
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
X-ray diffraction and high-resolution TEM observations of biopolymer nanoskin-covered metallic copper fine particles: preparative conditions and surface oxidation states
Tetsu
Yonezawa
*,
Yoshiki
Uchida
and
Hiroki
Tsukamoto
Division of Materials Science and Engineering, Faculty of Engineering, Hokkaido University, Kita 13 Nishi 8, Kita-ku, Sapporo, 060-8628, Japan. E-mail: tetsu@eng.hokudai.ac.jp
First published on 11th November 2015
Abstract
Metallic copper fine particles used for electro conductive pastes were prepared by the chemical reduction of cupric oxide microparticles in the presence of gelatin. After reduction, the fine particles were collected by decantation with pH control and washing, followed by drying at a moderate temperature. The surface oxidation state of the obtained copper fine particles could be considerably varied by altering the pH of the particle dispersion, as shown by X-ray diffraction and high-resolution transmission electron microscopy. Our results strongly indicate that decantation under a nitrogen atmosphere can prevent the oxidation of copper fine particles but a slight oxidation was found.
Introduction
Owing to their unique properties, metal nanoparticles and fine particles have great potential to be used in developing new products.1–4 Nanotechnology is currently used in diverse applications, including construction and composite materials,5 catalysts,6,7 antibacterial coatings,8 biosensing applications,9 plasmonic photo devices,10 electrical devices,11–13 and some consumer products. The steady increase in the use of nanoparticles and fine particles has resulted in their increased production and the development of handling and processing techniques for such materials.Additionally, the recent and remarkable progress in printed electronics is based on the development of useful materials, including nanomaterials.12–16 In particular, conductive inks and pastes consisting of metal nanoparticles and fine particles are key materials in printed electronics because they can be used in low-cost wet printing processes.12–15 Printing processes using these modern technologies are expected to facilitate the innovation of new manufacturing processes that will increase the productivity and reduce the required energy and resource consumption compared with the conventional processes. Indeed, to produce flexible, lightweight, cost-effective and eco-friendly electronic devices, such as smart labels using radiofrequency identification (RFID), organic light-emitting diodes (OLEDs), and flexible-displays, printed electronic technologies will be frequently used. Noble metals, especially silver nanoparticle inks, are highly stable and commonly used in conducting wires.17,18 However, high material costs limit their usage in large-scale printed electronic applications. The migration of silver is also an important issue that can degrade device performance. Conducting polymers may represent another possibility, but in many cases, their conductivity is not sufficient. In contrast, copper nanoparticles and fine particles are very attractive from the cost and anti-migratory perspectives. Copper is an ideal material for industrial-scale printed electronics. Therefore, in recent years, the synthesis of copper nanoparticles and fine particles has attracted substantial interest.19–22 However, they can be oxidized, which limits their wide use. Additionally, in printed electronic materials, the presence of copper oxides on the particle surface is undesirable, increasing the required sintering temperature and reducing the electro-conductivity.
To overcome the issue of copper nanoparticles and fine particle oxidation, many attempts have been made to improve the properties of the materials themselves and further develop appropriate sintering technologies. One promising approach to prevent the oxidation of the particle surface is the application of a protective coating19 of thin layers of polymers,14,23–25 metal oxides,26 or noble metals.27 For conductive inks and pastes, copper particles coated with organic polymers or molecules are highly useful because they can be readily dispersed in a suitable medium. The reported organic protective coating reagents, such as PVP,23 CTAB,28 oleic acids,29 and alkane thiols, etc.,19 form a nonconductive shell around the surface. Polymers are unique candidates for protective layers of copper fine particles because polymer molecules attach to the particle surfaces at multiple points. Therefore, because of multiple interactions between the polymer molecules and the metal surface, these polymer molecules cannot be easily detached from the surface. We previously described one interesting example of such particles, using the poly-ol reduction of copper ions to produce poly(ethylene glycol)-stabilized copper nanoparticles.30 Many chemical reduction processes have been performed using cupric salts, such as CuCl2 and CuSO4. However, for applications in electronics, the materials should not be contaminated by halogens and sulfur. Alternatively, organic copper compounds, such as copper acetylacetonate and the copper–AOT complex,19 can be used, although they are relatively expensive. Because copper is a low-cost metal, using expensive metal sources is inappropriate in industrial processes.
