J. Am. Chem. Soc., 2001, 123, 11519–11533 CrossRef PubMed.
- A. Lützen, A. R. Renslo, C. A. Schalley, B. M. O'Leary and J. Rebek, Jr., J. Am. Chem. Soc., 1999, 121, 7455–7456 CrossRef.
- C. A. Schalley, R. K. Castellano, M. S. Brody, D. M. Rudkevich, G. Siuzdak and J. Rebek, Jr., J. Am. Chem. Soc., 1999, 121, 4568–4579 CrossRef CAS.
- (a) M. Mäkinen, P. Vainiotalo, M. Nissinen and K. Rissanen, J. Am. Soc. Mass Spectrom., 2003, 14, 143–151 CrossRef; (b) H. Mansikkamäki, M. Nissinen, C. A. Schalley and K. Rissanen, New J. Chem., 2003, 27, 88–97 RSC; (c) H. Mansikkamäki, C. A. Schalley, M. Nissinen and K. Rissanen, New J. Chem., 2005, 29, 116–127 RSC.
- (a) L. Avram and Y. Cohen, J. Am. Chem. Soc., 2002, 124, 15148–15149 CrossRef CAS PubMed; (b) I. Philip and A. E. Kaifer, J. Org. Chem., 2005, 70, 1558–1564 CrossRef CAS PubMed.
- N. K. Beyeh, M. Kogej, A. Åhman, K. Rissanen and C. A. Schalley, Angew. Chem., Int. Ed., 2006, 45, 5214–5218 CrossRef CAS PubMed.
- B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef CAS PubMed.
- H. T. Baytekin, M. Sahre, A. Rang, M. Engeser, A. Schulz and C. A. Schalley, Small, 2008, 4, 1823–1834 CrossRef CAS PubMed.
- Y.-R. Zheng and P. J. Stang, J. Am. Chem. Soc., 2009, 131, 3487–3489 CrossRef CAS PubMed.
- S. Sato, Y. Ishido and M. Fujita, J. Am. Chem. Soc., 2009, 131, 6064–6065 CrossRef CAS PubMed.
- Y. Rudzevich, V. Rudzevich, F. Klautzsch, C. A. Schalley and V. Böhmer, Angew. Chem., Int. Ed., 2009, 48, 3867–3871 CrossRef CAS PubMed.
- L. Cera and C. A. Schalley, Chem. Soc. Rev., 2014, 43, 1800–1812 RSC.
- R. Zadmard, A. Kraft, T. Schrader and U. Linne, Chem. – Eur. J., 2004, 10, 4233–4239 CrossRef CAS PubMed.
- (a) J. Grunenberg and G. Barone, RSC Adv., 2013, 3, 4757–4762 RSC; (b) P. Ballester, Chem. Soc. Rev., 2010, 39, 3810–3830 RSC; (c) Z. R. Laughrey, T. G. Upton and B. C. Gibb, Chem. Commun., 2006, 970–972 RSC . For a review, see: R. K. Castellano, Curr. Org. Chem., 2004, 8, 845–865 Search PubMed.
- S. S. Zhu, H. Staats, K. Brandhorst, J. Grunenberg, F. Gruppi, E. Dalcanale, A. Lützen, K. Rissanen and C. A. Schalley, Angew. Chem., Int. Ed., 2008, 47, 788–792 CrossRef CAS PubMed.
- C. A. Schalley, T. Müller, P. Linnartz, M. Witt, M. Schäfer and A. Lützen, Chem. – Eur. J., 2002, 8, 3538–3551 CrossRef CAS.
- B. Brusilowskij, S. Neubacher and C. A. Schalley, Chem. Commun., 2009, 785–787 RSC.
- (a) T. Wyttenbach and M. T. Bowers, J. Am. Soc. Mass Spectrom., 1999, 10, 9–14 CrossRef CAS; (b) S. Campbell, M. T. Rodgers, E. M. Marzluff and J. L. Beauchamp, J. Am. Chem. Soc., 1995, 117, 12840–12854 CrossRef CAS.
- (a) E. Ventola, K. Rissanen and P. Vainiotalo, Chem. – Eur. J., 2004, 10, 6152–6162 CrossRef CAS PubMed; (b) E. Ventola, A. Hyyryläinen and P. Vainiotalo, Rapid Commun. Mass Spectrom., 2006, 20, 1218–1224 CrossRef CAS PubMed.
