
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAre the three hydroxyphenyl radical isomers created equal? – The role of the phenoxy radical –†
P.
Hemberger
*a,
G.
da Silva
b,
A. J.
Trevitt
c,
T.
Gerber
a and
A.
Bodi
a
aMolecular Dynamics Group, Paul Scherrer Institute, 5232 Villigen PSI, Switzerland. E-mail: patrick.hemberger@psi.ch
bDepartment of Chemical and Biomolecular Engineering, The University of Melbourne, Victoria 3010, Australia
cSchool of Chemistry, University of Wollongong, New South Wales 2522, Australia
First published on 21st October 2015
Abstract
We have investigated the thermal decomposition of the three hydroxyphenyl radicals (˙C6H4OH) in a heated microtubular reactor. Intermediates and products were identified isomer-selectively applying photoion mass-selected threshold photoelectron spectroscopy with vacuum ultraviolet synchrotron radiation. Similarly to the phenoxy radical (C6H5–O˙), hydroxyphenyl decomposition yields cyclopentadienyl (c-C5H5) radicals in a decarbonylation reaction at elevated temperatures. This finding suggests that all hydroxyphenyl isomers first rearrange to form phenoxy species, which subsequently decarbonylate, a mechanism which we also investigate computationally. Meta- and para-radicals were selectively produced and spectroscopically detectable, whereas the ortho isomer could not be traced due to its fast rethermalization and rapid decomposition in the reactor. A smaller barrier to isomerization to phenoxy was found to be the reason for this observation. Since hydroxyphenyl species may be present under typical sooting conditions in flames, the resonantly stabilized cyclopentadienyl radical adds to the hydrocarbon pool and can contribute to the formation of polycyclic aromatic hydrocarbons, which are precursors in soot formation.
Introduction
Oxygenated radicals, such as phenoxy (C6H5–O˙) play a large role in biological and environmental processes as transient species.1 Substituted or metal-complexed phenoxy radicals are commonly encountered in larger biomolecules such as microperoxidase and galactooxidase2 – important compounds in biocatalysis.3 In addition, phenoxy moieties are common linkages in lignin macromolecules, which can be cracked by fast-pyrolysis techniques to yield more valuable fine chemicals and fuels.4–9 Recently, we have investigated the thermal decomposition of diphenylether (DPE), a lignin model compound, and found that phenyl (C6H5) and phenoxy radicals are the major decomposition products at low temperatures.10 It is well known that resonantly-stabilized phenoxy radicals are common intermediates in combustion processes.11 Besides the reaction of OH radicals with benzene to form phenyl radicals (1) or phenol (2), it was found that the formation of phenoxy radicals (3) contributes significantly to the fuel consumption pathways in benzene/oxygen/argon flames.12–14| C6H6 + OH → C6H5 + H2O | (1) |
| C6H6 + OH → C6H5OH + H | (2) |
| C6H6 + O(3P) → C6H5O + H | (3) |
| C6H5 + O2 → C6H5–OO → C6H5O + O(3P) | (4) |
| C6H5O → C5H5 + CO | (5) |
| C6H5O + H → C6H5OH | (6) |
| C5H5 → C3H3 + C2H2 | (7) |
The oxidation of benzene by OH addition to yield phenol (2) is a key step in the oxidation chemistry of aromatic fuels.14 Hydrogen atom abstraction from the hydroxyl group can lead to phenoxy radicals, which are in turn unreactive towards oxidation, but decompose unimolecularly (5). However, it is also conceivable that H-abstraction from the ring or the fragmentation of larger aromatics leads to the formation of hydroxyphenyl radicals, ˙C6H4–OH. These radicals, which are another isomeric form of the composition C6H5O, are not very well understood in terms of their unimolecular and bimolecular reactivity and their ability to form larger PAHs and soot. This is mostly due to their relatively unknown electronic and structural properties, which makes their detection difficult. Nagata et al. measured the FT-IR spectrum of 2-hydroxyphenyl radicals in a low-temperature argon matrix after irradiation of 2-iodophenol with UV light.26 The electron spin resonance spectrum was measured by Kasai and McLeod Jr.28 in an argon matrix, and Schuler et al.29 investigated the conversion of 2-hydroxyphenyl into phenoxy in aqueous solution with the same technique. However, no ultraviolet, ionization, or photoelectron spectra have been measured so far to the best of our knowledge. The oxidation of ortho-hydroxyphenyl radicals has recently been investigated by time resolved VUV photoionization mass spectrometry, and cyclopentadienone and o-benzoquinone were identified as the dominant products.30 On the one hand, hydroxyphenyl radicals were hypothesized to be intermediates in 1,3-aryl shift chemistry of decomposing ortho-dimethoxybenzene, leading to cyclopentadienyl radicals.6 On the other, it is known that phenyl radicals decompose via hydrogen loss to yield benzyne (De = 78 kcal mol−1),31 which subsequently reacts in a retro-Diels–Alder fashion to acetylene and diacetylene.32 Substituting hydrogen with a hydroxyl group in phenyl could thus lead to a completely different deactivation pathway.
Our report deals with the generation, decomposition and detection of the three hydroxyphenyl radical isomers. In particular, we probe the reaction products of unimolecular processes, which can compete with the direct oxidation of substituted phenyl radicals in combustion systems.
Experimental and theoretical methods
Experiments were carried out at the VUV beamline of the Swiss Light Source in Villigen, Switzerland. Vacuum ultraviolet light is generated by a bending magnet and transferred to the experimental station by three optical elements.33 A 150 mm−1 gold-coated grating is used in a home-built monochromator, and provides a resolving power of 1500 at an exit slit of 200 μm. A differentially pumped gas filter with an inlet pressure of 10 mbar or an MgF2 window absorbs higher order radiation. The light is intersected with a molecular beam in the ionization region of the iPEPICO34 apparatus. A 120 V cm−1 field extracts the electrons, which are velocity map imaged (VMI) onto a Roentdek DLD40 delay line anode detector. The detection of an electron tags the time zero for the time-of-flight ion detection in a multiple-start multiple-stop scheme.35 Ions are detected by a Jordan-TOF MCP detector. Hydroxyphenyl radicals were produced from 2-, 3- and 4-iodophenol precursors, highly diluted in argon at a backing pressure of 1.3–1.5 bar, in a resistively heated SiC microtubular reactor (I.D. = 1 mm). It has been shown that iodophenols are excellent precursors to generate hydroxyphenyl radicals.36 The precursors were treated by several pump–vent cycles to remove volatile impurities. Due to the low vapor pressure, the radical precursors were heated to 25–35 °C in an in-vacuum sample container and expanded through a 100 μm nozzle directly into the heated microtubular reactor. The sample container temperature was accurately set in order to control the dilution of the precursor in argon to suppress bimolecular reactions. A temperature reduction of around 5 °C could reduce the amount of bimolecular products, e.g. hydrogen iodide and phenol, by almost one order of magnitude accompanied by a much smaller decrease in the unimolecular fragment signal. Thus the temperature of the sample container was set to minimize the amount of HI and other products from bimolecular chemistry, while being still high enough to obtain an acceptable signal-to-noise ratio in the unimolecular channels. The temperature of the reactor was measured by a type-C thermocouple at the surface of the SiC tube. According to the detailed study by Guan et al.,37 the centerline temperature in the reactor can be up to 25% less than the outside surface temperature of the reactor wall with argon bath gas. Based on the computational fluid dynamics simulations of Guan et al., we estimate the pressure at the entrance of the reactor after the flux-reducing 100 μm pinhole to be several tens of mbar. When the pyrolysed sample leaves the reactor ca. 10–100 μs later, the pressure drops by several orders of magnitude to 10−4 mbar.The molecular beam is skimmed by a 1 mm I.D. Beam Dynamics skimmer and intersected with the VUV light in the spectrometer chamber. We measured temperature dependent mass spectra at different photon energies to determine the reaction pathways. Photoion38,39 mass-selected threshold photoelectron spectra (ms-TPES) and photoionization (PI) spectra were obtained by scanning the photon energy in 10–20 meV steps and averaging for 120 to 300 s per point in order to identify different isomers. In case of PI spectra all kinetic energy electrons were selected in the coincidence scheme, whereas only near threshold energy electrons (Ekin < 5 meV) were selected in typical ms-TPE scans. The energetic (hot) electron contamination in the VMI images was subtracted by a procedure as suggested by Sztáray et al.40
Computations were performed utilizing the Gaussian0941 suite of programs. For the Franck–Condon simulations, we used ezSpectrum.OSX42 with geometries and force constant matrices obtained at the B3LYP/6-311++G(d,p) level of theory. Ionization energies were calculated applying the CBS-QB3 composite method, including zero-point corrections.43,44 For the potential energy surface of hydroxyphenyl radicals, the G3X-K theory was applied.45
Mass spectrometric analysis
In order to determine the decomposition pathways, we have measured mass spectra as a function of the reactor temperature at a photon energy of 9 eV as shown in Fig. 1(a) for 2-iodophenol. | ||
| Fig. 1 Mass spectra of 2- (a) and 3-iodophenol (b) as a function of the reactor temperature. The peak at m/z = 133 can be assigned to 1,2,3,4-tetrahydroquinoline, which remained in the ionization chamber from previous experiments and does not affect the discussion herein. The spectra are normalized to the most intense peak. | ||
The decomposition of the 2-iodophenol precursor to 2-hydroxyphenyl starts at around 600 °C. Complete decomposition is observed at temperatures above 700 °C. The appearance of m/z = 65 can be attributed to cyclopentadienyl (c-C5H5) and the trace amounts of m/z = 93 and 94, to 2-hydroxyphenyl (˙C6H4–OH) or phenoxy radicals, and phenol (C6H5OH), respectively. The m/z = 220 signal is attributed to the iodophenol radical precursor. It should be noted that the iodine loss at the β-carbon site of the precursor indeed results in 2-hydroxyphenyl, but its fast disappearance indicates subsequent rapid unimolecular decomposition to cyclopentadienyl. Above 700 °C, propargyl radical (m/z = 39) and acetylene (see ESI,† Fig. S1) signals appear, corresponding to fragments of the C5H5 radical. The overall reaction sequence is depicted in Scheme 1.
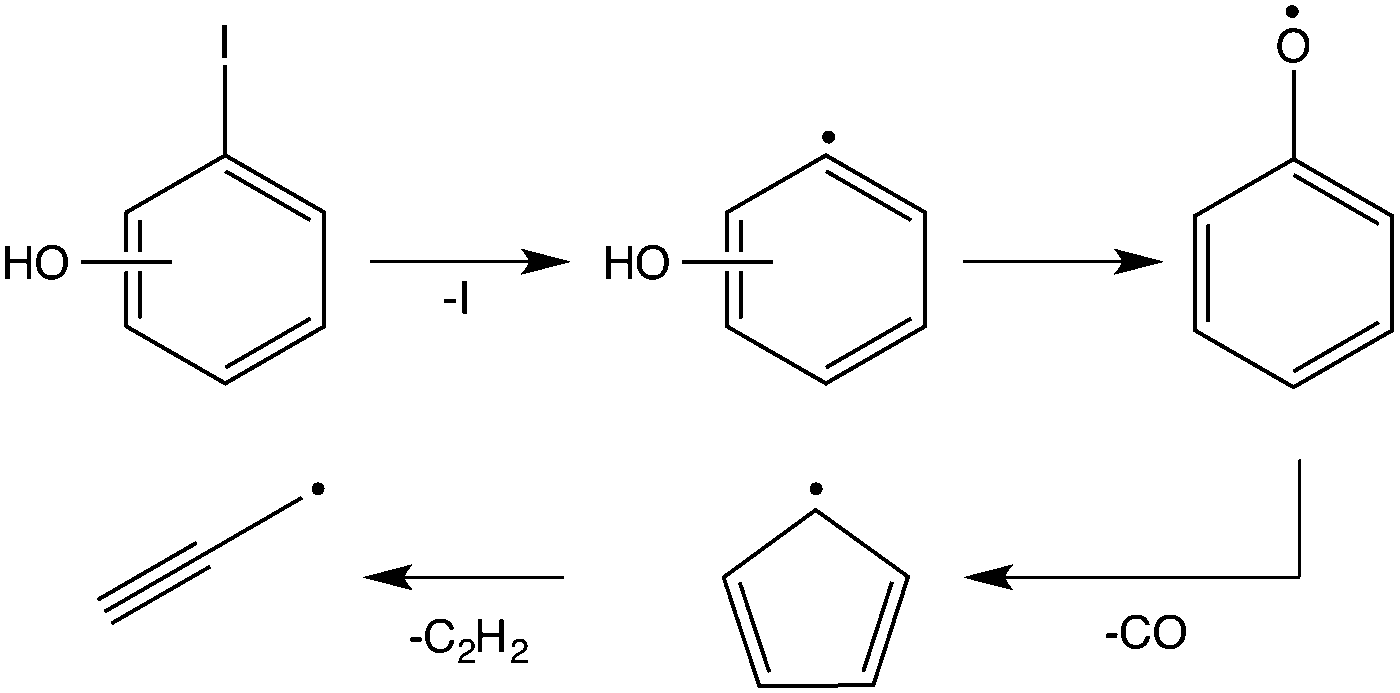 | ||
| Scheme 1 Decomposition pathway of n-iodophenol (n = 2–4). The hydroxyphenyl radicals rearrange to phenoxy and undergo decarbonylation to yield cyclopentadienyl radicals, and finally form propargyl radicals by acetylene loss. | ||
The PI-MS of meta-iodophenol, displayed in Fig. 1(b) as a function of the reactor temperature, shows some differences when compared to the ortho-iodophenol spectra. A greater m/z = 93 signal intensity points to an increased stability of 3-hydroxyphenyl towards both isomerization and decomposition (vide infra) compared to 2-hydroxyphenyl radicals. Here again, phenol at m/z = 94 is produced via bimolecular chemistry, when the highly reactive 3-hydroxyphenyl abstracts a hydrogen atom, e.g., from the non-pyrolyzed precursor. If the temperature is steadily increased, we observe the degradation of the C6H5O (m/z = 93) signal, while c-C5H5 (m/z = 65) is formed after CO loss. Indeed, both 2- and 3- and 4-hydroxyphenyl (see ESI,† Fig. S2) isomers follow the same decomposition pattern forming C5H5 and C3H3 species.46,47 This decomposition chemistry is different from the unsubstituted phenyl radical, which forms benzyne after a hydrogen loss, which in turn yields acetylene and diacetylene; hydroxyphenyl radicals do not show any convincing evidence for decomposition pathways leading to hydroxybenzyne (m/z = 92) or hydroxydiacetylene (m/z = 66).
The decomposition products and the more abundant intermediates can be assigned isomer specifically by their mass-selected threshold photoelectron spectra and, in some favourable cases, even by their photoionization spectra. Fig. 2 shows ms-TPE spectra of m/z = 65 and 39 as measured from decomposing 2-hydroxyphenyl radicals generated upon pyrolysis of 2-iodophenol. The feature at 8.43 eV of the black trace (m/z = 65) is in good agreement with the adiabatic ionization energy of cyclopentadienyl radicals, which is assigned to the ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 2E1′′→ 3A2′ transition. At 8.62 eV, a second ion state can be populated, which was assigned to the 2E1′′→ ã1E2′ transition by Wörner and Merkt.48 Trace amounts of l-C5H5 also contribute to the threshold ion signal between 8.0 and 8.2 eV, and their involvement in the reaction pathways might also be noticeable in light of recent findings.47,49 A mechanism for the formation of l-C5H5 and the PIE curve showing some evidence is displayed in the ESI† (Fig. S3 and S4). The propargyl radical (m/z = 39), is responsible for the feature arising at 8.70 eV (2B1 → 1A1).50–53 The ms-TPE spectra of propargyl and cyclopentadienyl from decomposing 3- and 4-hydroxyphenyl radicals are nearly identical to those shown in Fig. 2.
2E1′′→ 3A2′ transition. At 8.62 eV, a second ion state can be populated, which was assigned to the 2E1′′→ ã1E2′ transition by Wörner and Merkt.48 Trace amounts of l-C5H5 also contribute to the threshold ion signal between 8.0 and 8.2 eV, and their involvement in the reaction pathways might also be noticeable in light of recent findings.47,49 A mechanism for the formation of l-C5H5 and the PIE curve showing some evidence is displayed in the ESI† (Fig. S3 and S4). The propargyl radical (m/z = 39), is responsible for the feature arising at 8.70 eV (2B1 → 1A1).50–53 The ms-TPE spectra of propargyl and cyclopentadienyl from decomposing 3- and 4-hydroxyphenyl radicals are nearly identical to those shown in Fig. 2.
 | ||
| Fig. 2 ms-TPE spectra of 2-hydroxyphenyl decomposition products. The spectra are nearly identical for the isomers n = 2–4. The spectra can be assigned to propargyl (m/z = 39, red open circles), cyclopentadienyl (m/z = 65, black open squares). | ||
The above results indicate that, independently of the specific isomer formed from iodophenol, the final products of hydroxyphenyl radical decomposition are c-C5H5 and C3H3 radicals. Since the first reaction on the way to c-C5H5 and C3H3 is a CO loss, it is likely that the 2-, 3- and 4-hydroxyphenyl radicals first isomerize to the phenoxy radical and subsequently decompose according to Scheme 1.
Spectroscopic evidence for hydroxyphenyl radicals
The question now arises if one can disentangle the intermediate species in mass channel m/z = 93, which could correspond to one specific C6H5O isomer or a mixture of them. Therefore, we attempted to measure ms-TPE and PI spectra of m/z = 93 of all the three hydroxyphenyl isomers. 2-Hydroxphenyl derived from 2-iodophenol could not be detected in measurable amounts at any reactor temperature by PI spectra or ms-TPES, indicating a rapid decomposition directly or after a very fast isomerization to an unstable compound. Fig. 3 shows photoionization efficiency (PIE) curves, also called photoionization (PI) spectra, of m/z = 93 after pyrolysis of 4- (blue open circle) and 3-iodophenol (red open rectangles) at around 650 °C reactor temperature. | ||
| Fig. 3 PI spectra of m/z = 93 obtained by pyrolysis of 3- and 4-iodophenol at 650 °C and diphenylether (DPE) at 770 °C. The different inclination of the PI spectra obtained from iodophenol and diphenylether could infer different C6H5O isomers. | ||
The black curve in Fig. 3 was obtained from an earlier experiment where we investigated the decomposition of diphenylether (DPE), which, after a C–O cleavage reaction, yields phenyl and phenoxyl radicals as intermediates (Scheme 2).10
 | ||
| Scheme 2 Formation of phenoxy and phenyl radicals by decomposition of diphenylether. | ||
The reference PI spectrum for phenoxy (black triangles) exhibits a steeper slope between 8.55 and 8.8 eV than the curves for m/z = 93 obtained from 3-hydroxyphenol (red squares) and 4-hydroxyphenol (blue circles). The m/z = 93 isomers derived from the iodophenol precursors produced a higher ion signal than phenoxy alone at photon energies as low as 8.2 eV, suggesting that there are m/z = 93 species present with lower ionization energies than the phenoxy isomer. However, PI spectra are subject to temperature effects because of unresolved hot- and sequence bands that can shift the ionization onset to lower photon energies. On the other hand, ms-TPE spectroscopy can be used to address this issue, since vibronic transitions from the neutral into the ion's manifold can often be resolved. ms-TPE spectra of m/z = 93 obtained from 3-iodophenol pyrolysis at 650 °C are depicted in Fig. 4 along with the experimental spectrum of the phenoxy radical, showing marked differences between the two spectra.
 | ||
| Fig. 4 ms-TPE spectra and FC simulations of (a) phenoxy derived from diphenylether, and (b) 3-hydroxyphenyl derived from 3-iodophenol. A vibrational temperature of 500 K was assumed in the FC simulations. | ||
Indeed, these observations are supported by the calculated and experimental ionization energies of phenoxy and hydroxyphenyl radicals in Table 1. The relatively high ion yield at energies between 8.2 and 8.6 eV (Fig. 3) cannot be due to phenoxy alone but indicates the presence of sizable amounts of hydroxyphenyl isomers. On the other hand, the slow and structureless increase of the TPE signal (Fig. 4(b)) indicates small Franck–Condon factors close to the adiabatic ionization energy of hydroxyphenyl radicals.
| B3LYP/6-311++G(p,d) | CBS-QB3 (eV) | Exp. (eV) | |
|---|---|---|---|
| Phenoxy | 8.66 | 8.71 | 8.56,54 this work |
| 2-Hydroxyphenyl | 8.50 | 8.46 | — |
| 3-Hydroxyphenyl | 8.23 | 8.20 | 8.1 ± 0.2 |
| 4-Hydroxyphenyl | 8.30 | 8.27 | 8.1 ± 0.2 |
Quantum chemical calculations reveal that there is significant distortion of the C–C˙–C angle of the hydroxyphenyl radicals upon ionization, which results in poor Franck–Condon factors at the ionization threshold. Franck–Condon simulations of the TPE spectra of phenoxy (Fig. 4(a)) and 3-hydroxyphenyl (Fig. 4(b)) radicals are in accord with the experimental spectra. The spectrum of the phenoxy radical shows a well resolved peak at 8.56 eV, which can be assigned to the adiabatic ionization energy in agreement with Dewar and David.54 Due to the large step size of 50 meV the second transition at 8.62 eV is not resolved. Nonetheless, both the experimental and simulated spectra are in reasonable agreement and confirm the presence of phenoxy radicals upon pyrolysis of DPE.
The TPE spectrum in Fig. 4(b) is clearly different from the experiments with DPE as precursor. No pronounced feature at 8.56 eV is present, which should show up despite the rather low resolution of 20 meV. In addition, a FC simulation (sticks and Gaussian convolution FWHM = 20 meV) is also depicted, showing a large density of transitions from the neutral into the ion's ground state. However it does not reproduce the peak at around 8.75 eV, which could be caused by transitions into the triplet state of the 3-hydroxyphenyl ion, calculated to be at 8.71 eV by CBS-QB3. In summary the TPE and PI spectra of m/z = 93, as synthesized from 3- and 4-iodophenol (see ESI†), are clearly different from that of the phenoxy radical. This substantiates the assumption that the hydroxyphenyl radicals are formed as intermediate radicals on the way to cyclopentadienyl and propargyl.
Computed decomposition mechanism
Of particular interest is the finding that we can generate 3- and 4-hydrodroxyphenyl radicals from their corresponding iodophenols, however their decomposition products are ultimately the same as those of the phenoxy radical: cyclopentadienyl + CO and, at higher reactor temperatures, propargyl + acetylene.4,7 This points to phenoxy radical as a shared intermediate on the C6H5O potential energy surface. It is perhaps not surprising that, compared with the resonance-stabilized 2B1 phenoxy radical, the hydroxyphenyl radicals, which are σ-localized radicals, are only local minima on the potential energy surface. In order to yield CO and cyclopentadienyl radicals from hydroxyphenyl isomers, between one and three hydrogen migration steps must take place. The 4- and 3-hydroxyphenyl radicals (see Fig. 5) can interconvert to the 2-isomer after surmounting a barrier of roughly 60 kcal mol−1 (TS1 & TS2). A direct hydrogen loss yielding the hydroxysubstituted benzynes (m/z = 92) or OH radical loss to benzyne (m/z = 76) requires at least 78 kcal mol−1 and is thus less likely to occur.55 The 2-hydroxyphenyl radical is 25.3 kcal mol−1 less stable than phenoxy and can rearrange to phenoxy (the global minimum energy structure) after overcoming the TS3 barrier of only 38.9 kcal mol−1. This can be explained by simple steric considerations. While TS1 and TS2 are three-membered transition state structures TS3 possesses more flexibility and less strain. Interestingly, a state switch has to occur, since phenoxy and o-hydroxyphenyl radicals are of 2B1 (in C2v, corresponding to 2A′′ in Cs) and 2A′ ground state, respectively. Time-dependent density functional theory calculations show that the two states intersect below the barrier of the hydrogen shift at TS3, with the TS3 transition state ground state being of 2A′′ symmetry. In summary, when enough thermal energy is provided in the pyrolysis reactor all of the hydroxyphenyl radicals can interconvert to the phenoxy radical structure.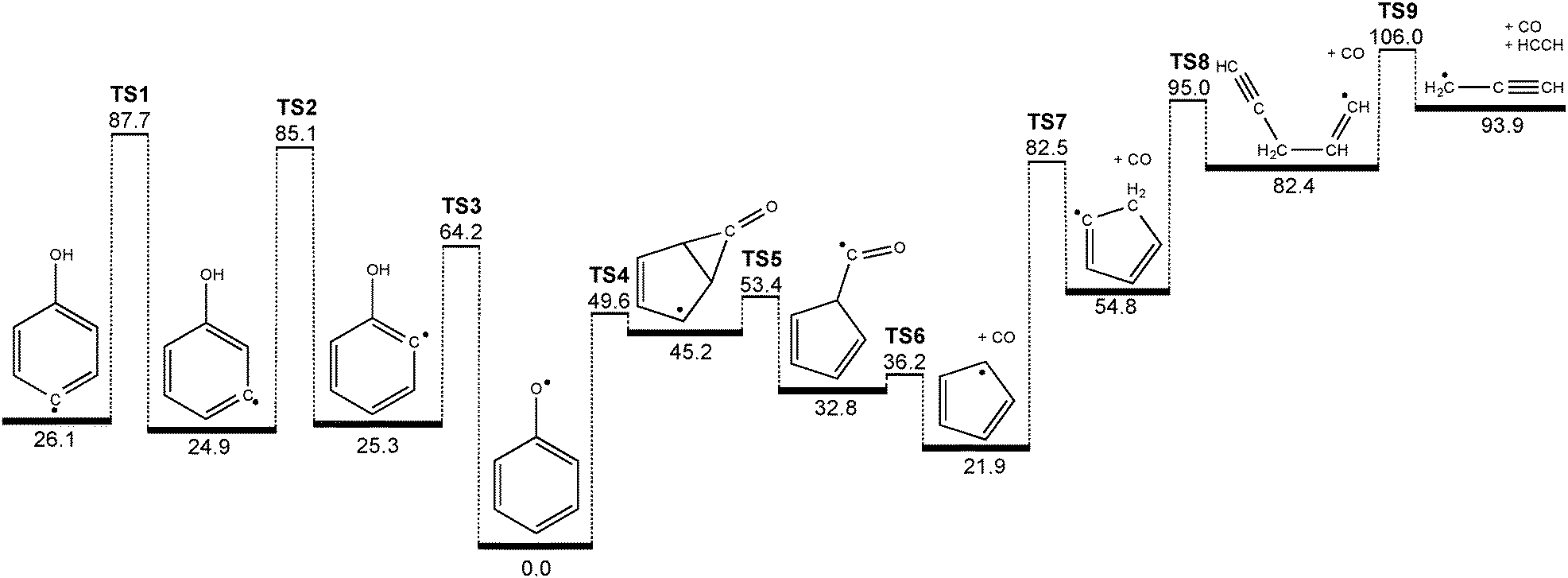 | ||
| Fig. 5 Decomposition mechanism of hydroxyphenyl radicals computed at the G3X-K level of theory. All values are given in kcal mol−1. | ||
Phenoxy itself can go over a 49.6 kcal mol−1 transition state (TS4) forming a CO bridged five membered ring intermediate, which is already lower than the barrier to rearrangement from 2-hydroxyphenyl (TS3). It can subsequently ring open (TS5) to form a cyclopentadiene-substituted formyl radical that is 32.8 kcal mol−1 less stable than phenoxy. The formyl radical site can lose the CO molecule and form cyclopentadienyl after surmounting a barrier (TS6) of 36.2 kcal mol−1 with respect to phenoxy. In a [1,5] sigmatropic H shift (TS7) cyclopentadienyl can generate a σ-radical, which subsequently performs a ring opening reaction (TS8) to form a propargyl substituted vinyl radical intermediate. This finally cleaves off acetylene, C2H2, while yielding propargyl radicals (C3H3) after overcoming TS9 (51.2 kcal mol−1 above c-C5H5). Although it is simple bond breaking, the involved barrier in this process is due to the linearization of the vinyl group coupled with the low endothermicity.
The potential energy surface shown in Fig. 5 holds the key to understanding why we found it impossible to measure the PI/ms-TPE spectrum of the 2-hydroxyphenyl radical. It is obvious that both 3- and 4-hydroxyphenyl possess deeper minima compared to the ortho isomer (≈60 vs. 40 kcal mol−1), due to steric effects when accessing the three- and four-membered transition states, as mentioned earlier. If we now neglect the influence of the hydroxyl group on the C–I bond dissociation energy of the iodophenol precursors, the bond dissociation energy should be comparable with that in phenyl iodide (67.4 kcal mol−1).56 In case of 2-hydroxyphenyl, the entrance barrier to form the radical (C–I bond cleavage) is larger than the activation energy to phenoxy isomer, and if rethermalized promptly at the corresponding temperature, 2-hydroxyphenyl can rearrange to phenoxy and subsequently dissociate.
This underlines the role of the apparently fast rethermalization occurring in the reactor. The C–I loss to form the 2-hydroxyphenyl radical takes place in the sweet spot of the reactor37 (relatively long residence times of several 100 μs) and the 2-hydroxyphenyl thus also completes the subsequent reaction steps, namely rearrangement to phenoxy and decomposition to cyclopentadienyl. We have found similarly fast re-thermalization in the decomposition of the flame retardant molecule dimethyl methyl phosphonate (DMMP).57
In contrast to the 2-hydroxyphenyl radical, nascent 3- and 4-hydroxyphenyl radicals are less prone to isomerization/decomposition, and can survive at the lower range of reactor temperatures required to achieve C–I bond homolysis in the parent compounds. Moreover, in all three cases the barriers required for hydroxyphenyl rearrangement to phenoxy (TS1, TS2, TS3) lie at energies above those required for further isomerization and decomposition to cyclopentadienyl + CO (TS4, TS5, TS6). We therefore expect thermal decomposition to proceed without the intermediacy of collisionally stabilized phenoxy (i.e., in a well-skipping process).
Conclusions
We have investigated the decomposition of the three hydroxyl-substituted phenyl radical isomers, synthesized from iodophenol precursors in a heated microtubular reactor. Using the iPEPICO technique in combination with VUV synchrotron radiation, we were able to unambiguously identify 3- and 4-hydroxyphenyl radicals by mass-selected threshold photoelectron spectra, in contrast with the more ambiguous interpretation of slight differences in the photoionization spectra. The ms-TPE spectra of 3- and 4-hydroxyphenyl radicals do not display vibrationally-resolved peaks, a fact that is attributed to the large change in geometry upon ionization. However, these spectra are significantly different from that of the phenoxy radical, the global minimum on the C6H5O potential energy surface. The three hydroxyphenyl radicals can isomerize by hydrogen-shifts towards the resonantly stabilized phenoxy radical, and decompose immediately at elevated temperatures. We have constructed a potential energy surface, which connects all hydroxyphenyl isomers over three and four-membered transition states to the phenoxy radical. Direct hydrogen abstraction reactions to yield hydroxysubstituted benzynes all lie higher in energy. The phenoxy radical can decompose to form cyclopentadienyl radicals and carbon monoxide at medium reactor temperatures. Due to the fact that the 2-hydroxyphenyl is less stable towards rearrangement to phenoxy than the 3- and 4-isomers, it does not survive when formed from its 2-iodophenol precursor.These rearrangement reactions are essential for the understanding of combustion processes, since there is another route to form phenoxy radicals apart from reactions (3) and (4), namely by rearrangement of hydroxyphenyl radicals. This formation can subsequently increase the amount of cyclopentadienyl, which dimerizes according to reaction (8) yielding naphthalene:
| c-C5H5 + c-C5H5 → C10H10 → C10H8 + H2 | (8) |
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Experiments were carried out at the VUV beamline of the Swiss Light Source, Paul Scherrer Institute. The work was financially supported by the Swiss Federal Office for Energy (BFE Contract Numbers 101969/152433 & SI/501269-01). A. J. T. and G. d. S. acknowledge funding support from the Australian Research Council (DP130100862, DP110103889, and FT130101304). The authors want to thank Victoria Custodis for providing the phenoxy spectrum, Matthew B. Predergast for useful remarks and proofreading this manuscript, and Jana Hemberger for the table of contents drawing.References
- J. Spanget-Larsen, M. Gil, A. Gorski, D. M. Blake, J. Waluk and J. G. Radziszewski, J. Am. Chem. Soc., 2001, 123, 11253–11261 CrossRef CAS PubMed.
- L. Casella, L. De Gioia, G. Frontoso Silvestri, E. Monzani, C. Redaelli, R. Roncone and L. Santagostini, J. Inorg. Biochem., 2000, 79, 31–40 CrossRef CAS.
- Y. Shimazaki, S. Huth, A. Odani and O. Yamauchi, Angew. Chem., Int. Ed., 2000, 39, 1666–1669 CrossRef CAS.
- A. V. Friderichsen, E.-J. Shin, R. J. Evans, M. R. Nimlos, D. C. Dayton and G. B. Ellison, Fuel, 2001, 80, 1747–1755 CrossRef CAS.
- M. W. Jarvis, J. W. Daily, H.-H. Carstensen, A. M. Dean, S. Sharma, D. C. Dayton, D. J. Robichaud and M. R. Nimlos, J. Phys. Chem. A, 2011, 115, 428–438 CrossRef CAS PubMed.
- D. J. Robichaud, A. M. Scheer, C. Mukarakate, T. K. Ormond, G. T. Buckingham, G. B. Ellison and M. R. Nimlos, J. Chem. Phys., 2014, 140, 234302 CrossRef PubMed.
- A. M. Scheer, C. Mukarakate, D. J. Robichaud, G. B. Ellison and M. R. Nimlos, J. Phys. Chem. A, 2010, 114, 9043–9056 CrossRef CAS PubMed.
- A. M. Scheer, C. Mukarakate, D. J. Robichaud, M. R. Nimlos, H.-H. Carstensen and G. Barney Ellison, J. Chem. Phys., 2012, 136, 044309 CrossRef PubMed.
- T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- V. B. F. Custodis, P. Hemberger, Z. Ma and J. A. van Bokhoven, J. Phys. Chem. B, 2014, 118, 8524–8531 CrossRef CAS PubMed.
- H. Richter and J. B. Howard, Phys. Chem. Chem. Phys., 2002, 4, 2038–2055 RSC.
- A. Y. Chang, J. W. Bozzelli and A. M. Dean, Z. Phys. Chem., 2000, 214, 1533 CrossRef CAS.
- S. Madronich and W. Felder, J. Phys. Chem., 1985, 89, 3556–3561 CrossRef CAS.
- R. A. Shandross, J. P. Longwell and J. B. Howard, Symp. (Int.) Combust., [Proc.], 1996, 26, 711–719 CrossRef.
- C. Barckholtz, M. J. Fadden and C. M. Hadad, J. Phys. Chem. A, 1999, 103, 8108–8117 CrossRef CAS.
- B. B. Kirk, D. G. Harman, H. I. Kenttamaa, A. J. Trevitt and S. J. Blanksby, Phys. Chem. Chem. Phys., 2012, 14, 16719–16730 RSC.
- D. S. N. Parker, R. I. Kaiser, T. P. Troy, O. Kostko, M. Ahmed and A. M. Mebel, J. Phys. Chem. A, 2015, 119, 7145–7154, DOI:10.1021/jp509170x.
- I. V. Tokmakov, G.-S. Kim, V. V. Kislov, A. M. Mebel and M. C. Lin, J. Phys. Chem. A, 2005, 109, 6114–6127 CrossRef CAS PubMed.
- P.-T. Howe and A. Fahr, J. Phys. Chem. A, 2003, 107, 9603–9610 CrossRef CAS.
- S. J. Klippenstein and J. A. Miller, J. Phys. Chem. A, 2002, 106, 9267–9277 CrossRef CAS.
- J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2001, 105, 7254–7266 CrossRef CAS.
- J. A. Miller and C. F. Melius, Combust. Flame, 1992, 91, 21–39 CrossRef CAS.
- E. Ikeda, R. S. Tranter, J. H. Kiefer, R. D. Kern, H. J. Singh and Q. Zhang, Proc. Combust. Inst., 2000, 28, 1725–1732 CrossRef CAS.
- A. M. Mebel and V. V. Kislov, J. Phys. Chem. A, 2009, 113, 9825–9833 CrossRef CAS PubMed.
- C. F. Melius, M. E. Colvin, N. M. Marinov, W. J. Pitz and S. M. Senkan, Proc. Combust. Inst., 1996, 26, 685–692 CrossRef.
- M. Nagata, Y. Futami, N. Akai, S. Kudoh and M. Nakata, Chem. Phys. Lett., 2004, 392, 259–264 CrossRef CAS PubMed.
- H. Richter, T. G. Benish, O. A. Mazyar, W. H. Green and J. B. Howard, Proc. Combust. Inst., 2000, 28, 2609–2618 CrossRef CAS.
- P. H. Kasai and D. McLeod, J. Am. Chem. Soc., 1974, 96, 2338–2342 CrossRef CAS.
- R. H. Schuler, P. Neta, H. Zemel and R. W. Fessenden, J. Am. Chem. Soc., 1976, 98, 3825–3831 CrossRef CAS.
- M. B. Prendergast, B. B. Kirk, J. D. Savee, D. L. Osborn, C. A. Taatjes, K.-S. Masters, S. J. Blanksby, G. d. Silva and A. J. Trevitt, Phys. Chem. Chem. Phys., 2015 10.1039/C5CP02953H.
- X. Zhang, A. T. Maccarone, M. R. Nimlos, S. Kato, V. M. Bierbaum, G. B. Ellison, B. Ruscic, A. C. Simmonett, W. D. Allen and H. F. Schaefer, J. Chem. Phys., 2007, 126, 044312 CrossRef PubMed.
- X. Zhang and P. Chen, J. Am. Chem. Soc., 1992, 114, 3147–3148 CrossRef CAS.
- M. Johnson, A. Bodi, L. Schulz and T. Gerber, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 610, 597–603 CrossRef CAS PubMed.
- A. Bodi, M. Johnson, T. Gerber, Z. Gengeliczki, B. Sztáray and T. Baer, Rev. Sci. Instrum., 2009, 80, 034101 CrossRef PubMed.
- A. Bodi, B. Sztáray, T. Baer, M. Johnson and T. Gerber, Rev. Sci. Instrum., 2007, 78, 084102 CrossRef PubMed.
- B. N. Moore, S. J. Blanksby and R. R. Julian, Chem. Commun., 2009, 5015–5017, 10.1039/B907833A.
- Q. Guan, K. N. Urness, T. K. Ormond, D. E. David, G. Barney Ellison and J. W. Daily, Int. Rev. Phys. Chem., 2014, 33, 447–487 CrossRef CAS PubMed.
- A. Bodi, P. Hemberger, D. L. Osborn and B. Sztáray, J. Phys. Chem. Lett., 2013, 4, 2948–2952 CrossRef CAS.
- P. Hemberger, A. J. Trevitt, E. Ross and G. da Silva, J. Phys. Chem. Lett., 2013, 4, 2546–2550 CrossRef CAS.
- B. Sztáray and T. Baer, Rev. Sci. Instrum., 2003, 74, 3763–3768 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Wallingford, CT, 2009, Key: citeulike:9096580.
- V. A. Mozhayskiy and A. I. Krylov, ezSpectrum, http://iopenshell.usc.edu/downloads.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822–2827 CrossRef CAS PubMed.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 2000, 112, 6532–6542 CrossRef CAS PubMed.
- G. da Silva, Chem. Phys. Lett., 2013, 558, 109–113 CrossRef CAS PubMed.
- N. Hansen, S. J. Klippenstein, J. A. Miller, J. Wang, T. A. Cool, M. E. Law, P. R. Westmoreland, T. Kasper and K. Kohse-Höinghaus, J. Phys. Chem. A, 2006, 110, 4376–4388 CrossRef CAS PubMed.
- B. Yang, C. Huang, L. Wei, J. Wang, L. Sheng, Y. Zhang, F. Qi, W. Zheng and W.-K. Li, Chem. Phys. Lett., 2006, 423, 321–326 CrossRef CAS PubMed.
- H. J. Wörner and F. Merkt, Angew. Chem., Int. Ed., 2006, 45, 293–296 CrossRef PubMed.
- J. D. Savee, T. M. Selby, O. Welz, C. A. Taatjes and D. L. Osborn, J. Phys. Chem. Lett., 2015, 4153–4158, DOI:10.1021/acs.jpclett.5b01896.
- P. Hemberger, M. Lang, B. Noller, I. Fischer, C. Alcaraz, B. K. Cunha de Miranda, G. A. Garcia and H. Soldi-Lose, J. Phys. Chem. A, 2011, 115, 2225–2230 CrossRef CAS PubMed.
- U. Jacovella, B. Gans and F. Merkt, J. Chem. Phys., 2013, 139, 084308 CrossRef CAS PubMed.
- J. D. Savee, S. Soorkia, O. Welz, T. M. Selby, C. A. Taatjes and D. L. Osborn, J. Chem. Phys., 2012, 136, 134307 CrossRef PubMed.
- P. Botschwina and R. Oswald, Chem. Phys., 2010, 378, 4–10 CrossRef CAS PubMed.
- M. J. S. Dewar and D. E. David, J. Am. Chem. Soc., 1980, 102, 7387–7389 CrossRef CAS.
- L. B. Harding, J. Am. Chem. Soc., 1981, 103, 7469–7475 CrossRef CAS.
- W. R. Stevens, B. Ruscic and T. Baer, J. Phys. Chem. A, 2010, 114, 13134–13145 CrossRef CAS PubMed.
- S. Liang, P. Hemberger, N. M. Neisius, A. Bodi, H. Grützmacher, J. Levalois-Grützmacher and S. Gaan, Chem. – Eur. J., 2015, 21, 1073–1080 CrossRef CAS PubMed.
- Z. Ma and J. A. vanBokhoven, ChemCatChem, 2012, 4, 2036–2044 CrossRef CAS PubMed.
- S. Shao, H. Zhang, Y. Wang, R. Xiao, L. Heng and D. Shen, Energy Fuels, 2015, 29, 1751–1757 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Mass spectra for 2-iodophenol (12 eV) and 4-iodophenol (9 eV) pyrolysis; photoionization spectrum of m/z 65 from 2-iodophenol pyrolysis; potential energy surface for c-C5H5 decomposition via l-C5H5; ms-TPE spectrum of m/z 93 from 4-iodophenol pyrolysis. See DOI: 10.1039/c5cp05346c |
| This journal is © the Owner Societies 2015 |