In our previous study, we proposed the use of gelatin as a stabilizer and an antioxidizing reagent for copper nano- and fine particles prepared via the chemical reduction of cupric oxide (CuO) and a copper salt.11,24,31,32 CuO is a solid copper source but contains only oxygen as the counter anion. Thus, in terms of contamination, CuO is an optimal candidate metal source for copper fine particles in conductive inks and pastes. Furthermore, this process can be achieved with a copper concentration of 1 mol dm−3 (64 g of Cu dm−3),24 which is much higher than that required in typical chemical reduction processes. Using the produced gelatin-stabilized copper fine particles, conductive pastes were prepared and used to generate the inner electrodes of multi-layered ceramic capacitors (MLCCs).11
Below, we describe the relationship between the surface oxidation states of the gelatin-stabilized copper fine particles and their preparative conditions. Following the reduction of CuO by hydrazine under an ambient atmosphere, the particles were collected by decantation and drying after adjusting the pH of the particle dispersion. We then controlled the experimental atmosphere to obtain metallic copper fine particles and determined their surface oxidation states by X-ray diffraction (XRD) and high-resolution transmission electron microscopy (HR-TEM).
Experimental
Materials
The metal source, CuO microparticles, was supplied by Nisshin Chemco (Japan), and the average diameter of the microparticles was approximately 4 μm. Hydrazine monohydrate, which was used as a reducing reagent, and aqueous ammonia (28%), which was used to adjust pH, were purchased from Wako or Junsei Chemicals (Japan). Gelatin (bovine origin) was obtained from Nitta Gelatin Inc. (Osaka, Japan). Water was purified using an Organo/ELGA Purelab system (>18 MΩ cm).Preparation of copper fine particles
The detailed preparative procedure is described in Fig. 1. The copper fine particles were prepared using a procedure similar to that described previously.31,32 First, 32 g of gelatin was dissolved in hot water (950 cm3). CuO microparticles (80 g) were introduced into the gelatin solution. Then, aqueous ammonia (28%) was introduced to adjust the pH of the dispersion at approximately 11. The reduction of CuO (80 g) to metallic copper by hydrazine monohydrate (120 cm3) was carried out under ambient conditions. Subsequently, the fine particles were collected by decantation after adding saturated aqueous citric acid to reduce the pH of the dispersion. Decantation was performed under ambient conditions at pH = 10 (Process A) and 8.5 (Process B) or under a nitrogen atmosphere at pH = 8.5 (Process C). The solvents used for decantation in Process C were treated with nitrogen gas by bubbling (2 dm3 min−1) for 5 min before use. The purification process was easily carried out in a large plastic bag under nitrogen (∼99.5%) flow. Then, the collected particles were dried at 60 °C under the nitrogen atmosphere.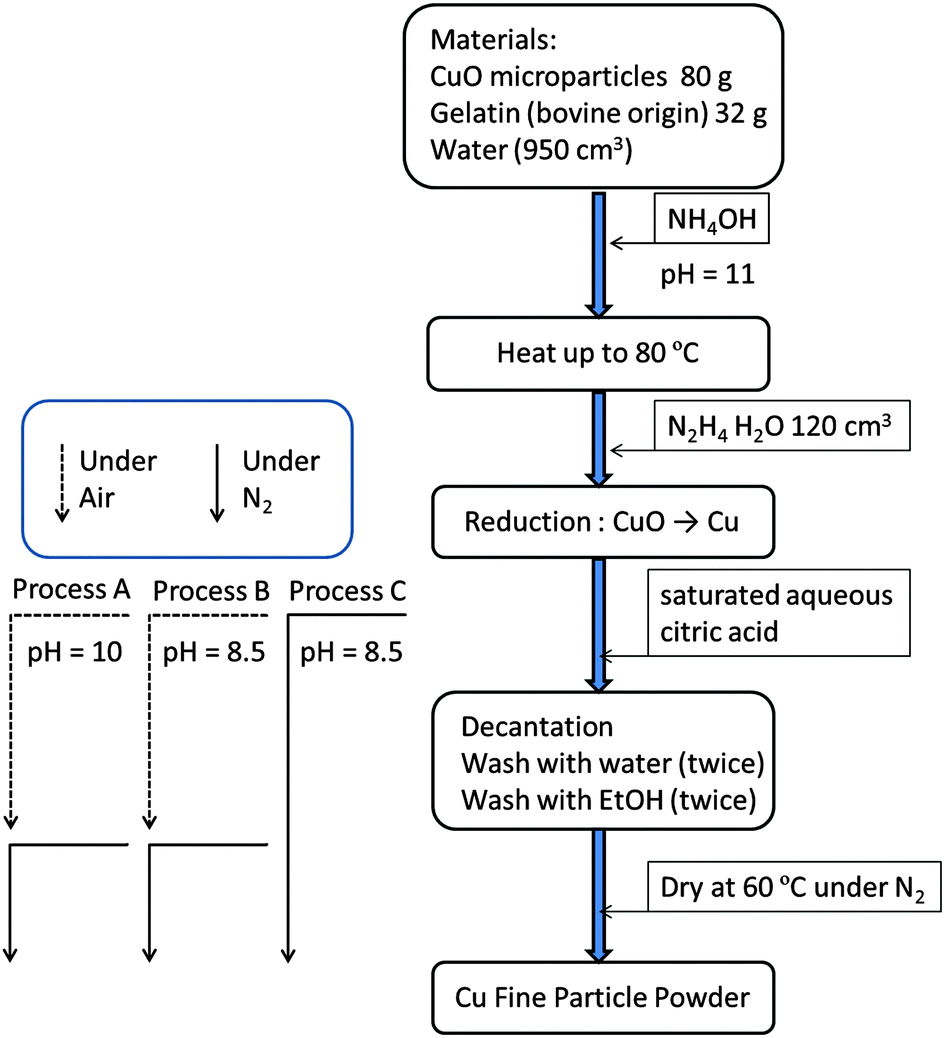 | ||
| Fig. 1 Preparation procedures to obtain gelatin covered copper fine particles from CuO microparticles by chemical reduction with hydrazine. After the reduction, three processes were performed separately. Process A: the pH of the obtained dispersion was decreased to 10 by adding saturated aqueous citric acid under ambient conditions. Then, the collected particles were washed with water and ethanol under ambient conditions. The washed particles were dried at 60 °C under the nitrogen atmosphere. Process B: the pH of the obtained dispersion was decreased to 8.5 by addition of saturated aqueous citric acid under ambient conditions. Then, the collected particles were washed with water and ethanol under ambient conditions. The washed particles were dried at 60 °C under the nitrogen atmosphere. Process C: the pH of the obtained dispersion was decreased to 8.5 by adding saturated aqueous citric acid in a plastic bag under nitrogen flow. Then, the collected particles were washed with water and ethanol under nitrogen flow. The washed particles were dried at 60 °C under the nitrogen atmosphere. | ||
Characterisation
Scanning electron microscopy (SEM) images of the particles were collected using a JEOL JSM-6701F with an acceleration voltage of 15 kV. XRD patterns were obtained using a Philips PANalytical Pro (Cu Kα). TEM images were collected using a Hitachi H-9500 with an acceleration voltage of 300 kV, and HR-TEM images were obtained using a JEOL JEM-2010F with an acceleration voltage of 200 kV. Before XRD measurements and TEM observations, we kept our samples under nitrogen in order to avoid oxidation in that period.Results and discussion
Preparation of gelatin nanoskin-covered copper fine particles
The copper fine particles were prepared by the chemical reduction of CuO microparticles by hydrazine, as previously reported.31,32 In the presence of gelatin, the reduction proceeds smoothly by the addition of hydrazine monohydrate under alkaline conditions. The injection of hydrazine monohydrate into the dispersion of CuO microparticles and gelatin instantly changed the color of the dispersion from black to red-brown, indicating that the reduction by hydrazine had occurred. As we added NH4OH in the preparation dispersion, copper(II) ions are extracted as copper–ammonium complexes into the solution. Then, these complexes are reduced to metallic copper by hydrazine. The prepared metallic copper particles were collected by decantation at pH = 10 (Process A) or 8.5 (Processes B and C) after the reduction. No filtration or centrifugation was required to collect the particles. Filtration and centrifugation are highly time-consuming processes in the preparation of nanoparticles and fine particles. The particles could be obtained readily and reproducibly. In all cases, red-brown copper fine particles were obtained using these preparation procedures. Following the reaction, the obtained copper fine particles were collected by decantation after adjusting the pH by adding saturated aqueous citric acid solution. The reaction was carried out at pH = 11 to facilitate hydrazine decomposition. Then, we adjusted the pH of the dispersion to collect the particles by decantation. The isoelectric point of gelatin (prepared under alkaline conditions) has been frequently reported to be 4.7, and the second critical point gelatin occurs at pH 7.7.33,34 The critical point at pH 7.7 is considered to be the isoelectric point of the sol forms of gelatin. When the pH value of the copper particle dispersion is lowered by adding saturated aqueous citric acid, the electrostatic repulsion forces between the particles decrease because of the ionic state of the gelatin molecules and the increasing ionic strength of the dispersion (the salting-out effect). Then, at a lower pH, the copper fine particles can aggregate and be readily collected by simple decantation. In these experiments, copper particles could be easily obtained by decantation at both pH = 10 and 8.5. Therefore, we used these two conditions in further experiments.Characterization of gelatin-layer covered copper fine particles
No obvious oxidation of the particles was noted, and no color change of the dry particles was observed for several weeks. CuO and cuprous oxide (Cu2O) are usually black and red, respectively. Fig. 2 presents the SEM images and size distributions of the obtained copper fine particles. The copper fine particles are not perfectly spherical but, rather, polyhedral, as reported previously.24,31,32 The particle surface was smoothly covered by gelatin and no obvious spots are observed on the particles' surfaces. This suggests that no obvious oxidation occurred in the ambient atmosphere. All samples exhibit fairly uniform particle sizes, and the size distributions are similar to normal distributions. The particle sizes did not change dramatically as the particle-collecting conditions after reduction were varied. The average particle sizes obtained in this study were ca. 120–130 nm.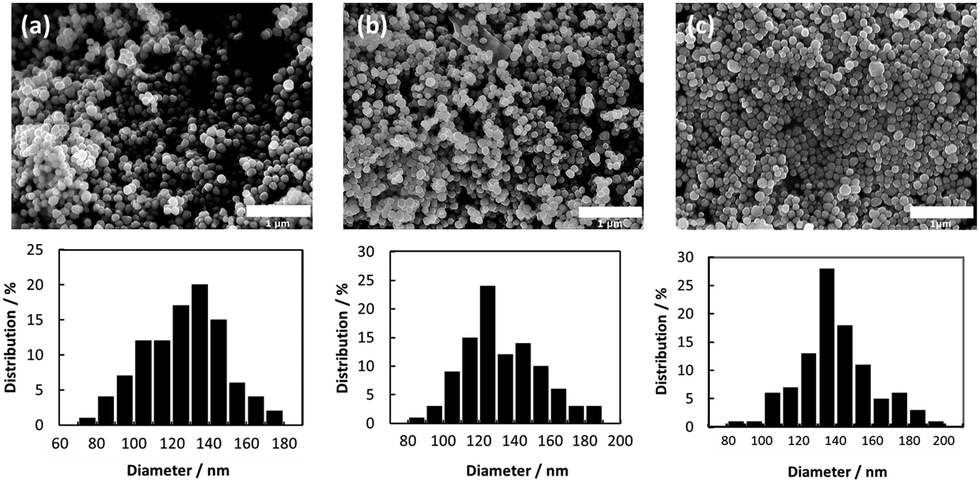 | ||
| Fig. 2 Scanning electron microscopy (SEM) images and size distributions of the obtained copper fine particles stabilized by a gelatin layer. (a) Process A: decantation was carried out at pH = 10 under an ambient atmosphere. (b) Process B: decantation was carried out at pH = 8.5 under an ambient atmosphere. (c) Process C: decantation was carried out at pH = 8.5 under a nitrogen atmosphere. | ||
TEM images of the surfaces of the copper fine particles prepared under different conditions are presented in Fig. 3. Thin smooth gelatin layers are readily evident on the particles' surfaces. Comparison of Fig. 3(a) (Process A, decantation at pH = 10) and Fig. 3(b) and (c) (Processes B and C, respectively: decantation at pH = 8.5,) reveals that the thickness of the gelatin layer in Fig. 3(a) is clearly smaller than those in Fig. 3(b) and (c). Adjusting the pH of the gelatin solution changes the thickness of the gelatin layer: approximately 2.7 nm (Process A), 5.0 nm (Process B), and 6.5 nm (Process C). These findings revealed that the thickness of the gelatin layer on the particle surface can be well controlled by adjusting the pH at decantation. Because the isoelectric point of gelatin in sol is ca. pH = 7.7, the gelatin molecules are almost neutral or slightly negative at pH = 8.5 and are more negatively charged at pH = 10. Therefore, the particles collected at pH = 8.5 are covered in a more dense and thicker layer of gelatin than those collected at pH = 10. TGA measurements of the copper particles obtained by Process C indicated that the amount of gelatin covering on the particle surface is about 2 wt% (data not shown).
 | ||
| Fig. 3 Transmission electron microscopy (TEM) images of the surfaces of the obtained copper fine particles stabilized by a gelatin layer: (a) prepared by Process A, (b) prepared by Process B, and (c) prepared by Process C. | ||
Fig. 4 presents the XRD patterns of the copper fine particles obtained via these three preparation procedures. Strong sharp peaks corresponding to fcc metallic copper can be clearly observed in all patterns.35 However, in Fig. 4(a) (Process A), two small peaks at 35.6° and 38.9°, which correspond to the (002) (35.495°) and (111) (38.726°) surfaces of CuO,36 respectively, are present. In the TEM image of the particles prepared by Process A (Fig. 3(a)), two small spots are evident on the particle surface (arrowed). In our previous studies, oxidative annealing of the gelatin-stabilized copper fine particles resulted in Cu2O spots on the surface.31,32 Cu2O is gradually oxidized to CuO under ambient conditions. The XRD pattern (Fig. 4(a)) and the TEM image (Fig. 3(a)) of the copper fine particles prepared by Process A strongly suggest that CuO crystalline layers are generated on the particle surface during the collection of the particles. In contrast, the XRD patterns of the copper fine particles collected at pH = 8.5 under ambient conditions (Fig. 4(b)) or the nitrogen atmosphere (Fig. 4(c)) do not show any peaks corresponding to copper oxides. Therefore, according to these XRD patterns, the surfaces of the copper particles obtained at pH = 10 are partially oxidized, whereas the surfaces of the particles obtained at pH = 8.5 under both air nitrogen do not appear to have been oxidized to CuO.
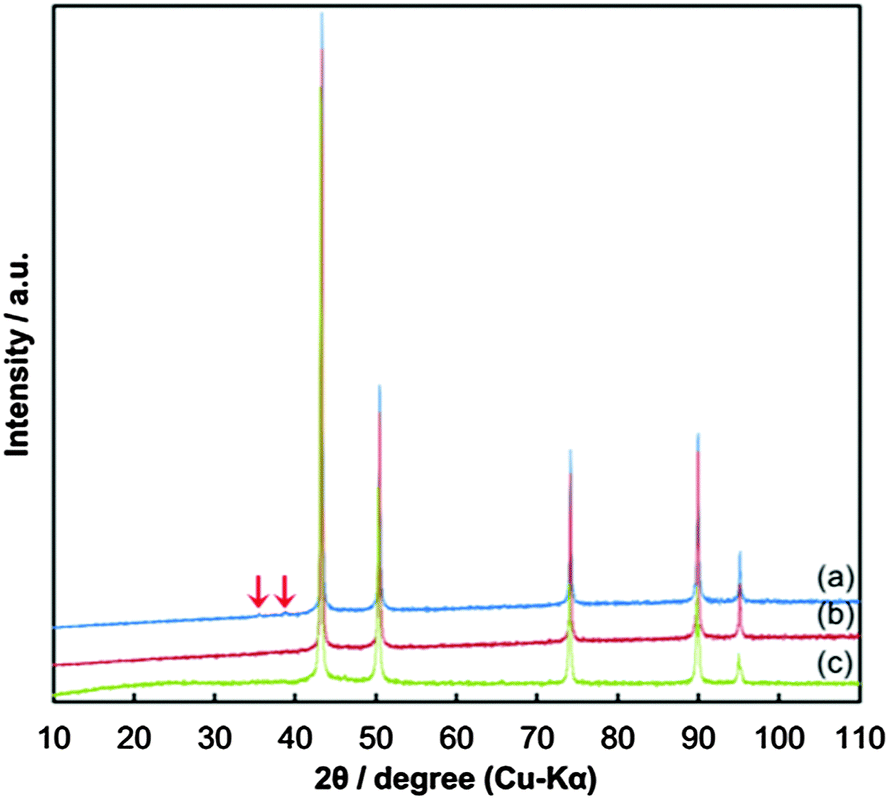 | ||
| Fig. 4 X-ray diffraction (XRD) patterns of the obtained copper fine particles stabilized by a gelatin layer: (a) prepared by Process A, (b) prepared by Process B, and (c) prepared by Process C. Arrows on the pattern (a) indicate the peaks corresponding to CuO(002) and CuO(111). | ||
However, if the oxidized grains are very small, the XRD peaks of the copper oxides become very broad and are difficult to identify. Therefore, other analytical methods should be applied to reveal the existence of such small oxidized grains. For this purpose, we chose HR-TEM to observe the surface of the copper fine particles. Alternatively, selected area electron diffraction (SAED) could be a good technique to confirm copper oxidation on the surface. However, a careful observation of lattice fringes can also provide useful information about oxidation, and the oxidized region can be directly identified in the images. The HR-TEM images of the obtained copper fine particles are presented in Fig. 5. The lattice fringes can be clearly observed in the images, indicating that all particles were well crystallized. Their surfaces were covered by gelatin, as seen in the images, especially in Fig. 5(a). Based on these images, we determined the distance between the two fringe lines, which corresponds to the lattice parameters of the stable surfaces of the samples. In the inner area of Fig. 5(a) (circle A), the average distance between the two fringe lines is 2.13 ± 0.11 Å. This distance corresponds to a Cu(1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1) lattice (2.087 Å)35 and shows that the particles have metallic cores. In contrast, in the surface region of circle B, the average distance between the two fringe lines is 2.52 ± 0.11 Å, which corresponds to CuO(002) (2.53 Å) or CuO(11) (2.526 Å) lattices.36 Based on the XRD pattern shown in Fig. 4(a) and the HR-TEM image (Fig. 5(a)) some areas on the surface of the particles obtained by decantation at pH = 10 under ambient conditions clearly exhibit oxidation.
1) lattice (2.087 Å)35 and shows that the particles have metallic cores. In contrast, in the surface region of circle B, the average distance between the two fringe lines is 2.52 ± 0.11 Å, which corresponds to CuO(002) (2.53 Å) or CuO(11) (2.526 Å) lattices.36 Based on the XRD pattern shown in Fig. 4(a) and the HR-TEM image (Fig. 5(a)) some areas on the surface of the particles obtained by decantation at pH = 10 under ambient conditions clearly exhibit oxidation.
 | ||
| Fig. 5 High-resolution transmission electron microscopic (HR-TEM) images of the surfaces of the gelatin-stabilized copper fine particles. (a) Prepared by Process A, (b) prepared by Process B, and (c) prepared by Process C. Circles A–E indicate the area where the distances of the fringe lattices were measured. | ||
We also obtained gelatin-stabilized copper fine particles by decantation under ambient conditions after decreasing the pH of the dispersion to 8.5. This pH value is near the isoelectric point of gelatin. The XRD pattern of these particles does not show clear peaks indicating CuO and Cu2O,37 as observed in Fig. 4(b). The oxide peaks were not observed after a couple of months keeping under an ambient atmosphere as a powder form. Fig. 5(b) presents the HR-TEM image of the copper fine particles collected by decantation at pH = 8.5. The gelatin layer on the surface of the particles is denser and thicker than that on the particles obtained at pH = 10. The surface of the layer is also smoother. Because the prepared copper fine particles were collected at pH = 8.5, the gelatin molecules on the particle surfaces exhibited stronger interactions with each other. Therefore, more gelatin molecules were adsorbed on the particle surface, forming a thicker gelatin layer. The lattice fringes of this sample were also evaluated. Inside area of Fig. 5(b) (circle C), the average distance between the two lattice fringe lines is 2.13 ± 0.04 Å, corresponding to a Cu(111) lattice (2.087 Å),30 indicating that the particles have metallic cores. The distance between the lattice fringes in three areas in the surface region was also determined. The average distances between the two fringes in circles D and F are 2.48 ± 0.07 Å and 2.55 ± 0.07 Å, respectively. These values also correspond to CuO(002) (2.53 Å) or CuO(11) (2.526 Å) lattices.36 However, the average distance of the two lattice fringes in circle E is 2.13 ± 0.02 Å, which corresponds to a Cu(111) lattice.35 Interestingly, the surface of the copper particles is perfectly metallic, although this area is simply covered by a polymer layer. The lattice fringes in the HR-TEM image strongly suggest that even at pH = 8.5, the oxidation of copper particles proceeded but was much slower than that at pH = 10 because of the difference in the thickness of the gelatin layer on the particle surface and also probably because of its density.
During the reduction step in the preparation process, the dispersion, which contained hydrazine, was under reductive conditions. CuO micro particles were reduced, resulting in metallic copper fine particles. Hydrazine produces nitrogen and hydrogen gases during the reduction reaction. Therefore, the surface of the copper metallic particles must not be oxidized after the formation of the copper fine particles. However, after the reduction, oxygen from the atmosphere began to dissolve in the dispersion, and the gelatin molecules began to swell. Therefore, the surface of the obtained copper fine particles can be gradually oxidized during decantation. The oxidation rate can be varied by adjusting the density and the thickness of the gelatin layer on the particle surface. Therefore, to prevent the surface oxidation of the copper fine particles, we investigated the effect of the atmosphere in contact with the dispersion during decantation. We added saturated aqueous citric acid to the particle dispersion and stirred the mixture under the nitrogen atmosphere. The subsequent decantation was also performed under nitrogen flow. For this purpose, the entire preparation system was covered with a plastic bag, and nitrogen gas was blown in from the top. Then, the obtained powder was dried under the nitrogen atmosphere at 60 °C.
The XRD pattern of the obtained copper fine particles is shown in Fig. 4(c). Clear peaks corresponding to metallic copper can be observed, but no peaks corresponding to copper oxides are present. This pattern suggests that the surface is covered by metallic copper. The HR-TEM image of these particles is shown in Fig. 5(c). The average distance between the two fringes in circle G in the inner region is 2.13 ± 0.09 Å, which suggests a Cu(111) lattice.35 In contrast, that in circle H in the particle's outermost region is 2.23 ± 0.07 Å. This value is slightly larger than that of a Cu(111) lattice but much smaller than those of CuO(002) (2.53 Å), CuO(11) (2.526 Å),36 or Cu2O(111) (2.413 Å).37 Other areas in the surface region also show small fringe distances. After a careful search on the literature it was found that the spacing of the Cu64O(404) face is 2.215 Å.38 Thus, it can be concluded that the particles obtained by decantation, washing, and drying under the nitrogen atmosphere have metallic structures or very slightly oxidized areas (Fig. 6). Additionally, it can be noted that further surface oxidation can be prevented by the dry gelatin layers.
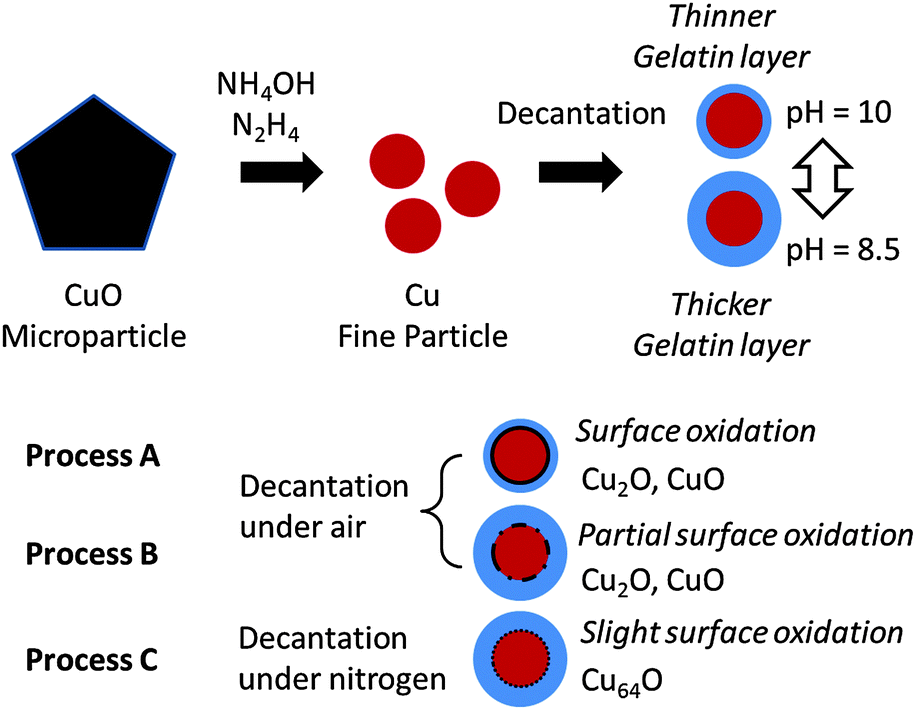 | ||
| Fig. 6 Schematic illustration of the relationship between the purification and collection procedure and surface oxidation of the particles. When collection of the particles was performed by decantation at a higher pH (10), the surface gelatin layer was thinner than that on the particles collected by decantation at a lower pH (8.5). The surface oxidation was observed on the particles collected under an ambient atmosphere. The particles collected at a lower pH showed partial surface oxidation. Slight surface oxidation was observed when the particles were collected under a nitrogen atmosphere. | ||
Two important results can be extracted from these experimental findings. First, surface oxidation occurs after reduction in the aqueous dispersion, likely because of the swelling of the gelatin layer in the aqueous solution. Second, the gelatin layer on the copper particles' surfaces strongly prevents the surface oxidation of the dry particles. We previously reported that gelatin-covered copper fine particles are quite stable and not easily oxidized in air.24 However, in this study, we found that non-oxidized copper particles can be obtained, as shown in Fig. 5(c), even if they are kept under ambient conditions, if they are dried.
Gelatin is a cost-effective and very useful polymer for nanoparticle synthesis. It is a polypeptide with amino groups, mercaptans, and carboxylic acid groups in the side chains. These functional groups can effectively coordinate with the particle surface. Therefore, the gold number of gelatin is extremely high, similar to that of poly(vinylpyrrolidone) (PVP), which is frequently used for nanoparticle stabilization.2,23 The inter molecular interactions between the gelatin polymer chains are also very strong because of the peptide groups in the main chain. Thus, the gelatin molecules should be densely adsorbed on the copper particle surface. This unique property of gelatin is indispensable for its anti-oxidant protection of dry copper fine particles under ambient conditions.
Conclusions
In conclusion, we prepared gelatin-coated copper fine particles via three procedures after reduction. After reduction, decantation, washing, and drying steps were performed. The copper fine particles obtained by decantation at pH = 10 under ambient conditions exhibit small CuO peaks in their XRD patterns, indicating that the particle surface was oxidized. In contrast, according to the lattice fringes observed by HR-TEM, decantation at pH = 8.5, washing, and drying under the nitrogen atmosphere resulted in copper fine particles stabilized by a gelatin layer with non-oxidized or slightly oxidized to Cu64O surfaces. The obtained particles were not readily further oxidized under ambient conditions in dry form. These copper fine particles are suitable for printed electronics, for which the surface oxidation state of the particles is very important. Our results also indicate that XRD analyses cannot perfectly reveal the surface oxidation of copper fine particles.Acknowledgements
This work is partially supported by NEDO and by KAKENHI (to TY, 24241041) from JSPS, Shinsei Foundation, and Murata Foundation. Authors thank Prof. N. Sakaguchi (Hokkaido Univ.) for his kind assistance in HR-TEM observation and Nitta Gelatin Inc. for gelatin supply.Notes and references
- Nanoparticles, ed. G. Schmid, Wiley-VCH, Weinheim, 2011 Search PubMed.
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179–1201 RSC.
- N. Li, P. Zhao and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 1756–1789 CrossRef CAS PubMed.
- T. Yonezawa, Kobunshi Ronbunshu, 2013, 70, 684–692 CrossRef CAS.
- S. Kango, S. Kalia, A. Celli, J. Njuguna, Y. Habibi and R. Kumar, Prog. Polym. Sci., 2013, 38, 1232–1261 CrossRef CAS.
- N. Yan, C. Xiao and Y. Kou, Coord. Chem. Rev., 2010, 254, 1179–1218 CrossRef CAS.
- F. Viñes, J. R. B. Gomes and F. Illas, Chem. Soc. Rev., 2014, 43, 4922–4939 RSC.
- S. Chernousova and M. Epple, Angew. Chem., Int. Ed., 2013, 52, 1636–1653 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim and X. Li, Chem. Rev., 2012, 112, 2379–2779 CrossRef PubMed.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Plasmonics, 2007, 2, 107–118 CrossRef CAS.
- T. Yonezawa, S. Takeoka, H. Kishi, K. Ida and M. Tomonari, Nanotechnology, 2008, 19, 145706 CrossRef PubMed.
- J. Perelaer, P. J. Smith, D. Mager, D. Soltman, S. K. Volkman, V. Subramanian, J. G. Korvink and U. S. Schubert, J. Mater. Chem., 2010, 20, 8446–8453 RSC.
- A. C. Arias, J. D. MacKenzie, I. MacCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- Y. Zhang, P. Zhu, G. Li, T. Zhao, X. Fu, R. Sun, F. Zhou and C.-p. Wong, ACS Appl. Mater. Interfaces, 2014, 6, 560–567 CAS.
- D. Deng, Y. Jin, Y. Cheng, T. Qi and F. Xiao, ACS Appl. Mater. Interfaces, 2013, 5, 3839–3846 CAS.
- Y.-C. Liao and Z.-K. Kao, ACS Appl. Mater. Interfaces, 2012, 4, 5109–5113 CAS.
- J. A. Lewis and B. Y. Ahn, Nature, 2015, 518, 42–43 CrossRef CAS PubMed.
- W. Shen, X. Zhang, Q. Huang and Q. Xu, Nanoscale, 2014, 6, 1622–1628 RSC.
- S. Magdassi, M. Grouchko and A. Kamyshny, Materials, 2010, 3, 4626–4638 CrossRef CAS.
- Y. Yong, T. Yonezawa, M. Matsubara and H. Tsukamoto, J. Mater. Chem. C, 2015, 3, 5890–5895 RSC.
- J. Xiong, Y. Wang, Q. Xue and X. Wu, Green Chem., 2011, 13, 900–904 RSC.
- D. Mott, J. Galkowski, L. Wang, J. Luo and C.-J. Zhong, Langmuir, 2007, 23, 5740–5745 CrossRef CAS PubMed.
- Y. Lee, J.-r. Choi, K. J. Lee, N. E. Stott and D. Kim, Nanotechnology, 2008, 19, 415604 CrossRef PubMed.
- M. Tomonari, K. Ida, T. Yamashita and T. Yonezawa, J. Nanosci. Nanotechnol., 2008, 8, 2468 CrossRef CAS PubMed.
- T. Yonezawa, N. Nishida and A. Hyono, Chem. Lett., 2010, 39, 548–549 CrossRef CAS.
- Y. Kobayashi and T. Sakuraba, Colloids Surf., A, 2008, 317, 756–759 CrossRef CAS.
- M. Grouchko, A. Kamyshny and S. Magdassi, J. Mater. Chem., 2009, 19, 3057–3062 RSC.
- X.-F. Tang, Z.-G. Yang and W.-J. Wang, Colloids Surf., A, 2010, 360, 99–104 CrossRef CAS.
- G.-J. Lee, C. K. Kim, M. K. Lee and C. K. Rhee, Powder Technol., 2014, 261, 143–146 CrossRef CAS.
- H. Kawasaki, Y. Kosaka, Y. Myojin, T. Narushima, T. Yonezawa and R. Arakawa, Chem. Commun., 2011, 47, 7740–7742 RSC.
- M. Matsubara, T. Yonezawa and H. Tsukamoto, Bull. Chem. Soc. Jpn. DOI:10.1246/bcsj.20150305.
- T. Yonezawa, H. Tsukamoto and M. Matsubara, RSC Adv., 2015, 5, 61290–61297 RSC.
- J. H. Highberger, J. Am. Chem. Soc., 1939, 61, 2302–2303 CrossRef CAS.
- B. Gaihre, M. S. Khil, D. R. Lee and H. K. Kim, Int. J. Pharmacol., 2009, 365, 180–189 CrossRef CAS PubMed.
- R. P. van Ingen, R. H. J. Fastenau and E. J. Mittemeijer, J. Appl. Phys., 1994, 76, 1871–1883 CrossRef CAS.
- S. Åsbrink and A. Waskowska, J. Phys.: Condens. Matter, 1991, 3, 8173–8180 CrossRef.
- A. Werner and H. D. Hochheimer, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 25, 5929–5934 CrossRef CAS.
- R. Guan, H. Hashimoto and K. H. Kuo, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 219–225 CrossRef.
| This journal is © the Owner Societies 2015 |