- (a) E. Kalenius, D. Moiani, E. Dalcanale and P. Vainiotalo, Chem. Commun., 2007, 3865–3867 RSC; (b) E. Kalenius, R. Neitola, M. Suman, E. Dalcanale and P. Vainiotalo, J. Am. Soc. Mass Spectrom., 2010, 21, 440–450 CrossRef CAS PubMed.
- (a) J.-P. Dutasta, Top. Curr. Chem., 2004, 232, 55–91 CrossRef CAS; (b) A. Irico, M. Vincenti and E. Dalcanale, Chem. – Eur. J., 2001, 7, 2034–2042 CrossRef CAS.
- (a) M. Mäkinen, P. Vainiotalo and K. Rissanen, J. Am. Soc. Mass Spectrom., 2002, 13, 851–861 CrossRef; (b) H. D. F. Winkler, E. V. Dzyuba, J. A. W. Sklorz, N. K. Beyeh, K. Rissanen and C. A. Schalley, Chem. Sci., 2011, 2, 615–624 RSC.
- C. A. Wraight, Biochim. Biophys. Acta, 2006, 1757, 886–912 CrossRef CAS PubMed.
- T. Wyttenbach, P. R. Kemper and M. T. Bowers, Int. J. Mass Spectrom., 2001, 212, 13–23 CrossRef CAS.
- See, for example: (a) A. E. Counterman and D. E. Clemmer, J. Phys. Chem. B, 2001, 105, 8092–8096 CrossRef CAS; (b) R. R. Julian, R. Hodyss, B. Kinnear, M. F. Jarrold and J. L. Beauchamp, J. Phys. Chem. B, 2002, 106, 1219–1228 CrossRef CAS; (c) C. A. Schalley, J. Hoernschemeyer, X. Y. Li, G. Silva and P. Weis, Int. J. Mass Spectrom., 2003, 228, 373–388 CrossRef CAS; (d) Y.-T. Chan, X. Li, M. Soler, J.-L. Wang, C. Wesdemiotis and G. R. Newkome, J. Am. Chem. Soc., 2009, 131, 16395–16397 CrossRef CAS PubMed.
- E. R. Brocker, S. E. Anderson, B. H. Northrop, P. J. Stang and M. T. Bowers, J. Am. Chem. Soc., 2010, 132, 13486–13494 CrossRef CAS PubMed.
- C. Wang, X.-Q. Hao, M. Wang, C. Guo, B. Xu, E. N. Tan, Y.-Y. Zhang, Y. Yu, Z.-Y. Li, H.-B. Yang, M.-P. Song and X. Li, Chem. Sci., 2014, 5, 1221–1226 RSC.
- J. Ujma, M. De Cecco, O. Chepelin, H. Levene, C. Moffat, S. J. Pike, P. J. Lusby and P. E. Barran, Chem. Commun., 2012, 48, 4423–4425 RSC.
- T.-C. Lee, E. Kalenius, A. I. Lazar, K. I. Assaf, N. Kuhnert, C. H. Grün, J. Jänis, O. A. Scherman and W. M. Nau, Nat. Chem., 2013, 5, 376–382 CrossRef CAS PubMed.
- See, for example: (a) W. Jiang, Q. Wang, I. Linder, F. Klautzsch and C. A. Schalley, Chem. – Eur. J., 2011, 17, 2344–2348 CrossRef CAS PubMed; (b) J. Černochová, P. Branná, M. Rouchal, P. Kulhánek, I. Kuřitka and R. Vícha, Chem. – Eur. J., 2012, 18, 13633–13637 CrossRef PubMed; (c) J. P. Da Silva, N. Jayaraj, S. Jockusch, N. J. Turro and V. Ramamurthy, Org. Lett., 2011, 13, 2410–2413 CrossRef CAS PubMed.
- For a discussion of optimal packing coefficients, see: (a) S. Mecozzi and J. Rebek, Jr., Chem. – Eur. J., 1998, 4, 1016–1022 CrossRef CAS; (b) S. Sánchez Carrera, J.-L. Kerdelhué, K. J. Langenwalter, N. Brown and R. Warmuth, Eur. J. Org. Chem., 2005, 2239–2249 CrossRef; (c) R. Warmuth, J.-L. Kerdelhué, S. Sánchez Carrera, K. J. Langenwalter and N. Brown, Angew. Chem., Int. Ed., 2002, 41, 96–99 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |